 Luyao Zhang
Luyao Zhang Zihua Chen
Zihua Chen Lanting Wang
Lanting Wang Xiaoqun Luo
Xiaoqun Luo- Department of Allergy and Immunology, Huashan Hospital, Fudan University, Shanghai, China
Bullous pemphigoid (BP) is an autoimmune disease that mainly occurs in the elderly, severely affecting their health and life quality. Traditional therapy for BP is mainly based on the systemic use of corticosteroids, but long-term use of corticosteroids results in a series of side effects. Type 2 inflammation is an immune response largely mediated by group 2 innate lymphoid cells, type 2 T helper cells, eosinophils, and inflammatory cytokines, such as interleukin (IL)-4, IL-5 and IL-13. Among patients with BP, the levels of immunoglobulin E and eosinophils are significantly increased in the peripheral blood and skin lesions, suggesting that the pathogenesis is tightly related to type 2 inflammation. To date, various targeted drugs have been developed to treat type 2 inflammatory diseases. In this review, we summarize the general process of type 2 inflammation, its role in the pathogenesis of BP and potential therapeutic targets and medications related to type 2 inflammation. The content of this review may contribute to the development of more effective drugs with fewer side effects for the treatment of BP.
1. Introduction
Bullous pemphigoid (BP) is one of the most frequent autoimmune bullous diseases that mainly occurs in the elderly and display no gender predilection. The cumulative incidence of BP is 8.2/million individuals, which is higher in Europe than in Asia (1). Although the incidence is generally low, the risk of developing BP increases with age. Studies have shown that people over 90 years of age have a 300-fold increased risk compared to people under 60 years of age (2). Moreover, compared to the general population of the same age, patients with BP have a 3.6 times increased risk of death (3). Although it is an autoimmune disease, it can be induced by multiple stimuli, including gliptin, COVID-19 vaccines, and programmed cell death-1/programmed cell death ligand-1 inhibitors (4–6), suggesting possible different biological underpinnings in these patients.
BP is predominantly evoked by autoantibodies against two types of hemidesmosomal proteins, BP180 (XVII collagen) and BP230, which are located in the basement membrane zone (BMZ) and responsible for the dermo-epidermal junction (7). During the formation of subepidermal blisters with negative Nikolsky sign, these autoantibodies and immune cells act together to destroy hemidesmosomes in keratinocytes (8). Histopathological examination frequently reveals separation of the dermis and epidermis, and inflammatory cell infiltration, mainly composed of lymphocytes and eosinophils (9).
BP cause a vast array of burdens to patients. Patients with BP usually develop pruritic, tense blisters or bullae locally or widespread on normal skin or erythematous background on the trunk and limbs (10). Some patients present with a non-bullous prodromic phase characterized by eczematous, excoriated, urticaria-like, or nodular lesions that varying in duration (11). Meanwhile, patients often suffer from comorbid health conditions, including neurological disorders, malignancies, and cardiovascular diseases. Specifically, it has been proven that multiple sclerosis, diabetes, hypertension, basal cell carcinoma of the skin, dementia, Parkinson’s disease, epilepsy, stroke, pneumonia, and pulmonary embolism have an increased prevalence among patients with BP (11–15). In addition to physical discomfort, BP could also contribute to decreased quality of life and increased psychological burden, such as anxiety and depression, because of the skin lesions, functional problems, pruritus, and disease chronicity (16, 17).
In terms of therapy, topical or systematic glucocorticosteroids are the primary treatment for BP, which can be supplemented by immunosuppressors such as methotrexate, azathioprine, and mycophenolate mofetil (2). However, long-term systemic use of corticosteroids may cause a variety of side effects, including hypertension, bone fracture, cataract, gastrointestinal discomfort and metabolic conditions, such as weight gain and hyperglycemia (18). Another key fact is that some patients with BP are resistant to traditional treatment. Currently, plasmapheresis, intravenous immunoglobulin, immunoadsorption, and rituximab can be administered to refractory patients (11). It has been demonstrated that more than 90% of patients with moderate-to-severe BP can get complete remission relatively safely using a combination therapy with rituximab and corticosteroids (19). However, some patients with BP are not sensitive to these therapies; thus, there is an urgent need for drugs with fewer side effects and superior efficacy.
Recently, an increasing number of studies have found that the pathogenesis of BP is closely related to type 2 inflammation, and the use of dupilumab, a monoclonal antibody against the type 2 inflammatory factors interleukin 4 (IL-4) and IL-13, seems to have a certain curative effect in patients with BP. Moreover, the European Academy of Dermatology and Venereology considers that dupilumab and omalizumab are optional treatments for refractory BP (20). These results provide a new direction for further exploration of the pathogenesis of BP and the search for more effective treatments. Here, we review the general process of type 2 inflammation, its role in the BP pathogenesis, and potential therapeutic targets and medications related to type 2 inflammation. Our aim is to provide new ideas and research directions for the development of more effective drugs with fewer side effects for the treatment of BP.
2. The general process of type 2 inflammation
Type 2 inflammation is an immune response which exerts an important role in host defense against parasites and is predominantly mediated by group 2 innate lymphoid cells (ILC2s), type 2 T helper (Th2) cells, eosinophils, and relevant cytokines, such as IL-4, IL-5, and IL-13 (21). A large number of stimuli can trigger type 2 inflammation, including helminths, various allergies, certain viral or bacterial infections, and endogenous molecules (22). The process involves both innate and adaptive immune responses.
After exposure of the epithelium to these stimuli, local tissue homeostasis is disrupted, and epithelial cells release IL-25, IL-33, and thymic stromal lymphopoietin (TSLP), which are termed alarmins. In general, epithelial tissues in different parts of the body release various alarmins. For example, in the lung, type 2 alveolar cells are a primary source of TSLP and IL-33 in the lung (23), whereas in the small intestine and skin, tuft cells and keratinocytes are a major source of IL-25, respectively (24, 25). In addition, other non-epithelial cells can also produce alarmins, with airway smooth muscle cells secreting TSLP (26) and fibroblasts producing IL-33 (27). Under the effect of these alarmins, tissue-resident ILC2s are activated and increase the production of cytokines of IL-4, IL-5, IL-9 and IL-13 (28), termed type 2 cytokines. ILC2 can also be mediated by other molecules released after the attack of exogenous stimuli, such as TL1A from TNF-family (29, 30), prostaglandin D2 and cysteinyl leukotriene (31, 32).
Meanwhile, in the presence of stimuli and cytokines produced by ILC2s, dendritic cells (DC) take up and transport parts of the antigens to local draining lymph nodes. Subsequently, the processed antigens induce naïve CD4+ T cells to activate the latter in the lymph nodes. The latter, with high GATA-binding protein 3 expression, can proliferate and differentiate into Th2 cells and subsequently produce type 2 cytokines with the help of DCs and the aforementioned type 2 cytokines, also including IL-33 and TSLP (21, 33, 34). After activation of ILC2s, this process generates as part of adaptive immunity. Furthermore, ILC2s have been shown to modulate the differentiation of naïve T cells into Th2 cells and the degree of Th2 response to a large extent. Once the stimulus is gone, ILC2s will be negatively regulated and decrease the support to Th2 cells, ultimately reducing the inflammation (35–37).
Generally, eosinophils, which develop in the bone marrow and circulate in the peripheral blood, play a vital role in type 2 inflammation. IL-5 is a crucial cytokine for the maturation, survival as well as recruitment of eosinophils and can prevent apoptosis (38). In type 2 inflammation, eosinophils are activated and recruited by IL-5, together with IL-33, TSLP, and eotaxins released by inflamed tissues and other chemoattractants, such as C3a and C5a (39–41). After activation, eosinophils can function as antigen-presenting cells for viral antigens (42) and upregulate associated molecules, including major histocompatibility complex (MHC) II, CD86, and CD40, ultimately leading to T cells activation and differentiation in draining lymph nodes (43). In the meantime, mature eosinophils can secrete IL-4, IL-25, and indoleamine 2,3-dioxygenase (44), which may cause selective differentiation of naïve T cells into Th2 cells instead of Th1 cells. Moreover, eosinophil-derived neurotoxin (EDN) and eosinophil peroxidase (EPO) released by eosinophils could strongly affect the maturation of DCs that migrate to lymph nodes, which can indirectly promote Th2 cell functions (45). Eosinophil-derived IL-25 can also promote the proliferation of and cytokine production by Th2 memory cells after antigen triggering (46). Besides their role as promoter for Th2 response, eosinophils can also act as an effector in type 2 inflammation. After recruitment into peripheral tissues, eosinophils cause tissue damage by releasing eosinophilic cationic protein (ECP), which can prompt cell cytotoxicity; EPO, which can generate oxidative stress; and major basic protein (MBP), which can destroy the lipid bilayer and increase cell permeability to damage the epithelium (43). Additionally, eosinophils are able to modulate classical and alternative pathways of complement activation as well as act through antibody-dependent cellular cytotoxicity against stimuli via their Fc receptors (43).
In the last phase of type 2 inflammation, the presence of stimuli and cytokines produced by both ILC2s and Th2 cells, especially IL-4 and IL-13, can facilitate the humoral immune response, activate B cells and cause rearrangement of the immunoglobulin heavy chain locus thus leading to immunoglobulin E (IgE) synthesis and differentiation into IgE-producing plasma cells (47, 48). IgE can bind to its high affinity receptor, FcϵRI, on basophils and mast cells to sensitize the body. When the body is exposed to the corresponding allergen again, the antigen recognition fragment of IgE binding to the cell surface binds to the allergen, resulting in cross-linking with the IgE-FcϵRI complex and degranulation of mast cells and basophils. The degranulation products, including histamine, proteases, prostaglandins and other cytokines (49, 50), lead to inflammatory reactions that manifest as erythema, itching, and edema on the skin; sneezing; cough; increased mucous secretions and bronchospasm in the respiratory tract; nausea, vomiting, and diarrhea in the digestive tract; and hypotension (49) (Figure 1)
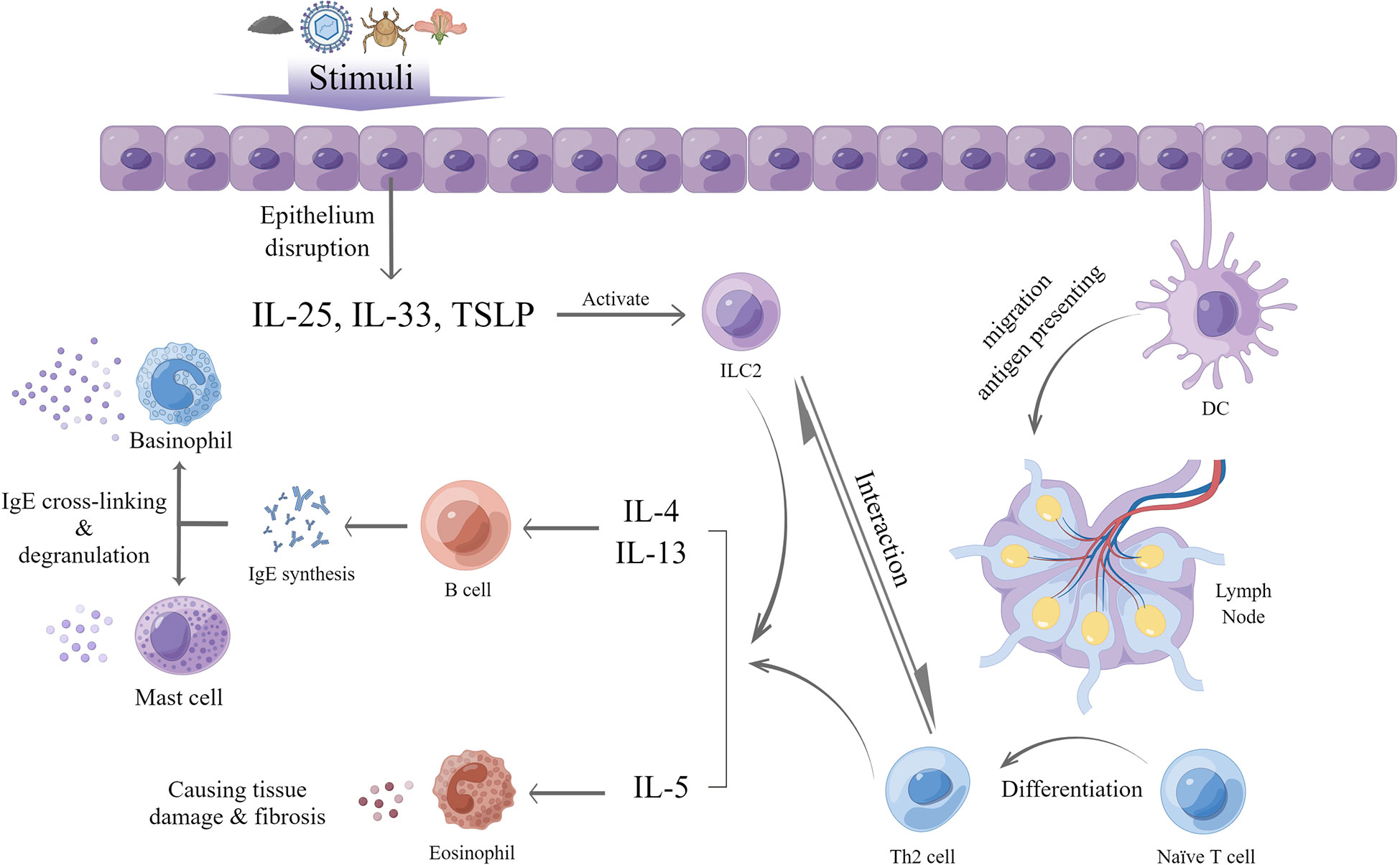
Figure 1 General process of type 2 inflammation (by Figdraw).
3. Type 2 inflammation in BP and potential therapeutic targets
As previously mentioned, BP is predominantly evoked by autoantibodies against BP180 and BP230. In these patients, BP180 and BP230 are recognized, ingested, processed by antigen presenting cells, and subsequently expressed on the cell surface combined with MHC II. After recognition of the antigen, naïve T cells are activated, differentiate to autoreactive Th2 cells and secrete specific cytokines, which in turn stimulate the differentiation and class-switch recombination of B cells. Plasmocytes produce autoantibodies IgG and IgE, leading to the deposition of autoantibodies in both the peripheral blood and the local basement membrane zone. Therefore, Th2 pathways are considered the primary triggers for antibody production in BP (51). It seems that anti-BP180 antibodies are more common than anti-BP230 antibodies in BP. One study showed that anti-BP180 IgG can be found in 95% of patients with BP, while anti-BP230 was found in 70% of patients in the same group (52). Autoantibody IgG can trigger complement activation, which can be proven by the deposition of complement C3 and C4 found in immunofluorescence examination, multiple immune cells recruitment, and proteases release, which can cause an inflammatory cascade reaction. Furthermore, it can directly target the corresponding antigens, causing hemidesmosome destruction and leading to the loss of adhesion to the BMZ and formation of subepidermal blisters (8). Among these autoantibodies, anti-BP180 antibodies are mainly targeted to the immunodominant region termed NC16A in the extracellular regions (53). These anti-BP180 NC16A IgG are positively correlated with the Autoimmune Bullous Skin Disorder Intensity Score and Bullous Pemphigoid Disease Area Index (BPDAI) in its subcomponents of erosion/blister, urticaria/erythema and pruritus scores, while the level of anti-BP230 IgG is not correlated with these scores, suggesting that anti-BP180 NC16A IgG can be a useful indicator of BP activity (54, 55). The above mechanism can partially explain the formation of blisters but fail to explain the manifestation of itching, erythema, and eosinophilia in BP. Elevated serum IgE levels are found in 70–85% patients with BP (56), and deposition of IgE can be seen in immunofluorescence examination, which is related to degranulation of mast cells and basophils, eosinophil recruitment, and manifestations of itch and blister formation (57). Therefore, abnormal T cell immune responses and autoantibodies together lead to the occurrence of BP. The pathogenesis of BP depends on a variety of immune cells, including Th2 cells and eosinophils; autoimmune antibodies, including IgE; and a variety of cytokines, such as IL-4, IL-5, and IL-13, which are all relevant to type 2 inflammation.
3.1. Role of type 2 cytokines in BP
A variety of studies have revealed that the levels of IL-4, IL-5, IL-6, IL-10, and IL-13, which are predominantly produced by ILC2 and Th2 cells, are elevated in the serum, blister fluid, and skin biopsies of patients with BP (58–60). Among them, IL-4 is considered to have the closest association with BP (61) because it is particularly essential for Th2 cell differentiation, class-switch recombination of B cells, and production of IgE while simultaneously suppressing Th1 and Th17 differentiation (62). Subsequently, Th2 cells continuously produce more IL-4, IL-5, and IL-13 to further enhance this process. IL-5 is a critical cytokine for the maturation, survival, and functional activity of eosinophils, and its level parallels the severity of BP (60). IL-13 is another crucial cytokine in BP and has some common features with IL-4. They share a common receptor subunit of IL-4 receptor α (IL-4Rα) and a common intracellular signaling pathway (63); thus, they can synergistically promote B cell differentiation and IgE production. IL-13 levels also have a positive correlation with the itch severity of BP (64). In addition, BP patients tend to show a lower frequency of the C allele in IL-13 gene variation (rs1800925) and the G-allele in IL-4R rs1805010 than healthy individuals, suggesting their protective effects to BP (65). However, A-allele in IL-13 rs20541 can function as a promoting factor to the susceptibility of BP (65).
Owing to their significance in BP, several targeted medications have emerged. Dupilumab is an IL-4Rα antagonist that can block the common IL-4Rα subunit to inhibit IL-4 and IL-13 signaling (66). It was approved for the treatment of atopic dermatitis (AD) in 2017 and is currently being studied for many other type 2 inflammatory diseases, including BP. Recently, some studies have shown that dupilumab exerts favorable therapeutic effects in patients with BP (67–74). It can result in rapid improvement of skin lesions and pruritus, leading to complete remission or a satisfactory treatment response in 92.3% of patients, including those who do not respond to traditional treatments (67). Additionally, a phase 2/3 randomized double-blind placebo-controlled trial for its use in BP is currently underway (NCT04206553).
Mepolizumab and reslizumab are humanized monoclonal antibodies targeting IL-5. Mepolizumab has been used to treat eosinophilic granulomatosis with polyangiitis, eosinophilic asthma, and chronic rhinosinusitis with nasal polyps in clinical trials, and has shown efficacy and safety (75–79). However, in a phase 2 pilot study of mepolizumab for BP, there was no significant difference in the cumulative rates of patients who achieved and maintained disease control between the mepolizumab and placebo groups, indicating that the primary endpoint was not met (80). Similar to mepolizumab, reslizumab has been approved as an add-on treatment for adults with severe eosinophilic asthma (81–83). A study of reslizumab use in BP reported that reslizumab can rapidly improve bullous skin lesion (84). Further research is needed to confirm the effects of anti-IL-5 treatment on BP.
Benralizumab is a monoclonal antibody against the IL-5-receptor licensed for treating severe eosinophilic asthma (85). It is also used in the treatment of eosinophilic granulomatosis with polyangiitis, and for the prevention of chronic obstructive pulmonary disease exacerbations (86, 87). Tralokinumab and lebrikizumab are monoclonal antibodies targeting IL-13. Tralokinumab can bind to IL-13, inhibiting its interaction with certain receptors and thereby neutralizing its biological activity (88). It has been approved for moderate to severe AD by the European Commission (89). Lebrikizumab is currently used for the treatment of asthma and AD (90–94). It has a satisfactory curative effect on asthma, especially in terms of the pulmonary function and exacerbation rates (93). Although it has not been studied whether benralizumab, tralokinumab, and lebrikizumab are effective for BP, based on the significance of IL-5 and IL-13 in its pathogenesis, they may potentially be used for the treatment of BP in the future (Table 1).
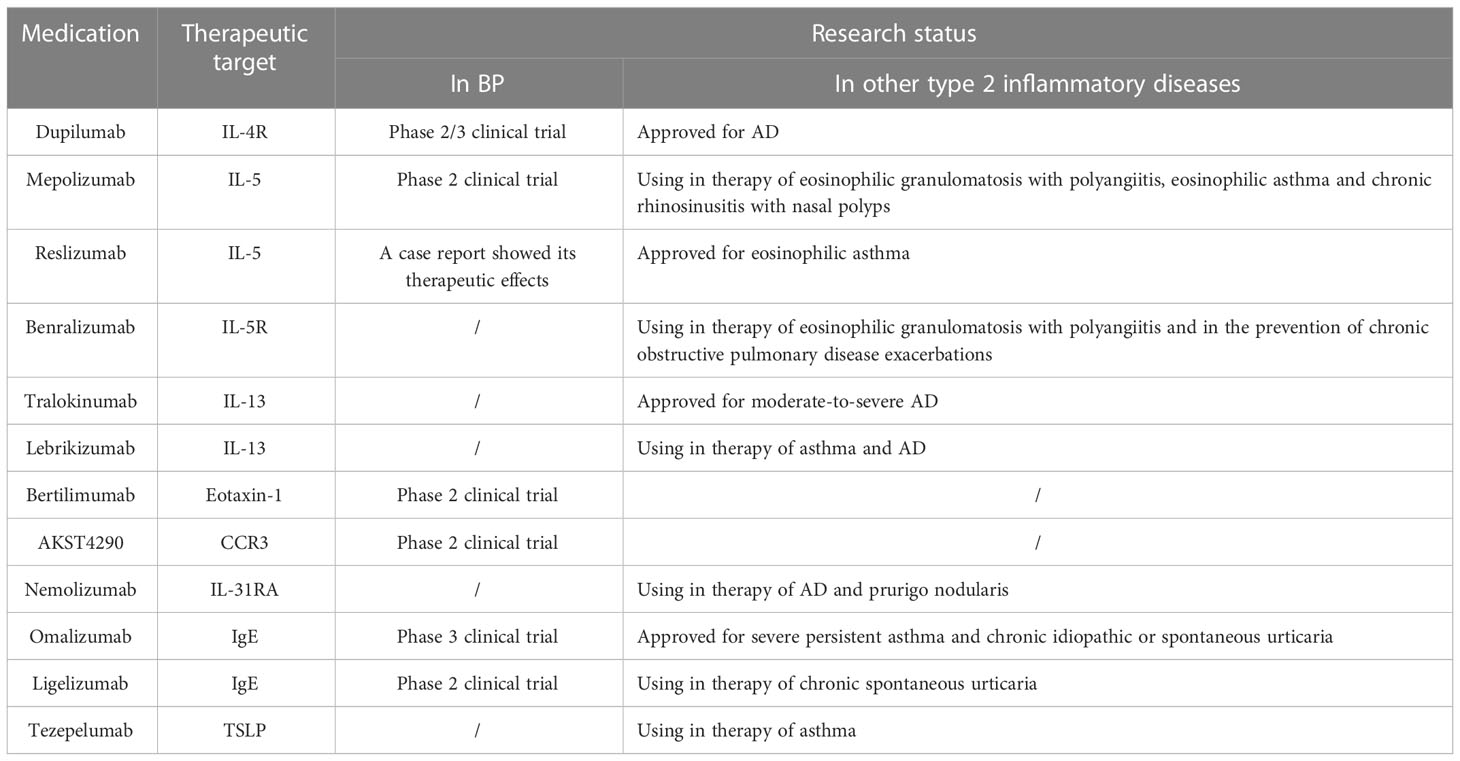
Table 1 Therapeutic targets and medication related to type 2 inflammation in BP.
3.2. Eosinophils and related molecules in BP
Eosinophil infiltration in skin lesions is a prominent feature of BP. They are recruited by many chemoattractants, including IL-5, eotaxin, and galectin-9, which are detected in the blister fluid (95). Studies have found that the level of eosinophils is elevated in the peripheral blood of over 50% of untreated patients with BP, which has been proven to be positively correlated with both disease and itch severity. Along with increased expression of CD69, eosinophils in both the blood and blister fluid are strongly activated (96–100). BP patients with eosinophilia tend to be older and have higher palmoplantar involvement than others (98). In patients with total IgE levels > 400 IU/mL, the level of eosinophils in peripheral blood is strongly related to the level of anti-BP180 IgE (101). The concentrations of ECP, EDN, and MBP, released by activated eosinophils, are higher in the serum and blister fluid of patients with BP than in the healthy controls (97). ECP and EDN can disrupt keratinocyte cell-matrix detachment, contributing to blister formation through ribonuclease activity (97, 102). ECP also affects the proliferation of T and B cells, promotes the degranulation of mast cells, and modulates the complement pathway (103). Eosinophils are considered the major source of IL-31 in BP, and the latter is a generally acknowledged pruritogen as well as a promoting factor for blister formation (102, 104, 105). The level of IL-31 in the serum is significantly associated with the level of anti-BP180 IgE (97). In addition, activated eosinophils can release metalloproteinase-9, which can lead to BP180 cleavage (106). In an in vitro study, eosinophils activated by IL-5 were shown to degranulate and directly cause blister formation in the presence of BP autoantibodies after adhesion to keratinocytes and FcγR activation (107). Furthermore, eosinophils have been proven to be correlated with blood coagulation in BP, since it has been proven that the level of ECP in blister fluid is elevated and paralleled to markers of coagulation activation (108). Eosinophils are considered a source of tissue factor and the latter can initiate blood coagulation cascade, manifested as elevated prothrombin fragment F1 + 2 and D-dimer levels in both the plasma and blister fluid, which may contribute to inflammation, tissue damage, blister formation, and thrombotic risk (109–111). Although in some studies, a lower eosinophil count is considered a potential risk factor for mucosal involvement (112, 113), elevated eosinophil levels are generally considered a marker of disease severity in BP, and targeting eosinophils may be a promising treatment for BP. Bertilimumab is a humanized monoclonal antibody targeting eotaxin-1. In a phase 2 clinical trial of BP, the disease severity decreased by 81% after 13 weeks of use of bertilimumab (114). AKST4290 is an antagonist of CCR3, the major receptor of eotaxin on eosinophils. In a phase 2 study of AKST4290, patients with BP were administered 400 mg AKST4290 twice together with mometasone furoate until the disease was under control (115). Nemolizumab, which is a monoclonal antibody against IL-31 receptor A, is licensed to treat AD and prurigo nodularis and can observably reduce pruritus (116). Based on the role of IL-31 in itching, nemolizumab may potentially be used in BP as an additional medication to control pruritus in the future (Table 1).
3.3. IgE in BP
As mentioned previously, IgE is critical for the occurrence and development of type 2 inflammation. In patients with BP, elevated IgE levels in the serum and deposition of IgE in the BMZ were first described in 1974 (117). Since then, an increasing amount of evidence has suggested that IgE is essential for the pathogenesis of BP. Later, researchers found that some IgE autoantibodies target the NC16A region of BP180, which is the same as anti-BP180 IgG (118), although the incidence of anti-BP180 IgE varies widely among patients. Other studies have detected BP230-specific IgE in BP (119, 120). The clinical features of BP are associated with IgE levels. The deposition of IgE in the BMZ is parallel to BPDAI scores and disease course (121). Patients with pathological findings of linear deposition of IgE in the BMZ tend to have higher levels of anti-BP180 IgE in the serum than patients without IgE deposition (121). Many studies have reported that the levels of total IgE in the serum are positively related to disease severity in patients with elevated IgE levels (122–124). Some studies have found that the level of anti-BP180 IgE in the serum is positively associated with BPDAI scores (124). It has also been reported that the level of anti-BP230 IgE is associated with disease activity or local eosinophil infiltration (125, 126). Moreover, anti-BP230 IgE is more common in patients resistant to topical corticosteroids, suggesting that it can be used as a marker for systemic corticosteroid therapy (127). In addition, in vitro experiments have shown that anti-BP180 IgE can cause a decline in keratinocyte adhesion and hemidesmosomal density (128), suggesting its function in blister formation. IgE receptors are increased as well in BP. It has been found that the expression of CD23, a receptor of IgE, is increased on peripheral B cells and correlates with IgE levels as well as disease severity (129, 130). Meanwhile, galectin-3, a soluble receptor for IgE, has lower expression around blisters in BP, which may contribute to the extension of blisters by disassembling the cell-extracellular matrix (131).
Recently, there have been several therapies aimed at increasing IgE levels. Omalizumab, a monoclonal anti-IgE antibody, has been approved for treatment of severe persistent asthma and chronic idiopathic or spontaneous urticaria (132, 133). It can bind to IgE with high affinity and block the binding site for FcϵRI to reduce the levels of both free IgE and peripheral eosinophils (134, 135). For patients with BP, omalizumab can reduce disease severity mainly by decreasing itching and blister counts, as well as the dose of systemic steroids (136–138). It has also shown promising therapeutic effects in refractory BP (137, 139). Ligelizumab (QGE031) is a second-generation monoclonal antibody with a higher affinity for IgE than omalizumab, which also has a significant therapeutic effect on chronic spontaneous urticaria (140). However, ligelizumab was discontinued due to insufficient efficacy in BP at phase 2 clinical trial (NCT01688882) (141). In view of the importance of IgE in the pathogenesis of BP, it will become a new target for treatment, and the research and development of more monoclonal antibodies against IgE will bring good news to patients with BP (Table 1).
3.4. Other molecule in BP
TSLP, predominantly produced by the epithelium and ILC2s when encountering stimuli, is an important initiator of type 2 inflammation and factor for itching. Multiple studies have found that the concentration of TSLP increases in skin lesions, blister fluid, and serum of patients with BP (142), and that it may be involved in the pathogenesis of BP through the direct activation of DCs (143). A study found that mice with BP180 dysfunction have increased the expression of TSLP and the latter is strongly correlated with itch severity (144). Tezepelumab, a human monoclonal antibody against TSLP, is used to treat severe and uncontrolled asthma in clinical trials and can significantly control the disease and improve quality of life (145, 146). Owing to its efficacy in asthma, tezepelumab has potential as an additive medication in BP therapy to relieve itching and improve patients’ quality of life (Table 1).
4. Discussion
BP is an autoimmune bullous disease that mainly occurs in the elderly, severely affecting their health and quality of life. Previous studies have demonstrated that BP is predominantly evoked by autoantibodies against BP180 and BP230, together with an abnormal T cell immune response, which results in the destruction of hemidesmosomes and local inflammation. Traditional therapies are mainly based on the systemic use of corticosteroids. However, long-term use of corticosteroids results in a series of side effects, such as hypertension, bone fracture, cataract, and hyperglycemia, and some patients are not sensitive to hormone therapy. Therefore, there is an urgent need for new medications with fewer side effects and superior therapeutic benefits. Type 2 inflammation is an immune response mainly mediated by ILC2s, Th2 cells, eosinophils, and inflammatory cytokines, such as IL-4, IL-5 and IL-13. As mentioned above, these immune cells and their related molecules play an essential role in the pathogenesis of BP. To date, a variety of monoclonal antibodies against the above factors have been used in the therapy of type 2 inflammation-related diseases and have achieved good clinical efficacy. From this perspective, these targeted drugs are expected to become a new and superior choice for patients with BP. However, the role and efficacy of these targeted medications in the treatment of BP is not completely clear yet; thus, more studies are needed to explore the efficacy of these targeted drugs on BP in the future to find more effective medications with fewer side effects that can benefit patients with BP.
Author contributions
Under the supervision of XL, the manuscript was written by LZ. ZC and LW provided critical evaluation of written content and contributed to manuscript revision. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by National Key Clinical Specialty Construction Project (Department of Allergy & Immunology, Huashan Hospital).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Persson MSM, Begum N, Grainge MJ, Harman KE, Grindlay D, Gran S. The global incidence of bullous pemphigoid: A systematic review and meta-analysis. Br J Dermatol (2022) 186(3):414–25. doi: 10.1111/bjd.20743
2. Bağcı IS, Horváth ON, Ruzicka T, Sárdy M. Bullous pemphigoid. Autoimmun Rev (2017) 16(5):445–55. doi: 10.1016/j.autrev.2017.03.010
3. Kridin K, Schwartz N, Cohen AD, Zelber-Sagi S. Mortality in bullous pemphigoid: A systematic review and meta-analysis of standardized mortality ratios. J Dermatol (2018) 45(9):1094–100. doi: 10.1111/1346-8138.14503
4. Maronese CA, Caproni M, Moltrasio C, Genovese G, Vezzoli P, Sena P, et al. Bullous pemphigoid associated with covid-19 vaccines: An Italian multicentre study. Front Med (2022) 9:841506. doi: 10.3389/fmed.2022.841506
5. Salemme A, Fania L, Scarabello A, Caproni M, Marzano AV, Cozzani E, et al. Gliptin-associated bullous pemphigoid shows peculiar features of anti-Bp180 and -Bp230 humoral response: Results of a multicenter study. J Am Acad Dermatol (2022) 87(1):56–63. doi: 10.1016/j.jaad.2022.02.036
6. Geisler AN, Phillips GS, Barrios DM, Wu J, Leung DYM, Moy AP, et al. Immune checkpoint inhibitor-related dermatologic adverse events. J Am Acad Dermatol (2020) 83(5):1255–68. doi: 10.1016/j.jaad.2020.03.132
7. Kasperkiewicz M, Zillikens D. The pathophysiology of bullous pemphigoid. Clin Rev Allergy Immunol (2007) 33(1-2):67–77. doi: 10.1007/s12016-007-0030-y
8. Hammers CM, Stanley JR. Mechanisms of disease: Pemphigus and bullous pemphigoid. Annu Rev Pathol (2016) 11:175–97. doi: 10.1146/annurev-pathol-012615-044313
9. Genovese G, Di Zenzo G, Cozzani E, Berti E, Cugno M, Marzano AV. New insights into the pathogenesis of bullous pemphigoid: 2019 update. Front Immunol (2019) 10:1506. doi: 10.3389/fimmu.2019.01506
10. Miyamoto D, Santi CG, Aoki V, Maruta CW. Bullous pemphigoid. Anais brasileiros dermatol (2019) 94(2):133–46. doi: 10.1590/abd1806-4841.20199007
11. Schmidt E, Zillikens D. Pemphigoid diseases. Lancet (London England) (2013) 381(9863):320–32. doi: 10.1016/s0140-6736(12)61140-4
12. Kibsgaard L, Rasmussen M, Lamberg A, Deleuran M, Olesen AB, Vestergaard C. Increased frequency of multiple sclerosis among patients with bullous pemphigoid: A population-based cohort study on comorbidities anchored around the diagnosis of bullous pemphigoid. Br J Dermatol (2017) 176(6):1486–91. doi: 10.1111/bjd.15405
13. Lee S, Rastogi S, Hsu DY, Nardone B, Silverberg JI. Association of bullous pemphigoid and comorbid health conditions: A case-control study. Arch Dermatol Res (2021) 313(5):327–32. doi: 10.1007/s00403-020-02100-2
14. Chai ZT, Tan C, MeiQi Liau M, Kaur H, Pang SM, Phoon YW, et al. Diabetes mellitus and hyperglycemic complications in bullous pemphigoid. J Am Acad Dermatol (2020) 82(5):1234–7. doi: 10.1016/j.jaad.2019.11.018
15. Albadri Z, Thorslund K, Häbel H, Seifert O, Grönhagen C. Increased risk of squamous cell carcinoma of the skin and lymphoma among 5,739 patients with bullous pemphigoid: A Swedish nationwide cohort study. Acta dermato-venereol (2020) 100(17):adv00289. doi: 10.2340/00015555-3622
16. Kouris A, Platsidaki E, Christodoulou C, Armyra K, Korkoliakou P, Stefanaki C, et al. Quality of life, depression, anxiety and loneliness in patients with bullous pemphigoid. a case control study. Anais brasileiros dermatol (2016) 91(5):601–3. doi: 10.1590/abd1806-4841.20164935
17. Briand C, Gourier G, Poizeau F, Jelti L, Bachelerie M, Quéreux G, et al. Characteristics of pruritus in bullous pemphigoid and impact on quality of life: A prospective cohort study. Acta dermato-venereol (2020) 100(18):adv00320. doi: 10.2340/00015555-3683
18. Rice JB, White AG, Scarpati LM, Wan G, Nelson WW. Long-term systemic corticosteroid exposure: A systematic literature review. Clin Ther (2017) 39(11):2216–29. doi: 10.1016/j.clinthera.2017.09.011
19. Cho YT, Chu CY, Wang LF. First-line combination therapy with rituximab and corticosteroids provides a high complete remission rate in moderate-to-Severe bullous pemphigoid. Br J Dermatol (2015) 173(1):302–4. doi: 10.1111/bjd.13633
20. Borradori L, Van Beek N, Feliciani C, Tedbirt B, Antiga E, Bergman R, et al. Updated S2 K guidelines for the management of bullous pemphigoid initiated by the European academy of dermatology and venereology (Eadv). J Eur Acad Dermatol Venereol JEADV (2022) 36(10):1689–704. doi: 10.1111/jdv.18220
21. Annunziato F, Romagnani C, Romagnani S. The 3 major types of innate and adaptive cell-mediated effector immunity. J Allergy Clin Immunol (2015) 135(3):626–35. doi: 10.1016/j.jaci.2014.11.001
22. Pulendran B, Artis D. New paradigms in type 2 immunity. Sci (New York NY) (2012) 337(6093):431–5. doi: 10.1126/science.1221064
23. Mohapatra A, Van Dyken SJ, Schneider C, Nussbaum JC, Liang HE, Locksley RM. Group 2 innate lymphoid cells utilize the Irf4-Il-9 module to coordinate epithelial cell maintenance of lung homeostasis. Mucosal Immunol (2016) 9(1):275–86. doi: 10.1038/mi.2015.59
24. von Moltke J, Ji M, Liang HE, Locksley RM. Tuft-Cell-Derived il-25 regulates an intestinal Ilc2-epithelial response circuit. Nature (2016) 529(7585):221–5. doi: 10.1038/nature16161
25. Leyva-Castillo JM, Galand C, Mashiko S, Bissonnette R, McGurk A, Ziegler SF, et al. Ilc2 activation by keratinocyte-derived il-25 drives il-13 production at sites of allergic skin inflammation. J Allergy Clin Immunol (2020) 145(6):1606–14.e4. doi: 10.1016/j.jaci.2020.02.026
26. Thompson M, Britt RD Jr., Pabelick CM, Prakash YS. Hypoxia and local inflammation in pulmonary artery structure and function. Adv Exp Med Biol (2017) 967:325–34. doi: 10.1007/978-3-319-63245-2_20
27. Mahapatro M, Foersch S, Hefele M, He GW, Giner-Ventura E, McHedlidze T, et al. Programming of intestinal epithelial differentiation by il-33 derived from pericryptal fibroblasts in response to systemic infection. Cell Rep (2016) 15(8):1743–56. doi: 10.1016/j.celrep.2016.04.049
28. McKenzie ANJ, Spits H, Eberl G. Innate lymphoid cells in inflammation and immunity. Immunity (2014) 41(3):366–74. doi: 10.1016/j.immuni.2014.09.006
29. Machida K, Aw M, Salter BMA, Ju X, Mukherjee M, Gauvreau GM, et al. The role of the Tl1a/Dr3 axis in the activation of group 2 innate lymphoid cells in subjects with eosinophilic asthma. Am J Respir Crit Care Med (2020) 202(8):1105–14. doi: 10.1164/rccm.201909-1722OC
30. Meylan F, Hawley ET, Barron L, Barlow JL, Penumetcha P, Pelletier M, et al. The tnf-family cytokine Tl1a promotes allergic immunopathology through group 2 innate lymphoid cells. Mucosal Immunol (2014) 7(4):958–68. doi: 10.1038/mi.2013.114
31. Doherty TA, Broide DH. Lipid regulation of group 2 innate lymphoid cell function: Moving beyond epithelial cytokines. J Allergy Clin Immunol (2018) 141(5):1587–9. doi: 10.1016/j.jaci.2018.02.034
32. Tojima I, Matsumoto K, Kikuoka H, Hara S, Yamamoto S, Shimizu S, et al. Evidence for the induction of Th2 inflammation by group 2 innate lymphoid cells in response to prostaglandin D(2) and cysteinyl leukotrienes in allergic rhinitis. Allergy (2019) 74(12):2417–26. doi: 10.1111/all.13974
33. Liang Y, Yu B, Chen J, Wu H, Xu Y, Yang B, et al. Thymic stromal lymphopoietin epigenetically upregulates fc receptor Γ subunit-related receptors on antigen-presenting cells and induces T(H)2/T(H)17 polarization through dectin-2. J Allergy Clin Immunol (2019) 144(4):1025–35.e7. doi: 10.1016/j.jaci.2019.06.011
34. Murakami-Satsutani N, Ito T, Nakanishi T, Inagaki N, Tanaka A, Vien PT, et al. Il-33 promotes the induction and maintenance of Th2 immune responses by enhancing the function of Ox40 ligand. Allergol Int (2014) 63(3):443–55. doi: 10.2332/allergolint.13-OA-0672
35. Mirchandani AS, Besnard AG, Yip E, Scott C, Bain CC, Cerovic V, et al. Type 2 innate lymphoid cells drive Cd4+ Th2 cell responses. J Immunol (Baltimore Md 1950) (2014) 192(5):2442–8. doi: 10.4049/jimmunol.1300974
36. Oliphant CJ, Hwang YY, Walker JA, Salimi M, Wong SH, Brewer JM, et al. Mhcii-mediated dialog between group 2 innate lymphoid cells and Cd4(+) T cells potentiates type 2 immunity and promotes parasitic helminth expulsion. Immunity (2014) 41(2):283–95. doi: 10.1016/j.immuni.2014.06.016
37. Drake LY, Iijima K, Kita H. Group 2 innate lymphoid cells and Cd4+ T cells cooperate to mediate type 2 immune response in mice. Allergy (2014) 69(10):1300–7. doi: 10.1111/all.12446
38. Ramirez GA, Yacoub MR, Ripa M, Mannina D, Cariddi A, Saporiti N, et al. Eosinophils from physiology to disease: A comprehensive review. BioMed Res Int (2018) 2018:9095275. doi: 10.1155/2018/9095275
39. Ziegler SF, Roan F, Bell BD, Stoklasek TA, Kitajima M, Han H. The biology of thymic stromal lymphopoietin (Tslp). Adv Pharmacol (San Diego Calif) (2013) 66:129–55. doi: 10.1016/b978-0-12-404717-4.00004-4
40. De Salvo C, Wang XM, Pastorelli L, Mattioli B, Omenetti S, Buela KA, et al. Il-33 drives eosinophil infiltration and pathogenic type 2 helper T-cell immune responses leading to chronic experimental ileitis. Am J Pathol (2016) 186(4):885–98. doi: 10.1016/j.ajpath.2015.11.028
41. Bass DA. Behavior of eosinophil leukocytes in acute inflammation. ii. eosinophil dynamics during acute inflammation. J Clin Invest (1975) 56(4):870–9. doi: 10.1172/jci108166
42. Handzel ZT, Busse WW, Sedgwick JB, Vrtis R, Lee WM, Kelly EA, et al. Eosinophils bind rhinovirus and activate virus-specific T cells. J Immunol (Baltimore Md 1950) (1998) 160(3):1279–84. doi: 10.4049/jimmunol.160.3.1279
43. Ravin KA, Loy M. The eosinophil in infection. Clin Rev Allergy Immunol (2016) 50(2):214–27. doi: 10.1007/s12016-015-8525-4
44. Esmaeili SA, Hajavi J. The role of indoleamine 2,3-dioxygenase in allergic disorders. Mol Biol Rep (2022) 49(4):3297–306. doi: 10.1007/s11033-021-07067-5
45. Spencer LA, Weller PF. Eosinophils and Th2 immunity: Contemporary insights. Immunol Cell Biol (2010) 88(3):250–6. doi: 10.1038/icb.2009.115
46. Wang YH, Angkasekwinai P, Lu N, Voo KS, Arima K, Hanabuchi S, et al. Il-25 augments type 2 immune responses by enhancing the expansion and functions of tslp-Dc-Activated Th2 memory cells. J Exp Med (2007) 204(8):1837–47. doi: 10.1084/jem.20070406
47. Akdis CA, Arkwright PD, Brüggen MC, Busse W, Gadina M, Guttman-Yassky E, et al. Type 2 immunity in the skin and lungs. Allergy (2020) 75(7):1582–605. doi: 10.1111/all.14318
48. Akdis M, Akdis CA. Ige class switching and cellular memory. Nat Immunol (2012) 13(4):312–4. doi: 10.1038/ni.2266
49. Stone KD, Prussin C, Metcalfe DD. Ige, mast cells, basophils, and eosinophils. J Allergy Clin Immunol (2010) 125(2 Suppl 2):S73–80. doi: 10.1016/j.jaci.2009.11.017
50. Kanagaratham C, El Ansari YS, Lewis OL, Oettgen HC. Ige and igg antibodies as regulators of mast cell and basophil functions in food allergy. Front Immunol (2020) 11:603050. doi: 10.3389/fimmu.2020.603050
51. Fang H, Li Q, Wang G. The role of T cells in pemphigus vulgaris and bullous pemphigoid. Autoimmun Rev (2020) 19(11):102661. doi: 10.1016/j.autrev.2020.102661
52. Iwata Y, Komura K, Kodera M, Usuda T, Yokoyama Y, Hara T, et al. Correlation of ige autoantibody to Bp180 with a severe form of bullous pemphigoid. Arch Dermatol (2008) 144(1):41–8. doi: 10.1001/archdermatol.2007.9
53. Di Zenzo G, Thoma-Uszynski S, Fontao L, Calabresi V, Hofmann SC, Hellmark T, et al. Multicenter prospective study of the humoral autoimmune response in bullous pemphigoid. Clin Immunol (Orlando Fla) (2008) 128(3):415–26. doi: 10.1016/j.clim.2008.04.012
54. Daneshpazhooh M, Ghiasi M, Lajevardi V, Nasiri N, Balighi K, Teimourpour A, et al. Bpdai and absis correlate with serum anti-Bp180 Nc16a igg but not with anti-Bp230 igg in patients with bullous pemphigoid. Arch Dermatol Res (2018) 310(3):255–9. doi: 10.1007/s00403-018-1817-9
55. Cai SC, Lim YL, Li W, Allen JC, Chua SH, Tan SH, et al. Anti-Bp180 Nc16a igg titres as an indicator of disease activity and outcome in Asian patients with bullous pemphigoid. Ann Acad Med Singapore (2015) 44(4):119–26. doi: 10.47102/annals-acadmedsg.V44N4p119
56. van Beek N, Schulze FS, Zillikens D, Schmidt E. Ige-mediated mechanisms in bullous pemphigoid and other autoimmune bullous diseases. Expert Rev Clin Immunol (2016) 12(3):267–77. doi: 10.1586/1744666x.2016.1123092
57. Lin L, Hwang BJ, Culton DA, Li N, Burette S, Koller BH, et al. Eosinophils mediate tissue injury in the autoimmune skin disease bullous pemphigoid. J Invest Dermatol (2018) 138(5):1032–43. doi: 10.1016/j.jid.2017.11.031
58. Kowalski EH, Kneibner D, Kridin K, Amber KT. Serum and blister fluid levels of cytokines and chemokines in pemphigus and bullous pemphigoid. Autoimmun Rev (2019) 18(5):526–34. doi: 10.1016/j.autrev.2019.03.009
59. Ameglio F, D'Auria L, Bonifati C, Ferraro C, Mastroianni A, Giacalone B. Cytokine pattern in blister fluid and serum of patients with bullous pemphigoid: Relationships with disease intensity. Br J Dermatol (1998) 138(4):611–4. doi: 10.1046/j.1365-2133.1998.02169.x
60. D'Auria L, Pietravalle M, Mastroianni A, Ferraro C, Mussi A, Bonifati C, et al. Il-5 levels in the serum and blister fluid of patients with bullous pemphigoid: Correlations with eosinophil cationic protein, rantes, ige and disease severity. Arch Dermatol Res (1998) 290(1-2):25–7. doi: 10.1007/s004030050272
61. Pickford WJ, Gudi V, Haggart AM, Lewis BJ, Herriot R, Barker RN, et al. T Cell participation in autoreactivity to Nc16a epitopes in bullous pemphigoid. Clin Exp Immunol (2015) 180(2):189–200. doi: 10.1111/cei.12566
62. Raphael I, Nalawade S, Eagar TN, Forsthuber TG. T Cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine (2015) 74(1):5–17. doi: 10.1016/j.cyto.2014.09.011
63. Harb H, Chatila TA. Mechanisms of dupilumab. Clin Exp Allergy (2020) 50(1):5–14. doi: 10.1111/cea.13491
64. Hashimoto T, Kursewicz CD, Fayne RA, Nanda S, Shah SM, Nattkemper L, et al. Pathophysiologic mechanisms of itch in bullous pemphigoid. J Am Acad Dermatol (2020) 83(1):53–62. doi: 10.1016/j.jaad.2019.07.060
65. Tabatabaei-Panah PS, Moravvej H, Alirajab M, Etaaty A, Geranmayeh M, Hosseine F, et al. Association between Th2 cytokine gene polymorphisms and risk of bullous pemphigoid. Immunol investig (2022) 51(2):343–56. doi: 10.1080/08820139.2020.1832113
66. Gooderham MJ, Hong HC, Eshtiaghi P, Papp KA. Dupilumab: A review of its use in the treatment of atopic dermatitis. J Am Acad Dermatol (2018) 78(3 Suppl 1):S28–s36. doi: 10.1016/j.jaad.2017.12.022
67. Abdat R, Waldman RA, de Bedout V, Czernik A, McLeod M, King B, et al. Dupilumab as a novel therapy for bullous pemphigoid: A multicenter case series. J Am Acad Dermatol (2020) 83(1):46–52. doi: 10.1016/j.jaad.2020.01.089
68. Geller S. Interleukin 4 and interleukin 13 inhibition: A promising therapeutic approach in bullous pemphigoid. J Am Acad Dermatol (2020) 83(1):37–8. doi: 10.1016/j.jaad.2020.03.017
69. Kaye A, Gordon SC, Deverapalli SC, Her MJ, Rosmarin D. Dupilumab for the treatment of recalcitrant bullous pemphigoid. JAMA Dermatol (2018) 154(10):1225–6. doi: 10.1001/jamadermatol.2018.2526
70. Seyed Jafari SM, Feldmeyer L, Bossart S, Simon D, Schlapbach C, Borradori L. Case report: Combination of omalizumab and dupilumab for recalcitrant bullous pemphigoid. Front Immunol (2020) 11:611549. doi: 10.3389/fimmu.2020.611549
71. Takamura S, Teraki Y. Treatment of bullous pemphigoid with dupilumab: Dupilumab exerts its effect by primarily suppressing T-helper 2 cytokines. J Dermatol (2022) 49(9):845–50. doi: 10.1111/1346-8138.16428
72. Wang SH, Zuo YG. Commentary: Efficacy and safety of dupilumab in moderate-to-Severe bullous pemphigoid. Front Immunol (2021) 12:800609. doi: 10.3389/fimmu.2021.800609
73. Yang J, Gao H, Zhang Z, Tang C, Chen Z, Wang L, et al. Dupilumab combined with low-dose systemic steroid therapy improves efficacy and safety for bullous pemphigoid. Dermatol Ther (2022) 35(8):e15648. doi: 10.1111/dth.15648
74. Zhang Y, Xu Q, Chen L, Chen J, Zhang J, Zou Y, et al. Efficacy and safety of dupilumab in moderate-to-Severe bullous pemphigoid. Front Immunol (2021) 12:738907. doi: 10.3389/fimmu.2021.738907
75. Bettiol A, Urban ML, Dagna L, Cottin V, Franceschini F, Del Giacco S, et al. Mepolizumab for eosinophilic granulomatosis with polyangiitis: A European multicenter observational study. Arthritis Rheumatol (Hoboken NJ) (2022) 74(2):295–306. doi: 10.1002/art.41943
76. Han JK, Bachert C, Fokkens W, Desrosiers M, Wagenmann M, Lee SE, et al. Mepolizumab for chronic rhinosinusitis with nasal polyps (Synapse): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Respir Med (2021) 9(10):1141–53. doi: 10.1016/s2213-2600(21)00097-7
77. Ortega HG, Liu MC, Pavord ID, Brusselle GG, FitzGerald JM, Chetta A, et al. Mepolizumab treatment in patients with severe eosinophilic asthma. New Engl J Med (2014) 371(13):1198–207. doi: 10.1056/NEJMoa1403290
78. Pavord ID, Korn S, Howarth P, Bleecker ER, Buhl R, Keene ON, et al. Mepolizumab for severe eosinophilic asthma (Dream): A multicentre, double-blind, placebo-controlled trial. Lancet (London England) (2012) 380(9842):651–9. doi: 10.1016/s0140-6736(12)60988-x
79. Wechsler ME, Akuthota P, Jayne D, Khoury P, Klion A, Langford CA, et al. Mepolizumab or placebo for eosinophilic granulomatosis with polyangiitis. New Engl J Med (2017) 376(20):1921–32. doi: 10.1056/NEJMoa1702079
80. Simon D, Yousefi S, Cazzaniga S, Bürgler C, Radonjic S, Houriet C, et al. Mepolizumab failed to affect bullous pemphigoid: A randomized, placebo-controlled, double-blind phase 2 pilot study. Allergy (2020) 75(3):669–72. doi: 10.1111/all.13950
81. Castro M, Zangrilli J, Wechsler ME, Bateman ED, Brusselle GG, Bardin P, et al. Reslizumab for inadequately controlled asthma with elevated blood eosinophil counts: Results from two multicentre, parallel, double-blind, randomised, placebo-controlled, phase 3 trials. Lancet Respir Med (2015) 3(5):355–66. doi: 10.1016/s2213-2600(15)00042-9
82. Deeks ED, Brusselle G. Reslizumab in eosinophilic asthma: A review. Drugs (2017) 77(7):777–84. doi: 10.1007/s40265-017-0740-2
83. Markham A. Reslizumab: First global approval. Drugs (2016) 76(8):907–11. doi: 10.1007/s40265-016-0583-2
84. Rhyou HI, Han SH, Nam YH. Successful induction treatment of bullous pemphigoid using reslizumab: A case report. Allergy asthma Clin Immunol (2021) 17(1):117. doi: 10.1186/s13223-021-00619-1
85. Kavanagh JE, Hearn AP, Dhariwal J, d'Ancona G, Douiri A, Roxas C, et al. Real-world effectiveness of benralizumab in severe eosinophilic asthma. Chest (2021) 159(2):496–506. doi: 10.1016/j.chest.2020.08.2083
86. Criner GJ, Celli BR, Brightling CE, Agusti A, Papi A, Singh D, et al. Benralizumab for the prevention of copd exacerbations. New Engl J Med (2019) 381(11):1023–34. doi: 10.1056/NEJMoa1905248
87. Guntur VP, Manka LA, Denson JL, Dunn RM, Dollin YT, Gill M, et al. Benralizumab as a steroid-sparing treatment option in eosinophilic granulomatosis with polyangiitis. J Allergy Clin Immunol In Pract (2021) 9(3):1186–93.e1. doi: 10.1016/j.jaip.2020.09.054
88. Wollenberg A, Blauvelt A, Guttman-Yassky E, Worm M, Lynde C, Lacour JP, et al. Tralokinumab for moderate-to-Severe atopic dermatitis: Results from two 52-week, randomized, double-blind, multicentre, placebo-controlled phase iii trials (Ecztra 1 and ecztra 2). Br J Dermatol (2021) 184(3):437–49. doi: 10.1111/bjd.19574
89. Duggan S. Tralokinumab: First approval. Drugs (2021) 81(14):1657–63. doi: 10.1007/s40265-021-01583-1
90. Antoniu SA. Lebrikizumab for the treatment of asthma. Expert Opin investig Drugs (2016) 25(10):1239–49. doi: 10.1080/13543784.2016.1227319
91. Bujarski S, Parulekar AD, Hanania NA. Lebrikizumab in the treatment of asthma. Expert Opin Biol Ther (2016) 16(6):847–52. doi: 10.1080/14712598.2016.1182152
92. Guttman-Yassky E, Blauvelt A, Eichenfield LF, Paller AS, Armstrong AW, Drew J, et al. Efficacy and safety of lebrikizumab, a high-affinity interleukin 13 inhibitor, in adults with moderate to severe atopic dermatitis: A phase 2b randomized clinical trial. JAMA Dermatol (2020) 156(4):411–20. doi: 10.1001/jamadermatol.2020.0079
93. Maselli DJ, Keyt H, Rogers L. Profile of lebrikizumab and its potential in the treatment of asthma. J Asthma Allergy (2015) 8:87–92. doi: 10.2147/jaa.S69932
94. Simpson EL, Flohr C, Eichenfield LF, Bieber T, Sofen H, Taïeb A, et al. Efficacy and safety of lebrikizumab (an anti-Il-13 monoclonal antibody) in adults with moderate-to-Severe atopic dermatitis inadequately controlled by topical corticosteroids: A randomized, placebo-controlled phase ii trial (Treble). J Am Acad Dermatol (2018) 78(5):863–71.e11. doi: 10.1016/j.jaad.2018.01.017
95. Pruessmann J, Pruessmann W, Holtsche MM, Linnemann B, Hammers CM, van Beek N, et al. Immunomodulator galectin-9 is increased in blood and skin of patients with bullous pemphigoid. Acta dermato-venereol (2021) 101(3):adv00419. doi: 10.2340/00015555-3771
96. Engmann J, Rüdrich U, Behrens G, Papakonstantinou E, Gehring M, Kapp A, et al. Increased activity and apoptosis of eosinophils in blister fluids, skin and peripheral blood of patients with bullous pemphigoid. Acta dermato-venereol (2017) 97(4):464–71. doi: 10.2340/00015555-2581
97. Liu Y, Wang Y, Chen X, Jin H, Li L. Factors associated with the activity and severity of bullous pemphigoid: A review. Ann Med (2020) 52(3-4):55–62. doi: 10.1080/07853890.2020.1742367
98. Kridin K. Peripheral eosinophilia in bullous pemphigoid: Prevalence and influence on the clinical manifestation. Br J Dermatol (2018) 179(5):1141–7. doi: 10.1111/bjd.16679
99. Park SH, Lee SH, Kim JH, Kim SC. Circulating eosinophil and neutrophil counts correlate with disease severity in bullous pemphigoid. Ann Dermatol (2018) 30(5):544–9. doi: 10.5021/ad.2018.30.5.544
100. Gore Karaali M, Koku Aksu AE, Cin M, Leblebici C, Kara Polat A, Gurel MS. Tissue eosinophil levels as a marker of disease severity in bullous pemphigoid. Australas J Dermatol (2021) 62(2):e236–e41. doi: 10.1111/ajd.13547
101. Messingham KN, Holahan HM, Frydman AS, Fullenkamp C, Srikantha R, Fairley JA. Human eosinophils express the high affinity ige receptor, fcϵri, in bullous pemphigoid. PloS One (2014) 9(9):e107725. doi: 10.1371/journal.pone.0107725
102. Amber KT, Chernyavsky A, Agnoletti AF, Cozzani E, Grando SA. Mechanisms of pathogenic effects of eosinophil cationic protein and eosinophil-derived neurotoxin on human keratinocytes. Exp Dermatol (2018) 27(12):1322–7. doi: 10.1111/exd.13782
103. Bystrom J, Amin K, Bishop-Bailey D. Analysing the eosinophil cationic protein–a clue to the function of the eosinophil granulocyte. Respir Res (2011) 12(1):10. doi: 10.1186/1465-9921-12-10
104. Kunsleben N, Rüdrich U, Gehring M, Novak N, Kapp A, Raap U. Il-31 induces chemotaxis, calcium mobilization, release of reactive oxygen species, and Ccl26 in eosinophils, which are capable to release il-31. J Invest Dermatol (2015) 135(7):1908–11. doi: 10.1038/jid.2015.106
105. Rüdrich U, Gehring M, Papakonstantinou E, Illerhaus A, Engmann J, Kapp A, et al. Eosinophils are a major source of interleukin-31 in bullous pemphigoid. Acta dermato-venereol (2018) 98(8):766–71. doi: 10.2340/00015555-2951
106. Ståhle-Bäckdahl M, Inoue M, Guidice GJ, Parks WC. 92-kd gelatinase is produced by eosinophils at the site of blister formation in bullous pemphigoid and cleaves the extracellular domain of recombinant 180-kd bullous pemphigoid autoantigen. J Clin Invest (1994) 93(5):2022–30. doi: 10.1172/jci117196
107. de Graauw E, Sitaru C, Horn M, Borradori L, Yousefi S, Simon HU, et al. Evidence for a role of eosinophils in blister formation in bullous pemphigoid. Allergy (2017) 72(7):1105–13. doi: 10.1111/all.13131
108. Tedeschi A, Marzano AV, Lorini M, Balice Y, Cugno M. Eosinophil cationic protein levels parallel coagulation activation in the blister fluid of patients with bullous pemphigoid. J Eur Acad Dermatol Venereol JEADV (2015) 29(4):813–7. doi: 10.1111/jdv.12464
109. Marzano AV, Tedeschi A, Fanoni D, Bonanni E, Venegoni L, Berti E, et al. Activation of blood coagulation in bullous pemphigoid: Role of eosinophils, and local and systemic implications. Br J Dermatol (2009) 160(2):266–72. doi: 10.1111/j.1365-2133.2008.08880.x
110. Marzano AV, Tedeschi A, Berti E, Fanoni D, Crosti C, Cugno M. Activation of coagulation in bullous pemphigoid and other eosinophil-related inflammatory skin diseases. Clin Exp Immunol (2011) 165(1):44–50. doi: 10.1111/j.1365-2249.2011.04391.x
111. Marzano AV, Genovese G, Cugno M. Venous thromboembolism in chronic inflammatory skin diseases-the need to consider bullous pemphigoid. JAMA Dermatol (2022) 158(3):330–1. doi: 10.1001/jamadermatol.2021.5662
112. Chen X, Zhao W, Jin H, Li L. Risk factors for mucosal involvement in bullous pemphigoid and the possible mechanism: A review. Front Med (2021) 8:680871. doi: 10.3389/fmed.2021.680871
113. Ständer S, Schmidt E, Zillikens D, Ludwig RJ, Kridin K. Immunological features and factors associated with mucocutaneous bullous pemphigoid - a retrospective cohort study. J der Deutschen Dermatologischen Gesellschaft (2021) 19(9):1289–95. doi: 10.1111/ddg.14494
114. Maglie R, Hertl M. Pharmacological advances in pemphigoid. Curr Opin Pharmacol (2019) 46:34–43. doi: 10.1016/j.coph.2018.12.007
115. Chu KY, Yu HS, Yu S. Current and innovated managements for autoimmune bullous skin disorders: An overview. J Clin Med (2022) 11(12):3528. doi: 10.3390/jcm11123528
116. Keam SJ. Nemolizumab: First approval. Drugs (2022) 82(10):1143–50. doi: 10.1007/s40265-022-01741-z
117. Provost TT, Tomasi TB Jr. Immunopathology of bullous pemphigoid. basement membrane deposition of ige, alternate pathway components and fibrin. Clin Exp Immunol (1974) 18(2):193–200.
118. Dresow SK, Sitaru C, Recke A, Oostingh GJ, Zillikens D, Gibbs BF. Ige autoantibodies against the intracellular domain of Bp180. Br J Dermatol (2009) 160(2):429–32. doi: 10.1111/j.1365-2133.2008.08858.x
119. Hashimoto T, Ohzono A, Teye K, Numata S, Hiroyasu S, Tsuruta D, et al. Detection of ige autoantibodies to Bp180 and Bp230 and their relationship to clinical features in bullous pemphigoid. Br J Dermatol (2017) 177(1):141–51. doi: 10.1111/bjd.15114
120. Fania L, Caldarola G, Müller R, Brandt O, Pellicano R, Feliciani C, et al. Ige recognition of bullous pemphigoid (Bp)180 and Bp230 in bp patients and elderly individuals with pruritic dermatoses. Clin Immunol (Orlando Fla) (2012) 143(3):236–45. doi: 10.1016/j.clim.2012.02.003
121. Kamata A, Kurihara Y, Funakoshi T, Takahashi H, Kuroda K, Hachiya T, et al. Basement membrane zone ige deposition is associated with bullous pemphigoid disease severity and treatment results. Br J Dermatol (2020) 182(5):1221–7. doi: 10.1111/bjd.18364
122. Kalowska M, Ciepiela O, Kowalewski C, Demkow U, Schwartz RA, Wozniak K. Enzyme-linked immunoassay index for anti-Nc16a igg and ige auto-antibodies correlates with severity and activity of bullous pemphigoid. Acta dermato-venereol (2016) 96(2):191–6. doi: 10.2340/00015555-2101
123. Asbrink E, Hovmark A. Serum ige levels in patients with bullous pemphigoid and its correlation to the activity of the disease and anti-basement membrane zone antibodies. Acta dermato-venereol (1984) 64(3):243–6.
124. Saniklidou AH, Tighe PJ, Fairclough LC, Todd I. Ige autoantibodies and their association with the disease activity and phenotype in bullous pemphigoid: A systematic review. Arch Dermatol Res (2018) 310(1):11–28. doi: 10.1007/s00403-017-1789-1
125. Ishiura N, Fujimoto M, Watanabe R, Nakashima H, Kuwano Y, Yazawa N, et al. Serum levels of ige anti-Bp180 and anti-Bp230 autoantibodies in patients with bullous pemphigoid. J Dermatol Sci (2008) 49(2):153–61. doi: 10.1016/j.jdermsci.2007.08.008
126. Cozzani E, Micalizzi C, Parodi A, Rebora A. Anti-230 kda circulating ige in bullous pemphigoid: Relationship with disease activity. Acta dermato-venereol (1997) 77(3):236. doi: 10.2340/0001555577236
127. Shih YC, Yuan H, Shen J, Zheng J, Pan M. Bp230 ige autoantibodies in topical-Steroid-Resistant bullous pemphigoid. J Dermatol (2021) 48(9):1372–80. doi: 10.1111/1346-8138.15952
128. Messingham KA, Onoh A, Vanderah EM, Giudice GJ, Fairley JA. Functional characterization of an ige-class monoclonal antibody specific for the bullous pemphigoid autoantigen, Bp180. Hybridoma (2005) (2012) 31(2):111–7. doi: 10.1089/hyb.2011.0102
129. Inaoki M, Sato S, Takehara K. Elevated expression of Cd23 on peripheral blood b lymphocytes from patients with bullous pemphigoid: Correlation with increased serum ige. J Dermatol Sci (2004) 35(1):53–9. doi: 10.1016/j.jdermsci.2004.03.009
130. Selb R, Eckl-Dorna J, Neunkirchner A, Schmetterer K, Marth K, Gamper J, et al. Cd23 surface density on b cells is associated with ige levels and determines ige-facilitated allergen uptake, as well as activation of allergen-specific T cells. J Allergy Clin Immunol (2017) 139(1):290–9.e4. doi: 10.1016/j.jaci.2016.03.042
131. Aghighi M, Smoller BR. Diminished expression of galectin-3 around blisters in bullous pemphigoid: An immunohistochemistry study. Dermatol Pract conceptual (2020) 10(4):e2020106. doi: 10.5826/dpc.1004a106
132. Maurer M, Rosén K, Hsieh HJ, Saini S, Grattan C, Gimenéz-Arnau A, et al. Omalizumab for the treatment of chronic idiopathic or spontaneous urticaria. New Engl J Med (2013) 368(10):924–35. doi: 10.1056/NEJMoa1215372
133. Humbert M, Beasley R, Ayres J, Slavin R, Hébert J, Bousquet J, et al. Benefits of omalizumab as add-on therapy in patients with severe persistent asthma who are inadequately controlled despite best available therapy (Gina 2002 step 4 treatment): Innovate. Allergy (2005) 60(3):309–16. doi: 10.1111/j.1398-9995.2004.00772.x
134. Holgate S, Casale T, Wenzel S, Bousquet J, Deniz Y, Reisner C. The anti-inflammatory effects of omalizumab confirm the central role of ige in allergic inflammation. J Allergy Clin Immunol (2005) 115(3):459–65. doi: 10.1016/j.jaci.2004.11.053
135. Seyed Jafari SM, Gadaldi K, Feldmeyer L, Yawalkar N, Borradori L, Schlapbach C. Effects of omalizumab on fcϵri and ige expression in lesional skin of bullous pemphigoid. Front Immunol (2019) 10:1919. doi: 10.3389/fimmu.2019.01919
136. James T, Salman S, Stevenson B, Bundell C, Kelly G, Nolan D, et al. Ige blockade in autoimmunity: Omalizumab induced remission of bullous pemphigoid. Clin Immunol (Orlando Fla) (2019) 198:54–6. doi: 10.1016/j.clim.2018.12.015
137. Balakirski G, Alkhateeb A, Merk HF, Leverkus M, Megahed M. Successful treatment of bullous pemphigoid with omalizumab as corticosteroid-sparing agent: Report of two cases and review of literature. J Eur Acad Dermatol Venereol JEADV (2016) 30(10):1778–82. doi: 10.1111/jdv.13758
138. Yu KK, Crew AB, Messingham KA, Fairley JA, Woodley DT. Omalizumab therapy for bullous pemphigoid. J Am Acad Dermatol (2014) 71(3):468–74. doi: 10.1016/j.jaad.2014.04.053
139. Sarrazin M, Jouen F, Duvert-Lehembre S. Refractory bullous pemphigoid with ige anti-Bp230 and igg anti-P200 antibodies successfully treated with omalizumab. Annales dermatol venereol (2021) 148(1):60–2. doi: 10.1016/j.annder.2020.08.053
140. Maurer M, Giménez-Arnau AM, Sussman G, Metz M, Baker DR, Bauer A, et al. Ligelizumab for chronic spontaneous urticaria. New Engl J Med (2019) 381(14):1321–32. doi: 10.1056/NEJMoa1900408
141. Messingham KN, Crowe TP, Fairley JA. The intersection of ige autoantibodies and eosinophilia in the pathogenesis of bullous pemphigoid. Front Immunol (2019) 10:2331. doi: 10.3389/fimmu.2019.02331
142. Wang SH, Zuo YG. Thymic stromal lymphopoietin in cutaneous immune-mediated diseases. Front Immunol (2021) 12:698522. doi: 10.3389/fimmu.2021.698522
143. Li SZ, Jin XX, Ge XL, Zuo YG, Jin HZ. Thymic stromal lymphopoietin is implicated in the pathogenesis of bullous pemphigoid by dendritic cells. J Immunol Res (2020) 2020:4594630. doi: 10.1155/2020/4594630
144. Zhang Y, Hwang BJ, Liu Z, Li N, Lough K, Williams SE, et al. Bp180 dysfunction triggers spontaneous skin inflammation in mice. Proc Natl Acad Sci United States America (2018) 115(25):6434–9. doi: 10.1073/pnas.1721805115
145. Menzies-Gow A, Corren J, Bourdin A, Chupp G, Israel E, Wechsler ME, et al. Tezepelumab in adults and adolescents with severe, uncontrolled asthma. New Engl J Med (2021) 384(19):1800–9. doi: 10.1056/NEJMoa2034975
Keywords: type 2 inflammation, bullous pemphigoid, immunoglobulin E, eosinophils, targeted therapy
Citation: Zhang L, Chen Z, Wang L and Luo X (2023) Bullous pemphigoid: The role of type 2 inflammation in its pathogenesis and the prospect of targeted therapy. Front. Immunol. 14:1115083. doi: 10.3389/fimmu.2023.1115083
Received: 03 December 2022; Accepted: 06 February 2023;
Published: 16 February 2023.
Edited by:
Gaetano Isola, University of Catania, ItalyReviewed by:
Christoph M. Hammers, University of Kiel, GermanyAngelo Valerio Marzano, University of Milan, Italy
Copyright © 2023 Zhang, Chen, Wang and Luo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoqun Luo, bHVveGlhb3F1bjkxM0AxMjYuY29t