 Katharina Pracht
Katharina Pracht Jens Wittner
Jens Wittner Fritz Kagerer
Fritz Kagerer Hans-Martin Jäck
Hans-Martin Jäck Wolfgang Schuh
Wolfgang Schuh
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 16 February 2023
Sec. B Cell Biology
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1114348
This article is part of the Research Topic Expert Opinions in Plasma Cell Differentiation and Spatial Arrangement - 2022 View all 5 articles
To achieve longevity, IgA plasma cells require a sophisticated anatomical microenvironment that provides cytokines, cell-cell contacts, and nutrients as well as metabolites. The intestinal epithelium harbors cells with distinct functions and represents an important defense line. Anti-microbial peptide-producing paneth cells, mucus-secreting goblet cells and antigen-transporting microfold (M) cells cooperate to build a protective barrier against pathogens. In addition, intestinal epithelial cells are instrumental in the transcytosis of IgA to the gut lumen, and support plasma cell survival by producing the cytokines APRIL and BAFF. Moreover, nutrients are sensed through specialized receptors such as the aryl hydrocarbon receptor (AhR) by both, intestinal epithelial cells and immune cells. However, the intestinal epithelium is highly dynamic with a high cellular turn-over rate and exposure to changing microbiota and nutritional factors. In this review, we discuss the spatial interplay of the intestinal epithelium with plasma cells and its potential contribution to IgA plasma cell generation, homing, and longevity. Moreover, we describe the impact of nutritional AhR ligands on intestinal epithelial cell-IgA plasma cell interaction. Finally, we introduce spatial transcriptomics as a new technology to address open questions in intestinal IgA plasma cell biology.
The intestine consists of the small and the large intestine. The small intestine starts at the pylorus and is subdivided into three main parts: the duodenum, the jejunum, and the ileum. The large intestine consists of the caecum, the proximal colon, the transverse colon, the distal colon, the rectum, and ends at the anus. The cellular composition of the epithelium as well as that of the lamina propria (LP) differs along the segments of the intestine, concomitant with the different physiological functions and the different bacterial densities of the small intestine and the colon. The primary function of the small intestine is digestion and the absorption of nutrients. To increase the surface for food absorption, the small intestine is characterized by the presence of villi, whereas villi cannot be found in the caecum and the colon. The main function of the colon is water reabsorption and removal of undigested food. The colon contains the highest density of commensal bacteria (1, 2). Despite their anatomical and functional differences, all segments of the intestine are lined by the mono-layered intestinal epithelium. The intestinal epithelial cells (IECs) and the immune cell composition differs along the segments of the intestine, concurrent with the different physiological functions and the bacterial load of the small intestine and the colon.
Mucosal surfaces are the most critical entry sites for pathogens into our body. Therefore, a sophisticated mucosal defense system evolved that combines chemical, physical, and cellular barriers. The mucosal immune system in the intestine consists of immune cells and the intestinal epithelium that orchestrates innate as well as adaptive immune responses. The epithelium constitutes the interface between the gut lumen and the LP. Its functions include the uptake of nutrients and antigens on the one hand, as well as microbial sensing and exclusion of pathogens on the other hand. The intestinal epithelium consists of an epithelial cell monolayer, the LP and the muscularis mucosae. It can be subdivided into the crypt area where stem cells are located and the villus area (Figure 1). IECs are connected by tight junctions and are attached to a basement membrane that consists of laminin, collagen, fibronectin and other extracellular matrix (ECM) components. The basement membrane provides a platform for cell adhesion, migration, differentiation, and functions as a barrier (3, 4). In addition, it harbors pores of various sizes that allow immune cells (e.g., intra-epithelial T cells) to physically interact with epithelial cells (4, 5).
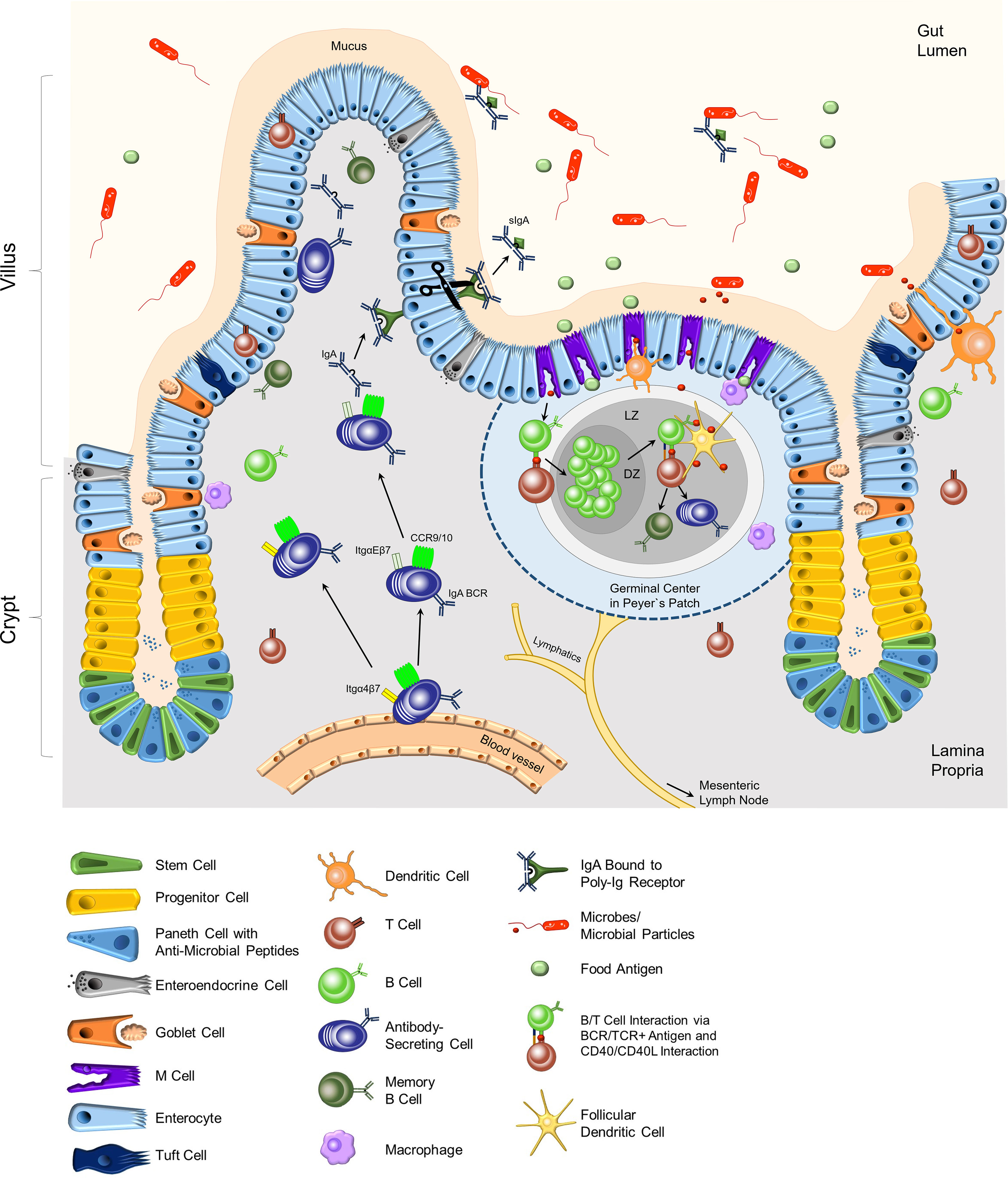
Figure 1 Composition of the epithelium and the lamina propria of the small intestine. In the crypt region, paneth cells secrete anti-microbial peptides into the mucus. Stem cells differentiate into the specialized cells of the intestinal epithelial layer in the villi. Goblet cells secrete mucus, which keeps microbes at bay. Immune cells, mostly T cells, can be located as intra-epithelial lymphocytes (IEL). M cells are located adjacent to Peyer`s patches (PPs), which are part of the lymphoid organs of the intestinal tract. M cells transport food antigens or microbial particles to DCs, macrophages, and B and T lymphocytes in the PPs. Antigen-specific B and T lymphocytes induce a germinal center with the dark zone (DZ) and light zone (LZ) in the PPs. Here, activated B cells undergo affinity maturation and class switch recombination with the help of T cells and follicular dendritic cells, resulting in mainly IgA-class-switched, antigen-specific antibody-secreting cells and memory B cells. Dendritic cells support the activation of adaptive immune cells by acquiring antigens through the epithelial layer and transporting them to the PPs or the mLNs. IgA-secreting cells migrate from the blood vessels to the epithelial layer, attracted by cytokine gradients (CCR9/10) and guided by integrins. Dimeric IgA that binds to p-Ig receptors on epithelial cells is transported through the epithelium. Secreted IgA (sIgA) in the lumen binds to specific antigens and regulates the intestinal microbiota composition. BCR, B cell receptor; Itg, integrin.
IECs originate from Lgr5+ stem cells in the crypts and differentiate into specialized epithelial cells (6). Enterocytes represent the majority of IECs and their primary function is the absorption of nutrients. Besides enterocytes, the epithelium contains specialized tuft cells, enteroendocrine cells, goblet cells, paneth cells, and microfold (M) cells (Figure 1). The intestinal epithelium is characterized by a high turn-over with an average turn-over time of 4-5 days. Stem cells in the crypts constantly divide and give rise to so-called transit-amplifying cells or progenitor cells (6–8). These cells further differentiate into specialized IECs. During this process, newly formed cells move from the crypt towards the villus tip. IECs that reach the villus tip undergo cell death, and are shed off and replaced. The proliferation, differentiation, and cell death processes of IECs are regulated by gradients of ligands of the Wnt, the BMP, the Notch, and the ephrin signaling pathways (6–8). The following paragraphs will briefly describe the various cell epithelial types and their biological functions (Figure 1).
Tuft cells are chemosensory cells characterized by their unique bottle-shaped morphology with brush-like apical microvilli. Tuft cells express taste receptors on their surface, such as TRPM-5. They utilize components of the “taste receptor” signaling cascades, and are the source of IL-25, a cytokine that acts on innate lymphoid cells (ILC) 2 and natural killer T (NKT) cells, and contributes to anti-helminth immune responses (9). Enteroendocrine cells (EECs) produce a multitude of hormones, neurotransmitters and neuropeptides that in turn regulate gut motility, digestion, food absorption, and insulin secretion. The function of EECs is modulated by nutrients and microbiota metabolites, such as short-chain fatty acids (SCFAs) (10). Mucin secretion by goblet cells is the source of mucus production (11, 12). Mucins are glycoproteins consisting of a core protein and O-linked glycans and can be subdivided in gel-forming mucins and transmembrane mucins. Gel-forming mucins are characterized by homo-dimerization and the formation of networks. The predominant component of mucus in the intestine is the gel-forming mucin Muc2. The biological relevance of Muc2 was demonstrated in Muc2-knockout mice which develop adenocarcinomas and colorectal cancer (13). The frequencies of goblet cells increase from the small intestine to the colon, where approximately 25% of all IECs are goblet cells. Consequently, the mucus thickness differs between the small intestine and the colon, with a thickening of the mucus in the colon (14). Paneth cells are localized in the crypts of the small intestine but are absent in the colon. Their biological function is the secretion of anti-microbial peptides (AMP) to the gut lumen. AMPs include, amongst others, defensins, lysozyme, secretory phospholipase A2, and RegIII (15, 16). Defensins can insert into the bacterial membranes where they form pores and thereby, disrupt the membrane or metabolic processes (16). RegIII proteins bind peptidoglycans on gram-positive bacteria and damage their cell wall (17).
The intestinal epithelium constitutes the interface between the gut lumen which contains bacteria, their metabolites, nutrients as well as food antigens on one side, and the LP containing immune cells on the other side. IECs together with intra-epithelial lymphocytes (IELs) and immune cells within the LP are involved in sensing and transporting antigens to the Peyer`s patches (PPs) and the mesenteric lymph nodes (mLNs) and maintaining epithelial integrity (18). The majority of IELs are specialized T cells that are localized within the epithelium. IELs express C-C chemokine receptor (CCR) 9 as well as integrin αE (CD103) chain in combination with the integrin β7 chain on their cell surface (19). Integrin αEβ7 mediates the binding to E-Cadherin and contributes to the retention of IELs in the epithelium. IELs function as sentinels and support the homeostasis of the epithelium and its integrity. Moreover, CD103+ dendritic cells (DCs) are recruited to the epithelium and are instrumental in antigen sampling by forming protrusions through the epithelium to capture antigen from the gut lumen. Antigens are subsequently processed, and peptide fragments derived from the antigen are presented on MHC II molecules on the cell surface. Antigen-presenting DCs migrate to the PPs and mLNs to activate antigen-specific T cells (20).
Antigen transport to the LP is also mediated by M cells in collaboration with DCs as well as Macrophages that closely interact with them. M cells are located in so-called follicle-associated epithelium adjacent to the PPs and isolated lymphocyte follicles (ILFs) (21). M cells are instrumental in luminal antigen sampling and transport. Their specific structure enables the close contact with DCs and macrophages which take up and process antigens, and subsequently present antigen peptide fragments of the antigen on their MHC II molecules. Antigen-presenting CD103+ DCs migrate to the PP or mLNs to prime antigen-specific T cells. PPs as well as mLNs are secondary lymphoid organs and are structurally divided into a B cell and a T cell zone. B cells residing in PPs and mLNs bind antigen via their B cell receptor (BCR) and are activated upon cognate interaction with antigen-specific T cells. As a result, activated B cells proliferate, undergo class switch recombination (CSR) to Immunoglobulin (Ig)A and somatic hypermutation (SHM) induced by Activation-induced cytidine deaminase (AID) within the germinal center (GC) reaction in PPs and mLNs (Figure 1) (22–24). IgA plasma cells and IgA memory B cells derive from activated B cells in the GC reaction. Furthermore, IgA plasma cell generation also occurs in isolated lymphoid follicles (ILFs) in the LP (25, 26). Class switch to IgA is triggered by the cytokine transforming growth factor-β (TGF-β) in cooperation with the vitamin A metabolite retinoic acid (RA). TGF-β is produced by various cell types, including regulatory T cells (Tregs), follicular T helper (TFH) cells, DCs, eosinophils, and also B cells (23, 27–29). Furthermore, DCs located in the PPs and mLNs express the enzymes ALDH1 and ALDH2 that are involved in RA generation (29, 30). Moreover, IL-21 provided by TFH cells enhances the CSR to IgA (27). Besides T cell-dependent IgA class switch, T cell-independent CSR to IgA has also been described (22, 31). Grasset and colleagues demonstrated that signals triggered by the transmembrane activator and CAML interactor (TACI) receptor on B cells induce CSR to IgA in the absence of T cells (32).
Microbial sensing by IECs is mediated by multiple surface and intracellular Toll-like receptors (TLR) (33). In response to microbial sensing through TLRs, IECs promote the homeostasis of the epithelium and its integrity by promoting cell survival and repair. Furthermore, the microbiota – IEC interplay regulates mucus and AMP-secretion. Moreover, upon TLR activation, IECs produce crucial cytokines and chemokines that orchestrate immune responses, such as chemokine (C-C motif) ligand (CCL) 25, CCL28, a proliferation-inducing ligand (APRIL), B cell-activating factor (BAFF), IL-25, RA, TGF-β as well as thymic stromal lymphopoietin (TSLP) (34, 35). Furthermore, the cytokines TSLP, TGF-β and RA induce DCs and macrophages to provide tolerogenic signals, such as the secretion of IL-10 (36, 37). Homing of immune cells is promoted by TGF-β and RA as both cytokines were shown to be implicated in the upregulation of integrins β7 and αE on immune cells. Moreover, RA was also shown to upregulate the gut-homing receptor CCR9 on T and B cells (38, 39). In addition, TGF-β induces integrin αE expression in T cells, especially in CD8+ tissue-resident T cells (TRM) (40–45). Moreover, TGF-β, RA, and nitric-oxid (NO) produced by IECs are key cytokines for the induction of IgA CSR of B cells (39, 46–54). Importantly, BAFF and APRIL produced by IECs support B cell and plasma cell survival, respectively (55–57). In this context, IEC-derived TSLP also fosters additional APRIL and BAFF production by DCs (58).
Immune cell homing to the intestinal LP is controlled by the timely and spatially coordinated action of specific selectins, chemokine receptors and integrins (59). Gut homing requires the expression and activation of integrin α4β7 which binds to its ligand mucosal addressin cell adhesion molecule-1 (MadCAM-1) on endothelial cells in the high endothelial venules (HEVs) in the PPs and the mLNs, as well as on post-capillary venules in the intestinal LP (60–64). MadCAM-1 is also expressed in lactating mammary glands, the spleen and the bone marrow (65–72). In addition to integrin α4β7 expression, homing to the PPs and the mLNs requires L-Selectin (CD62L) (60, 73). Homing to the gut LP is orchestrated by the chemokines CCL25 and CCL28 and their corresponding chemokine receptors CCR9 and CCR10 on immune cells (Figure 2) (35, 74). IECs are the source of the chemokines CCL25 and CCL28 and are therefore crucial for the recruitment of immune cells to the LP, including IgA plasmablasts (35, 74). The critical role of CCR9 was demonstrated in CCR9-deficient mice, in which a severe impact on IgA plasmablast homing to the small intestine was observed (75). In addition to CCR9, CCR10 and its ligand CCL28 contribute to IgA B cell and IgA plasmablast homing. Surprisingly, CCR10-deficient mice had normal serum and fecal IgA levels and exhibited only slight alterations in IgA-secreting cell numbers in the gut LP. However, a striking reduction of IgA-secreting cells was detected in the lactating mammary gland, demonstrating that CCR10 plays a critical role in mammary gland plasmablast homing (76).
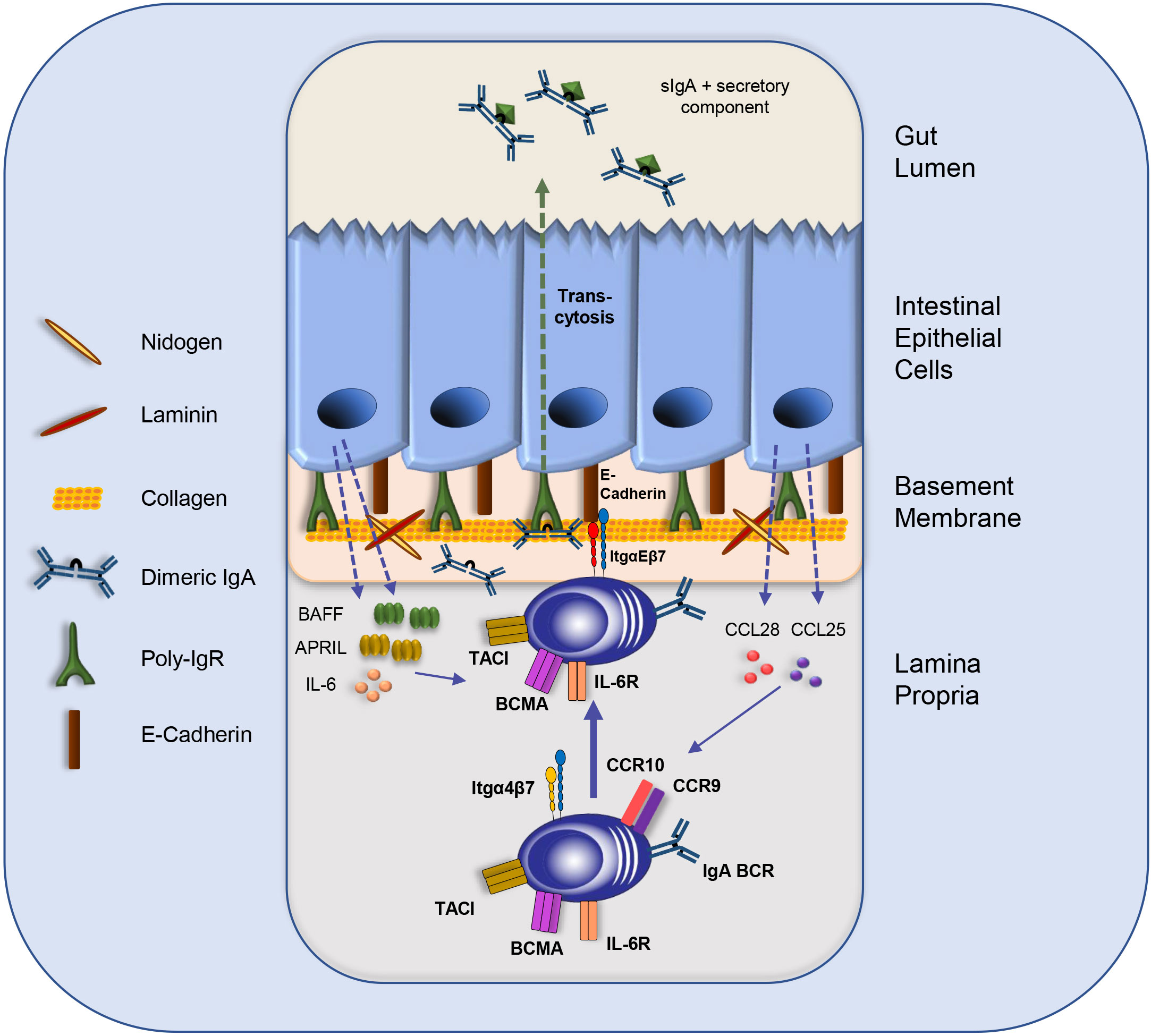
Figure 2 The intestinal IEC-IgA plasma cell niche. IgA plasmablast homing to the LP is directed by integrin α4β7 and the chemokine receptors CCR9 and CCR10. Intestinal epithelial cells (IECs) secrete CCL25 and CCL28 to attract IgA plasma cells and other immune cells. IECs produce the survival cytokines IL-6, BAFF and APRIL. Moreover, the direct interaction of IgA plasma cells with IECs is mediated by the binding of integrin αE to E-Cadherin, which might support plasma cell retention and facilitate the transcytosis of dimeric IgA to the gut lumen via binding to the p-IgR on IECs. Furthermore, IgA-secreting plasma cells might interact with components of the basement membrane, such as Collagen type IV and other extra cellular matrix (ECM) compounds. BCR: B cell receptor; IL: Interleukin; Itg: integrin; sIgA: secretory IgA.
We have identified the transcription factor Krüppel-like factor 2 (KLF2) as a key regulator of integrin α4β7 and CCR9 expression and consequently as an essential regulator of IgA plasmablast homing to the intestinal LP. B cell-specific deletion of KLF2 resulted in reduced integrin β7 and CCR9 expression and subsequently in a severe reduction of IgA plasmablasts/plasma cells in the LP concomitant with reduced IgA in the serum and in the gut lumen (77, 78). Besides CCR9 and integrin β7, KLF2 also activates L-Selectin (CD62L) expression in B cells and IgA plasma cells (77–79). Of note, KLF2 directly binds to the promoter of the integrin β7 gene and activates its expression (80). Deletion of integrin β7 in mice resulted in smaller PPs and fewer IELs, reduced numbers of IgA B cells, IgA plasma cells and CD4+ T cells in the LP due to impaired homing (81). Moreover, deregulation of the integrin β7 chain was also found in Kmt2d-defienct mice (82), a model for the Kabuki syndrome. The abundance of integrin β7 was reduced on Kmt2d-deficient B cells and consequently, Kmt2d-deficient mice displayed decreased serum IgA levels, smaller PPs and reduced numbers of IgA-secreting cells (82).
The intestinal epithelium is the source of the key chemokines CCL25 and CCL28. Both are crucial for attracting immune cells, including B cells and plasmablasts, to the LP (Figure 2). Furthermore, direct interactions between epithelial cells and plasma cells were observed. IgA plasmablasts in the LP can be divided into different subsets according to their expression of α4β7 and αE(CD103)β7 integrins. In a recent study, Guzman and colleagues revealed that a subpopulation of IgA plasmablasts in the LP express integrin αEβ7 allowing them to interact physically with IECs via E-Cadherin (Figure 2). This intimate plasma cell-IEC interaction allows the efficient transcytosis of dimeric IgA via the poly-IgR (p-IgR) on IECs to the gut lumen (83). Dimeric IgA consist of two IgA monomers that are covalently connected by the joining chain (J-chain) (84, 85). P-IgR binds dimeric IgA via its J-chain and facilitates its transport through epithelial cells by a process which is called “transcytosis” (Figures 1, 2) (22, 23, 84, 85). Upon transport to the luminal side, proteases cleave the ectodomain of the p-IgR. The ectodomain (secretory component) is released together with dimeric, secretory IgA (sIgA). The secretory component protects IgA from degradation and has immunomodulatory functions [Figure 2 (86)].
Upregulation of integrin αE is a common mechanism applied by immune cells to achieve tissue residency (e.g., DCs, IELs, tissue-resident memory T cells (18–20, 87). Induction of integrin αE expression in IgA plasma cells might be triggered by TGF-ß and RA, both of which are secreted by the IECs.
Therefore, IECs do not only orchestrate the recruitment of IgA plasmablasts to the LP, but also foster the establishment of an intimate IgA plasma cell - IEC interaction. Tissue-residency of immune cells is also regulated by inhibition of sphingosine-1-phosphate (S1P)-mediated migration to blood vessels and lymph. Tissue-resident cells upregula te CD69, which binds to the S1P-receptor 1 (S1PR1) resulting in S1PR1 inhibition and degradation (88); a mechanism that might also apply to IgA plasma cells in their intestinal niche. However, it remains unclear how IgA plasma cells establish the direct interaction with E-Cadherin on IECs in the presence of the basement membrane. Pore sizes in the basement membranes vary and can reach a diameter of up to 8 µm. Pores with sizes of 1-5 µM were frequently found in the follicle-associated epithelium, whereas large pores were mainly found in the crypt region (4). Intra-epithelial lymphocytes can migrate through these pores due to their small cell size (i.e., IELs) or form protrusion (i.e., DCs). If and how plasma cells with their cell sizes ranging from 15-20 µM can migrate through the basement membrane pores remains unclear. One possible scenario is that plasma cells form protrusions through the pores in the basement membrane to establish the binding of integrin αEβ7 to E-Cadherin.
Thus, the β7 integrin chain plays a central role in IgA plasma cells homeostasis in the gut by controlling the migration from IgA plasmablasts from the blood to the LP when bound to the α4 integrin chain, and by enabling the direct interaction of IgA plasma cells with the epithelium when paired with the αE integrin chain (Figure 2). How the expression of the αE integrin chain is induced in IgA plasma cells remains unclear. Studies with T cells suggested a role of TGF-β in inducing integrin αE expression (89).
In addition to the supportive function of dimeric IgA transport to the gut lumen, IECs also promote the survival of plasma cells. IECs and smooth muscle cells in the LP were shown to produce IL-6 (90). The role of IL-6 for plasma cell survival is controversially discussed. On the one hand IL-6 supports plasma cell generation (91–94), on the other hand, plasma cell short-term survival after transfer into IL-6-deficient mice is not impaired (95). For their generation and survival, plasma cells require the expression of cytokine receptors TACI and B cell maturation antigen (BCMA) on their cell surface (57, 96–100). BCMA might exert its pro-survival effect by upregulating the anti-apoptotic factor Mcl-1 as shown for bone marrow plasma cells (101). IECs are one source of the ligands for these survival receptors, i.e., APRIL and BAFF (56, 58). Thus, regarding their biological properties, IECs are the central component of the intestinal plasma cell niche, comparable to the stromal cells in the plasma cell survival niche in the bone marrow (55, 64). Furthermore, IEC-derived TSLP fostered production and secretion of APRIL and BAFF by myeloid cells, such as DCs (29, 58). APRIL production by intestinal eosinophils was also reported and might contribute to IgA plasma cell survival (102). Thus, IECs together with various immune cells, such as myeloid cells, T cells and eosinophils provide cytokines as well as cell-cell contact that allow IgA plasma cells to persist for many decades in the human intestine and to become long-lived in the murine intestine (103–105). Therefore, LP cells in cooperation with epithelial cells may constitute a plasma cell survival niche similar to the one described for the bone marrow. In both microenvironments, plasma cells are provided with the survival factors APRIL and IL-6 by neighboring cells. Furthermore, the integrin-mediated cell-cell contact of plasma cells with bone marrow stromal cells is a major aspect contributing to the survival of long-lived bone marrow plasma cells (55, 64, 106, 107). In the intestinal niche, IgA plasma cells also express integrins that mediate their homing to the LP as well as their adhesion to e.g., IECs via integrin αEβ7; an interaction that might support the persistence of IgA plasma cells and the long-lasting secretion of protective IgA. However, due to the high turn-over rate of IECs, the constant process of epithelial cell death, and their dynamic replacement, it remains unclear how IgA plasma cells in the proximity or in direct contact with the epithelium achieve longevity. Furthermore, it remains elusive how the plasma cell survival niche in this highly dynamic environment is preserved. Hence, kinetic studies combined with spatial analyses of IgA plasma cells in their intestinal survival niches and in different sections of the intestine (i.e., small intestine, colon, and cecum) and within different areas along the crypt-villus axis need to be performed.
The intestinal immune system can be influenced by various external factors, e.g., the diet (108). Therefore, it is more than likely that the modeling of the intestinal plasma cell survival niche and the support of plasma cell function does not depend only on the intestinal milieu itself. One transcription factor, the Aryl hydrocarbon receptor (AhR), has been described to connect nutritional intake with the microbiota composition and the immune response (109). The AhR was first discovered as a sensor for xenobiotics like polycyclic aromatic hydrocarbons (PAHs) and dioxins (110). The treatment of Hepa-1 cells with 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) induces the translocation of the AhR from the cytosol to the nucleus, followed by the induction of drug-metabolizing enzymes (111). Therefore, AhR was revealed to function as a transcription factor involved in metabolic processes and detoxification early after its discovery. Today, we know that the AhR is a member of a transcription factor superfamily mainly characterized by two motifs, a basic N-terminal helix-loop-helix (bHLH) and a Per-Arnt-Sim (PAS) domain with two subunits (PAS-A and PAS-B) (112). While the bHLH domain allows dimerization of the protein and its binding to DNA, the PAS domain located at the C-terminal end of the bHLH domain is necessary to bind other PAS-proteins like the aryl hydrocarbon receptor nuclear translocator (ARNT) and the chaperone family member heat-shock protein 90 (HSP90) (113, 114). Essential and unique for AhR among the bHLH/PAS superfamily members, its PAS-B subdomain encodes for a ligand binding side specific for “all classes of dioxin receptor ligands”, which partially overlaps with the HSP90 binding side (115). The nuclear localization and the nuclear export signal can be found at the N-terminal side of the AhR protein, respectively (116). In contrast, the C-terminal region contains a glutamine-rich transactivation domain (TAD), which is essential for binding the AhR to its transcriptional co-activators and, therefore, for the induction of its target genes transcription (117). In the absence of AhR ligands, HSP90 binds to the PAS domain, thereby restricting its entrance to the nucleus and retaining AhR in an inactive conformation in the cytosol. Ligand binding to the PAS-B domain of the AhR in the cytosol results in the dissociation of HSP90, where after the AhR ligand complex translocates into the nucleus, binds to various transcriptional co-activators and induces its target gene expression (118). Important AhR-targets are the enzyme Cyp1a1 that degrades xenobiotics serving as AhR ligands (119), and the AhR repressor (AhRR) that suppresses AhR-activity by competing for the binding to the AhR co-activator ARNT (120). Both mechanisms result in a feedback loop regulating AhR activity. AhR-ARNT complex binding to the xenobiotic response element (XRE; also dioxin response element, DRE) in the target gene promotor region and its mediated gene expression is defined as the canonical AhR signaling (121–123). However, more recent studies demonstrated that AhR can interact with a variety of co-activators in a non-canonical signaling pathway, depending on the co-activator’s availability, and presumably the nature of the activating ligand (124, 125). Therefore, all studies addressing the AhR function by activation or inhibition through specific ligands must be compared carefully to allow reliable conclusions.
Among the initially described toxic chemicals functioning as AhR ligands, a vast number of naturally occurring (126) and synthesized AhR agonists and antagonists have been described (125, 127). The number of the latter is steadily increasing with the rising interest in AhR as a drug target in various diseases, including cancer, rheumatoid arthritis and inflammatory bowel disease (125). The interplay between the dietary composition, the variety of diet-derived AhR ligands generated by individual members of the intestinal microbiota or liver enzymes and the transport of these compounds through the organism to the site of action is incredibly complex and differs between individuals. Therefore, the prospect of treating diseases by directly manipulating AhR-activity through specifically designed ligands administrable as drugs seems promising. However, the possibility of treating or preventing diseases or even supporting our immune defenses with a diet rich or low in AhR ligands is tempting, especially as a mixture of AhR ligands can result in an altered AhR-functionality compared to their individual activity (128).
A wide range of naturally occurring, exogenous AhR ligands are generated during the metabolism of tryptophan or glucobrassicin found in vegetables (129–131). Metabolism of tryptophan by individual microorganisms of the intestinal microbiota results in the production of the AhR agonists indole acetic acid (IAA), indole-3-acetaldehyde and indole-3-aldehyde (IAld) (132–134). In addition, a very potent AhR agonist is the tryptophan photoproduct 6-formylindolo [3,2-b] carbazole (FICZ), which is generated by UV-radiation (135). In contrast to tryptophan, glucobrassicin from cruciferous vegetables of the family Brassicaceae is already metabolized in the oral cavity by myrosinases, resulting in the production of the AhR agonist precursor indole-3-carbinol (I3C) and indole-3-acetonitrile (I3ACN) (136). In the stomach, non-enzymatic acid condensation of I3C and I3ACN produces chemical compounds, such as 3,3’ diindolylmethane (DIM), 2-(indol-3-ylmethyl)-3,3’ diindolylmethane and indolo[3,2-b]carbazole (ICZ), that can function as AhR ligands (131, 137–139).
Diet-derived short-chain fatty acids (SCFAs) like butyrate can also induce AhR and AhR target gene expression in human IECs (140). This is particularly interesting as SCFAs are discussed for preventing or curing different diseases (141). In addition, by consuming stimulants like coffee (caffeine) or tobacco (nicotine), the AhR can be activated in the corresponding organs, while flavonoids found in, e.g., tea function mainly as AhR antagonists (142–144).
Even though studies of human and murine blood plasma showed biologically active concentrations of diet-derived AhR ligands that can potentially control AhR activity in distinct organs (145–148), the digestive tract is the first responding organ that is in contact with the highest concentrations of AhR ligands due to the primarily oral uptake of AhR ligands or their precursors.
AhR protein abundance has been identified in various cell types, including a wide range of immune cells as well as cells functioning at barrier sites like the lung, skin or intestine (149). Upholding intestinal homeostasis requires a delicate interplay between the microbiota commensals, the IECs, and immune cells found in the LP or IELs, with the majority expressing AhR in varying abundance (150). The analysis of an AhR-reporter mouse model revealed widespread expression of AhR in the intestinal epithelium with a proximal-distal gradient in the small intestine (151). A weaker and mosaic-structured AhR expression was detected in the colon of the same mice, indicating a more cell-type selective AhR function. Of note, the AhR gradient in the intestinal tract is comparable to the observed IgA gradient (1, 2). Therefore, AhR signaling may play a critical role in the intestinal plasma cell niche.
Of note, depending on the sensitivity of the used method, a gene expression analysis in whole organs does not always allow reliable distinguishing between different cell types. For this reason, more specific and detailed studies are required to identify the cell types in the intestinal tract that express AhR and to reveal how these cells are influenced by AhR activity. Therefore, advanced technologies like spatial transcriptomics will help to solve the remaining questions about the intestinal plasma cell niche and the functional relevance of AhR signaling therein.
However, several studies addressed the role of AhR, or more precisely of individual AhR ligands, for the intestinal epithelial barrier. One study revealed that FICZ supplementation prevents the intestinal permeability caused by damage of the epithelial layer due to decreased tight junction stability between epithelial cells during dextran sodium sulfate (DSS)–induced murine colitis (152). The authors demonstrated that FICZ-induced AhR signaling in a Caco-2 cell monolayer suppressed NF-kB p65 signaling and thereby protected against the tumor necrosis factor (TNF)-α/IFN-γ-mediated reduction in tight junction protein. However, FICZ-mediated rescue of DSS-colitis in vivo may not exclusively be attributed to the intrinsic role of AhR in IECs, as the activation of AhR also affects immune cells in the intestinal tissue (153). Supporting the importance of AhR in IECs, AhR-deficient mice or mice lacking AhR specifically in IECs were more susceptible to infection with the gram-negative bacterium Citrobacter rodentium (154–156). This phenotype could be re-produced by constitutive expression of the AhR-target Cyp1a1 (R26Cyp1a1-mice) (109). In this mouse model, constant degradation of diet-derived AhR ligands by Cyp1a1-overexpression efficiently inhibited AhR signaling in IECs, resulting in an impaired immunity to C. rodentium and, thereby, an increasing systemic burden. However, the dietary supply of the AhR agonist precursor I3C enabled sufficient clearance of C. rodentium and rescued R26Cyp1a1-mice.
Bacterial invasion of the LP does depend on the permeability of the IECs. Still, it is also controlled by the thickness of the intestinal mucus layer and its abundance of AMPs (Figure 1). Therefore, an increased bacterial burden in AhR-deficient mice during a C. rodentium infection may also be caused by a diminished intestinal mucus layer. This hypothesis is supported by the observation that AhR is required for the differentiation of secretory cells in the intestinal epithelium, thereby enhancing the resistance to enteropathogenic Escherichia coli (157). In detail, activation of AhR by L-Kynurenine represses Notch1 signaling in murine intestinal cells, thereby allowing indoleamine 2,3-dioxygenase 1 (IDO1)-mediated promotion of differentiation into goblet and paneth cells. Furthermore, IEC-specific AhR-deficiency restricted stem cell differentiation to epithelial cells, e.g., goblet cells, while stem cells proliferated uncontrollably, resulting in an increased tendency to malignant transformation (154).
Studies in human colorectal cancer showed that upregulation of Wnt signaling at the bottom of the intestinal crypts through mutations in the Wnt-pathway component genes adenomatous polyposis coli (APC), β-catenin and/or AXIN2 causes this pathogenicity (158). Therefore, it is of particular interest that, besides its functions as a transcription factor, AhR was shown to support ubiquitination and, thereby, degradation of β-catenin by participating in an E3 ubiquitin ligase complex (159, 160). In addition, analyzing mice lacking AhR specifically in IECs demonstrated that dietary-induced AhR signaling prevents intestinal tumorigenesis by inhibiting the Wnt/β-catenin signaling in intestinal stem cells through induction of the transcription of E3 ubiquitin ligases (154). Furthermore, AhR was shown to be indispensable for the termination of the regenerative response in IECs after an injury (161). Here, the authors analyzed colonic organoid cultures under specific media conditions that either simulate differentiating or regenerative conditions. They demonstrated by RNAseq analysis that AhR regulates critical factors involved in the regenerative response or the reacquisition of intestinal identity post-injury. Interestingly, AhR signaling was also shown to control the differentiation of hematopoietic stem cells into lymphocytes (162). In addition, AhR can directly function as a tumor suppressor of acute myeloid leukemia by suppression of self-renewing of leukemia stem cells (163).
PCB 126, formerly produced as a lubricant in electronic equipment (164), is a very toxic AhR ligand (165). Treatment of human breast epithelial cell cultures with a very low dose of PCB 126 resulted in a significant reduction of CCL28 mRNA levels (166). As the parallel treatment with an AhR inhibitor nullified the observed phenotype, the authors concluded that AhR activation by PCB 126 inhibits CCL28 expression in an AhR-dependent manner. CCL28 secretion by IECs is associated with the homing of IgA plasmablasts to the small intestine (64, 74) (Figure 2). Therefore, it should be considered as an AhR-controlled mechanism associated with antibody-secreting cell (ASC) migration in the gut. Of note, PCB126 treatment also altered the microbial community structure in the murine intestines (167).
Additional studies in AhR- or IL-10-deficient mice demonstrated that treatment with the Braccicacea-originating AhR ligand indole-3-carboxaldehyde (ICA) promotes goblet cell differentiation and proliferation of IECs through IL-10 in an AhR-dependent manner (168). It is well known that aging results in changes in the intestinal epithelium composition that involves the reduction of goblet cells, thereby, a decline in the epithelial barrier integrity and increased risk of inflammation. However, early colonization with bacteria known for their capacity to produce indoles, dampens this age-related phenotype. Furthermore, another xenobiotic AhR ligand called 2,3,7,8-tetrachlorodibenzofuran (TCDF) shifted the ratio of Firmicutes to Bacteroidetes, a change in the microbial community which was associated with the development of intestinal diseases (169). As the described phenotypes did not occur in AhR-deficient mice, the authors concluded that they are AhR-dependent.
In another study, the analysis of liver cells from TCDD-treated mice showed that AhR can potentially induce the expression of E-Cadherin when interacting with KLF6 as a co-activator (170). As E-cadherin may be necessary for cell-cell interaction between IECs and lymphocytes in the intestine (83), this mechanism has to be considered when analyzing the role of AhR in the intestinal plasma cell niche. However, functional studies are still missing, and it must be determined whether AhR interacts with KLF6 to induce E-Cadherin in IECs.
Interestingly, mice suffering from 2,4,6-trinitrobenzenesulfonic acid (TNBS)-induced colitis showed significantly less damage in colonic tissue and reduced infiltration of inflammatory cells when treated with the AhR ligand TCDD prior to disease onset (171). Surprisingly, the treatment with TCDD led to an increased mRNA abundance of the plasma cell survival factor APRIL in colonic tissue three days after TNBS-induction. As the colon harbors a vast variance of cell types, the localization of the exact APRIL source seems challenging and could be addressed by spatial transcriptomics. However, in vitro LPS-activated IECs showed a profound increase in APRIL and BAFF mRNA when treated with TCDD. Therefore, TCDD-induced AhR signaling in IECs may support the viability of ASCs in the intestinal survival niche by inducing the secretion of the survival factors APRIL and BAFF (Figure 2).
Based on these data, it is out of the question that AhR signaling plays a vital role in IECs. However, if AhR-regulated processes in IECs are essential for the intestinal plasma cell niche and healthy IgA secretion remains unanswered.
Mice with a CD11c-Cre-mediated specific deletion of AhR in DCs suffer as much from DSS-induced colitis as AhR-/- mice (172). Surprisingly, the development of the intestinal epithelium was disrupted in these mice, resulting in shorter villi, reduction in paneth cells, but an increase of goblet cell numbers per villus. Therefore, AhR signaling in DCs may be essential for the intestinal plasma cell niche. Interestingly, AhR-deficient CD11c+MHCIIhigh DCs in the mLNs expressed less integrin αE (CD103), potentially affecting their migration behavior in the intestinal tissue and indicating that CD103 is a potential AhR target gene. As CD103 might be involved in IgA plasma cell homing to the intestinal tissue and their interaction with IECs, this mechanism is of potential interest when determining their intestinal survival niche. Of note, IgA levels did not differ between the feces of mice lacking AhR in DCs and of control animals.
Culturing human monocytes in a specific differentiation culture system was shown to induce differentiation into monocyte-derived (mo-) macrophages and mo-DCs which resemble those found in vivo (173). Interestingly, under these culture conditions FICZ-induced AhR signaling favored the differentiation into mo-DCs by directly inducing the expression of the transcription factor Blimp-1. As Blimp-1 is the key transcription factor driving B cell differentiation into ASCs, an AhR-Blimp-1 axis would be particularly interesting for the formation of IgA plasma cells.
AhR-deficient LPS-activated macrophages secreted elevated amounts of IL-6, IL-12, and TNF-α (174). In detail, AhR mediates inhibition of IL-6 promotor activity by forming complexes with NFκB and signal transducer and activator of transcription 1 (Stat1). Furthermore, LPS-induced inflammation of bone marrow stromal cells in vitro results in an increase of IL-6 (175). However, this was abolished when cells were pre-treated with the AhR ligand TCDD. As IL-6 supports human long-lived plasma cells, AhR-controlled IL-6 production in the intestinal environment could play an important role in the intestinal plasma cell survival niche (93).
Nutrition studies demonstrated that a dietary lack of the AhR ligand precursor tryptophan alters the gut microbiota and results in an impaired intestinal immune system (176). Especially tryptophan degradation to indole-3-aldehyde by Lactobacilli induces AhR signaling in ILC3s, thereby promoting the production of IL-22 (132), an essential mechanism described in the immune response to Citrobacter rodentium (177). Furthermore, IL-22 is indispensable for a functional intestinal epithelial barrier, as it mediates the differentiation of intestinal stem cells into IECs and supports their viability (178). Thereby, IL-22 guarantees mucosal protection from damage and the repair of defects in the epithelial barrier integrity. Interestingly, IL-22 is expressed in the small intestine but barely in the colon (179), forming a gradient along the intestinal tract comparable to the one for AhR expression and IgA density. Thus, AhR-mediated IL-22 production may be beneficial for IgA plasma cell longevity in the intestine.
In contrast, the numbers of IL-5 and IL-13-secreting ILC2 cells were significantly elevated in AhR-deficient mice (180). Authors claimed that the AhR is the regulating factor that balances the intestinal ILC2 and ILC3 populations and, thereby, the intestinal immune homeostasis with an appropriate response to acute infections. As the function of ILCs in the intestinal tract is out of the question, it seems obvious to assume that they are also an AhR-regulated part of the intestinal plasma cell niche.
A variety of T cell subpopulations express AhR (181). However, there are contradicting reports about the abundance and importance of AhR in the same T cell subpopulations. AhR was shown to induce IL-22 production in CD4+ T cells by interacting with Stat3 as a co-activator (182). IL-22 is secreted by Th17 and Th22 cells, both of which require the activity of the transcription factor RORγt for differentiation (183–185). Of note, more recent studies revealed that AhR and RORγt can directly interact as a transcription factor complex (186) and are a potential target for treating lupus erythematosus (187). However, in vitro-activated T cells, primed for differentiation into Th17 cells, showed even further elevation of IL-22 production when treated with the AhR agonists β-naphthoflavone and FICZ (188). In strict contrast, AhR-/- mice showed increased numbers of Th17 cells and elevated secretion of the cytokines IL-22 and IL-17 compared to AhR+/- mice after imiquimod-induced psoriasiform skin inflammation (189). With IL-22 being a critical cytokine in the intestinal environment, the AhR-T cell interaction must be kept in mind when analyzing the role of nutritional AhR ligands and their impact on the intestinal IgA plasma cells niche.
Analysis of a Foxp3Yfp-Cre reporter mouse demonstrated that Tregs located in the spleen and mLNs showed less AhR expression than intestinal Treg cells (190). The authors of this manuscript also pointed out that AhR has a cell-intrinsic role in intestinal Tregs. They demonstrated that AhR-deficient intestinal CD4+TCRβ+Foxp3+ Tregs lack the expression of CD103 and the chemokine CCL20, which is involved in cell migration. CD103 expression in Tregs is important for their activation and retention at the site of inflammation and CD103 can also be found on IgA plasma cells (83). Furthermore, mice treated with the AhR agonist FICZ expressed more CD103 in intestinal Tregs than control-treated animals, implying CD103 as a potential AhR target gene. As AhR-deficient intestinal Tregs express more inflammatory cytokines like IL-17 and IFN-γ, this study demonstrated the importance of AhR signaling in Treg cells for intestinal homing, and its requirement for their immunosuppressive function in colitis. T cells as a part of the intestinal micro-milieu regulate inflammatory responses and also the differentiation of epithelial cells. Therefore, T cells may undoubtedly be an AhR-controlled part of the intestinal plasma cell niche.
Various studies addressed the effect of AhR ligand treatment on B cells (153). Few of them focused on the intrinsic function of AhR in B cell activation and differentiation, and none discussed the role of AhR in intestinal plasma cells. However, the results of these studies are controversial, claiming either a supportive function of AhR during B cell activation or the exact opposite, i.e., inhibition of the B cell response. A study comparing activated human and murine B cell cultures claimed that TCDD treatment diminished IgM secretion in vitro, independent of the species (191). However, intracellular IgM abundance was reduced in activated murine B cells, but increased in human B cells after TCDD treatment. This indicates that TCDD: AhR inhibits the formation of IgM-secreting cells, even though the data are partially contradicting. There may also be ligand- or species-depending differences in AhR function in B cells.
Investigations of the role of AhR signaling in the B cell response in vivo using an adoptive transfer model demonstrated that AhR-/- mature B cells are outcompeted by AhR+/+ cells in the bone marrow and mLNs of mixed bone marrow chimeras (192). Even more interesting, challenging a recipient mouse reconstituted with AhR-/- and AhR+/+ splenocytes revealed that AhR-deficient B cells expanded less. Of note, B cell-specific AhR-deficient mice showed altered steady state serum Ig titers compared to controls, with elevated IgM but diminished IgG1 concentrations. IgA serum titers were unaltered. ASC numbers were reduced in the spleen of the same mice, but remained unchanged in their bone marrow. These data indicate a function of AhR in ASC formation, but very likely not in their maintenance in the bone marrow. Unfortunately, this study failed to determine ASC formation and maintenance in other organs or niches, e.g., the intestinal tract or their migration behavior. Even though IgA serum titers were unvaried in these mice, changes in intestinal plasma cells or the amount of mucus secreted IgA was not addressed and remains unclarified. However, this study indicates that AhR signaling may also be necessary for the formation of intestinal plasma cells.
One study analyzing B cell-specific AhR-deficient mice demonstrated that AhR signaling in regulatory B cells (Bregs) induces IL-10 secretion, thereby, dampening inflammatory Th1 and Th17 responses in a murine rheumatoid arthritis model (193). A more recent study of the same group indicates that AhR signaling induced by dietary SCFA butyrate supplementation favors the formation of Bregs during GC response and, thereby, alleviating inflammation in the cause of rheumatoid arthritis (194). In sharp contrast to the previously mentioned study (192), the authors claimed that butyrate-induced AhR signaling in B cells suppresses GC formation and plasmablast differentiation. But even though butyrate treatment reduced the numbers of CD19+ plasmablasts in the spleen of control animals, there was a significant increase in this population detected in butyrate-fed B cell-specific AhR-deficient mice. In contrast, numbers were unaltered between standard-fed knock-out and control animals. As B cells lack AhR in this model, why did butyrate still affect plasmablast numbers in the knock-out mouse? In addition, the population defined as Bregs showed a severe upregulation of the plasma cell signature transcription factor Blimp-1 (prdm-1 gene) when lacking AhR and butyrate feeding markedly reduced Blimp-1 expression independent of the genotype. This gene expression profile questions the nature of the analyzed cell population, especially as plasma cells can also secrete IL-10 in autoimmune inflammation (195) and express high amounts of CD24 (196, 197). Of note, the analysis of the B cell response in the intestinal tract was missing in this study.
Due to contradicting data, the role of AhR signaling in B cell activation and plasma cell differentiation is still unresolved. Especially the function of AhR in the intestinal ASC pool, and their migration behavior remains completely unclear.
In addition to several studies that described a potential function of AhR signaling in B cells, there are also analyses addressing a connection between AhR activity and IgA production.
Analyzing the effect of TCDD treatment during viral airway infection with Influenza A demonstrated an overall immunosuppressive phenotype (198). Interestingly, virus-specific IgG and IgM levels were reduced in the blood plasma of TCDD-treated infected mice, while antigen-specific IgA was 4-fold increased. In contrast, analyzing broncho-alveolar lavage fluid revealed that IgA was unaltered at the side of infection, even so, IgG levels were again diminished after TCDD application. Of note, all mice in this study were female and observations may reflect a gender-specific phenotype. Even though this data may indicate a connection between AhR signaling and IgA production, the authors failed to determine whether this phenotype was associated with a B cell-intrinsic AhR signaling or a secondary effect due to the overall observed alterations in T cells and cytokine production. Furthermore, organ-specific alterations in IgA levels indicate a differential role of AhR activity for secreted, mucus-associated IgA and systemic IgA in the blood. This contradicts a potential function of AhR signaling in p-IgR-mediated IgA transport through lung epithelial cells into the mucus. However, whether the same is true for the intestinal tract remains unanswered.
Research focusing on the formation of IgA-secreting cells in the intestine revealed that feeding wildtype C57Bl/6 mice with the AhR ligand TCDD alters fecal IgA concentrations independent of the circadian rhythm (199). However, the authors did not clarify whether the observed phenotype is mediated by a B cell-intrinsic AhR function or associated with AhR activity in the surrounding cells e.g., IECs. Furthermore, TCDD treatment elevated fecal and serum IgA levels in male mice independent of the analyzed dose. However, female mice showed reduced IgA levels when treated with low doses of TCDD, while results were comparable to their male counterparts during high-dose TCDD treatment. Notably, hormonal changes can affect IgA secretion (200). Regarding the female menstrual cycle, the AhR can recruit the estrogen receptor α to the promotor region of AhR-regulated genes (201) or even to the estrogen response elements (202). Therefore, gender-specific variances in AhR ligand-induced IgA secretion are not surprising and must be addressed in future experiments.
In support, AhR ligand 2,3,7,8-tetrachlorodibenzofuran (TCDF)-fed male wildtype mice showed increased fecal IgA abundance and elevated concentrations of inflammatory cytokines in the ileum (169). Surprisingly, LPS abundance in the blood serum was significantly increased in TCDF-treated mice, indicating diminished intestinal barrier integrity and increased translocation of bacteria from the gut lumen into the LP and adjacent organs. These data oppose previously described studies which showed that FICZ-mediated AhR activity in IECs strengthened intestinal barrier function by increasing tight junction protein production (152). This conflicting data may be caused by the authors missing to address the question if the TCDF-mediated disruption of the intestinal barrier is AhR-dependent or caused by the very toxic nature of the ligand. In addition, it has to be considered that altered IgA levels in TCDF-fed mice could be triggered by increasing bacterial infiltration and, therefore, may not depend on TCDF-induced AhR activity in ASCs.
A study assessing TCDD-mediated AhR signaling and its influence on the course of Crohn`s disease demonstrated faster recovery of TCDD-fed mice from 2,4,6-trinitrobenzenesulfonic acid (TNBS)-induced colitis, which was associated with increased numbers of Foxp3+ Tregs in the intestines (171). Interestingly, IgA concentrations significantly increased in feces and in homogenized colon tissue after TCDD treatment. This phenotype was independent of TNBS-induced colitis but mediated by AhR activity, as IgA levels were comparable between TCDD- and control-fed AhR-/- mice. As mentioned before, TCDD treatment of LPS-activated IECs led to elevated APRIL and BAFF mRNA abundance, cytokines known to support B cell and ASC viability, respectively. In addition, TCDD-induced AhR signaling in IECs or intestinal immune cells could trigger the production of cytokines that induce or support ASC differentiation. Therefore, augmentation of IgA levels in TCDD-treated mice may rather be attributed to AhR signaling in other cell types and not B cell or ASC intrinsic.
A unique study among AhR-focused research projects determined the influence of AhR activity on IgA production in neonatal mice (203). Therefore, researchers compared AhR-/- mice and wildtype mice whose parents were already fed with a standard diet or an AhR ligand-free diet, which was continued after birth. Two-week-old AhR-/- mice showed no fecal IgA, while their serum IgA levels were comparable to wildtype mice fed with a standard diet. In contrast, an AhR ligand-free diet abolished serum IgA in wildtype mice but did not affect fecal IgA concentrations. Furthermore, the authors claimed that IgA abundance in the gastric content were comparable between animals during the nursing period, indicating no influence of maternal IgA on the observed phenotype. However, a direct diet-mediated effect on 2 week-old suckling mice is hard to imagine. Therefore, it has to be considered that other components in the maternal milk besides IgA itself may contribute to the observed differences in the IgA concentrations in the different groups of neonatal mice. Interestingly, depending on the available diet for nutrient intake, eight-week-old weaned mice did not show any differences in serum IgA concentrations. In contrast, only mice fed with an AhR ligand-free diet lacked fecal IgA, likely caused by reduced CD19+B220+ B cell numbers in the GALT. Even though the authors provided a unique set of data analyzing the influence of a diet or AhR signaling in neonatal mice, the results of this study are contradicting. As the observed alterations in IgA concentrations differed between AhR ligand-free diet-fed mice and AhR-/- mice, these results implicate an AhR-independent effect of the diet on the intestinal immune system. As obviously all cells of an organism are affected by the deficiency in AhR-/- mice or mice fed with a specific diet, their analysis does not allow conclusions about the B cell-intrinsic role of AhR in the formation of IgA-secreting cells or IgA-secretion itself. However, the differences between fecal and serum IgA concentrations in mice of the same genotype or diet-fed group imply a differential AhR function for secreted mucus-associated IgA and serum IgA, again.
Regarding the significant number of studies addressing the AhR function, it was often neglected that TCDD is a toxic substance causing oxidative stress, DNA damage and endocrine disruption amongst other symptoms (204–207). Until now, it is just partially known which of these mechanisms are caused by TCDD : AhR or an AhR-independent pathway. Therefore, studies analyzing the effect of AhR by simply adding a chemical like, e.g., TCDD to the system must be viewed with caution.
Novel approaches utilizing spatial transcriptomics (ST) will help to understand how plasma cells differ in their phenotypes and functions in different anatomical locations. ST allows the comparative analysis of gene-expression profiles of IgA plasmablasts and plasma cells in their anatomical and physiological environment. In this context, comparing the transcriptomes of IgA plasma cells in the villus tips to those located next to the crypts in human and murine small intestines will be of interest. ST of intestinal stromal cells along the small intestine villi - crypt axis have already been successfully conducted and resulted in the identification of four spatially distinct mesenchymal cell populations (208). The characterization of the gene expression profiles of IgA plasma cells of the small intestine, the colon, and the cecum under normal and disease conditions, after oral immunizations or upon exposure to specialized diets will provide important insights into the plasticity and heterogeneity of IgA plasma cells and their response to environmental factors. Furthermore, the combination of genetic models (e.g., conditional knockout mice) with ST will allow us to identify the impact of gene function on IgA plasma cell heterogeneity. Hence, ST combined with functional analyses will help to answer the following open questions in IgA plasma cell biology:
1) How can IgA plasma cells persist for decades in the LP in close proximity to the epithelium while IECs are constantly replaced and renewed? The intestinal epithelium is characterized by a high turn-over rate of 3-5 days. Stem cells in the crypts continually divide and give rise to transit-amplifying cells which then differentiate into specialized IECs. During this process, the newly formed cells move from the crypt towards the villus tip. As soon as they reach the villus tip, they undergo cell death and are shed off. The processes of proliferation, differentiation and cell death are regulated by gradients of ligands of the Wnt, the BMP, the Notch, and the Ephrin signaling pathways (7, 8), some of them controlled by AhR signaling. As dividing and differentiating IECs are moving towards the villus tip, it remains elusive how the interaction between IgA plasma cells and IECs is maintained. Moreover, the impact of the Wnt, Notch, BMP, Ephrin or AhR ligand gradients on IgA plasma cell survival and function is unknown. Transcriptional regulation of components of the above-mentioned signaling pathways by e.g., AhR signaling, can be identified by applying ST to IgA plasma cells dissected from different regions within the crypt-villus axis.
2) How does the interaction with the basement membrane and the ECM affect IgA plasma cell survival and functions? IECs reside on a basement membrane consisting of a dense network of ECM components. Therefore, the interaction of IgA plasma cells with ECM components might be crucial for retaining IgA plasma cells in close proximity to the epithelial layer. The basement membrane has been shown to contribute to cell migration and differentiation. In addition, the basement membrane exerts barrier functions (209). It contains pores that allow immune cells (e.g., IELs or protrusions of DCs) to interact directly with epithelial cells and to reside within the epithelial layer. As shown for the rat intestine, pore sizes in the basement membrane are variable. Their numbers differ in areas of the villous structures and in proximity to lymphocyte follicles and M cells (4). How plasma cell homeostasis and plasma cell contact with the components of the basement membrane, the ECM, and the epithelium impacts their survival, their regulatory functions, and IgA secretion is poorly understood and needs to be investigated.
3) How does compartmentalization affect IgA plasma cell and epithelial cell function? The small intestine consists of the duodenum, jejunum, and ileum. The duodenum is characterized by a high concentration of food ligands and a low bacterial content. Along the duodenum–ileum axis, the concentrations of food ligands decrease, the bacterial concentration, however, increases (1, 2). Concomitantly, CCL25 production is higher in the duodenum compared to the ileum, enabling the recruitment of more immune cells. Furthermore, higher numbers of IgA cells are located in the duodenum. Moreover, the expression of the p-IgR is higher in the duodenum resulting in higher luminal sIgA. However, the numbers of IELs decrease from the duodenum to the ileum (1, 2, 210). In addition, expression of AhR also shows a proximal-distal gradient in the small intestine, indicating changes in the impact of food compounds on the transcriptional program of epithelial and immune cells. Hence, IgA plasma cells in the proximal and the distal parts of the small intestine are confronted with varying environmental signals. To determine the impact of these changing conditions, comparative spatial analyses of IgA plasma cells in duodenum, jejunum and ileum are required. Moreover, IgA plasma cells can reside in different locations along the crypt-villus axis within the villi. Depending on their location, they are exposed to gradients of Wnt, BMP, Notch, and Ephrin factors and to a constant change of their neighboring IECs as these cells move towards the villus tip. ST of IgA plasma cells from the crypt, the mid-villus and villus tip area will identify how the anatomical location modulates IgA plasma cell survival and function (i.e., cytokine release). In addition, mucosal surfaces in the small intestine and the colon are structurally very different. In the colon, villi are absent and the epithelial surface is flattened. However, the mucus layer is thicker due to a higher frequency of goblet cells within the colonic epithelium (14). Paneth cells can only be found in the small intestine but are absent in the colon. The bacterial density peaks in the colon concomitant with higher numbers of IgA-secreting cells and higher p-IgR expression compared to the small intestine (1, 2, 210). In humans, IgA-secreting cells can be subdivided in IgA1- and IgA2-secreting cells. IgA1 and IgA2 antibodies differ in their hinge regions (1, 211–213). The shorter hinge region of IgA2 might be more resistant to degradation by bacterial enzymes, and explain the predominant occurrence of IgA2 in the colon and the enrichment of IgA1 in the small intestine. Characterizing the gene expression profiles of human IgA1- and IgA2-secreting plasma cells will identify differences in their homing, survival, and functional abilities. Therefore, comparing IgA plasma cell subsets in different regions of the small intestine to those residing in the colon will provide a deeper insight into the heterogeneity of the IgA plasma cell pool.
4) How do signals mediated by the IgA surface BCR on plasma cells affect their localization, survival, and function? IgA plasmablasts and, more importantly, mature IgA plasma cells still express IgA-BCRs on their cell surface (77, 214–216). However, the biological function of their IgA-BCR expression remains elusive. One could envision that IgA-BCRs on plasma cells initiate tonic signals similar to the BCR on B cells (217). IgA-BCR signals might also be triggered by antigen and continuous or repetitive antigen-binding might be crucial for survival and/or inducing regulatory functions of plasma cells, such as cytokine production. For IgM plasma cells it was demonstrated that antigen binding to the surface IgM-BCR resulted in transcriptional changes and altered cytokine profiles (218). Signals mediated via the IgA-BCR might also contribute to the localization of IgA plasma cells within the villus.
Hence, new genetic models in combination with ST need to be developed to investigate the biological relevance of the surface IgA-BCR and its signals in the context of IgA plasma cell function and longevity.
KP and WS conceptualized and wrote the manuscript. JW, FK, and H-MJ revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was in part funded by Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) TRR130 project 09 (GEPRIS DFG project number: 215346292) to H-MJ and WS, the DFG GRK2599 to H-MJ and intramural funding by the Interdisziplinäres Zentrum für Klinische Forschung (IZKF), Friedrich-Alexander-Universität Erlangen-Nürnberg to KP. We acknowledge financial support by Deutsche Forschungsgemeinschaft and Friedrich-Alexander-Universität Erlangen-Nürnberg within the funding programme “Open Access Publication Funding".
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Mowat AM, Agace WW. Regional specialization within the intestinal immune system. Nat Rev Immunol (2014) 14(10):667–85. doi: 10.1038/nri3738
2. Agace WW, McCoy KD. Regionalized development and maintenance of the intestinal adaptive immune landscape. Immunity (2017) 46(4):532–48. doi: 10.1016/j.immuni.2017.04.004
3. Pompili S, Latella G, Gaudio E, Sferra R, Vetuschi A. The charming world of the extracellular matrix: A dynamic and protective network of the intestinal wall. Front Med (Lausanne). (2021) 8:610189. doi: 10.3389/fmed.2021.610189
4. Takeuchi T, Gonda T. Distribution of the pores of epithelial basement membrane in the rat small intestine. J Vet Med Sci (2004) 66(6):695–700. doi: 10.1292/jvms.66.695
5. Rowe RG, Weiss SJ. Breaching the basement membrane: Who, when and how? Trends Cell Biol (2008) 18(11):560–74. doi: 10.1016/j.tcb.2008.08.007
6. Spit M, Koo BK, Maurice MM. Tales from the crypt: Intestinal niche signals in tissue renewal, plasticity and cancer. Open Biol (2018) 8(9). doi: 10.1098/rsob.180120
7. Kurokawa K, Hayakawa Y, Koike K. Plasticity of intestinal epithelium: Stem cell niches and regulatory signals. Int J Mol Sci (2020) 22(1). doi: 10.3390/ijms22010357
8. Barker N. Adult intestinal stem cells: Critical drivers of epithelial homeostasis and regeneration. Nat Rev Mol Cell Biol (2014) 15(1):19–33. doi: 10.1038/nrm3721
9. Schneider C, O'Leary CE, Locksley RM. Regulation of immune responses by tuft cells. Nat Rev Immunol (2019) 19(9):584–93. doi: 10.1038/s41577-019-0176-x
10. Gribble FM, Reimann F. Enteroendocrine cells: Chemosensors in the intestinal epithelium. Annu Rev Physiol (2016) 78:277–99. doi: 10.1146/annurev-physiol-021115-105439
11. Pelaseyed T, Bergstrom JH, Gustafsson JK, Ermund A, Birchenough GM, Schutte A, et al. The mucus and mucins of the goblet cells and enterocytes provide the first defense line of the gastrointestinal tract and interact with the immune system. Immunol Rev (2014) 260(1):8–20. doi: 10.1111/imr.12182
12. Grondin JA, Kwon YH, Far PM, Haq S, Khan WI. Mucins in intestinal mucosal defense and inflammation: Learning from clinical and experimental studies. Front Immunol (2020) 11:2054. doi: 10.3389/fimmu.2020.02054
13. Velcich A, Yang W, Heyer J, Fragale A, Nicholas C, Viani S, et al. Colorectal cancer in mice genetically deficient in the mucin Muc2. Science (2002) 295(5560):1726–9. doi: 10.1126/science.1069094
14. Johansson ME, Phillipson M, Petersson J, Velcich A, Holm L, Hansson GC. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci U S A. (2008) 105(39):15064–9. doi: 10.1073/pnas.0803124105
15. Porter EM, Bevins CL, Ghosh D, Ganz T. The multifaceted paneth cell. Cell Mol Life Sci (2002) 59(1):156–70. doi: 10.1007/s00018-002-8412-z
16. Lueschow SR, McElroy SJ. The paneth cell: The curator and defender of the immature small intestine. Front Immunol (2020) 11:587. doi: 10.3389/fimmu.2020.00587
17. Shin JH, Seeley RJ. Reg3 proteins as gut hormones? Endocrinology (2019) 160(6):1506–14. doi: 10.1210/en.2019-00073
18. Ma H, Qiu Y, Yang H. Intestinal intraepithelial lymphocytes: Maintainers of intestinal immune tolerance and regulators of intestinal immunity. J Leukoc Biol (2021) 109(2):339–47. doi: 10.1002/JLB.3RU0220-111
19. Olivares-Villagomez D, Van Kaer L. Intestinal intraepithelial lymphocytes: Sentinels of the mucosal barrier. Trends Immunol (2018) 39(4):264–75. doi: 10.1016/j.it.2017.11.003
20. del Rio ML, Bernhardt G, Rodriguez-Barbosa JI, Forster R. Development and functional specialization of CD103+ dendritic cells. Immunol Rev (2010) 234(1):268–81. doi: 10.1111/j.0105-2896.2009.00874.x
21. Dillon A, Lo DD. M cells: Intelligent engineering of mucosal immune surveillance. Front Immunol (2019) 10:1499. doi: 10.3389/fimmu.2019.01499
22. Pabst O. New concepts in the generation and functions of IgA. Nat Rev Immunol (2012) 12(12):821–32. doi: 10.1038/nri3322
23. Cerutti A. The regulation of IgA class switching. Nat Rev Immunol (2008) 8(6):421–34. doi: 10.1038/nri2322
24. Reboldi A, Cyster JG. Peyer's patches: Organizing b-cell responses at the intestinal frontier. Immunol Rev (2016) 271(1):230–45. doi: 10.1111/imr.12400
25. Keppler SJ, Goess MC, Heinze JM. The wanderings of gut-derived IgA plasma cells: Impact on systemic immune responses. Front Immunol (2021) 12:670290. doi: 10.3389/fimmu.2021.670290
26. Gommerman JL, Rojas OL, Fritz JH. Re-thinking the functions of IgA(+) plasma cells. Gut Microbes (2014) 5(5):652–62. doi: 10.4161/19490976.2014.969977
27. Fagarasan S, Kawamoto S, Kanagawa O, Suzuki K. Adaptive immune regulation in the gut: T cell-dependent and T cell-independent IgA synthesis. Annu Rev Immunol (2010) 28:243–73. doi: 10.1146/annurev-immunol-030409-101314
28. Mackay F, Schneider P. Cracking the BAFF code. Nat Rev Immunol (2009) 9(7):491–502. doi: 10.1038/nri2572
29. Tezuka H, Ohteki T. Regulation of IgA production by intestinal dendritic cells and related cells. Front Immunol (2019) 10:1891. doi: 10.3389/fimmu.2019.01891
30. Seo GY, Jang YS, Kim HA, Lee MR, Park MH, Park SR, et al. Retinoic acid, acting as a highly specific IgA isotype switch factor, cooperates with TGF-beta1 to enhance the overall IgA response. J Leukoc Biol (2013) 94(2):325–35. doi: 10.1189/jlb.0313128
31. Bergqvist P, Stensson A, Lycke NY, Bemark M. T Cell-independent IgA class switch recombination is restricted to the GALT and occurs prior to manifest germinal center formation. J Immunol (2010) 184(7):3545–53. doi: 10.4049/jimmunol.0901895
32. Grasset EK, Chorny A, Casas-Recasens S, Gutzeit C, Bongers G, Thomsen I, et al. Gut T cell-independent IgA responses to commensal bacteria require engagement of the TACI receptor on b cells. Sci Immunol (2020) 5(49). doi: 10.1126/sciimmunol.aat7117
33. Abreu MT. Toll-like receptor signalling in the intestinal epithelium: How bacterial recognition shapes intestinal function. Nat Rev Immunol (2010) 10(2):131–44. doi: 10.1038/nri2707
34. Peterson LW, Artis D. Intestinal epithelial cells: Regulators of barrier function and immune homeostasis. Nat Rev Immunol (2014) 14(3):141–53. doi: 10.1038/nri3608
35. Svensson M, Agace WW. Role of CCL25/CCR9 in immune homeostasis and disease. Expert Rev Clin Immunol (2006) 2(5):759–73. doi: 10.1586/1744666X.2.5.759
36. Rimoldi M, Chieppa M, Salucci V, Avogadri F, Sonzogni A, Sampietro GM, et al. Intestinal immune homeostasis is regulated by the crosstalk between epithelial cells and dendritic cells. Nat Immunol (2005) 6(5):507–14. doi: 10.1038/ni1192
37. Zeuthen LH, Fink LN, Frokiaer H. Epithelial cells prime the immune response to an array of gut-derived commensals towards a tolerogenic phenotype through distinct actions of thymic stromal lymphopoietin and transforming growth factor-beta. Immunology (2008) 123(2):197–208. doi: 10.1111/j.1365-2567.2007.02687.x
38. Iwata M, Hirakiyama A, Eshima Y, Kagechika H, Kato C, Song SY. Retinoic acid imprints gut-homing specificity on T cells. Immunity (2004) 21(4):527–38. doi: 10.1016/j.immuni.2004.08.011
39. Mora JR, Iwata M, Eksteen B, Song SY, Junt T, Senman B, et al. Generation of gut-homing IgA-secreting b cells by intestinal dendritic cells. Science (2006) 314(5802):1157–60. doi: 10.1126/science.1132742
40. Casey KA, Fraser KA, Schenkel JM, Moran A, Abt MC, Beura LK, et al. Antigen-independent differentiation and maintenance of effector-like resident memory T cells in tissues. J Immunol (2012) 188(10):4866–75. doi: 10.4049/jimmunol.1200402
41. Kilshaw PJ, Murant SJ. A new surface antigen on intraepithelial lymphocytes in the intestine. Eur J Immunol (1990) 20(10):2201–7. doi: 10.1002/eji.1830201008
42. Kilshaw PJ, Murant SJ. Expression and regulation of beta 7(beta p) integrins on mouse lymphocytes: Relevance to the mucosal immune system. Eur J Immunol (1991) 21(10):2591–7. doi: 10.1002/eji.1830211041
43. Mackay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon ML, et al. The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat Immunol (2013) 14(12):1294–301. doi: 10.1038/ni.2744
44. Sheridan BS, Pham QM, Lee YT, Cauley LS, Puddington L, Lefrancois L. Oral infection drives a distinct population of intestinal resident memory CD8(+) T cells with enhanced protective function. Immunity (2014) 40(5):747–57. doi: 10.1016/j.immuni.2014.03.007
45. Thom JT, Weber TC, Walton SM, Torti N, Oxenius A. The salivary gland acts as a sink for tissue-resident memory CD8(+) T cells, facilitating protection from local cytomegalovirus infection. Cell Rep (2015) 13(6):1125–36. doi: 10.1016/j.celrep.2015.09.082
46. Mora JR, von Andrian UH. Differentiation and homing of IgA-secreting cells. Mucosal Immunol (2008) 1(2):96–109. doi: 10.1038/mi.2007.14
47. van Ginkel FW, Wahl SM, Kearney JF, Kweon MN, Fujihashi K, Burrows PD, et al. Partial IgA-deficiency with increased Th2-type cytokines in TGF-beta 1 knockout mice. J Immunol (1999) 163(4):1951–7. doi: 10.4049/jimmunol.163.4.1951
48. Cazac BB, Roes J. TGF-beta receptor controls b cell responsiveness and induction of IgA in vivo. Immunity (2000) 13(4):443–51. doi: 10.1016/S1074-7613(00)00044-3
49. Borsutzky S, Cazac BB, Roes J, Guzman CA. TGF-beta receptor signaling is critical for mucosal IgA responses. J Immunol (2004) 173(5):3305–9. doi: 10.4049/jimmunol.173.5.3305
50. Tezuka H, Abe Y, Iwata M, Takeuchi H, Ishikawa H, Matsushita M, et al. Regulation of IgA production by naturally occurring TNF/iNOS-producing dendritic cells. Nature (2007) 448(7156):929–33. doi: 10.1038/nature06033
51. Glick AB, McCune BK, Abdulkarem N, Flanders KC, Lumadue JA, Smith JM, et al. Complex regulation of TGF beta expression by retinoic acid in the vitamin a-deficient rat. Development (1991) 111(4):1081–6. doi: 10.1242/dev.111.4.1081
52. Tokuyama H, Tokuyama Y. Retinoids enhance IgA production by lipopolysaccharide-stimulated murine spleen cells. Cell Immunol (1993) 150(2):353–63. doi: 10.1006/cimm.1993.1203
53. Glick AB, Flanders KC, Danielpour D, Yuspa SH, Sporn MB. Retinoic acid induces transforming growth factor-beta 2 in cultured keratinocytes and mouse epidermis. Cell Regul (1989) 1(1):87–97. doi: 10.1091/mbc.1.1.87
54. Tokuyama H, Tokuyama Y. Endogenous cytokine expression profiles in retinoic acid-induced IgA production by LPS-stimulated murine splenocytes. Cell Immunol (1995) 166(2):247–53. doi: 10.1006/cimm.1995.9973
55. Schuh W, Mielenz D, Jack HM. Unraveling the mysteries of plasma cells. Adv Immunol (2020) 146:57–107. doi: 10.1016/bs.ai.2020.01.002
56. He B, Xu W, Santini PA, Polydorides AD, Chiu A, Estrella J, et al. Intestinal bacteria trigger T cell-independent immunoglobulin A(2) class switching by inducing epithelial-cell secretion of the cytokine APRIL. Immunity (2007) 26(6):812–26. doi: 10.1016/j.immuni.2007.04.014
57. Bossen C, Schneider P. BAFF. APRIL and their receptors: Structure, function and signaling. Semin Immunol (2006) 18(5):263–75. doi: 10.1016/j.smim.2006.04.006
58. Xu W, He B, Chiu A, Chadburn A, Shan M, Buldys M, et al. Epithelial cells trigger frontline immunoglobulin class switching through a pathway regulated by the inhibitor SLPI. Nat Immunol (2007) 8(3):294–303. doi: 10.1038/ni1434
59. Wittner J, Schuh W. Kruppel-like factor 2 (KLF2) in immune cell migration. Vaccines (Basel). (2021) 9(10). doi: 10.3390/vaccines9101171
60. Gorfu G, Rivera-Nieves J, Ley K. Role of beta7 integrins in intestinal lymphocyte homing and retention. Curr Mol Med (2009) 9(7):836–50. doi: 10.2174/156652409789105525
61. Hamann A, Andrew DP, Jablonski-Westrich D, Holzmann B, Butcher EC. Role of alpha 4-integrins in lymphocyte homing to mucosal tissues in vivo. J Immunol (1994) 152(7):3282–93. doi: 10.4049/jimmunol.152.7.3282
62. Berlin C, Berg EL, Briskin MJ, Andrew DP, Kilshaw PJ, Holzmann B, et al. Alpha 4 beta 7 integrin mediates lymphocyte binding to the mucosal vascular addressin MAdCAM-1. Cell (1993) 74(1):185–95. doi: 10.1016/0092-8674(93)90305-A
63. Hu MC, Crowe DT, Weissman IL, Holzmann B. Cloning and expression of mouse integrin beta p(beta 7): A functional role in peyer's patch-specific lymphocyte homing. Proc Natl Acad Sci U S A. (1992) 89(17):8254–8. doi: 10.1073/pnas.89.17.8254
64. Lindquist RL, Niesner RA, Hauser AE. In the right place, at the right time: Spatiotemporal conditions determining plasma cell survival and function. Front Immunol (2019) 10:788. doi: 10.3389/fimmu.2019.00788
65. Nishimura T, Koike R, Miyasaka M. Mammary glands of aly mice: Developmental changes and lactation-related expression of specific proteins, alpha-casein, GLyCAM-1 and MAdCAM-1. Am J Reprod Immunol (2000) 43(6):351–8. doi: 10.1111/j.8755-8920.2000.430604.x
66. Tanneau GM, Hibrand-Saint Oyant L, Chevaleyre CC, Salmon HP. Differential recruitment of T- and IgA b-lymphocytes in the developing mammary gland in relation to homing receptors and vascular addressins. J Histochem Cytochem (1999) 47(12):1581–92. doi: 10.1177/002215549904701210
67. Kraal G, Schornagel K, Streeter PR, Holzmann B, Butcher EC. Expression of the mucosal vascular addressin, MAdCAM-1, on sinus-lining cells in the spleen. Am J Pathol (1995) 147(3):763–71.
68. Streeter PR, Berg EL, Rouse BT, Bargatze RF, Butcher EC. A tissue-specific endothelial cell molecule involved in lymphocyte homing. Nature (1988) 331(6151):41–6. doi: 10.1038/331041a0
69. Briskin MJ, McEvoy LM, Butcher EC. MAdCAM-1 has homology to immunoglobulin and mucin-like adhesion receptors and to IgA1. Nature (1993) 363(6428):461–4. doi: 10.1038/363461a0
70. Nakache M, Berg EL, Streeter PR, Butcher EC. The mucosal vascular addressin is a tissue-specific endothelial cell adhesion molecule for circulating lymphocytes. Nature (1989) 337(6203):179–81. doi: 10.1038/337179a0
71. Katayama Y, Hidalgo A, Peired A, Frenette PS. Integrin alpha4beta7 and its counterreceptor MAdCAM-1 contribute to hematopoietic progenitor recruitment into bone marrow following transplantation. Blood (2004) 104(7):2020–6. doi: 10.1182/blood-2003-12-4157
72. Tada T, Inoue N, Widayati DT, Fukuta K. Role of MAdCAM-1 and its ligand on the homing of transplanted hematopoietic cells in irradiated mice. Exp Anim. (2008) 57(4):347–56. doi: 10.1538/expanim.57.347
73. Wagner N, Lohler J, Tedder TF, Rajewsky K, Muller W, Steeber DA. L-selectin and beta7 integrin synergistically mediate lymphocyte migration to mesenteric lymph nodes. Eur J Immunol (1998) 28(11):3832–9. doi: 10.1002/(SICI)1521-4141(199811)28:11<3832::AID-IMMU3832>3.0.CO;2-J
74. Pan J, Kunkel EJ, Gosslar U, Lazarus N, Langdon P, Broadwell K, et al. A novel chemokine ligand for CCR10 and CCR3 expressed by epithelial cells in mucosal tissues. J Immunol (2000) 165(6):2943–9. doi: 10.4049/jimmunol.165.6.2943
75. Pabst O, Ohl L, Wendland M, Wurbel MA, Kremmer E, Malissen B, et al. Chemokine receptor CCR9 contributes to the localization of plasma cells to the small intestine. J Exp Med (2004) 199(3):411–6. doi: 10.1084/jem.20030996
76. Morteau O, Gerard C, Lu B, Ghiran S, Rits M, Fujiwara Y, et al. An indispensable role for the chemokine receptor CCR10 in IgA antibody-secreting cell accumulation. J Immunol (2008) 181(9):6309–15. doi: 10.4049/jimmunol.181.9.6309
77. Wittner J, Schulz SR, Steinmetz TD, Berges J, Hauke M, Channell WM, et al. Kruppel-like factor 2 controls IgA plasma cell compartmentalization and IgA responses. Mucosal Immunol (2022) 15(4):668–82. doi: 10.1038/s41385-022-00503-0
78. Winkelmann R, Sandrock L, Porstner M, Roth E, Mathews M, Hobeika E, et al. B cell homeostasis and plasma cell homing controlled by kruppel-like factor 2. Proc Natl Acad Sci U S A. (2011) 108(2):710–5. doi: 10.1073/pnas.1012858108
79. Hart GT, Wang X, Hogquist KA, Jameson SC. Kruppel-like factor 2 (KLF2) regulates b-cell reactivity, subset differentiation, and trafficking molecule expression. Proc Natl Acad Sci U S A. (2011) 108(2):716–21. doi: 10.1073/pnas.1013168108
80. Alles M, Turchinovich G, Zhang P, Schuh W, Agenes F, Kirberg J. Leukocyte beta7 integrin targeted by kruppel-like factors. J Immunol (2014) 193(4):1737–46. doi: 10.4049/jimmunol.1302613
81. Wagner N, Lohler J, Kunkel EJ, Ley K, Leung E, Krissansen G, et al. Critical role for beta7 integrins in formation of the gut-associated lymphoid tissue. Nature (1996) 382(6589):366–70. doi: 10.1038/382366a0
82. Pilarowski GO, Cazares T, Zhang L, Benjamin JS, Liu K, Jagannathan S, et al. Abnormal peyer patch development and b-cell gut homing drive IgA deficiency in kabuki syndrome. J Allergy Clin Immunol (2020) 145(3):982–92. doi: 10.1016/j.jaci.2019.11.034
83. Guzman M, Lundborg LR, Yeasmin S, Tyler CJ, Zgajnar NR, Taupin V, et al. An integrin alphaEbeta7-dependent mechanism of IgA transcytosis requires direct plasma cell contact with intestinal epithelium. Mucosal Immunol (2021) 14(6):1347–57. doi: 10.1038/s41385-021-00439-x
84. Mestecky J, Zikan J, Butler WT. Immunoglobulin m and secretory immunoglobulin a: Presence of a common polypeptide chain different from light chains. Science (1971) 171(3976):1163–5. doi: 10.1126/science.171.3976.1163
85. Mostov KE, Deitcher DL. Polymeric immunoglobulin receptor expressed in MDCK cells transcytoses IgA. Cell (1986) 46(4):613–21. doi: 10.1016/0092-8674(86)90887-1
86. Kaetzel CS. The polymeric immunoglobulin receptor: Bridging innate and adaptive immune responses at mucosal surfaces. Immunol Rev (2005) 206:83–99. doi: 10.1111/j.0105-2896.2005.00278.x
87. Szabo PA, Miron M, Farber DL. Location, location, location: Tissue resident memory T cells in mice and humans. Sci Immunol (2019) 4(34). doi: 10.1126/sciimmunol.aas9673
88. Mackay LK, Braun A, Macleod BL, Collins N, Tebartz C, Bedoui S, et al. Cutting edge: CD69 interference with sphingosine-1-phosphate receptor function regulates peripheral T cell retention. J Immunol (2015) 194(5):2059–63. doi: 10.4049/jimmunol.1402256
89. Hardenberg JB, Braun A, Schon MP. A yin and yang in epithelial immunology: The roles of the alpha(E)(CD103)beta(7) integrin in T cells. J Invest Dermatol (2018) 138(1):23–31. doi: 10.1016/j.jid.2017.05.026
90. Ng EK, Panesar N, Longo WE, Shapiro MJ, Kaminski DL, Tolman KC, et al. Human intestinal epithelial and smooth muscle cells are potent producers of IL-6. Mediators Inflamm (2003) 12(1):3–8. doi: 10.1080/0962935031000096917
91. Minges Wols HA, Underhill GH, Kansas GS, Witte PL. The role of bone marrow-derived stromal cells in the maintenance of plasma cell longevity. J Immunol (2002) 169(8):4213–21. doi: 10.4049/jimmunol.169.8.4213
92. Cassese G, Arce S, Hauser AE, Lehnert K, Moewes B, Mostarac M, et al. Plasma cell survival is mediated by synergistic effects of cytokines and adhesion-dependent signals. J Immunol (2003) 171(4):1684–90. doi: 10.4049/jimmunol.171.4.1684
93. Jourdan M, Cren M, Robert N, Bollore K, Fest T, Duperray C, et al. IL-6 supports the generation of human long-lived plasma cells in combination with either APRIL or stromal cell-soluble factors. Leukemia (2014) 28(8):1647–56. doi: 10.1038/leu.2014.61
94. Kopf M, Baumann H, Freer G, Freudenberg M, Lamers M, Kishimoto T, et al. Impaired immune and acute-phase responses in interleukin-6-deficient mice. Nature (1994) 368(6469):339–42. doi: 10.1038/368339a0
95. Belnoue E, Tougne C, Rochat AF, Lambert PH, Pinschewer DD, Siegrist CA. Homing and adhesion patterns determine the cellular composition of the bone marrow plasma cell niche. J Immunol (2012) 188(3):1283–91. doi: 10.4049/jimmunol.1103169
96. O'Connor BP, Raman VS, Erickson LD, Cook WJ, Weaver LK, Ahonen C, et al. BCMA is essential for the survival of long-lived bone marrow plasma cells. J Exp Med (2004) 199(1):91–8. doi: 10.1084/jem.20031330
97. Xu S, Lam KP. B-cell maturation protein, which binds the tumor necrosis factor family members BAFF and APRIL, is dispensable for humoral immune responses. Mol Cell Biol (2001) 21(12):4067–74. doi: 10.1128/MCB.21.12.4067-4074.2001
98. von Bulow GU, Bram RJ. NF-AT activation induced by a CAML-interacting member of the tumor necrosis factor receptor superfamily. Science (1997) 278(5335):138–41. doi: 10.1126/science.278.5335.138
99. von Bulow GU, van Deursen JM, Bram RJ. Regulation of the T-independent humoral response by TACI. Immunity (2001) 14(5):573–82. doi: 10.1016/S1074-7613(01)00130-3
100. Yan M, Wang H, Chan B, Roose-Girma M, Erickson S, Baker T, et al. Activation and accumulation of b cells in TACI-deficient mice. Nat Immunol (2001) 2(7):638–43. doi: 10.1038/89790
101. Peperzak V, Vikstrom I, Walker J, Glaser SP, LePage M, Coquery CM, et al. Mcl-1 is essential for the survival of plasma cells. Nat Immunol (2013) 14(3):290–7. doi: 10.1038/ni.2527
102. Chu VT, Beller A, Rausch S, Strandmark J, Zanker M, Arbach O, et al. Eosinophils promote generation and maintenance of immunoglobulin-a-expressing plasma cells and contribute to gut immune homeostasis. Immunity (2014) 40(4):582–93. doi: 10.1016/j.immuni.2014.02.014
103. Landsverk OJ, Snir O, Casado RB, Richter L, Mold JE, Reu P, et al. Antibody-secreting plasma cells persist for decades in human intestine. J Exp Med (2017) 214(2):309–17. doi: 10.1084/jem.20161590
104. Lemke A, Kraft M, Roth K, Riedel R, Lammerding D, Hauser AE. Long-lived plasma cells are generated in mucosal immune responses and contribute to the bone marrow plasma cell pool in mice. Mucosal Immunol (2016) 9(1):83–97. doi: 10.1038/mi.2015.38
105. Hapfelmeier S, Lawson MA, Slack E, Kirundi JK, Stoel M, Heikenwalder M, et al. Reversible microbial colonization of germ-free mice reveals the dynamics of IgA immune responses. Science (2010) 328(5986):1705–9. doi: 10.1126/science.1188454
106. Nutt SL, Hodgkin PD, Tarlinton DM, Corcoran LM. The generation of antibody-secreting plasma cells. Nat Rev Immunol (2015) 15(3):160–71. doi: 10.1038/nri3795
107. Khodadadi L, Cheng Q, Radbruch A, Hiepe F. The maintenance of memory plasma cells. Front Immunol (2019) 10:721. doi: 10.3389/fimmu.2019.00721
108. Childs CE, Calder PC, Miles EA. Diet and immune function. Nutrients (2019) 11(8). doi: 10.3390/nu11081933
109. Schiering C, Wincent E, Metidji A, Iseppon A, Li Y, Potocnik AJ, et al. Feedback control of AHR signalling regulates intestinal immunity. Nature (2017) 542(7640):242–5. doi: 10.1038/nature21080
110. Lubet RA, Connolly G, Kouri RE, Nebert DW, Bigelow SW. Biological effects of the Sudan dyes. Role Ah cytosolic receptor. Biochem Pharmacol (1983) 32(20):3053–8. doi: 10.1016/0006-2952(83)90248-4
111. Okey AB, Bondy GP, Mason ME, Nebert DW, Forster-Gibson CJ, Muncan J, et al. Temperature-dependent cytosol-to-nucleus translocation of the ah receptor for 2,3,7,8-tetrachlorodibenzo-p-dioxin in continuous cell culture lines. J Biol Chem (1980) 255(23):11415–22. doi: 10.1016/S0021-9258(19)70307-X
112. Fukunaga BN, Probst MR, Reisz-Porszasz S, Hankinson O. Identification of functional domains of the aryl hydrocarbon receptor. J Biol Chem (1995) 270(49):29270–8. doi: 10.1074/jbc.270.49.29270
113. Denis M, Cuthill S, Wikstrom AC, Poellinger L, Gustafsson JA. Association of the dioxin receptor with the Mr 90,000 heat shock protein: A structural kinship with the glucocorticoid receptor. Biochem Biophys Res Commun (1988) 155(2):801–7. doi: 10.1016/S0006-291X(88)80566-7
114. Perdew GH. Association of the ah receptor with the 90-kDa heat shock protein. J Biol Chem (1988) 263(27):13802–5. doi: 10.1016/S0021-9258(18)68314-0
115. Coumailleau P, Poellinger L, Gustafsson JA, Whitelaw ML. Definition of a minimal domain of the dioxin receptor that is associated with Hsp90 and maintains wild type ligand binding affinity and specificity. J Biol Chem (1995) 270(42):25291–300. doi: 10.1074/jbc.270.42.25291
116. Ikuta T, Eguchi H, Tachibana T, Yoneda Y, Kawajiri K. Nuclear localization and export signals of the human aryl hydrocarbon receptor. J Biol Chem (1998) 273(5):2895–904. doi: 10.1074/jbc.273.5.2895
117. Rowlands JC, McEwan IJ, Gustafsson JA. Trans-activation by the human aryl hydrocarbon receptor and aryl hydrocarbon receptor nuclear translocator proteins: Direct interactions with basal transcription factors. Mol Pharmacol (1996) 50(3):538–48.
118. Abel J, Haarmann-Stemmann T. An introduction to the molecular basics of aryl hydrocarbon receptor biology. Biol Chem (2010) 391(11):1235–48. doi: 10.1515/bc.2010.128
119. Wincent E, Bengtsson J, Mohammadi Bardbori A, Alsberg T, Luecke S, Rannug U, et al. Inhibition of cytochrome P4501-dependent clearance of the endogenous agonist FICZ as a mechanism for activation of the aryl hydrocarbon receptor. Proc Natl Acad Sci U S A. (2012) 109(12):4479–84. doi: 10.1073/pnas.1118467109
120. Evans BR, Karchner SI, Allan LL, Pollenz RS, Tanguay RL, Jenny MJ, et al. Repression of aryl hydrocarbon receptor (AHR) signaling by AHR repressor: Role of DNA binding and competition for AHR nuclear translocator. Mol Pharmacol (2008) 73(2):387–98. doi: 10.1124/mol.107.040204
121. Murre C, Mccaw PS, Vaessin H, Caudy M, Jan LY, Jan YN, et al. Interactions between heterologous helix-Loop-Helix proteins generate complexes that bind specifically to a common DNA-sequence. Cell (1989) 58(3):537–44. doi: 10.1016/0092-8674(89)90434-0
122. Swanson HI, Chan WK, Bradfield CA. DNA Binding specificities and pairing rules of the ah receptor, ARNT, and SIM proteins. J Biol Chem (1995) 270(44):26292–302. doi: 10.1074/jbc.270.44.26292
123. Bacsi SG, Hankinson O. Functional characterization of DNA-binding domains of the subunits of the heterodimeric aryl hydrocarbon receptor complex imputing novel and canonical basic helix-loop-helix protein-DNA interactions. J Biol Chem (1996) 271(15):8843–50. doi: 10.1074/jbc.271.15.8843
124. Wright EJ, De Castro KP, Joshi AD, Elferink CJ. Canonical and non-canonical aryl hydrocarbon receptor signaling pathways. Curr Opin Toxicol (2017) 2:87–92. doi: 10.1016/j.cotox.2017.01.001
125. Lin L, Dai Y, Xia Y. An overview of aryl hydrocarbon receptor ligands in the last two decades (2002-2022): A medicinal chemistry perspective. Eur J Med Chem (2022) 244:114845. doi: 10.1016/j.ejmech.2022.114845
126. Ashida H, Nishiumi S, Fukuda I. An update on the dietary ligands of the AhR. Expert Opin Drug Metab Toxicol (2008) 4(11):1429–47. doi: 10.1517/17425255.4.11.1429
127. Safe S, Jin UH, Park H, Chapkin RS, Jayaraman A. Aryl hydrocarbon receptor (AHR) ligands as selective AHR modulators (SAhRMs). Int J Mol Sci (2020) 21(18). doi: 10.3390/ijms21186654
128. Vrzalova A, Pecinkova P, Illes P, Gurska S, Dzubak P, Szotkowski M, et al. Mixture effects of tryptophan intestinal microbial metabolites on aryl hydrocarbon receptor activity. Int J Mol Sci (2022) 23(18). doi: 10.3390/ijms231810825
129. Perdew GH, Babbs CF. Production of ah receptor ligands in rat fecal suspensions containing tryptophan or indole-3-carbinol. Nutr Cancer (1991) 16(3-4):209–18. doi: 10.1080/01635589109514159
130. Heath-Pagliuso S, Rogers WJ, Tullis K, Seidel SD, Cenijn PH, Brouwer A, et al. Activation of the ah receptor by tryptophan and tryptophan metabolites. Biochemistry (1998) 37(33):11508–15. doi: 10.1021/bi980087p
131. Bjeldanes LF, Kim JY, Grose KR, Bartholomew JC, Bradfield CA. Aromatic hydrocarbon responsiveness-receptor agonists generated from indole-3-carbinol in vitro and in vivo: Comparisons with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Proc Natl Acad Sci U S A. (1991) 88(21):9543–7. doi: 10.1073/pnas.88.21.9543
132. Zelante T, Iannitti RG, Cunha C, De Luca A, Giovannini G, Pieraccini G, et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity (2013) 39(2):372–85. doi: 10.1016/j.immuni.2013.08.003
133. Fujioka M, Wada H. The bacterial oxidation of indole. Biochim Biophys Acta (1968) 158(1):70–8. doi: 10.1016/0304-4165(68)90073-1
134. Lamas B, Natividad JM, Sokol H. Aryl hydrocarbon receptor and intestinal immunity. Mucosal Immunol (2018) 11(4):1024–38. doi: 10.1038/s41385-018-0019-2
135. Rannug A, Fritsche E. The aryl hydrocarbon receptor and light. Biol Chem (2006) 387(9):1149–57. doi: 10.1515/BC.2006.143
136. Agerbirk N, De Vos M, Kim JH, Jander G. Indole glucosinolate breakdown and its biological effects. Phytochem Rev (2009) 8(1):101–20. doi: 10.1007/s11101-008-9098-0
137. Ito S, Chen C, Satoh J, Yim S, Gonzalez FJ. Dietary phytochemicals regulate whole-body CYP1A1 expression through an arylhydrocarbon receptor nuclear translocator-dependent system in gut. J Clin Invest. (2007) 117(7):1940–50. doi: 10.1172/JCI31647
138. Shapiro TA, Fahey JW, Wade KL, Stephenson KK, Talalay P. Chemoprotective glucosinolates and isothiocyanates of broccoli sprouts: Metabolism and excretion in humans. Cancer Epidemiol Biomarkers Prev (2001) 10(5):501–8.
139. Loub WD, Wattenberg LW, Davis DW. Aryl hydrocarbon hydroxylase induction in rat tissues by naturally occurring indoles of cruciferous plants. J Natl Cancer Inst (1975) 54(4):985–8.
140. Marinelli L, Martin-Gallausiaux C, Bourhis JM, Beguet-Crespel F, Blottiere HM, Lapaque N. Identification of the novel role of butyrate as AhR ligand in human intestinal epithelial cells. Sci Rep (2019) 9(1):643. doi: 10.1038/s41598-018-37019-2
141. Tan J, McKenzie C, Potamitis M, Thorburn AN, Mackay CR, Macia L. The role of short-chain fatty acids in health and disease. Adv Immunol (2014) 121:91–119. doi: 10.1016/B978-0-12-800100-4.00003-9
142. Goasduff T, Dreano Y, Guillois B, Menez JF, Berthou F. Induction of liver and kidney CYP1A1/1A2 by caffeine in rat. Biochem Pharmacol (1996) 52(12):1915–9. doi: 10.1016/S0006-2952(96)00522-9
143. Ashida H, Fukuda I, Yamashita T, Kanazawa K. Flavones and flavonols at dietary levels inhibit a transformation of aryl hydrocarbon receptor induced by dioxin. FEBS Lett (2000) 476(3):213–7. doi: 10.1016/S0014-5793(00)01730-0
144. Iba MM, Scholl H, Fung J, Thomas PE, Alam J. Induction of pulmonary CYP1A1 by nicotine. Xenobiotica (1998) 28(9):827–43. doi: 10.1080/004982598239083
145. Manach C, Williamson G, Morand C, Scalbert A, Remesy C. Bioavailability and bioefficacy of polyphenols in humans. I. Rev 97 bioavailability Stud Am J Clin Nutr (2005) 81(1 Suppl):230S–42S. doi: 10.1093/ajcn/81.1.230S
146. Amakura Y, Tsutsumi T, Nakamura M, Kitagawa H, Fujino J, Sasaki K, et al. Preliminary screening of the inhibitory effect of food extracts on activation of the aryl hydrocarbon receptor induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Biol Pharm Bull (2002) 25(2):272–4. doi: 10.1248/bpb.25.272
147. Paganga G, Rice-Evans CA. The identification of flavonoids as glycosides in human plasma. FEBS Lett (1997) 401(1):78–82. doi: 10.1016/S0014-5793(96)01442-1
148. Belghasem M, Roth D, Richards S, Napolene MA, Walker J, Yin W, et al. Metabolites in a mouse cancer model enhance venous thrombogenicity through the aryl hydrocarbon receptor-tissue factor axis. Blood (2019) 134(26):2399–413. doi: 10.1182/blood.2019001675
149. Pernomian L, Duarte-Silva M, de Barros Cardoso CR. The aryl hydrocarbon receptor (AHR) as a potential target for the control of intestinal inflammation: Insights from an immune and bacteria sensor receptor. Clin Rev Allergy Immunol (2020) 59(3):382–90. doi: 10.1007/s12016-020-08789-3
150. Stockinger B, Shah K, Wincent E. AHR in the intestinal microenvironment: safeguarding barrier function. Nat Rev Gastroenterol Hepatol (2021) 18(8):559–70. doi: 10.1038/s41575-021-00430-8
151. Ireland H, Kemp R, Houghton C, Howard L, Clarke AR, Sansom OJ, et al. Inducible cre-mediated control of gene expression in the murine gastrointestinal tract: Effect of loss of beta-catenin. Gastroenterology (2004) 126(5):1236–46. doi: 10.1053/j.gastro.2004.03.020
152. Yu M, Wang Q, Ma Y, Li L, Yu K, Zhang Z, et al. Aryl hydrocarbon receptor activation modulates intestinal epithelial barrier function by maintaining tight junction integrity. Int J Biol Sci (2018) 14(1):69–77. doi: 10.7150/ijbs.22259
153. Trikha P, Lee DA. The role of AhR in transcriptional regulation of immune cell development and function. Biochim Biophys Acta Rev Canc (2020) 1873(1):188335. doi: 10.1016/j.bbcan.2019.188335
154. Metidji A, Omenetti S, Crotta S, Li Y, Nye E, Ross E, et al. The environmental sensor AHR protects from inflammatory damage by maintaining intestinal stem cell homeostasis and barrier integrity. Immunity (2018) 49(2):353–62 e5. doi: 10.1016/j.immuni.2018.07.010
155. Lee JS, Cella M, McDonald KG, Garlanda C, Kennedy GD, Nukaya M, et al. AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of notch. Nat Immunol (2011) 13(2):144–51. doi: 10.1038/ni.2187
156. Kiss EA, Vonarbourg C, Kopfmann S, Hobeika E, Finke D, Esser C, et al. Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science (2011) 334(6062):1561–5. doi: 10.1126/science.1214914
157. Alvarado DM, Chen B, Iticovici M, Thaker AI, Dai N, VanDussen KL, et al. Epithelial indoleamine 2,3-dioxygenase 1 modulates aryl hydrocarbon receptor and notch signaling to increase differentiation of secretory cells and alter mucus-associated microbiota. Gastroenterology (2019) 157(4):1093–108 e11. doi: 10.1053/j.gastro.2019.07.013
158. Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature (2012) 487(7407):330–7. doi: 10.1038/nature11252
159. Luecke-Johansson S, Gralla M, Rundqvist H, Ho JC, Johnson RS, Gradin K, et al. A molecular mechanism to switch the aryl hydrocarbon receptor from a transcription factor to an E3 ubiquitin ligase. Mol Cell Biol (2017) 37(13). doi: 10.1128/MCB.00630-16
160. Kawajiri K, Kobayashi Y, Ohtake F, Ikuta T, Matsushima Y, Mimura J, et al. Aryl hydrocarbon receptor suppresses intestinal carcinogenesis in ApcMin/+ mice with natural ligands. Proc Natl Acad Sci U S A. (2009) 106(32):13481–6. doi: 10.1073/pnas.0902132106
161. Shah K, Maradana MR, Joaquina Delas M, Metidji A, Graelmann F, Llorian M, et al. Cell-intrinsic aryl hydrocarbon receptor signalling is required for the resolution of injury-induced colonic stem cells. Nat Commun (2022) 13(1):1827. doi: 10.1038/s41467-022-29098-7
162. Boitano AE, Wang J, Romeo R, Bouchez LC, Parker AE, Sutton SE, et al. Aryl hydrocarbon receptor antagonists promote the expansion of human hematopoietic stem cells. Science (2010) 329(5997):1345–8. doi: 10.1126/science.1191536
163. Ly M, Rentas S, Vujovic A, Wong N, Moreira S, Xu J, et al. Diminished AHR signaling drives human acute myeloid leukemia stem cell maintenance. Cancer Res (2019) 79(22):5799–811. doi: 10.1158/0008-5472.CAN-19-0274
164. Faroon O, Ruiz P. Polychlorinated biphenyls: New evidence from the last decade. Toxicol Ind Health (2016) 32(11):1825–47. doi: 10.1177/0748233715587849
165. Safe SH. Polychlorinated biphenyls (PCBs): Environmental impact, biochemical and toxic responses, and implications for risk assessment. Crit Rev Toxicol (1994) 24(2):87–149. doi: 10.3109/10408449409049308
166. Morin SM, Majhi PD, Crisi GM, Gregory KJ, Franca R, Schalet B, et al. Interindividual variation contributes to differential PCB 126 induced gene expression in primary breast epithelial cells and tissues. Ecotoxicol Environ Saf. (2022) 241:113722. doi: 10.1016/j.ecoenv.2022.113722
167. Petriello MC, Brandon JA, Hoffman J, Wang C, Tripathi H, Abdel-Latif A, et al. Dioxin-like PCB 126 increases systemic inflammation and accelerates atherosclerosis in lean LDL receptor-deficient mice. Toxicol Sci (2018) 162(2):548–58. doi: 10.1093/toxsci/kfx275
168. Powell DN, Swimm A, Sonowal R, Bretin A, Gewirtz AT, Jones RM, et al. Indoles from the commensal microbiota act via the AHR and IL-10 to tune the cellular composition of the colonic epithelium during aging. Proc Natl Acad Sci U S A. (2020) 117(35):21519–26. doi: 10.1073/pnas.2003004117
169. Zhang L, Nichols RG, Correll J, Murray IA, Tanaka N, Smith PB, et al. Persistent organic pollutants modify gut microbiota-host metabolic homeostasis in mice through aryl hydrocarbon receptor activation. Environ Health Perspect (2015) 123(7):679–88. doi: 10.1289/ehp.1409055
170. Jackson DP, Joshi AD, Elferink CJ. Ah receptor pathway intricacies; signaling through diverse protein partners and DNA-motifs. Toxicol Res (Camb). (2015) 4(5):1143–58. doi: 10.1039/C4TX00236A
171. Benson JM, Shepherd DM. Aryl hydrocarbon receptor activation by TCDD reduces inflammation associated with crohn's disease. Toxicol Sci (2011) 120(1):68–78. doi: 10.1093/toxsci/kfq360
172. Chng SH, Kundu P, Dominguez-Brauer C, Teo WL, Kawajiri K, Fujii-Kuriyama Y, et al. Ablating the aryl hydrocarbon receptor (AhR) in CD11c+ cells perturbs intestinal epithelium development and intestinal immunity. Sci Rep (2016) 6:23820. doi: 10.1038/srep23820
173. Goudot C, Coillard A, Villani AC, Gueguen P, Cros A, Sarkizova S, et al. Aryl hydrocarbon receptor controls monocyte differentiation into dendritic cells versus macrophages. Immunity (2017) 47(3):582–96 e6. doi: 10.1016/j.immuni.2017.08.016
174. Kimura A, Naka T, Nakahama T, Chinen I, Masuda K, Nohara K, et al. Aryl hydrocarbon receptor in combination with Stat1 regulates LPS-induced inflammatory responses. J Exp Med (2009) 206(9):2027–35. doi: 10.1084/jem.20090560
175. Jensen BA, Leeman RJ, Schlezinger JJ, Sherr DH. Aryl hydrocarbon receptor (AhR) agonists suppress interleukin-6 expression by bone marrow stromal cells: an immunotoxicology study. Environ Health (2003) 2(1):16. doi: 10.1186/1476-069X-2-16
176. Hashimoto T, Perlot T, Rehman A, Trichereau J, Ishiguro H, Paolino M, et al. ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature (2012) 487(7408):477–81. doi: 10.1038/nature11228
177. Qiu J, Heller JJ, Guo X, Chen ZM, Fish K, Fu YX, et al. The aryl hydrocarbon receptor regulates gut immunity through modulation of innate lymphoid cells. Immunity (2012) 36(1):92–104. doi: 10.1016/j.immuni.2011.11.011
178. Patnaude L, Mayo M, Mario R, Wu X, Knight H, Creamer K, et al. Mechanisms and regulation of IL-22-mediated intestinal epithelial homeostasis and repair. Life Sci (2021) 271:119195. doi: 10.1016/j.lfs.2021.119195
179. Mizoguchi A, Yano A, Himuro H, Ezaki Y, Sadanaga T, Mizoguchi E. Clinical importance of IL-22 cascade in IBD. J Gastroenterol (2018) 53(4):465–74. doi: 10.1007/s00535-017-1401-7
180. Li S, Bostick JW, Ye J, Qiu J, Zhang B, Urban JF Jr., et al. Aryl hydrocarbon receptor signaling cell intrinsically inhibits intestinal group 2 innate lymphoid cell function. Immunity (2018) 49(5):915–28 e5. doi: 10.1016/j.immuni.2018.09.015
181. Zhou L. AHR function in lymphocytes: Emerging concepts. Trends Immunol (2016) 37(1):17–31. doi: 10.1016/j.it.2015.11.007
182. Yeste A, Mascanfroni ID, Nadeau M, Burns EJ, Tukpah AM, Santiago A, et al. IL-21 induces IL-22 production in CD4+ T cells. Nat Commun (2014) 5:3753. doi: 10.1038/ncomms4753
183. Plank MW, Kaiko GE, Maltby S, Weaver J, Tay HL, Shen W, et al. Th22 cells form a distinct Th lineage from Th17 cells In vitro with unique transcriptional properties and tbet-dependent Th1 plasticity. J Immunol (2017) 198(5):2182–90. doi: 10.4049/jimmunol.1601480
184. Ruan Q, Kameswaran V, Zhang Y, Zheng S, Sun J, Wang J, et al. The Th17 immune response is controlled by the rel-RORgamma-RORgamma T transcriptional axis. J Exp Med (2011) 208(11):2321–33. doi: 10.1084/jem.20110462
185. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell (2006) 126(6):1121–33. doi: 10.1016/j.cell.2006.07.035
186. Gutierrez-Vazquez C, Quintana FJ. Regulation of the immune response by the aryl hydrocarbon receptor. Immunity (2018) 48(1):19–33. doi: 10.1016/j.immuni.2017.12.012
187. Chuang HC, Chen YM, Chen MH, Hung WT, Yang HY, Tseng YH, et al. AhR-ROR-gammat complex is a therapeutic target for MAP4K3/GLK(high)IL-17A(high) subpopulation of systemic lupus erythematosus. FASEB J (2019) 33(10):11469–80. doi: 10.1096/fj.201900105RR
188. Trifari S, Kaplan CD, Tran EH, Crellin NK, Spits H. Identification of a human helper T cell population that has abundant production of interleukin 22 and is distinct from T(H)-17, T(H)1 and T(H)2 cells. Nat Immunol (2009) 10(8):864–71. doi: 10.1038/ni.1770
189. Di Meglio P, Duarte JH, Ahlfors H, Owens ND, Li Y, Villanova F, et al. Activation of the aryl hydrocarbon receptor dampens the severity of inflammatory skin conditions. Immunity (2014) 40(6):989–1001. doi: 10.1016/j.immuni.2014.04.019
190. Ye J, Qiu J, Bostick JW, Ueda A, Schjerven H, Li S, et al. The aryl hydrocarbon receptor preferentially marks and promotes gut regulatory T cells. Cell Rep (2017) 21(8):2277–90. doi: 10.1016/j.celrep.2017.10.114
191. Zhou J, Henriquez J, Crawford R, Kaminski N. Suppression of the IgM response by aryl hydrocarbon receptor activation in human primary b cells involves impairment of immunoglobulin secretory processes. Toxicol Sci (2018) 163(1):319–29. doi: 10.1093/toxsci/kfy036
192. Villa M, Gialitakis M, Tolaini M, Ahlfors H, Henderson CJ, Wolf CR, et al. Aryl hydrocarbon receptor is required for optimal b-cell proliferation. EMBO J (2017) 36(1):116–28. doi: 10.15252/embj.201695027
193. Rosser EC, Piper CJM, Matei DE, Blair PA, Rendeiro AF, Orford M, et al. Microbiota-derived metabolites suppress arthritis by amplifying aryl-hydrocarbon receptor activation in regulatory b cells. Cell Metab (2020) 31(4):837–51 e10. doi: 10.1016/j.cmet.2020.03.003
194. Piper CJM, Rosser EC, Oleinika K, Nistala K, Krausgruber T, Rendeiro AF, et al. Aryl hydrocarbon receptor contributes to the transcriptional program of IL-10-Producing regulatory b cells. Cell Rep (2019) 29(7):1878–92 e7. doi: 10.1016/j.celrep.2019.10.018
195. Matsumoto M, Baba A, Yokota T, Nishikawa H, Ohkawa Y, Kayama H, et al. Interleukin-10-producing plasmablasts exert regulatory function in autoimmune inflammation. Immunity (2014) 41(6):1040–51. doi: 10.1016/j.immuni.2014.10.016
196. Immunological Genome P. ImmGen at 15. Nat Immunol (2020) 21(7):700–3. doi: 10.1038/s41590-020-0687-4
197. Gross Even-Zohar N, Pick M, Hofstetter L, Shaulov A, Nachmias B, Lebel E, et al. CD24 is a prognostic marker for multiple myeloma progression and survival. J Clin Med (2022) 11(10). doi: 10.3390/jcm11102913
198. Warren TK, Mitchell KA, Lawrence BP. Exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) suppresses the humoral and cell-mediated immune responses to influenza a virus without affecting cytolytic activity in the lung. Toxicol Sci (2000) 56(1):114–23. doi: 10.1093/toxsci/56.1.114
199. Foxx CL, Nagy MR, King AE, Albin D, DeKrey GK. TCDD exposure alters fecal IgA concentrations in male and female mice. BMC Pharmacol Toxicol (2022) 23(1):25. doi: 10.1186/s40360-022-00563-9
200. Gomez E, Ortiz V, Saint-Martin B, Boeck L, Diaz-Sanchez V, Bourges H. Hormonal regulation of the secretory IgA (sIgA) system: Estradiol- and progesterone-induced changes in sIgA in parotid saliva along the menstrual cycle. Am J Reprod Immunol (1993) 29(4):219–23. doi: 10.1111/j.1600-0897.1993.tb00590.x
201. Matthews J, Gustafsson JA. Estrogen receptor and aryl hydrocarbon receptor signaling pathways. Nucl Recept Signal (2006) 4:e016. doi: 10.1621/nrs.04016
202. Ohtake F, Takeyama K, Matsumoto T, Kitagawa H, Yamamoto Y, Nohara K, et al. Modulation of oestrogen receptor signalling by association with the activated dioxin receptor. Nature (2003) 423(6939):545–50. doi: 10.1038/nature01606
203. Culbreath C, Tanner SM, Yeramilli VA, Berryhill TF, Lorenz RG, Martin CA. Environmental-mediated intestinal homeostasis in neonatal mice. J Surg Res (2015) 198(2):494–501. doi: 10.1016/j.jss.2015.04.002
204. Pesatori AC, Consonni D, Rubagotti M, Grillo P, Bertazzi PA. Cancer incidence in the population exposed to dioxin after the "Seveso accident": Twenty years of follow-up. Environ Health (2009) 8:39. doi: 10.1186/1476-069X-8-39
205. Dragan YP, Schrenk D. Animal studies addressing the carcinogenicity of TCDD (or related compounds) with an emphasis on tumour promotion. Food Addit Contam. (2000) 17(4):289–302. doi: 10.1080/026520300283360
206. Viluksela M, Bager Y, Tuomisto JT, Scheu G, Unkila M, Pohjanvirta R, et al. Liver tumor-promoting activity of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in TCDD-sensitive and TCDD-resistant rat strains. Cancer Res (2000) 60(24):6911–20.
207. Knerr S, Schrenk D. Carcinogenicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin in experimental models. Mol Nutr Food Res (2006) 50(10):897–907. doi: 10.1002/mnfr.200600006
208. Bahar Halpern K, Massalha H, Zwick RK, Moor AE, Castillo-Azofeifa D, Rozenberg M, et al. Lgr5+ telocytes are a signaling source at the intestinal villus tip. Nat Commun (2020) 11(1):1936. doi: 10.1038/s41467-020-15714-x
209. Vllasaliu D, Falcone FH, Stolnik S, Garnett M. Basement membrane influences intestinal epithelial cell growth and presents a barrier to the movement of macromolecules. Exp Cell Res (2014) 323(1):218–31. doi: 10.1016/j.yexcr.2014.02.022
210. Brandtzaeg P. Function of mucosa-associated lymphoid tissue in antibody formation. Immunol Invest (2010) 39(4-5):303–55. doi: 10.3109/08820131003680369
211. Plaut AG, Wistar R Jr., Capra JD. Differential susceptibility of human IgA immunoglobulins to streptococcal IgA protease. J Clin Invest. (1974) 54(6):1295–300. doi: 10.1172/JCI107875
212. Kilian M, Reinholdt J, Lomholt H, Poulsen K, Frandsen EV. Biological significance of IgA1 proteases in bacterial colonization and pathogenesis: Critical evaluation of experimental evidence. APMIS (1996) 104(5):321–38. doi: 10.1111/j.1699-0463.1996.tb00724.x
213. Lin M, Du L, Brandtzaeg P, Pan-Hammarstrom Q. IgA subclass switch recombination in human mucosal and systemic immune compartments. Mucosal Immunol (2014) 7(3):511–20. doi: 10.1038/mi.2013.68
214. Kamata T, Nogaki F, Fagarasan S, Sakiyama T, Kobayashi I, Miyawaki S, et al. Increased frequency of surface IgA-positive plasma cells in the intestinal lamina propria and decreased IgA excretion in hyper IgA (HIGA) mice, a murine model of IgA nephropathy with hyperserum IgA. J Immunol (2000) 165(3):1387–94. doi: 10.4049/jimmunol.165.3.1387
215. Pinto D, Montani E, Bolli M, Garavaglia G, Sallusto F, Lanzavecchia A, et al. A functional BCR in human IgA and IgM plasma cells. Blood (2013) 121(20):4110–4. doi: 10.1182/blood-2012-09-459289
216. Pracht K, Meinzinger J, Daum P, Schulz SR, Reimer D, Hauke M, et al. A new staining protocol for detection of murine antibody-secreting plasma cell subsets by flow cytometry. Eur J Immunol (2017) 47(8):1389–92. doi: 10.1002/eji.201747019
217. Lam KP, Kuhn R, Rajewsky K. In vivo ablation of surface immunoglobulin on mature b cells by inducible gene targeting results in rapid cell death. Cell (1997) 90(6):1073–83. doi: 10.1016/S0092-8674(00)80373-6
Keywords: IgA, IgA plasma cells, intestinal epithelial cell, survival niche, intestinal epithelial barrier, Aryl hydrocarbon (Ah) receptor, plasma cell
Citation: Pracht K, Wittner J, Kagerer F, Jäck H-M and Schuh W (2023) The intestine: A highly dynamic microenvironment for IgA plasma cells. Front. Immunol. 14:1114348. doi: 10.3389/fimmu.2023.1114348
Received: 02 December 2022; Accepted: 23 January 2023;
Published: 16 February 2023.
Edited by:
Peter Dion Pioli, University of Saskatchewan, CanadaReviewed by:
Claude-Agnes Reynaud, U1151 Institut Necker Enfants Malades (INSERM), FranceCopyright © 2023 Pracht, Wittner, Kagerer, Jäck and Schuh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wolfgang Schuh, d29sZmdhbmcuc2NodWhAdWstZXJsYW5nZW4uZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.