Corrigendum: RIPK1 in the inflammatory response and sepsis: recent advances, drug discovery and beyond
 Xiaoyu Liu
Xiaoyu Liu A-Ling Tang
A-Ling Tang Jie Chen1,2
Jie Chen1,2 Nan Gao
Nan Gao Guoqiang Zhang
Guoqiang Zhang Cheng Xiao
Cheng Xiao- 1Department of Emergency, China-Japan Friendship Hospital, Beijing, China
- 2China-Japan Friendship Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China
- 3Graduate School, Beijing University of Chinese Medicine, Beijing, China
Cytokine storms are an important mechanism of sepsis. TNF-α is an important cytokine. As a regulator of TNF superfamily receptors, RIPK1 not only serves as the basis of the scaffold structure in complex I to promote the activation of the NF-κB and MAPK pathways but also represents an important protein in complex II to promote programmed cell death. Ubiquitination of RIPK1 is an important regulatory function that determines the activation of cellular inflammatory pathways or the activation of death pathways. In this paper, we introduce the regulation of RIPK1, RIPK1 PANoptosome’s role in Inflammatory and sepsis, and perspectives.
1 Introduction
Sepsis is a life-threatening condition characterized by organ dysfunction resulting from a dysregulated host response to infection. Cytokine storm, caused by imbalances in cytokines such as IL-1β, IL-6, and TNF-α, disrupts the normal immune balance, ultimately leading to organ dysfunction. TNF-α, a well-studied cytokine, is secreted by macrophages within 30 minutes after infection (1) and activates inflammatory pathways such as NF-κB, MAPK, and JNK signaling, or programmed cell death (2). RIPK1, a nodal protein in the TNF pathway, binds to TNFR1 at its intracellular terminus after TNF-α binds to TNFR1, subsequently recruiting TRAF1, cIAP1/2, LUBIC, and other E3 ubiquitinates to form proinflammatory complex I, thereby activating inflammation-related pathways. Deubiquitination of RIPK1 by CYLD, OTULIN, and A20 leads to the phosphorylation of RIPK1 and formation of death complex II, which mediates programmed cell death (3). RIPK1 has been implicated in TNF-induced cell death and proposed as a target for cancer therapy to induce cancer cell death (4, 5). Recent studies have elucidated the important role of TNF-mediated necroptosis in the inflammatory response, and RIPK1 inhibition has shown therapeutic effects on various rheumatic immune diseases as well as infections. However, the mechanism regulating RIPK1 in sepsis remains unclear. This manuscript centers on the impact of RIPK1 regulation induced by sepsis on the inflammatory response, and the current state of medical research.
2 Structure and function of RIPK1
RIPK1, the first member of the receptor-interacting Ser/Thr kinase (RIPK) family, is a crucial protein involved in the cell death process (1, 2). Its structure (Figure 1) comprises the N-terminal kinase domain, the C-terminal death domain that mediates death signaling, the bridging intermediate domain, and the RIP homotypic interaction motif (RHIM) (3–5). RIPK1 binds to the DD domains of TRADD and TNFRSF to form a trimeric structure, and TRADD then recruits E3 ubiquitin ligases such as cIAP1/2 and LUBAC to form a complex I scaffold structure, exerting proinflammatory effects (6). When the deubiquitinating enzymes A20, CYLD, and OTULIN degrade the ubiquitin chain of RIPK1, complex I dissociate, and the released RIPK1 forms the cytoplasmic death-induced signaling complex (DISC), Complex IIa, or complex IIb, inducing programmed cell death (7). RIPK1 has complex functions in cells, including the regulation of cell death associated with its kinase activity and regulation of pro-survival inflammatory pathways associated with its scaffold structure independent of its kinase activity. RIPK1 is constitutively located in the cytoplasm of cells (8). However, nuclear translocation of RIPK1 has also been reported (9–12), although its function in the nucleus has not been clarified.
FIGURE 1
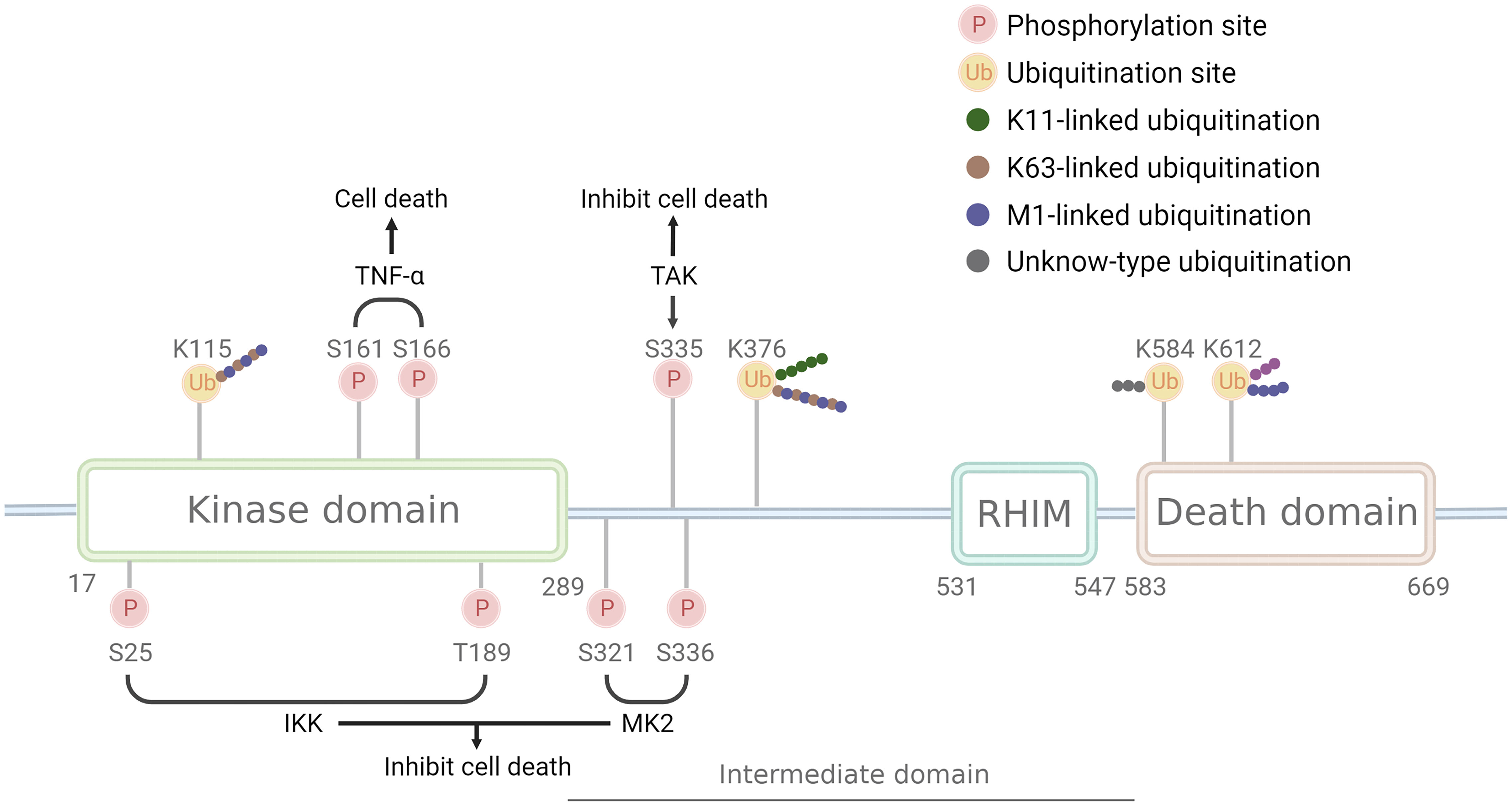
Figure 1 Structure and modification sites of RIPK1. RIPK1 comprises the N-terminal kinase domain, the C-terminal death domain that mediates death signaling, the bridging intermediate domain, and the RIP homotypic interaction motif (RHIM). Phosphorylation sites have two opposite effects: S161 and S166 have a pro-death effect and are phosphorylated after TNF-αstimulate, while S25, T189, S321, S335, and S336 have a pro-survival effect and are phosphorylated by IKK, MK2, and TAK. Ubiquitination sites can be linked by different Ub chains: K115 can be linked by K63 and M1 chain, K376 linked by K11, K63, and M1 chain, K612 linked by M1 chain, while K584 remains unknown. Pink circle with P indicates phosphorylation sites, a yellow circle with Ub indicates ubiquitination sites, green dots indicate K11 ub chain, brown dots indicates K63 ub chain, purple dots indicate M1 ub chain, grey indicates unknown ub chain.
In recent years, the inflammatory response and necroptosis induced by RIPK1 have been extensively studied and are involved in the pathogenesis of various autoimmune and chronic inflammatory diseases, such as neurodegeneration, ischemia-reperfusion injury, severe infections, cardiovascular disease, chronic obstructive pulmonary disease, skin inflammation, systemic lupus erythematosus, inflammatory bowel disease, rheumatoid arthritis, and psoriasis (13–22). Meanwhile, inhibiting RIPK1 by necrostatin-1 reduced TNF-α-affected model in mouse mortality, cell death, and persistent inflammatory responses (23). But interestedly, Patients with biallelic inactivating mutations in RIPK1, including missense, nonsense, and frameshift mutations, exhibit symptoms of inflammatory bowel disease and combined immunodeficiency, develop lymphopenia, and display increased sensitivity to TNFR1-mediated cell death (21, 22). RIPK1 knockout mice are embryonic lethal and exhibit severe in vivo inflammatory responses that activate apoptosis and necroptosis pathways (24). In summary, RIPK1 both triggers cytokine release and cell death. However, the underlying mechanism by which RIPK1 leads to changes in cytokine release remains unclear.
3 Regulation of RIPK1
RIPK1 is subject to regulation by a variety of enzymes and modifications, including ubiquitination and phosphorylation (Table 1). Ubiquitination can activate multiple inflammatory pathways and inhibits kinase activity, thereby reducing cell death. The kinase activity of RIPK1 is responsible for inducing programmed cell death (25). Mutations at the K45A site, which result in a loss of kinase activity, protect cells from TNF-α-stimulated necroptosis (26). Similarly, the D138N mutation, which also causes a loss of kinase activity, confers resistance to TNF-α induced necroptosis (27), and rats carrying this mutation are resistant to TNF-α-induced shock (28), however, Newton et.al (29). found RIPK1 D138N catalytically inactive mice were as sensitive to LPS challenge as wildtype mice, but were resistant to body temperature loss after TNF challenge. Numerous studies have identified S161 and S166 as important phosphorylation sites in RIPK1, and the crystal structure of RIPK1 indicates that S161 is located in the T-loop structure in an open conformation, suggesting that phosphorylation at this site promotes interaction between RIPK1 and RIPK3 and initiates the occurrence of necroptosis (30). Autophosphorylation at S166 induces TNF-mediated inflammatory responses in a variety of animal models (31). Mutations at the aforementioned sites (K45, S166, S161, and D138) all reside in the kinase domain of RIPK1, underscoring the critical role of RIPK1 kinase activity in necroptosis, and these mutations promote the systemic inflammatory response both in vivo and in vitro. Therefore, RIPK1 kinase activity may contribute to TNF-α-induced necroptosis and exacerbation of the inflammatory response.
TABLE 1
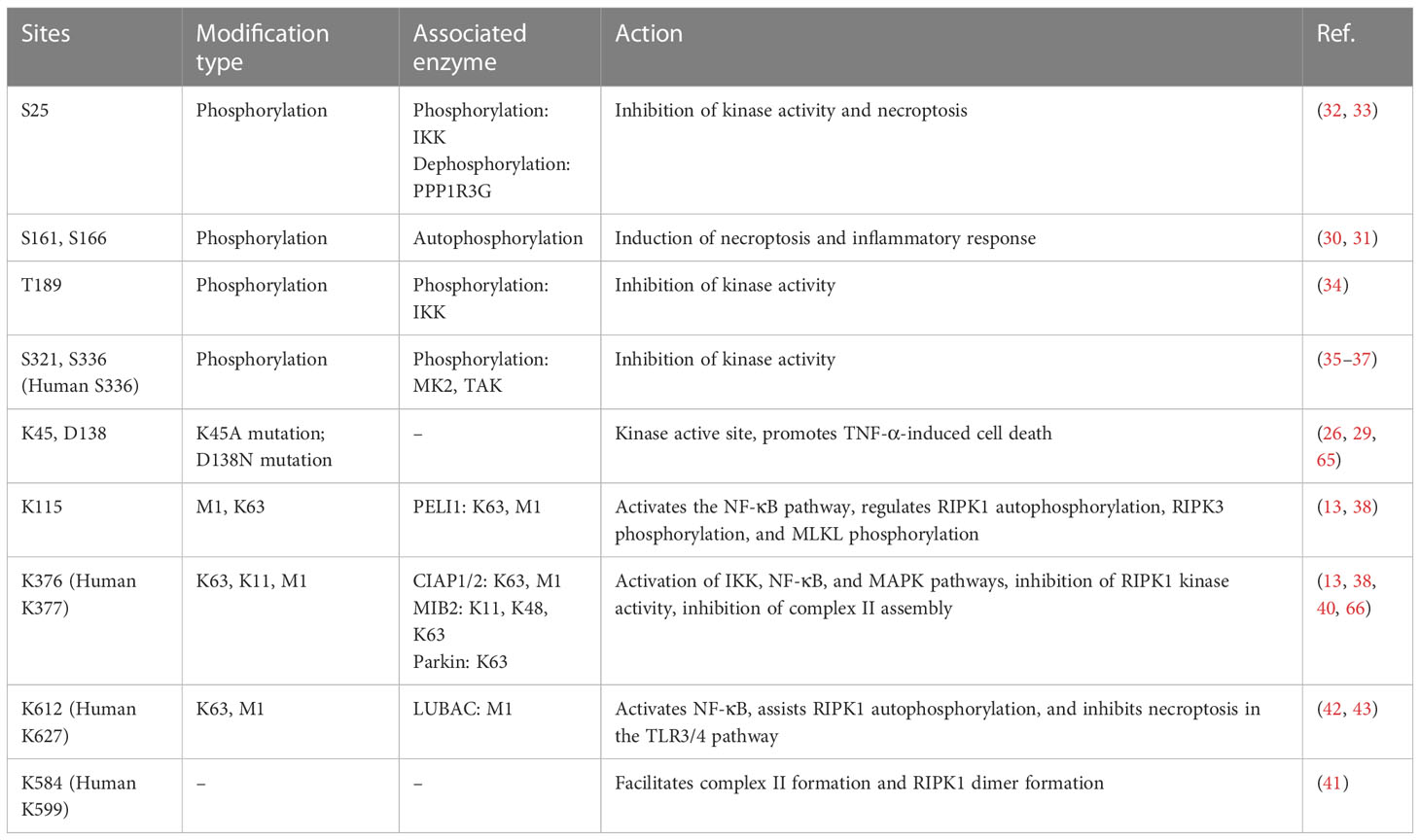
Table 1 Ubiquitin and phosphoryl sites of RIPK1.
In addition, RIPK1 also contains inhibitory phosphorylation sites. Phosphorylation of the S25 site inhibits RIPK1 kinase activity, IKKs phosphorylate the S25 site in complex I and protect cells deficient in the ubiquitinating enzyme SHARPIN from death (32), and dephosphorylation of the S25 site by PPP1R3G promotes the kinase activity of RIPK1, inducing necroptosis (33). T189 also inhibits RIPK1 kinase activity through phosphorylation by IKKs (34), while the S321 and S336 (human S335) sites are phosphorylated by MK2 and TAK to inhibit its kinase activity (35–37), and phosphorylation of the aforementioned inhibitory sites requires the activation of the NF-kB or MAPK pathway. This may be a negative feedback regulation mechanism of RIPK1-associated inflammatory factor transcription.
RIPK1 possesses multiple ubiquitination sites, with K11, K48, K63, and M1 ubiquitin chains being identified in this protein. K63 ubiquitin chain of RIPK1 is crucial for complex I formation and activation of inflammatory factor transcription, such as NF-κB signaling, whereas deubiquitinated RIPK1 leads to the formation of complex II and cell death. K115 is located in the kinase domain of RIPK1, compared with the wild type, the K115R mutation showed a decrease in the synthesis of K63 and M1 ubiquitin chains, and K115R significantly decreased the phosphorylation of RIPK1, RIPK3, and MLKL, ultimately inhibiting the occurrence of necroptosis (38, 39). K376 (human K377) is located in the bridging domain, mutation of K376 promotes necroptosis and apoptosis, reduces the ubiquitination of K11, K63, and M1, hinders TNFR1 complex formation, and causes embryonic lethality (38). Mutation of K376 in mice significantly enhances TNF-α-stimulated cell death, alters TNF-induced activation of the NF-kB and MAPK pathways, accompanied by increased phosphorylation of RIPK1, RIPK3, and MLKL, resulting in embryonic death, which was reversed by inhibition of RIPK1 kinase activity (38). The K377 site is a critical ubiquitylation site for NF-κB activation and the inhibition of open reading frame 3 (ORF3) in humans (40). Mutation of K584 (human K599), located in the DD domain of RIPK1, impedes homodimerization of RIPK1 through the DD domain, inhibiting complex II formations (41). The K612R mutation inhibits RIPK1 phosphorylation, leading to the inhibition of caspase-3, caspase-8, and RIPK1 cleavage, reduced ubiquitination of complex I, attenuated NF-κB activation, and impaired complex IIa formation, ultimately inhibiting MLKL phosphorylation. The K612R mutation inhibited RIPK1 kinase activity to a greater extent than NF-κB activity, resulting in an intestinal inflammatory response and splenomegaly in K612R mice (42). LPS stimulation of bone marrow-derived macrophages expressing the K612R mutant showed increased necroptosis, reflecting the inhibitory effect of the K612 site on necroptosis in the TLR3/4 pathway (42). Recent studies show that K612 site mutations in mice reduce the length of ubiquitin chains attached to the M1 site, causing systemic inflammatory responses and emergency hematopoietic myelograms. This change resulted from damage-associated molecular patterns (DAMPs) induced by TNFR activation. K612R mice transplanted with WT mouse bone marrow or K612R + TNF-/- mice showed improved outcomes (43).
In conclusion, the scaffold structure of complex I that binds initiator proteins, promotes the activation of the NF-kB, MAPK, and JNK pathways, and inhibits the kinase activity of RIPK1. whereas the ubiquitination at the K115, K376 (human K377), K612 (human K627), and K584 (human K599) sites, leads to the activation of the NF-kB pathway. Differences between these sites are mainly due to different stimuli or ubiquitin chain lengths. Mutations in RIPK1 ubiquitination sites, which normally enhance the kinase activity of RIPK1, lead to the activation of programmed cell death.
4 Regulation of RIPK1 with Inflammatory and Rheumatic Immune Diseases
RIPK1 has been implicated in the regulation of rheumatic immune diseases and chronic inflammatory responses, with its scaffold structure and kinase activity serving as key factors. Excessive ubiquitination can lead to the overactivation of the inflammatory response (44), while excessive deubiquitination can trigger necroptosis and subsequently elicit an excessive inflammatory response (45). Thus, the normal function of RIPK1 may hinge on maintaining a delicate balance between its scaffold structure and kinase activity. Specifically, deubiquitination is necessary for RIPK1’s kinase activity to manifest, and as such, the activity of the ubiquitin ligase that regulates RIPK1 may represent a crucial factor for controlling this balance (Table 2).
TABLE 2
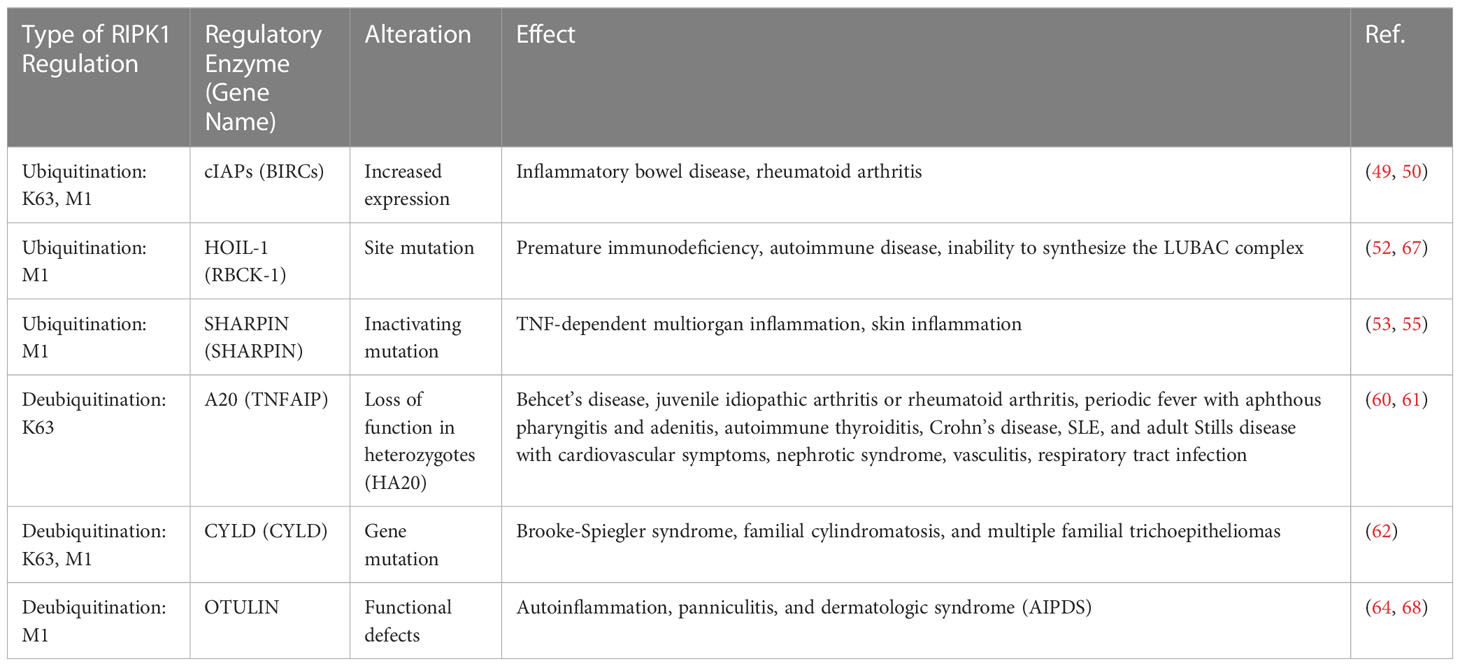
Table 2 Ubiquitinases and deubiquitinases of RIPK1 and its effect.
cIAP1/2, an E3 ubiquitin ligase, plays a critical role in regulating NF-κB signaling and modifying the K63 and M1 ubiquitin chains of RIPK1. As a result, it regulates not only caspases and apoptosis but also numerous cellular processes, including inflammatory signaling and immunity, mitogenic kinase signaling and cell proliferation, as well as cell invasion and metastasis (46–48). Mutations in cIAP (BIRC2/3) are linked to various diseases, including tumors, autoimmunity, and inflammatory disorder (49–51). A clinical study involving individuals with inflammatory bowel disease (IBD) demonstrated nonclassical NF-κB activation with reduced responses to infliximab and adalimumab therapy, accompanied by increased expression of cIAP1/2 and various related enzymes (50). In addition, gene expression analysis revealed a significant upregulation of BIRC2 expression in individuals with rheumatoid arthritis, with cIAP1 being a central protein identified in protein-protein interaction networks linked to rheumatoid arthritis (49).
The LUBAC complex comprises HOIL-1, HOIP, and SHARPIN, with the corresponding genes being RBCK1, RNF31, and SHARPIN, respectively. This complex encodes E3 ubiquitin enzyme complexes that selectively elongate the M1 chain in RIPK1. In 2019, Oda et al. (52) identified a patient with a HOIP protein deficiency who presented with premature immunodeficiency and autoimmune inflammation. The patient carried two mutations in the RNF31 gene, and the analysis of PBMC lysates revealed that the HOIP defect resulted in secondary abnormalities in SHARPIN and HOIL-1, which prevented the formation of LUBAC complexes. Stimulation of TNF-α in PBMCs from this patient delayed activation of the NF-κB pathway, whereas IL-1β stimulation excessively activated monocytes to produce high levels of IL-6 and IL-1β. In addition, LUBAC-deficient mice exhibited enhanced sensitivity to apoptosis or necroptosis (53–56).
A20 is a deubiquitinating enzyme that targets the K63 site of RIPK1. Genome-wide association studies (GWAS) have identified single nucleotide polymorphisms (SNPs) at A20 sites that are associated with various autoimmune diseases, and it has been suggested that these SNPs may lead to reduced A20 expression (57–59). Deletion of enhancers in the A20 gene in humanized mice has been shown to increase autoantibody production, inflammation, and arthritis. Heterozygous loss-of-function mutations in A20, referred to as haploinsufficient for A20 (HA20), were first reported by Zhou et al. in 2016 (60), and subsequently, a large number of HA20 patients have been identified. HA20 patients present with variable clinical manifestations, even among family members carrying the same variant. These patients are initially diagnosed with diseases such as Behcet’s disease, juvenile idiopathic arthritis, rheumatoid arthritis, periodic fever with aphthous pharyngitis and adenitis, autoimmune thyroiditis, Crohn’s disease, SLE, or adult Still’s disease, along with cardiovascular symptoms, nephrotic syndrome, vasculitis, respiratory tract infections, and other diseases caused by autoantibodies (61).
Compared to other genes, CYLD variants have thus far only been linked to Brooke-Spiegler syndrome, familial cylindromatosis, and multiple familial piloepitheliomas (62). Patients with Brooke-Spiegler syndrome are predisposed to a range of skin adnexal tumors, such as cylindroma, trichoepithelioma, and spiradenoma. Familial cylindromatosis and multiple familial piloepitheliomas are also part of Brooke-Spiegler syndrome, with single lesions being the primary manifestation. However, due to the rarity of this disease, its mechanism has not been extensively studied.
OTULIN is a deubiquitinating enzyme that specifically hydrolyzes the M1 ubiquitin chain in RIPK1. Defects in the OTULIN gene have been shown to cause autoinflammation, panniculitis, and dermatological syndrome (AIPDS) (45). AIPDS is manifested as neutrophil infiltration and recurrent nodular panniculitis with recurrent fever, as well as increased immunoglobulin levels and autoantibody titers in the blood. OTULIN is the major deubiquitinating enzyme of the M1 ubiquitin chain and negatively regulates the NF-κB pathway (63). Stimulation of fibroblasts and PBMCs from OTULIN-deficient patients with TNF-a results in increased activation of NF-kB and MAPK and increased ubiquitination of M1 in RIPK1, suggesting that loss-of-function mutations in OTULIN lead to increased M1 ubiquitination, activation of TNF pathway and NF-κB-dependent inflammation (64).
5 Roles and mechanism of RIPK1 in the development of sepsis
Studies have shown that both the kinase activity and scaffold structure of RIPK1 can induce inflammation, but while the scaffold structure only induces the transcription of cytokines, the kinase activity of RIPK1 induces cell death and a more intense inflammatory response (Figure 2). Consequently, the role of RIPK1 kinase activity in the systemic inflammatory response and sepsis has garnered increased attention. Table 3 lists some animal experiments related to RIPK1 in sepsis. Caspase-8 inhibition is a crucial factor for RIPK1-induced necroptosis (69), and a variety of pathogens inhibit caspase-8 activity: Macrophages respond to infections with certain pathogens such as Serratia marcescens, Staphylococcus aureus, Streptococcus pneumoniae, Streptococcus monocytogenes, and uropathogenic E. coli by inhibiting caspase-8 activity, thereby inducing necrosome formation and necroptosis (70). Necroptosis, a complementary programmed death pathway that is activated when apoptosis and pyroptosis are inhibited. This process exposes immature intracellular pathogens to the tissue interstitial space and releases various chemokines that activate various immune cells for defense, which is beneficial for intracellular infections (71). However, severe infections can result in a large number of necroptosis-induced DAMPs and chemotaxis-induced neutrophil traps (NETs) that promote the release of cytokines, forming a positive feedback loop of the inflammatory response that ultimately leads to a systemic inflammatory response and sepsis. Recent studies have highlighted the role of RIPK1 kinase activity-induced necroptosis, which has been detected in vascular endothelial cells In recent years, the impact of RIPK1 kinase activity-induced necroptosis has gradually emerged in research. Studies have found that RIPK1-dependent necroptosis occurs in pulmonary vascular endothelial cells, and inhibition of necroptosis can alleviate the injury of ARDS induced by a large amount of LPS in a mouse model (72–74). Furthermore, RIPK3 knockdown can also reduce the severity of LPS-induced mouse ARDS (75).
FIGURE 2
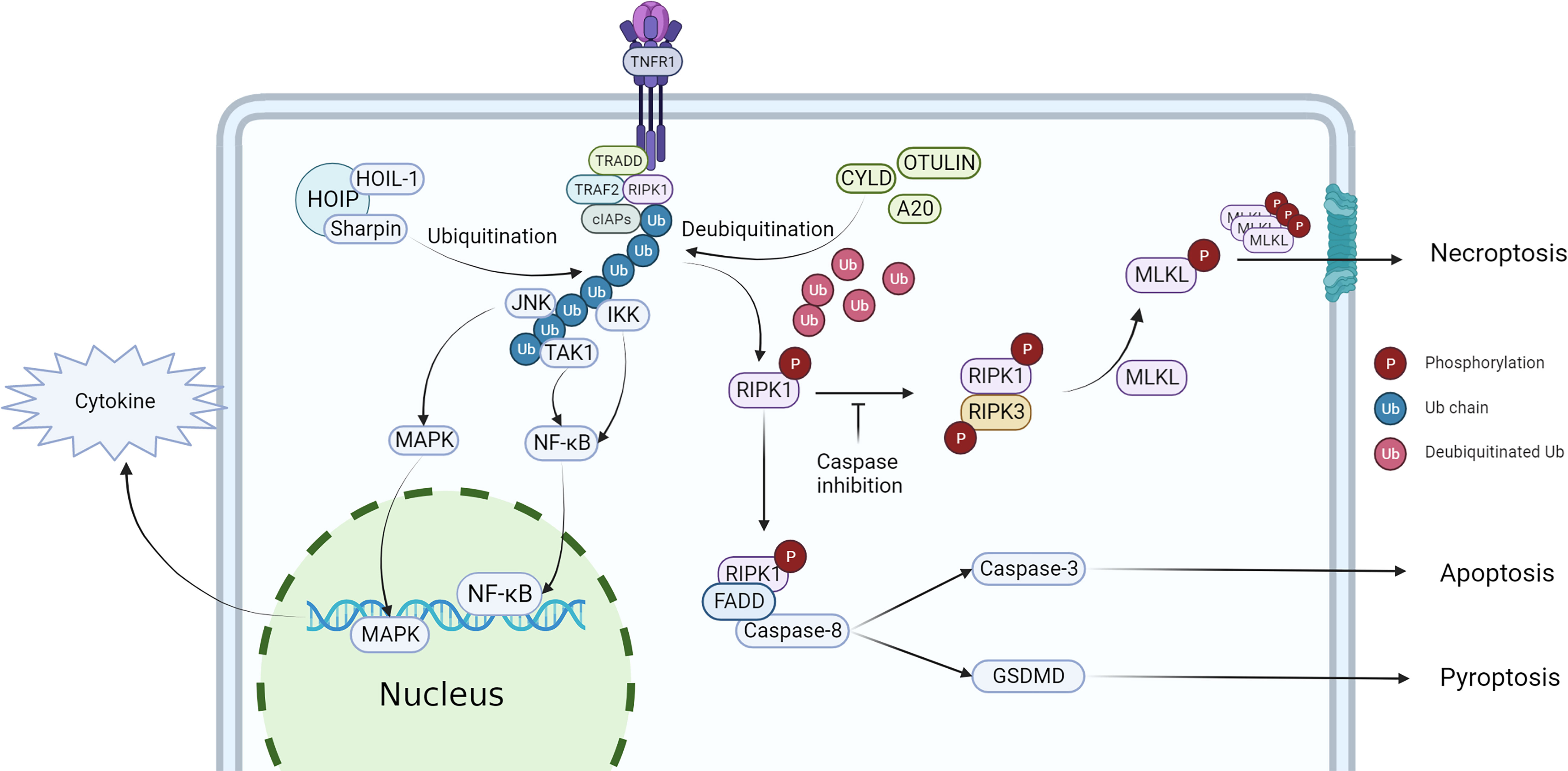
Figure 2 Regulation of RIPK1 and downstream effect. After stimulated by TNF-α, TRADD, TRAF2, RIPK1 and cIAPs would recruit to the intracellular terminus of TNFR1, then RIPK1 is ubiquitinated to form complex I, activating NF-κB and MAPK pathway. When RIPK1 has been deubiquitinated, the cell death pathway would be activated, triggering necroptosis, apoptosis and pyroptosis.
TABLE 3

Table 3 Study on RIPK1 and downstream sites in sepsis model.
Lipopolysaccharide (LPS) is a large molecule that is found in the outer membrane of gram-negative bacteria. The LPS-induced inflammatory response also contributes to the pathogenesis of sepsis. Toll-like receptor 4 (TLR4) recognizes LPS and activates downstream inflammatory response pathways (76), such as the MAPK and NF-κB pathways, through MyD88-dependent and MyD88-independent pathways. The MyD88-independent pathway requires the participation of RIPK1, which is recruited after the intracellular segment of TLR4 interacts with TRAF and immediately activates the inflammatory response pathway (77). The production of cytokines and DAMPs exacerbates the inflammatory response, ultimately inducing sepsis and organ dysfunction. Regulation of RIPK1 ubiquitination has potential value for the treatment of bacterial infections, but few studies have examined this mechanism (Table 3). In an LPS-induced septic piglet model, inhibition of RIPK1 attenuated intestinal epithelial injury, accompanied by an increase in the level of MLKL phosphorylation, suggesting that necroptosis affects intestinal epithelial cell death during sepsis (13). A similar result was observed in an LPS-induced liver injury model (78). In addition, necroptosis predominated in mice stimulated with high doses of LPS compared with low doses of LPS (75), and stimulation with various PAMPs enhanced necroptosis in macrophages expressing the RIPK1 K45A mutation (79). Wang et al. (80) found that upregulation of the deubiquitinating enzyme Cezanne in BV2 cells inhibited the NF-κB pathway, and inhibition of Cezanne was accompanied by increased levels of RIPK1 K63 ubiquitination. The importance of cell death in regulating systemic inflammation is highlighted by the RIPK1 knockout embryonic mice, which die during the embryonic stage due to systemic inflammation, and by blocking the necroptosis pathway and the MyD88 pathway reduce inflammation, but only mice with both RIPK3 and caspase-8 knockout survive after weaning (81).
In certain cases of pathogen infection, caspase8 activation can trigger pyroptosis by activating GSDMD (82). When caspase8 activity is inhibited, RIPK1 is phosphorylated and binds with RIPK3 to initiate necroptosis, providing an alternative route when pyroptosis is inhibited (69). For many years, caspase8 has been considered a key protein regulating cell death pathways, including apoptosis, pyroptosis, and necroptosis (69, 83). However, recent research has led to propose the concept of “inflammatory cell death” which integrates these pathways into a single entity known as “PANoptosis” (18, 84). At the core of PANoptosis is the PANoptosome, a complex of proteins comprising RIPK1, RIPK3, and caspase8 that form interactions through RHIM and DD domains with other related proteins to mediate pyroptosis and necroptosis (85). Prior research has shown that inhibition of RIPK1 can reduce inflammation, likely by disrupting PANoptosome formation. Therefore, the PANoptosome may serve as a critical complex in regulating inflammatory cell death (86, 87). However, the molecular mechanisms underlying its regulation of the inflammatory response are not yet fully understood, and further experimentation is needed to elucidate its therapeutic potential.
Despite the growing interest in sepsis models that focus on necroptosis or downstream regulators, the upstream pathways that regulate RIPK1 and its modifications have received comparatively less attention. Although several studies suggest that the deubiquitination of RIPK1 is a critical factor in the occurrence of necroptosis, direct evidence supporting this hypothesis remains limited. Recently, it has been proposed that cIAP2 downregulation and decreased ubiquitination of RIPK1 are evident in patients with H7N9-induced ARDS; however, changes in the ubiquitination and phosphorylation of RIPK1, or alterations in complex I, have yet to be fully elucidated (88).
6 Inhibitors of RIPK1 and its effects on sepsis
In clinical research, there has been relatively little investigation into the association between RIPK1 and sepsis (Table 4). Previous studies have suggested that serum RIPK1 does not differ significantly in relation to the severity of sepsis (95). Therefore, RIPK1 may not be suitable for determining the severity of sepsis. However, this research was based on the earlier sepsis 2.0 criteria, which differ from the currently widely used sepsis 3.0 criteria. Additionally, it did not evaluate other differences such as the degree of inflammatory response and organ function. Subsequent research has focused more on differences in indicators such as RIPK3 and MLKL (96, 97), but this does not necessarily indicate that inhibition of RIPK1 is ineffective in treating sepsis. The differences observed in RIPK3 and MLKL may be due to their location downstream in the pathway, which amplifies signals and makes them more likely to show differences. In recent years, the COVID-19 pandemic, with its prominent inflammatory response, has led some researchers to focus on RIPK1. In one study, respiratory epithelial cells collected from throat smears of symptomatic patients who had tested positive for SARS-CoV-2 by PCR showed positive results for phosphorylated RIPK1, while no phosphor-RIPK1-positive cells were detected in control samples from healthy individuals (91). Another study of critically ill COVID-19 patients found significantly higher levels of serum RIPK1 than in healthy controls, with differences observed in relation to disease severity (98).
TABLE 4

Table 4 clinical trial of RIPK1.
Given the exploration of RIPK1 in rheumatoid immune diseases and inflammatory reactions, there have been studies using RIPK1 inhibitors to regulate the inflammatory response in sepsis (Table 5). The traditional RIPK1 inhibitors, necrostatin-1 (NEC-1) and RIPA56 have been widely used in various animal inflammation models, both of which can reduce the inflammatory response and decrease necrotic apoptosis (23, 89, 90): in the TNF-α-induced SIRS model, the use of NEC-1 was found to alleviate the inflammatory response and reduce the mortality rate of mice (23).
TABLE 5
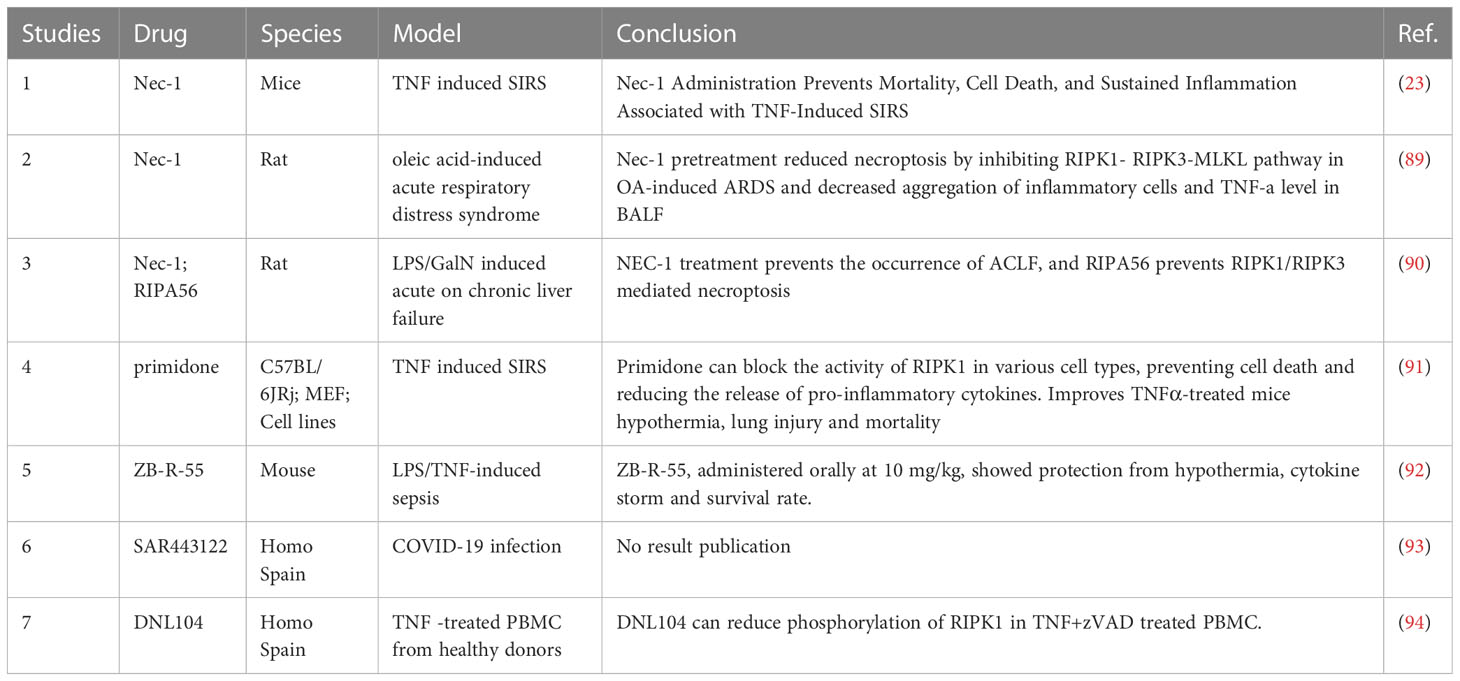
Table 5 RIPK1 inhibitors and their effect.
Subsequently, in the oleic acid-induced ARDS model, NEC-1 was also found to improve the oxygenation index of ARDS mice (89). In the mouse model of acute on chronic liver failure, using LPS to stimulate chronic liver failure mice, it was found that RIPK1-mediated cell death in liver cells could be induced, and the use of NEC-1 could alleviate the degree of acute liver failure. Furthermore, the use of the RIPK1 inhibitor, RIIPA56, which is more selective for RIPK1, led to a decrease in the expression of RIPK3 and the phosphorylation of MLKL, significantly reducing necrotic apoptosis in liver cells (90). Moreover, primidone, a drug previously used to treat epilepsy, has been found to have an inhibitory effect on RIPK1. By inhibiting the formation of complex II, primidone can reduce the occurrence of necrotic apoptosis in cells under TNF stimulation. Further animal experiments have found that primidone can alleviate renal ischemia-reperfusion injury, reduce necrotic apoptosis in the renal outer medulla, protect renal function, and also reduce the mortality rate of mice mediated by TNF-α (91). In terms of new drug development, ZB-R-55 has improved its molecular structure, pharmacokinetics, and other aspects, achieving a more efficient RIPK1 inhibitory effect. In inhibiting the cytokine storm, it has shown a similar effect to dexamethasone (92). In addition, there is a clinical trial using RIPK1 inhibitors for COVID-19 infected patients. The trial has collected data, but the results have not yet been published (93).
7 Discussion and perspectives
Sepsis is one of the most difficult clinical challenges. The mechanism by which the initial infection leads to excessive inflammatory responses remains unclear, but interventions targeting cytokine storms may be used as one of the treatments to reduce organ damage in individuals with sepsis, such as the use of glucocorticoids to reduce inflammatory responses, but the benefits are limited (100). Hemoperfusion and other approaches do not produce the ideal therapeutic effect, suggesting that perhaps interventions targeting cytokines at more upstream sites in the pathway will achieve better results. The initial immune response after pathogen invasion that causes the subsequent exacerbation of the inflammatory response is one of the theoretical bases of the cytokine storm, and the blockade of waterfall activation of the cytokine storm may be an ideal target, while the regulation of RIPK1 controls the direction of cellular outcomes after TNF-a stimulation.
Previous clinical studies have primarily focused on measuring the concentration of serum RIPK1, providing a reference for predicting the occurrence and development of sepsis. However, the advantages of RIPK1 in this direction seem to be less significant compared to downstream proteins such as RIPK3 and MLKL. From another perspective, it may be more suitable as an intervention target. As a critical node in necroptosis, apoptosis, and necroptosis, RIPK1 regulates the process of inflammatory cell death and thereby affects the course of the inflammatory response. One clear thing is that inhibiting RIPK1 can reduce RIPK3 and MLKL phosphorylation, as well as decrease the release of inflammatory factors such as HMGB-1, thus reducing the degree of the inflammatory response. In fact, it even has an effect comparable to that of dexamethasone (92). Therefore, RIPK1 inhibition may be another means of alleviating the cytokine storm associated with sepsis.
However, studies of RIPK1 in sepsis are still lacking. The effect of treatments targeting RIPK1 on the development of sepsis has not been clarified. Fortunately, there has been a growing body of research aimed at exploring the role of RIPK1 in regulating the inflammatory response in sepsis. This shift towards targeting RIPK1 function has spurred the development of numerous drugs, which are currently being evaluated in clinical trials. Thus, studies examining the regulation of RIPK1 may hopefully provide a better understanding of the mechanism of sepsis and directions for the treatment of sepsis in the future.
Author contributions
XL: Investigation, writing - original drift. A-LT: Writing – review. JC: Resources, visualization. NG: Acquisition of data. GZ: Conceptualization. CX: Supervision. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China [No. 82272196].
Acknowledgments
The authors sincerely thank the funding of the National Natural Science Foundation of China [No. 82272196].
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol (2005) 1:112–9. doi: 10.1038/nchembio711
2. Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, et al. Fas triggers an alternative, caspase-8–independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol (2000) 1:489–95. doi: 10.1038/82732
3. He S, Wang X. RIP kinases as modulators of inflammation and immunity. Nat Immunol (2018) 19:912–22. doi: 10.1038/s41590-018-0188-x
4. Ofengeim D, Yuan J. Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat Rev Mol Cell Biol (2013) 14:727–36. doi: 10.1038/nrm3683
5. Sun X, Yin J, Starovasnik MA, Fairbrother WJ, Dixit VM. Identification of a novel homotypic interaction motif required for the phosphorylation of receptor-interacting protein (RIP) by RIP3 *. J Biol Chem (2002) 277:9505–11. doi: 10.1074/jbc.M109488200
6. Hsu H, Huang J, Shu H-B, Baichwal V, Goeddel DV. TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity (1996) 4:387–96. doi: 10.1016/S1074-7613(00)80252-6
7. Berghe TV, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol (2014) 15:135–47. doi: 10.1038/nrm3737
8. Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell (2003) 114:181–90. doi: 10.1016/S0092-8674(03)00521-X
9. Jang K-H, Jang T, Son E, Choi S, Kim E. Kinase-independent role of nuclear RIPK1 in regulating parthanatos through physical interaction with PARP1 upon oxidative stress. Biochim Biophys Acta BBA - Mol Cell Res (2018) 1865:132–41. doi: 10.1016/j.bbamcr.2017.10.004
10. Ramnarain DB, Paulmurugan R, Park S, Mickey BE, Asaithamby A, Saha D, et al. RIP1 links inflammatory and growth factor signaling pathways by regulating expression of the EGFR. Cell Death Differ (2008) 15:344–53. doi: 10.1038/sj.cdd.4402268
11. Weber K, Roelandt R, Bruggeman I, Estornes Y, Vandenabeele P. Nuclear RIPK3 and MLKL contribute to cytosolic necrosome formation and necroptosis. Commun Biol (2018) 1:6. doi: 10.1038/s42003-017-0007-1
12. Yoon S, Bogdanov K, Kovalenko A, Wallach D. Necroptosis is preceded by nuclear translocation of the signaling proteins that induce it. Cell Death Differ (2016) 23:253–60. doi: 10.1038/cdd.2015.92
13. Liu Y, Xu Q, Wang Y, Liang T, Li X, Wang D, et al. Necroptosis is active and contributes to intestinal injury in a piglet model with lipopolysaccharide challenge. Cell Death Dis (2021) 12:62. doi: 10.1038/s41419-020-03365-1
14. Feoktistova M, Makarov R, Brenji S, Schneider AT, Hooiveld GJ, Luedde T, et al. A20 promotes ripoptosome formation and TNF-induced apoptosis via cIAPs regulation and NIK stabilization in keratinocytes. Cells (2020) 9:E351. doi: 10.3390/cells9020351
15. Yuan J, Amin P, Ofengeim D. Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat Rev Neurosci (2019) 20:19–33. doi: 10.1038/s41583-018-0093-1
16. DeRoo E, Zhou T, Liu B. The role of RIPK1 and RIPK3 in cardiovascular disease. Int J Mol Sci (2020) 21:E8174. doi: 10.3390/ijms21218174
17. Ofengeim D, Ito Y, Najafov A, Zhang Y, Shan B, DeWitt JP, et al. Activation of necroptosis in multiple sclerosis. Cell Rep (2015) 10:1836–49. doi: 10.1016/j.celrep.2015.02.051
18. Malireddi RKS, Gurung P, Kesavardhana S, Samir P, Burton A, Mummareddy H, et al. Innate immune priming in the absence of TAK1 drives RIPK1 kinase activity-independent pyroptosis, apoptosis, necroptosis, and inflammatory disease. J Exp Med (2020) 217:e20191644. doi: 10.1084/jem.20191644
19. Slebos D-J, Ryter SW, van der Toorn M, Liu F, Guo F, Baty CJ, et al. Mitochondrial localization and function of heme oxygenase-1 in cigarette smoke-induced cell death. Am J Respir Cell Mol Biol (2007) 36:409–17. doi: 10.1165/rcmb.2006-0214OC
20. Mizumura K, Cloonan SM, Nakahira K, Bhashyam AR, Cervo M, Kitada T, et al. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J Clin Invest (2014) 124:3987–4003. doi: 10.1172/JCI74985
21. Li Y, Führer M, Bahrami E, Socha P, Klaudel-Dreszler M, Bouzidi A, et al. Human RIPK1 deficiency causes combined immunodeficiency and inflammatory bowel diseases. Proc Natl Acad Sci USA. (2019) 116:970–5. doi: 10.1073/pnas.1813582116
22. Cuchet-Lourenço D, Eletto D, Wu C, Plagnol V, Papapietro O, Curtis J, et al. Biallelic RIPK1 mutations in humans cause severe immunodeficiency, arthritis, and intestinal inflammation. Science (2018) 361:810–3. doi: 10.1126/science.aar2641
23. Duprez L, Takahashi N, Van Hauwermeiren F, Vandendriessche B, Goossens V, Vanden Berghe T, et al. RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity (2011) 35:908–18. doi: 10.1016/j.immuni.2011.09.020
24. Dillon CP, Weinlich R, Rodriguez DA, Cripps JG, Quarato G, Gurung P, et al. RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell (2014) 157:1189–202. doi: 10.1016/j.cell.2014.04.018
25. Wang Q, Fan D, Xia Y, Ye Q, Xi X, Zhang G, et al. The latest information on the RIPK1 post-translational modifications and functions. BioMed Pharmacother (2021) 142:112082. doi: 10.1016/j.biopha.2021.112082
26. Kaiser WJ, Daley-Bauer LP, Thapa RJ, Mandal P, Berger SB, Huang C, et al. RIP1 suppresses innate immune necrotic as well as apoptotic cell death during mammalian parturition. Proc Natl Acad Sci U.S.A. (2014) 111:7753–8. doi: 10.1073/pnas.1401857111
27. Newton K, Dugger DL, Wickliffe KE, Kapoor N, de Almagro MC, Vucic D, et al. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science (2014) 343:1357–60. doi: 10.1126/science.1249361
28. Stark K, Goncharov T, Varfolomeev E, Xie L, Ngu H, Peng I, et al. Genetic inactivation of RIP1 kinase activity in rats protects against ischemic brain injury. Cell Death Dis (2021) 12:1–15. doi: 10.1038/s41419-021-03651-6
29. Newton K, Dugger DL, Maltzman A, Greve JM, Hedehus M, Martin-McNulty B, et al. RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Differ (2016) 23:1565–76. doi: 10.1038/cdd.2016.46
30. Zhang Y, Su SS, Zhao S, Yang Z, Zhong C-Q, Chen X, et al. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat Commun (2017) 8:14329. doi: 10.1038/ncomms14329
31. Laurien L, Nagata M, Schünke H, Delanghe T, Wiederstein JL, Kumari S, et al. Autophosphorylation at serine 166 regulates RIP kinase 1-mediated cell death and inflammation. Nat Commun (2020) 11:1747. doi: 10.1038/s41467-020-15466-8
32. Dondelinger Y, Delanghe T, Priem D, Wynosky-Dolfi MA, Sorobetea D, Rojas-Rivera D, et al. Serine 25 phosphorylation inhibits RIPK1 kinase-dependent cell death in models of infection and inflammation. Nat Commun (2019) 10:1729. doi: 10.1038/s41467-019-09690-0
33. Du J, Xiang Y, Liu H, Liu S, Kumar A, Xing C, et al. RIPK1 dephosphorylation and kinase activation by PPP1R3G/PP1γ promote apoptosis and necroptosis. Nat Commun (2021) 12:7067. doi: 10.1038/s41467-021-27367-5
34. Lafont E, Draber P, Rieser E, Reichert M, Kupka S, de Miguel D, et al. TBK1 and IKKϵ prevent TNF-induced cell death by RIPK1 phosphorylation. Nat Cell Biol (2018) 20:1389–99. doi: 10.1038/s41556-018-0229-6
35. Jaco I, Annibaldi A, Lalaoui N, Wilson R, Tenev T, Laurien L, et al. MK2 phosphorylates RIPK1 to prevent TNF-induced cell death. Mol Cell (2017) 66:698–710.e5. doi: 10.1016/j.molcel.2017.05.003
36. Menon MB, Gropengießer J, Fischer J, Novikova L, Deuretzbacher A, Lafera J, et al. p38MAPK/MK2-dependent phosphorylation controls cytotoxic RIPK1 signalling in inflammation and infection. Nat Cell Biol (2017) 19:1248–59. doi: 10.1038/ncb3614
37. Geng J, Ito Y, Shi L, Amin P, Chu J, Ouchida AT, et al. Regulation of RIPK1 activation by TAK1-mediated phosphorylation dictates apoptosis and necroptosis. Nat Commun (2017) 8:359. doi: 10.1038/s41467-017-00406-w
38. Kist M, Kőműves LG, Goncharov T, Dugger DL, Yu C, Roose-Girma M, et al. Impaired RIPK1 ubiquitination sensitizes mice to TNF toxicity and inflammatory cell death. Cell Death Differ (2021) 28:985–1000. doi: 10.1038/s41418-020-00629-3
39. de Almagro MC, Goncharov T, Izrael-Tomasevic A, Duttler S, Kist M, Varfolomeev E, et al. Coordinated ubiquitination and phosphorylation of RIP1 regulates necroptotic cell death. Cell Death Differ (2017) 24:26–37. doi: 10.1038/cdd.2016.78
40. He M, Wang M, Huang Y, Peng W, Zheng Z, Xia N, et al. The ORF3 protein of genotype 1 hepatitis e virus suppresses TLR3-induced NF-κB signaling via TRADD and RIP1. Sci Rep (2016) 6:27597. doi: 10.1038/srep27597
41. Meng H, Liu Z, Li X, Wang H, Jin T, Wu G, et al. Death-domain dimerization-mediated activation of RIPK1 controls necroptosis and RIPK1-dependent apoptosis. Proc Natl Acad Sci (2018) 115:E2001–9. doi: 10.1073/pnas.1722013115
42. Li X, Zhang M, Huang X, Liang W, Li G, Lu X, et al. Ubiquitination of RIPK1 regulates its activation mediated by TNFR1 and TLRs signaling in distinct manners. Nat Commun (2020) 11:6364. doi: 10.1038/s41467-020-19935-y
43. Tu H, Tang Y, Zhang J, Cheng L, Joo D, Zhao X, et al. Linear ubiquitination of RIPK1 on Lys612 regulates systemic inflammation via preventing cell death. J Immunol (2021) 207:602–12. doi: 10.4049/jimmunol.2100299
44. Keusekotten K, Elliott PR, Glockner L, Fiil BK, Damgaard RB, Kulathu Y, et al. OTULIN antagonizes LUBAC signaling by specifically hydrolyzing Met1-linked polyubiquitin. Cell (2013) 153:1312–26. doi: 10.1016/j.cell.2013.05.014
45. Damgaard RB, Walker JA, Marco-Casanova P, Morgan NV, Titheradge HL, Elliott PR, et al. The deubiquitinase OTULIN is an essential negative regulator of inflammation and autoimmunity. Cell (2016) 166:1215–1230.e20. doi: 10.1016/j.cell.2016.07.019
46. Mahoney DJ, Cheung HH, Mrad RL, Plenchette S, Simard C, Enwere E, et al. Both cIAP1 and cIAP2 regulate TNFalpha-mediated NF-kappaB activation. Proc Natl Acad Sci U.S.A. (2008) 105:11778–83. doi: 10.1073/pnas.0711122105
47. Zhang J, Webster JD, Dugger DL, Goncharov T, Roose-Girma M, Hung J, et al. Ubiquitin ligases cIAP1 and cIAP2 limit cell death to prevent inflammation. Cell Rep (2019) 27:2679–2689.e3. doi: 10.1016/j.celrep.2019.04.111
48. Wong WW-L, Vince JE, Lalaoui N, Lawlor KE, Chau D, Bankovacki A, et al. cIAPs and XIAP regulate myelopoiesis through cytokine production in an RIPK1- and RIPK3-dependent manner. Blood (2014) 123:2562–72. doi: 10.1182/blood-2013-06-510743
49. Liu F-Q. Analysis of differentially expressed genes in rheumatoid arthritis and osteoarthritis by integrated microarray analysis. J Cell Biochem (2019) 120:12653–64. doi: 10.1002/jcb.28533
50. Nguyen VQ, Eden K, Morrison HA, Sammons MB, Knight KK, Sorrentino S, et al. Noncanonical NF-κB signaling upregulation in inflammatory bowel disease patients is associated with loss of response to anti-TNF agents. Front Pharmacol (2021) 12:655887. doi: 10.3389/fphar.2021.655887
51. Ekedahl J, Bertrand J, Grigoriev MY, Muller M, Magnusson C, Lewensohn R, et al. Expression of inhibitor of apoptosis proteins in small- and non-small-cell lung carcinoma cells. Exp Cell Res (2002) 279:277–90. doi: 10.1006/excr.2002.5608
52. Oda H, Beck DB, Kuehn HS, Sampaio Moura N, Hoffmann P, Ibarra M, et al. Second case of HOIP deficiency expands clinical features and defines inflammatory transcriptome regulated by LUBAC. Front Immunol (2019) 10:479. doi: 10.3389/fimmu.2019.00479
53. Rickard JA, Anderton H, Etemadi N, Nachbur U, Darding M, Peltzer N, et al. TNFR1-dependent cell death drives inflammation in sharpin-deficient mice. eLife (2014) 3:e03464. doi: 10.7554/eLife.03464
54. Peltzer N, Darding M, Montinaro A, Draber P, Draberova H, Kupka S, et al. LUBAC is essential for embryogenesis by preventing cell death and enabling haematopoiesis. Nature (2018) 557:112–7. doi: 10.1038/s41586-018-0064-8
55. Kumari S, Redouane Y, Lopez-Mosqueda J, Shiraishi R, Romanowska M, Lutzmayer S, et al. Sharpin prevents skin inflammation by inhibiting TNFR1-induced keratinocyte apoptosis. eLife (2014) 3:e03422. doi: 10.7554/eLife.03422
56. Peltzer N, Rieser E, Taraborrelli L, Draber P, Darding M, Pernaute B, et al. HOIP deficiency causes embryonic lethality by aberrant TNFR1-mediated endothelial cell death. Cell Rep (2014) 9:153–65. doi: 10.1016/j.celrep.2014.08.066
57. Adrianto I, Wen F, Templeton A, Wiley G, King JB, Lessard CJ, et al. Association of a functional variant downstream of TNFAIP3 with systemic lupus erythematosus. Nat Genet (2011) 43:253–8. doi: 10.1038/ng.766
58. Wang S, Wen F, Tessneer KL, Gaffney PM. TALEN-mediated enhancer knockout influences TNFAIP3 gene expression and mimics a molecular phenotype associated with systemic lupus erythematosus. Genes Immun (2016) 17:165–70. doi: 10.1038/gene.2016.4
59. Wang S, Wen F, Wiley GB, Kinter MT, Gaffney PM. An enhancer element harboring variants associated with systemic lupus erythematosus engages the TNFAIP3 promoter to influence A20 expression. PloS Genet (2013) 9:e1003750. doi: 10.1371/journal.pgen.1003750
60. Zhou Q, Wang H, Schwartz DM, Stoffels M, Park YH, Zhang Y, et al. Loss-of-function mutations in TNFAIP3 leading to A20 haploinsufficiency cause an early-onset autoinflammatory disease. Nat Genet (2016) 48:67–73. doi: 10.1038/ng.3459
61. Yu M-P, Xu X-S, Zhou Q, Deuitch N, Lu M-P. Haploinsufficiency of A20 (HA20): updates on the genetics, phenotype, pathogenesis and treatment. World J Pediatr WJP (2020) 16:575–84. doi: 10.1007/s12519-019-00288-6
62. Young AL, Kellermayer R, Szigeti R, Tészás A, Azmi S, Celebi JT. CYLD mutations underlie brooke-spiegler, familial cylindromatosis, and multiple familial trichoepithelioma syndromes. Clin Genet (2006) 70:246–9. doi: 10.1111/j.1399-0004.2006.00667.x
63. Zhao M, Song K, Hao W, Wang L, Patil G, Li Q, et al. Non-proteolytic ubiquitination of OTULIN regulates NF-κB signaling pathway. J Mol Cell Biol (2020) 12:163–75. doi: 10.1093/jmcb/mjz081
64. Zhou Q, Yu X, Demirkaya E, Deuitch N, Stone D, Tsai WL, et al. Biallelic hypomorphic mutations in a linear deubiquitinase define otulipenia, an early-onset autoinflammatory disease. Proc Natl Acad Sci U.S.A. (2016) 113:10127–32. doi: 10.1073/pnas.1612594113
65. Polykratis A, Hermance N, Zelic M, Roderick J, Kim C, Van T-M, et al. RIPK1 kinase inactive mice are viable and protected from TNF-induced necroptosis in vivo. J Immunol Baltim Md 1950 (2014) 193:1539–43. doi: 10.4049/jimmunol.1400590
66. Tang Y, Tu H, Zhang J, Zhao X, Wang Y, Qin J, et al. K63-linked ubiquitination regulates RIPK1 kinase activity to prevent cell death during embryogenesis and inflammation. Nat Commun (2019) 10:4157. doi: 10.1038/s41467-019-12033-8
67. Boisson B, Laplantine E, Dobbs K, Cobat A, Tarantino N, Hazen M, et al. Human HOIP and LUBAC deficiency underlies autoinflammation, immunodeficiency, amylopectinosis, and lymphangiectasia. J Exp Med (2015) 212:939–51. doi: 10.1084/jem.20141130
68. Damgaard RB, Elliott PR, Swatek KN, Maher ER, Stepensky P, Elpeleg O, et al. OTULIN deficiency in ORAS causes cell type-specific LUBAC degradation, dysregulated TNF signalling and cell death. EMBO Mol Med (2019) 11:e9324. doi: 10.15252/emmm.201809324
69. Fritsch M, Günther SD, Schwarzer R, Albert M-C, Schorn F, Werthenbach JP, et al. Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature (2019) 575:683–7. doi: 10.1038/s41586-019-1770-6
70. González-Juarbe N, Gilley RP, Hinojosa CA, Bradley KM, Kamei A, Gao G, et al. Pore-forming toxins induce macrophage necroptosis during acute bacterial pneumonia. PloS Pathog (2015) 11:e1005337. doi: 10.1371/journal.ppat.1005337
71. Weng D, Marty-Roix R, Ganesan S, Proulx MK, Vladimer GI, Kaiser WJ, et al. Caspase-8 and RIP kinases regulate bacteria-induced innate immune responses and cell death. Proc Natl Acad Sci U.S.A. (2014) 111:7391–6. doi: 10.1073/pnas.1403477111
72. Zelic M, Roderick JE, O’Donnell JA, Lehman J, Lim SE, Janardhan HP, et al. RIP kinase 1-dependent endothelial necroptosis underlies systemic inflammatory response syndrome. J Clin Invest (2018) 128:2064–75. doi: 10.1172/JCI96147
73. Wang L, Zhou L, Zhou Y, Liu L, Jiang W, Zhang H, et al. Necroptosis in pulmonary diseases: A new therapeutic target. Front Pharmacol (2021) 12:737129. doi: 10.3389/fphar.2021.737129
74. Yu X, Mao M, Liu X, Shen T, Li T, Yu H, et al. A cytosolic heat shock protein 90 and co-chaperone p23 complex activates RIPK3/MLKL during necroptosis of endothelial cells in acute respiratory distress syndrome. J Mol Med Berl Ger (2020) 98:569–83. doi: 10.1007/s00109-020-01886-y
75. Wang L, Wang T, Li H, Liu Q, Zhang Z, Xie W, et al. Receptor interacting protein 3-mediated necroptosis promotes lipopolysaccharide-induced inflammation and acute respiratory distress syndrome in mice. PloS One (2016) 11:e0155723. doi: 10.1371/journal.pone.0155723
76. Murad S. Toll-like receptor 4 in inflammation and angiogenesis: A double-edged sword. Front Immunol (2014) 5:313. doi: 10.3389/fimmu.2014.00313
77. Bohannon JK, Hernandez A, Enkhbaatar P, Adams WL, Sherwood ER. The immunobiology of toll-like receptor 4 agonists: From endotoxin tolerance to immunoadjuvants. Shock (2013) 40:451–62. doi: 10.1097/SHK.0000000000000042
78. Xu Q, Guo J, Li X, Wang Y, Wang D, Xiao K, et al. Necroptosis underlies hepatic damage in a piglet model of lipopolysaccharide-induced sepsis. Front Immunol (2021) 12:633830. doi: 10.3389/fimmu.2021.633830
79. Shutinoski B, Alturki NA, Rijal D, Bertin J, Gough PJ, Schlossmacher MG, et al. K45A mutation of RIPK1 results in poor necroptosis and cytokine signaling in macrophages, which impacts inflammatory responses. vivo. Cell Death Differ (2016) 23:1628–37. doi: 10.1038/cdd.2016.51
80. Wang Z-C, Chen Q, Wang J, Yu L-S, Chen L-W. Sulforaphane mitigates LPS-induced neuroinflammation through modulation of Cezanne/NF-κB signalling. Life Sci (2020) 262:118519. doi: 10.1016/j.lfs.2020.118519
81. Rickard JA, O’Donnell JA, Evans JM, Lalaoui N, Poh AR, Rogers T, et al. RIPK1 regulates RIPK3-MLKL-driven systemic inflammation and emergency hematopoiesis. Cell (2014) 157:1175–88. doi: 10.1016/j.cell.2014.04.019
82. Zheng Z, Deng W, Bai Y, Miao R, Mei S, Zhang Z, et al. The lysosomal rag-ragulator complex licenses RIPK1 and caspase-8-mediated pyroptosis by yersinia. Science (2021) 372:eabg0269. doi: 10.1126/science.abg0269
83. Bertheloot D, Latz E, Franklin BS. Necroptosis, pyroptosis and apoptosis: an intricate game of cell death. Cell Mol Immunol (2021) 18:1106–21. doi: 10.1038/s41423-020-00630-3
84. Malireddi RKS, Kesavardhana S, Kanneganti T-D. ZBP1 and TAK1: Master regulators of NLRP3 Inflammasome/Pyroptosis, apoptosis, and necroptosis (PAN-optosis). Front Cell Infect Microbiol (2019) 9:406. doi: 10.3389/fcimb.2019.00406
85. Malireddi RKS, Kesavardhana S, Karki R, Kancharana B, Burton AR, Kanneganti T-D. RIPK1 distinctly regulates yersinia-induced inflammatory cell death, PANoptosis. ImmunoHorizons (2020) 4:789–96. doi: 10.4049/immunohorizons.2000097
86. Samir P, Malireddi RKS, Kanneganti T-D. The PANoptosome: A deadly protein complex driving pyroptosis, apoptosis, and necroptosis (PANoptosis). Front Cell Infect Microbiol (2020) 10:238. doi: 10.3389/fcimb.2020.00238
87. Wang Y, Kanneganti T-D. From pyroptosis, apoptosis and necroptosis to PANoptosis: A mechanistic compendium of programmed cell death pathways. Comput Struct Biotechnol J (2021) 19:4641–57. doi: 10.1016/j.csbj.2021.07.038
88. Qin C, Sai X-Y, Qian X-F, Wu Y, Zou L-F, Wang H-M, et al. Close relationship between cIAP2 and human ARDS induced by severe H7N9 infection. BioMed Res Int (2019) 2019:2121357. doi: 10.1155/2019/2121357
89. Pan L, Yao D-C, Yu Y-Z, Li S-J, Chen B-J, Hu G-H, et al. Necrostatin-1 protects against oleic acid-induced acute respiratory distress syndrome in rats. Biochem Biophys Res Commun (2016) 478:1602–8. doi: 10.1016/j.bbrc.2016.08.163
90. Kondo T, Macdonald S, Engelmann C, Habtesion A, Macnaughtan J, Mehta G, et al. The role of RIPK1 mediated cell death in acute on chronic liver failure. Cell Death Dis (2021) 13:1–12. doi: 10.1038/s41419-021-04442-9
91. Riebeling T, Jamal K, Wilson R, Kolbrink B, von Samson-Himmelstjerna FA, Moerke C, et al. Primidone blocks RIPK1-driven cell death and inflammation. Cell Death Differ (2021) 28:1610–26. doi: 10.1038/s41418-020-00690-y
92. Yang X, Lu H, Xie H, Zhang B, Nie T, Fan C, et al. Potent and selective RIPK1 inhibitors targeting dual-pockets for the treatment of systemic inflammatory response syndrome and sepsis. Angew Chem Int Ed Engl (2022) 61:e202114922. doi: 10.1002/anie.202114922
93. Clot P-F, Farenc C, Suratt B, Krahnke T, Tardat A, Florian P, et al. Late breaking abstract - immunomodulatory and clinical effects of RIPK1 inhibitor SAR443122 in patients with severe COVID-19. Eur Respir J (2021) 58:PA2363. doi: 10.1183/13993003.congress-2021.PA2363
94. Grievink HW, Heuberger JAAC, Huang F, Chaudhary R, Birkhoff WAJ, Tonn GR, et al. DNL104, a centrally penetrant RIPK1 inhibitor, inhibits RIP1 kinase phosphorylation in a randomized phase I ascending dose study in healthy volunteers. Clin Pharmacol Ther (2020) 107:406–14. doi: 10.1002/cpt.1615
95. Wang B, Li J, Gao H-M, Xing Y-H, Lin Z, Li H-J, et al. Necroptosis regulated proteins expression is an early prognostic biomarker in patient with sepsis: a prospective observational study. Oncotarget (2017) 8:84066–73. doi: 10.18632/oncotarget.21099
96. Mallarpu CS, Ponnana M, Prasad S, Singarapu M, Kim J, Haririparsa N, et al. Distinct cell death markers identified in critical care patient survivors diagnosed with sepsis. Immunol Lett (2021) 231:1–10. doi: 10.1016/j.imlet.2020.12.009
97. Yoo H, Im Y, Ko R-E, Lee JY, Park J, Jeon K. Association of plasma level of high-mobility group box-1 with necroptosis and sepsis outcomes. Sci Rep (2021) 11:9512. doi: 10.1038/s41598-021-88970-6
98. Ruskowski K, Neb H, Talbot SR, Choorapoikayil S, Adam EH, von Knethen A, et al. Persistently elevated plasma concentrations of RIPK3, MLKL, HMGB1, and RIPK1 in patients with COVID-19 in the intensive care unit. Am J Respir Cell Mol Biol (2022) 67:405–8. doi: 10.1165/rcmb.2022-0039LE
99. Siempos II, Ma KC, Imamura M, Baron RM, Fredenburgh LE, Huh J-W, et al. RIPK3 mediates pathogenesis of experimental ventilator-induced lung injury. JCI Insight (2018) 3:e97102. doi: 10.1172/jci.insight.97102
100. Dellinger RP, Bagshaw SM, Antonelli M, Foster DM, Klein DJ, Marshall JC, et al. Effect of targeted polymyxin b hemoperfusion on 28-day mortality in patients with septic shock and elevated endotoxin level: The EUPHRATES randomized clinical trial. JAMA (2018) 320:1455–63. doi: 10.1001/jama.2018.14618
Keywords: RIPK1, sepsis, cytokine storm, inflammatory response, PANoptosis
Citation: Liu X, Tang A-L, Chen J, Gao N, Zhang G and Xiao C (2023) RIPK1 in the inflammatory response and sepsis: Recent advances, drug discovery and beyond. Front. Immunol. 14:1114103. doi: 10.3389/fimmu.2023.1114103
Received: 22 December 2022; Accepted: 24 March 2023;
Published: 05 April 2023.
Edited by:
Jianke Zhang, Thomas Jefferson University, United StatesReviewed by:
Chunlin Zhuang, Second Military Medical University, ChinaDomagoj Vucic, Genentech Inc., United States
Chunxia Wang, Shanghai Children’s Hospital, China
Copyright © 2023 Liu, Tang, Chen, Gao, Zhang and Xiao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guoqiang Zhang, emhhbmdjaG9uZzIwMDNAdmlwLnNpbmEuY29t; Cheng Xiao, eGMyMDAyODEyQDEyNi5jb20=