 Alexandra T. Matola1,2
Alexandra T. Matola1,2 Angéla Fülöp3Bernadette Rojkovich3
Angéla Fülöp3Bernadette Rojkovich3 György Nagy4,5,6,7
György Nagy4,5,6,7 Gabriella Sármay1
Gabriella Sármay1 Mihály Józsi1,2*†
Mihály Józsi1,2*† Barbara Uzonyi1,2†
Barbara Uzonyi1,2†- 1Department of Immunology, ELTE Eötvös Loránd University, Budapest, Hungary
- 2MTA-ELTE Complement Research Group, Eötvös Loránd Research Network (ELKH) at the Department of Immunology, ELTE Eötvös Loránd University, Budapest, Hungary
- 3Buda Hospital of the Hospitaller Order of Saint John of God, Budapest, Hungary
- 4Department of Rheumatology and Clinical Immunology, Semmelweis University, Budapest, Hungary
- 5Department of Internal Medicine and Oncology, Semmelweis University, Budapest, Hungary
- 6Heart and Vascular Center, Faculty of Medicine, Semmelweis University, Budapest, Hungary
- 7Department of Genetics, Cell- and Immunobiology, Faculty of Medicine, Semmelweis University, Budapest, Hungary
Rheumatoid arthritis (RA) is a chronic inflammatory autoimmune disorder affecting the joints. Many patients carry anti-citrullinated protein autoantibodies (ACPA). Overactivation of the complement system seems to be part of the pathogenesis of RA, and autoantibodies against the pathway initiators C1q and MBL, and the regulator of the complement alternative pathway, factor H (FH), were previously reported. Our aim was to analyze the presence and role of autoantibodies against complement proteins in a Hungarian RA cohort. To this end, serum samples of 97 ACPA-positive RA patients and 117 healthy controls were analyzed for autoantibodies against FH, factor B (FB), C3b, C3-convertase (C3bBbP), C1q, MBL and factor I. In this cohort, we did not detect any patient with FH autoantibodies but detected C1q autoantibodies in four patients, MBL autoantibodies in two patients and FB autoantibodies in five patients. Since the latter autoantibodies were previously reported in patients with kidney diseases but not in RA, we set out to further characterize such FB autoantibodies. The isotypes of the analyzed autoantibodies were IgG2, IgG3, IgGκ, IgGλ and their binding site was localized in the Bb part of FB. We detected in vivo formed FB–autoanti-FB complexes by Western blot. The effect of the autoantibodies on the formation, activity and FH-mediated decay of the C3 convertase in solid phase convertase assays was determined. In order to investigate the effect of the autoantibodies on complement functions, hemolysis assays and fluid phase complement activation assays were performed. The autoantibodies partially inhibited the complement-mediated hemolysis of rabbit red blood cells, inhibited the activity of the solid phase C3-convertase and C3 and C5b-9 deposition on complement activating surfaces. In summary, in ACPA-positive RA patients we identified FB autoantibodies. The characterized FB autoantibodies did not enhance complement activation, rather, they had inhibitory effect on complement. These results support the involvement of the complement system in the pathomechanism of RA and raise the possibility that protective autoantibodies may be generated in some patients against the alternative pathway C3 convertase. However, further analyses are needed to assess the exact role of such autoantibodies.
Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory autoimmune disorder targeting typically the peripheral joints of the hands and feet, leading to persistent inflammation. With time it may cause the formation of a new tissue with infiltrating inflammatory immune cells called pannus and the destruction of the bones and cartilage which may give rise to deformities (1, 2). RA affects approximately 0.5-1% of the population (3) with slightly different frequencies between geographical regions and populations (4–6). RA affects mainly women with rising prevalence with age (7, 8). Though the cause of RA is unknown, several predisposing factors may play a role in disease development: environmental exposures such as smoking (9), infection and genetic factors (3, 10, 11). Presumably, hormonal factors also play a role in the mechanism causing the overrepresentation of women in the disease (8, 12).
Most of the patients develop rheumatoid factor or anti-modified protein antibodies. The classical serological marker of RA was rheumatoid factor, an autoantibody targeting the Fc region of IgG. It has relatively low specificity as it can be found in other pathological conditions and healthy individuals (8, 13, 14). By contrast, autoantibodies against modified (e.g., carbamylated, citrullinated, acetylated) self-proteins are more specific for RA and can be detected in patients before clinical onset, in the subclinical stage (8, 15). The most frequently detected anti-citrullinated protein antibody (ACPA) is a diagnostic criterion of RA (16) and the presence of ACPA can predict the development of the disease (17–19).
The above-mentioned autoantibodies can form immune complexes with self-antigens in the patients and activate complement, which plays a remarkable role in the pathogenesis of RA (20). The complement system as part of the innate immune system has a crucial role in host defense including elimination of microbes, apoptotic and necrotic cells, and the clearance of immune complexes (21). The activation trigger defines by which of the three pathways will complement be activated. The classical pathway is initiated by the binding of its pattern recognition molecule C1q to IgG- or IgM-containing immune complexes. The lectin pathway is induced by sensing microbial carbohydrate structures, and the alternative pathway is constantly activated on a low level (C3 tick over mechanism) due to the spontaneous hydrolysis of the thioester group of the central C3 molecule (C3(H2O)). Thus, factor B (FB) bound to C3(H2O) and cleaved by factor D (FD), forms the fluid phase C3 convertase of the alternative pathway, C3(H2O)Bb. This in turn cleaves C3, generating C3b which can covalently bind to target surfaces and together with FB (again, cleaved by FD) and the stabilizing properdin forms the C3 convertase of the alternative pathway C3bBbP. The activation of the cascades leads to the generation of small anaphylatoxins (e.g., C3a, C5a) and larger fragments which opsonize the target (20–23). To avoid activation on host surfaces, complement is tightly regulated at several points by membrane-bound and fluid-phase regulators (24). In case of mutations or presence of autoantibodies against complement components and/or regulators, the system turns defective, and plays an important role in the pathomechanism of several autoimmune diseases (20, 25).
The involvement of the complement system in RA has long been studied. Decreased levels of C3 and C4 proteins (26) and elevated levels of complement activation products C3a, C5a, Bb and soluble C5b-9 (sC5b-9) in the synovial fluid of patients suggested the overactivation of the complement system (27–29). Elevated levels of sC5b-9 in patients’ plasma (30, 31) indicate that the complement activation in RA is not limited to the joints but may affect other organs and tissues. Furthermore, treatment with anti-rheumatic drugs can decrease the elevated sC5b-9 levels (31). Autoantibodies against complement components were found in RA patients, including C1q (32, 33), factor H (FH) (34) and mannose binding lectin (MBL) (35), further confirming the involvement of the three complement pathways in the pathomechanism of RA, although the functions and relevance of these autoantibodies were not investigated and are still unknown.
To assess the role of anti-complement autoantibodies in RA, the aim of this study was to investigate whether the previously reported autoantibodies can be detected and/or other, novel autoantibodies can be found in a Hungarian cohort of RA patients. Here, we report the identification and characterization of FB autoantibodies in RA.
Materials and methods
Proteins and antibodies
Complement proteins FH (#341274), FB (#341262), C3b (#204860), C3 (#204885), factor I (FI) (#341280), C1q (#204876), goat anti-FB (#341272) and goat anti-FH (#341276) antiserum were obtained from Merck (Darmstadt, Germany; obtained via Merck Life Science Kft., Budapest, Hungary). FD (#A409), properdin (#A412), mouse anti-C5b-9 (#A239), anti-FB (Bb) (#A227), anti-FB (Ba) (#A225) and C3a EIA kit (#A032) were from Quidel (obtained via Biomedica, Budapest, Hungary). Purified human IgG (#I2511), human serum albumin (#A3782), human alpha-1 antitrypsin (#SRP6312), antibodies against C1q (#234390), human IgG (#A6029), IgM (#A0420), IgA (#A0295), IgG1 (clone HP-6001, #I9388), IgG2 (clone HP-6002, #I9513), IgG3 (clone HP-6050, #I7260), IgG4 (clone HP-6025, #I7385), IgGκ (clone KP-53, #K4377), IgGλ (clone HP-6054, #L6522), and avidin-HRP (#A7419) were purchased from Merck. Mouse IgG1 (clone MOPC-21, #BZ-400101) was obtained from BioLegend (San Diego, CA). MBL (#9086-MB) and biotin-conjugated goat anti-MBL (#BAF2307) were from R&D Systems (Minneapolis, MN), HRP-conjugated goat anti-mouse (#P0447), rabbit anti-goat (#P0449) and swine anti-rabbit (#P0217) antibodies were from DAKO (Hamburg, Germany). Goat anti-C3 F(ab’)2 (#55062) and HRP-conjugated goat anti-C3 F(ab’)2 (#55237) were from MP Biomedicals (Solon, OH).
Serum samples
Serum samples were collected from Hungarian rheumatoid arthritis patients (n=97) and from healthy individuals (n=117) after informed consent in accordance with the Declaration of Helsinki. The study was approved by the National Ethical Committee (33986-1/2018 EKU). Characteristics of the patients and healthy donors are shown in Table 1.
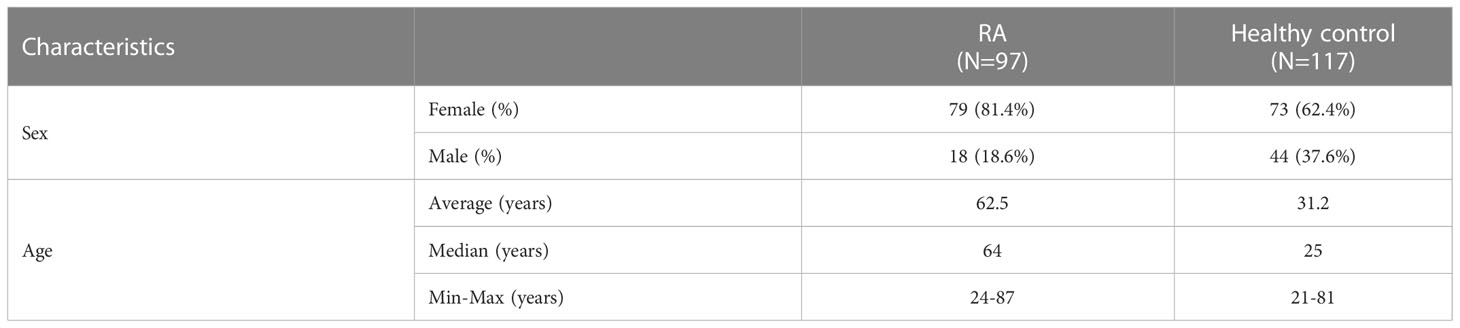
Table 1 Characteristics of patients with RA and healthy controls.
Autoantibody detection
Autoantibodies were detected using ELISA method, as described by Józsi and Uzonyi (36). Briefly, FH, FB, C3b, FI in 5 µg/ml, MBL and C1q in 2 µg/ml and HSA or alpha-1 antitrypsin as negative controls were immobilized on microtiter plates. After blocking with 5% BSA in DPBS-0.1% Tween-20, serum samples were added in DPBS-0.1% Tween-20 at a dilution of 1:50 for 1 hour at RT. To detect autoantibodies against solid phase C3 convertase of the alternative pathway (C3bBbP), the convertase was built up (see below) and incubated with serum samples diluted 1:50. The bound autoantibodies were detected with HRP-conjugated goat anti-human IgG, anti-human IgA or anti-human IgM. Color reaction was developed by adding TMB solution (BioLegend). After stopping with H2SO4, absorbance was measured at 450 nm and at 620 nm as reference wavelength. To detect anti-MBL autoantibodies, serum samples were diluted in DPBS containing 10 mM EDTA. In the case of detecting anti-C1q autoantibodies, serum samples were diluted in DPBS containing 1 M NaCl to avoid interaction of C1q globular heads with IgG Fc parts. Samples were accepted as positive if the mean OD value for the investigated protein was at least two-fold greater than the mean OD value for the negative control protein alpha-1 antitrypsin. The isotypes of the autoantibodies were determined using monoclonal antibodies specific for IgG1, IgG2, IgG3, IgG4, IgGκ and IgGλ.
IgG isolation
IgG fraction of the serum samples were isolated by Protein G column (GE Healthcare) according to the manufacturer’s instructions. For functional analyses, the in vivo formed FB–FB-autoantibody complexes were removed. To this end, FB-specific antibodies isolated by Protein G column from goat anti-FB antiserum were immobilized on NHS column (GE Healthcare). The patient’s and control’s IgG fractions were loaded onto the anti-FB column, the flow-through fraction was collected and tested for the presence of FB.
Western blot
To analyze FHR1- and/or FHR3-deficiency, serum samples at a dilution of 1:50 were separated on 10% SDS-PAGE under non-reducing conditions and transferred to nitrocellulose membrane (BioRad). After blocking with 1% BSA, 4% non-fat milk powder in DPBS-0.05% Tween-20, FHR3 and FHR1 were detected with rabbit anti-FHR3 (kindly provided by Dr. Ilse Jongerius, Sanquin Health Solutions, Amsterdam, the Netherlands) and goat anti-FH antisera and the corresponding HRP-conjugated secondary antibodies, respectively. The blots were developed with the ECL detection kit (Merck).
To detect in vivo formed immune complexes, IgG fractions of the autoanti-FB positive patient and healthy controls were separated on 10% SDS-PAGE under non-reducing conditions and blotted onto nitrocellulose membrane. After blocking with 1% BSA, 4% non-fat milk powder in DPBS-0.05% Tween-20, the membranes were incubated with goat anti-FB antiserum and HRP-conjugated rabbit anti-goat antibodies. These immune complexes were removed before further experiments to prevent their interference with the assays.
Convertase assays
The solid phase AP C3 convertase was built up as described by Hourcade et al. in 2002 (37) with modifications. C3b was immobilized at 5 µg/ml on microplate wells (Maxisorp, NUNC). After washing, 2 µg/ml FB, 4 µg/ml properdin and 0.1 µg/ml FD were added in convertase buffer (DPBS containing 4% BSA, 2 mM NiCl2, 0.1% Tween-20) for 1 hour at 37°C. Convertase formation was detected with goat anti-FB and HRP-conjugated rabbit anti-goat antibodies. Convertase activity was determined by adding 10 µg/ml C3 for 1 hour at 37°C. The generated C3a was measured by the C3a EIA kit (Quidel). In some assays, patient’s or control IgG was added together with convertase components to analyze their effect on convertase formation. Convertase decay was determined after incubation of the formed convertase with the isolated IgG fractions alone or in the presence of 1 µg/ml FH for 1 hour at 37°C. Remaining convertase was detected with goat anti-FB antiserum.
Complement activation assay
LPS (#L4524, Merck) was immobilized at 10 µg/ml on Maxisorp microplate wells. Wells were washed with DPBS and blocked with 2% BSA. 10% complement active normal human serum (Quidel) was preincubated with the FB-free autoantibodies or with 5×106 rabbit red blood cells (RRBC, Culex Bt.) as positive control for 15 min at 37°C. The serum was then added to the LPS-coated wells for 1 h at 37°C to determine the remaining complement activity. Complement deposition was measured with HRP-conjugated goat anti-C3 or mouse anti-C5b-9 and HRP-conjugated goat anti-mouse IgG.
Hemolysis assay
5×106 RRBC were incubated with 7% complement active normal human serum (Quidel) in HEPES buffer (20 mM HEPES, 7 mM MgCl2, 10 mM EGTA, 144 mM NaCl, 1% BSA, pH 7.4) with serial dilutions of purified IgG for 30 min at 37°C. After centrifugation, optical density of the supernatants was measured at 414 nm.
Statistical analysis
Statistical analyses were performed with GraphPad Prism version 6.01 for Windows (GraphPad Software, San Diego, CA). A p value < 0.05 was considered statistically significant. Comparison of gender distribution between RA patients and healthy controls were determined by χ2 test. To compare continuous variables (age), Mann–Whitney U test was performed. Comparison of FHR1- and/or FHR3-deficiency distribution between RA patients and healthy donors were performed by χ2 test. For comparison of the effects of IgG isolated from RA547 or healthy controls, one-way ANOVA was used with Dunnett’s multiple comparison test.
Results
Screening for anti-complement autoantibodies
To investigate the presence and role of complement-specific autoantibodies in RA, we screened a Hungarian RA cohort for autoantibodies against FH, as well as C1q and MBL which were described as autoantibody targets in RA (33–35). In addition, we looked for autoantibodies against FB, FI, C3b and the solid phase C3 convertase C3bBbP, and analyzed the cohort for deficiency of the complement proteins FHR1 and FHR3. Serum samples of RA patients and healthy controls were tested for IgG and IgM antibodies against the above-mentioned complement components in ELISA.
Healthy control samples were negative for all tested antibodies, and we did not detect autoantibodies against FH, FI, C3b or the C3bBbP convertase in this RA cohort (Supplementary Figures 1C, D and data not shown). Four patients (RA240, RA268, RA557, RA587) were positive for autoantibodies against C1q (Figure 1A; Supplementary Figure 1A), two patients were positive for autoantibodies against MBL (RA277, RA348) (Figure 1B; Supplementary Figure 1B) and five patients (RA223, RA243, RA274, RA282, RA547) were positive for autoanti-FB IgG (Figure 2A; Supplementary Figure 1C). The serum of patient RA282 showed high signal on alpha-1 antitrypsin, and it turned out that this patient had autoantibodies against alpha-1 antitrypsin. Positivity for FB-autoantibodies was determined using other inert proteins as negative controls (Figure 2B). In two patients (RA288, RA547) autoanti-FB IgA was detected (Figure 2C). Characteristics of the autoantibody positive patients are shown in Table 2. The titer and isotype were determined for all the five autoanti-FB IgG positive samples. We could detect in all samples both light chains and various combinations of heavy chains, pointing to an oligoclonal origin of the autoantibodies (Table 3). Based on the amount of serum available, RA547 sample was selected and further characterized in detail.
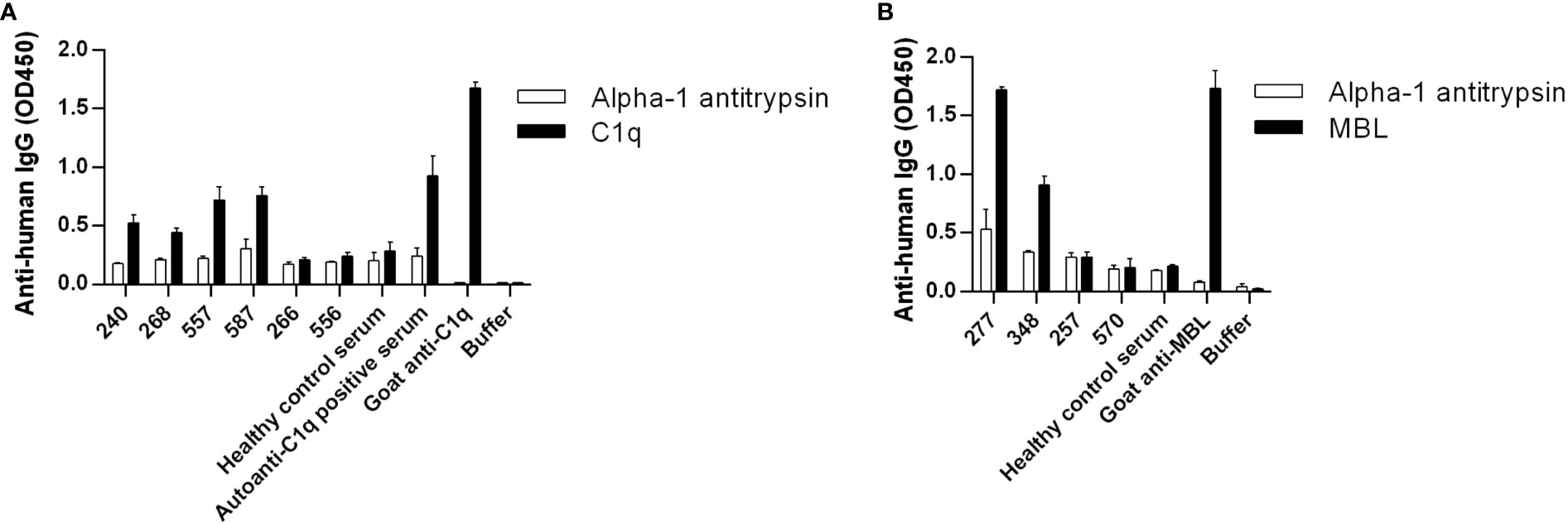
Figure 1 Detection of autoantibodies against C1q and MBL in RA patients. (A) C1q, (B) MBL, and alpha-1 antitrypsin as negative control were immobilized on microtiter plate wells. After blocking, serum samples diluted 1:50 were added. In the case of C1q, high salt concentration buffer was used and in the case of MBL 10 mM EDTA was used to exclude unspecific binding, as described in Materials and methods. Bound IgG was detected by HRP-conjugated anti-human IgG. Selected samples are shown: positive patient samples, some negative patient samples and controls. For full datasets please refer to Supplementary Figure 1. Data are mean ± SEM of two experiments.
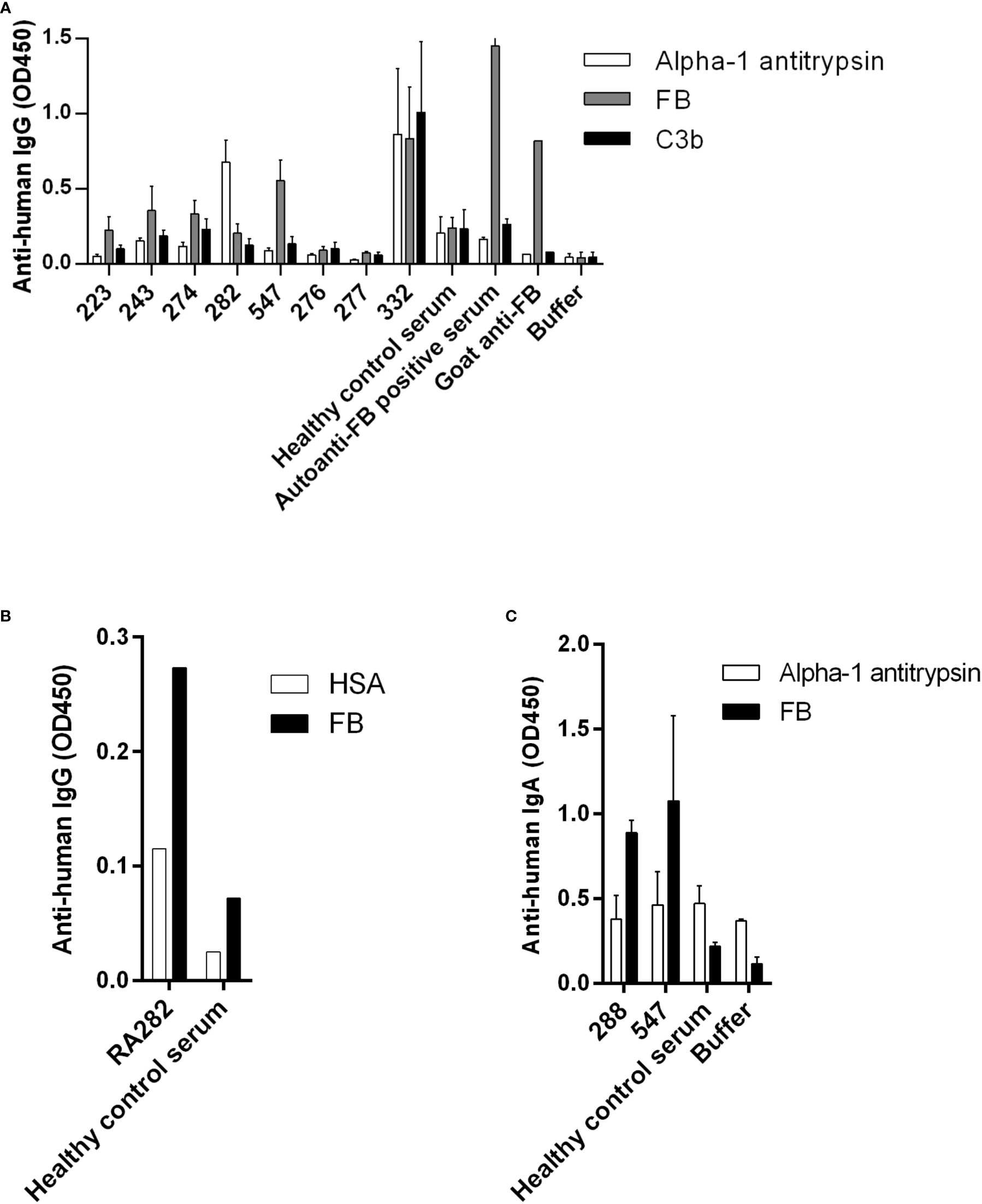
Figure 2 Autoanti-FB in RA patients. (A) FB, C3b and alpha-1 antitrypsin as negative control were immobilized on microtiter plate wells. After blocking, serum samples diluted 1:50 were added. Bound IgG was detected by HRP-conjugated anti-human IgG. Data are mean ± SEM of three experiments. (B) RA282 autoanti-FB was analyzed and confirmed using another negative control protein, human serum albumin (HSA). Data from a single experiment are shown. (C) IgA autoantibodies against FB were detected using HRP-conjugated anti-human IgA. Data are mean ± SD of two experiments. In all three panels selected samples are shown: positive patient samples, some negative patient samples and controls. For full datasets please refer to Supplementary Figure 1.
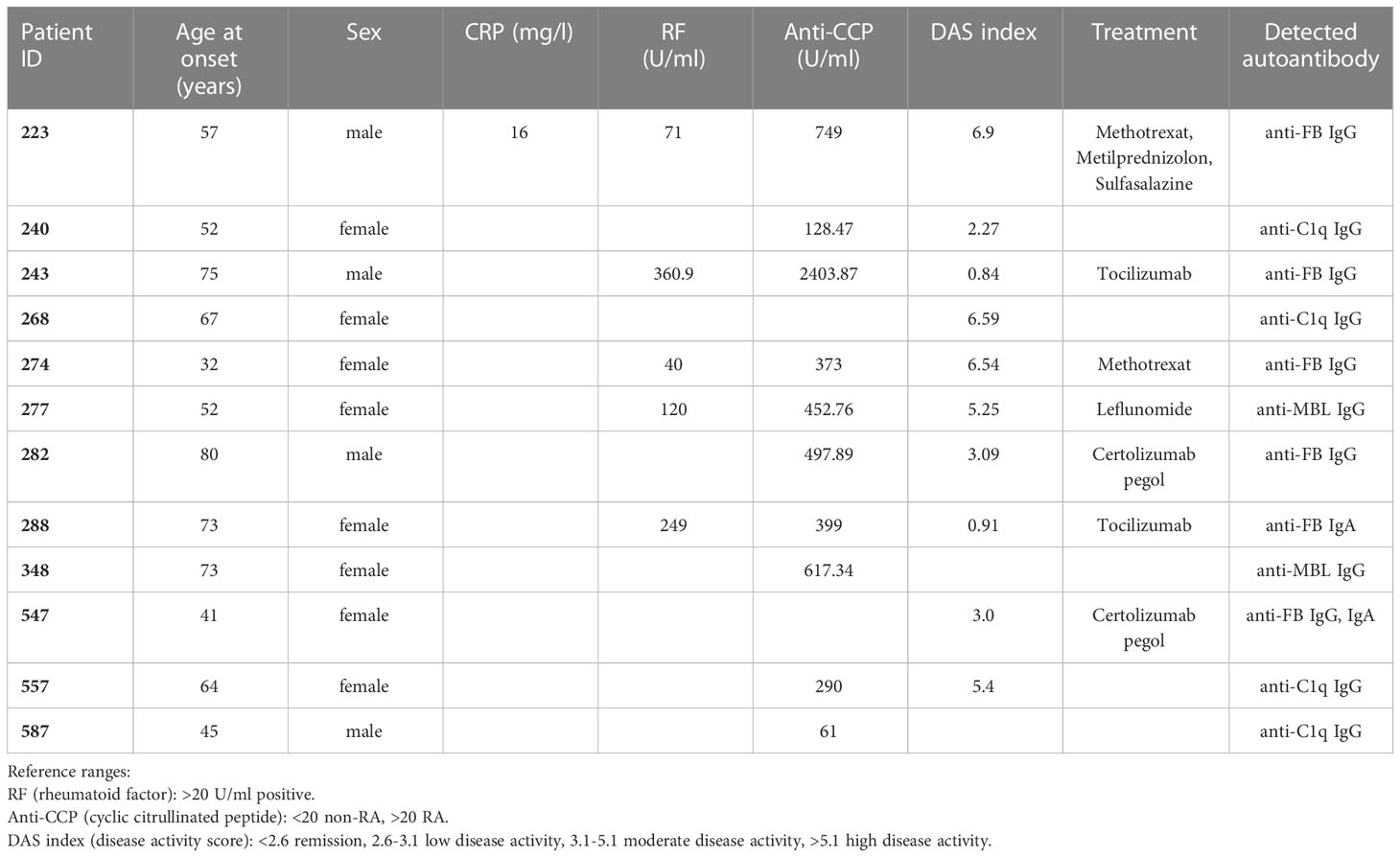
Table 2 Characteristics of the autoantibody positive patients.
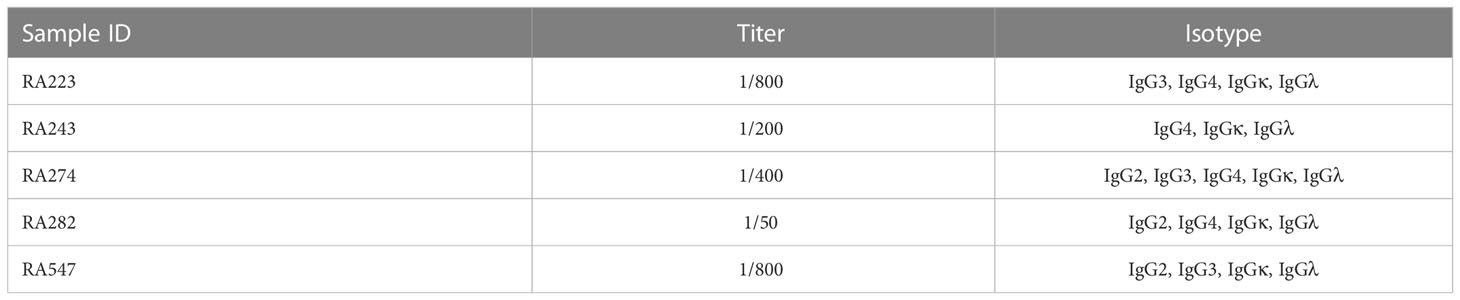
Table 3 Titer and isotype of the autoanti-FB IgG antibodies.
IgG of RA547 and that of healthy controls was isolated from serum by Protein G column. IgG-depleted serum of RA547 did not show binding to FB (Figure 3A) while the isolated IgG fraction showed dose-dependent binding to FB (Figure 3B), and the antibody also bound to FB as part of the solid phase C3 convertase (C3bBbP) (Figure 3C). To test whether the antibody binds FB in its native form in vivo, the isolated IgG fraction was analyzed by Western blot. A strong band corresponding to FB was detected in the RA547 sample, indicating the formation of FB–anti-FB immune complexes in vivo. (Figure 3D).
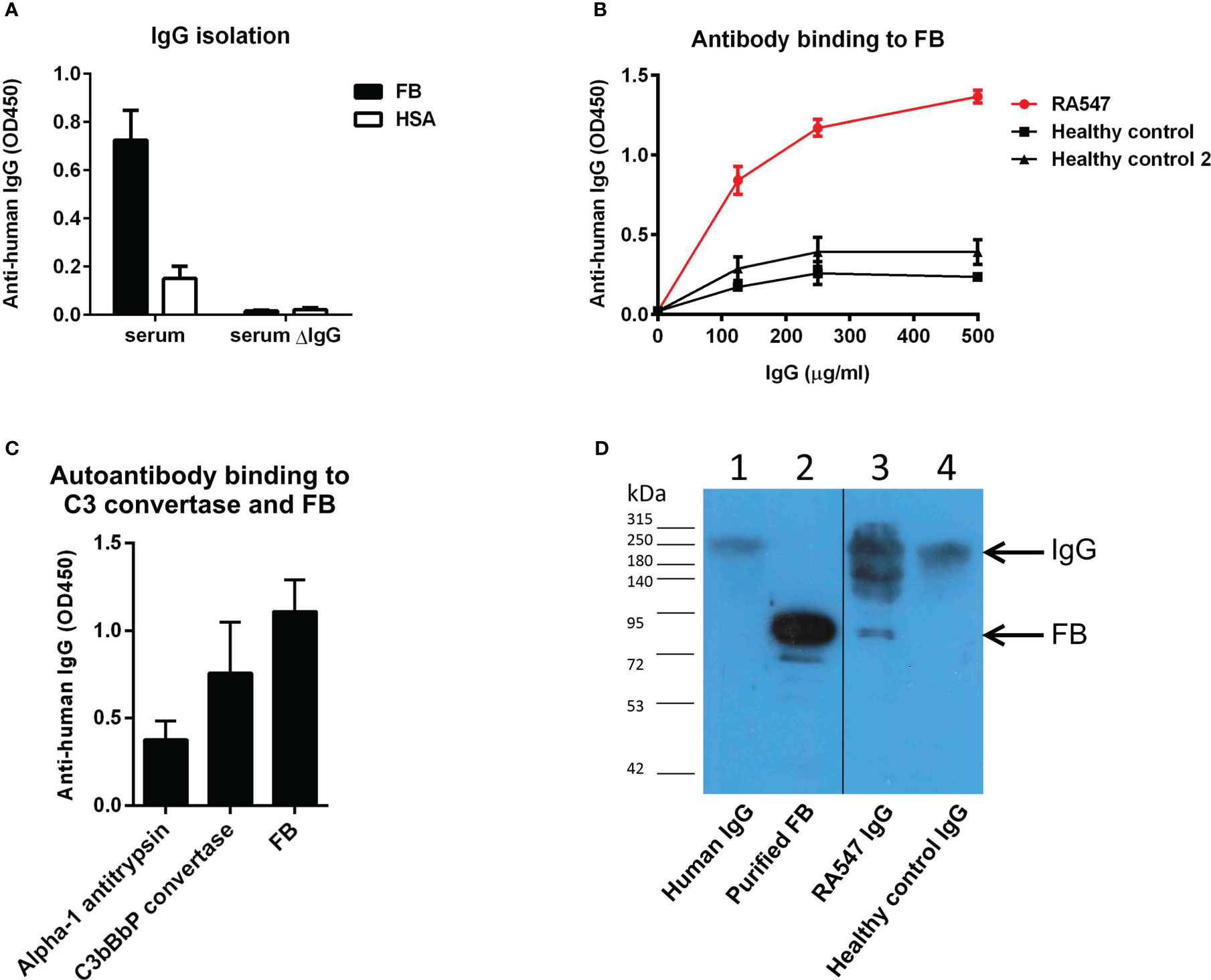
Figure 3 Anti-FB autoantibody characterization. (A) IgG was isolated with Protein G column from RA547 patient’s serum and healthy controls’ sera. The IgG depleted serum lost its ability to bind FB. (B) Serial dilutions from RA patient’s IgG showed dose-dependent binding to FB in contrast to healthy control IgG. Data are mean ± SD from three independent experiments. (C) Autoanti-FB bound to FB as part of the C3 convertase of the alternative pathway (C3bBbP). (D) In vivo formed IgG-FB complexes were detected in the patient’s IgG fraction with polyclonal anti-FB antibody in Western blot. Lanes: 1: purified IgG, 2: purified FB, 3: RA547 IgG, 4: Healthy control IgG. 25 µg isolated IgG was run on SDS-PAGE, blotted onto nitrocellulose membrane, and developed by anti-FB antibody. Blot is representative of three experiments.
RA547 autoanti-FB characterization
Characteristics as titer, isotype, and binding site of the autoanti-FB in RA547 were determined in ELISA-based assays (Table 3 and Figure 4). The titer and isotype were determined in the third available sample (Figures 4B, C). The presence of anti-FB autoantibodies was monitored in serial samples; over time, probably due to therapy, the anti-FB autoantibody levels decreased, and in the last sample it was not detectable anymore (Figure 4A). To determine the binding site of autoantibodies on FB, immobilized FB was preincubated with monoclonal antibodies specific for either the Ba or Bb part of FB, as well as with anti-FB antiserum. The anti-Bb and the polyclonal anti-FB reduced the binding of the autoantibodies to FB, while anti-Ba did not significantly influence the binding, suggesting that the autoantibody binding site is on the enzymatically active Bb part of FB (Figure 4D). Functional effects of the antibodies were analyzed in further assays.
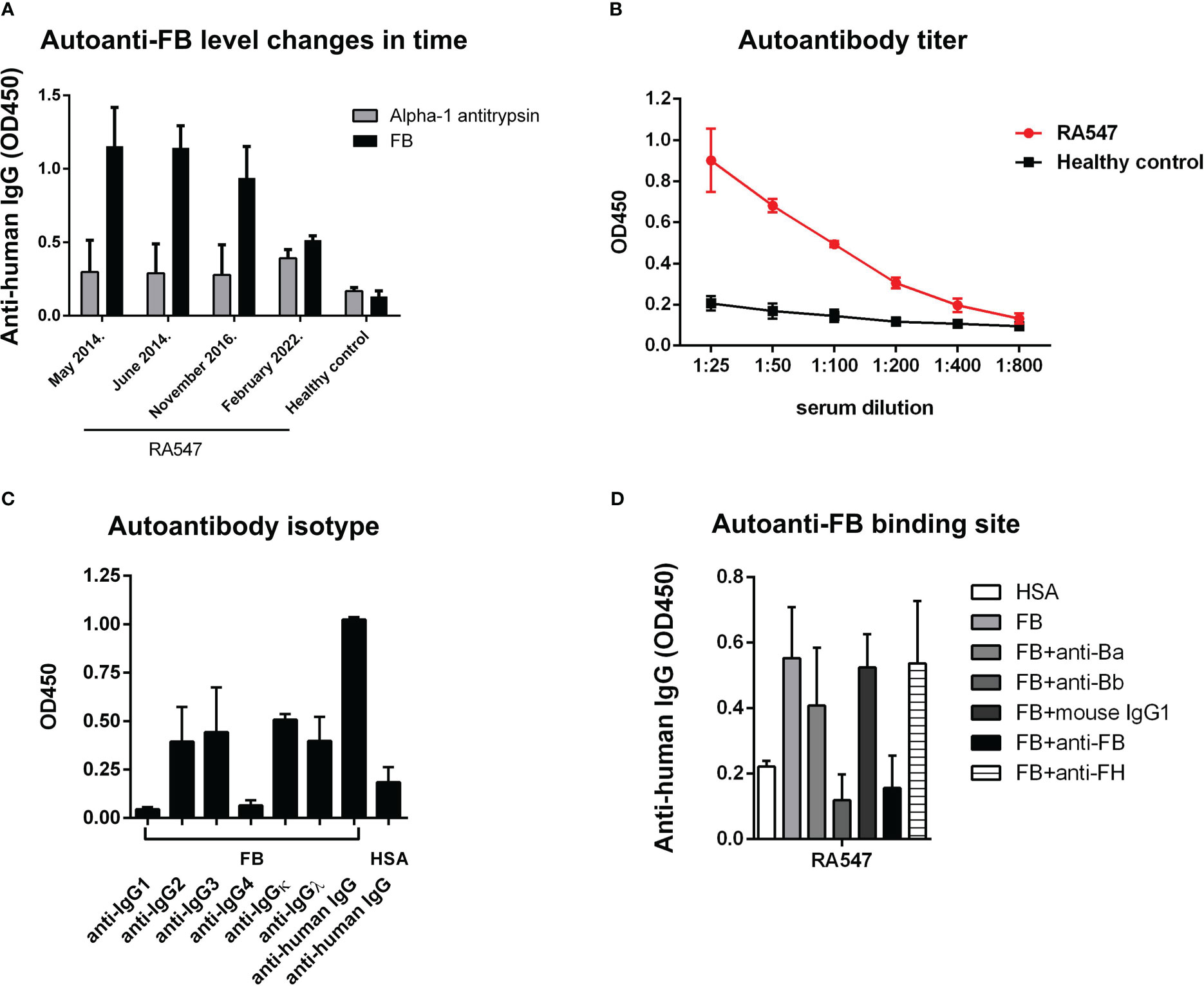
Figure 4 Autoantibody characterization. (A) We had serum samples from four time points to analyze the presence of autoanti-FB in RA547. Data are results ± SD of two experiments. (B) Titer of autoanti-FB was determined in ELISA, with serial dilutions of the patient’s serum. (C) Autoantibodies were added to immobilized FB, and the isotypes were determined with monoclonal antibodies against the IgG subclasses. Data are mean ± SD from two independent experiments. Values obtained with an autoantibody negative healthy control as a background were subtracted from the values obtained with RA547 before plotting. (D) Autoantibody binding to immobilized FB was decreased by the preincubation of FB with monoclonal anti-Bb antibody and with polyclonal anti-FB antiserum. Data are mean ± SEM from three independent experiments. Values obtained with an autoantibody negative healthy control as a background were subtracted from the values obtained with RA547.
FB autoantibodies influence the activation of the alternative pathway and affect the terminal pathway
To measure the effect of the purified, patient-derived anti-FB autoantibodies on the activation of the alternative pathway, rabbit red blood cells (RRBCs), which are sensitive to lysis in normal human serum, were used. RRBCs were mixed with normal human serum in a buffer containing Mg2+-EGTA, which allows only the activation of the alternative pathway, and supplemented with serial dilutions of IgG isolated from the serum of the patient or healthy controls. After incubation, lysis was determined by measuring the OD of released hemoglobin in the supernatant. Compared to the control IgGs, patient IgG inhibited the hemolysis in a dose-dependent manner (Figure 5).
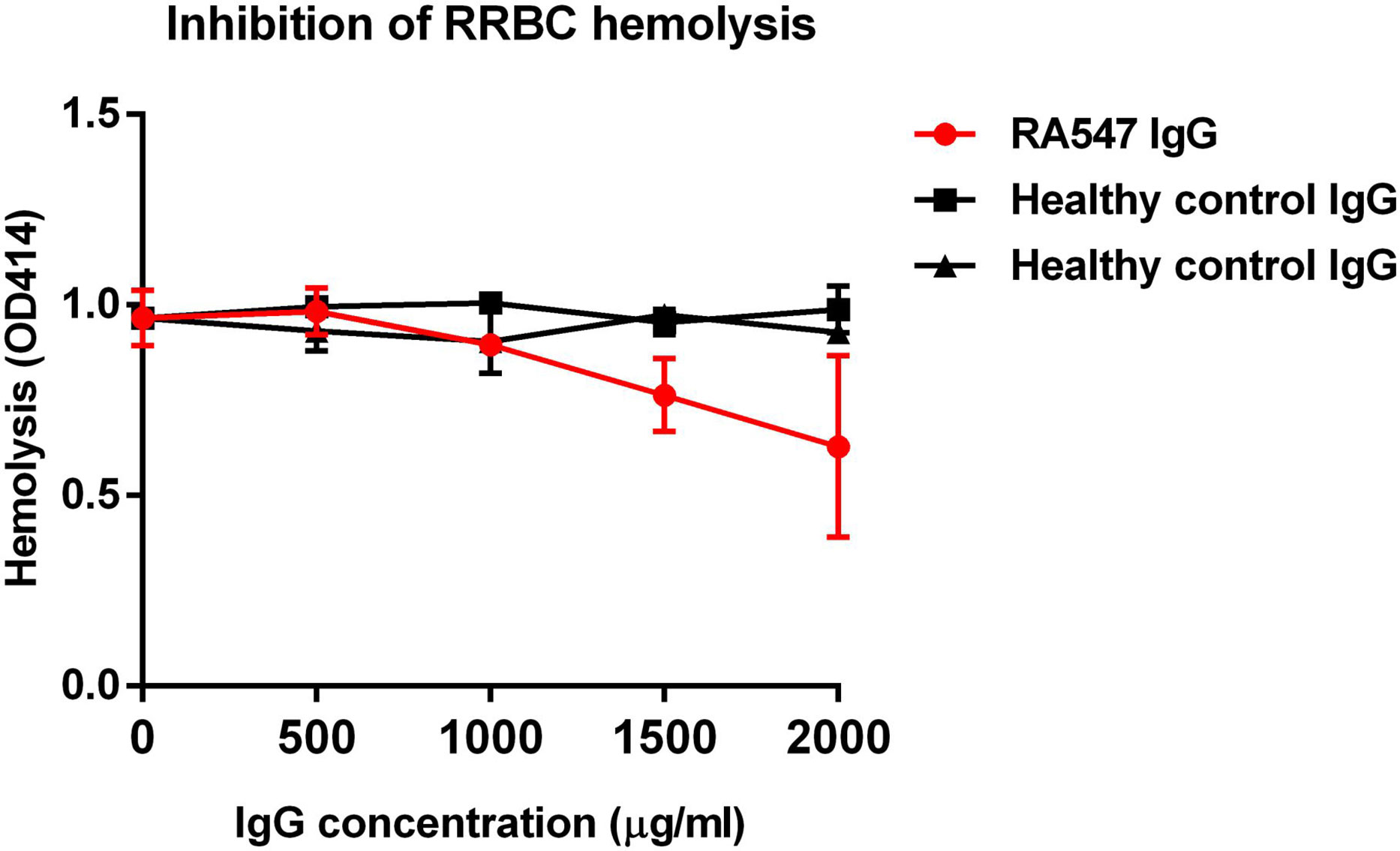
Figure 5 RA547 patient’s IgG dose-dependently inhibited RRBC hemolysis. Complement active normal human serum was mixed with RRBCs and IgG from patient or healthy controls, then incubated for 30 min at 37°C. Samples were centrifuged, and the OD of the supernatant was measured at 414 nm. Data are mean ± SD from two independent experiments.
Next, we investigated how the autoantibodies influence complement activation in serum in the fluid phase as described by Zhao et al. (38). To this end, normal human serum was mixed and incubated with IgG from RA547 or healthy controls and with RRBC as positive control. After incubation, the residual complement activity was measured on LPS-coated surface in the form of C3- and C5b-9-deposition (Figure 6). Serum alone retains its complement activity and when added to LPS, C3- and C5b-9-depostion can be detected. RRBC depletes complement during incubation, so no complement deposition can be measured on LPS, similar to the EDTA-treated serum sample. Incubation of the serum with IgG generally led to activity loss to some extent, probably due to activation via immune complexes present in the IgG preparations. However, compared to the IgG of healthy individuals, the autoanti-FB-containing patient IgG further decreased both C3- and C5b-9-deposition, presumably via inhibitory effects of the anti-FB autoantibodies (Figure 6).
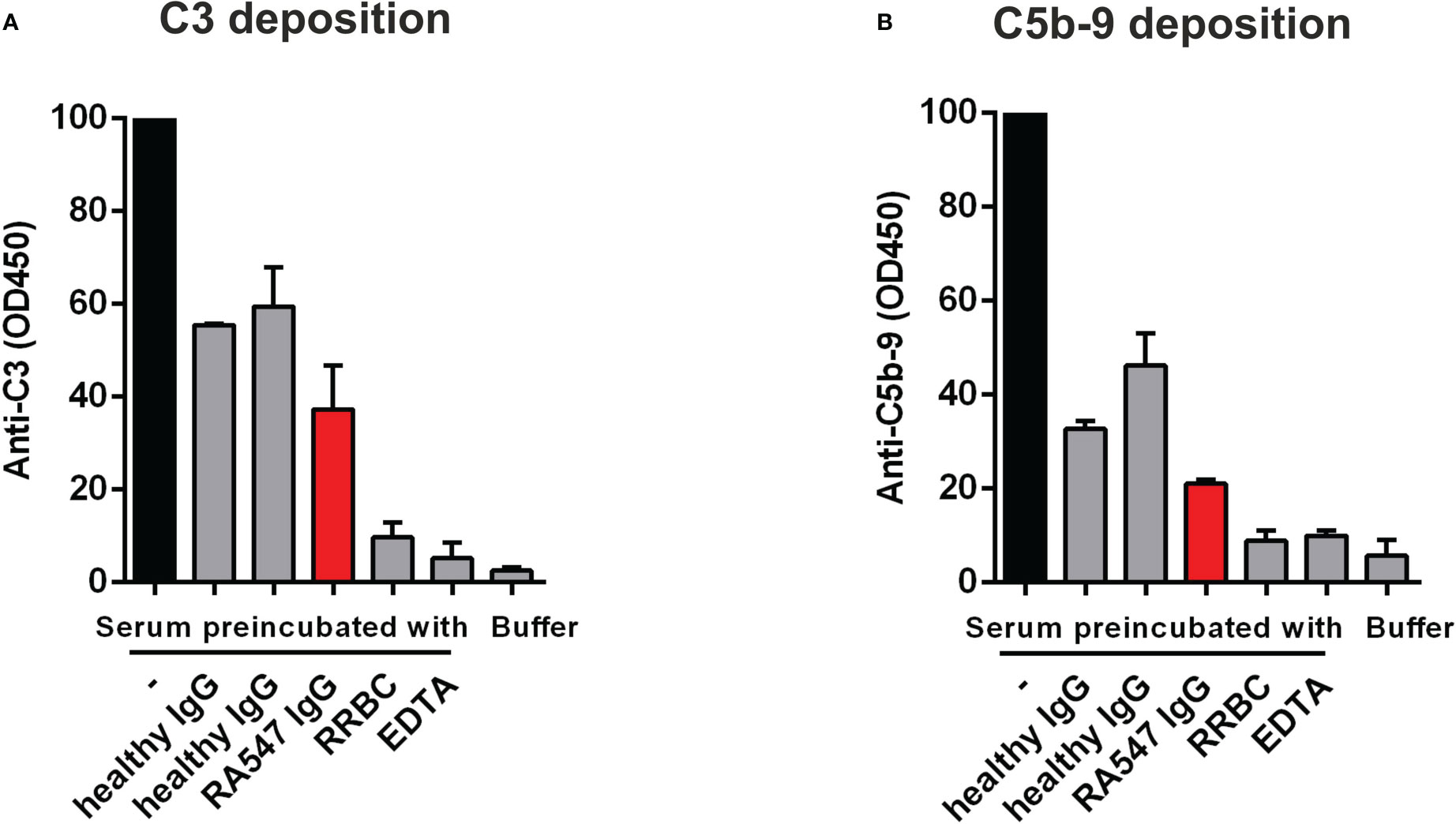
Figure 6 Anti-FB autoantibodies decrease complement activation-derived deposition of C3 and C5b-9 on LPS-coated surface. Normal human serum was preincubated with PBS, patient- or healthy control-derived IgG, and RRBC as a positive control, then added to LPS-coated wells. EDTA was used to inhibit complement activation on LPS. The deposition of (A) C3 and (B) C5b-9 was detected with anti-C3 and anti-C5b-9 antibodies, respectively. Data are mean ± SD from two independent experiments. RRBC: Rabbit Red Blood Cell.
FB-autoantibodies impair the activity of the solid phase C3 convertase of the alternative pathway
To decipher the mechanism behind the inhibitory effects measured using whole serum, next we analyzed the effect of the autoantibodies on the C3 convertase. C3bBbP was generated on microplates using purified proteins as described by Hourcade et al. (37) and Strobel et al. (39) with modifications. Convertase formation was detected with polyclonal goat anti-FB, while convertase activity was determined by adding C3 and measuring the generated C3a (Figure 7A).
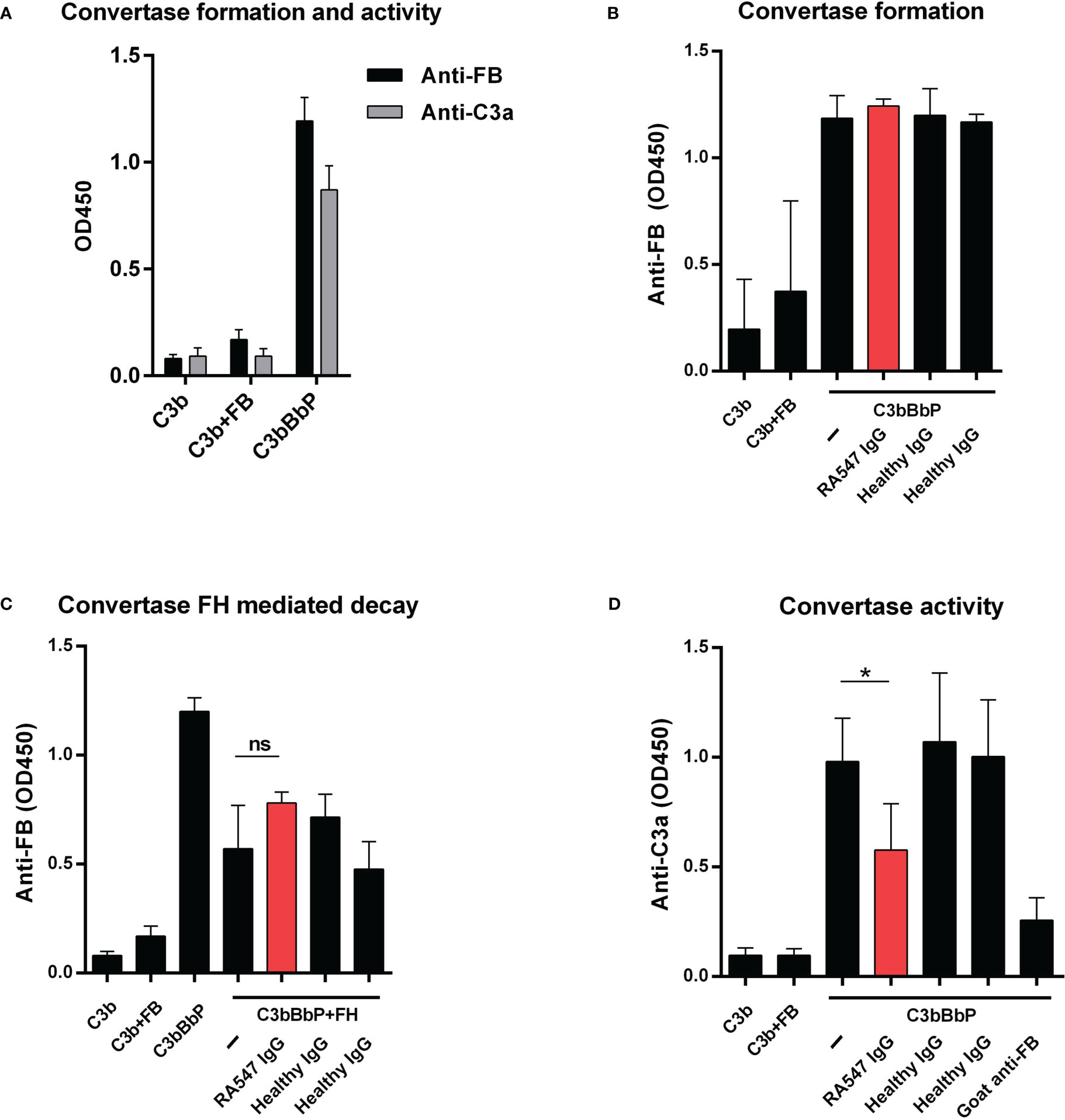
Figure 7 Effect of the patient’s IgG on alternative pathway C3 convertase. (A) Generation of solid phase C3 convertase in microplate wells. We added FB, FD and properdin together to the immobilized C3b and, after incubation, detected the convertase formation by polyclonal anti-FB. To confirm the activity of the convertase, C3 was added in the fluid phase, and we measured the released C3a from the supernatant with commercially available C3a EIA (Quidel). (B) To test the autoantibodies’ effect on convertase formation, we added the patient’s or healthy controls’ IgG together with FB, FD and properdin to immobilized C3b. The assembly of the convertase was detected with polyclonal goat anti-FB antibody. (C) Autoantibodies had no effect on the FH mediated decay of the C3 convertase. The convertase was incubated with IgG and FH and the remaining convertase was detected with polyclonal anti-FB antibody. (D) To investigate the effect of the patient’s autoantibodies on the activity of the convertase, the convertase was incubated with purified IgG and, after washing, C3 was added. Supernatants were collected and the C3 cleavage product C3a was measured using C3a EIA. Data are mean ± SD of three (A, C, D) or two (B) independent experiments. *p<0.05, One-Way ANOVA.
First, we investigated the effects of autoantibodies on convertase formation. IgG from the patient and healthy controls were added to immobilized C3b together with the components of the convertase and, after incubation, convertase formation was measured. The anti-FB autoantibodies did not influence the assembly of the C3bBbP convertase (Figure 7B).
Next, we measured the effects of the autoantibodies on the FH-mediated decay of the convertase. The generated convertase was preincubated with IgG from the patient or healthy controls, then FH was added without removing the antibodies. After incubation, the remaining convertase was detected with goat anti-FB. The anti-FB autoantibodies had no effect in this assay; they did not protect the convertase from FH-mediated decay (Figure 7C).
Lastly, the effect of autoantibodies on convertase activity was measured. The generated convertase was preincubated with IgG from the patient and healthy controls, then C3 was added and the released C3a was measured from the supernatant. When added to the convertase, IgG of healthy controls had no effect, whereas, anti-FB autoantibodies significantly reduced the amount of generated C3a, similarly to that observed with the polyclonal anti-FB, used as positive control (Figure 7D).
FHR1 and FHR3 deficiency
Deficiency of FHR1 and/or FHR3 is associated with various conditions, it can be both a protective and a predisposing factor depending on the disease (22, 40). We investigated the frequency of FHR1 and FHR3 deficiency in this RA cohort. Genomic DNA was not available; therefore, serum samples were screened for the presence of the FHR1 and FHR3 proteins by Western blot. To our knowledge, none of the patients or controls received plasmapheresis or fresh frozen plasma, thus the detected proteins have no ex vivo origin. We found individuals lacking only FHR1, only FHR3 or both proteins, both among the RA patients and the healthy controls, and the differences between the two groups were not significant (Table 4; Supplementary Figure 2).
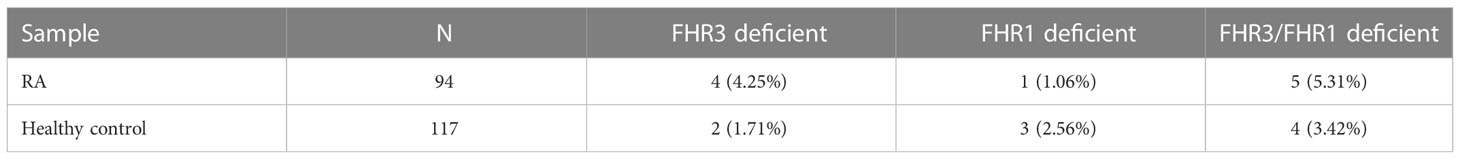
Table 4 Overview of FHR1- and FHR3-deficiency in the RA patients and healthy controls.
Discussion
In this study we analyzed the presence of autoantibodies against complement components in a Hungarian RA cohort. We found anti-C1q and anti-MBL autoantibodies (Figure 1.), in accordance with previous reports (32, 33, 35). No antibodies against FH, FI, C3b and the alternative pathway C3 convertase (C3bBbP) were detected in these samples. In the case of FH, our results contradict the findings of Foltyn Zadura et al., who found FH-autoantibodies in 16.5% and 9.2% of the analyzed samples (34). While high IgG signals on FH-coated wells in 6.9% of samples were detected in our assays, these serum samples gave high signal on inert control protein as well, thus we considered them false positives. In addition to anti-C1q and anti-MBL autoantibodies, we found anti-FB autoantibodies in 5 patients, which have not been described in RA so far. The anti-FB IgG titer varied between patients; however, the presence of both light chains and various heavy chains (Table 3) was a common feature, suggesting an oligoclonal origin of the antibodies in all cases. Since the RA group and the healthy control group significantly differed regarding age and gender in our study, we cannot state that the identified autoantibodies are RA-specific. However, to our knowledge, no anti-FB autoantibodies have been described in healthy people at any age.
The analyzed anti-FB autoantibodies bind FB both in its native form and as part of the C3 convertase (Figure 3C). In contrast to C3Nefs that bind to and stabilize the C3 convertase, increase its activity and enhance terminal pathway activity (41), the analyzed anti-FB autoantibody did not influence solid-phase C3 convertase stability but impaired its activity (Figure 7). This is also different from the characteristics of the anti-FB autoantibody described in dense deposit disease (DDD) by Strobel et al. that stabilized the C3 convertase, increased its activity but inhibited the C5 convertase (39). FB autoantibodies described by Chen et al. in DDD and Marinozzi et al. in C3G also appeared pathogenic by enhancing C3 turnover (42, 43). Here, the anti-FB autoantibody binding site was mapped to the enzymatically active Bb part on FB (Figure 4D). Presumably, the autoantibody binding to Bb in the C3 convertase leads to impairment of C3 convertase activity measured in ELISA and in hemolysis assay (Figures 7D, 5).
Since our Western blot analysis demonstrated the presence of FB within the isolated patient IgG fraction (Figure 3D), we conclude that the autoanti-FB antibodies formed immune complexes in the patient in vivo. We observed impaired complement activation in the form of reduced C3 and C5b-9 deposition on LPS-coated surface after preincubation of the serum with purified patient IgG, containing the FB–autoanti-FB immune complexes (Figure 6). This could be explained by activation of the classical pathway by the immune complexes and/or by the inhibitory effect of the anti-FB autoantibodies on C3 convertases formed on the LPS surface.
The production of such autoantibodies may be triggered by the prolonged presence of the complement protein activation products and complexes due to overactivation of the system. Our data demonstrate that not all the complement specific autoantibodies are pathogenic and contribute to complement activation in RA. Autoantibodies may be beneficial for patients, as it was shown for autoantibodies against FH in non-small cell lung cancer where anti-FH blocked FH activity at tumor cells (44) or in lupus patients (45). The autoanti-FB in RA reported here also demonstrates that some autoantibodies could hold back the overwhelming complement activation which may be beneficial for the patients. This suggests that the generation of such non-activating autoantibodies is part of a protective response and contributes to the prevention of the tissue damage caused by excessive complement activation.
The data also highlight that the presence/level of autoanti-FB in RA can be transient (Figure 4A) probably depending on the actual state of the disease (remission, or active phase), and the received treatment. In patient RA547 the autoanti-FB levels were mostly constant between May 2014 and November 2016, during which the patient had anti-TNFα therapy (certolizumab pegol). Later the therapy was changed (from 2020) to adalimumab which is another kind of anti-TNFα treatment. In the most recent serum sample of the patient (February 2022) autoanti-FB was not detectable, presumably due to the change in therapy or disease activity.
Autoantibodies against the components of the convertase can disturb the balance of complement activation, contributing to autoimmune diseases (39, 46). In many cases, autoantibodies have diagnostic and prognostic value, therefore their detection is coming to fore (47). Though in RA autoantibodies against the early components of the complement system (32, 33, 35) as well as autoantibodies against the complement regulator FH (34) were described years ago, their role in disease mechanism has not been investigated. Our results further support that the pathophysiology of RA implies the complement system and its dysfunction. These data also draw attention to the importance of characterization and functional analysis of autoantibodies, because simply detecting autoantibodies can be misleading and reporting their presence may misinform physicians. FB autoantibodies may be pathogenic in certain conditions, such as likely in DDD, but may be non-pathogenic or even protective in other diseases.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving human participants were reviewed and approved by National Ethical Committee (33986-1/2018 EKU). The patients/participants provided their written informed consent to participate in this study.
Author contributions
ATM and BU performed experiments, analyzed the data, prepared the figures. AF, BR, GN took care of the patients and provided serum samples. GS, MJ and BU initiated the study, supervised the research. ATM, BU and MJ wrote the manuscript with help of the other authors. All authors read and approved the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The work of the authors was supported in part by the National Research, Development and Innovation Fund of Hungary (grant K 125219), the Institutional Excellence Program to ELTE (NKFIH-1157/8/2019, D11206), the Eötvös Loránd Research Network and the Hungarian Academy of Sciences (0106307), and by project no. FIEK_16-1-2016-0005, implemented with the support provided from the National Research, Development and Innovation Fund of Hungary, financed under the FIEK_16 funding scheme.
Acknowledgments
We thank Dr. Ilse Jongerius (Sanquin Health Solutions, Amsterdam, the Netherlands) for providing the rabbit anti-FHR3 antiserum.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1113015/full#supplementary-material
References
1. Okroj M, Heinegård D, Holmdahl R, Blom AM. Rheumatoid arthritis and the complement system. Ann Med (2007) 39(7):517–30. doi: 10.1080/07853890701477546
2. Firestein GS. Evolving concepts of rheumatoid arthritis. Nature (2003) 423(6937):356–61. doi: 10.1038/nature01661
3. Silman AJ, Pearson JE. Epidemiology and genetics of rheumatoid arthritis. Arthritis Res (2002) 4(Suppl 3):S265–72. doi: 10.1186/ar578
4. Bernatsky S, Dekis A, Hudson M, Pineau CA, Boire G, Fortin PR, et al. Rheumatoid arthritis prevalence in Quebec. BMC Res Notes. (2014) 7:937. doi: 10.1186/1756-0500-7-937
5. Kuo CF, Luo SF, See LC, Chou IJ, Chang HC, Yu KH. Rheumatoid arthritis prevalence, incidence, and mortality rates: A nationwide population study in Taiwan. Rheumatol Int (2013) 33(2):355–60. doi: 10.1007/s00296-012-2411-7
6. Arima H, Koirala S, Nema K, Nakano M, Ito H, Poudel KM, et al. High prevalence of rheumatoid arthritis and its risk factors among Tibetan highlanders living in tsarang, mustang district of Nepal. J Physiol Anthropol. (2022) 41(1):12. doi: 10.1186/s40101-022-00283-3
7. Scott DL, Wolfe F, Huizinga TWJ. Rheumatoid arthritis. Lancet (2010) 376(9746):1094–108. doi: 10.1016/S0140-6736(10)60826-4
8. van Delft MAM, Huizinga TWJ. An overview of autoantibodies in rheumatoid arthritis. J Autoimmunity. (2020) 110:102392. doi: 10.1016/j.jaut.2019.102392
9. Di Giuseppe D, Discacciati A, Orsini N, Wolk A. Cigarette smoking and risk of rheumatoid arthritis: A dose-response meta-analysis. Arthritis Res Ther (2014) 16(2):R61. doi: 10.1186/ar4498
10. Chen HH, Huang N, Chen YM, Chen TJ, Chou P, Lee YL, et al. Association between a history of periodontitis and the risk of rheumatoid arthritis: A nationwide, population-based, case–control study. Ann Rheum Dis (2013) 72(7):1206–11. doi: 10.1136/annrheumdis-2012-201593
11. Busch R, Kollnberger S, Mellins ED. HLA associations in inflammatory arthritis: Emerging mechanisms and clinical implications. Nat Rev Rheumatol (2019) 15(6):364–81. doi: 10.1038/s41584-019-0219-5
12. Alpízar-Rodríguez D, Pluchino N, Canny G, Gabay C, Finckh A. The role of female hormonal factors in the development of rheumatoid arthritis. Rheumatology (2017) 56(8):1254–63. doi: 10.1093/rheumatology/kew318
13. Sokolova MV, Schett G, Steffen U. Autoantibodies in rheumatoid arthritis: Historical background and novel findings. Clin Rev Allergy Immunol (2022) 63(2):138–51. doi: 10.1007/s12016-021-08890-1
14. Newkirk MM. Rheumatoid factors: Host resistance or autoimmunity? Clin Immunol (2002) 104(1):1–13. doi: 10.1006/clim.2002.5210
15. Kwon EJ, Ju JH. Impact of posttranslational modification in pathogenesis of rheumatoid arthritis: Focusing on citrullination, carbamylation, and acetylation. Int J Mol Sci (2021) 22(19):10576. doi: 10.3390/ijms221910576
16. Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO, et al. 2010 Rheumatoid arthritis classification criteria: An American college of Rheumatology/European league against rheumatism collaborative initiative. Arthritis Rheum (2010) 62(9):2569–81. doi: 10.1002/art.27584
17. Hensvold AH, Frisell T, Magnusson PKE, Holmdahl R, Askling J, Catrina AI. How well do ACPA discriminate and predict RA in the general population: A study based on 12 590 population-representative Swedish twins. Ann Rheum Dis (2017) 76(1):119–25. doi: 10.1136/annrheumdis-2015-208980
18. Seri A, Mohamed HKA, Essa MEA, Elagib EM, Eltahirm NIA, Mansour SMA, et al. Analysis of serum immune markers in seropositive and seronegative rheumatoid arthritis among sudanese patients and the relation between the serotype and joint involvement: A cohort study. Open Access Rheumatol (2021) 13:325–32. doi: 10.2147/OARRR.S339134
19. Carbonell-Bobadilla N, Soto-Fajardo C, Amezcua-Guerra LM, Batres-Marroquín AB, Vargas T, Hernández-Diazcouder A, et al. Patients with seronegative rheumatoid arthritis have a different phenotype than seropositive patients: A clinical and ultrasound study. Front Med (Lausanne) (2022) 9:978351. doi: 10.3389/fmed.2022.978351
20. Galindo-Izquierdo M, Pablos Alvarez JL. Complement as a therapeutic target in systemic autoimmune diseases. Cells (2021) 10(1):148. doi: 10.3390/cells10010148
21. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: A key system for immune surveillance and homeostasis. Nat Immunol (2010) 11(9):785–97. doi: 10.1038/ni.1923
22. Cserhalmi M, Papp A, Brandus B, Uzonyi B, Józsi M. Regulation of regulators: Role of the complement factor H-related proteins. Semin Immunol (2019) 45:101341. doi: 10.1016/j.smim.2019.101341
23. Ricklin D, Reis ES, Mastellos DC, Gros P, Lambris JD. Complement component C3 – the “Swiss army knife” of innate immunity and host defense. Immunol Rev (2016) 274(1):33–58. doi: 10.1111/imr.12500
24. Schmidt CQ, Lambris JD, Ricklin D. Protection of host cells by complement regulators. Immunol Rev (2016) 274(1):152–71. doi: 10.1111/imr.12475
25. Matola AT, Józsi M, Uzonyi B. Overview on the role of complement-specific autoantibodies in diseases. Mol Immunol (2022) 151:52–60. doi: 10.1016/j.molimm.2022.08.011
26. Swaak AJG, van Rooyen A, Planten O, Han H, Hattink O, Hack E. An analysis of the levels of complement components in the synovial fluid in rheumatic diseases. Clin Rheumatol (1987) 6(3):350–7. doi: 10.1007/BF02206833
27. Moxley G, Ruddy S. Elevated C3 anaphylatoxin levels in synovial fluids from patients with rheumatoid arthritis. Arthritis Rheumatol (1985) 28(10):1089–95. doi: 10.1002/art.1780281003
28. Hornum L, Hansen AJ, Tornehave D, Fjording MS, Colmenero P, Wätjen IF, et al. C5a and C5aR are elevated in joints of rheumatoid and psoriatic arthritis patients, and C5aR blockade attenuates leukocyte migration to synovial fluid. PloS One (2017) 12(12):e0189017. doi: 10.1371/journal.pone.0189017
29. Brodeur JP, Ruddy S, Schwartz LB, Moxley G. Synovial fluid levels of complement SC5b-9 and fragment bb are elevated in patients with rheumatoid arthritis. Arthritis Rheumatol (1991) 34(12):1531–7. doi: 10.1002/art.1780341209
30. Corallini F, Bossi F, Gonelli A, Tripodo C, Castellino G, Mollnes TE, et al. The soluble terminal complement complex (SC5b-9) up-regulates osteoprotegerin expression and release by endothelial cells: Implications in rheumatoid arthritis. Rheumatology (2008) 48(3):293–8. doi: 10.1093/rheumatology/ken495
31. Nguyen THP, Hokstad I, Fagerland MW, Mollnes TE, Hollan I, Feinberg MW, et al. Antirheumatic therapy is associated with reduced complement activation in rheumatoid arthritis. PloS One (2022) 17(2):e0264628. doi: 10.1371/journal.pone.0264628
32. Siegert CE, Daha MR, Halma C, van der Voort EA, Breedveld FC. IgG and IgA autoantibodies to C1q in systemic and renal diseases. Clin Exp Rheumatol (1992) 10(1):19–23.
33. Liaskos C, Rentouli S, Simopoulou T, Gkoutzourelas A, Norman GL, Brotis A, et al. Anti-C1q autoantibodies are frequently detected in patients with systemic sclerosis associated with pulmonary fibrosis. Br J Dermatol (2019) 181(1):138–46. doi: 10.1111/bjd.17886
34. Foltyn Zadura A, Zipfel PF, Bokarewa MI, Sturfelt G, Jönsen A, Nilsson SC, et al. Factor h autoantibodies and deletion of complement factor h-related protein-1 in rheumatic diseases in comparison to atypical hemolytic uremic syndrome. Arthritis Res Ther (2012) 14(4):R185. doi: 10.1186/ar4016
35. Gupta B, Raghav SK, Agrawal C, Chaturvedi VP, Das RH, Das HR. Anti-MBL autoantibodies in patients with rheumatoid arthritis: Prevalence and clinical significance. J Autoimmun (2006) 27(2):125–33. doi: 10.1016/j.jaut.2006.07.002
36. Józsi M, Uzonyi B. Detection of complement factor b autoantibodies by ELISA. Methods Mol Biol (2021) 2227:141–5. doi: 10.1007/978-1-0716-1016-9_14
37. Hourcade DE, Mitchell L, Kuttner-Kondo LA, Atkinson JP, Medof ME. Decay-accelerating factor (DAF), complement receptor 1 (CR1), and factor h dissociate the complement AP C3 convertase (C3bBb) via sites on the type a domain of bb. J Biol Chem (2002) 277(2):1107–12. doi: 10.1074/jbc.M109322200
38. Zhao F, Afonso S, Lindner S, Hartmann A, Löschmann I, Nilsson B, et al. C3-glomerulopathy autoantibodies mediate distinct effects on complement C3- and C5-convertases. Front Immunol (2019) 10:1030. doi: 10.3389/fimmu.2019.01030
39. Strobel S, Zimmering M, Papp K, Prechl J, Józsi M. Anti-factor b autoantibody in dense deposit disease. Mol Immunol (2010) 47(7–8):1476–83. doi: 10.1016/j.molimm.2010.02.002
40. Medjeral-Thomas N, Pickering MC. The complement factor h-related proteins. Immunol Rev (2016) 274(1):191–201. doi: 10.1111/imr.12477
41. Corvillo F, Okrój M, Nozal P, Melgosa M, Sánchez-Corral P, López-Trascasa M. Nephritic factors: An overview of classification, diagnostic tools and clinical associations. Front Immunol (2019) 10:886. doi: 10.3389/fimmu.2019.00886
42. Marinozzi MC, Roumenina LT, Chauvet S, Hertig A, Bertrand D, Olagne J, et al. Anti-factor b and anti-C3b autoantibodies in C3 glomerulopathy and ig-associated membranoproliferative GN. J Am Soc Nephrol (2017) 28(5):1603–13. doi: 10.1681/ASN.2016030343
43. Chen Q, Müller D, Rudolph B, Hartmann A, Kuwertz-Bröking E, Wu K, et al. Combined C3b and factor b autoantibodies and MPGN type II. New Engl J Med (2011) 365(24):2340–2. doi: 10.1056/NEJMc1107484
44. Campa MJ, Gottlin EB, Bushey RT, Patz EF. Complement factor h antibodies from lung cancer patients induce complement-dependent lysis of tumor cells, suggesting a novel immunotherapeutic strategy. Cancer Immunol Res (2015) 3(12):1325–32. doi: 10.1158/2326-6066.CIR-15-0122
45. Li LL, Tan Y, Song D, Li YZ, Yu F, Chen M, et al. Anti-complement factor h autoantibodies may be protective in lupus nephritis. Clinica Chimica Acta (2020) 508:1–8. doi: 10.1016/j.cca.2020.05.005
46. Chauvet S, Berthaud R, Devriese M, Mignotet M, Vieira Martins P, Robe-Rybkine T, et al. Anti-factor b antibodies and acute postinfectious GN in children. J Am Soc Nephrology. (2020) 31(4):829–40. doi: 10.1681/ASN.2019080851
Keywords: complement, alternative pathway, C3 convertase, autoantibody, rheumatoid arthritis, factor B (FB)
Citation: Matola AT, Fülöp A, Rojkovich B, Nagy G, Sármay G, Józsi M and Uzonyi B (2023) Autoantibodies against complement factor B in rheumatoid arthritis. Front. Immunol. 14:1113015. doi: 10.3389/fimmu.2023.1113015
Received: 30 November 2022; Accepted: 23 January 2023;
Published: 20 February 2023.
Edited by:
Lillemor Skattum, Lund University, SwedenReviewed by:
Maciej Cedzynski, Institute for Medical Biology, Polish Academy of Sciences, PolandRafael Bayarri-Olmos, Rigshospitalet, Denmark
Copyright © 2023 Matola, Fülöp, Rojkovich, Nagy, Sármay, Józsi and Uzonyi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mihály Józsi, bWloYWx5LmpvenNpQHR0ay5lbHRlLmh1
†These authors have contributed equally to this work and share last authorship