 Yi Wang1†
Yi Wang1† Cheng-long Zhu1†
Cheng-long Zhu1† Peng Li1
Peng Li1 Qiang Liu1,2
Qiang Liu1,2 Hui-ru Li1,3
Hui-ru Li1,3 Chang-meng Yu1,2
Chang-meng Yu1,2 Xiao-ming Deng1,2,3*
Xiao-ming Deng1,2,3* Jia-feng Wang1*
Jia-feng Wang1*- 1Faculty of Anesthesiology, Changhai Hospital, Naval Medical University, Shanghai, China
- 2Jiangsu Province Key Laboratory of Anesthesiology, Xuzhou Medical University, Xuzhou, Jiangsu, China
- 3Faculty of Anesthesiology, Weifang Medical University, Weifang, Shandong, China
Sepsis is defined as a life-threatening dysfunction due to a dysregulated host response to infection. It is a common and complex syndrome and is the leading cause of death in intensive care units. The lungs are most vulnerable to the challenge of sepsis, and the incidence of respiratory dysfunction has been reported to be up to 70%, in which neutrophils play a major role. Neutrophils are the first line of defense against infection, and they are regarded as the most responsive cells in sepsis. Normally, neutrophils recognize chemokines including the bacterial product N-formyl-methionyl-leucyl-phenylalanine (fMLP), complement 5a (C5a), and lipid molecules Leukotriene B4 (LTB4) and C-X-C motif chemokine ligand 8 (CXCL8), and enter the site of infection through mobilization, rolling, adhesion, migration, and chemotaxis. However, numerous studies have confirmed that despite the high levels of chemokines in septic patients and mice at the site of infection, the neutrophils cannot migrate to the proper target location, but instead they accumulate in the lungs, releasing histones, DNA, and proteases that mediate tissue damage and induce acute respiratory distress syndrome (ARDS). This is closely related to impaired neutrophil migration in sepsis, but the mechanism involved is still unclear. Many studies have shown that chemokine receptor dysregulation is an important cause of impaired neutrophil migration, and the vast majority of these chemokine receptors belong to the G protein-coupled receptors (GPCRs). In this review, we summarize the signaling pathways by which neutrophil GPCR regulates chemotaxis and the mechanisms by which abnormal GPCR function in sepsis leads to impaired neutrophil chemotaxis, which can further cause ARDS. Several potential targets for intervention are proposed to improve neutrophil chemotaxis, and we hope that this review may provide insights for clinical practitioners.
Introduction
Sepsis is a severe life-threatening inflammatory response syndrome secondary to an infection, which is often characterized by multiple organ dysfunction (1, 2). The treatment of sepsis is still dominated by antibiotic therapy, respiratory support, fluid therapy, and organ function support since there are no approved specific drugs for sepsis currently (3). This kind of non-specific treatment prolongs the survival time of sepsis patients, and sepsis may develop into a critical illness lasting several weeks or months, further increasing the financial burden on patients’ families (4). Even with the use of these extrinsic supports, sepsis survivors often have other complications such as cognitive dysfunction, immune dysfunction, and neuromuscular disorders (5). It remains the leading cause of death in hospitalized patients, resulting in a heavy economic burden of medical care. In 2016, a systematic review and meta-analysis estimated that there are 31.5 million sepsis cases worldwide each year, with potentially 5.3 million deaths annually, based on data from seven high-income countries on four continents over the past decade (6). Given the lack of epidemiological studies on sepsis in low- and middle-income countries (LMICs), the actual global cumulative incidence may be higher. In 2020, based on data obtained in more LMICs from 1990 to 2017, an institute estimated that there are 48.9 million sepsis cases worldwide each year, with 11 million sepsis-related deaths annually, accounting for 19.7% of global deaths (7). According to the same study, approximately 50 percent of sepsis cases occurred in children and adolescents (7). Similarly, a recent international multicenter prospective observational study found that neonatal sepsis is the leading cause of neonatal death in LMICs (8). Therefore, sepsis is not only a fatal global syndrome but also a serious public health issue.
The third international consensus definition of Sepsis 3 defines sepsis as a life-threatening organ dysfunction caused by the host’s dysregulated response to infection, which distinguishes sepsis from the infection itself (1). It lies not only in the existence of infections caused by bacteria, fungi, viruses, or parasites but also in organ dysfunction caused by the dysfunctional response of the host to infections. In sepsis caused by secondary infection, the host recognizes microbial-derived pathogen-related molecular patterns (PAMPs) or endogenous damage-related molecular patterns (DAMPs) through a series of pattern recognition receptors (PRRs) at the cell membrane or in the cells, thereby activating innate immune cells to produce cytokines that mediate downstream signaling pathways (9–11). In the development of sepsis, the immune system is over-activated initially, characterized by overstimulated neutrophils and overload of the inflammatory cascade, and followed by immunosuppression, as shown by neutrophil dysfunction and increased apoptosis of lymphocytes (11–13). The phenomenon of early immune overactivation and late immune suppression in sepsis is called immune imbalance. As the most abundant innate immune cells at the locus of infection, neutrophils are the initial defense line of the host against infection and play a crucial role in eliminating local infection and injury healing (1). This cytokine storm is one of the important reasons why sepsis is difficult to control and the mortality remains high.
As a normal response to infection, neutrophils recognize chemokines including the bacterial product N-formyl-methionyl-leucyl-phenylalanine (fMLP), complement 5a (C5a), and lipid molecules Leukotriene B4 (LTB4) and C-X-C motif chemokine ligand 8 (CXCL8), and then enter the site of infection through mobilization, rolling, adhesion, migration, and chemotaxis (14). However, sepsis may induce dysfunction in neutrophil chemotaxis, characterized as compromised migration of neutrophils targeting infected organs (15), while a large number of neutrophils accumulate in the lung. The inflammatory mediators released by neutrophils will trigger an overwhelming cascade of inflammatory responses, further exacerbating the activation of other innate immune cells (16), leading to severe acute lung injury (ALI) and even acute respiratory distress syndrome (ARDS) (4). In non-sepsis conditions, neutrophils in the lung may even undergo reverse migration, returning to the vasculature after local infiltration at the site of inflammation (17). They will then return to the bone marrow to undergo apoptosis via C-X-C motif chemokine receptor 4 (CXCR4) reacts to CXCL12 (stromal cell-derived factor-1, SDF-1) (18). But in sepsis, neutrophils will be seized in the lungs and their apoptosis processes are significantly inhibited (18). Experiments using a murine model of abdominal infection showed that the reversing migration of neutrophils in the blood circulation produces excessive inducible Nitric Oxide Synthase (iNOS) and neutrophil extracellular traps (NETs), and promotes tissue inflammation and damage, which is positively correlated with the degree of lung injury (19–21).
In recent years, although some progress has been made in the identification and management of sepsis in terms of microbial pathogenicity and host reactivity, the physiological and pathological mechanisms leading to sepsis-induced lung injury are still not fully elucidated. Previous studies have shown that neutrophil migration dysfunction is associated with sepsis prognosis (22–24), providing a new insight that targeting the disturbance in neutrophil chemotaxis might be promising in reversing sepsis-induced organ dysfunction (25, 26), and the receptors responsible for chemotaxis might be important therapeutic targets in sepsis (27). It has been reported that 18 out of 23 human chemokine receptors belong to G protein-coupled receptors (GPCRs), whose expression and downstream signaling pathways are precisely regulated (28). The typical function of these chemokine receptors is to coordinate cell polarization and induce the directed migration of leukocytes to their chemokine ligands (29, 30). Furthermore, neutrophils in higher vertebrates respond to more than 30 GPCR signals corresponding to a range of chemotactic agents that affect cell polarization and thus migration in tissues (31). As the most abundant and diverse group of eukaryotic cell surface receptors, with more than 800 forms of human expression (32, 33), GPCRs expressed on various cell surfaces mediate neutrophils to monitor the danger signals generated by various external stimuli such as hormones, pathogens, inflammatory factors, and control their chemotaxis to infection foci to play an immune role (34). Approximately 34% of drugs approved by the US Food and Drug Administration (FDA) are GPCR-related (35), and these receptors are the largest family of pharmaceutically available proteins in the human genome (36), demonstrating their importance as key therapeutic targets. The purpose of this review is to elucidate the role of GPCRs in neutrophil dysfunction during sepsis-induced acute respiratory distress syndrome (ARDS) to provide therapeutic targets for the corresponding clinical treatment.
The role of neutrophils in health
Neutrophils, as the main effector cells of innate immunity and involved in the initiation, diffusion, and resolution of inflammation, account for 50~70% of all circulating white blood cells (37). They are generally regarded as the prototype of fast amoeboid-migrating cells, which means they have plastic deformability that enables them to take distinct migration strategies to move optimally in different microenvironments (17, 38). Normally, neutrophils reside in three different pools, known as the proliferative, circulating, and marginating pools (39). The number of neutrophils in each pool is affected by individual health state and the maturational development of cells. The proliferative or mitotic pool is composed of early neutrophil precursors such as myeloblasts, promyelocytes, and young myelocytes, which are located in the bone marrow and replenish the neutrophil population through mitosis (40). An adult produces about 1011 neutrophils per day, with an estimated 109 cells/kg of body weight leaving the bone marrow (41–43). Granulocyte colony-stimulating factor (G-CSF) and granulocyte-macrophage colony-stimulating factor (GM-CSF) have the potential to promote motivation, maturation, and activation of them (44), and to extend their lifespan (45). The release of neutrophils from the bone marrow is positively regulated by CXCR2 signaling and negatively regulated by the CXCR4-CXCL12 axis (Figure 1), keeping neutrophils in balance in the circulating and marginating pools (46). To avoid altering the activity of neutrophils in vitro, recent in vivo isotope or nanotechnology labeling experiments have shown that neutrophils are normally short-lived, with the mean half-life of inactive neutrophils ranging from 7 to 12 hours (47–50). Specifically, human neutrophils originate from granulocyte-macrophage progenitor (GMP) cells, which produce neutrophil promyelocytes that proliferate and differentiate into myelocytes (51–54). Following the myelocyte stage, the neutrophil progenitors lose their capacity to divide and take 4-6 days to mature (49, 55). Under noninfectious conditions, mature neutrophils are released into intravascular circulating pools and finally enter the marginating pools of reticuloendothelial organs such as the liver, spleen, and lung, or even return to the bone marrow for apoptosis (56, 57). Apoptotic neutrophils are phagocytosed by macrophages to limit inflammatory responses (58).
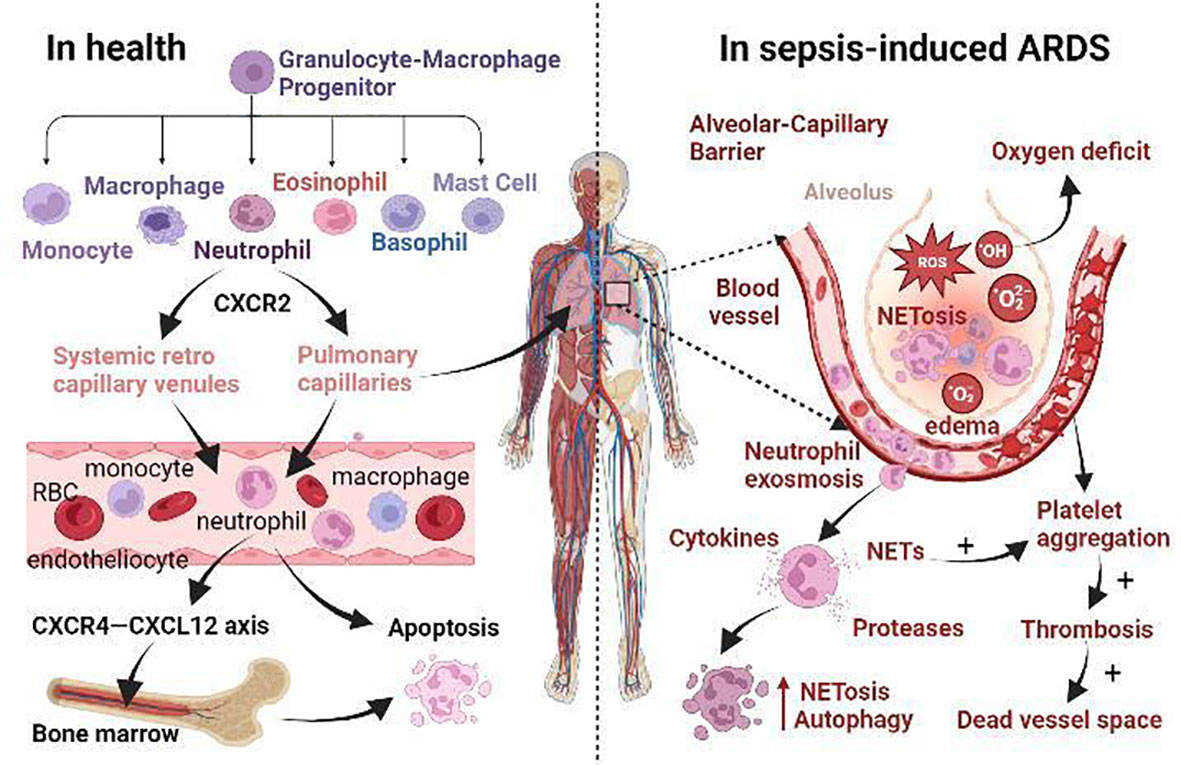
Figure 1 The role of neutrophils in health and the mechanism of sepsis-induced ARDS. (i) Normally, neutrophils originate from granulocyte-macrophage progenitor (GMP) in the bone marrow. When neutrophils mature, CXCR2 promotes their release from the bone marrow. Then they enter the systemic circulation via systemic retro capillary venules and into the pulmonary circulation via pulmonary capillaries. Finally, they enter the liver, spleen and other reticuloendothelial organs, or even return to the bone marrow via CXCR4-CXCL12 axis for apoptosis. (ii) In sepsis-induced ARDS, neutrophil apoptosis is delayed and migration is impaired, and a large number of neutrophils accumulate in the lungs Neutrophils promote self-aggregation and platelet aggregation by releasing ROS, NETs, protease, and other substances, leading to pulmonary ischemia and hypoxia, tissue edema, and micro thrombosis. Prolonged retention of late apoptotic neutrophils in pulmonary capillaries leads to the formation of a vascular dead lumen. Intense inflammatory response, endothelial barrier breakdown, alveolar edema, neutrophil dysfunction, and microcirculation disturbance are the main pathological changes of septic ARDS.
The role of neutrophils in general infection
During general infection, neutrophils in the circulating blood must first adhere to the blood vessel walls before being recruited to the infection site in response to the chemokine gradient. The upregulation of endothelial adhesion molecules and the loosening of vascular tight connections facilitate the migration of neutrophils to the target tissue (11). Activated endothelial cells upregulated the prestored P-selectin (CD62P) from Weibel-Palade bodies within minutes and the de novo synthesized E-selectin (CD62E) within 90 minutes (59, 60). These two selectins on endothelial cells bind to glycosylated ligands such as CD44, E-selectin ligand 1 (ESL1), and p-selectin glycoprotein ligand 1 (PSGL1) on neutrophils (61), trapping endovascular free-flowing neutrophils to the surface of endothelial cells, where they roll along the blood vessels toward the bloodstream. Rolling neutrophils recognize CD34, PSGL1, and glycosylation-dependent cell adhesion molecule (GlyCAM) on the surface of endothelial cells via L-selectin (CD62L) to be captured by endothelial cells again (62, 63). Bacterial components and pro-inflammatory cytokines promote L-selectin shedding but increase β2-integrins expression on neutrophils such as LFA-1 (lymphocyte function-associated antigen 1, CD11a/CD18) and MAC1 (CD11b/CD18) (64). These β2-integrins adhere to endothelial cells by binding to intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) with high affinity (65). Therefore, it has been suggested that the migration of neutrophils depends more on integrins than selectin (63). However, both selectin and integrin-mediated adhesion are brief and weak, promoting neutrophils to roll along endothelial cells and inducing their exosmosis to inflamed sites (66, 67).
Subsequently, neutrophils respond to bacterial toxins such as lipopolysaccharide (LPS) carried by the infectious agent, bacterial products such as fMLP, and chemokines produced by immune system activation such as C5a, LTB4, platelet activator (PAF), and interleukin (IL) -8 (CXCL8), all of which are effective activators of neutrophils (68–70). They are usually positively charged molecules that bind to negatively charged heparan sulfates and thus anchor to endothelial cells to form a certain intravascular chemotactic gradient (71). The rolling of neutrophils helps them to contact chemokine-decorate endothelial cells, thereby inducing neutrophilic activation. These chemo-attractants also induce the re-localization of integrins such as MAC1 stored intracellularly to the surface of neutrophils (72). The cytoskeletal protein talin1 binds to the β subunit of the activated integrin cytoplasmic tail to induce the extension of LFA1, thereby reducing the affinity of neutrophils to endothelial cells through conformational changes and promoting the slow rolling of neutrophils on endothelial cells (14). Another protein containing the FERM domain, kindlin 3 (also known as fermitin family homologue 3), binds to the same site on integrin and induces LFA1 to adopt a high-affinity conformation that promotes neutrophil stasis on endothelial cells (73). Due to fluid shear stress, endothelial cell structures are more elongated in the direction of blood flow, and neutrophils tend to crawl vertically at the endothelium-cell junctions (74). Neutrophils preferentially select endothelial tricellular corners with fewer junctional proteins and less orderly arrangement for paracellular migration (between endothelial cells) (75) over less efficient transcellular migration (through endothelial cells) (76).
When they finally migrate to the source of infection or damaged tissue, neutrophils emit pseudopods through membrane invaginations that envelop cytotoxic proteins, peptides, and enzymes in the phagosomes (40). The formation of the phagosomes attracts neutrophilic granulocyte particles to bind to them and degranulate themselves (77). At the same time, NADPH oxidases located on the phagosome membrane are activated to produce superoxide anion (O2-) and metabolize into hydrogen peroxide (H2O2) and other reactive oxygen species (ROS) (27). ROS, in turn, induce NETs release from activated neutrophils, which trap and kill pathogenic microorganisms to prevent their spread. They are a network of chromatin fibers that consist of DNA, citrullinated histone 3 (Cit-H3), myeloperoxidase (MPO), neutrophil elastase (NE), cathepsin G (CatG), proteinase-3 (PR-3), and other granular proteins (78). When the NETs formation is associated with cell death, neutrophils undergo suicidal dissolution of NETosis, a process that may release inflammatory mediators (79). Specifically, NETosis is a programmed neutrophil death distinct from apoptosis or necroptosis that facilitates host defense against pathogens and is characterized by nuclear delobulation, histone citrullination, chromatin decondensation, membrane permeabilization, and NETs release (80, 81). It was first described in 2004, where NET release and cell death were observed in response to high doses of phorbol-12-myristate-13-acetate (PMA) (82).
To date, three forms of NETosis are known, including suicidal, noncanonical, and vital NETosis (83). Suicidal NETosis mediated ROS production, MPO activation, and cytoplasmic release of NE by NADPH oxidase (83). In the presence of H2O2, MPO catalyzes the oxidation of chloride to hydrochloric acid and induces neutrophils to release NE, which together mediate chromatin decondensation during NETosis (84). In the cytoplasm, NE binds to the actin skeleton and disintegrates actin by degrading F-actin, thereby inhibiting neutrophil movement (85). In the nucleus, NE cleaves and inactivates histones, resulting in chromatin relaxation and DNA decondensation (84). In addition, peptidyl arginine deiminase 4 (PAD4), activated by high intracellular calcium concentrations induced by ionic carriers or bacterial products, mediates chromatin decondensation by histones citrullination (86, 87). Gasdermin D (GSDMD) is an executor of suicidal and noncanonical NETosis (81, 88). During suicidal NETosis, GSDMD is cleaved by NE and GSDMD-p30 pores are formed in the nuclear, granular, and plasma membranes (81). In turn, the breakdown of granules further promotes the release of NE in the cytoplasm, thus further cleaving GSDMD (88). Decondensated chromatin is released extracellularly through GSDMD-p30 pores or GSDMD-driven membrane rupture, ultimately resulting in neutrophil death (82, 88). Noncanonical NETosis is a novel suicidal NETosis pathway, which relies on the induction of non-classical inflammasomes to gram-negative bacterial LPS and activates mouse caspase-11 and human caspase-4/5 to cleave GSDMD to produce GSDMD-p30 pores, ultimately resulting in NETs release and cell death (81, 89). At this point, the release of NETs is dependent on Toll-like receptor 4 (TLR4) expression on platelets and P-selectin-mediated platelet-neutrophil interactions (90).
Suicidal and noncanonical NETosis release NETs relatively slowly, taking about 3 hours, while vital NETosis releases NETs only half an hour, and neutrophils that produce vital NETosis do not die immediately but remain phagocytic, migratory, and bactericidal (91). Vital NETosis is stimulated by bacteria, bacterial products, activated platelets, or complement proteins to induce the rapid release of NETs, and does not necessarily rely on NADPH oxidase to induce NE cleavage (87, 92). Moreover, the key features of vital NETosis are rapid histone citrullination, nuclear blebbing, and the vesicular transport of nuclear blebs to the plasma membrane, in which the nuclear membrane is swollen but not ruptured (83). An electron microscopy study showed that these vesicles of Vital NETosis exocytosis on the plasma membrane after budding from the outer nuclear membrane (ONM), thus ensuring the integrity of the nuclear and plasma membrane during NETs release (93). Studies have shown that neutrophil activation is involved in the initiation of NETosis through bacterial toxins or surface receptors. GPCRs (94), tumor necrosis factor (TNF) (95), Fc receptors (94), and TLR4 (96) on neutrophils have been reported to bind to ligands that trigger the release of calcium stored in the endoplasmic reticulum and induce NETosis by activating PAD4 to make histones citrullinated (97, 98). Among several chemokine receptors, CXCR1, CXCR2, and CXCR4 are involved in the formation of NETs (99). On the one hand, CXCR2 induces NETs formation by cooperating with PSGL1 (100). On the other hand, CXCR2 is also involved in neutrophil circadian rhythm regulation, altering the ability of NETs formation by disarming processes involving the neutrophil proteome (101). This circadian pattern is characterized by functional and phenotypic changes from neutrophil release to clearance, and this process is described as neutrophil aging (102). CXCR4 acts by releasing macrophage migration inhibitory factor (MIF), which in turn leads to NETs formation (103).
Additionally, the migration of activated neutrophils also depends on cytoskeletal dynamics mediated by molecular motor (myosin, kinesin, and dynein) drives (104, 105). Myosin is mainly involved in vesicle transport on actin filaments, while kinesin and dynein contribute to transport along microtubules. Kinesin moves intracellular cargo toward the plus-ends of the microtubules, while dynein moves toward the minus-ends of them (106). Inflammatory chemokines can act on intracellular signaling pathways through corresponding receptors (mostly GPCRs) to induce neutrophils to migrate by a polymeric contractile balance between anterior branch actin and posterior myosin II-dependent actin (107–110). Studies of zebrafish larvae have presented cytoskeletal requirements for neutrophil redirection (111, 112) Firstly, actin-related protein 2/3 (Arp 2/3)-mediated dendritic actin networks determine the extensive search phase of neutrophils, followed by rapid actin flow that accelerates cell migration to the source of infection. Formyl peptide receptors (FPRs) are G-protein-coupled receptors that are expressed on the surface of neutrophils that recognize peptides containing N-formylated methionine, such as fMLP. FPRs transmit chemotactic signals that mediate host defense and inflammatory responses such as cell adhesion, directed migration, particle release, and superoxide production. The human FPR family consists of three members, in which FPR3 is expressed in monocytes but only FPR1 and FPR2 are expressed in neutrophils (Table 1) (113). Notably, a variety of non-GPCRS receptors are expressed on the surface of neutrophils, including cytokine receptors, integrins, Fc-receptors, and other innate immune receptors (Table 1), which are also critical for the differentiation, adhesion, recruitment, and phagocytosis of neutrophils (114).
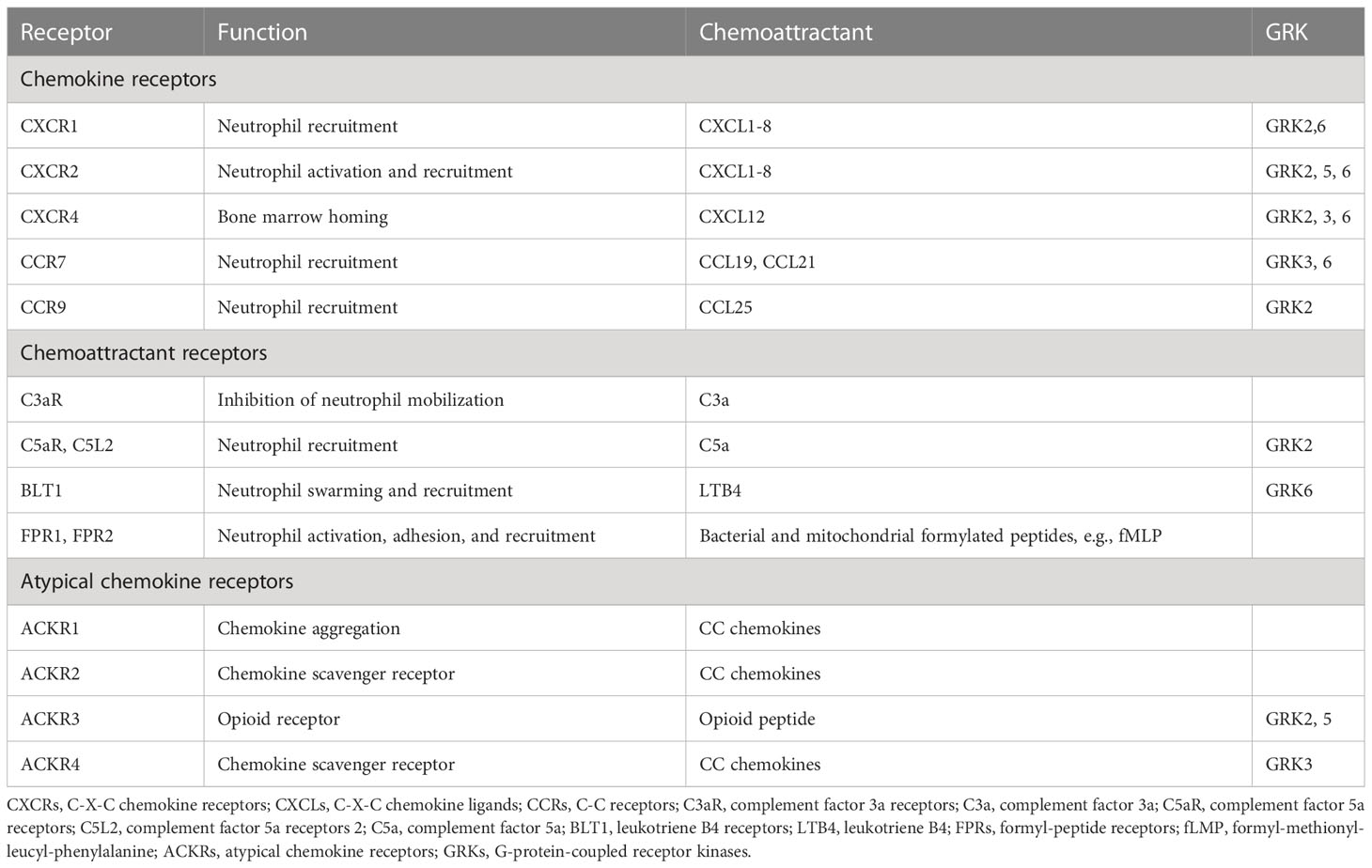
Table 1 Common receptors and their corresponding chemo-attractants and GRKs.
The contribution of neutrophils and GPCRs in sepsis
It is well-accepted that neutrophil dysfunction occurs in sepsis and has been considered the main cause of organ failure (27, 115–117). Neutrophil degranulation and endothelial dysfunction are shown to be the core events of the pathophysiology of sepsis (77, 118). In the early stage of sepsis, mature neutrophils in the bone marrow are rapidly released, increasing 10-fold in circulation within hours, and largely accumulate in the lungs, according to autopsy reports (119). Bacterial products and over-release of pro-inflammatory cytokines such as TNF-α, IL-1b, IL-6, and IL-17 increase G-CSF expression and indirectly mobilize neutrophils by altering the balance between CXCR4 and CXCR2 ligands in bone marrow, affecting their release, activation, and migration (120, 121). During sepsis, the spontaneous apoptosis of neutrophils is delayed, but other types of death such as necrosis, necroptosis, pyroptosis, NETosis, and autophagy may happen (10, 116, 122). It is well established that the proportion of neutrophil NETosis (123) and autophagy (124) increases in sepsis with reduced apoptosis (125), but the other three types of neutrophil death remain poorly understood (Figure 1). A study showed that 50% of neutrophils were apoptotic after 24 hours of in vitro culture, compared with only 5-10% in sepsis (125). In models of ALI caused by cecal ligation puncture (CLP) or endotoxemia, the proportion of lung neutrophils undergoing apoptosis within 24 hours was significantly reduced (126). Moreover, septic animals and patients showed a reduction in neutrophil migration (127, 128). Due to migration dysfunction, neutrophils with delayed apoptosis cannot effectively reach the site of infection to eliminate pathogenic bacteria and their phagocytic activity is decreased. Instead, most of them are detained in the lung, liver, and other organs, mediating nonspecific organ damage through the release of ROS, NETs, protease, and other cytotoxic substances (129–131).
In sepsis, the complement system is activated, releasing small fragments such as C3a and C5a, which have potent pro-inflammatory effects (132). C5a binds to its receptors (C5aR and C5L2) on the surface of activated neutrophils to promote the release of NETs, in which DNA fibers and histone (H3 and H4) networks provide scaffolds for the aggregation, relocation, and activation of platelets, neutrophils, and red blood cells (133). TLR4-mediated platelet activation increases endothelial cell adhesion at the site of inflammation (66) and promotes neutrophil activation, exosmosis, and aggregation by expressing higher levels of P-selectin (Figure 1) (134). Platelet-derived chemokine heterodimers (of CXCL4 and CXCL5) are also important for neutrophil recruitment (135), and endothelial presentation of CXCL5 depends on platelets (136). Activated platelets interact with neutrophils through β2-integrin LFA-1, thereby enhancing neutrophilic activation and lowering the threshold for NETs release (137, 138). Interactions between activated endothelial cells further promote the release of NETs (Figure 1) (139–141). Many components of NETs, such as DNA, histones, and granular proteins, all have procoagulant activity (142). DNA initiates the intrinsic clotting cascade and nucleic acid enhances serine protease thrombin activity (143). Histones promote thrombin production by inhibiting anticoagulants such as antithrombin (AT) and activating protein C (APC) (144). Granular proteins, especially neutrophil elastase, promote thrombosis by inhibiting tissue factor pathway inhibitors (TFPI) and anticoagulants (145). At the same time, coagulation factors, thrombin, plasmin, and APC in the blood induce downstream signaling pathway transduction by activating protease-activating receptors (PARs, a small family of GPCRs) expressed on platelets, endothelial cells, and vascular smooth muscle cells (VSMC) (118), further aggravating tissue and organ damage. There are currently four types of PARs in the human genome, among which platelet expresses PAR1 and PAR4 (146), endothelial cells express PAR1, PAR2, and PAR4 (147), VSMC expresses PAR1 and PAR2 (148, 149), and PAR3 is mainly expressed in bone marrow (150). Of the four major Gα subfamilies, PAR1 and PAR2 mediate signaling via Gq, Gi, and G12/13, PAR3 via Gq-mediated signaling, and PAR4 via Gq and G12/13 (151). In sepsis, these reactions lead to increased vascular permeability, endothelial barrier breakdown, abnormal accumulation of neutrophils, and excessive inflammatory responses, which further aggravate ischemia, hypoxia, tissue hypoperfusion, and microcirculation disorders, eventually leading to organ failure, shock, and even death (152, 153).
The contribution of neutrophils and GPCRs in sepsis-induced ARDS
Among all the damaged organs, the lung is the first and most frequent organ to be compromised, and ARDS is one of the key prognostic factors for the mortality of septic patients (154). In systemic circulation, neutrophils enter the tissue through systemic retro capillary venules, but in pulmonary circulation, neutrophils migrate through pulmonary capillaries (Figure 1) (155, 156). Neutrophils must undergo deformation to pass through the pulmonary capillaries because of their larger diameter, a time-consuming process called neutrophil sequestration, which was first described in 1993 and has been observed using macroscopic radiolabeling imaging devices (157, 158). In contrast to the doughnut-shaped nuclei of mice, human neutrophils are leafy, increasing the flexibility of mature neutrophils to navigate tissue space (159, 160). Nevertheless, the physiological and pathological mechanism of how neutrophil sequestration in sepsis causes ARDS is still poorly understood (161, 162). An uncontrolled immune inflammatory response caused by sepsis eventually leads to multiple organ dysfunction syndromes (MODS) of the heart, brain, liver, lung, kidney, etc (63, 153). Autopsy of these patients with MODS in sepsis revealed abundant neutrophils in the kidneys and lungs (163). It has been found that the functional capillary ratio of the pulmonary microcirculation is decreased in sepsis-induced ALI (164), which represents the occurrence of abnormal tissue perfusion, and organ dysfunction may be caused solely by neutrophils sequestered in the microvascular system (165). In the early stage of sepsis, neutrophils are activated and migrate in the pulmonary capillaries. In the late stage, neutrophils with delayed apoptosis are trapped in the pulmonary capillaries for a long time, resulting in the formation of a vascular dead lumen (Figure 1), which further triggers the aggregation of neutrophils and leads to microcirculation disturbance, exacerbating the hypoxia caused by sepsis-induces ARDS (164). This suggests that neutrophil sequestration may be a critical stage in the initiation of multiple organ failure (166) and that damage in one organ by a large accumulation of neutrophils may trigger the same aggregation effect in other organs (167).
In addition, the severity of septic ARDS is positively correlated with the degree of neutrophil infiltration and the intensity of its derived proteolytic enzymes in bronchoalveolar lavage fluid (168). Results from the bronchoalveolar lavage tests showed that IL-2 concentrations were associated with delayed apoptosis of neutrophils (169), and IL-8 (170), and IL-18 concentrations were associated with poor outcomes in patients (171). IL-8 binds to CXCR1 and CXCR2, which are the high-affinity receptors of CXCL. But only CXCR2 rather than CXCR1 is reduced on neutrophils in patients with sepsis, and the interleukin-8 mediated chemotaxis is impaired (172). Xu et al. had shown that an increase in C5a during sepsis inhibits neutrophil IL-8 secretion, resulting in neutrophil migration dysfunction through downstream signaling pathways mediated by C5aR and C5L2 (173). It is concluded that GPCR-mediated aggregation, activation, and apoptosis of the accumulated neutrophils in the lung of septic patients are important causes of ARDS and are related to the release of tissue-destructive immune mediators (174, 175).
GPCR-mediated positive and negative feedback regulates neutrophil aggregation
During sepsis, the first neutrophils detecting the local tissue infection rapidly release LTB4 within minutes. Other activators such as C5a, LPS, and fMLP, also promote LTB4 release, with a cascade of amplification reactions mediating neutrophils aggregation (176, 177). Neutrophils can sense and monitor the relevant danger signals released by tissues through GPCRs, which promote the release of attractants for communication between cells. The GPCR-mediated positive feedback is the basis of neutrophil aggregation, and its downstream molecular pathways are major triggers of neutrophil polar movement (38).
When GPCR is activated, the Gα subunit of the G protein binds to GTP and dissociates from the Gβγ dimer. The activated Gα induces the generation of a second messenger such as cAMP (178), while the dissociated Gβγ dimer binds to downstream signaling molecules and activates downstream signaling pathways such as phospholipase C β2/3 (PLCβ2/3) and phosphatidylinositol 3-kinase (PI3K) (179, 180). Activation of PLCβ in neutrophils can promote the release of Ca2+ from Ca2+ pools in the endoplasmic reticulum, leading to an increase in Ca2+ concentration and providing power for the secretion of intracellular vesicles or granulosa proteins (40). The human genome encodes four distinct Gα subunits, including Gαs, Gαi, Gαq, and Gα12/13 (181). Gαs activates adenylate cyclase (AC) to catalyze the conversion of ATP to cyclic adenosine monophosphates (cAMP) (182), which is the second messenger that regulates many downstream signaling pathways of GPCRs, while Gαi inversely regulates cAMP concentration by inhibiting AC activity or activating phosphodiesterase (PDE). Gαq activates PLC to hydrolyze phosphatidylinositol 4, 5-diphosphate (PIP2) to inositol triphosphate (IP3) and diacylglycerol (DAG), which acts as a protein kinase C (PKC) activator, while Gα12/13 regulates small GTPases that affect the cytoskeleton of actin and tubulin (183). Despite their differences in function, both the Gαs and Gαi subunits activate the GTPase activity of tubulin to disrupt the stability of microtubules (183). In neutrophils, different Gαi play different roles in GPCR signal transduction, and Gαi does not internalize in response to activation compared to Gαs (184). Gαi2 is responsible for vascular Ca2+ flux and neutrophil stasis, while Gαi3 is responsible for neutrophil migration and activation of the PI3K/Akt pathway (185, 186). The helical H5 structure in Gα has been shown to play a key role in mediating the allosteric regulation of GPCRs (187).
Studies have shown that GPCR desensitization is an intrinsic negative feedback mechanism for neutrophils and plays a key role in preventing excessive aggregation of neutrophils to maintain a balance between tissue destruction and host protection (176, 188, 189). Hidalgo and col. showed that neutrophils acquire different phenotypes and functional properties in normal tissues, and they enter these tissues under the action of chemokines, such as CXCR4 signaling to chemotactic neutrophils into the lung in response to CXCL12 stimulation (190, 191). Kinase omics analysis of septic neutrophils revealed impaired activity, indicating an immunosuppressed neutrophilic phenotype (192). C5a down-regulates CXCR4 expression on neutrophils and promotes protease release, which degrades matrix proteins and inhibits the homing effect of CXCL12, ultimately leading to neutrophil phenotype changes (193). At the same time, high levels of C5a may lead to reduced targeted migration of neutrophils (194). However, C5a receptor expression on neutrophils peaks at the first 24 hours after sepsis initiation and gradually declines thereafter (195). PI3K can control the aggregation effect of C5a-mediated neutrophils by regulating the oxidative burst and phagocytosis activity of neutrophils. Inhibition of PI3K in vitro up-regulates TLR4-mediated pro-inflammatory cytokine expression in neutrophils (196) and activated TLR4 enhances neutrophils’ chemokine reactivity by down-regulating the expression of G-protein-coupled receptor kinases (GRKs) involved in GPCRs desensitization (197). Therefore, C5a and PI3K interact with TLR during sepsis to upregulate GRKs expression, thereby internalizing and desensitizing GPCRs to reduce neutrophil chemotaxis and negatively regulate neutrophil aggregation. In PI3Kc-/- mice with sepsis, the expression of GRK2 was decreased while that of CXCR2 was increased, and the survival rate was higher, which was consistent with the above conclusion (198). Besides, TLR4-deficient mice did not develop neutrophil migration dysfunction, suggesting that the phenomenon is TLR4-dependent.
The bias of GPCR expression: G protein or GRK-arrestin pathway
After GPCR is stimulated by extracellular signals, activated GPCR can induce structural rearrangement of its cytoplasmic region (199) and induce intracellular signal transduction through classical G protein or GRK-arrestin pathway (Figure 2) (200, 201), and this signal transduction can be biased in three ways. System bias refers to the differential expression of signaling pathways of the same receptor or ligand in a different time, space, and cell type. Receptor bias refers to the selective action of the same agonist on different receptors to mediate their downstream pathways. Ligand bias refers to the agonist recognized by a single receptor that prefers a certain downstream pathway, which is generally thought to be related to the conformation of the GPCR-arrestin complex (114, 202, 203). Residues near the GPCR binding site are much less conserved than critical residues on the intracellular membrane of the cells, resulting in complexes that have both “clingy” and “hanging” conformations, with the former critical for desensitization of G protein signaling and the latter promoting G protein activation or G protein independent signaling (204, 205). In most cases, G protein signaling is the cause of adverse drug reactions, and ligand bias can be used to reduce such adverse reactions, leading to the development of more promising and safer drugs (206, 207). How these ligands reconfigure cytoplasmic regions of the GPCR to selectively promote G protein, GRK, or arrestin binding is not yet known.
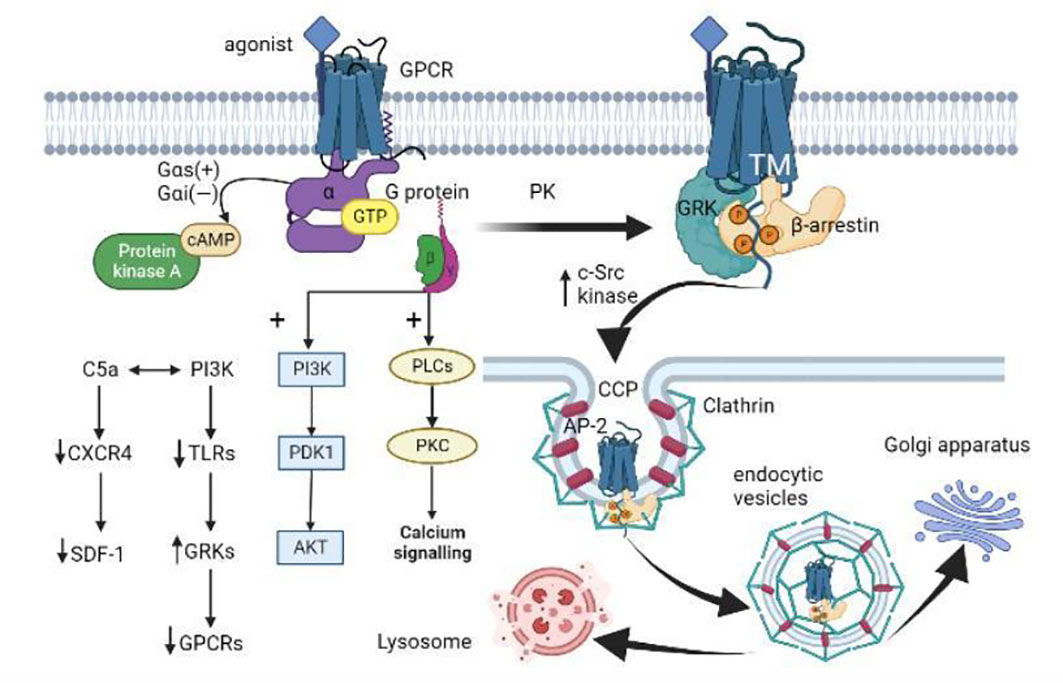
Figure 2 GPCR activation, internalization, and desensitization. (i) When GPCR is activated by an agonist, the Gα subunit of the G protein binds to GTP and dissociates from the Gβγ dimer. Gαs activates adenylate cyclase to catalyze the conversion of ATP to cAMP, while Gαi inversely regulates cAMP concentration. The dissociated Gβγ dimer activates the PLC and PI3K downstream pathways. (ii) GPCR is subsequently phosphorylated by different protein kinases (PKs) like GRK. Then β-arrestin binds to the GPCR-activated transmembrane (TM) core, allosterically modulates its three proline regions (PRs) to induce conformational changes, and spatially impedes the interaction between GPCR and G proteins. β-arrestin induces the aggregation of clathrin and adaptor protein 2 (AP-2) by increasing the activity of c-Src kinases, and targets the receptor on clathrin-coated pits (CCP). (iii) The local membrane invagination promotes the formation of endocytic vesicles. The endocytic vesicles target the Golgi apparatus or lysosomes under the action of Rab proteins and cytoskeleton. (iiii) In addition, activated PI3K promotes GRK activity by down-regulating TLRs expression, thus mediating GPCR desensitization. C5a interacts with PI3K to regulate neutrophil aggregation by down-regulating the expression of CXCR4 in neutrophils and inhibiting the homing effect of SDF-1 (CXCL12).
There are more than 286 GPCR structures that can combine with G proteins (208), but only five of them can bind to arrestin and activate GPCR, including two neurotensin receptor 1 (NTSR1) (209), one M2 muscarinic receptor (M2R) (210), one β1 adrenergic receptor (β1AR) (211), and one V2 vasopressin receptor (V2R) (212). Angiotensin II (Ang II), as a biomarker of sepsis severity, is associated with the progression of septic ARDS (213, 214). Modified angiotensin II type 1 receptor (AT1R) can preferentially stimulate the G-protein-mediated or arrestin-mediated signaling pathway after binding to Ang II, and this model system is often used to study the biased signaling of GPCR (215, 216). The GPCR-arrestin interaction has a shorter duration on receptors such as β1AR, but a longer duration on V2R and AT1R (217). GRK and arrestin are important for regulating the duration and amplitude of GPCR signaling (218). Two of the four arrestin subtypes, β-arrestin 1 and β-arrestin 2, are expressed in almost all cells of vertebrates and interact with different GPCRs on the cell surface to induce their downstream non-classical signaling pathways (219).
The continuous activation of GPCR during inflammation leads to the activation of GRK, which in turn negatively regulates the phosphorylation of C-terminal clusters of GPCR, and then the recruitment of inhibitory protein β-arrestin to bind to these clusters (220). The increased β-arrestin also sometimes binds to the activated transmembrane (TM) core of GPCR, allosterically modulates its proline regions (PRs) (221), causing specific conformational changes, and impeding the interaction between GPCR and G proteins in space, further attenuating GPCR signaling (Figure 2). Furthermore, β-arrestins that bind to GPCRs influence the activity of regulatory ubiquitin E3 ligase and a stress-related JNK3 kinase (222–224), but they remained in this active conformation after dissociation from the receptor (225–228). β-arrestin conjugated GPCR to SH3-domain proteins (SH3-CPs) via three proline domains (P1, P2, P3), each with different affinity to the receptors and downstream effectors (Figure 2). Specifically, phosphoric acid sites 5 on GPCRs were coupled to P1 and P2, phosphoric acid sites 2-3 were coupled to P3, and phosphoric acid binding sites 5 induced conformational changes in regions P1 and P2 through the propagation paths of V40→P114→F87 and P36→V34→F123, respectively (221). The different GPCRs phosphorylation sites regulated by different GRKs lead to different β-arrestin conformations and thus different signal transduction functions, which is known as the “phosphorylation barcode hypothesis” (229–231).
GPCRs internalization/desensitization: A key factor for neutrophil migration dysfunction
It has been shown that GPCRs desensitization, internalization, and signal transduction are mediated by GRKs and the inhibitory protein β-arrestins (Figure 2) (232). There are two forms of GPCRs desensitization, including homologous and heterologous desensitization. Heterologous desensitization means that one GPCR signal can act as desensitizing signal of multiple other GPCR types, causing immune cells to develop resistance to several chemotactic stimuli (38). However, in vitro studies have shown that human neutrophils experience homologous desensitization initiated by GRKs, as shown by no response to continuous or repeated stimuli (233, 234). GRKs are key serine/threonine protein kinases in which GRK2-3 and GRK5-6 are commonly expressed in cells and tissues, while GRK1 and GRK7 are localized in the retina and GRK4 is localized mainly in the testis (235). There are four GRKs expressed on neutrophils (GRK2, GRK3, GRK5, GRK6), with different target GPCRs. According to the references, GRK2 binds to CXCR1 (236) and CXCR2 (197, 237, 238), GRK5 to CXCR2 (197), and GRK6 to LTB4R1 (239) and CXCR2 (236). Phosphorylation of GRKs at serine (Ser) or threonine (Thr) residues facilitates the high-affinity binding of GPCR-β-arrestin, which partly explains the desensitization of G protein signal transduction (233). After CXCR4 agonist stimulation, GRK2/3 phosphorylates the distal Ser/Thr locus on the C-tail, while GRK6 phosphorylates more at the proximal Ser/Thr locus (240). In addition, GRK2 also promotes the formation of the GRK2-tubulin complex and tubulin phosphorylation, which may be involved in receptor transport (241). GRK2 also phosphorylates ezrin, a protein involved in cortical actin cross-linking with the plasma membrane (242), which may mediate GPCRs internalization and actin cytoskeletal recombination (243). This difference in the phosphorylation site of GRKs may mediate different signaling pathways, which partly explains the different pharmacodynamics of agonists.
β-arrestin recruited by phosphorylated GPCRs through Clathrin-mediated endocytosis is the main pathway for the internalization of GPCRs, which is important for both receptor recycling and intracellular signal transduction (244). Src are a kind of kinases that contain the SH3 domain and recognize the specific PRs conformation in β-arrestin without recognizing the C-terminal shift of GPCRs (221). The binding of β-arrestin to GPCR can also induce clathrin and adaptor protein 2 (AP-2) aggregation by increasing c-Src kinase activity (245–247). Thus, receptors can be targeted at clathrin-coated pits (CCPs) to make GPCRs locally invaginate to promote ingestion in vivo (248). Although AP-2 is not the key to forming the CCPs (249), it can recruit transmembrane cargo and auxiliary proteins via a specific motif of clathrin (Figure 2) (250). These cargo proteins need to contain specific motifs, such as YXXΦ motifs to be recognized by AP-2 (251). S-nitrosylation of β-arrestin by receptor activation or direct modification promotes GPCRs desensitization and concave endogenization, while β-arrestin lacking such post-translational modification prolongs the signaling time of cAMP, extracellular signal-regulated kinase (ERK), and nitric oxide (NO) (252). In this regard, some scholars believe that the desensitization of GPCRs is not signal shutdown, but signal switching, that is, from G protein signal transduction to G protein independent signal transduction, and this G protein independent signal transduction may be mainly mediated by β-arrestin (253, 254). In addition to mediating the internalization of GPCRs, another key role of β-arrestin is to stimulate mitogen-activated protein kinase (MAPK) cascades. As reported, p38 and ERK induce GRK2 phosphorylation at Ser670 and inhibit GRK2 translocation and the following GPCRs internalization (255), except that p38 promotes monocyte migration while ERK inhibits monocyte migration. The anaplastic lymphoma kinase inhibitor LDK378 inhibits Ser670 phosphorylation on GRK2 by inhibiting p38 activation, resulting in enhanced GRK2 translocation and CXCR2 internalization, thus inhibiting the recruitment of myeloid suppressor cells (MDSCs) to the spleen (11).
Subcellular localization is one of the mechanisms regulating GRKs’ activity and GPCRs’ desensitization (256). Furthermore, the movement of GPCRs in different cellular compartments during internalization occurs in vesicles and is regulated by synergistic interaction with cytoskeletal elements (217). Normally, GPCRs are synthesized in the rough endoplasmic reticulum (ER) and move from the Golgi apparatus to the plasma membrane in the form of vesicles, which is a down-direction transport, while the vesicles formed after the internalization of GPCRs are directed to the lysosome or proteasome, which is a reverse transport (217). Biased agonists stimulate GPCRs in the plasma membrane with a sustained and strong effect, but not in the intracellular compartment, suggesting that the production of second messengers such as Ca2+, cAMP, IP3, and DAG is essential for subcellular localization (257). The production of free vesicles is dependent on dynamin, which exists in three subtypes in mammals and binds to acidic phospholipids on the cytoplasmic side of the plasma membrane via the pleckstrin homology (PH) domain (Figure 2). During the internalization of GPCRs, the dynamin is located at the neck of the budding vesicles and relies on the hydrolysis of GTP to form a helical polymer conformation that promotes the fission of the underlying tubular membrane section, resulting in the generation of endocytic vesicles. This dynamin is also present in the actin reticulum associated with the Arp 2/3 complex (258, 259). Some desensitized neutrophil GPCRs can be reactivated by cytoskeletal destruction (114). Rab proteins are a kind of monomer GTP proteases, which are expressed differently on different membranes and can regulate vesicle docking and fusion. Their activities are partly regulated by the concentration of PI3K (260). A few studies have demonstrated the interaction between GPCRs internalization and various Rab proteins in different vesicular compartments. Among them, Rab5a mainly binds to the last 10 amino acids of AT1AR and participates in the receptor cycle of reverse transport (261), while Rab11 is associated with the circulation of receptors that are transported downstream and is located in the trans-Golgi network (TGN), post-Golgi vesicles, and recycling endosomes (262, 263). This suggests that GPCRs can regulate the activity of intracellular transport components through vesicle cargo proteins to control the targeting of receptors to specific cellular compartments after internalization (264, 265).
CXCR2 is essential for GPCRs internalization and neutrophil migration
Currently, 46 human chemokines are known to activate 20 chemokine receptors (266, 267) and another four atypical chemokine receptors (ACKRs) (268) (Table 1). CXCR2 is a receptor for eight CXC chemokines (CXCL1-8) (269, 270), which can drive innate immune cells to infiltrate remote organs in sepsis (127, 271, 272) and seems to have a synergistic effect with lipid signaling. It can make neutrophils move from random movement to directed migration, thus forming local cell clusters (176, 188). Non-survivors of sepsis showed higher CXCR2 expression on neutrophils and more neutrophil migration dysfunction than survivors (273). CXCR2 is internalized faster than CXCR1 but takes longer to be recycled (274). Studies have shown that reverse-migrating neutrophils have reduced CXCR1 expression (275). CXCR1 desensitization in zebrafish is associated with neutrophil diffusion to sites far from the wound, and this transition involves CXCR2 signaling (189). GRK2 controls the internalization of CXCR2 while desensitized LTB4R1 remains in the plasma membrane (38), suggesting that CXCR2 but not LTB4R1 is involved in receptor internalization.
In addition, CXCR2 sometimes dimerizes chemokine receptors, forming homologous dimers through the Ala106-Lys163 region even in the absence of ligands (276). When neutrophils express both CXCR1 and CXCR2, they may also form heterodimers (277). As a ligand of the two receptors, CXCL8 can deactivate the source dimer and stabilize the homologous dimer to promote the internalization of the receptor (278). It can be seen that the activation of different ligands leads to the selective expression of GPCRs dimerization, thus regulating receptor desensitization, which may be one of the ways to fine-regulate neutrophil chemotaxis, but the specific mechanism is still unclear.
The C-terminal of CXCR2 contains a leucine-rich domain (325-329aa), the LLKIL motif (279). CXCR2 binds to a variety of proteins through this LLKIL motif, including LIM and SH3 protein 1 (LASP-1), Hsc70 interacting protein (Hip), and AP-2 (279, 280). The pathway involved in the regulation of LASP-1 affects cytoskeletal rearrangement and thus neutrophil migration (281). The Hsc70 complex formed by the binding of CXCR2 to the Hip is related to the internalization of CXCR2 (282). By binding to membrane phosphatidylinositol (PI), AP-2 is docked in the plasma membrane and causes microtubule acetylation, which is essential for the targeted movement of neutrophils (283). GRK2-phosphorylated CXCR2 can also internalize the receptor by binding β-arrestin independently of clathrin and AP-2, but this is dependent on Lys327 ubiquitination on CXCR2 (284).
CXCR2 is induced on the surface of neutrophils in a TLR2 or TLR4-dependent manner in patients with sepsis and mediates neutrophil infiltration into important organs such as the lungs, hearts, and kidneys (127). TLRs, a homolog of the Drosophila protein Toll, are a kind of PRRs that control innate immunity and contribute to the inflammatory response of DAMPs. TLRs currently have 11 forms, among which TLR4 is the signal transduction receptor of LPS (285). High mobility group box 1 (HMGB1) acts as a DAMP that interacts with TLR9 to regulate the neutrophil NET formation, thereby mediating inflammatory tissue damage (286). The activation of TLRs or PI3K on activated neutrophils can induce the expression of TNF-α and iNOS. During sepsis, iNOS inhibited the expression of selectin, integrin, and cell adhesion factor -1 and promoted the production of NO, which can not only dilate pulmonary vessels and increase vascular permeability to aggravate pulmonary vascular injury but also reduce heme oxygenase-1 to inhibit neutrophil rolling and adhesion (287–290). TLRs, TNF-α, and NO can all lead to the up-regulation of GRK2, which can be used as a negative feedback mechanism to induce CXCR2 desensitization (272, 291–293). IL-33 inhibits TLR-mediated up-regulation of GRK2 expression and CXCR2 internalization by binding to its heterodimer receptor complex signal transducer 2(ST2), thereby reducing neutrophil migration dysfunction and enhancing their recruitment to the infection sites (140, 237, 294).
However, experiments showed that CXCR2 desensitization controlled by GRK2 could not prevent the migration of neutrophils to the aggregation site after GRK2 depletion, despite the desensitization resistance, and its bacterial clearance ability was impaired, indicating that the internalization of CXCR2 was not the only factor leading to the excessive migration of neutrophils (176). In addition, miRNA may also affect CXCR2 activity. MiR-K12-3, a miRNA-125 from Kaposi sarcoma-associated herpes virus (KSHV) (295), can desensitize and internalize CXCR2 by directly reducing GRK2 expression and phosphorylating GPCRs (237). A fungus containing miRNA-1321 and miRNA-3188 expressed in vitro even directly reduced the microRNA expressed by CXCR2 (296).
Potential therapeutic targets for septic ARDS
The lungs are the most common source of sepsis, accounting for 64%, which easily leads to the occurrence of ARDS (297). There are currently no specific drugs for sepsis, and the only drug approved for severe sepsis, recombinant Human (rh) APC, has been shown not significantly reduced sepsis mortality (298, 299). Similarly, many pharmacological therapies have been used to treat ARDS, including aspirin, β-2 agonists, statins, and corticosteroids, but none of them have a significant effect (300). Mechanistically, NETs are involved in the progression of ARDS induced by sepsis, and inhibition of neutrophil NETs formation may be a potential target to prevent ARDS. Aspirin inhibits neutrophil migration and thromboxane-dependent NETs formation by inhibiting platelet-neutrophil interactions (301). High-dose intravenous vitamin C (HDIVC) improves ARDS by reducing the occurrence of NETosis and the shedding of the vascular endothelial glycocalyx by down-regulating the expression of cell-free DNA in neutrophils and proteoglycan syndecan-1 in vascular endothelial cells (302). α1-antitrypsin (AAT), as a protease inhibitor, especially a NE inhibitor, inhibits neutrophil migration and NETs formation by binding to IL-8 to inhibit CXCR1/CXCL8 signaling axis (303). Sivelestat (ONO-5046), another selective NE inhibitor, also improved neutrophil-mediated vascular endothelial injury and increased vascular permeability (304). Moreover, zinc-dependent histone deacetylases (HDAC) mediate the deacetylation of histone H3, which is a necessary step to allow histone citrullination and NETs formation (305). Ricolinostat (ACY-1215), an HDAC6 inhibitor currently in Phase II clinical trials, can improve lung function by inhibiting NETosis (305). In addition, glycyrrhizin, the active component of traditional Chinese medicine, reduces neutrophil NETs formation in sepsis by inhibiting the activation of the HMGB1/TLR9/MyD88 (myeloid differentiation primary response 88) pathway (286). Inhibitors of protein tyrosine phosphatase-1B (PTP1B) reduce neutrophil chemotaxis and NETs formation by inhibiting the PI3γ/AKT/mTOR pathway downstream of CXCR4 signaling (306). Interestingly, administration of DNase-I-coated melanin-like nanospheres (DNase-I pMNSs) mitigates sepsis-associated NETosis, thereby preventing the further progression of ARDS (307). The oleic acid-based nanosystems dose-dependently inhibited the production of ROS, O2-, and NE by activated neutrophils, n, thereby reducing NETs formation, and larger nanocarriers showed greater efficiency in improving ARDS (308). The drugs coated by lipid nanocarriers have the characteristics of prolonging circulatory half-life, increasing drug stability, and enhancing focus targeting ability (309), and are especially suitable for directional lung delivery in sepsis (310). The pulmonary capillary network enables larger nanoparticles to deposit in the lung (311), where anionic nanoparticles are well tolerated (312), which may also be a new route for treating septic ARDS.
One of the signs of ARDS in patients with sepsis is increased pulmonary microvascular endothelial permeability and abnormal accumulation of neutrophils. Lung endothelial barrier damage is mediated by cell contact breakdown and actin remodeling, and the accumulation of neutrophils in the lungs is mainly driven by cytoskeletal rearrangement (155). In the early stage of sepsis, excessive inflammatory responses activate neutrophils and promote their migration to the lungs. Subsequent neutrophil sequestration in the lungs leads to abnormal tissue perfusion, which in turn leads to pulmonary capillary microcirculation disturbance. Then, neutrophils with delayed apoptosis cause lung tissue damage through the release of ROS, protease, NETs, etc., eventually leading to the occurrence of severe hypoxemia and pulmonary edema. Studies showed that the peripheral miRNA may be the key epigenetic factor of endothelial activation and neutrophil dysfunction in ARDS. MiR50-887-3 expression increased on the endothelial cells of septic patients with ARDS, promoting neutrophil trans-endothelial migration by increasing VCAM-1, TLR2, CCL5, and CXCL10 expression (313). Inhibition of miR50-887-3 expression on vascular endothelium may be a potential target for septic ARDS. Sphingosine-1-phosphate (S1P) in plasma, a lipid recognized by GPCRs (S1Pr1, S1Pr2, S1Pr3) on endothelial cells, regulates neutrophil leakage triggered by lung inflammation in mice (314). As an analog of S1P, FTY720 and its analog FTY720 s-phosphonate (Tys) enhance the barrier protection function of lung endothelial cells by activating S1Pr1 (315, 316). The binding of S1P to S1Pr1 induces actin cytoskeletal recombination, adhesion junction assembly, and VE-cadherin localization to intercellular contact areas, but S1Pr1 also undergoes time-dependent desensitization (317). Fingolimod, an S1Pr agonist with a bias toward receptor internalization and degradation, is effective in treating multiple sclerosis (318–320) and may be used to control neutrophil aggregation. In addition, the sweet taste receptor T1R3 is also a kind of G-protein-coupled receptor. Activated T1R3 may protect the lung endothelial barrier by eliminating Src/P21-activated kinase (PAK)/p110αPI3K-mediated cell contact breakdown and Src/myosin light chain 2 (MLC2)/heat shock protein 27 (HSP27) -mediated actin remodeling (321). In vitro studies have shown that exogenous prostaglandin F2α (PGF2α) promotes neutrophil migration in endometrial carcinoma by up-regulating CXCR2/CXCL1 expression through PGF2α receptors (also known as FP receptors, a type of GPCRs) (322, 323). However, FP receptor antagonist AL8810 promotes neutrophil migration to BAFL by increasing adhesion molecules expression on the vascular endothelium such as ICAM-1 and E-selectin, while decreasing the gene expression of alveolar surfactant protein and aggravating pulmonary edema (324). FP receptor agonists may have the potential to treat septic ARDS, but this needs further study. The administration of ligands binding to GPCRs on endothelial cells to activate intracellular signal transduction pathways mediates cytoskeletal recombination to inhibit neutrophil aggregation to the lung, which may be a new therapeutic target for lung endothelial cell protection in septic ARDS.
On the other hand, modifying the chemotaxis of neutrophils by regulating the internalization of GPCRs may improve the inflammatory response in the lungs during sepsis. Barbadin is a novel inhibitor that blocks the interaction of β-arrestin and AP-2, reducing receptor desensitization by inhibiting clathrin-mediated internalization of GPCRs, thus allowing neutrophils to continuously respond to external stimuli (325, 326). Furthermore, LMBD1, the first protein was shown to be involved in the regulation of insulin receptor (IR) internalization (327), is hypothesized to contain nine transmembrane domains that subcellular localize in multiple membrane-bound organelles, including lysosomes and plasma membranes (328). LMBD1 can interact with AP-2 and selectively participate in clathrin-mediated IR endocytosis (327). When LMBD1 is deficient, IR is retained in the plasma membrane, suggesting that LMBRD1 may be an important molecule mediating GPCRs internalization. In addition, administration of the glycolytic inhibitor 2-deoxyglucose (2-DG) restored the ability of neutrophils to cluster to infected abdominal lesions in sepsis mice, partially reversed glycolytic-induced neutrophils migration dysfunction and down-regulated CXCR2 expression, suggesting that 2-DG may be a potential therapeutic strategy (329).
Conclusion
ARDS is an important cause of acute respiratory failure, which is usually associated with multiple organ failure, and sepsis is one of its common causes (330). The incidence of sepsis-induced ARDS remains high despite the clinical efficacy of antibiotic therapy during infection (1). The lung is usually the first target organ attacked by multiple organ dysfunction in sepsis (331, 332). The main pathophysiological changes are the destruction of the alveoli-capillary barrier (333), which leads to increased permeability of pulmonary blood vessels, increased migration of neutrophils to the lungs, and accumulation of protein-rich edema fluid in the alveoli, thus causing tissue and organ damage (334). Due to the difficulty in balancing the specificity and sensitivity of currently available biomarkers, finding new targeted therapeutic markers is still crucial to prevent ARDS in patients with sepsis.
The excessive inflammatory response shown in the early stage of sepsis leads to a large accumulation of neutrophils in the lungs, excessive activation, and delayed apoptosis, which is an important cause of acute lung injury or acute respiratory distress syndrome. In sepsis, the impaired migration of neutrophils to the pulmonary tissues may be due to excessive binding to vascular endothelial cells during initial adhesion, followed by reduced chemotactic reactivity due to GPCR desensitization and internalization, resulting in strong infiltration of these cells into the infected lung tissue. Therefore, the ideal treatment strategy for septic ARDS is to reduce the accumulation of neutrophils in the lungs without impairing its microbial clearance and regenerative capacity (335, 336) Cytoskeleton-regulated subcellular localization and conformational regulation such as phosphorylation and dimerization are important mechanisms of GPCRs desensitization. According to the ligand-bias and phosphoryl-barcoding hypothesis, different ligands preferentially promote different downstream GPCR pathways, so it is important to develop allosteric regulators to precisely control the effects of activated neutrophils. At present, the main treatment method for septic ARDS is still a lung-supporting ventilation strategy (337). Targeting GPCRs to regulate neutrophil chemotaxis to improve pulmonary inflammation and microcirculation may be a promising field for the treatment of septic ARDS. On the other hand, targeting the interaction between neutrophils and GPCRs on pulmonary vascular endothelial cells may reduce the over-aggregation of neutrophils in the lungs, which may also be an effective strategy. Although other pathways inhibit neutrophil migration, such as activating proteins C (338), ROS (339), and NETs (340), this review focuses on the physiological mechanism of how GPCRs expressed in sepsis patients cause ARDS by influencing neutrophil migration to improve the prognosis of patients and prevent the occurrence of irreversible MODS, providing more valuable reference targets for the development of new therapies.
Author contributions
YW, C-LZ, and J-FW reviewed the literature, wrote drafts of the manuscript, and prepared figures. X-MD, PL, QL, H-RL, and C-MY helped in the evaluation of the literature and the submission of the manuscript. J-FW and X-MD designed and supervised the work. All authors contributed to the review and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (No. 82072147, No. 82272214).
Acknowledgments
All the figures in this review are made by BioRender (https://app.biorender.com/).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA. (2016) 315(8):801. doi: 10.1001/jama.2016.0287
2. Seree-aphinan C, Vichitkunakorn P, Navakanitworakul R, Khwannimit B. Distinguishing sepsis from infection by neutrophil dysfunction: A promising role of CXCR2 surface level. Front Immunol (2020) 11:608696. doi: 10.3389/fimmu.2020.608696
3. Evans L, Rhodes A, Alhazzani W, Antonelli M, Coopersmith CM, French C, et al. Surviving sepsis campaign: International guidelines for management of sepsis and septic shock 2021. Intensive Care Med (2021) 47(11):1181–247. doi: 10.1007/s00134-021-06506-y
4. Deutschman CS, Tracey KJ. Sepsis: Current dogma and new perspectives. Immunity. (2014) 40(4):463–75. doi: 10.1016/j.immuni.2014.04.001
5. Buchman TG, Simpson SQ, Sciarretta KL, Finne KP, Sowers N, Collier M, et al. Sepsis among Medicare beneficiaries. Crit Care Med (2020) 48(3):302–18. doi: 10.1097/CCM.0000000000004225
6. Fleischmann C, Scherag A, Adhikari NKJ, Hartog CS, Tsaganos T, Schlattmann P, et al. Assessment of global incidence and mortality of hospital-treated sepsis. current estimates and limitations. Am J Respir Crit Care Med (2016) 193(3):259–72. doi: 10.1164/rccm.201504-0781OC
7. Rudd KE, Johnson SC, Agesa KM, Shackelford KA, Tsoi D, Kievlan DR, et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: analysis for the global burden of disease study. Lancet (2020) 395(10219):200–11. doi: 10.1016/S0140-6736(19)32989-7
8. Milton R, Gillespie D, Dyer C, Taiyari K, Carvalho MJ, Thomson K, et al. Neonatal sepsis and mortality in low-income and middle-income countries from a facility-based birth cohort: an international multisite prospective observational study. Lancet Global Health (2022) 10(5):e661–72. doi: 10.1016/S2214-109X(22)00043-2
9. Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A. Neutrophil function: From mechanisms to disease. Annu Rev Immunol (2012) 30(1):459–89. doi: 10.1146/annurev-immunol-020711-074942
10. Iba T, Hashiguchi N, Nagaoka I, Tabe Y, Murai M. Neutrophil cell death in response to infection and its relation to coagulation. J Intensive Care (2013) 1(1):13. doi: 10.1186/2052-0492-1-13
11. Zhang YY, Ning BT. Signaling pathways and intervention therapies in sepsis. Sig Transduct Target Ther (2021) 6(1):407. doi: 10.1038/s41392-021-00471-0
12. van der Poll T, van de Veerdonk FL, Scicluna BP, Netea MG. The immunopathology of sepsis and potential therapeutic targets. Nat Rev Immunol (2017) 17(7):407–20. doi: 10.1038/nri.2017.36
13. Lee MJ, Bae J, Lee JH, Park YJ, Lee HAR, Mun S, et al. Serial change of endotoxin tolerance in a polymicrobial sepsis model. IJMS. (2022) 23(12):6581. doi: 10.3390/ijms23126581
14. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol (2013) 13(3):159–75. doi: 10.1038/nri3399
15. Reddy RC, Standiford TJ. Effects of sepsis on neutrophil chemotaxis. Curr Opin Hematology. (2010) 17(1):18–24. doi: 10.1097/MOH.0b013e32833338f3
16. de Oliveira S, Rosowski EE, Huttenlocher A. Neutrophil migration in infection and wound repair: going forward in reverse. Nat Rev Immunol (2016) 16(6):378–91. doi: 10.1038/nri.2016.49
17. Nourshargh S, Renshaw SA, Imhof BA. Reverse migration of neutrophils: Where, when, how, and why? Trends Immunol (2016) 37(5):273–86. doi: 10.1016/j.it.2016.03.006
18. Wang J, Hossain M, Thanabalasuriar A, Gunzer M, Meininger C, Kubes P. Visualizing the function and fate of neutrophils in sterile injury and repair. Science. (2017) 358(6359):111–6. doi: 10.1126/science.aam9690
19. Li B, Han X, Ye X, Ni J, Wu J, Dai J, et al. Substance p-regulated leukotriene B4 production promotes acute pancreatitis-associated lung injury through neutrophil reverse migration. Int Immunopharmacology. (2018) 57:147–56. doi: 10.1016/j.intimp.2018.02.017
20. Ode Y, Aziz M, Wang P. CIRP increases ICAM-1 + phenotype of neutrophils exhibiting elevated iNOS and NETs in sepsis. J Leukoc Biol (2018) 103(4):693–707. doi: 10.1002/JLB.3A0817-327RR
21. Jin H, Aziz M, Ode Y, Wang P. CIRP induces neutrophil reverse transendothelial migration in sepsis. Shock. (2019) 51(5):548–56. doi: 10.1097/SHK.0000000000001257
22. Dellinger R, Vincent J. The surviving sepsis campaign sepsis change bundles and clinical practice. Crit Care (2005) 9(6):653–4. doi: 10.1186/cc3952
23. Kaukonen KM, Bailey M, Suzuki S, Pilcher D, Bellomo R. Mortality related to severe sepsis and septic shock among critically ill patients in Australia and new Zealand, 2000-2012. JAMA. (2014) 311(13):1308. doi: 10.1001/jama.2014.2637
24. Margaria JP, Moretta L, Alves-Filho JC, Hirsch E. PI3K signaling in mechanisms and treatments of pulmonary fibrosis following sepsis and acute lung injury. Biomedicines. (2022) 10(4):756. doi: 10.3390/biomedicines10040756
25. Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med (2000) 342(18):1334–49. doi: 10.1056/NEJM200005043421806
26. Yuan Y, Alwis I, Wu MCL, Kaplan Z, Ashworth K, Bark D, et al. Neutrophil macroaggregates promote widespread pulmonary thrombosis after gut ischemia. Sci Transl Med (2017) 9(409):eaam5861. doi: 10.1126/scitranslmed.aam5861
27. Shen XF, Cao K, Jiang JP, Guan WX, Du JF. Neutrophil dysregulation during sepsis: an overview and update. J Cell Mol Med (2017) 21(9):1687–97. doi: 10.1111/jcmm.13112
28. Murdoch C, Finn A. Chemokine receptors and their role in inflammation and infectious diseases. Blood. (2000) 95(10):3032–43. doi: 10.1182/blood.V95.10.3032
29. Weiner OD, Servant G, Welch MD, Mitchison TJ, Sedat JW, Bourne HR. Spatial control of actin polymerization during neutrophil chemotaxis. Nat Cell Biol (1999) 1(2):75–81. doi: 10.1038/10042
30. Berzat A, Hall A. Cellular responses to extracellular guidance cues. EMBO J (2010) 29(16):2734–45. doi: 10.1038/emboj.2010.170
31. Lämmermann T, Germain RN. The multiple faces of leukocyte interstitial migration. Semin Immunopathol (2014) 36(2):227–51. doi: 10.1007/s00281-014-0418-8
32. Lee Y, Basith S, Choi S. Recent advances in structure-based drug design targeting class a G protein-coupled receptors utilizing crystal structures and computational simulations. J Med Chem (2017) 61(1):1–46. doi: 10.1021/acs.jmedchem.6b01453
33. Fredriksson R, Schiöth HB. The repertoire of G-Protein–coupled receptors in fully sequenced genomes. Mol Pharmacol (2005) 67(5):1414–25. doi: 10.1124/mol.104.009001
34. Trzaskowski B, Latek D, Yuan S, Ghoshdastider U, Debinski A, Filipek S. Action of molecular switches in GPCRs - theoretical and experimental studies. CMC. (2012) 19(8):1090–109. doi: 10.2174/092986712799320556
35. Wettschureck N, Offermanns S. Mammalian G proteins and their cell type specific functions. Physiol Rev (2005) 85(4):1159–204. doi: 10.1152/physrev.00003.2005
36. Rask-Andersen M, Almén MS, Schiöth HB. Trends in the exploitation of novel drug targets. Nat Rev Drug Discovery (2011) 10(8):579–90. doi: 10.1038/nrd3478
37. Mestas J, Hughes CCW. Of mice and not men: Differences between mouse and human immunology. J Immunol (2004) 172(5):2731–8. doi: 10.4049/jimmunol.172.5.2731
38. Mihlan M, Glaser KM, Epple MW, Lämmermann T. Neutrophils: Amoeboid migration and swarming dynamics in tissues. Front Cell Dev Biol (2022) 10:871789. doi: 10.3389/fcell.2022.871789
39. Hidalgo A, Chilvers ER, Summers C, Koenderman L. The neutrophil life cycle. Trends Immunol (2019) 40(7):584–97. doi: 10.1016/j.it.2019.04.013
40. Cowland JB, Borregaard N. Granulopoiesis and granules of human neutrophils. Immunol Rev (2016) 273(1):11–28. doi: 10.1111/imr.12440
41. Cronkite EP, Fliedner TM. Granulocytopoiesis. N Engl J Med (1964) 270(25):1347–52. doi: 10.1056/NEJM196406182702506
42. Manz MG, Boettcher S. Emergency granulopoiesis. Nat Rev Immunol (2014) 14(5):302–14. doi: 10.1038/nri3660
43. Jiao J, Dragomir AC, Kocabayoglu P, Rahman AH, Chow A, Hashimoto D, et al. Central role of conventional dendritic cells in regulation of bone marrow release and survival of neutrophils. JI. (2014) 192(7):3374–82. doi: 10.4049/jimmunol.1300237
44. Lieschke G, Grail D, Hodgson G, Metcalf D, Stanley E, Cheers C, et al. Mice lacking granulocyte colony-stimulating factor have chronic neutropenia, granulocyte and macrophage progenitor cell deficiency, and impaired neutrophil mobilization. Blood. (1994) 84(6):1737–46. doi: 10.1182/blood.V84.6.1737.1737
45. Lee A, Whyte MKB, Haslett C. Inhibition of apoptosis and prolongation of neutrophil functional longevity by inflammatory mediators. J Leukoc Biol (1993) 54(4):283–8. doi: 10.1002/jlb.54.4.283
46. Devi S, Wang Y, Chew WK, Lima R, A-González N, Mattar CNZ, et al. Neutrophil mobilization via plerixafor-mediated CXCR4 inhibition arises from lung demargination and blockade of neutrophil homing to the bone marrow. J Exp Med (2013) 210(11):2321–36. doi: 10.1084/jem.20130056
47. Fox S, Leitch AE, Duffin R, Haslett C, Rossi AG. Neutrophil apoptosis: Relevance to the innate immune response and inflammatory disease. J Innate Immun (2010) 2(3):216–27. doi: 10.1159/000284367
48. Tak T, Tesselaar K, Pillay J, Borghans JAM, Koenderman L. Whatˈs your age again? determination of human neutrophil half-lives revisited. J Leukocyte Biol (2013) 94(4):595–601. doi: 10.1189/jlb.1112571
49. Lahoz-Beneytez J, Elemans M, Zhang Y, Ahmed R, Salam A, Block M, et al. Human neutrophil kinetics: modeling of stable isotope labeling data supports short blood neutrophil half-lives. Blood. (2016) 127(26):3431–8. doi: 10.1182/blood-2016-03-700336
50. Liu X, Wu M, Wang M, Duan Y, Phan CU, Chen H, et al. AIEgen-lipid conjugate for rapid labeling of neutrophils and monitoring of their behavior. Angew Chem Int Ed (2020) 60(6):3175–81. doi: 10.1002/anie.202012594
51. Cowland JB, Borregaard N. Isolation of neutrophil precursors from bone marrow for biochemical and transcriptional analysis. J Immunol Methods (1999) 232(1–2):191–200. doi: 10.1016/S0022-1759(99)00176-3
52. Kawamura S, Onai N, Miya F, Sato T, Tsunoda T, Kurabayashi K, et al. Identification of a human clonogenic progenitor with strict monocyte differentiation potential: A counterpart of mouse cMoPs. Immunity. (2017) 46(5):835–848.e4. doi: 10.1016/j.immuni.2017.04.019
53. Ramírez C, Mendoza L. Phenotypic stability and plasticity in GMP-derived cells as determined by their underlying regulatory network. Bioinformatics. (2017) 34(7):1174–82. doi: 10.1093/bioinformatics/btx736
54. Zhu YP, Padgett L, Dinh HQ, Marcovecchio P, Blatchley A, Wu R, et al. Identification of an early unipotent neutrophil progenitor with pro-tumoral activity in mouse and human bone marrow. Cell Rep (2018) 24(9):2329–2341.e8. doi: 10.1016/j.celrep.2018.07.097
55. Summers C, Rankin SM, Condliffe AM, Singh N, Peters AM, Chilvers ER. Neutrophil kinetics in health and disease. Trends Immunol (2010) 31(8):318–24. doi: 10.1016/j.it.2010.05.006
56. Suratt BT, Young SK, Lieber J, Nick JA, Henson PM, Worthen GS. Neutrophil maturation and activation determine anatomic site of clearance from circulation. Am J Physiology-Lung Cell Mol Physiol (2001) 281(4):L913–21. doi: 10.1152/ajplung.2001.281.4.L913
57. Martin C, Burdon PCE, Bridger G, Gutierrez-Ramos JC, Williams TJ, Rankin SM. Chemokines acting via CXCR2 and CXCR4 control the release of neutrophils from the bone marrow and their return following senescence. Immunity. (2003) 19(4):583–93. doi: 10.1016/S1074-7613(03)00263-2
58. Savill J, Dransfield I, Gregory C, Haslett C. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat Rev Immunol (2002) 2(12):965–75. doi: 10.1038/nri957
59. Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol (2007) 7(9):678–89. doi: 10.1038/nri2156
60. Petri B, Phillipson M, Kubes P. The physiology of leukocyte recruitment: An In vivo perspective. J Immunol (2008) 180(10):6439–46. doi: 10.4049/jimmunol.180.10.6439
61. Zarbock A, Ley K, McEver RP, Hidalgo A. Leukocyte ligands for endothelial selectins: specialized glycoconjugates that mediate rolling and signaling under flow. Blood. (2011) 118(26):6743–51. doi: 10.1182/blood-2011-07-343566
62. Bargatze RF, Kurk S, Butcher EC, Jutila MA. Neutrophils roll on adherent neutrophils bound to cytokine-induced endothelial cells via l-selectin on the rolling cells. J Exp Med (1994) 180(5):1785–92. doi: 10.1084/jem.180.5.1785
63. Brown KA, Brain SD, Pearson JD, Edgeworth JD, Lewis SM, Treacher DF. Neutrophils in development of multiple organ failure in sepsis. Lancet (2006) 368(July 8, 2006):157–69. doi: 10.1016/S0140-6736(06)69005-3
64. Phillipson M, Heit B, Colarusso P, Liu L, Ballantyne CM, Kubes P. Intraluminal crawling of neutrophils to emigration sites: a molecularly distinct process from adhesion in the recruitment cascade. J Exp Med (2006) 203(12):2569–75. doi: 10.1084/jem.20060925
65. Sanchez-Madrid F, González-Amaro R. Cell adhesion molecules: Selectins and integrins. Crit Rev Immunol (1999) 19(5–6):41. doi: 10.1615/CritRevImmunol.v19.i5-6.20
66. Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med (2007) 13(4):463–9. doi: 10.1038/nm1565
67. Filippi MD. Neutrophil transendothelial migration: updates and new perspectives. Blood. (2019) 133(20):2149–58. doi: 10.1182/blood-2018-12-844605
68. Guthrie LA, McPhail LC, Henson PM, Johnston RB. Priming of neutrophils for enhanced release of oxygen metabolites by bacterial lipopolysaccharide. evidence for increased activity of the superoxide-producing enzyme. J Exp Med (1984) 160(6):1656–71. doi: 10.1084/jem.160.6.1656
69. Daniels R, Finnen M, Hill M, Lackie J. Recombinant human monocyte IL-8 primes NADPH-oxidase and phospholipase A2 activation in human neutrophils. Immunology. (1992) 75(1):157–63.
70. Ward PA. The dark side of C5a in sepsis. Nat Rev Immunol (2004) 4(2):133–42. doi: 10.1038/nri1269
71. Massena S, Christoffersson G, Hjertström E, Zcharia E, Vlodavsky I, Ausmees N, et al. A chemotactic gradient sequestered on endothelial heparan sulfate induces directional intraluminal crawling of neutrophils. Blood. (2010) 116(11):1924–31. doi: 10.1182/blood-2010-01-266072
72. Jones DH, Anderson DC, Burr BL, Rudioff HE, Smith CW, Krater SS, et al. Quantitation of intracellular mac-1 (CD11b/CD18) pools in human neutrophils. J Leukoc Biol (1988) 44(6):535–44. doi: 10.1002/jlb.44.6.535
73. Lefort CT, Rossaint J, Moser M, Petrich BG, Zarbock A, Monkley SJ, et al. Distinct roles for talin-1 and kindlin-3 in LFA-1 extension and affinity regulation. Blood. (2012) 119(18):4275–82. doi: 10.1182/blood-2011-08-373118
74. Dewey CF, Bussolari SR, Gimbrone MA, Davies PF. The dynamic response of vascular endothelial cells to fluid shear stress. J Biomechanical Engineering. (1981) 103(3):177–85. doi: 10.1115/1.3138276
75. Woodfin A, Voisin MB, Beyrau M, Colom B, Caille D, Diapouli FM, et al. The junctional adhesion molecule JAM-c regulates polarized transendothelial migration of neutrophils in vivo. Nat Immunol (2011) 12(8):761–9. doi: 10.1038/ni.2062
76. Phillipson M, Kaur J, Colarusso P, Ballantyne CM, Kubes P. Endothelial domes encapsulate adherent neutrophils and minimize increases in vascular permeability in paracellular and transcellular emigration. PLoS One (2008) 3(2):e1649. doi: 10.1371/journal.pone.0001649
77. Martín-Fernández M, Tamayo-Velasco Á, Aller R, Gonzalo-Benito H, Martínez-Paz P, Tamayo E. Endothelial dysfunction and neutrophil degranulation as central events in sepsis physiopathology. IJMS. (2021) 22(12):6272. doi: 10.3390/ijms22126272
78. Delgado-Rizo V, Martínez-Guzmán MA, Iñiguez-Gutierrez L, García-Orozco A, Alvarado-Navarro A, Fafutis-Morris M. Neutrophil extracellular traps and its implications in inflammation: An overview. Front Immunol (2017) 8:81. doi: 10.3389/fimmu.2017.00081
79. Burn GL, Foti A, Marsman G, Patel DF, Zychlinsky A. The neutrophil. Immunity. (2021) 54(7):1377–91. doi: 10.1016/j.immuni.2021.06.006
80. Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol (2007) 176(2):231–41. doi: 10.1083/jcb.200606027
81. Chen KW, Monteleone M, Boucher D, Sollberger G, Ramnath D, Condon ND, et al. Noncanonical inflammasome signaling elicits gasdermin d–dependent neutrophil extracellular traps. Sci Immunol (2018) 3(26):eaar6676. doi: 10.1126/sciimmunol.aar6676
82. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. (2004) 303(5663):1532–5. doi: 10.1126/science.1092385
83. Burgener SS, Schroder K. Neutrophil extracellular traps in host defense. Cold Spring Harb Perspect Biol (2019) 12(7):a037028. doi: 10.1101/cshperspect.a037028
84. Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol (2010) 191(3):677–91. doi: 10.1083/jcb.201006052
85. Metzler KD, Goosmann C, Lubojemska A, Zychlinsky A. Papayannopoulos v. a myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Rep (2014) 8(3):883–96. doi: 10.1016/j.celrep.2014.06.044
86. Neeli I, Khan SN, Radic M. Histone deimination as a response to inflammatory stimuli in neutrophils. J Immunol (2008) 180(3):1895–902. doi: 10.4049/jimmunol.180.3.1895
87. Douda DN, Khan MA, Grasemann H, Palaniyar N. SK3 channel and mitochondrial ROS mediate NADPH oxidase-independent NETosis induced by calcium influx. Proc Natl Acad Sci USA (2015) 112(9):2817–22. doi: 10.1073/pnas.1414055112
88. Sollberger G, Choidas A, Burn GL, Habenberger P, Di Lucrezia R, Kordes S, et al. Gasdermin d plays a vital role in the generation of neutrophil extracellular traps. Sci Immunol (2018) 3(26):eaar6689. doi: 10.1126/sciimmunol.aar6689
89. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin d causes pyroptosis by forming membrane pores. Nature. (2016) 535(7610):153–8. doi: 10.1038/nature18629
90. Pieterse E, Rother N, Yanginlar C, Hilbrands LB, van der Vlag J. Neutrophils discriminate between lipopolysaccharides of different bacterial sources and selectively release neutrophil extracellular traps. Front Immunol (2016) 7. doi: 10.3389/fimmu.2016.00484
91. Yipp BG, Petri B, Salina D, Jenne CN, Scott BNV, Zbytnuik LD, et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat Med (2012) 18(9):1386–93. doi: 10.1038/nm.2847
92. Pilsczek FH, Salina D, Poon KKH, Fahey C, Yipp BG, Sibley CD, et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response toStaphylococcus aureus. JI. (2010) 185(12):7413–25. doi: 10.4049/jimmunol.1000675
93. Thiam HR, Wong SL, Wagner DD, Waterman CM. Cellular mechanisms of NETosis. Annu Rev Cell Dev Biol (2020) 36(1):191–218. doi: 10.1146/annurev-cellbio-020520-111016
94. Rossaint J, Herter JM, Van Aken H, Napirei M, Döring Y, Weber C, et al. Synchronized integrin engagement and chemokine activation is crucial in neutrophil extracellular trap–mediated sterile inflammation. Blood. (2014) 123(16):2573–84. doi: 10.1182/blood-2013-07-516484
95. Keshari RS, Jyoti A, Dubey M, Kothari N, Kohli M, Bogra J, et al. Cytokines induced neutrophil extracellular traps formation: Implication for the inflammatory disease condition. PLoS One (2012) 7(10):e48111. doi: 10.1371/journal.pone.0048111
96. Schappe MS, Szteyn K, Stremska ME, Mendu SK, Downs TK, Seegren PV, et al. Chanzyme TRPM7 mediates the Ca2+ influx essential for lipopolysaccharide-induced toll-like receptor 4 endocytosis and macrophage activation. Immunity. (2018) 48(1):59–74.e5. doi: 10.1016/j.immuni.2017.11.026
97. Kandasamy K, Bezavada L, Escue RB, Parthasarathi K. Lipopolysaccharide induces endoplasmic store Ca2+-dependent inflammatory responses in lung microvessels. PLoS One (2013) 8(5):e63465. doi: 10.1371/journal.pone.0063465
98. Immler R, Simon SI, Sperandio M. Calcium signalling and related ion channels in neutrophil recruitment and function. Eur J Clin Invest. (2018) 48:e12964. doi: 10.1111/eci.12964
99. Chen T, Li Y, Sun R, Hu H, Liu Y, Herrmann M, et al. Receptor-mediated NETosis on neutrophils. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.775267
100. Yago T, Liu Z, Ahamed J, McEver RP. Cooperative PSGL-1 and CXCR2 signaling in neutrophils promotes deep vein thrombosis in mice. Blood. (2018) 132(13):1426–37. doi: 10.1182/blood-2018-05-850859
101. Adrover JM, Aroca-Crevillén A, Crainiciuc G, Ostos F, Rojas-Vega Y, Rubio-Ponce A, et al. Programmed ‘disarming’ of the neutrophil proteome reduces the magnitude of inflammation. Nat Immunol (2020) 21(2):135–44. doi: 10.1038/s41590-019-0571-2
102. Adrover JM, Nicolás-Ávila JA, Hidalgo A. Aging: A temporal dimension for neutrophils. Trends Immunol (2016) 37(5):334–45. doi: 10.1016/j.it.2016.03.005
103. Rodrigues DAS, Prestes EB, Gama AMS, Silva L de S, Pinheiro AAS, Ribeiro JMC, et al. CXCR4 and MIF are required for neutrophil extracellular trap release triggered by plasmodium-infected erythrocytes. PLoS Pathog (2020) 16(8):e1008230. doi: 10.1371/journal.ppat.1008230
104. Lämmermann T, Sixt M. Mechanical modes of a’moeboid’ cell migration. Curr Opin Cell Biol (2009) 21(5):636–44. doi: 10.1016/j.ceb.2009.05.003
105. Salvermoser M, Pick R, Weckbach LT, Zehrer A, Löhr P, Drechsler M, et al. Myosin 1f is specifically required for neutrophil migration in 3D environments during acute inflammation. Blood. (2018) 131(17):1887–98. doi: 10.1182/blood-2017-10-811851
106. Gennerich A, Vale RD. Walking the walk: how kinesin and dynein coordinate their steps. Curr Opin Cell Biol (2009) 21(1):59–67. doi: 10.1016/j.ceb.2008.12.002
107. Lämmermann T, Bader BL, Monkley SJ, Worbs T, Wedlich-Söldner R, Hirsch K, et al. Rapid leukocyte migration by integrin-independent flowing and squeezing. Nature. (2008) 453(7191):51–5. doi: 10.1038/nature06887
108. Fritz-Laylin LK K, Riel-Mehan M, Chen BC, Lord SJ, Goddard TD, Ferrin TE, et al. Actin-based protrusions of migrating neutrophils are intrinsically lamellar and facilitate direction changes. eLIFE. (2017) 6:e26990. doi: 10.7554/eLife.26990
109. Zehrer A, Pick R, Salvermoser M, Boda A, Miller M, Stark K, et al. A fundamental role of Myh9 for neutrophil migration in innate immunity. JI. (2018) 201(6):1748–64. doi: 10.4049/jimmunol.1701400
110. Graziano BR, Town JP, Sitarska E, Nagy TL, Fošnarič M, Penič S, et al. Cell confinement reveals a branched-actin independent circuit for neutrophil polarity. PLoS Biol (2019) 17(10):e3000457. doi: 10.1371/journal.pbio.3000457
111. Barros-Becker F, Lam PY, Fisher R, Huttenlocher A. Live imaging reveals distinct modes of neutrophil and macrophage migration within interstitial tissues. J Cell Sci (2017) 130(22):3801–8. doi: 10.1242/jcs.206128
112. Georgantzoglou A, Poplimont H, Lämmermann T, Sarris M. Interstitial leukocyte navigation through a search and run response to gradients. Cold Spring Harbor Lab (2021) 00:1–30. doi: 10.1101/2021.03.03.433706
113. He HQ, Ye R. The formyl peptide receptors: Diversity of ligands and mechanism for recognition. Molecules. (2017) 22(3):455. doi: 10.3390/molecules22030455
114. Dahlgren C, Lind S, Mårtensson J, Björkman L, Wu Y, Sundqvist M, et al. G Protein coupled pattern recognition receptors expressed in neutrophils : Recognition, activation/modulation, signaling and receptor regulated functions. Immunol Rev (2022) 00:1–24. doi: 10.1111/imr.13151
115. Zhang F, Liu AL, Gao S, Ma S, Guo SB. Neutrophil dysfunction in sepsis. Chin Med J (2016) 129(22):2741–4. doi: 10.4103/0366-6999.193447
116. Zhu CL, Wang Y, Liu Q, Li HR, Yu CM, Li P, et al. Dysregulation of neutrophil death in sepsis. Front Immunol (2022) 13. doi: 10.3389/fimmu.2022.963955
117. Zhou YY, Sun BW. Recent advances in neutrophil chemotaxis abnormalities during sepsis. Chin J Traumatology. (2022) 25(6):317–24. doi: 10.1016/j.cjtee.2022.06.002
118. Alberelli MA, De Candia E. Functional role of protease activated receptors in vascular biology. Vasc Pharmacol (2014) 62(2):72–81. doi: 10.1016/j.vph.2014.06.001
119. Torgersen C, Moser P, Luckner G, Mayr V, Jochberger S, Hasibeder WR, et al. Macroscopic postmortem findings in 235 surgical intensive care patients with sepsis. Anesth Analgesia. (2009) 108(6):1841–7. doi: 10.1213/ane.0b013e318195e11d
120. Oberholzer C, Oberholzer A, Bahjat FR, Minter RM, Tannahill CL, Abouhamze A, et al. Targeted adenovirus-induced expression of IL-10 decreases thymic apoptosis and improves survival in murine sepsis. Proc Natl Acad Sci (2001) 98(20):11503–8. doi: 10.1073/pnas.181338198
121. Paudel S, Baral P, Ghimire L, Bergeron S, Jin L, DeCorte JA, et al. CXCL1 regulates neutrophil homeostasis in pneumonia-derived sepsis caused by streptococcus pneumoniae serotype 3. Blood. (2019) 133(12):1335–45. doi: 10.1182/blood-2018-10-878082
122. Kovach MA, Standiford TJ. The function of neutrophils in sepsis. Curr Opin Infect Diseases. (2012) 25(3):321–7. doi: 10.1097/QCO.0b013e3283528c9b
123. Silva CMS, Wanderley CWS, Veras FP, Sonego F, Nascimento DC, Gonçalves AV, et al. Gasdermin d inhibition prevents multiple organ dysfunction during sepsis by blocking NET formation. Blood. (2021) 138(25):2702–13. doi: 10.1182/blood.2021011525
124. Park SY, Shrestha S, Youn YJ, Kim JK, Kim SY, Kim H, et al. Autophagy primes neutrophils for neutrophil extracellular trap formation during sepsis. Am J Respir Crit Care Med (2017) 196(5):577–89. doi: 10.1164/rccm.201603-0596OC
125. Taneja R, Parodo J, Jia SH, Kapus A, Rotstein OD, Marshall JC. Delayed neutrophil apoptosis in sepsis is associated with maintenance of mitochondrial transmembrane potential and reduced caspase-9 activity*. Crit Care Med (2004) 32(7):1460–9. doi: 10.1097/01.CCM.0000129975.26905.77
126. Wang JF, Wang YP, Xie J, Zhao ZZ, Gupta S, Guo Y, et al. Upregulated PD-L1 delays human neutrophil apoptosis and promotes lung injury in an experimental mouse model of sepsis. Blood. (2021) 138(9):806–10. doi: 10.1182/blood.2020009417
127. Souto FO, Alves-Filho JC, Turato WM, Auxiliadora-Martins M, Basile-Filho A, Cunha FQ. Essential role of CCR2 in neutrophil tissue infiltration and multiple organ dysfunction in sepsis. Am J Respir Crit Care Med (2011) 183(2):234–42. doi: 10.1164/rccm.201003-0416OC
128. Martin EL, Souza DG, Fagundes CT, Amaral FA, Assenzio B, Puntorieri V, et al. Phosphoinositide-3 kinase γ activity contributes to sepsis and organ damage by altering neutrophil recruitment. Am J Respir Crit Care Med (2010) 182(6):762–73. doi: 10.1164/rccm.201001-0088OC
129. Marshall JC. Neutrophils in the pathogenesis of sepsis. Crit Care Med (2005) 33(Suppl):S502–5. doi: 10.1097/01.CCM.0000186266.34541.5F
130. Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol (2011) 11(8):519–31. doi: 10.1038/nri3024
131. Hazeldine J, Hampson P, Lord JM. The impact of trauma on neutrophil function. Injury. (2014) 45(12):1824–33. doi: 10.1016/j.injury.2014.06.021
132. Merle NS, Noe R, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, Roumenina LT. Complement system part II: Role in immunity. Front Immunol (2015) 6. doi: 10.3389/fimmu.2015.00257
133. Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci (2010) 107(36):15880–5. doi: 10.1073/pnas.1005743107
134. Ostrovsky L, King AJ, Bond S, Mitchell D, Lorant DE, Zimmerman GA, et al. A juxtacrine mechanism for neutrophil adhesion on platelets involves platelet-activating factor and a selectin-dependent activation process. Blood. (1998) 91(8):3028–36. doi: 10.1182/blood.V91.8.3028.3028_3028_3036
135. Grommes J, Alard JE, Drechsler M, Wantha S, Mörgelin M, Kuebler WM, et al. Disruption of platelet-derived chemokine heteromers prevents neutrophil extravasation in acute lung injury. Am J Respir Crit Care Med (2012) 185(6):628–36. doi: 10.1164/rccm.201108-1533OC
136. Drechsler M, Megens RTA, van Zandvoort M, Weber C, Soehnlein O. Hyperlipidemia-triggered neutrophilia promotes early atherosclerosis. Circulation. (2010) 122(18):1837–45. doi: 10.1161/CIRCULATIONAHA.110.961714
137. McDonald B, Urrutia R, Yipp BG, Jenne CN, Kubes P. Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell Host Microbe (2012) 12(3):324–33. doi: 10.1016/j.chom.2012.06.011
138. Jenne CN, Wong CHY, Zemp FJ, McDonald B, Rahman MM, Forsyth PA, et al. Neutrophils recruited to sites of infection protect from virus challenge by releasing neutrophil extracellular traps. Cell Host Microbe (2013) 13(2):169–80. doi: 10.1016/j.chom.2013.01.005
139. Gupta AK, Joshi MB, Philippova M, Erne P, Hasler P, Hahn S, et al. Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. FEBS Lett (2010) 584(14):3193–7. doi: 10.1016/j.febslet.2010.06.006
140. Tanaka K, Koike Y, Shimura T, Okigami M, Ide S, Toiyama Y, et al. In vivo characterization of neutrophil extracellular traps in various organs of a murine sepsis model. PLoS One (2014) 9(11):e111888. doi: 10.1371/journal.pone.0111888
141. Gao X, Hao S, Yan H, Ding W, Li K, Li J. Neutrophil extracellular traps contribute to the intestine damage in endotoxemic rats. J Surg Res (2015) 195(1):211–8. doi: 10.1016/j.jss.2014.12.019
142. von Brühl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med (2012) 209(4):819–35. doi: 10.1084/jem.20112322
143. Kannemeier C, Shibamiya A, Nakazawa F, Trusheim H, Ruppert C, Markart P, et al. Extracellular RNA constitutes a natural procoagulant cofactor in blood coagulation. Proc Natl Acad Sci (2007) 104(15):6388–93. doi: 10.1073/pnas.0608647104
144. Liaw PC, Ito T, Iba T, Thachil J, Zeerleder S. DAMP and DIC: The role of extracellular DNA and DNA-binding proteins in the pathogenesis of DIC. Blood Rev (2016) 30(4):257–61. doi: 10.1016/j.blre.2015.12.004
145. Massberg S, Grahl L, von Bruehl ML, Manukyan D, Pfeiler S, Goosmann C, et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat Med (2010) 16(8):887–96. doi: 10.1038/nm.2184
146. Coughlin SR. Thrombin signalling and protease-activated receptors. Nature. (2000) 407(6801):258–64. doi: 10.1038/35025229
147. Kataoka H, Hamilton JR, McKemy DD, Camerer E, Zheng YW, Cheng A, et al. Protease-activated receptors 1 and 4 mediate thrombin signaling in endothelial cells. Blood. (2003) 102(9):3224–31. doi: 10.1182/blood-2003-04-1130
148. McNamara CA, Sarembock IJ, Gimple LW, Fenton JW, Coughlin SR, Owens GK. Thrombin stimulates proliferation of cultured rat aortic smooth muscle cells by a proteolytically activated receptor. J Clin Invest. (1993) 91(1):94–8. doi: 10.1172/JCI116206
149. Bono F, Lamarche I, Herbert JM. Induction of vascular smooth muscle cell growth by selective activation of the proteinase activated receptor-2 (PAR-2). Biochem Biophys Res Commun (1997) 241(3):762–4. doi: 10.1006/bbrc.1997.7847
150. Kahn ML, Zheng YW, Huang W, Bigornia V, Zeng D, Moff S, et al. A dual thrombin receptor system for platelet activation. Nature. (1998) 394(6694):690–4. doi: 10.1038/29325
151. Soh UJ, Dores MR, Chen B, Trejo J. Signal transduction by protease-activated receptors. Br J Pharmacol (2010) 160(2):191–203. doi: 10.1111/j.1476-5381.2010.00705.x
152. Crouser ED, Matthay MA. Endothelial damage during septic shock. Chest. (2017) 152(1):1–3. doi: 10.1016/j.chest.2017.02.016
153. Pool R, Gomez H, Kellum JA. Mechanisms of organ dysfunction in sepsis. Crit Care Clinics. (2018) 34(1):63–80. doi: 10.1016/j.ccc.2017.08.003
154. Lagu T, Rothberg MB, Shieh MS, Pekow PS, Steingrub JS, Lindenauer PK. Hospitalizations, costs, and outcomes of severe sepsis in the united states 2003 to 2007. Crit Care Med (2012) 40(3):754–61. doi: 10.1097/CCM.0b013e318232db65
155. Rossaint J, Zarbock A. Tissue-specific neutrophil recruitment into the lung, liver, and kidney. J Innate Immun (2013) 5(4):348–57. doi: 10.1159/000345943
156. Maas SL, Soehnlein O, Viola JR. Organ-specific mechanisms of transendothelial neutrophil migration in the lung, liver, kidney, and aorta. Front Immunol (2018) 9:02739. doi: 10.3389/fimmu.2018.02739
157. MacNee W, Selby C. New perspectives on basic mechanisms in lung disease. 2. neutrophil traffic in the lungs: role of haemodynamics, cell adhesion, and deformability. Thorax. (1993) 48(1):79–88. doi: 10.1136/thx.48.1.79
158. Doerschuk CM. Mechanisms of leukocyte sequestration in inflamed lungs. Microcirculation. (2001) 8(2):71–88. doi: 10.1111/j.1549-8719.2001.tb00159.x
159. Wolf K, te Lindert M, Krause M, Alexander S, te Riet J, Willis AL, et al. Physical limits of cell migration: Control by ECM space and nuclear deformation and tuning by proteolysis and traction force. J Cell Biol (2013) 201(7):1069–84. doi: 10.1083/jcb.201210152
160. Manley HR, Keightley MC, Lieschke GJ. The neutrophil nucleus: An important influence on neutrophil migration and function. Front Immunol (2018) 9:02867. doi: 10.3389/fimmu.2018.02867
161. Kuebler WM, Borges J, Borges J, Kuhnle GE, Bergh K, Messmer K, et al. Role of l-selectin in leukocyte sequestration in lung capillaries in a rabbit model of endotoxemia. Am J Respir Crit Care Med (2000) 161(1):36–43. doi: 10.1164/ajrccm.161.1.9901039
162. Lien D, Henson P, Capen R, Henson J, Hanson W, Wagner W, et al. Neutrophil kinetics in the pulmonary microcirculation during acute inflammation. Lab investigation; J Tech Methods Pathol (1991) 65(2):145–59. doi: 10.1186/s13054-016-1208-6
163. Brealey D, Singer M. Multi-organ dysfunction in the critically ill: effects on different organs. J R Coll Physicians Lond (2000) 34(5):428–31.
164. Park I, Kim M, Choe K, Song E, Seo H, Hwang Y, et al. Neutrophils disturb pulmonary microcirculation in sepsis-induced acute lung injury. Eur Respir J (2019) 53(3):1800786. doi: 10.1183/13993003.00786-2018
165. Kelly FJ, Postle AD, Phillips GJ. Neutrophils and oxygen-induced lung injury: a case of when a few is still too many. Redox Rep (1994) 1(1):37–44. doi: 10.1080/13510002.1994.11746954
166. Botha AJ, Moore FA, Moore EE, Sauaia A, Banerjee A, Peterson VM. Early neutrophil sequestration after injury. J Trauma: Injury Infection Crit Care (1995) 39(3):411–7. doi: 10.1097/00005373-199509000-00003
167. Zhou MY, Lo SK, Bergenfeldt M, Tiruppathi C, Jaffe A, Xu N, et al. In vivo expression of neutrophil inhibitory factor via gene transfer prevents lipopolysaccharide-induced lung neutrophil infiltration and injury by a beta2 integrin-dependent mechanism. J Clin Invest. (1998) 101(11):2427–37. doi: 10.1172/JCI407
168. Windsor ACJ, Mullen PG, Fowler AA, Sugerman HJ. Role of the neutrophil in adult respiratory distress syndrome. Br J Surg (1993) 80(1):10–7. doi: 10.1002/bjs.1800800106
169. Lesur O, Kokis A, Hermans C, Fülöp T, Bernard A, Lane D. Interleukin-2 involvement in early acute respiratory distress syndrome: Relationship with polymorphonuclear neutrophil apoptosis and patient survival. Crit Care Med (2000) 28(12):3814–22. doi: 10.1097/00003246-200012000-00010
170. Donnelly SC, Haslett C, Strieter RM, Kunkel SL, Walz A, Robertson CR, et al. Interleukin-8 and development of adult respiratory distress syndrome in at-risk patient groups. Lancet (1993) 341(8846):643–7. doi: 10.1016/0140-6736(93)90416-E
171. Dolinay T, Kim YS, Howrylak J, Hunninghake GM, An CH, Fredenburgh L, et al. Inflammasome-regulated cytokines are critical mediators of acute lung injury. Am J Respir Crit Care Med (2012) 185(11):1225–34. doi: 10.1164/rccm.201201-0003OC
172. Chishti AD, Shenton BK, Kirby JA, Baudouin SV. Neutrophil chemotaxis and receptor expression in clinical septic shock. Intensive Care Med (2004) 30(4):605–11. doi: 10.1007/s00134-004-2175-y
173. Xu R, Lin F, Bao C, Huang H, Ji C, Wang S, et al. Complement 5a receptor-mediated neutrophil dysfunction is associated with a poor outcome in sepsis. Cell Mol Immunol (2015) 13(1):103–9. doi: 10.1038/cmi.2014.136
174. Yu WY, Gao CX, Zhang HH, Wu YG, Yu CH. Herbal active ingredients: Potential for the prevention and treatment of acute lung injury. BioMed Res Int (2021) 2021:1–19. doi: 10.1155/2021/5543185
175. Conrad C, Yildiz D, Cleary SJ, Margraf A, Cook L, Schlomann U, et al. ADAM8 signaling drives neutrophil migration and ARDS severity. JCI Insight (2022) 7(3):e149870. doi: 10.1172/jci.insight.149870
176. Kienle K, Glaser KM, Eickhoff S, Mihlan M, Knöpper K, Reátegui E, et al. Neutrophils self-limit swarming to contain bacterial growth in vivo. Science (2021) 372(6548):eabe7729. doi: 10.1126/science.abe7729
177. Golenkina EA, Galkina SI, Pletjushkina O, Chernyak B, Gaponova TV, Romanova YM, et al. Gram-negative bacteria salmonella typhimurium boost leukotriene synthesis induced by chemoattractant fMLP to stimulate neutrophil swarming. Front Pharmacol (2022) 12:814113. doi: 10.3389/fphar.2021.814113
178. Weis WI, Kobilka BK. The molecular basis of G protein–coupled receptor activation. Annu Rev Biochem (2018) 87(1):897–919. doi: 10.1146/annurev-biochem-060614-033910
179. Wacker D, Stevens RC, Roth BL. How ligands illuminate GPCR molecular pharmacology. Cell. (2017) 170(3):414–27. doi: 10.1016/j.cell.2017.07.009
180. Lämmermann T, Kastenmüller W. Concepts of GPCR -controlled navigation in the immune system. Immunol Rev (2019) 289(1):205–31. doi: 10.1111/imr.12752
181. Hurowitz EH, Melnyk JM, Chen YJ, Kouros-Mehr H, Simon MI, Shizuya H. Genomic characterization of the human heterotrimeric G protein α, β, and γ subunit genes. DNA Res (2000) 7:111–20. doi: 10.1093/dnares/7.2.111
182. Altarejos JY, Montminy M. CREB and the CRTC co-activators: sensors for hormonal and metabolic signals. Nat Rev Mol Cell Biol (2011) 12(3):141–51. doi: 10.1038/nrm3072
183. Schappi JM, Krbanjevic A, Rasenick MM. Tubulin, actin and heterotrimeric G proteins: Coordination of signaling and structure. Biochim Biophys Acta (BBA) - Biomembranes. (2014) 1838(2):674–81. doi: 10.1016/j.bbamem.2013.08.026
184. Allen JA, Yu JZ, Donati RJ, Rasenick MM. β-adrenergic receptor stimulation promotes gαs internalization through lipid rafts: A study in living cells. Mol Pharmacol (2005) 67(5):1493–504. doi: 10.1124/mol.104.008342
185. Zarbock A, Deem TL, Burcin TL, Ley K. Gαi2 is required for chemokine-induced neutrophil arrest. Blood. (2007) 110(10):3773–9. doi: 10.1182/blood-2007-06-094565
186. Kuwano Y, Adler M, Zhang H, Groisman A, Ley K. Gαi2 and Gαi3 differentially regulate arrest from flow and chemotaxis in mouse neutrophils. J Immunol (2016) 196(9):3828–33. doi: 10.4049/jimmunol.1500532
187. Flock T, Ravarani CNJ, Sun D, Venkatakrishnan AJ, Kayikci M, Tate CG, et al. Universal allosteric mechanism for gα activation by GPCRs. Nature. (2015) 524(7564):173–9. doi: 10.1038/nature14663
188. Lämmermann T, Afonso PV, Angermann BR, Wang JM, Kastenmüller W, Parent CA, et al. Neutrophil swarms require LTB4 and integrins at sites of cell death in vivo. Nature (2013) 498(7454):371–5. doi: 10.1038/nature12175
189. Coombs C, Georgantzoglou A, Walker HA, Patt J, Merten N, Poplimont H, et al. Chemokine receptor trafficking coordinates neutrophil clustering and dispersal at wounds in zebrafish. Nat Commun (2019) 10(1):5166. doi: 10.1038/s41467-019-13107-3
190. Casanova-Acebes M, Nicolás-Ávila JA, Li JL, García-Silva S, Balachander A, Rubio-Ponce A, et al. Neutrophils instruct homeostatic and pathological states in naive tissues. J Exp Med (2018) 215(11):2778–95. doi: 10.1084/jem.20181468
191. Ballesteros I, Rubio-Ponce A, Genua M, Lusito E, Kwok I, Fernández-Calvo G, et al. Co-Option of neutrophil fates by tissue environments. Cell. (2020) 183(5):1282–1297.e18. doi: 10.1016/j.cell.2020.10.003
192. Hoogendijk AJ, van Vught LA, Wiewel MA, Fuhler GM, Belkasim-Bohoudi H, Horn J, et al. Kinase activity is impaired in neutrophils of sepsis patients. Haematologica. (2018) 104(6):e233–5. doi: 10.3324/haematol.2018.201913
193. Borkowska S, Suszynska M, Mierzejewska K, Ismail A, Budkowska M, Salata D, et al. Novel evidence that crosstalk between the complement, coagulation and fibrinolysis proteolytic cascades is involved in mobilization of hematopoietic stem/progenitor cells (HSPCs). Leukemia. (2014) 28(11):2148–54. doi: 10.1038/leu.2014.115
194. Jones CN, Moore M, Dimisko L, Alexander A, Ibrahim A, Hassell BA, et al. Spontaneous neutrophil migration patterns during sepsis after major burns. PLoS One (2014) 9(12):e114509. doi: 10.1371/journal.pone.0114509
195. Dong YL, Abdullah K, Yan TZ, Rutan T, Broemeling L, Robson M, et al. Effect of thermal injury and sepsis on neutrophil function. J Trauma: Injury Infection Crit Care (1993) 34(3):417–21. doi: 10.1097/00005373-199303000-00019
196. Wrann CD, Tabriz NA, Barkhausen T, Klos A, van Griensven M, Pape HC, et al. The phosphatidylinositol 3-kinase signaling pathway exerts protective effects during sepsis by controlling C5a-mediated activation of innate immune functions. J Immunol (2007) 178(9):5940–8. doi: 10.4049/jimmunol.178.9.5940
197. Fan J, Malik AB. Toll-like receptor-4 (TLR4) signaling augments chemokine-induced neutrophil migration by modulating cell surface expression of chemokine receptors. Nat Med (2003) 9(3):315–21. doi: 10.1038/nm832
198. Sakai K, Suzuki H, Oda H, Akaike T, Azuma Y, Murakami T, et al. Phosphoinositide 3-kinase in nitric oxide synthesis in macrophage. J Biol Chem (2006) 281(26):17736–42. doi: 10.1074/jbc.M601896200
199. Venkatakrishnan AJ, Deupi X, Lebon G, Heydenreich FM, Flock T, Miljus T, et al. Diverse activation pathways in class a GPCRs converge near the G-protein-coupling region. Nature. (2016) 536(7617):484–7. doi: 10.1038/nature19107
200. Wootten D, Christopoulos A, Marti-Solano M, Babu MM, Sexton PM. Mechanisms of signalling and biased agonism in G protein-coupled receptors. Nat Rev Mol Cell Biol (2018) 19(10):638–53. doi: 10.1038/s41580-018-0049-3
201. Tokmakova A, Kim D, Goddard WA, Liggett SB. Biased β-agonists favoring gs over β-arrestin for individualized treatment of obstructive lung disease. JPM. (2022) 12(3):331. doi: 10.3390/jpm12030331
202. Violin JD, Crombie AL, Soergel DG, Lark MW. Erratum: Biased ligands at G protein-coupled receptors: promise and progress. Trends Pharmacol Sci (2014) 35(9):489. doi: 10.1016/j.tips.2014.07.005
203. Slosky LM, Caron MG, Barak LS. Biased allosteric modulators: New frontiers in GPCR drug discovery. Trends Pharmacol Sci (2021) 42(4):283–99. doi: 10.1016/j.tips.2020.12.005
204. Kumari P, Srivastava A, Banerjee R, Ghosh E, Gupta P, Ranjan R, et al. Functional competence of a partially engaged GPCR–β-arrestin complex. Nat Commun (2016) 7(1):13416. doi: 10.1038/ncomms13416
205. Cahill TJ, Thomsen ARB, Tarrasch JT, Plouffe B, Nguyen AH, Yang F, et al. Distinct conformations of GPCR–β-arrestin complexes mediate desensitization, signaling, and endocytosis. Proc Natl Acad Sci USA (2017) 114(10):2562–7. doi: 10.1073/pnas.1701529114
206. Urban JD, Clarke WP, von Zastrow M, Nichols DE, Kobilka B, Weinstein H, et al. Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther (2006) 320(1):1–13. doi: 10.1124/jpet.106.104463
207. Whalen EJ, Rajagopal S, Lefkowitz RJ. Therapeutic potential of β-arrestin- and G protein-biased agonists. Trends Mol Med (2011) 17(3):126–39. doi: 10.1016/j.molmed.2010.11.004
208. Kooistra AJ, Mordalski S, Pándy-Szekeres G, Esguerra M, Mamyrbekov A, Munk C, et al. GPCRdb in 2021: integrating GPCR sequence, structure and function. Nucleic Acids Res (2020) 49(D1):D335–43. doi: 10.1093/nar/gkaa1080
209. Yin W, Li Z, Jin M, Yin YL, de Waal PW, Pal K, et al. A complex structure of arrestin-2 bound to a G protein-coupled receptor. Cell Res (2019) 29(12):971–83. doi: 10.1038/s41422-019-0256-2
210. Staus DP, Hu H, Robertson MJ, Kleinhenz ALW, Wingler LM, Capel WD, et al. Structure of the M2 muscarinic receptor–β-arrestin complex in a lipid nanodisc. Nature. (2020) 579(7798):297–302. doi: 10.1038/s41586-020-1954-0
211. Lee Y, Warne T, Nehmé R, Pandey S, Dwivedi-Agnihotri H, Chaturvedi M, et al. Molecular basis of β-arrestin coupling to formoterol-bound β1-adrenoceptor. Nature. (2020) 583(7818):862–6. doi: 10.1038/s41586-020-2419-1
212. Bous J, Fouillen A, Orcel H, Trapani S, Cong X, Fontanel S, et al. Structure of the vasopressin hormone–V2 receptor–β-arrestin1 ternary complex. Sci Adv (2022) 8(35):eabo7761. doi: 10.1126/sciadv.abo7761
213. Kumpers P, van Meurs M, David S, Molema G, Bijzet J, Lukasz A, et al. Time course of angiopoietin-2 release during experimental human endotoxemia and sepsis. Crit Care (2009) 13(3):R64. doi: 10.1186/cc7866
214. Davis JS, Yeo TW, Piera KA, Woodberry T, Celermajer DS, Stephens DP, et al. Angiopoietin-2 is increased in sepsis and inversely associated with nitric oxide-dependent microvascular reactivity. Crit Care (2010) 14(3):R89. doi: 10.1186/cc9020
215. Rajagopal S, Ahn S, Rominger DH, Gowen-MacDonald W, Lam CM, DeWire SM, et al. Quantifying ligand bias at seven-transmembrane receptors. Mol Pharmacol (2011) 80(3):367–77. doi: 10.1124/mol.111.072801
216. Strachan RT, Sun JP, Rominger DH, Violin JD, Ahn S, Rojas Bie Thomsen A, et al. Divergent transducer-specific molecular efficacies generate biased agonism at a G protein-coupled receptor (GPCR). J Biol Chem (2014) 289(20):14211–24. doi: 10.1074/jbc.M114.548131
217. Martínez-Morales JC, Solís KH, Romero-Ávila MT, Reyes-Cruz G, García-Sáinz JA. Cell trafficking and function of G protein-coupled receptors. Arch Med Res (2022) 53(5):451–60. doi: 10.1016/j.arcmed.2022.06.008
218. Reiter E, Lefkowitz RJ. GRKs and β-arrestins: roles in receptor silencing, trafficking and signaling. Trends Endocrinol Metab (2006) 17(4):159–65. doi: 10.1016/j.tem.2006.03.008
219. Chen Q, Iverson TM, Gurevich VV. Structural basis of arrestin-dependent signal transduction. Trends Biochem Sci (2018) 43(6):412–23. doi: 10.1016/j.tibs.2018.03.005
220. Su Y, Raghuwanshi SK, Yu Y, Nanney LB, Richardson RM, Richmond A. Altered CXCR2 signaling in β-Arrestin-2-Deficient mouse models. J Immunol (2005) 175(8):5396–402. doi: 10.4049/jimmunol.175.8.5396
221. Yang F, Xiao P, Qu CX, Liu Q, Wang LY, Liu ZX, et al. Allosteric mechanisms underlie GPCR signaling to SH3-domain proteins through arrestin. Nat Chem Biol (2018) 14(9):876–86. doi: 10.1038/s41589-018-0115-3
222. Mayor F, Cruces-Sande M, Arcones AC, Vila-Bedmar R, Briones AM, Salaices M, et al. G Protein-coupled receptor kinase 2 (GRK2) as an integrative signalling node in the regulation of cardiovascular function and metabolic homeostasis. Cell Signalling. (2018) 41:25–32. doi: 10.1016/j.cellsig.2017.04.002
223. Mores KL, Cassell RJ, van Rijn RM. Arrestin recruitment and signaling by G protein-coupled receptor heteromers. Neuropharmacology. (2019) 152:15–21. doi: 10.1016/j.neuropharm.2018.11.010
224. Bonifacio L, Dodds M, Prohaska D, Moss A, Giaccia A, Tabibiazar R, et al. Target-mediated drug disposition Pharmacokinetic/Pharmacodynamic model-informed dose selection for the first-in-Human study of AVB-S6-500. Clin Transl Sci (2019) 13(1):204–11. doi: 10.1111/cts.12706
225. Shukla AK, Violin JD, Whalen EJ, Gesty-Palmer D, Shenoy SK, Lefkowitz RJ. Distinct conformational changes in -arrestin report biased agonism at seven-transmembrane receptors. Proc Natl Acad Sci (2008) 105(29):9988–93. doi: 10.1073/pnas.0804246105
226. Yang F, Yu X, Liu C, Qu CX, Gong Z, Liu HD, et al. Phospho-selective mechanisms of arrestin conformations and functions revealed by unnatural amino acid incorporation and 19F-NMR. Nat Commun (2015) 6(1):8202. doi: 10.1038/ncomms9202
227. Nuber S, Zabel U, Lorenz K, Nuber A, Milligan G, Tobin AB, et al. β-arrestin biosensors reveal a rapid, receptor-dependent activation/deactivation cycle. Nature. (2016) 531(7596):661–4. doi: 10.1038/nature17198
228. Lee MH, Appleton KM, Strungs EG, Kwon JY, Morinelli TA, Peterson YK, et al. The conformational signature of β-arrestin2 predicts its trafficking and signalling functions. Nature. (2016) 531(7596):665–8. doi: 10.1038/nature17154
229. Hernández-Espinosa DA, Carmona-Rosas G, Alfonzo-Méndez MA, Alcántara-Hernández R, García-Sáinz JA. Sites phosphorylated in human α1B-adrenoceptors in response to noradrenaline and phorbol myristate acetate. Biochim Biophys Acta (BBA) - Mol Cell Res (2019) 1866(10):1509–19. doi: 10.1016/j.bbamcr.2019.07.006
230. Carmona-Rosas G, Hernández-Espinosa DA, Alcántara-Hernández R, Alfonzo-Méndez MA, García-Sainz JA. Distinct phosphorylation sites/clusters in the carboxyl terminus regulate α1D-adrenergic receptor subcellular localization and signaling. Cell Signalling. (2019) 53:374–89. doi: 10.1016/j.cellsig.2018.11.003
231. Divorty N, Jenkins L, Ganguly A, Butcher AJ, Hudson BD, Schulz S, et al. Agonist-induced phosphorylation of orthologues of the orphan receptor GPR35 functions as an activation sensor. J Biol Chem (2022) 298(3):101655. doi: 10.1016/j.jbc.2022.101655
232. Zhuo Y, Crecelius JM, Marchese A. G Protein–coupled receptor kinase phosphorylation of distal c-tail sites specifies βarrestin1-mediated signaling by chemokine receptor CXCR4. J Biol Chem (2022) 298(9):102351. doi: 10.1016/j.jbc.2022.102351
233. Gurevich VV, Gurevich EV. GPCR signaling regulation: The role of GRKs and arrestins. Front Pharmacol (2019) 10:00125. doi: 10.3389/fphar.2019.00125
234. Komolov KE, Benovic JL. G Protein-coupled receptor kinases: Past, present and future. Cell Signalling. (2018) 41:17–24. doi: 10.1016/j.cellsig.2017.07.004
235. Gurevich EV, Tesmer JJG, Mushegian A, Gurevich VV. G Protein-coupled receptor kinases: More than just kinases and not only for GPCRs. Pharmacol Ther (2012) 133(1):40–69. doi: 10.1016/j.pharmthera.2011.08.001
236. Raghuwanshi SK, Su Y, Singh V, Haynes K, Richmond A, Richardson RM. The chemokine receptors CXCR1 and CXCR2 couple to distinct G protein-coupled receptor kinases to mediate and regulate leukocyte functions. JI. (2012) 189(6):2824–32. doi: 10.4049/jimmunol.1201114
237. Alves-Filho JC, Sônego F, Souto FO, Freitas A, Verri WA, Auxiliadora-Martins M, et al. Interleukin-33 attenuates sepsis by enhancing neutrophil influx to the site of infection. Nat Med (2010) 16(6):708–12. doi: 10.1038/nm.2156
238. Lee SK, Kim SD, Kook M, Lee HY, Ghim J, Choi Y, et al. Phospholipase D2 drives mortality in sepsis by inhibiting neutrophil extracellular trap formation and down-regulating CXCR2. J Exp Med (2015) 212(9):1381–90. doi: 10.1084/jem.20141813
239. Kavelaars A, Vroon A, Raatgever RP, Fong AM, Premont RT, Patel DD, et al. Increased acute inflammation, leukotriene B4-induced chemotaxis, and signaling in mice deficient for G protein-coupled receptor kinase 6. J Immunol (2003) 171(11):6128–34. doi: 10.4049/jimmunol.171.11.6128
240. Busillo JM, Armando S, Sengupta R, Meucci O, Bouvier M, Benovic JL. Site-specific phosphorylation of CXCR4 is dynamically regulated by multiple kinases and results in differential modulation of CXCR4 signaling. J Biol Chem (2010) 285(10):7805–17. doi: 10.1074/jbc.M109.091173
241. Pitcher JA, Hall RA, Daaka Y, Zhang J, Ferguson SSG, Hester S, et al. The G protein-coupled receptor kinase 2 is a microtubule-associated protein kinase that phosphorylates tubulin. J Biol Chem (1998) 273(20):12316–24. doi: 10.1074/jbc.273.20.12316
242. Bretscher A. Rapid phosphorylation and reorganization of ezrin and spectrin accompany morphological changes induced in a-431 cells by epidermal growth factor. J Cell Biol (1989) 108(3):921–30. doi: 10.1083/jcb.108.3.921
243. Cant SH, Pitcher JA. G Protein-coupled receptor kinase 2–mediated phosphorylation of ezrin is required for G protein-coupled receptor–dependent reorganization of the actin cytoskeleton. MBoC. (2005) 16(7):3088–99. doi: 10.1091/mbc.e04-10-0877
244. Ceresa BP, Kao AW, Santeler SR, Pessin JE. Inhibition of clathrin-mediated endocytosis selectively attenuates specific insulin receptor signal transduction pathways. Mol Cell Biol (1998) 18(7):3862–70. doi: 10.1128/MCB.18.7.3862
245. Goodman O, Krupnick J, Santini F, Gurevich V, Penn R, Gagnon A, et al. Beta-arrestin acts as a clathrin adaptor in endocytosis of the beta2-adrenergic receptor. Nature (1996) 383(6599):447–50. doi: 10.1038/383447a0
246. Laporte SA, Oakley RH, Holt JA, Barak LS, Caron MG. The interaction of β-arrestin with the AP-2 adaptor is required for the clustering of β2-adrenergic receptor into clathrin-coated pits. J Biol Chem (2000) 275(30):23120–6. doi: 10.1074/jbc.M002581200
247. Fessart D, Simaan M, Laporte SA. C-src regulates clathrin adapter protein 2 interaction with β-arrestin and the angiotensin II type 1 receptor during clathrin- mediated internalization. Mol Endocrinology. (2005) 19(2):491–503. doi: 10.1210/me.2004-0246
248. Goodman OB, Krupnick JG, Santini F, Gurevich VV, Penn RB, Gagnon AW, et al. β-arrestin acts as a clathrin adaptor in endocytosis of the β2-adrenergic receptor. Nature. (1996) 383(6599):447–50. doi: 10.1038/383447a0
249. Henne WM, Boucrot E, Meinecke M, Evergren E, Vallis Y, Mittal R, et al. FCHo proteins are nucleators of clathrin-mediated endocytosis. Science. (2010) 328(5983):1281–4. doi: 10.1126/science.1188462
250. Kelly BT, McCoy AJ, Späte K, Miller SE, Evans PR, Höning S, et al. A structural explanation for the binding of endocytic dileucine motifs by the AP2 complex. Nature. (2008) 456(7224):976–9. doi: 10.1038/nature07422
251. Jackson LP, Kelly BT, McCoy AJ, Gaffry T, James LC, Collins BM, et al. A Large-scale conformational change couples membrane recruitment to cargo binding in the AP2 clathrin adaptor complex. Cell. (2010) 141(7):1220–9. doi: 10.1016/j.cell.2010.05.006
252. Fonseca FV, Raffay TM, Xiao K, McLaughlin PJ, Qian Z, Grimmett ZW, et al. S-nitrosylation is required for β2AR desensitization and experimental asthma. Mol Cell (2022) 82(16):3089–3102.e7. doi: 10.1016/j.molcel.2022.06.033
253. DeFea KA, Zalevsky J, Thoma MS, Déry O, Mullins RD, Bunnett NW. β-Arrestin–dependent endocytosis of proteinase-activated receptor 2 is required for intracellular targeting of activated Erk1/2. J Cell Biol (2000) 148(6):1267–82. doi: 10.1083/jcb.148.6.1267
254. McDonald PH, Chow CW, Miller WE, Laporte SA, Field ME, Lin FT, et al. β-arrestin 2: A receptor-regulated MAPK scaffold for the activation of JNK3. Science. (2000) 290(5496):1574–7. doi: 10.1126/science.290.5496.1574
255. Ribas C, Penela P, Murga C, Salcedo A, García-Hoz C, Jurado-Pueyo M, et al. The G protein-coupled receptor kinase (GRK) interactome: Role of GRKs in GPCR regulation and signaling. Biochim Biophys Acta (BBA) - Biomembranes. (2007) 1768(4):913–22. doi: 10.1016/j.bbamem.2006.09.019
256. Evron T, Daigle TL, Caron MG. GRK2: multiple roles beyond G protein-coupled receptor desensitization. Trends Pharmacol Sci (2012) 33(3):154–64. doi: 10.1016/j.tips.2011.12.003
257. White AD, Peña KA, Clark LJ, Maria CS, Liu S, Jean-Alphonse FG, et al. Spatial bias in cAMP generation determines biological responses to PTH type 1 receptor activation. Sci Signal (2021) 14(703):eabc5944. doi: 10.1126/scisignal.abc5944
258. Ferguson SM, De Camilli P. Dynamin, a membrane-remodelling GTPase. Nat Rev Mol Cell Biol (2012) 13(2):75–88. doi: 10.1038/nrm3266
259. Kalia R, Frost A. Open and cut: allosteric motion and membrane fission by dynamin superfamily proteins. MBoC. (2019) 30(17):2097–104. doi: 10.1091/mbc.E16-10-0709
260. Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol (2010) 11(5):329–41. doi: 10.1038/nrm2882
261. Seachrist JL, Laporte SA, Dale LB, Babwah AV, Caron MG, Anborgh PH, et al. Rab5 association with the angiotensin II type 1A receptor promotes Rab5 GTP binding and vesicular fusion. J Biol Chem (2002) 277(1):679–85. doi: 10.1074/jbc.M109022200
262. Welz T, Wellbourne-Wood J, Kerkhoff E. Orchestration of cell surface proteins by Rab11. Trends Cell Biol (2014) 24(7):407–15. doi: 10.1016/j.tcb.2014.02.004
263. Homma Y, Hiragi S, Fukuda M. Rab family of small GTPases: an updated view on their regulation and functions. FEBS J (2020) 288(1):36–55. doi: 10.1111/febs.15453
264. Seachrist JL, Ferguson SSG. Regulation of G protein-coupled receptor endocytosis and trafficking by rab GTPases. Life Sci (2003) 74(2–3):225–35. doi: 10.1016/j.lfs.2003.09.009
265. Shenoy SK, Lefkowitz RJ. β-arrestin-mediated receptor trafficking and signal transduction. Trends Pharmacol Sci (2011) 32(9):521–33. doi: 10.1016/j.tips.2011.05.002
266. Hughes CE, Nibbs RJB. A guide to chemokines and their receptors. FEBS J (2018) 285(16):2944–71. doi: 10.1111/febs.14466
267. Korbecki J, Kupnicka P, Chlubek M, Gorący J, Gutowska I, Baranowska-Bosiacka I. CXCR2 receptor: Regulation of expression, signal transduction, and involvement in cancer. IJMS. (2022) 23(4):2168. doi: 10.3390/ijms23042168
268. Graham GJ, Locati M, Mantovani A, Rot A, Thelen M. The biochemistry and biology of the atypical chemokine receptors. Immunol Lett (2012) 145(1–2):30–8. doi: 10.1016/j.imlet.2012.04.004
269. Ahuja SK, Murphy PM. The CXC chemokines growth-regulated oncogene (GRO) α, GROβ, GROγ, neutrophil-activating peptide-2, and epithelial cell-derived neutrophil-activating peptide-78 are potent agonists for the type b, but not the type a, human interleukin-8 receptor. J Biol Chem (1996) 271(34):20545–50. doi: 10.1074/jbc.271.34.20545
270. Bachelerie F, Ben-Baruch A, Burkhardt AM, Combadiere C, Farber JM, Graham GJ, et al. International union of basic and clinical pharmacology. LXXXIX. update on the extended family of chemokine receptors and introducing a new nomenclature for atypical chemokine receptors. Pharmacol Rev (2013) 66(1):1–79. doi: 10.1124/pr.114.009647
271. Sônego F, Alves-Filho JC, Cunha FQ. Targeting neutrophils in sepsis. Expert Rev Clin Immunol (2014) 10(8):1019–28. doi: 10.1586/1744666X.2014.922876
272. Sônego F, Castanheira FV e S, Ferreira RG, Kanashiro A, Leite CAVG, Nascimento DC, et al. Paradoxical roles of the neutrophil in sepsis: Protective and deleterious. Front Immunol (2016) 7:00155. doi: 10.3389/fimmu.2016.00155
273. Tavares-Murta BM, Zaparoli M, Ferreira RB, Silva-Vergara ML, B. Oliveira CH, Murta EFC, et al. Failure of neutrophil chemotactic function in septic patients*. Crit Care Med (2002) 30(5):1056–61. doi: 10.1097/00003246-200205000-00017
274. Richardson RM, Pridgen BC, Haribabu B, Ali H, Snyderman R. Differential cross-regulation of the human chemokine receptors CXCR1 and CXCR2. J Biol Chem (1998) 273(37):23830–6. doi: 10.1074/jbc.273.37.23830
275. Buckley CD, Ross EA, McGettrick HM, ChloeE O, Haworth O, Schmutz C, et al. Identification of a phenotypically and functionally distinct population of long-lived neutrophils in a model of reverse endothelial migration. J Leukocyte Biol (2005) 79(2):303–11. doi: 10.1189/jlb.0905496
276. Trettel F, Di Bartolomeo S, Lauro C, Catalano M, Ciotti MT, Limatola C. Ligand-independent CXCR2 dimerization. J Biol Chem (2003) 278(42):40980–8. doi: 10.1074/jbc.M306815200
277. Wilson S, Wilkinson G, Milligan G. The CXCR1 and CXCR2 receptors form constitutive homo- and heterodimers selectively and with equal apparent affinities. J Biol Chem (2005) 280(31):28663–74. doi: 10.1074/jbc.M413475200
278. Martínez Muñoz L, Lucas P, Navarro G, Checa AI, Franco R, Martínez-A C, et al. Dynamic regulation of CXCR1 and CXCR2 homo- and heterodimers. J Immunol (2009) 183(11):7337–46. doi: 10.4049/jimmunol.0901802
279. Sai J, Walker G, Wikswo J, Richmond A. The IL sequence in the LLKIL motif in CXCR2 is required for full ligand-induced activation of erk, akt, and chemotaxis in HL60 cells. J Biol Chem (2006) 281(47):35931–41. doi: 10.1074/jbc.M605883200
280. Sai J. The c-terminal domain LLKIL motif of CXCR2 is required for ligand-mediated polarization of early signals during chemotaxis. J Cell Science. (2004) 117(23):5489–96. doi: 10.1242/jcs.01398
281. Raman D, Sai J, Neel NF, Chew CS, Richmond A. LIM and SH3 protein -1 modulates CXCR2-mediated cell migration. PLoS One (2010) 5(4):e10050. doi: 10.1371/journal.pone.0010050
282. Fan GH, Yang W, Sai J, Richmond A. Hsc/Hsp70 interacting protein (Hip) associates with CXCR2 and regulates the receptor signaling and trafficking. J Biol Chem (2002) 277(8):6590–7. doi: 10.1074/jbc.M110588200
283. Montagnac G, Meas-Yedid V, Irondelle M, Castro-Castro A, Franco M, Shida T, et al. αTAT1 catalyses microtubule acetylation at clathrin-coated pits. Nature. (2013) 502(7472):567–70. doi: 10.1038/nature12571
284. Leclair HM, Dubois SM, Azzi S, Dwyer J, Bidère N, Gavard J. Control of CXCR2 activity through its ubiquitination on K327 residue. BMC Cell Biol (2014) 15(1):38. doi: 10.1186/s12860-014-0038-0
285. Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, et al. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. (1999) 11(4):443–51. doi: 10.1016/S1074-7613(00)80119-3
286. Gu J, Ran X, Deng J, Zhang A, Peng G, Du J, et al. Glycyrrhizin alleviates sepsis-induced acute respiratory distress syndrome via suppressing of HMGB1/TLR9 pathways and neutrophils extracellular traps formation. Int Immunopharmacology. (2022) 108:108730. doi: 10.1016/j.intimp.2022.108730
287. Naughton P, Hoque M, Green C, Foresti R, Motterlini R. Interaction of heme with nitroxyl or nitric oxide amplifies heme oxygenase-1 induction: involvement of the transcription factor Nrf2. Cell Mol Biol (2002) 48(8):885–94.
288. Freitas A, Alves-Filho JC, Secco DD, Neto AF, Ferreira SH, Barja-Fidalgo C, et al. Heme oxygenase/carbon monoxide-biliverdin pathway down regulates neutrophil rolling, adhesion and migration in acute inflammation. Br J Pharmacol (2006) 149(4):345–54. doi: 10.1038/sj.bjp.0706882
289. Fortin CF, McDonald PP, Fülöp T, Lesur O. Sepsis, leukocytes, and nitric oxide (no). Shock. (2010) 33(4):344–52. doi: 10.1097/SHK.0b013e3181c0f068
290. Ince C, Mayeux PR, Nguyen T, Gomez H, Kellum JA, Ospina-Tascón GA, et al. The endothelium in sepsis. Shock. (2016) 45(3):259–70. doi: 10.1097/SHK.0000000000000473
291. Arraes SMA, Freitas MS, da Silva SV, de Paula Neto HA, Alves-Filho JC, Martins MA, et al. Impaired neutrophil chemotaxis in sepsis associates with GRK expression and inhibition of actin assembly and tyrosine phosphorylation. Blood. (2006) 108(9):2906–13. doi: 10.1182/blood-2006-05-024638
292. Reddy RC, Narala VR, Keshamouni VG, Milam JE, Newstead MW, Standiford TJ. Sepsis-induced inhibition of neutrophil chemotaxis is mediated by activation of peroxisome proliferator-activated receptor-γ. Blood. (2008) 112(10):4250–8. doi: 10.1182/blood-2007-12-128967
293. Takatani Y, Ono K, Suzuki H, Inaba M, Sawada M, Matsuda N. Inducible nitric oxide synthase during the late phase of sepsis is associated with hypothermia and immune cell migration. Lab Invest. (2018) 98(5):629–39. doi: 10.1038/s41374-018-0021-z
294. Le HT, Tran VG, Kim W, Kim J, Cho HR, Kwon B. IL-33 priming regulates multiple steps of the neutrophil-mediated anti-candida albicansResponse by modulating TLR and dectin-1 signals. JI. (2012) 189(1):287–95. doi: 10.4049/jimmunol.1103564
295. Li W, Jia X, Shen C, Zhang M, Xu J, Shang Y, et al. A KSHV microRNA enhances viral latency and induces angiogenesis by targeting GRK2 to activate the CXCR2/AKT pathway. Oncotarget. (2016) 7(22):32286–305. doi: 10.18632/oncotarget.8591
296. Liu S, Tang J, Huang L, Xu Q, Ling X, Liu J. Cordyceps militaris alleviates severity of murine acute lung injury through miRNAs-mediated CXCR2 inhibition. Cell Physiol Biochem (2015) 36(5):2003–11. doi: 10.1159/000430168
297. Font MD, Thyagarajan B, Khanna AK. Sepsis and septic shock – basics of diagnosis, pathophysiology and clinical decision making. Med Clinics North America. (2020) 104(4):573–85. doi: 10.1016/j.mcna.2020.02.011
298. Ranieri VM, Thompson BT, Barie PS, Dhainaut JF, Douglas IS, Finfer S, et al. Drotrecogin Alfa (Activated) in adults with septic shock. N Engl J Med (2012) 366(22):2055–64. doi: 10.1056/NEJMoa1202290
299. Holder AL, Huang DT. A dream deferred: the rise and fall of recombinant activated protein c. Crit Care (2013) 17(2):309. doi: 10.1186/cc12533
300. Matthay MA, Zemans RL, Zimmerman GA, Arabi YM, Beitler JR, Mercat A, et al. Acute respiratory distress syndrome. Nat Rev Dis Primers. (2019) 5(1):1469–70. doi: 10.1038/s41572-019-0069-0
301. Hamid U, Krasnodembskaya A, Fitzgerald M, Shyamsundar M, Kissenpfennig A, Scott C, et al. Aspirin reduces lipopolysaccharide-induced pulmonary inflammation in human models of ARDS. Thorax. (2017) 72(11):971–80. doi: 10.1136/thoraxjnl-2016-208571
302. Qiao X, Kashiouris MG, L’Heureux M, Fisher BJ, Leichtle SW, Truwit JD, et al. Biological effects of intravenous vitamin c on neutrophil extracellular traps and the endothelial glycocalyx in patients with sepsis-induced ARDS. Nutrients. (2022) 14(20):4415. doi: 10.3390/nu14204415
303. Hogan G, Geoghegan P, Carroll TP, Clarke J, McElvaney OF, McElvaney OJ, et al. α1-antitrypsin: Key player or bystander in acute respiratory distress syndrome? Anesthesiology (2021) 134(5):792–808. doi: 10.1097/ALN.0000000000003727
304. Sahebnasagh A, Saghafi F, Safdari M, Khataminia M, Sadremomtaz A, Talaei Z, et al. Neutrophil elastase inhibitor (sivelestat) may be a promising therapeutic option for management of acute lung injury/acute respiratory distress syndrome or disseminated intravascular coagulation in COVID-19. J Clin Pharm Ther (2020) 45(6):1515–9. doi: 10.1111/jcpt.13251
305. Poli V, Pui-Yan Ma V, Di Gioia M, Broggi A, Benamar M, Chen Q, et al. Zinc-dependent histone deacetylases drive neutrophil extracellular trap formation and potentiate local and systemic inflammation. iScience. (2021) 24(11):103256. doi: 10.1016/j.isci.2021.103256
306. Song D, Adrover JM, Felice C, Christensen LN, He XY, Merrill JR, et al. PTP1B inhibitors protect against acute lung injury and regulate CXCR4 signaling in neutrophils. JCI Insight (2022) 7(14):e158199. doi: 10.1172/jci.insight.158199
307. Park HH, Park W, Lee YY, Kim H, Seo HS, Choi DW, et al. Bioinspired DNase-I-Coated melanin-like nanospheres for modulation of infection-associated NETosis dysregulation. Adv Sci (2020) 7(23):2001940. doi: 10.1002/advs.202001940
308. Yu HP, Liu FC, Umoro A, Lin ZC, Elzoghby AO, Hwang TL, et al. Oleic acid-based nanosystems for mitigating acute respiratory distress syndrome in mice through neutrophil suppression: how the particulate size affects therapeutic efficiency. J Nanobiotechnol (2020) 18(1):25. doi: 10.1186/s12951-020-0583-y
309. Lin MH, Lin CF, Yang SC, Hung CF, Fang JY. The interplay between nanoparticles and neutrophils. J BioMed nanotechnol. (2018) 14(1):66–85. doi: 10.1166/jbn.2018.2459
310. Weber S, Zimmer A, Pardeike J. Solid lipid nanoparticles (SLN) and nanostructured lipid carriers (NLC) for pulmonary application: A review of the state of the art. Eur J Pharmaceutics Biopharmaceutics. (2014) 86(1):7–22. doi: 10.1016/j.ejpb.2013.08.013
311. Javidi J, Haeri A, Nowroozi F, Dadashzadeh S. Pharmacokinetics, tissue distribution and excretion of Ag2S quantum dots in mice and rats: the effects of injection dose, particle size and surface charge. Pharm Res (2019) 36(3):46. doi: 10.1007/s11095-019-2571-1
312. Fromen CA, Rahhal TB, Robbins GR, Kai MP, Shen TW, Luft JC, et al. Nanoparticle surface charge impacts distribution, uptake and lymph node trafficking by pulmonary antigen-presenting cells. Nanomedicine: Nanotechnology Biol Med (2016) 12(3):677–87. doi: 10.1016/j.nano.2015.11.002
313. Goodwin AJ, Li P, Halushka PV, Cook JA, Sumal AS, Fan H. Circulating miRNA 887 is differentially expressed in ARDS and modulates endothelial function. Am J Physiology-Lung Cell Mol Physiol (2020) 318(6):L1261–9. doi: 10.1152/ajplung.00494.2019
314. Watkins MT. Sphingosine-1-phosphate in the plasma compartment regulates basal and inflammation-induced vascular leak in mice. Yearbook Vasc Surgery. (2010) 2010:32–3. doi: 10.1016/S0749-4041(10)79323-4
315. Wang L, Sammani S, Moreno-Vinasco L, Letsiou E, Wang T, Camp SM, et al. FTY720 (S)-phosphonate preserves sphingosine 1-phosphate receptor 1 expression and exhibits superior barrier protection to FTY720 in acute lung injury. Crit Care Med (2014) 42(3):e189–99. doi: 10.1097/CCM.0000000000000097
316. Wang L, Letsiou E, Wang H, Belvitch P, Meliton LN, Brown ME, et al. MRSA-induced endothelial permeability and acute lung injury are attenuated by FTY720 s-phosphonate. Am J Physiology-Lung Cell Mol Physiol (2022) 322(1):L149–61. doi: 10.1152/ajplung.00100.2021
317. Obinata H, Hla T. Sphingosine 1-phosphate in coagulation and inflammation. Semin Immunopathol (2011) 34(1):73–91. doi: 10.1007/s00281-011-0287-3
318. Brinkmann V, Billich A, Baumruker T, Heining P, Schmouder R, Francis G, et al. Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nat Rev Drug Discovery (2010) 9(11):883–97. doi: 10.1038/nrd3248
319. Oo ML, Chang SH, Thangada S, Wu MT, Rezaul K, Blaho V, et al. Engagement of S1P1-degradative mechanisms leads to vascular leak in mice. J Clin Invest. (2011) 121(6):2290–300. doi: 10.1172/JCI45403
320. Martínez-Morales JC, Romero-Ávila MT, Reyes-Cruz G, García-Sáinz JA. S1P1 receptor phosphorylation, internalization, and interaction with rab proteins: effects of sphingosine 1-phosphate, FTY720-p, phorbol esters, and paroxetine. Bioscience Rep (2018) 38(6):BSR20181612. doi: 10.1042/BSR20181612
321. Harrington EO, Vang A, Braza J, Shil A, Chichger H. Activation of the sweet taste receptor, T1R3, by the artificial sweetener sucralose regulates the pulmonary endothelium. Am J Physiology-Lung Cell Mol Physiol (2018) 314(1):L165–76. doi: 10.1152/ajplung.00490.2016
322. Narumiya S. Physiology and pathophysiology of prostanoid receptors. Proc Japan Acad Ser B Phys Biol Sci (2007) 83(9-10):296–319. doi: 10.2183/pjab.83.296
323. Wallace AE, Sales KJ, Catalano RD, Anderson RA, Williams ARW, Wilson MR, et al. Prostaglandin F2α-F-Prostanoid receptor signaling promotes neutrophil chemotaxis via chemokine (C-X-C motif) ligand 1 in endometrial adenocarcinoma. Cancer Res (2009) 69(14):5726–33. doi: 10.1158/0008-5472.CAN-09-0390
324. Maehara T, Fujimori K. Inhibition of prostaglandin F2α receptors exaggerates HCl-induced lung inflammation in mice. IJMS. (2021) 22(23):12843. doi: 10.3390/ijms222312843
325. Beautrait A, Paradis JS, Zimmerman B, Giubilaro J, Nikolajev L, Armando S, et al. A new inhibitor of the β-arrestin/AP2 endocytic complex reveals interplay between GPCR internalization and signalling. Nat Commun (2017) 8(1):15054. doi: 10.1038/ncomms15054
326. Sundqvist M, Holdfeldt A, Wright SC, Møller TC, Siaw E, Jennbacken K, et al. Barbadin selectively modulates FPR2-mediated neutrophil functions independent of receptor endocytosis. Cold Spring Harbor Lab (2020) 1867(12):118849. doi: 10.1101/2020.04.30.070011
327. Tseng LTL, Lin CL, Tzen KY, Chang SC, Chang MF. LMBD1 protein serves as a specific adaptor for insulin receptor internalization. J Biol Chem (2013) 288(45):32424–32. doi: 10.1074/jbc.M113.479527
328. Rutsch F, Gailus S, Miousse IR, Suormala T, Sagné C, Toliat MR, et al. Identification of a putative lysosomal cobalamin exporter altered in the cblF defect of vitamin B12 metabolism. Nat Genet (2009) 41(2):234–9. doi: 10.1038/ng.294
329. Tan C, Gu J, Chen H, Li T, Deng H, Liu K, et al. Inhibition of aerobic glycolysis promotes neutrophil to influx to the infectious site Via CXCR2 in sepsis. Shock. (2020) 53(1):114–23. doi: 10.1097/SHK.0000000000001334
330. Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Invest. (2012) 122(8):2731–40. doi: 10.1172/JCI60331
331. Wu R, Dong W, Zhou M, Zhang F, Marini CP, Ravikumar TS, et al. Ghrelin attenuates sepsis-induced acute lung injury and mortality in rats. Am J Respir Crit Care Med (2007) 176(8):805–13. doi: 10.1164/rccm.200604-511OC
332. Wong JJM, Jit M, Sultana R, Mok YH, Yeo JG, Koh JWJC, et al. Mortality in pediatric acute respiratory distress syndrome: A systematic review and meta-analysis. J Intensive Care Med (2017) 34(7):563–71. doi: 10.1177/0885066617705109
333. Kumar V. Pulmonary innate immune response determines the outcome of inflammation during pneumonia and sepsis-associated acute lung injury. Front Immunol (2020) 11:01722. doi: 10.3389/fimmu.2020.01722
334. Zhang P, Liu B, Zheng W, Chen Y, Wu Z, Lu Y, et al. Pulmonary microbial composition in sepsis-induced acute respiratory distress syndrome. Front Mol Biosci (2022) 9:862570. doi: 10.3389/fmolb.2022.862570
335. Steinberg KP, Milberg JA, Martin TR, Maunder RJ, Cockrill BA, Hudson LD. Evolution of bronchoalveolar cell populations in the adult respiratory distress syndrome. Am J Respir Crit Care Med (1994) 150(1):113–22. doi: 10.1164/ajrccm.150.1.8025736
336. Peiseler M, Kubes P. More friend than foe: the emerging role of neutrophils in tissue repair. J Clin Invest. (2019) 129(7):2629–39. doi: 10.1172/JCI124616
337. Thompson BT, Chambers RC, Liu KD. Acute respiratory distress syndrome. N Engl J Med (2017) 377(6):562–72. doi: 10.1056/NEJMra1608077
338. Nick JA, Coldren CD, Geraci MW, Poch KR, Fouty BW, O’Brien J, et al. Recombinant human activated protein c reduces human endotoxin-induced pulmonary inflammation via inhibition of neutrophil chemotaxis. Blood. (2004) 104(13):3878–85. doi: 10.1182/blood-2004-06-2140
339. Wang G, Cao L, Liu X, Sieracki NA, Di A, Wen X, et al. Oxidant sensing by TRPM2 inhibits neutrophil migration and mitigates inflammation. Dev Cell (2016) 38(5):453–62. doi: 10.1016/j.devcel.2016.07.014
Keywords: sepsis, ARDS, neutrophil, GPCR, desensitization, internalization
Citation: Wang Y, Zhu C-l, Li P, Liu Q, Li H-r, Yu C-m, Deng X-m and Wang J-f (2023) The role of G protein-coupled receptor in neutrophil dysfunction during sepsis-induced acute respiratory distress syndrome. Front. Immunol. 14:1112196. doi: 10.3389/fimmu.2023.1112196
Received: 30 November 2022; Accepted: 07 February 2023;
Published: 20 February 2023.
Edited by:
Sjoerd Schetters, Flanders Institute for Biotechnology, BelgiumReviewed by:
Helena Aegerter, Flemish Institute for Biotechnology, BelgiumRebecca Kelley Martin, Virginia Commonwealth University, United States
Copyright © 2023 Wang, Zhu, Li, Liu, Li, Yu, Deng and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jia-feng Wang, amZ3YW5nQHNtbXUuZWR1LmNu; Xiao-ming Deng, ZGVuZ3BoZEBzbW11LmVkdS5jbg==
†These authors have contributed equally to this work