 Kyler S. Crawford
Kyler S. Crawford Brian F. Volkman
Brian F. Volkman- Department of Biochemistry, Medical College of Wisconsin, Milwaukee, WI, United States
The chemokine network is comprised of a family of signal proteins that encode messages for cells displaying chemokine G-protein coupled receptors (GPCRs). The diversity of effects on cellular functions, particularly directed migration of different cell types to sites of inflammation, is enabled by different combinations of chemokines activating signal transduction cascades on cells displaying a combination of receptors. These signals can contribute to autoimmune disease or be hijacked in cancer to stimulate cancer progression and metastatic migration. Thus far, three chemokine receptor-targeting drugs have been approved for clinical use: Maraviroc for HIV, Plerixafor for hematopoietic stem cell mobilization, and Mogalizumab for cutaneous T-cell lymphoma. Numerous compounds have been developed to inhibit specific chemokine GPCRs, but the complexity of the chemokine network has precluded more widespread clinical implementation, particularly as anti-neoplastic and anti-metastatic agents. Drugs that block a single signaling axis may be rendered ineffective or cause adverse reactions because each chemokine and receptor often have multiple context-specific functions. The chemokine network is tightly regulated at multiple levels, including by atypical chemokine receptors (ACKRs) that control chemokine gradients independently of G-proteins. ACKRs have numerous functions linked to chemokine immobilization, movement through and within cells, and recruitment of alternate effectors like β-arrestins. Atypical chemokine receptor 1 (ACKR1), previously known as the Duffy antigen receptor for chemokines (DARC), is a key regulator that binds chemokines involved in inflammatory responses and cancer proliferation, angiogenesis, and metastasis. Understanding more about ACKR1 in different diseases and populations may contribute to the development of therapeutic strategies targeting the chemokine network.
Introduction
Chemokine receptors (CKRs) are specialized seven-transmembrane domain surface receptors in the class A subfamily of the G-protein coupled receptor (GPCR) superfamily. Chemokine ligands are small, structurally-conserved proteins categorized by the configuration of a cysteine motif (CXC, CC, CX3C, C) in the N-terminus (1). The classical function of chemokine GPCRs is to activate leukocyte migration along increasing chemokine concentration gradients towards their source, with different tissues producing distinct combinations of chemokines to attract specific cell types. Chemokine messages elicit complex, multicellular responses encoded in the combinatorial diversity of overlapping ligand-receptor specificities and dynamic membrane interactions. Receptor stimulation recruits β-arrestins, an intracellular effector that decreases activation of heterotrimeric G-proteins, scaffolds cytoskeletal adaptors that internalize surface receptors, and signals through distinct pathways (2). The chemokine network is tightly regulated with overlapping mechanisms to amplify, diversify, and resolve cellular signals (3). One arm of chemokine control is exerted through expression of atypical chemokine receptors (ACKRs), dedicated chemokine receptors uncoupled from G-protein cascades that regulate chemokine patterning and GPCR sensitivity (4). CKRs and ACKRs have complementary roles in exerting and modulating chemokine function. ACKRs have an independent role to bind, scavenge, and traffic chemokine ligands and maintain gradients so that cells are directed to their functional compartments (5). ACKRs can also directly regulate GPCR signaling through ligand depletion or resolution of activated intracellular cascades.
Chemokine signals are crucial for immune cell recruitment, embryonic development, and retention of discrete cellular niches. Consequently, dysregulation of the chemokine network can contribute to a multitude of disease and CKRs are appealing therapeutic drug targets. GPCRs are the target of a third or more of all drugs, but chemokine GPCRs present unique challenges to drug design that prevent compounds from progressing to approved therapeutics (6, 7). Inhibitors of individual GPCRs can have deleterious side effects by perturbing the balance of these signal pathways and interfering in unrelated physiological functions that involve the target GPCR. A druggable chemokine network becomes more achievable when the interplay of signaling and regulatory components in the system is well-understood. Here we discuss the role of ACKR1/DARC in disease and potential therapeutic strategies.
The atypical chemokine receptor family
The four known atypical chemokine receptors, ACKR1-4, exhibit distinct expression patterns, chemokine-binding profiles, and cellular effects. The chemokine ligands of the atypical receptors are shown in Figure 1. ACKR1 is a promiscuous receptor for chemokines involved in diverse functions including angiogenesis, chemotaxis, and cellular retention signals. Expression is restricted to erythroid cells, cerebellar Purkinje neurons and the endothelial cell lining of capillary-draining venules, where ACKR1 binds and transports chemokines. ACKR2 binds the second-most chemokines and was thought to be restricted to binding CC-class chemokines until recent reports have described interactions with CXCL10 and CXCL14 (8–10). ACKR2 is primarily found in the lymphatic, not vascular, endothelium but it is also expressed in certain B-lymphocytes, myeloid immune cells, and developing trophoblasts (11–13). ACKR2 serves as a chemokine scavenger that constitutively recycles from membrane to endosome through a pathway involving β-arrestin (14). ACKR3 is a high affinity receptor for several proteins including endogenous opioid peptides and viral chemokine vCCL2/vMIP-II, but only binds two human chemokines, CXCL11 and CXCL12 (15, 16). ACKR3 expression has been described in a diversity of cell types with increasing evidence of ligand-specific, β-arrestin-mediated signaling pathways and multiple internalization mechanisms (17–19). ACKR4 binds CCL19, CCL20, CCL21, CCL22 and CCL25, a subset of chemokines associated with spatial organization of T-cells and dendritic cells (20). Knowledge of ACKR4 expression is incomplete, but it has been characterized as a component of endothelial barriers in tissues including the skin, spleen, and lymphatic vasculature and as a scavenger on fibroblasts in the dermis and intestinal submucosa (21–23). ACKR4 scavenging uses a similar internalization mechanism to ACKR2 involving β-arrestin recruitment, but without the downstream ERK1/2, Akt, or Src kinase activation attributed to ACKR3 (24). Candidate members of the ACKR family include CC chemokine receptor-like 2 (CCRL2/ACKR5) as a receptor for the chemotactic protein chemerin, and membrane-associated phosphatidylinositol transfer protein 3 (PITPNM3/ACKR6) as a receptor for CCL18 (25, 26). Overall, ACKRs bind the majority of CC and CXC chemokines and expression is spatially organized in tissues to maintain functional chemokine gradients and regulate GPCR signaling. ACKR1 has several advantages as a potential drug target because it is promiscuous and encompasses multiple important chemokine-induced pathways, while being uncoupled from direct signal transduction and exhibiting restricted tissue expression.
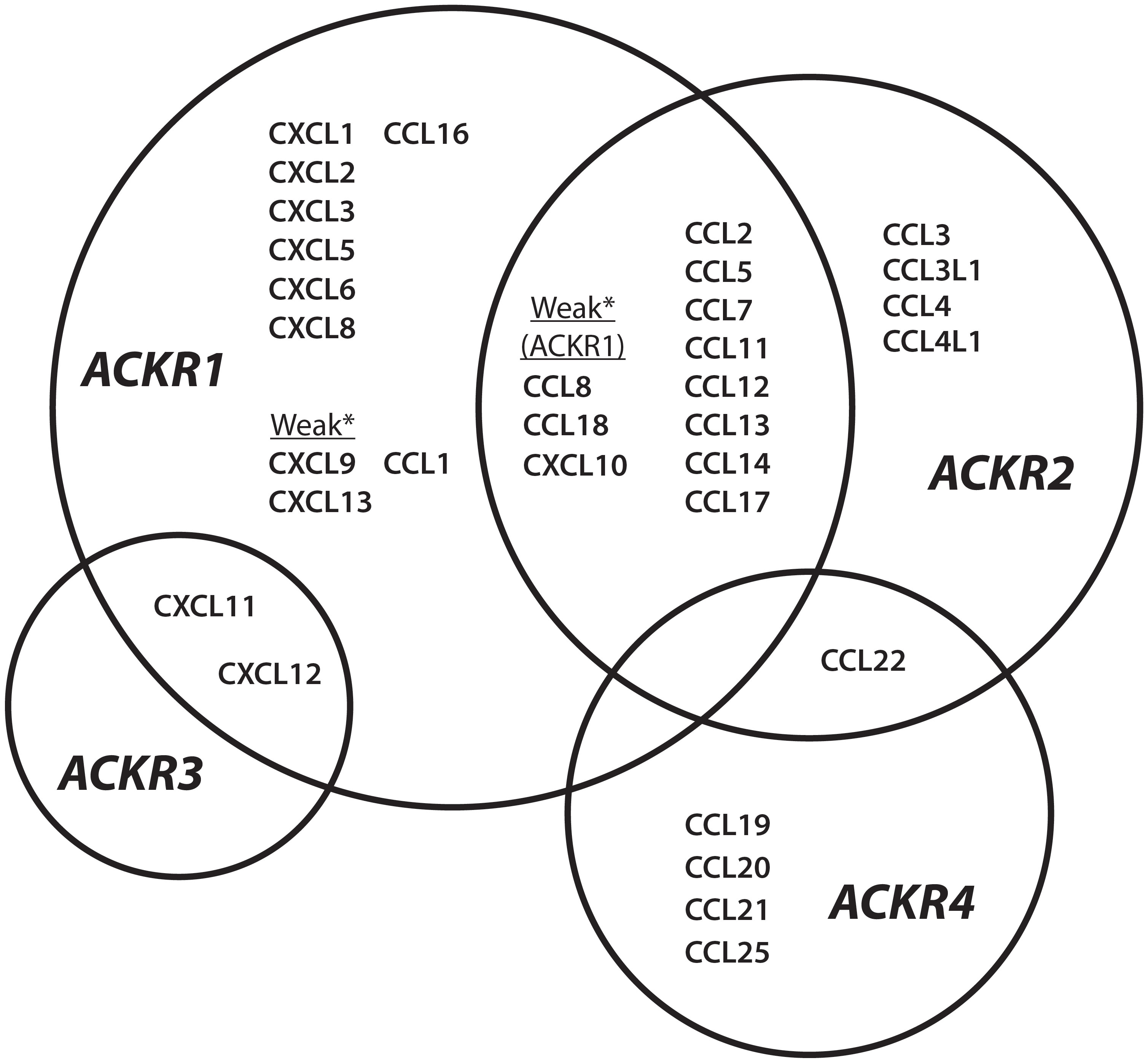
Figure 1 ACKR1-4 chemokine interaction network Chemokine ligands described for atypical chemokine receptors ACKR1, ACKR2, ACKR3, and ACKR4. *Chemokines are described as weak binders to ACKR1.
ACKR1 genetics
ACKR1 expression in humans was initially described as the “Duffy” or “Fy” blood group after a hemophiliac patient who developed hemolytic reactions from mismatched blood (27). The recognition sites of the “Fy-reactive” antibodies were mapped to distinct erythrocyte surface antigens, later revealed to correspond to regions of ACKR1. These include a conformational epitope (Fy3) capturing the extracellular loops, a linear pentapeptide sequence in the N-terminus (Fy6), and allelic N-terminal single nucleotide polymorphism (SNP) variants (FyA and FyB). Multiple ACKR1 phenotypes arise from SNPs in the upstream promoter and coding sequence of the ACKR1 gene (28). The major isoform of ACKR1 is a 336 amino acid protein with two common alleles FyA (42Gly), FyB(42Asp), and the less common FyX, most associated with R89C (29).
A unique selective pressure from malaria parasites contributes to distinct population-specific and geographic patterns of ACKR1 expression (30). The N-terminus of ACKR1 is a recognition site for Plasmodium vivax and P. knowlesi, which invade erythrocytes during blood infection (31). Malarial resistance is conferred by the “Duffy-negative” or “erythrocyte silent” (FyES) single nucleotide polymorphism (SNP), that alters the GATA1 transcription factor binding site in the ACKR1 promoter, ceasing erythroid, but not endothelial, expression (32). The coevolutionary history of Plasmodia parasites and FyES phenotype is complex, but the current evidence indicates that African P. vivax selected the “erythroid silent” polymorphism in the FyB allele in endemic regions. FyBES is now the prevalent phenotype of people in Africa, regions within the Arabian Peninsula, and with African ancestry (33, 34). The ancestral form of ACKR1 may have been FyB, which then adapted through the FyA variation (42G) conferring diminished susceptibility to P. vivax or the silencing polymorphism FyBES (rs2814778) (35, 36). The FyX variant is linked to both R89C and A100T mutations and decreases detection of ACKR1 expression (37). This effect may arise from a disruption in the first intracellular loop between the first and second transmembrane domains, and may interrupt trafficking to the membrane, impede protein folding, or cause formation of destabilizing inter/intra-molecular disulfide bonds (38, 39). The amino acid sequence of ACKR1 is depicted in Figure 2. Current understanding is that the primary drivers of differentiation of ACKR1 expression and the molecular basis of the Duffy blood group are the FyA/FyB alleles encoding Gly42 or Asp42 in the N-terminus and the FyES SNP, which determines if ACKR1 is present on erythrocyte surfaces to display epitopes like Fy3 or Fy6. These genetic variations that alter ACKR1 expression and N-terminal sequence may have a significant impact on disease by changing the abundance and distribution of ACKR1 ligands (40, 41).
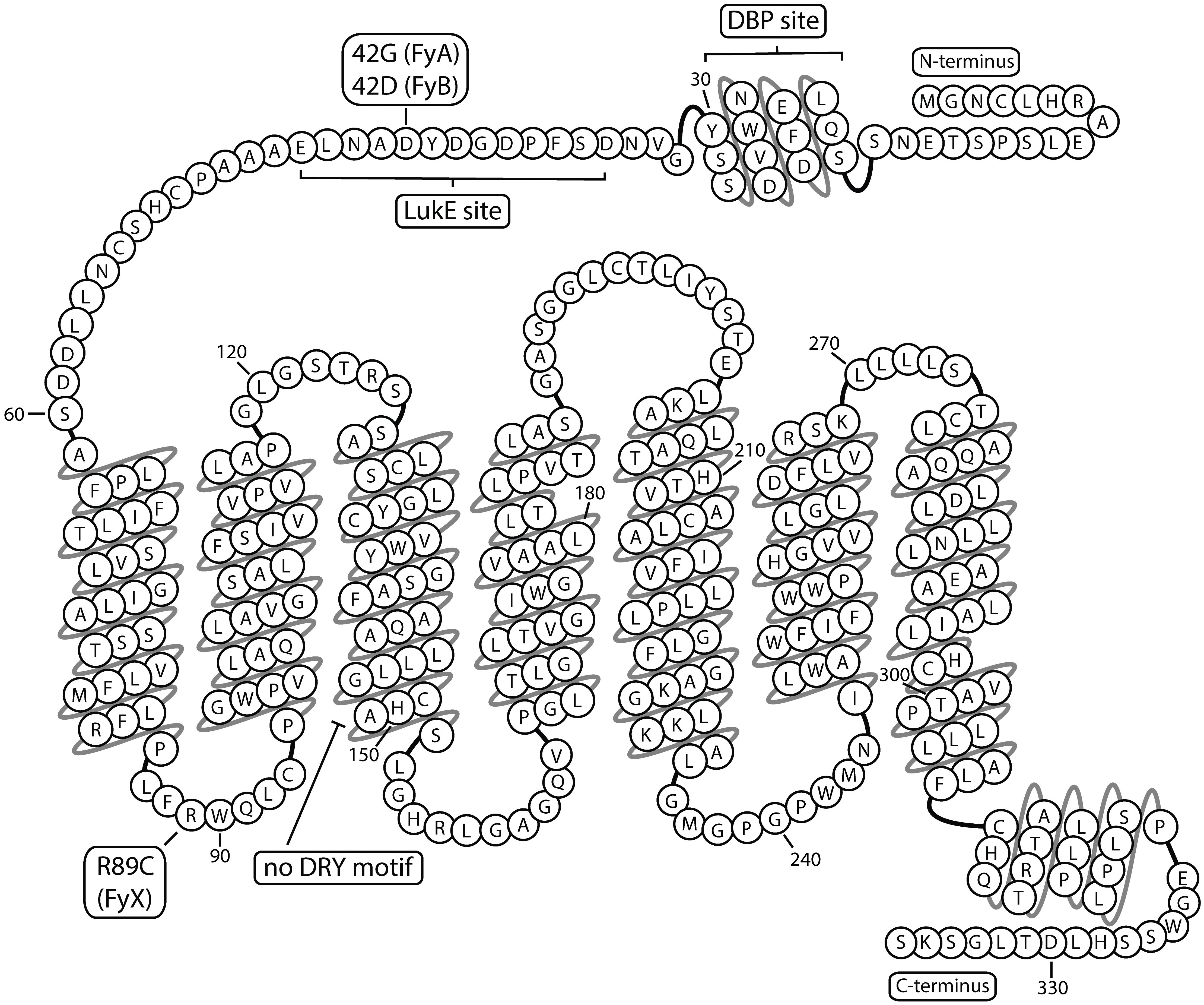
Figure 2 ACKR1 snake plot Atypical chemokine receptor 1 has seven transmembrane domains and multiple binding sites in the extracellular N-terminus. Residue 42 is depicted as aspartic acid corresponding to FyB variant. DBP, Duffy Binding Protein; LukE, Leukocidin E.
ACKR1 structure and function
Chemokine receptors are activated after binding ligands in a multi-step interaction using the receptor N-terminus that extends from the first α-helical transmembrane domain. The chemokine binding pocket is formed within the transmembrane helices and the extracellular connecting loop regions. Engagement of a typical chemokine receptor triggers conserved microswitches and conformational changes in the transmembrane helices followed by activation of intracellular secondary messengers (42). G-protein coupling occurs at a conserved “DRYLAIV” sequence motif found at the intracellular end of transmembrane helix 3. However, atypical receptors have sequence modifications at this position that prevent G-protein mediated signaling. While ACKR1 has no homologous motifs at this position, ACKR2 has DKYLEIV, ACKR3 has DRYLSIT, and ACKR4 DRYVAVT. Another common feature of GPCRs is a feedback inhibition mechanism wherein sustained receptor activation leads to phosphorylation of the C-terminus by G-protein coupled receptor kinases (GRKs). GRK activity supports association with β-arrestins, causing receptor internalization and alternative signaling. Both CKRs and ACKRs have serine and threonine-rich sequences in the intracellular C-terminal domain that are substrates for GRK-mediated phosphorylation. β-arrestin recruitment has been described for ACKR2-4, but while ACKR1 has analogous sites encoded in the C-terminus, investigation of GRK interactions has yet to be thoroughly explored (43). Thus, ACKR1 with the lowest sequence similarity to the other chemokine receptors, seems to have a distinct activation mechanism and network of intracellular interactions that is distinct from other ACKRs (44–46).
Solved structures of chemokine receptors are limited in the resolution of receptor N-terminal interactions, but studies support the importance of this domain for atypical chemokine receptor function (47). The ACKR2 N-terminus is selective for CC-type chemokines, and a protein derived from the critical domains has been proposed as an anti-inflammatory chemokine sink (48). The N-terminus of ACKR1 is among the longest of any chemokine receptors and contains extended regions of amino acids modeled to form electrostatic interactions with the basic and positively charged surfaces characteristic of chemokines (49). A distinguishing feature of ACKR1 is the capacity to bind multiple CXC and CC class chemokines, and the flexibility of this mostly disordered region allows for variable configurations to dock many different ligands (50). The binding interactions at the N-termini of ACKRs are shown in Table 1. Discrete ACKR1 N-terminal residues determine ligand affinity and different segments have been successfully engaged by antibodies or antibody-derived fragments to prohibit ligand binding (51, 52). A chimeric construct with the N-terminus of ACKR1 and the transmembrane domains and extracellular loops of CXCR2 retained the binding profile of full-length ACKR1, with high affinity for non-CXCR2 ligands CCL5 and N-terminally modified CXCL1 (53). The independence of the N-terminus for certain ligands also suggests utility of a soluble platform with the binding affinity of ACKR1, for example as a decoy for pathogens targeting erythrocytes. Additional detailed structural data describing interactions between the ACKR1 N-terminus and different chemokine ligands will contribute to understanding conserved and chemokine-specific binding mechanisms.
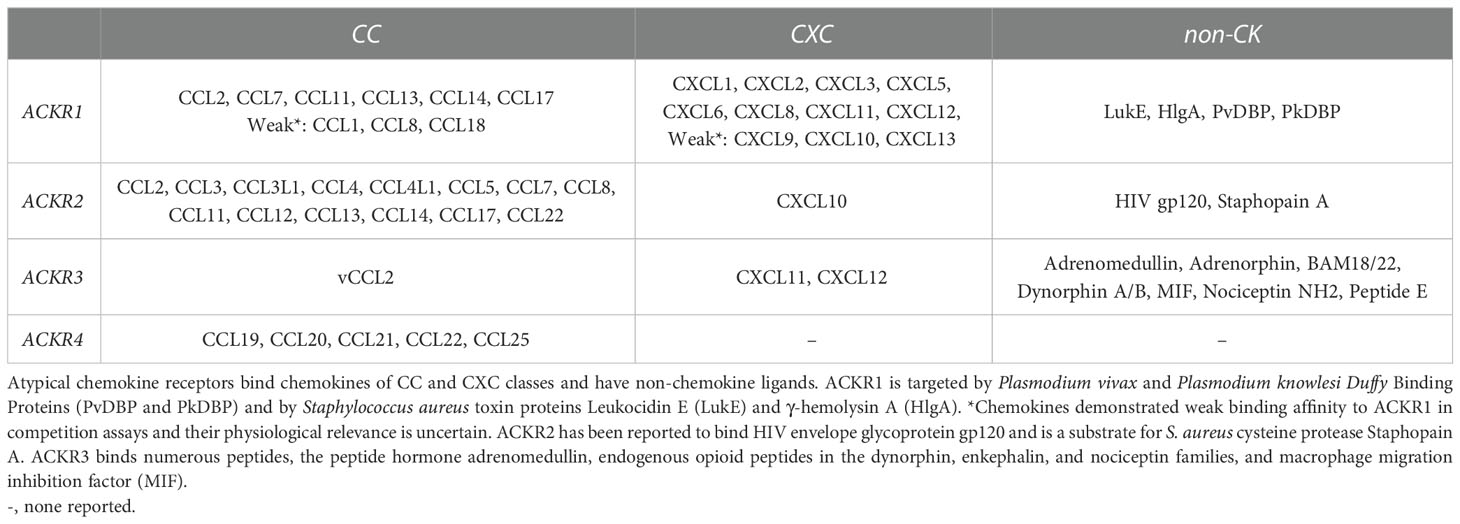
Table 1 Ligands of atypical chemokine receptors 1-4.
Initial surveys of ACKR1 functions suggested a binding preference for chemokines containing the sequence motif “ELR” in the N-terminus, a subgroup of CXC chemokines distinguished for its capacity for angiogenesis and inflammatory signaling through neutrophil receptors CXCR1 and CXCR2 (54, 55). One of the first reported angiogenic chemokines was CXCL8, and a model of neovascularization emerged with ELR+ CXCR2 ligands stimulating endothelial migration and tube formation countered by ELR- CXCR3 ligands. Angiogenic effects have since been ascribed to non ELR+ CXCL12 and other CC chemokines, particularly CCL2, suggesting a multifactorial system of CXC and CC chemokine receptors on endothelial cells and other immune cell types (56, 57). Evidence for the anti-angiogenic properties of ACKR1 was initially shown in a mouse by overexpressing ACKR1, decreasing CXCR2-mediated corneal angiogenesis in response to CXCL2 stimulation (58). Further investigation using radioligand displacement supported strong binding of ACKR1 to ELR+ chemokines like CXCL5 and CXCL8 that signal through CXCR2, but highest binding affinities were calculated for CCL5, CCL7, and non-ELR+ CXCL11 (59). The next functional categorization was regulation of “inflammatory” chemokines over “homeostatic” chemokines since chemokines CXCL12 and CCL21 showed weak ability to displace CXCL8 bound to ACKR1 (59). However, studies have since described many roles for both chemokines in inflammation and binding interactions have been reported between ACKR1 and CXCL12 (60, 61). ACKR1 binds most chemokines including the ELR+ CXC subfamily, and chemokines CXCL10, CXCL13, and CCL1 that were reported as non-binders were found to have weak but sub-micromolar affinities for ACKR1 on human erythrocytes (59). ACKR1 does not bind every chemokine, for example CXCL4 and several lymphoid CC chemokines have been shown not to bind ACKR1-expressing cells (59, 62).
The binding profile of ACKR1 has been primarily surveyed using radioligand displacement assays with pre-bound, high-affinity ligands and erythrocyte ACKR1 that may underrepresent lower-affinity interactions with chemokines or the influence of other mediators on endothelial surfaces like glycosaminoglycans. This selectivity was reported to play a role in filtering chemokines at high endothelial venules (HEVs), where ACKR1 may restrict inflammatory chemokines from entering secondary lymphoid organs and interfering with chemokine sensitivity (62).
While ACKR1 is most readily detected on mature erythrocytes, ACKR1 expression is highest in the bone marrow on progenitor nucleated erythroid cells (NECs), where key cell contacts are made with hematopoietic stem cells (HSCs) (63). The erythroid silent variant (FyES), though providing malarial protection, loses this developmental cue, resulting in a neutrophil phenotype with altered surface markers and increased propensity to leave circulation (64, 65). The observed neutropenia, historically called “benign ethnic neutropenia” and now more accurately “Duffy-associated neutrophil count” (DANC), does not eliminate effective inflammatory immune responses and is hypothesized to be asymptomatic in otherwise-healthy patients (66–68).
Outside of the erythroid lineage, ACKR1 is expressed on endothelial cells of post-capillary venules, where affinity for certain chemokines results in immobilized gradients that direct cell migration (69–71). A hallmark of tissue inflammation is increased chemokine production, but chemokines must be concentrated and displayed in the vascular compartment with a coordinated gradient to effectively direct immune responses. Endothelial ACKR1 function involves a combination of chemokine retention, presentation to circulating leukocytes, and trafficking from tissues to the luminal surface (72). ACKR1 is distinguished from the other ACKRs by ligand-triggered chemokine transcytosis through venular endothelial cells. ACKR1 has been shown to transport chemokines from basolateral to luminal sides of endothelial cells and retain chemokines on the apical surface promoting signaling through GPCRs (73–76). One demonstration of this function is neutrophil diapedesis, where ACKR1 concentrated at endothelial junctions binds and exchanges CXCL1 and CXCL2 chemokines to direct neutrophils and prevent reverse migration (77). These functions at the endothelium have been shown to modulate neuroinflammation as well, by trafficking chemokines and immune cells across the blood-brain barrier (78, 79). ACKR expression is detected in the brain on cerebellar Purkinje cells, where it may regulate cellular excitation for smooth motor control (53, 80). Further studies of ACKR1 in different tissues, including neurons, and with non-chemokine ligands may reveal additional complexity and specialized functions.
ACKR1 and infectious disease
The extracellular domain of ACKR1 is a potential target to inhibit pathogenicity mechanisms of atypical malaria, S. aureus, and HIV. Plasmodia malarial parasites replicate and mature inside human reticulocytes and erythrocytes, and the “atypical” P. vivax and P. knowlesi parasites identify these targets by secreting Duffy Binding Protein (DBP), which binds to and oligomerizes around the N-terminal domain of ACKR1 (81). While P. falciparum secretes multiple soluble factors, atypical malaria invasion can be avoided with the erythroid silent polymorphism or by blocking the DBP-ACKR1 binding interface with inhibitory chemokines or antibodies (51, 82, 83). Crystal structures have been solved showing a dimer of PvDBP dimers binding a peptide corresponding to ACKR1 residues 14-43. The receptor peptide could be resolved between residues 19-30 as an amphipathic α-helix structure with Y30 oriented towards a positively charged pocket (84). An ACKR1 mimetic was designed from this N-terminal helix, with the DBP-binding residues grafted onto a stable scaffold (85). The engineered protein could successfully inhibit DBP dimerization and binding to erythrocytes. Non-falciparum malaria, particularly from P. vivax, is an increasingly widespread disease that can cause severe or fatal illness, and the dependence on ACKR1-mediated invasion provides a prime therapeutic target (86).
A role for ACKR1 has been proposed in HIV pathogenesis, however the potential mechanisms of interaction are unclear. HIV uses chemokine receptors CXCR4 or CCR5 as co-receptors for targeting leukocytes, and the CCR5 inhibitor Maraviroc can successfully prevent binding by viral glycoproteins (87). Some studies have proposed ACKR1 is involved in HIV interactions with erythrocytes that promote infection of other blood cells or maintain a viral reservoir (88–90). However, the FyES phenotype was not confirmed to alter HIV susceptibility or disease progression (91, 92).
ACKR1 is also a target for Staphylococcus aureus toxins LukED and HlgAB (93). S. aureus bacteremia is particularly dangerous because these pore-forming, bicomponent toxin systems cause hemolysis and vascular leakage when they engage ACKR1 on red blood cells and endothelial junctions (94, 95). A crystal structure of the LukE toxin protein and the ACKR1 N-terminus resolved residues 34-46 of the receptor with Y41 stabilized in a lysine and arginine-enriched viral pocket, similar to the mechanism of interaction observed in the crystal structure of PvDBP and ACKR1 (96). Further analysis using time-resolved mass spectrometry and resonance energy transfer from a C-terminal bioluminescent tag suggests toxin binding may modulate receptor conformation to form ACKR1 homodimers and even alter interactions with intracellular Gαi1 subunits (97). Structure-guided strategies targeting ACKR1 could be useful to address pathogenicity mechanisms of significant infectious agents.
ACKR1 and pathoinflammation
Immune dysregulation involves an excess of chemokines and other soluble inflammatory mediators and can incur tissue damage from resultant immune cell infiltrates. Modulation of the chemokine network to treat autoimmune disease has yielded promising leads, but few have shown clinical effectiveness and safety (98, 99). Currently trials are ongoing for a CCR9 antagonist for Crohn’s disease and a CCR1 antagonist for rheumatoid arthritis (100, 101). Reparixin, an allosteric CXCR1 and CXCR2 blocker, did not progress past a phase 3 trial as a drug adjuvant for pancreatic islet allotransplantation to treat type 1 diabetes, but it is still a candidate for ongoing trials for metastatic breast cancer and COVID-19 related acute lung injury (102–104). Alternatively, blocking chemokines may decrease autoinflammation, and an antibody drug bertilimumab targeting CCL11 was designed to prevent eosinophil-mediated autoimmune damage in bullous pemphigoid skin disorder and inflammatory bowel disease (105, 106). Administration of anti-CXCL10 antibody was a promising strategy to limit cytotoxic T-cell liver damage, but clinical utility was hindered by continuous CXCL10 secretion and retention on endothelial cells (107, 108).
Controlling chemokine concentrations via ACKR1 could contribute to the success of these drug strategies or offer new avenues for regulating immune responses. ACKR1 regulation may contribute to resolution of chemokine-driven inflammation. ACKR1 binds chemokines at the inflamed synovial endothelium, and diminished expression of ACKR1 may be associated with rheumatoid arthritis (109). People with the FyES phenotype that decreases erythrocyte ACKR1 were observed to have increased IgE in serum samples and higher susceptibility for asthma (110). Knocking out all ACKR1 expression in an endotoxin-induced mouse model of inflammation was shown to increase lung and liver damage from granulocytic infiltrates (111). These studies support a protective role for ACKR1 by decreasing circulating chemokine levels, particularly through expression on erythrocytes.
However, ACKR1 on the endothelial surface may have separate functions in chemokine retention and has been observed to increase leukocyte recruitment and activity (112). Endothelial ACKR1 expression may potentiate respiratory distress, as seen in patients with suppurative pneumonia, and require balance from erythrocyte ACKR1 to avoid acute lung injury (113, 114). This finding has been reinforced in mouse models of lung inflammation, where studies show that ACKR1 knockout mice are protected from neutrophil-mediated tissue damage (115, 116). ACKR1 receptors supporting chemokine-mediated leukocyte infiltration have also been reported to contribute to patient lesions of giant cell/temporal arteritis and nephrotoxicity in a mouse model of renal failure (117, 118).
ACKR1 can also facilitate neutrophil reverse transendothelial migration and indirectly cause systemic inflammation (119). Using aged mice subjected to IL-1 stimulation, ACKR1 was shown to concentrate mast cell derived CXCL1 at endothelial junctions, causing desensitization of CXCR2 on circulating neutrophils and dysregulated chemotaxis. Without tight regulation of chemokine patterns, the activated neutrophils migrated to the lung leading to vascular leakage, which could be a targetable mechanism for aging-related inflammation or acute lung injury such as COVID-19 pneumonia (120, 121). An increase in ACKR1 expression was also detected in humoral and cellular rejection of renal allografts, but it remains unclear if upregulation is induced by an inflammatory program, or which component of graft rejection would be influenced (122, 123).
Chemokines are also important mediators of chronic inflammatory damage in cardiovascular disease, including atherosclerosis, where chemokine concentrations, combinations, and oligomerization all contribute to initiation and progression of vascular lesions (124). ACKR1 involvement and targeting to treat atherosclerosis was initially proposed because endothelial dysfunction and chemokines like CXCL8 immobilized on erythrocyte membranes contribute to plaque formation and coronary artery disease (125, 126). In an atherosclerosis mouse model, knocking out ACKR1 led to diminished plaque formation, cellular infiltrate in the vessel walls, and activation of macrophages (127). As the chemokine network is further studied in the context of cardiovascular diseases, ACKR1 binding inflammatory chemokines may become a relevant drug target. More detailed investigation is required to discern the role of ACKR1 in acute and chronic phases of inflammation and what changes in cellular immune responses may be feasible by targeting ACKR1.
Cancer angiogenesis, metastasis, prognostics
Therapeutic cancer interventions include drugs to attack primary tumors or alter pro-metastatic signals and biomarkers for prognostic screening. Chemokine patterning and chemokine receptor signaling are integral to the proliferation and spread of tumor cells (128). A challenge to targeting CKRs in cancer is that the same chemokines that stimulate tumor growth and neovascularization can also activate and direct tumor-killing immune cells. For example, CCL5 signaling through CCR5 supports recruitment of anti-tumor natural killer cells and cytotoxic T cells, but also stimulates pro-tumor, tissue-resident myeloid cells and lymphocytes (129). Nevertheless, the chemokine receptor drugs that have demonstrated promising anti-cancer activity in clinical trials, particularly antagonizing CCR2, CCR4, CXCR2, and CXCR4, emphasizes the importance of studying chemokine regulation and receptor mechanisms (130).
Neovascularization of an emerging tumor is an essential process to tumor growth and vascular access that involves distorting the balance of pro and anti-angiogenic chemokines (131). Angiogenesis is difficult to target because it can be triggered by tumor cells through an increase in CXCR2 agonism, or by a change in the cellular tumor infiltrate that favor tumor-associated macrophages (132). The mechanism of ACKR1 regulating pro-cancer chemokine signaling involves interplay between endothelial cells and erythrocytes that influences the activation of GPCRs CXCR2 and CXCR3. ACKR1 and the ACKR subfamily may balance chemokine abundance and patterning to benefit host immune cell recruitment that is lost in unregulated, aggressive cancer types (133, 134).
Studies show that when ACKR1 is expressed on malignant cells it is protective against tumor angiogenesis and subsequent metastasis. Proposed contributions of ACKR1 are shown in Figure 3. When transgenic ACKR1+ non-small cell lung cancer cells were implanted in SCID mice, the resulting tumors had decreased vascularization, and metastatic potential (135). Immunoassay for chemokines secreted by ACKR1+ tumor cells showed a decrease in CXCL5 and CXCL8, and chemokine detection suggested the chemokines were bound by ACKR1 and internalized or immobilized on the cell surface rather than removed from the tumor microenvironment. Another study injected mice with different cancer cell lines that expressed high or low levels of ACKR1 levels to show that cancer invasiveness was inversely related to ACKR1 activity (136). MDA-MB-231 breast adenocarcinoma cells were used to represent aggressive breast cancer with low endogenous ACKR1 expression, and MDA-MB-435 melanoma cells were used to model an ACKR1-expressing tumor (137, 138). Testing in either cell culture or the tumor xenografts showed that ACKR1 expression could prevent the spike of CCL2 and CXCL8 released into the growth media or tumor microenvironment. These findings were correlated with a breast cancer clinical cohort, where patients with higher levels of detectable ACKR1 had less invasive cancers and lower mortality rates. Altering the global ACKR1 expression also changes the tumor microenvironment. ACKR1 global knockout in a spontaneous murine prostate cancer model resulted in less dense, more necrotic tumors with increased intratumor concentrations of CXCL1 and CXCL2 (139). Overexpression of the endothelial ACKR1 in mice implanted with melanoma tumors demonstrated inhibition of tumor growth and vascularity and showed an increase in CD4+ and CD8+ T-cell and macrophage infiltration (140).
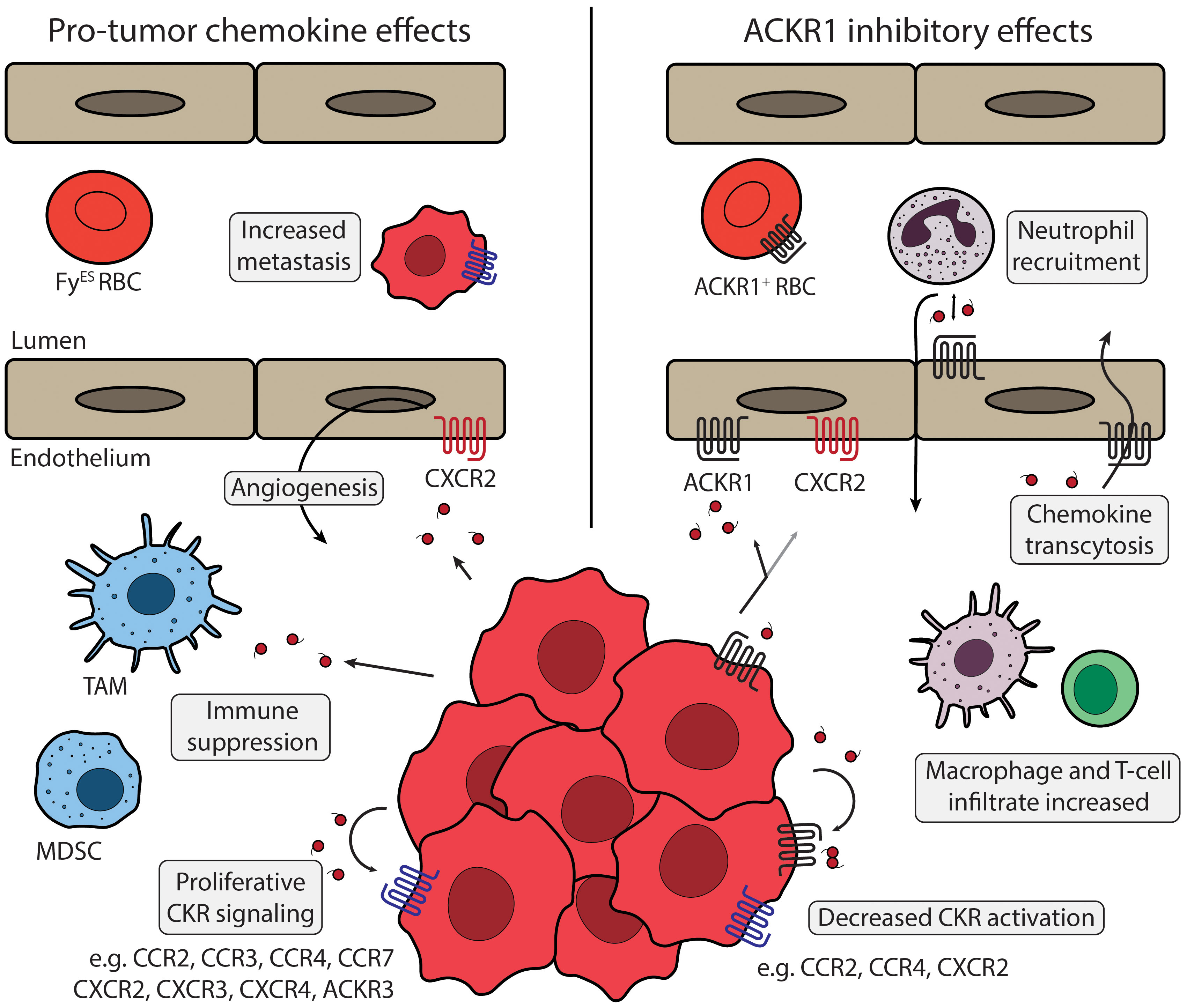
Figure 3 ACKR1 and tumor microenvironment Chemokine signaling in the tumor microenvironment is regulated by ACKR1 expression. Left panel describes chemokine effects that promote tumor phenotypes. ACKR1 (black) expression can be diminished on tumor cells or by the FyES polymorphism. Angiogenesis can be triggered by chemokines secreted from TAMs, stromal cells, or by cancer cells themselves via activation of endothelial CXCR2 (red). Cancer cells release numerous chemokines, including CCL2, CCL5, CXCL8, and others that can act to suppress anti-tumor immunity. Various cancer types express a panel of CKRs (blue) including CCR1, CCR2, CXCR2, CXCR4, and others that support tumor proliferation and metastasis. Primary tumors can silence expression of chemokines like CXCL12 and increase expression of CKRs like CXCR4 to promote metastasis. Right panel shows proposed mechanisms of ACKR1 regulation. ACKR1 receptors on erythrocytes can act as a sink to buffer chemokine levels and may have interactions with ACKR1 expressed on endothelial cells. ACKR1 enrichment at endothelial junctions promotes neutrophil diapedesis via CXCL1 and CXCL2 exchange, and increased endothelial ACKR1 improves recruitment of macrophages, CD4+ and CD8+ T-cells. Expression of ACKR1 in cancer models or patient tumor samples has been shown to modulate CCL2 and CXCL8, ligands of CCR2, CCR4, and CXCR2. ACKR1 modulates many chemokines and regulation of multiple CKRs may contribute to the improved clinical outcomes observed. TAM, Tumor associated macrophage; MDSC, Myeloid-derived suppressor cell; FyES RBC, “Erythroid-silent” erythrocyte; CKR, chemokine receptor.
Angiogenesis is a continual process in healthy tissue that involves migration, proliferation, and differentiation and ACKR1 could influence feedback mechanisms triggered by CXCR2 signaling pathways. A study investigated how ACKR1 expression on non-malignant endothelial cells could decrease capillary formation and detected an upregulation of senescence biomarkers (141). In pancreatic cancer cells lines, co-expression of ACKR1 in CXCR2+ tumors was sufficient to inhibit CXCL8-triggered activation of STAT3 and mediators of epithelial-mesenchymal transition (142, 143). Blocking these oncogenic pathways is an important strategy to induce cellular senescence and restore the anti-tumor effects of immune defenses (144, 145). CXCR2 has a complex role in tumor progression, as receptor overstimulation and autocrine activation may also trigger and sustain a p53-mediated cellular senescence (146). Furthermore, it is possible ACKR1 could contribute cell cycle regulation through other interactions including the tumor suppressor CD82/KAI1, a multifunctional surface tetraspanin. A study found that CD82+ cancer cells have increased adherence to ACKR1+ vascular endothelial cells and suggested that a direct interaction leads to p21 cyclin-dependent kinase inhibition and prevention of metastatic escape (147). A follow-up study also detected p21 upregulation connected to CD82 and potentially ACKR1, and implied that CD82 opposes CXCL8 effects by downregulating secretion from tumors and displacing CXCL8 from endothelial ACKR1 (148). The data interpretation from these reports is limited without testing CXCR2 signaling or reliable antibody detection of ACKR1.
Another important target of anti-cancer therapeutics is metastasis, the major cause of cancer mortality (149). Blocking chemokine signaling is an appealing strategy because metastatic invasion of susceptible cellular niches is inefficient without chemokine-directed migration and often characterized by chemotactic GPCR overexpression (150). ACKR1 may play a role in fine-tuning the complex chemokine patterns that are hijacked by migrating cancer cells. Many of the studies that observed an inverse correlation between the proliferative potential of primary tumors and ACKR1 expression also reported a decrease in metastatic phenotype. Another possible mechanism is alteration of the chemokine oligomeric equilibrium. Chemokine dimers elicit distinct signaling from monomeric chemokines, potentially representing feedback inhibition that could be used as an antimetastatic cue (5, 151, 152). Multiple factors increase the propensity of chemokine dimerization, including GAGs and interactions with the N-termini of GPCRs (153, 154). ACKR1 also shows similar activity by binding preferentially to the dimeric form of CXCL12 (155). Improved quantitation of chemokine concentrations in different cellular compartments and the relation between dimerization and chemotaxis are needed to predict the effects of ACKR1 preferentially binding certain chemokines as dimers.
Testing ACKR1 genotype and expression in tumor biopsies may be a clinically useful cancer biomarker. Multiple studies have indicated that higher ACKR1 expression levels in breast cancer tumors improve relapse-free patient survival, while loss of ACKR1 expression, frequently in patients with African ancestry, is an indicator of increased tumor aggressiveness, metastatic propensity, and mortality (156–162). Detailed analysis is warranted for different cancer types, since comparing prostate cancer incidence within patient groups did not detect a strong correlation between the FyES polymorphism and increased cancer risk (163, 164). Additionally, blood typing to discern ACKR1 phenotype could be an effective, low-cost way to inform cancer treatment. ACKR1-mediated DANC neutropenia affects patient care by impeding administration of drugs like clozapine or azathioprine and leading to potentially unwarranted bone marrow biopsies (165–167). Patients with FyES phenotype are at increased risk of side effects from chemotherapy but using the same neutropenic cutoff values may unnecessarily delay initiation and prolong duration of cancer treatment (168–172). Adapting standard of care for patients with DANC could provide an opportunity to address disparate treatment outcomes with a precision medicine approach. Overall, a cancer-protective role for ACKR1 is supported by cell culture, mouse models, and genetic associations, and independent anti-angiogenic properties for endothelial, erythroid, and tumor ACKR1 expression can contribute to improved patient outcomes.
Discussion
ACKR1 exhibits favorable structural features, expression profile, and biological activity for development of therapeutic interventions. More investigation is needed to determine the extent of control over chemokine scaffolding by ACKR1 that can be attained by different classes of molecules. Antibodies binding to different ACKR1 epitopes do not uniformly inhibit chemokine binding, suggesting some capacity to alter ACKR1 specificity. Development of screening readouts for binding that can supplement competition assays will facilitate identification of small molecules. The independence of chemokine-binding and DBP recognition sites located in the extended N-terminus indicates that this domain could be isolated to provide an effective ACKR1 decoy, similar to a strategy proposed for the ACKR2 N-terminus. The positioning and functions of ACKR1 receptors in the hematopoietic compartment, on the surface of erythrocytes, and at the junctions of endothelial regions specialized for cell trafficking provide an opportunity to control immune cell migration into tissues. Additionally, further exploration of the impact of ACKR1 expressed at the blood-brain barrier and on different neuronal cell types may reveal a targetable role in regulating neuroinflammation. Still, the mechanisms of ACKR1 retaining or sequestering different chemokines have yet to be elucidated in detail, particularly in the context of the tumor microenvironment. Assigning ACKR1 expression to specific cell types within and around tumors of different origins will be needed to understand the correlation observed in experimental models between ACKR1 expression and decreased malignant phenotypes.
Targeting ACKR1 is an appealing approach for new compounds that modulate chemokine biology without interfering with the chemokine sensitivity and signaling functions of immune cell CKRs. ACKR1 in circulation is only reliably found in post-capillary venules and erythrocytes rather than myeloid or lymphoid cells, suggesting targeting ACKR1 would not directly impact immune effector function. While some studies report ACKR1 detection on other cells like bone marrow macrophages, these reports use a polyclonal antibody which has been shown to recognize non-ACKR1 surface markers (173, 174). Furthermore, unlike the other ACKRs, ACKR1 functions seem independent of G-protein or β-arrestin signaling pathways (175). The restricted tissue and signaling capabilities suggest side effects of ACKR1 inhibition may be modest compared to the signaling GPCRs or other ACKRs. As ACKR1 biology and molecular pharmacology are examined in greater detail, development of new ligands to alter its function will be useful as research tools and may enable amelioration of specific disease pathologies.
Current opportunities for intervention should include shielding extracellular ACKR1 residues from virulence factors of important human pathogens. This approach may have multiple benefits, including preventing erythrocytic replication of Plasmodia and maintaining the integrity of endothelial junctions during S. aureus infections. Additionally, animal models, cancer cell experiments, ACKR1 biochemistry, and meta-analysis of clinical cohorts all indicate ACKR1 activity impedes cancer progression. This underscores the importance of elucidating ACKR1 chemokine-binding mechanisms and the impact on immune cell responses to tumors to take steps towards enhancement or reconstitution of ACKR1-mediated protection in cancer therapy. Until then, ACKR1 may be used as a prognostic indicator for the aggressiveness of different cancer types and may be inform treatment regimens for patients with different patterns of ACKR1 expression. The next steps include detailing the binding interactions of different chemokines to ACKR1 and the mechanisms that alter receptor expression and enable chemokine trafficking through cells. Future development and implementation of therapeutics that target the chemokine network should consider the role of ACKR1 in patient physiology and the possibility of targeting ACKR1 itself.
Author contributions
Manuscript written and edited by KC and BV. All authors contributed to the article and approved the submitted version.
Funding
This study was funded by NIH R37 AI058072. KC was funded by MCW Center for Immunology and MSTP NIGMS T32-GM080202.
Conflict of interest
Author BV has ownership interests in Protein Foundry, LLC and XLock Biosciences, LLC.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Miller MC, Mayo KH. Chemokines from a structural perspective. Int J Mol Sci (2017) 18(10). doi: 10.3390/ijms18102088
2. Amarandi RM, Hjorto GM, Rosenkilde MM, Karlshoj S. Probing biased signaling in chemokine receptors. Methods Enzymol (2016) 570:155–86. doi: 10.1016/bs.mie.2015.09.001
3. Hughes CE, Nibbs RJB. A guide to chemokines and their receptors. FEBS J (2018) 285(16):2944–71. doi: 10.1111/febs.14466
4. Stone MJ, Hayward JA, Huang C, EH Z, Sanchez J. Mechanisms of regulation of the chemokine-receptor network. Int J Mol Sci (2017) 18(2). doi: 10.3390/ijms18020342
5. Lau S, Feitzinger A, Venkiteswaran G, Wang J, Lewellis SW, Koplinski CA, et al. A negative-feedback loop maintains optimal chemokine concentrations for directional cell migration. Nat Cell Biol (2020) 22(3):266–73. doi: 10.1038/s41556-020-0465-4
6. Sriram K, Insel PA. G Protein-coupled receptors as targets for approved drugs: How many targets and how many drugs? Mol Pharmacol (2018) 93(4):251–8. doi: 10.1124/mol.117.111062
7. Solari R, Pease JE, Begg M. Chemokine receptors as therapeutic targets: Why aren't there more drugs? Eur J Pharmacol (2015) 746:363–7. doi: 10.1016/j.ejphar.2014.06.060
8. Fra AM, Locati M, Otero K, Sironi M, Signorelli P, Massardi ML, et al. Cutting edge: scavenging of inflammatory CC chemokines by the promiscuous putatively silent chemokine receptor D6. J Immunol (2003) 170(5):2279–82. doi: 10.4049/jimmunol.170.5.2279
9. Chevigne A, Janji B, Meyrath M, Reynders N, D'Uonnolo G, Uchanski T, et al. CXCL10 is an agonist of the CC family chemokine scavenger receptor ACKR2/D6. Cancers (Basel) (2021) 13(5). doi: 10.3390/cancers13051054
10. Sjoberg E, Meyrath M, Milde L, Herrera M, Lovrot J, Hagerstrand D, et al. A novel ACKR2-dependent role of fibroblast-derived CXCL14 in epithelial-to-Mesenchymal transition and metastasis of breast cancer. Clin Cancer Res (2019) 25(12):3702–17. doi: 10.1158/1078-0432.CCR-18-1294
11. Teoh PJ, Menzies FM, Hansell CA, Clarke M, Waddell C, Burton GJ, et al. Atypical chemokine receptor ACKR2 mediates chemokine scavenging by primary human trophoblasts and can regulate fetal growth, placental structure, and neonatal mortality in mice. J Immunol (2014) 193(10):5218–28. doi: 10.4049/jimmunol.1401096
12. Nibbs RJ, Kriehuber E, Ponath PD, Parent D, Qin S, Campbell JD, et al. The beta-chemokine receptor D6 is expressed by lymphatic endothelium and a subset of vascular tumors. Am J Pathol (2001) 158(3):867–77. doi: 10.1016/S0002-9440(10)64035-7
13. McKimmie CS, Fraser AR, Hansell C, Gutierrez L, Philipsen S, Connell L, et al. Hemopoietic cell expression of the chemokine decoy receptor D6 is dynamic and regulated by GATA1. J Immunol (2008) 181(5):3353–63. doi: 10.4049/jimmunol.181.5.3353
14. Vacchini A, Cancellieri C, Milanesi S, Badanai S, Savino B, Bifari F, et al. Control of cytoskeletal dynamics by beta-Arrestin1/Myosin vb signaling regulates endosomal sorting and scavenging activity of the atypical chemokine receptor ACKR2. Vaccines (Basel) (2020) 8(3). doi: 10.3390/vaccines8030542
15. Meyrath M, Szpakowska M, Zeiner J, Massotte L, Merz MP, Benkel T, et al. The atypical chemokine receptor ACKR3/CXCR7 is a broad-spectrum scavenger for opioid peptides. Nat Commun (2020) 11(1):3033. doi: 10.1038/s41467-020-16664-0
16. Szpakowska M, Dupuis N, Baragli A, Counson M, Hanson J, Piette J, et al. Human herpesvirus 8-encoded chemokine vCCL2/vMIP-II is an agonist of the atypical chemokine receptor ACKR3/CXCR7. Biochem Pharmacol (2016) 114:14–21. doi: 10.1016/j.bcp.2016.05.012
17. Saaber F, Schutz D, Miess E, Abe P, Desikan S, Ashok Kumar P, et al. ACKR3 regulation of neuronal migration requires ACKR3 phosphorylation, but not beta-arrestin. Cell Rep (2019) 26(6):1473–88 e9. doi: 10.1016/j.celrep.2019.01.049
18. Zarca A, Perez C, van den Bor J, Bebelman JP, Heuninck J, de Jonker RJF, et al. Differential involvement of ACKR3 c-tail in beta-arrestin recruitment, trafficking and internalization. Cells (2021) 10(3). doi: 10.3390/cells10030618
19. Koenen J, Bachelerie F, Balabanian K, Schlecht-Louf G, Gallego C. Atypical chemokine receptor 3 (ACKR3): A comprehensive overview of its expression and potential roles in the immune system. Mol Pharmacol (2019) 96(6):809–18. doi: 10.1124/mol.118.115329
20. Meyrath M, Reynders N, Uchanski T, Chevigne A, Szpakowska M. Systematic reassessment of chemokine-receptor pairings confirms CCL20 but not CXCL13 and extends the spectrum of ACKR4 agonists to CCL22. J Leukoc Biol (2021) 109(2):373–6. doi: 10.1002/JLB.2AB0520-275R
21. Werth K, Hub E, Gutjahr JC, Bosjnak B, Zheng X, Bubke A, et al. Expression of ACKR4 demarcates the "peri-marginal sinus," a specialized vascular compartment of the splenic red pulp. Cell Rep (2021) 36(2):109346. doi: 10.1016/j.celrep.2021.109346
22. Bastow CR, Bunting MD, Kara EE, McKenzie DR, Caon A, Devi S, et al. Scavenging of soluble and immobilized CCL21 by ACKR4 regulates peripheral dendritic cell emigration. Proc Natl Acad Sci USA (2021) 118(17):e2025763118. doi: 10.1073/pnas.2025763118
23. Thomson CA, van de Pavert SA, Stakenborg M, Labeeuw E, Matteoli G, Mowat AM, et al. Expression of the atypical chemokine receptor ACKR4 identifies a novel population of intestinal submucosal fibroblasts that preferentially expresses endothelial cell regulators. J Immunol (2018) 201(1):215–29. doi: 10.4049/jimmunol.1700967
24. Matti C, Salnikov A, Artinger M, D'Agostino G, Kindinger I, Uguccioni M, et al. ACKR4 recruits GRK3 prior to beta-arrestins but can scavenge chemokines in the absence of beta-arrestins. Front Immunol (2020) 11:720. doi: 10.3389/fimmu.2020.00720
25. Chen J, Yao Y, Gong C, Yu F, Su S, Chen J, et al. CCL18 from tumor-associated macrophages promotes breast cancer metastasis via PITPNM3. Cancer Cell (2011) 19(4):541–55. doi: 10.1016/j.ccr.2011.02.006
26. Del Prete A, Bonecchi R, Vecchi A, Mantovani A, Sozzani S. CCRL2, a fringe member of the atypical chemoattractant receptor family. Eur J Immunol (2013) 43(6):1418–22. doi: 10.1002/eji.201243179
27. Cutbush M, Mollison PL. The Duffy blood group system. Heredity (Edinb). (1950) 4(3):383–9. doi: 10.1038/hdy.1950.31
28. Hoher G, Fiegenbaum M, Almeida S. Molecular basis of the Duffy blood group system. Blood Transfus. (2018) 16(1):93–100. doi: 10.2450/2017.0119-16
29. Iwamoto S, Li J, Omi T, Ikemoto S, Kajii E. Identification of a novel exon and spliced form of Duffy mRNA that is the predominant transcript in both erythroid and postcapillary venule endothelium. Blood (1996) 87(1):378–85. doi: 10.1182/blood.V87.1.378.378
30. McManus KF, Taravella AM, Henn BM, Bustamante CD, Sikora M, Cornejo OE. Population genetic analysis of the DARC locus (Duffy) reveals adaptation from standing variation associated with malaria resistance in humans. PloS Genet (2017) 13(3):e1006560. doi: 10.1371/journal.pgen.1006560
31. Chaudhuri A, Polyakova J, Zbrzezna V, Williams K, Gulati S, Pogo AO. Cloning of glycoprotein d cDNA, which encodes the major subunit of the Duffy blood group system and the receptor for the plasmodium vivax malaria parasite. Proc Natl Acad Sci U S A. (1993) 90(22):10793–7. doi: 10.1073/pnas.90.22.10793
32. Tournamille C, Colin Y, Cartron JP, Le Van Kim C. Disruption of a GATA motif in the Duffy gene promoter abolishes erythroid gene expression in Duffy-negative individuals. Nat Genet (1995) 10(2):224–8. doi: 10.1038/ng0695-224
33. Liu W, Li Y, Shaw KS, Learn GH, Plenderleith LJ, Malenke JA, et al. African Origin of the malaria parasite plasmodium vivax. Nat Commun (2014) 5:3346. doi: 10.1038/ncomms4346
34. Howes RE, Patil AP, Piel FB, Nyangiri OA, Kabaria CW, Gething PW, et al. The global distribution of the Duffy blood group. Nat Commun (2011) 2:266. doi: 10.1038/ncomms1265
35. King CL, Adams JH, Xianli J, Grimberg BT, McHenry AM, Greenberg LJ, et al. Fy(a)/Fy(b) antigen polymorphism in human erythrocyte Duffy antigen affects susceptibility to plasmodium vivax malaria. Proc Natl Acad Sci U S A. (2011) 108(50):20113–8. doi: 10.1073/pnas.1109621108
36. Tournamille C, Blancher A, Le Van Kim C, Gane P, Apoil PA, Nakamoto W, et al. Sequence, evolution and ligand binding properties of mammalian Duffy antigen/receptor for chemokines. Immunogenetics (2004) 55(10):682–94. doi: 10.1007/s00251-003-0633-2
37. Gassner C, Kraus RL, Dovc T, Kilga-Nogler S, Utz I, Mueller TH, et al. Fyx is associated with two missense point mutations in its gene and can be detected by PCR-SSP. Immunohematology (2000) 16(2):61–7. doi: 10.21307/immunohematology-2019-579
38. Tournamille C, Le Van Kim C, Gane P, Le Pennec PY, Roubinet F, Babinet J, et al. Arg89Cys substitution results in very low membrane expression of the Duffy antigen/receptor for chemokines in fy(x) individuals. Blood (1998) 92(6):2147–56. doi: 10.1182/blood.V92.6.2147
39. Ansart-Pirenne H, Martin-Blanc S, Le Pennec PY, Rouger P, Cartron JP, Tournamille C. FY*X real-time polymerase chain reaction with melting curve analysis associated with a complete one-step real-time FY genotyping. Vox Sang. (2007) 92(2):142–7. doi: 10.1111/j.1423-0410.2006.00872.x
40. Van Alsten SC, Aversa JG, Santo L, Camargo MC, Kemp T, Liu J, et al. Association between ABO and Duffy blood types and circulating chemokines and cytokines. Genes Immun (2021) 22(3):161–71. doi: 10.1038/s41435-021-00137-5
41. Jilma-Stohlawetz P, Homoncik M, Drucker C, Marsik C, Rot A, Mayr WR, et al. Fy phenotype and gender determine plasma levels of monocyte chemotactic protein. Transfusion (2001) 41(3):378–81. doi: 10.1046/j.1537-2995.2001.41030378.x
42. Nygaard R, Frimurer TM, Holst B, Rosenkilde MM, Schwartz TW. Ligand binding and micro-switches in 7TM receptor structures. Trends Pharmacol Sci (2009) 30(5):249–59. doi: 10.1016/j.tips.2009.02.006
43. Lagana M, Schlecht-Louf G, Bachelerie F. The G protein-coupled receptor kinases (GRKs) in chemokine receptor-mediated immune cell migration: From molecular cues to physiopathology. Cells (2021) 10(1). doi: 10.3390/cells10010075
44. Arimont M, Sun SL, Leurs R, Smit M, de Esch IJP, de Graaf C. Structural analysis of chemokine receptor-ligand interactions. J Med Chem (2017) 60(12):4735–79. doi: 10.1021/acs.jmedchem.6b01309
45. Nomiyama H, Yoshie O. Functional roles of evolutionary conserved motifs and residues in vertebrate chemokine receptors. J Leukoc Biol (2015) 97(1):39–47. doi: 10.1189/jlb.2RU0614-290R
46. Chakera A, Seeber RM, John AE, Eidne KA, Greaves DR. The duffy antigen/receptor for chemokines exists in an oligomeric form in living cells and functionally antagonizes CCR5 signaling through hetero-oligomerization. Mol Pharmacol (2008) 73(5):1362–70. doi: 10.1124/mol.107.040915
47. Gustavsson M, Dyer DP, Zhao C, Handel TM. Kinetics of CXCL12 binding to atypical chemokine receptor 3 reveal a role for the receptor n terminus in chemokine binding. Sci Signal (2019) 12(598):eaaw3657. doi: 10.1126/scisignal.aaw3657
48. Hewit KD, Fraser A, Nibbs RJ, Graham GJ. The n-terminal region of the atypical chemokine receptor ACKR2 is a key determinant of ligand binding. J Biol Chem (2014) 289(18):12330–42. doi: 10.1074/jbc.M113.534545
49. de Brevern AG, Autin L, Colin Y, Bertrand O, Etchebest C. In silico studies on DARC. Infect Disord Drug Targets (2009) 9(3):289–303. doi: 10.2174/1871526510909030289
50. de Brevern AG, Wong H, Tournamille C, Colin Y, Le Van Kim C, Etchebest C. A structural model of a seven-transmembrane helix receptor: the Duffy antigen/receptor for chemokine (DARC). Biochim Biophys Acta (2005) 1724(3):288–306. doi: 10.1016/j.bbagen.2005.05.016
51. Smolarek D, Hattab C, Hassanzadeh-Ghassabeh G, Cochet S, Gutierrez C, de Brevern AG, et al. A recombinant dromedary antibody fragment (VHH or nanobody) directed against human Duffy antigen receptor for chemokines. Cell Mol Life Sci (2010) 67(19):3371–87. doi: 10.1007/s00018-010-0387-6
52. Tournamille C, Le Van Kim C, Gane P, Blanchard D, Proudfoot AE, Cartron JP, et al. Close association of the first and fourth extracellular domains of the Duffy antigen/receptor for chemokines by a disulfide bond is required for ligand binding. J Biol Chem (1997) 272(26):16274–80. doi: 10.1074/jbc.272.26.16274
53. Horuk R, Martin A, Hesselgesser J, Hadley T, Lu ZH, Wang ZX, et al. The Duffy antigen receptor for chemokines: structural analysis and expression in the brain. J Leukoc Biol (1996) 59(1):29–38. doi: 10.1002/jlb.59.1.29
54. Strieter RM, Polverini PJ, Kunkel SL, Arenberg DA, Burdick MD, Kasper J, et al. The functional role of the ELR motif in CXC chemokine-mediated angiogenesis. J Biol Chem (1995) 270(45):27348–57. doi: 10.1074/jbc.270.45.27348
55. Szabo MC, Soo KS, Zlotnik A, Schall TJ. Chemokine class differences in binding to the Duffy antigen-erythrocyte chemokine receptor. J Biol Chem (1995) 270(43):25348–51. doi: 10.1074/jbc.270.43.25348
56. Stamatovic SM, Keep RF, Mostarica-Stojkovic M, Andjelkovic AV. CCL2 regulates angiogenesis via activation of ets-1 transcription factor. J Immunol (2006) 177(4):2651–61. doi: 10.4049/jimmunol.177.4.2651
57. Salcedo R, Oppenheim JJ. Role of chemokines in angiogenesis: CXCL12/SDF-1 and CXCR4 interaction, a key regulator of endothelial cell responses. Microcirculation (2003) 10(3-4):359–70. doi: 10.1080/mic.10.3-4.359.370
58. Du J, Luan J, Liu H, Daniel TO, Peiper S, Chen TS, et al. Potential role for Duffy antigen chemokine-binding protein in angiogenesis and maintenance of homeostasis in response to stress. J Leukoc Biol (2002) 71(1):141–53. doi: 10.1189/jlb.71.1.141
59. Gardner L, Patterson AM, Ashton BA, Stone MA, Middleton J. The human Duffy antigen binds selected inflammatory but not homeostatic chemokines. Biochem Biophys Res Commun (2004) 321(2):306–12. doi: 10.1016/j.bbrc.2004.06.146
60. Klei TRL, Aglialoro F, Mul FPJ, Tol S, Ligthart PC, Seignette IM, et al. Differential interaction between DARC and SDF-1 on erythrocytes and their precursors. Sci Rep (2019) 9(1):16245. doi: 10.1038/s41598-019-52186-6
61. Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol (2014) 32:659–702. doi: 10.1146/annurev-immunol-032713-120145
62. Kashiwazaki M, Tanaka T, Kanda H, Ebisuno Y, Izawa D, Fukuma N, et al. A high endothelial venule-expressing promiscuous chemokine receptor DARC can bind inflammatory, but not lymphoid, chemokines and is dispensable for lymphocyte homing under physiological conditions. Int Immunol (2003) 15(10):1219–27. doi: 10.1093/intimm/dxg121
63. Duchene J, Novitzky-Basso I, Thiriot A, Casanova-Acebes M, Bianchini M, Etheridge SL, et al. Atypical chemokine receptor 1 on nucleated erythroid cells regulates hematopoiesis. Nat Immunol (2017) 18(7):753–61. doi: 10.1038/ni.3763
64. Reich D, Nalls MA, Kao WH, Akylbekova EL, Tandon A, Patterson N, et al. Reduced neutrophil count in people of African descent is due to a regulatory variant in the Duffy antigen receptor for chemokines gene. PloS Genet (2009) 5(1):e1000360. doi: 10.1371/journal.pgen.1000360
65. Palmblad J, Hoglund P. Ethnic benign neutropenia: A phenomenon finds an explanation. Pediatr Blood Cancer. (2018) 65(12):e27361. doi: 10.1002/pbc.27361
66. Merz LE, Story CM, Osei MA, Ren S, Park HS, Jolley K, et al. Absolute neutrophil count by Duffy status among healthy black and African American adults. Blood Adv (2023) 7(3):317–20. doi: 10.1182/bloodadvances.2022007679
67. Rappoport N, Simon AJ, Amariglio N, Rechavi G. The Duffy antigen receptor for chemokines, ACKR1,- 'Jeanne DARC' of benign neutropenia. Br J Haematol (2019) 184(4):497–507. doi: 10.1111/bjh.15730
68. Ortiz MV, Meier ER, Hsieh MM. Identification and clinical characterization of children with benign ethnic neutropenia. J Pediatr Hematol Oncol (2016) 38(3):e140-3. doi: 10.1097/MPH.0000000000000528
69. Hadley TJ, Lu ZH, Wasniowska K, Martin AW, Peiper SC, Hesselgesser J, et al. Postcapillary venule endothelial cells in kidney express a multispecific chemokine receptor that is structurally and functionally identical to the erythroid isoform, which is the Duffy blood group antigen. J Clin Invest. (1994) 94(3):985–91. doi: 10.1172/JCI117465
70. Rot A. In situ binding assay for studying chemokine interactions with endothelial cells. J Immunol Methods (2003) 273(1-2):63–71. doi: 10.1016/S0022-1759(02)00502-1
71. Thiriot A, Perdomo C, Cheng G, Novitzky-Basso I, McArdle S, Kishimoto JK, et al. Differential DARC/ACKR1 expression distinguishes venular from non-venular endothelial cells in murine tissues. BMC Biol (2017) 15(1):45. doi: 10.1186/s12915-017-0381-7
72. Rot A. Contribution of Duffy antigen to chemokine function. Cytokine Growth Factor Rev (2005) 16(6):687–94. doi: 10.1016/j.cytogfr.2005.05.011
73. Middleton J, Neil S, Wintle J, Clark-Lewis I, Moore H, Lam C, et al. Transcytosis and surface presentation of IL-8 by venular endothelial cells. Cell (1997) 91(3):385–95. doi: 10.1016/S0092-8674(00)80422-5
74. Middleton J, Patterson AM, Gardner L, Schmutz C, Ashton BA. Leukocyte extravasation: chemokine transport and presentation by the endothelium. Blood (2002) 100(12):3853–60. doi: 10.1182/blood.V100.12.3853
75. Pruenster M, Mudde L, Bombosi P, Dimitrova S, Zsak M, Middleton J, et al. The Duffy antigen receptor for chemokines transports chemokines and supports their promigratory activity. Nat Immunol (2009) 10(1):101–8. doi: 10.1038/ni.1675
76. Zhao Y, Mangalmurti NS, Xiong Z, Prakash B, Guo F, Stolz DB, et al. Duffy Antigen receptor for chemokines mediates chemokine endocytosis through a macropinocytosis-like process in endothelial cells. PloS One (2011) 6(12):e29624. doi: 10.1371/journal.pone.0029624
77. Girbl T, Lenn T, Perez L, Rolas L, Barkaway A, Thiriot A, et al. Distinct compartmentalization of the chemokines CXCL1 and CXCL2 and the atypical receptor ACKR1 determine discrete stages of neutrophil diapedesis. Immunity (2018) 49(6):1062–76 e6. doi: 10.1016/j.immuni.2018.09.018
78. Minten C, Alt C, Gentner M, Frei E, Deutsch U, Lyck R, et al. DARC shuttles inflammatory chemokines across the blood-brain barrier during autoimmune central nervous system inflammation. Brain (2014) 137(Pt 5):1454–69. doi: 10.1093/brain/awu045
79. Marchetti L, Francisco D, Soldati S, Haghayegh Jahromi N, Barcos S, Gruber I, et al. ACKR1 favors transcellular over paracellular T-cell diapedesis across the blood-brain barrier in neuroinflammation in vitro. Eur J Immunol (2022) 52(1):161–77. doi: 10.1002/eji.202149238
80. Schneider EH, Fowler SC, Lionakis MS, Swamydas M, Holmes G, Diaz V, et al. Regulation of motor function and behavior by atypical chemokine receptor 1. Behav Genet (2014) 44(5):498–515. doi: 10.1007/s10519-014-9665-7
81. Batchelor JD, Zahm JA, Tolia NH. Dimerization of plasmodium vivax DBP is induced upon receptor binding and drives recognition of DARC. Nat Struct Mol Biol (2011) 18(8):908–14. doi: 10.1038/nsmb.2088
82. Hesselgesser J, Chitnis CE, Miller LH, Yansura DG, Simmons LC, Fairbrother WJ, et al. A mutant of melanoma growth stimulating activity does not activate neutrophils but blocks erythrocyte invasion by malaria. J Biol Chem (1995) 270(19):11472–6. doi: 10.1074/jbc.270.19.11472
83. Urusova D, Carias L, Huang Y, Nicolete VC, Popovici J, Roesch C, et al. Structural basis for neutralization of plasmodium vivax by naturally acquired human antibodies that target DBP. Nat Microbiol (2019) 4(9):1486–96. doi: 10.1038/s41564-019-0461-2
84. Batchelor JD, Malpede BM, Omattage NS, DeKoster GT, Henzler-Wildman KA, Tolia NH. Red blood cell invasion by plasmodium vivax: structural basis for DBP engagement of DARC. PloS Pathog (2014) 10(1):e1003869. doi: 10.1371/journal.ppat.1003869
85. Tobin AR, Crow R, Urusova DV, Klima JC, Tolia NH, Strauch E-M. Inhibition of a malaria host-pathogen interaction by a computationally designed inhibitor. Protein Sci (2023) 32(1):e4507. doi: 10.1002/pro.4507
86. Howes RE, Battle KE, Mendis KN, Smith DL, Cibulskis RE, Baird JK, et al. Global epidemiology of plasmodium vivax. Am J Trop Med Hyg (2016) 95(6 Suppl):15–34. doi: 10.4269/ajtmh.16-0141
87. Tan Q, Zhu Y, Li J, Chen Z, Han GW, Kufareva I, et al. Structure of the CCR5 chemokine receptor-HIV entry inhibitor maraviroc complex. Science (2013) 341(6152):1387–90. doi: 10.1126/science.1241475
88. Lachgar A, Jaureguiberry G, Le Buenac H, Bizzini B, Zagury JF, Rappaport J, et al. Binding of HIV-1 to RBCs involves the Duffy antigen receptors for chemokines (DARC). BioMed Pharmacother. (1998) 52(10):436–9. doi: 10.1016/S0753-3322(99)80021-3
89. He W, Neil S, Kulkarni H, Wright E, Agan BK, Marconi VC, et al. Duffy Antigen receptor for chemokines mediates trans-infection of HIV-1 from red blood cells to target cells and affects HIV-AIDS susceptibility. Cell Host Microbe (2008) 4(1):52–62. doi: 10.1016/j.chom.2008.06.002
90. Kulkarni H, Marconi VC, He W, Landrum ML, Okulicz JF, Delmar J, et al. The Duffy-null state is associated with a survival advantage in leukopenic HIV-infected persons of African ancestry. Blood (2009) 114(13):2783–92. doi: 10.1182/blood-2009-04-215186
91. Julg B, Reddy S, van der Stok M, Kulkarni S, Qi Y, Bass S, et al. Lack of Duffy antigen receptor for chemokines: no influence on HIV disease progression in an African treatment-naive population. Cell Host Microbe (2009) 5(5):413–5. doi: 10.1016/j.chom.2009.04.009
92. Mpofu R, Otwombe K, Mlisana K, Nchabeleng M, Allen M, Kublin J, et al. Benign ethnic neutropenia in a south African population, and its association with HIV acquisition and adverse event reporting in an HIV vaccine clinical trial. PloS One (2021) 16(1):e0241708. doi: 10.1371/journal.pone.0241708
93. Spaan AN, Reyes-Robles T, Badiou C, Cochet S, Boguslawski KM, Yoong P, et al. Staphylococcus aureus targets the Duffy antigen receptor for chemokines (DARC) to lyse erythrocytes. Cell Host Microbe (2015) 18(3):363–70. doi: 10.1016/j.chom.2015.08.001
94. Lubkin A, Lee WL, Alonzo F 3rd, Wang C, Aligo J, Keller M, et al. Staphylococcus aureus leukocidins target endothelial DARC to cause lethality in mice. Cell Host Microbe (2019) 25(3):463–70 e9. doi: 10.1016/j.chom.2019.01.015
95. Spaan AN, van Strijp JAG, Torres VJ. Leukocidins: staphylococcal bi-component pore-forming toxins find their receptors. Nat Rev Microbiol (2017) 15(7):435–47. doi: 10.1038/nrmicro.2017.27
96. Lambey P, Otun O, Cong X, Hoh F, Brunel L, Verdie P, et al. Structural insights into recognition of chemokine receptors by staphylococcus aureus leukotoxins. Elife (2022) 11:e72555. doi: 10.7554/eLife.72555.sa2
97. Grison CM, Lambey P, Jeannot S, Del Nero E, Fontanel S, Peysson F, et al. Molecular insights into mechanisms of GPCR hijacking by staphylococcus aureus. Proc Natl Acad Sci USA (2021) 118(42):e2108856118. doi: 10.1073/pnas.2108856118
98. Lebre MC, Vergunst CE, Choi IY, Aarrass S, Oliveira AS, Wyant T, et al. Why CCR2 and CCR5 blockade failed and why CCR1 blockade might still be effective in the treatment of rheumatoid arthritis. PloS One (2011) 6(7):e21772. doi: 10.1371/journal.pone.0021772
99. Neighbour H, Boulet LP, Lemiere C, Sehmi R, Leigh R, Sousa AR, et al. Safety and efficacy of an oral CCR3 antagonist in patients with asthma and eosinophilic bronchitis: a randomized, placebo-controlled clinical trial. Clin Exp Allergy (2014) 44(4):508–16. doi: 10.1111/cea.12244
100. Keshav S, Vanasek T, Niv Y, Petryka R, Howaldt S, Bafutto M, et al. A randomized controlled trial of the efficacy and safety of CCX282-b, an orally-administered blocker of chemokine receptor CCR9, for patients with crohn's disease. PloS One (2013) 8(3):e60094. doi: 10.1371/journal.pone.0060094
101. Tak PP, Balanescu A, Tseluyko V, Bojin S, Drescher E, Dairaghi D, et al. Chemokine receptor CCR1 antagonist CCX354-c treatment for rheumatoid arthritis: CARAT-2, a randomised, placebo controlled clinical trial. Ann Rheum Dis (2013) 72(3):337–44. doi: 10.1136/annrheumdis-2011-201605
102. Maffi P, Lundgren T, Tufveson G, Rafael E, Shaw JAM, Liew A, et al. Targeting CXCR1/2 does not improve insulin secretion after pancreatic islet transplantation: A phase 3, double-blind, randomized, placebo-controlled trial in type 1 diabetes. Diabetes Care (2020) 43(4):710–8. doi: 10.2337/dc19-1480
103. Landoni G, Piemonti L, Monforte AD, Grossi P, Zangrillo A, Bucci E, et al. A multicenter phase 2 randomized controlled study on the efficacy and safety of reparixin in the treatment of hospitalized patients with COVID-19 pneumonia. Infect Dis Ther (2022) 11(4):1559–74. doi: 10.1007/s40121-022-00644-6
104. Mulnaes D, Schott-Verdugo S, Koenig F, Gohlke H. TopProperty: Robust metaprediction of transmembrane and globular protein features using deep neural networks. J Chem Theory Comput (2021) 17(11):7281–9. doi: 10.1021/acs.jctc.1c00685
105. Zhou T, Peng B, Geng S. Emerging biomarkers and therapeutic strategies for refractory bullous pemphigoid. Front Immunol (2021) 12:718073. doi: 10.3389/fimmu.2021.718073
106. ImmunePharmaceuticals. Evaluation of safety, efficacy and pharmacodynamic effect of bertilimumab in patients with bullous pemphigoid (2016). Available at: https://ClinicalTrials.gov/show/NCT02226146.
107. Bonvin P, Gueneau F, Buatois V, Charreton-Galby M, Lasch S, Messmer M, et al. Antibody neutralization of CXCL10 in vivo is dependent on binding to free and not endothelial-bound chemokine: Implications for the design of a new generation of anti-chemokine therapeutic antibodies. J Biol Chem (2017) 292(10):4185–97. doi: 10.1074/jbc.M116.745877
108. de Graaf KL, Lapeyre G, Guilhot F, Ferlin W, Curbishley SM, Carbone M, et al. NI-0801, an anti-chemokine (C-X-C motif) ligand 10 antibody, in patients with primary biliary cholangitis and an incomplete response to ursodeoxycholic acid. Hepatol Commun (2018) 2(5):492–503. doi: 10.1002/hep4.1170
109. Patterson AM, Siddall H, Chamberlain G, Gardner L, Middleton J. Expression of the duffy antigen/receptor for chemokines (DARC) by the inflamed synovial endothelium. J Pathol (2002) 197(1):108–16. doi: 10.1002/path.1100
110. Vergara C, Tsai YJ, Grant AV, Rafaels N, Gao L, Hand T, et al. Gene encoding Duffy antigen/receptor for chemokines is associated with asthma and IgE in three populations. Am J Respir Crit Care Med (2008) 178(10):1017–22. doi: 10.1164/rccm.200801-182OC
111. Dawson TC, Lentsch AB, Wang Z, Cowhig JE, Rot A, Maeda N, et al. Exaggerated response to endotoxin in mice lacking the Duffy antigen/receptor for chemokines (DARC). Blood (2000) 96(5):1681–4. doi: 10.1182/blood.V96.5.1681
112. Novitzky-Basso I, Rot A. Duffy Antigen receptor for chemokines and its involvement in patterning and control of inflammatory chemokines. Front Immunol (2012) 3:266. doi: 10.3389/fimmu.2012.00266
113. Lee JS, Frevert CW, Thorning DR, Segerer S, Alpers CE, Cartron JP, et al. Enhanced expression of Duffy antigen in the lungs during suppurative pneumonia. J Histochem Cytochem (2003) 51(2):159–66. doi: 10.1177/002215540305100204
114. Kangelaris KN, Sapru A, Calfee CS, Liu KD, Pawlikowska L, Witte JS, et al. The association between a darc gene polymorphism and clinical outcomes in African American patients with acute lung injury. Chest (2012) 141(5):1160–9. doi: 10.1378/chest.11-1766
115. Zarbock A, Bishop J, Muller H, Schmolke M, Buschmann K, Van Aken H, et al. Chemokine homeostasis vs. chemokine presentation during severe acute lung injury: the other side of the Duffy antigen receptor for chemokines. Am J Physiol Lung Cell Mol Physiol (2010) 298(3):L462–71. doi: 10.1152/ajplung.00224.2009
116. Reutershan J, Harry B, Chang D, Bagby GJ, Ley K. DARC on RBC limits lung injury by balancing compartmental distribution of CXC chemokines. Eur J Immunol (2009) 39(6):1597–607. doi: 10.1002/eji.200839089
117. Bruhl H, Vielhauer V, Weiss M, Mack M, Schlondorff D, Segerer S. Expression of DARC, CXCR3 and CCR5 in giant cell arteritis. Rheumatol (Oxford). (2005) 44(3):309–13. doi: 10.1093/rheumatology/keh485
118. Zarbock A, Schmolke M, Bockhorn SG, Scharte M, Buschmann K, Ley K, et al. The Duffy antigen receptor for chemokines in acute renal failure: A facilitator of renal chemokine presentation. Crit Care Med (2007) 35(9):2156–63. doi: 10.1152/ajplung.00224.2009
119. Barkaway A, Rolas L, Joulia R, Bodkin J, Lenn T, Owen-Woods C, et al. Age-related changes in the local milieu of inflamed tissues cause aberrant neutrophil trafficking and subsequent remote organ damage. Immunity (2021) 54(7):1494–510 e7. doi: 10.1016/j.immuni.2021.04.025
120. Nourshargh S, Renshaw SA, Imhof BA. Reverse migration of neutrophils: Where, when, how, and why? Trends Immunol (2016) 37(5):273–86. doi: 10.1016/j.it.2016.03.006
121. Loyer C, Lapostolle A, Urbina T, Elabbadi A, Lavillegrand JR, Chaigneau T, et al. Impairment of neutrophil functions and homeostasis in COVID-19 patients: association with disease severity. Crit Care (2022) 26(1):155. doi: 10.1186/s13054-022-04002-3
122. Segerer S, Bohmig GA, Exner M, Colin Y, Cartron JP, Kerjaschki D, et al. When renal allografts turn DARC. Transplantation (2003) 75(7):1030–4. doi: 10.1097/01.TP.0000054679.91112.6F
123. Klager J, Eskandary F, Bohmig GA, Kozakowski N, Kainz A, Colin Y, et al. Renal allograft DARCness in subclinical acute and chronic active ABMR. Transpl Int (2021) 34(8):1494–505. doi: 10.1111/tri.13904
124. Zernecke A, Weber C. Chemokines in the vascular inflammatory response of atherosclerosis. Cardiovasc Res (2010) 86(2):192–201. doi: 10.1093/cvr/cvp391
125. Tziakas DN, Chalikias GK, Tentes IK, Stakos D, Chatzikyriakou SV, Mitrousi K, et al. Interleukin-8 is increased in the membrane of circulating erythrocytes in patients with acute coronary syndrome. Eur Heart J (2008) 29(22):2713–22. doi: 10.1093/eurheartj/ehn382
126. Apostolakis S, Chalikias GK, Tziakas DN, Konstantinides S. Erythrocyte Duffy antigen receptor for chemokines (DARC): diagnostic and therapeutic implications in atherosclerotic cardiovascular disease. Acta Pharmacol Sin (2011) 32(4):417–24. doi: 10.1038/aps.2011.13
127. Wan W, Liu Q, Lionakis MS, Marino AP, Anderson SA, Swamydas M, et al. Atypical chemokine receptor 1 deficiency reduces atherogenesis in ApoE-knockout mice. Cardiovasc Res (2015) 106(3):478–87. doi: 10.1093/cvr/cvv124
128. Balkwill F. Cancer and the chemokine network. Nat Rev Cancer. (2004) 4(7):540–50. doi: 10.1038/nrc1388
129. Seo W, Shimizu K, Kojo S, Okeke A, Kohwi-Shigematsu T, Fujii SI, et al. Runx-mediated regulation of CCL5 via antagonizing two enhancers influences immune cell function and anti-tumor immunity. Nat Commun (2020) 11(1):1562. doi: 10.1038/s41467-020-15375-w
130. Mollica Poeta V, Massara M, Capucetti A, Bonecchi R. Chemokines and chemokine receptors: New targets for cancer immunotherapy. Front Immunol (2019) 10:379. doi: 10.3389/fimmu.2019.00379
131. Albini A, Bruno A, Noonan DM, Mortara L. Contribution to tumor angiogenesis from innate immune cells within the tumor microenvironment: Implications for immunotherapy. Front Immunol (2018) 9:527. doi: 10.3389/fimmu.2018.00527
132. Mehraj U, Qayoom H, Mir MA. Prognostic significance and targeting tumor-associated macrophages in cancer: new insights and future perspectives. Breast Cancer. (2021) 28(3):539–55. doi: 10.1007/s12282-021-01231-2
133. Jin J, Lin J, Xu A, Lou J, Qian C, Li X, et al. CCL2: An important mediator between tumor cells and host cells in tumor microenvironment. Front Oncol (2021) 11:722916. doi: 10.3389/fonc.2021.722916
134. Comerford I, Nibbs RJ. Post-translational control of chemokines: a role for decoy receptors? Immunol Lett (2005) 96(2):163–74. doi: 10.1016/j.imlet.2004.08.018
135. Addison CL, Belperio JA, Burdick MD, Strieter RM. Overexpression of the duffy antigen receptor for chemokines (DARC) by NSCLC tumor cells results in increased tumor necrosis. BMC Cancer. (2004) 4:28. doi: 10.1186/1471-2407-4-28
136. Wang J, Ou ZL, Hou YF, Luo JM, Shen ZZ, Ding J, et al. Enhanced expression of Duffy antigen receptor for chemokines by breast cancer cells attenuates growth and metastasis potential. Oncogene (2006) 25(54):7201–11. doi: 10.1038/sj.onc.1209703
137. Ellison G, Klinowska T, Westwood RF, Docter E, French T, Fox JC. Further evidence to support the melanocytic origin of MDA-MB-435. Mol Pathol (2002) 55(5):294–9. doi: 10.1136/mp.55.5.294
138. Rae JM, Creighton CJ, Meck JM, Haddad BR, Johnson MD. MDA-MB-435 cells are derived from M14 melanoma cells–a loss for breast cancer, but a boon for melanoma research. Breast Cancer Res Treat (2007) 104(1):13–9. doi: 10.1007/s10549-006-9392-8
139. Shen H, Schuster R, Stringer KF, Waltz SE, Lentsch AB. The Duffy antigen/receptor for chemokines (DARC) regulates prostate tumor growth. FASEB J (2006) 20(1):59–64. doi: 10.1096/fj.05-4764com
140. Horton LW, Yu Y, Zaja-Milatovic S, Strieter RM, Richmond A. Opposing roles of murine duffy antigen receptor for chemokine and murine CXC chemokine receptor-2 receptors in murine melanoma tumor growth. Cancer Res (2007) 67(20):9791–9. doi: 10.1158/0008-5472.CAN-07-0246
141. Xu L, Ashkenazi A, Chaudhuri A. Duffy Antigen/receptor for chemokines (DARC) attenuates angiogenesis by causing senescence in endothelial cells. Angiogenesis (2007) 10(4):307–18. doi: 10.1007/s10456-007-9084-y
142. Maeda S, Kuboki S, Nojima H, Shimizu H, Yoshitomi H, Furukawa K, et al. Duffy Antigen receptor for chemokines (DARC) expressing in cancer cells inhibits tumor progression by suppressing CXCR2 signaling in human pancreatic ductal adenocarcinoma. Cytokine (2017) 95:12–21. doi: 10.1016/j.cyto.2017.02.007
143. Mrouj K, Andres-Sanchez N, Dubra G, Singh P, Sobecki M, Chahar D, et al. Ki-67 regulates global gene expression and promotes sequential stages of carcinogenesis. Proc Natl Acad Sci USA (2021) 118(10):e2026507118. doi: 10.1073/pnas.2026507118
144. Tkach M, Coria L, Rosemblit C, Rivas MA, Proietti CJ, Diaz Flaque MC, et al. Targeting Stat3 induces senescence in tumor cells and elicits prophylactic and therapeutic immune responses against breast cancer growth mediated by NK cells and CD4+ T cells. J Immunol (2012) 189(3):1162–72. doi: 10.4049/jimmunol.1102538
145. Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer (2009) 9(11):798–809. doi: 10.1038/nrc2734
146. Acosta JC, O'Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell (2008) 133(6):1006–18. doi: 10.1016/j.cell.2008.03.038
147. Bandyopadhyay S, Zhan R, Chaudhuri A, Watabe M, Pai SK, Hirota S, et al. Interaction of KAI1 on tumor cells with DARC on vascular endothelium leads to metastasis suppression. Nat Med (2006) 12(8):933–8. doi: 10.1038/nm1444
148. Khanna P, Chung CY, Neves RI, Robertson GP, Dong C. CD82/KAI expression prevents IL-8-mediated endothelial gap formation in late-stage melanomas. Oncogene (2014) 33(22):2898–908. doi: 10.1038/onc.2013.249
149. Esposito M, Ganesan S, Kang Y. Emerging strategies for treating metastasis. Nat Cancer. (2021) 2(3):258–70. doi: 10.1038/s43018-021-00181-0
150. Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. (2002) 2(8):563–72. doi: 10.1038/nrc865
151. Drury LJ, Ziarek JJ, Gravel S, Veldkamp CT, Takekoshi T, Hwang ST, et al. Monomeric and dimeric CXCL12 inhibit metastasis through distinct CXCR4 interactions and signaling pathways. Proc Natl Acad Sci U S A. (2011) 108(43):17655–60. doi: 10.1073/pnas.1101133108
152. Takekoshi T, Ziarek JJ, Volkman BF, Hwang ST. A locked, dimeric CXCL12 variant effectively inhibits pulmonary metastasis of CXCR4-expressing melanoma cells due to enhanced serum stability. Mol Cancer Ther (2012) 11(11):2516–25. doi: 10.1158/1535-7163.MCT-12-0494
153. Veldkamp CT, Seibert C, Peterson FC, Sakmar TP, Volkman BF. Recognition of a CXCR4 sulfotyrosine by the chemokine stromal cell-derived factor-1alpha (SDF-1alpha/CXCL12). J Mol Biol (2006) 359(5):1400–9. doi: 10.1016/j.jmb.2006.04.052
154. Poluri KM, Joseph PRB, Sawant KV, Rajarathnam K. Molecular basis of glycosaminoglycan heparin binding to the chemokine CXCL1 dimer. J Biol Chem (2013) 288(35):25143–53. doi: 10.1074/jbc.M113.492579
155. Gutjahr JC, Crawford KS, Jensen DR, Naik P, Peterson FC, Samson GPB, et al. The dimeric form of CXCL12 binds to atypical chemokine receptor 1. Sci Signal (2021) 14(696):eabc9012. doi: 10.1126/scisignal.abc9012
156. Jenkins BD, Martini RN, Hire R, Brown A, Bennett B, Brown I, et al. Atypical chemokine receptor 1 (DARC/ACKR1) in breast tumors is associated with survival, circulating chemokines, tumor-infiltrating immune cells, and African ancestry. Cancer Epidemiol Biomarkers Prev (2019) 28(4):690–700. doi: 10.1158/1055-9965.EPI-18-0955
157. Zeng XH, Ou ZL, Yu KD, Feng LY, Yin WJ, Li J, et al. Coexpression of atypical chemokine binders (ACBs) in breast cancer predicts better outcomes. Breast Cancer Res Treat (2011) 125(3):715–27. doi: 10.1007/s10549-010-0875-2
158. Yang C, Yu KD, Xu WH, Chen AX, Fan L, Ou ZL, et al. Effect of genetic variants in two chemokine decoy receptor genes, DARC and CCBP2, on metastatic potential of breast cancer. PloS One (2013) 8(11):e78901. doi: 10.1371/journal.pone.0078901
159. Davis MB, Walens A, Hire R, Mumin K, Brown AM, Ford D, et al. Distinct transcript isoforms of the atypical chemokine receptor 1 (ACKR1)/Duffy antigen receptor for chemokines (DARC) gene are expressed in lymphoblasts and altered isoform levels are associated with genetic ancestry and the Duffy-null allele. PloS One (2015) 10(10):e0140098. doi: 10.1371/journal.pone.0140098
160. Martini R, Chen Y, Jenkins BD, Elhussin IA, Cheng E, Hoda SA, et al. Investigation of triple-negative breast cancer risk alleles in an international African-enriched cohort. Sci Rep (2021) 11(1):9247. doi: 10.1038/s41598-021-88613-w
161. Newman LA, Jenkins B, Chen Y, Oppong JK, Adjei E, Jibril AS, et al. Hereditary susceptibility for triple negative breast cancer associated with Western Sub-Saharan African ancestry: Results from an international surgical breast cancer collaborative. Ann Surg (2019) 270(3):484–92. doi: 10.1097/SLA.0000000000003459
162. Liu XF, Li LF, Ou ZL, Shen R, Shao ZM. Correlation between Duffy blood group phenotype and breast cancer incidence. BMC Cancer. (2012) 12:374. doi: 10.1186/1471-2407-12-374
163. Elson JK, Beebe-Dimmer JL, Morgenstern H, Chilkuri M, Blanchard J, Lentsch AB. The Duffy Antigen/Receptor for chemokines (DARC) and prostate-cancer risk among Jamaican men. J Immigr Minor Health (2011) 13(1):36–41. doi: 10.1007/s10903-010-9330-z
164. Nemesure B, Wu SY, Hennis A, Leske MC. Distribution of Duffy antigen receptor for chemokines (DARC) and risk of prostate cancer in Barbados, West indies. J Immigr Minor Health (2015) 17(3):679–83. doi: 10.1007/s10903-013-9970-x
165. Oloyede E, Dzahini O, Barnes N, Mijovic A, Gandhi S, Stuart-Smith S, et al. Benign ethnic neutropenia: an analysis of prevalence, timing and identification accuracy in two large inner-city NHS hospitals. BMC Psychiatry (2021) 21(1):502. doi: 10.1186/s12888-021-03514-6
166. Dickson AL, Daniel LL, Jackson E, Zanussi J, Yang W, Plummer WD, et al. Race, genotype, and azathioprine discontinuation : A cohort study. Ann Intern Med (2022) 175(8):1092–9. doi: 10.7326/M21-4675
167. Van Driest SL, Abul-Husn NS, Glessner JT, Bastarache L, Nirenberg S, Schildcrout JS, et al. Association between a common, benign genotype and unnecessary bone marrow biopsies among African American patients. JAMA Intern Med (2021) 181(8):1100–5. doi: 10.1001/jamainternmed.2021.3108
168. Atallah-Yunes SA, Ready A, Newburger PE. Benign ethnic neutropenia. Blood Rev (2019) 37:100586. doi: 10.1016/j.blre.2019.06.003
169. Glisovic SJ, Pastore YD, Gagne V, Plesa M, Laverdiere C, Leclerc JM, et al. Impact of genetic polymorphisms determining leukocyte/neutrophil count on chemotherapy toxicity. Pharmacogenomics J (2018) 18(2):270–4. doi: 10.1038/tpj.2017.16
170. Hershman D, Weinberg M, Rosner Z, Alexis K, Tiersten A, Grann VR, et al. Ethnic neutropenia and treatment delay in African American women undergoing chemotherapy for early-stage breast cancer. J Natl Cancer Inst (2003) 95(20):1545–8. doi: 10.1093/jnci/djg073
171. Hsieh MM, Tisdale JF, Rodgers GP, Young NS, Trimble EL, Little RF. Neutrophil count in African americans: lowering the target cutoff to initiate or resume chemotherapy? J Clin Oncol (2010) 28(10):1633–7. doi: 10.1200/JCO.2009.24.3881
172. Mantzaris I, Yu Y, Msaouel P, Lam AP, Janakiram M, Friedman EW, et al. Analysis of overall survival in a large multiethnic cohort reveals absolute neutrophil count of 1,100 as a novel prognostic cutoff in African americans. Oncotarget (2016) 7(42):67948–55. doi: 10.18632/oncotarget.8996
173. Rot A, Gutjahr JC, Biswas A, Aslani M, Hub E, Thiriot A, et al. Murine bone marrow macrophages and human monocytes do not express atypical chemokine receptor 1. Cell Stem Cell (2022) 29(7):1013–5. doi: 10.1016/j.stem.2021.11.010
174. Kwon YW, Chae CW, Lee H, Shin D, Yoo H, Lee CS, et al. A subset of macrophages and monocytes in the mouse bone marrow express atypical chemokine receptor 1. Cell Stem Cell (2022) 29(7):1016–7. doi: 10.1016/j.stem.2022.06.011
Keywords: ACKR1, DARC, chemokine, cancer, inflammation
Citation: Crawford KS and Volkman BF (2023) Prospects for targeting ACKR1 in cancer and other diseases. Front. Immunol. 14:1111960. doi: 10.3389/fimmu.2023.1111960
Received: 30 November 2022; Accepted: 03 March 2023;
Published: 15 March 2023.
Edited by:
Vadim Gaponenko, University of Illinois at Chicago, United StatesReviewed by:
Cristina Tecchio, University of Verona, ItalyIain Comerford, University of Adelaide, Australia
Copyright © 2023 Crawford and Volkman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kyler S. Crawford, a3NjcmF3Zm9yZEBtY3cuZWR1