 Xinyi Liu
Xinyi Liu Guanglei Li
Guanglei Li Yin Liu
Yin Liu Fuling Zhou
Fuling Zhou Xingxu Huang
Xingxu Huang Kui Li
Kui Li
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 27 March 2023
Sec. Primary Immunodeficiencies
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1111777
This article is part of the Research Topic Advances in Therapeutic Strategies of Inborn Errors of Immunity View all 9 articles
Inborn errors of immunity (IEIs) are a group of inherited disorders caused by mutations in the protein-coding genes involved in innate and/or adaptive immunity. Hematopoietic stem cell transplantation (HSCT) is a mainstay definitive therapy for many severe IEIs. However, the lack of HLA-matched donors increases the risk of developing severe immunological complications. Gene therapy provides long-term clinical benefits and could be an attractive therapeutic strategy for IEIs. In this review, we describe the development and evolution of clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated proteins (Cas) gene-editing systems, including double-strand break (DSB)-based gene editing and DSB-free base editing or prime editing systems. Here, we discuss the advances in and issues associated with CRISPR/Cas gene editing tools and their potential as therapeutic alternatives for IEIs. We also highlight the progress of preclinical studies for the treatment of human genetic diseases, including IEIs, using CRISR/Cas and ongoing clinical trials based on this versatile technology.
Inborn errors of immunity (IEIs), also known as primary immunodeficiencies (PIDs), are a group of inherited disorders caused by monogenic mutations in encoded proteins involved in immune responses. IEIs have been considered rare diseases with an overall estimated prevalence of approximately 1/10,000 to 1/50,000; however, along with the continued discovery of novel IEIs, the prevalence of these diseases appears to have been underestimated by 10-fold, with IEIs actually affecting more than 1 in 1000 to 1 in 5000 births (1). The advent and application of next-generation sequencing has expedited the discovery of novel gene defects that affect the development and function of the innate or adaptive immune system (2). Currently, 430 distinct immunological disorders caused by known genetic defects have been identified, and they have diverse underlying manifestations, such as susceptibility to infections, allergy, autoimmunity, autoinflammation, and malignancy (1, 3).
Patients bearing a disease-causing mutation that disables the immune system usually suffering from life-threatening infectious disease, early diagnosis and proper management are of paramount importance to prevent the development of complications, especially for those with severe complications. The development of high throughput sequencing technologies and advances in diagnostic methods have resulted in a growing number of IEIs could be identified in pediatric age (4). The treatment options for IEIs include small-molecule inhibitor administration, immunoglobulin/enzyme replacement therapy, hematopoietic stem cell transplantation (HSCT), and gene therapy. These treatments have developed rapidly based on remarkable biotechnological advances capable of characterizing their molecular mechanisms (5). Small molecule inhibitors like ruxolitinib, a Janus kinase (JAK) family protein tyrosine kinase inhibitor, has been successfully applied in treatment of infections caused by gain of function (GOF) mutations of signal transducer and activator of transcription (STAT1) (6, 7). Intravenous or subcutaneous injection of immunoglobulin improves the immune phenotype of patients with absence of B cells or deficient antibody production (8–10). Enzyme replacement therapy (ERT) with the form of polyethylene-glycol-modified ADA (PEG-ADA) administration displayed significant clinical improvement in adenosine deaminase-deficient severe combined immunodeficiency (ADA-SCID) patients (11–13). Targeted drugs ameliorate the clinical symptoms of an IEI, however, the effects are often poorly sustained and substantial treatment burdens are heavy over life-long medication. Recently, HSCT has been the mainstay definitive therapy for severe combined immunodeficiency (SCID) and other severe forms of IEIs. HSCT from a well-matched donor can lead to a lifelong cure for more than 90% of SCID patients, who subsequently present very robust and long-lasting immune reconstitution (14, 15). However, the availability of HLA-matched donors is limited for a significant number of patients. Moreover, HSCT using an HLA-mismatched donor increases the risk of developing severe immunological complications, such as graft-versus-host disease (GvHD) and infections (16–18). Considering the pathogenic mutations of coding genes, gene therapy can provide long-term clinical benefits and thus represents an attractive therapeutic strategy.
Gene therapy for IEIs has been developed to provide an autologous HSCT option by adding a normal copy of the responsible disease-related gene or correcting the mutation in the patient’s own HSCs. For loss-of-function mutations, viral vector-mediated gene augmentation, which provides the correct copy of defective genes, can be used for gene therapy. Gamma retroviral vectors were used as delivery vectors in early clinical trials for gene therapy of IEIs, including ADA-SCID, X-linked SCID (X-SCID), Wiskott–Aldrich syndrome (WAS), and X-linked chronic granulomatous disease (X-CGD) (19–27). To prevent insertional oncogenic mutagenesis, self-inactivating (SIN) lentiviral vectors have been developed and become the most commonly used vectors for insertion of therapeutic genes into hematopoietic stem and progenitor cells (HSPCs) in both research and clinical trials for some IEIs, including X-SCID, ADA-SCID, X-CGD, and WAS (18, 28, 29). For gain-of-function (GOF) mutations or genes that require precise regulation, in situ correction of the pathogenic mutations is needed and gene editing systems provide useful tools to revise GOF mutations. Here, we describe advances in precise genome editing using the lustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated proteins (Cas) gene editing system and reveal the great potential of CRISPR/Cas-based gene therapy for the treatment of IEIs. In this review, we focus on DNA editing using CRISPR/Cas; thus, RNA editing will not be included.
Targeted genome editing introduces stable genetic modifications at sites of interest using engineered nucleases. Methods for eukaryotic genomic manipulation have evolved rapidly over the past decade. Programmable nucleases, including zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and CRISPR/Cas, are three popular nucleases that have been engineered to induce double-strand breaks (DSBs) at specific endogenous gene loci to enable targeted genome manipulation. DSBs can be repaired by the error-prone non-homologous end joining (NHEJ) pathway, which often results in nucleotide insertions or deletions (indels), thus inactivating the gene, or by the homology-directed repair (HDR) pathway, which enables gene correction or insertion of a transgene when a homologous donor DNA template is provided (Figure 1). Compared with ZFNs and TALENs, which recognize target sequences through protein-DNA interactions, CRISPR/Cas genome editing systems are RNA-guided nuclease target sequences based on RNA-DNA base pairing. For each new target site, ZFN and TALEN require the engineering of a new protein, whereas the CRISPR/Cas system can be reprogrammed simply by RNA redesign, thus offering distinct advantages.
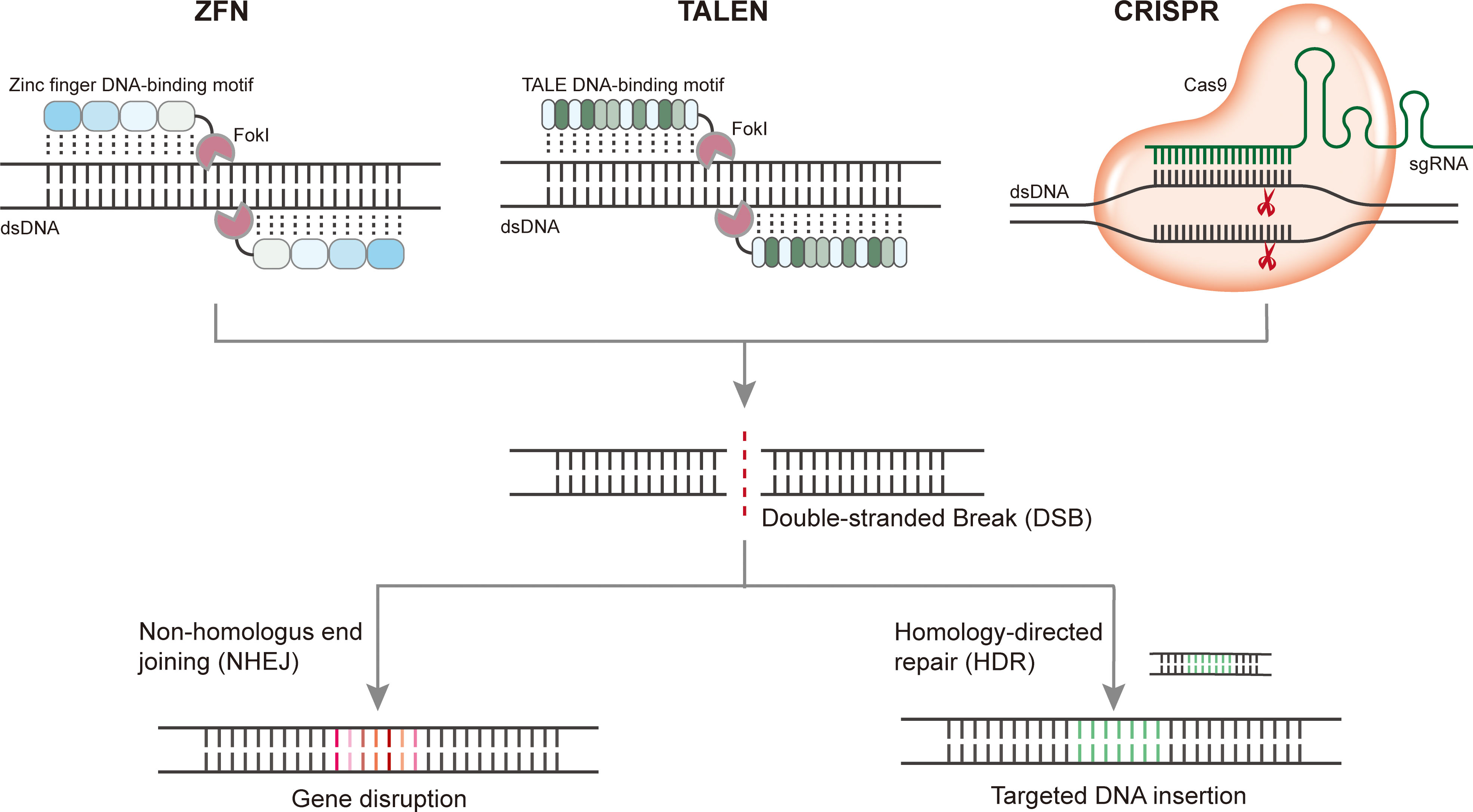
Figure 1 Programmable gene editing using ZFN, TALEN, and CRISPR. Upon cleavage of target DNA, DSBs induced by nuclease can be repaired by the error-prone nonhomologous end joining (NHEJ) pathway which often resulting in nucleotide insertions or deletions (indels) thus disturb the target gene, or by homology-directed repair (HDR) pathway which enables gene correction or insertion of a transgene when a homologous donor DNA template are provided.
CRISPR/Cas systems function as adaptive immune systems in bacteria and archaea and have been exploited for biological research and translational applications. The widely used CRISPR/Cas9 and CRISPR/Cas12 systems belong to the class 2 CRISPR/Cas system, which employs single large effector proteins with multiple domains for nucleic acid cleavage (30). In the CRISPR/Cas9 system, target DNA is cleaved by the formation of ribonucleoprotein complexes with CRISPR RNAs (crRNAs) and trans-activating crRNAs (tracrRNAs) in their native context, whereas in genome editing applications, the tracrRNA and crRNA complexes have been engineered as single guide RNA (sgRNA) chimeras to facilitate simple and versatile genome manipulation (31). Two distinct nuclease domains of Cas9, the HNH nuclease domain and RuvC-like nuclease domain, are responsible for the cleavage of the guide RNA–bound target DNA strand and the protospacer adjacent motif (PAM)-containing non-target DNA strand, respectively (31, 32). Inactivation of either domain abolishes the nuclease activity of Cas9 and generates a nickase, while the simultaneous inactivation of both domains results in a catalytically dead Cas9 (dCas9) (3). Cas9 nickase (nCas9) has been applied to improve the targeting precision of Cas9 by using paired nickases together with two sgRNAs (33, 34), and precision genome editing tools have been developed, such as base editors (BEs) and prime editors (35–37). The sequence-specific DNA-binding property of dCas9 allows for transcriptional regulation and epigenetic modification without genetically altering the DNA sequence (38, 39) or chromosome imaging in live cells (40). Moreover, dCas9 was harnessed to increase the specificity of CRISPR/Cas9-mediated DSB formation by fusing to the catalytic domain of Fok1 (41, 42). The presence of a PAM sequence is critical for Cas protein binding, and a number of Cas9 protein orthologs have been discovered to expand targeting scope. For example, SaCas9 recognizes the NNGRRT PAM sequence (43), CjCas9 recognizes the NNNNACA PAM sequence (44), NmCas9 recognizes the NNNNGATT PAM sequence (45), and ScCas9 recognizes the NGN PAM sequence (46). Compared with Cas9, Cas12 possesses a single RuvC-like domain that cleaves both the guide RNA complementary DNA strand and the noncomplementary DNA strand (30). The CRISPR/Cas12 system encompasses several sub-type effectors, many of which are guided by a single crRNA, although some (such as Cas12b, Cas12c2, and Cas12e) require both crRNA and tracrRNA for activation (47, 48). Cas12a (formerly Cpf1) and Cas12b (formerly C2c1) are two Cas12 effectors that have been studied in detail. Cas12a is a single RNA-guided endonuclease that recognizes a T-rich PAM sequence and mediates robust DNA interference via a single RuvC catalytic domain, and it has been widely used for genome editing applications (49). Compared with Cas12a, Cas12b requires both tracrRNA and crRNA for DNA cleavage. Guided by chimeric sgRNA, engineered Cas12b facilitates robust genome editing in human cells and mice with high specificity (50, 51). Recently, miniature Cas12f has been revealed as a compact nuclease with high efficiency that offers more options for therapeutic applications (52–54). For several Cas12 variants, RNA-guided target recognition and binding unleash their robust, indiscriminate single-stranded DNA or RNA cleavage activity (55, 56). This collateral cleavage activity has facilitated the development of numerous strategies for rapid nucleic acid detection and portable diagnosis (55, 57, 58).
Upon nuclease cleavage, two main DNA repair pathways, NHEJ and HDR, are involved in the repair of nuclease-induced DSBs (Figure 2, left panel). In most mammalian cells, error-prone NHEJ is the predominant repair pathway, which often introduces indels at sites of DSBs, resulting in the disruption of target gene sequences or regulatory elements (59, 60). In some cases, indels resulting from NHEJ are not sufficient to disrupt the function of gene clusters or regulatory sequences; thus, large deletions are more desirable. Using dual guide RNAs targeting adjacent regions of a chromosome sequence, Cas9 nuclease can introduce two DSBs simultaneously, which often leads to the deletion or inversion of the intervening sequence (61, 62). By the delivery of Cas9 nuclease coupled with paired sgRNAs flanking the mutated Dmd exon23, efficient target sequence excision, and phenotypic restoration were achieved in the mdx mouse model of Duchenne muscular dystrophy (DMD) (63). CRISPR/Cas-mediated NHEJ exhibits great potential for the correction of dominant-negative mutations underlying diseases. Delivery of the CRISPR/Cas9:sgRNA complex through cationic lipids into a mouse model of human genetic deafness disrupted the dominant inherited mutation in Tmc1 and ameliorated the pathogenic phenotype (64). The NHEJ repair process is fast and flexible, and frameshifts resulting from indels are likely to generate premature termination codons, which might lead to nonsense-mediated decay of mRNA and prevent protein translation (65). Disruption of multiple genes simultaneously could be achieved by simultaneously targeting multiple genomic loci (66, 67). Although NHEJ mediates efficient gene disruption, the editing results are usually uncontrollable due to the formation of indels at sites of DSBs, which has hindered its broad application in clinical transformation.
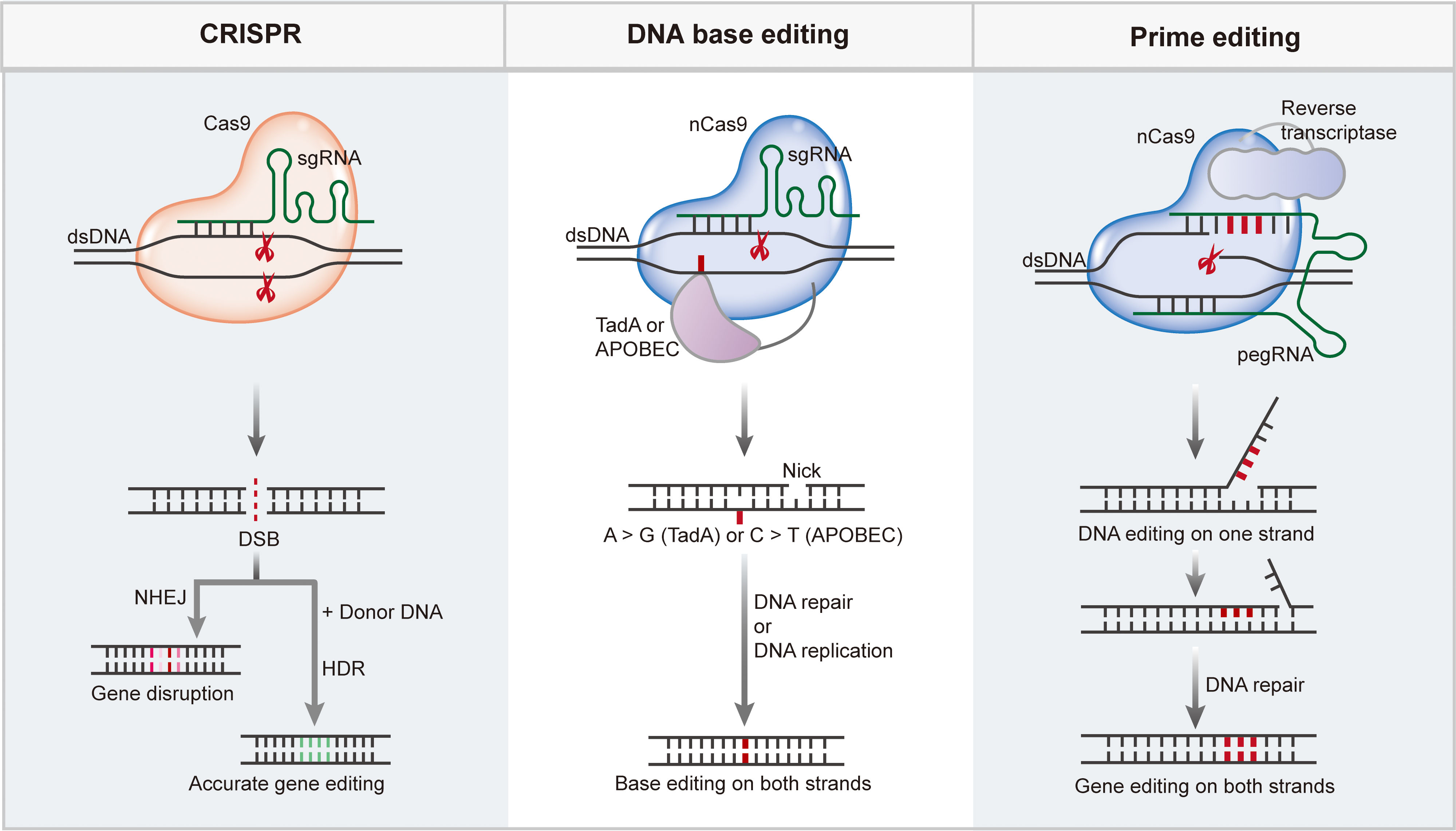
Figure 2 CRISPR/Cas mediate efficient and precise genome editing. CRISPR/Cas nucleases generate DSBs upon target recognition, DSBs can be repaired through NHEJ pathway to disturb the target gene, or through HDR pathway to enable targeted gene insertion or replacement. Base editing is performed by a fusion complex of catalytically impaired Cas nuclease and a single strand DNA (ssDNA) deaminase enzyme. Base editing enabling efficient and precisely targeted base conversion at single base resolution without producing DSBs or requiring a donor DNA template. Prime editing requires a complex of Cas9 H840A nickase fused to an engineered reverse transcriptase enzyme as well as a pegRNA, enables base conversions, insertions or deletions in a precise way.
In the presence of a homologous donor template, the HDR repair pathway can be initiated to insert a fragment of interest through homologous recombination at a precisely targeted genomic location. The HDR pathway competes with NHEJ for the repair of nuclease-induced DSBs, and selection of a repair pathway is influenced by many factors, such as the structure and sequence context of cutting ends, the format of the donor template, the state of the cell cycle, etc. (65). The donor template for HDR could be a double-stranded DNA (dsDNA)-like plasmid donor or single-stranded DNA (ssDNA), such as ssODN. ssDNA usually mediates a more efficient HDR process, whereas dsDNA allows for larger DNA fragment knock-in (68). With a proper donor, the repair of CRISPR/Cas-induced DSBs by HDR can introduce a variety of genome edits, including gene insertion or replacement (69, 70). Compared with the NHEJ process, which is efficient in most mammalian cells, HDR is inefficient and generally active only in dividing cells; moreover, the frequency of intended sequence insertion can be improved by synchronizing cells at the S and G2 phases using chemical factors (71). Efforts to improve the efficiency of HDR include modifying the length and symmetry of homologous arms, inhibiting the NHEJ pathway, or enhancing the HDR pathway by manipulating core factors (72, 73).
CRISPR/Cas nuclease-mediated gene disruption is widely used in biomedical research. Efficient and specific abrogation of protein function is of particular interest in the study and treatment of genetic disorders caused by dominant-negative mutations. HDR can be used to either insert a gene fragment into a specific site or replace defective genes in situ. However, CRISPR/Cas9-induced DSBs at a target locus often result in undesired mutations and HDR appears to be inefficient in quiescent cell types. Moreover, more than half of the known human pathogenic mutations are point mutations (74); thus, alternative approaches that efficiently and precisely install or reverse pathogenic mutations are highly desirable.
DNA BEs are mainly composed of a catalytically impaired Cas nuclease and ssDNA deaminase enzyme. They enable efficient and precise targeted base conversions at a single-base resolution without producing DSBs or requiring a donor DNA template (74). DNA BEs have been categorized into two major classes: cytosine BEs (CBEs) and adenine BEs (ABEs), of which CBEs mediate C-G to T-A base conversions using naturally existing cytidine deaminases while ABEs mediate A-T to G-C base conversions through revolutionized Escherichia coli tRNA adenosine deaminase TadA (Figure 2, middle panel) (35, 36).
CBEs were first reported by Liu and co-workers in 2016 (35), and base editor 1 (BE1) was developed by fusing a naturally existed cytidine deaminase rAPOBEC1 to dCas9 (Cas9 variants containing both D10A and H840A mutations). rAPOBEC1 catalyzes cytosine (C) deamination into uracil (U), which is recognized as thymine (T) in base-pairing process; this results in the conversion of a C-G base pair to T-A base pair. To elevate base editing efficiencies, uracil DNA glycosylase inhibitor (UGI) was introduced into BE2 to inhibit uracil N-glycosylase (UNG), which mediates base excision repair (BER) of U-G mismatch, usually resulting in the reversion of the U-G intermediate back to a C-G base pair. BE3 was generated by replacing dCas9 of BE2 with an nCas9 (D10A mutation), which specifically nicks the non-deaminated strand to induce mismatch repair (MMR) process resulting in U-G mismatch transformation into the desired T-A base-pair. BE3 is the first reported CBE that enabled efficient C-G to T-A base conversions in mammalian cells with an editing window at positions 4–8 (counting the first nucleotide of the protospacer as position 1 with the PAM at positions 21–23); a small frequency of indels was also observed (35). Similar to cytosine that deaminates into uracil, deamination of adenosine (A) yields inosine, which pairs with C and therefore is recognized as guanine (G). To achieve A-T to G-C base conversion, Liu’s group performed protein evolution and engineering of an adenosine deaminase TadA due to the lack of any known natural enzymes capable of acting on ssDNA (36). Following several rounds of evolution, a TadA mutation (TadA*) was identified and fused to nCas9 in a heterodimeric form (wtTadA-TadA* complex) to generate ABE7.10. ABE7.10 enables efficient A-T to G-C base conversion in mammalian cells with very high product purity and very low rates of indels within an active editing window of positions 4–7 (counting the first nucleotide of the protospacer as position 1 with the PAM at positions 21–23) (36).
Both CBE and ABE are powerful tools for irreversible DNA base transitions (A-G, C-T, G-A, and T-C) in various cell types. The editing efficiency of BEs was continuously increased through linker selection, additional UGI appending, nuclear localization sequence modification, codon optimization, and Cas protein or deaminase engineering (75–78). For example, BE4max and AncBE4max were developed by optimization of the nuclear localization sequence and codon usage, and ancestral reconstruction of the deaminase, respectively (76). Joung and co-workers developed miniABEmax by removing wild type TadA monomer and demonstrated that this component is unnecessary for ABE activity (77). Further, ABEs were considered to have poor compatibility with some Cas homologs (such as Cas12). In contrast to CBEs, the developed ABE8e through deaminase evolution exhibits substantially improved editing efficiencies especially when paired with a variety of Cas homologs (78). The invention of CBEs and ABEs has enabled efficient base transitions (A-G, C-T, G-A, and T-C). Despite being valuable, the utility of these two category BEs in correcting pathogenic variants is hindered due to the inability of base transversions. To achieve C to G base transversion in mammalian cells, cytosine deaminase rAPOBEC1 and uracil excision-related protein UNG were fused with nCas9 by researchers (79, 80). Another group developed CGBE by the fusion of nCas9 with rAPOBEC1 and base excision repair protein rXRCC1 to enable efficient C-G base transversion in WCW, ACC, or GCT sequence contexts with a precise editing window (81). To further expand the editing competency of the system, dual-function BEs which enable concurrent cytosine and adenine editing were developed by simultaneously fusing cytosine and adenine deaminases into Cas9 variants and applied in mammalian cells and plants (82–87). Most recently, an adenine transversion base editor, AYBE, was developed for A-C and A-T transversion editing in mammalian cells by fusing an ABE with hypoxanthine excision protein N-methylpurine DNA glycosylase (MPG) (88).
The improvements of BEs for precise base editing have mainly focused on several aspects, such as broadening the targeting scope, enhancing product purity, reducing off-target activity, and others. As mentioned above, BEs employ Cas proteins to localize deaminases to the target regions, and target bases must be positioned in the editing window to ensure efficient base editing. Thus, the PAM availability together with the width of the editing window determines the targeting scope of base editing. SpCas9 requires an NGG PAM for target DNA recognition and binding; this PAM requirement restricts the target space of SpCas9 to every 8 bp on average in the human genome (66). To broaden the targeting scope of BEs, other Cas orthologs and engineered Cas variants recognizing different PAMs have been incorporated into BEs (75, 89–94). Among Cas orthologs, SaCas9 and ScCas9 are two smaller Cas9 proteins, and BEs developed with these homologs should be easier to deliver in vivo (75, 93). Engineered SpCas9 variants VQR-Cas9, EQR-Cas9, VRER-Cas9, xCas9, SpCas9-NG, and Cas9-SpRY with alternative PAMs have been employed to develop BEs (89–92). In addition to pursuing PAM availability, altering the location and/or width of the editing window by replacing Cas and/or deaminase of BEs also expands the number of targetable sites. BEs with different Cas proteins or deaminases possess different editing windows. By combining pmCDA1 with nCas9, Nishida et al. developed a cytosine base editing system named ‘Target-AID’ that enabled targeted C-G to T-A base conversion in yeast and mammalian cells with an editing window shift comparable to that of BE3 (95). BEs with AID and A3A deaminases typically have wider windows, and CBEs containing highly active deaminases, such as human AID and some APOBEC3 family members, have been developed to mediate base conversions with high efficiency in different editing contexts (96, 97). The editing window of BEs generated with circularly permuted Cas9 expanded from ~4–5 nucleotides to ~8–9 nucleotides while also retaining product purity (98). Except for SpCas9 and engineered variants, the editing efficiency of ABE variants developed with other Cas enzymes is modest. The poor compatibility of ABEs has been overcome through the evolution of ABE8, which exhibits substantially improved editing efficiencies when paired with a variety of Cas homologs, including Cas12 enzymes (78). By combining the rapidly expanding Cas effectors with the growing set of deaminases, the targeting scope of BEs has been substantially expanded.
The product purity of base editing is a critical aspect that should be considered when optimizing BEs, especially for therapeutic applications. Ideal DNA BEs enable efficient and precise target-base conversion without generating excess by-products, unexpected edits, or detectable indels. Bystander editing occurs when multiple editable nucleotides are present within the editing window because the deaminase of BEs can deaminate all cytosines or adenines within the activity window. Due to degeneracy in the genetic code, some bystander edits will result in a synonymous mutation, whereas in some other cases, bystander editing results in amino acid alteration and is often problematic. Engineered deaminase variants with narrow activity windows or high sequence context dependence have been integrated into BEs to reduce or avoid bystander editing (77, 89, 97). The presence of unexpected edits caused by UNG-mediated uracil removing reduces the product purity of CBEs; additional expression or recruitment of UGI copies to the BE3 construct has been demonstrated to decrease unexpected editing and increase editing efficiency (75, 99, 100). Editing by BEs can also yield detectable indels due to the nicks formed on the deaminated strand by DNA-(apurinic or apyrimidinic site) lyase (AP lyase) and non-deaminated strand by Cas9 nickase; the two nicks may result in a DSB which is likely to be repaired by indel-prone NHEJ process. The Mu bacteriophage-derived Gam protein (Mu-GAM) binds to the free ends of DSBs and protects them from degradation (101), and fusing CBEs with Mu-GAM leads to substantial reduction of indels in human cells, rabbit embryos, and human tripronuclear (3PN) zygotes (75, 102, 103). Compared to CBEs, ABEs typically exhibit high product purity as well as very low and often undetectable indel frequencies, presumably because the removal of inosine is substantially inefficient and causes fewer nicks in the deaminated strand, resulting in fewer DSBs and indels (74).
DNA BE-induced off-target mutations have been reported (104–106), and genome-wide and transcriptome-wide off-target effects of BEs can occur in a Cas-dependent or Cas-independent manner. The former is usually caused by the Cas effector and sgRNA binding to sequences that are similar to the on-target locus, and the latter is due to the overexpression of deaminases that can randomly deaminate accessible nucleotides. To combat Cas-dependent off-target effects, engineered Cas9 variants with high fidelity, such as Cas9-HF and sniper-Cas9, have been incorporated into CBE structures (107, 108). SgRNA truncation and deaminase embedding have also been used to reduce the off-target editing of BEs (96, 109). Cas-independent DNA off-target as well as RNA off-target effects of CBEs can be ameliorated by engineering cytosine deaminase to generate various high-specific deaminase variants. For example, engineered rAPOBEC1 and human APOBEC3 family members have been incorporated into CBEs to reduce Cas-independent off-target effects (89, 110, 111). SECURE-BE3, another CBE variant using engineered rAPOBEC1 to deaminate cytosines, has been reported to reduce RNA off-target edits and narrow the editing window (77). Additional deaminases have also been identified and employed to generate next-generation CBEs with minimal Cas-independent DNA and RNA off-target edits (112). Although no genome-wide off-target A-T to G-C base editing has been observed, ABE can create substantial off-target edits on RNA (104, 105). Similar to CBEs, rational engineering of adenine deaminase can minimize the Cas9-independent RNA editing activity of ABEs (77, 111, 113, 114). Additionally, delivering BEs as purified ribonucleoprotein (RNP) complexes instead of DNA constructs to limit the exposure time of BEs greatly decreases off-target editing while maintaining comparable on-target editing (115).
BEs utilize Cas effector-tethered nucleotide deaminases to induce efficient target base substitutions at a single-base resolution without introducing DSBs, which enables the use of base editing as a new therapeutic option for disorders caused by point mutations. Numerous studies have demonstrated the ability of BEs to correct pathogenic mutations underlying various human genetic diseases, including Duchenne muscular dystrophy (116), metabolic liver diseases (117, 118), hereditary deafness (64), progeria (23), Marfan syndrome (119), sickle cell disease (120), β-thalassemia (121), among others. Efficient modeling of pathogenic point mutations using BEs has also been performed in cultured human primary cells (122), embryos (123, 124), and various organisms, including mice (125), rabbits (102), pigs (126), and non-human primates (127–129). In addition to modeling or correcting human pathogenic point mutations, BEs can be used to regulate gene expression in a more precise manner. By targeting CAA, CAG, CGA, and TGG, CBEs can introduce premature stop codons, which usually result in the degradation of the target mRNA via nonsense-mediated decay (NMD), thus silencing gene expression (130, 131). Gene expression can also be shut down by ABE-mediated destruction of the start codon, ATG (132). Decreased abundance of abnormally expressed proteins could palliate the disease progress of these incurable disorders. In addition, modification of alternative splicing sites (donor or acceptor) using BEs facilitates selective skipping of mutation-containing exons while maintaining the preferred normal isoforms for gene therapy (133).
A major application of BEs is modeling or correcting point mutations underlying human genetic diseases along with the engineering of deaminase enzymes as well as Cas effectors. The editing characteristics of BEs, including the targeting scope, product purity, and potential off-target edits, have been improved to a large extent; thus, more BE variants with therapeutic potential are available.
Although BEs have been considered a safer and more precise genome editing tool, they can only mediate very limited editing types. Prime editors (PEs) have emerged as a genome-editing approach to enable all 12 types of base conversions, base pair insertions or deletions, and even combinations without the generation of DSBs in a targeted way (37). PEs are composed of Cas9 H840A nickase fused to an engineered reverse transcriptase (RTase) enzyme as well as a prime-editing guide RNA (pegRNA) (Figure 2, right panel). The pegRNA contains an sgRNA sequence for Cas9 targeting, a primer binding site (PBS) for RT initiation, and an RT template with the desired edits. To achieve more efficient and precise editing, an additional sgRNA (nicking sgRNA) has been introduced into the PE3 and PE3b systems (37). Although PEs enable precise genome editing with great versatility, the editing efficiency among endogenous sites varies (134, 135). The design of optimal pegRNAs plays a critical role in PE with high efficiency, and numerous efforts have been made to improve the editing efficiency of PE by modifying pegRNAs. For example, ePE was developed by introducing multiple modifications to pegRNAs, which resulted in a marked increase in editing efficiency (136). Nelson et al. reported pegRNAs with enhanced stability and improved editing efficiency, and they did not show increased off-target editing activity (137). xrPE presents substantially enhanced editing efficiency based on appending an RNA motif to prevent pegRNA degradation (138). Li et al. developed spegRNA and apegRNA to increase the PE base-editing and indel editing efficiency by introducing same-sense mutations into the RT template of pegRNA and altering the pegRNA secondary structure, respectively (139). Modification of pegRNAs with G-quadruplexes also effectively improved the editing efficiency of Pes (140). PE4 and PE5 were developed by transient co-expression of an engineered DNA mismatch repair inhibiting protein MLH1 with PE2 and PE3, respectively (141), and further optimization resulted in PEmax with enhanced PE performance. Employing paired pegRNAs that target opposite DNA strands, PRIME-Del has been reported to enable precise genomic deletions of up to 10 kb, and extended expression of PE components can substantially enhance efficiency without compromising precision (142). Zhuang et al. reported an approach named HOPE in which paired pegRNAs encoding the same edits in both sense and antisense DNA strands were used to achieve high editing efficiency as well as high editing purity (143). Using a PE protein and two pegRNAs, twinPE enables efficient gene replacement or excision, large DNA plasmid (>5,000 bp) integration, and targeted sequence inversions (144). A similar strategy has been implemented to enable the donor-free insertion of large DNA sequences by GRAND editing (145). PE-Cas9-based deletion and repair (PEDAR) has been reported to enable precise genomic deletion and replacement of genomic fragments (146). Replacing SpCas9 H840A nickase with PAM-flexible Cas9 variants has been reported to expand the editing modality of PE (147, 148). By generating an all-in-one PE system and a Cas9 nuclease instead of a nickase, Adikusuma et al. showed improved editing efficiency in cultured cells and mouse zygotes (149). Furthermore, for efficient and simple manipulation, researchers have developed computational tools to design pegRNAs or predict pegRNA efficiency (150–152). As a versatile genome editing tool, PEs have been used in cultured mammalian cells (37), plants (135), and model animals (134, 153, 154) to enable the introduction of any small genetic mutation. The off-target effect of PE is relatively low at the same target sites compared with that of Cas9, which may be due to the two additional nucleic acid hybridization steps (nicked target strand–PBS hybridization and 3′ flap–target strand hybridization) that are required for PE (96). Using genomic and transcriptomic sequencing analyses, researchers demonstrated that PE3 did not induce any pegRNA-independent off-target mutations in mammalian cells or zygotes (155–157).
CRISPR/Cas systems enable targeted genome manipulation in a programmable and efficient manner and have remarkable potential for correcting mutations underlying human genetic diseases. Recent progress in the development of advanced gene editing approaches with high activity and precision has paved the way for theories to enter practice.
CRISPR/Cas-mediated gene disruption may be useful for removing disease-causing replicated fragments or GOF mutants. Huntington’s disease (HD) is caused by a dominantly inherited CAG repeat expansion in exon 1 of the huntingtin gene (HTT), which can be removed through CRISPR-Cas9-mediated fragment deletion in patient-derived fibroblasts and mouse models (158, 159). Mutations in the hemoglobin subunit beta gene (HBB), which encodes β-globin, is related to hereditary anemias such as sickle cell disease (SCD) and β-thalassemia. Re-expression of the paralogous γ-globin genes (HBG1/2) could ameliorate the severe β-globin disorders. Diminishing the expression of the γ-globin transcriptional repressor or attenuating γ-globin-to-β-globin switching through CRISPR/Cas9-mediated gene disruption resulted in increased γ-globin expression, which ameliorated the pathogenic phenotype (160–166). Clinical trials for the treatment of patients with β-thalassemia and severe sickle cell disease by transfusion of CRISPR/Cas-edited HSCs have been initiated and have displayed exciting results (167, 168). Leber congenital amaurosis 10 (LCA10) is a severe rare genetic eye disease caused by mutations in the CEP290 gene, and SaCas9 was delivered in conjunction with dual gRNAs into a human CEP290 IVS26 knock-in mouse model by a single adeno-associated virus (AAV) to remove the aberrant splice donor generated by the IVS26 mutation of the CEP290 gene (169). The genome editing therapeutic EDIT-101 was initiated by Allergan and Editas Medicine for the treatment of LCA10. By targeting misfolded transthyretin (TTR), CRISPR/Cas9 has been used in the treatment of transthyretin (ATTR) amyloidosis by targeting misfolded transthyretin (TTR), and the safety and pharmacodynamic effects of NTLA-2001 have been evaluated in an ongoing clinical study (170). Splicing mutations responsible for cystic fibrosis (CF) have been successfully repaired using CRISPR/Cas-mediated sequence cleavage (171). CRISPR/Cas gene disruption has also been applied in the treatment of inherited liver diseases. Hereditary tyrosinemia type I (HTI) is a severe inherited metabolic disorder caused by loss-of-function mutation of FAH. Knocking out hydroxyphenylpyruvate dioxygenase (HPD, an upstream enzyme of FAH) has been demonstrated to prevent toxic metabolite accumulation and has been used to treat HTI metabolic disease in Fah−/−mice (172). Disruption of GOF mutations in alpha-1 antitrypsin (AAT) by CRISPR/Cas could ameliorate the pathologic liver phenotype (173). Disruption of proprotein convertase subtilisin/kexin type 9 (PCSK9) by delivering CRISPR/Cas into the mouse liver resulted in significant reductions in serum Pcsk9 and total cholesterol levels has been observed (43, 174, 175). CRISPR/Cas-mediated frameshifts have also been exploited to treat DMD, a progressive muscular dystrophy caused by mutations in the dystrophin gene. Dystrophin expression was rescued and the pathogenic phenotype was partially recovered in DMD animals after local or systemic delivery of CRISPR/Cas components (176–179). Transmembrane channel-like 1 (Tmc1) mutation causes hearing loss, and cationic lipid-mediated in vivo delivery of Cas9:gRNA RNP complex in the humanized transmembrane channel-like 1 (Tmc1) Beethoven (Bth) mouse model ameliorated the pathogenic phenotype (64).
Replacement of a mutated sequence with the correct fragment via the HDR pathway is an attractive strategy for gene therapy. HDR-mediated gene replacement has been used to correct disease-causing point mutations in the β-globin (HBB) gene in HSCs and patient-derived HSPCs to treat β-thalassemia and sickle cell disease (SCD) (63). Martyn et al. reported that the precise insertion of a point mutation via CRISPR/Cas-mediated HDR creates a de novo binding site for the transcriptional activator GATA1, which drives γ-globin expression and HbF production, thus providing a therapeutic strategy for β-thalassemia (180). Successful modeling and correction of human familial hypercholesterolemia, which causes point mutations, through the CRISPR/Cas9-mediated HDR pathway have been achieved (181). Using ssODNs as a homology donor, gene correction was achieved in Cockayne syndrome (CS) or amyotrophic lateral sclerosis (ALS) patient-derived induced pluripotent stem cells (iPSCs) via the CRISPR/Cas9-mediated HDR pathway (182, 183). Furthermore, HDR-mediated mutant gene correction has also been reported in inherited liver diseases, such as HTI and AATD (184–186). CRISPR/Cas HDR-mediated corrections of pathogenic mutations in pre-implantation human embryos have also been reported (187). However, the application of HDR-mediated gene therapy is limited because this method is confined to dividing cells and has a low editing rate and co-delivering donor DNA with CRISPR/Cas components is difficult (188).
Compared with HDR, BE-mediated editing can be applied to both dividing cells and terminally differentiated cell types in a predictable manner. Researchers have delivered ABEs mRNA and sgRNAs targeting Pcsk9 in vivo using lipid nanoparticles (LNPs) to introduce PCSK9 loss-of-function mutations in living cynomolgus monkeys, which resulted in significant reductions in the blood levels of PCSK9 and LDL cholesterol (118, 189). Zeng et al. performed base editing using purified A3A (N57Q)-BE3 protein with chemically modified synthetic sgRNAs as RNP complexes targeting the BCL11A erythroid enhancer for fetal hemoglobin (HbF) induction in human CD34 HSPCs (190). By utilizing ABE8e-NRCH, Newby et al. successfully converted an SCD-causing mutation into a nonpathogenic variant in HSPCs derived from patients or humanized SCD mice (120). Villiger et al. delivered SaKKH-BE3 through intravenous injection of dual-AAV plasmids into a PKU mouse model to restore phenylalanine hydroxylase (PAH) enzyme activity and revert the light fur phenotype in these animals (117). Viral delivery of BE3 in utero resulted in long-term postnatal persistence of edited cells and reduced plasma PCSK9 and cholesterol levels (191). Song et al. delivered ABE into an adult mouse model of HTI in a non-viral manner to correct a FAH point mutation (192). Using a dual-AAV system, ABE was delivered to muscle cells in a DMD mouse model to correct a nonsense mutation in the dystrophin gene (116). Splice donor sites of the dystrophin gene have been targeted using ABE in human iPSCs and transgenic mice, and this process restores dystrophin expression (193). Efficient and precise correction of pathogenic point mutations involving Marfan syndrome, β-thalassemia, and lamellar ichthyosis has also been performed in human embryos, thus demonstrating the feasibility of curing genetic diseases in human embryos by BEs (104, 119, 194). Delivery of ABEs using a lentivirus to patient-derived fibroblasts or administering AAV to a transgenic mice resulted in the substantial direct correction of the pathogenic mutation underlying Hutchinson-Gilford progeria syndrome (HGPS or progeria) (23). Delivery of BEs into the mouse brain using dual AAVs successfully corrected a mutation that causes Niemann-Pick disease type C, which slows down neurodegeneration and increases lifespan (195). CRISPR DNA BEs were designed to mediate irreversible base substitutions without generating DSBs or requiring donor DNA; thus, in theory, they could enable the manipulation of genomes in a safer and more precise manner. However, BEs can only mediate limited types of editing, while most known disease-associated genetic variants are specific transversions, deletions, insertions, and others (37). Thus, genome engineering tools that can install more types of genetic changes are aspired.
In theory, PEs can correct up to 89% of pathogenic human genetic variants reported in ClinVar (196). Schwank’s al. delivered PE into a mouse model of phenylketonuria (PKU) to repair the disease-causing Pahenu2 mutation via the human adenoviral vector 5 (AdV) (197). Utilizing a dual-AAV delivery system, Liu et al. delivered a split-intein PE to enable the correction of AATD pathogenic mutations in mouse liver (198). Zhi et al. developed a dual-AAV delivery strategy that successfully delivered PEs into adult mouse retina for Dnmt1 editing in vivo (199). Jang et al. reported that PEs delivered by hydrodynamic injection and AAV enable precise correction of disease-causing mutations and amelioration of disease phenotypes in mouse models of HT1 and LCA without detectable off-target edits (200). By introducing insertions, a PE-mediated reframing strategy has been exploited to restore dystrophin expression in human cardiomyocytes (193). PE-mediated correction of pathogenic point mutations could also be performed in chemically derived hepatic progenitors (CdHs) through a non-viral delivery method to treat genetic liver disease (201). Schene et al. demonstrated that PEs could functionally recover disease-causing mutations in intestinal organoids from patients with DGAT1-deficiency and liver organoids from a patient with Wilson disease (ATP7B) (202). Using optimized PEs, Jiang et al. removed a 1.38-kb pathogenic insertion within the Fah gene and precisely repaired the deletion junction to restore FAH expression in the liver of a tyrosinemia mouse model (146). PEs have also been utilized to correct COL7A1 mutations in recessive dystrophic epidermolysis bullosa (RDEB) patient-derived fibroblasts, and functional rescue was observed (203). Correction of a disease-related mutation in α 1-antitrypsin (A1AT)-deficient patient-derived induced pluripotent stem cells (iPSCs) has been achieved, and guide RNA-independent off-target mutations have not been detected in the genome (155). Due to the large size of PEs, delivery efficiency as well as editing efficiency may represent limitations that significantly impact the therapeutic application of this technology.
As most IEIs are caused by loss-of-function mutations, a viral-mediated gene addition strategy has been developed for T-cell therapies. A retroviral strategy was the first class used, and although long-term immune reconstitution was achieved in patients subjected to retroviral gene transfer, insertional mutagenesis and oncogene transactivation associated with the use of retroviral vectors were observed in gene therapy trials for X-SCID, CGD, and WAS (204–207). To circumvent these adverse events, an improved generation of gene delivery vectors, referred to as SIN vectors, has been developed. Compared with gammaretroviral vectors, which integrate primarily within transcriptional regulatory elements, lentiviral vectors preferentially integrate into active transcription units and, hence, have a better safety profile. Furthermore, lentiviral vectors can be produced at much higher titers and may enable effective transduction in a wide range of cell types; thus, lentiviral vectors have become the most commonly used vectors for the insertion of therapeutic genes into HSPCs in both research and clinical trials (18, 28, 29). Lentiviral vector-mediated gene transfer has been successfully applied in gene therapy for some IEIs, including X-SCID, ADA-SCID, X-CGD, and WAS; moreover, effective immune reconstitution has been observed and no adverse events related to the vector have been reported in these clinical trials (208–213). These positive results suggest that lentiviral-based gene addition therapy provides sustained clinical benefits for patients with IEIs. However, for genes involved in some IEIs caused by dominant-negative mutations or those that require precise regulation (e.g., forkhead box P3, FOXP3; Bruton tyrosine kinase, BTK; CD40 ligand, CD40LG), gene augmentation therapy is not appropriate; however, in situ correction of the endogenous defective gene would allow for physiological regulation of gene transcription and protein expression.
As a promising therapeutic approach for treating monogenic hematological diseases, such as IEIs, CRISPR/Cas-mediated gene disruption has been employed to generate animal models while CRISPR/Cas-mediated gene replacement through HDR enables precise correction of disease-causing genes. Targeted integration of one correct copy of a gene of interest in its physiological transcriptional regulation context is particularly important for the treatment of disorders caused by mutations in tightly regulated genes. The CRISPR/Cas-based gene therapy strategy has been applied to the modeling and correction of many forms of IEIs, including SCID, immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX), X-linked MAGT1 deficiency with increased susceptibility to EBV-infection and N-linked glycosylation defect (XMEN), X-CGD, and WAS. Table 1 listed known IEIs with CRISPR/Cas technology has been applied in their studies.
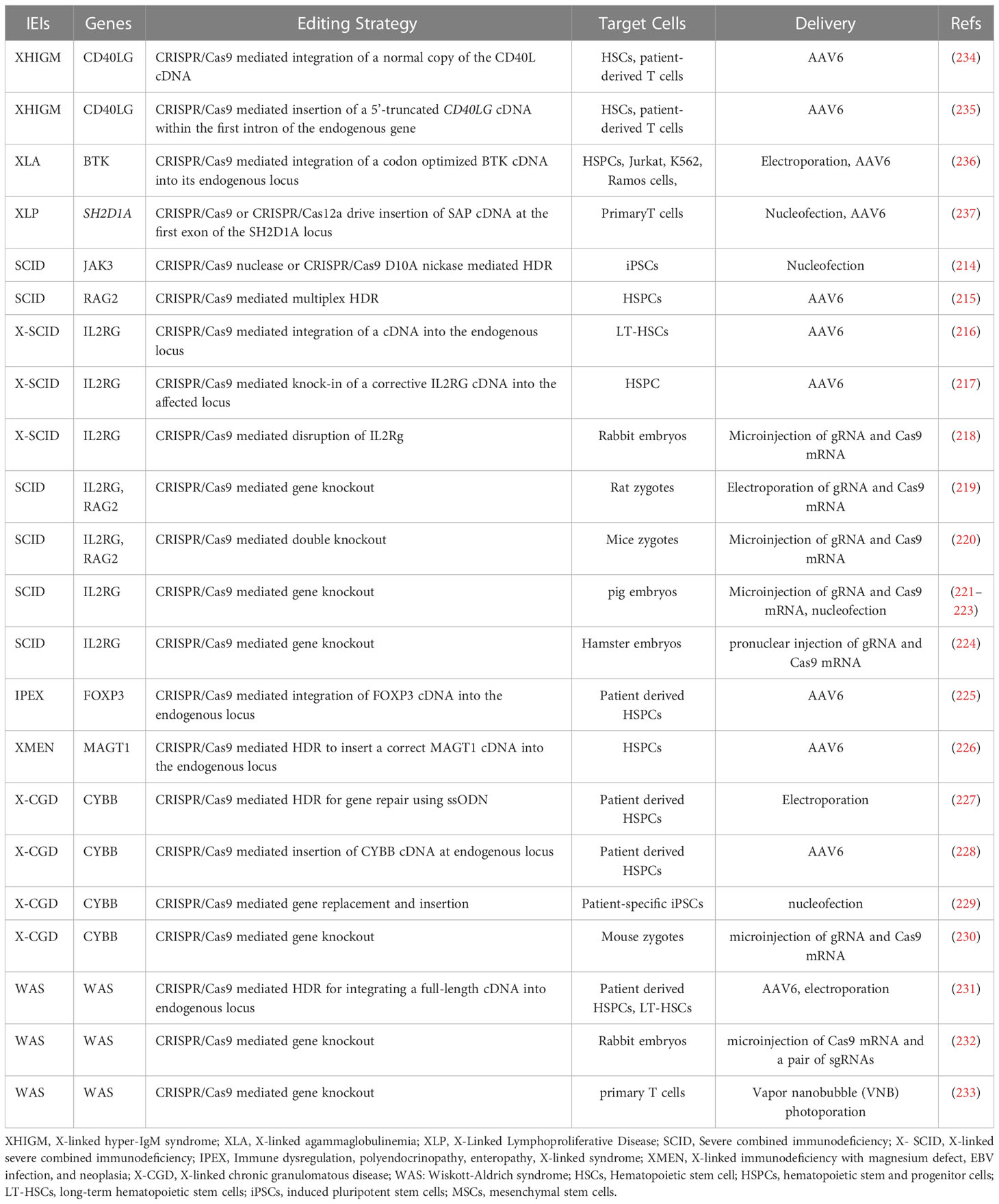
Table 1 List of the known IEIs with CRISPR-Cas technology has been applied in their studies.
SCID is the most profound IEI affecting cellular and humoral immunity, and comprises a group of disorders caused by mutations in genes involved in lymphocyte development. By employing Cas9-mediated HDR strategy, Chang et al. achieved correction of JAK3 mutations in SCID patient-specific induced pluripotent stem cells (iPSCs) and observed restoration of T cell development (214). Recently, Iancu et al. reported a CRISPR-mediated multiplex HDR strategy for disease modeling and correction in SCID patient-derived HSPCs; biallelic knockout of genes was utilized to generate a disease model while knockin/knockout strategy was used for single-allelic gene correction (215). The most common form of SCID, X-SCID, is caused by mutations in the genes encoding interleukin 2 receptor gamma (IL2RG). Integration of IL2RG cDNA into the endogenous start site in human long-term hematopoietic stem cells (LT-HSCs) using the CRISPR/Cas9-mediated HDR strategy has led to the functional correction of disease-causing mutations throughout the gene (216). In a preclinical study, functional integration of IL2RG was achieved by integrase-defective lentiviral vector (IDLVs)-mediated delivery of a donor DNA template and sgRNA in SCID-X1–Cas9+/− HSPCs with low cytotoxicity (217). In addition to treatment, CRISPR/Cas technology has been used to generate SCID animal models, including mice, rabbits, rats, pigs, and hamsters, for basic and translational research (218–224).
IPEX syndrome is a monogenic immune disease caused by mutations in FOXP3 that selectively affect the function of regulatory T cells (Tregs). By combining the CRISPR/Cas9 system with a DNA repair homology donor, Goodwin et al. enabled efficient and precise insertion of the FOXP3 cDNA into the endogenous locus of patient derived HSPCs, and the normal differentiation potential of edited HSPCs was maintained both in vitro and in vivo in immunodeficient mice (225).
XMEN is a recently described IEI caused by an X-linked MAGT1 deficiency, and it is marked by defective T cells and natural killer (NK) cells. Brault et al. reported an ex vivo targeted gene therapy approach using an optimized CRISPR/Cas HDR strategy to insert a therapeutic MAGT1 gene into the endogenous locus, and it presented high genetic correction efficiency and functional rescue in engrafted mice (226).
X-CGD is an immunodeficiency caused by mutations in the CYBB gene, which encodes the gp91phox protein. CRISPR/Cas9-mediated HDR was employed for CYBB gene correction in HSPCs and functional restoration was achieved (227–229). Sweeney et al. reported that targeted insertion of CYBB exon 2-13 by CRISPR/Cas HDR at the endogenous CYBB exon 2 site in patient HSPCs fully restored gp91phox expression and transient inhibition of the NHEJ pathway increased the gene correction rate (228). De Ravin et al. repaired a frequent mutation of the CYBB gene in HSPCs from X-CGD patients through CRISPR/Cas9-mediated HDR with an ssODN donor and achieved functional restoration up to 5 months after transplantation of the corrected HSPCs into NSG immunodeficient mice (227). Gene replacement of CYBB exon 5 mutations using CRISPR/Cas HDR has also been administered administrated in patient-derived iPSCs (229). To enable in vivo assessments of gene and cell therapy strategies for treating CGD, a mouse model of X-CGD was established using the CRISPR/Cas system by targeting CYBB exon 1 or exon 3 (230).
Wiskott–Aldrich syndrome (WAS) is an X-linked recessive immunodeficiency disease caused by mutations in the WAS gene encoding WAS protein (WASp). Rai et el. inserted a therapeutic WAS cDNA in-frame with its endogenous translation start codon in patient-derived HSPCs via CRISPR/Cas9-mediated HDR, and full rescue of WASp expression and correction of functional defects were observed (231). Microinjection of Cas9 mRNA and a pair of sgRNAs targeting exons 2 and 7 of WAS into rabbit embryos was performed to generate a model exhibiting symptoms similar to those of WAS patients for preclinical studies (232). In a proof-of-concept study, the WAS gene was disrupted in human primary T cells using the CRISPR/Cas9 system through an advanced delivery method (233).
X-linked hyper-immunoglobulin M (hyper-IgM) syndrome (XHIGM) is an IEI caused by mutations in CD40 ligand (CD40L) that affect immunoglobulin class-switch recombination and somatic hypermutation. Utilizing either the TALEN or CRISPR/Cas9 platforms, Kuo et al. integrated a normal copy of the CD40L cDNA with the endogenous CD40L promoter to maintain the physiological expression of CD40L in primary human HSCs and XHIM-patient-derived T cells (234). In a preclinical study, Vavassori et al. developed a universal CRISPR/Cas editing strategy for effective integration of a corrective CD40L cDNA within its endogenous locus and achieved effective T cell correction and selection by optimizing the editing strategy. The therapeutic potential of T-cell and HSPC therapies was further investigated in the HIGM1 mouse model (235).
X-linked agammaglobulinemia (XLA) is a monogenic IEI caused by a loss-of-function mutation in Bruton’s tyrosine kinase (BTK) gene, and it is characterized by reduced mature B lymphocytes. Gray and colleagues reported that precise integration of a corrective, codon-optimized cDNA along with a truncated terminal intron of the BTK gene into its endogenous locus by CRISPR/Cas-mediated HDR substantially elevated BTK expression to potentially therapeutic levels in cell lines and human HSPCs (236).
X-Linked Lymphoproliferative Disease (XLP) is a rare IEI that is caused by a mutation in the signaling lymphocyte activation molecule (SLAM)-associated protein (SAP) protein-coding gene SH2D1A. Targeted insertion of SAP cDNA at the first exon of the SH2D1A locus was efficiently achieved using TALEN, CRISPR/Cas9, or CRISPR/Cas12a, and SAP protein expression and SAP-dependent immune function were restored in patient-derived T cells (237).
Compared with the inefficient HDR-mediated site-specific integration of target genes, NHEJ-based homology-independent targeted integration (HITI) with improved integration frequencies has been utilized to achieve robust site-specific transgene integration at a clinically relevant genetic locus in human mobilized peripheral blood HSPCs (238). This strategy provides an alternative to HDR-based gene integration in human HSPCs. Together, these studies demonstrated that CRISPR/Cas gene editing in HSCs is highly promising for correcting disease-causing mutations in IEIs. (Figure 3)

Figure 3 Work flow of CRISPR/Cas gene editing in gene therapy of IEIs. Autologous HSCs collected from patients undergo ex vivo culture and CRISPR/Cas editing, after screening and expansion, therapeutic edited cells are transfusion into conditioned patients for immune system reconstruction. CRISPR/Cas gene editing agents could be delivered into HSCs in forms of RNP, “all RNA”, or AAV vector to enable efficient pathogenic gene correction.
BEs enable single-base substitutions without producing DSBs, while PEs enable precise base conversions, insertions, deletions, and even combinations without inducing DSBs, and both of these editors are promising in genetic disease modeling and correction. Compared with BEs, PEs induce more accurate and precise on-target editing, and more importantly, PEs enable multiple types of editing. Gene therapy studies based on BEs or PEs for IEI treatment have not been reported yet. Thus, to identify potential targets for BE- and/or PE-mediated gene correction, disease-causing genes need to be carefully sequenced and sgRNAs and pegRNAs should be designed appropriately to enable efficient editing.
The targeted and efficient delivery of editing reagents plays an important role in gene therapy. Mainstream methods for delivering editing components into HSCs are based on electroporation, viral vectors, or nanoparticles (239). Electroporation is an effective method for delivering editing reagents into a large number of HSCs; however, voltage-induced cell toxicity should be considered (240). Lentiviruses and AAVs are mainly used to deliver editing elements into HSCs, with lentiviruses integrating DNA fragments into genome and AAV presenting a limited cargo capacity for CRISPR/Cas system packaging (239). Developing non-integrating viral vectors and compact CRISPR/Cas systems could provide alternatives to overcome these challenges. Nanoparticles are a class of materials that can encapsulate therapeutic nucleic acids for targeted delivery, and lipid nanoparticles (LNPs) are the most common and have been investigated for their ability to deliver CRISPR/Cas systems for therapeutic purposes (241). A protein-based nanoparticle system (Selective Endogenous eNcapsidation for cellular Delivery, SEND) has been developed to enable efficient delivery of CRISPR/Cas editing reagents into mammalian cells (242). The recently developed delivery strategy named SORT (selective ORgan targeting (SORT) allows lipid nanoparticles to encapsulate gene therapy reagents to be accurately delivered in an organ-specific manner (243). Compared with electroporation, delivery strategies based on viral vectors and nanoparticles can be utilized in both ex vivo and in vivo gene therapy.
CRISPR/Cas components are usually delivered into HSCs in two major forms: the RNP complex of purified Cas protein precomplexed with sgRNAs and “all RNA” delivery with Cas mRNA with or without chemically modified sgRNAs. Hendel et al. reported that chemically modified sgRNAs could enhance genome editing in both human primary T cells and HSPCs; use of RNA- or RNP-based delivery method further improved editing (244). Protection of both sgRNA termini with chemically modified nucleotides increases the stability of sgRNA and renders the ‘all RNA’-based CRISPR/Cas9 system highly effective in primary HSPCs and T cells (245). By optimizing and comparing several delivery strategies, Lattanzi et al. demonstrated that the RNP-based delivery of CRISPR/Cas editing reagents exhibited a good balance between cytotoxicity and genomic rearrangement efficiency (246). The donor template for HDR-mediated gene correction can be delivered to HSCs with AAV6-based vectors or ssODNs. Co-delivery of an AAV6 donor in combination with CRISPR/Cas RNP enabled the precise correction of WAS mutations in up to 60% of human HSPCs (231). A recent study reported that FOXP3 cDNA was delivered by AAV6 with the high-fidelity Cas9 variant HiFi-Cas9:sgRNA RNP into HSPCs to correct pathogenic mutations with high specificity (225). AAV6 donor templates can mediate transgene integration at up to ~4 kb in size, thus encompassing an expression cassette for a reporter gene, which is important for the enrichment of HSCs with precisely targeted integration (245). In comparison to AAV6, ssODNs are usually ~50–200 bp in total size and can be useful donor templates for single point mutation correction. Phosphorothioate-modified ssODNs of the CYBB gene fragment have been delivered with the CRISPR/Cas RNP complex to patient-derived HSPCs to repair a mutation causing X-CGD. Transplanting gene-repaired X-CGD HSPCs into NSG mice resulted in efficient engraftment and production of functional mature human myeloid and lymphoid cells for up to five months (227).
In terms of safety, CRISPR/Cas gene therapy avoids the need for semi-random integration of viral vectors, thereby minimizing the risk of integration-activated oncogenesis. One of the major concerns associated with CRISPR/Cas gene therapy is the potential off-target effects; many efforts have been made for off-target prediction, characterization, and reduction in both cell lines and human HSPCs (247, 248). Off-target prediction by in silico predictive algorithms is powerful and useful for potential genome-wide off-target prediction, and cell-based and cell-free detection methods have been developed to determine the distribution and frequency of off-target events. Protein engineering, gRNA modification, and delivery methods have been exploited to limit the off-target effect (247). CRISPR/Cas systems introduce DSBs in the DNA and thus induce HDR, and indels induced by DSBs at the target site can be minimized by inhibiting the NHEJ pathway to enhance HDR (72, 73). BEs and PEs are DSB-independent editing strategies, and Cas-independent genomic and transcriptomic off-targets of BEs have been reported (104–106). Rational nucleotide deaminase engineering could circumvent this and provide BEs with reduced off-target editing activity, while maintaining robust on-target editing efficiency (77, 89). Additionally, delivering BEs, such as RNPs, could also greatly decrease off-target editing while maintaining comparable on-target editing (115). PEs were demonstrated to have high specificity since neither off-target DNA nor off-target RNA was detected in mammalian cells or zygotes (155–157).
CRISPR/Cas gene therapy has been considered a promising strategy to treat many incurable genetic and nongenetic disorders, including hereditary blood diseases, ocular diseases, neuromuscular disorders, metabolic disorders, cancers, and acquired immunodeficiency syndrome. Currently initiated clinical trials based on CRISPR/Cas technology are both ex vivo and in vivo. Ex vivo gene therapy infuses cultured cells into patients after editing by CRISPR/Cas, and in vivo gene therapy delivers CRISPR/Cas gene editing reagents into patients to facilitate gene modification at pathogenic sequences. The current active CRISPR/Cas-based clinical trials are listed in Table 2.
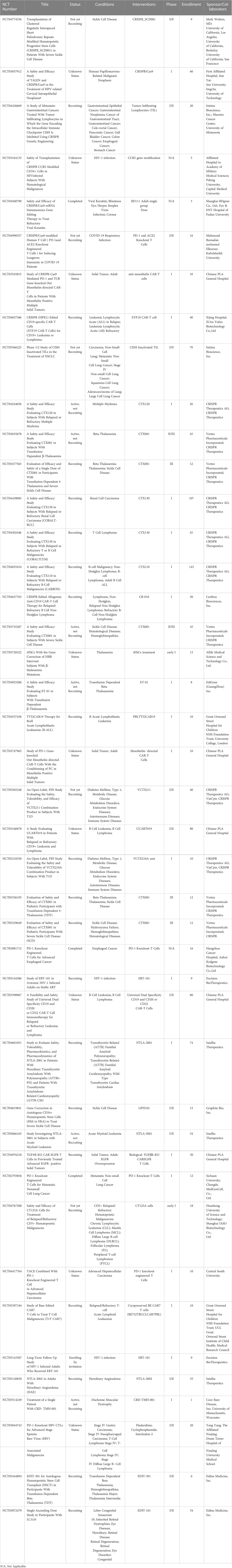
Table 2 Currently active clinical trials based on CRISPR/Cas technology.
The first in-human CRISPR/Cas clinical trial was performed at Sichuan University’s West China Hospital to treat advanced non-small cell lung cancer by targeting the PD-1 gene using CRISPR/Cas9 (ClinicalTrials.gov NCT02793856). According to the reported results, the safety and feasibility were proven, and they demonstrated that a more advanced gene editing strategy to improve efficacy is required in future trials (249). Several phase I/II clinical trials to treat carcinoma by targeting PD-1 with CRISPR/Cas have been initiated (ClinicalTrials.gov NCT02793856, NCT03747965, NCT04417764, and NCT03545815). CRISPR/Cas has also been used to generate chimeric antigen receptor (CAR) T-cells, and clinical trials have been initiated for the immunotherapy of hematologic malignancies and solid tumors (ClinicalTrials.gov NCT03166878, NCT03747965). CRISPR/Cas-mediated gene disruption can be used to alter virus host receptor expression, and clinical trials for antiviral therapeutics against infectious diseases AIDs and COVID-19 have also been registered by targeting CCR5 and ACE2, respectively (ClinicalTrials.gov NCT03164135, NCT04990557).
CRISPR/Cas-based clinical trials have been initiated for the treatment of genetic diseases. The first Cas9-mediated in vivo gene therapy trial was initiated by Editas Medicine and Allergan to treat genetic ocular disease LCA10 by eliminating the disease-causing CEP290 mutant (ClinicalTrials.gov NCT03872479). In another CRISPR/Cas-based in vivo clinical trial (ClinicalTrials.gov NCT04601051), NTLA-2001 comprised LNP-encapsulated CRISPR/Cas9 elements targeting TTR to treat ATTR amyloidosis by reducing the concentration of TTR in serum. The safety and pharmacodynamic effects of single escalating doses of NTLA-2001 were evaluated in six patients with hereditary ATTR amyloidosis, and the results demonstrated mild adverse events and dose-dependent pharmacodynamics, and a significant reduction in serum TTR protein was observed (170). In addition, a phase I clinical study was initiated by curing rare diseases to assess the safety and efficacy of a CRISPR-based therapy for the treatment of DMD (ClinicalTrials.gov NCT05514249). CRISPR/Cas-based clinical trial for another genetic disease Hereditary Angioedema have also been initiated (ClinicalTrials.gov NCT05120830). Clinical trials to treat metabolic disorder Diabetes Mellitus, Type 1 (T1D) using CRISPR/Cas edited allogeneic pancreatic endoderm cells have been registered (ClinicalTrials.gov NCT05210530, NCT05565248). Several CRISPR/Cas gene therapy companies like Editas Medicine, CRISPR Therapeutics, EdiGene, and Allife Medical Science and Technology have initiated clinical trials to treat transfusion dependent β-Thalassemia (TDT) (ClinicalTrials.gov NCT05444894, NCT05356195, NCT04925206, NCT03655678, NCT05477563, NCT03728322), and some of these clinical trials have entered phase III. Similar to TDT, CRISPR/Cas gene therapy drugs to treat another severe hematological disease sickle cell disease (SCD) have also been used in clinical trials (ClinicalTrials.gov NCT04774536, NCT05477563, NCT03745287, NCT05329649, NCT04819841) CRISPR/Cas-based gene therapy for IEIs has not yet reached clinical trials and preclinical studies involving CRISPR/Cas-edited human HSCs engrafted into mice to correct mutations of IEIs indicated safety and efficacy. With the development of safer and more advanced gene editing tools, as well as versatile delivery methods, an increasing number of CRISPR/Cas-based clinical trials will be initiated for incurable disorders.
The positive results of clinical trials of viral-based gene addition therapy suggest that autologous HSC gene therapy could provide sustained clinical benefits in patients with IEIs. However, gene addition approaches are not amenable for cases where the physiological regulation of genes is required to avoid transgene-related toxicity and genes with large sizes are difficult to deliver. The CRISPR/Cas gene editing system enables permanent, precise, and flexible gene editing without the drawbacks of semi-random genomic insertion, thus emerging as an important new tool for genetic manipulation of HSCs (28). Advances in CRISPR/Cas gene-editing technology have led to new therapeutic options for a wide range of genetic and nongenetic diseases. CRISPR/Cas-mediated HDR has been applied in the gene correction of some IEIs, including X-SCID, IPEX, X-CGD, and WAS (216, 225, 229, 231).
Although clinical trials of CRISPR/Cas gene therapy are in progress, their application for IEIs is still in its infancy, and many issues still need to be addressed to enable safe and effective clinical application. A critical limitation of the gene therapy approach for IEIs is the broad spectrum of disease categories as well as the varying mutations, which require the development of personalized medicine approaches depending on the specific mutation a patient carries (250). To ensure successful clinical outcomes for gene therapy of IEIs, a sufficient number of gene-corrected HSCs with engraftment capabilities following CRISPR/Cas9 gene editing is of paramount importance (251). HDR occurs most efficiently in dividing cells, and the low efficiency of HDR in HSCs can be improved by manipulating critical factors of DNA repair mechanisms to inhibit the NHEJ pathway (72, 73). Optimizing CRISPR/Cas genome editing tools to maximize editing efficiency while minimizing off-target effects presents directions for advancing genome editing agents (247). The engraftment capability and self-renewal ability of gene-corrected HSCs are critical factors that should be taken into account because manipulating and expanding gene-corrected cells in vitro increases the risk of losing the multilineage potential of HSCs (251). Exposure to nucleases and the toxic effects caused by condition agents largely affect the self-renew ability of gene corrected HSCs, implementation of “hit-and-run” strategy to reduce nucleases exposure time and development of advanced conditioning regimens could ameliorate these harmfulness to enhance HSC self-renewal ability (14, 251).
The advent and evolution of BE and PE variants with efficient and precise editing holds tremendous promise for the development of novel gene therapies for IEIs. Moreover, the implementation of decentralized manufacturing will enable the translation of gene-modified cell therapies from basic research to hospital-based clinical trials.
XL, GL, XH designed the manuscript, XL drafted the manuscript, XL, YL prepared figures for the manuscript, FZ, GL, KL, and XH reviewed and revised the manuscript. All authors contributed to the article and approved the submitted version.
This work is supported by grants from National Key R&D Program of China (2021YFF100604) and National Natural Science Foundation of China (32202647).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human inborn errors of immunity: 2019 update on the classification from the international union of immunological societies expert committee. J Clin Immunol (2020) 40(1):24–64. doi: 10.1007/s10875-019-00737-x
2. Meyts I, Bosch B, Bolze A, Boisson B, Itan Y, Belkadi A, et al. Exome and genome sequencing for inborn errors of immunity. J Allergy Clin Immunol (2016) 138(4):957–69. doi: 10.1016/j.jaci.2016.08.003
3. Bousfiha A, Jeddane L, Picard C, Al-Herz W, Ailal F, Chatila T, et al. Human inborn errors of immunity: 2019 update of the IUIS phenotypical classification. J Clin Immunol (2020) 40(1):66–81. doi: 10.1007/s10875-020-00758-x
4. Booth C, Romano R, Roncarolo MG, Thrasher AJ. Gene therapy for primary immunodeficiency. Hum Mol Genet (2019) 28(R1):R15–23. doi: 10.1093/hmg/ddz170
5. Notarangelo LD. Primary immunodeficiencies. J Allergy Clin Immunol (2010) 125(2 Suppl 2):S182–94. doi: 10.1016/j.jaci.2009.07.053
6. Weinacht KG, Charbonnier LM, Alroqi F, Plant A, Qiao Q, Wu H, et al. Ruxolitinib reverses dysregulated T helper cell responses and controls autoimmunity caused by a novel signal transducer and activator of transcription 1 (STAT1) gain-of-function mutation. J Allergy Clin Immunol (2017) 139(5):1629–1640.e2. doi: 10.1016/j.jaci.2016.11.022
7. Higgins E, Al Shehri T, McAleer MA, Conlon N, Feighery C, Lilic D, et al. Use of ruxolitinib to successfully treat chronic mucocutaneous candidiasis caused by gain-of-function signal transducer and activator of transcription 1 (STAT1) mutation. J Allergy Clin Immunol (2015) 135(2):551–3. doi: 10.1016/j.jaci.2014.12.1867
8. Wiesik-Szewczyk E, Sołdacki D, Paczek L, Jahnz-Różyk K. Facilitated subcutaneous immunoglobulin replacement therapy in clinical practice: A two center, long-term retrospective observation in adults with primary immunodeficiencies. Front Immunol (2020) 11:981. doi: 10.3389/fimmu.2020.00981
9. Vultaggio A, Azzari C, Ricci S, Martire B, Palladino V, Gallo V, et al. Biweekly hizentra® in primary immunodeficiency: A multicenter, observational cohort study (IBIS). J Clin Immunol (2018) 38(5):602–9. doi: 10.1007/s10875-018-0528-5
10. Jolles S, Rojavin MA, Lawo JP, Nelson R Jr, Wasserman RL, Borte M, et al. Long-term efficacy and safety of hizentra® in patients with primary immunodeficiency in Japan, Europe, and the united states: A review of 7 phase 3 trials. J Clin Immunol (2018) 38(8):864–75. doi: 10.1007/s10875-018-0560-5
11. Booth C, Gaspar HB. Pegademase bovine (PEG-ADA) for the treatment of infants and children with severe combined immunodeficiency (SCID). Biologics (2009) 3:349–58.
12. Nakazawa Y, Kawai T, Uchiyama T, Goto F, Watanabe N, Maekawa T, et al. Effects of enzyme replacement therapy on immune function in ADA deficiency patient. Clin Immunol (2015) 161(2):391–3. doi: 10.1016/j.clim.2015.06.011
13. Serana F, Sottini A, Chiarini M, Zanotti C, Ghidini C, Lanfranchi A, et al. The different extent of b and T cell immune reconstitution after hematopoietic stem cell transplantation and enzyme replacement therapies in SCID patients with adenosine deaminase deficiency. J Immunol (2010) 185(12):7713–22. doi: 10.4049/jimmunol.1001770
14. Ferrari G, Thrasher AJ, Aiuti A. Gene therapy using haematopoietic stem and progenitor cells. Nat Rev Genet (2021) 22(4):216–34. doi: 10.1038/s41576-020-00298-5
15. Castagnoli R, Delmonte OM, Calzoni E, Notarangelo LD. Hematopoietic stem cell transplantation in primary immunodeficiency diseases: Current status and future perspectives. Front Pediatr (2019) 7:295. doi: 10.3389/fped.2019.00295
16. Pessach IM, Notarangelo LD. Gene therapy for primary immunodeficiencies: Looking ahead, toward gene correction. J Allergy Clin Immunol (2011) 127(6):1344–50. doi: 10.1016/j.jaci.2011.02.027
17. Kohn DB, Kuo CY. New frontiers in the therapy of primary immunodeficiency: From gene addition to gene editing. J Allergy Clin Immunol (2017) 139(3):726–32. doi: 10.1016/j.jaci.2017.01.007
18. Buckland KF, Bobby Gaspar H. Gene and cell therapy for children–new medicines, new challenges? Adv Drug Delivery Rev (2014) 73:162–9. doi: 10.1016/j.addr.2014.02.010
19. Cicalese MP, Ferrua F, Castagnaro L, Pajno R, Barzaghi F, Giannelli S, et al. Update on the safety and efficacy of retroviral gene therapy for immunodeficiency due to adenosine deaminase deficiency. Blood (2016) 128(1):45–54. doi: 10.1182/blood-2016-01-688226
20. Candotti F, Shaw KL, Muul L, Carbonaro D, Sokolic R, Choi C, et al. Gene therapy for adenosine deaminase-deficient severe combined immune deficiency: Clinical comparison of retroviral vectors and treatment plans. Blood (2012) 120(18):3635–46. doi: 10.1182/blood-2012-02-400937
21. Gaspar HB, Parsley KL, Howe S, King D, Gilmour KC, Sinclair J, et al. Gene therapy of X-linked severe combined immunodeficiency by use of a pseudotyped gammaretroviral vector. Lancet (2004) 364(9452):2181–7. doi: 10.1016/S0140-6736(04)17590-9
22. Koblan LW, Arbab M, Shen MW, Hussmann JA, Anzalone AV, Doman JL, et al. Efficient C*G-to-G*C base editors developed using CRISPRi screens, target-library analysis, and machine learning. Nat Biotechnol (2021), 39(11):1414–25. doi: 10.1038/s41587-021-00938-z
23. Koblan LW, Erdos MR, Wilson C, Cabral WA, Levy JM, Xiong ZM, et al. In vivo base editing rescues Hutchinson-gilford progeria syndrome in mice. Nature (2021) 589(7843):608–14. doi: 10.1038/s41586-020-03086-7
24. Huang TP, Newby GA, Liu DR. Precision genome editing using cytosine and adenine base editors in mammalian cells. Nat Protoc (2021) 16(2):1089–128. doi: 10.1038/s41596-020-00450-9
25. Kang HJ, Bartholomae CC, Paruzynski A, Arens A, Kim S, Yu SS, et al. Retroviral gene therapy for X-linked chronic granulomatous disease: Results from phase I/II trial. Mol Ther (2011) 19(11):2092–101. doi: 10.1038/mt.2011.166
26. Ott MG, Schmidt M, Schwarzwaelder K, Stein S, Siler U, Koehl U, et al. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat Med (2006) 12(4):401–9. doi: 10.1038/nm1393
27. Aiuti A, Roncarolo MG, Naldini L. Gene therapy for ADA-SCID, the first marketing approval of an ex vivo gene therapy in Europe: Paving the road for the next generation of advanced therapy medicinal products. EMBO Mol Med (2017) 9(6):737–40. doi: 10.15252/emmm.201707573
28. Cavazzana M, Bushman FD, Miccio A, Andre-Schmutz I, Six E. Gene therapy targeting haematopoietic stem cells for inherited diseases: Progress and challenges. Nat Rev Drug Discovery (2019) 18(6):447–62. doi: 10.1038/s41573-019-0020-9
29. Milone MC, O’Doherty U. Clinical use of lentiviral vectors. Leukemia (2018) 32(7):1529–41. doi: 10.1038/s41375-018-0106-0
30. Makarova KS, Wolf YI, Iranzo J, Shmakov SA, Alkhnbashi OS, Brouns SJJ, et al. Evolutionary classification of CRISPR-cas systems: A burst of class 2 and derived variants. Nat Rev Microbiol (2020) 18(2):67–83. doi: 10.1038/s41579-019-0299-x
31. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science (2012) 337(6096):816–21. doi: 10.1126/science.1225829
32. Gasiunas G, Barrangou R, Horvath P, Siksnys V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci U.S.A. (2012) 109(39):E2579–86. doi: 10.1073/pnas.1208507109
33. Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell (2013) 154(6):1380–9. doi: 10.1016/j.cell.2013.08.021
34. Gopalappa R, Suresh B, Ramakrishna S, Kim HH. Paired D10A Cas9 nickases are sometimes more efficient than individual nucleases for gene disruption. Nucleic Acids Res (2018) 46(12):e71. doi: 10.1093/nar/gky222
35. Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature (2016) 533(7603):420–4. doi: 10.1038/nature17946
36. Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI, et al. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature (2017) 551(7681):464–71. doi: 10.1038/nature24644
37. Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature (2019) 576(7785):149–57. doi: 10.1038/s41586-019-1711-4
38. Thakore PI, Black JB, Hilton IB, Gersbach CA. Editing the epigenome: Technologies for programmable transcription and epigenetic modulation. Nat Methods (2016) 13(2):127–37. doi: 10.1038/nmeth.3733
39. Dominguez AA, Lim WA, Qi LS. Beyond editing: Repurposing CRISPR-Cas9 for precision genome regulation and interrogation. Nat Rev Mol Cell Biol (2016) 17(1):5–15. doi: 10.1038/nrm.2015.2
40. Ma H, Tu LC, Naseri A, Huisman M, Zhang S, Grunwald D, et al. Multiplexed labeling of genomic loci with dCas9 and engineered sgRNAs using CRISPRainbow. Nat Biotechnol (2016) 34(5):528–30. doi: 10.1038/nbt.3526
41. Guilinger JP, Thompson DB, Liu DR. Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nat Biotechnol (2014) 32(6):577–82. doi: 10.1038/nbt.2909
42. Tsai SQ, Wyvekens N, Khayter C, Foden JA, Thapar V, Reyon D, et al. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat Biotechnol (2014) 32(6):569–76. doi: 10.1038/nbt.2908
43. Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, et al. In vivo genome editing using staphylococcus aureus Cas9. Nature (2015) 520(7546):186–91. doi: 10.1038/nature14299
44. Kim E, Koo T, Park SW, Kim D, Kim K, Cho HY, et al. In vivo genome editing with a small Cas9 orthologue derived from campylobacter jejuni. Nat Commun (2017) 8:14500. doi: 10.1038/ncomms14500
45. Hou Z, Zhang Y, Propson NE, Howden SE, Chu LF, Sontheimer EJ, et al. Efficient genome engineering in human pluripotent stem cells using Cas9 from neisseria meningitidis. Proc Natl Acad Sci U.S.A. (2013) 110(39):15644–9. doi: 10.1073/pnas.1313587110
46. Chatterjee P, Jakimo N, Jacobson JM. Minimal PAM specificity of a highly similar SpCas9 ortholog. Sci Adv (2018) 4(10):eaau0766. doi: 10.1126/sciadv.aau0766
47. Murugan K, Babu K, Sundaresan R, Rajan R, Sashital DG. The revolution continues: Newly discovered systems expand the CRISPR-cas toolkit. Mol Cell (2017) 68(1):15–25. doi: 10.1016/j.molcel.2017.09.007
48. Kurihara N, Nakagawa R, Hirano H, Okazaki S, Tomita A, Kobayashi K, et al. Structure of the type V-c CRISPR-cas effector enzyme. Mol Cell (2022), 82(10):1865–77.e4. doi: 10.1016/j.molcel.2022.03.006
49. Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS, Essletzbichler P, et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-cas system. Cell (2015) 163(3):759–71. doi: 10.1016/j.cell.2015.09.038
50. Strecker J, Jones S, Koopal B, Schmid-Burgk J, Zetsche B, Gao L, et al. Engineering of CRISPR-Cas12b for human genome editing. Nat Commun (2019) 10(1):212. doi: 10.1038/s41467-018-08224-4
51. Teng F, Cui T, Feng G, Guo L, Xu K, Gao Q, et al. Repurposing CRISPR-Cas12b for mammalian genome engineering. Cell Discovery (2018) 4:63. doi: 10.1038/s41421-018-0069-3
52. Wu Z, Zhang Y, Yu H, Pan D, Wang Y, Wang Y, et al. Programmed genome editing by a miniature CRISPR-Cas12f nuclease. Nat Chem Biol (2021) 17(11):1132–8. doi: 10.1038/s41589-021-00868-6
53. Kim DY, Lee JM, Moon SB, Chin HJ, Park S, Lim Y, et al. Efficient CRISPR editing with a hypercompact Cas12f1 and engineered guide RNAs delivered by adeno-associated virus. Nat Biotechnol (2022) 40(1):94–102. doi: 10.1038/s41587-021-01009-z
54. Bigelyte G, Young JK, Karvelis T, Budre K, Zedaveinyte R, Djukanovic V, et al. Miniature type V-f CRISPR-cas nucleases enable targeted DNA modification in cells. Nat Commun (2021) 12(1):6191. doi: 10.1038/s41467-021-26469-4
55. Chen JS, Ma E, Harrington LB, Da Costa M, Tian X, Palefsky JM, et al. CRISPR-Cas12a target binding unleashes indiscriminate singlestranded DNase activity. Science (2018) 360(6387):436–9. doi: 10.1126/science.aar6245
56. Winston X, Yan PH, Alfonse LE, Carte JM, Keston-Smith SSE, Garrity AJ, et al. Functionally diverse type V CRISPR-cas systems. Science (2019) p:88–91. doi: 10.1126/science.aav7271
57. Wang X, Shang X, Huang X. Next-generation pathogen diagnosis with CRISPR/Cas-based detection methods. Emerg Microbes Infect (2020) 9(1):1682–91. doi: 10.1080/22221751.2020.1793689
58. Wang M, Zhang R, Li J. CRISPR/cas systems redefine nucleic acid detection: Principles and methods. Biosens Bioelectron (2020) 165:112430. doi: 10.1016/j.bios.2020.112430
59. Korkmaz G, Lopes R, Ugalde AP, Nevedomskaya E, Han R, Myacheva K, et al. Functional genetic screens for enhancer elements in the human genome using CRISPR-Cas9. Nat Biotechnol (2016) 34(2):192–8. doi: 10.1038/nbt.3450
60. Doench JG, Hartenian E, Graham DB, Tothova Z, Hegde M, Smith I, et al. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat Biotechnol (2014) 32(12):1262–7. doi: 10.1038/nbt.3026
61. Choi PS, Meyerson M. Targeted genomic rearrangements using CRISPR/Cas technology. Nat Commun (2014) 5:3728. doi: 10.1038/ncomms4728
62. Park CY, Kim DH, Son JS, Sung JJ, Lee J, Bae S, et al. Functional correction of Large factor VIII gene chromosomal inversions in hemophilia a patient-derived iPSCs using CRISPR-Cas9. Cell Stem Cell (2015) 17(2):213–20. doi: 10.1016/j.stem.2015.07.001
63. Dever DP, Bak RO, Reinisch A, Camarena J, Washington G, Nicolas CE, et al. CRISPR/Cas9 beta-globin gene targeting in human haematopoietic stem cells. Nature (2016) 539(7629):384–9. doi: 10.1038/nature20134
64. Gao X, Tao Y, Lamas V, Huang M, Yeh WH, Pan B, et al. Treatment of autosomal dominant hearing loss by in vivo delivery of genome editing agents. Nature (2018) 553(7687):217–21. doi: 10.1038/nature25164
65. Sledzinski P, Dabrowska M, Nowaczyk M, Olejniczak M. Paving the way towards precise and safe CRISPR genome editing. Biotechnol Adv (2021) 49:107737. doi: 10.1016/j.biotechadv.2021.107737
66. Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, et al. Multiplex genome engineering using CRISPR/Cas systems. Science (2013) 339:819–23. doi: 10.1126/science.1231143
67. Niu Y, Shen B, Cui Y, Chen Y, Wang J, Wang L, et al. Generation of gene-modified cynomolgus monkey via Cas9/RNA-mediated gene targeting in one-cell embryos. Cell (2014) 156(4):836–43. doi: 10.1016/j.cell.2014.01.027
68. Zhang X, Li T, Ou J, Huang J, Liang P. Homology-based repair induced by CRISPR-cas nucleases in mammalian embryo genome editing. Protein Cell (2022) 13(5):316–35. doi: 10.1007/s13238-021-00838-7
69. Byrne SM, Ortiz L, Mali P, Aach J, Church GM. Multi-kilobase homozygous targeted gene replacement in human induced pluripotent stem cells. Nucleic Acids Res (2015) 43(3):e21. doi: 10.1093/nar/gku1246
70. Findlay GM, Boyle EA, Hause RJ, Klein JC, Shendure J. Saturation editing of genomic regions by multiplex homology-directed repair. Nature (2014) 513(7516):120–3. doi: 10.1038/nature13695
71. Yang D, Scavuzzo MA, Chmielowiec J, Sharp R, Bajic A, Borowiak M. Enrichment of G2/M cell cycle phase in human pluripotent stem cells enhances HDR-mediated gene repair with customizable endonucleases. Sci Rep (2016) 6:21264. doi: 10.1038/srep21264
72. Richardson CD, Ray GJ, DeWitt MA, Curie GL, Corn JE. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat Biotechnol (2016) 34(3):339–44. doi: 10.1038/nbt.3481
73. Chu VT, Weber T, Wefers B, Wurst W, Sander S, Rajewsky K, et al. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat Biotechnol (2015) 33(5):543–8. doi: 10.1038/nbt.3198
74. Rees HA, Liu DR. Base editing: Precision chemistry on the genome and transcriptome of living cells. Nat Rev Genet (2018) 19(12):770–88. doi: 10.1038/s41576-018-0059-1
75. Driehuis E, Clevers H. CRISPR/Cas 9 genome editing and its applications in organoids. Am J Physiol Gastrointest Liver Physiol (2017) 312(3):G257–65. doi: 10.1152/ajpgi.00410.2016
76. Koblan LW, Doman JL, Wilson C, Levy JM, Tay T, Newby GA, et al. Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat Biotechnol (2018) 36(9):843–6. doi: 10.1038/nbt.4172
77. Grunewald J, Zhou R, Iyer S, Lareau CA, Garcia SP, Aryee MJ, et al. CRISPR DNA base editors with reduced RNA off-target and self-editing activities. Nat Biotechnol (2019) 37(9):1041–8. doi: 10.1038/s41587-019-0236-6
78. Richter MF, Zhao KT, Eton E, Lapinaite A, Newby GA, Thuronyi BW, et al. Phage-assisted evolution of an adenine base editor with improved cas domain compatibility and activity. Nat Biotechnol (2020) 38(7):883–91. doi: 10.1038/s41587-020-0453-z
79. Zhao D, Li J, Li S, Xin X, Hu M, Price MA, et al. Glycosylase base editors enable c-to-A and c-to-G base changes. Nat Biotechnol (2021) 39(1):35–40. doi: 10.1038/s41587-020-0592-2
80. Kurt IC, Zhou R, Iyer S, Garcia SP, Miller BR, Langner LM, et al. CRISPR c-to-G base editors for inducing targeted DNA transversions in human cells. Nat Biotechnol (2021) 39(1):41–6. doi: 10.1038/s41587-020-0609-x
81. Chen L, Park JE, Paa P, Rajakumar PD, Prekop HT, Chew YT, et al. Programmable C:G to G:C genome editing with CRISPR-Cas9-directed base excision repair proteins. Nat Commun (2021) 12(1):1384. doi: 10.1038/s41467-021-21559-9
82. Li C, Zhang R, Meng X, Chen S, Zong Y, Lu C, et al. Targeted, random mutagenesis of plant genes with dual cytosine and adenine base editors. Nat Biotechnol (2020) 38(7):875–82. doi: 10.1038/s41587-019-0393-7
83. Grunewald J, Zhou R, Lareau CA, Garcia SP, Iyer S, Miller BR, et al. A dual-deaminase CRISPR base editor enables concurrent adenine and cytosine editing. Nat Biotechnol (2020) 38(7):861–4. doi: 10.1038/s41587-020-0535-y
84. Tao W, Liu Q, Huang S, Wang X, Qu S, Guo J, et al. CABE-RY: A PAM-flexible dual-mutation base editor for reliable modeling of multi-nucleotide variants. Mol Ther Nucleic Acids (2021) 26:114–21. doi: 10.1016/j.omtn.2021.07.016
85. Zhang X, Zhu B, Chen L, Xie L, Yu W, Wang Y, et al. Dual base editor catalyzes both cytosine and adenine base conversions in human cells. Nat Biotechnol (2020) 38(7):856–60. doi: 10.1038/s41587-020-0527-y
86. Xie J, Huang X, Wang X, Gou S, Liang Y, Chen F, et al. ACBE, a new base editor for simultaneous c-to-T and a-to-G substitutions in mammalian systems. BMC Biol (2020) 18(1):131. doi: 10.1186/s12915-020-00866-5
87. Liang Y, Xie J, Zhang Q, Wang X, Gou S, Lin L, et al. AGBE: A dual deaminase-mediated base editor by fusing CGBE with ABE for creating a saturated mutant population with multiple editing patterns. Nucleic Acids Res (2022) 50(9):5384–99. doi: 10.1093/nar/gkac353
88. Tong H, Wang X, Liu Y, Liu N, Li Y, Luo J, et al. Programmable a-to-Y base editing by fusing an adenine base editor with an n-methylpurine DNA glycosylase. Nat Biotechnol (2023). doi: 10.1038/s41587-022-01595-6
89. Kim YB, Komor AC, Levy JM, Packer MS, Zhao KT, Liu DR. Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat Biotechnol (2017) 35(4):371–6. doi: 10.1038/nbt.3803
90. Nishimasu H, Shi X, Ishiguro S, Gao L, Hirano S, Okazaki S, et al. Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science (2018) 361(6408):1259–62. doi: 10.1126/science.aas9129
91. Hu JH, Miller SM, Geurts MH, Tang W, Chen L, Sun N, et al. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature (2018) 556(7699):57–63. doi: 10.1038/nature26155
92. Walton RT, Christie KA, Whittaker MN, Kleinstiver BP. Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science (2020) 368(6488):290–6. doi: 10.1126/science.aba8853
93. Li G, Li X, Zhuang S, Wang L, Zhu Y, Chen Y, et al. Gene editing and its applications in biomedicine. Sci China Life Sci (2022) 65(4):660–700. doi: 10.1007/s11427-021-2057-0
94. Li X, Wang Y, Liu Y, Yang B, Wang X, Wei J, et al. Base editing with a Cpf1-cytidine deaminase fusion. Nat Biotechnol (2018) 36(4):324–7. doi: 10.1038/nbt.4102
95. Nishida K, Arazoe T, Yachie N, Banno S, Kakimoto M, Tabata M, et al. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science (2016) 353(6305). doi: 10.1126/science.aaf8729
96. Anzalone AV, Koblan LW, Liu DR. Genome editing with CRISPR-cas nucleases, base editors, transposases and prime editors. Nat Biotechnol (2020) 38(7):824–44. doi: 10.1038/s41587-020-0561-9
97. Yu W, Li J, Huang S, Li X, Li P, Li G, et al. Harnessing A3G for efficient and selective c-to-T conversion at c-rich sequences. BMC Biol (2021) 19(1):34. doi: 10.1186/s12915-020-00879-0
98. Huang TP, Zhao KT, Miller SM, Gaudelli NM, Oakes BL, Fellmann C, et al. Circularly permuted and PAM-modified Cas9 variants broaden the targeting scope of base editors. Nat Biotechnol (2019) 37(6):626–31. doi: 10.1038/s41587-019-0134-y
99. Jiang W, Feng S, Huang S, Yu W, Li G, Yang G, et al. BE-PLUS: A new base editing tool with broadened editing window and enhanced fidelity. Cell Res (2018) 28(8):855–61. doi: 10.1038/s41422-018-0052-4
100. Wang L, Xue W, Yan L, Li X, Wei J, Chen M, et al. Enhanced base editing by co-expression of free uracil DNA glycosylase inhibitor. Cell Res (2017) 27(10):1289–92. doi: 10.1038/cr.2017.111
101. Komor AC, Zhao KT, Packer MS, Gaudelli NM, Waterbury AL, Koblan LW, et al. Improved base excision repair inhibition and bacteriophage mu gam protein yields C:G-to-T:A base editors with higher efficiency and product purity. Sci Adv (2017) 3(8):eaao4774. doi: 10.1126/sciadv.aao4774
102. Liu Z, Chen M, Chen S, Deng J, Song Y, Lai L, et al. Highly efficient RNA-guided base editing in rabbit. Nat Commun (2018) 9(1):2717. doi: 10.1038/s41467-018-05232-2
103. Liu X, Li G, Zhou X, Qiao Y, Wang R, Tang S, et al. Improving editing efficiency for the sequences with NGH PAM using xCas9-derived base editors. Mol Ther Nucleic Acids (2019) 17:626–35. doi: 10.1016/j.omtn.2019.06.024
104. Dang L, Zhou X, Zhong X, Yu W, Huang S, Liu H, et al. Correction of the pathogenic mutation in TGM1 gene by adenine base editing in mutant embryos. Mol Ther (2022) 30(1):175–83. doi: 10.1016/j.ymthe.2021.05.007
105. Xie SS, Zhang Y, Zhang LS, Li GL, Zhao CZ, Ni P, et al. [sgRNA design for the CRISPR/Cas9 system and evaluation of its off-target effects]. Yi Chuan (2015) 37(11):1125–36. doi: 10.16288/j.yczz.15-093
106. Lee HK, Smith HE, Liu C, Willi M, Hennighausen L. Cytosine base editor 4 but not adenine base editor generates off-target mutations in mouse embryos. Commun Biol (2020) 3(1):19. doi: 10.1038/s42003-019-0745-3
107. Rees HA, Komor AC, Yeh WH, Caetano-Lopes J, Warman M, Edge ASB, et al. Improving the DNA specificity and applicability of base editing through protein engineering and protein delivery. Nat Commun (2017) 8:15790. doi: 10.1038/ncomms15790
108. Lee JK, Jeong E, Lee J, Jung M, Shin E, Kim YH, et al. Directed evolution of CRISPR-Cas9 to increase its specificity. Nat Commun (2018) 9(1):3048. doi: 10.1038/s41467-018-05477-x
109. Liu Y, Zhou C, Huang S, Dang L, Wei Y, He J, et al. A cas-embedding strategy for minimizing off-target effects of DNA base editors. Nat Commun (2020) 11(1):6073. doi: 10.1038/s41467-020-19690-0
110. Gehrke JM, Cervantes O, Clement MK, Wu Y, Zeng J, Bauer DE, et al. An APOBEC3A-Cas9 base editor with minimized bystander and off-target activities. Nat Biotechnol (2018) 36(10):977–82. doi: 10.1038/nbt.4199
111. Zhou C, Sun Y, Yan R, Liu Y, Zuo E, Gu C, et al. Off-target RNA mutation induced by DNA base editing and its elimination by mutagenesis. Nature (2019) 571(7764):275–8. doi: 10.1038/s41586-019-1314-0
112. Yu Y, Leete TC, Born DA, Young L, Barrera LA, Lee SJ, et al. Cytosine base editors with minimized unguided DNA and RNA off-target events and high on-target activity. Nat Commun (2020) 11(1):2052. doi: 10.1038/s41467-020-15887-5
113. Jeong YK, Lee S, Hwang GH, Hong SA, Park SE, Kim JS, et al. Adenine base editor engineering reduces editing of bystander cytosines. Nat Biotechnol (2021) 39(11):1426–33. doi: 10.1038/s41587-021-00943-2
114. Li J, Yu W, Huang S, Wu S, Li L, Zhou J, et al. Structure-guided engineering of adenine base editor with minimized RNA off-targeting activity. Nat Commun (2021) 12(1):2287. doi: 10.1038/s41467-021-22519-z
115. Li J, Liu Z, Huang S, Wang X, Li G, Xu Y, et al. Efficient base editing in G/C-rich regions to model androgen insensitivity syndrome. Cell Res (2019) 29(2):174–6. doi: 10.1038/s41422-018-0133-4
116. Ryu SM, Koo T, Kim K, Lim K, Baek G, Kim ST, et al. Adenine base editing in mouse embryos and an adult mouse model of duchenne muscular dystrophy. Nat Biotechnol (2018) 36(6):536–9. doi: 10.1038/nbt.4148
117. Villiger L, Grisch-Chan HM, Lindsay H, Ringnalda F, Pogliano CB, Allegri G, et al. Treatment of a metabolic liver disease by in vivo genome base editing in adult mice. Nat Med (2018) 24(10):1519–25. doi: 10.1038/s41591-018-0209-1
118. Musunuru K, Chadwick AC, Mizoguchi T, Garcia SP, DeNizio JE, Reiss CW, et al. In vivo CRISPR base editing of PCSK9 durably lowers cholesterol in primates. Nature (2021) 593(7859):429–34. doi: 10.1038/s41586-021-03534-y
119. Zeng Y, Li J, Li G, Huang S, Yu W, Zhang Y, et al. Correction of the marfan syndrome pathogenic FBN1 mutation by base editing in human cells and heterozygous embryos. Mol Ther (2018) 26(11):2631–7. doi: 10.1016/j.ymthe.2018.08.007
120. Newby GA, Yen JS, Woodard KJ, Mayuranathan T, Lazzarotto CR, Li Y, et al. Base editing of haematopoietic stem cells rescues sickle cell disease in mice. Nature (2021) 595(7866):295–302. doi: 10.1038/s41586-021-03609-w
121. Liang P, Ding C, Sun H, Xie X, Xu Y, Zhang X, et al. Correction of β-thalassemia mutant by base editor in human embryos. Protein Cell (2017) 8(11):811–22. doi: 10.1007/s13238-017-0475-6
122. Jalil S, Keskinen T, Maldonado R, Sokka J, Trokovic R, Otonkoski T, et al. Simultaneous high-efficiency base editing and reprogramming of patient fibroblasts. Stem Cell Rep (2021) 16(12):3064–75. doi: 10.1016/j.stemcr.2021.10.017
123. Li G, Liu X, Huang S, Zeng Y, Yang G, Lu Z, et al. Efficient generation of pathogenic a-to-G mutations in human tripronuclear embryos via ABE-mediated base editing. Mol Ther Nucleic Acids (2019) 17:289–96. doi: 10.1016/j.omtn.2019.05.021
124. Zhou C, Zhang M, Wei Y, Sun Y, Sun Y, Pan H, et al. Highly efficient base editing in human tripronuclear zygotes. Protein Cell (2017) 8(10):772–5. doi: 10.1007/s13238-017-0459-6
125. Liu Z, Lu Z, Yang G, Huang S, Li G, Feng S, et al. Efficient generation of mouse models of human diseases via ABE- and BE-mediated base editing. Nat Commun (2018) 9(1):2338. doi: 10.1038/s41467-018-04768-7
126. Xie J, Ge W, Li N, Liu Q, Chen F, Yang X, et al. Efficient base editing for multiple genes and loci in pigs using base editors. Nat Commun (2019) 10(1):2852. doi: 10.1038/s41467-019-10421-8
127. Reddy P, Shao Y, Hernandez-Benitez R, Nuñez Delicado E, Izpisua Belmonte JC. First progeria monkey model generated using base editor. Protein Cell (2020) 11(12):862–5. doi: 10.1007/s13238-020-00765-z
128. Wang F, Zhang W, Yang Q, Kang Y, Fan Y, Wei J, et al. Generation of a Hutchinson-gilford progeria syndrome monkey model by base editing. Protein Cell (2020) 11(11):809–24. doi: 10.1007/s13238-020-00740-8
129. Lu Z, He S, Jiang J, Zhuang L, Wang Y, Yang G, et al. Base-edited cynomolgus monkeys mimic core symptoms of STXBP1 encephalopathy. Mol Ther (2022) 30(6):2163–75. doi: 10.1016/j.ymthe.2022.03.001
130. Billon P, Bryant EE, Joseph SA, Nambiar TS, Hayward SB, Rothstein R, et al. CRISPR-mediated base editing enables efficient disruption of eukaryotic genes through induction of STOP codons. Mol Cell (2017) 67(6):1068–1079.e4. doi: 10.1016/j.molcel.2017.08.008
131. Kuscu C, Parlak M, Tufan T, Yang J, Szlachta K, Wei X, et al. CRISPR-STOP: Gene silencing through base-editing-induced nonsense mutations. Nat Methods (2017) 14(7):710–2. doi: 10.1038/nmeth.4327
132. Wang X, Liu Z, Li G, Dang L, Huang S, He L, et al. Efficient gene silencing by adenine base Editor-mediated start codon mutation. Mol Ther (2020) 28(2):431–40. doi: 10.1016/j.ymthe.2019.11.022
133. Gapinske M, Luu A, Winter J, Woods WS, Kostan KA, Shiva N, et al. CRISPR-SKIP: programmable gene splicing with single base editors. Genome Biol (2018) 19(1):107–7. doi: 10.1186/s13059-018-1482-5
134. Liu Y, Li X, He S, Huang S, Li C, Chen Y, et al. Efficient generation of mouse models with the prime editing system. Cell Discovery (2020) 6(1):27. doi: 10.1038/s41421-020-0165-z
135. Lin Q, Zong Y, Xue C, Wang S, Jin S, Zhu Z, et al. Prime genome editing in rice and wheat. Nat Biotechnol (2020) 38(5):582–5. doi: 10.1038/s41587-020-0455-x
136. Liu Y, Yang G, Huang S, Li X, Wang X, Li G, et al. Enhancing prime editing by Csy4-mediated processing of pegRNA. Cell Res (2021) 31(10):1134–6. doi: 10.1038/s41422-021-00520-x
137. Nelson JW, Randolph PB, Shen SP, Everette KA, Chen PJ, Anzalone AV, et al. Engineered pegRNAs improve prime editing efficiency. Nat Biotechnol (2022) 40(3):402–10. doi: 10.1038/s41587-021-01039-7
138. Zhang G, Liu Y, Huang S, Qu S, Cheng D, Yao Y, et al. Enhancement of prime editing via xrRNA motif-joined pegRNA. Nat Commun (2022) 13(1):1856. doi: 10.1038/s41467-022-29507-x
139. Li X, Zhou L, Gao BQ, Li G, Wang X, Wang Y, et al. Highly efficient prime editing by introducing same-sense mutations in pegRNA or stabilizing its structure. Nat Commun (2022) 13(1):1669. doi: 10.1038/s41467-022-29339-9
140. Li X, Wang X, Sun W, Huang S, Zhong M, Yao Y, et al. Enhancing prime editing efficiency by modified pegRNA with RNA G-quadruplexes. J Mol Cell Biol (2022) 14(4). doi: 10.1093/jmcb/mjac022
141. Chen PJ, Hussmann JA, Yan J, Knipping F, Ravisankar P, Chen PF, et al. Enhanced prime editing systems by manipulating cellular determinants of editing outcomes. Cell (2021) 184(22):5635–5652 e29. doi: 10.1016/j.cell.2021.09.018
142. Choi J, Chen W, Suiter CC, Lee C, Chardon FM, Yang W, et al. Precise genomic deletions using paired prime editing. Nat Biotechnol (2022) 40(2):218–26. doi: 10.1038/s41587-021-01025-z
143. Zhuang Y, Liu J, Wu H, Zhu Q, Yan Y, Meng H, et al. Increasing the efficiency and precision of prime editing with guide RNA pairs. Nat Chem Biol (2022) 18(1):29–37. doi: 10.1038/s41589-021-00889-1
144. Anzalone AV, Gao XD, Podracky CJ, Nelson AT, Koblan LW, Raguram A, et al. Programmable deletion, replacement, integration and inversion of large DNA sequences with twin prime editing. Nat Biotechnol (2021), 40(5):731–40. doi: 10.1101/2021.11.01.466790
145. Wang J, He Z, Wang G, Zhang R, Duan J, Gao P, et al. Efficient targeted insertion of large DNA fragments without DNA donors. Nat Methods (2022) 19(3):331–40. doi: 10.1038/s41592-022-01399-1
146. Jiang T, Zhang XO, Weng Z, Xue W. Deletion and replacement of long genomic sequences using prime editing. Nat Biotechnol (2022) 40(2):227–34. doi: 10.1038/s41587-021-01026-y
147. Oh Y, Lee WJ, Hur JK, Song WJ, Lee Y, Kim H, et al. Expansion of the prime editing modality with Cas9 from francisella novicida. Genome Biol (2022) 23(1):92. doi: 10.1186/s13059-022-02644-8
148. Kweon J, Yoon JK, Jang AH, Shin HR, See JE, Jang G, et al. Engineered prime editors with PAM flexibility. Mol Ther (2021) 29(6):2001–7. doi: 10.1016/j.ymthe.2021.02.022
149. Adikusuma F, Lushington C, Arudkumar J, Godahewa GI, Chey YCJ, Gierus L, et al. Optimized nickase- and nuclease-based prime editing in human and mouse cells. Nucleic Acids Res (2021) 49(18):10785–95. doi: 10.1093/nar/gkab792
150. Chow RD, Chen JS, Shen J, Chen S.. A web tool for the design of prime-editing guide RNAs. Nat BioMed Eng (2021) 5(2):190–4. doi: 10.1038/s41551-020-00622-8
151. Kim HK, Yu G, Park J, Min S, Lee S, Yoon S, et al. Predicting the efficiency of prime editing guide RNAs in human cells. Nat Biotechnol (2021) 39(2):198–206. doi: 10.1038/s41587-020-0677-y
152. Hwang GH, Jeong YK, Habib O, Hong SA, Lim K, Kim JS, et al. PE-Designer and PE-analyzer: web-based design and analysis tools for CRISPR prime editing. Nucleic Acids Res (2021) 49(W1):W499–w504. doi: 10.1093/nar/gkab319
153. Qian Y, Zhao D, Sui T, Chen M, Liu Z, Liu H, et al. Efficient and precise generation of Tay-Sachs disease model in rabbit by prime editing system. Cell Discovery (2021) 7(1):50. doi: 10.1038/s41421-021-00276-z
154. Lin J, Liu X, Lu Z, Huang S, Wu S, Yu W, et al. Modeling a cataract disorder in mice with prime editing. Mol Ther Nucleic Acids (2021) 25:494–501. doi: 10.1016/j.omtn.2021.06.020
155. Habib O, Habib G, Hwang GH, Bae S. Comprehensive analysis of prime editing outcomes in human embryonic stem cells. Nucleic Acids Res (2022) 50(2):1187–97. doi: 10.1093/nar/gkab1295
156. Gao R, Fu ZC, Li X, Wang Y, Wei J, Li G, et al. Genomic and transcriptomic analyses of prime editing guide RNA-independent off-target effects by prime editors. CRISPR J (2022) 5(2):276–93. doi: 10.1089/crispr.2021.0080
157. Aida T, Wilde JJ, Yang L, Hou Y, Li M, Xu D, et al. Prime editing in mice reveals the essentiality of a single base in driving tissue-specific gene expression. Genome Biol (2020) 22:83. doi: 10.1186/s13059-021-02304-3
158. Monteys AM, Ebanks SA, Keiser MS, Davidson BL. CRISPR/Cas9 editing of the mutant huntingtin allele in vitro and In vivo. Mol Ther (2017) 25(1):12–23. doi: 10.1016/j.ymthe.2016.11.010
159. Wild EJ, Tabrizi SJ. Therapies targeting DNA and RNA in huntington’s disease. Lancet Neurol (2017) 16(10):837–47. doi: 10.1016/S1474-4422(17)30280-6
160. Wu Y, Zeng J, Roscoe BP, Liu P, Yao Q, Lazzarotto CR, et al. Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat Med (2019) 25(5):776–83. doi: 10.1038/s41591-019-0401-y
161. Canver MC, Smith EC, Sher F, Pinello L, Sanjana NE, Shalem O, et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature (2015) 527(7577):192–7. doi: 10.1038/nature15521
162. Traxler EA, Yao Y, Wang YD, Woodard KJ, Kurita R, Nakamura Y, et al. A genome-editing strategy to treat β-hemoglobinopathies that recapitulates a mutation associated with a benign genetic condition. Nat Med (2016) 22(9):987–90. doi: 10.1038/nm.4170
163. Antoniani C, Meneghini V, Lattanzi A, Felix T, Romano O, Magrin E, et al. Induction of fetal hemoglobin synthesis by CRISPR/Cas9-mediated editing of the human β-globin locus. Blood (2018) 131(17):1960–73. doi: 10.1182/blood-2017-10-811505
164. Topfer SK, Feng R, Huang P, Ly LC, Martyn GE, Blobel GA., et al. Disrupting the adult globin promoter alleviates promoter competition and reactivates fetal globin gene expression. Blood (2022) 139(14):2107–18. doi: 10.1182/blood.2021014205
165. Ye L, Wang J, Tan Y, Beyer AI, Xie F, Muench MO, et al. Genome editing using CRISPR-Cas9 to create the HPFH genotype in HSPCs: An approach for treating sickle cell disease and β-thalassemia. Proc Natl Acad Sci U.S.A. (2016) 113(38):10661–5. doi: 10.1073/pnas.1612075113
166. Li C, Psatha N, Sova P, Gil S, Wang H, Kim J, et al. Reactivation of γ-globin in adult β-YAC mice after ex vivo and in vivo hematopoietic stem cell genome editing. Blood (2018) 131(26):2915–28. doi: 10.1182/blood-2018-03-838540
167. Frangoul H, Altshuler D, Cappellini MD, Chen YS, Domm J, Eustace BK, et al. CRISPR-Cas9 gene editing for sickle cell disease and β-thalassemia. N Engl J Med (2021) 384(3):252–60. doi: 10.1056/NEJMoa2031054
168. Fu B, Liao J, Chen S, Li W, Wang Q, Hu J, et al. CRISPR-Cas9-mediated gene editing of the BCL11A enhancer for pediatric β(0)/β(0) transfusion-dependent β-thalassemia. Nat Med (2022) 28(8):1573–80. doi: 10.1038/s41591-022-01906-z
169. Maeder ML, Stefanidakis M, Wilson CJ, Baral R, Barrera LA, Bounoutas GS, et al. Development of a gene-editing approach to restore vision loss in leber congenital amaurosis type 10. Nat Med (2019) 25(2):229–33. doi: 10.1038/s41591-018-0327-9
170. Gillmore JD, Gane E, Taubel J, Kao J, Fontana M, Maitland ML, et al. CRISPR-Cas9 In vivo gene editing for transthyretin amyloidosis. N Engl J Med (2021) 385(6):493–502. doi: 10.1056/NEJMoa2107454
171. Maule G, Casini A, Montagna C, Ramalho AS, De Boeck K, Debyser Z, et al. Allele specific repair of splicing mutations in cystic fibrosis through AsCas12a genome editing. Nat Commun (2019) 10(1):3556. doi: 10.1038/s41467-019-11454-9
172. Pankowicz FP, Barzi M, Legras X, Hubert L, Mi T, Tomolonis JA, et al. Reprogramming metabolic pathways in vivo with CRISPR/Cas9 genome editing to treat hereditary tyrosinaemia. Nat Commun (2016) 7:12642. doi: 10.1038/ncomms12642
173. Bjursell M, Porritt MJ, Ericson E, Taheri-Ghahfarokhi A, Clausen M, Magnusson L, et al. Therapeutic genome editing with CRISPR/Cas9 in a humanized mouse model ameliorates alpha1-antitrypsin deficiency phenotype. EBioMedicine (2018) 29:104–11. doi: 10.1016/j.ebiom.2018.02.015
174. Ding Q, Strong A, Patel KM, Ng SL, Gosis BS, Regan SN, et al. Permanent alteration of PCSK9 with in vivo CRISPR-Cas9 genome editing. Circ Res (2014) 115(5):488–92. doi: 10.1161/CIRCRESAHA.115.304351
175. Zhang L, Wang L, Xie Y, Wang P, Deng S, Qin A, et al. Triple-targeting delivery of CRISPR/Cas9 to reduce the risk of cardiovascular diseases. Angew Chem Int Ed Engl (2019) 58(36):12404–8. doi: 10.1002/anie.201903618
176. Tabebordbar M, Zhu K, Cheng JKW, Chew WL, Widrick JJ, Yan WX, et al. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science (2016) 351(6271):407–11. doi: 10.1126/science.aad5177
177. Nelson CE, Hakim CH, Ousterout DG, Thakore PI, Moreb EA, Castellanos Rivera RM, et al. In vivo genome editing improves muscle function in a mouse model of duchenne muscular dystrophy. Science (2016) 351(6271):403–7. doi: 10.1126/science.aad5143
178. Long C, Amoasii L, Mireault AA, McAnally JR, Li H, Sanchez-Ortiz E, et al. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science (2016) 351(6271):400–3. doi: 10.1126/science.aad5725
179. Amoasii L, Hildyard JCW, Li H, Sanchez-Ortiz E, Mireault A, Caballero D, et al. Gene editing restores dystrophin expression in a canine model of duchenne muscular dystrophy. Science (2018) 362(6410):86–91. doi: 10.1126/science.aau1549
180. Martyn GE, Wienert B, Kurita R, Nakamura Y, Quinlan KGR, Crossley M. A natural regulatory mutation in the proximal promoter elevates fetal globin expression by creating a de novo GATA1 site. Blood (2019) 133(8):852–6. doi: 10.1182/blood-2018-07-863951
181. Zhao H, Li Y, He L, Pu W, Yu W, Li Y, et al. In vivo AAV-CRISPR/Cas9-Mediated gene editing ameliorates atherosclerosis in familial hypercholesterolemia. Circulation (2020) 141(1):67–79. doi: 10.1161/CIRCULATIONAHA.119.042476
182. Wang S, Min Z, Ji Q, Geng L, Su Y, Liu Z, et al. Rescue of premature aging defects in cockayne syndrome stem cells by CRISPR/Cas9-mediated gene correction. Protein Cell (2020) 11(1):1–22. doi: 10.1007/s13238-019-0623-2
183. Wang L, Yi F, Fu L, Yang J, Wang S, Wang Z, et al. CRISPR/Cas9-mediated targeted gene correction in amyotrophic lateral sclerosis patient iPSCs. Protein Cell (2017) 8(5):365–78. doi: 10.1007/s13238-017-0397-3
184. Song CQ, Wang D, Jiang T, O'Connor K, Tang Q, Cai L, et al. In vivo genome editing partially restores Alpha1-antitrypsin in a murine model of AAT deficiency. Hum Gene Ther (2018) 29(8):853–60. doi: 10.1089/hum.2017.225
185. Yin H, Song CQ, Dorkin JR, Zhu LJ, Li Y, Wu Q, et al. Therapeutic genome editing by combined viral and non-viral delivery of CRISPR system components in vivo. Nat Biotechnol (2016) 34(3):328–33. doi: 10.1038/nbt.3471
186. Yin H, Xue W, Chen S, Bogorad RL, Benedetti E, Grompe M, et al. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat Biotechnol (2014) 32(6):551–3. doi: 10.1038/nbt.2884
187. Ma H, Marti-Gutierrez N, Park SW, Wu J, Lee Y, Suzuki K, et al. Correction of a pathogenic gene mutation in human embryos. Nature (2017) 548(7668):413–9. doi: 10.1038/nature23305
188. Wu SS, Li QC, Yin CQ, Xue W, Song CQ. Advances in CRISPR/Cas-based gene therapy in human genetic diseases. Theranostics (2020) 10(10):4374–82. doi: 10.7150/thno.43360
189. Rothgangl T, Dennis MK, Lin PJC, Oka R, Witzigmann D, Villiger L, et al. In vivo adenine base editing of PCSK9 in macaques reduces LDL cholesterol levels. Nat Biotechnol (2021) 39(8):949–57. doi: 10.1038/s41587-021-00933-4
190. Zeng J, Wu Y, Ren C, Bonanno J, Shen AH, Shea D, et al. Therapeutic base editing of human hematopoietic stem cells. Nat Med (2020) 26(4):535–41. doi: 10.1038/s41591-020-0790-y
191. Rossidis AC, Stratigis JD, Chadwick AC, Hartman HA, Ahn NJ, Li H, et al. In utero CRISPR-mediated therapeutic editing of metabolic genes. Nat Med (2018) 24(10):1513–8. doi: 10.1038/s41591-018-0184-6
192. Song CQ, Jiang T, Richter M, Rhym LH, Koblan LW, Zafra MP, et al. Adenine base editing in an adult mouse model of tyrosinaemia. Nat BioMed Eng (2020) 4(1):125–30. doi: 10.1038/s41551-019-0357-8
193. Chemello F, Chai AC, Li H, Rodriguez-Caycedo C, Sanchez-Ortiz E, Atmanli A, et al. Precise correction of duchenne muscular dystrophy exon deletion mutations by base and prime editing. Sci Adv (2021) 7(18). doi: 10.1126/sciadv.abg4910
194. Liang P, Ding C, Sun H, Xie X, Xu Y, Zhang X, et al. Correction of beta-thalassemia mutant by base editor in human embryos. Protein Cell (2017) 8(11):811–22. doi: 10.1007/s13238-017-0475-6
195. Levy JM, Yeh WH, Pendse N, Davis JR, Hennessey E, Butcher R, et al. Cytosine and adenine base editing of the brain, liver, retina, heart and skeletal muscle of mice via adeno-associated viruses. Nat BioMed Eng (2020) 4(1):97–110. doi: 10.1038/s41551-019-0501-5
196. Kantor A, McClements ME, MacLaren RE. CRISPR-Cas9 DNA base-editing and prime-editing. Int J Mol Sci (2020) 21(17). doi: 10.3390/ijms21176240
197. Böck D, Rothgangl T, Villiger L, Schmidheini L, Matsushita M, Mathis N, et al. In vivo prime editing of a metabolic liver disease in mice. Sci Transl Med (2022) 14(636):eabl9238. doi: 10.1126/scitranslmed.abl9238
198. Liu P, Liang SQ, Zheng C, Mintzer E, Zhao YG, Ponnienselvan K, et al. Improved prime editors enable pathogenic allele correction and cancer modelling in adult mice. Nat Commun (2021) 12(1):2121. doi: 10.1038/s41467-021-22295-w
199. Zhi S, Chen Y, Wu G, Wen J, Wu J, Liu Q, et al. Dual-AAV delivering split prime editor system for in vivo genome editing. Mol Ther (2022) 30(1):283–94. doi: 10.1016/j.ymthe.2021.07.011
200. Jang H, Jo DH, Cho CS, Shin JH, Seo JH, Yu G, et al. Application of prime editing to the correction of mutations and phenotypes in adult mice with liver and eye diseases. Nat BioMed Eng (2022) 6(2):181–94. doi: 10.1038/s41551-021-00788-9
201. Kim Y, Hong SA, Yu J, Eom J, Jang K, Yoon S, et al. Adenine base editing and prime editing of chemically derived hepatic progenitors rescue genetic liver disease. Cell Stem Cell (2021) 28(9):1614–1624.e5. doi: 10.1016/j.stem.2021.04.010
202. Schene IF, Joore IP, Oka R, Mokry M, van Vugt AHM, van Boxtel R, et al. Prime editing for functional repair in patient-derived disease models. Nat Commun (2020) 11(1):5352. doi: 10.1038/s41467-020-19136-7
203. Hong SA, Kim SE, Lee AY, Hwang GH, Kim JH, Iwata H, et al. Therapeutic base editing and prime editing of COL7A1 mutations in recessive dystrophic epidermolysis bullosa. Mol Ther (2022) 30(8):2664–79. doi: 10.1016/j.ymthe.2022.06.005
204. Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science (2003) 302(5644):415–9. doi: 10.1126/science.1088547
205. Hacein-Bey-Abina S, von Kalle C, Schmidt M, Le Deist F, Wulffraat N, McIntyre E, et al. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N Engl J Med (2003) 348(3):255–6. doi: 10.1056/NEJM200301163480314
206. Stein S, Ott MG, Schultze-Strasser S, Jauch A, Burwinkel B, Kinner A, et al. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat Med (2010) 16(2):198–204. doi: 10.1038/nm.2088
207. Braun CJ, Boztug K, Paruzynski A, Witzel M, Schwarzer A, Rothe M, et al. Gene therapy for wiskott-Aldrich syndrome–long-term efficacy and genotoxicity. Sci Transl Med (2014) 6(227):227ra33. doi: 10.1126/scitranslmed.3007280
208. Magnani A, Semeraro M, Adam F, Booth C, Dupre L, Morris EC, et al. Long-term safety and efficacy of lentiviral hematopoietic stem/progenitor cell gene therapy for wiskott-Aldrich syndrome. Nat Med (2022) 28(1):71–80. doi: 10.1038/s41591-021-01641-x
209. Kohn DB, Booth C, Shaw KL, Xu-Bayford J, Garabedian E, Trevisan V, et al. Autologous ex vivo lentiviral gene therapy for adenosine deaminase deficiency. N Engl J Med (2021) 384(21):2002–13. doi: 10.1056/NEJMoa2027675
210. Kohn DB, Booth C, Kang EM, Pai SY, Shaw KL, Santilli G, et al. Lentiviral gene therapy for X-linked chronic granulomatous disease. Nat Med (2020) 26(2):200–6. doi: 10.1038/s41591-019-0735-5
211. Mamcarz E, Zhou S, Lockey T, Abdelsamed H, Cross SJ, Kang G, et al. Lentiviral gene therapy combined with low-dose busulfan in infants with SCID-X1. N Engl J Med (2019) 380(16):1525–34. doi: 10.1056/NEJMoa1815408
212. Hacein-Bey Abina S, Gaspar HB, Blondeau J, Caccavelli L, Charrier S, Buckland K, et al. Outcomes following gene therapy in patients with severe wiskott-Aldrich syndrome. JAMA (2015) 313(15):1550–63. doi: 10.1001/jama.2015.3253
213. Aiuti A, Biasco L, Scaramuzza S, Ferrua F, Cicalese MP, Baricordi C, et al. Lentiviral hematopoietic stem cell gene therapy in patients with wiskott-Aldrich syndrome. Science (2013) 341(6148):1233151. doi: 10.1126/science.1233151
214. Chang CW, Lai YS, Westin E, Khodadadi-Jamayran A, Pawlik KM, Lamb LS Jr, et al. Modeling human severe combined immunodeficiency and correction by CRISPR/Cas9-enhanced gene targeting. Cell Rep (2015) 12(10):1668–77. doi: 10.1016/j.celrep.2015.08.013
215. Iancu O, Allen D, Knop O, Zehavi Y, Breier D, Arbiv A, et al. Multiplex HDR for disease and correction modeling of SCID by CRISPR genome editing in human HSPCs. Mol Ther - Nucleic Acids (2023) 31:105–21. doi: 10.1016/j.omtn.2022.12.006
216. Pavel-Dinu M, Wiebking V, Dejene BT, Srifa W, Mantri S, Nicolas CE, et al. Gene correction for SCID-X1 in long-term hematopoietic stem cells. Nat Commun (2019) 10(1):1634. doi: 10.1038/s41467-019-09614-y
217. Schiroli G, Ferrari S, Conway A, Jacob A, Capo V, Albano L, et al. Preclinical modeling highlights the therapeutic potential of hematopoietic stem cell gene editing for correction of SCID-X1. Sci Transl Med (2017) 9(411). doi: 10.1126/scitranslmed.aan0820
218. Hashikawa Y, Hayashi R, Tajima M, Okubo T, Azuma S, Kuwamura M, et al. Generation of knockout rabbits with X-linked severe combined immunodeficiency (X-SCID) using CRISPR/Cas9. Sci Rep (2020) 10(1):9957. doi: 10.1038/s41598-020-66780-6
219. Miyasaka Y, Wang J, Hattori K, Yamauchi Y, Hoshi M, Yoshimi K, et al. A high-quality severe combined immunodeficiency (SCID) rat bioresource. PloS One (2022) 17(8):e0272950. doi: 10.1371/journal.pone.0272950
220. Zhao Y, Liu P, Xin Z, Shi C, Bai Y, Sun X, et al. Biological characteristics of severe combined immunodeficient mice produced by CRISPR/Cas9-mediated Rag2 and IL2rg mutation. Front Genet (2019) 10:401. doi: 10.3389/fgene.2019.00401
221. Kang JT, Cho B, Ryu J, Ray C, Lee EJ, Yun YJ, et al. Biallelic modification of IL2RG leads to severe combined immunodeficiency in pigs. Reprod Biol Endocrinol (2016) 14(1):74. doi: 10.1186/s12958-016-0206-5
222. Boettcher AN, Li Y, Ahrens AP, Kiupel M, Byrne KA, Loving CL, et al. Novel engraftment and T cell differentiation of human hematopoietic cells in ART(-/-)IL2RG(-/Y) SCID pigs. Front Immunol (2020) 11:100. doi: 10.3389/fimmu.2020.00100
223. Ren J, Yu D, Fu R, An P, Sun R, Wang Z, et al. IL2RG-deficient minipigs generated via CRISPR/Cas9 technology support the growth of human melanoma-derived tumours. Cell Prolif (2020) 53(10):e12863. doi: 10.1111/cpr.12863
224. Li R, Ying B, Liu Y, Spencer JF, Miao J, Tollefson AE, et al. Generation and characterization of an Il2rg knockout Syrian hamster model for XSCID and HAdV-C6 infection in immunocompromised patients. Dis Model Mech (2020) 13(8). doi: 10.1242/dmm.044602
225. Goodwin M, Lee E, Lakshmanan U, Shipp S, Froessl L, Barzaghi F, et al. CRISPR-based gene editing enables FOXP3 gene repair in IPEX patient cells. Sci Adv (2020) 6(19):eaaz0571. doi: 10.1126/sciadv.aaz0571
226. Brault J, Liu T, Bello E, Liu S, Sweeney CL, Meis RJ, et al. CRISPR-targeted MAGT1 insertion restores XMEN patient hematopoietic stem cells and lymphocytes. Blood (2021) 138(26):2768–80. doi: 10.1182/blood.2021011192
227. De Ravin SS, Li L, Wu X, Choi U, Allen C, Koontz S, et al. CRISPR-Cas9 gene repair of hematopoietic stem cells from patients with X-linked chronic granulomatous disease. Sci Transl Med (2017) 9(372). doi: 10.1126/scitranslmed.aah3480
228. Sweeney CL, Pavel-Dinu M, Choi U, Brault J, Liu T, Koontz S, et al. Correction of X-CGD patient HSPCs by targeted CYBB cDNA insertion using CRISPR/Cas9 with 53BP1 inhibition for enhanced homology-directed repair. Gene Ther (2021) 28(6):373–90. doi: 10.1038/s41434-021-00251-z
229. Sweeney CL, Zou J, Choi U, Merling RK, Liu A, Bodansky A, et al. Targeted repair of CYBB in X-CGD iPSCs requires retention of intronic sequences for expression and functional correction. Mol Ther (2017) 25(2):321–30. doi: 10.1016/j.ymthe.2016.11.012
230. Sweeney CL, Choi U, Liu C, Koontz S, Ha SK, Malech HL. CRISPR-mediated knockout of cybb in NSG mice establishes a model of chronic granulomatous disease for human stem-cell gene therapy transplants. Hum Gene Ther (2017) 28(7):565–75. doi: 10.1089/hum.2017.005
231. Rai R, Romito M, Rivers E, Turchiano G, Blattner G, Vetharoy W, et al. Targeted gene correction of human hematopoietic stem cells for the treatment of wiskott - Aldrich syndrome. Nat Commun (2020) 11(1):4034. doi: 10.1038/s41467-020-17626-2
232. Zhou J, Yan Q, Tang C, Liao Y, Zhang Q, Wang X, et al. Development of a rabbit model of wiskott-Aldrich syndrome. FASEB J (2021) 35(2):e21226. doi: 10.1096/fj.202002118RR
233. Raes L, Pille M, Harizaj A, Goetgeluk G, Van Hoeck J, Stremersch S, et al. Cas9 RNP transfection by vapor nanobubble photoporation for ex vivo cell engineering. Mol Ther Nucleic Acids (2021) 25:696–707. doi: 10.1016/j.omtn.2021.08.014
234. Kuo CY, Long JD, Campo-Fernandez B, de Oliveira S, Cooper AR, Romero Z, et al. Site-specific gene editing of human hematopoietic stem cells for X-linked hyper-IgM syndrome. Cell Rep (2018) 23(9):2606–16. doi: 10.1016/j.celrep.2018.04.103
235. Vavassori V, Mercuri E, Marcovecchio GE, Castiello MC, Schiroli G, Albano L, et al. Modeling, optimization, and comparable efficacy of T cell and hematopoietic stem cell gene editing for treating hyper-IgM syndrome. EMBO Mol Med (2021) 13(3):e13545. doi: 10.15252/emmm.202013545
236. Gray DH, Villegas I, Long J, Santos J, Keir A, Abele A, et al. Optimizing integration and expression of transgenic bruton’s tyrosine kinase for CRISPR-Cas9-Mediated gene editing of X-linked agammaglobulinemia. Crispr J (2021) 4(2):191–206. doi: 10.1089/crispr.2020.0080
237. Houghton BC, Panchal N, Haas SA, Chmielewski KO, Hildenbeutel M, Whittaker T, et al. Genome editing with TALEN, CRISPR-Cas9 and CRISPR-Cas12a in combination with AAV6 homology donor restores T cell function for XLP. Front Genome Ed (2022) 4:828489. doi: 10.3389/fgeed.2022.828489
238. Bloomer H, Smith RH, Hakami W, Larochelle A. Genome editing in human hematopoietic stem and progenitor cells via CRISPR-Cas9-mediated homology-independent targeted integration. Mol Ther (2021) 29(4):1611–24. doi: 10.1016/j.ymthe.2020.12.010
239. Drysdale CM, Nassehi T, Gamer J, Yapundich M, Tisdale JF, Uchida N. Hematopoietic-Stem-Cell-Targeted gene-addition and gene-editing strategies for β-hemoglobinopathies. Cell Stem Cell (2021) 28(2):191–208. doi: 10.1016/j.stem.2021.01.001
240. Bolhassani A, Khavari A, Orafa Z. Electroporation: Advantages and drawbacks for delivery of drug, gene and vaccine. Appl Nanotechnol Drug Delivery (2014) p:369–97. doi: 10.5772/58376
241. Liu C, Zhang L, Liu H, Cheng K. Delivery strategies of the CRISPR-Cas9 gene-editing system for therapeutic applications. J Control Release (2017) 266:17–26. doi: 10.1016/j.jconrel.2017.09.012
242. Segel M, Lash B, Song J, Ladha A, Liu CC, Jin X, et al. Mammalian retrovirus-like protein PEG10 packages its own mRNA and can be pseudotyped for mRNA delivery. Science (2021) 373(6557):882–9. doi: 10.1126/science.abg6155
243. Cheng Q, Wei T, Farbiak L, Johnson LT, Dilliard SA, Siegwart DJ. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR-cas gene editing. Nat Nanotechnol (2020) 15(4):313–20. doi: 10.1038/s41565-020-0669-6
244. Hendel A, Bak RO, Clark JT, Kennedy AB, Ryan DE, Roy S, et al. Chemically modified guide RNAs enhance CRISPR-cas genome editing in human primary cells. Nat Biotechnol (2015) 33(9):985–9. doi: 10.1038/nbt.3290
245. Bak RO, Dever DP, Porteus MH. CRISPR/Cas9 genome editing in human hematopoietic stem cells. Nat Protoc (2018) 13(2):358–76. doi: 10.1038/nprot.2017.143
246. Lattanzi A, Meneghini V, Pavani G, Amor F, Ramadier S, Felix T, et al. Optimization of CRISPR/Cas9 delivery to human hematopoietic stem and progenitor cells for therapeutic genomic rearrangements. Mol Ther (2019) 27(1):137–50. doi: 10.1016/j.ymthe.2018.10.008
247. Kim D, Luk K, Wolfe SA, Kim JS. Evaluating and enhancing target specificity of gene-editing nucleases and deaminases. Annu Rev Biochem (2019) 88:191–220. doi: 10.1146/annurev-biochem-013118-111730
248. Allen D, Weiss LE, Saguy A, Rosenberg M, Iancu O, Matalon O, et al. High-throughput imaging of CRISPR- and recombinant adeno-associated virus–induced DNA damage response in human hematopoietic stem and progenitor cells. CRISPR J (2022) 5(1):80–94. doi: 10.1089/crispr.2021.0128
249. Lu Y, Xue J, Deng T, Zhou X, Yu K, Deng L, et al. Safety and feasibility of CRISPR-edited T cells in patients with refractory non-small-cell lung cancer. Nat Med (2020) 26(5):732–40. doi: 10.1038/s41591-020-0840-5
250. Sipe CJ, Claudio Vazquez PN, Skeate JG, McIvor RS, Moriarity BS. Targeted genome editing for the correction or alleviation of primary immunodeficiencies. Prog Mol Biol Transl Sci (2021) 182:111–51. doi: 10.1016/bs.pmbts.2021.03.001
Keywords: inborn errors of immunity (IEIs), Primary Immunodeficiencies (PIDs), CRISPR/Cas, gene therapy, gene editing, base editing, prime editing
Citation: Liu X, Li G, Liu Y, Zhou F, Huang X and Li K (2023) Advances in CRISPR/Cas gene therapy for inborn errors of immunity. Front. Immunol. 14:1111777. doi: 10.3389/fimmu.2023.1111777
Received: 30 November 2022; Accepted: 10 March 2023;
Published: 27 March 2023.
Edited by:
Huawei Mao, Beijing Children’s Hospital, Capital Medical University, ChinaCopyright © 2023 Liu, Li, Liu, Zhou, Huang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xingxu Huang, aHVhbmd4eEBzaGFuZ2hhaXRlY2guZWR1LmNu; Kui Li, bGlrdWlAY2Fhcy5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.