 Susana Lechuga
Susana Lechuga Manuel B. Braga-Neto
Manuel B. Braga-Neto Nayden G. Naydenov
Nayden G. Naydenov Florian Rieder
Florian Rieder Andrei I. Ivanov
Andrei I. Ivanov- 1Department of Inflammation and Immunity, Lerner Research Institute, Cleveland Clinic Foundation, Cleveland, OH, United States
- 2Department of Gastroenterology, Hepatology and Nutrition, Digestive Diseases and Surgery Institute, Cleveland Clinic Foundation, Cleveland, OH, United States
Disruption of the intestinal epithelial barrier is a hallmark of mucosal inflammation. It increases exposure of the immune system to luminal microbes, triggering a perpetuating inflammatory response. For several decades, the inflammatory stimuli-induced breakdown of the human gut barrier was studied in vitro by using colon cancer derived epithelial cell lines. While providing a wealth of important data, these cell lines do not completely mimic the morphology and function of normal human intestinal epithelial cells (IEC) due to cancer-related chromosomal abnormalities and oncogenic mutations. The development of human intestinal organoids provided a physiologically-relevant experimental platform to study homeostatic regulation and disease-dependent dysfunctions of the intestinal epithelial barrier. There is need to align and integrate the emerging data obtained with intestinal organoids and classical studies that utilized colon cancer cell lines. This review discusses the utilization of human intestinal organoids to dissect the roles and mechanisms of gut barrier disruption during mucosal inflammation. We summarize available data generated with two major types of organoids derived from either intestinal crypts or induced pluripotent stem cells and compare them to the results of earlier studies with conventional cell lines. We identify research areas where the complementary use of colon cancer-derived cell lines and organoids advance our understanding of epithelial barrier dysfunctions in the inflamed gut and identify unique questions that could be addressed only by using the intestinal organoid platforms.
Introduction
The intestinal epithelium plays crucial homeostatic roles by forming the protective barrier that separates the internal organs from the luminal microbiota and regulates bidirectional fluxes of water, solutes, nutrients, and waste. These functions are enabled by structural adaptations of intestinal epithelial cells (IEC) that include extensive intercellular contacts and the apico-basal cell polarity (1, 2). Both the intercellular contacts and the polarized architecture of IEC are mediated by elaborate epithelial junctions (3, 4). The two most apically located junctional complexes, tight junctions (TJs) and adherens junctions (AJs), are known to be critical for controlling permeability of the gut barrier, whereas the role of the third junctional complex, desmosomes, remain less understood (3, 4). TJs and AJs represent multiprotein platforms assembled at the plasma membrane that contain transmembrane adhesive and cytoplasmic scaffolding proteins (Figure 1A) (3–5). Transmembrane components of TJs, such as claudins, occludin, and junctional adhesion molecule A (JAM-A), form adhesive bonds with their partners on the opposing cell membrane, whereas on the cytoplasmic side of the membrane, they interact with different scaffolds, most notably members of a ‘zonula occludens’ (ZO) protein family (5, 6). A predominant transmembrane AJ protein, E-cadherin, participates in homotypic interactions with other E-cadherin molecules at the cell surface and makes complexes with cytoplasmic β-catenin, p120-catenin, and α-catenin proteins (7–9).
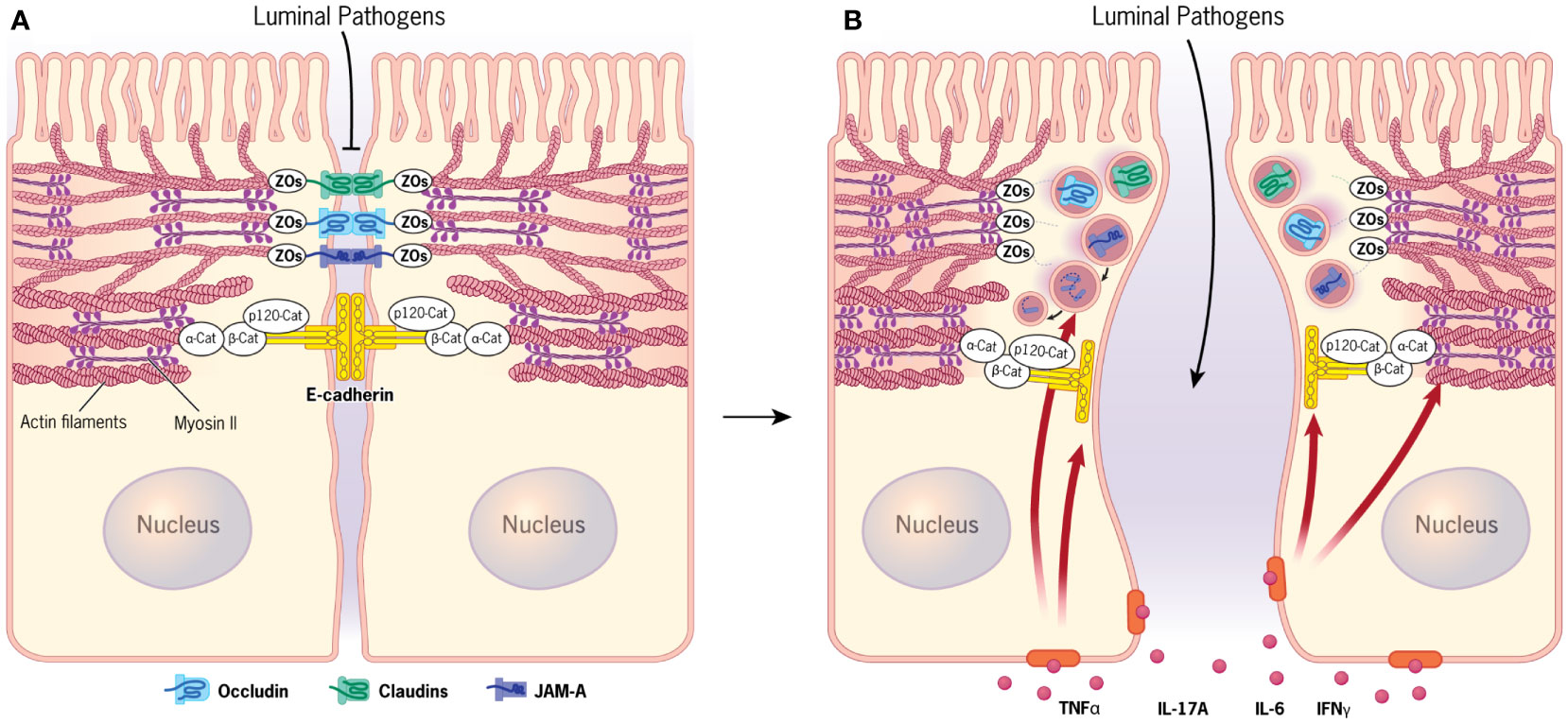
Figure 1 Apical junctions in normal and inflamed intestinal epithelium. (A) Normal intestinal epithelial barrier is established by assembly of tight junctions and adherens junctions. Both junctional complexes are physically associated with the circumferential cytoskeletal belt containing actin bundles and myosin II filaments. (B) Signaling by different cytokines accumulating in the inflamed intestinal mucosa triggers disruption of the epithelial barrier that is driven by removal of junctional proteins from the intercellular contacts and remodeling of the perijunctional actomyosin cytoskeleton. Figure 1 by Gwendolyn Fuller, MFA. Reprinted with the permission of the Cleveland Clinic Center for Medical Art & Photography © 2023. All Rights Reserved.
Proper plasma membrane assembly of different TJ and AJ components is necessary, but not sufficient, to create and maintain the functional intestinal epithelial barrier. An additional mechanism critical for barrier regulation involves the coupling of epithelial junctions to the underlying cortical actin cytoskeleton. A circumferential actin filament belt enriched with an actin motor, non-muscle myosin II (NM II), is a prominent structural feature of well-differentiated IEC (Figure 1A) (10–15). This perijunctional actomyosin belt generates mechanical forces, which stabilize and remodel both AJs and TJs. It also transduces and integrates signaling from different extracellular stimuli and intracellular molecular pathways to modulate tightness of the gut barrier under homeostatic and disease states (10–15).
Disruption of the intestinal epithelial barrier is a well-recognized hallmark of mucosal inflammation. This phenomenon is best studied in chronic autoimmune disorders, such as inflammatory bowel diseases that include Crohn’s disease (CD) and ulcerative colitis (UC), as well as celiac disease (16–20). Additionally, gut barrier leakiness appears to be a common feature for many extraintestinal and systemic disorders with inflammatory etiology, such as asthma, sepsis, diabetes, multiple sclerosis, etc. (21–23). It should be noted that increased permeability of IEC barrier in CD and UC patients develops in parallel to other defects of the intestinal host defense. Such defects can include dysfunctions of Paneth cells leading to diminished secretion of antimicrobial peptides (24–27) and also abnormal mucin production by Goblet cells (24, 28–30). The combined impairment of the cellular and secretory epithelial defense mechanisms in the gut result in excessive exposure to luminal microbiota, leading to activation of the immune response and release of various inflammatory mediators in the intestinal mucosa (17, 19, 21, 23, 31). These inflammatory mediators, including tumor necrosis factor (TNF)-α, interleukins, and interferon (IFN)-γ, act on IEC to accelerate TJ/AJ disassembly and barrier leakiness, thereby accelerating mucosal inflammation (Figure 1B) (31–35).
Multiple mechanisms could contribute to the junctional disassembly and disruption of the epithelial barrier in the inflamed intestinal mucosa (17–19, 36). These mechanisms include the decreased expression of different AJ and TJ proteins, dysregulated vesicular trafficking of junctional components, as well as altered assembly and contractility of junction-associated actomyosin cytoskeleton (17, 18, 36, 37). Such complexity of molecular triggers and mechanisms requires careful selection of appropriate in vitro models to recapitulate and dissect the signaling and cellular responses characteristic of the inflamed human intestine in vivo. Until recently, the experimental toolbox for in vitro studies of the human intestinal epithelial barrier included just a handful of well-differentiated colon cancer-derived cell lines (38). While studies of these model cell lines provided a crucial foundation for our understanding of the structure and function of IEC junctions, whether obtained data faithfully reflect the dynamics and regulation of the gut barrier in vivo is a lingering question.
Development of a new technology allowing ex vivo growth and differentiation of primary intestinal organoids provided a major methodological breakthrough in investigations of gastrointestinal physiology and diseases. This technology has been increasingly applied to study structure and regulation of the intestinal epithelial barrier, resulting in the accumulation of a new wealth of important experimental data while also revealing some limitations of this methodology. It is necessary, therefore, to understand how well the newest data obtained with primary intestinal organoids align with older studies that utilized conventional colonic epithelial cell lines and, in addition, what unique aspects of gut barrier regulation during intestinal inflammation could be discovered using primary human organoids. This review attempts to address these important questions. In order to provide a logical in-depth description of the data, we will focus on human intestinal organoids and colonic epithelial cell lines and minimizing discussions of the data obtained using rodent models. Furthermore, the attention will be limited to responses of primary organoid and conventional IEC to the most studied inflammatory cytokines and some bacterial compounds, such as lipopolysaccharide (LPS), but will exclude studies of intestinal epithelial interactions with bacteria and viruses. Such a rapidly expanding field is largely focused on examining the inflammatory responses and has been recently summarized in several excellent reviews (39–41).
Conventional cell culture models for in vitro studies of human intestinal epithelial barrier
The vast majority of studies that model disruption of the intestinal epithelial barrier during mucosal inflammation in vitro have been performed using a limited set of colonic epithelial cells lines that include T84, Caco-2, and HT-29 cells. All these cell lines originate from human colon carcinoma samples. T84 cells were derived from lung metastasis, and this cell line was established after serial passaging of the tumor specimens in nude mice (42). Caco-2 and HT-29 cells were derived from primary colon tumors (43). When growing on permeable membrane support, T84 cells form well-polarized cell monolayers with developed junctional complexes and a tight paracellular barrier (44–49). Caco-2 cell monolayers also assemble robust apical junctions and establish the paracellular barrier, although not as tight as developed by T84 monolayers (50). Both parental Caco-2 cells and its more differentiated clone, Caco-2BBE, have been used to study IEC junctions (51–54). Unlike T84 and Caco-2 cells, the parental HT-29 cell line is poorly differentiated, and these cells do not form a tight paracellular barrier. However, several well-differentiated clones of HT-29 cells, most notably HT-29/B6 (55–57) and HT-29cf8 (58–60) have been used to study barrier properties and the molecular organization of IEC junctions.
Despite the fact that T84, Caco-2 cells, and HT-29 clones have the morphological characteristics of mature IEC, establish the epithelial barrier, and respond to inflammatory cytokines with barrier disruption, there are several reasons why these cell lines cannot faithfully recapitulate the structural/molecular features and regulation of a normal intestinal epithelium. First, the colonic cancer cell lines have multiple karyotypic abnormalities that include chromosome loss and amplification (61). Consistently, a whole genome transcriptomic and proteomic analyses observes large differences in gene and protein expression profiles between all three model IEC lines and a normal human intestinal epithelium (62, 63). Second, T84, Caco-2, and HT-29 cells possess several oncogenic mutations that could affect junctional integrity and inflammatory signaling. Most common are mutations of the tumor suppressors, p53 and adenomatous polyposis coli (APC), as well as mutations of KRAS, PIK3CA, BRAF, and CTNNB1 genes (64–68). APC and CTNNB1 mutations could directly affect the structure and adhesive properties of AJs by modulating the junctional recruitment of β-catenin (69, 70). Mutations of p53, KRAS, PIK3CA, and BRAF could have indirect effects on the IEC barrier and apical junctions by altering the architecture and dynamics of the perijunctional actin cytoskeleton (71–74). Likewise, extensive pro-oncogenic changes in the genetic landscape of colon cancer cell lines could affect their responses to various inflammatory stimuli, thereby adding concerns about utilizing these cell lines to model molecular pathway characteristics for the inflamed intestinal mucosa in vivo. Finally, well-differentiated T84, Caco-2, and HT-29 monolayers are composed of a relatively homogenous cell population resembling colonic enterocytes and are devoid of the complexity of the gut epithelium that also contains other cellular types, such as Goblet, Paneth, and enteroendocrine cells.
Development of primary human intestinal organoids and measuring integrity of their epithelial barrier
Intestinal organoids represent ex vivo self-organizing cellular structures that mimic the complex organization of gut tissue. Development of the organoids depends on the establishment and maintenance of the intestinal stem cell ‘niche’ with stem cell differentiation into various gut-like structures depending on the chemical composition and mechanical properties of the environment (75). Initially established with mouse small intestinal organoids (76), this technology has been successfully applied to mimic different segments of the human gastrointestinal system (77). Organoid cultures derived from the small intestine are referred to as ‘enteroids’, whereas colon-derived structures are called ‘colonoids’ (78). A comprehensive description of organoid development, features, and applications is provided by several excellent recent reviews (75, 79–82) and will not be detailed here. We will briefly describe major types of human intestinal organoids and outline methodological approaches commonly used to measure epithelial barrier permeability in these model systems.
Human intestinal organoids could be generated from either somatic or pluripotent stem cells (Figure 2). Somatic cell-derived organoids are driven by adult stem cells that populate isolated small intestinal or colonic crypts. The isolated crypts are mounted into the extracellular matrix (ECM) scaffold, such as Matrigel, and supplied with stem cell niche factors that for human organoids include R-spondin, Noggin, EGF, and Wnt3a (75, 80). ECM-embedded organoids initially form spherical cyst-like structures with central lumen and depending on the environment could acquire more complex morphology, including crypt-like protrusions in small intestinal enteroids (Figure 2). The organoids could be propagated and studied in the 3-D culture or dissociated and plated on permeable membrane supports where they form polarized 2-D monolayers (79, 83). The adult stem cell-derived organoids more closely recapitulate the intestinal epithelial layer in vivo since they do not possess either chromosomal abnormalities or oncogenic mutations typical for colon cancer cells. Furthermore, they contain different IEC lineages, which proportions could be experimentally modulated by altering stem cell niche factor signaling (84). This attractive experimental system, however, has its own limitations. One is the lack of the mesenchymal/stromal compartment and immune cells, which precludes studying the regulatory cross-talks between IEC and other mucosal/submucosal cell types. Loss of intestinal intraepithelial lymphocytes (IEL) in crypt derived organoids can be considered as a particular shortcoming of this experimental system. IEL, especially TCRγδ and TCRαβ T cells, are emerging regulators of intestinal homeostasis, host-microbiota interactions and mucosal inflammation (85, 86). Importantly, IEL directly interact with different IEC lineages and regulate barrier integrity, repair, and host antimicrobial defenses (87–89). Other limitations of adult stem cell-derived intestinal organoids are the epigenetic and phenotypic changes that may occur following multiple passages ex vivo, including DNA methylation and cell senescence (90, 91). Finally, there is an issue with data reproducibility since organoids are currently derived from a limited number of different individuals and samples obtained from varying locations within the gut. Future comprehensive and better documented studies of human intestinal organoids with large numbers of individuals are required to separate meaningful population data variability from the experimental noise.
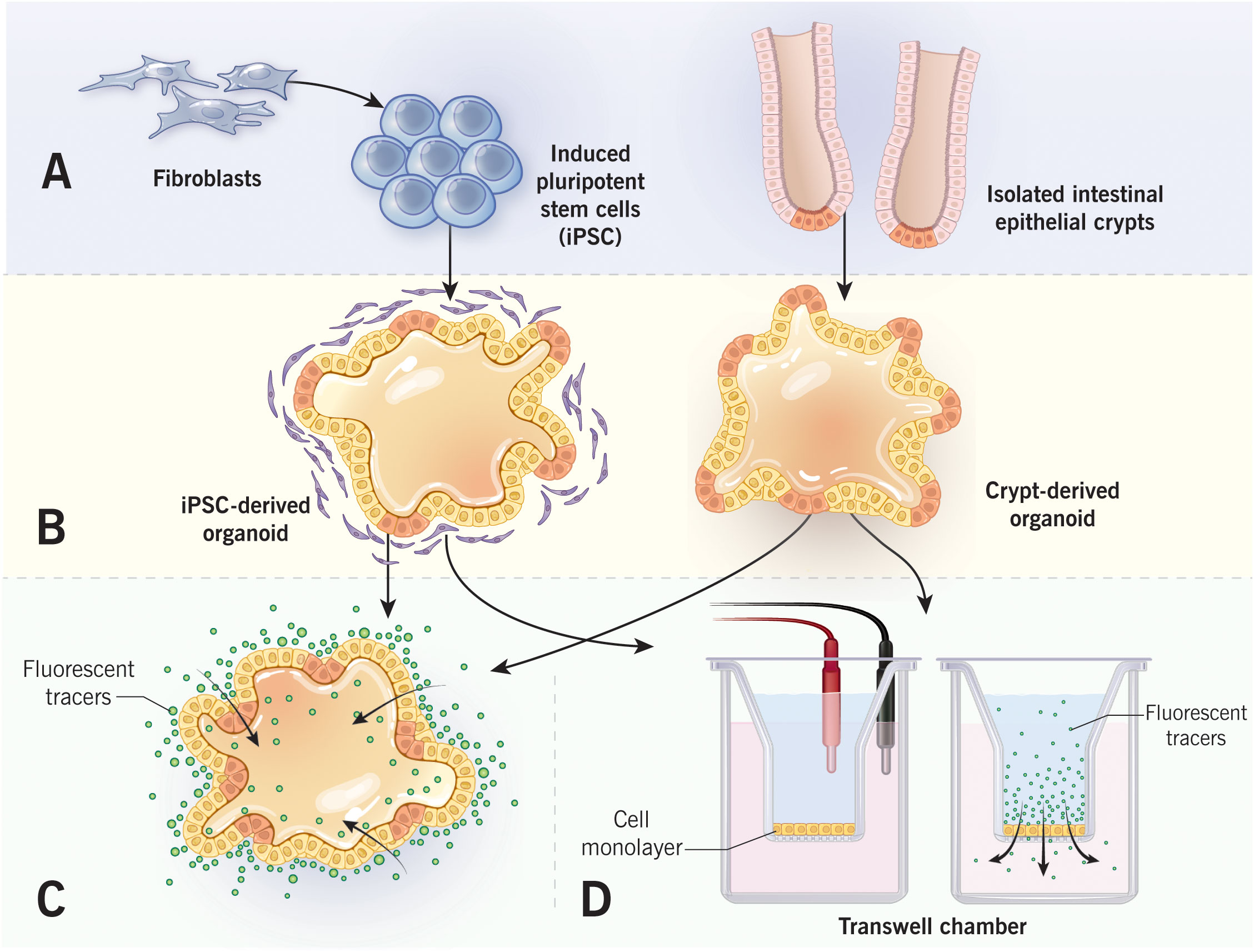
Figure 2 Measurements of epithelial barrier permeability in human intestinal organoids. Intestinal organoids that are derived from either induced pluripotent stem cells or isolated intestinal crypts (A) form cyst-like structure after embedding into 3-D extracellular matrix (B). Epithelial permeability could be evaluated in a 3-D spherical organoids by monitoring the passage of fluorescent markers into organoid lumen (C). Alternatively, organoids could be cultured as 2-D epithelial monolayers in Transwell chambers (D). Permeability of organoid-derived monolayers could be examined by measuring either transepithelial electrical resistance, or transmonolayer flux of fluorescent markers. Figure 2 by Gwendolyn Fuller, MFA. Reprinted with the permission of the Cleveland Clinic Center for Medical Art & Photography © 2022. All Rights Reserved.
An alternative approach involves generating intestinal organoids from human pluripotent stem cells (PSC). The PSC could be established from either pluripotent embryonic stem cells or by inducing differentiated cells, such as fibroblasts, to convert into the pluripotent stage (92, 93). Human intestinal organoids could be generated from PSC via a multistep process involving initial stem cell conversion into appropriate germ layers with subsequent differentiation into tissue-specific organoids (92, 94). Importantly, organoids resembling either the small intestine or colon could be generated by manipulating bone morphogenic protein signaling (92, 94). Unlike somatic stem cell-derived intestinal organoids that contain only epithelial cells, PSC-derived organoids contain both epithelial and mesenchymal compartments, thereby better representing the complexity of the gut tissue. A major drawback for this experimental system, however, is that generation of PSC-derived organoids is a complex multistep process that requires significant expertise and resources. Furthermore, PSC-derived organoids do not differentiate in vitro and need to be transplanted in the kidney capsule of immunocompromised mice in order to achieve complete differentiation (94). As any labor-intensive sophisticated technology generation of PSC-derived organoids is difficult to standardize, and data reproducibility becomes a problem.
Differentiated intestinal organoids provide a unique opportunity to study the assembly and regulation of the gut epithelial barrier either in heterogeneous populations of primary epithelial cells or under more complex conditions involving epithelial-stromal interactions. Different experimental approaches have been developed to measure barrier permeability at two distinct morphological states of the organoids (3-D spheroids versus 2-D monolayers) (38). Permeability of 3-D organoids has been evaluated by measuring transepithelial flux of marker molecules between the organoid lumen and the environment (Figure 2). Two commonly used permeability markers are FITC-Dextran (4,000 Da) and a much smaller fluorescent dye, Lucifer Yellow (95–98). These markers are most frequently added to the cell culture medium, and their accumulation in the organoid’s lumen is measured by fluorescence microscopy (95, 97, 98). Alternatively, the fluorescent marker could be injected into the organoid lumen with subsequent measuring of the decrease in its luminal fluorescence intensity over time (96, 99). When intestinal organoids are cultured as 2-D monolayers on permeable membrane supports, their barrier permeability could be measured using the same experimental approaches developed for conventional epithelial cell monolayers (38, 100). Specifically, transepithelial electrical resistance (TEER) and apical-to-basal flux of FITC dextran or other fluorescent markers are used to measure paracellular ionic permeability and the passage of large uncharged molecules, respectively (Figure 2) (38, 100). Finally, junctional structure could be studied in both 2-D and 3-D organoids by using either transmission electron microscopy or by immunolabeling and confocal microscopy of different TJ and AJ proteins.
Barrier-disrupting effects of inflammatory mediators in human intestinal organoids and conventional IEC lines
Intestinal crypt-derived organoids. Since the introduction of primary human intestinal organoids, they have been increasingly used for understanding the effects of inflammatory cytokines on barrier properties as well as the structure of epithelial junctions either in 3-D spheroids or 2-D organoid-derived monolayers. Most studies focused on the effects of TNFα and IFNγ because these cytokines are essential for IBD pathogenesis and have been extensively investigated using conventional human IEC lines (Table 1) (11, 31–33, 36).
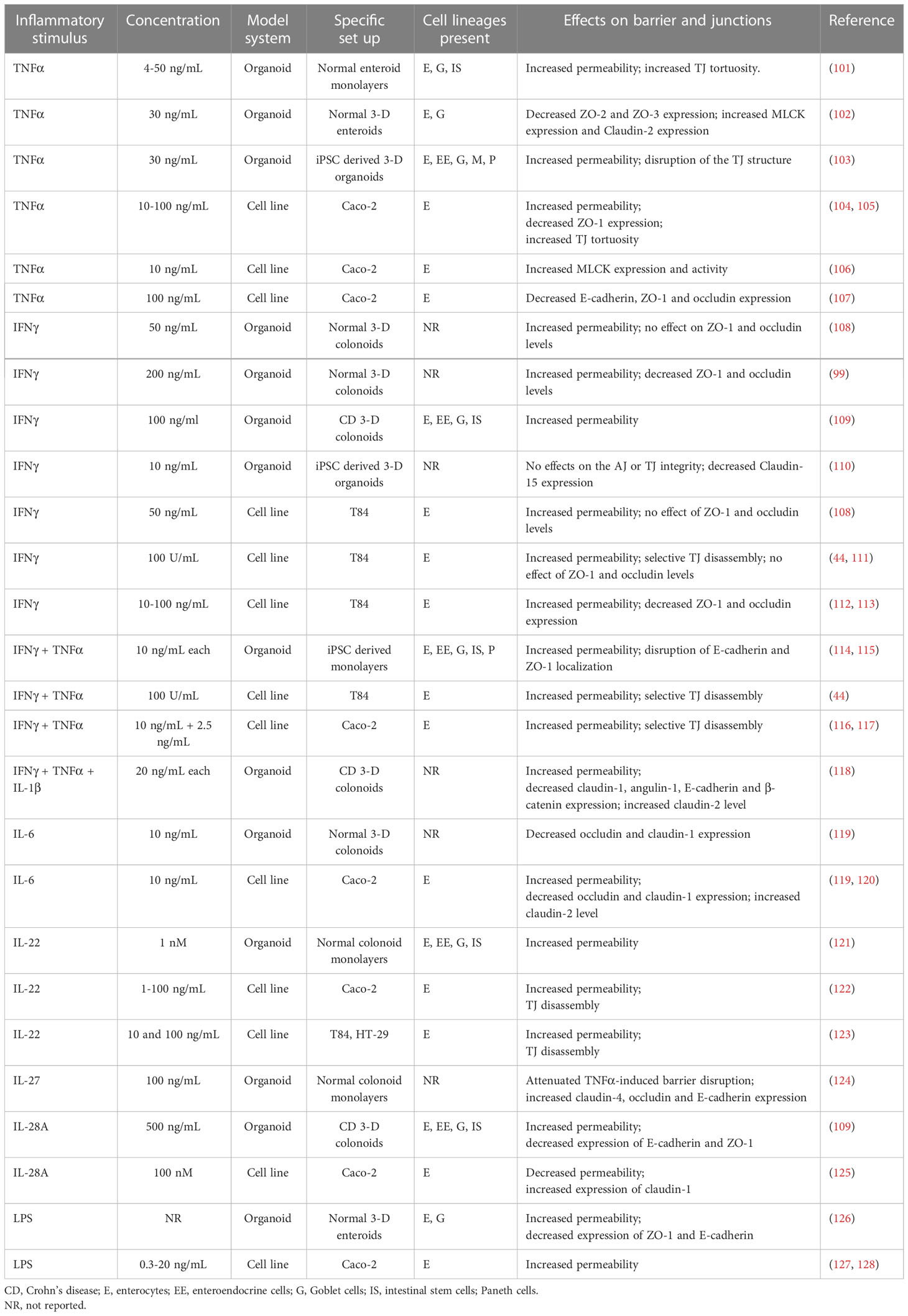
Table 1 Effects of inflammatory mediators on barrier properties of human intestinal organoids and conventional IEC lines.
Primary IEC monolayers derived from human duodenal biopsies developed well-defined TJs and a tight paracellular barrier with TEER ~ 1,000 Ohm x cm2, which significantly exceeded the barrier tightness of Caco-2 cell monolayers used as a control (101). Treatment of these enteroid-derived monolayers with different concentrations of TNFα caused a dose-dependent increase in barrier permeability manifested by decreased TEER and increased Lucifer Yellow flux (101). While TNFα triggered IEC apoptosis, it did not enhance transmonolayer passage of a large tracer, 70 kDa dextran, which indicates a lack of significant epithelial cell loss and acellular gaps in the cytokine-exposed monolayers. Immunofluorescence labeling of ZO-1 in enteroid-derived monolayers did not show gross disruption of the TJ integrity by TNFα; however, it did reveal deformation of intercellular junctions manifested by their tortuous morphology (101). Since the increased TJ tortuosity has been previously attributed to the activation of junction-associated NM II (104), it is likely that TNFα disrupted TJ integrity in organoid-derived IEC monolayers by stimulating actomyosin contractility. This suggestion is consistent with another study that examined the effects of TNFα on 3-D small intestinal enteroids (102). TNFα treatment induced global transcriptional alterations in the enteroids. One of the most interesting effects of TNFα was the expressional upregulation of a key NM II activator, myosin light chain kinase (MLCK), although functional consequences of such MLCK induction was not investigated in this report (102). In addition to MLCK upregulation, TNFα had variable effects on TJ proteins by decreasing ZO-2 and ZO-3 expression and increasing mRNA levels for several other junctional proteins, including a ‘leaky’ claudin-2 (102). These data exemplify multiple mechanisms of TNFα-dependent barrier disruption in primary intestinal organoids that involve the altered expression of junctional components and increased contractility of the junction-associated actomyosin cytoskeleton.
Several recent reports investigated the effects of IFNγ treatment of barrier properties of normal human colonoids (99, 108, 109). All of those studies were performed with 3-D spherical colonoids, by measuring FITC-dextran passage into the spheroid’s lumen to evaluate epithelial barrier integrity. While all studies consistently observed the increased dextran permeability in IFNγ-treated colonoids (99, 108, 109), some variability in possible mechanisms of IEC barrier disruption has been reported. For example, IFNγ exposure decreased ZO-1 and occludin expression in primary colonoids, according to one study (99), but did not change expression of these TJ proteins in a similar experimental system in another report (108). These differences may be due to different cytokine concentrations used in the studies, since a higher IFNγ concentration (200 ng/mL) (99) induced more pronounced junctional damage as compared to its lower concentration (50 ng/mL) (108). A combination of TNFα, IFNγ, and IL-1β caused a marked (~ 2 fold) increase in dextran permeability in CD patient-derived colonoids, which was accompanied by alterations in junctional composition (118). Such alterations involved decreased expression claudin-1, angulin-1, E-cadherin, and β-catenin, as well as upregulation of claudin-2 mRNA levels (118). Furthermore, the combined cytokine treatment markedly activated MLCK and STAT1. Importantly, the cytokine-induced barrier leakiness and junctional abnormalities in CD colonoids were reversed by glucocorticoid treatment, thereby highlighting the colonoid model as an attractive ex vivo system for testing the barrier protective effects of anti-inflammatory drugs (118).
In addition to TNFα and IFNγ, the effects of few other inflammatory cytokines on assembly and permeability of epithelial junctions in human intestinal organoids have been investigated. Thus, IL-6 treatment was shown to downregulate occludin and claudin-1 mRNA expression in normal colonoids by increasing repressive histone methylation at the promoter regions of these transcripts (119). It is unclear, however, if such transcriptional downregulation resulted in any defects in colonoid permeability or their TJ structure. Another study manufactured an ‘intestinal chip’ based on normal colonoid-derived monolayers and exposed these monolayers on the chip to IL-22, a cytokine relevant to IBD pathogenesis (121). IL-22 treatment disrupted epithelial barrier integrity according to the FITC-dextran flux assay by yet to be defined mechanisms. Similar barrier disruptive effects were attributed to IL-28A, which is a newly-identified member of the interferon family (109). A relatively high concentration of IL-28A (500 ng/mL) increased permeability of 3-D colonoids obtained from CD patients that involved activation of the JAK-STAT signaling pathway and was accompanied by decreased expression of ZO-1 and E-cadherin (109). An interesting example of the barrier protective function has been recently described for IL-27, an immunomodulatory cytokine with anti-inflammatory properties (124). Specifically, exposure of colonoid-derived epithelial monolayers to IL-27 markedly attenuated TNFα-induced barrier disruption and also restored diminished expressions of claudin-4, occludin, and E-cadherin in the monolayers exposed to a combination of TNFα and bacterial lipopolysaccharide (LPS) (124).
While several recent reports describe the effects of inflammatory cytokines on barrier permeability in human intestinal organoids, very little is known about the barrier-disruptive actions of bacterial factors under these experimental conditions. One example is a study aimed at modeling necrotizing enterocolitis by examining responses of 3-D neonatal ileal enteroids to bacterial LPS (126). LPS caused a marked increase in enteroid permeability accompanied by the disruption of both AJs and TJs and diminished mRNA expression of occludin and E-cadherin. By contrast, claudin-2 was significantly upregulated by the LPS treatment (126). Furthermore, LPS stimulated transcription of inflammatory cytokines, TNFα and IL-1β; however, it is unclear if such upregulated cytokine expression was responsible for the described barrier-disruptive effects of LPS.
Inducible pluripotent stem cell-derived organoids. Since intestinal organoids generated from human inducible pluripotent stem cells (iPSC) represent more complex structures, retaining both epithelial and mesenchymal compartments, they should more closely recapitulate epithelial responses characteristic of the complex multicellular environment of the intestinal tissue (92–94). Depending on cultured conditions, iPSC-derived intestinal organoids developed either 3-D epithelial structures (103, 110, 129) or 2-D monolayers with well-defined AJs and TJs (114, 115, 130). The epithelial compartment of these organoid was shown to contain all major mucosal cell types, such as enterocytes, Goblet cells, Paneth cells, and enteroendocrine cells, identified by the presence of their lineage-specific protein markers. Interestingly, iPSC differentiated into either colonic or small intestinal monolayers displayed different barrier properties (114). Thus, the TEER values of ~ 1300 Ohm X cm2 reported for colon-like monolayers were significantly higher than the TEER of small intestinal-like monolayers (~ 480 Ohm X cm2) (114). This is consistent with the known differences in permeability of distinct intestinal segments in vivo with the colonic epithelium developing a tighter paracellular barrier as compared to the small intestine (131). Regardless of their morphological appearance (spheres or monolayers), iPSC-derived intestinal organoids responded to inflammatory cytokines with epithelial barrier disruption. Thus, in iPSC-derived intestinal monolayers, exposure to IFNγ and TNFα resulted in decreased TEER and increased transmonolayer FITC-dextran flux (114). Such barrier leakiness was accompanied by the internalization of E-cadherin, disruption of the continuous ZO-1 labeling pattern at TJs, and decreased expression of E-cadherin, ZO-1, and JAM-A (114). Interestingly, in iPSC-derived 3-D intestinal organoids, IFNγ alone did not alter localization of E-cadherin and ZO-1 but caused a selective downregulation of claudin-15 expression (110). By contrast, TNFα treatment of iPSC-derived 3-D organoids resulted in marked disassembly of ZO-1 and occludin-based TJs and increased barrier permeability evident from the enhanced FITC-dextran passage into the spheroids lumen (103).
A unique response of iPSC-derived intestinal organoids to inflammatory stimuli includes the induction of fibrogenic molecular pathways. For example, organoid treatment with TNFα and IL-1α stimulated expression of extracellular matrix proteins, fibronectin and collagen, along with upregulation of a mesenchymal marker α-smooth muscle actin (α-SMA) (132). Furthermore, exposure of iPSC-derived intestinal organoids to a key profibrotic factor, TGF-β, triggered the epithelial-to-mesenchymal transition (EMT) manifested by the increased expression of EMT-related transcriptional factors and the appearance of mesenchymal markers, α-SMA and vimentin, in the epithelial compartment (103, 129).
Overall, the described studies provided solid evidence that the integrity of epithelial barriers developed by either intestinal crypt-derived or iPSC-derived human intestinal organoids become compromised during exposure to key inflammatory cytokines characteristic for IBD mucosa. This highlights intestinal organoids as an attractive model system to evaluate the impact of inflammatory mediators on the intestinal epithelial barrier, investigate their mechanisms of action, and develop novel pharmacological approaches to prevent or reverse compromised epithelial barrier integrity in the inflamed intestinal mucosa.
Comparison of intestinal organoids with conventional IEC lines. A large number of studies focusing on the disruption of the gut barrier during mucosal inflammation have been previously performed using conventional IEC lines (17, 32, 33, 36). More recently, these studies are being replicated using human intestinal organoids; hence, there is a need to reconcile the data obtained using new and old experimental systems. It would allow us to answer several important questions. First, do the previous hypotheses and paradigms regarding barrier disruption in the inflamed gut obtained with colon cancer-derived cell lines still hold true, or they need to be replaced with new paradigms? Second, what are the unique responses and mechanisms that could be examined in organoids but not in IEC lines? And finally, could the old and the new experimental systems be used together, or should we abandon the conventional IEC lines and focus exclusively on the primary intestinal organoids?
So far, published studies describe remarkably similar effects of inflammatory stimuli on the barrier properties and junctional integrity in primary human organoids as well as conventional IEC lines. This is particularly evident for the most investigated responses to TNFα and IFNγ. Indeed, earlier studies that exposed Caco-2 and HT-29 cell monolayers with different concentrations of TNFα (10-100 ng/mL) documented the increased barrier permeability (55, 105, 106, 133) and decreased expression of several TJ proteins (105, 107), which resemble the recently-described effects of TNFα in human intestinal organoids (Table 1). Furthermore, TNFα similarly increased the expression of the ‘leaky’ claudin-2 in Caco-2 or HT-29 cell monolayers (55, 134) and human enteroids (102), which could contribute to cytokine-induced increase in epithelial permeability. Another common mechanism of TNFα-induced barrier disruption involves increased contractility of the perijunctonal actomyosin ring. This mechanism is supported by the upregulated MLCK expression (102, 106) as well as increased TJ tortuosity (101, 104, 105) observed both in TNFα-treated Caco-2 cells and enteroid-derived cell monolayers.
IFNγ alone or in the combination with TNFα was previously identified as a potent disruptor of epithelial barrier integrity in T84 and Caco-2 cell lines (44, 49, 108, 111–113, 116, 117, 133, 135). This is consistent with the barrier-disrupting effects of this cytokine observed in human intestinal organoids (Table 1). Studies addressing the mechanisms of IFNγ-induced disruption of intestinal epithelial barrier yield somewhat controversial results even in similar cellular systems. For example, several reports found decreased expression of the TJ proteins, occludin, and ZO-1, in IFNγ-challenged T84 cells (49, 112, 113), whereas other studies revealed cytokine-induced internalization but not expressional downregulation of these junctional proteins in the same IEC line and using similar IFNγ concentrations (10-100 mg/mL) (44, 108, 111, 136). Likewise, a significant variability of the effects of IFNγ on junctional protein expression was reported by different studies utilizing human intestinal organoids (Table 1). A key mechanism responsible for the IFNγ-induced disruption of epithelial junctions in colonic epithelial cell lines involves increased contractility of the actomyosin cytoskeleton that provides driving forces for junctional disassembly. Early studies performed with T84 and Caco-2 cells revealed such increased contractility is driven by activation of the perijunctional NM II motor by its upstream kinases MLCK and Rho-associated kinase (ROCK) (111, 112, 116, 117, 135). Activation of actomyosin contractility was also observed in primary intestinal organoids exposed to the mixture of IFNγ, TNFα and IL-1β, based on a marked upregulation of myosin light chain phosphorylation (118). However, causal roles of MLCK or ROCK-driven activation of perijunctional actomyosin in cytokine-induced barrier disruption in human intestinal organoids have not been yet demonstrated.
In addition to the described actions by TNFα, and IFNγ, other inflammatory mediators appear to have consistent effects on the barrier properties of human intestinal organoids and conventional IEC lines (Table 1). For instance, potent barrier-disruptive effects of IL-22 were reported in both Caco-2 monolayers and human colonoids (121–123). Additionally, LPS treatment disrupted TJ barrier in neonatal ileal enteroids and Caco-2 cells (126–128). The only notable example of inconsistent response is the opposite effect of IL-28A in human colonoids and Caco-2 cells (109, 125). Specifically, this cytokine disrupted barrier integrity and decreased AJ/TJ protein expression in colonoids (109) but tightened the paracellular barrier and increased claudin-1 expression in Caco-2 monolayers (125). There are several possible reasons for such contrasting responses. They could be related to different epithelial architectures (3-D spheroids versus Caco-2 monolayers) or the fact that colonoids were derived from CD patients and may display unique, disease-dependent responses. Overall, the described data indicate that the disruptive effects of inflammatory mediators on the epithelial barrier of human primary organoids faithfully reproduced barrier dysfunctions previously described in colon cancer-derived IEC lines. Furthermore, associative evidence suggests that mechanisms underlying epithelial barrier disruption could also be similar in primary intestinal organoids and conventional IEC lines. In this specific field, the utilization of organoids neither resulted in the paradigm shift, nor provided major advances in understanding disruption of the gut barrier during intestinal inflammation.
Inflammatory mediator induced programmed cell death in intestinal organoids
One of the most obvious benefits of using intestinal organoids is an opportunity to study the roles and mechanisms of the programmed cell death in the inflamed mucosa. Cell death is an important homeostatic process that mediates self-renewal of the normal intestinal epithelium, where terminally-differentiated cells undergo constant apoptosis and shedding from the gut surface into the lumen (137, 138). This homeostatic cell death is known to be dysregulated in the inflamed intestinal epithelium of UC and CD patients (139–141). Active inflammation stimulates several major cell death pathways in IBD mucosa, including apoptosis, necroptosis and pyroptosis (139–141). One could suggest such excessive IEC death would lead to the disruption of the intestinal epithelial barrier by either destabilizing junctional complexes or creating cell-free mucosal wounds. Surprisingly, the causal role of cell death in compromising the IEC barrier remains underappreciated. One reason is studies performed with T84 and Caco-2 epithelial cells concluding that apoptosis does not play a major role in IEC barrier disruption caused by TNFα and IFNγ (44, 116, 133, 142). This conclusion was based on either a lack of apoptotic marker induction in cytokine-treated IEC or the inefficiency of pharmacological inhibition of apoptosis in preventing cytokine-induced barrier breakdown (44, 116, 133, 142). Subsequent studies with murine intestinal organoids, however, convincingly demonstrated TNFα, IFNs, and IL-22 induce strong programmed cell death responses in IEC, primarily via the apoptotic and necroptotic mechanisms (143–148). Furthermore, a direct comparison of primary murine organoids and Caco-2 cells exposed to different cytotoxic agents, such as TNFα, chemotherapeutic drugs, and X-ray irradiation demonstrated a high magnitude of cell death in primary organoids and blunted cytotoxic responses of Caco-2 cells (149). This important study provides direct evidence that colon cancer-derived IEC lines could be generally resistant to cell death, presumably due to the activation of pro-survival signaling pathways by their oncogenic mutations.
Several recent studies with human intestinal organoids also suggest that IEC death is a common response to different inflammatory cytokines (Table 2). Thus, apoptosis was detected in human duodenal enteroid-monolayers and 3-D colonoids treated with either TNFα alone or in a combination with IL-1β and flagellin (101, 150). Likewise, apoptosis was induced in normal 3-D colonoids and ileal enteroids by a combination of types I, II, and III interferons, and was significantly exaggerated by the co-treatment of interferons and TNFα (152). Another study reported that TNFα induced the necroptotic cell death in small intestinal enteroids derived from CD patients and non-IBD controls by stimulating the expression of several necroptosis mediators including the mixed-lineage kinase domain-like pseudokinase (102). Yet another report suggested the combination of TNFα and IFNγ triggers a non-canonical cell death in colonoids derived from CD patients and non-IBD controls (151). Such non-canonical cell death does not involve apoptotic and necroptotic signaling, but it depends on the JAK1/2-STAT1 activation and non-enzymatic scaffolding activity of caspase 8 (151). In addition to the described cytotoxic effects of TNFα and interferons, other cytokines such as IL-17 and IL-22 were shown to induce different cell death pathways in human primary intestinal organoids (121, 153) (Table 2). For example, IL-17A triggered pyroptotic cell death in small intestinal enteroids detected by the cleavage of Gasdermin D and reversed by caspase-1 inhibition (153). On the other hand, IL-22 was shown to induce a classical caspase-3 dependent apoptosis in human colonoid monolayers grown on the intestinal chips (121).
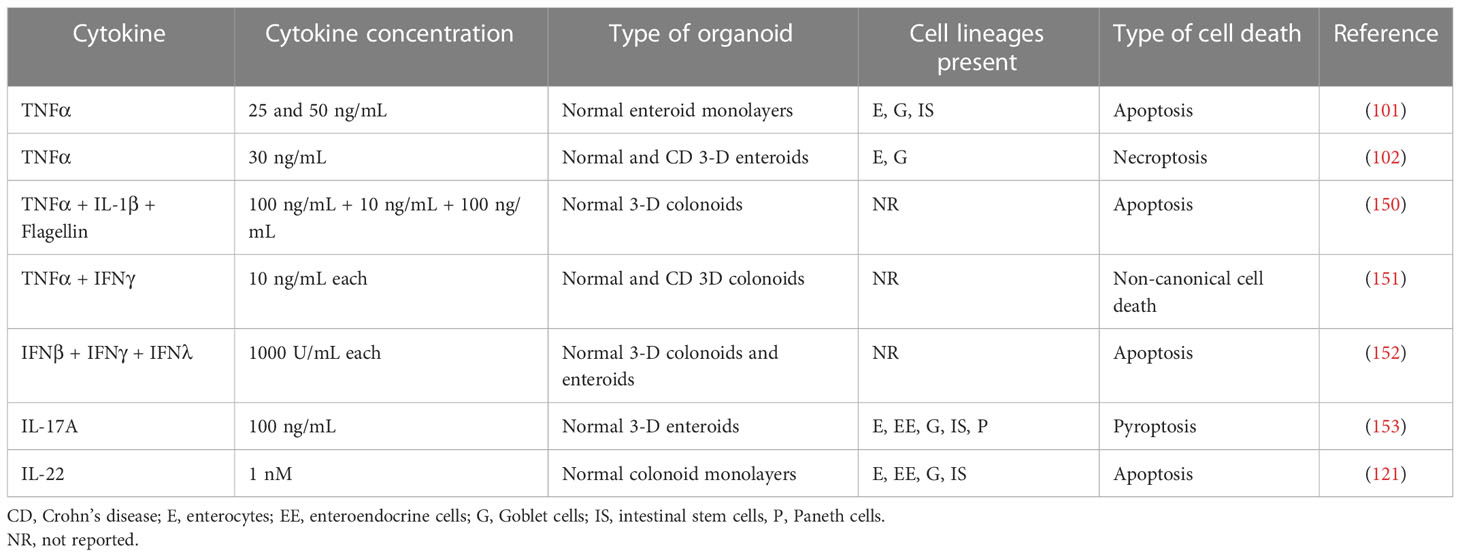
Table 2 Inflammatory cytokine-induced cell death in human intestinal organoids.
It should be noted that while the described studies demonstrated the induction of cell death in cytokine-exposed human intestinal organoids, which in some instances was accompanied by increased barrier permeability and the loss of epithelial junctions (101, 102, 121), a causal link between IEC death and barrier disruption is yet to be established. Specifically, there is no data to demonstrate either the pharmacological or genetic inhibition of apoptosis, necroptosis, or pyroptosis could attenuate the barrier breakdown in cytokine-treated organoids. It could be, however, a challenge to prove such a causal relationship, given a recently-proposed concept of the integrated inflammatory cell death program called PANoptosis (154, 155). This concept implies that key programmed cell death pathways could be simultaneously activated by inflammatory stimuli, and they are commonly controlled by a cytoplasmic multiprotein complex, PANoptosome. The individual inhibition of apoptosis, necroptosis, or pyroptosis may not be sufficient to prevent inflammatory cell death that will proceed via alternative pathways (154, 155). PANoptosis induction has been recently described in cancer and immune cells treated by the combination of TNFα and IFNγ (156, 157). Such an integrated response could explain the diversity of cell death pathways reported in the cytokine-treated human intestinal organoids (Table 2) as well as the inefficiency of pharmacological inhibition of apoptosis in restoring barrier integrity of TNFα/IFNγ treated T84 monolayers (44). We conclude this part by highlighting intestinal organoids as preferred models to study the roles and mechanisms of IEC death in inflamed human intestinal mucosa. Future studies using these experimental systems should shed light on the contribution of the inflammatory cell death pathway in the disruption of the gut barrier in IBD and other inflammatory disorders.
Compromised barrier integrity and altered composition of apical junctions in intestinal organoids derived from IBD and celiac disease patients
Several recent studies demonstrated that organoids generated from either intestinal crypts or iPSC of IBD and celiac disease patients preserve unique transcriptomic signatures and inflammatory features described for the intestinal epithelium in these diseases (110, 158–162). Such sustained genetic reprogramming has been linked to epigenetic mechanisms, such as changes in DNA methylation patterns (160). One could suggest, therefore, the intestinal organoids also preserve defects in the epithelial barrier integrity and junctional structure characteristics for IBD and other intestinal disorders. Several recent studies tested this idea by investigating the barrier properties and molecular composition of apical junctions in intestinal organoids generated from IBD mucosa (Table 3).
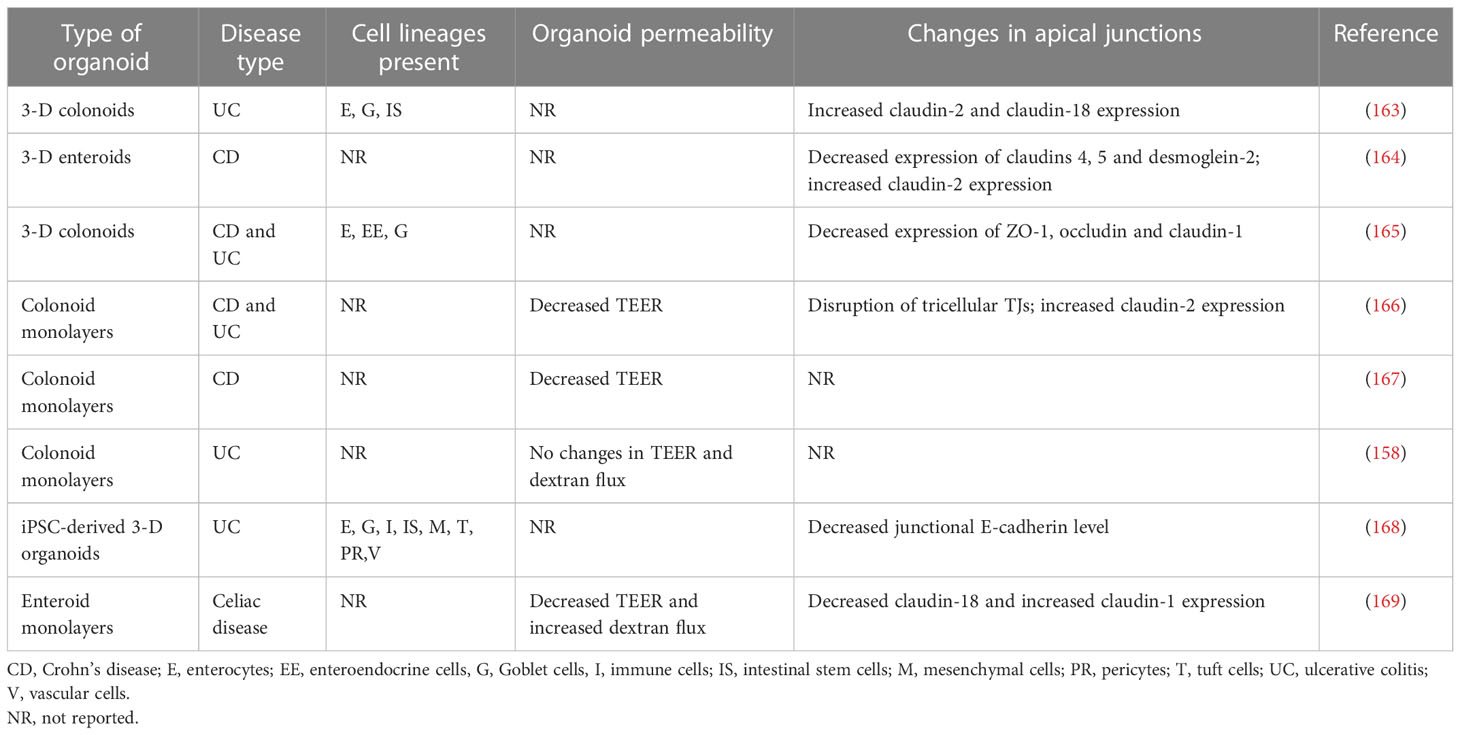
Table 3 Increased barrier permeability and altered apical junction composition in intestinal organoids of IBD and celiac disease patients.
The reported data remain fragmented and contradictory. For example, a study utilizing colonoid-derived epithelial monolayers obtained from actively inflamed tissue biopsies of UC and CD patients reported significant increases in their ionic permeability when compared to the monolayers obtained from non-IBD controls (166). Such barrier leakiness of IBD-derived monolayers was associated with a selective disruption of tricellular TJs and was reversible by pharmacological activation of the AMP-dependent protein kinase (166). Another study reported the increased ionic permeability of colonoid-derived IEC monolayers obtained from non-inflamed CD crypts (167). By contrast, colonic cell monolayers obtained from UC patients had unaltered barrier properties according to TEER and transmonolayer FITC-dextran flux measurements in comparison to the permeability of non-IBD colonoid derived monolayers (158). According to this report, UC colonoids still displayed a diseases-specific transcriptional signature distinct from normal mucosa-derived colonoids, which was not sufficient to cause epithelial barrier disruption (158).
Not only barrier leakiness but also altered molecular composition of epithelial junctions observed in the intestinal mucosa of IBD patients was shown to be preserved in cultured intestinal organoids. For example, a recent study performed a side-by side comparison of the expressional profiles of different junctional proteins in surgically resected ileal samples of CD patients and non-IBD controls to enteroids generated from these tissue samples (164). Importantly, enteroids produced from both severely inflamed and non-inflamed segments of the same tissue samples were examined. This study revealed the decreased protein expression of major TJ, AJ, and desmosomal molecules—such as claudins 1, 4, and 5, E-cadherin, desmoglein-2, and desmocolin-2—both in the non-inflamed and inflamed tissue segments of CD mucosa in comparison to non-IBD controls. By contrast, the expression of claudin-2 protein was upregulated. Interestingly, many of these molecular alterations, including the downregulation of claudins 4 and 5, desmoglein-2, and the upregulation of claudin-2 protein expression were preserved in enteroids generated from the inflamed CD ileum (164). On the other hand, the downregulation of claudin-1, E-cadherin, and desmocolin-2 was not preserved in inflamed CD enteroids. This study also reported an interesting mechanistic observation that the described alterations in junctional composition were at the protein, not mRNA levels. Expectedly, studies examining the expression of junctional proteins in IBD-derived intestinal organoids showed some data variability. Thus, the decreased expression of claudin-1, occludin, and ZO-1 proteins was observed in another cohort of IBD-derived colonoids (165). This contradicts the results obtained with small intestinal enteroids of CD patients, which did not show altered claudin-1 and occludin expression not only in the cultured organoids but even in the original patient tissues (164). Furthermore, while increased expression of claudin-2 protein was consistently found in IBD organoids, such increased protein level was associated with the enhanced claudin-2 mRNA transcription in UC, but not in CD colonoids (163, 164). Possible reasons for such variable results could include differential mucosal responses to inflammation in either of the different IBD types (CD versus UC) or in distinct gut regions (ileum versus colon).
It remains unknown if the altered structure and permeability of epithelial junctions found in IBD intestinal mucosa could be recapitulated in human organoids generated from iPSCs of these patients. Only one study demonstrated concordant decreased expression of E-cadherin along with increased level of RhoA, an important junctional regulator, both in colonic mucosa of UC and paired iPSC-derived intestinal organoids (168). Another study, while demonstrating that iPSC-derived organoids of IBD patients and non-IBD controls respond to proinflammatory cytokines with barrier disruption, did not include sufficient number of samples to detect significant changes in baseline barrier properties of IBD versus non-IBD organoids (114).
Defects in intestinal epithelial barrier integrity also appear to be preserved in enteroids generated from celiac disease patients. Indeed, a recent study that examined duodenal enteroid-derived IEC monolayers reported leakier barrier in celiac disease monolayers as compared to non-celiac controls (169). Such barrier leakiness was manifested by lower TEER and higher transmonolayer FITC-dextran flux. Furthermore, the altered molecular composition of TJs was detected in celiac patient derived IEC monolayers that involved decreased junctional recruitment of ZO-1 and dysregulated expression of several claudin proteins (169). Interestingly, exposure to gliadin, a major trigger of mucosal inflammation in celiac disease, resulted in additional barrier leakiness in celiac, but not control, monolayers that was accompanied by the robust release of several inflammatory cytokines (169). Overall, the described studies identified another valuable application of human intestinal organoids, which provides the unique opportunity to characterize barrier defects of patients with IBD, celiac disease, and other inflammatory disorders as well as understand the molecular mechanisms that underline such barrier dysfunction.
Conclusion
Human intestinal organoids provide a robust, physiologically-relevant experimental platform to study gastrointestinal homeostasis and diseases. A cadre of recent studies used this novel methodology to model the homeostatic development of the intestinal epithelium and characterize epithelial abnormalities in intestinal disorders (79, 83, 170–172). One of the most attractive applications of intestinal organoid research is to understand the role and mechanisms of intestinal barrier disruption during gut inflammation. There are, however, intrinsic problems and limitations of the organoid utilization, such as their significant heterogeneity, alterations in long-term culture, high data variability, the significant cost and labor intensity of the technology (81, 92, 93, 173). Furthermore, the most frequently used adult stem cell-derived organoids still do not recapitulate the complexity of intestinal tissue due to lack of intraepithelial immune cells and the stromal compartment. Because of these limitations it is important to understand which unique aspect of gut barrier regulation should be investigated using the organoid technology and which could be addressed by working with conventional IEC lines. So far, studies of epithelial barrier disruption in human intestinal organoids recapitulated many phenomena already described in conventional IEC lines and did not discover major novel responses or mechanisms. This does not undermine the value of intestinal organoid research, but, rather, reflects the fact that conventional IEC lines remain valuable models to study intestinal barrier integrity and dynamics under homeostatic conditions and in diseases. There are, however, several particular aspects of intestinal barrier regulation that should be investigated using primary intestinal organoids. Thus, intestinal organoids allow to examine barrier permeability and apical junction integrity in a complex system composed of different intestinal epithelial cell lineages, such as enterocytes, Goblet and Paneth cells. More importantly, primary enteroids represent the only experimental system to study structure and regulation of human small intestinal epithelial barrier due to lack of well-differentiated human small intestinal epithelial cell lines. Likewise, human intestinal organoids represent a superb experimental platform to examine the mechanism of programmed cell death and its effect on intestinal epithelial barrier integrity. Furthermore, patient-derived intestinal organoids provide a unique opportunity to investigate the mechanisms underlying leaky gut barrier in various diseases such as IBD and to develop barrier protecting pharmacological therapies. Finally, iPSC-derived intestinal organoids allow to investigate complex epithelial-stromal interactions and fibrogenic pathways induced by TGF-β and other profibrotic factors in the gut. A particularly important advancement provided by iPSC-derived organoids is the ability to study EMT, junctional disassembly, and tumor metastasis in human colon cancers. In summary, it would be wise to give a negative answer to the question presented in the title of this manuscript. While studying disruption of the gut barrier during mucosal inflammation we should use advances of both traditional IEC cell lines and intestinal organoids in order to understand the roles and mechanisms of this key manifestation of different gastrointestinal disorders.
Author contributions
Analyzed data and participated in writing, SL, MBN, NN, FR, and AI. Supervision, AI. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Institute Of Diabetes And Digestive And Kidney Diseases of the National Institutes of Health under Award Numbers R01DK126702 and R01DK131550, to A.I.I.
Acknowledgments
We thank Mr. Kyle Bandy for the excellent editorial assistance.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Markov AG, Aschenbach JR, Amasheh S. The epithelial barrier and beyond: Claudins as amplifiers of physiological organ functions. IUBMB Life (2017) 69:290–6. doi: 10.1002/iub.1622
2. Snoeck V, Goddeeris B, Cox E. The role of enterocytes in the intestinal barrier function and antigen uptake. Microbes Infect (2005) 7:997–1004. doi: 10.1016/j.micinf.2005.04.003
3. Garcia MA, Nelson WJ, Chavez N. Cell-cell junctions organize structural and signaling networks. Cold Spring Harb Perspect Biol (2018) 10:a029181. doi: 10.1101/cshperspect.a029181
4. Rubsam M, Broussard JA, Wickstrom SA, Nekrasova O, Green KJ, Niessen CM. Adherens junctions and desmosomes coordinate mechanics and signaling to orchestrate tissue morphogenesis and function: An evolutionary perspective. Cold Spring Harb Perspect Biol (2018) 10:a029207. doi: 10.1101/cshperspect.a029207
5. Van Itallie CM, Anderson JM. Architecture of tight junctions and principles of molecular composition. Semin Cell Dev Biol (2014) 36:157–65. doi: 10.1016/j.semcdb.2014.08.011
6. Suzuki T. Regulation of intestinal epithelial permeability by tight junctions. Cell Mol Life Sci (2013) 70:631–59. doi: 10.1007/s00018-012-1070-x
7. Ivanov AI, Naydenov NG. Dynamics and regulation of epithelial adherens junctions: recent discoveries and controversies. Int Rev Cell Mol Biol (2013) 303:27–99. doi: 10.1016/B978-0-12-407697-6.00002-7
8. Takeichi M. Dynamic contacts: rearranging adherens junctions to drive epithelial remodelling. Nat Rev Mol Cell Biol (2014) 15:397–410. doi: 10.1038/nrm3802
9. Troyanovsky S. Adherens junction assembly. Sub-cellular Biochem (2012) 60:89–108. doi: 10.1007/978-94-007-4186-7_5
10. Charras G, Yap AS. Tensile forces and mechanotransduction at cell-cell junctions. Curr Biol (2018) 28:R445–57. doi: 10.1016/j.cub.2018.02.003
11. He WQ, Wang J, Sheng JY, Zha JM, Graham WV, Turner JR. Contributions of myosin light chain kinase to regulation of epithelial paracellular permeability and mucosal homeostasis. Int J Mol Sci (2020) 21:993. doi: 10.3390/ijms21030993
12. Ivanov AI. Actin motors that drive formation and disassembly of epithelial apical junctions. Front bioscience (2008) 13:6662–81. doi: 10.2741/3180
13. Ivanov AI, Lechuga S, Marino-Melendez A, Naydenov NG. Unique and redundant functions of cytoplasmic actins and nonmuscle myosin II isoforms at epithelial junctions. Ann N Y Acad Sci (2022) 1515:61–74. doi: 10.1111/nyas.14808
14. Mege RM, Ishiyama N. Integration of cadherin adhesion and cytoskeleton at adherens junctions. Cold Spring Harb Perspect Biol (2017) 9:a028738. doi: 10.1101/cshperspect.a028738
15. Varadarajan S, Stephenson RE, Miller AL. Multiscale dynamics of tight junction remodeling. J Cell Sci (2019) 132:jcs229286. doi: 10.1242/jcs.229286
16. Barone MV, Auricchio R, Nanayakkara M, Greco L, Troncone R, Auricchio S. Pivotal role of inflammation in celiac disease. Int J Mol Sci (2022) 23:7177. doi: 10.3390/ijms23137177
17. Buckley A, Turner JR. Cell biology of tight junction barrier regulation and mucosal disease. Cold Spring Harb Perspect Biol (2018) 10:a029314. doi: 10.1101/cshperspect.a029314
18. Guo C, Shen J. Cytoskeletal organization and cell polarity in the pathogenesis of crohn's disease. Clin Rev Allergy Immunol (2021) 60:164–74. doi: 10.1007/s12016-020-08795-5
19. Laukoetter MG, Bruewer M, Nusrat A. Regulation of the intestinal epithelial barrier by the apical junctional complex. Curr Opin Gastroenterol (2006) 22:85–9. doi: 10.1097/01.mog.0000203864.48255.4f
20. Michielan A, D'Inca R. Intestinal permeability in inflammatory bowel disease: Pathogenesis, clinical evaluation, and therapy of leaky gut. Mediators Inflammation (2015) 2015:628157. doi: 10.1155/2015/628157
21. Fasano A. All disease begins in the (leaky) gut: role of zonulin-mediated gut permeability in the pathogenesis of some chronic inflammatory diseases. F1000Res (2020) 9:69. doi: 10.12688/f1000research.20510.1
22. Pellegrini C, Fornai M, D'Antongiovanni V, Antonioli L, Bernardini N, Derkinderen P. The intestinal barrier in disorders of the central nervous system. Lancet Gastroenterol Hepatol (2023) 8:66–80. doi: 10.1016/S2468-1253(22)00241-2
23. Sugita K, Kabashima K. Tight junctions in the development of asthma, chronic rhinosinusitis, atopic dermatitis, eosinophilic esophagitis, and inflammatory bowel diseases. J Leukoc Biol (2020) 107:749–62. doi: 10.1002/JLB.5MR0120-230R
24. Antoni L, Nuding S, Wehkamp J, Stange EF. Intestinal barrier in inflammatory bowel disease. World J Gastroenterol (2014) 20:1165–79. doi: 10.3748/wjg.v20.i5.1165
25. Liu TC, Gurram B, Baldridge MT, Head R, Lam V, Luo C, et al. Paneth cell defects in crohn's disease patients promote dysbiosis. JCI Insight (2016) 1:e86907. doi: 10.1172/jci.insight.86907
26. Salzman NH, Underwood MA, Bevins CL. Paneth cells, defensins, and the commensal microbiota: a hypothesis on intimate interplay at the intestinal mucosa. Semin Immunol (2007) 19:70–83. doi: 10.1016/j.smim.2007.04.002
27. Wehkamp J, Salzman NH, Porter E, Nuding S, Weichenthal M, Petras RE, et al. Reduced paneth cell alpha-defensins in ileal crohn's disease. Proc Natl Acad Sci United States America (2005) 102:18129–34. doi: 10.1073/pnas.0505256102
28. Kang Y, Park H, Choe BH, Kang B. The role and function of mucins and its relationship to inflammatory bowel disease. Front Med (Lausanne) (2022) 9:848344. doi: 10.3389/fmed.2022.848344
29. Nystrom EEL, Martinez-Abad B, Arike L, Birchenough GMH, Nonnecke EB, Castillo PA, et al. An intercrypt subpopulation of goblet cells is essential for colonic mucus barrier function. Science (2021) 372:eabb1590. doi: 10.1126/science.abb1590
30. Singh V, Johnson K, Yin J, Lee S, Lin R, Yu H, et al. Chronic inflammation in ulcerative colitis causes long-term changes in goblet cell function. Cell Mol Gastroenterol Hepatol (2022) 13:219–32. doi: 10.1016/j.jcmgh.2021.08.010
31. Abraham C, Abreu MT, Turner JR. Pattern recognition receptor signaling and cytokine networks in microbial defenses and regulation of intestinal barriers: Implications for inflammatory bowel disease. Gastroenterology (2022) 162:1602–16. doi: 10.1053/j.gastro.2021.12.288
32. Andrews C, McLean MH, Durum SK. Cytokine tuning of intestinal epithelial function. Front Immunol (2018) 9:1270. doi: 10.3389/fimmu.2018.01270
33. Capaldo CT, Nusrat A. Cytokine regulation of tight junctions. Biochim Biophys Acta (2009) 1788:864–71. doi: 10.1016/j.bbamem.2008.08.027
34. Kaminsky LW, Al-Sadi R, Ma TY. IL-1beta and the intestinal epithelial tight junction barrier. Front Immunol (2021) 12:767456. doi: 10.3389/fimmu.2021.767456
35. Ruder B, Atreya R, Becker C. Tumour necrosis factor alpha in intestinal homeostasis and gut related diseases. Int J Mol Sci (2019) 20:1887. doi: 10.3390/ijms20081887
36. Lechuga S, Ivanov AI. Disruption of the epithelial barrier during intestinal inflammation: Quest for new molecules and mechanisms. Biochim Biophys Acta Mol Cell Res (2017) 1864:1183–94. doi: 10.1016/j.bbamcr.2017.03.007
37. Ivanov AI, Parkos CA, Nusrat A. Cytoskeletal regulation of epithelial barrier function during inflammation. Am J Pathol (2010) 177:512–24. doi: 10.2353/ajpath.2010.100168
38. Schoultz I, Keita AV. The intestinal barrier and current techniques for the assessment of gut permeability. Cells (2020) 9:1909. doi: 10.3390/cells9081909
39. Aguilar-Rojas A, Olivo-Marin JC, Guillen N. Human intestinal models to study interactions between intestine and microbes. Open Biol (2020) 10:200199. doi: 10.1098/rsob.200199
40. Bozzetti V, Senger S. Organoid technologies for the study of intestinal microbiota-host interactions. Trends Mol Med (2022) 28:290–303. doi: 10.1016/j.molmed.2022.02.001
41. Small JT, Weiss AA. Intestinal enteroid monolayers model the human intestinal environment for escherichia coli infection. J Bacteriol (2022) 204:e0062021. doi: 10.1128/jb.00620-21
42. Murakami H, Masui H. Hormonal control of human colon carcinoma cell growth in serum-free medium. Proc Natl Acad Sci U S A (1980) 77:3464–8. doi: 10.1073/pnas.77.6.3464
43. Fogh J, Wright WC, Loveless JD. Absence of HeLa cell contamination in 169 cell lines derived from human tumors. J Natl Cancer Inst (1977) 58:209–14. doi: 10.1093/jnci/58.2.209
44. Bruewer M, Luegering A, Kucharzik T, Parkos CA, Madara JL, Hopkins AM, et al. Proinflammatory cytokines disrupt epithelial barrier function by apoptosis-independent mechanisms. J Immunol (2003) 171:6164–72. doi: 10.4049/jimmunol.171.11.6164
45. Dharmsathaphorn K, McRoberts JA, Mandel KG, Tisdale LD, Masui H. A human colonic tumor cell line that maintains vectorial electrolyte transport. Am J Physiol (1984) 246:G204–208. doi: 10.1152/ajpgi.1984.246.2.G204
46. Ivanov AI, Hunt D, Utech M, Nusrat A, Parkos CA. Differential roles for actin polymerization and a myosin II motor in assembly of the epithelial apical junctional complex. Mol Biol Cell (2005) 16:2636–50. doi: 10.1091/mbc.e05-01-0043
47. Ivanov AI, McCall IC, Parkos CA, Nusrat A. Role for actin filament turnover and a myosin II motor in cytoskeleton-driven disassembly of the epithelial apical junctional complex. Mol Biol Cell (2004) 15:2639–51. doi: 10.1091/mbc.e04-02-0163
48. Madara JL, Dharmsathaphorn K. Occluding junction structure-function relationships in a cultured epithelial monolayer. J Cell Biol (1985) 101:2124–33. doi: 10.1083/jcb.101.6.2124
49. Scharl M, Paul G, Barrett KE, McCole DF. AMP-activated protein kinase mediates the interferon-gamma-induced decrease in intestinal epithelial barrier function. J Biol Chem (2009) 284:27952–63. doi: 10.1074/jbc.M109.046292
50. Lea T. Caco-2 cell line. In: Verhoeckx K, Cotter P, Lopez-Exposito I, Kleiveland C, Lea T, Mackie A, Requena T, Swiatecka D, Wichers H, editors. The impact of food bioactives on health: In vitro and ex vivo models. New York, Dordrecht, London: Springer Cham Heidelberg (2015). p. 103–11.
51. Devriese S, Van den Bossche L, Van Welden S, Holvoet T, Pinheiro I, Hindryckx P, et al. T84 monolayers are superior to caco-2 as a model system of colonocytes. Histochem Cell Biol (2017) 148:85–93. doi: 10.1007/s00418-017-1539-7
52. Kennedy M, Denenberg AG, Szabo C, Salzman AL. Poly(ADP-ribose) synthetase activation mediates increased permeability induced by peroxynitrite in caco-2BBe cells. Gastroenterology (1998) 114:510–8. doi: 10.1016/S0016-5085(98)70534-7
53. Sambuy Y, De Angelis I, Ranaldi G, Scarino ML, Stammati A, Zucco F. The caco-2 cell line as a model of the intestinal barrier: influence of cell and culture-related factors on caco-2 cell functional characteristics. Cell Biol Toxicol (2005) 21:1–26. doi: 10.1007/s10565-005-0085-6
54. Spalinger MR, Sayoc-Becerra A, Ordookhanian C, Canale V, Santos AN, King SJ, et al. The JAK inhibitor tofacitinib rescues intestinal barrier defects caused by disrupted epithelial-macrophage interactions. J Crohns Colitis (2021) 15:471–84. doi: 10.1093/ecco-jcc/jjaa182
55. Amasheh M, Fromm A, Krug SM, Amasheh S, Andres S, Zeitz M, et al. TNFalpha-induced and berberine-antagonized tight junction barrier impairment via tyrosine kinase, akt and NFkappaB signaling. J Cell Sci (2010) 123:4145–55. doi: 10.1242/jcs.070896
56. Bucker R, Krug SM, Rosenthal R, Gunzel D, Fromm A, Zeitz M, et al. Aerolysin from aeromonas hydrophila perturbs tight junction integrity and cell lesion repair in intestinal epithelial HT-29/B6 cells. J Infect Dis (2011) 204:1283–92. doi: 10.1093/infdis/jir504
57. Heller F, Florian P, Bojarski C, Richter J, Christ M, Hillenbrand B, et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology (2005) 129:550–64. doi: 10.1016/j.gastro.2005.05.002
58. Lechuga S, Cartagena-Rivera AX, Khan A, Crawford BI, Narayanan V, Conway DE, et al. A myosin chaperone, UNC-45A, is a novel regulator of intestinal epithelial barrier integrity and repair. FASEB J (2022) 36:e22290. doi: 10.1096/fj.202200154R
59. Mitchell DM, Ball JM. Characterization of a spontaneously polarizing HT-29 cell line, HT-29/cl.f8. In Vitro Cell Dev Biol Anim (2004) 40:297–302. doi: 10.1290/04100061.1
60. Wang D, Naydenov NG, Feygin A, Baranwal S, Kuemmerle JF, Ivanov AI. Actin-depolymerizing factor and cofilin-1 have unique and overlapping functions in regulating intestinal epithelial junctions and mucosal inflammation. Am J Pathol (2016) 186:844–58. doi: 10.1016/j.ajpath.2015.11.023
61. Chen TR, Drabkowski D, Hay RJ, Macy M, Peterson W Jr. WiDr is a derivative of another colon adenocarcinoma cell line, HT-29. Cancer Genet Cytogenet (1987) 27:125–34. doi: 10.1016/0165-4608(87)90267-6
62. Bourgine J, Billaut-Laden I, Happillon M, Lo-Guidice JM, Maunoury V, Imbenotte M, et al. Gene expression profiling of systems involved in the metabolism and the disposition of xenobiotics: comparison between human intestinal biopsy samples and colon cell lines. Drug Metab Dispos (2012) 40:694–705. doi: 10.1124/dmd.111.042465
63. Lenaerts K, Bouwman FG, Lamers WH, Renes J, Mariman EC. Comparative proteomic analysis of cell lines and scrapings of the human intestinal epithelium. BMC Genomics (2007) 8:91. doi: 10.1186/1471-2164-8-91
64. Ahmed D, Eide PW, Eilertsen IA, Danielsen SA, Eknaes M, Hektoen M, et al. Epigenetic and genetic features of 24 colon cancer cell lines. Oncogenesis (2013) 2:e71. doi: 10.1038/oncsis.2013.35
65. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature (2002) 417:949–54. doi: 10.1038/nature00766
66. Ikediobi ON, Davies H, Bignell G, Edkins S, Stevens C, O'Meara S, et al. Mutation analysis of 24 known cancer genes in the NCI-60 cell line set. Mol Cancer Ther (2006) 5:2606–12. doi: 10.1158/1535-7163.MCT-06-0433
67. Ilyas M, Tomlinson IP, Rowan A, Pignatelli M, Bodmer WF. Beta-catenin mutations in cell lines established from human colorectal cancers. Proc Natl Acad Sci United States America (1997) 94:10330–4. doi: 10.1073/pnas.94.19.10330
68. Mouradov D, Sloggett C, Jorissen RN, Love CG, Li S, Burgess AW, et al. Colorectal cancer cell lines are representative models of the main molecular subtypes of primary cancer. Cancer Res (2014) 74:3238–47. doi: 10.1158/0008-5472.CAN-14-0013
69. Hankey W, Frankel WL, Groden J. Functions of the APC tumor suppressor protein dependent and independent of canonical WNT signaling: Implications for therapeutic targeting. Cancer Metastasis Rev (2018) 37:159–72. doi: 10.1007/s10555-017-9725-6
70. Juanes MA. Cytoskeletal control and wnt signaling-APC's dual contributions in stem cell division and colorectal cancer. Cancers (Basel) (2020) 12:3811. doi: 10.3390/cancers12123811
71. Araki K, Ebata T, Guo AK, Tobiume K, Wolf SJ, Kawauchi K. p53 regulates cytoskeleton remodeling to suppress tumor progression. Cell Mol Life Sci (2015) 72:4077–94. doi: 10.1007/s00018-015-1989-9
72. Jiang Y, Wang Y, Wang T, Hawke DH, Zheng Y, Li X, et al. PKM2 phosphorylates MLC2 and regulates cytokinesis of tumour cells. Nat Commun (2014) 5:5566. doi: 10.1038/ncomms6566
73. Reichert F, Rotshenker S. Galectin-3 (MAC-2) controls microglia phenotype whether amoeboid and phagocytic or branched and non-phagocytic by regulating the cytoskeleton. Front Cell Neurosci (2019) 13:90. doi: 10.3389/fncel.2019.00090
74. Schafer C, Mohan A, Burford W, Driscoll MK, Ludlow AT, Wright WE, et al. Differential Kras(V12) protein levels control a switch regulating lung cancer cell morphology and motility. Converg Sci Phys Oncol (2016) 2:035004. doi: 10.1088/2057-1739/2/3/035004
75. Fujii M, Sato T. Somatic cell-derived organoids as prototypes of human epithelial tissues and diseases. Nat Mater (2021) 20:156–69. doi: 10.1038/s41563-020-0754-0
76. Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature (2009) 459:262–5. doi: 10.1038/nature07935
77. Sato T, Stange DE, Ferrante M, Vries RG, Van Es JH, Van den Brink S, et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and barrett's epithelium. Gastroenterology (2011) 141:1762–72. doi: 10.1053/j.gastro.2011.07.050
78. Stelzner M, Helmrath M, Dunn JC, Henning SJ, Houchen CW, Kuo C, et al. A nomenclature for intestinal in vitro cultures. Am J Physiol Gastrointestinal liver Physiol (2012) 302:G1359–1363. doi: 10.1152/ajpgi.00493.2011
79. Gomez DP, Boudreau F. Organoids and their use in modeling gut epithelial cell lineage differentiation and barrier properties during intestinal diseases. Front Cell Dev Biol (2021) 9:732137. doi: 10.3389/fcell.2021.732137
80. Gunther C, Winner B, Neurath MF, Stappenbeck TS. Organoids in gastrointestinal diseases: from experimental models to clinical translation. Gut (2022) 71:1892–908. doi: 10.1136/gutjnl-2021-326560
81. He J, Zhang X, Xia X, Han M, Li F, Li C, et al. Organoid technology for tissue engineering. J Mol Cell Biol (2020) 12:569–79. doi: 10.1093/jmcb/mjaa012
82. Shin YC, Shin W, Koh D, Wu A, Ambrosini YM, Min S, et al. Three-dimensional regeneration of patient-derived intestinal organoid epithelium in a physiodynamic mucosal interface-on-a-Chip. Micromachines (Basel) (2020) 11:663. doi: 10.3390/mi11070663
83. Poletti M, Arnauts K, Ferrante M, Korcsmaros T. Organoid-based models to study the role of host-microbiota interactions in IBD. J Crohns Colitis (2021) 15:1222–35. doi: 10.1093/ecco-jcc/jjaa257
84. Pearce SC, Al-Jawadi A, Kishida K, Yu S, Hu M, Fritzky LF, et al. Marked differences in tight junction composition and macromolecular permeability among different intestinal cell types. BMC Biol (2018) 16:19. doi: 10.1186/s12915-018-0481-z
85. Gui Y, Cheng H, Zhou J, Xu H, Han J, Zhang D. Development and function of natural TCR(+) CD8alphaalpha(+) intraepithelial lymphocytes. Front Immunol (2022) 13:1059042. doi: 10.3389/fimmu.2022.1059042
86. Lutter L, Hoytema van Konijnenburg DP, Brand EC, Oldenburg B, van Wijk F. The elusive case of human intraepithelial T cells in gut homeostasis and inflammation. Nat Rev Gastroenterol Hepatol (2018) 15:637–49. doi: 10.1038/s41575-018-0039-0
87. Hoytema van Konijnenburg DP, Reis BS, Pedicord VA, Farache J, Victora GD, Mucida D. Intestinal epithelial and intraepithelial T cell crosstalk mediates a dynamic response to infection. Cell (2017) 171:783–794 e713. doi: 10.1016/j.cell.2017.08.046
88. Jia L, Wu G, Alonso S, Zhao C, Lemenze A, Lam YY, et al. A transmissible gammadelta intraepithelial lymphocyte hyperproliferative phenotype is associated with the intestinal microbiota and confers protection against acute infection. Mucosal Immunol (2022) 15:772–82. doi: 10.1038/s41385-022-00522-x
89. Matsuzawa-Ishimoto Y, Yao X, Koide A, Ueberheide BM, Axelrad JE, Reis BS, et al. The gammadelta IEL effector API5 masks genetic susceptibility to paneth cell death. Nature (2022) 610:547–54. doi: 10.1038/s41586-022-05259-y
90. Chesnokova V, Zonis S, Apostolou A, Estrada HQ, Knott S, Wawrowsky K, et al. Local non-pituitary growth hormone is induced with aging and facilitates epithelial damage. Cell Rep (2021) 37:110068. doi: 10.1016/j.celrep.2021.110068
91. Edgar RD, Perrone F, Foster AR, Payne F, Lewis S, Nayak KM, et al. Culture-associated DNA methylation changes impact on cellular function of human intestinal organoids. Cell Mol Gastroenterol Hepatol (2022) 14:1295–310. doi: 10.1016/j.jcmgh.2022.08.008
92. Frum T, Spence JR. hPSC-derived organoids: models of human development and disease. J Mol Med (Berl) (2021) 99:463–73. doi: 10.1007/s00109-020-01969-w
93. Hautefort I, Poletti M, Papp D, Korcsmaros T. Everything you always wanted to know about organoid-based models (and never dared to ask). Cell Mol Gastroenterol Hepatol (2022) 14:311–31. doi: 10.1016/j.jcmgh.2022.04.012
94. Daoud A, Munera JO. Insights into human development and disease from human pluripotent stem cell derived intestinal organoids. Front Med (Lausanne) (2019) 6:297. doi: 10.3389/fmed.2019.00297
95. Bardenbacher M, Ruder B, Britzen-Laurent N, Schmid B, Waldner M, Naschberger E, et al. Permeability analyses and three dimensional imaging of interferon gamma-induced barrier disintegration in intestinal organoids. Stem Cell Res (2019) 35:101383. doi: 10.1016/j.scr.2019.101383
96. Hanyu H, Yokoi Y, Nakamura K, Ayabe T, Tanaka K, Uno K, et al. Mycotoxin deoxynivalenol has different impacts on intestinal barrier and stem cells by its route of exposure. Toxins (Basel) (2020) 12:610. doi: 10.3390/toxins12100610
97. Liebe H, Schlegel C, Cai X, Golubkova A, Leiva T, Berry WL, et al. Determining intestinal permeability using Lucifer yellow in an apical-out enteroid model. J Vis Exp (2022) 185. doi: 10.3791/64215
98. Xu P, Becker H, Elizalde M, Masclee A, Jonkers D. Intestinal organoid culture model is a valuable system to study epithelial barrier function in IBD. Gut (2018) 67:1905–6. doi: 10.1136/gutjnl-2017-315685
99. Han X, Lee A, Huang S, Gao J, Spence JR, Owyang C. Lactobacillus rhamnosus GG prevents epithelial barrier dysfunction induced by interferon-gamma and fecal supernatants from irritable bowel syndrome patients in human intestinal enteroids and colonoids. Gut Microbes (2019) 10:59–76. doi: 10.1080/19490976.2018.1479625
100. Bednarek R. In vitro methods for measuring the permeability of cell monolayers. Methods Protoc (2022) 5:17. doi: 10.3390/mps5010017
101. Hosic S, Lake W, Stas E, Koppes R, Breault DT, Murthy SK, et al. Cholinergic activation of primary human derived intestinal epithelium does not ameliorate TNF-alpha induced injury. Cell Mol Bioeng (2020) 13:487–505. doi: 10.1007/s12195-020-00633-0
102. Lee C, An M, Joung JG, Park WY, Chang DK, Kim YH, et al. TNFalpha induces LGR5+ stem cell dysfunction in patients with crohn's disease. Cell Mol Gastroenterol Hepatol (2022) 13:789–808. doi: 10.1016/j.jcmgh.2021.10.010
103. Onozato D, Akagawa T, Kida Y, Ogawa I, Hashita T, Iwao T, et al. Application of human induced pluripotent stem cell-derived intestinal organoids as a model of epithelial damage and fibrosis in inflammatory bowel disease. Biol Pharm Bull (2020) 43:1088–95. doi: 10.1248/bpb.b20-00088
104. Grosheva I, Zheng D, Levy M, Polansky O, Lichtenstein A, Golani O, et al. High-throughput screen identifies host and microbiota regulators of intestinal barrier function. Gastroenterology (2020) 159:1807–23. doi: 10.1053/j.gastro.2020.07.003
105. Ma TY, Iwamoto GK, Hoa NT, Akotia V, Pedram A, Boivin MA, et al. TNF-alpha-induced increase in intestinal epithelial tight junction permeability requires NF-kappa b activation. Am J Physiol Gastrointestinal liver Physiol (2004) 286:G367–376. doi: 10.1152/ajpgi.00173.2003
106. Ma TY, Boivin MA, Ye D, Pedram A, Said HM. Mechanism of TNF-{alpha} modulation of caco-2 intestinal epithelial tight junction barrier: Role of myosin light-chain kinase protein expression. Am J Physiol Gastrointestinal liver Physiol (2005) 288:G422–430. doi: 10.1152/ajpgi.00412.2004
107. Tak LJ, Kim HY, Ham WK, Agrahari G, Seo Y, Yang JW, et al. Superoxide dismutase 3-transduced mesenchymal stem cells preserve epithelial tight junction barrier in murine colitis and attenuate inflammatory damage in epithelial organoids. Int J Mol Sci (2021) 22:6431. doi: 10.3390/ijms22126431
108. Sayoc-Becerra A, Krishnan M, Fan S, Jimenez J, Hernandez R, Gibson K, et al. The JAK-inhibitor tofacitinib rescues human intestinal epithelial cells and colonoids from cytokine-induced barrier dysfunction. Inflammatory bowel Dis (2020) 26:407–22. doi: 10.1093/ibd/izz266
109. Xu P, Becker H, Elizalde M, Pierik M, Masclee A, Jonkers D. Interleukin-28A induces epithelial barrier dysfunction in CD patient-derived intestinal organoids. Am J Physiol Gastrointestinal liver Physiol (2021) 320:G689–99. doi: 10.1152/ajpgi.00064.2020
110. Workman MJ, Troisi E, Targan SR, Svendsen CN, Barrett RJ. Modeling intestinal epithelial response to interferon-gamma in induced pluripotent stem cell-derived human intestinal organoids. Int J Mol Sci (2020) 22:288. doi: 10.3390/ijms22010288
111. Bruewer M, Utech M, Ivanov AI, Hopkins AM, Parkos CA, Nusrat A. Interferon-gamma induces internalization of epithelial tight junction proteins via a macropinocytosis-like process. FASEB J (2005) 19:923–33. doi: 10.1096/fj.04-3260com
112. Boivin MA, Roy PK, Bradley A, Kennedy JC, Rihani T, Ma TY. Mechanism of interferon-gamma-induced increase in T84 intestinal epithelial tight junction. J Interferon Cytokine Res (2009) 29:45–54. doi: 10.1089/jir.2008.0128
113. Youakim A, Ahdieh M. Interferon-gamma decreases barrier function in T84 cells by reducing ZO-1 levels and disrupting apical actin. Am J Physiol (1999) 276:G1279–1288. doi: 10.1152/ajpgi.1999.276.5.G1279
114. Gleeson JP, Estrada HQ, Yamashita M, Svendsen CN, Targan SR, Barrett RJ. Development of physiologically responsive human iPSC-derived intestinal epithelium to study barrier dysfunction in IBD. Int J Mol Sci (2020) 21:1438. doi: 10.3390/ijms21041438
115. Workman MJ, Gleeson JP, Troisi EJ, Estrada HQ, Kerns SJ, Hinojosa CD, et al. Enhanced utilization of induced pluripotent stem cell-derived human intestinal organoids using microengineered chips. Cell Mol Gastroenterol Hepatol (2018) 5:669–77. doi: 10.1016/j.jcmgh.2017.12.008
116. Wang F, Graham WV, Wang Y, Witkowski ED, Schwarz BT, Turner JR. Interferon-gamma and tumor necrosis factor-alpha synergize to induce intestinal epithelial barrier dysfunction by up-regulating myosin light chain kinase expression. Am J Pathol (2005) 166:409–19. doi: 10.1016/S0002-9440(10)62264-X
117. Zolotarevsky Y, Hecht G, Koutsouris A, Gonzalez DE, Quan C, Tom J, et al. A membrane-permeant peptide that inhibits MLC kinase restores barrier function in in vitro models of intestinal disease. Gastroenterology (2002) 123:163–72. doi: 10.1053/gast.2002.34235
118. Xu P, Elizalde M, Masclee A, Pierik M, Jonkers D. Corticosteroid enhances epithelial barrier function in intestinal organoids derived from patients with crohn's disease. J Mol Med (Berl) (2021) 99:805–15. doi: 10.1007/s00109-021-02045-7
119. Wiley JW, Zong Y, Zheng G, Zhu S, Hong S. Histone H3K9 methylation regulates chronic stress and IL-6-induced colon epithelial permeability and visceral pain. Neurogastroenterol Motil (2020) 32:e13941. doi: 10.1111/nmo.13941
120. Al-Sadi R, Ye D, Boivin M, Guo S, Hashimi M, Ereifej L, et al. Interleukin-6 modulation of intestinal epithelial tight junction permeability is mediated by JNK pathway activation of claudin-2 gene. PLoS One (2014) 9:e85345. doi: 10.1371/journal.pone.0085345
121. Apostolou A, Panchakshari RA, Banerjee A, Manatakis DV, Paraskevopoulou MD, Luc R, et al. A novel microphysiological colon platform to decipher mechanisms driving human intestinal permeability. Cell Mol Gastroenterol Hepatol (2021) 12:1719–41. doi: 10.1016/j.jcmgh.2021.07.004
122. Wang Y, Mumm JB, Herbst R, Kolbeck R, Wang Y. IL-22 increases permeability of intestinal epithelial tight junctions by enhancing claudin-2 expression. J Immunol (2017) 199:3316–25. doi: 10.4049/jimmunol.1700152
123. Delbue D, Lebenheim L, Cardoso-Silva D, Dony V, Krug SM, Richter JF, et al. Reprogramming intestinal epithelial cell polarity by interleukin-22. Front Med (Lausanne) (2021) 8:656047. doi: 10.3389/fmed.2021.656047
124. Brice DP, Murray GI, Wilson HM, Porter RJ, Berry S, Durum SK, et al. Interleukin-27 regulates the function of the gastrointestinal epithelial barrier in a human tissue-derived organoid model. Biol (Basel) (2022) 11:427. doi: 10.3390/biology11030427
125. Li L, Zhou C, Li T, Xiao W, Yu M, Yang H. Interleukin-28A maintains the intestinal epithelial barrier function through regulation of claudin-1. Ann Transl Med (2021) 9:365. doi: 10.21037/atm-20-5494
126. Buonpane C, Ares G, Yuan C, Schlegel C, Liebe H, Hunter CJ. Experimental modeling of necrotizing enterocolitis in human infant intestinal enteroids. J Invest Surg (2022) 35:111–8. doi: 10.1080/08941939.2020.1829755
127. Guo S, Al-Sadi R, Said HM, Ma TY. Lipopolysaccharide causes an increase in intestinal tight junction permeability in vitro and in vivo by inducing enterocyte membrane expression and localization of TLR-4 and CD14. Am J Pathol (2013) 182:375–87. doi: 10.1016/j.ajpath.2012.10.014
128. Nighot M, Al-Sadi R, Guo S, Rawat M, Nighot P, Watterson MD, et al. Lipopolysaccharide-induced increase in intestinal epithelial tight permeability is mediated by toll-like receptor 4/Myeloid differentiation primary response 88 (MyD88) activation of myosin light chain kinase expression. Am J Pathol (2017) 187:2698–710. doi: 10.1016/j.ajpath.2017.08.005
129. Estrada HQ, Patel S, Rabizadeh S, Casero D, Targan SR, Barrett RJ. Development of a personalized intestinal fibrosis model using human intestinal organoids derived from induced pluripotent stem cells. Inflammatory bowel Dis (2022) 28:667–79. doi: 10.1093/ibd/izab292
130. Yoshida S, Honjo T, Iino K, Ishibe R, Leo S, Shimada T, et al. Generation of human-induced pluripotent stem cell-derived functional enterocyte-like cells for pharmacokinetic studies. Stem Cell Rep (2021) 16:295–308. doi: 10.1016/j.stemcr.2020.12.017
131. Artursson P, Ungell AL, Lofroth JE. Selective paracellular permeability in two models of intestinal absorption: Cultured monolayers of human intestinal epithelial cells and rat intestinal segments. Pharm Res (1993) 10:1123–9. doi: 10.1023/A:1018903931777
132. Kandilogiannakis L, Filidou E, Drygiannakis I, Tarapatzi G, Didaskalou S, Koffa M, et al. Development of a human intestinal organoid model for In vitro studies on gut inflammation and fibrosis. Stem Cells Int (2021) 2021:9929461. doi: 10.1155/2021/9929461
133. Wang F, Schwarz BT, Graham WV, Wang Y, Su L, Clayburgh DR, et al. IFN-gamma-induced TNFR2 expression is required for TNF-dependent intestinal epithelial barrier dysfunction. Gastroenterology (2006) 131:1153–63. doi: 10.1053/j.gastro.2006.08.022
134. Zhang C, Yan J, Xiao Y, Shen Y, Wang J, Ge W, et al. Inhibition of autophagic degradation process contributes to claudin-2 expression increase and epithelial tight junction dysfunction in TNF-alpha treated cell monolayers. Int J Mol Sci (2017) 18:157. doi: 10.3390/ijms18010157
135. Utech M, Ivanov AI, Samarin SN, Bruewer M, Turner JR, Mrsny RJ, et al. Mechanism of IFN-gamma-induced endocytosis of tight junction proteins: myosin II-dependent vacuolarization of the apical plasma membrane. Mol Biol Cell (2005) 16:5040–52. doi: 10.1091/mbc.e05-03-0193
136. Smyth D, Leung G, Fernando M, McKay DM. Reduced surface expression of epithelial e-cadherin evoked by interferon-gamma is fyn kinase-dependent. PloS One (2012) 7:e38441. doi: 10.1371/journal.pone.0038441
137. Koch S, Nusrat A. The life and death of epithelia during inflammation: lessons learned from the gut. Annu Rev Pathol (2012) 7:35–60. doi: 10.1146/annurev-pathol-011811-120905
138. Potten CS, Loeffler M. Stem cells: attributes, cycles, spirals, pitfalls and uncertainties. Lessons crypt. Dev (1990) 110:1001–20. doi: 10.1242/dev.110.4.1001
139. Blander JM. On cell death in the intestinal epithelium and its impact on gut homeostasis. Curr Opin Gastroenterol (2018) 34:413–9. doi: 10.1097/MOG.0000000000000481
140. Nunes T, Bernardazzi C, de Souza HS. Cell death and inflammatory bowel diseases: apoptosis, necrosis, and autophagy in the intestinal epithelium. BioMed Res Int (2014) 2014:218493. doi: 10.1155/2014/218493
141. Patankar JV, Becker C. Cell death in the gut epithelium and implications for chronic inflammation. Nat Rev Gastroenterol Hepatol (2020) 17:543–56. doi: 10.1038/s41575-020-0326-4
142. Chotikatum S, Naim HY, El-Najjar N. Inflammation induced ER stress affects absorptive intestinal epithelial cells function and integrity. Int Immunopharmacol (2018) 55:336–44. doi: 10.1016/j.intimp.2017.12.016
143. Aden K, Tran F, Ito G, Sheibani-Tezerji R, Lipinski S, Kuiper JW, et al. ATG16L1 orchestrates interleukin-22 signaling in the intestinal epithelium via cGAS-STING. J Exp Med (2018) 215:2868–86. doi: 10.1084/jem.20171029
144. Jones LG, Vaida A, Thompson LM, Ikuomola FI, Caamano JH, Burkitt MD, et al. NF-kappaB2 signalling in enteroids modulates enterocyte responses to secreted factors from bone marrow-derived dendritic cells. Cell Death Dis (2019) 10:896. doi: 10.1038/s41419-019-2129-5
145. Matsuzawa-Ishimoto Y, Shono Y, Gomez LE, Hubbard-Lucey VM, Cammer M, Neil J, et al. Autophagy protein ATG16L1 prevents necroptosis in the intestinal epithelium. J Exp Med (2017) 214:3687–705. doi: 10.1084/jem.20170558
146. Pott J, Kabat AM, Maloy KJ. Intestinal epithelial cell autophagy is required to protect against TNF-induced apoptosis during chronic colitis in mice. Cell Host Microbe (2018) 23:191–202. doi: 10.1016/j.chom.2017.12.017
147. Stolzer I, Dressel A, Chiriac MT, Neurath MF, Gunther C. An IFN-STAT axis augments tissue damage and inflammation in a mouse model of crohn's disease. Front Med (Lausanne) (2021) 8:644244. doi: 10.3389/fmed.2021.644244
148. Van Winkle JA, Constant DA, Li L, Nice TJ. Selective interferon responses of intestinal epithelial cells minimize tumor necrosis factor alpha cytotoxicity. J Virol (2020) 94:e00603–20. doi: 10.1128/JVI.00603-20
149. Grabinger T, Luks L, Kostadinova F, Zimberlin C, Medema JP, Leist M, et al. Ex vivo culture of intestinal crypt organoids as a model system for assessing cell death induction in intestinal epithelial cells and enteropathy. Cell Death Dis (2014) 5:e1228. doi: 10.1038/cddis.2014.183
150. Watanabe S, Nishimura R, Shirasaki T, Katsukura N, Hibiya S, Kirimura S, et al. Schlafen 11 is a novel target for mucosal regeneration in ulcerative colitis. J Crohns Colitis (2021) 15:1558–72. doi: 10.1093/ecco-jcc/jjab032
151. Woznicki JA, Saini N, Flood P, Rajaram S, Lee CM, Stamou P, et al. TNF-alpha synergises with IFN-gamma to induce caspase-8-JAK1/2-STAT1-dependent death of intestinal epithelial cells. Cell Death Dis (2021) 12:864. doi: 10.1038/s41419-021-04151-3
152. Constant DA, Van Winkle JA, VanderHoek E, Dekker SE, Sofia MA, Regner E, et al. Transcriptional and cytotoxic responses of human intestinal organoids to IFN types I, II, and III. Immunohorizons (2022) 6:416–29. doi: 10.4049/immunohorizons.2200025
153. Lee C, Song JH, Cha YE, Chang DK, Kim YH, Hong SN. Intestinal epithelial responses to IL-17 in adult stem cells-derived human intestinal organoids. J Crohns Colitis (2022) 16:1911–23. doi: 10.1093/ecco-jcc/jjac101
154. Gullett JM, Tweedell RE, Kanneganti TD. It's all in the PAN: Crosstalk, plasticity, redundancies, switches, and interconnectedness encompassed by PANoptosis underlying the totality of cell death-associated biological effects. Cells (2022) 11:1495. doi: 10.3390/cells11091495
155. Samir P, Malireddi RKS, Kanneganti TD. The PANoptosome: A deadly protein complex driving pyroptosis, apoptosis, and necroptosis (PANoptosis). Front Cell Infect Microbiol (2020) 10:238. doi: 10.3389/fcimb.2020.00238
156. Karki R, Sharma BR, Tuladhar S, Williams EP, Zalduondo L, Samir P, et al. Synergism of TNF-alpha and IFN-gamma triggers inflammatory cell death, tissue damage, and mortality in SARS-CoV-2 infection and cytokine shock syndromes. Cell (2021) 184:149–68. doi: 10.1016/j.cell.2020.11.025
157. Malireddi RKS, Karki R, Sundaram B, Kancharana B, Lee S, Samir P, et al. Inflammatory cell death, PANoptosis, mediated by cytokines in diverse cancer lineages inhibits tumor growth. Immunohorizons (2021) 5:568–80. doi: 10.4049/immunohorizons.2100059
158. Arnauts K, Sudhakar P, Verstockt S, Lapierre C, Potche S, Caenepeel C, et al. Microbiota, not host origin drives ex vivo intestinal epithelial responses. Gut Microbes (2022) 14:2089003. doi: 10.1080/19490976.2022.2089003
159. Arnauts K, Verstockt B, Ramalho AS, Vermeire S, Verfaillie C, Ferrante M. Ex vivo mimicking of inflammation in organoids derived from patients with ulcerative colitis. Gastroenterology (2020) 159:1564–7. doi: 10.1053/j.gastro.2020.05.064
160. Howell KJ, Kraiczy J, Nayak KM, Gasparetto M, Ross A, Lee C, et al. DNA Methylation and transcription patterns in intestinal epithelial cells from pediatric patients with inflammatory bowel diseases differentiate disease subtypes and associate with outcome. Gastroenterology (2018) 154:585–98. doi: 10.1053/j.gastro.2017.10.007
161. Niklinska-Schirtz BJ, Venkateswaran S, Anbazhagan M, Kolachala VL, Prince J, Dodd A, et al. Ileal derived organoids from crohn's disease patients show unique transcriptomic and secretomic signatures. Cell Mol Gastroenterol Hepatol (2021) 12:1267–80. doi: 10.1016/j.jcmgh.2021.06.018
162. Porpora M, Conte M, Lania G, Bellomo C, Rapacciuolo L, Chirdo FG, et al. Inflammation is present, persistent and more sensitive to proinflammatory triggers in celiac disease enterocytes. Int J Mol Sci (2022) 23:1973. doi: 10.3390/ijms23041973
163. Dotti I, Mora-Buch R, Ferrer-Picon E, Planell N, Jung P, Masamunt MC, et al. Alterations in the epithelial stem cell compartment could contribute to permanent changes in the mucosa of patients with ulcerative colitis. Gut (2017) 66:2069–79. doi: 10.1136/gutjnl-2016-312609
164. Meir M, Salm J, Fey C, Schweinlin M, Kollmann C, Kannapin F, et al. Enteroids generated from patients with severe inflammation in crohn's disease maintain alterations of junctional proteins. J Crohns Colitis (2020) 14:1473–87. doi: 10.1093/ecco-jcc/jjaa085
165. d'Aldebert E, Quaranta M, Sebert M, Bonnet D, Kirzin S, Portier G, et al. Characterization of human colon organoids from inflammatory bowel disease patients. Front Cell Dev Biol (2020) 8:363. doi: 10.3389/fcell.2020.00363
166. Sahoo D, Swanson L, Sayed IM, Katkar GD, Ibeawuchi SR, Mittal Y, et al. Artificial intelligence guided discovery of a barrier-protective therapy in inflammatory bowel disease. Nat Commun (2021) 12:4246. doi: 10.1038/s41467-021-24470-5
167. Angus HC, Urbano PC, Laws GA, Fan S, Gadeock S, Schultz M, et al. An autologous colonic organoid-derived monolayer model to study immune: bacterial interactions in crohn's disease patients. Clin Transl Immunol (2022) 11:e1407. doi: 10.1002/cti2.1407
168. Sarvestani SK, Signs S, Hu B, Yeu Y, Feng H, Ni Y, et al. Induced organoids derived from patients with ulcerative colitis recapitulate colitic reactivity. Nat Commun (2021) 12:262. doi: 10.1038/s41467-020-20351-5
169. Freire R, Ingano L, Serena G, Cetinbas M, Anselmo A, Sapone A, et al. Human gut derived-organoids provide model to study gluten response and effects of microbiota-derived molecules in celiac disease. Sci Rep (2019) 9:7029. doi: 10.1038/s41598-019-43426-w
170. Antfolk M, Jensen KB. A bioengineering perspective on modelling the intestinal epithelial physiology in vitro. Nat Commun (2020) 11:6244. doi: 10.1038/s41467-020-20052-z
171. Ingber DE. Human organs-on-chips for disease modelling, drug development and personalized medicine. Nat Rev Genet (2022) 23:467–91. doi: 10.1038/s41576-022-00466-9
172. Ojo BA, VanDussen KL, Rosen MJ. The promise of patient-derived colon organoids to model ulcerative colitis. Inflammatory bowel Dis (2022) 28:299–308. doi: 10.1093/ibd/izab161
Keywords: actin cytoskeleton, adherens junctions, colonoids, cytokines, enteroids, epithelial barrier, inflammatory bowel diseases, tight junctions
Citation: Lechuga S, Braga-Neto MB, Naydenov NG, Rieder F and Ivanov AI (2023) Understanding disruption of the gut barrier during inflammation: Should we abandon traditional epithelial cell lines and switch to intestinal organoids?. Front. Immunol. 14:1108289. doi: 10.3389/fimmu.2023.1108289
Received: 25 November 2022; Accepted: 01 February 2023;
Published: 16 February 2023.
Edited by:
Arnaud Besson, MCD UMR5077 CNRS/University Toulouse III Paul Sabatier, FranceReviewed by:
Edith Porter, California State University, Los Angeles, United StatesBert Devriendt, Ghent University, Belgium
Copyright © 2023 Lechuga, Braga-Neto, Naydenov, Rieder and Ivanov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrei I. Ivanov, ivanova2@ccf.org