
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 09 February 2023
Sec. Autoimmune and Autoinflammatory Disorders : Autoimmune Disorders
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1107670
This article is part of the Research Topic Mitochondrial Dysfunction in Inflammation and Autoimmunity View all 5 articles
 Weiyao Jing1†
Weiyao Jing1† Cui Liu1†
Cui Liu1† Chenghong Su1
Chenghong Su1 Limei Liu1Ping Chen2Xiangjun Li2
Limei Liu1Ping Chen2Xiangjun Li2 Xinghua Zhang3
Xinghua Zhang3 Bo Yuan4
Bo Yuan4 Haidong Wang2*
Haidong Wang2* Xiaozheng Du1*
Xiaozheng Du1*Rheumatoid arthritis (RA) is an autoimmune disease characterized by synovial inflammation, pannus formation, and bone and cartilage damage. It has a high disability rate. The hypoxic microenvironment of RA joints can cause reactive oxygen species (ROS) accumulation and mitochondrial damage, which not only affect the metabolic processes of immune cells and pathological changes in fibroblastic synovial cells but also upregulate the expression of several inflammatory pathways, ultimately promoting inflammation. Additionally, ROS and mitochondrial damage are involved in angiogenesis and bone destruction, thereby accelerating RA progression. In this review, we highlighted the effects of ROS accumulation and mitochondrial damage on inflammatory response, angiogenesis, bone and cartilage damage in RA. Additionally, we summarized therapies that target ROS or mitochondria to relieve RA symptoms and discuss the gaps in research and existing controversies, hoping to provide new ideas for research in this area and insights for targeted drug development in RA.
Rheumatoid arthritis (RA) is an autoimmune disease characterized by synovial inflammation and pannus, bone, and cartilage damage. It has a global prevalence of approximately 0.5–1% and occurs more commonly in women than in men (1). Genetics is a key factor in the development of RA, and sex, smoking, and environmental factors influence the development of RA (2). In RA, permanent T-cell and monocyte-mediated synovial inflammation are the underlying cause of disease progression. Pannus is a characteristic pathological product of RA, with tumor-like properties that drive synovial proliferation and bone erosion, which can eventually lead to disability and seriously affect patients’ quality of life (3). The onset of RA involves not only the joints but also cardiovascular disease and interstitial lung disease, which are serious complications that result in a shorter life expectancy in patients with RA (4, 5). Mitochondria are an important source of reactive oxygen species (ROS) within most mammalian cells. However, ROS accumulation may activate mitochondrial permeability transition pore (mPTP) and inner membrane anion channel (IMAC) opening. Longer mPTP openings may release a burst of ROS, which contributes to mitochondrial damage (6). ROS and mitochondrial damage are inextricably linked to several key pathological processes in RA (7–10), and the regulation of mitochondrial function, clearance of ROS, and alleviation of oxidative stress are currently popular targets for RA treatment (11, 12). In this review, we describe the role of ROS and mitochondrial damage in the major pathological changes that occur in RA, summarize the drugs targeting ROS or mitochondria for RA treatment, and suggest their relatively weakly studied but value-rich directions in RA.
Mitochondria are organelles with a bilayer membrane structure that supply the organism with adenosine triphosphate mainly through the oxidative phosphorylation process. This process comprises five mitochondrial respiratory chain enzyme complexes; complexes I–IV constitute the electron transport chain, and complex V is ATP synthase (13). The electron transport chain oxidizes nicotinamide adenine dinucleotide and flavin adenine dinucleotide produced by glycolysis and the tricarboxylic acid (TCA) cycle and pumps protons out of the mitochondrial inner membrane to produce a proton gradient. Thus, mitochondria can transfer electrons and regulate the body’s oxidation/reduction (redox) reactions (14). ROS mainly include free radicals, such as superoxide anions () and hydroxyl radicals (OH-), and non-radical oxidants, such as hydrogen peroxide (H2O2) and singlet oxygen (1O2) (15). ROS are mainly derived from the process of electron transfer from the mitochondrial electron transport chain complex to O2, with complexes I and II producing in the mitochondrial matrix and complex III producing in the matrix and membrane interstitium, which is the precursor of most ROS. in the mitochondrial matrix is converted to H2O2 by manganese superoxide dismutase, and copper- and zinc-containing superoxide dismutase mainly converts in the membrane interstitium and cytoplasm, which is eventually catabolized to H2O. Under normal conditions, ROS production and elimination are balanced and play a role in promoting immunity and regulating the cell cycle (16).
Redox homeostasis is determined by the balance between ROS generation and ROS quenching capacity. When the equilibrium is tilted toward ROS production, conditions are created for oxidative stress. Mild ROS cumulation can cause oxidation of essential mitochondrial components; in extreme cases, it can irreversibly cause mitochondrial damage. Cardiolipin (CL) is a dimeric phospholipid with a high content of unsaturated fatty acids mainly distributed in the inner mitochondrial membrane. CL is particularly prone to ROS-induced oxidative attacks. Oxidized CL redistributes from the inner mitochondrial membrane to the outer membrane (17, 18). The accumulation of oxidized CL on the OMM results in mPTP formation. These mPTP changes result in more ROS formation, known as “ROS-induced ROS release” (RIRR), and cause an oxidative stress response (6). In fact, mitochondria-derived ROS can self-destruct mitochondria. ROS in the matrix can cause oxidative damage to mtDNA, leading to mutation of mtDNA, inhibition of mitochondrial aerobic respiration, reduction of ATP production, and disruption of the mitochondrial membrane potential (ΔΨm). These changes eventually lead to mitochondrial depolarization. Recent studies have shown that persistent deletion of mtDNA leads to irreversible mitochondrial damage (19). Studies have shown that glutathione (GSH) peroxidase 4 inhibitor (RSL3) induces high ROS expression in mouse embryonic fibroblasts, enhanced mitochondrial fragmentation, mitochondrial membrane potential loss, reduced mitochondrial respiration, and ROS scavenger mitoquinone (MitoQ)-preserved mitochondrial integrity and function (20) (Figure 1).
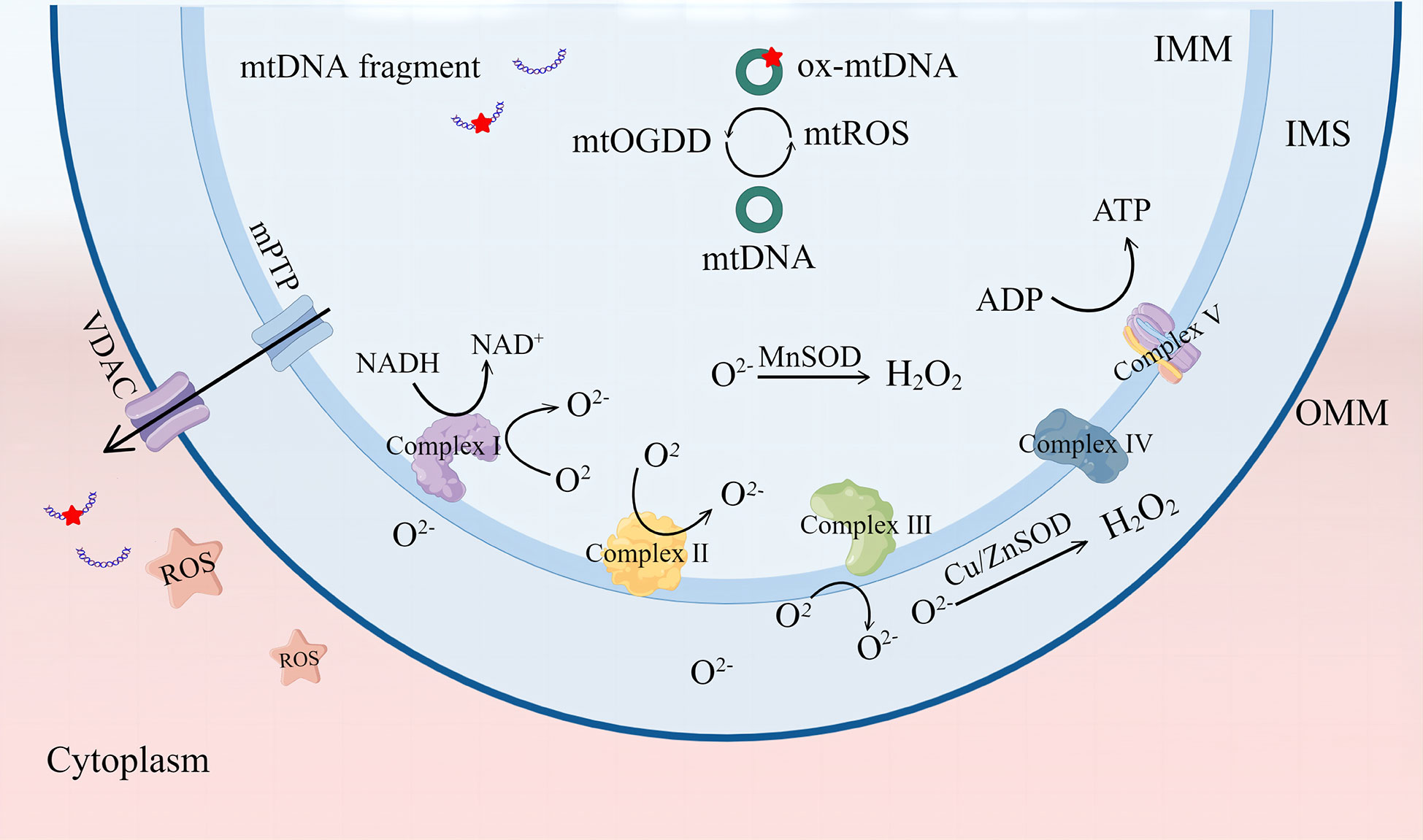
Figure 1 The mitochondrial respiratory chain complex is the main site for ROS production. ROS can be catalyzed as H2O2 by MnSOD or Cu/ZnSOD, and ROS accumulation causes mtDNA damage and leakage into the cytoplasm through mPTP and VDAC channels. VDAC, voltage-dependent anion channel. mPTP, mitochondrial permeability transition pore. ROS, reactive oxygen species. SOD, superoxide dismutase; IMM, inner mitochondrial membrane; IMS, intermembrane space; OMM, mitochondrial outer membrane. By Figdraw.
In patients with RA, tPO2 in the joint cavity is significantly lower than that in normal tissues (21), and joint cavity hypoxia is the underlying condition for ROS accumulation and mitochondrial damage in synovial tissue (22). ROS have a strong correlation with the level of disease activity in patients with RA. They are positively correlated with C-reactive protein (CRP) and anti-cyclic peptide-containing citrulline levels in patients’ blood and can be used as an indirect assessment indicator of the degree of synovial inflammation in patients with RA (23–26). Previous studies have reported that 18 mitochondria-related proteins were upregulated and four proteins were downregulated in patients with RA, with significant mtDNA damage and reduced mitochondrial membrane potential, superoxide, and cellular ATP levels. These findings indicate that mitochondrial damage plays an important role in RA (27–29). In fact, ROS can modulate immune cell function and activation processes (30, 31). Mitochondria affect their cell cycle and inflammation tendency by adjusting metabolism or cell death and participate in RA inflammation (32, 33). Fibroblast-like synoviocytes (FLS) are the key effector cells in RA inflammation. ROS and mitochondrial damage regulate FLS proliferation, invasion, and production of inflammatory factors and can affect FLS survival by affecting apoptosis, autophagy, and other processes mediated by FLS (34). ROS can cause mtDNA accumulation in the cytoplasm and in turn act as pathogen-associated molecular patterns (PAMPs) to activate multiple inflammatory pathways and mediate inflammatory responses (19). In synovial tissue, ROS promote pannus formation by inducing angiogenesis and angiogenesis by upregulating HIF, VEGF, and Notch expression (35–37). Changes in mitochondrial membrane proteins and abnormal mitochondrial respiration affect this process (38). Besides, due to the different responsiveness of osteoclasts and osteoblasts to ROS, ROS induce the differentiation and activation of osteoclasts and mitochondrial damage and apoptosis of osteoblasts. This favors the balance toward the process of bone resorption, causing bone destruction, while ROS affect the release of MMPs and activity of chondrocytes, exacerbating cartilage damage (39–41). Therefore, both ROS and mitochondrial damage are involved in the development of RA (42, 43). In a randomized controlled trial, Yoga was shown to enhance mitochondrial quality, reduce oxidative stress marker production, and improve the Disease Activity Score-Erythrocyte Sedimentation Rate and Health Assessment Questionnaire-Disability Index scores in patients with RA, which may be a beneficial adjunct to training for RA (44).
Synovial inflammatory response is the central mechanism of RA lesions and main factor leading to pannus (45, 46) and cartilage destruction (47, 48). The subsynovial and lining layers are altered in patients with RA, and T-cells, B cells, and dendritic cells are widely distributed in the subsynovial region (49). They drive synovial inflammation together (50, 51). The synovial lining in RA is dominated by synovial macrophages and FLSs, which are highly activated and produce a large number of pro-inflammatory factors, chemokines, and growth factors (52). These factors can activate FLSs and mediate proliferation, anti-apoptosis, erosion, migration, and other pathological behaviors (53, 54). Mitochondrial damage and oxidative stress can affect immune cell metabolic processes and promote inflammatory behavior (55, 56). Additionally, ROS and the products of mitochondrial damage are good activators of inflammatory response.
In RA, immune cells function abnormally, producing a large number of inflammatory factors, such as tumor necrosis factor (TNF)-α, interleukin (IL)-6, and IL-17, which are distributed in clusters in the diseased joints, causing an inflammatory storm (57–59). Mitochondria-derived ROS assist in antigen presentation and are important regulators of the T-cell cycle and function (30, 60). Studies have shown that ROS promote Th17 differentiation and increase IL-17 production (61). The use of ROS scavengers significantly inhibits Th17 differentiation (62). The IEX-1 gene plays a key role in this process, and IEX-1 overexpression significantly inhibits mtROS production. Collagen-induced arthritis (CIA) mice with knocked-out IEX-1 have higher amounts of Th17 and exhibit more severe joint inflammation (63). Additionally, hypoxia not only induces ROS accumulation but also induces high expression of HIF-1α (64). Hyperbaric oxygen therapy reduces the levels of IL-17a, CRP, and rheumatoid factor in CIA mice by regulating the expression of HIF-1α and promotes the differentiation of Tregs, alleviating oxidative stress and inflammatory response (65). CD3+ T-cells and CD68 macrophages cultured in a hypoxic environment exhibit a stronger inflammatory response (21), confirming the critical role of hypoxia in immune inflammation in RA (27). The hypoxic environment in the RA joint cavity leads to altered immune cell metabolism, impaired oxidative phosphorylation processes, and significantly increased aerobic glycolytic activity in organisms with RA (66–68). This process causes lactate accumulation and promotes the inflammatory behavior of immune cells (69, 70). T-cells in RA prefer the glycolytic pathway to break down glucose into ATP; however, the underlying mechanism remains unknown, and its relationship with ROS is not yet clear (71, 72). Indeed, the role of ROS in T-cells remains paradoxical, as ROS can activate the expression of nuclear factor of activated T-cells (NFAT) and induce c-MYC transcription, contributing to T-cell activation (73). The accumulation of ROS leads to the upregulation of GSH expression due to the presence of an oxidative coordination system. GSH can inhibit NFAT and MYC activation, whereas GSH production protects the integrity of T-cell metabolism (74).
In RA, reduced mitochondrial DNA biostability and mtDNA leakage into the cytoplasm due to the lack of DNA repair nuclease (MRE11A) increase the pro-inflammatory tendency of T-cells, whereas upregulation of MRE11A expression reduces mitochondrial damage and has an inhibitory effect on T-cell scorching and immune inflammation (32, 75). The expression of malondialdehyde (MDA) H2O2 was significantly increased in the paw tissue of CIA mice, and intervention with curarelinone, the active ingredient of bitter ginseng, significantly inhibited oxidative damage and decreased the phosphorylation levels of signal transducer and activator of transcription (STAT)1, STAT3, and ratio of Th1 and Th17 cells in lymph nodes (62). Yun et al. (76) used rosmarinic acid intervention to induce cytochrome C release from the mitochondria and induce apoptosis in activated T-cell subsets in patients with RA by blocking mitochondrial depolarization. Mesenchymal stem cells (MSCs) have potential in RA treatment (77, 78). Th17 cells co-cultured with bone marrow-derived MSCs (BM-MSCs) could reduce TNF-α and IL-17 production and restore T-cell oxidative phosphorylation activity in a contact-dependent manner through a mechanism related to mitochondrial transfer. Th17 cells could reduce IL-17 production through the uptake of healthy mitochondria in BM-MSCs with immunomodulatory effects. RA-synovium-derived MSCs have impaired mitochondrial transfer to Th17 cells, which may be a key reason for the persistent inflammatory response in RA synovial tissues (79, 80). Furthermore, mitochondrial transfer treatment corrects cellular metabolic defects, increases ATP production, and decreases ROS levels. Therefore, mitochondrial transfer has therapeutic potential in regulating immune cell function in RA (81–83).
B cells play an important role in the immune response to RA. On the one hand, B cells can produce autoantibodies, such as rheumatoid factor (rheumatoid factor, RF) and anti-citrulline protein antibody (ACPA), which can form immune complexes that are deposited in joints, and promote the inflammatory process through complement and cell activation. On the other hand, B cells, as potent antigen presenting cells (APC), activate T cells through the expression of co-stimulatory molecules (84). B cell depletion therapy highlights some advantages in RA therapy. Rituximab (RTX) is a human-mouse chimeric monoclonal antibody targeting the B cell-specific antigen, CD20, that induces B cell death through antibody-dependent cytotoxicity and phagocytosis mediated by Fc receptor γ. In anti-TNF inadequate responder patients with RA, RTX can reduce the levels of ESR, CRP, and RF, and improve clinical symptoms (85). However, the therapeutic effect of RTX is seemingly limited by the number of B cells in the synovial tissue.
BCR signaling is a critical step in controlling B cell maturation and differentiation. Endogenous ROS can regulate the level of BCR signaling through a reversible inhibition of protein tyrosine phosphatase activity. This process is associated with a reduced activation threshold of the spleen tyrosine kinase (SYK) (86), removing ROS by a scavenger, N-acetylcysteine, and resulting in impaired BCR-induced activation (87). ROS affects the proliferation of B cells and participates in the CD47-mediated G1 phase arrest of B cells, and clearing ROS can effectively eliminate this response (31). Additionally, the fate of B cells is greatly correlated with mitochondrial function. According to studies, mitochondrial mass and membrane potential were significantly higher in B cells of class-switch recombination (CSR) type, whereas B cells of plasma cell differentiation (PCD) type showed a decrease in mitochondrial mass and membrane potential (33). New studies have revealed that mitochondrial fission factor can specifically bind to TRAF 3 to regulate the progression of B cell apoptosis (88). These studies confirmed the association between ROS and mitochondria and B cells. In a mouse model of RA, ROS mediated the tolerance of B cells to autoantigens, and the mutated NCF 1 gene caused ROS deficiency, disrupting resistance to arthritis (89).
Neutrophils represent about 60% of the total leukocytes. They are the first cells to migrate to the site of inflammation and infection. Neutrophils exhibit multifunctional heterogeneity in orchestrating adaptive immune responses. Through exudation, neutrophils migrate from the bloodstream to the involved tissues and release degrading enzymes and ROS to play a cytotoxic role during infection (90). Evidence suggests that neutrophil extracellular trap (NET), peptidylarginine deiminase (PAD) activation, and citrullinated peptide generation are the crux of RA pathogenesis. Activated neutrophils release PAD enzymes that promote citrullination of synovial tissue. Recognition of a citrullinated peptide by MHC II promotes T cell activation and autoantibody production. Neutrophils accumulate heavily in RA synovial fluid and synovial tissue, and RA-FLS have the ability to internalize NET-associated citrullinated peptides, acquire antigen-presenting cells (APC), and present them to CD4+ T cells to induce an autoimmune response (91).
The level of neutrophil ROS in the synovial tissue of patients with RA is significantly higher than that in patients with other forms of arthritis. The ROS produced by neutrophil degranulation may affect the degree of oxidative stress in RA (92). A clinical study observed an increase in the intracellular and mitochondrial oxidative stress and decreased antioxidant enzymes. The intracellular level of ROS in polymorphonuclear neutrophils (PMNs) were positively correlated with inflammatory response and disease severity (93). Furthermore, RA synovial tissue highly expressed neutrophil chemokines and ROS. They lost their migratory properties and remained resident in joints to cause inflammation and bone destruction by recruiting and activating immune cells (94). In RA, neutrophils form a vicious cycle with oxidative stress and inflammation. The proinflammatory microenvironment in RA synovial tissue combined with high concentrations of ROS has been shown to jointly induce neutrophils to neutrophil-dendritic (N-DC) differentiation and show more ROS generation and inflammation tendency (95). Methotrexate reduces the expression of ROS, CD177, and CD11b in circulating neutrophils of patients with RA, which may be one of the mechanisms underlying its treatment of RA (96). mitochondrial formyl peptides (mtNFPs) is one of the key molecular patterns associated with mitochondrial damage, with increased circulating mtNFPs in RA patients, associated with disease activity.mtNFPs Neutrophil activation can be induced via formyl peptide receptor 1 (FPR 1) (97).Plastoquinonyl-Decyl-triphenylphosphonium bromide (SkQ1), an antioxidant targeting mitochondria, continuously removes ROS from mitochondria and protects cardiolipin on the inner mitochondrial membrane from oxidisal damage. SkQ1 intervention improved arthritis index and pathological injury severity in RA rats and promoted apoptosis of neutrophils in vitro. This mechanism may be one of the mechanisms by which SkQ1 exerts its pharmacological activity (98).
RA-FLSs have a distinctly aggressive nature; its cause is related to synovial inflammatory stimulation and epigenetic modifications (99, 100). The hypoxic environment of synovial tissue and mitochondrial damage are responsible for synovial inflammation and oxidative DNA damage, which increase the aggressiveness of FLSs (101). Microsatellite instability was reportedly significantly higher in RA synovial tissues than in osteoarthritis (OA) synovial tissues, indicating a decreased DNA mismatch repair (MMR) capacity and severe DNA damage. Oxidative stress can downregulate the DNA MMR system in RA-FLSs by inhibiting hMSH6. The oxidative stress environment can interfere with the repair process of single-base mutations and DNA damage by inhibiting hMSH6 (102, 103), and mutations in genes, such as P53 and LBH, can lead to pathological behaviors, such as invasion and proliferation of RA-FLSs (104, 105). ROS accumulation leads to metabolic abnormalities in RA-FLSs, with decreased mitochondrial oxidative phosphorylation and ATP reserve capacity but increased glycolytic activity in RA-FLSs, promoting RA synovial inflammation (106). High expression of HIF-2α in RA joints induces the secretion of multiple chemokines, promotes FLS migration and invasion, induces pannus formation, and aggravates bone destruction (107, 108). Additionally, ROS and HIF-2α can enhance the migration of RA-FLSs by regulating CD70 expression, whereas reduced oxidative damage can inhibit its migration (109). The level of mtROS in Treg cells of patients with RA increases with disease activity, and peripheral blood mononuclear cells (PBMCs) cultured with ROS inhibitors significantly reduce RA-FLS inflammation (110). This appears to be a possible reason for the ozone treatment of RA (111).
ROS accumulation leads to the corresponding activation of the Keap1/Nrf2 pathway, and Nrf2 transcribes various antioxidant enzymes, including superoxide dismutase (SOD), heme oxygenase-1 (HO-1), and GSH. It is a key pathway in the fight against oxidative damage (112). Knockdown of Nrf2 leads to RA-FLS activation and promotes its proliferation (113). The intervention of RA-FLSs with resveratrol significantly increases the expression of Nrf2 and HO-1, reduces the production of ROS and MDA and activation of nuclear factor kappa-B (NF-κB) p65, inhibits the proliferation and migration of RA-FLSs, and promotes its apoptosis (114, 115). Similarly, mitochondrial damage plays a key role in the pathological behavior of FLSs, and studies have shown that healthy mitochondrial transfer inhibits LPS-induced FLS proliferation and migration and promotes apoptosis, which can reduce the inflammatory response (80). In contrast, inducing mitochondrial damage in normal FLSs can promote NF-κB pathway activation and ROS production and increase the secretion of inflammatory factors (116). Adenosine 5’-monophosphate (AMP)-activated protein kinase (AMPK) is a key regulatory protein of mitochondrial mass (117), and its activation significantly inhibits the activation and proliferation of RA-FLSs (118). In fact, the high expression of glycogen synthase-1 in RA-FLSs can lead to the excessive accumulation of glycogen and inhibit AMPK expression, leading to the high expression of matrix metallopeptidase (MMP)-1, MMP-9, IL-6, and CCL-2, along with increased proliferation and migration of FLSs. Intervention with the AMPK-specific agonist, AICAR, blocked RA-FLS activity and improved symptoms in CIA rats (119). AMPK can remove damaged mitochondria by inducing mitochondrial autophagy and regulating mitochondrial quality, which reduces ROS production while removing damaged mitochondria. However, the effect of mitochondrial autophagy has two sides (120, 121). On the one hand, promoting mitophagy can inhibit multiple pathological behaviors of FLSs and reduce the inflammatory response induced by mtDNA and ROS (122–124). On the other hand, mitochondrial autophagy, as a means for cells to deal with stress, may contribute to cell survival. Inhibition of mitochondrial autophagy has been proposed to have the ability to contribute to FLS apoptosis, and the effects mediated by the different periods of mitochondrial autophagy vary (125–127). Therefore, the role and mechanisms underlying mitochondrial autophagy in RA-FLSs and RA need to be explored further.
Lipid peroxidation caused by ROS is involved in apoptosis, autophagy, and ferroptosis (128). However, it remains controversial in regulating RA-FLS cell death. H2O2 reduced mitochondrial membrane potential and increased ROS production in treated RA-FLS and activation of caspase-3, caspase-9, and Bax to induce FLS apoptosis, a process associated with oxidative stress-mediated activation of macrophage stimulating 1 (Mst1) and inhibition of the AMPK-Sirt1 signaling pathway (129). Exposure to Mitomycin C (MMC) has been shown to increase ROS production in RA-FLS and disrupt ΔΨ m, increasing the release of mitochondrial cytochrome c and the ratio of Bax/Bcl-2 and inducing apoptosis in RA-FLS (130). Therefore, ROS play a promoting role in the apoptotic process of RA-FLS. Conversely, induction of ROS production in FLS increases the level of cellular autophagy, thereby protecting FLS from apoptosis (131). Interestingly, knockdown of Atg 5 promoted the expression of RA-FLS inflammatory factors and transcriptional activity of NF-κB, which inhibited its secretion by activation of RA-FLS-specific autophagy (132). Ferroptosis is a type of cell death caused by iron-dependent lipid peroxidation (133). The mitochondrial TCA cycle and electron transport chain also promote ferroptosis progression by acting as a major source of cellular lipid peroxide production (134). Evidence has shown that ferroptosis is strongly linked to the pathological process of RA, and that induction of FLS ferroptosis helps to delay arthritis progression in CIA mice (135).
RA-FLSs have tumor cell-like anti-apoptotic characteristics, and inhibition of their proliferation and migration has been the focus of attention in RA therapy. ROS and mitochondrial damage play an important role in the proliferation and migration of FLSs, and they have emerged as important targets in RA therapy. Fang et al. (136) developed an ROS-responsive berberine polymer micelle based on the abnormal elevation of ROS in RA-FLSs, which could increase the uptake of berberine by RA-FLSs, inhibit synovial tissue proliferation, and attenuate the inflammatory response by recognizing ROS and mitochondrial superoxide. The highly effective and targeted mode of action of berberine undoubtedly provides a new direction for RA treatment.
ROS can disrupt mitochondrial lipid membrane integrity and lead to mPTP abnormalities, resulting in oxidative damage and leakage of mtDNA (137, 138). ROS and mtDNA are PAMPs that are good activators of several inflammatory pathways and an important bridge between oxidative stress and inflammatory responses (139, 140) (Figure 2).
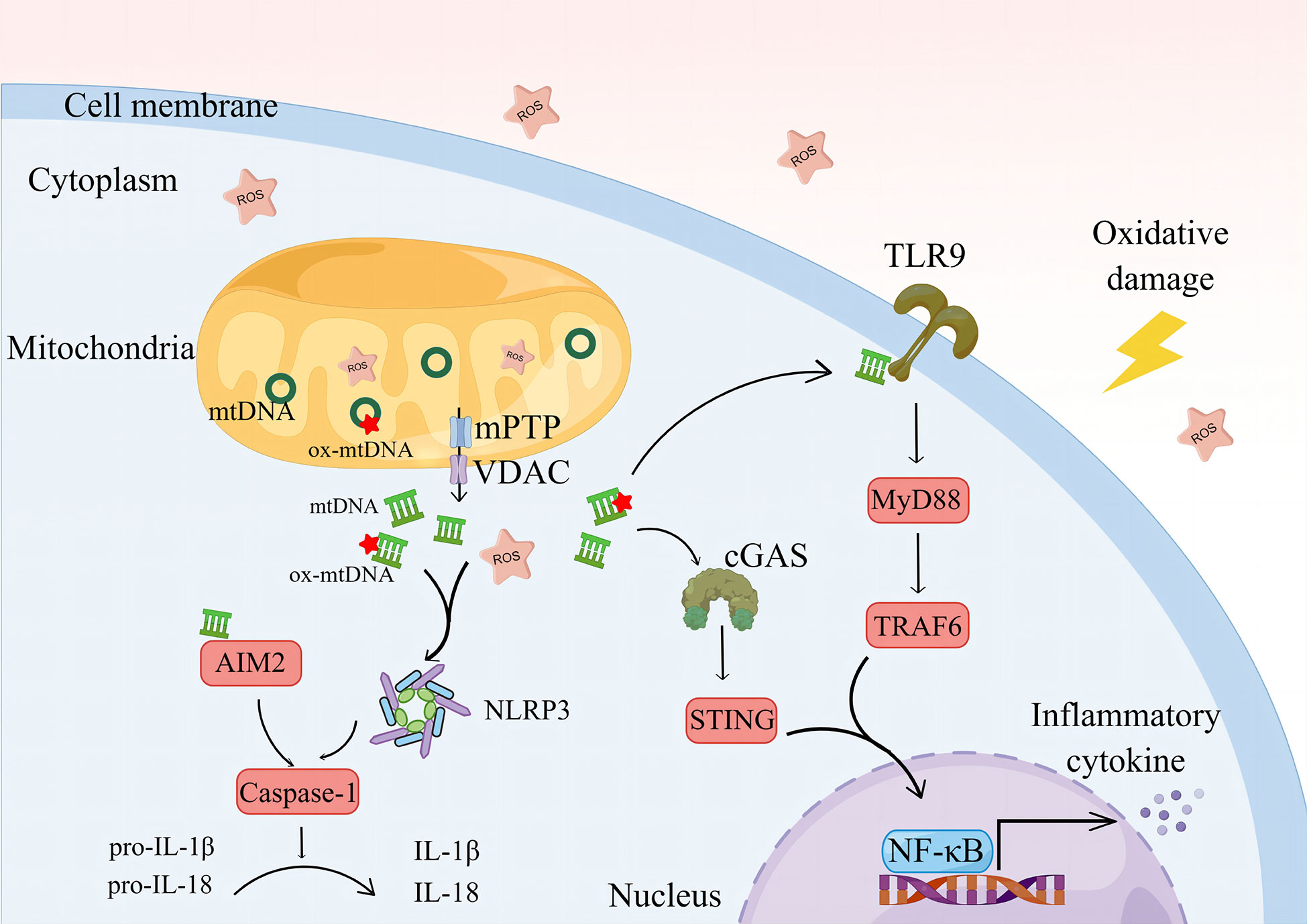
Figure 2 The oxidative damage environment and ROS cause mitochondrial damage, prompting mtDNA leakage and a further increase in ROS. mtDNA is oxidized by ROS to form ox-mtDNA, co-activating NLRP3 and prompted casape-1 maturation, activated IL-1β and IL-18. The cytoplasmic mtDNA can be recognized by AIM2, mediating the activation of AIM2 and promoting the cleavage of casape-1, mediating the inflammatory response. By recognizing the free mtDNA, cGAS activates the STING/NF-κB pathway in the cytoplasm.NLRP3, NOD-like receptor protein 3. IL-1β, interleukin-1β. IL-18, interleukin-18. AIM2, Absent in melanoma-2; cGAS, cyclic GMP-AMP synthase; STING, stimulator of interferon genes; TLR9, Toll-like receptors 9; MyD88, myeloid differentiation factor 88; TRAF6, TNF receptor associated factor 6; NF-κB, nuclear factor kappa-B. By Figdraw.
NOD-like receptor protein (NLRP)3 is a multiprotein complex that functions to activate IL-1β (141). It is associated with RA activity and inflammatory responses (142, 143). Studies have shown that tofacitinib regulates Treg/Th17 cell homeostasis by inhibiting NLRP3 inflammatory vesicle activity during RA treatment (144). ROS can promote Th17 differentiation by activating NLRP3 (145). ROS and mtDNA play a key role in the assembly of NLRP3 (146), and LPS-induced inflammatory response in macrophages involves the activation of NLRP3, a process that requires the involvement of mtROS. The removal of mtROS using molecular hydrogen (H2) significantly reduces NLRP3 activation and inflammatory factor production (147). The accumulation of mtROS leads to oxidative damage of mtDNA and formation of oxidized mitochondrial genes (ox-mtDNA). ox-mtDNA fragments escape into the cytoplasm via the mPTP and voltage-dependent anion channel (VDAC), which, in turn, initiates the assembly of NLRP3. Interestingly, mtROS do not induce VDAC oligomerization. Additionally, the escaped ox-mtDNA are recognized by cyclic GMP-AMP synthase (cGAS)/stimulator of interferon genes (STING) signaling and mediate the inflammatory response in RA. A crosstalk may occur between the cGAS/STING pathway and NLRP3 (148). In the mitochondria, the DNA glycosylase, OGG1 (mt-OGG1), can deoxidize ox-mtDNA, thereby maintaining mtDNA quality. mt-OGG1 overexpression significantly reduces ox-mtDNA content in the cytoplasm and mitochondria and inhibits NLRP3 activation (149), indicating the lack of DNA repair capacity in PBMCs of patients with RA (150). In the future, repairing specific DNA damage using clustered regularly interspaced short palindromic repeats (CRISPR) technology may produce a cure for patients with RA (151).
Additionally, NLRP3 is an important protein in mediating cell pyroptosis. Caspase-1 is a key effector protein of NLRP3. Activated caspase-1 enables the N-terminal sequence of cleaved gasdermin D (GSDMD) to bind to the cell membrane to produce membrane pores, leading to cell pyroptosis and release of a large number of inflammatory factors (152). Effectively, the GSDMD-mediated pyroptosis process promotes the release of mtDNA. Gasdermin targeted at the plasma membrane promotes mitochondrial collapse and leads to the initial accumulation of mtDNA in the cytosol (153). In a study, ROS promoted the progression of pyroptosis mediated by NLRP 3. Oxidative stress resulted in the oxidation of four amino acid residues of GSDMD in macrophages and significantly improved the cutting efficiency of caspase-1 on GSDMD (154). In RA-FLS, ROS can increase the level of caspase-1 by activating G protein-coupled receptor kinase 2 (GRK 2)/HIF-1α/NLRP 3, increase the cleavage of GSDMD, and promote the pyroptosis of FLS. Using monomeric derivatives of paeoniflorin (MDP) or removing ROS can reduce the phosphorylation of GRK 2 and inhibit FLS pyroptosis (155).
NLRP3 has now become a focus in RA research, NLRP3 inhibitors highlight the therapeutic potential. MCC950 is a small-molecule inhibitor targeting NLRP3. MCC950 intervention in CIA mice inhibited NLRP3 activation in the synovium, reduced the production of IL-1β, and alleviated joint inflammation and bone destruction (143). The JAK pathway inhibitor, tofacitinib, regulated the Treg/Th17 cell ratio in CIA mice and suppressed NLRP3 activation, whereas administration of NLRP3 abrogated this effect of tofacitinib, suggesting that NLRP3 played a pivotal role in the process of tofacitinib-mediated Th17 cell activation (144). The NLRP3 inhibitor, OLT1177, has been clinically studied in gouty arthritis and knee osteoarthritis. It has a significant effect in improving the pain of joint inflammation. However, its application is still lacking in RA (156).
The cGAS/STING pathway is mainly found in the cytoplasm. It is a key pathway mediating autoimmunity, sterile inflammation, and cellular senescence by recognizing free DNA in the cytoplasm and activating inflammatory responses (157, 158). The cGAS/STING pathway has become a hot topic in cancer research (159). TNF is a core factor mediating inflammation in RA and reduces mitophagy by inhibiting PTEN-induced putative kinase 1 (PINK1), leading to mitochondrial damage. This, in turn, increases the level of mtDNA in the cytoplasm and directly activates the cGAS/STING pathway, promoting the production of inflammatory factors. Knockdown of cGAS can significantly reduce the expression of multiple chemokines and attenuate toe joint swelling in CIA mice (160). The activation of the cGAS/STING pathway causes ROS accumulation and mitochondrial damage and promotes the migration and invasion of RA-FLSs, a process associated with the activation of the Hippo pathway. The knockdown of FOXO1 and MST1, key genes of the Hippo pathway, significantly inhibits the migration and invasion of RA-FLSs (161). The cGAS/STING pathway plays a key role in the chronic inflammatory network by activating NF-κB and NLRP3 to promote the production of multiple inflammatory factors (162). Additionally, inhibition of pathway activation can play a therapeutic role in RA (163). However, its role in RA needs to be explored further.
Toll-like receptor (TLR) is an early discovered class of pattern recognition receptors that play an important role in the inflammatory response to RA (164, 165). TLR9 is highly expressed in the PBMCs of patients with RA and positively correlates with the levels of inflammatory factors, such as IL-6 and TNF-α. TLR9 plays a key role in the interaction between FLSs and neutrophils (166, 167). Neutrophil extracellular traps (NETs) are a major source of guanylated autoantibodies. NET-containing guanylated peptides can be internalized by FLSs through the TLR9 pathway, elevating the inflammatory phenotype of FLSs and upregulating the expression of major histocompatibility complex class II molecules, which subsequently produce autoantibodies by presentation to Ag-specific T-cells (168). Hydroxychloroquine is a classical therapeutic agent for RA, and its therapeutic mechanism is related to the inhibition of dendritic cell (DC) activation by blocking TLR9 activation (169). The hypomethylated CpG sequence in mtDNA binds specifically to the N-terminal part of the C-shaped leucine-rich repeat region of TLR9, mediating TLR9 activation (170, 171). TLR9 activation can mediate NF-κB phosphorylation through myeloid differentiation factor 88 (MyD88) and then translate various factors, including IL-1, IL-6, and TNF-α, to mediate the inflammatory cascade (172). Additionally, studies have shown that ROS play a key role as a “secondary messenger” in regulating B-cell maturation and lgG and lgM production, which require the involvement of TLR9 (173). However, this aspect of the study has not yet been reported in RA.
Absent in melanoma-2 (AIM2) inflammasome is a member of the innate immune sensor, which can detect double-stranded DNA (dsDNA), including mtDNA, in the cytoplasm independent of sequence. dsDNA can form PYD domain helical filament with AIM 2, nucleate ASC, mediate the activation of caspase-1, and induce the pyroptosis process or release active IL-1β and IL-18, which have great inflammatory potential (174). AIM2, ASC, and caspase-1 were more expressed in the knee synovium of patients with RA than those with OA. They were positively correlated with ESR and CRP levels, which may be associated with high mtDNA expression in the synovial fluid of patients with RA. Inhibition of AIM2 expression or transfection of AIM2 siRNA can significantly inhibit the proliferation and inflammatory behavior of FLS (175). ROS is a key factor leading to mtDNA leakage, and inhibition of oxidative stress contributes to reduction of mitochondrial damage, release of dsDNA, and activation of AIM2 (176). Additionally, ROS assisted in the process of AIM2 activation during bacterial infection (177).
Pannus is a characteristic pathological product of RA and consists of neovascularization, inflammatory cells, proliferating synovial cells, and mechanized fibrin (178). Synovitis is the pathological basis for pannus. Persistent chronic synovitis leads to synovial congestion and edema and gradual accumulation of neutrophils and various immune cells in synovial tissue, whereas the proliferation of FLSs and active immune cells in synovial tissue increases the demand for oxygen and nutrient supply, forcing microangiogenesis in synovial tissue and eventually leading to a dysregulated neovascular network and the formation of villi-like proliferating granulation tissue (179–181). Therefore, the key step in the formation of pannus is angiogenesis, which provides a resupply for proliferating and migrating synovial cells and aggravates cartilage destruction and erosion (182). Mitochondrial damage in synovial tissue and oxidative stress environment are key factors in the induction of angiogenesis (38, 183). Repairing mitochondrial damage or scavenging ROS can inhibit angiogenesis (10, 184).
HIFs are major regulators that respond to ROS and mediate angiogenesis. HIFs consist of two subunits, α and β. Under normal conditions, HIFs are hydroxylated by the hydroxylase family in an oxygen-dependent manner, which leads to a substantial reduction in the transcriptional activity of HIFs, whereas under hypoxic conditions, the hydroxylate activity is inhibited, and HIFs accumulate in the cytoplasm. Activated HIFs translocate to the nucleus and rapidly transcribe various metabolic enzymes and vascular-related reactive substances to adapt to the hypoxic environment (185, 186). ROS accumulation in the RA joints prompts the high expression of HIFs, including HIF-1α and HIF-2α (35), and increase the expression of MMP-1, MMP-13, and IL-1β in FLSs. Silencing HIF-1α with siRNA significantly reduces the expression of these factors (187). HIF-1α perpetuates the interaction between synoviocytes and T and B cells, which in turn induces persistent production of inflammatory factors and autoantibodies (188). Moreover, HIF-1α can crosstalk with the TLR pathway to drive RA inflammatory response (64). Although HIF-2α shares many similarities with HIF-1α, HIF-2α and HIF-1α have been shown to differ in their sensitivities to hypoxic signaling and inflammation and can play a catabolic role in RA (189). However, both HIFs can accelerate cartilage destruction in RA (190). In addition to increasing the activity of MMPs, HIFs increase the production of chondrocyte glycolysis and the mitochondrial activity of chondrocytes under hypoxia, but ultimately leads to chondrocyte death (191). Mitochondrial ROS production in neutrophils increases the stability of HIF-1α and plays an important role in chronic inflammatory diseases (192). Nicotinamide adenine dinucleotide phosphate oxidase 4 (NOX4) increases ROS production, and stimulation of FLSs with NOX4 elevates the expression of vascular cell adhesion molecule 1 (VCAM1) and VEGF, contributing to vascular neogenesis and proliferation and migration of FLSs (193). Calreticulin (CRT) has been shown to be related to the pathogenesis of RA. CRT stimulation increases synovial NO production and phosphorylation levels of nitric oxide synthase in human umbilical vein endothelial cells (HUVECs) and promotes the proliferation, migration, and angiogenesis of HUVECs (194).
Additionally, the Notch signaling pathway responds to the regulation of ROS and has a close association with RA angiogenesis (37). In a study, Notch 1 and Notch 3 were highly expressed in RA synovial tissue. Notch 3 signaling from the vascular endothelium drove FLS activation, and mice with genetic deletion of Notch3 were resistant to serum-induced joint inflammatory responses. The Notch pathway inhibitor, LY411575, attenuated joint destruction and pannus severity in CIA rats (195, 196). Another study showed that cyclic, uniaxial stretch of human VSMCs increased Nox derived-ROS formation and Notch3 activation. Using Catalase to clear H2O2 prevented the stretch-induced translocation of Notch3 to the nucleus and decreased the Notch3 extracellular domain (197). Notch1 mediates VEGF/Ang2-induced angiogenesis and EC invasion in RA synovial tissue (198). Clearing of ROS from HUVECs inhibits Notch-induced HUVEC proliferation, migration, and adhesion (199). Besides, the Notch pathway is regulated by HIF. Notch1, Notch3 intracellular domain (N1ICD, N3ICD), and HIF-1 α were highly expressed in RASFC. Hypoxia-induced N1ICD and N3ICD expression in RASFC was blocked by siHIF-1α. Concurrently, siNotch1 and siNotch3 inhibited hypoxia-induced RASFC invasion and angiogenesis in vitro, whereas N1ICD and N3ICD overexpression promoted these processes (200).
ROS can transcribe VEGF through the activation of the NF-κB pathway and participate in processes, such as microvascular neogenesis and proliferation (36). In an oxidative stress environment, VEGF increases plasminogen activator (PA) and PA inhibitor-l (PAI-1) mRNA expression, increases plasminogen activator activity, hydrolyzes extracellular proteins, and thus promotes neocapillary formation (201, 202). By binding to its receptor, VEGF induces VEGF receptor phosphorylation and activates mitogen-activated protein kinase (MAPK), which induces vascular endothelial cell proliferation (203, 204). Studies have shown that VEGF gene polymorphisms are associated with RA susceptibility and activity and can be used for the clinical diagnosis and treatment of RA. High expression of VEGF can increase small vessel density in synovial inflammatory areas and elevate the levels of inflammatory factors, such as TNF-α and IL-1β (205–207).
Additionally, mitochondria play a regulatory role in angiogenesis (38). Mitochondrial thioredoxin reductase 2 (TrxR2), uncoupling protein 2 (UCP2), and panthenol-cytochrome c reductase-binding protein (UQCRB) can regulate VEGF activity and vascular endothelial activity (208). Among them, UQCRB is one of the subunits of the mitochondrial respiratory chain complex III, and mutations in UQCRB increase mtROS production and activate HIF-1 transactivation, promoting vascular neovascularization, a process that can be regulated by UQCRB inhibitors (209). FUN14 domain-containing protein 1 (FUNDC1), a protein localized on the outer mitochondrial membrane, is associated with mitophagy and mediates the formation of mitochondria-associated endoplasmic reticulum membranes, which can lead to increased cytoplasmic levels of Ca2+. This promotes serum response factor (SRF) phosphorylation and enhances SRF binding to the VEGFR2 promoter and leads to increased VEGFR2 transcription, leading to angiogenesis. In contrast, silencing of FUNDC1 can reverse the above process (210). Glucose-6-phosphate isomerase (GPI) is closely related to RA activity (211) and a key enzyme involved in the “Warburg effect” of RA. The accumulation of GPI is associated with abnormal mitochondrial respiratory processes (212). Hypoxic conditions can upregulate GPI activity, and in RA synovial tissue cells, upregulated GPI can induce RA angiogenesis by increasing the expression of HIF-1α and VEGF (213, 214).
Articular damage is a serious complication of RA that can lead to irreversible joint deformity, severely limiting joint mobility and affecting the quality of life of patients (1). The main cause of joint damage in patients with RA is the imbalance between osteoblasts and osteoclasts, which is characterized by increased bone resorption by osteoclasts and decreased bone formation by osteoblasts, accompanied by apoptosis of chondrocytes (215, 216). Previous studies have shown that an active immune response in synovial tissue is a key factor affecting RA joint damage. Recent studies have shown that ROS and mitochondrial damage similarly modulate RA joint damage and play an important role.
Bone destruction in RA joints presents as localized bone loss, initially involving cortical bone, disrupting the natural barrier between the external bony tissue and trabecular space of the marrow cavity. When the pannus invades the cortical bone, subchondral bone, and adjacent bone marrow cavity, eventually the trabecular bone disappears (217, 218). The tilt of RA bone metabolic balance towards bone resorption is a main factor causing bone destruction and leads to decreased bone mineral density and increased bone fragility. Therefore, patients with RA have a higher risk of fracture (219). Osteoclasts are the main players in RA bone destruction and cartilage damage. Osteoclasts are huge multinucleated cells derived from monocyte/macrophage cell lines, filling between inflammatory synovial tissue and the surface of bone joints. Through various proteases, such as cathepsin K, MMPs, and tartarate hydrochloric acid phosphatase (TRAP), they produce a local acidic environment, initiate calcium lysis, and degrade bone matrix.
Receptor activator of NF-κB ligand (RANKL) is a peptide type II transmembrane protein of the TNF superfamily that is associated with osteoclast differentiation and development, increases osteoclast bone resorption, and regulates its fate (220). Two receptors are available for RANKL; one is RANK, which is present on the cell membrane surface of osteoclast precursor cells. The binding of RANKL to RANK can promote the differentiation and maturation of osteoclasts, increase bone resorption, and delay osteoclast apoptosis. The other is osteoprotegerin (OPG), a member of the tumor necrosis factor receptor (TNFR) superfamily, with a stronger affinity to RANKL than RANK, which can competitively prevent RANKL from binding to RANK, thus inhibiting osteoclast differentiation and bone resorption activity and inducing its apoptosis. Denosumab is a monoclonal anti-RANKL antibody, which inhibits osteoclastic formation by binding to RANKL on osteoblasts. In patients with RA undergoing long-term denosumab treatment, denosumab effectively inhibited the progression of joint destruction and was generally well tolerated (221).
One study showed that radiation therapy in patients with malignant tumors easily leads to damage to the skeletal system. In vitro experiments showed that radiation can induce the ratio of ROS levels and RANKL to OPG in osteoclast precursor cells (RAW 264.7), prompting the differentiation of RAW 264.7 into osteoclasts. Intervention with the therapeutic drug, amifostine (AMI), can reduce DNA damage and ROS levels in cells and the ratio of RANKL to OPG, inhibit the maturation and differentiation of osteoclasts, and has a bone protective effect (222).Osteoclasts cause ROS accumulation during bone resorption or increased RANKL expression (223). ROS are key factors in the regulation of osteoclast differentiation (Figure 3). On the one hand, ROS, as a second messenger, can activate MAPK or NF-κB pathway and mediate granulocyte macrophage colony-stimulating factor (GM-CSF) production (224), which can interact with RNAKL and M-CSF to promote osteoclast differentiation (225). On the other hand, ROS induce the binding of Src homology 2 domain-containing phosphatase 1 (SHP1) to c-Src and the oxidation of c-Src and SHP-1, which lead to SHP-1 inactivation and activation of c-Src via phosphorylation of Tyr416, contributing to osteoclast survival and increasing bone loss (226). Additionally, ROS can upregulate HIF-1α expression, leading to activation of the Janus kinase (JAK) 2/STAT3 pathway, which promotes high RANKL expression and induces osteoclast differentiation (227). Additionally, ROS upregulation of HIF-1α can increase angiopoietin-like 4 expression in osteoclasts and enhance osteoclast activity (228, 229). Ni et al. (230) demonstrated that HIF-1α inhibits osteoclast ferritin phagocytosis and autophagocytosis in a hypoxic environment, reducing osteoclast ferroptosis. They further showed that the use of specific inhibitors of HIF-1α is effective in preventing bone loss.
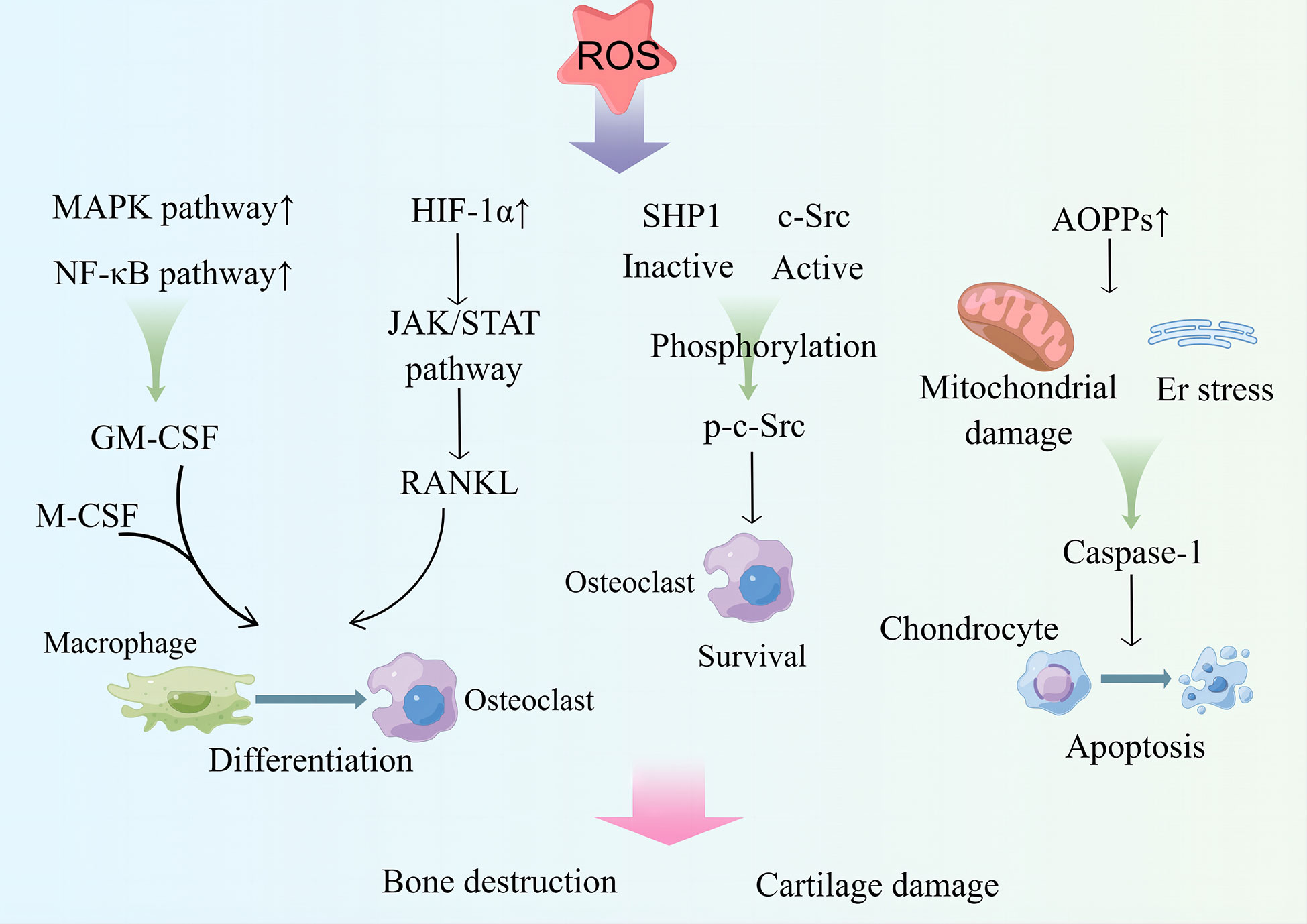
Figure 3 ROS upregulates MAPK and NF-κB pathway activities and transcribes GM-CSF, which acts in conjunction with M-CSF to induce osteoclast differentiation. ROS can upregulate the expression of HIF-1α, activate the JAK/STAT pathway, induce RANKL production, and promote osteoclast differentiation. ROS activate c-Src for osteoclast survival by inactivating SHP1. ROS drive the production of AOPPs, causing ER stress and mitochondrial damage; produce caspase-1; and drive chondrocyte apoptosis. These processes play an important role in bone destruction and cartilage destruction in RA. MAPK, mitogen-activated protein kinase. GM-CSF, granulocyte macrophage colony-stimulating factor. HIF-1α, hypoxia-inducible factor. JAK, Janus kinase; STAT, signal transducer and activator of transcription; RANKL, receptor activator of NF-κB ligand; SHP1, Src homology 2 domain-containing phosphatase 1; AOPPs, advanced oxidation production products. By Figdraw.
Additionally, mtROS affect osteoclast differentiation. Targeted removal of mtROS using MitoQ reverses hypoxia-induced calcineurin activity and NF-κB activity and inhibits the differentiation of RAW 264.7 macrophages into osteoclasts (231). Glucose metabolism by mitochondrial oxidative phosphorylation is the main bioenergetic pathway that supports osteoclast differentiation, but increased glycolytic activity promotes osteoclast differentiation (232). The hypoxic environment of the RA joint cavity and abnormal mitochondrial respiration of FLSs lead to an increase in glycolytic activity and “Warburg effect” (70). This leads to the accumulation of lactic acid.
Cartilage is mainly composed of chondrocytes, outer matrix proteoglycans, and type II collagen (233). The extracellular matrix of cartilage can be degraded by MMPs. MMP-1 and MMP-13 are mainly degradable-type collagens, and non-collagen matrix protein components, such as MMP-3, are degradable proteoglycans (234). Synovial inflammation is a key driver of cartilage damage in RA, and inflammatory stimuli contribute to the high expression of multiple MMPs and RANKL in the synovial tissues of patients with RA (48). The pannus attached to the cartilage surface exacerbates inflammation and hypoxia, thereby promoting bone erosion (235). Previous studies have confirmed the above view that in the post-arthroplasty tissues of patients with RA, RNAKL is mainly expressed at the endothelial-bone interface and subchondral bone erosion sites, the site of contact between the pannus and cartilage (236). TNF-α and IL-6 promote the conversion of RANKL-induced PBMCs into osteoclasts, and PBMCs of patients with RA show a higher differentiation potential (237). Therefore, regulating the differentiation of monocytes into osteoclasts in patients with RA is an attractive target.
Cartilage destruction in RA is closely linked to the oxidative stress environment, and the hypoxic environment increases monocyte differentiation toward osteoclasts and elevates osteoclast activity (39) (Figure 3). ROS affect the progression of cartilage damage by regulating chondrocyte life cycle and metabolism of cartilage matrix. As a signaling intermediate, elevated ROS levels can affect growth factor bioavailability by preventing extracellular matrix (ECM) synthesis, affecting bioavailability, participating in degradation of ECM components, promoting MMP production, and inducing chondrocyte death (40). On the one hand, excessive ROS generation is involved in the process of chondrocyte growth inhibition and apoptosis promotion through signaling pathways, such as PI3K/AKT and p38 pathways (238, 239). On the other hand, ROS increases the sensitivity of chondrocytes to ROS-mediated chondrocyte death through dysregulation of GSH antioxidant system, and ROS clearance reduces chondrocyte death and enhances chondrocyte viability (41, 240).
The increased expression of advanced oxidation production products (AOPPs) in patients with RA is associated with the process of bone destruction. AOPPs can induce apoptosis in chondrocytes by triggering mitochondrial dysfunction and endoplasmic reticulum stress, leading to caspase activation, which can be blocked by the use of antioxidants (241). Furthermore, high levels of 3-nitrotyrosine in the cartilage of patients with RA induce cellular mitochondrial dysfunction and chondrocyte apoptosis through a calcium-dependent process (242). Additionally, chondrocyte death is difficult to repair, and a degree of mitochondrial autophagy is necessary for chondrocyte protection when stress occurs (243). Intervention of experimental arthritic mice with the autophagic agonist, rapamycin, reduces the severity of arthritis (244). Mitophagy is a way to repair mitochondrial damage and maintain mitochondrial homeostasis. AMPK and sirtuin (SIRT)3 are key proteins that regulate mitochondrial homeostasis. They have been shown to exert a potential protective effect on chondrocytes by maintaining mitochondrial homeostasis (245, 246). However, excessive mitochondrial autophagy may induce apoptosis in chondrocytes (247). Uncontrolled mitophagy may lead to an imbalance in cellular homeostasis and requires further investigation in RA.
Inhibition of inflammatory response is critical in the course of RA treatment, and biological agents, such as TNF-α monoclonal antibodies, IL-6 monoclonal antibodies, and JAK pathway inhibitors, have been developed for inflammatory factors and have achieved significant clinical efficacy. However, their prolonged application increases the risk of viral infection and immune suppression (248). Adalimumab is a recombinant, fully human, IgG1 monoclonal antibody. It binds specifically to TNF-α and blocks their interaction with p55 and p75 cell surface TNF receptors. It is the major drug in RA therapy (249). Some studies have explored the effects of adalimumab treatment on the global gene expression profile in PBMCs of responder patients with RA. The results showed that immune response and regulation of mitochondrial redox are the key therapeutic mechanisms of adalimumab (250). Tocilizumab is a humanized anti-IL-6 receptor monoclonal antibody. Tocilizumab with methotrexate is effective for improving the symptoms of RA in patients with inadequate response to TNFi (251). In a study on systemic juvenile idiopathic arthritis (sJIA), tocilizumab significantly altered genes regulating mitochondrial dysfunction and oxidative stress in patient neutrophils (252). Anakinra is an IL-1 receptor antagonist used for treating moderate-to-severe RA that has been unresponsive to initial disease-modifying anti-rheumatic drug (DMARD) therapy. The study showed that anakinra promotes the binding of SOD2 to the deubiquitinase, ubiquitin specific peptidase 36 (USP36), and constitutive photomorphogenesis 9 (COP9) signalosome, thus increasing SOD2 protein longevity. This effect could mediate the clearance of ROS and inhibit NLRP3 activation (253). Furthermore, in cystic fibrosis, anakinra improved the proteostatic network by coupling the mitochondrial redox balance to autophagy (254).
Therefore, exploring new therapeutic targets remains a challenge. Several studies have shown that ROS and mitochondria can be used as targets to inhibit inflammation in RA. Many active ingredients of herbal medicines have been shown to improve inflammation in RA by scavenging ROS or regulating mitochondrial function, which can provide a basis for the development of natural botanicals.
The active ingredient of leigongteng, a herbal medicine commonly used in RA treatment, has been developed as a leigongteng polyglucoside, which can be used in the clinical treatment of RA (255). Celastrol (Cel) is a quinone-methylated triterpenoid extracted from Tripterygium wilfordii that has been shown to alleviate inflammatory response in RA by inhibiting the ROS/NF-κB/NLRP3 axis (256). ROS-sensitive polymer micelles have been developed for Cel delivery, which can overcome the disadvantages of poor water solubility and short half-life of Cel. These micelles can alleviate RA synovial inflammation by inhibiting macrophage M1 polarization (257). Salicin from Alangium chinense has anti-inflammatory effects, reduces ROS production by activating Nrf2/HO-1, and inhibits inflammatory factor secretion by FLSs in vivo and in vitro (258).
Several natural drugs inhibit inflammatory responses associated with the regulation of mitochondrial homeostasis in RA. Quercetin (Que) is a major active flavonoid component isolated from Herba taxilli. It activates the SIRT1/peroxisome proliferator-activated receptor-gamma coactivator 1 α (PGC-1α) pathway to promote mitochondrial biogenesis, regulate mitochondrial homeostasis, and inhibit the high mobility group protein (HMGB)1/TLR4/p38/extracellular regulated protein kinases (ERK)1/2 pathway to reduce inflammatory responses in CIA mice (259). The combination of Cornus officinalis and Paeonia lactiflora was effective in ameliorating oxidative stress and inflammation in CIA rats, a process associated with the regulation of AMPK-mediated mitochondrial homeostasis. Apoptosis of synovial cells may be involved in the treatment (260). Mitochondria are closely related to the cell cycle and play key roles in apoptosis (261, 262). Many natural drugs can regulate the cell cycle and promote apoptosis in FLSs through the mitochondrial pathway (263, 264). However, unlike previous findings, shikonin, icariin, and other drugs induce mitochondrial dysfunction by increasing ROS levels, decreasing mitochondrial membrane potential, and elevating the release of cytochrome C and pro-apoptotic proteins, such as caspase-3 and caspase-9, to induce apoptosis in FLSs to suppress inflammatory response (265, 266). However, this seems to be a manifestation of drug cytotoxicity. Therefore, toxic effects should be considered when studying plant drugs.
Synovial inflammatory response in RA is dependent on angiogenesis, which is mutually reinforcing and central to the progressive development of pannus. Current studies have shown that several DMARDs, such as methotrexate, can inhibit angiogenesis. Methotrexate inhibits angiogenesis in a three-dimensional co-culture model (containing synovial fibroblasts and vascular endothelial cells) and inhibits the formation of pannus (267). Leflunomide has been shown to inhibit angiogenesis-related endothelial function, and novel biologics, such as the JAK pathway inhibitors, peficitinib and tofacitinib, have been shown to treat RA by inhibiting VEGF expression and angiogenesis (268–270). The VEGF monoclonal antibody, ranibizumab, significantly improved synovial inflammation in CIA rats and was superior to the IL-6 monoclonal antibody, tocilizumab, in terms of anti-bone destruction (271). Currently, DMARDs, in combination with angiogenesis inhibitors, are considered a potential strategy for RA treatment (272). However, only a few clinical studies have reported on this treatment strategy for RA.
Abatacept (ABT) is a co-stimulation inhibitor that can bind to CD80 and CD86, preventing CD28-mediated T cell activation by blocking costimulatory signaling. In patients with RA, ABT produced significant clinical and functional benefits. Moreover, VEGF was significantly decreased in the serum of patients with RA receiving ABT (273), while transcriptomics showed that the mechanism of action of ABT is associated with improved antioxidative damage and regulation of the ETC pathway (274, 275).
Accumulation of ROS and upregulation of HIF-1α contribute to M1 cell polarization and cause inflammatory responses, whereas knockdown of HIF-1α facilitates M2 polarization (276). Kim et al. (277) developed a biocompatible therapeutic agent for ROS accumulation using manganese ferrite and ceria nanoparticle-anchored mesoporous silica nanoparticles (MFC-MSNs) joint cavity injection, which can actively clear ROS and produce O2. MFC-MSNs can be used as drug delivery vehicles to enhance therapeutic effects through sustained release of methotrexate (MTX). Li et al. (278) prepared ROS-responsive artesunate (ART) and dexamethasone (DEX) as a prodrug micellar nanosystem (DEX/HTA), which can effectively accumulate ART and DEX in AIA rats with arthritis. It can be specifically internalized by M1 cells, release ART and DEX, scavenge ROS, inhibit the HIF-1α/NF-κB pathway, and mediate repolarization of macrophages.
Resveratrol has been intensively investigated in several aspects of RA treatment, and it can delay the progression of RA by scavenging ROS and reducing angiogenesis by blocking the MAPK pathway (279). Liquiritin, a natural extract of Glycyrrhiza uralensis, has been found to inhibit RA angiogenesis. Liquiritin can promote apoptosis by regulating changes in mitochondrial membrane potential and inhibit the expression of p38 and VEGF (280). AMPK is a key protein that senses oxidative stress and regulates the body’s antioxidant activity. AMPK can improve oxidative stress by regulating SOD (SOD2) expression and mitochondrial superoxide levels and is involved in VEGF expression and angiogenesis (281). In conclusion, mitochondria are considered to be a key target for angiogenesis inhibition. However, more studies on RA are needed.
Clinical trials have shown that the use of tocilizumab in combination with methotrexate in patients with moderate-to-severe RA can significantly decrease the level of the bone remodeling markers, C-terminal cross-linked telopeptide of type I collagen and MMP-degraded type II collagen, and inhibit the bone remodeling process (282). Therefore, early and regular drug administration is the key to reduce the disability rate of RA. A double-blind randomized controlled trial showed that treatment with the anti-RANKL antibody, denosumab, significantly inhibited the progression of joint destruction and was well tolerated, which is expected to become the clinical treatment for RA (283). Curculigoside is a glycoside in polyphenols obtained from roots and exhibit antioxidant effects. It regulates cartilage destruction, enhances osteoblast differentiation, reduces osteoclast differentiation, and inhibits osteolytic progression (284). Therefore, curculigoside has been studied extensively in bone destruction-related diseases, such as osteoporosis and RA. Network pharmacology analysis has shown that the phosphoinositide 3 kinase/protein kinase B (PI3K/AKT) pathway and proteins, such as epidermal growth factor receptor, recombinant MAPK kinase 1 (MAP2K1), and MMP-2, are key targets for curculigoside therapy (285). In vitro studies have shown that curculigoside inhibits tartrate-resistant acid phosphatase activity in osteoblasts induced by RANKL or H2O2 and reduces the expression of cathepsin K (Ctsk) and MMP-9. Its mechanism of action is closely related to the regulation of the Nrf2/NF-κB pathway and reduction of ROS levels (286). Sanguis draconis is a traditional Chinese herb, and its active ingredient, loureirin B (LrB), is widely used in the treatment of inflammatory and immune diseases. LrB can reduce RANKL-induced osteoclastogenesis by inhibiting recombinant NFAT (NFATC1) and ROS activity. It is a potential drug for the treatment of osteoporosis (287). LrB inhibits Ca2+ influx and IL-2 secretion in Jurkat T-cells by inhibiting the KV1.3 and stromal interaction molecule 1 (STIM1)/Orai1 pathways, which can induce immunosuppressive effects (288). It is a potential drug for the treatment of autoimmune diseases. However, it has not yet been studied in RA.
Several DMARDs have been shown to slow the progression of cartilage damage in patients with RA. Both in vivo and in vitro studies have shown that methotrexate inhibits FLS invasion and reduces cartilage degradation (289). Additionally, leflunomide treatment decreased the levels of MMP-1, MMP-9, and cartilage oligomeric matrix protein (COMP) in the serum of patients with RA (290). Meanwhile, studies have designed loaded PEI-SS-IND-MTX-MMP-9 siRNA nanoparticles for RA cartilage damage for the delivery of indindexin (IND), MTX, and MMP-9 siRNA, which significantly downregulated the expression of MMP-9 and various inflammatory factors in Raw-264.7 cells and showed anti-inflammatory activity and reversal of bone destruction in RA mice (291). Postprandial selenium supplementation can inhibit ROS and RANKL levels and reduce cartilage destruction in CIA mice. However, the optimal dose of selenium supplementation has not yet been determined, and relevant clinical studies are underway (292).
As mentioned above, multiple factors, such as ROS accumulation and mitochondria, can promote articular cartilage destruction and accelerate joint deformation in RA. Many drugs have been shown to slow the process of bone destruction by targeting ROS or mitochondrial damage. Diosmin is an unsaturated glycoside with antioxidant and anti-inflammatory properties. Intervention with diosmin and trolox (a water-soluble vitamin E that can be used to scavenge ROS) in rats administered complete freund’s adjuvant (CFA) reduced the levels of various peroxidation products and production of various MMPs and elevated the Nrf2 activity, inflammatory response, and cartilage destruction in CFA-administered rats (293). Mitochondria are equally attractive targets for stopping the process of bone destruction in RA. Estrogen levels are associated with several bone damage-phase diseases and can affect chondrocyte metabolism and cell cycle (294). Some studies have shown that 17b-estradiol (17b-E2) promotes mitophagy and enhances chondrocyte viability by elevating AMPK/mammalian target of rapamycin (mTOR) pathway activity (295). However, a cohort study showed that the role of estrogen in RA remains controversial (296). Urolithin A (UA), a natural metabolite produced by intestinal bacteria and mainly found in fruits, such as pomegranate, can improve mitochondrial function. UA reduces disease progression, cartilage degeneration, synovial inflammation, and pain symptoms in OA mouse models and may play a therapeutic role in RA, suggesting dietary advice for patients with RA (297). The Chinese herbs, turmeric, yujin, and curcumin, are commonly used in RA treatment. Their common active ingredient, curcumin, can mediate mitophagy in OA chondrocytes by promoting AMPK/PINK1/Parkin, scavenging ROS, elevating mitochondrial membrane potential, and exerting an inhibitory effect on cartilage destruction (298). Although many similarities exist between OA and RA, many differences exist because mitochondrial damage and oxidative stress are more pronounced in RA than in OA (299). However, studies on targeting mitochondria for RA treatment remain inadequate.
In recent years, studies have provided new insights into the development of RA. Oxidative stress and mitochondrial damage are inextricably linked to RA development, and our focus was on the mitochondrial regulation of metabolism affecting the disease process of RA. However, mitochondria, as a signaling center, affect RA in multiple ways. Here, we summarized the role of the vicious cycle of mitochondrial damage and ROS accumulation in RA and introduced potential drugs that target mitochondria or scavenge ROS for RA treatment, with the aim of providing some help for the clinical treatment of RA (Table 1).
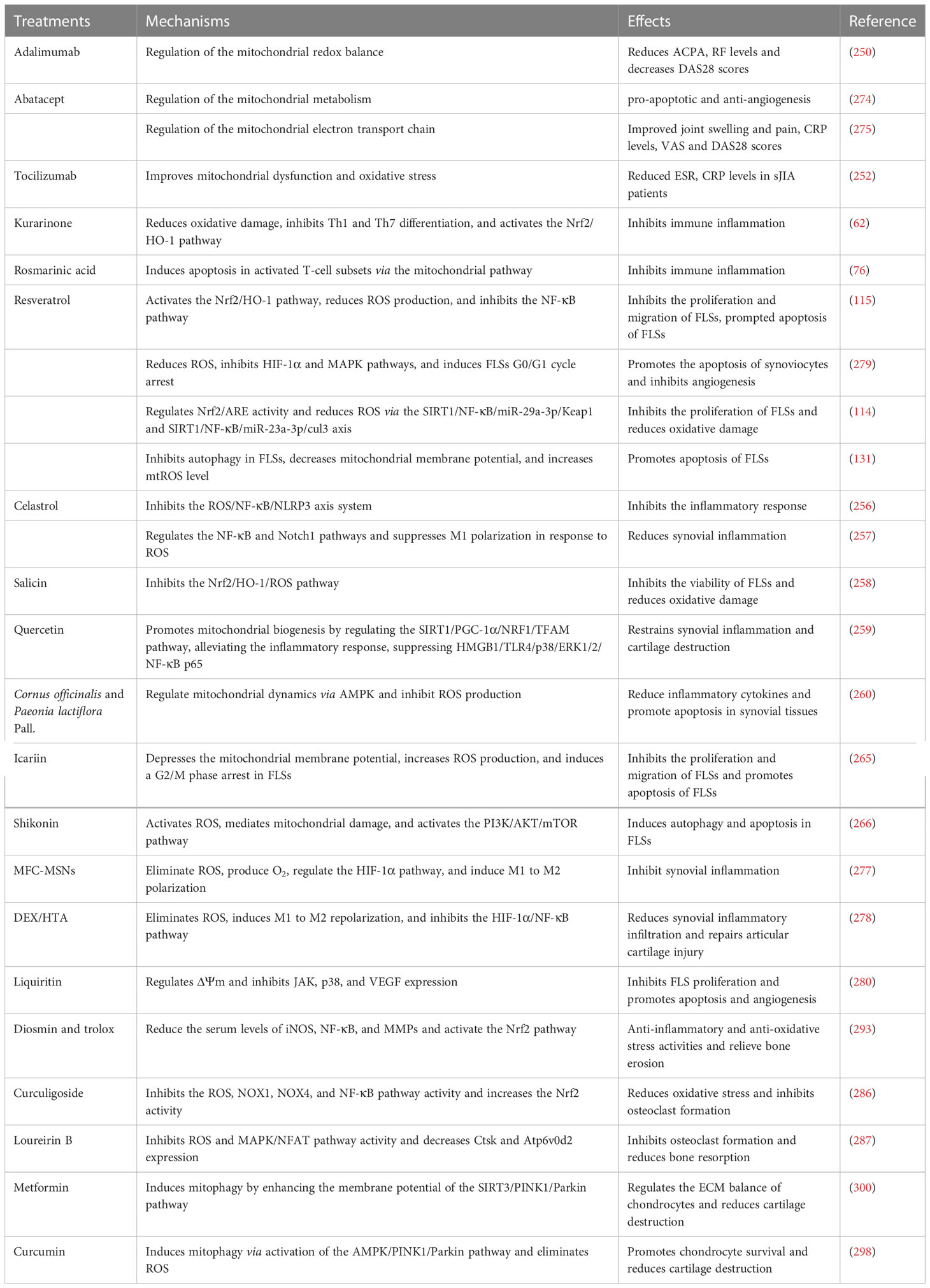
Table 1 Drugs used in RA treatment that act by regulating ROS or the mitochondria.
Although progress has been made in the study of mitochondria in RA, many questions remain unanswered. Mitochondrial damage induces mitochondrial division, which in turn promotes mitochondrial autophagic behavior to remove damaged mitochondria and induce mitochondrial regeneration. However, different studies have reported different results, with some showing that inhibition of dynamin 1-like protein expression and mitochondrial division can inhibit mitophagy and alleviate inflammatory response in RA (301). However, some studies have reported contrasting results (260). This may be related to the diversity of mitochondrial division forms and great variations in the outcomes caused by different division forms (302). Additionally, the roles of ROS and mitophagy in RA are complex and diverse, with ROS inducing mitochondrial autophagic behavior and enhancing mitochondrial autophagic activity, possibly contributing to the survival of FLSs and increasing the inflammatory response, and inhibition of autophagy inducing apoptosis of FLSs (303). Elevation of ROS levels to promote apoptosis has been widely studied in cancer research (304). Therefore, the two-fold nature of mitophagy and its role in RA need to be studied further (305).
We have summarized many potential therapeutic agents for mitochondrial damage and ROS accumulation. Although ROS or mitochondria is a reliable target for RA treatment, many therapeutic agents that are being used in the clinics can play a role in repairing mitochondrial damage or scavenging ROS. However, the advantages and differences in these potential therapeutic agents, compared with existing drugs, require further research and exploration. Much work is needed before potential drugs can be used in clinical treatment.
WJ wrote the manuscript, CL proofread the manuscript. XD and HW revised the manuscript. All authors contributed to the article and approved the submitted version.
The National Natural Science Foundation of China (No. 82060891), Natural Science Foundation of Gansu Province (No. 21JR7RA568, No. 22JR5RA637), Project of Zheng’s Acupuncture Academic Schools of Heritage Studio, Gansu Province, State Administration of TCM (No. 2305135901), and Gansu Province Youth Science and Technology Fund (No. 20JR10RA344) funded the study.
We are grateful to the editors and reviewers for the painstaking care taken in helping improve the clarity of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet (2016) 388:2023–38. doi: 10.1016/S0140-6736(16)30173-8
2. Scherer HU, Häupl T, Burmester GR. The etiology of rheumatoid arthritis. J Autoimmun (2020) 110:102400. doi: 10.1016/j.jaut.2019.102400
3. Lin Y-J, Anzaghe M, Schülke S. Update on the pathomechanism, diagnosis, and treatment options for rheumatoid arthritis. Cells (2020) 9:E880. doi: 10.3390/cells9040880
4. Liao KP. Cardiovascular disease in patients with rheumatoid arthritis. Trends Cardiovasc Med (2017) 27:136–40. doi: 10.1016/j.tcm.2016.07.006
5. Kadura S, Raghu G. Rheumatoid arthritis-interstitial lung disease: manifestations and current concepts in pathogenesis and management. Eur Respir Rev (2021) 30:210011. doi: 10.1183/16000617.0011-2021
6. Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med (2000) 192:1001–14. doi: 10.1084/jem.192.7.1001
7. Bolduc JA, Collins JA, Loeser RF. Reactive oxygen species, aging and articular cartilage homeostasis. Free Radic Biol Med (2019) 132:73–82. doi: 10.1016/j.freeradbiomed.2018.08.038
8. Kan S, Duan M, Liu Y, Wang C, Xie J. Role of mitochondria in physiology of chondrocytes and diseases of osteoarthritis and rheumatoid arthritis. Cartilage (2021) 13:1102S–21S. doi: 10.1177/19476035211063858
9. Fearon U, Canavan M, Biniecka M, Veale DJ. Hypoxia, mitochondrial dysfunction and synovial invasiveness in rheumatoid arthritis. Nat Rev Rheumatol (2016) 12:385–97. doi: 10.1038/nrrheum.2016.69
10. Konisti S, Kiriakidis S, Paleolog EM. Hypoxia–a key regulator of angiogenesis and inflammation in rheumatoid arthritis. Nat Rev Rheumatol (2012) 8:153–62. doi: 10.1038/nrrheum.2011.205
11. Clayton SA, MacDonald L, Kurowska-Stolarska M, Clark AR. Mitochondria as key players in the pathogenesis and treatment of rheumatoid arthritis. Front Immunol (2021) 12:673916. doi: 10.3389/fimmu.2021.673916
12. Phull A-R, Nasir B, Haq IU, Kim SJ. Oxidative stress, consequences and ROS mediated cellular signaling in rheumatoid arthritis. Chem Biol Interact (2018) 281:121–36. doi: 10.1016/j.cbi.2017.12.024
13. Guo R, Gu J, Zong S, Wu M, Yang M. Structure and mechanism of mitochondrial electron transport chain. BioMed J (2018) 41:9–20. doi: 10.1016/j.bj.2017.12.001
14. Zhao R-Z, Jiang S, Zhang L, Yu Z-B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int J Mol Med (2019) 44:3–15. doi: 10.3892/ijmm.2019.4188
15. Li R, Jia Z, Trush MA. Defining ROS in biology and medicine. React Oxyg Species (Apex) (2016) 1:9–21. doi: 10.20455/ros.2016.803
16. Sarniak A, Lipińska J, Tytman K, Lipińska S. Endogenous mechanisms of reactive oxygen species (ROS) generation. Postepy Hig Med Dosw (Online) (2016) 70:1150–65. doi: 10.5604/17322693.1224259
17. Korytowski W, Basova LV, Pilat A, Kernstock RM, Girotti AW. Permeabilization of the mitochondrial outer membrane by bax/truncated bid (tBid) proteins as sensitized by cardiolipin hydroperoxide translocation: mechanistic implications for the intrinsic pathway of oxidative apoptosis. J Biol Chem (2011) 286:26334–43. doi: 10.1074/jbc.M110.188516
18. Tajeddine N. How do reactive oxygen species and calcium trigger mitochondrial membrane permeabilisation? Biochim Biophys Acta (2016) 1860:1079–88. doi: 10.1016/j.bbagen.2016.02.013
19. Stenberg S, Li J, Gjuvsland AB, Persson K, Demitz-Helin E, González Peña C, et al. Genetically controlled mtDNA deletions prevent ROS damage by arresting oxidative phosphorylation. Elife (2022) 11:e76095. doi: 10.7554/eLife.76095
20. Jelinek A, Heyder L, Daude M, Plessner M, Krippner S, Grosse R, et al. Mitochondrial rescue prevents glutathione peroxidase-dependent ferroptosis. Free Radic Biol Med (2018) 117:45–57. doi: 10.1016/j.freeradbiomed.2018.01.019
21. Ng CT, Biniecka M, Kennedy A, McCormick J, Fitzgerald O, Bresnihan B, et al. Synovial tissue hypoxia and inflammation in vivo. Ann Rheum Dis (2010) 69:1389–95. doi: 10.1136/ard.2009.119776
22. Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev (2014) 94:909–50. doi: 10.1152/physrev.00026.2013
23. Fuchs K, Kuehn A, Mahling M, Guenthoer P, Hector A, Schwenck J, et al. In vivo hypoxia PET imaging quantifies the severity of arthritic joint inflammation in line with overexpression of hypoxia-inducible factor and enhanced reactive oxygen species generation. J Nucl Med (2017) 58:853–60. doi: 10.2967/jnumed.116.185934
24. Khojah HM, Ahmed S, Abdel-Rahman MS, Hamza A-B. Reactive oxygen and nitrogen species in patients with rheumatoid arthritis as potential biomarkers for disease activity and the role of antioxidants. Free Radic Biol Med (2016) 97:285–91. doi: 10.1016/j.freeradbiomed.2016.06.020
25. Oğul Y, Gür F, Cengiz M, Gür B, Sarı RA, Kızıltunç A. Evaluation of oxidant and intracellular anti-oxidant activity in rheumatoid arthritis patients: In vivo and in silico studies. Int Immunopharmacol (2021) 97:107654. doi: 10.1016/j.intimp.2021.107654
26. Kardeş S, Karagülle M, Durak İ, Avcı A, Karagülle MZ. Association of oxidative stress with clinical characteristics in patients with rheumatoid arthritis. Eur J Clin Invest (2018) 48(1):e12858–e12858. doi: 10.1111/eci.12858
27. Khanna S, Padhan P, Jaiswal KS, Jain AP, Ghosh A, Tripathy A, et al. Altered mitochondrial proteome and functional dynamics in patients with rheumatoid arthritis. Mitochondrion (2020) 54:8–14. doi: 10.1016/j.mito.2020.06.005
28. Jaiswal KS, Khanna S, Ghosh A, Padhan P, Raghav SK, Gupta B. Differential mitochondrial genome in patients with rheumatoid arthritis. Autoimmunity (2021) 54:1–12. doi: 10.1080/08916934.2020.1846182
29. Panga V, Kallor AA, Nair A, Harshan S, Raghunathan S. Mitochondrial dysfunction in rheumatoid arthritis: A comprehensive analysis by integrating gene expression, protein-protein interactions and gene ontology data. PloS One (2019) 14:e0224632. doi: 10.1371/journal.pone.0224632
30. Padgett LE, Tse HM. NADPH oxidase-derived superoxide provides a third signal for CD4 T cell effector responses. J Immunol (2016) 197:1733–42. doi: 10.4049/jimmunol.1502581
31. Kim E-K, Seo H-S, Chae M-J, Jeon I-S, Song B-Y, Park Y-J, et al. Enhanced antitumor immunotherapeutic effect of b-cell-based vaccine transduced with modified adenoviral vector containing type 35 fiber structures. Gene Ther (2014) 21:106–14. doi: 10.1038/gt.2013.65
32. Li Y, Shen Y, Jin K, Wen Z, Cao W, Wu B, et al. The DNA repair nuclease MRE11A functions as a mitochondrial protector and prevents T cell pyroptosis and tissue inflammation. Cell Metab (2019) 30:477–492.e6. doi: 10.1016/j.cmet.2019.06.016
33. Jang K-J, Mano H, Aoki K, Hayashi T, Muto A, Nambu Y, et al. Mitochondrial function provides instructive signals for activation-induced b-cell fates. Nat Commun (2015) 6:6750. doi: 10.1038/ncomms7750
34. Zhao J, Jiang P, Guo S, Schrodi SJ, He D. Apoptosis, autophagy, NETosis, necroptosis, and pyroptosis mediated programmed cell death as targets for innovative therapy in rheumatoid arthritis. Front Immunol (2021) 12:809806. doi: 10.3389/fimmu.2021.809806
35. Giatromanolaki A, Sivridis E, Maltezos E, Athanassou N, Papazoglou D, Gatter KC, et al. Upregulated hypoxia inducible factor-1alpha and -2alpha pathway in rheumatoid arthritis and osteoarthritis. Arthritis Res Ther (2003) 5:R193–201. doi: 10.1186/ar756
36. Wang Z, Castresana MR, Newman WH. Reactive oxygen and NF-kappaB in VEGF-induced migration of human vascular smooth muscle cells. Biochem Biophys Res Commun (2001) 285:669–74. doi: 10.1006/bbrc.2001.5232
37. Gao W, Sweeney C, Connolly M, Kennedy A, Ng CT, McCormick J, et al. Notch-1 mediates hypoxia-induced angiogenesis in rheumatoid arthritis. Arthritis Rheum (2012) 64:2104–13. doi: 10.1002/art.34397
38. Reichard A, Asosingh K. The role of mitochondria in angiogenesis. Mol Biol Rep (2019) 46:1393–400. doi: 10.1007/s11033-018-4488-x
39. Knowles HJ. Hypoxic regulation of osteoclast differentiation and bone resorption activity. Hypoxia (Auckl) (2015) 3:73–82. doi: 10.2147/HP.S95960
40. Li J, Dong S. The signaling pathways involved in chondrocyte differentiation and hypertrophic differentiation. Stem Cells Int (2016) 2016:2470351. doi: 10.1155/2016/2470351
41. Khan NM, Haseeb A, Ansari MY, Devarapalli P, Haynie S, Haqqi TM. Wogonin, a plant derived small molecule, exerts potent anti-inflammatory and chondroprotective effects through the activation of ROS/ERK/Nrf2 signaling pathways in human osteoarthritis chondrocytes. Free Radic Biol Med (2017) 106:288–301. doi: 10.1016/j.freeradbiomed.2017.02.041
42. López-Armada MJ, Fernández-Rodríguez JA, Blanco FJ. Mitochondrial dysfunction and oxidative stress in rheumatoid arthritis. Antioxidants (Basel) (2022) 11:1151. doi: 10.3390/antiox11061151
43. Biniecka M, Fox E, Gao W, Ng CT, Veale DJ, Fearon U, et al. Hypoxia induces mitochondrial mutagenesis and dysfunction in inflammatory arthritis. Arthritis Rheum (2011) 63:2172–82. doi: 10.1002/art.30395
44. Gautam S, Kumar U, Kumar M, Rana D, Dada R. Yoga improves mitochondrial health and reduces severity of autoimmune inflammatory arthritis: A randomized controlled trial. Mitochondrion (2021) 58:147–59. doi: 10.1016/j.mito.2021.03.004
45. Balogh E, Biniecka M, Fearon U, Veale DJ, Szekanecz Z. Angiogenesis in inflammatory arthritis. Isr Med Assoc J (2019) 21:345–52.
46. Maruotti N, Cantatore FP, Crivellato E, Vacca A, Ribatti D. Angiogenesis in rheumatoid arthritis. Histol Histopathol (2006) 21:557–66. doi: 10.14670/HH-21.557
47. Tateiwa D, Yoshikawa H, Kaito T. Cartilage and bone destruction in arthritis: Pathogenesis and treatment strategy: A literature review. Cells (2019) 8:E818. doi: 10.3390/cells8080818
48. Komatsu N, Takayanagi H. Mechanisms of joint destruction in rheumatoid arthritis - immune cell-fibroblast-bone interactions. Nat Rev Rheumatol (2022) 18:415–29. doi: 10.1038/s41584-022-00793-5
49. Floudas A, Neto N, Orr C, Canavan M, Gallagher P, Hurson C, et al. Loss of balance between protective and pro-inflammatory synovial tissue T-cell polyfunctionality predates clinical onset of rheumatoid arthritis. Ann Rheum Dis (2022) 81:193–205. doi: 10.1136/annrheumdis-2021-220458
50. Bartok B, Firestein GS. Fibroblast-like synoviocytes: key effector cells in rheumatoid arthritis. Immunol Rev (2010) 233:233–55. doi: 10.1111/j.0105-2896.2009.00859.x
51. Kemble S, Croft AP. Critical role of synovial tissue-resident macrophage and fibroblast subsets in the persistence of joint inflammation. Front Immunol (2021) 12:715894. doi: 10.3389/fimmu.2021.715894
52. Huang Q-Q, Doyle R, Chen S-Y, Sheng Q, Misharin AV, Mao Q, et al. Critical role of synovial tissue-resident macrophage niche in joint homeostasis and suppression of chronic inflammation. Sci Adv (2021) 7:eabd0515. doi: 10.1126/sciadv.abd0515
53. Feng L-J, Jiang T-C, Zhou C-Y, Yu C-L, Shen Y-J, Li J, et al. Activated macrophage-like synoviocytes are resistant to endoplasmic reticulum stress-induced apoptosis in antigen-induced arthritis. Inflamm Res (2014) 63:335–46. doi: 10.1007/s00011-013-0705-1
54. Tu J, Hong W, Zhang P, Wang X, Körner H, Wei W. Ontology and function of fibroblast-like and macrophage-like synoviocytes: How do they talk to each other and can they be targeted for rheumatoid arthritis therapy? Front Immunol (2018) 9:1467. doi: 10.3389/fimmu.2018.01467
55. Cui L, Weiyao J, Chenghong S, Limei L, Xinghua Z, Bo Y, et al. Rheumatoid arthritis and mitochondrial homeostasis: The crossroads of metabolism and immunity. Front Med (Lausanne) (2022) 9:1017650. doi: 10.3389/fmed.2022.1017650
56. Wójcik P, Gęgotek A, Žarković N, Skrzydlewska E. Oxidative stress and lipid mediators modulate immune cell functions in autoimmune diseases. Int J Mol Sci (2021) 22:E723. doi: 10.3390/ijms22020723
57. Kondo N, Kuroda T, Kobayashi D. Cytokine networks in the pathogenesis of rheumatoid arthritis. Int J Mol Sci (2021) 22:10922. doi: 10.3390/ijms222010922
58. Hou S-M, Chen P-C, Lin C-M, Fang M-L, Chi M-C, Liu J-F. CXCL1 contributes to IL-6 expression in osteoarthritis and rheumatoid arthritis synovial fibroblasts by CXCR2, c-raf, MAPK, and AP-1 pathway. Arthritis Res Ther (2020) 22:251. doi: 10.1186/s13075-020-02331-8
59. Matsumoto H, Fujita Y, Asano T, Matsuoka N, Temmoku J, Sato S, et al. Association between inflammatory cytokines and immune-checkpoint molecule in rheumatoid arthritis. PloS One (2021) 16:e0260254. doi: 10.1371/journal.pone.0260254
60. Chávez MD, Tse HM. Targeting mitochondrial-derived reactive oxygen species in T cell-mediated autoimmune diseases. Front Immunol (2021) 12:703972. doi: 10.3389/fimmu.2021.703972
61. Abimannan T, Peroumal D, Parida JR, Barik PK, Padhan P, Devadas S. Oxidative stress modulates the cytokine response of differentiated Th17 and Th1 cells. Free Radic Biol Med (2016) 99:352–63. doi: 10.1016/j.freeradbiomed.2016.08.026
62. Tang K-T, Lin C-C, Lin S-C, Wang J-H, Tsai S-W. Kurarinone attenuates collagen-induced arthritis in mice by inhibiting Th1/Th17 cell responses and oxidative stress. Int J Mol Sci (2021) 22:4002. doi: 10.3390/ijms22084002
63. Zhi L, Ustyugova IV, Chen X, Zhang Q, Wu MX. Enhanced Th17 differentiation and aggravated arthritis in IEX-1-deficient mice by mitochondrial reactive oxygen species-mediated signaling. J Immunol (2012) 189:1639–47. doi: 10.4049/jimmunol.1200528
64. Hu F, Mu R, Zhu J, Shi L, Li Y, Liu X, et al. Hypoxia and hypoxia-inducible factor-1α provoke toll-like receptor signalling-induced inflammation in rheumatoid arthritis. Ann Rheum Dis (2014) 73:928–36. doi: 10.1136/annrheumdis-2012-202444
65. Harnanik T, Soeroso J, Suryokusumo MG, Juliandhy T. Effects of hyperbaric oxygen on T helper 17/regulatory T polarization in antigen and collagen-induced arthritis: Hypoxia-inducible factor-1α as a target. Oman Med J (2020) 35:e90. doi: 10.5001/omj.2020.08
66. Weyand CM, Wu B, Goronzy JJ. The metabolic signature of T cells in rheumatoid arthritis. Curr Opin Rheumatol (2020) 32:159–67. doi: 10.1097/BOR.0000000000000683
67. Andonian BJ, Koss A, Koves TR, Hauser ER, Hubal MJ, Pober DM, et al. Rheumatoid arthritis T cell and muscle oxidative metabolism associate with exercise-induced changes in cardiorespiratory fitness. Sci Rep (2022) 12:7450. doi: 10.1038/s41598-022-11458-4
68. Balogh E, Veale DJ, McGarry T, Orr C, Szekanecz Z, Ng C-T, et al. Oxidative stress impairs energy metabolism in primary cells and synovial tissue of patients with rheumatoid arthritis. Arthritis Res Ther (2018) 20:95. doi: 10.1186/s13075-018-1592-1
69. Pucino V, Certo M, Bulusu V, Cucchi D, Goldmann K, Pontarini E, et al. Lactate buildup at the site of chronic inflammation promotes disease by inducing CD4+ T cell metabolic rewiring. Cell Metab (2019) 30:1055–1074.e8. doi: 10.1016/j.cmet.2019.10.004
70. Souto-Carneiro MM, Klika KD, Abreu MT, Meyer AP, Saffrich R, Sandhoff R, et al. Effect of increased lactate dehydrogenase a activity and aerobic glycolysis on the proinflammatory profile of autoimmune CD8+ T cells in rheumatoid arthritis. Arthritis Rheumatol (2020) 72:2050–64. doi: 10.1002/art.41420
71. Harshan S, Dey P, Raghunathan S. Altered transcriptional regulation of glycolysis in circulating CD8+ T cells of rheumatoid arthritis patients. Genes (Basel) (2022) 13:1216. doi: 10.3390/genes13071216
72. Qiu J, Wu B, Goodman SB, Berry GJ, Goronzy JJ, Weyand CM. Metabolic control of autoimmunity and tissue inflammation in rheumatoid arthritis. Front Immunol (2021) 12:652771. doi: 10.3389/fimmu.2021.652771
73. Gambhir L, Sharma V, Kandwal P, Saxena S. Perturbation in cellular redox homeostasis: Decisive regulator of T cell mediated immune responses. Int Immunopharmacol (2019) 67:449–57. doi: 10.1016/j.intimp.2018.12.049
74. Mak TW, Grusdat M, Duncan GS, Dostert C, Nonnenmacher Y, Cox M, et al. Glutathione primes T cell metabolism for inflammation. Immunity (2017) 46:1089–90. doi: 10.1016/j.immuni.2017.06.009
75. Li Y, Shen Y, Hohensinner P, Ju J, Wen Z, Goodman SB, et al. Deficient activity of the nuclease MRE11A induces T cell aging and promotes arthritogenic effector functions in patients with rheumatoid arthritis. Immunity (2016) 45:903–16. doi: 10.1016/j.immuni.2016.09.013
76. Hur Y-G, Suh C-H, Kim S, Won J. Rosmarinic acid induces apoptosis of activated T cells from rheumatoid arthritis patients via mitochondrial pathway. J Clin Immunol (2007) 27:36–45. doi: 10.1007/s10875-006-9057-8
77. Lopez-Santalla M, Fernandez-Perez R, Garin MI. Mesenchymal Stem/Stromal cells for rheumatoid arthritis treatment: An update on clinical applications. Cells (2020) 9:E1852. doi: 10.3390/cells9081852
78. Luque-Campos N, Contreras-López RA, Jose Paredes-Martínez M, Torres MJ, Bahraoui S, Wei M, et al. Mesenchymal stem cells improve rheumatoid arthritis progression by controlling memory T cell response. Front Immunol (2019) 10:798. doi: 10.3389/fimmu.2019.00798
79. Luz-Crawford P, Hernandez J, Djouad F, Luque-Campos N, Caicedo A, Carrère-Kremer S, et al. Mesenchymal stem cell repression of Th17 cells is triggered by mitochondrial transfer. Stem Cell Res Ther (2019) 10:232. doi: 10.1186/s13287-019-1307-9
80. Kornicka-Garbowska K, Groborz S, Lynda B, Galuppo L, Marycz K. Mitochondria transfer restores fibroblasts-like synoviocytes (FLS) plasticity in LPS-induced, in vitro synovitis model. Cell Commun Signal (2022) 20:137. doi: 10.1186/s12964-022-00923-2
81. Giwa R, Brestoff JR. Mitochondria transfer to CD4+ T cells may alleviate rheumatoid arthritis by suppressing pro-inflammatory cytokine production. Immunometabolism (2022) 4:e220009. doi: 10.20900/immunometab20220009
82. Lightowlers RN, Chrzanowska-Lightowlers ZM, Russell OM. Mitochondrial transplantation-a possible therapeutic for mitochondrial dysfunction?: mitochondrial transfer is a potential cure for many diseases but proof of efficacy and safety is still lacking. EMBO Rep (2020) 21:e50964. doi: 10.15252/embr.202050964
83. Kim MJ, Hwang JW, Yun C-K, Lee Y, Choi Y-S. Delivery of exogenous mitochondria via centrifugation enhances cellular metabolic function. Sci Rep (2018) 8:3330. doi: 10.1038/s41598-018-21539-y
84. Kristyanto H, Blomberg NJ, Slot LM, van der Voort EIH, Kerkman PF, Bakker A, et al. Persistently activated, proliferative memory autoreactive b cells promote inflammation in rheumatoid arthritis. Sci Transl Med (2020) 12:eaaz5327. doi: 10.1126/scitranslmed.aaz5327
85. Tavakolpour S, Alesaeidi S, Darvishi M, GhasemiAdl M, Darabi-Monadi S, Akhlaghdoust M, et al. A comprehensive review of rituximab therapy in rheumatoid arthritis patients. Clin Rheumatol (2019) 38:2977–94. doi: 10.1007/s10067-019-04699-8
86. Polikowsky HG, Wogsland CE, Diggins KE, Huse K, Irish JM. Cutting edge: Redox signaling hypersensitivity distinguishes human germinal center b cells. J Immunol (2015) 195:1364–7. doi: 10.4049/jimmunol.1500904
87. Wheeler ML, Defranco AL. Prolonged production of reactive oxygen species in response to b cell receptor stimulation promotes b cell activation and proliferation. J Immunol (2012) 189:4405–16. doi: 10.4049/jimmunol.1201433
88. Liu Y, Gokhale S, Jung J, Zhu S, Luo C, Saha D, et al. Mitochondrial fission factor is a novel interacting protein of the critical b cell survival regulator TRAF3 in b lymphocytes. Front Immunol (2021) 12:670338. doi: 10.3389/fimmu.2021.670338
89. Khmaladze I, Saxena A, Nandakumar KS, Holmdahl R. B-cell epitope spreading and inflammation in a mouse model of arthritis is associated with a deficiency in reactive oxygen species production. Eur J Immunol (2015) 45:2243–51. doi: 10.1002/eji.201545518
90. Wright HL, Moots RJ, Edwards SW. The multifactorial role of neutrophils in rheumatoid arthritis. Nat Rev Rheumatol (2014) 10:593–601. doi: 10.1038/nrrheum.2014.80
91. O’Neil LJ, Kaplan MJ. Neutrophils in rheumatoid arthritis: Breaking immune tolerance and fueling disease. Trends Mol Med (2019) 25:215–27. doi: 10.1016/j.molmed.2018.12.008
92. Bedouhène S, Dang PM-C, Hurtado-Nedelec M, El-Benna J. Neutrophil degranulation of azurophil and specific granules. Methods Mol Biol (2020) 2087:215–22. doi: 10.1007/978-1-0716-0154-9_16
93. Kaushal J, Kamboj A, Anupam K, Tandon A, Sharma A, Bhatnagar A. Interplay of redox imbalance with matrix gelatinases in neutrophils and their association with disease severity in rheumatoid arthritis patients. Clin Immunol (2022) 237:108965. doi: 10.1016/j.clim.2022.108965
94. Wright HL, Lyon M, Chapman EA, Moots RJ, Edwards SW. Rheumatoid arthritis synovial fluid neutrophils drive inflammation through production of chemokines, reactive oxygen species, and neutrophil extracellular traps. Front Immunol (2020) 11:584116. doi: 10.3389/fimmu.2020.584116
95. Bagchi A, Ghosh P, Ghosh A, Chatterjee M. Role of oxidative stress in induction of trans-differentiation of neutrophils in patients with rheumatoid arthritis. Free Radic Res (2022) 56:290–302. doi: 10.1080/10715762.2022.2089567
96. Kaundal U, Khullar A, Leishangthem B, Jain S, Dhooria A, Saikia B, et al. The effect of methotrexate on neutrophil reactive oxygen species and CD177 expression in rheumatoid arthritis. Clin Exp Rheumatol (2021) 39:479–86. doi: 10.55563/clinexprheumatol/4h5onh
97. Duvvuri B, Baddour AA, Deane KD, Feser ML, Nelson JL, Demoruelle MK, et al. Mitochondrial n-formyl methionine peptides associate with disease activity as well as contribute to neutrophil activation in patients with rheumatoid arthritis. J Autoimmun (2021) 119:102630. doi: 10.1016/j.jaut.2021.102630
98. Andreev-Andrievskiy AA, Kolosova NG, Stefanova NA, Lovat MV, Egorov MV, Manskikh VN, et al. Efficacy of mitochondrial antioxidant plastoquinonyl-decyl-triphenylphosphonium bromide (SkQ1) in the rat model of autoimmune arthritis. Oxid Med Cell Longev (2016) 2016:8703645. doi: 10.1155/2016/8703645
99. Svensson MND, Zoccheddu M, Yang S, Nygaard G, Secchi C, Doody KM, et al. Synoviocyte-targeted therapy synergizes with TNF inhibition in arthritis reversal. Sci Adv (2020) 6:eaba4353. doi: 10.1126/sciadv.aba4353
100. McHugh J. SUMOylation links metabolic and aggressive phenotype of RA FLS. Nat Rev Rheumatol (2020) 16:668. doi: 10.1038/s41584-020-00526-6
101. Souliotis VL, Vlachogiannis NI, Pappa M, Argyriou A, Sfikakis PP. DNA Damage accumulation, defective chromatin organization and deficient DNA repair capacity in patients with rheumatoid arthritis. Clin Immunol (2019) 203:28–36. doi: 10.1016/j.clim.2019.03.009
102. Lee S-H, Chang DK, Goel A, Boland CR, Bugbee W, Boyle DL, et al. Microsatellite instability and suppressed DNA repair enzyme expression in rheumatoid arthritis. J Immunol (2003) 170:2214–20. doi: 10.4049/jimmunol.170.4.2214
103. Kullmann F, Widmann T, Kirner A, Jüsten HP, Wessinghage D, Dietmaier W, et al. Microsatellite analysis in rheumatoid arthritis synovial fibroblasts. Ann Rheum Dis (2000) 59:386–9. doi: 10.1136/ard.59.5.386
104. Zhang T, Li H, Shi J, Li S, Li M, Zhang L, et al. p53 predominantly regulates IL-6 production and suppresses synovial inflammation in fibroblast-like synoviocytes and adjuvant-induced arthritis. Arthritis Res Ther (2016) 18:271. doi: 10.1186/s13075-016-1161-4
105. Ekwall A-KH, Whitaker JW, Hammaker D, Bugbee WD, Wang W, Firestein GS. The rheumatoid arthritis risk gene LBH regulates growth in fibroblast-like synoviocytes. Arthritis Rheumatol (2015) 67:1193–202. doi: 10.1002/art.39060
106. Garcia-Carbonell R, Divakaruni AS, Lodi A, Vicente-Suarez I, Saha A, Cheroutre H, et al. Critical role of glucose metabolism in rheumatoid arthritis fibroblast-like synoviocytes. Arthritis Rheumatol (2016) 68:1614–26. doi: 10.1002/art.39608
107. Huh YH, Lee G, Song W-H, Koh J-T, Ryu J-H. Crosstalk between FLS and chondrocytes is regulated by HIF-2α-mediated cytokines in arthritis. Exp Mol Med (2015) 47:e197. doi: 10.1038/emm.2015.88
108. Huh YH, Lee G, Lee K-B, Koh J-T, Chun J-S, Ryu J-H. HIF-2α-induced chemokines stimulate motility of fibroblast-like synoviocytes and chondrocytes into the cartilage-pannus interface in experimental rheumatoid arthritis mouse models. Arthritis Res Ther (2015) 17:302. doi: 10.1186/s13075-015-0816-x
109. Yoo S-J, Lee H-R, Kim J, Yoo IS, Park CK, Kang SW. Hypoxia-inducible factor-2 alpha regulates the migration of fibroblast-like synoviocytes via oxidative stress-induced CD70 expression in patients with rheumatoid arthritis. Int J Mol Sci (2022) 23:2342. doi: 10.3390/ijms23042342
110. Lee H-R, Yoo S-J, Kim J, Park CK, Kang SW. Reduction of oxidative stress in peripheral blood mononuclear cells attenuates the inflammatory response of fibroblast-like synoviocytes in rheumatoid arthritis. Int J Mol Sci (2021) 22:12411. doi: 10.3390/ijms222212411
111. León Fernández OS, Viebahn-Haensler R, Cabreja GL, Espinosa IS, Matos YH, Roche LD, et al. Medical ozone increases methotrexate clinical response and improves cellular redox balance in patients with rheumatoid arthritis. Eur J Pharmacol (2016) 789:313–8. doi: 10.1016/j.ejphar.2016.07.031
112. Kaur G, Sharma A, Bhatnagar A. Role of oxidative stress in pathophysiology of rheumatoid arthritis: insights into NRF2-KEAP1 signalling. Autoimmunity (2021) 54:385–97. doi: 10.1080/08916934.2021.1963959
113. Du Y, Wang Q, Tian N, Lu M, Zhang X-L, Dai S-M. Knockdown of nrf2 exacerbates TNF-α-Induced proliferation and invasion of rheumatoid arthritis fibroblast-like synoviocytes through activating JNK pathway. J Immunol Res (2020) 2020:6670464. doi: 10.1155/2020/6670464
114. Wang G, Xie X, Yuan L, Qiu J, Duan W, Xu B, et al. Resveratrol ameliorates rheumatoid arthritis via activation of SIRT1-Nrf2 signaling pathway. Biofactors (2020) 46:441–53. doi: 10.1002/biof.1599
115. Zhang Y, Wang G, Wang T, Cao W, Zhang L, Chen X. Nrf2-Keap1 pathway-mediated effects of resveratrol on oxidative stress and apoptosis in hydrogen peroxide-treated rheumatoid arthritis fibroblast-like synoviocytes. Ann N Y Acad Sci (2019) 1457:166–78. doi: 10.1111/nyas.14196
116. Valcárcel-Ares MN, Riveiro-Naveira RR, Vaamonde-García C, Loureiro J, Hermida-Carballo L, Blanco FJ, et al. Mitochondrial dysfunction promotes and aggravates the inflammatory response in normal human synoviocytes. Rheumatol (Oxford) (2014) 53:1332–43. doi: 10.1093/rheumatology/keu016
117. Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol (2018) 19:121–35. doi: 10.1038/nrm.2017.95
118. Yan H, Zhou H-F, Hu Y, Pham CTN. Suppression of experimental arthritis through AMP-activated protein kinase activation and autophagy modulation. J Rheum Dis Treat (2015) 1:5. doi: 10.23937/2469-5726/1510005
119. Shi M, Wang J, Xiao Y, Wang C, Qiu Q, Lao M, et al. Glycogen metabolism and rheumatoid arthritis: The role of glycogen synthase 1 in regulation of synovial inflammation via blocking AMP-activated protein kinase activation. Front Immunol (2018) 9:1714. doi: 10.3389/fimmu.2018.01714
120. Li Y, Meng W, Hou Y, Li D, Wang X, Wu K, et al. Dual role of mitophagy in cardiovascular diseases. J Cardiovasc Pharmacol (2021) 78:e30–9. doi: 10.1097/FJC.0000000000001046
121. Yan C, Li T-S. Dual role of mitophagy in cancer drug resistance. Anticancer Res (2018) 38:617–21. doi: 10.21873/anticanres.12266
122. Nam J-H, Lee J-H, Choi H-J, Choi S-Y, Noh K-E, Jung N-C, et al. TNF-α induces mitophagy in rheumatoid arthritis synovial fibroblasts, and mitophagy inhibition alleviates synovitis in collagen antibody-induced arthritis. Int J Mol Sci (2022) 23:5650. doi: 10.3390/ijms23105650
123. Jiang Y, Krantz S, Qin X, Li S, Gunasekara H, Kim Y-M, et al. Caveolin-1 controls mitochondrial damage and ROS production by regulating fission - fusion dynamics and mitophagy. Redox Biol (2022) 52:102304. doi: 10.1016/j.redox.2022.102304
124. Jannat A, John P, Bhatti A, Hayat MQ. Tomorou attenuates progression of rheumatoid arthritis through alteration in ULK-1 independent autophagy pathway in collagen induced arthritis mice model. Cell Death Discov (2019) 5:142. doi: 10.1038/s41420-019-0222-2
125. Zhu J, Wang KZQ, Chu CT. After the banquet: mitochondrial biogenesis, mitophagy, and cell survival. Autophagy (2013) 9:1663–76. doi: 10.4161/auto.24135
126. Li S, Zhang J, Liu C, Wang Q, Yan J, Hui L, et al. The role of mitophagy in regulating cell death. Oxid Med Cell Longev (2021) 2021:6617256. doi: 10.1155/2021/6617256
127. Deng R, Wang Y, Bu Y, Wu H. BNIP3 mediates the different adaptive responses of fibroblast-like synovial cells to hypoxia in patients with osteoarthritis and rheumatoid arthritis. Mol Med (2022) 28:64. doi: 10.1186/s10020-022-00490-9
128. Su L-J, Zhang J-H, Gomez H, Murugan R, Hong X, Xu D, et al. Reactive oxygen species-induced lipid peroxidation in apoptosis, autophagy, and ferroptosis. Oxid Med Cell Longev (2019) 2019:5080843. doi: 10.1155/2019/5080843
129. Wang Y, Yang Q, Shen S, Zhang L, Xiang Y, Weng X. Mst1 promotes mitochondrial dysfunction and apoptosis in oxidative stress-induced rheumatoid arthritis synoviocytes. Aging (Albany NY) (2020) 12:16211–23. doi: 10.18632/aging.103643
130. Yan C, Kong D, Ge D, Zhang Y, Zhang X, Su C, et al. Mitomycin c induces apoptosis in rheumatoid arthritis fibroblast-like synoviocytes via a mitochondrial-mediated pathway. Cell Physiol Biochem (2015) 35:1125–36. doi: 10.1159/000373938
131. Zhang J, Song X, Cao W, Lu J, Wang X, Wang G, et al. Autophagy and mitochondrial dysfunction in adjuvant-arthritis rats treatment with resveratrol. Sci Rep (2016) 6:32928. doi: 10.1038/srep32928
132. Lee H, Kang SW, Byun HS, Jeon J, Park KA, Kang K, et al. Brazilin limits inflammatory responses through induction of prosurvival autophagy in rheumatoid fibroblast-like synoviocytes. PloS One (2015) 10:e0136122. doi: 10.1371/journal.pone.0136122
133. Zheng J, Conrad M. The metabolic underpinnings of ferroptosis. Cell Metab (2020) 32:920–37. doi: 10.1016/j.cmet.2020.10.011
134. Gao M, Yi J, Zhu J, Minikes AM, Monian P, Thompson CB, et al. Role of mitochondria in ferroptosis. Mol Cell (2019) 73:354–63.e3. doi: 10.1016/j.molcel.2018.10.042
135. Wu J, Feng Z, Chen L, Li Y, Bian H, Geng J, et al. TNF antagonist sensitizes synovial fibroblasts to ferroptotic cell death in collagen-induced arthritis mouse models. Nat Commun (2022) 13:676. doi: 10.1038/s41467-021-27948-4
136. Fan X-X, Xu M-Z, Leung EL-H, Jun C, Yuan Z, Liu L. ROS-responsive berberine polymeric micelles effectively suppressed the inflammation of rheumatoid arthritis by targeting mitochondria. Nanomicro Lett (2020) 12:76. doi: 10.1007/s40820-020-0410-x
137. Huang Z, Chen Y, Zhang Y. Mitochondrial reactive oxygen species cause major oxidative mitochondrial DNA damages and repair pathways. J Biosci (2020) 45:84.
138. West AP. Mitochondrial dysfunction as a trigger of innate immune responses and inflammation. Toxicology (2017) 391:54–63. doi: 10.1016/j.tox.2017.07.016
139. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature (2010) 464:104–7. doi: 10.1038/nature08780
140. Patel S. Danger-associated molecular patterns (DAMPs): the derivatives and triggers of inflammation. Curr Allergy Asthma Rep (2018) 18:63. doi: 10.1007/s11882-018-0817-3
141. Haneklaus M, O’Neill LAJ. NLRP3 at the interface of metabolism and inflammation. Immunol Rev (2015) 265:53–62. doi: 10.1111/imr.12285
142. Choulaki C, Papadaki G, Repa A, Kampouraki E, Kambas K, Ritis K, et al. Enhanced activity of NLRP3 inflammasome in peripheral blood cells of patients with active rheumatoid arthritis. Arthritis Res Ther (2015) 17:257. doi: 10.1186/s13075-015-0775-2
143. Guo C, Fu R, Wang S, Huang Y, Li X, Zhou M, et al. NLRP3 inflammasome activation contributes to the pathogenesis of rheumatoid arthritis. Clin Exp Immunol (2018) 194:231–43. doi: 10.1111/cei.13167
144. Yang X, Zhan N, Jin Y, Ling H, Xiao C, Xie Z, et al. Tofacitinib restores the balance of γδTreg/γδT17 cells in rheumatoid arthritis by inhibiting the NLRP3 inflammasome. Theranostics (2021) 11:1446–57. doi: 10.7150/thno.47860
145. Zhao C, Gu Y, Zeng X, Wang J. NLRP3 inflammasome regulates Th17 differentiation in rheumatoid arthritis. Clin Immunol (2018) 197:154–60. doi: 10.1016/j.clim.2018.09.007
146. Heid ME, Keyel PA, Kamga C, Shiva S, Watkins SC, Salter RD. Mitochondrial reactive oxygen species induces NLRP3-dependent lysosomal damage and inflammasome activation. J Immunol (2013) 191:5230–8. doi: 10.4049/jimmunol.1301490
147. Ren J-D, Wu X-B, Jiang R, Hao D-P, Liu Y. Molecular hydrogen inhibits lipopolysaccharide-triggered NLRP3 inflammasome activation in macrophages by targeting the mitochondrial reactive oxygen species. Biochim Biophys Acta (2016) 1863:50–5. doi: 10.1016/j.bbamcr.2015.10.012
148. Xian H, Watari K, Sanchez-Lopez E, Offenberger J, Onyuru J, Sampath H, et al. Oxidized DNA fragments exit mitochondria via mPTP- and VDAC-dependent channels to activate NLRP3 inflammasome and interferon signaling. Immunity (2022) 55:1370–1385.e8. doi: 10.1016/j.immuni.2022.06.007
149. Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin X-J, et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature (2018) 560:198–203. doi: 10.1038/s41586-018-0372-z
150. Galita G, Brzezińska O, Gulbas I, Sarnik J, Poplawska M, Makowska J, et al. Increased sensitivity of PBMCs isolated from patients with rheumatoid arthritis to DNA damaging agents is connected with inefficient DNA repair. J Clin Med (2020) 9:E988. doi: 10.3390/jcm9040988
151. Nambiar TS, Baudrier L, Billon P, Ciccia A. CRISPR-based genome editing through the lens of DNA repair. Mol Cell (2022) 82:348–88. doi: 10.1016/j.molcel.2021.12.026
152. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature (2015) 526:660–5. doi: 10.1038/nature15514
153. de Torre-Minguela C, Gómez AI, Couillin I, Pelegrín P. Gasdermins mediate cellular release of mitochondrial DNA during pyroptosis and apoptosis. FASEB J (2021) 35:e21757. doi: 10.1096/fj.202100085R
154. Wang Y, Shi P, Chen Q, Huang Z, Zou D, Zhang J, et al. Mitochondrial ROS promote macrophage pyroptosis by inducing GSDMD oxidation. J Mol Cell Biol (2019) 11:1069–82. doi: 10.1093/jmcb/mjz020
155. Hong Z, Zhang X, Zhang T, Hu L, Liu R, Wang P, et al. The ROS/GRK2/HIF-1α/NLRP3 pathway mediates pyroptosis of fibroblast-like synoviocytes and the regulation of monomer derivatives of paeoniflorin. Oxid Med Cell Longev (2022) 2022:4566851. doi: 10.1155/2022/4566851
156. Olatec Therapeutics LLC. A phase 2b randomized, double-blind, vehicle-controlled, repeat-dose, multi- center, efficacy and safety clinical trial of topically applied OLT1177 gel in subjects with moderate to severe pain associated with osteoarthritis of the knee following cessation of pain therapy (2017). Available at: https://clinicaltrials.gov/ct2/show/NCT02104050 (Accessed January 5, 2023).
157. Decout A, Katz JD, Venkatraman S, Ablasser A. The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat Rev Immunol (2021) 21:548–69. doi: 10.1038/s41577-021-00524-z
158. Hopfner K-P, Hornung V. Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat Rev Mol Cell Biol (2020) 21:501–21. doi: 10.1038/s41580-020-0244-x
159. Jiang M, Chen P, Wang L, Li W, Chen B, Liu Y, et al. cGAS-STING, an important pathway in cancer immunotherapy. J Hematol Oncol (2020) 13:81. doi: 10.1186/s13045-020-00916-z
160. Willemsen J, Neuhoff M-T, Hoyler T, Noir E, Tessier C, Sarret S, et al. TNF leads to mtDNA release and cGAS/STING-dependent interferon responses that support inflammatory arthritis. Cell Rep (2021) 37:109977. doi: 10.1016/j.celrep.2021.109977
161. Li R, Lin W, Kuang Y, Wang J, Xu S, Shen C, et al. cGAS/STING signaling in the regulation of rheumatoid synovial aggression. Ann Transl Med (2022) 10:431. doi: 10.21037/atm-21-4533
162. Guo Q, Chen X, Chen J, Zheng G, Xie C, Wu H, et al. STING promotes senescence, apoptosis, and extracellular matrix degradation in osteoarthritis via the NF-κB signaling pathway. Cell Death Dis (2021) 12:13. doi: 10.1038/s41419-020-03341-9
163. Xu A, Yang R, Zhang M, Wang X, Di Y, Jiang B, et al. Macrophage targeted triptolide micelles capable of cGAS-STING pathway inhibition for rheumatoid arthritis treatment. J Drug Target (2022) 30:961–72. doi: 10.1080/1061186X.2022.2070173
164. Chen J-Q, Szodoray P, Zeher M. Toll-like receptor pathways in autoimmune diseases. Clin Rev Allergy Immunol (2016) 50:1–17. doi: 10.1007/s12016-015-8473-z
165. Clanchy FIL, Borghese F, Bystrom J, Balog A, Penn H, Hull DN, et al. TLR expression profiles are a function of disease status in rheumatoid arthritis and experimental arthritis. J Autoimmun (2021) 118:102597. doi: 10.1016/j.jaut.2021.102597
166. Lacerte P, Brunet A, Egarnes B, Duchêne B, Brown JP, Gosselin J. Overexpression of TLR2 and TLR9 on monocyte subsets of active rheumatoid arthritis patients contributes to enhance responsiveness to TLR agonists. Arthritis Res Ther (2016) 18:10. doi: 10.1186/s13075-015-0901-1
167. Fischer A, Abdollahi-Roodsaz S, Böhm C, Niederreiter B, Meyer B, Yau ACY, et al. The involvement of toll-like receptor 9 in the pathogenesis of erosive autoimmune arthritis. J Cell Mol Med (2018) 22:4399–409. doi: 10.1111/jcmm.13735
168. Carmona-Rivera C, Carlucci PM, Moore E, Lingampalli N, Uchtenhagen H, James E, et al. Synovial fibroblast-neutrophil interactions promote pathogenic adaptive immunity in rheumatoid arthritis. Sci Immunol (2017) 2:eaag3358. doi: 10.1126/sciimmunol.aag3358
169. Han J, Li X, Luo X, He J, Huang X, Zhou Q, et al. The mechanisms of hydroxychloroquine in rheumatoid arthritis treatment: Inhibition of dendritic cell functions via toll like receptor 9 signaling. BioMed Pharmacother (2020) 132:110848. doi: 10.1016/j.biopha.2020.110848
170. Pohar J, Lainšček D, Ivičak-Kocjan K, Cajnko M-M, Jerala R, Benčina M. Short single-stranded DNA degradation products augment the activation of toll-like receptor 9. Nat Commun (2017) 8:15363. doi: 10.1038/ncomms15363
171. Ohto U, Ishida H, Shibata T, Sato R, Miyake K, Shimizu T. Toll-like receptor 9 contains two DNA binding sites that function cooperatively to promote receptor dimerization and activation. Immunity (2018) 48:649–658.e4. doi: 10.1016/j.immuni.2018.03.013
172. Ma L, Geng J, Chen W, Qin M, Wang L, Zeng Y. Effects of TLR9/NF-κB on oxidative stress and inflammation in IPEC-J2 cells. Genes Genomics (2022) 44:1149–58. doi: 10.1007/s13258-022-01271-8
173. Gilljam KM, Holm KL, Zahoor M, Centonze FG, Farhan H, Blomhoff HK. Differential effects of reactive oxygen species on IgG versus IgM levels in TLR-stimulated b cells. J Immunol (2020) 204:2133–42. doi: 10.4049/jimmunol.1901131
174. Fernandes-Alnemri T, Yu J-W, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature (2009) 458:509–13. doi: 10.1038/nature07710
175. Chen Y, Fujuan Q, Chen E, Yu B, Zuo F, Yuan Y, et al. Expression of AIM2 in rheumatoid arthritis and its role on fibroblast-like synoviocytes. Mediators Inflammation (2020) 2020:1693730. doi: 10.1155/2020/1693730
176. Wang Y, Chen C, Chen J, Sang T, Peng H, Lin X, et al. Overexpression of NAG-1/GDF15 prevents hepatic steatosis through inhibiting oxidative stress-mediated dsDNA release and AIM2 inflammasome activation. Redox Biol (2022) 52:102322. doi: 10.1016/j.redox.2022.102322
177. Crane DD, Bauler TJ, Wehrly TD, Bosio CM. Mitochondrial ROS potentiates indirect activation of the AIM2 inflammasome. Front Microbiol (2014) 5:438. doi: 10.3389/fmicb.2014.00438
178. Xue C, Takahashi M, Hasunuma T, Aono H, Yamamoto K, Yoshino S, et al. Characterisation of fibroblast-like cells in pannus lesions of patients with rheumatoid arthritis sharing properties of fibroblasts and chondrocytes. Ann Rheum Dis (1997) 56:262–7. doi: 10.1136/ard.56.4.262
179. Kim J-W, Kong J-S, Lee S, Yoo S-A, Koh JH, Jin J, et al. Angiogenic cytokines can reflect the synovitis severity and treatment response to biologics in rheumatoid arthritis. Exp Mol Med (2020) 52:843–53. doi: 10.1038/s12276-020-0443-8
180. Avouac J, Pezet S, Vandebeuque E, Orvain C, Gonzalez V, Marin G, et al. Semaphorins: From angiogenesis to inflammation in rheumatoid arthritis. Arthritis Rheumatol (2021) 73:1579–88. doi: 10.1002/art.41701
181. Taylor PC, Sivakumar B. Hypoxia and angiogenesis in rheumatoid arthritis. Curr Opin Rheumatol (2005) 17:293–8. doi: 10.1097/01.bor.0000155361.83990.5b
182. Yoo S-A, Park J-H, Hwang S-H, Oh S-M, Lee S, Cicatiello V, et al. Placental growth factor-1 and -2 induce hyperplasia and invasiveness of primary rheumatoid synoviocytes. J Immunol (2015) 194:2513–21. doi: 10.4049/jimmunol.1402900
183. Kim Y-W, Byzova TV. Oxidative stress in angiogenesis and vascular disease. Blood (2014) 123:625–31. doi: 10.1182/blood-2013-09-512749
184. Park D, Dilda PJ. Mitochondria as targets in angiogenesis inhibition. Mol Aspects Med (2010) 31:113–31. doi: 10.1016/j.mam.2009.12.005
185. Hua S, Dias TH. Hypoxia-inducible factor (HIF) as a target for novel therapies in rheumatoid arthritis. Front Pharmacol (2016) 7:184. doi: 10.3389/fphar.2016.00184
186. Imtiyaz HZ, Simon MC. Hypoxia-inducible factors as essential regulators of inflammation. Curr Top Microbiol Immunol (2010) 345:105–20. doi: 10.1007/82_2010_74
187. Lee Y-A, Choi HM, Lee S-H, Hong S-J, Yang H-I, Yoo MC, et al. Hypoxia differentially affects IL-1β-stimulated MMP-1 and MMP-13 expression of fibroblast-like synoviocytes in an HIF-1α-dependent manner. Rheumatol (Oxford) (2012) 51:443–50. doi: 10.1093/rheumatology/ker327
188. Hu F, Liu H, Xu L, Li Y, Liu X, Shi L, et al. Hypoxia-inducible factor-1α perpetuates synovial fibroblast interactions with T cells and b cells in rheumatoid arthritis. Eur J Immunol (2016) 46:742–51. doi: 10.1002/eji.201545784
189. Ryu J-H, Chae C-S, Kwak J-S, Oh H, Shin Y, Huh YH, et al. Hypoxia-inducible factor-2α is an essential catabolic regulator of inflammatory rheumatoid arthritis. PloS Biol (2014) 12:e1001881. doi: 10.1371/journal.pbio.1001881
190. Knowles HJ. Distinct roles for the hypoxia-inducible transcription factors HIF-1α and HIF-2α in human osteoclast formation and function. Sci Rep (2020) 10:21072. doi: 10.1038/s41598-020-78003-z
191. Morten KJ, Badder L, Knowles HJ. Differential regulation of HIF-mediated pathways increases mitochondrial metabolism and ATP production in hypoxic osteoclasts. J Pathol (2013) 229:755–64. doi: 10.1002/path.4159
192. Willson JA, Arienti S, Sadiku P, Reyes L, Coelho P, Morrison T, et al. Neutrophil HIF-1α stabilization is augmented by mitochondrial ROS produced via the glycerol 3-phosphate shuttle. Blood (2022) 139:281–6. doi: 10.1182/blood.2021011010
193. Lee H-R, Yoo S-J, Kim J, Yoo IS, Park CK, Kang SW. The effect of nicotinamide adenine dinucleotide phosphate oxidase 4 on migration and invasion of fibroblast-like synoviocytes in rheumatoid arthritis. Arthritis Res Ther (2020) 22:116. doi: 10.1186/s13075-020-02204-0
194. Ding H, Hong C, Wang Y, Liu J, Zhang N, Shen C, et al. Calreticulin promotes angiogenesis via activating nitric oxide signalling pathway in rheumatoid arthritis. Clin Exp Immunol (2014) 178:236–44. doi: 10.1111/cei.12411
195. Wei K, Korsunsky I, Marshall JL, Gao A, Watts GFM, Major T, et al. Notch signalling drives synovial fibroblast identity and arthritis pathology. Nature (2020) 582:259–64. doi: 10.1038/s41586-020-2222-z
196. Chen J, Li J, Chen J, Cheng W, Lin J, Ke L, et al. Treatment of collagen-induced arthritis rat model by using notch signalling inhibitor. J Orthop Translat (2021) 28:100–7. doi: 10.1016/j.jot.2021.01.003
197. Zhu J-H, Chen C-L, Flavahan S, Harr J, Su B, Flavahan NA. Cyclic stretch stimulates vascular smooth muscle cell alignment by redox-dependent activation of Notch3. Am J Physiol Heart Circ Physiol (2011) 300:H1770–80. doi: 10.1152/ajpheart.00535.2010
198. Ma S, Wang J, Lin J, Jin S, He F, Mei J, et al. Survivin promotes rheumatoid arthritis fibroblast-like synoviocyte cell proliferation, and the expression of angiogenesis-related proteins by activating the NOTCH pathway. Int J Rheum Dis (2021) 24:922–9. doi: 10.1111/1756-185X.14150
199. Cai W-X, Liang L, Wang L, Han J-T, Zhu X-X, Han H, et al. Inhibition of notch signaling leads to increased intracellular ROS by up-regulating Nox4 expression in primary HUVECs. Cell Immunol (2014) 287:129–35. doi: 10.1016/j.cellimm.2013.12.009
200. Chen J, Cheng W, Li J, Wang Y, Chen J, Shen X, et al. Notch-1 and notch-3 mediate hypoxia-induced activation of synovial fibroblasts in rheumatoid arthritis. Arthritis Rheumatol (2021) 73:1810–9. doi: 10.1002/art.41748
201. Görlach A, Diebold I, Schini-Kerth VB, Berchner-Pfannschmidt U, Roth U, Brandes RP, et al. Thrombin activates the hypoxia-inducible factor-1 signaling pathway in vascular smooth muscle cells: Role of the p22(phox)-containing NADPH oxidase. Circ Res (2001) 89:47–54. doi: 10.1161/hh1301.092678
202. Ismail AA, Shaker BT, Bajou K. The plasminogen-activator plasmin system in physiological and pathophysiological angiogenesis. Int J Mol Sci (2021) 23:337. doi: 10.3390/ijms23010337
203. Doanes AM, Hegland DD, Sethi R, Kovesdi I, Bruder JT, Finkel T. VEGF stimulates MAPK through a pathway that is unique for receptor tyrosine kinases. Biochem Biophys Res Commun (1999) 255:545–8. doi: 10.1006/bbrc.1999.0227
204. Yashima R, Abe M, Tanaka K, Ueno H, Shitara K, Takenoshita S, et al. Heterogeneity of the signal transduction pathways for VEGF-induced MAPKs activation in human vascular endothelial cells. J Cell Physiol (2001) 188:201–10. doi: 10.1002/jcp.1107
205. Lee YH, Bae S-C. Correlation between circulating VEGF levels and disease activity in rheumatoid arthritis: a meta-analysis. Z Rheumatol (2018) 77:240–8. doi: 10.1007/s00393-016-0229-5
206. Zhang Y-F, Gao S-S, Li J-L, Zuo W-S, Qiu Y-W, Xiao Y-C. Comparison and correlation study of synovial ultrasound indices and serum VEGF in rheumatoid wrist arthritis before and after treatment. Clin Rheumatol (2022) 41:2677–83. doi: 10.1007/s10067-022-06213-z
207. Jia W, Wu W, Yang D, Xiao C, Huang M, Long F, et al. GATA4 regulates angiogenesis and persistence of inflammation in rheumatoid arthritis. Cell Death Dis (2018) 9:503. doi: 10.1038/s41419-018-0570-5
208. Marcu R, Zheng Y, Hawkins BJ. Mitochondria and angiogenesis. Adv Exp Med Biol (2017) 982:371–406. doi: 10.1007/978-3-319-55330-6_21
209. Chang J, Jung HJ, Jeong SH, Kim HK, Han J, Kwon HJ. A mutation in the mitochondrial protein UQCRB promotes angiogenesis through the generation of mitochondrial reactive oxygen species. Biochem Biophys Res Commun (2014) 455:290–7. doi: 10.1016/j.bbrc.2014.11.005
210. Wang C, Dai X, Wu S, Xu W, Song P, Huang K. FUNDC1-dependent mitochondria-associated endoplasmic reticulum membranes are involved in angiogenesis and neoangiogenesis. Nat Commun (2021) 12:2616. doi: 10.1038/s41467-021-22771-3
211. Xu J, Zhang X-Y, Li R, Liu J, Ye H, Zhang X-W, et al. Glucose-6-phosphate isomerase is associated with disease activity and declines in response to infliximab treatment in rheumatoid arthritis. Chin Med J (Engl) (2020) 133:886–91. doi: 10.1097/CM9.0000000000000750
212. Han S, Auger C, Thomas SC, Beites CL, Appanna VD. Mitochondrial biogenesis and energy production in differentiating murine stem cells: a functional metabolic study. Cell Reprogram (2014) 16:84–90. doi: 10.1089/cell.2013.0049
213. Lu Y, Yu S-S, Zong M, Fan S-S, Lu T-B, Gong R-H, et al. Glucose-6-Phosphate isomerase (G6PI) mediates hypoxia-induced angiogenesis in rheumatoid arthritis. Sci Rep (2017) 7:40274. doi: 10.1038/srep40274
214. Naughton DP. Hypoxia-induced upregulation of the glycolytic enzyme glucose-6-phosphate isomerase perpetuates rheumatoid arthritis. Med Hypotheses (2003) 60:332–4. doi: 10.1016/s0306-9877(02)00396-1
215. Schett G, Gravallese E. Bone erosion in rheumatoid arthritis: mechanisms, diagnosis and treatment. Nat Rev Rheumatol (2012) 8:656–64. doi: 10.1038/nrrheum.2012.153
216. Mueller A-L, Payandeh Z, Mohammadkhani N, Mubarak SMH, Zakeri A, Alagheband Bahrami A, et al. Recent advances in understanding the pathogenesis of rheumatoid arthritis: New treatment strategies. Cells (2021) 10:3017. doi: 10.3390/cells10113017
217. Panagopoulos PK, Lambrou GI. Bone erosions in rheumatoid arthritis: recent developments in pathogenesis and therapeutic implications. J Musculoskelet Neuronal Interact (2018) 18:304–19.
218. Li G, Ma Y, Cheng TS, Landao-Bassonga E, Qin A, Pavlos NJ, et al. Identical subchondral bone microarchitecture pattern with increased bone resorption in rheumatoid arthritis as compared to osteoarthritis. Osteoarthritis Cartilage (2014) 22:2083–92. doi: 10.1016/j.joca.2014.08.015
219. Wysham KD, Baker JF, Shoback DM. Osteoporosis and fractures in rheumatoid arthritis. Curr Opin Rheumatol (2021) 33:270–6. doi: 10.1097/BOR.0000000000000789
220. Tanaka S, Tanaka Y. RANKL as a therapeutic target of rheumatoid arthritis. J Bone Miner Metab (2021) 39:106–12. doi: 10.1007/s00774-020-01159-1
221. Tanaka Y, Takeuchi T, Soen S, Yamanaka H, Yoneda T, Tanaka S, et al. Effects of denosumab in Japanese patients with rheumatoid arthritis treated with conventional antirheumatic drugs: 36-month extension of a phase III study. J Rheumatol (2021) 48:1663–71. doi: 10.3899/jrheum.201376
222. Zhang L, Huang B, Tang H, Ye X, Yao Y, Gong P, et al. Amifostine inhibited the differentiation of RAW264.7 cells into osteoclasts by reducing the production of ROS under 2 Gy radiation. J Cell Biochem (2020) 121:497–507. doi: 10.1002/jcb.29247
223. Ashtar M, Tenshin H, Teramachi J, Bat-Erdene A, Hiasa M, Oda A, et al. The roles of ROS generation in RANKL-induced osteoclastogenesis: Suppressive effects of febuxostat. Cancers (Basel) (2020) 12:E929. doi: 10.3390/cancers12040929
224. Sheng K-C, Pietersz GA, Tang CK, Ramsland PA, Apostolopoulos V. Reactive oxygen species level defines two functionally distinctive stages of inflammatory dendritic cell development from mouse bone marrow. J Immunol (2010) 184:2863–72. doi: 10.4049/jimmunol.0903458
225. Momiuchi Y, Motomura Y, Suga E, Mizuno H, Kikuta J, Morimoto A, et al. Group 2 innate lymphoid cells in bone marrow regulate osteoclastogenesis in a reciprocal manner via RANKL, GM-CSF and IL-13. Int Immunol (2021) 33:573–85. doi: 10.1093/intimm/dxab062
226. Ke K, Sul O-J, Choi E-K, Safdar AM, Kim E-S, Choi H-S. Reactive oxygen species induce the association of SHP-1 with c-src and the oxidation of both to enhance osteoclast survival. Am J Physiol Endocrinol Metab (2014) 307:E61–70. doi: 10.1152/ajpendo.00044.2014
227. Zhu J, Tang Y, Wu Q, Ji Y-C, Feng Z-F, Kang F-W. HIF-1α facilitates osteocyte-mediated osteoclastogenesis by activating JAK2/STAT3 pathway in vitro. J Cell Physiol (2019) 234:21182–92. doi: 10.1002/jcp.28721
228. Swales C, Athanasou NA, Knowles HJ. Angiopoietin-like 4 is over-expressed in rheumatoid arthritis patients: association with pathological bone resorption. PloS One (2014) 9:e109524. doi: 10.1371/journal.pone.0109524
229. Knowles HJ, Cleton-Jansen A-M, Korsching E, Athanasou NA. Hypoxia-inducible factor regulates osteoclast-mediated bone resorption: role of angiopoietin-like 4. FASEB J (2010) 24:4648–59. doi: 10.1096/fj.10-162230
230. Ni S, Yuan Y, Qian Z, Zhong Z, Lv T, Kuang Y, et al. Hypoxia inhibits RANKL-induced ferritinophagy and protects osteoclasts from ferroptosis. Free Radic Biol Med (2021) 169:271–82. doi: 10.1016/j.freeradbiomed.2021.04.027
231. Srinivasan S, Koenigstein A, Joseph J, Sun L, Kalyanaraman B, Zaidi M, et al. Role of mitochondrial reactive oxygen species in osteoclast differentiation. Ann N Y Acad Sci (2010) 1192:245–52. doi: 10.1111/j.1749-6632.2009.05377.x
232. Li B, Lee W-C, Song C, Ye L, Abel ED, Long F. Both aerobic glycolysis and mitochondrial respiration are required for osteoclast differentiation. FASEB J (2020) 34:11058–67. doi: 10.1096/fj.202000771R
233. Carballo CB, Nakagawa Y, Sekiya I, Rodeo SA. Basic science of articular cartilage. Clin Sports Med (2017) 36:413–25. doi: 10.1016/j.csm.2017.02.001
234. Burrage PS, Mix KS, Brinckerhoff CE. Matrix metalloproteinases: role in arthritis. Front Biosci (2006) 11:529–43. doi: 10.2741/1817
235. Ainola MM, Mandelin JA, Liljeström MP, Li TF, Hukkanen MVJ, Konttinen YT. Pannus invasion and cartilage degradation in rheumatoid arthritis: involvement of MMP-3 and interleukin-1beta. Clin Exp Rheumatol (2005) 23:644–50.
236. Pettit AR, Walsh NC, Manning C, Goldring SR, Gravallese EM. RANKL protein is expressed at the pannus-bone interface at sites of articular bone erosion in rheumatoid arthritis. Rheumatol (Oxford) (2006) 45:1068–76. doi: 10.1093/rheumatology/kel045
237. Yokota K, Sato K, Miyazaki T, Aizaki Y, Tanaka S, Sekikawa M, et al. Characterization and function of tumor necrosis factor and interleukin-6-Induced osteoclasts in rheumatoid arthritis. Arthritis Rheumatol (2021) 73:1145–54. doi: 10.1002/art.41666
238. Rao Z, Wang S, Wang J. Protective effects of psoralidin on IL−1β−induced chondrocyte apoptosis. Mol Med Rep (2018) 17:3418–24. doi: 10.3892/mmr.2017.8248
239. Yu S-M, Kim S-J. Withaferin a-caused production of intracellular reactive oxygen species modulates apoptosis via PI3K/Akt and JNKinase in rabbit articular chondrocytes. J Korean Med Sci (2014) 29:1042–53. doi: 10.3346/jkms.2014.29.8.1042
240. Del Carlo M, Loeser RF. Nitric oxide-mediated chondrocyte cell death requires the generation of additional reactive oxygen species. Arthritis Rheum (2002) 46:394–403. doi: 10.1002/art.10056
241. Ye W, Zhu S, Liao C, Xiao J, Wu Q, Lin Z, et al. Advanced oxidation protein products induce apoptosis of human chondrocyte through reactive oxygen species-mediated mitochondrial dysfunction and endoplasmic reticulum stress pathways. Fundam Clin Pharmacol (2017) 31:64–74. doi: 10.1111/fcp.12229
242. Whiteman M, Armstrong JS, Cheung NS, Siau J-L, Rose P, Schantz J-T, et al. Peroxynitrite mediates calcium-dependent mitochondrial dysfunction and cell death via activation of calpains. FASEB J (2004) 18:1395–7. doi: 10.1096/fj.03-1096fje
243. Wang F-S, Kuo C-W, Ko J-Y, Chen Y-S, Wang S-Y, Ke H-J, et al. Irisin mitigates oxidative stress, chondrocyte dysfunction and osteoarthritis development through regulating mitochondrial integrity and autophagy. Antioxidants (Basel) (2020) 9:E810. doi: 10.3390/antiox9090810
244. Caramés B, Hasegawa A, Taniguchi N, Miyaki S, Blanco FJ, Lotz M. Autophagy activation by rapamycin reduces severity of experimental osteoarthritis. Ann Rheum Dis (2012) 71:575–81. doi: 10.1136/annrheumdis-2011-200557
245. Chen Y, Wu Y-Y, Si H-B, Lu Y-R, Shen B. Mechanistic insights into AMPK-SIRT3 positive feedback loop-mediated chondrocyte mitochondrial quality control in osteoarthritis pathogenesis. Pharmacol Res (2021) 166:105497. doi: 10.1016/j.phrs.2021.105497
246. He Y, Wu Z, Xu L, Xu K, Chen Z, Ran J, et al. The role of SIRT3-mediated mitochondrial homeostasis in osteoarthritis. Cell Mol Life Sci (2020) 77:3729–43. doi: 10.1007/s00018-020-03497-9
247. Kim D, Song J, Jin E-J. BNIP3-dependent mitophagy via PGC1α promotes cartilage degradation. Cells (2021) 10:1839. doi: 10.3390/cells10071839
248. Burmester GR, Pope JE. Novel treatment strategies in rheumatoid arthritis. Lancet (2017) 389:2338–48. doi: 10.1016/S0140-6736(17)31491-5
249. Law ST, Taylor PC. Role of biological agents in treatment of rheumatoid arthritis. Pharmacol Res (2019) 150:104497. doi: 10.1016/j.phrs.2019.104497
250. Meugnier E, Coury F, Tebib J, Ferraro-Peyret C, Rome S, Bienvenu J, et al. Gene expression profiling in peripheral blood cells of patients with rheumatoid arthritis in response to anti-TNF-alpha treatments. Physiol Genomics (2011) 43:365–71. doi: 10.1152/physiolgenomics.00127.2010
251. Humby F, Durez P, Buch MH, Lewis MJ, Rizvi H, Rivellese F, et al. Rituximab versus tocilizumab in anti-TNF inadequate responder patients with rheumatoid arthritis (R4RA): 16-week outcomes of a stratified, biopsy-driven, multicentre, open-label, phase 4 randomised controlled trial. Lancet (2021) 397:305–17. doi: 10.1016/S0140-6736(20)32341-2
252. Omoyinmi E, Hamaoui R, Bryant A, Jiang MC, Athigapanich T, Eleftheriou D, et al. Mitochondrial and oxidative stress genes are differentially expressed in neutrophils of sJIA patients treated with tocilizumab: a pilot microarray study. Pediatr Rheumatol Online J (2016) 14:7. doi: 10.1186/s12969-016-0067-7
253. Pariano M, Pieroni S, De Luca A, Iannitti RG, Borghi M, Puccetti M, et al. Anakinra activates superoxide dismutase 2 to mitigate inflammasome activity. Int J Mol Sci (2021) 22:6531. doi: 10.3390/ijms22126531
254. van de Veerdonk FL, Renga G, Pariano M, Bellet MM, Servillo G, Fallarino F, et al. Anakinra restores cellular proteostasis by coupling mitochondrial redox balance to autophagy. J Clin Invest (2022) 132:e144983. doi: 10.1172/JCI144983
255. Liu L, Zan J, Zhang Y. Clinical analysis of tripterygium wlfordii polyglycosides combined with immunosuppressant in the treatment of rheumatoid arthritis. Panminerva Med (2021). doi: 10.23736/S0031-0808.21.04483-9
256. Jing M, Yang J, Zhang L, Liu J, Xu S, Wang M, et al. Celastrol inhibits rheumatoid arthritis through the ROS-NF-κB-NLRP3 inflammasome axis. Int Immunopharmacol (2021) 98:107879. doi: 10.1016/j.intimp.2021.107879
257. An L, Li Z, Shi L, Wang L, Wang Y, Jin L, et al. Inflammation-targeted celastrol nanodrug attenuates collagen-induced arthritis through NF-κB and Notch1 pathways. Nano Lett (2020) 20:7728–36. doi: 10.1021/acs.nanolett.0c03279
258. Zhai K-F, Duan H, Khan GJ, Xu H, Han F-K, Cao W-G, et al. Salicin from alangium chinense ameliorates rheumatoid arthritis by modulating the Nrf2-HO-1-ROS pathways. J Agric Food Chem (2018) 66:6073–82. doi: 10.1021/acs.jafc.8b02241
259. Shen P, Lin W, Ba X, Huang Y, Chen Z, Han L, et al. Quercetin-mediated SIRT1 activation attenuates collagen-induced mice arthritis. J Ethnopharmacol (2021) 279:114213. doi: 10.1016/j.jep.2021.114213
260. Huang L, Hu S, Shao M, Wu X, Zhang J, Cao G. Combined cornus officinalis and paeonia lactiflora pall therapy alleviates rheumatoid arthritis by regulating synovial apoptosis via AMPK-mediated mitochondrial fission. Front Pharmacol (2021) 12:639009. doi: 10.3389/fphar.2021.639009
261. Heinke L. Mitochondrial ROS drive cell cycle progression. Nat Rev Mol Cell Biol (2022) 23:581. doi: 10.1038/s41580-022-00523-5
262. Horbay R, Bilyy R. Mitochondrial dynamics during cell cycling. Apoptosis (2016) 21:1327–35. doi: 10.1007/s10495-016-1295-5
263. Jie L, Du H, Huang Q, Wei S, Huang R, Sun W. Tanshinone IIA induces apoptosis in fibroblast-like synoviocytes in rheumatoid arthritis via blockade of the cell cycle in the G2/M phase and a mitochondrial pathway. Biol Pharm Bull (2014) 37:1366–72. doi: 10.1248/bpb.b14-00301
264. Zheng M, Kuang N, Zeng X, Wang J, Zou Y, Fu Y. Daphnetin induces apoptosis in fibroblast-like synoviocytes from collagen-induced arthritic rats mainly via the mitochondrial pathway. Cytokine (2020) 133:155146. doi: 10.1016/j.cyto.2020.155146
265. Pu L, Meng Q, Li S, Liu B, Li F. Icariin arrests cell cycle progression and induces cell apoptosis through the mitochondrial pathway in human fibroblast-like synoviocytes. Eur J Pharmacol (2021) 912:174585. doi: 10.1016/j.ejphar.2021.174585
266. Li J, Pang J, Liu Z, Ge X, Zhen Y, Jiang CC, et al. Shikonin induces programmed death of fibroblast synovial cells in rheumatoid arthritis by inhibiting energy pathways. Sci Rep (2021) 11:18263. doi: 10.1038/s41598-021-97713-6
267. Lin J, Sun AR, Li J, Yuan T, Cheng W, Ke L, et al. A three-dimensional Co-culture model for rheumatoid arthritis pannus tissue. Front Bioeng Biotechnol (2021) 9:764212. doi: 10.3389/fbioe.2021.764212
268. Di Benedetto P, Ruscitti P, Berardicurti O, Panzera N, Grazia N, Di Vito Nolfi M, et al. Blocking Jak/STAT signalling using tofacitinib inhibits angiogenesis in experimental arthritis. Arthritis Res Ther (2021) 23:213. doi: 10.1186/s13075-021-02587-8
269. Waldman WJ, Bickerstaff A, Gordillo G, Orosz K, Knight DA, Orosz CG. Inhibition of angiogenesis-related endothelial activity by the experimental immunosuppressive agent leflunomide. Transplantation (2001) 72:1578–82. doi: 10.1097/00007890-200111150-00018
270. Ishikawa G, Kwon C, Fujii Y. Peficitinib inhibits fibroblast-like synoviocyte activation and angiogenic vascular endothelial tube formation via inhibitory effects on PDGF and VEGF signaling in addition to JAK. J Pharmacol Sci (2022) 150:74–80. doi: 10.1016/j.jphs.2022.07.002
271. Abdel-Maged AE-S, Gad AM, Abdel-Aziz AK, Aboulwafa MM, Azab SS. Comparative study of anti-VEGF ranibizumab and interleukin-6 receptor antagonist tocilizumab in adjuvant-induced arthritis. Toxicol Appl Pharmacol (2018) 356:65–75. doi: 10.1016/j.taap.2018.07.014
272. Wang Y, Wu H, Deng R. Angiogenesis as a potential treatment strategy for rheumatoid arthritis. Eur J Pharmacol (2021) 910:174500. doi: 10.1016/j.ejphar.2021.174500
273. Kanbe K, Chiba J, Nakamura A. Immunohistological analysis of synovium treated with abatacept in rheumatoid arthritis. Rheumatol Int (2013) 33:1883–7. doi: 10.1007/s00296-011-2326-8
274. Derambure C, Dzangue-Tchoupou G, D’Agostino MA, Lequerré T, Vittecoq O. Gene expression regulated by abatacept associated with methotrexate and correlation with disease activity in rheumatoid arthritis. PloS One (2020) 15:e0237143. doi: 10.1371/journal.pone.0237143
275. Derambure C, Dzangue-Tchoupou G, Berard C, Vergne N, Hiron M, D’Agostino MA, et al. Pre-silencing of genes involved in the electron transport chain (ETC) pathway is associated with responsiveness to abatacept in rheumatoid arthritis. Arthritis Res Ther (2017) 19:109. doi: 10.1186/s13075-017-1319-8
276. Guo X, Chen G. Hypoxia-inducible factor is critical for pathogenesis and regulation of immune cell functions in rheumatoid arthritis. Front Immunol (2020) 11:1668. doi: 10.3389/fimmu.2020.01668
277. Kim J, Kim HY, Song SY, Go S-H, Sohn HS, Baik S, et al. Synergistic oxygen generation and reactive oxygen species scavenging by manganese Ferrite/Ceria Co-decorated nanoparticles for rheumatoid arthritis treatment. ACS Nano (2019) 13:3206–17. doi: 10.1021/acsnano.8b08785
278. Li Y, Liang Q, Zhou L, Cao Y, Yang J, Li J, et al. An ROS-responsive artesunate prodrug nanosystem co-delivers dexamethasone for rheumatoid arthritis treatment through the HIF-1α/NF-κB cascade regulation of ROS scavenging and macrophage repolarization. Acta Biomater (2022) 152:406–24. doi: 10.1016/j.actbio.2022.08.054
279. Yang G, Chang C-C, Yang Y, Yuan L, Xu L, Ho C-T, et al. Resveratrol alleviates rheumatoid arthritis via reducing ROS and inflammation, inhibiting MAPK signaling pathways, and suppressing angiogenesis. J Agric Food Chem (2018) 66:12953–60. doi: 10.1021/acs.jafc.8b05047
280. Zhai K-F, Duan H, Cui C-Y, Cao Y-Y, Si J-L, Yang H-J, et al. Liquiritin from glycyrrhiza uralensis attenuating rheumatoid arthritis via reducing inflammation, suppressing angiogenesis, and inhibiting MAPK signaling pathway. J Agric Food Chem (2019) 67:2856–64. doi: 10.1021/acs.jafc.9b00185
281. Zippel N, Malik RA, Frömel T, Popp R, Bess E, Strilic B, et al. Transforming growth factor-β-activated kinase 1 regulates angiogenesis via AMP-activated protein kinase-α1 and redox balance in endothelial cells. Arterioscler Thromb Vasc Biol (2013) 33:2792–9. doi: 10.1161/ATVBAHA.113.301848
282. Bay-Jensen AC, Platt A, Byrjalsen I, Vergnoud P, Christiansen C, Karsdal MA. Effect of tocilizumab combined with methotrexate on circulating biomarkers of synovium, cartilage, and bone in the LITHE study. Semin Arthritis Rheum (2014) 43:470–8. doi: 10.1016/j.semarthrit.2013.07.008
283. Takeuchi T, Tanaka Y, Soen S, Yamanaka H, Yoneda T, Tanaka S, et al. Effects of the anti-RANKL antibody denosumab on joint structural damage in patients with rheumatoid arthritis treated with conventional synthetic disease-modifying antirheumatic drugs (DESIRABLE study): a randomised, double-blind, placebo-controlled phase 3 trial. Ann Rheum Dis (2019) 78:899–907. doi: 10.1136/annrheumdis-2018-214827
284. Zhu F, Wang J, Ni Y, Yin W, Hou Q, Zhang Y, et al. Curculigoside protects against titanium particle-induced osteolysis through the enhancement of osteoblast differentiation and reduction of osteoclast formation. J Immunol Res (2021) 2021:5707242. doi: 10.1155/2021/5707242
285. Han J, Wan M, Ma Z, Hu C, Yi H. Prediction of targets of curculigoside a in osteoporosis and rheumatoid arthritis using network pharmacology and experimental verification. Drug Des Devel Ther (2020) 14:5235–50. doi: 10.2147/DDDT.S282112
286. Liu M, Liu S, Zhang Q, Fang Y, Yu Y, Zhu L, et al. Curculigoside attenuates oxidative stress and osteoclastogenesis via modulating Nrf2/NF-κB signaling pathway in RAW264.7 cells. J Ethnopharmacol (2021) 275:114129. doi: 10.1016/j.jep.2021.114129
287. Liu Y, Wang C, Wang G, Sun Y, Deng Z, Chen L, et al. Loureirin b suppresses RANKL-induced osteoclastogenesis and ovariectomized osteoporosis via attenuating NFATc1 and ROS activities. Theranostics (2019) 9:4648–62. doi: 10.7150/thno.35414
288. Shi S, Zhao Q, Ke C, Long S, Zhang F, Zhang X, et al. Loureirin b exerts its immunosuppressive effects by inhibiting STIM1/Orai1 and KV1.3 channels. Front Pharmacol (2021) 12:685092. doi: 10.3389/fphar.2021.685092
289. Fiehn C, Neumann E, Wunder A, Krienke S, Gay S, Müller-Ladner U. Methotrexate (MTX) and albumin coupled with MTX (MTX-HSA) suppress synovial fibroblast invasion and cartilage degradation. vivo Ann Rheum Dis (2004) 63:884–6. doi: 10.1136/ard.2003.013748
290. Kullich WC, Mur E, Aglas F, Niksic F, Czerwenka C. Inhibitory effects of leflunomide therapy on the activity of matrixmetalloproteinase-9 and the release of cartilage oligomeric matrix protein in patients with rheumatoid arthritis. Clin Exp Rheumatol (2006) 24:155–60.
291. Yin N, Tan X, Liu H, He F, Ding N, Gou J, et al. A novel indomethacin/methotrexate/MMP-9 siRNA in situ hydrogel with dual effects of anti-inflammatory activity and reversal of cartilage disruption for the synergistic treatment of rheumatoid arthritis. Nanoscale (2020) 12:8546–62. doi: 10.1039/d0nr00454e
292. Qin J, Huang X, Wang N, Zhou P, Zhang H, Chen Z, et al. Supranutritional selenium suppresses ROS-induced generation of RANKL-expressing osteoclastogenic CD4+ T cells and ameliorates rheumatoid arthritis. Clin Transl Immunol (2021) 10:e1338. doi: 10.1002/cti2.1338
293. Shaaban HH, Hozayen WG, Khaliefa AK, El-Kenawy AE, Ali TM, Ahmed OM. Diosmin and trolox have anti-arthritic, anti-inflammatory and antioxidant potencies in complete freund’s adjuvant-induced arthritic Male wistar rats: Roles of NF-κB, iNOS, Nrf2 and MMPs. Antioxidants (Basel) (2022) 11:1721. doi: 10.3390/antiox11091721
294. Bertoldo E, Adami G, Rossini M, Giollo A, Orsolini G, Viapiana O, et al. The emerging roles of endocrine hormones in different arthritic disorders. Front Endocrinol (Lausanne) (2021) 12:620920. doi: 10.3389/fendo.2021.620920
295. Dupuis ML, Conti F, Maselli A, Pagano MT, Ruggieri A, Anticoli S, et al. The natural agonist of estrogen receptor β silibinin plays an immunosuppressive role representing a potential therapeutic tool in rheumatoid arthritis. Front Immunol (2018) 9:1903. doi: 10.3389/fimmu.2018.01903
296. Salliot C, Nguyen Y, Gusto G, Gelot A, Gambaretti J, Mariette X, et al. Female hormonal exposures and risk of rheumatoid arthritis in the French E3N-EPIC cohort study. Rheumatol (Oxford) (2021) 60:4790–800. doi: 10.1093/rheumatology/keab101
297. D’Amico D, Olmer M, Fouassier AM, Valdés P, Andreux PA, Rinsch C, et al. Urolithin a improves mitochondrial health, reduces cartilage degeneration, and alleviates pain in osteoarthritis. Aging Cell (2022) 21:e13662. doi: 10.1111/acel.13662
298. Jin Z, Chang B, Wei Y, Yang Y, Zhang H, Liu J, et al. Curcumin exerts chondroprotective effects against osteoarthritis by promoting AMPK/PINK1/Parkin-mediated mitophagy. BioMed Pharmacother (2022) 151:113092. doi: 10.1016/j.biopha.2022.113092
299. Mishra R, Singh A, Chandra V, Negi MPS, Tripathy BC, Prakash J, et al. A comparative analysis of serological parameters and oxidative stress in osteoarthritis and rheumatoid arthritis. Rheumatol Int (2012) 32:2377–82. doi: 10.1007/s00296-011-1964-1
300. Wang C, Yang Y, Zhang Y, Liu J, Yao Z, Zhang C. Protective effects of metformin against osteoarthritis through upregulation of SIRT3-mediated PINK1/Parkin-dependent mitophagy in primary chondrocytes. Biosci Trends (2019) 12:605–12. doi: 10.5582/bst.2018.01263
301. Wang X, Chen Z, Fan X, Li W, Qu J, Dong C, et al. Inhibition of DNM1L and mitochondrial fission attenuates inflammatory response in fibroblast-like synoviocytes of rheumatoid arthritis. J Cell Mol Med (2020) 24:1516–28. doi: 10.1111/jcmm.14837
302. Kleele T, Rey T, Winter J, Zaganelli S, Mahecic D, Perreten Lambert H, et al. Distinct fission signatures predict mitochondrial degradation or biogenesis. Nature (2021) 593:435–9. doi: 10.1038/s41586-021-03510-6
303. Kim EK, Kwon J-E, Lee S-Y, Lee E-J, Kim DS, Moon S-J, et al. IL-17-mediated mitochondrial dysfunction impairs apoptosis in rheumatoid arthritis synovial fibroblasts through activation of autophagy. Cell Death Dis (2017) 8:e2565. doi: 10.1038/cddis.2016.490
304. Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discovery (2013) 12:931–47. doi: 10.1038/nrd4002
305. Martelli-Palomino G, Paoliello-Paschoalato AB, Crispim JCO, Rassi DM, Oliveira RD, Louzada P, et al. DNA Damage increase in peripheral neutrophils from patients with rheumatoid arthritis is associated with the disease activity and the presence of shared epitope. Clin Exp Rheumatol (2017) 35:247–54.
Keywords: rheumatoid arthritis, reactive oxygen species, mitochondrial damage, targeted drugs, oxidative stress
Citation: Jing W, Liu C, Su C, Liu L, Chen P, Li X, Zhang X, Yuan B, Wang H and Du X (2023) Role of reactive oxygen species and mitochondrial damage in rheumatoid arthritis and targeted drugs. Front. Immunol. 14:1107670. doi: 10.3389/fimmu.2023.1107670
Received: 25 November 2022; Accepted: 30 January 2023;
Published: 09 February 2023.
Edited by:
Jens Staal, Ghent University, BelgiumReviewed by:
Ping Jiang, Shanghai University of Traditional Chinese Medicine, ChinaCopyright © 2023 Jing, Liu, Su, Liu, Chen, Li, Zhang, Yuan, Wang and Du. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Haidong Wang, d2hhaWRvbmc1ODk1QDE2My5jb20=; Xiaozheng Du, bHotZHV4aWFvemhlbmdAMTYzLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.