 Debanjan Goswamy
Debanjan Goswamy Xavier Gonzalez
Xavier Gonzalez Sid A. Labed†
Sid A. Labed† Javier E. Irazoqui
Javier E. Irazoqui
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 13 February 2023
Sec. Cytokines and Soluble Mediators in Immunity
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1094145
This article is part of the Research Topic Community Series in Immunoregulation at Mucosal Surfaces Volume II View all 7 articles
In recent years, transcription factors of the Microphthalmia-TFE (MiT) family, including TFEB and TFE3 in mammals and HLH-30 in Caenorhabditis elegans, have emerged as important regulators of innate immunity and inflammation in invertebrates and vertebrates. Despite great strides in knowledge, the mechanisms that mediate downstream actions of MiT transcription factors in the context of innate host defense remain poorly understood. Here, we report that HLH-30, which promotes lipid droplet mobilization and host defense, induces the expression of orphan nuclear receptor NHR-42 during infection with Staphylococcus aureus. Remarkably, NHR-42 loss of function promoted host infection resistance, genetically defining NHR-42 as an HLH-30-controlled negative regulator of innate immunity. During infection, NHR-42 was required for lipid droplet loss, suggesting that it is an important effector of HLH-30 in lipid immunometabolism. Moreover, transcriptional profiling of nhr-42 mutants revealed wholesale activation of an antimicrobial signature, of which abf-2, cnc-2, and lec-11 were important for the enhanced survival of infection of nhr-42 mutants. These results advance our knowledge of the mechanisms by which MiT transcription factors promote host defense, and by analogy suggest that TFEB and TFE3 may similarly promote host defense via NHR-42-homologous nuclear receptors in mammals.
Recent years have witnessed the emergence of MiT transcription factors as key regulators of innate immunity genes during a wide range of disease conditions, including infections by bacteria and viruses and chronic inflammation (1). Mostly thought of as master regulators of lysosomal biogenesis and autophagy, their functions include positive regulation of lipid mobilization and cytokine induction (2–9). While overall the genes that MiT factors control in different scenarios are becoming better understood, their mechanisms of regulation and action are much less so.
To address these knowledge gaps, we apply the model organism C. elegans in the context of intestinal infection by S. aureus and other human and nematode pathogens (8, 10). In C. elegans, S. aureus infects the intestinal lumen by the natural oral route and causes a 7-fold reduction in host lifespan, which is the result of a pathogenic process that involves enterocyte effacement, intestinal epithelial destruction, and organ consumption within the span of 48 h (10). In previous studies, we identified two main genetic pathways that promote the induction of an infection-specific gene expression signature that promotes host survival (11): the acetylcholine-Wnt pathway (12, 13) and the TFEB/HLH-30 pathway (8, 14). Like its human and mouse counterparts, HLH-30 resides in the cytoplasm in homeostasis and accumulates in the nucleus of cells throughout the animal during S. aureus infection (8) and other stresses (4, 5, 15–19). We also understand that HLH-30 activation by infection requires the activity of an upstream genetic pathway that involves EGL-30/Gαq and DKF-1/Protein kinase D, which is conserved in murine macrophages (14).
Although 95% of the infection-specific signature is HLH-30 dependent (11), a vast majority of its genes lack HLH-30 cognate motifs in their regulatory sequences (8, 20). This raises the possibility that HLH-30 may exert its pro-host defense action through intermediate regulators of gene expression, likely transcription factors themselves. Indeed, we showed that HLH-30 induces the expression of 17 transcription factors during S. aureus infection (8), of which the majority are orphan nuclear hormone receptors (NHR).
The C. elegans genome encodes an expanded family of 284 nhr genes, compared to 48 in humans and 49 in mice, but only around 20 of these genes have assigned functions (21). In invertebrates and vertebrates, nuclear receptors can induce or repress target gene transcription depending on their structure, protein-protein interactions, and ligand binding (21). In C. elegans, specific nuclear receptors have been showed to regulate metabolism, reproduction, lifespan, and development (22). Recent studies have begun to uncover important roles for NHR in C. elegans host defense against infection (11, 23–29).
Here we report the unexpected finding that infection-induced HLH-30-dependent gene nhr-42 is an important negative regulator of host infection resistance, which functions at the site of infection, i.e. the intestinal epithelium. During infection, we find that NHR-42 is essential for lipid droplet loss, suggesting that rather than through direct induction of lipid mobilizing genes, HLH-30 functions through NHR-42. In noninfected animals, NHR-42 represses specific genes that encode antimicrobial peptides and limits host survival in infected animals. These observations support the view that HLH-30 (and, by analogy, possibly TFEB and TFE3) induces transcription factors that are capable of repressing the host response and promoting the mobilization of lipids from intestinal lipid stores, with important implications for the resolution of infectious inflammation and for immunometabolism.
Strains used in this study are detailed in Table 1. C. elegans was grown on nematode-growth media (NGM) plates seeded with E. coli OP50 at 20°C, according to standard procedures (30), unless mentioned otherwise.
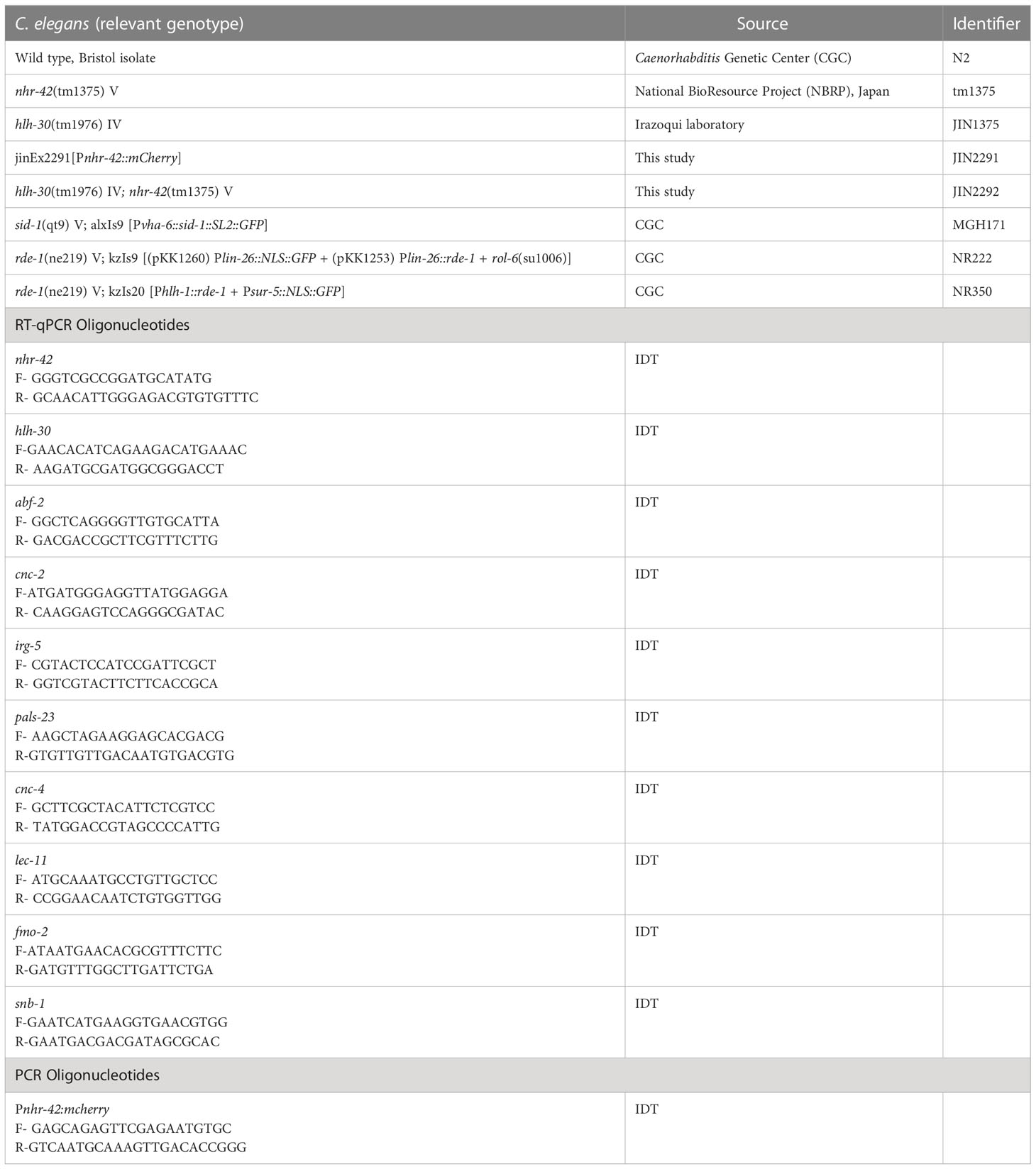
Table 1 List of C. elegans strains and oligonucleotides.
A single colony of SH1000 strain was grown in tryptic soy broth (TSB) supplemented with 50 µg/ml kanamycin overnight at 37°C shaking at 200-220 RPM. 10 µl of the overnight culture was spread on tryptic soy agar (TSA plates) supplemented with 10 ug/ml kanamycin. These plates were grown at 37°C for 5 h and then shifted to 25°C overnight. C. elegans were treated on solid media with 100 μg/ml 5-fluoro-2′-deoxyuridine (FUDR) at L4 larval stage overnight at 15°C before transfer to S. aureus TSA plates with full lawns of bacteria. Three TSA plates each containing 30-40 animals were examined for each genotype of C. elegans. Infection assays were carried out at 25°C as described (30). For RNAi experiments animals were grown to L4 larval stage on E. coli strain HT115 expressing double-stranded RNA for the target genes at 20°C, treated with 100 μg/ml FUDR and transferred to 15°C overnight and subsequently used for infection assays. Survival was quantified using standard methods (30). Animals dying of disrupted vulva or crawling on walls were censored for analysis, which was performed using Prism 9 (Graphpad Inc.). Infection assays were performed 2-3 independent times.
Slow killing plates were made using single colony of PA14 strain was grown overnight in LB broth. 10 μl of overnight culture was spread onto slow killing assay plates and incubated at 37°C for 24 h. Plates were then placed at 25°C for a day to obtain a full bacterial lawn. Animals were grown to L4 stage on OP50 in NGM plates at 20°C. 100 μg/ml FUDR was added at L4 stage. 30-40 L4 animals of all genotypes were transferred to infection plates. 200 μg/ml FUDR was added to the infection plates. Animals were scored according to established protocols (30). Animals dying of disrupted vulva or crawling on walls were censored for analysis, which was performed using Prism 9 (Graphpad Inc.). Infection assays were performed 3 independent times.
A single colony of V583 strain of E. faecalis was grown in BHI broth at 37°C for 6-8 h. 10 μl of this culture was spread onto BHI agar plates containing 10 μg/ml kanamycin and incubated overnight at 37°C 32. 30-40 L4 animals grown at 20°C were then transferred onto infection plates as described (30). Animals dying of disrupted vulva or crawling on walls were censored for analysis, which was performed using Prism 9 (Graphpad Inc.). Infection assays were performed 3 independent times.
C. elegans was grown at 20°C until L4 stage on NGM plates seeded with OP50 and then 100 μg/ml of FUDR was added. Animals were transferred to 25°C. Dead animals were counted every 24-48 hours. Animals that did not respond to touch with a pick were considered dead. 2-3 replicates with 20-30 animals on each plate were used for each experiment. Animals dying of disrupted vulva or crawling on walls were censored for analysis, which was performed using Prism 9 (Graphpad Inc.). Longevity assays were performed 3 independent times.
Animals were washed off growth or infection plates twice using sterile water and collected in 1.5 ml tubes containing 1,000 μl TRIzol (Sigma-Aldrich) for lysis. 100 μl chloroform was added and centrifuged at 12,000 rpm to separate the aqueous phase containing RNA. The aqueous phase was transferred to Purelink RNA mini kit (ThermoFisher Scientific) columns and purified total RNA was obtained following manufacturer’s instructions. cDNA was made using iScript cDNA synthesis kit (Bio-Rad). RT-qPCR was performed using SYBR Green Super mix (Bio-Rad) using a ViiA7 Real-Time qPCR system (Applied Biosystems). Test genes were normalized to neuronal reference gene snb-1. Fold change was calculated using the Pfaffl method (31).
Total RNA was collected from C. elegans using Purelink RNA kit as described above. Total RNA from 4 biological replicates for each condition was sent to BGI Genomics for library preparation and sequencing using BGI-seq 500. BGI provided clean reads in FASTQ format. FASTQ files were analyzed by DolphinNext high-throughput sequence analysis software (32). Steps followed in DolphinNext were as follows: FastQC was used to create quality control outputs. FASTQ files were aligned to the reference C. elegans genome using STAR. STAR was used to count or filter out common RNAs (eg. rRNA, miRNA, tRNA, piRNA). RSEM was used to align reads to reference transcripts and estimate gene and isoform expression levels. Genome-wide Bam analysis was done by RseQC. DEBrowser v1.22.5 was used for batch effect correction and normalization. Data analysis including generation of volcano plots and heat maps was done using DE analysis within DEBrowser v1.22.5. Adjusted p-value (Padj) ≤ 0.01 was considered significant for differential gene expression. Gene Ontology representation analysis was performed using the enrichment analysis tool in Wormbase (33) and g:Profiler (34).
To generate Pnhr-42::mCherry, the nhr-42 promoter region (2 kbp upstream of the transcription start site) was amplified and cloned into entry vector pTOPO for Gateway® recombination (ThermoFisher Sci.). This amplified region was recombined into destination vector pDest-16 (generous gift by Dr. Michael Francis, UMass Chan Medical School) expressing mCherry to generate the expression vector. The expression vector was sequenced to verify proper insertion of the nhr-42 promoter upstream of the mCherry coding sequence. Transformation was carried out by gonad microinjection following standard procedures (35).
Images were taken using a Lionheart FX automatic microscope (BioTek Instruments) at 4x and 20x magnification. 30-50 C. elegans were used for each experimental condition. 10-20 animals at a time were anesthetized using 100 mM NaN3 on a 2% agar pad for imaging. All images were captured at the same exposure and intensity. Greyscale images were used for image analysis. A region of interest (ROI) was drawn around the pharynx, which showed fluorescent expression of mCherry. Mean fluorescence intensity (MFI) of the ROI was determined by using the Analyze>Measure function in ImageJ (NIH). MFI values were plotted in Prism 9 for statistical analysis. Fluorescence microscopy experiments were repeated 2 independent times.
C. elegans was grown at 20°C until L4 stage on NGM plates seeded with OP50. 20-30 animals were placed on TSA plates containing a full lawn of S. aureus or BHI plates for E. faecalis infection. 8 h after infection, animals were washed off with water and collected in centrifuge tubes. Lipid staining using Oil Red O was performed as described (36). 10-20 animals were used for imaging. Animals were mounted on agarose pads and visualized using a Lionheart microscope at 4x and 20x magnification and the staining intensity was determined using ImageJ. Greyscale.tiff images were used for quantification. A relative threshold for the detection of red color staining was set manually (Image>Threshold>). The set threshold was constant for all images quantified. A region of interest (ROI) was drawn around each whole animal. The percent area stained inside the ROI by Oil Red O for each whole animal was calculated (Analyze>Measure>Area%). For intestine-specific quantification, the intestine (visualized by brightfield microscopy) was used as the ROI. Percent of area (Area%) stained by Oil Red O per animal was plotted and statistical analysis were performed in Prism.
C. elegans was grown at 20°C until L4 stage on NGM plates seeded with OP50 or RNAi clones as mentioned. Thereafter, 30-40 animals were transferred to TSA plates containing a full lawn of S. aureus. At the desired timepoint 10 live animals were collected in centrifuge tubes containing autoclaved water. Colony forming units were determined as in (8). 5-7 biological replicates (10 animals per replicate) were used for each condition.
GraphPad Prism 9 was used for statistical analysis. Survival curves were compared using Log-Rank (Mantel-Cox test). Unpaired, two-sample two-tailed t-tests were performed to compare with a single reference. For multiple comparisons, one-way ANOVA was used to establish significance followed by Šidák’s post-hoc test. P ≤ 0.05 was considered significant.
We previously showed that hlh-30 induces around 600 genes during S. aureus infection (8). However, HLH-30 is predicted to induce just 10% of these genes by directly binding their promoters (Figure 1A) (8). We hypothesized that indirect regulation of key host defense genes by hlh-30 could be mediated by the 17 transcription factors it induces during infection (8). To identify which are important in the intestinal epithelium for host defense, we performed intestine-specific silencing by RNAi. As expected, we found genes whose knockdown impaired host survival (e.g., nhr-55, Figure S1A) suggesting that they may be important for inducing the intestinal host response to infection. However, we were surprised to identify one gene (nhr-42), whose knockdown significantly promoted host survival (Figure S1D), suggesting that nhr-42 may function as a host response repressor.
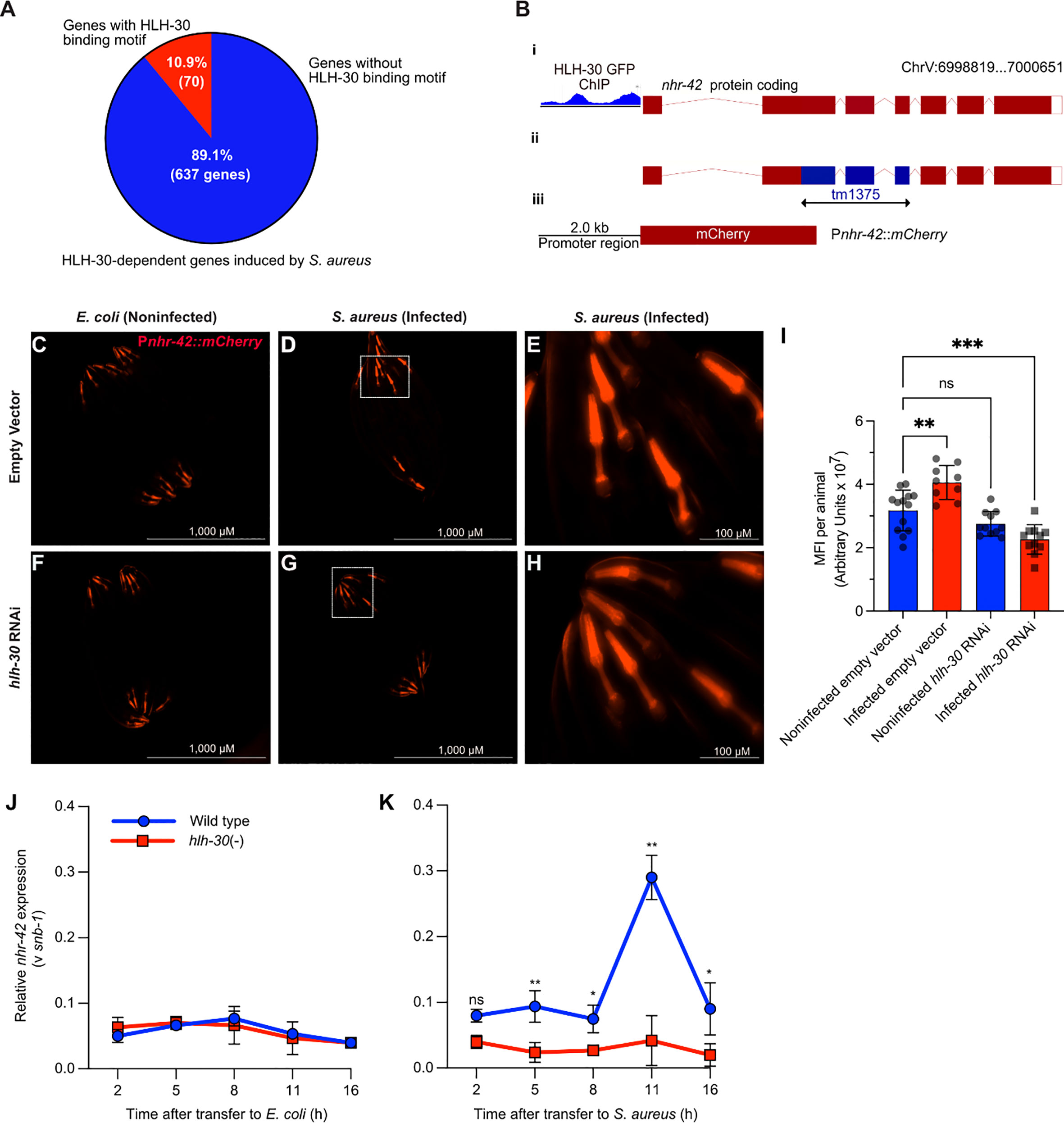
Figure 1 S. aureus induces nhr-42 in an hlh-30-dependent manner (A) Pie chart depicting predicted genes directly induced by HLH-30 during S. aureus infection [Original data are from (8)]. (B) Cartoon depicting the nhr-42 locus on Chromosome V, highlighting i) promoter region containing HLH-30 ChIP-seq peaks within 2 kbp of the transcription start site [original data from (37), ii) tm1375 deletion of 451 bp spanning exons 2-4,and iii) Pnhr-42::mCherry transcriptional reporter construct from this study. (C–H) Epifluorescence micrographs of Pnhr-42::mCherry animals fed E. coli, expressing RNAi empty vector or hlh-30 dsRNA, and then exposed to E. coli OP50 (C, F) or to S. aureus SH1000 (D, G) for 24 h. E and H show magnified areas of D and G, respectively. Scale bars: 1,000 µm for images (C, D, F, G), and 100 µm for E and (H). Data are representative of 2 independent replicates. (I) Quantification of mean fluorescence intensity (MFI) per animal from (C–H). n = 10 -20. **P ≤ 0.01, ****P ≤ 0.0001. ns, not significant. One-way ANOVA with Šídák’s post-hoc test. (J–K) RT-qPCR of nhr-42 transcript in wild type and hlh-30(tm1978) mutants fed E. coli OP50 (J) or infected with S. aureus (K). *P ≤ 0.05, **P ≤ 0.01. ns, not significant. 2- sided 2-sample unpaired t-test. 2-4 biological replicates. N = ~1,000 animals per replicate.
Public datasets contain evidence that HLH-30 may bind directly to the nhr-42 promoter (Figure 1B) (37–39), suggesting that nhr-42 may be a direct target of HLH-30. A fluorescent nhr-42 transcriptional reporter showed expression mainly in the pharynx of noninfected animals (Figure 1C), in agreement with a previous report (40). The same reporter exhibited increased activity in the pharynx after S. aureus infection (Figures 1D, E, I), suggesting that nhr-42 expression may be induced transcriptionally. In contrast, silencing of hlh-30 abrogated induced reporter activity after infection (Figures 1F–I). These results suggested that the nhr-42 promoter is active in the pharynx, and that its activity increases during infection, in the same tissue, in an HLH-30-dependent manner. This result is consistent with our prior RNA-seq data (8).
To gain better understanding of the timing of nhr-42 induction, we performed time-resolved infection followed by RT-qPCR. This revealed that nhr-42 expression is relatively stable in noninfected animals, and independent of HLH-30 (Figure 1J). During infection, nhr-42 expression was elevated throughout the experiment and peaked around 11 h in wild type animals, but in hlh-30 (–) mutants nhr-42 expression remained low (Figure 1K). These data showed that in noninfected animals nhr-42 expression does not require HLH-30, but that its early induction and peak about 11 h during infection are HLH-30-dependent.
Although we observed nhr-42 promoter activity mainly in the pharynx, nhr-42 could function in other tissues to repress host defense. To systematically identify the tissues in which nhr-42 functions, we performed tissue-specific knockdown of nhr-42 by RNAi (Figure 2). nhr-42 knockdown in the whole animal strongly enhanced host survival (Figure 2A), providing a benchmark. As previously, intestinal knockdown enhanced survival of infection (Figure 2B), as also did epidermal (hypodermal) silencing (Figure 2C). In contrast, muscle knockdown showed no significant effect (Figure 2D). These data suggest that nhr-42 represses host survival of infection mainly from the intestinal epithelium, with contributions from the epidermis.
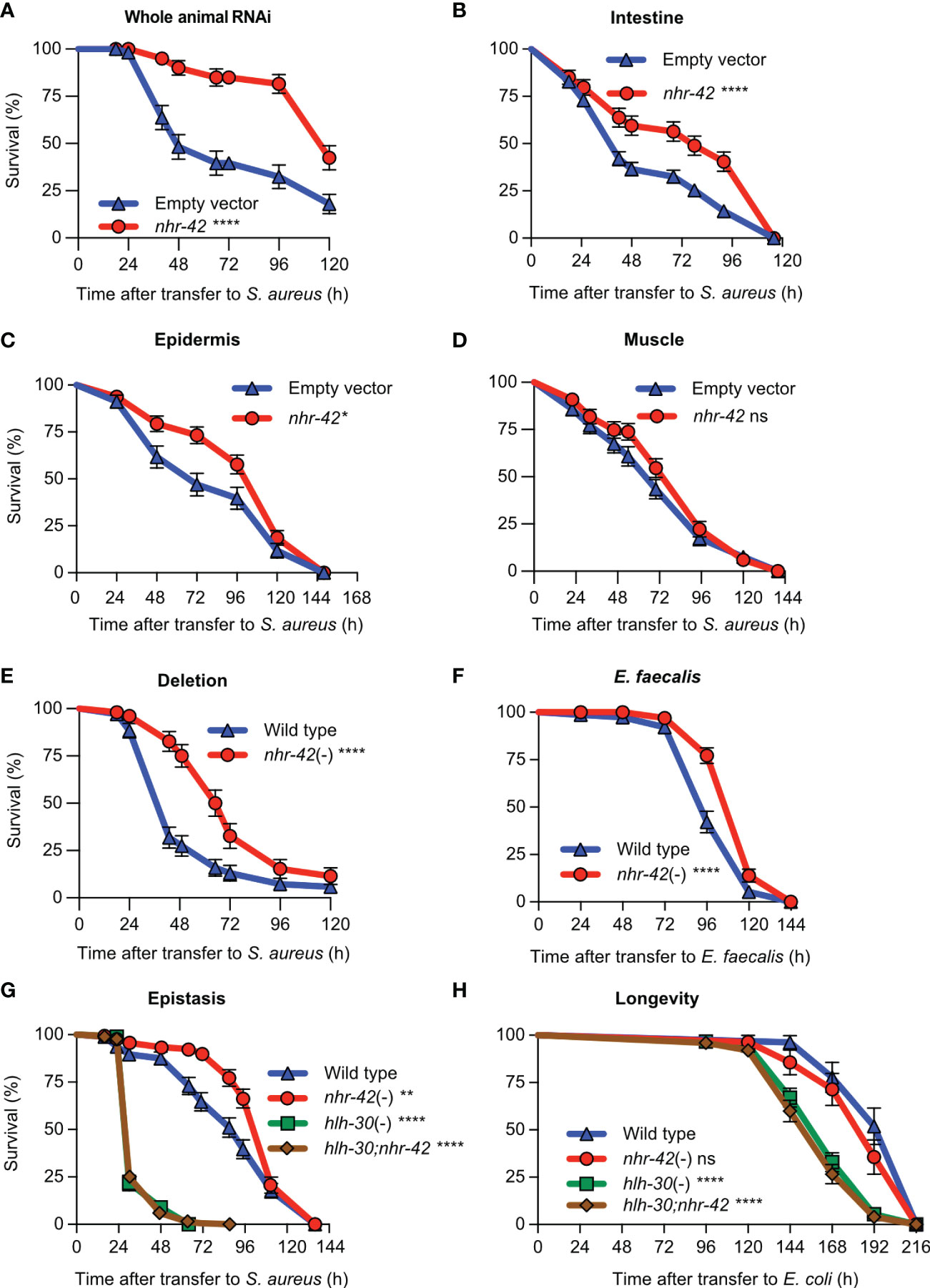
Figure 2 nhr-42 represses infection survival (A) Survival of wild type animals fed E. coli HT115 expressing RNAi empty vector or nhr-42 dsRNA during S. aureus SH1000 infection. Data are representative of two independent replicates. (B) Survival of intestine-specific MGH171 RNAi animals fed E. coli expressing RNAi empty vector or nhr-42 dsRNA during S. aureus SH1000 infection. Data are representative of two independent replicates. (C) Survival of hypodermis-specific RNAi NR222 animals fed E. coli expressing RNAi empty vector or nhr-42 dsRNA during S. aureus SH1000 infection. Data are representative of two independent replicates. (D) Survival of muscle-specific RNAi NR350 animals fed E. coli expressing RNAi empty vector or nhr-42 dsRNA during S. aureus SH1000 infection. Data are representative of two independent replicates. (E) Survival of wild type and nhr-42(tm1375) animals during S. aureus SH1000 infection. Data are representative of 3 independent replicates. (F) Survival of wild type and nhr-42(tm1375) animals during E. faecalis infection. Data are representative of 3 independent replicates. (G) Survival of wild type, nhr-42(tm1375), hlh-30(tm1978), and hlh-30;nhr-42 double mutants during S. aureus infection. Data are representative of 3 independent replicates. (H) Lifespan of wild type, nhr-42(tm1375), hlh-30(tm1978), and hlh-30;nhr-42 double mutants fed E. coli OP50 (25°C). Data are representative of 3 independent replicates.*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001. ns, not significant. Log-Rank (Kruskal-Wallis) test. N = 90-135 animals per trial.
Consistently, a total knockout of nhr-42 provided strong protection against S. aureus (Figure 2E) and Enterococcus faecalis, a distantly related Gram-positive human pathogen (Figure 2F). However, loss of nhr-42 did not affect defense against Pseudomonas aeruginosa, a Gram-negative human pathogen (Figure S2). These results show remarkable specificity of nhr-42 function.
Because nhr-42 induction by S. aureus is hlh-30-dependent, we hypothesized that they might function in the same genetic pathway. hlh-30;nhr-42 double mutants exhibited the same susceptibility to S. aureus as hlh-30 single mutants, completely suppressing the enhanced survival phenotype of nhr-42 mutants (Figure 2G). Remarkably, nhr-42 loss did not affect lifespan on nonpathogenic E. coli, and again hlh-30 was epistatic to nhr-42 (Figure 2H). In a linear genetic pathway model, these results would imply that hlh-30 functions downstream or parallel to nhr-42; however, hlh-30 expression was unaffected in nhr-42 mutants (see below). Given the results so far, we rather favored a model in which hlh-30 functions upstream of nhr-42 to induce host defense genes that nhr-42 represses.
To gain further insight into the function of nhr-42, we performed whole-animal RNA-seq of noninfected and infected animals (Figure 3A). We compared the transcriptomes of mutants to wild type under both conditions to identify differentially expressed genes (Figures 3, S3; Table S1).
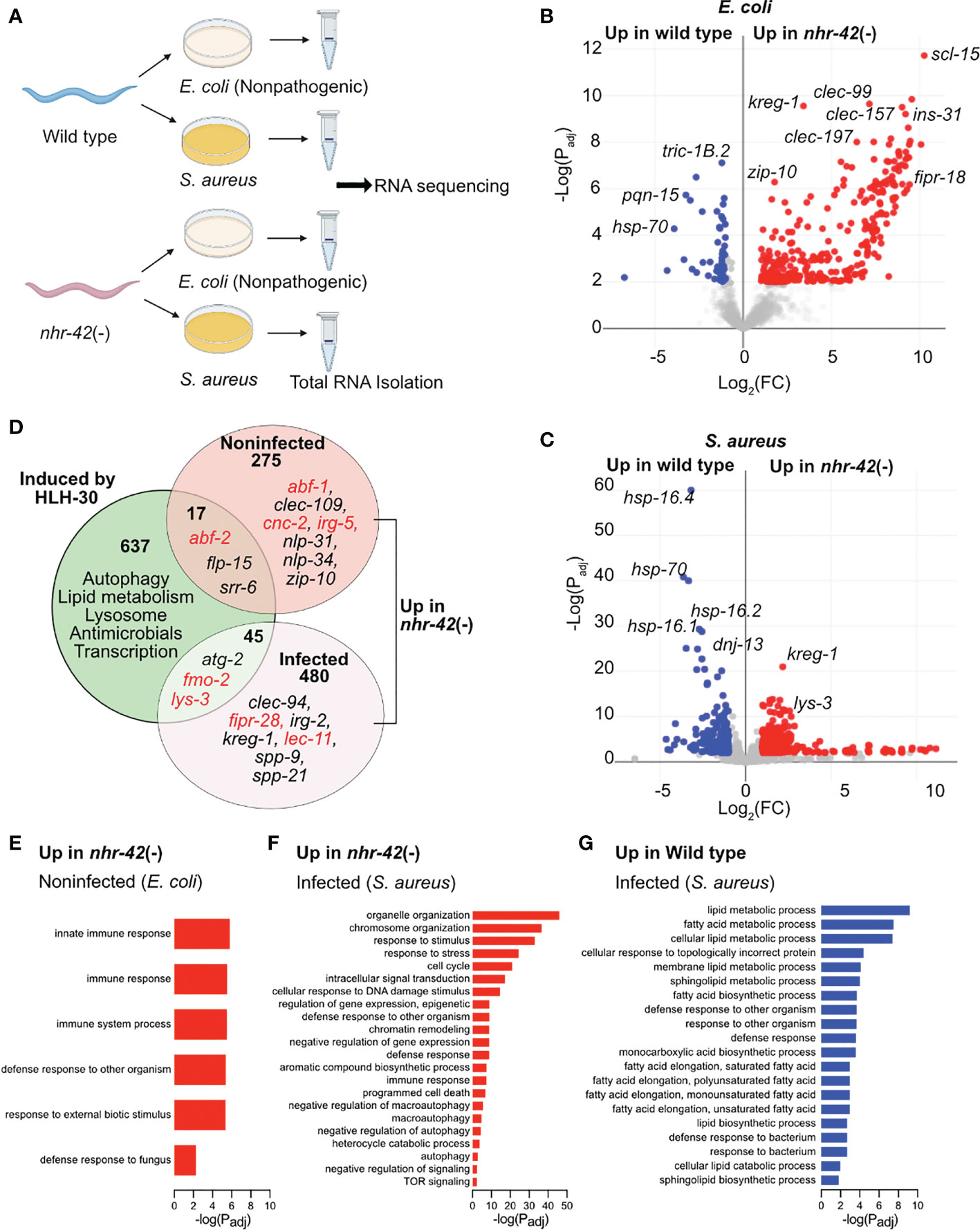
Figure 3 nhr-42 regulates host defense and metabolism genes (A) Experimental design for RNA-seq of wild type and nhr-42(tm1375) animals fed E. coli OP50 or infected with S. aureus SH1000 for 5 h. (B) Representation of differentially expressed genes in noninfected nhr-42(tm1375) animals compared to wild type. Red, genes upregulated in nhr-42(tm1375); blue, genes upregulated in wild type. FC, fold change. Padj, adjusted P value. (C) Representation of differentially expressed genes in infected nhr-42(tm1375) animals compared to wild type. Red, genes upregulated in nhr-42(tm1375); blue, genes upregulated in wild type. (D) Summary of genes induced by HLH-30 during S. aureus infection (8) and genes that are upregulated in noninfected and infected nhr-42(tm1375) animals. Numbers indicate total number of differentially expressed genes in each sector. Specific examples of antimicrobial genes are provided in each sector, and relevant genes are highlighted in red. (E) GO over-representation among upregulated genes in noninfected nhr-42(tm1375) animals relative to wild type. (F) GO over-representation among upregulated genes in infected nhr-42(tm1375) animals relative to wild type. (G) GO over-representation among upregulated genes in infected wild type animals relative to nhr-42(tm1375).
292 transcripts were expressed more highly in noninfected nhr-42 mutants compared to wild type (Figures 3B; Table S1). We found only minor overlap between this set of genes overexpressed in nhr-42 mutants and HLH-30-induced genes in infected animals (8)(Figure 3D). Nonetheless, noninfected nhr-42 mutants showed patterns that were easily identified by gene ontology (GO) over-representation analysis (Table S2). Specifically, relative to wild type, nhr-42 mutants showed increased expression of genes related to various innate immunity and host defense categories (including Defense Response to Fungus) and decreased expression of genes related to the response to unfolded protein (Figure 3E; Table S2). This remarkable enrichment for host defense genes reveals that NHR-42 functions to repress host defense in noninfected animals, which is consistent with the observed enhanced survival of infection that is caused by nhr-42 loss. Together, these results suggest that, in noninfected animals, NHR-42 represses host defense genes that are mostly HLH-30-independent.
During infection, a more complex picture emerged. nhr-42 mutants showed increased expression of 525 transcripts (Figure 3C; Table S1), enriched for genes related to reproduction (including cell cycle and embryo development) and DNA damage response, of unclear relevance to infection biology (Table S2; Figure 3F). Perhaps easier to rationalize, they also expressed higher levels of genes related to signal transduction, response to stress, defense response (including innate immune response), apoptosis, and autophagy (including macroautophagy, negative regulation of autophagy, and TOR signaling), which are all processes that have been linked to host defense in C. elegans by prior research.
In contrast, infected nhr-42 mutants showed decreased expression of 218 transcripts relative to wild type (Table S2). The most outstanding over-represented categories were related to lipid metabolism, including fatty acid biosynthesis and lipid catabolism (Figure 3G). This result prompted us to investigate the relationships among infection, lipid stores, and nhr-42.
The RNA-seq analysis showed that nhr-42 mutants expressed lower levels of seven known or proposed lipid catabolism genes (Figure 4A). These include predicted enoyl-CoA hydratase ech-7 and acyl-CoA oxidases acox1.3 and acox1.4, which are potentially involved in fatty acid β-oxidation. Also downregulated in nhr-42 mutants was Abhydrolase domain-containing homolog abhd-3.2, which is predicted to be a phospholipase involved in catabolism of medium chain fatty acids. However, genes involved in lipid biosynthesis were also repressed in infected nhr-42 mutants (Table S2). To determine the biological significance of these observations, we assessed the lipid status of nhr-42 mutants by neutral lipid droplet staining with Oil Red O (ORO) (41, 42).
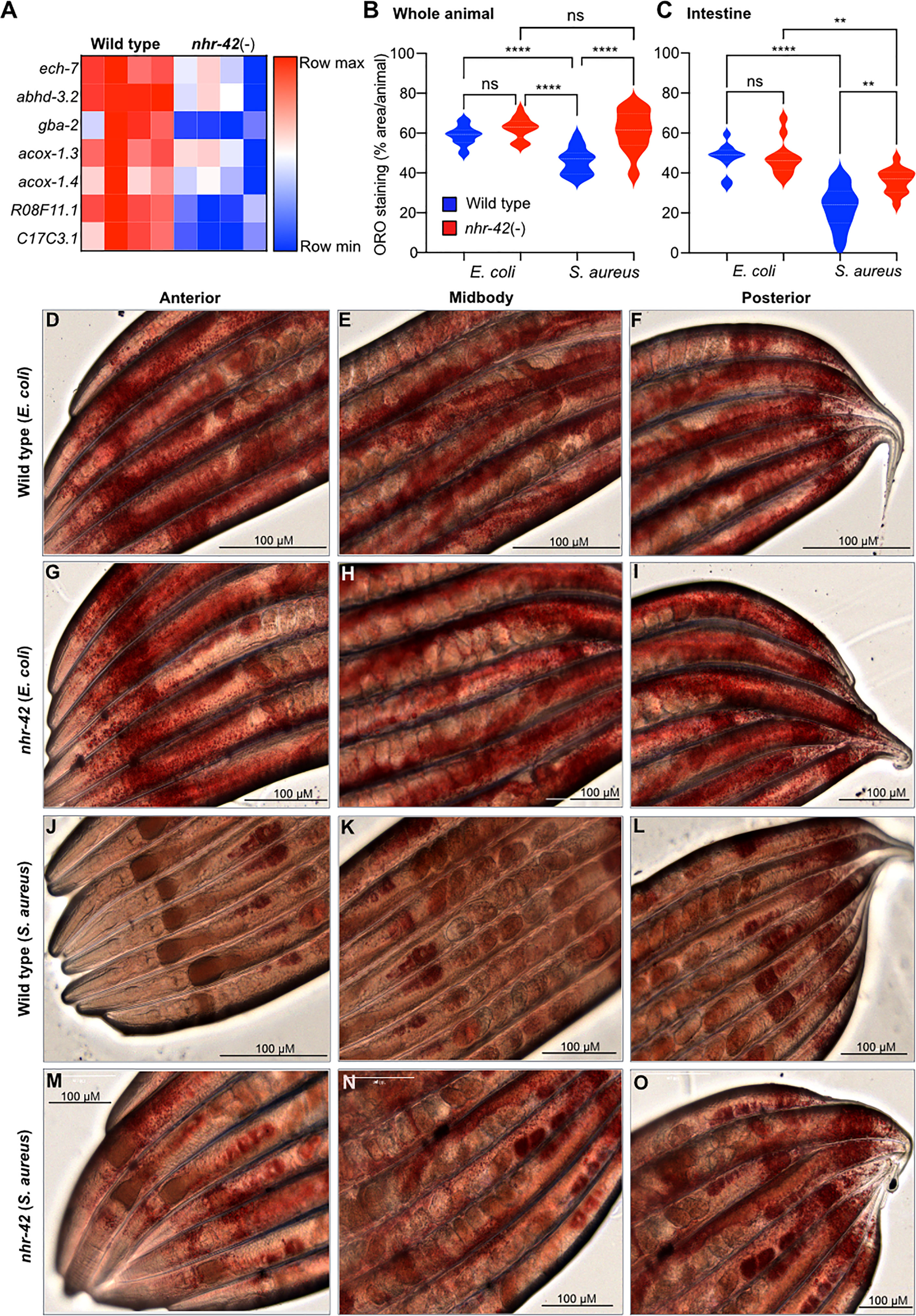
Figure 4 nhr-42 is required for lipid loss during infection (A) Lipid catabolism genes downregulated in noninfected nhr-42(tm1375) mutants relative to wild type. Row-normalized FPKM values. (B) Quantification of Oil Red O staining in whole wild type and nhr-42(tm1375) animals. ****P ≤ 0.0001. ns, not significant. One-way ANOVA with Šídák’s post-hoc test. Representative of 3 independent replicates, 10-20 animals per condition. (C) Quantification of Oil Red O staining in the intestines of wild type and nhr-42(tm1375) animals. **P ≤ 0.01. One-way ANOVA with Šídák’s post-hoc test. Representative of 3 independent replicates, 10-20 animals per condition (D–F) ORO staining of wild type animals fed with E. coli OP50 for 10 h (G–I) ORO staining of nhr-42(tm1375) animals fed with E. coli OP50 for 10 h (J–L) ORO staining of wild type animals infected with S. aureus SH1000 for 10 h (M–O) ORO staining of nhr-42(tm1375) animals infected with S. aureus SH1000 for 10 h. Scale bars: 100 µm.
Noninfected nhr-42 mutants showed a similar level of ORO staining as wild type (Figure 4B–I), indicating that nhr-42 does not affect the overall levels of lipid droplets at baseline. After infection, wild type animals showed strongly decreased ORO staining (Figures 4B, C, J–L), whereas in nhr-42 mutants staining was significantly preserved (Figures 4B, C, M–O). This indicated that nhr-42 is required for full lipid loss during infection with S. aureus. Furthermore, we observed a similar defect during infection with E. faecalis (Figure S4), showing that nhr-42 is partially required for lipid loss during infection by two archetypal Gram-positive pathogens.
We and others showed that hlh-30 is required for lipid loss during starvation (5, 6) and for host defense against S. aureus infection (8), suggesting that the two functions of HLH-30 may be related. Indeed, it was proposed that HLH-30 induces the expression of lipid catabolism genes that may be responsible for lipid droplet loss, at least during starvation (5). Recall that hlh-30 was epistatic to nhr-42 for infection survival (Figure 2G) i.e., hlh-30;nhr-42 double mutants are defective in infection survival, like the hlh-30 single mutants. To determine the relationship between this phenotype and lipid loss, we performed ORO staining of the hlh-30;nhr-42 double mutants (Figure 5). hlh-30;nhr-42 double mutants were not significantly different from wild type, hlh-30, or nhr-42 animals at baseline (Figures 5A–F, M). After infection with S. aureus, the hlh-30 single mutants showed a much smaller drop in ORO staining compared to wild type, to a level similar to the nhr-42 single mutants (Figures 5G–I, M). Moreover, the hlh-30;nhr-42 double mutants were statistically indistinguishable from the single mutants (Figures 5J–M). Therefore, hlh-30 is also required for lipid loss during S. aureus infection, and by epistasis participates in the same genetic pathway as nhr-42. However, loss of hlh-30 impairs host infection survival while loss of nhr-42 promotes it; therefore, these mutations have opposite effects on infection survival but the same effect on lipid loss. These data support the notion that nhr-42 mediates the lipid loss function of hlh-30, and thus that HLH-30 does not directly drive lipid loss during infection. In further support of this notion, of the lipase-like (lipl) genes that were proposed to function downstream of HLH-30, only lipl-2 was differentially expressed by RNA-seq, and only in one condition: infected nhr-42 mutants expressed more lipl-2 compared with infected wild type (Table S1). Therefore, the defective lipid loss of nhr-42 mutants is likely not due to decreased lipl gene expression. It follows that infection survival and lipid loss have distinct genetic requirements, and that lipid droplet loss does not explain the enhanced resistance to infection of nhr-42 mutants.
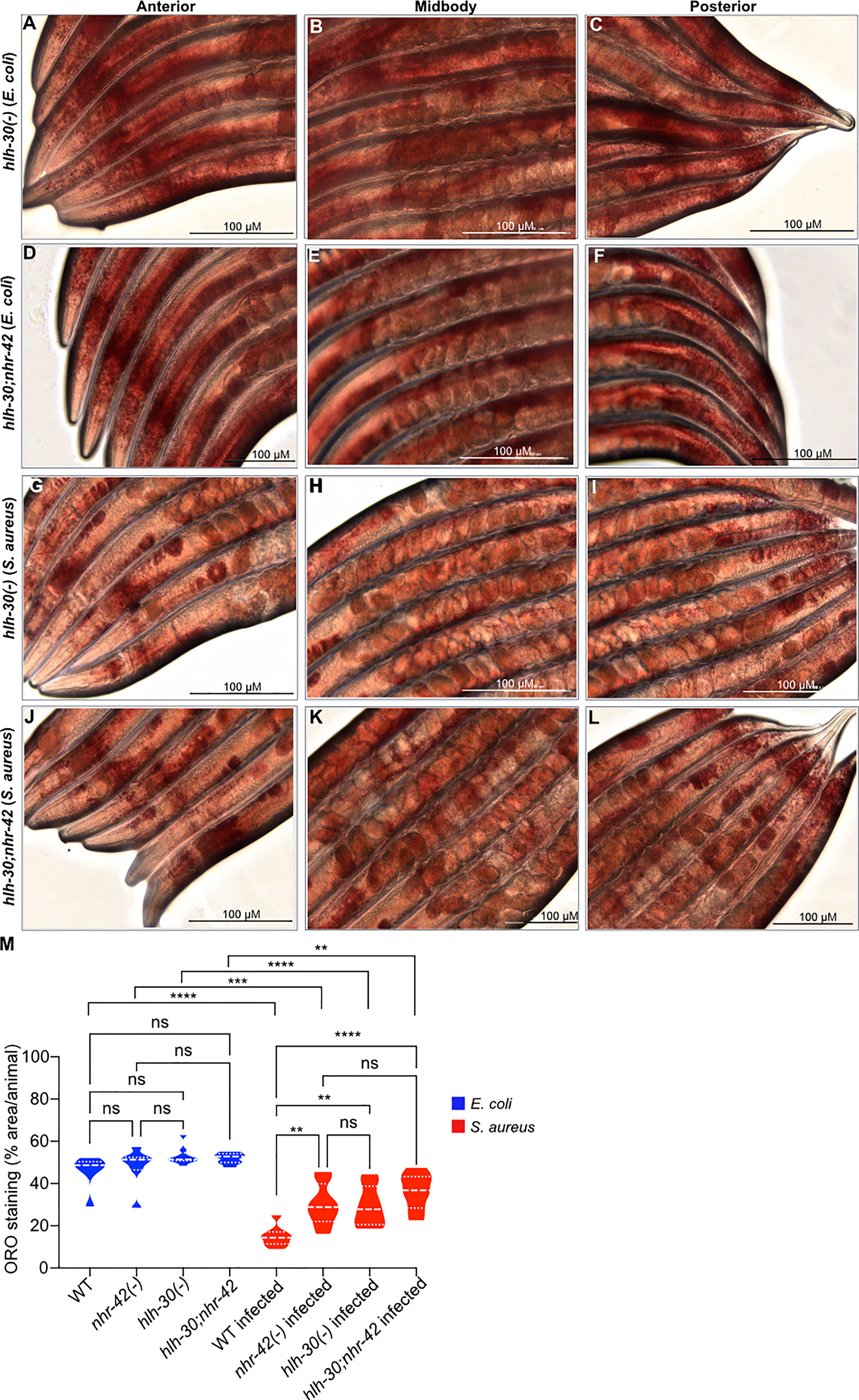
Figure 5 Loss of lipid droplets in hlh-30 mutants (A–C) ORO staining of hlh-30(tm1978) animals fed with E. coli OP50 for 10 h. (D–F) ORO staining of hlh-30(tm1978)IV;nhr-42(tm1375)V animals fed with E. coli OP50 for 10 h. (G–I) ORO staining of hlh-30(tm1978) animals infected with S. aureus SH1000 for 10 h. (J–L) ORO staining of hlh-30(tm1978)IV;nhr-42(tm1375)V animals infected with S. aureus SH1000 for 10 h. (M) Quantification of ORO staining. 7-10 animals per condition. **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001. ns, not significant. One-way ANOVA with Šídák’s post-hoc test.
As mentioned, noninfected nhr-42 mutants displayed a strong signature of upregulated host defense genes (Figure 3E). Such upregulated genes include antibacterial factor-related genes abf-1 and abf-2, antimicrobial caenacins cnc-2, cnc-4, and cnc-6, and immune response gene irg-5, as well as lec-11 and pals-23, which were previously implicated in host defense (Figure 6A) (43–50). RT-qPCR independently confirmed their upregulation, except for irg-5 (Figure 6B). To test the hypothesis that antimicrobial gene upregulation may be a major contributor to the enhanced infection survival of nhr-42 mutants, we performed RNAi-mediated knockdown. Silencing of cnc-4, irg-5, and pals-23 had no significant effect in nhr-42 mutants, whereas silencing of abf-2, cnc-2, and lec-11 significantly decreased their enhanced survival (Figures 6C–H). abf-2 and cnc-2 knockdowns showed the strongest effects in nhr-42 mutants, but did not affect the wild type (Figures 6C, D). Curiously, silencing of lec-11 (which reduced the survival of nhr-42) and of pals-23 (which did not), both enhanced the survival of wild type (Figures 6G, H). Thus, specific antimicrobial effectors that are constitutively induced in nhr-42 mutants are required for their enhanced infection survival.
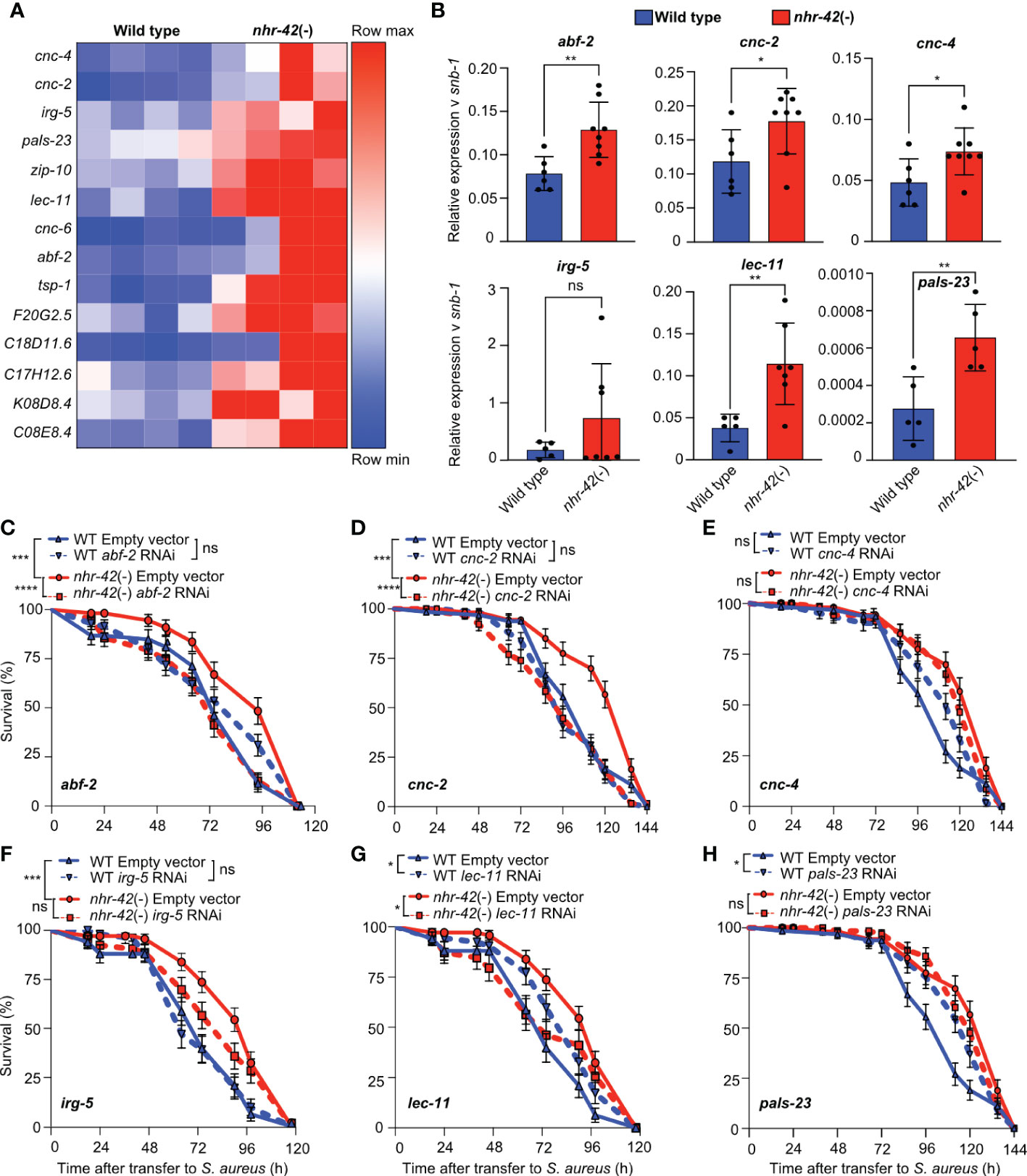
Figure 6 Antimicrobial genes required for enhanced infection survival of nhr-42 mutants (A) Host defense genes upregulated in noninfected nhr-42(tm1375) mutants relative to wild type. Row-normalized FPKM values. (B)RT-qPCR of transcripts for select antimicrobial genes in noninfected wild type and nhr-42(tm1375) mutants. 5-8 biological replicates. N = ~1,000 animals per replicate. *P ≤ 0.05, **P ≤ 0.01. ns, not significant. t-test. (C) Infection survival of wild type and nhr-42(tm1375) animals fed E. coli HT115 expressing RNAi empty vector or abf-2 dsRNA DNA prior to infection with S. aureus SH1000. Data are representative of 3 independent replicates. (D) Infection survival of wild type and nhr-42(tm1375) animals fed E. coli HT115 expressing RNAi empty vector or cnc-2 dsRNA prior to infection with S. aureus SH1000. Data are representative of 2 independent replicates. (E) Infection survival of wild type and nhr-42(tm1375) animals fed E. coli HT115 expressing RNAi empty vector or irg-5 dsRNA prior to infection with S. aureus SH1000. Data are representative of 2 independent replicates. (F) Infection survival of wild type and nhr-42(tm1375) animals fed E. coli HT115 expressing RNAi empty vector or pals-23 dsRNA prior to infection with S. aureus SH1000. Data are representative of 2 independent replicates. (G) Infection survival of wild type and nhr-42(tm1375) animals fed E. coli HT115 expressing RNAi empty vector or cnc-4 dsRNA prior to infection with S. aureus SH1000. Data are representative of 2 independent replicates. (H) Infection survival of wild type and nhr-42(tm1375) animals fed E. coli HT115 expressing RNAi empty vector or lec-11 dsRNA prior to infection with S. aureus SH1000. Data are representative of 2 independent replicates. *P ≤ 0.05, ***P ≤ 0.001, ****P ≤ 0.0001. ns, not significant. Log-Rank (Kruskal-Wallis test). The Empty Vector controls for (D, E, H) are the same. The Empty Vector controls for F and G are the same. In (C–H) N = 90-135 animals per trial.
To determine if nhr-42 mutants show enhanced infection survival because of enhanced infection resilience (i.e., same colonization as wild type) or resistance (i.e., lower colonization than wild type), we measured S. aureus accumulation in the C. elegans gut over time. Whereas by 24 h of infection the nhr-42 mutants accumulated significantly less S. aureus than wild type (Figure 7A), they tended to accumulate less at earlier time points as well. Control experiments showed that the pharynxes of nhr-42 mutants pumped at rates that are indistinguishable from wild type (Figure 7B), suggesting that the decreased accumulation of S. aureus in nhr-42 mutants was not due to decreased oral intake. Of the genes that were required for enhanced survival of nhr-42 mutants, abf-2 is the only one whose product has demonstrated antibacterial activity in vitro (44), and therefore we chose it for further study. Animals treated with abf-2 RNAi showed increased S. aureus load, comparable to wild type (Figure 7C), suggesting that abf-2 mediates infection resistance in nhr-42 mutants. As mentioned, noninfected nhr-42 mutants exhibit higher expression of abf-2 than wild type counterparts (Figures 6A, B). However, abf-2 expression at 5 h infection was similar in wild type and nhr-42 mutant animals (Figure 7D), suggesting that abf-2 may be overexpressed only in noninfected nhr-42 mutants. However, these data do not rule out potentially increased abf-2 expression in nhr-42 mutants at other times of infection.

Figure 7 abf-2 mediates infection resistance in nhr-42 mutants (A) S. aureus SH1000 colony forming units (CFU) from wild type and nhr-42(tm1375) animals after 6, 12, and 24 h of infection. Each dot represents a single animal (N = 6 per time point). Means ± SD. (B) Pumping rates of noninfected wild type and nhr-42(tm1375) animals. Each dot represents a single animal (N = 10). Means ± SD. (C) S. aureus SH1000 CFU from wild type and nhr-42(tm1375) animals fed E. coli HT115 expressing RNAi empty vector or abf-2 dsRNA prior to infection for 24 h. Each dot represents a single animal (N = 5-7). Means ± SD. (D) RT-qPCR of abf-2 transcript in infected wild type and nhr-42(tm1375) animals. Each dot represents a single biological replicate of ~1,000 animals. Means ± SD. * P≤ 0.05, ** P≤ 0.01. ns, not significant. 2-sided 2-sample unpaired t-test.
We describe studies that implicate the HLH-30-dependent transcription factor gene nhr-42 as an important modulator of host defense and of lipid droplet metabolism. Although the exact molecular and organismal relationships between these two processes requires further study, our data demonstrate that downstream regulatory target genes that are induced by HLH-30 play central roles in infection survival and organismal lipid metabolism.
nhr-42 is a previously uncharacterized member of the nuclear receptor family, which is amplified in C. elegans (22). For this reason and because of evolutionary drift, establishing clear one-to-one orthology with human genes is challenging. Nonetheless, the presence of a coiled-coil domain N-terminal to the ligand binding domain and homology-based predictions by Ensembl Compara suggest that NHR-42 may be related to mammalian NR1D1 (also known as REV-ERBα). In mammalian innate immune cells, NR1D1 represses TLR4 and pro-inflammatory genes, such as CCL-2 and IL-6 (51–53). Pharmacological NR1D1 activation represses pro-inflammatory cytokine secretion and promotes IL-10 in primary rat microglia (54), in murine models of colitis (55), in the lungs of murine models of inhaled LPS (56), in the livers of murine models of fulminant hepatitis, and in murine and human macrophages (57). In murine and human gastric epithelial cells, NR1D1 represses antimicrobial peptides and CCL-21, repressing host defense and promoting Helicobacter pylori infection (58). Nr1d1-/- mice show spontaneous microglial activation and enhanced hippocampal neuroinflammatory responses to systemic LPS (59). Moreover, recent work showed that murine TFEB/TFE3 induce Nr1d1 expression during fasting in the liver (60) and that NR1D1 induces the expression of TFEB in human macrophage-like THP-1 cells (61). Thus, although published precedent supports NHR-42 as a functional homolog of NR1D1, the regulation of NHR-42/NR1D1 by HLH-30/TFEB during infection is a novel and important interaction that could be exploited for therapeutic purposes. As a nuclear receptor, NHR-42 is expected to bind to lipophilic ligands, but their identity and biological functions remain unknown. Moreover, promoter reporters show that nhr-42 and its repression target abf-2 are expressed in the pharynx. It is unclear why HLH-30, which is expressed throughout the soma (8), induces nhr-42 only in this tissue. One possibility is that a second necessary factor, possibly a transcriptional co-regulator, is pharynx-restricted.
ABF-2 is a 62-residue peptide homologous to Ascaris suum antibacterial factor (ASABF) and carries a conserved arthropod defensin consensus motif (44). It is one of a remarkably small number of C. elegans antimicrobial effectors with experimentally demonstrated bactericidal activity. In vitro, recombinant ABF-2 exhibits antimicrobial activity against Gram-positive and -negative bacteria, as well as yeasts (44). The expression of gene abf-2, which curiously resides together with paralog abf-1 within an intron belonging to uncharacterized gene C50F2.2, is induced by Salmonella-infected animals in a manner that requires the sole C. elegans toll-like receptor gene, tol-1 (62, 63). Additionally, abf-2 is upregulated in long-lived age-1 and immune-deficient dbl-1 mutants, in a manner that requires daf-16 and nsy-1 (of the insulin signaling and p38 MAPK pathways, respectively) (64). Additionally, abf-2 is induced by S. aureus and is HLH-30-dependent (8), which illustrates how NHR-42 can repress antimicrobial genes that are HLH-30 targets. Remarkably, loss of nhr-42 did not protect animals from P. aeruginosa. This may reflect how remarkably different the host responses to S. aureus and P aeruginosa are (10). Alternatively, loss of nhr-42 may better counteract the pathologies caused by S. aureus, which are markedly different those caused by P. aeruginosa (10). It is worth noting that ABF-2 has not been shown to exert antibacterial activity against P. aeruginosa (44). Nevertheless, our results highlight how constitutively increased expression of abf-2 may protect C. elegans from S. aureus in nhr-42 mutants. To our knowledge, this is the first report of the in vivo importance of ABF-2 in host infection resistance.
In addition to its host defense gene repressive role, we discovered that nhr-42 promotes the loss of lipid stores during infection. Previously, HLH-30 was proposed to induce lipid mobilization by direct induction of lipase genes (5). However, our results suggest that this mechanism may be secondary to another, which is nhr-42-dependent. What this nhr-42-dependent mechanism might be is speculation. Whether lipids are lost in wild type animals due to increased consumption (mobilization) or decreased biosynthesis remains an open question. Silencing of lipid catabolic genes that are downregulated in nhr-42 mutants did not prevent lipid droplet loss in infected wild type animals (not shown), thereby not supporting the hypothesis that downregulation of these genes individually does not account for decreased lipid droplets in nhr-42 mutants. Moreover, RNA-seq showed that several genes related to lipid biosynthesis were downregulated in infected nhr-42 mutants compared to wild type, which would be expected to deplete lipid droplets rather than preserve them.
Equally enigmatic is the role that lipid droplet depletion plays in host defense. Although lipid droplet depletion has been observed during infection with other model pathogens (65, 66), whether it aids or hinders host defense is unknown. Additionally, mutants in which the stress-response transcription factor SKN-1/NRF2 is hyperactivated exhibit lipid depletion and enhanced infection survival (66). However, the mechanistic relationship between the two correlated phenomena is currently unknown. Lipid species can perform a broad range of biological functions, including energy storage, building blocks for biosynthesis of metabolites and organelles, membrane and cellular repair, antibacterial activity, and signaling as regulatory ligands for enzymes and transcriptional regulators. Our previous data show that S. aureus causes wholesale disruption of the apical domain of the intestinal epithelium, followed by cytolysis of the epithelium and underlying tissues (10). We also have found that autophagy, which requires membrane rearrangements and is involved in membrane repair and damaged organelle recycling, is required for defense against S. aureus (8). Thus, existing evidence supports a role of lipids in host cytoprotection. However, other roles (energy, signaling, bactericide) cannot be ruled out and deserve further exploration. Lipid stores serve as sources of energy and as building blocks for oogenesis (67). In absence of lipid mobilization, nhr-42 mutants are expected to obtain energy for host defense from alternative sources: one possibility could be autophagy-mediated degradation of macromolecules and organelles. Remarkably, reduced lipid mobilization in nhr-42 mutants did affect their brood sizes (not shown). Therefore, nhr-42 mutants may be adapted to obtain energy for biosynthesis and cytoprotection from sources other than lipid droplets, and their enhanced infection resistance does not appear to be related to reproduction.
There remain many plausible answers to what purpose induction of nhr-42 by HLH-30 may serve during infection. If we assume that NHR-42 mainly functions as a direct repressor of gene expression, its induction may help terminate the host response after infection clearance. This is not allowed to happen by design in our infection assays to facilitate experimental study, but in the wild animals are free to deploy behavioral host defense and thus physically avoid ingesting pathogenic bacteria. The hypothetical direct repressor activity of NHR-42 may (or may not) be regulated by yet unidentified lipophilic ligands, as is the case for better understood nuclear receptors. Alternatively, NHR-42 may function indirectly, by inducing the expression of an unknown and intermediate repressor of gene expression. In either scenario, the net effects of NHR-42 activity would be to suppress host defense. Future work must examine these mechanistic scenarios and their physiological significance.
The RNA-seq data are deposited in the Sequence Read Archive of the National Library of Medicine of the USA, accession number PRJNA929270 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA929270/).
Experimental conception and design: JI, DG, SL. Data analysis: DG, XG, SL, JI. Experimentation: DG, XG, SL. Writing and editing: DG, XG, SL, JI. All authors contributed to the article and approved the submitted version.
This work was partially funded by grants R01GM101056 (JI), T32AI095213 (XG), T32AI132152 (XG), and T32GM135751 (XG) from the National Institutes of Health of the USA and the Dr. Marcellette Williams Memorial Fund (JI). Some C. elegans strains were provided by the CGC, which is funded by the NIH Office of Research Infrastructure Programs (P40OD010440). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The authors thank the members of the Irazoqui laboratory and Department of Microbiology and Physiological Systems for helpful insights and discussions, Dr. Michael Francis for vector pDest-16, Michael Francis and Craig Mello laboratory members Christopher Lambert, Krishna Ghanta, and Daniel Durning for their help in providing and generating reagents, Dr. Alper Kucukural for his help with RNA-sequencing data analysis, and Amy Parker, Annette Bohigian, Richard Fish, Tracey Rae, and Dhruti Desai for critical administrative support.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1094145/full#supplementary-material
1. Irazoqui JE. Key roles of MiT transcription factors in innate immunity and inflammation. Trends Immunol (2020) 41:157–71. doi: 10.1016/j.it.2019.12.003
2. Li M, Wang Z, Wang P, Li H, Yang L. TFEB: A emerging regulator in lipid homeostasis for atherosclerosis. Front Physiol (2021) 12:639920. doi: 10.3389/fphys.2021.639920
3. Keerti R, Aarti P, Ravi M. TFEB – at the crossroads of host–pathogen interactions. J Cell Sci (2021) 134:jcs252981. doi: 10.1242/jcs.252981
4. Lapierre LR, Filho CDDM, McQuary PR, Chu C-C, Visvikis O, Chang JT, et al. The TFEB orthologue HLH-30 regulates autophagy and modulates longevity in caenorhabditis elegans. Nat Commun (2013) 4:2267. doi: 10.1038/ncomms3267
5. O’Rourke EJ, Ruvkun G. MXL-3 and HLH-30 transcriptionally link lipolysis and autophagy to nutrient availability. Nat Cell Biol (2013) 15:668–76. doi: 10.1038/ncb2741
6. Settembre C, Cegli RD, Mansueto G, Saha PK, Vetrini F, Visvikis O, et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat Cell Biol (2013) 15:647–58. doi: 10.1038/ncb2718
7. Kim S, Song H-S, Yu J, Kim Y-M. MiT family transcriptional factors in immune cell functions. Mol Cells (2021) 44:342–55. doi: 10.14348/molcells.2021.0067
8. Visvikis O, Ihuegbu N, Labed SA, Luhachack LG, Alves A-MF, Wollenberg AC, et al. Innate host defense requires TFEB-mediated transcription of cytoprotective and antimicrobial genes. Immunity (2014) 40:896–909. doi: 10.1016/j.immuni.2014.05.002
9. Pastore N, Brady OA, Diab HI, Martina JA, Sun L, Huynh T, et al. TFEB and TFE3 cooperate in the regulation of the innate immune response in activated macrophages. Autophagy (2016) 12:1–19. doi: 10.1080/15548627.2016.1179405
10. Irazoqui JE, Troemel ER, Feinbaum RL, Luhachack LG, Cezairliyan BO, Ausubel FM. Distinct pathogenesis and host responses during infection of c. elegans by p. aeruginosa and s. aureus. PloS Pathog (2010) 6:e1000982. doi: 10.1371/journal.ppat.1000982
11. Wani KA, Goswamy D, Taubert S, Ratnappan R, Ghazi A, Irazoqui JE. NHR-49/PPAR-α and HLH-30/TFEB cooperate for c. elegans host defense via a flavin-containing monooxygenase. Elife (2021) 10:e62775. doi: 10.7554/elife.62775
12. Irazoqui JE, Ng A, Xavier RJ, Ausubel FM. Role for β-catenin and HOX transcription factors in caenorhabditis elegans and mammalian host epithelial-pathogen interactions. Proc Natl Acad Sci (2008) 105:17469–74. doi: 10.1073/pnas.0809527105
13. Labed SA, Wani KA, Jagadeesan S, Hakkim A, Najibi M, Irazoqui JE. Intestinal epithelial wnt signaling mediates acetylcholine-triggered host defense against infection. Immunity (2018) 48:963–978.e3. doi: 10.1016/j.immuni.2018.04.017
14. Najibi M, Labed SA, Visvikis O, Irazoqui JE. An evolutionarily conserved PLC-PKD-TFEB pathway for host defense. Cell Rep (2016) 15:1728–42. doi: 10.1016/j.celrep.2016.04.052
15. Nakamura S, Karalay Ö, Jäger PS, Horikawa M, Klein C, Nakamura K, et al. Mondo complexes regulate TFEB via TOR inhibition to promote longevity in response to gonadal signals. Nat Commun (2016) 7:10944. doi: 10.1038/ncomms10944
16. Bennett CF, Kwon JJ, Chen C, Russell J, Acosta K, Burnaevskiy N, et al. Transaldolase inhibition impairs mitochondrial respiration and induces a starvation-like longevity response in caenorhabditis elegans. PloS Genet (2017) 13:e1006695. doi: 10.1371/journal.pgen.1006695
17. Lee S-H, Omi S, Thakur N, Taffoni C, Belougne J, Engelmann I, et al. Modulatory upregulation of an insulin peptide gene by different pathogens in c. elegans. Virulence (2018) 9:648–58. doi: 10.1080/21505594.2018.1433969
18. Lin X-X, Sen I, Janssens GE, Zhou X, Fonslow BR, Edgar D, et al. DAF-16/FOXO and HLH-30/TFEB function as combinatorial transcription factors to promote stress resistance and longevity. Nat Commun (2018) 9:4400. doi: 10.1038/s41467-018-06624-0
19. Butler VJ, Gao F, Corrales CI, Cortopassi WA, Caballero B, Vohra M, et al. Age- and stress-associated c. elegans granulins impair lysosomal function and induce a compensatory HLH-30/TFEB transcriptional response. Plos Genet (2019) 15:e1008295. doi: 10.1371/journal.pgen.1008295
20. Grove CA, Masi FD, Barrasa MI, Newburger DE, Alkema MJ, Bulyk ML, et al. A multiparameter network reveals extensive divergence between c. elegans bHLH transcription factors. Cell (2009) 138:314–27. doi: 10.1016/j.cell.2009.04.058
21. Hoffmann JM, Partridge L. Nuclear hormone receptors: Roles of xenobiotic detoxification and sterol homeostasis in healthy aging. Crit Rev Biochem Mol (2015) 50:380–92. doi: 10.3109/10409238.2015.1067186
22. Antebi A. Nuclear receptor signal transduction in c. elegans. Wormbook (2015), 1–49. doi: 10.1895/wormbook.1.64.2
23. Sim S, Hibberd ML. Caenorhabditis elegans susceptibility to gut enterococcus faecalis infection is associated with fat metabolism and epithelial junction integrity. BMC Microbiol (2016) 16:6. doi: 10.1186/s12866-016-0624-8
24. Rajan M, Anderson CP, Rindler PM, Romney SJ, Santos MCFD, Gertz J, et al. NHR-14 loss of function couples intestinal iron uptake with innate immunity in c. elegans through PQM-1 signaling. Elife (2019) 8:e44674. doi: 10.7554/elife.44674
25. Park MR, Ryu S, Maburutse BE, Oh NS, Kim SH, Oh S, et al. Probiotic lactobacillus fermentum strain JDFM216 stimulates the longevity and immune response of caenorhabditis elegans through a nuclear hormone receptor. Sci Rep-uk (2018) 8:7441. doi: 10.1038/s41598-018-25333-8
26. Otarigho B, Aballay A. Cholesterol regulates innate immunity via nuclear hormone receptor NHR-8. Iscience (2020) 23:101068. doi: 10.1016/j.isci.2020.101068
27. Hummell NA, Revtovich AV, Kirienko NV. Novel immune modulators enhance caenorhabditis elegans resistance to multiple pathogens. Msphere (2021) 6:e00950-20. doi: 10.1128/msphere.00950-20
28. Naim N, Amrit FRG, Ratnappan R, DelBuono N, Loose JA, Ghazi A. Cell nonautonomous roles of NHR-49 in promoting longevity and innate immunity. Aging Cell (2021) 20:e13413. doi: 10.1111/acel.13413
29. Peterson ND, Icso JD, Salisbury JE, Rodríguez T, Thompson PR, Pukkila-Worley R. Pathogen infection and cholesterol deficiency activate the c. elegans p38 immune pathway through a TIR-1/SARM1 phase transition. Elife (2022) 11:e74206. doi: 10.7554/elife.74206
30. Powell JR, Ausubel FM. Innate immunity. Methods Mol Biol Clifton N J (2008) 415:403–27. doi: 10.1007/978-1-59745-570-1_24
31. Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res (2001) 29:45e–45. doi: 10.1093/nar/29.9.e45
32. Yukselen O, Turkyilmaz O, Ozturk AR, Garber M, Kucukural A. DolphinNext: a distributed data processing platform for high throughput genomics. BMC Genomics (2020) 21:310. doi: 10.1186/s12864-020-6714-x
33. Angeles-Albores D, Lee RYN, Chan J, Sternberg PW. Tissue enrichment analysis for c. elegans genomics. BMC Bioinf (2016) 17:366. doi: 10.1186/s12859-016-1229-9
34. Reimand J, Arak T, Adler P, Kolberg L, Reisberg S, Peterson H, et al. g:Profiler–a web server for functional interpretation of gene lists (2016 update). Nucleic Acids Res (2016) 44:W83–9. doi: 10.1093/nar/gkw199
35. Kadandale P, Chatterjee I, Singson A. Germline transformation of caenorhabditis elegans by injection. Methods Mol Biol (Clifton NJ) (2009) 518:123 133. doi: 10.1007/978-1-59745-202-1_10
36. Nhan JD, Curran SP. Aging, methods and protocols. Methods Mol Biol (2020) 2144:103–10. doi: 10.1007/978-1-0716-0592-9_9
37. Niu W, Lu ZJ, Zhong M, Sarov M, Murray JI, Brdlik CM, et al. Diverse transcription factor binding features revealed by genome-wide ChIP-seq in c. elegans. Genome Res (2011) 21:245–54. doi: 10.1101/gr.114587.110
38. Kudron MM, Victorsen A, Gevirtzman L, Hillier LW, Fisher WW, Vafeados D, et al. The modERN resource: Genome-wide binding profiles for hundreds of drosophila and caenorhabditis elegans transcription factors. Genetics (2017) 208:937–49. doi: 10.1534/genetics.117.300657
39. Gerstein MB, Lu ZJ, Nostrand ELV, Cheng C, Arshinoff BI, Liu T, et al. Integrative analysis of the caenorhabditis elegans genome by the modENCODE project. Science (2010) 330:1775–87. doi: 10.1126/science.1196914
40. Reece-Hoyes JS, Shingles J, Dupuy D, Grove CA, Walhout AJ, Vidal M, et al. Insight into transcription factor gene duplication from caenorhabditis elegans promoterome-driven expression patterns. BMC Genomics (2007) 8:27. doi: 10.1186/1471-2164-8-27
41. Yen K, Le TT, Bansal A, Narasimhan SD, Cheng J-X, Tissenbaum HA. A comparative study of fat storage quantitation in nematode caenorhabditis elegans using label and label-free methods. PloS One (2010) 5:e12810. doi: 10.1371/journal.pone.0012810
42. O’Rourke EJ, Soukas AA, Carr CE, Ruvkun GC. Elegans major fats are stored in vesicles distinct from lysosome-related organelles. Cell Metab (2009) 10:430–5. doi: 10.1016/j.cmet.2009.10.002
43. Zehrbach AMD, Rogers AR, Tarr DEK. An investigation of the potential antifungal properties of CNC-2 in caenorhabditis elegans. J Nematol (2017) 49:472–6.
44. Kato Y, Aizawa T, Hoshino H, Kawano K, Nitta K, Zhang H. Abf-1 and abf-2, ASABF-type antimicrobial peptide genes in caenorhabditis elegans. Biochem J (2002) 361:221–30. doi: 10.1042/0264-6021:3610221
45. Nandakumar M, Tan M-W. Gamma-linolenic and stearidonic acids are required for basal immunity in caenorhabditis elegans through their effects on p38 MAP kinase activity. PloS Genet (2008) 4:e1000273. doi: 10.1371/journal.pgen.1000273
46. Reddy KC, Dror T, Sowa JN, Panek J, Chen K, Lim ES, et al. An intracellular pathogen response pathway promotes proteostasis in c. elegans. Curr Biol (2017) 27:3544–3553.e5. doi: 10.1016/j.cub.2017.10.009
47. Panek J, Gang SS, Reddy KC, Luallen RJ, Fulzele A, Bennett EJ, et al. A cullin-RING ubiquitin ligase promotes thermotolerance as part of the intracellular pathogen response in caenorhabditis elegans. P Natl Acad Sci USA (2020) 117:7950–60. doi: 10.1073/pnas.1918417117
48. Zugasti O, Ewbank JJ. Neuroimmune regulation of antimicrobial peptide expression by a noncanonical TGF-β signaling pathway in caenorhabditis elegans epidermis. Nat Immunol (2009) 10:249–56. doi: 10.1038/ni.1700
49. Pujol N, Zugasti O, Wong D, Couillault C, Kurz CL, Schulenburg H, et al. Anti-fungal innate immunity in c. elegans is enhanced by evolutionary diversification of antimicrobial peptides. PloS Pathog (2008) 4:e1000105. doi: 10.1371/journal.ppat.1000105
50. Pujol N, Cypowyj S, Ziegler K, Millet A, Astrain A, Goncharov A, et al. Distinct innate immune responses to infection and wounding in the c. elegans epidermis. Curr Biol Cb (2008) 18:481–9. doi: 10.1016/j.cub.2008.02.079
51. Fontaine C, Rigamonti E, Pourcet B, Duez H, Duhem C, Fruchart J-C, et al. The nuclear receptor rev-erbα is a liver X receptor (LXR) target gene driving a negative feedback loop on select LXR-induced pathways in human macrophages. Mol Endocrinol (2008) 22:1797–811. doi: 10.1210/me.2007-0439
52. Gibbs JE, Blaikley J, Beesley S, Matthews L, Simpson KD, Boyce SH, et al. The nuclear receptor REV-ERBα mediates circadian regulation of innate immunity through selective regulation of inflammatory cytokines. P Natl Acad Sci USA (2011) 109:582–7. doi: 10.1073/pnas.1106750109
53. Curtis AM, Bellet MM, Sassone-Corsi P, O’Neill LAJ. Circadian clock proteins and immunity. Immunity (2014) 40:178–86. doi: 10.1016/j.immuni.2014.02.002
54. Wolff SEC, Wang X-L, Jiao H, Sun J, Kalsbeek A, Yi C-X, et al. The effect of rev-erbα agonist SR9011 on the immune response and cell metabolism of microglia. Front Immunol (2020) 11:550145. doi: 10.3389/fimmu.2020.550145
55. Wang S, Lin Y, Yuan X, Li F, Guo L, Wu B. REV-ERBα integrates colon clock with experimental colitis through regulation of NF-κB/NLRP3 axis. Nat Commun (2018) 9:4246. doi: 10.1038/s41467-018-06568-5
56. Pariollaud M, Gibbs J, Hopwood T, Brown S, Begley N, Vonslow R, et al. Circadian clock component REV-ERBα controls homeostatic regulation of pulmonary inflammation. J Clin Invest (2018) 128:2281–96. doi: 10.1172/jci93910
57. Pourcet B, Zecchin M, Ferri L, Beauchamp J, Sitaula S, Billon C, et al. Nuclear receptor subfamily 1 group d member 1 regulates circadian activity of NLRP3 inflammasome to reduce the severity of fulminant hepatitis in mice. Gastroenterology (2018) 154:1449–1464.e20. doi: 10.1053/j.gastro.2017.12.019
58. Mao F-Y, Lv Y-P, Hao C-J, Teng Y-S, Liu Y-G, Cheng P, et al. Helicobacter pylori-induced rev-erbα fosters gastric bacteria colonization by impairing host innate and adaptive defense. Cell Mol Gastroenterol Hepatol (2021) 12:395–425. doi: 10.1016/j.jcmgh.2021.02.013
59. Griffin P, Dimitry JM, Sheehan PW, Lananna BV, Guo C, Robinette ML, et al. Circadian clock protein rev-erbα regulates neuroinflammation. Proc Natl Acad Sci (2019) 116:201812405. doi: 10.1073/pnas.1812405116
60. Pastore N, Vainshtein A, Herz NJ, Huynh T, Brunetti L, Klisch TJ, et al. Nutrient-sensitive transcription factors TFEB and TFE3 couple autophagy and metabolism to the peripheral clock. EMBO J (2019) 38:e101347. doi: 10.15252/embj.2018101347
61. Chandra V, Bhagyaraj E, Nanduri R, Ahuja N, Gupta P. NR1D1 ameliorates mycobacterium tuberculosis clearance through regulation of autophagy. Autophagy (2015) 11:1987–97. doi: 10.1080/15548627.2015.1091140
62. Sahu SN, Anriany Y, Grim CJ, Kim S, Chang Z, Joseph SW, et al. Identification of virulence properties in salmonella typhimurium DT104 using caenorhabditis elegans. PloS One (2013) 8:e76673. doi: 10.1371/journal.pone.0076673
63. Tenor JL, Aballay A. A conserved toll-like receptor is required for caenorhabditis elegans innate immunity. EMBO Rep (2008) 9:103–9. doi: 10.1038/sj.embor.7401104
64. Zhou M, Liu X, Yu H, Yin X, Nie S-P, Xie M-Y, et al. Cell signaling of caenorhabditis elegans in response to enterotoxigenic escherichia coli infection and lactobacillus zeae protection. Front Immunol (2018) 9:1745. doi: 10.3389/fimmu.2018.01745
65. Dasgupta M, Shashikanth M, Gupta A, Sandhu A, De A, Javed S, et al. NHR-49 transcription factor regulates immunometabolic response and survival of caenorhabditis elegans during enterococcus faecalis infection. Infect Immun (2020) 88:e00130-20. doi: 10.1128/iai.00130-20
66. Nhan JD, Turner CD, Anderson SM, Yen C-A, Dalton HM, Cheesman HK, et al. Redirection of SKN-1 abates the negative metabolic outcomes of a perceived pathogen infection. P Natl Acad Sci USA (2019) 116:22322–30. doi: 10.1073/pnas.1909666116
Keywords: Caenorhabditis elegans, Staphylococcus aureus, nuclear receptor (NR), TFEB, host response, infection, innate immunity, lipid droplets (LD)
Citation: Goswamy D, Gonzalez X, Labed SA and Irazoqui JE (2023) C. elegans orphan nuclear receptor NHR-42 represses innate immunity and promotes lipid loss downstream of HLH-30/TFEB. Front. Immunol. 14:1094145. doi: 10.3389/fimmu.2023.1094145
Received: 10 November 2022; Accepted: 27 January 2023;
Published: 13 February 2023.
Edited by:
J. Louise Lines, Dartmouth College, United StatesReviewed by:
Tania Del Rivero, Organicell Regenerative Medicine Inc, United StatesCopyright © 2023 Goswamy, Gonzalez, Labed and Irazoqui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Javier E. Irazoqui, amF2aWVyLmlyYXpvcXVpQHVtYXNzbWVkLmVkdQ==
†Present address: Sid A. Labed, EvoDenovo Inc., Woburn, MA, United States
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.