
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 15 February 2023
Sec. B Cell Biology
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1093208
This article is part of the Research TopicRegulatory B cells in gastrointestinal diseasesView all 4 articles
 Luyao Wen1
Luyao Wen1 Bei Zhang1Xinfeng Wu1
Bei Zhang1Xinfeng Wu1 Rongzeng Liu2Hua Fan3Lei Han1Zhibo Zhang1Xin Ma1Cong-Qiu Chu4Xiaofei Shi1*
Rongzeng Liu2Hua Fan3Lei Han1Zhibo Zhang1Xin Ma1Cong-Qiu Chu4Xiaofei Shi1*Systemic lupus erythematosus (SLE) is an autoimmune illness marked by the loss of immune tolerance and the production of autoantibodies against nucleic acids and other nuclear antigens (Ags). B lymphocytes are important in the immunopathogenesis of SLE. Multiple receptors control abnormal B-cell activation in SLE patients, including intrinsic Toll-like receptors (TLRs), B-cell receptors (BCRs), and cytokine receptors. The role of TLRs, notably TLR7 and TLR9, in the pathophysiology of SLE has been extensively explored in recent years. When endogenous or exogenous nucleic acid ligands are recognized by BCRs and internalized into B cells, they bind TLR7 or TLR9 to activate related signalling pathways and thus govern the proliferation and differentiation of B cells. Surprisingly, TLR7 and TLR9 appear to play opposing roles in SLE B cells, and the interaction between them is still poorly understood. In addition, other cells can enhance TLR signalling in B cells of SLE patients by releasing cytokines that accelerate the differentiation of B cells into plasma cells. Therefore, the delineation of how TLR7 and TLR9 regulate the abnormal activation of B cells in SLE may aid the understanding of the mechanisms of SLE and provide directions for TLR-targeted therapies for SLE.
Systemic lupus erythematosus (SLE) is a common autoimmune disease that is defined by the inappropriate activation of self-reactive T and B cells and the production of autoantibodies and immune complexes that can cause irreparable organ damage (1). The variety of SLE symptoms make both diagnosis and treatment difficult. Consequently, it is of great significance to further explore the mechanism underlying the pathogenesis of SLE.
B lymphocytes perform various functions in SLE, including autoantibody production, presentation of antigen to T cells, and activation of myeloid cells to produce various cytokines (2–4). B cells can be activated by diverse factors, including Toll-like receptors (TLRs). TLRs are pathogen pattern recognition receptors that sense conserved microbial components (5). TLRs are crucial for both the development of innate immune responses and adaptive immunity, and they are thought to bridge the innate and adaptive immune systems (6). Studies show that overactivation or impaired function of the TLRs that detect nucleic acids, TLR7 and TLR9, induces dysfunction of innate immunity and breakdown of immune tolerance, which can lead to the occurrence of autoimmunity (7, 8). Both TLR7 and TLR9 are strongly associated with interferon (IFN) production by plasmacytoid dendritic cells (pDCs) (9). In addition, they are also expressed in B cells, wherein they play crucial functions (10). Surprisingly, TLR7 and TLR9 appear to play opposing roles in SLE B cells (11). TLR7 contributes to the loss of germinal centre (GC) tolerance and drives the extrafollicular B-cell response implicated in SLE (12). Critically, TLR9 in B cells seems to protect against SLE even though it is needed to produce anti-double-stranded DNA (dsDNA) antibodies (13). In fact, TLR9 has a complex dual regulatory mechanism in B cells, which can both maintain immune tolerance and participate in autoimmunity (14). This may be due to the specific presence of signalling pathways in B cells (such as BCR, CD19 and related molecules)that play a role in these processes (14, 15). In addition, the participation of co-stimulatory molecules, pro-inflammatory factors and MyD88 (which is a key molecule downstream of TLR9) also determines the fate of B cells (16, 17). Additionally, pDCs and T cells can accelerate the progression of SLE by releasing cytokines to reinforce the sensitivity of TLR7 in B cells (18–21). These findings suggest that the onset of SLE is likely significantly influenced by dysregulation of TLR7 and TLR9 signalling in B cells. However, the findings of functional and genetic studies of TLR7 and TLR9 in SLE remain controversial.
In this review, the contribution of intracellular TLR signalling in B lymphocytes to SLE is discussed. First, we present the expression levels and functions of TLR7 and TLR9 in B cells during the development of SLE. Second, we emphasize that downstream signalling pathways connected to B cell proliferation and differentiation in SLE are activated by TLR7 and TLR9 in B cells. Third, we discuss the opposing effects of TLR7 and TLR9 in B cells and their interactions in SLE. Fourth, we discuss how pDCs and T cells accelerate the conversion of B cells into plasma cells in patients with SLE through TLR signalling. Finally, we look ahead to potential strategies for targeting TLRs to treat SLE.
Earlier studies have shown that the TLR7 gene is involved in SLE (22–24). The TLR7 gene is located on human chromosome Xp22.3. Interestingly, unlike other genes on the X chromosome, TLR7 is able to evade silencing in females (25). Therefore, it has a higher level of expression in monocytes, pDCs and B cells in females than in males (26). Similar to the case in humans, this female-biased increased expression of X-linked genes is also present in mice susceptible to autoimmune diseases (27). In addition, TLR7 may be coexpressed with other X-linked genes (e.g., CXorf21) to jointly promote the occurrence of autoimmune diseases with gender bias (28). With the increased use of genome-wide association studies (GWAS), researchers have found that SLE risk and clinical phenotype and the production of autoantibodies in SLE are influenced by TLR7 single-nucleotide polymorphisms (SNPs) (29, 30). This finding partly explains the heterogeneity of SLE. With the rapid development of gene sequencing technologies, TLR7 mRNA has been found to be upregulated in the peripheral blood mononuclear cells of SLE patients by using bioinformatics analysis (31). Another investigation found that the expression of TLR7 in CD19+ B cells in SLE patients was markedly elevated compared to that in healthy individuals (32). Another study showed that SLE patients had expansion of CD19+CD24hiCD38hi transitional B cells and increased release of autoantibodies due to overexpression of TLR7 (33). Similarly, transitional T1 B cells expansion and autoantibodies production can be found when overexpression of TLR7 in transgenic mice (34). In addition, Tlr7 transgenic mice spontaneously manifest SLE-like illnesses in a B-cell-dependent manner (35). Furthermore, Hwang et al. (36) demonstrated that upregulation of TLR7 in SLE-prone mice induces B-cell marginal zone impairment, elevated levels of antibodies targeting RNA/protein complexes in B cells, and progression of disease. Brown et al. (37) reported that a child with serious lupus had a novel de novo missense mutation in TLR7 (TLR7Y264H). The variant selectively enhances sensing of guanosine and 2’,3’-cyclic guanosine monophosphate (2’,3’-cGMP) and triggers lupus-like disease when introduced in mice (generating so-called kika mice). Further studies have found that the Age-associated B cells (ABCs) of kika mice have higher TLR7 expression than those of WT mice (but lower TLR7 expression than Tlr7-double-positive Yaa mice). Nevertheless, the effects of the Y264H mutation were more serious than those of mutations of the Yaa allele, with kika mice showing higher levels of autoantibodies (37). Moreover, when wild-type (WT) mice of diverse genetic backgrounds were treated with the TLR7 agonists imiquimod and R848 for 4 weeks, they developed systemic autoimmune disease accompanied by an increased titre of autoantibodies against nucleic acids and multiple organ damage, including glomerulonephritis, carditis, hepatitis, and photosensitivity (38). According to additional research, focusing on the TLR7 pathway in lupus-prone models may provide insights for preventing the onset of serious disease (39). The above studies show that TLR7 promotes the occurrence and development of SLE at the genetic and molecular levels. These findings suggests that TLR7 is a candidate for targeted therapy for SLE.
TLR7 contributes to the loss of germinal centre tolerance and drives the extrafollicular B-cell response implicated in SLE (Figure 1). In the periphery, GCs are a crucial place where B-cell–tolerance is assessed (40). Studies have shown that the GC response and production of antibodies are significantly decreased in Tlr7-null mice B cells upon RNA virus infection (41). Consistent with these findings, TLR7 in B cells is essential for GC production in the BWAS−/− model, in which B cells lack the Wiskott-Aldrich syndrome protein (42). In another mouse model with two copies of Tlr7, GCs fail to eliminate markedly autoreactive B cells, and these cells undergo subsequent clonal expansion and somatic mutation and then enter the plasma-cell compartment (43). This finding suggests that B-cell-intrinsic TLR7 can disrupt GC tolerance. Another study showed that B6.Sle1b mice treated with a TLR7 agonist have increased numbers of spontaneous germinal centres (Spt-GCs) (44). Conversely, Tlr7 deficiency prevented the formation of Spt-GCs in autoimmune B6.Sle1b mice and reduced the production of autoantibodies (44). Additionally, a lack of TLR7/MyD88 impairs B-cell proliferation and survival after stimulation compared to B6 controls (44). These results demonstrate that the development of Spt-GCs and elevated autoantibody production in SLE are mediated by B-cell-intrinsic TLR7 signalling.
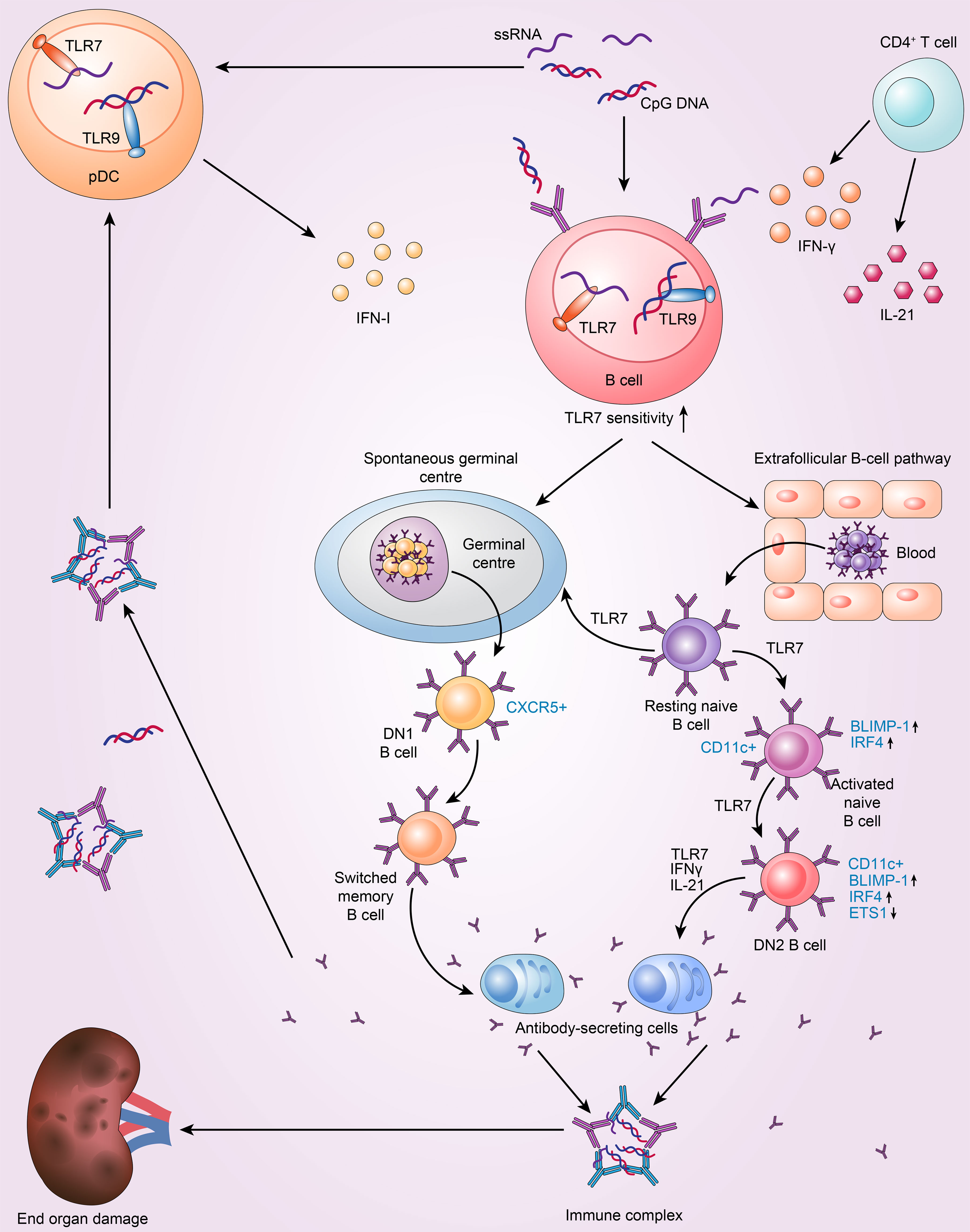
Figure 1 TLR7 drives the extrafollicular B-cell response and the loss of germinal centre (GC) tolerance, which is implicated in the pathogenesis of SLE. 1. Resting naive B cells enter the germinal centre pathway, which generates DN1 B cells and memory B cells, and then differentiate into antibody-secreting cells. The above processes are dependent on TLR7. 2. TLR7 drives the extrafollicular pathway, in which resting naive B cells successively become activated naive B cells, DN2 subset B cells and finally antibody-secreting cells. 3. pDC and T cells intensify TLR7 sensitivity to drive autoreactive B-cell development into antibody-secreting cells by secreting IFN-I, IFN-γ and IL-21.
Furthermore, in cases of active SLE, TLR7 can promote extrafollicular B-cell differentiation, causing resting naive B cells to constantly differentiate into activated naive B cells, DN2 B cells, and ultimately Ab-secreting cells (12). Individuals with SLE have higher levels of activated naive B cells(CD11c+IgD+CD27−CD21−MTG+CD23−) and the DN2 subset of IgD−CD27− double-negative B cells (DN2 B cells; IgD−CD27−CD11c+T-Bet+CD69+CD21−CD24−CD38−CXCR5−FCRL4−FCRL5+) (45). Moreover, SLE patients with increased abundance of DN2 B cells have more anti-RNA and anti-Sm/RNP autoantibodies and exhibit higher disease activity (12). DN2 cells are the predominant B-cell population in active SLE; they tend to develop into plasma cells and exhibit high levels of CD11c and T-Bet expression but low levels of CXCR5 and CD62L expression (12). Interestingly, the phenotype of DN2 cells is distinct from that of activated naive B cells. The two cell types share a similar transcriptional profile, with only 42 differentially expressed genes between them (12, 46). Furthermore, DN2 B cells are thought to be derived from activated naïve B cells (12). B lymphocyte-induced maturation protein 1 (BLIMP-1) and IFN regulatory factor 4 (IRF4) levels are higher in these two types of cells and control plasma cell differentiation and the expression of transcription factors (TFs) related to plasmablasts (47). DN2 cells express low levels of ETS proto-oncogene 1 (ETS1), a CD22-regulated TF that inhibits plasma cell differentiation (48). These features result in the accumulation of extrafollicular autoreactive B cells (49). SLE DN2 cells are hyperresponsive to TLR7 agonists, and TLR7-mediated upregulation of costimulatory molecule expression and suppression of the expression of CD72 (a receptor) inhibit the activation of endosomal TLR7 (50). Moreover, chronic TLR7 activation in lupus models is sufficient to cause CD11c+ B cells to differentiate and produce anti-Sm/RNP autoantibodies, whereas B-cell intrinsic TLR7 deficiency reduces the number of CD11c+ B cells and ameliorates lupus-like conditions (42). Indeed, TLR7 possesses stimulatory action in these autoreactive B-cell subsets and, in both humans and animals, can cause naive B-cell differentiation towards DN2 cells and plasma cells with the help of IL-21 and IFNγ (47, 51). In SLE patients, IL-21 promotes the growth of CD11c+T-Bet+CD21−CD38−B cells, which resemble DN2 B cells and are autoreactive and associated with clinical symptoms (18). Furthermore, TLR7 and B-cell receptor (BCR) agonists cooperate with IFNγ, IL-2, IL-21 and B-lymphocyte stimulator (BlyS) to stimulate naive human B cells in SLE, and an in vitro study reported that the produced IgD−CD27−CD11c+T-BethiCD21−CXCR5−IRF4intFcRL5+ B cells resemble DN2 B cells (52). Age-associated B cells (ABCs), the mouse equivalent of DN2 B cells, are another crucial B-cell subpopulation in SLE. These CD11b+CD11c+T-Bet + cells are increased in various mouse models of lupus, including Mer−/− and SWEF-deficient mice; these cells are also especially sensitive to TLR7 and differentiate into autoantibody-secreting cells (53–55). Another study found that ABCs in C57BL/6 mice can also present antigens to T cells (56). Specifically, the Tlr7-null lupus-prone mouse model lacks CD11c+T-Bet+CD21− B cells. Moreover, with repeated TLR7 agonist stimulation, ABCs accumulate rapidly in mice based on a specific TLR7 signalling pathway in B cells (51). Brown et al. (37) identified that even though there was marked Spt-GC formation in Tlr7Y264H mice, the lupus-like symptoms did not resolve with GC formation deficiency, thus indicating that extrafollicular ABCs may be the main reason for pathogenicity (37). In summary, the GC reactions and extrafollicular responses lead to B cell activation and the formation of plasma cells via B-cell-intrinsic TLR7 signalling in SLE. Additionally, TLR7 signalling restricts the proliferation of B10 cells, a regulatory B-cell subpopulation that generates the immunosuppressive cytokine IL-10, in an IFNγ-dependent manner (57).
TLR9 is a receptor that recognizes viral and bacterial DNA containing unmethylated cytosine–phosphate–guanine (CpG) motifs (58). One study showed that anti-dsDNA antibody levels and proteinuria in childhood-onset lupus nephritis (LN) are associated with the expression of TLR9. This result suggests that TLR9 is involved in the pathophysiology of LN (59). Consistent with these results, investigation has demonstrated that whole blood TLR9 mRNA levels are considerably greater in SLE patients than in healthy controls and are associated with the severity and type of renal pathology (60). Furthermore, the levels are positively correlated with SLEDAI grade, anti-dsDNA antibody titre, and interleukin-6 (IL-6) and other cytokine levels but negatively correlated with complement C3 levels (61). Notably, during a two-year follow-up period, patients with poor prognoses still had elevated TLR9 mRNA levels (62). Compared with the levels at SLE onset, the levels of TLR9 mRNA in patients with a favourable prognosis were obviously decreased (62). Strikingly, studies show that Tlr9 deficiency in B cells in murine models of lupus is sufficient to accelerate renal disease even though deplete anti-nucleosome antibodies. Moreover, overexpression of B-cell–specific Tlr9 induces remission of nephritis (13). In contrast, Tlr9 deficiency in other cell types, including pDCs, dendritic cells, and neutrophils, has no obvious effect on LN (13). These lines of evidence suggest that TLR9 in B cells plays a specialized role in restricting the formation of autoantibodies and preventing the onset of disease. That the expression of TLR9 in SLE is proportional to disease activity appears to contradict its protective function, suggesting a unique and complex role of TLR9 in SLE B cells.
Although B cells are crucial to the pathophysiology of SLE, the aberrant pathways that result in the loss of B-cell tolerance are unknown. A significant fraction of autoreactive B cells are eliminated throughout development (63), but self-reactive and polyreactive clonotypes still make up a significant portion of mature B-cell pools (40, 64). This indicates that there may be activation-associated checkpoints that can prevent these cells from differentiating into memory B cells and producing antibodies targeting self-antigens. However, patients with SLE likely have impaired peripheral and central B-cell tolerance checkpoints, leading to an accumulation of autoreactive B cells in their blood (65). TLR9 is expressed in the spleen and skeletal muscle in mammals and in peripheral blood mononuclear cells, mainly in pDCs and B cells. However, the biological effects are different in these cells. Endogenous or exogenous nonmethylated CpG-DNA can be recognized by BCR and internalized into B cells, and the process may involve phosphatidylinositol 3-kinases (PI3Ks) (66). Subsequently, the CpG-DNA is transported into early endosomes and ultimately transported into late endosomes containing TLR9, by which it is specifically identified, activating downstream signalling pathways and triggering inflammatory responses and immune effects (66). TLR9 can also promote B cell differentiation into plasma cells through germinal centre (GC) formation and extrafollicular B-cell response. However, unlike TLR7, TLR9 has a complex role in B cell differentiation and is affected by multiple elements. This results in TLR9 acting as both an immune checkpoint to maintain peripheral and central tolerance and to participate in autoimmunity (14). Rookhuizen and DeFranco discovered that TLR9-MyD88-dependent signalling in B cells increased GC output by increasing affinity maturation, the production of memory antibodies, a transition to the IgG2a subclass of antibodies and by specifically producing high-affinity antibodies (67). Additionally, the combined ligation of TLR9 and BCR has also been demonstrated to activate autoreactive B cells in previous studies (68–71). Moreover, these activated autoreactive B cells develop into extrafollicular plasma cells that can facilitate the generation of IgG2 isotype autoantibodies (72, 73). Eckl-Dorna and Batista (74) found that for both in vitro and in vivo studies, Ag-CpG conjugates are effective at promoting B cell proliferation and differentiation into short-lived extrafollicular plasma cells via intrinsic TLR9. In addition, ABCs do not respond to BCR ligation alone, but they quickly multiply when TLR9 is activated in aged mice (75). Activated B cells are prepared for an ABC fate by B cell-intrinsic TLR9 signals, but they also initiate a cell death program (53). Cancro’s group (14, 76) have posited that antigen taken up by the BCR is internalized and interacts with innate molecular pattern sensors. If TLR9 is activated (either alone or in conjunction with TLR7), the cell is also primed to adopt an effector or memory T-Bet+ ABC fate. However, the default pathway is cell cycle arrest and apoptosis, unless it is avoided by prosurvival signals or costimulation via cognate T cell assistance. In reality, studies have revealed that BCR can bind to TLR9 agonists on all premature murine B-cell subsets, and CD27– human B cells can undergo apoptotic death after an initial proliferative burst that involves hyperphosphorylation of nuclear factor-κB (NF-κB) and p38 mitogen-activated protein kinase (MAPK) (16, 77). Moreover, p38 inhibits the persistent expansion and the survival of autoreactive B cells by inducing G1 cell cycle arrest and mitochondrial apoptosis (78, 79). However, B cells going through this stage can be saved, and the way they are saved influences what happens to them thereafter (16). In fact, impaired TLR9 function, BlyS stimulation, cognate T-cell-mediated effects or CD40 costimulation with IL-21 or IFN-γ can prolong the lifespan of autoreactive B cells (80, 81). Interestingly, a study using CpG to stimulate B cells and pDCs isolated from SLE patients who had not received hydroxychloroquine treatment found decreased expression of both B-cell activation molecules and numerous cytokines in B cells (15). However, normal reactions appear in pDCs from SLE patients; therefore, it appears that there is defective TLR9 function in SLE B cells (15). This impaired TLR9 function might prevent SLE-related autoreactive B-cell death and, consequently, harmful autoantibody generation.
All TLRs in B cells trigger similar signalling pathways that in turn activate NF-κB, MAPKs, extracellular signal-modulating kinase (ERK), p38, and Jun N-terminal kinase (JNK) (82). The common linker protein used by almost all TLRs is MyD88, which has death and toll interleukin 1 receptor (TIR) domains (83). When MyD88 binds to a TLR, it recruits members of the IL-1 receptor-associated kinase (IRAK) family, IRAK1 and IRAK4, by interacting with the death domains, thereby phosphorylating TNF receptor correlator 6 (TRAF6) (84). TRAF6 is phosphorylated by IRAKs and binds to Ubc13 and Uev1A to form a complex that activates a mitogen-activating protein kinase kinase (MAPKKK) called transforming growth factor β-activating kinase-1 (TAK-1). Activated TAK-1 phosphorylates MKKs, which are upstream kinases of p38 MAPKs and JNK (85). p38 is involved in many cellular physiological and pathological processes, including apoptosis, cellular stress, the cell cycle, and the inflammatory response (86). In addition, TAK-1 can activate the IκBα kinase (IKK) complex, consisting of the IKKα and IKKβ kinases and the hinge protein IKKγ (85). Phosphorylation of IκBα leads to its own degradation and the release of NF-κB (Figure 2). The NF-κB signalling pathway is critical in innate and adaptive immunity because it modulates the transcription of various immune-inflammatory genes, such as TNF, IL-1, and IL-6, resulting in chemotaxis, granulocyte and macrophage aggregation, and lymphocyte infiltration (84, 87, 88). Although the downstream pathways triggered by many TLRs are similar, TLR7 and TLR9 perform different functions in controlling the behavior of SLE B cells. We therefore hypothesize that in SLE, B-cell-specific signalling impacts the response of TLRs.
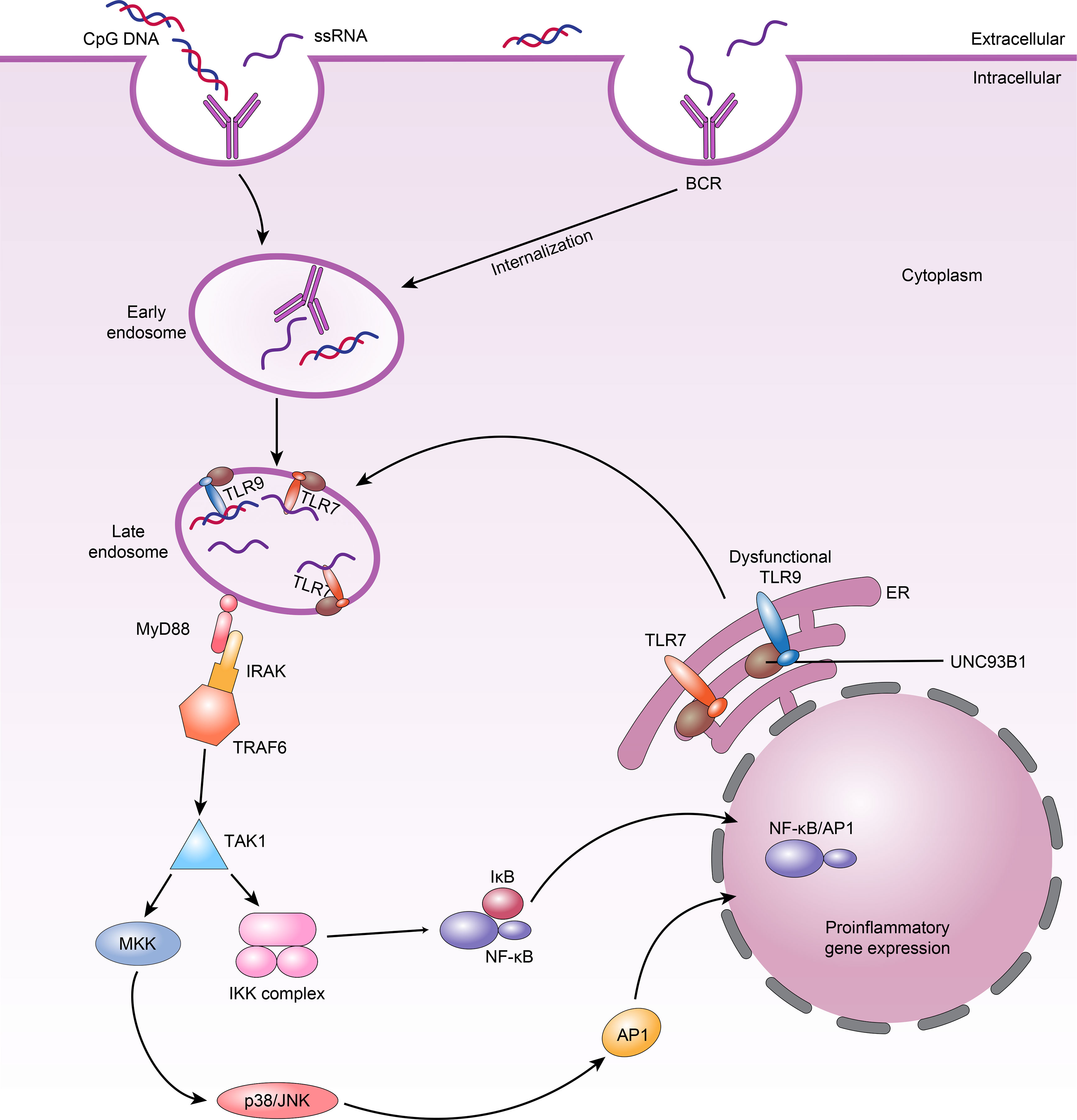
Figure 2 TLRs signalling in B cells and TLR7 and dysfunctional TLR9 compete for UNC93B1-dependent trafficking in SLE B cells. 1.When nucleic acid ligands are recognized by BCRs and internalized into B cells, they bind TLR7 or TLR9 in the late endosome; TLR7 or TLR9 then binds MyD88. MyD88 subsequently recruits the IRAK–TRAF6 complex, ultimately leading to activation of downstream NF-κB and AP1 signalling. 2.Dysfunctional TLR9 can lead to weakened competition of these TLRs for binding to UNC93B1 in endosomes, increasing TLR7 availability and resulting in higher TLR7 trafficking and responses, which manifest as autoimmune phenotypes.
B cells, in contrast to dendritic cells (DC), do not internalize external substances via microcytosis or endocytosis, so natural extracellular nucleic acids cannot directly contact TLR7 and TLR9 inside B cells. Instead, these TLRs are activated by BCR-antigen complexes that are delivered to late endosomes, where they trigger the costimulation of B cells (74, 89). This suggests that specific TLR-driven cell activation is associated with the expression of unique BCRs on B cells. In addition, mouse B cells that lack the BCR are unable to proliferate upon TLR agonist stimulation (90). Moreover, LAG-3+CD138hi natural regulatory plasma cells, which produce IL-10 upon exposure to TLR agonists in mice, also grow in a BCR-dependent manner (91). These studies reaffirm that BCR activation facilitates endosomal TLR signalling in B cells, enabling these cells to react to TLR agonists (92).
The molecular changes responsible for impaired TLR9 activity in SLE B cells are unclear. TLR9 gene transcription is similar in B cells of both healthy individuals and SLE patients (15, 93). Hence, poor TLR9 responses in SLE B cells is unlikely due to decreased TLR9 expression (93). In SLE patients, TLR9 function is impaired in B cells but not in pDCs, indicating that B-cell-specific receptors may disturb the TLR9 response in SLE B cells. Several TLR9 actions in human B cells are mediated by CD19, which is expressed in B cells but not pDCs (15). CD19 is a transmembrane protein found on B cells that amplifies proximal BCR signalling by successively recruiting and activating LYN, PI3K, Bruton tyrosine kinase (BTK), and protein kinase B (AKT) (94). Likewise, B cells from individuals with one or two CD19 allele variants exhibit lower or defective activation, respectively, in response to TLR9 agonist stimulation (94). This suggests that CD19 is indispensable for the control of TLR9 function in human B cells. Human SLE B cells have been shown to express less CD19 according to prior studies (95–97). In conjunction with this observation, Gies et al. (15) found that CD19 expression was lower on B cells from quiescent and active SLE patients. Furthermore, it has been demonstrated that the transitional B cells from individuals with SLE have abnormal phenotypes, including lower CD19 expression and impaired sensitivity to TLR9 stimuli (98). Previous studies have also shown that heterozygous and homozygous CD19 gene mutations in B cells may be associated with the onset of SLE (99). However, such mutations are unlikely to frequently occur in SLE to alter the expression of CD19. Gies et al. (15) discovered that decreased CD19 expression on B cells from SLE patients normalized after 48 hours of in vitro culture, therefore, it is assumed that external factors are responsible for CD19 downregulation in vivo. Subsequent research has revealed that downregulation of CD19 on B cells is associated with the existence of anti-dsDNA antibodies, suggesting that immune complexes and apoptotic bodies containing DNA are involved in CD19 and TLR9 regulation in B cells in the bone marrow (BM) (98). By contrast, type I and II interferons, which are linked to the immunopathogenesis of SLE, are unable to modify CD19 (15). Primary Sjögren’s syndrome, a condition marked by the generation of anti-single-stranded RNA (ssRNA) antibodies but not anti-dsDNA antibodies, is characterized by ordinary CD19 expression on patient B cells (15). Consistent with this finding, DNA is found on the surface of B cells from SLE patients who are actively ill (but not on the surface of healthy donor cells) (100). However, whether CD19 downregulation in SLE B cells is a necessary factor for impaired TLR9 responses and leads to a break in immune tolerance in SLE B cells remains to be determined. Although both TLR7 and TLR9 activate downstream NF-κB and p38 MAPK signalling through MyD88 in B cells, data suggest that TLR7 does not promote B-cell death after proliferation in SLE in vivo and in vitro, but it does promote plasma cell differentiation (77). Interestingly, TLR7 function is preserved in SLE B cells, and the reasons for this effect are worth further investigations (98).
TLR9 dysfunction leads to a decrease in the differentiation of B cells into regulatory B cells (Bregs). Bregs are a class of immunomodulatory B cells that do not rely on the secretion of immunoglobulins (101). Upon stimulation by the TLR9 agonist CpG-DNA, these cells can secrete the cytokine IL-10 and effectively induce apoptosis of mouse and human effector T cells in an IL-10-dependent manner, which has a protective effect on the body under physiological conditions (57, 102). The TLR9 hyporesponsiveness of SLE B cells may underlie their inability to differentiate into IL-10-secreting Bregs (103). Diminished IL-10 secretion may then promote an increase in IFN-α production by pDCs from SLE patients. These pDCs are probably activated through their functional TLR9 by DNA-containing immune complexes, and this step may favour the progression of immature SLE B cells into plasmablasts, which frequently create autoreactive antibodies (15, 104). On the other hand, TLR9-deficient cells tend to differentiate into autoantibody-producing cells with expression of CD138 or other molecular markers of plasma cells in vivo and in vitro (77).
Nündel et al. (77) found that even though Tlr9-deficient autoimmune disease-prone mice fail to generate an autoantibody response to dsDNA, the Tlr9-deficient cells continue to differentiate along the plasma cell lineage, and these mice develop more serious clinical disorders and have shorter lifespans. However, Tlr7-null and Tlr7/9 double-null autoimmune disease-prone mice exhibit less severe disease. Furthermore, Tlr9−/− B cells predominantly become IgG autoantibody-producing cells in vivo when BCR/TLR7 is activated. Additionally, the functions of BCR/TLR7 may be at least partially limited by BCR/TLR9 activation (77). The dependence of protection on TLR7 elimination indicates that there may be direct interaction between TLR7 and TLR9 and that the negative influence of TLR7 on health is greater than the positive influence of TLR9. In fact, the transmembrane protein UNC93B1 plays a key role during the activation of TLR7 and TLR9 by trafficking them from the endoplasmic reticulum (ER) to late endosomes, where they communicate with their ligands (105). B cells from patients with active SLE have considerably higher UNC93B1 mRNA and protein levels than B cells from patients with inactive SLE and healthy controls (106). Further research found that knocking out Unc93b1 extinguishes the response from TLR7 and TLR9 in B cells, and the levels of antibodies against Ro-52/60, La, DNA and cardiolipin are all apparently decreased in the KO mice (10, 107, 108). In addition, there is competition between TLR7 and TLR9 for UNC93B1 trafficking in B cells, in which TLR9 predominates due to its higher affinity for UNC93B1 (109). Thus, overactivation of TLR7 is regulated by UNC93B1 through balancing of TLR9 to TLR7 trafficking. Desnues et al. (110) showed that dysfunctional TLR9 can lead to weakened competition of these TLRs for binding to UNC93B1 in endosomes. This improves TLR7 availability and causes increased TLR7 trafficking and response, which can emerge as autoimmune symptoms (Figure 2). Significantly, it has also recently been shown that UNC93B1 can specifically suppress TLR7 signalling in B cells by recruiting syntenin-1 (111). Syntenin-1 binds to UNC93B1, causing TLR7 to move to intraluminal vesicles of multivesicular bodies and preventing TLR7 signalling (111).
However, impaired TLR9 does not seem to explain the exacerbation of the disease caused by Tlr9-KO, suggesting that the protective mechanisms of TLR9 in SLE involve non-classical pathways. Recent genetic studies of TLR9 functions in lupus uncovered complex regulatory and unclear proinflammatory functions of TLR9. Leibler et al. (17) constructed a murine model of lupus with TLR9 point mutations that either hinder ligand binding(Tlr9K51E) or MyD88 signalling (Tlr9P915H) and subsequently detected the effects of these mutations on the lupus phenotype. A scaffold protective role of TLR9 was identified in Tlr9K51E mice, which showed less severe disease than Tlr9−/−control mice. This scaffolding role is dependent on the protein’s presence and supports the UNC93B1 hypothesis, which suggests that TLR7 endosomal localization should differ in Tlr9−/− and Tlr9WT cells (13, 112). However, there was not a marked difference in TLR7 endosomal or lysosomal localization, NF-κB signalling or TLR7-driven gene expression in mice with or without TLR9 (17). Despite the findings of Leibler et al. (17), studies have been unable to exclude a subtle impact of TLR9 expression on TLR7 activation and signalling, though this impact may not be a major reason why TLR9 restrains lupus. This calls into question the traditional conception that TLR9 only competitively restrains the signalling and expression of TLR7. Unexpectedly, compared with Tlr9K51E and Tlr9WT mice, Tlr9P915H mice exhibited greater protection, demonstrating that TLR9 also exerts a ligand-dependent but MyD88-independent control effect. Additionally, neither of the two abovementioned suppressive, MyD88-independent regulatory roles of TLR9 rely on the absence of anti-DNA antibodies, as neither of these mice produce anti-DNA antibodies, yet lupus is substantially ameliorated in Tlr9K51E and Tlr9P915H mice compared with Tlr9−/− mice (17). Furthermore, studies of triple chimaeras with WT, KO and mutant TLR9 haematopoietic cells in the bone marrow uncovered TLR9-MyD88-independent regulatory functions that were intrinsic to B cells and constrained the development of pathogenic ABCs and plasma cells (17). In addition, although Leibler et al. (17) were unable to exclude the possibility of regulatory anti-inflammatory functions of TLR9–MyD88 signalling, they did identify that the major impact of TLR9–MyD88 signalling is to accelerate disease through proinflammatory signalling. That TLR9 possesses unique regulatory functions and operates via proinflammatory and protective mechanisms that could explain why it differs from TLR7. In addition, BCR/TLR co-signalling, in which both ligands can be present in a single molecular complex, suggests that there are different roles for TLR7 and TLR9, and TLR9 may have unique roles in regulating BCR/TLR co-signals (16, 77). Ultimately, TLR9 plays a more complex role than TLR7 in SLE B cells, but further exploration is required.
pDCs are a dominant producer of IFN-I. The secretion of IFN-I by pDCs involves the TLR7/9-MyD88 signalling pathway (19). SLC15A4, which is a histidine transporter that controls lysosomal pH, is necessary for this process and impacts the production of autoantibodies (113–116). IFN-I signalling has a crucial role in SLE pathogenesis and specifically in the germinal centre response (117). By specifically upregulating TLR7 expression, pDC-derived IFN-I increases the TLR7 sensitivity of human naive B cells. The level of TLR9 expression, however, is unaltered (118). Soni et al. (119) experiments with mice showed that endosomal TLR7/TLR9 and IFN-I derived from pDCs promote the transformation of extrafollicular B-cells into short-lived antibody-forming cells (AFCs). Moreover, the optimal expansion of extrafollicular (ExFO) CD138+ B cells and ICOShiExFO-Th cells requires IFN-I (119). In addition, T cells participate in the proliferation and differentiation of SLE B cells. CD4+ effector T cells, including follicular helper T (Tfh) cells, are the main sources of TLR7-induced IFN-γ. Furthermore, IFN-γ signalling in SLE plays an indispensable role in TLR7-driven autoreactive B-cell development into antibody-secreting cells via germinal centre signalling (20, 21). The differentiation of SLE B cells into plasma cells is facilitated by IL-21 secreted by Tfh cells (18, 120).Additionally, T-Bet and CD11c expression in TLR-activated B cells is controlled by an interaction between IL-4, IL-21, and IFN-γ (121).. In addition, Soni et al. (119) showed that extrafollicular plasmablasts drive the autoantibody response in a T-cell-dependent manner. These autoantibodies bind to antigens to form immune complexes. On the one hand, immune complexes can cause damage to organs; on the other hand, they can be recognized by pDCs to produce IFN-I, thus forming a vicious cycle (119). Taken together, these findings suggest connections between pDCs, T cells, B cells, and endosomal TLRs (Figure 1).
TLR7 is extremely important in the pathogenesis of SLE, so inhibitors of TLR7 may be a crucial component of SLE treatment. Indeed, some promising therapies for inhibiting signalling downstream of TLR7 are under clinical assessment. For example, several studies have demonstrated the safe and efficient use of an IRAK4 inhibitor in suppressing inflammatory gene expression and cytokine secretion in mouse and human peripheral blood mononuclear cells (122–124). Inhibitors of MyD88, IRAK1, TRAF6, BANK1 and TAK1, which are TLR7 signalling-related molecules, are also being developed (84, 125, 126). Notably, these approaches not only lack specificity for TLR7 over other endosomal TLRs but also may block the activation of various cell types in addition to B cells upon formation of SLE patient immune complexes or TLR7 ligand stimulation, including monocytes and pDCs. Therefore, it is essential to carefully assess the safety of these inhibitors. Recently, a new oral TLR7/8 inhibitor developed by Merck, KGaA in Germany known as enpatoran (M5049) was able to inhibit the production of IL-6 and IFN-α and is well tolerated in a dose-dependent manner in phase 1 clinical trials (127). It is expected that this drug can be used to treat SLE (128). Hydroxychloroquine (HCQ) plays an important role as an immunosuppressant in SLE and is widely used in the clinical treatment of autoimmune diseases. The mechanism includes direct binding to nucleic acids when they become concentrated in the endosome, which limits ligand binding to TLR7/9 (129). This leads to an impaired ability of pDCs from patients with SLE to produce IFN-α and TNF-α (130). However, given the therapeutic range and target population, HCQ may not be able to suppress immune storms once they progress because SLE is linked to multiple downstream pathways of TLR7/9 (131). In SLE, dysfunction of B-cell-intrinsic TLR9 prevents apoptosis of autoreactive B cells, so enhancing the TLR9 signal by upregulating CD19 may be a novel strategy for targeting TLR9 to treat SLE (13, 16, 98).
In addition to UNC93B1 and syntenin-1, the MHCII-associated invariant chain has been shown to have a more significant impact in restricting TLR7 signalling by regulating its trafficking (132). In addition, studies have found that in mice, B cells with αv or β3 integrin deficiency display hyporesponsiveness to TLR signalling in the marginal zone and B-1a B-cell subsets and a reduction in GC formation upon immunization with TLR7 stimuli (133, 134). Hence, modulation of the function of UNC93B1, syntenin-1, the MHCII-associated invariant chain or αvβ3 integrin to affect TLR7 trafficking mechanisms in SLE B cells is an interesting strategy for treatment (111, 132, 135).
TLRs were originally identified as pattern recognition receptors and are now considered an important hub connecting innate and adaptive immunity. SLE is characterized by successive stimulation of the innate and adaptive immune systems by endogenous nucleic acids released by necrotic or apoptotic cells. TLR7 and TLR9, whose ligands are ssRNA and dsDNA, respectively, work as innate sensors for detecting viral infections. Endosomal TLR recognition of self-nucleic acids in B cells is considered a crucial step in the progression of SLE and leads to the formation of antinuclear antibodies. Both TLR7 and TLR9 mRNA are expressed to varying degrees in SLE B cells, but the two play divergent roles. TLR7 is a significant contributor to the pathogenesis of SLE in B cells, mediating GC responses and extrafollicular B-cell responses to promote the expansion of antibody-secreting cells and accelerate disease progression. However, B-cell-specific TLR9 appears to play a protective role in SLE. Moreover, the conventional views hold that impaired TLR9 function in SLE B cells leads to a breakdown of the balance of the TLR7 and TLR9 pathways, which manifests as enhanced TLR7 signalling, impaired apoptotic death after an initial proliferative burst, and increased conversion of B cells to plasma cells instead of Bregs, which eventually worsens the disease. However, recent research has found that B-cell-intrinsic TLR9 seems to have complex bidirectional regulatory roles in SLE, and its expression does not affect TLR7 signalling (17). These novel findings reveal unprecedented insights into TLR9 biology in environments beyond SLE. This unique dual regulatory role of TLR9 may be related to Toll–IL-1R (TIR) domain signalling from IL-1R family members, which have both proinflammatory and anti-inflammatory functions (136). Only 46% homology exists between the TIR domains of TLR7 and TLR9, explaining why these receptors have different functions. This may partly explain why TLR7 and TLR9 have different roles in SLE B cells. Ultimately, genetic assessment of the TIR domains in each protein and exploration at the biochemical level are needed to determine such differences.
Infection is known to be an important trigger exacerbating SLE, and TLR-induced innate immunity can kill pathogens to maintain homeostasis (137, 138). However, abnormal activation of TLR7 or impaired TLR9 function in B cells in patients with SLE leads to disruption of immune homeostasis and adaptive immune activation upon exposure to pathogens containing ssRNA and dsDNA. Therefore, therapy targeting B-cell TLR7/TLR9 has potential for SLE. Some drugs targeting TLR7 have been developed and applied in the clinic; however, drugs targeting B-cell TLR9 are still relatively lacking. We propose a method to enhance the function of TLR9 by upregulating the expression of CD19 in B cells, thereby restoring the postproliferative apoptosis pathway of TLR9. This is expected to prevent disruption of immune homeostasis in SLE patients upon infection with pathogens containing ssRNA and dsDNA, providing new ways to target B-cell TLR9 to treat SLE. Curiously, the expression of TLR9 in B cells of patients with SLE is upregulated and positively correlated with SLE disease activity. This seems to contradict the protective effects of TLR9 and may be the result of a feedback mechanism that upregulates the expression of TLR9 to induce protection. However, this hypothesis needs to be validated. Likewise, whether TLR9 expression and/or function differs between humans and mice remains unclear.
It should be noted that SLE is a disease involving multiple factors, so the study of SLE pathogenesis cannot be limited to B cells. A large number of studies have indicated that cytokines such as IFN and ILs derived from pDCs and T cells, the expression of CD19 and BCR, and the interaction of transporters such as UNC93B1 and SLC15A4 with TLRs operate in a complex network that controls autoreactive antibody responses to nucleic acid antigens and results in the complex pathogenesis and symptom heterogeneity of SLE. However, the interactions of these factors are still unclear and need to be further explored.
LW drafted the manuscript. BZ revised and created some of the figures. XW, RL, HF, and LH read and revised the manuscript. ZZ and XM performed the literature search. C-QC participated in the revision of words and sentences. XS designed, revised and approved the final manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Key Project of Medical Science and Technology Research Program of Henan Province (SBGJ202002098).
We thank the Key Project of Medical Science and Technology Research Program of Henan Province for funding support.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Tsokos GC. Systemic lupus erythematosus. N Engl J Med (2011) 365:2110–21. doi: 10.1056/NEJMra1100359
2. Ruprecht CR, Lanzavecchia A. Toll-like receptor stimulation as a third signal required for activation of human naive b cells. Eur J Immunol (2006) 36:810–6. doi: 10.1002/eji.200535744
3. Jiang W, Lederman MM, Harding CV, Rodriguez B, Mohner RJ, Sieg SF. TLR9 stimulation drives naïve b cells to proliferate and to attain enhanced antigen presenting function. Eur J Immunol (2007) 37:2205–13. doi: 10.1002/eji.200636984
4. Lampropoulou V, Hoehlig K, Roch T, Neves P, Gómez EC, Sweenie CH, et al. TLR-activated b cells suppress T cell-mediated autoimmunity. J Immunol (2008) 180:4763–73. doi: 10.4049/jimmunol.180.7.4763
5. Radakovics K, Battin C, Leitner J, Geiselhart S, Paster W, Stöckl J, et al. A highly sensitive cell-based TLR reporter platform for the specific detection of bacterial TLR ligands. Front Immunol (2022) 12:817604. doi: 10.3389/fimmu.2021.817604
6. Fitzgerald KA, Kagan JC. Toll-like receptors and the control of immunity. Cell (2020) 180:1044–66. doi: 10.1016/j.cell.2020.02.041
7. Krieg AM, Vollmer J. Toll-like receptors 7, 8, and 9: linking innate immunity to autoimmunity. Immunol Rev (2007) 220:251–69. doi: 10.1111/j.1600-065X.2007.00572.x
8. Green NM, Marshak-Rothstein A. Toll-like receptor driven b cell activation in the induction of systemic autoimmunity. Semin Immunol (2011) 23:106–12. doi: 10.1016/j.smim.2011.01.016
9. Szumilas N, Corneth OBJ, Lehmann CHK, Schmitt H, Cunz S, Cullen JG, et al. Siglec-h-deficient mice show enhanced type I IFN responses, but do not develop autoimmunity after influenza or LCMV infections. Front Immunol (2021) 12:698420. doi: 10.3389/fimmu.2021.698420
10. Garimella MG, He C, Chen G, Li QZ, Huang X, Karlsson MCI. The b cell response to both protein and nucleic acid antigens displayed on apoptotic cells are dependent on endosomal pattern recognition receptors. J Autoimmun (2021) 117:102582. doi: 10.1016/j.jaut.2020.102582
11. Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity (2006) 25:417–28. doi: 10.1016/j.immuni.2006.07.013
12. Jenks SA, Cashman KS, Zumaquero E, Marigorta UM, Patel AV, Wang X, et al. Distinct effector b cells induced by unregulated toll-like receptor 7 contribute to pathogenic responses in systemic lupus erythematosus. Immunity (2018) 49:725–39.e6. doi: 10.1016/j.immuni.2018.08.015
13. Tilstra JS, John S, Gordon RA, Leibler C, Kashgarian M, Bastacky S, et al. B cell-intrinsic TLR9 expression is protective in murine lupus. J Clin Invest (2020) 130:3172–87. doi: 10.1172/jci132328
14. Johnson JL, Scholz JL, Marshak-Rothstein A, Cancro MP. Molecular pattern recognition in peripheral b cell tolerance: lessons from age-associated b cells. Curr Opin Immunol (2019) 61:33–8. doi: 10.1016/j.coi.2019.07.008
15. Gies V, Schickel JN, Jung S, Joublin A, Glauzy S, Knapp AM, et al. Impaired TLR9 responses in b cells from patients with systemic lupus erythematosus. JCI Insight (2018) 3:e96795. doi: 10.1172/jci.insight.96795
16. Sindhava VJ, Oropallo MA, Moody K, Naradikian M, Higdon LE, Zhou L, et al. A TLR9-dependent checkpoint governs b cell responses to DNA-containing antigens. J Clin Invest (2017) 127:1651–63. doi: 10.1172/jci89931
17. Leibler C, John S, Elsner RA, Thomas KB, Smita S, Joachim S, et al. Genetic dissection of TLR9 reveals complex regulatory and cryptic proinflammatory roles in mouse lupus. Nat Immunol (2022) 23:1457–69. doi: 10.1038/s41590-022-01310-2
18. Wang S, Wang J, Kumar V, Karnell JL, Naiman B, Gross PS, et al. IL-21 drives expansion and plasma cell differentiation of autoreactive CD11chiT-bet+ b cells in SLE. Nat Commun (2018) 9:1758. doi: 10.1038/s41467-018-03750-7
19. Barrat FJ, Meeker T, Gregorio J, Chan JH, Uematsu S, Akira S, et al. Nucleic acids of mammalian origin can act as endogenous ligands for toll-like receptors and may promote systemic lupus erythematosus. J Exp Med (2005) 202:1131–9. doi: 10.1084/jem.20050914
20. Reynolds JM, Dong C. Toll-like receptor regulation of effector T lymphocyte function. Trends Immunol (2013) 34:511–9. doi: 10.1016/j.it.2013.06.003
21. Chodisetti SB, Fike AJ, Domeier PP, Singh H, Choi NM, Corradetti C, et al. Type II but not type I IFN signaling is indispensable for TLR7-promoted development of autoreactive b cells and systemic autoimmunity. J Immunol (2020) 204:796–809. doi: 10.4049/jimmunol.1901175
22. Pisitkun P, Deane JA, Difilippantonio MJ, Tarasenko T, Satterthwaite AB, Bolland S. Autoreactive b cell responses to RNA-related antigens due to TLR7 gene duplication. Science (2006) 312:1669–72. doi: 10.1126/science.1124978
23. Mohan C, Putterman C. Genetics and pathogenesis of systemic lupus erythematosus and lupus nephritis. Nat Rev Nephrol (2015) 11:329–41. doi: 10.1038/nrneph.2015.33
24. Subramanian S, Tus K, Li QZ, Wang A, Tian XH, Zhou J, et al. A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proc Natl Acad Sci USA (2006) 103:9970–5. doi: 10.1073/pnas.0603912103
25. Souyris M, Cenac C, Azar P, Daviaud D, Canivet A, Grunenwald S, et al. TLR7 escapes X chromosome inactivation in immune cells. Sci Immunol (2018) 3:eaap8855. doi: 10.1126/sciimmunol.aap8855
26. Souyris M, Mejía JE, Chaumeil J, Guéry JC. Female predisposition to TLR7-driven autoimmunity: gene dosage and the escape from X chromosome inactivation. Semin Immunopathol (2019) 41:153–64. doi: 10.1007/s00281-018-0712-y
27. Syrett CM, Sierra I, Beethem ZT, Dubin AH, Anguera MC. Loss of epigenetic modifications on the inactive X chromosome and sex-biased gene expression profiles in b cells from NZB/W F1 mice with lupus-like disease. J Autoimmun (2020) 107:102357. doi: 10.1016/j.jaut.2019.102357
28. Odhams CA, Roberts AL, Vester SK, Duarte CST, Beales CT, Clarke AJ, et al. Interferon inducible X-linked gene CXorf21 may contribute to sexual dimorphism in systemic lupus erythematosus. Nat Commun (2019) 10:2164. doi: 10.1038/s41467-019-10106-2
29. Raafat II, El Guindy N, Shahin RMH, Samy LA, El Refai RM. Toll-like receptor 7 gene single nucleotide polymorphisms and the risk for systemic lupus erythematosus: a case-control study. Z Rheumatol (2018) 77:416–20. doi: 10.1007/s00393-017-0283-7
30. García-Ortiz H, Velázquez-Cruz R, Espinosa-Rosales F, Jiménez-Morales S, Baca V, Orozco L. Association of TLR7 copy number variation with susceptibility to childhood-onset systemic lupus erythematosus in Mexican population. Ann Rheum Dis (2010) 69:1861–5. doi: 10.1136/ard.2009.124313
31. Song W, Qiu J, Yin L, Hong X, Dai W, Tang D, et al. Integrated analysis of competing endogenous RNA networks in peripheral blood mononuclear cells of systemic lupus erythematosus. J Transl Med (2021) 19:362. doi: 10.1186/s12967-021-03033-8
32. Klonowska-Szymczyk A, Wolska A, Robak T, Cebula-Obrzut B, Smolewski P, Robak E. Expression of toll-like receptors 3, 7, and 9 in peripheral blood mononuclear cells from patients with systemic lupus erythematosus. Med Inflamm (2014) 2014:381418. doi: 10.1155/2014/381418
33. Wang T, Marken J, Chen J, Tran VB, Li QZ, Li M, et al. High TLR7 expression drives the expansion of CD19+CD24hiCD38hi transitional b cells and autoantibody production in SLE patients. Front Immunol (2019) 10:1243. doi: 10.3389/fimmu.2019.01243
34. Giltiay NV, Chappell CP, Sun X, Kolhatkar N, Teal TH, Wiedeman AE, et al. Overexpression of TLR7 promotes cell-intrinsic expansion and autoantibody production by transitional T1 b cells. J Exp Med (2013) 210:2773–89. doi: 10.1084/jem.20122798
35. Walsh ER, Pisitkun P, Voynova E, Deane JA, Scott BL, Caspi RR, et al. Dual signaling by innate and adaptive immune receptors is required for TLR7-induced b-cell-mediated autoimmunity. Proc Natl Acad Sci USA (2012) 109:16276–81. doi: 10.1073/pnas.1209372109
36. Hwang SH, Lee H, Yamamoto M, Jones LA, Dayalan J, Hopkins R, et al. B cell TLR7 expression drives anti-RNA autoantibody production and exacerbates disease in systemic lupus erythematosus-prone mice. J Immunol (2012) 189:5786–96. doi: 10.4049/jimmunol.1202195
37. Brown GJ, Cañete PF, Wang H, Medhavy A, Bones J, Roco JA, et al. TLR7 gain-of-function genetic variation causes human lupus. Nature (2022) 605:349–56. doi: 10.1038/s41586-022-04642-z
38. Yokogawa M, Takaishi M, Nakajima K, Kamijima R, Fujimoto C, Kataoka S, et al. Epicutaneous application of toll-like receptor 7 agonists leads to systemic autoimmunity in wild-type mice: A new model of systemic lupus erythematosus. Arthritis Rheumatol (2014) 66:694–706. doi: 10.1002/art.38298
39. Celhar T, Lu HK, Benso L, Rakhilina L, Lee HY, Tripathi S, et al. TLR7 protein expression in mild and severe lupus-prone models is regulated in a leukocyte, genetic, and IRAK4 dependent manner. Front Immunol (2019) 10:1546. doi: 10.3389/fimmu.2019.01546
40. Meffre E, Wardemann H. B-cell tolerance checkpoints in health and autoimmunity. Curr Opin Immunol (2008) 20:632–8. doi: 10.1016/j.coi.2008.09.001
41. Clingan JM, Matloubian M. B cell-intrinsic TLR7 signaling is required for optimal b cell responses during chronic viral infection. J Immunol (2013) 191:810–8. doi: 10.4049/jimmunol.1300244
42. Jackson SW, Scharping NE, Kolhatkar NS, Khim S, Schwartz MA, Li QZ, et al. Opposing impact of b cell-intrinsic TLR7 and TLR9 signals on autoantibody repertoire and systemic inflammation. J Immunol (2014) 192:4525–32. doi: 10.4049/jimmunol.1400098
43. Boneparth A, Huang W, Bethunaickan R, Woods M, Sahu R, Arora S, et al. TLR7 influences germinal center selection in murine SLE. PloS One (2015) 10:e0119925. doi: 10.1371/journal.pone.0119925
44. Soni C, Wong EB, Domeier PP, Khan TN, Satoh T, Akira S, et al. B cell-intrinsic TLR7 signaling is essential for the development of spontaneous germinal centers. J Immunol (2014) 193:4400–14. doi: 10.4049/jimmunol.1401720
45. Wei C, Anolik J, Cappione A, Zheng B, Pugh-Bernard A, Brooks J, et al. A new population of cells lacking expression of CD27 represents a notable component of the b cell memory compartment in systemic lupus erythematosus. J Immunol (2007) 178:6624–33. doi: 10.4049/jimmunol.178.10.6624
46. Jenks SA, Cashman KS, Woodruff MC, Lee FE, Sanz I. Extrafollicular responses in humans and SLE. Immunol Rev (2019) 288:136–48. doi: 10.1111/imr.12741
47. Nutt SL, Hodgkin PD, Tarlinton DM, Corcoran LM. The generation of antibody-secreting plasma cells. Nat Rev Immunol (2015) 15:160–71. doi: 10.1038/nri3795
48. Luo W, Mayeux J, Gutierrez T, Russell L, Getahun A, Müller J, et al. A balance between b cell receptor and inhibitory receptor signaling controls plasma cell differentiation by maintaining optimal Ets1 levels. J Immunol (2014) 193:909–20. doi: 10.4049/jimmunol.1400666
49. Russell L, John S, Cullen J, Luo W, Shlomchik MJ, Garrett-Sinha LA. Requirement for transcription factor Ets1 in b cell tolerance to self-antigens. J Immunol (2015) 195:3574–83. doi: 10.4049/jimmunol.1500776
50. Akatsu C, Shinagawa K, Numoto N, Liu Z, Ucar AK, Aslam M, et al. CD72 negatively regulates b lymphocyte responses to the lupus-related endogenous toll-like receptor 7 ligand Sm/RNP. J Exp Med (2016) 213:2691–706. doi: 10.1084/jem.20160560
51. Rubtsov AV, Rubtsova K, Fischer A, Meehan RT, Gillis JZ, Kappler JW, et al. Toll-like receptor 7 (TLR7)-driven accumulation of a novel CD11c+ b-cell population is important for the development of autoimmunity. Blood (2011) 118:1305–15. doi: 10.1182/blood-2011-01-331462
52. Zumaquero E, Stone SL, Scharer CD, Jenks SA, Nellore A, Mousseau B, et al. IFNγ induces epigenetic programming of human T-bet(hi) b cells and promotes TLR7/8 and IL-21 induced differentiation. Elife (2019) 8:e41641. doi: 10.7554/eLife.41641
53. Cancro MP. Age-associated b cells. Annu Rev Immunol (2020) 38:315–40. doi: 10.1146/annurev-immunol-092419-031130
54. Rubtsov AV, Rubtsova K, Kappler JW, Marrack P. TLR7 drives accumulation of ABCs and autoantibody production in autoimmune-prone mice. Immunol Res (2013) 55:210–6. doi: 10.1007/s12026-012-8365-8
55. Ricker E, Manni M, Flores-Castro D, Jenkins D, Gupta S, Rivera-Correa J, et al. Altered function and differentiation of age-associated b cells contribute to the female bias in lupus mice. Nat Commun (2021) 12:4813. doi: 10.1038/s41467-021-25102-8
56. Rubtsov AV, Rubtsova K, Kappler JW, Jacobelli J, Friedman RS, Marrack P. CD11c-expressing b cells are located at the T cell/B cell border in spleen and are potent APCs. J Immunol (2015) 195:71–9. doi: 10.4049/jimmunol.1500055
57. Chodisetti SB, Fike AJ, Domeier PP, Choi NM, Soni C, Rahman ZSM. TLR7 negatively regulates B10 cells predominantly in an IFNγ signaling dependent manner. Front Immunol (2020) 11:1632. doi: 10.3389/fimmu.2020.01632
58. Ohto U, Shibata T, Tanji H, Ishida H, Krayukhina E, Uchiyama S, et al. Structural basis of CpG and inhibitory DNA recognition by toll-like receptor 9. Nature (2015) 520:702–5. doi: 10.1038/nature14138
59. Machida H, Ito S, Hirose T, Takeshita F, Oshiro H, Nakamura T, et al. Expression of toll-like receptor 9 in renal podocytes in childhood-onset active and inactive lupus nephritis. Nephrol Dial Transplant (2010) 25:2530–537. doi: 10.1093/ndt/gfq058
60. Elloumi N, Fakhfakh R, Abida O, Ayadi L, Marzouk S, Hachicha H, et al. Relevant genetic polymorphisms and kidney expression of toll-like receptor (TLR)-5 and TLR-9 in lupus nephritis. Clin Exp Immunol (2017) 190:328–39. doi: 10.1111/cei.13022
61. Rao H, Zeng Q, Liang Y, Xiao C, Xie S, Xu X. Correlation between TLR9 expression and cytokine secretion in the clinical diagnosis of systemic lupus erythematosus. Mediators Inflammation (2015) 2015:710720. doi: 10.1155/2015/710720
62. Yuan Y, Zhao L, Ye Z, Ma H, Wang X, Jiang Z. Association of toll-like receptor 9 expression with prognosis of systemic lupus erythematosus. Exp Ther Med (2019) 17:3247–54. doi: 10.3892/etm.2019.7290
63. Nemazee DA, Bürki K. Clonal deletion of b lymphocytes in a transgenic mouse bearing anti-MHC class I antibody genes. Nature (1989) 337:562–6. doi: 10.1038/337562a0
64. Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC. Predominant autoantibody production by early human b cell precursors. Science (2003) 301:1374–7. doi: 10.1126/science.1086907
65. Yurasov S, Wardemann H, Hammersen J, Tsuiji M, Meffre E, Pascual V, et al. Defective b cell tolerance checkpoints in systemic lupus erythematosus. J Exp Med (2005) 201:703–11. doi: 10.1084/jem.20042251
66. Klinman DM. Immunotherapeutic uses of CpG oligodeoxynucleotides. Nat Rev Immunol (2004) 4:249–58. doi: 10.1038/nri1329
67. Rookhuizen DC, DeFranco AL. Toll-like receptor 9 signaling acts on multiple elements of the germinal center to enhance antibody responses. Proc Natl Acad Sci USA (2014) 111:E3224–33. doi: 10.1073/pnas.1323985111
68. Busconi L, Bauer JW, Tumang JR, Laws A, Perkins-Mesires K, Tabor AS, et al. Functional outcome of b cell activation by chromatin immune complex engagement of the b cell receptor and TLR9. J Immunol (2007) 179:7397–405. doi: 10.4049/jimmunol.179.11.7397
69. Christensen SR, Kashgarian M, Alexopoulou L, Flavell RA, Akira S, Shlomchik MJ. Toll-like receptor 9 controls anti-DNA autoantibody production in murine lupus. J Exp Med (2005) 202:321–31. doi: 10.1084/jem.20050338
70. Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A. Chromatin-IgG complexes activate b cells by dual engagement of IgM and toll-like receptors. Nature (2002) 416:603–7. doi: 10.1038/416603a
71. Viglianti GA, Lau CM, Hanley TM, Miko BA, Shlomchik MJ, Marshak-Rothstein A. Activation of autoreactive b cells by CpG dsDNA. Immunity (2003) 19:837–47. doi: 10.1016/s1074-7613(03)00323-6
72. Ehlers M, Fukuyama H, McGaha TL, Aderem A, Ravetch JV. TLR9/MyD88 signaling is required for class switching to pathogenic IgG2a and 2b autoantibodies in SLE. J Exp Med (2006) 203:553–61. doi: 10.1084/jem.20052438
73. Herlands RA, William J, Hershberg U, Shlomchik MJ. Anti-chromatin antibodies drive in vivo antigen-specific activation and somatic hypermutation of rheumatoid factor b cells at extrafollicular sites. Eur J Immunol (2007) 37:3339–51. doi: 10.1002/eji.200737752
74. Eckl-Dorna J, Batista FD. BCR-mediated uptake of antigen linked to TLR9 ligand stimulates b-cell proliferation and antigen-specific plasma cell formation. Blood (2009) 113:3969–77. doi: 10.1182/blood-2008-10-185421
75. Hao Y, O’Neill P, Naradikian MS, Scholz JL, Cancro MP. A b-cell subset uniquely responsive to innate stimuli accumulates in aged mice. Blood (2011) 118:1294–304. doi: 10.1182/blood-2011-01-330530
76. Naradikian MS, Hao Y, Cancro MP. Age-associated b cells: key mediators of both protective and autoreactive humoral responses. Immunol Rev (2016) 269:118–29. doi: 10.1111/imr.12380
77. Nündel K, Green NM, Shaffer AL, Moody KL, Busto P, Eilat D, et al. Cell-intrinsic expression of TLR9 in autoreactive b cells constrains BCR/TLR7-dependent responses. J Immunol (2015) 194:2504–12. doi: 10.4049/jimmunol.1402425
78. Cai B, Chang SH, Becker EB, Bonni A, Xia Z. p38 MAP kinase mediates apoptosis through phosphorylation of BimEL at ser-65. J Biol Chem (2006) 281:25215–22. doi: 10.1074/jbc.M512627200
79. Thornton TM, Rincon M. Non-classical p38 map kinase functions: cell cycle checkpoints and survival. Int J Biol Sci (2009) 5:44–51. doi: 10.7150/ijbs.5.44
80. Nickerson KM, Christensen SR, Cullen JL, Meng W, Prak ETL, Shlomchik MJ. TLR9 promotes tolerance by restricting survival of anergic anti-DNA b cells, yet is also required for their activation. J Immunol (2013) 190:1447–56. doi: 10.4049/jimmunol.1202115
81. Hondowicz BD, Alexander ST, Quinn WJ 3rd, Pagán AJ, Metzgar MH, Cancro MP, et al. The role of BLyS/BLyS receptors in anti-chromatin b cell regulation. Int Immunol (2007) 19:465–75. doi: 10.1093/intimm/dxm011
82. Takeda K, Akira S. TLR signaling pathways. Semin Immunol (2004) 16:3–9. doi: 10.1016/j.smim.2003.10.003
83. Qian C, Cao X. Regulation of toll-like receptor signaling pathways in innate immune responses. Ann N Y Acad Sci (2013) 1283:67–74. doi: 10.1111/j.1749-6632.2012.06786.x
84. Georg I, Díaz-Barreiro A, Morell M, Pey AL, Alarcón-Riquelme ME. BANK1 interacts with TRAF6 and MyD88 in innate immune signaling in b cells. Cell Mol Immunol (2020) 17:954–65. doi: 10.1038/s41423-019-0254-9
85. Etoh T, Kim YP, Ohsaki A, Komiyama K, Hayashi M. Inhibitory effect of erythraline on toll-like receptor signaling pathway in RAW264.7 cells. Biol Pharm Bull (2013) 36:1363–9. doi: 10.1248/bpb.b12-00910
86. Liu Z, Liu Y, Li T, Wang P, Mo X, Lv P, et al. CMTM7 plays key roles in TLR-induced plasma cell differentiation and p38 activation in murine b-1 b cells. Eur J Immunol (2020) 50:809–21. doi: 10.1002/eji.201948363
87. Jabara HH, McDonald DR, Janssen E, Massaad MJ, Ramesh N, Borzutzky A, et al. DOCK8 functions as an adaptor that links TLR-MyD88 signaling to b cell activation. Nat Immunol (2012) 13:612–20. doi: 10.1038/ni.2305
88. Yee NK, Hamerman JA. β(2) integrins inhibit TLR responses by regulating NF-κB pathway and p38 MAPK activation. Eur J Immunol (2013) 43:779–92. doi: 10.1002/eji.201242550
89. Lau CM, Broughton C, Tabor AS, Akira S, Flavell RA, Mamula MJ, et al. RNA-Associated autoantigens activate b cells by combined b cell antigen receptor/Toll-like receptor 7 engagement. J Exp Med (2005) 202:1171–7. doi: 10.1084/jem.20050630
90. Otipoby KL, Waisman A, Derudder E, Srinivasan L, Franklin A, Rajewsky K. The b-cell antigen receptor integrates adaptive and innate immune signals. Proc Natl Acad Sci U.S.A. (2015) 112:12145–50. doi: 10.1073/pnas.1516428112
91. Lino AC, Dang VD, Lampropoulou V, Welle A, Joedicke J, Pohar J, et al. LAG-3 inhibitory receptor expression identifies immunosuppressive natural regulatory plasma cells. Immunity (2018) 49:120–33.e9. doi: 10.1016/j.immuni.2018.06.007
92. Bernasconi NL, Onai N, Lanzavecchia A. A role for toll-like receptors in acquired immunity: up-regulation of TLR9 by BCR triggering in naive b cells and constitutive expression in memory b cells. Blood (2003) 101:4500–4. doi: 10.1182/blood-2002-11-3569
93. Papadimitraki ED, Choulaki C, Koutala E, Bertsias G, Tsatsanis C, Gergianaki I, et al. Expansion of toll-like receptor 9-expressing b cells in active systemic lupus erythematosus: implications for the induction and maintenance of the autoimmune process. Arthritis Rheum (2006) 54:3601–11. doi: 10.1002/art.22197
94. Morbach H, Schickel JN, Cunningham-Rundles C, Conley ME, Reisli I, Franco JL, et al. CD19 controls toll-like receptor 9 responses in human b cells. J Allergy Clin Immunol (2016) 137:889–98.e6. doi: 10.1016/j.jaci.2015.08.040
95. Korganow AS, Knapp AM, Nehme-Schuster H, Soulas-Sprauel P, Poindron V, Pasquali JL, et al. Peripheral b cell abnormalities in patients with systemic lupus erythematosus in quiescent phase: decreased memory b cells and membrane CD19 expression. J Autoimmun (2010) 34:426–34. doi: 10.1016/j.jaut.2009.11.002
96. Sato S, Hasegawa M, Fujimoto M, Tedder TF, Takehara K. Quantitative genetic variation in CD19 expression correlates with autoimmunity. J Immunol (2000) 165:6635–43. doi: 10.4049/jimmunol.165.11.6635
97. Culton DA, Nicholas MW, Bunch DO, Zhen QL, Kepler TB, Dooley MA, et al. Similar CD19 dysregulation in two autoantibody-associated autoimmune diseases suggests a shared mechanism of b-cell tolerance loss. J Clin Immunol (2007) 27:53–68. doi: 10.1007/s10875-006-9051-1
98. Dieudonné Y, Gies V, Guffroy A, Keime C, Bird AK, Liesveld J, et al. Transitional b cells in quiescent SLE: an early checkpoint imprinted by IFN. J Autoimmun (2019) 102:150–8. doi: 10.1016/j.jaut.2019.05.002
99. van Zelm MC, Reisli I, van der Burg M, Castaño D, van Noesel CJ, van Tol MJ, et al. An antibody-deficiency syndrome due to mutations in the CD19 gene. N Engl J Med (2006) 354:1901–12. doi: 10.1056/NEJMoa051568
100. Kang S, Rogers JL, Monteith AJ, Jiang C, Schmitz J, Clarke SH, et al. Apoptotic debris accumulates on hematopoietic cells and promotes disease in murine and human systemic lupus erythematosus. J Immunol (2016) 196:4030–9. doi: 10.4049/jimmunol.1500418
101. Mauri C, Bosma A. Immune regulatory function of b cells. Annu Rev Immunol (2012) 30:221–41. doi: 10.1146/annurev-immunol-020711-074934
102. Brosseau C, Durand M, Colas L, Durand E, Foureau A, Cheminant MA, et al. CD9(+) regulatory b cells induce T cell apoptosis via IL-10 and are reduced in severe asthmatic patients. Front Immunol (2018) 9:3034. doi: 10.3389/fimmu.2018.03034
103. Miles K, Heaney J, Sibinska Z, Salter D, Savill J, Gray D, et al. A tolerogenic role for toll-like receptor 9 is revealed by b-cell interaction with DNA complexes expressed on apoptotic cells. Proc Natl Acad Sci USA (2012) 109:887–92. doi: 10.1073/pnas.1109173109
104. Menon M, Blair PA, Isenberg DA, Mauri C. A regulatory feedback between plasmacytoid dendritic cells and regulatory b cells is aberrant in systemic lupus erythematosus. Immunity (2016) 44:683–97. doi: 10.1016/j.immuni.2016.02.012
105. Pelka K, Bertheloot D, Reimer E, Phulphagar K, Schmidt SV, Christ A, et al. The chaperone UNC93B1 regulates toll-like receptor stability independently of endosomal TLR transport. Immunity (2018) 48:911–22.e7. doi: 10.1016/j.immuni.2018.04.011
106. Nakano S, Morimoto S, Suzuki S, Watanabe T, Amano H, Takasaki Y. Up-regulation of the endoplasmic reticulum transmembrane protein UNC93B in the b cells of patients with active systemic lupus erythematosus. Rheumatol (Oxford) (2010) 49:876–81. doi: 10.1093/rheumatology/keq001
107. Kono DH, Haraldsson MK, Lawson BR, Pollard KM, Koh YT, Du X, et al. Endosomal TLR signaling is required for anti-nucleic acid and rheumatoid factor autoantibodies in lupus. Proc Natl Acad Sci U.S.A. (2009) 106:12061–6. doi: 10.1073/pnas.0905441106
108. Koh YT, Scatizzi JC, Gahan JD, Lawson BR, Baccala R, Pollard KM, et al. Role of nucleic acid-sensing TLRs in diverse autoantibody specificities and anti-nuclear antibody-producing b cells. J Immunol (2013) 190:4982–90. doi: 10.4049/jimmunol.1202986
109. Fukui R, Saitoh S, Kanno A, Onji M, Shibata T, Ito A, et al. Unc93B1 restricts systemic lethal inflammation by orchestrating toll-like receptor 7 and 9 trafficking. Immunity (2011) 35:69–81. doi: 10.1016/j.immuni.2011.05.010
110. Desnues B, Macedo AB, Roussel-Queval A, Bonnardel J, Henri S, Demaria O, et al. TLR8 on dendritic cells and TLR9 on b cells restrain TLR7-mediated spontaneous autoimmunity in C57BL/6 mice. Proc Natl Acad Sci U.S.A. (2014) 111:1497–502. doi: 10.1073/pnas.1314121111
111. Majer O, Liu B, Kreuk LSM, Krogan N, Barton GM. UNC93B1 recruits syntenin-1 to dampen TLR7 signalling and prevent autoimmunity. Nature (2019) 575:366–70. doi: 10.1038/s41586-019-1612-6
112. Kim YM, Brinkmann MM, Paquet ME, Ploegh HL. UNC93B1 delivers nucleotide-sensing toll-like receptors to endolysosomes. Nature (2008) 452:234–8. doi: 10.1038/nature06726
113. Heinz LX, Lee J, Kapoor U, Kartnig F, Sedlyarov V, Papakostas K, et al. TASL is the SLC15A4-associated adaptor for IRF5 activation by TLR7-9. Nature (2020) 581:316–22. doi: 10.1038/s41586-020-2282-0
114. Kobayashi T, Shimabukuro-Demoto S, Yoshida-Sugitani R, Furuyama-Tanaka K, Karyu H, Sugiura Y, et al. The histidine transporter SLC15A4 coordinates mTOR-dependent inflammatory responses and pathogenic antibody production. Immunity (2014) 41:375–88. doi: 10.1016/j.immuni.2014.08.011
115. Blasius AL, Arnold CN, Georgel P, Rutschmann S, Xia Y, Lin P, et al. Slc15a4, AP-3, and hermansky-pudlak syndrome proteins are required for toll-like receptor signaling in plasmacytoid dendritic cells. Proc Natl Acad Sci USA (2010) 107:19973–8. doi: 10.1073/pnas.1014051107
116. Baccala R, Gonzalez-Quintial R, Blasius AL, Rimann I, Ozato K, Kono DH, et al. Essential requirement for IRF8 and SLC15A4 implicates plasmacytoid dendritic cells in the pathogenesis of lupus. Proc Natl Acad Sci USA (2013) 110:2940–5. doi: 10.1073/pnas.1222798110
117. Domeier PP, Chodisetti SB, Schell SL, Kawasawa YI, Fasnacht MJ, Soni C, et al. B-cell-intrinsic type 1 interferon signaling is crucial for loss of tolerance and the development of autoreactive b cells. Cell Rep (2018) 24:406–18. doi: 10.1016/j.celrep.2018.06.046
118. Bekeredjian-Ding IB, Wagner M, Hornung V, Giese T, Schnurr M, Endres S, et al. Plasmacytoid dendritic cells control TLR7 sensitivity of naive b cells via type I IFN. J Immunol (2005) 174:4043–50. doi: 10.4049/jimmunol.174.7.4043
119. Soni C, Perez OA, Voss WN, Pucella JN, Serpas L, Mehl J, et al. Plasmacytoid dendritic cells and type I interferon promote extrafollicular b cell responses to extracellular self-DNA. Immunity (2020) 52:1022–38.e7. doi: 10.1016/j.immuni.2020.04.015
120. Cui C, Wang J, Fagerberg E, Chen PM, Connolly KA, Damo M, et al. Neoantigen-driven b cell and CD4 T follicular helper cell collaboration promotes anti-tumor CD8 T cell responses. Cell (2021) 184:6101–18.e13. doi: 10.1016/j.cell.2021.11.007
121. Naradikian MS, Myles A, Beiting DP, Roberts KJ, Dawson L, Herati RS, et al. Cutting edge: IL-4, IL-21, and IFN-γ interact to govern T-bet and CD11c expression in TLR-activated b cells. J Immunol (2016) 197:1023–8. doi: 10.4049/jimmunol.1600522
122. Hjorton K, Hagberg N, Israelsson E, Jinton L, Berggren O, Sandling JK, et al. Cytokine production by activated plasmacytoid dendritic cells and natural killer cells is suppressed by an IRAK4 inhibitor. Arthritis Res Ther (2018) 20:238. doi: 10.1186/s13075-018-1702-0
123. Corzo CA, Varfolomeev E, Setiadi AF, Francis R, Klabunde S, Senger K, et al. The kinase IRAK4 promotes endosomal TLR and immune complex signaling in b cells and plasmacytoid dendritic cells. Sci Signal (2020) 13:eaaz1053. doi: 10.1126/scisignal.aaz1053
124. Winkler A, Sun W, De S, Jiao A, Sharif MN, Symanowicz PT, et al. The interleukin-1 receptor-associated kinase 4 inhibitor PF-06650833 blocks inflammation in preclinical models of rheumatic disease and in humans enrolled in a randomized clinical trial. Arthritis Rheumatol (2021) 73:2206–18. doi: 10.1002/art.41953
125. Nanda SK, Lopez-Pelaez M, Arthur JS, Marchesi F, Cohen P. Suppression of IRAK1 or IRAK4 catalytic activity, but not type 1 IFN signaling, prevents lupus nephritis in mice expressing a ubiquitin binding-defective mutant of ABIN1. J Immunol (2016) 197:4266–73. doi: 10.4049/jimmunol.1600788
126. Scarneo SA, Hughes PF, Yang KW, Carlson DA, Gurbani D, Westover KD, et al. A highly selective inhibitor of interleukin-1 receptor-associated kinases 1/4 (IRAK-1/4) delineates the distinct signaling roles of IRAK-1/4 and the TAK1 kinase. J Biol Chem (2020) 295:1565–74. doi: 10.1074/jbc.RA119.011857
127. Port A, Shaw JV, Klopp-Schulze L, Bytyqi A, Vetter C, Hussey E, et al. Phase 1 study in healthy participants of the safety, pharmacokinetics, and pharmacodynamics of enpatoran (M5049), a dual antagonist of toll-like receptors 7 and 8. Pharmacol Res Perspect (2021) 9:e00842. doi: 10.1002/prp2.842
128. Klopp-Schulze L, Shaw JV, Dong JQ, Khandelwal A, Vazquez-Mateo C, Goteti K. Applying modeling and simulations for rational dose selection of novel toll-like receptor 7/8 inhibitor enpatoran for indications of high medical need. Clin Pharmacol Ther (2022) 112:297–306. doi: 10.1002/cpt.2606
129. Lamphier M, Zheng W, Latz E, Spyvee M, Hansen H, Rose J, et al. Novel small molecule inhibitors of TLR7 and TLR9: mechanism of action and efficacy in vivo. Mol Pharmacol (2014) 85:429–40. doi: 10.1124/mol.113.089821
130. Sacre K, Criswell LA, McCune JM. Hydroxychloroquine is associated with impaired interferon-alpha and tumor necrosis factor-alpha production by plasmacytoid dendritic cells in systemic lupus erythematosus. Arthritis Res Ther (2012) 14:R155. doi: 10.1186/ar3895
131. Clancy RM, Markham AJ, Buyon JP. Endosomal toll-like receptors in clinically overt and silent autoimmunity. Immunol Rev (2016) 269:76–84. doi: 10.1111/imr.12383
132. Tohme M, Maisonneuve L, Achour K, Dussiot M, Maschalidi S, Manoury B. TLR7 trafficking and signaling in b cells is regulated by the MHCII-associated invariant chain. J Cell Sci (2020) 133:jcs236711. doi: 10.1242/jcs.236711
133. Acharya M, Sokolovska A, Tam JM, Conway KL, Stefani C, Raso F, et al. αv integrins combine with LC3 and atg5 to regulate toll-like receptor signalling in b cells. Nat Commun (2016) 7:10917. doi: 10.1038/ncomms10917
134. Raso F, Sagadiev S, Du S, Gage E, Arkatkar T, Metzler G, et al. αv integrins regulate germinal center b cell responses through noncanonical autophagy. J Clin Invest (2018) 128:4163–78. doi: 10.1172/jci99597
135. Majer O, Liu B, Woo BJ, Kreuk LSM, Van Dis E, Barton GM. Release from UNC93B1 reinforces the compartmentalized activation of select TLRs. Nature (2019) 575:371–4. doi: 10.1038/s41586-019-1611-7
136. Dinarello CA. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol Rev (2018) 281:8–27. doi: 10.1111/imr.12621
137. Kaul A, Gordon C, Crow MK, Touma Z, Urowitz MB, van Vollenhoven R, et al. Systemic lupus erythematosus. Nat Rev Dis Primers (2016) 2:16039. doi: 10.1038/nrdp.2016.39
Keywords: systemic lupus erythematosus, B cells, TLR signaling, TLR7, TLR9, targeted therapies
Citation: Wen L, Zhang B, Wu X, Liu R, Fan H, Han L, Zhang Z, Ma X, Chu C-Q and Shi X (2023) Toll-like receptors 7 and 9 regulate the proliferation and differentiation of B cells in systemic lupus erythematosus. Front. Immunol. 14:1093208. doi: 10.3389/fimmu.2023.1093208
Received: 08 November 2022; Accepted: 02 February 2023;
Published: 15 February 2023.
Edited by:
Hong Zan, The University of Texas Health Science Center at San Antonio, United StatesReviewed by:
Roberto Baccala, San Diego Biomedical Research Institute, United StatesCopyright © 2023 Wen, Zhang, Wu, Liu, Fan, Han, Zhang, Ma, Chu and Shi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaofei Shi, eGlhb2ZlaXNAaGF1c3QuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.