
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Immunol., 25 January 2023
Sec. Molecular Innate Immunity
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1090898
This article is part of the Research TopicNew Functional Separation and Analysis Technologies Utilizing Human Fc ReceptorsView all 6 articles
 Saeko Yanaka1,2,3,4
Saeko Yanaka1,2,3,4 Rina Yogo1,2,3†
Rina Yogo1,2,3† Hirokazu Yagi1,3Masayoshi Onitsuka5Natsumi Wakaizumi6
Hirokazu Yagi1,3Masayoshi Onitsuka5Natsumi Wakaizumi6 Yuki Yamaguchi6Susumu Uchiyama6
Yuki Yamaguchi6Susumu Uchiyama6 Koichi Kato1,2,3*
Koichi Kato1,2,3*Although interactions of small molecular drugs with serum proteins have been widely studied from pharmacokinetic and pharmacodynamic perspectives, there have been few reports on the effects of serum components on therapeutic antibody functions. This study reports the effect of abundant serum proteins on antibody-dependent cellular cytotoxicity (ADCC) mediated by rituximab and Fcγ receptor III (FcγRIII). Human serum albumin (HSA) and the Fab fragment from the pooled serum polyclonal IgG were found to compromise ADCC as non-competitive inhibitors. Our nuclear magnetic resonance data provided direct evidence for the interactions of HSA with both the Fab and Fc regions of rituximab and also with the extracellular region of FcγRIII (sFcγRIII). The degree of involvement in the interaction decreased in the order of rituximab-Fab > rituximab-Fc > sFcγRIII, suggesting preferential binding of HSA to net positively charged proteins. Although much less pronounced than the effect of HSA, polyclonal IgG-Fab specifically interacted with rituximab-Fc. The NMR data also showed that the serum protein interactions cover the Fc surface extensively, suggesting that they can act as pan-inhibitors against various Fc receptor-mediated functions and pharmacokinetics. Our findings highlight the importance of considering serum–protein interactions in the design and application of antibody-based drugs with increased efficacy and safety.
Most drugs enter the blood after administration and are distributed to various tissues. In the bloodstream, they bind to serum proteins, such as albumin. The interactions of drugs with serum proteins significantly affect their pharmacokinetics and pharmacodynamics and are therefore carefully evaluated during drug discovery and development (1–3). Many of the currently used therapeutic antibodies are administered intravenously and exert their effect in the blood (4). However, limited studies have been conducted on the effects of interactions between therapeutic antibodies and serum proteins.
A previous study found that serum polyclonal antibodies had a negative effect on antibody-dependent cellular cytotoxicity (ADCC) (5). This may be the result of their competitive inhibition of the binding of therapeutic antibodies to Fcγ receptor IIIa (FcγRIIIa) through their Fc region. In this regard, the binding target of the serum protein is not the therapeutic antibody, but rather its cognate receptor. Such Fc-mediated competitive binding unavoidably occurs with various Fc receptors, including FcRn, thereby affecting other effector functions and pharmacokinetics of therapeutic antibodies, such as blood half-life (6). On the other hand, several studies have indicated that human serum components can interact with immunoglobulin G (IgG). For instance, analytical ultracentrifugation indicated that human serum albumin (HSA) interacts with human monoclonal IgG1 (7, 8), whereas our nuclear magnetic resonance (NMR) studies demonstrated that mouse IgG2b semi-specifically interacts with the Fab region of pooled human serum polyclonal antibody but barely with HSA (9, 10). However, the functional effects of these interactions remain unexplored.
Given this situation, we investigated the effects of the interaction between serum proteins and human IgG1 on its ADCC function mediated by FcγRIIIa. We used rituximab, an anti-CD20 mouse/human-chimeric IgG1 (11), along with HSA and polyclonal antibody, which together account for >70% of the serum proteins (12). Additionally, we characterized the interactions using stable-isotope-assisted NMR spectroscopy.
Pooled off-the-clot human serum was purchased from Access Biologicals. Human serum polyclonal IgG and HSA were purchased from Sigma-Aldrich. The Fab fragment of the polyclonal IgG was digested using papain (13), purified using a Protein G Sepharose column (Cytiva) to remove Fc fragments, and then applied to a Superdex 200 16/60 gel filtration column (Cytiva) as previously described (9). Rituximab without isotope labeling was purchased from Zenyaku Kogyo.
Metabolic isotope labeling of antibodies was performed as previously described (14). A Chinese hamster ovary cell line producing rituximab (14), grown in a modified Nissui NYSF-404 medium containing appropriate stable isotope-labeled metabolic precursors was used. The amino acid components in the medium were substituted by uniformly 15N-labeled algal amino acid mixture supplemented with 15N-labeled analogs of the following amino acids: L-leucine, L-histidine, L-cysteine, and L-asparagine. The Fab and Fc fragments of isotope-labeled IgG1 were prepared through proteolytic digestion using papain as described above and subjected to NMR measurements. Digestion products were separated into Fab and Fc fragments using a protein A affinity column (GE Healthcare) and further purified through gel filtration using a HiLoad 16/60 Superdex 200 pg column (GE Healthcare) equilibrated with 50 mM Tris–HCl (pH 8.0) containing 150 mM NaCl (9). The rituximab-Fc fragment was treated with recombinant β1,4-galactosidase from Streptococcus pneumoniae (New England Biolabs), producing a uniformly fucosylated, nongalactosylated glycoform as previously described (14). A soluble form of human FcγRIIIb (NA2 form) composed of the extracellular domains (simply designated as sFcγRIIIb) was bacterially expressed with a C-terminal hexahistidine tag and uniform 15N-labeling and purified as previously described (15).
NMR measurements, the concentrations of the 15N-labeled forms of rituximab-Fc, rituximab-Fab, and sFcγRIIIb were set to 120 μM, 240 μM, and 120 μM, respectively, in 5 mM sodium phosphate buffer containing 50 mM NaCl. Serum polyclonal IgG, its Fab fragment, and HSA were added at final concentrations of 120 μM, 240 μM and 600 μM, respectively. The pH and temperature of the solutions were set to pH 7.4 and 37°C, respectively. 1H-15N heteronuclear single-quantum coherence (HSQC) peaks originating from the backbone of Fc were assigned based on the previous assignment in the BioMagResBank database (http://www.bmrb.wisc.edu) under the accession number 25224 (16). A series of NMR spectra were obtained using AVANCE 800 and AVANCEIII 900 spectrometers (Bruker BioSpin). The obtained data were processed using the NMRpipe software (17).
An Octet HTX system (Sartorius) was used for biolayer interferometry (BLI) measurements of rituximab-FcγRIIIa interaction using the extracellular region of human FcγRIIIa (158V allele) in which Asn45 and Asn162 were N-glycosylated while the remaining N-glycosylation sites were substituted by glutamine. This bis-glycosylated receptor was expressed by CHO/dhFr- cells (ATCC® CRL-9096) with a C-terminal hexahistidine tag, purified using cOmplete His-Tag Purification Resin column (Roche), and biotinylated as previously described (13, 18, 19). Before each assay, SAX (High Precision Streptavidin) biosensor tips were pre-wetted in 200 µL of HBS-P+ buffer (Cytiva, BR100827; 0.01 HEPES, 0.15 M NaCl and 0.05% (v/v) Surfactant P20) for at least 10 min. The measurements were performed at 30°C. First, a baseline was established using the buffer for 90 s, followed by the capture of the biotinylated receptor. Subsequently, a second baseline was obtained using HBS-P+, followed by the association and dissociation of rituximab with or without HSA and polyclonal IgG-Fab. The two-fold dilution series of rituximab starting at 2.0 µM were used for the assay. The regeneration step was performed using 1 M MgCl2 (Fujifilm Wako Pure Chemical Corp., 136–03995) after the cycle, and the same samples were measured thrice (n = 3).
Cell-based ADCC assays of rituximab were performed using a nuclear factor of activated T cell (NFAT)-driven luciferase reporter system (recombinant Jurkat T cells expressing firefly luciferase gene under the control of NFAT response elements with constitutive expression of human FcγRIIIa, high affinity (V158) variant and FcR γ chain) as previously described (20). Briefly, Jurkat/FcγRIIIa/NFAT-Luc cells (effector cells) were seeded at an effector with a target ratio of 10:1 and cultured with serially diluted rituximab mixed with polyclonal IgG-Fab or HSA at a final concentration of 240 μM or 600 μM, respectively. After incubation at 37°C for 4 h in a 5% CO2 atmosphere, luciferase activity was evaluated using the ONE-Glo Luciferase Assay System (Promega).
Protein isoelectric point was calculated using Compute pI/Mw (21).
Rituximab is clinically used to treat certain cancers, including chronic lymphocytic leukemia and non-Hodgkin’s lymphoma (11). The anticancer activity of this therapeutic antibody is based on ADCC through its interaction with FcγRIIIa, which is typically expressed on natural killer cells. We evaluated ADCC activity using a reporter cell line expressing human FcγRIIIa and NFAT-driven luciferase reporter gene (20). The EC50 obtained from the present ADCC reporter assay was approximately 20 ng/mL, which is consistent with the previous report (22). The serum concentrations of HSA and polyclonal IgG have been reported as 35-50 mg/mL and 4.1-21.7 mg/mL, respectively (23, 24). Taking this into account, 600 μM of HSA or 240 μM of polyclonal human IgG-Fab were added to this cell-based assay system. However, an equimolar amount of their Fab fragment was used instead of polyclonal IgG antibodies because full-length IgG was expected to be competitive with rituximab regarding the Fc-mediated binding to FcγRIIIa (5). The assay results revealed that ADCC was moderately compromised by the polyclonal IgG-Fab fragments, while HSA negatively affected ADCC more intensely (Figure 1). These serum proteins little affected EC50, but rather reduced the magnitude of the maximum response, indicating their non-competitive inhibition.
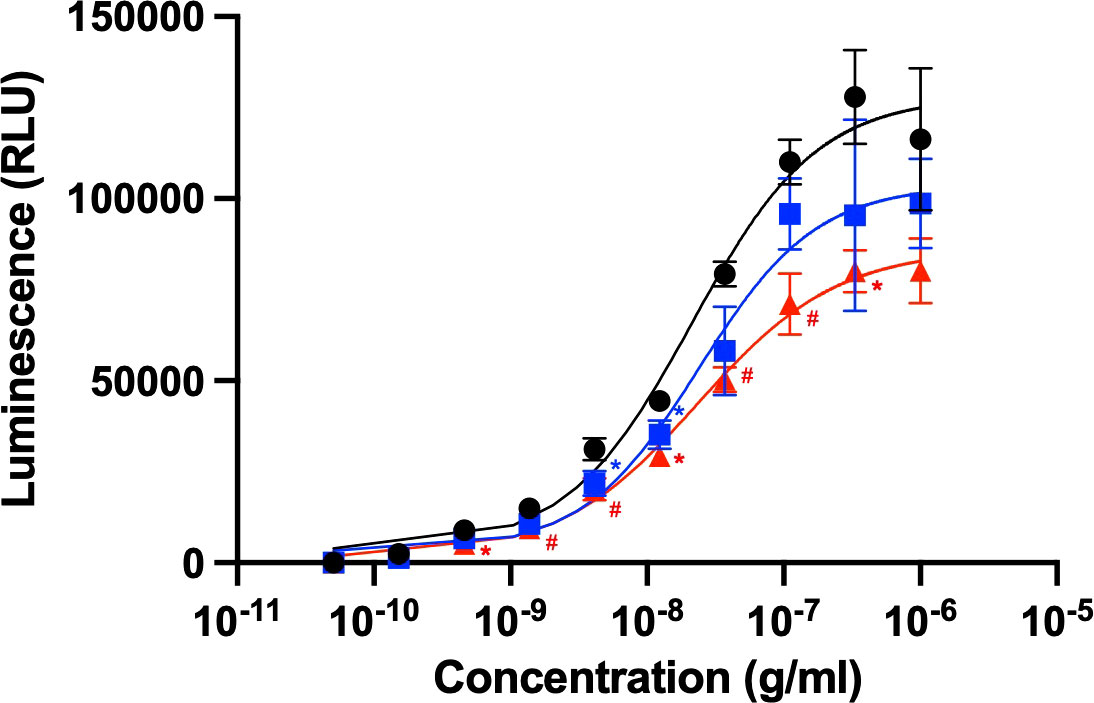
Figure 1 Rituximab-mediated ADCC activity influenced by the presence of serum components. ADCC activity of rituximab was measured in the absence (black circle) or presence of HSA (red triangle) or polyclonal IgG-Fab (blue rectangle). Rituximab concentration was prepared as a series of 3-fold dilutions from 1 μg/mL. Final concentrations of HSA and polyclonal IgG-Fab were 600 μM and 240 μM, respectively. Data represent the mean ± SD (n = 3). Significant p-values (*p < 0.05, #p < 0.01) are compared to the control by two-way ANOVA followed by Tukey’s multiple comparisons.
Next, the effects of these serum proteins on IgG1–FcγRIIIa interaction were examined via BLI analysis. The extracellular region of FcγRIIIa was immobilized onto a sensor, which was subjected to rituximab solutions in the presence or absence of HSA or polyclonal IgG-Fab. The results showed that HSA negatively affected their interaction, while no significant inhibition was observed by polyclonal IgG-Fab (Figure 2). Hence, we attempted to use a more sensitive technique for detecting weak interactions involving the serum proteins.
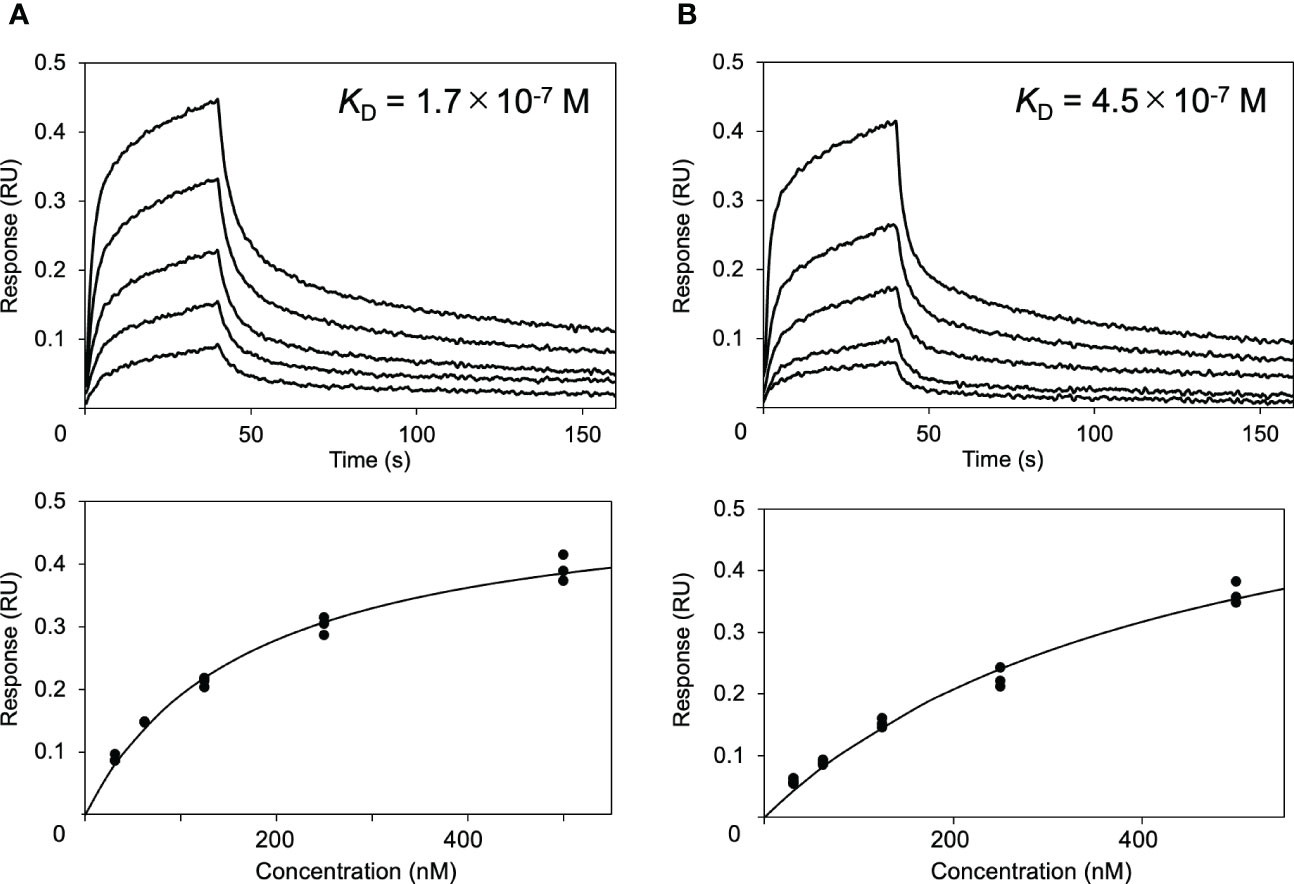
Figure 2 Effect of HSA on the interaction between rituximab and FcγRIIIa. Interaction of rituximab with the immobilized form of the extracellular region of human FcγRIIIa was observed by BLI in the (A) absence or (B) presence of HSA.
NMR can be a valuable tool for detecting weak interactions while dealing with heterogeneous systems. For selective observation of antibody NMR signals, we have established stable-isotope-labeling techniques of IgG glycoproteins using various eukaryotic expression systems (10, 14, 25, 26). In the present study, we prepared uniformly 15N-labeled rituximab, which was cleaved into Fab and Fc fragments for HSQC spectral measurements, to assess their interactions with HSA or polyclonal IgG-Fab. Additionally, we subjected uniformly 15N-labeled sFcγRIIIb to HSQC spectral measurements. Upon addition of HSA, the HSQC peaks of these 15N-labeled proteins exhibited attenuation of intensity (Figure 3), which was quantified for each peak according to the equation (Io-Ip)/Io, where Io and Ip are original peak intensity and intensity after perturbation, respectively. The degree of impact by HSA decreased in the order of rituximab-Fab > rituximab-Fc > sFcγRIIIb (Figure 4). The serum polyclonal IgG-Fab induced significant intensity attenuation for the peaks originating from rituximab-Fc, but the impact was smaller than that caused by HSA (Figures 3, 4). We have established assignments of the backbone HSQC peaks originating from Fc (16) and sFcγRIIIb (15), enabling us to map the spectral perturbation on their crystal structures (27, 28). The results showed that the interactions with HSA and polyclonal IgG-Fab cover these molecular surfaces extensively (Figure 5). Thus, the NMR data provide valuable information regarding the interactions of rituximab with the serum proteins, although the method required much higher concentrations of 15N-labeled proteins as compared with the expected concentration in the blood for rituximab treatment (>10 μg/mL), due to the sensitivity limitations of the NMR method (30). Human serum itself and the full-length form of serum polyclonal IgG caused more enhanced spectral changes of rituximab-Fc than the polyclonal IgG-Fab and HSA (Figure 6).
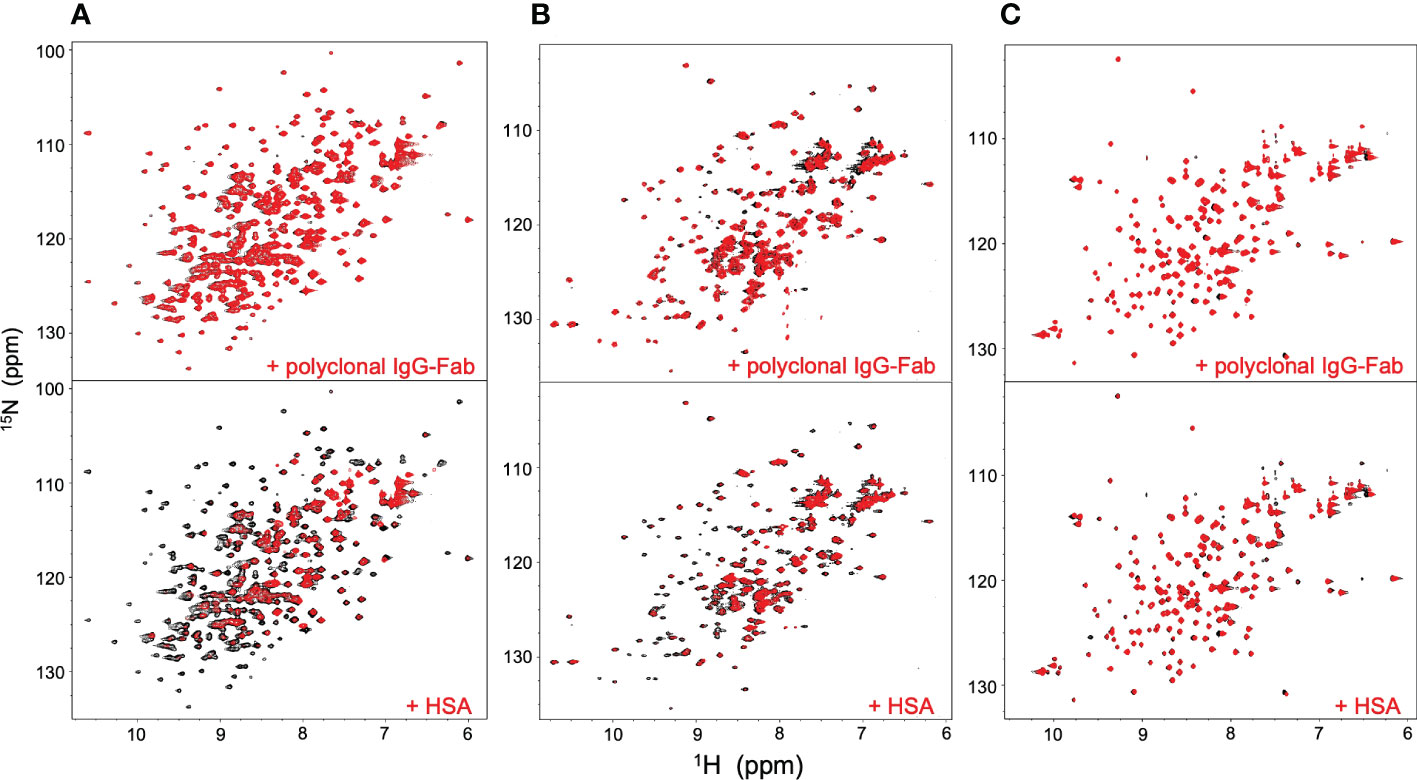
Figure 3 HSQC spectral changes of rituximab-Fc, rituximab-Fab, and sFcγRIIIb induced by the serum components. HSQC spectra of uniformly 15N-labeled rituximab-Fab (A), rituximab-Fc (B), and sFcγRIIIb (C) were measured in the absence (black) or presence (red) of HSA (lower) or polyclonal IgG-Fab (upper).
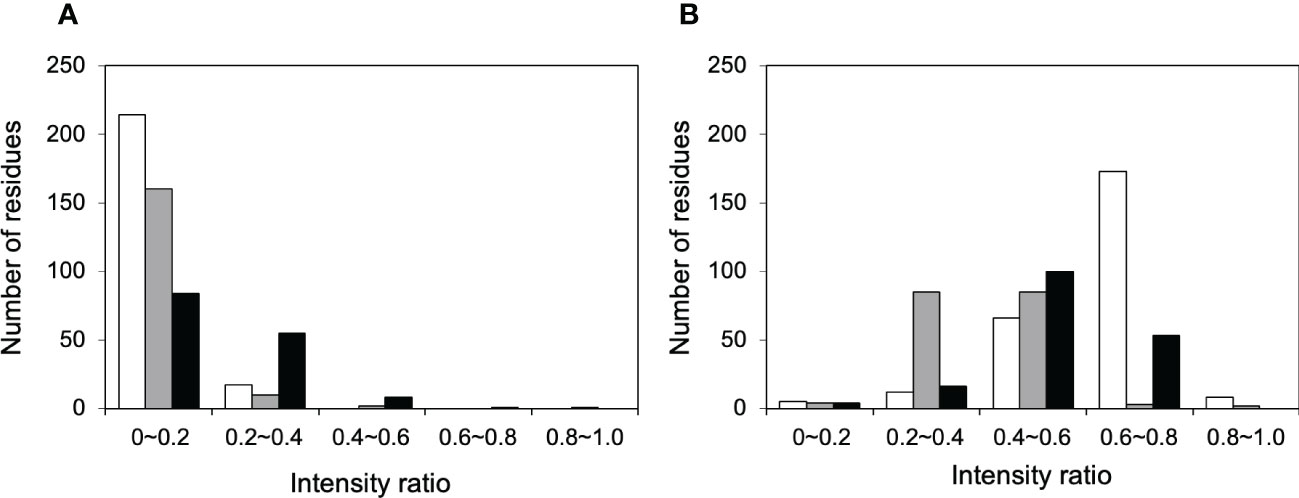
Figure 4 Histograms of the 1H-15N HSQC peaks exhibiting attenuation in intensity upon addition of serum proteins. The residues showing different degree of peak attenuation by the addition of (A) polyclonal IgG-Fab and (B) HSA were counted for 15N-labeled forms of rituximab-Fab (white), rituximab-Fc (black), and sFcγRIIIb (gray). The data were calculated from the spectra shown in Figure 3. The attenuation in intensity [(Io-Ip)/Io, where Io and Ip are original peak intensity and intensity after perturbation, respectively] was calculated for all observable 1H-15N HSQC peaks. The average levels of peak intensity attenuation caused by HSA were 0.60, 0.51, and 0.41 for rituximab-Fab, rituximab-Fc, and sFcγRIIIb, respectively, and those by polyclonal IgG-Fab are 0.33, 0.07, and 0.11 for rituximab-Fab, rituximab-Fc, and sFcγRIIIb, respectively.
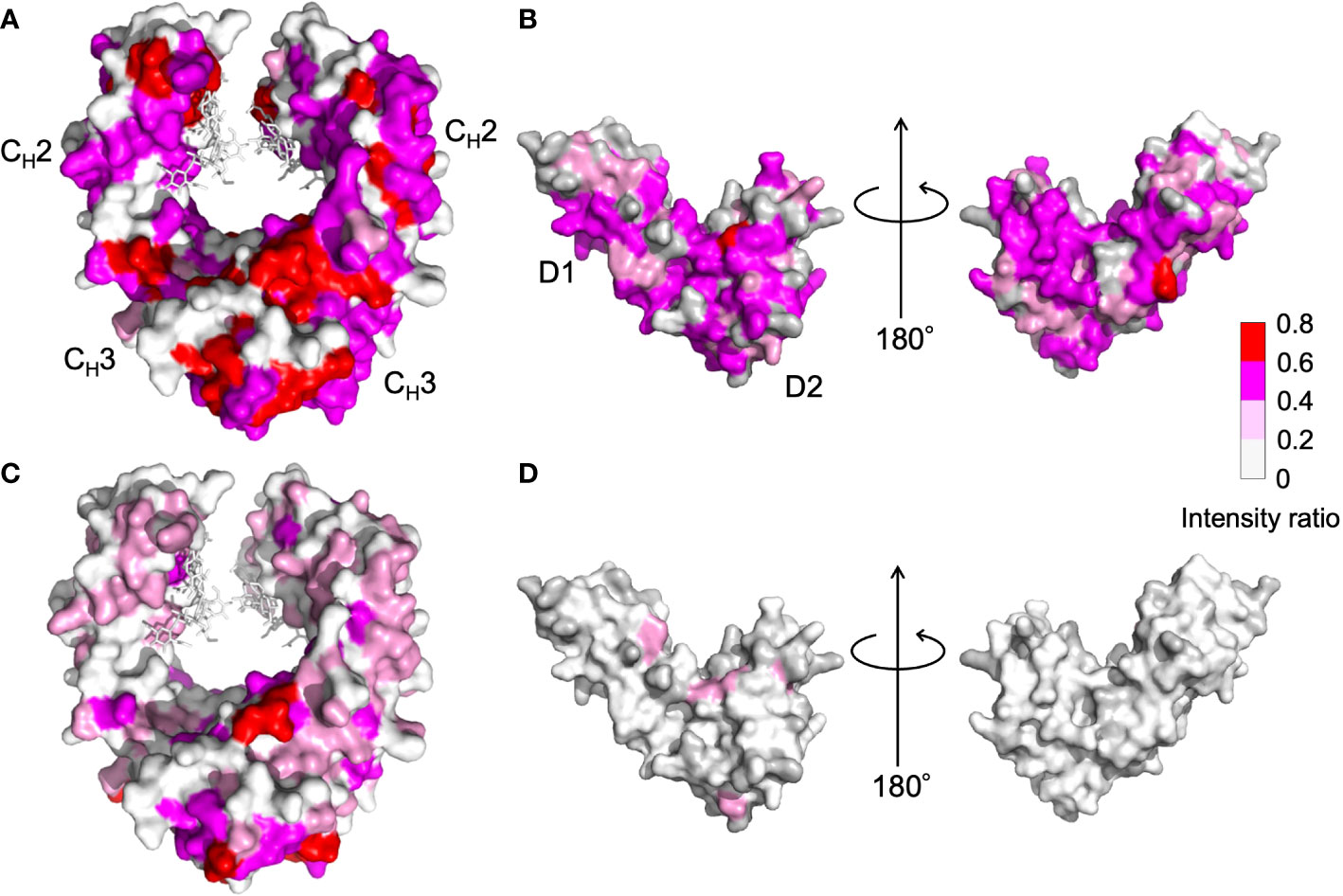
Figure 5 Mapping on the crystal structure of (A, C) human IgG1-Fc [PDB code: 3ave (27)] and (B, D) sFcγRIIIb [PDB code: 1T89 (28)] with the observed spectral perturbations by addition of (A, B) HSA, and (C, D) polyclonal IgG-Fab. The attenuation in intensity of the HSQC peaks originating from the backbones of human IgG1-Fc and sFcγRIIIb was calculated as (Io-Ip)/Io, where Io and Ip are original peak intensity and intensity after perturbation, respectively, and mapped on their crystal structures. The proline residues and the residues whose 1H-15N HSQC peaks could not be observed as probe because of broadening and/or overlapping are shown in gray. The N-glycans of IgG1-Fc are shown as stick models. The molecular graphics were generated using PyMOL (29).
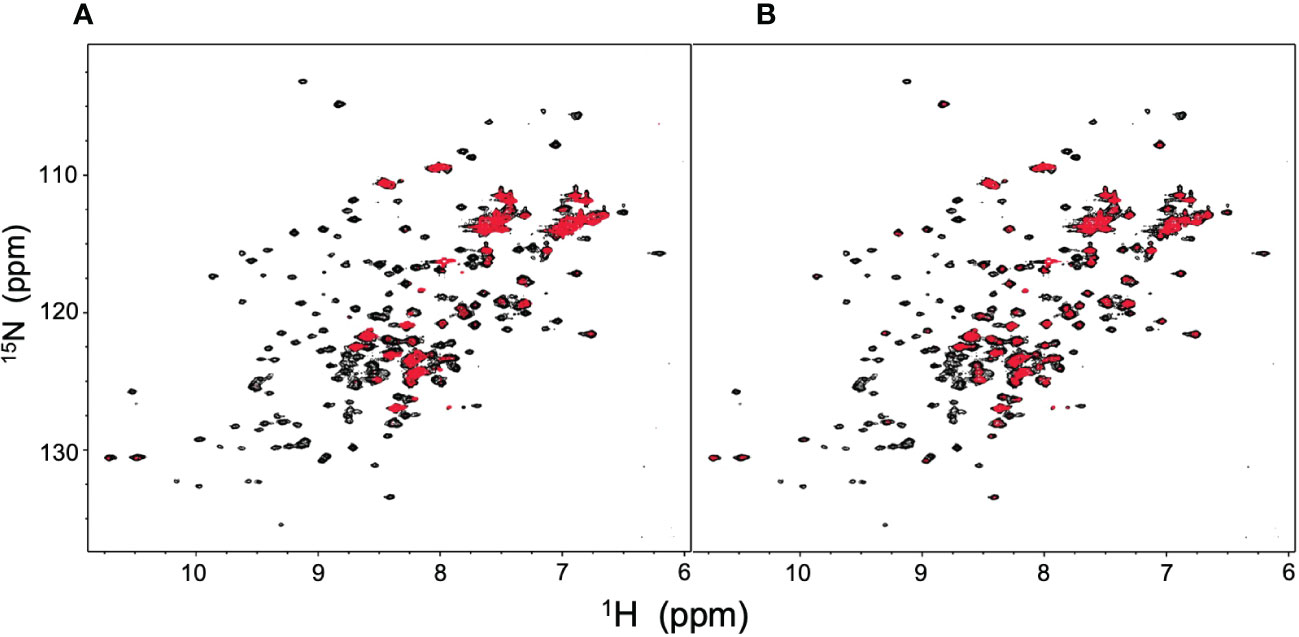
Figure 6 HSQC spectral changes of rituximab-Fc induced by human serum and human serum polyclonal IgG. HSQC spectra of uniformly 15N-labeled rituximab-Fc measured (A) in human serum or (B) in the presence of polyclonal IgG (red) are superposed on the spectrum of rituximab-Fc alone (black).
This study demonstrated that HSA, the most abundant serum protein, negatively affects the interaction between rituximab and FcγRIIIa and compromises ADCC. The NMR data provide direct evidence for the interaction of HSA with rituximab, and to a lesser extent, with the extracellular region of FcγRIIIb. The extracellular region of FcγRIIIa shares 96% amino acid identity with that of FcγRIIIb and therefore possibly interacts with HSA to the same extent as FcγRIIIb in this condition, although their N-glycans may affect the interaction. HSA interacted with both the Fab and Fc regions of rituximab, but to a greater extent with Fab, which is not only responsible for antigen binding but also directly involved in the interaction with FcγRIIIa (13, 19, 31). Hence, HSA may interfere at various points in ADCC, at least for interaction with FcγRIIIa mediated by both Fab and Fc, and possibly for antigen recognition.
Residues perturbed by HSA were widely distributed in the Fc and sFcγRIIIb molecules, including their binding sites, suggesting that the interaction is not mediated by specific sites (Figure 5). Our previous NMR study demonstrated that mouse IgG2b-Fc interacted with polyclonal IgG-Fab from pooled human serum but to a considerably weaker extent with HSA (9). In contrast, analytical centrifugation detected weak reversible interactions between HSA and adalimumab, a human IgG1 monoclonal antibody antagonizing tumor necrosis factor (7). Thus, the interaction with HSA exhibits specificity for human IgG1 to a certain extent.
Although HSA harbors versatile binding pockets that accommodate basic, acidic, or neutral small molecules (32), the overall physical properties, such as net charges, rather than local structural features, may govern its interactions with macromolecules. Indeed, charge variation in IgG affects the binding to FcγRIIIa (33). In a neutral solution, HSA (pI = 5.7) is net negatively charged and therefore interacts preferably with positively charged proteins. This consistently explains the NMR observations, i.e., extensive, moderate, and undetectable interactions with rituximab-Fab (pI = 8.8), rituximab-Fc (pI = 7.1), and sFcγRIIIb (pI = 6.2), respectively. This may also explain why mouse IgG2b-Fc (pI = 6.3) shows little binding to HSA. This means that charge variations of IgG1, such as deamination, affect its interaction with HSA. According to this theory, an Fab with a λ chain would be more intensively interact with HSA than rituximab-Fab. This is because the Cλ domains have a higher isoelectric point (pI = 6.9 – 9.1) than that of the Cκ domain (pI = 5.6), which is harbored in rituximab-Fab.
Although much less pronounced than the effect of HSA, the Fab fragment from the pooled serum polyclonal IgG specifically interacted with the Fc region of rituximab and significantly affected ADCC. It was unexpected that polyclonal IgG-Fab interacted with the Fc region of rituximab, i.e. human IgG1-Fc, rather than the more heterologous rituximab-Fab harboring mouse-derived variable region. As in the HSA interaction, this significant preference may be attributed to the difference in net charge between Fab and Fc regions in rituximab because polyclonal IgG-Fab isolated from human serum generally has a pI of >7 (34). However, polyclonal IgG-Fab barely interacts with sFcγRIIIb, which has the lower pI value than rituximab-Fc, suggesting that some unknown factors determine the reactivity of polyclonal IgG-Fab. Anyway, the negative effect on ADCC is plausibly enhanced in the full-length form of serum polyclonal IgG with bivalency and bulkiness in addition to the Fc-mediated competition for FcγRIIIa (5). Indeed, the intact form of polyclonal IgG and human serum containing it had greater impacts on rituximab-Fc than the Fab fragment derived it (Figure 6).
Because many therapeutic antibodies and Fc-fusion therapeutics share the common Fc region derived from human IgG1 (35), serum polyclonal IgG as well as HSA are likely to interact with these protein drugs affecting their interactions with FcγRIIIa and functional efficacy. The interactions with the serum proteins cover the Fc surface so extensively that they can act pan-inhibitory on various Fc receptor-mediated functions and pharmacokinetics. Furthermore, the non-competitive inhibition of ADCC by HSA and polyclonal IgG-Fab suggests that they can be allosteric inhibitors of Fc affecting its dynamic structure (36). Therefore, controlling the interactions with the serum proteins is an essential factor to consider in the development of therapeutic antibodies and Fc-fusion therapeutics. Their interactions are likely to depend on artificial and naturally occurring modifications, such as drug conjugation and glycosylation, and are presumably controllable through mutational charge modifications. Serum components, including HSA and polyclonal antibodies, can differ qualitatively and quantitatively depending on the disease state and drug administration history (37). In this regard, methods to assess interactions with serum components are valuable for the development and application of antibody-based drugs with increased efficacy and safety.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
SY: Conceptualization; data curation; funding acquisition; formal analysis; investigation; writing – original draft preparation; writing – review and editing. RY: Conceptualization; data curation; writing – review and editing. HY: data curation; writing – review and editing. MO: data curation; writing – review and editing. NW: data curation; writing – review and editing. YY: data curation; writing – review and editing. SU: data curation; investigation; writing – review and editing. KK: Conceptualization; funding acquisition; investigation; project administration; supervision; writing – original draft preparation; writing – review and editing. All authors contributed to the article and approved the submitted version.
This study was supported in part by the Nanotechnology Platform Program (Molecule and Material Synthesis) and by the Advanced NMR Application and Platform (program No. PF17-01-R-008 and PF19-01-R-022) of the Ministry of Education, Culture, Sports, Science and Technology (MEXT), and by Joint Research of the Exploratory Research Center on Life and Living Systems (ExCELLS) (ExCELLS program No.20-308 and 21-308 to HY). This study was also supported by Japan and MEXT/JSPS Grants-in-Aid for Scientific Research (Grant Numbers, JP19J15602 to RY; JP17H05893, JP20K15981, and JP22H02755 to SY; and JP19H01017 to KK) as well as AMED (Grant Numbers JP21ae0121020h0001 to SY and JP21ae0121013h0301 to KK).
We thank Kiyomi Senda (Nagoya City University) and Yukiko Isono (Institute for Molecular Science) for their assistance in the preparation of the recombinant proteins.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Vallner JJ. Binding of drugs by albumin and plasma-protein. J Pharm Sci (1977) 66(4):447–65. doi: 10.1002/jps.2600660402
2. Meyer MC, Guttman DE. The binding of drugs by plasma proteins. J Pharm Sci (1968) 57(6):895–918. doi: 10.1002/jps.2600570601
3. Jusko WJ, Gretch M. Plasma and tissue protein-binding of drugs in pharmacokinetics. Drug Metab Rev (1976) 5(1):43–140. doi: 10.3109/03602537608995839
4. Jin JF, Zhu LL, Chen M, Xu HM, Wang HF, Feng XQ, et al. The optimal choice of medication administration route regarding intravenous, intramuscular, and subcutaneous injection. Patient Prefer Adherence (2015) 9:923–42. doi: 10.2147/Ppa.S87271
5. Preithner S, Elm S, Lippold S, Locher M, Wolf A, da Silva AJ, et al. High concentrations of therapeutic IgG1 antibodies are needed to compensate for inhibition of antibody-dependent cellular cytotoxicity by excess endogenous immunoglobulin G. Mol Immunol (2006) 43(8):1183–93. doi: 10.1016/j.molimm.2005.07.010
6. Saunders KO. Conceptual approaches to modulating antibody effector functions and circulation half-life. Front Immunol (2019) 10:1296. doi: 10.3389/fimmu.2019.01296
7. Krayukhina E, Noda M, Ishii K, Maruno T, Wakabayashi H, Tada M, et al. Analytical ultracentrifugation with fluorescence detection system reveals differences in complex formation between recombinant human TNF and different biological TNF antagonists in various environments. MAbs (2017) 9(4):664–79. doi: 10.1080/19420862.2017.1297909
8. Wright RT, Hayes DB, Stafford WF, Sherwood PJ, Correia JJ. Characterization of therapeutic antibodies in the presence of human serum proteins by AU-FDS analytical ultracentrifugation. Anal Biochem (2018) 550:72–83. doi: 10.1016/j.ab.2018.04.002
9. Yanaka S, Yamazaki T, Yogo R, Noda M, Uchiyama S, Yagi H, et al. NMR detection of semi-specific antibody interactions in serum environments. Molecules (2017) 22(10). doi: 10.3390/molecules22101619
10. Yanaka S, Yagi H, Yogo R, Yagi-Utsumi M, Kato K. Stable isotope labeling approaches for NMR characterization of glycoproteins using eukaryotic expression systems. J Biomol NMR (2018) 71(3):193–202. doi: 10.1007/s10858-018-0169-2
11. Marshall MJE, Stopforth RJ, Cragg MS. Therapeutic antibodies: What have we learnt from targeting CD20 and where are we going? Front Immunol (2017) 8:1245. doi: 10.3389/fimmu.2017.01245
12. Tirumalai RS, Chan KC, Prieto DA, Issaq HJ, Conrads TP, Veenstra TD. Characterization of the low molecular weight human serum proteome. Mol Cell Proteomics (2003) 2(10):1096–103. doi: 10.1074/mcp.M300031-MCP200
13. Yogo R, Yamaguchi Y, Watanabe H, Yagi H, Satoh T, Nakanishi M, et al. The fab portion of immunoglobulin G contributes to its binding to Fcγ receptor III. Sci Rep (2019) 9:11957. doi: 10.1038/s41598-019-48323-w
14. Yanaka S, Yagi H, Yogo R, Onitsuka M, Kato K. Glutamine-free mammalian expression of recombinant glycoproteins with uniform isotope labeling: An application for NMR analysis of pharmaceutically relevant Fc glycoforms of human immunoglobulin G1. J Biomol NMR (2022) 76(1-2):17–22. doi: 10.1007/s10858-021-00387-5
15. Yogo R, Yanaka S, Kato K. Backbone 1H, 13C, and 15N assignments of the extracellular region of human Fcγ receptor IIIb. Biomol NMR Assign (2018) 12(1):201–4. doi: 10.1007/s12104-018-9809-4
16. Yagi H, Zhang Y, Yagi-Utsumi M, Yamaguchi T, Iida S, Yamaguchi Y, et al. Backbone 1H, 13C, and 15N resonance assignments of the Fc fragment of human immunoglobulin G glycoprotein. Biomol NMR Assign (2015) 9(2):257–60. doi: 10.1007/s12104-014-9586-7
17. Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J Biomol NMR (1995) 6(3):277–93. doi: 10.1007/BF00197809
18. Shibata-Koyama M, Iida S, Okazaki A, Mori K, Kitajima-Miyama K, Saitou S, et al. The n-linked oligosaccharide at FcγRIIIa asn-45: An inhibitory element for high FcγRIIIa binding affinity to IgG glycoforms lacking core fucosylation. Glycobiology (2009) 19(2):126–34. doi: 10.1093/glycob/cwn110
19. Yamaguchi Y, Wakaizumi N, Irisa M, Maruno T, Shimada M, Shintani K, et al. The fab portion of immunoglobulin G has sites in the CL domain that interact with Fcγ receptor IIIa. MAbs (2022) 14(1):2038531. doi: 10.1080/19420862.2022.2038531
20. Tada M, Ishii-Watabe A, Suzuki T, Kawasaki N. Development of a cell-based assay measuring the activation of Fcγ RIIa for the characterization of therapeutic monoclonal antibodies. PloS One (2014) 9(4):e95787. doi: 10.1371/journal.pone.0095787
21. Bjellqvist B, Basse B, Olsen E, Celis JE. Reference points for comparisons of two-dimensional maps of proteins from different human cell types defined in a pH scale where isoelectric points correlate with polypeptide compositions. Electrophoresis (1994) 15(3-4):529–39. doi: 10.1002/elps.1150150171
22. Kiyoshi M, Caaveiro JMM, Tada M, Tamura H, Tanaka T, Terao Y, et al. Assessing the heterogeneity of the Fc-glycan of a therapeutic antibody using an engineered Fcγ Receptor IIIa-immobilized column. Sci Rep (2018) 8(1):3955. doi: 10.1038/s41598-018-22199-8
23. Gonzalez-Quintela A, Alende R, Gude F, Campos J, Rey J, Meijide LM, et al. Serum levels of immunoglobulins (IgG, IgA, IgM) in a general adult population and their relationship with alcohol consumption, smoking and common metabolic abnormalities. Clin Exp Immunol (2008) 151(1):42–50. doi: 10.1111/j.1365-2249.2007.03545.x
24. Peter T. All about albumin: Biochemistry, genetics, and medical applications London. UK: Academic Press (1996).
25. Yamaguchi Y, Yagi H, Kato K. Stable isotope labeling of glycoproteins for NMR study. In: Kato K, Peters T, editors. NMR in glycoscience and glycotechnology. (Cambridge: RSC Publishing) (2017). p. 194–205.
26. Kato K, Yanaka S, Yagi H. Technical basis for nuclear magnetic resonance approach for glycoproteins. In: Naito A, Asakura T, Shimada I, Takegoshi K, Yamamoto Y, editors. Experimental approaches of NMR spectroscopy-methodology and application to life science and materials science. Japan: Springer (2018). p. 415–38.
27. Matsumiya S, Yamaguchi Y, Saito J, Nagano M, Sasakawa H, Otaki S, et al. Structural comparison of fucosylated and nonfucosylated Fc fragments of human immunoglobulin G1. J Mol Biol (2007) 368(3):767–79. doi: 10.1016/j.jmb.2007.02.034
28. Radaev S, Motyka S, Fridman WH, Sautes-Fridman C, Sun PD. The structure of a human type III Fcγ receptor in complex with Fc. J Biol Chem (2001) 276(19):16469–77. doi: 10.1074/jbc.M100350200
30. Berinstein NL, Grillo-Lopez AJ, White CA, Bence-Bruckler I, Maloney D, Czuczman M, et al. Association of serum rituximab (IDEC-C2B8) concentration and anti-tumor response in the treatment of recurrent low-grade or follicular non-hodgkin's lymphoma. Ann Oncol (1998) 9(9):995–1001. doi: 10.1023/A:1008416911099
31. Anderson KW, Bergonzo C, Scott K, Karageorgos IL, Gallagher ES, Tayi VS, et al. HDX-MS and MD simulations provide evidence for stabilization of the IgG1-Fcγ RIa (CD64a) immune complex through intermolecular glycoprotein bonds. J Mol Biol (2022) 434(2):167391. doi: 10.1016/j.jmb.2021.167391
32. He XM, Carter DC. Atomic structure and chemistry of human serum albumin. Nature (1992) 358(6383):209–15. doi: 10.1038/358209a0
33. Hintersteiner B, Lingg N, Zhang P, Woen S, Hoi KM, Stranner S. Et al, charge heterogeneity: Basic antibody charge variants with increased binding to Fc receptors. MAbs (2016) 8(8):1548–60. doi: 10.1080/19420862.2016.1225642
34. Harada S, Nishimura S, Hosoi S, Mikawa H. Purification of polyclonal human IgG of four subclasses from pooled human γ globulin. Seibutsubutsurikagaku (1988) 32:285–93. doi: 10.2198/sbk.32.285
35. Lu RM, Hwang YC, Liu IJ, Lee CC, Tsai HZ, Li HJ, et al. Development of therapeutic antibodies for the treatment of diseases. J BioMed Sci (2020) 27(1):1. doi: 10.1186/s12929-019-0592-z
36. Yanaka S, Yogo R, Inoue R, Sugiyama M, Itoh SG, Okumura H, et al. Dynamic views of the Fc region of immunoglobulin G provided by experimental and computational observations. Antib (Basel) (2019) 8(3). doi: 10.3390/antib8030039
Keywords: human serum albumin, therapeutic antibody, antibody-dependent cellular cytotoxicity, NMR, stable isotope labeling
Citation: Yanaka S, Yogo R, Yagi H, Onitsuka M, Wakaizumi N, Yamaguchi Y, Uchiyama S and Kato K (2023) Negative interference with antibody-dependent cellular cytotoxicity mediated by rituximab from its interactions with human serum proteins. Front. Immunol. 14:1090898. doi: 10.3389/fimmu.2023.1090898
Received: 06 November 2022; Accepted: 03 January 2023;
Published: 25 January 2023.
Edited by:
Tilman Schlothauer, Roche Innovation Center Munich, GermanyReviewed by:
Aaron Wecksler, Genentech Inc., United StatesCopyright © 2023 Yanaka, Yogo, Yagi, Onitsuka, Wakaizumi, Yamaguchi, Uchiyama and Kato. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Koichi Kato, a2thdG9ubXJAaW1zLmFjLmpw
†Present address: Rina Yogo, Biomedical Research Centre, School of Biomedical Engineering, The University of British Columbia, Vancouver, Canada
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.