 Shuang Yang1
Shuang Yang1 Ming Zhao
Ming Zhao Sujie Jia
Sujie Jia
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 14 February 2023
Sec. Inflammation
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1080310
This article is part of the Research TopicPlasticity of monocytes/macrophages: phenotypic changes during disease progressionView all 17 articles
The macrophage is an essential part of the innate immune system and also serves as the bridge between innate immunity and adaptive immune response. As the initiator and executor of the adaptive immune response, macrophage plays an important role in various physiological processes such as immune tolerance, fibrosis, inflammatory response, angiogenesis and phagocytosis of apoptotic cells. Consequently, macrophage dysfunction is a vital cause of the occurrence and development of autoimmune diseases. In this review, we mainly discuss the functions of macrophages in autoimmune diseases, especially in systemic lupus erythematosus (SLE), rheumatic arthritis (RA), systemic sclerosis (SSc) and type 1 diabetes (T1D), providing references for the treatment and prevention of autoimmune diseases.
According to whether tissues and organs are targeted by the damaging immune response, autoimmune diseases classified into systemic autoimmune disease, such as systemic lupus erythematosus (SLE) and systemic sclerosis (SSc) and rheumatoid arthritis (RA), or organ-specific autoimmune diseases, such as thyroid disease, type 1 diabetes (T1D), myasthenia gravis and multiple sclerosis (1, 2). The autoimmune diseases are clinically diverse but share a fundamental etiology: the form of self-reactive antibodies, presence of self-reactive T cells, and activation of the innate immune system (3). Although the exact pathogenesis remains unclear, it is interesting to note that genetic, immunological, hormonal and environmental factors are important triggers for autoimmune diseases (4).
However, it is difficult to precisely inhibit the abnormal immunity activation triggered by pathogenic factors. The current treatment of autoimmune diseases is limited and relatively conservative, which mainly depends on the overall inhibition of the immune response. However, blindly suppressing the immune response can cause inevitable side effects such as infection. Therefore, there is an urgent need to understand the pathological mechanism that causes the initiation and development of autoimmune diseases so as to provide new ideas for the prevention and treatment of autoimmune diseases.
The innate immune system exerts immune function independently of antigens, which form the body’s immune defense system interacting with the adaptive immune system. Abnormal innate immune response is a significant reason for the breakdown of autoimmune tolerance, which is closely related to the occurrence and development of autoimmune diseases (5). Macrophage is a crucial part of the innate immune system and participates in almost every biological process such as tissue homeostasis, resisting infection, repairing after infection, metabolism and inflammation, affecting the body’s development and immune response (6, 7). This review summarizes the impaired functions and abnormal macrophage activation and their roles in the pathogenesis of autoimmune diseases showed in Figure 1, especially in SLE, RA, SSc and T1D. In addition, the potential value of macrophages in the treatment and prevention of autoimmune diseases is also summarized.
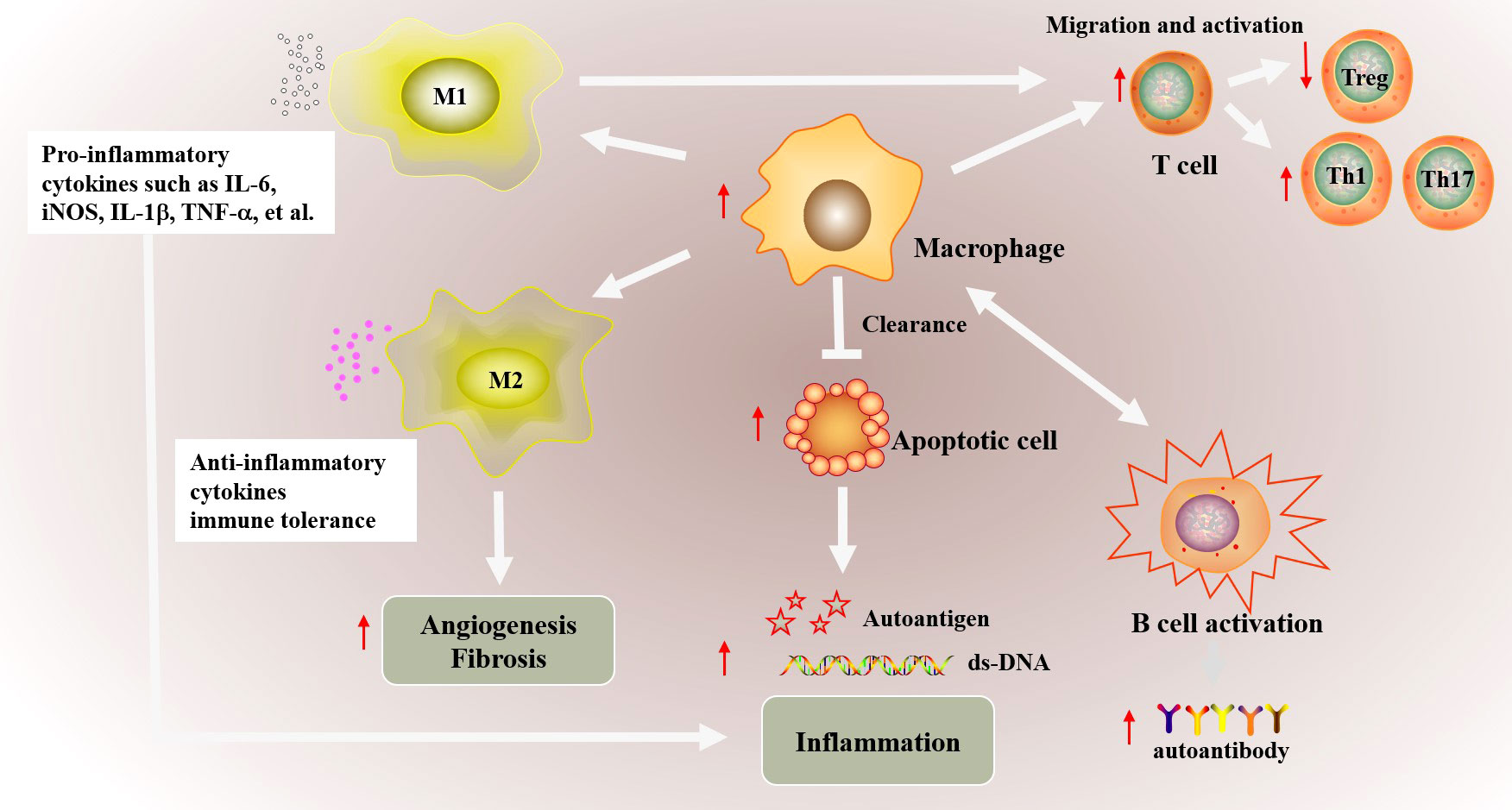
Figure 1 The possible abnormal macrophage activation in autoimmune diseases. The phagocytic function of macrophages is weakened in autoimmune diseases, which inhibits the clearance of apoptotic cells. Increased apoptotic cells promotes the production of autoimmune antigens and antibody, and further exacerbates inflammatory inflammation. In addition, macrophages promote the migration and abnormal activation of T cells including increased Th1/Th17 differentiation and downregulated Treg differentiation, and ultimately cause abnormal activation of B cells. Besides, the imbalance of M1/M2 macrophages also involved in autoimmune. Abnormal M1 macrophage activation promotes the production of proinflammatory cytokines such as IL-6, iNOS, TNF-α and IL-1β, which promote inflammation in targeted organs. Decreased M2 polarization inhibited the production of anti-inflammatory cytokines and the immune tolerance. Besides, abnormal M2 macrophage polarization also affects vascular proliferation, fibrosis in autoimmune disease such as SSc.
It has been universally accepted that macrophages in tissues are differentiated from monocytes that originate in bone marrow (8). However, studies in recent years have found that monocytes are not the only source of macrophages. Tissue macrophages are also derived from the yolk sac and fetal liver, which have self-renewal properties independent of monocyte recruitment (9, 10). According to the tissue distribution, macrophages can be divided into alveolar macrophages, intestinal macrophages, osteoclasts in bone, microglia in the brain, Kupffer cells in the liver, Langerhans cells in the epidermis (10). Secondary lymphoid organs also have distinct macrophages, including marginal zone macrophages (MZMs) and metallophilic macrophages in the spleen, which involved in clearance of apoptotic cell and tolerance to auto-antigens (11). It is worth noting that microglia and partial Langerhans cells are derived from yolk sac progenitor cells as shown by pedigree tracing experiments. In contrast, macrophages in other tissues, such as intestinal lamina propria and dermis, are mainly derived from hematopoietic stem cells (12–16). Macrophages are, therefore, key tissue sentinel cells that react to tissue-specific signals, while retaining the ability to execute physiological functions such as phagocytes. During chronic inflammation such as autoimmune diseases, tissue-resident macrophages fail to solve aggravated inflammation that leads to immune system abnormal activation and damage. And peripheral monocytes are recruited and differentiated into macrophages non-homeostatically in combination with injury-associated signals including pro-inflammatory cytokines, which are further activated and participated in the body’s immune responses (17, 18). The tissue-resident macrophages participate jointly in protecting tissue homeostasis, and form the first line of defense against invading pathogens. Miriam. et al. considered that embryonically derived and monocyte-derived tissue-resident macrophages are likely to promote the development of the disease through the maintenance of tissue homeostasis through phagocytosis of cell fragments, resistance to pathogen invasion, while recruited monocyte-derived macrophages by disease-associated signals drives disease progression (19). Similarly, recruited monocyte-derived macrophage also plays an important role in autoimmune related diseases. For example, infiltrated macrophages, especially proliferating macrophages was seen in glomerulonephritis from patients with lupus, which may be a potential diagnostic and prognostic indicator for renal injury (20). Ly6C is a marker for circulating monocytes in mice. Different monocyte subpopulations Ly6Chi and Ly6Clo exist in mice, which express different adhesion molecule and chemokine receptor and gene expression profile. Response to inflammatory signals Ly6Chi monocytes could rapidly infiltrate in inflamed tissues mostly dependent on chemokine receptors C-C motif chemokine receptor 2 (CCR2), CCR6 and CCR8 and results in enhanced liver fibrosis (21). Inhibiting migration of blood monocyte into liver alleviated macrophage infiltration in liver, and decreased pro-inflammatory cytokines such as interferon gamma (IFNγ), IL-6 expression in chronic hepatic injury (22). Besides, inhibiting monocyte recruitment by blocking C-C motif chemokine ligand 24 (CCL24) or CCL2 may be an appealing novel therapy to limit fibrotic manifestations of SSc (23).
The complex origin of macrophages has caused great difficulties in the study of macrophage functions in autoimmune diseases. Although the construction of mice with myeloid knockout has brought a lot of convenience for the study of macrophages in vivo, it also has certain limitations. On one hand, there are many kinds of myeloid cells, and it is difficult to accurately study the function of a single macrophage. On the other hand, macrophages in different tissue are heterogeneous and plastic, showing different morphologies and surface molecules. With the development of scientific research, especially in flow cytometry and single cell sequencing technology, the macrophage markers in different tissues are gradually discovered, and the research of macrophages ushers in new opportunities and challenges. At present, the studies about macrophage from different sources is limited, and mostly current studies focus on the abnormal function and mechanism of macrophages infiltrated in targeted organs and tissues. How to specifically distinguish unbalanced macrophages, specially manufacture macrophages that promote disease, and supplement and maintain tissue stable macrophages are the key and difficult points in autoimmune disease research.
Macrophages are vital participant of innate immunity, which recognize and effectively respond to invading pathogens, thus providing an early defense against external attack. Pattern recognition receptors (PRRs) on the surfaces of macrophage including toll-like receptors (TLRs) and the NOD-like receptors (NLRs) recognize pathogen-associated molecular patterns (PAMPs) and endogenous danger-associated molecular patterns (DAMPs) presented in the invaders and promote macrophage activation. Macrophages further release antimicrobial mediators to target the invading pathogen, chemokines to recruit immune cells to the inflammatory site, and pro-inflammatory cytokines to aggravate further inflammation, and even induce the adaptive immune response for the particular invading pathogen. Besides, macrophage forms a bridge connecting innate and adaptive immunity by presenting endogenous or exogenous antigen. It has been well known that antigen cross-presentation is crucial for initiating of adaptive immune responses against cancer, infection and immune tolerance. During this process, antigen-presenting cells (APCs) present intracellular and extracellular peptides derived from ingested antigens on primary histocompatibility complex class I (MHCI) protein complex to T lymphocytes (24). Although the cross-presentation of antigens by macrophages is not understood as well as that by dendritic cells (DCs), it is becoming clear that the cross-presentation by macrophages especially in spleen, liver and lymph nodes may help activate CD8+ T lymphocytes (25).
Macrophage can participate in antigen presentation to Th1 cells and proliferation of T cells by surface co-expression molecules CD86 and MHCII, which indicate the significant role of the macrophage in the development of cancer, autoimmunity and viral infections (26–29). The CD8+ T cells in mice with spontaneous autoimmune peripheral neuropathy (APN) exhibit an effector/memory phenotype required for the disease initiation. However, only effector/memory CD8+ T (CD8+ TEM) cells are not sufficient to induce autoimmune-mediated peripheral neuropathy and macrophages are additionally required (30). The early depletion of regulatory T cells (Tregs) in mice with acute cardiac injury enhances the inflammatory activation of macrophages by increasing the production of IFN-γ, which restrains muscle regeneration (31). Human macrophages activated by C1q can inhibit the T helper (Th)17 and Th1 but promote Treg proliferation, orchestrating the adaptive immune system to avoid autoimmunity (32). Hence, the role of macrophage in connecting innate/adaptive immunity provides opportunities to prevent disease onset, reduce relapses and develop new therapeutic strategies. Intervening macrophage-T cell communication signals to prevent excessive activation of T cells may be an important research direction in the treatment of autoimmune diseases
Phagocytosis is an essential process for the uptake of particulate matter, including microbes and dying cells. Dying cells can expose and secrete signals that attract phagocytes and promote their phagocytosis. Several studies have shown that macrophage phagocytosis is affected by a variety of signaling pathways including TLRs (26). Reactive oxygen species (ROS) generated by the nicotinamide adenine dinucleotide phosphate oxidases (NADPH oxidase-2, also known as NOX2) in macrophages is dispensable for phagocytosis (33). The liver X receptors (LXRs) and the peroxisome proliferator-activated receptors (PPARs), nuclear receptor families that regulate genes involved in lipid metabolism and transport are important components of macrophage phagocytosis (34). The phagocytosis of dead and dying cells is a process known as efferocytosis, which is performed by macrophages, other immune phagocytes such as monocyte and DCs and non-phagocytes including epithelial cells. Efficient efferocytosis limits the release of intracellular PAMPs that drive inflammation and disrupting homeostatic efferocytosis can also lead to accumulation of uncleared apoptotic cells in autoimmune diseases. Efferocytosis mechanisms depends on the signaling programs depicted: chemoattractant-mediated recruitment of phagocytes, receptor-mediated recognition such as PtdSer receptor cell immunoglobulin mucin receptor 4 (TIM4), TAM family receptor tyrosine kinase receptor, engulfment of apoptotic cells, and the processing of engulfed cellular material (35). Disrupted efferocytosis of macrophage promoted the accumulation of uncleared apoptotic or necroptosis cells in autoimmune, which is a universal feature of damaged tissues (36).
In response to exogenous danger signals or exogenous signals recognized by pattern-recognition receptors (PRRs), macrophages undergo physiological changes to initiate signal transduction cascades and result in abnormal production of chemokines, cytokines and toxic mediators, which can further enhance inflammation and contribute to autoimmune pathologies (37). The anti-inflammatory mediators by macrophages contribute to the dissolution of the inflammatory response. Cytokines such as tumor necrosis factor (TNF)-α, Interleukin (IL)-6, IL-1β, IL-12, IL-18, IL-23 and chemokines such as CXC chemokine ligands (CXCL)1, CXCL3 are secreted by macrophages, which are essential mediators and drivers of chronic inflammation and autoimmune diseases (38–40). Besides, macrophages contribute to angiogenesis by secreting proangiogenic proteases such as matrix metalloproteinases (MMP)-9 and MMP-12 (41–43). TNF-α occupies a pivotal position in RA pathogenesis. The TNF blockade reduced stromal cell activation, angiogenesis, and sustain regulatory pathways by mediating cytokine and chemokine and MMPs expression. And IL-6 signaling pathway promotes T cell activation and migration by regulating chemokine expression (44). In addition to clearing dead cells, macrophages significantly mediate wound healing and tissue homeostasis by producing anti-inflammatory molecules and tissue remodeling growth factors like IL-10 and transforming growth factor beta(TGF-β) (45). Cytokines including IL-6, IL-23, IL-10 and TGF-β all shaped Th17 cell differentiation placed at the center of autoimmune inflammation (46). IL-18 contributes to Th1/2 differentiation, participate in cytotoxic T cells (CTLs) and natural killer (NK) cells activation, and ultimately IgE production from B cells (47). Besides, tissue macrophages synthesize chemokines CXCL1/CXCL2 to increase neutrophil recruitment, which is an important early step in controlling tissue infections or injury (48). Islet-resident and islet-infiltrating macrophages can exacerbate β-cell destruction by synthesizing TNF-α, IL-12, IL-1β, and NOX2-derived ROS, which mature autoreactive CD4 and CD8 T cell effector responses (49).
Macrophages are also involved in a variety of metabolic processes, including arginine metabolism and glucose metabolism, which was indicated in Figure 2. The M1 macrophages express nitric oxide synthase (NOS) to metabolize arginine into NO and citrulline, which further promotes the synthesis of downstream active nitrogen, finally facilitating inflammatory response (50, 51). In addition, M2 macrophages regulate arginine metabolism and thus regulate cell proliferation, tissue repair and inhibited inflammation by medicating polyamine/proline synthesis (52). Macrophages maintain adaptive responses to oxygen gradients and hypoxia by regulating their glucose oxidative phosphorylation, glycolysis and fatty acid oxidation (53). Based on the demands for energy and the production of specific functional-associated factors, pro-inflammatory macrophages and anti-inflammatory macrophages opt for distinct metabolic pathways upon activation. Instead of M2 macrophage, M1 macrophages carry out glycolysis and rely on fatty acid biosynthesis, and increased glycolysis causes succinate accumulation and promote inflammation by ROS/(hypoxia-inducible factor-1α) HIF-1α/IL1β pathway (54, 55). On the other hand, M2 macrophages possess a high basal mitochondrial oxygen consumption rate (OCR), carry out oxidative phosphorylation (OXPHOS), and require the induction of fatty acid oxidation (55, 56). Different fatty acid metabolism, particularly mitochondrial fatty acid oxidation in macrophage modulates inflammatory signatures and macrophage phenotype, which indicated the vital function of macrophage in hyperlipidemia-associated autoimmune diseases include psoriasis, RA, and SLE. Programmed macrophages by setting metabolic commitment for OXPHOS increased programmed death ligand 1 (PD-L1) expression, decreased IL-1β after pro-inflammatory activation in macrophage, and promoted Treg differentiation to increase the regulatory function in immune system (57). Moreover, macrophages resist parasite infection by regulating glutathione and redox metabolism and participate in tissue repair, tumor growth and anti-inflammatory response by regulating iron metabolism (58–60). Furthermore, the metabolites, in turn, mediate the macrophage response to inflammation. Recent study has found that citrulline levels in lipopolysaccharide (LPS) and IFNγ-stimulated macrophages are significantly reduced, which promotes inflammatory signals by activating Janus kinase 2 (JAK2)- signal transducer and activator of transcription 1 (STAT1) pathway (61). Citrulline can inhibit bacterial load in the spleen and liver of Listeria monocytogenes-infected mice by impeding pro-inflammatory macrophage activation (61).
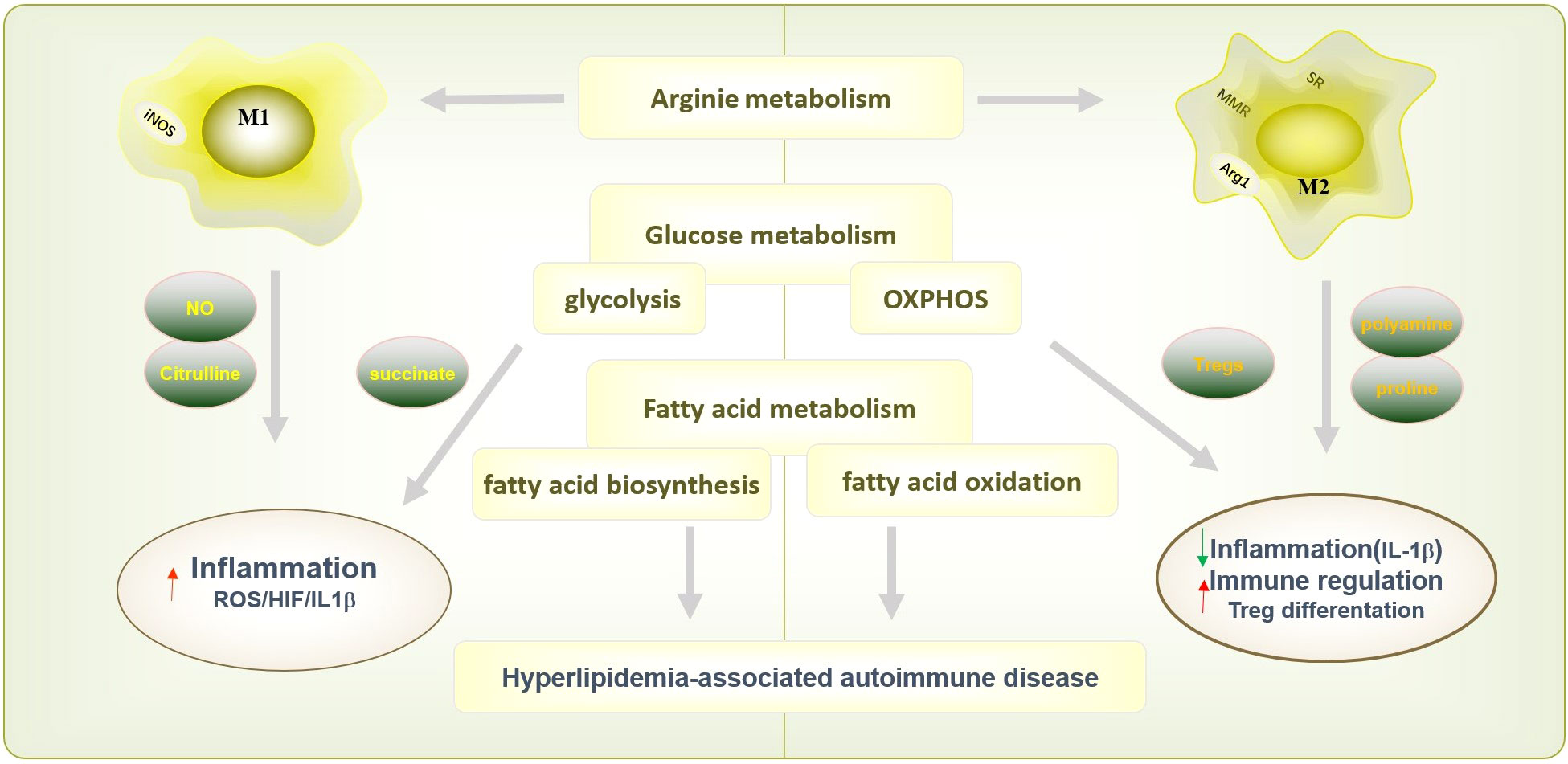
Figure 2 Macrophages polarization and function in metabolism processes. Macrophage are also involved in a variety of metabolic processes, including arginine metabolism glucose metabolism and fatty acid metabolism, to exert different immunological functions. The M1 macrophages express nitric oxide synthase (NOS) to metabolize arginine into NO and citrulline, which further promotes inflammatory response. Instead, M2 macrophages inhibited inflammation by medicating polyamine/proline synthesis. M1 macrophages carry out glycolysis, and causes succinate accumulation and promote inflammation by ROS/HIF-1α/IL1β pathway. And M2 macrophages carry out oxidative phosphorylation (OXPHOS) could decrease IL-1β after pro-inflammatory activation in macrophage, and promoted Treg differentiation to increase the regulatory function in immune system. M1 macrophage rely on fatty acid biosynthesis and M2 macrophage require fatty acid oxidation. The function of macrophage on fatty acid metabolism modulates inflammatory signatures and involved in hyperlipidemia-associated autoimmune diseases include psoriasis, RA, and SLE.
Macrophages display specific phenotypes and rapidly change their functions under the local microenvironment, called macrophage polarization (62). The phenotypes of macrophage polarization are generally divided into two types: one is classically activated macrophages (M1), which are pro-inflammatory and involved in the elimination of pathogens and resist infection. The other is alternative activation macrophages (M2) that are anti-inflammatory and involved in tissue repair and reconstruction (63). The Th1 cytokine, such as IFNγ or LPS, can induce M1 polarization, while Th2 cytokines, such as IL-4, can induce M2 polarization. Intracellular metabolite profiles of each macrophage activation state presented a unique metabolic signature. The 1D 1H NMR-based metabolomics identified increased adenosine triphosphate (ATP) and decreased intracellular nicotinamide adenine dinucleotide (NAD+) in M1 macrophage, and increased adenosine diphosphate (ADP), guanosine triphosphate (GTP), adenosine monophosphate (AMP) in M2 macrophage (64). The M1 macrophages express high levels of pro-inflammatory cytokines, active nitrogen and oxygen intermediates, promote the responses of Th1 and Th17 by secreting IL12 and IL23, and have strong bactericidal and tumor-killing activity (63, 65). However, the M2 macrophages indicate high phagocytic activity and high expression of scavenger receptor (SR), macrophage mannose receptor (MMR), arginase-1(Arg-1), IL10, TGF-β, which are mainly involved in parasite containment, phagocytosis, promote tissue repair, wound healing, angiogenesis, fibrosis and immune regulation (66, 67). In fact, depending on induced agents, expressed markers, secreted mediators and functions, M2 macrophages are further classified as M2a, M2b, M2c, as well as M2d macrophages. The M2a macrophages induced by IL-4 or IL-13, also known as wound healing macrophages, can increase endocytosis activity and have immunity to parasites, tissue repair, collagen formation and fibrogenesis (68). M2b macrophages stimulated by immune complexes, TLR ligands or IL-1β, also known as regulatory macrophages, have strong anti-inflammatory and immunosuppressive effects (69). M2c macrophages induced by glucocorticoids, IL-10 or TGF-β promote phagocytosis and clearance of dead cells (70). M2d macrophages induced mainly by TLR antagonists, also known as tumor associated macrophages (TAM), can promote angiogenesis and tumor progression (71). However, M1 and M2 macrophages are the two extremes of the activation state of macrophages which cannot fully represent macrophages in the complex microenvironment in vivo. The dynamic balance of M1/M2 is crucial to maintain homeostasis. Response to foreign stimulation such as microbial infection or tumor, M1 macrophage is activated and promote inflammation to perform robust antimicrobial and anti-tumoral function. And to protect against the chronic inflammatory response, M1 macrophage is inhibited by regulatory mechanisms driven by anti-inflammatory function of enhanced M2 macrophages differentiation and promote tissue regeneration, angiogenesis and wound healing (72). And the imbalance contributes to the occurrence and development of many diseases including infection, tumor and autoimmune diseases (73–75). Fortunately, the high degree of plasticity allows macrophage switch from one phenotype to another depending on encountered micro-environment signals in each specific tissue, which providing a potential treatment target for autoimmune disease.
SLE is a chronic systemic autoimmune disease with diverse clinical manifestations characterized by immune system infiltration and inflammation in damaged organs covering skin, lungs, joints, kidneys and central nervous system (76). The abnormalities in the activation state of circulating and tissue macrophages in patients with SLE are crucial factors in the occurrence and development of the disease (77, 78). Depleting macrophage attenuated skin and kidney disease severity, which suggested the vital function in SLE pathogenesis (79, 80). The pro-inflammatory patrolling monocytes (PMOS) accumulated in the glomeruli in SLE patients and lupus mice are the main components of lupus glomerular or kidney inflammation (81). Emerging evidence has demonstrated that macrophage infiltration is associated with lupus nephritis in mice and humans (82, 83). Renal macrophage infiltration appears in spontaneous NZB/W nephritis and IFN-accelerated models of lupus nephritis (84). The function and numbers of MZMs are also reduced in autoimmune BXD2 mice (85). The absence of MZMs results in retention of apoptotic cell debris within the marginal zone and drives follicular Ag-transportation by marginal zone B (MZB) cells to stimulate an autoimmune response (85). The abnormal functions of macrophage in SLE are indicated in Figure 3.
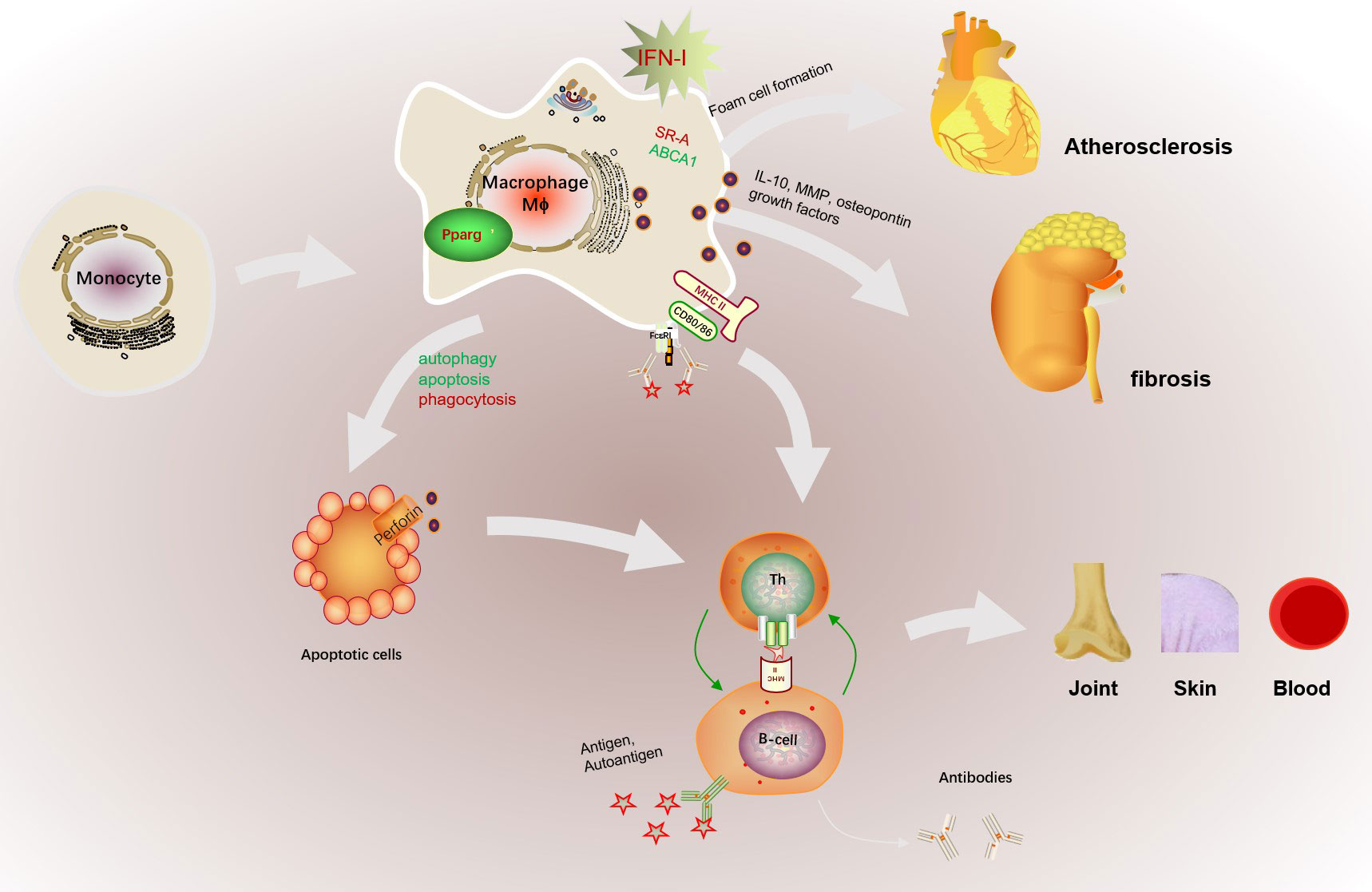
Figure 3 The abnormal activation of macrophage in SLE. The abnormal microenvironment in SLE patients, such as the high expressed IFN-I, promotes monocyte to recruit and differentiate into macrophages. In addition, increased antigen presentation of macrophage promotes B cell activation assisted with Th cells and further promotes the production of autoantibodies. Macrophages can also cause cardiovascular risk by promoting foam cell formation with increased SR-A and decreased ABCA1 expression. The dysfunction of macrophage phagocytosis may increase the gathered apoptotic cells and results in retention of apoptotic cell debris in SLE patients. Increased macrophage apoptosis and autophagy could contribute to autoantibody formation and organ damage by increased apoptotic load and impaired clearance of apoptotic material, finally exacerbated the production of autoantigens. Besides, macrophage infiltration the kidney promoted glomerular cell proliferation and early fibrosis by IL-10, MMP, osteopontin and growth factors. Besides, increased apoptotic cells serves as autoantigen to aggravate autoimmune reaction and may cause multiple targeted organs such as skin, joint, or blood.
The phagocytic ability of macrophages from SLE patients is weakened, which results in the production of autoantibodies and SLE-like autoimmune nephropahy (78, 85). Hence the dysfunction of macrophage phagocytosis may partly explain the gathering of apoptotic cells in the germinal center of lymph nodes in SLE patients (86). The mechanism response to the reduced clearance rate of macrophages has been widely demonstrated. It has been shown that the absence of PPARγ in macrophage cannot obtain an anti-inflammatory phenotype in the presence of apoptotic cells, finally resulting in glomerulonephritis and the autoantibodies production of nuclear Ags (87). The transcription factors Kruppel-like factor 2 (KLF2) and KLF4 also control apoptotic cells clearance program in tissue macrophage and maintain the homeostasis (88). Moreover, the increased autophagy and apoptosis in macrophage also contribute to the pathogenesis of SLE. The autophagy-related genes (Atg5, Atg12 and Beclin 1) were significantly upregulated in the splenic and renal macrophages in activated lymphocytes-derived DNA (ALD-DNA) induced lupus mice and in the peripheral blood mononuclear cells from SLE patients. And adoptive transfer of autophagy-suppressed macrophages alleviated lupus symptoms in SLE mice (89). Increased monocyte/macrophage apoptosis could contribute to autoantibody formation and organ damage by increased apoptotic load and impaired clearance of apoptotic material, finally exacerbated the autoimmune phenotype in NZB x SWR lupus-prone mice (90). Besides, research about lupus nephritis in NZB/W mice suggested that macrophage infiltration in the kidney promoted glomerular cell proliferation and early fibrosis by IL-10, MMP, osteopontin and growth factors (91).
SLE is a prototype autoimmune disease in which genetics play a major role. Researchers have identified many new loci which are attributed to the pathogenesis of SLE by genomewide association studies (GWAS). SLE susceptibility loci related to macrophages are mainly concentrated in genes that affect type I interferon (IFNI) signaling, NFκB activation, TLR signaling, phagocytosis and immune tolerance. Currently, more than 100 genetic risk sites related to SLE and more than half of them are closely related to the production or response of IFNI (92). IFNI promotes monocyte differentiation and the expression of MHCII and costimulatory molecules (such as CD40, CD80 and CD86) of macrophages to promote T cell activation (93, 94). Besides, increased IFNI levels in SLE patients can further promote the recruitment and adhesion of monocytes, and accumulation of macrophages in kidney and vascular lesions of SLE patients (95–97). In addition, IFNI enhances scavenger receptor SR-A and reduces ATP binding cassette subfamily A 1 (ABCA1) expression to promote cholesterol efflux, oxidation low lipoprotein (ox-LDL) uptake in macrophage and foam cell formation, which increasing the risk of cardiovascular diseases (96). Abnormal increased IFNI promotes the translocation of MZB cells to the follicular region of the spleen and disrupts the interaction between MZBs and MZMs, preventing clearance of apoptotic cells debris and follicular entry deterrence of apoptotic cells by MZMs (98, 99). The amplified TLR7 signaling in macrophage activation during antiviral responses and autoimmune diseases can occur product IFNI in turn by promoting phosphorylation and activation of MAP kinase p38 and transcription factor STAT1 (100). TNIP1(TNFAIP3-interacting protein 1, also known as ABIN1), a characteristic susceptibility gene for SLE identified by GWAS can regulate IFN-I production in DCs and macrophages through the TLR7 pathway (101). Large numbers of renal myeloid cells in patients with lupus nephritis, including macrophages, are activated. Almost all known susceptibility genes that affect innate immune signals may potentially affect the progression of lupus nephritis by activating myeloid cells in the kidney (102). Some genes such as ITGAM and FCR can potentially affect the recruitment of myeloid cells to the glomerular matrix by binding to the immune complexes in the glomerulus (103). DCs, macrophages and endothelial cells engulf C1q-coated apoptotic cells, and deficient in the complement protein C1q inhibit the clearance of apoptotic material and intensify lupus-like skin manifestations in mice and humans (100). ITGAM is an established SLE susceptibility locus, which impairs phagocytosis of complement-opsonized targets in monocytes, neutrophils and macrophages. In conclusion, these susceptible genes promote SLE pathogenesis through IFN-I-macrophage immune axis, and rebalancing macrophage functions may resist the damage of highly expressed IFN-I.
Abnormal macrophage polarization also has been identified in the occurrence and development of SLE. The overwhelming M1 macrophages promote the exposure of autoantigens and the occurrence of autoimmune reactions (104, 105). The gene expression profiles of myeloid cells from active SLE patients expressed higher M1-related genes and tend to promote inflammation. In comparison, myeloid cells from inactive SLE patients expressed higher M2-related genes and participated in immune repair (106). Aberrantly expanding M1 macrophages were dominating in MRL-Fas(Lpr) mice, hastened the onset of lupus nephritis, mediated defective renal repair and non-resolving inflammation (107). In the early stage of apoptosis, M2 macrophages can promote the production of anti-inflammatory factors and phagocytize apoptotic cells in an anti-inflammatory way called “bubble drink” (108, 109). Increased M2 macrophages reduced pro-inflammatory cytokines expression and increased the secretion of anti-inflammatory cytokines, which could be used for anti-inflammatory therapy in SLE (110). TIPE2 overexpression by AAV-TIPE2 induced M2 macrophage polarization, induced serum anti-dsDNA autoantibody and pathological renal damage, increased urine protein levels in the ALD-induced SLE mice (111). Adoptive transplantation of M2 macrophages or stimulating monocytes to differentiate into M2-like macrophages significantly reduced the severity of SLE, while M1 macrophage metastasis aggravated the development of SLE (112, 113). Virgin olive oil and its phenolic components have been shown to prevent various inflammatory and immune diseases, which may be related to inhibiting M1 and promoting M2 macrophage polarization (114, 115). The above studies show that the abnormal polarization of macrophages plays a vital role in SLE, which will be a potential target for SLE therapy.
Current therapies for SLE are designed to resolve inflammation with the goal of preventing permanent organ injury, and reduce clinical symptoms. Mycophenolate mofetil (MMF), an inhibitor of purine synthesis, inhibits the recruitment of monocytes and the production of nitric oxide and superoxide in activated macrophages to restrain tissue damage (116). The heterogeneity of disease mechanisms in SLE suggests that cell- and cytokine- or pathway-specific therapies for macrophage would be effective in treatment for SLE.
RA is an autoimmune disease characterized by chronic inflammation that eventually results in joint damage and even joint dysfunction. It has been found that macrophage infiltration is positively correlated with the degree of joint erosion, and increased synovial macrophage infiltration in synovial tissue is an early sign of RA (117–119). Clodronate could reduce knee swelling, inflammation and joint destruction by eliminating synovial macrophages in rats with antigen-induced arthritis (AIA) (120). Various mechanisms generally lead to increased macrophage infiltration in inflammatory sites, such as facilitating the expression of chemokines and pro-inflammatory cytokines, local survival rate/reducing apoptosis (121). Inhibited macrophage infiltration in synovial tissue may be a protential target for RA treatment. Increased apoptosis of Ly6C+ monocyte derived macrophages, reduced monocyte migration into the ankles and enhanced macrophage migration from the inflamed synovial tissue to the draining lymph nodes are responsible for the reduction of macrophages in synovial tissue after infliximab treatment alleviated disease progress in hTNF-Tg mice (122).
The infiltrated macrophage further mediated various inflammatory cell states, significantly contributing to the initiation and perpetuation of synovitis in RA by orchestrating cytokine network (123), as shown in Figure 4. Macrophages expedite inflammation by promoting the production of Th17 cells and stimulating osteoclast differentiation by secreting cytokines including IL-26 (124, 125). Besides, macrophages in synovial tissue and synovial fluid mediate the chemotaxis and proliferation of endothelial cells, promote the formation of pannus and infiltration of inflammatory cells, and further expand the inflammatory response in RA by producing vascular endothelial growth factor (VEGF) (126, 127). And macrophage-derived IL-8 also promote angiogenesis disorder in RA (128). The mechanism of abnormal activation of macrophages is not clear at present. Burbano. et al. found that increased circulating microparticles (MP) forming immune complexes in SLE and RA patients favored the polarization of monocyte-derived macrophages into a proinflammatory profile, which promoted T and B cell activation, and B-cell survival (129). Transcriptome profiles of highly inflamed RA synovial tissue (RA-ST) also demonstrated that monocytes/macrophages show similar gene patterns induced by bacterial and fungal, and activated B or T cells also activate monocytes/macrophages (130).
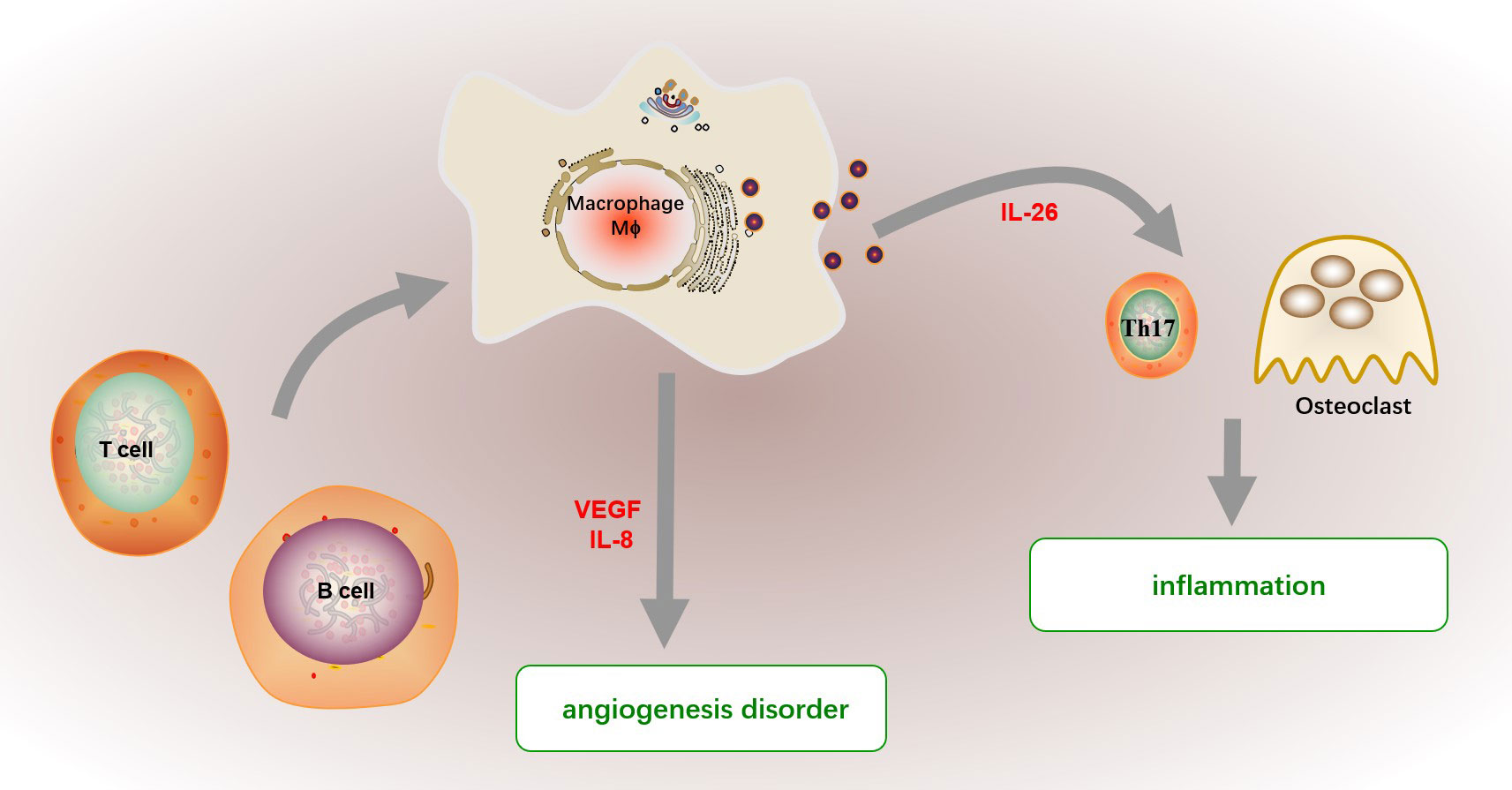
Figure 4 The function of macrophage in RA. The infiltrated macrophage further mediated various inflammatory cell states in synovitis by orchestrating cytokine network. Macrophages promote inflammation by promoting Th17 cell differentiation and stimulating osteoclast differentiation by secreting cytokines including IL-26. Besides, macrophages in synovial tissue and fluid mediate the chemotaxis and proliferation of endothelial cells, promote the formation of pannus and infiltration of inflammatory cells, and further expand the inflammation in RA by producing endothelial growth factor (VEGF). And macrophage-derived IL-8 also promote angiogenesis disorder in RA. The abnormally activated macrophage in RA patients show a proinflammatory profile, which may be supported by activated B or T cells.
Current conventional synthetic and biologic disease-modifying anti-rheumatic drugs (DMARDs) used in the clinic to treat of RA are related to adjusting macrophage activation and reducing synovial macrophage infiltration. Methotrexate, leflunomide or sulfasalazine reduces macrophage accumulation by promoting apoptosis and inhibiting Th1 response (123). Besides, the anti-TNF biological anti-rheumatic drugs such as etanercept, adalimumab decreased inflammatory cytokines production and increased phagocytosis in monocyte derived macrophages, which all alleviated inflammatory reactions (131). In addition, various monoclonal antibodies targeting biomolecules produced by macrophages are available for the therapeutic options of RA. The therapeutic efficacy of blocking granulocyte-macrophage colony stimulating factor receptor (GM-CSF) pathway like anti-GM-CSFR monoclonal antibody mavrilimumab is linked to inhibited production of pro-inflammatory mediators such as VICM (citrullinated and MMP degraded vimentin fragment) biomarker released by activated macrophages (132, 133). A monoclonal antibody to folate receptor β (FR-β) produced by macrophages specifically accumulates in inflamed lesions of murine RA and peritonitis disease models, facilitating immune cells, including T cells, B cells, neutrophils and DCs, to exit from the inflamed lesions and allative disease processes (134).
The imbalance of macrophage polarization also occurs in RA. The blood monocytes from RA patients had a propensity for preferential differentiate toward M1-like macrophages that contributed to synovial inflammation (135). Transcriptional omics study showed that synovial macrophages facilitate the expression of pro-inflammatory genes (INHBA, FCER1A, SLC2A1, MMP12, EGLN3, NOS and CCR2) but restrain anti-inflammatory genes (IGF1, HTR2B, FOLR2 and CD36) expression (136, 137). Besides, M1 macrophages are characterized by decreased heme uptake and iron output but increased iron storage, which could partly explain the phenomenon of anemia in RA patients (138). The M1-to-M2 macrophage re-polarization can also serve as a promising treatment for RA. Targeted biologics that selectively regulate the function of macrophages have broad research prospects for the treatment of RA and also could solve the adverse effects of non-targeted drugs to a certain extent. Interfering with glycolytic pathways activated in M1 macrophages can reduce pro-inflammatory factors production and IgG antibodies, finally alleviating joint inflammation and damage in CIA mice (139). The administration of Wilforlide A reduced clinical scores, joint swelling and histological damage of collagen-induced RA mice by inhibiting the secretion of pro-inflammatory factors (MCP1, GM-CSF and M-CSF) and iNOS in the synovium (140). Angiotensin II type 2 receptor (AT2R) activation and a developed triamcinolone-gold nanoparticle (Triam-AuNP) complex promotes proinflammatory synovial macrophages to differentiate into the tolerogenic macrophage, finally attenuating the joint pathology in a rat model of collagen-induced RA (141, 142). The above studies have shown that M1 macrophages are dominant in RA synovium, regulating abnormal macrophage polarization is one of the important therapies of RA. Many new drug vectors and targets have been found to regulate macrophage function selectively. Encapsulated plasmid DNA encoding IL-10 and the chemotherapeutic drug betamethasone sodium phosphate (BSP) in biomimetic vector M2 exosomes derived from M2 macrophages, folate-modified triptolide liposomes (FA-TP-Lips) and folic acid modified silver nanoparticles(FA-AgNPs) all serve as a promising biocompatible drug to facilitate M2 macrophages polarization selectively, thereby treating RA safely and effectively (143–145). However, macrophages are incredibly heterogeneous. The focus and difficulty of RA drug development will be how to distinguish, identify and act on specific activated pathogenic macrophages.
Systemic sclerosis (SSc) is a chronic multi-system disease characterized by autoimmunity, immune cell infiltration and activation, fibrosis and vascular lesions, often accompanied by skin involvement and visceral dysfunction including heart and lungs caused by fibrosis (146, 147). Vascular complications such as pulmonary hypertension and scleroderma renal crisis have become the leading causes of disability and death of SSc (148, 149). The infiltrating inflammatory leukocytes in the new affected skin from SSc patients are mainly CD14+ monocytes/macrophages (150). Transcriptomics analysis found that monocytes continuously migrated and differentiated into alveolar macrophages to promote fibrosis during pulmonary fibrosis and selectively targeting the differentiation of alveolar macrophages in the lung may improve fibrosis (151). These researches suggested that monocytes/macrophages play an essential role in the early pathogenesis of SSc, which was displayed in Figure 5.
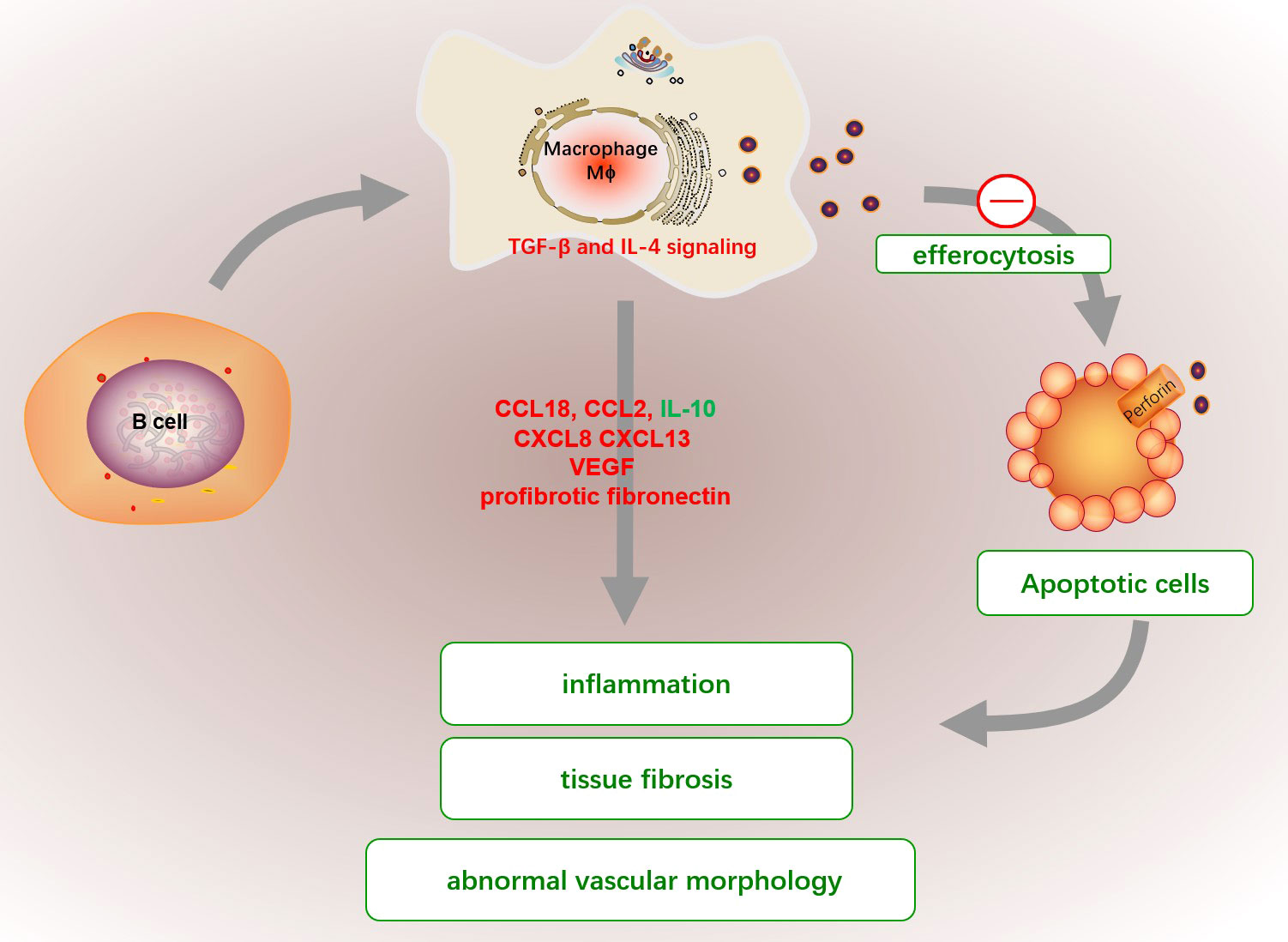
Figure 5 The abnormal functions of macrophage in SSc. The efferocytosis capacities of macrophage in SSc patients are significantly reduced, which cause the emergence of circulating nuclear antigens and promote proinflammatory fibroblasts. Activated macrophages produced a variety of cytokines, such as high levels of CCL18, CCL2, and CXCL8 but low IL-10 expression, which enriched in perivascular regions of highly fibrotic SSc skin to favor pro-inflammatory fibroblasts. Additionally, the excessive production of CXCL13, elevated profibrotic fibronectin and VEGF by macrophages can also promote tissue fibrosis, immune activation and abnormal vascular morphology in SSc. The fibrotic macrophage might be activated by a dysfunctional B cell or dysregulation of TGF-β and IL-4.
Apoptotic cell clearance (efferocytosis) capacities of monocyte-induced macrophage from SSc patients are significantly lower than those in healthy donors, which partly explains the emergence of circulating nuclear antigens (152). Besides, macrophage is a main contributor for fibirosis. The CD14+ monocytes and CD14+ pulmonary macrophages in SSc patients have elevated profibrotic fibronectin production and are considered extracellular matrix producers (153). Activated macrophages produced a variety of cytokines, such as high levels of CCL18, CCL2, and CXCL8 but low IL-10 expression, which enriched in perivascular regions of highly fibrotic SSc skin to favor pro-inflammatory fibroblasts (154, 155). Additionally, the excessive production of CXCL13 and vascular VEGF by macrophages can also promote tissue fibrosis, immune activation and abnormal vascular morphology in SSc (156, 157). The formation mechanism of fibrogenic macrophages is still unclear. It has been demonstrated that fibrotic macrophage might be activated by a dysfunctional B cell in mice with bleomycin-induced SSc, and correlated with the severity of fibrosis in SSc patients (158). Besides, Dysregulation of TGF-β and IL-4 signaling may also be responsible for the pro-fibrotic function in SSc macrophages (159).
The abnormal polarization of macrophages in SSc is relatively complex. Studies have found that crystalline silica SiO or response gene to complement 32 (RGC32) can promote macrophages to form an M1-like phenotype and reduce M2 polarization, which caused the reduction of macrophages efferocytosis in SSc (160, 161). However, the gene expression profiles of affected skin, lung, esophagus and peripheral blood in patients with SSc showed that the expression of M2-related genes was significantly up-regulated in macrophages with pronounced fibrogenic effect (162). Infiltrated macrophages in skin lesions from SSc and local scleroderma were found to highly express CD163 (163–165), indicating that M2 macrophage may also involve in skin fibrosis. Besides, studies have found that some biological agents can inhibit the process of SSc by reversing the polarization of M2 macrophages. The PDE4 inhibition induced by nintedanib, rolipram and apremilast and glycyrrhizin all ameliorate the fibroblast activation by impeding M2 macrophage function in SSc-related mice (166–168). All the above studies indicate that M2 macrophage infiltration may be a target for SSc treatment. However, researchers had found the number of M1 and M2 macrophages in the skin of SSc patients was significantly increased, indicating that macrophages in different polarized states might synergistically promote the pathogenesis of SSc (169). Skin biopsy RNA examined by next-generation RNA sequencing suggested that most early diffuse SSc patients had a concomitant M1 and/or M2 macrophage signature, suggesting co-occurrence of dysregulated fibroblast and macrophage polarization (169). Studies about TLR signaling in fibrosis in SSC and other fibrotic diseases hinted that the conflicting results may be related to long-term inflammatory stimulation (170). Furthermore, macrophages can acquire memory-like characteristics to copy with antigen exposure, protection against re-infection and more efficient vaccine strategies. Recent research found that trained macrophage acquired memory-like characteristics in response to antigen exposure can be targeted to SSc treatment. Low-dose LPS training and adoptive transfer alleviated fibrosis and inflammation in SSc mice, while BCG-training aggravated disease in this model (171). The long-term and complex in vivo microenvironment may be an essential promoter of macrophage activation that is unique to SSc patients. However, the function and mechanism need to be further explored.
T1D is an autoimmune disease characterized by the continuous destruction of islet cells caused by islet leukocyte infiltration (172). The loss of pancreatic β cells can lead to uncontrolled blood glucose and various complications such as cardiovascular disease, nephropathy, retinopathy, heart attack and stroke, which require lifelong dependence on exogenous insulin (173). Islet inflammation is one of the main mechanisms of pancreatic β-cell injury and the development of T1D. In diabetes-prone biological breeding rats (DP-BB), it has been demonstrated that macrophages are the first immune cells to infiltrate into islets (174). Furthermore, there were no lymphocytes in the islets when macrophage infiltration was prevented (175), suggesting that lymphocyte recruitment in islets depends on the macrophage. In addition, the immunohistochemical results of pancreatic specimens from newly diagnosed T1D patients confirmed the presence of macrophages in early and advanced inflammation (176). Various research about spontaneous T1D animal models has shown that specific clearance of macrophages in vivo can significantly inhibit Th1 but increase Th2 immune response induced mainly by IL-12, and inhibited cytotoxic effector of CD8+ T, even remaining selective acceleration of the recruitment of CD8+ T cells into the islets (177–179). Depleting macrophage by liposomes containing clodronate also selectly abolished diabetogenic CD4+ T cells induced diabetes even with inflammation existence (180).
The microenvironment in T1D pancreas promote the recruitment of macrophages and abnormal functions. It was found that islet resident macrophages of non-autoimmune mice had immunomodulatory phenotype and could promote Treg cell differentiation in vitro (181). Deficiency of immunomodulatory function in macrophages may be an essential mechanism of pathogenesis of T1D (181). In addition, the migration and phagocytosis to target inflammatory cells of macrophage in the streptozotocin (STZ) -induced T1D model weakened islet cell immune defense (182). Diabetgenic CD4 T cells produce a variety of inflammatory cytokines and chemokines such as CCL1, resulting in the recruitment of macrophages into pancreas (183). Reduced integrin-associated surface factor CD47 on islet cells promoted macrophage migration and phagocytosis of endogenous cells (182). Instead of clearing apoptotic cells silently without production of pro-inflammatory cytokines, macrophages in T1D secret inappropriately high amounts of IL-1β and TNF-α to contribute to the initiation or continuation of an immune attack towards the pancreatic beta-cells (184). Besides, previously research also showed that macrophages from non-obese diabetic (NOD) mice are activated and engulf apoptotic cells at a lower rate, which might result in secondary necrosis, inflammation and self-antigen presentation in T1D (185).
And increased macrophage-derived cytokines including IL-12, TNF-α and IL-1β selectively in spleen lymphocytes and pancreatic islet are responsible for the inflammatory cascade of events leading to the destruction of pancreatic β cells (186). Macrophages are involved in regulating the infiltration and functions of immune cells in T1D. Recruited macrophages in the pancreas by diabetes-derived T cell produce IL-1β, TNF-α and NO, and express chemokine receptors CCR5, CXCR3 and CCR8 to further recruit and active other inflammatory cells (183). The interaction between inflammatory macrophages and β-cells promote the production of CXCR2 ligands (CXCL1 and CXCL2) in the pancreas of T1D mice, which further recruit diabetogenic CXCR2+ neutrophils from the blood into the pancreatic islets (187). Autoreactive CD4+ T cells destroyed β cells through a Fas-dependent mechanism that was assisted by cytokines IL-1α, IL-1β, and IFN-γ (188). Besides, macrophge derived IL-12 might contribute to the development and activation of β cell–cytotoxic Th1 and CD8 cells in NOD mice (189). And macrophages selectively traffick autoimmune cytotoxic T cells into the islets via IFN-I signaling even without entering the islets, and ablation of IFN-I signaling on macrophages limits the onset of T1D (190). The role of macrophage on the pathological process of T1D was shown in Figure 6.
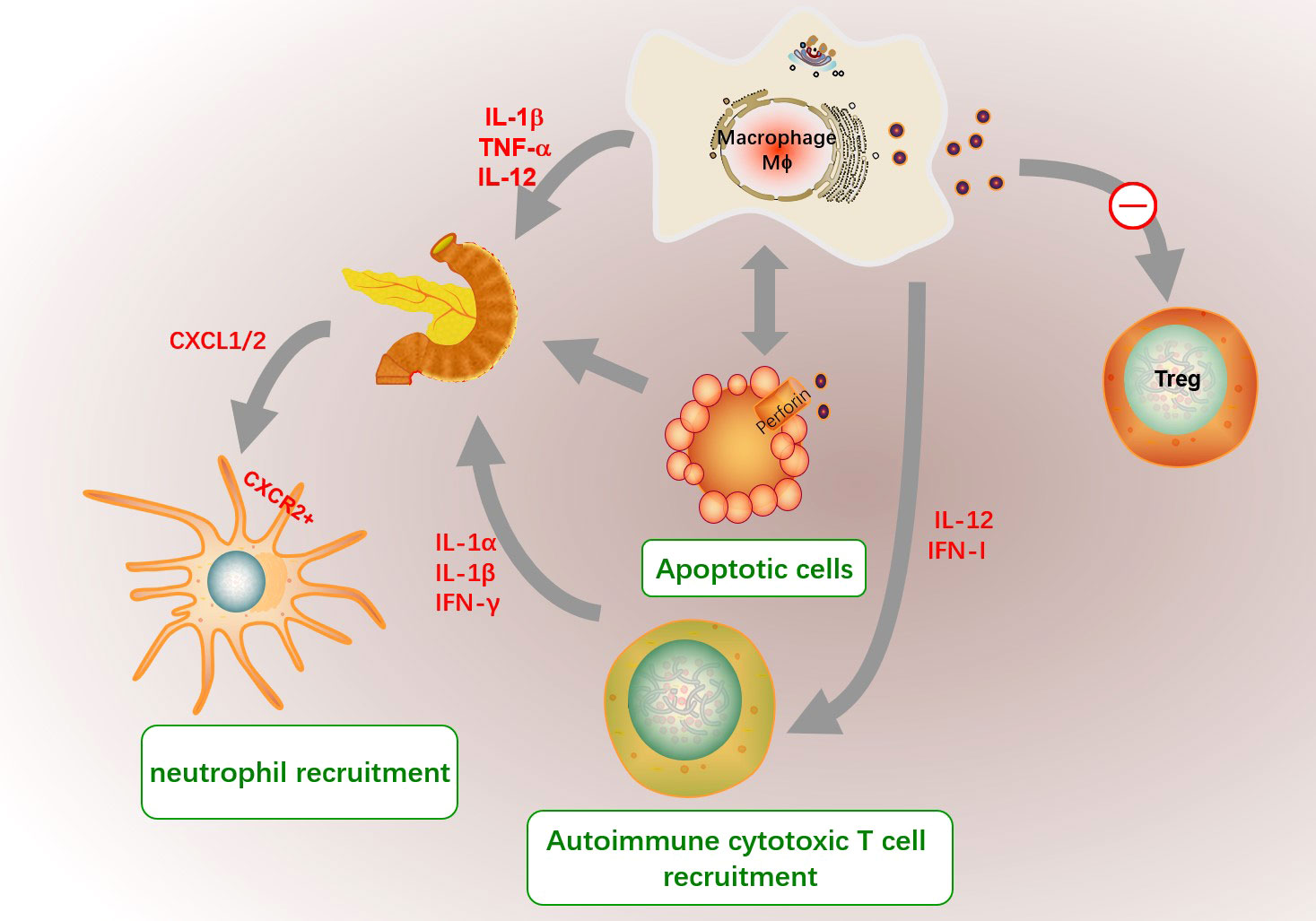
Figure 6 The role of macrophages in the pathogenesis of T1D. The microenvironment in T1D pancreas promote the recruitment of macrophages and abnormal functions. Islet resident macrophages in T1D had defected immunomodulatory phenotype and might inhibit Treg cell differentiation. In addition, the migration and phagocytosis to target inflammatory cells such as apoptotic cells of macrophage in T1D are decrease. And macrophage secret inappropriately high amounts of IL1β and TNFα to contribute to the initiation of an immune attack towards the pancreatic beta-cells. The unengulfed apoptotic cells might result in secondary necrosis, inflammation and self-antigen presentation in islet. And increased macrophage-derived cytokines including IL-12, TNF-α and IL-1β are responsible for the inflammatory cascade of events leading to the destruction of pancreatic β cells. The interaction between inflammatory macrophages and β-cells promote the production of CXCR2 ligands (CXCL1 and CXCL2), which further recruit diabetogenic CXCR2+ neutrophils. Autoreactive CD4+ T cells destroyed β cells assisted by cytokines IL-1α, IL-1β, and IFN-γ. Besides, macrophage derived IL-12 and IFN-I signaling might contribute to the development and activation of β cell–cytotoxic Th1 and CD8 cells.
Macrophage polarization may act as a potential therapeutic agent for T1D. M2 macrophages explicitly located in the inflammatory pancreas could significantly inhibit the proliferation of T cells and promote the survival of β cells after adoptive transfer into spontaneous T1D mice, resulting in resistance to T1D in non-obese resistant (NOR) mice (191). In addition, the survival of transplanted islets was partly dependent on the content of M2 macrophages (192, 193). The early glycosylation products (EGPs) produced in the first step of Maillard reaction/glycosylation alleviated insulin resistance and pancreatic immune infiltration by increasing the M2/M1 ratio (194). Macrophage-specific knockout ubiquitin coupling enzyme E2 can weaken the energy metabolism and M2 type polarization of macrophages, thus increasing the risk of diabetes T1D induced by STZ (195). Hence, promoting M2 but inhibiting M1 macrophage polarization may be an important target for preventing and treating T1D.
Macrophage-derived proinflammatory cytokines, chemokines and their receptors were identified the suitable targets for the therapeutic interventions of T1D. The TNF-α inhibitor infliximab could alleviate T1D, which might be related with the reduced presentation of islet antigen to both effector CD4+ and CD8+ T cells (196, 197). And IL-6 has also been suggested as a target for T1D treatment (198). Multiple strategies blocking the CXCR1/2 pathway main expressed in macrophage inhibited leucocyte recruitment and prevent inflammation and autoimmune mediated islet damage, which was new interventional approach for T1D (199).
The possible functions of macrophages in autoimmune diseases as described in Table 1. In brief, the scavenging ability of macrophages was destroyed, leading to the accumulation of autoimmune complexes in local tissues. Besides, the abnormal macrophage activation induced a series of irrepressible pro-inflammatory responses, and promoted the activation and recruitment of lymphocytes in local tissues, resulting in tissue damage. In addition, the aberrant polarization of macrophages has been identified to contribute to the pathogenesis of autoimmune diseases. However, due to the significant heterogeneity of macrophages, the polarization of macrophages varies significantly in different tissues and even in different phases of the same disease. Systematically and comprehensively understanding the polarization of macrophages in autoimmune diseases will conduce to the prevention and treatment of autoimmune diseases.
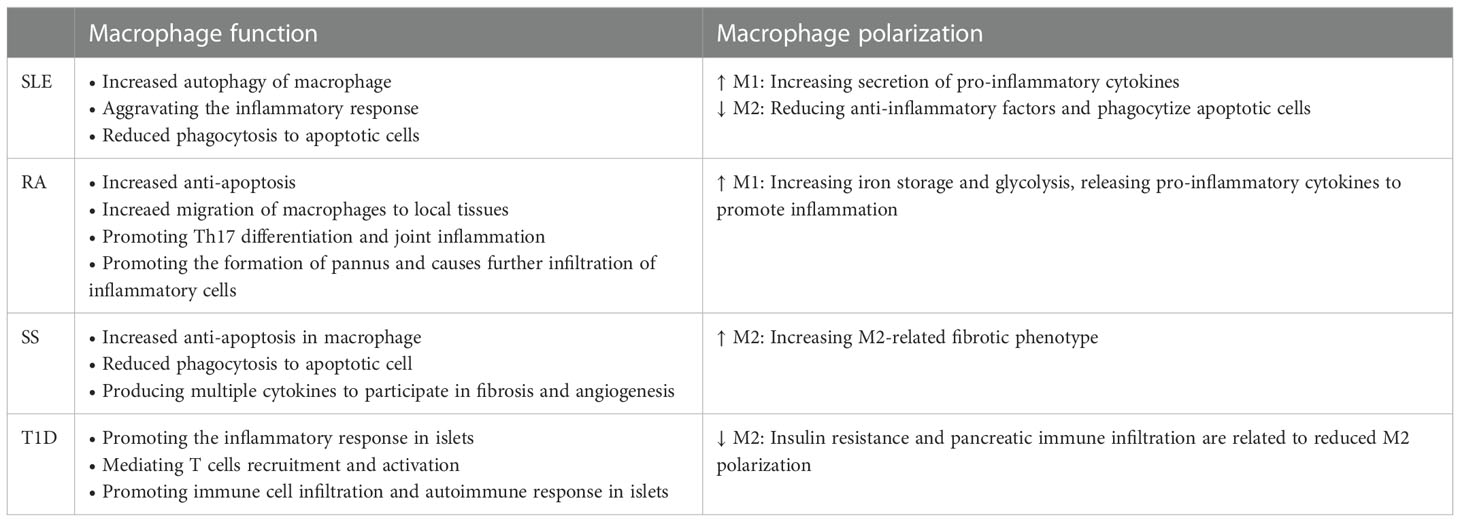
Table 1 Possible function of macrophages in autoimmune diseases.
Currently anti-macrophage therapy in autoimmune diseases mainly focuses on down-regulation the production of abnormal macrophage-derived pro-inflammatory cytokines production, elimination of dysfunctional macrophage from the inflammatory regions such as inhibiting monocyte recruitment and differentiation, and upregulation of anti-inflammatory cytokines. In recent years, macrophage-derived extracellular vesicles composed of microvesicles and exosomes have aroused increased interest in the treatment for autoimmune disease. The macrophage-derived extracellular vesicles are considered as optimal delivery vehicles for the minimal toxicity and specific target effect. Macrophage-derived microvesicle-coated poly (lactic-co-glycolic acid) (PLGA) nanoparticles to encapsulate tacrolimus significant suppress the progression of RA in mice, which is an efficient biomimetic vehicle for RA targeted treatment (200). Besides, macrophage-derived extracellular vesicles efficiently delivered dexamethasone into inflamed kidney and effectively suppress inflammation and fibrosis in kidney (201).
Various new techniques such as single-cell sequencing, metabolomics and other multi-omics research methods have been applied in autoimmune diseases research and have achieved considerable achievements. A single-cell sequencing result of a mixed lung cell sample from bleomycin-induced lung injury mice found a group of disease-related transitional macrophages that specifically express CX3CR1 and PDGF-AA and are located in fibrotic scars to promote fibrosis (202). This study provides an effective target for preventing and treating pulmonary fibrosis-related diseases. In addition, single-cell pseudo-time analysis infer the transcription trajectory of macrophages when they gradually change their gene expression profile during autoimmunity, suggesting that we can find the molecular changes in the early stage of the disease and the most decisive target. The application of multi-omics methods at the single-cell level will provide an effective means for exploring the potential mechanisms of abnormal macrophage phenotypes and offer a solid theoretical basis for preventing and treating autoimmune diseases.
SY wrote the main manuscript. SJ, MZ revised the manuscript. SJ had primary responsibility for the final content. All authors agree to be accountable for the content of the work. All authors contributed to the article and approved the submitted version.
This work was supported by the Natural Science Foundation of Hunan province, China (Grant No.2020JJ4837), the Health Commission of Hunan province (Grant No.202202044436), the Project for leading talents in science and technology in Hunan province (2019RS3003) and National Natural Science Foundation of China (No. 82030097).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Juarranz Y. Molecular and cellular basis of autoimmune diseases. Cells. (2021) 10(2):474. doi: 10.3390/cells10020474
2. Marrack P, Kappler J, Kotzin BL. Autoimmune disease: why and where it occurs. Nat Med (2001) 7(8):899–905. doi: 10.1038/90935
3. Rose NR. Prediction and prevention of autoimmune disease in the 21st century: A review and preview. Am J Epidemiol. (2016) 183(5):403–6. doi: 10.1093/aje/kwv292
4. Molina V, Shoenfeld Y. Infection, vaccines and other environmental triggers of autoimmunity. Autoimmunity. (2005) 38(3):235–45. doi: 10.1080/08916930500050277
5. Saferding V, Blüml S. Innate immunity as the trigger of systemic autoimmune diseases. J Autoimmun (2020) 110:102382. doi: 10.1016/j.jaut.2019.102382
6. Pollard JW. Trophic macrophages in development and disease. Nat Rev Immunol (2009) 9(4):259–70. doi: 10.1038/nri2528
7. Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. (2013) 496(7446):445–55. doi: 10.1038/nature12034
8. van FR, Cohn ZA, Hirsch JG, Humphrey JH, Spector WG, Langevoort HL. The mononuclear phagocyte system: A new classification of macrophages, monocytes, and their precursor cells. Bull World Health Organ (1972) 46(6):845.
9. Ginhoux F, Guilliams M. Tissue-resident macrophage ontogeny and homeostasis. Immunity. (2016) 44(3):439–49. doi: 10.1016/j.immuni.2016.02.024
10. Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol (2005) 5(12):953–64. doi: 10.1038/nri1733
11. Den Haan JM, Kraal G. Innate immune functions of macrophage subpopulations in the spleen. J Innate Immun (2012) 4(5-6):437–45. doi: 10.1159/000335216
12. Hoeffel G, Wang Y, Greter M, See P, Teo P, Malleret B, et al. Adult langerhans cells derive predominantly from embryonic fetal liver monocytes with a minor contribution of yolk sac-derived macrophages. J Exp Med (2012) 209(6):1167–81. doi: 10.1084/jem.20120340
13. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. (2010) 330(6005):841–5. doi: 10.1126/science.1194637
14. Sheng J, Ruedl C, Karjalainen K. Most tissue-resident macrophages except microglia are derived from fetal hematopoietic stem cells. Immunity. (2015) 43(2):382–93. doi: 10.1016/j.immuni.2015.07.016
15. Bain CC, Bravo-Blas A, Scott CL, Perdiguero EG, Geissmann F, Henri S, et al. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat Immunol (2014) 15(10):929–37. doi: 10.1038/ni.2967
16. Tamoutounour S, Guilliams M, Montanana Sanchis F, Liu H, Terhorst D, Malosse C, et al. Origins and functional specialization of macrophages and of conventional and monocyte-derived dendritic cells in mouse skin. Immunity. (2013) 39(5):925–38. doi: 10.1016/j.immuni.2013.10.004
17. Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. (2016) 44(3):450–62. doi: 10.1016/j.immuni.2016.02.015
18. Gordon S, Plüddemann A, Martinez Estrada F. Macrophage heterogeneity in tissues: phenotypic diversity and functions. Immunol Rev (2014) 262(1):36–55. doi: 10.1111/imr.12223
19. Park MD, Silvin A, Ginhoux F, Merad M. Macrophages in health and disease. Cell. (2022) 185(23):4259–79. doi: 10.1016/j.cell.2022.10.007
20. Yang N, Isbel NM, Nikolic-Paterson DJ, Li Y, Ye R, Atkins RC, et al. Local macrophage proliferation in human glomerulonephritis. Kidney Int (1998) 54(1):143–51. doi: 10.1046/j.1523-1755.1998.00978.x
21. Tacke F. Functional role of intrahepatic monocyte subsets for the progression of liver inflammation and liver fibrosis In vivo. Fibrogenesis Tissue Repair (2012) 5(Suppl 1):S27. doi: 10.1186/1755-1536-5-S1-S27
22. Baeck C, Wehr A, Karlmark KR, Heymann F, Vucur M, Gassler N, et al. Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut. (2012) 61(3):416–26. doi: 10.1136/gutjnl-2011-300304
23. Lescoat A, Lecureur V, Varga J. Contribution of monocytes and macrophages to the pathogenesis of systemic sclerosis: recent insights and therapeutic implications. Curr Opin Rheumatol (2021) 33(6):463–70. doi: 10.1097/BOR.0000000000000835
24. Schuppan D. Current concepts of celiac disease pathogenesis. Gastroenterology (2000) 119(1):234–42. doi: 10.1053/gast.2000.8521
25. Fujiwara N, Kobayashi K. Macrophages in inflammation. Curr Drug Targets Inflammation Allergy (2005) 4(3):281–6. doi: 10.2174/1568010054022024
26. Celhar T, Pereira-Lopes S, Thornhill SI, Lee HY, Dhillon MK, Poidinger M, et al. TLR7 and TLR9 ligands regulate antigen presentation by macrophages. Int Immunol (2016) 28(5):223–32. doi: 10.1093/intimm/dxv066
27. Frascaroli G, Lecher C, Varani S, Setz C, van der Merwe J, Brune W, et al. Human macrophages escape inhibition of major histocompatibility complex-dependent antigen presentation by cytomegalovirus and drive proliferation and activation of memory CD4+ and CD8+ T cells. Front Immunol (2018) 9:1129. doi: 10.3389/fimmu.2018.01129
28. Wang J, Yang L, Mao X, Li Z, Lin X, Jiang C. Streptococcus salivarius-mediated CD8+ T cell stimulation required antigen presentation by macrophages in oral squamous cell carcinoma. Exp Cell Res (2018) 366(2):121–6. doi: 10.1016/j.yexcr.2018.03.007
29. Ulfig A, Bader V, Varatnitskaya M, Lupilov N, Winklhofer KF, Leichert LI. Hypochlorous acid-modified human serum albumin suppresses MHC class II - dependent antigen presentation in pro-inflammatory macrophages. Redox Biol (2021) 43:101981. doi: 10.1016/j.redox.2021.101981
30. Yang M, Shi XQ, Peyret C, Oladiran O, Wu S, Chambon J, et al. Effector/memory CD8+ T cells synergize with co-stimulation competent macrophages to trigger autoimmune peripheral neuropathy. Brain Behav Immun (2018) 71:142–57. doi: 10.1016/j.bbi.2018.04.001
31. Panduro M, Benoist C, Mathis D. Treg cells limit IFN-γ production to control macrophage accrual and phenotype during skeletal muscle regeneration. Proc Natl Acad Sci U S A. (2018) 115(11):E2585–93. doi: 10.1073/pnas.1800618115
32. Clarke EV, Weist BM, Walsh CM, Tenner AJ. Complement protein C1q bound to apoptotic cells suppresses human macrophage and dendritic cell-mediated Th17 and Th1 T cell subset proliferation. J Leukoc Biol (2015) 97(1):147–60. doi: 10.1189/jlb.3A0614-278R
33. Martinez J, Malireddi RK, Lu Q, Cunha LD, Pelletier S, Gingras S, et al. Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat Cell Biol (2015) 17(7):893–906. doi: 10.1038/ncb3192
34. Boada-Romero E, Martinez J, Heckmann BL, Green DR. The clearance of dead cells by efferocytosis. Nat Rev Mol Cell Biol (2020) 21(7):398–414. doi: 10.1038/s41580-020-0232-1
35. Nishi C, Yanagihashi Y, Segawa K, Nagata S. MERTK tyrosine kinase receptor together with TIM4 phosphatidylserine receptor mediates distinct signal transduction pathways for efferocytosis and cell proliferation. J Biol Chem (2019) 294(18):7221–30. doi: 10.1074/jbc.RA118.006628
36. Kawano M, Nagata S. Efferocytosis and autoimmune disease. Int Immunol (2018) 30(12):551–8. doi: 10.1093/intimm/dxy055
37. Zhang X, Mosser DM. Macrophage activation by endogenous danger signals. J Pathol (2008) 214(2):161–78. doi: 10.1002/path.2284
38. Cuda CM, Pope RM, Perlman H. The inflammatory role of phagocyte apoptotic pathways in rheumatic diseases. Nat Rev Rheumatol (2016) 12(9):543–58. doi: 10.1038/nrrheum.2016.132
39. Udalova IA, Mantovani A, Feldmann M. Macrophage heterogeneity in the context of rheumatoid arthritis. Nat Rev Rheumatol (2016) 12(8):472–85. doi: 10.1038/nrrheum.2016.91
40. Jiang Z, Jiang JX, Zhang GX. Macrophages: a double-edged sword in experimental autoimmune encephalomyelitis. Immunol Lett (2014) 160(1):17–22. doi: 10.1016/j.imlet.2014.03.006
41. Giraudo E, Inoue M, Hanahan D. An amino-bisphosphonate targets MMP-9-expressing macrophages and angiogenesis to impair cervical carcinogenesis. J Clin Invest. (2004) 114(5):623–33. doi: 10.1172/JCI22087
42. Aurora AB, Porrello ER, Tan W, Mahmoud AI, Hill JA, Bassel-Duby R, et al. Macrophages are required for neonatal heart regeneration. J Clin Invest. (2014) 124(3):1382–92. doi: 10.1172/JCI72181
43. Guan C, Xiao Y, Li K, Wang T, Liang Y, Liao G. MMP-12 regulates proliferation of mouse macrophages via the ERK/P38 MAPK pathways during inflammation. Exp Cell Res (2019) 378(2):182–90. doi: 10.1016/j.yexcr.2019.03.018
44. McInnes IB, Schett G. Pathogenetic insights from the treatment of rheumatoid arthritis. Lancet. (2017) 389(10086):2328–37. doi: 10.1016/S0140-6736(17)31472-1
45. Kim SY, Nair MG. Macrophages in wound healing: activation and plasticity. Immunol Cell Biol (2019) 97(3):258–67. doi: 10.1111/imcb.12236
46. Huang G, Wang Y, Chi H. Regulation of TH17 cell differentiation by innate immune signals. Cell Mol Immunol (2012) 9(4):287–95. doi: 10.1038/cmi.2012.10
47. Esmailbeig M, Ghaderi A. Interleukin-18: a regulator of cancer and autoimmune diseases. Eur Cytokine Netw (2017) 28(4):127–40. doi: 10.1684/ecn.2018.0401
48. De Filippo K, Dudeck A, Hasenberg M, Nye E, van Rooijen N, Hartmann K, et al. Mast cell and macrophage chemokines CXCL1/CXCL2 control the early stage of neutrophil recruitment during tissue inflammation. Blood. (2013) 121(24):4930–7. doi: 10.1182/blood-2013-02-486217
49. Feduska JM, Tse HM. The proinflammatory effects of macrophage-derived NADPH oxidase function in autoimmune diabetes. Free Radic Biol Med (2018) 125:81–89. doi: 10.1016/j.freeradbiomed.2018.04.581
50. Marletta MA, Yoon PS, Iyengar R, Leaf CD, Wishnok JS. Macrophage oxidation of l-arginine to nitrite and nitrate: nitric oxide is an intermediate. Biochemistry. (1988) 27(24):8706–11. doi: 10.1021/bi00424a003
51. Palmer RM, Ashton DS, Moncada S. Vascular endothelial cells synthesize nitric oxide from l-arginine. Nature. (1988) 333(6174):664–6. doi: 10.1038/333664a0
52. Rath M, Müller I, Kropf P, Closs EI, Munder M. Metabolism via arginase or nitric oxide synthase: Two competing arginine pathways in macrophages. Front Immunol (2014) 5:532. doi: 10.3389/fimmu.2014.00532
53. Viola A, Munari F, Sánchez-Rodríguez R, Scolaro T, Castegna A. The metabolic signature of macrophage responses. Front Immunol (2019) 10:1462. doi: 10.3389/fimmu.2019.01462
54. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. (2013) 496(7444):238–42. doi: 10.1038/nature11986
55. Batista-Gonzalez A, Vidal R, Criollo A, Carreño LJ. New insights on the role of lipid metabolism in the metabolic reprogramming of macrophages. Front Immunol (2020) 10:2993. doi: 10.3389/fimmu.2019.02993
56. Huang SC, Smith AM, Everts B, Colonna M, Pearce EL, Schilling JD, et al. Metabolic reprogramming mediated by the mTORC2-IRF4 signaling axis is essential for macrophage alternative activation. Immunity. (2016) 45(4):817–30. doi: 10.1016/j.immuni.2016.09.016
57. Du L, Lin L, Li Q, Liu K, Huang Y, Wang X, et al. IGF-2 preprograms maturing macrophages to acquire oxidative phosphorylation-dependent anti-inflammatory properties. Cell Metab (2019) 29(6):1363–75. doi: 10.1016/j.cmet.2019.01.006
58. Biswas SK, Mantovani A. Orchestration of metabolism by macrophages. Cell Metab (2012) 15(4):432–7. doi: 10.1016/j.cmet.2011.11.013
59. Orihuela R, McPherson CA, Harry GJ. Microglial M1/M2 polarization and metabolic states. Br J Pharmacol (2016) 173(4):649–65. doi: 10.1111/bph.13139
60. Cairo G, Recalcati S, Mantovani A, Locati M. Iron trafficking and metabolism in macrophages: contribution to the polarized phenotype. Trends Immunol (2011) 32(6):241–7. doi: 10.1016/j.it.2011.03.007
61. Mao Y, Shi D, Li G, Jiang P. Citrulline depletion by ASS1 is required for proinflammatory macrophage activation and immune responses. Mol Cell (2022) 82(3):527–41. doi: 10.1016/j.molcel.2021.12.006
62. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas[J]. J Clin Invest (2012) 122(3):787–95. doi: 10.1172/JCI59643
63. Murray PJ. Macrophage polarization. Annu Rev Physiol (2017) 79:541–66. doi: 10.1146/annurev-physiol-022516-034339
64. Fuchs AL, Schiller SM, Keegan WJ, Ammons MCB, Eilers B, Tripet B, et al. Quantitative 1H NMR metabolomics reveal distinct metabolic adaptations in human macrophages following differential activation. Metabolites. (2019) 9(11):248. doi: 10.3390/metabo9110248
65. Draijer C, Boorsma CE, Robbe P, Timens W, Hylkema MN, Ten Hacken NH, et al. Human asthma is characterized by more IRF5+ M1 and CD206+ M2 macrophages and less IL-10+ M2-like macrophages around airways compared with healthy airways. J Allergy Clin Immunol (2017) 140(1):280–283.e3. doi: 10.1016/j.jaci.2016.11.020
66. Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. (2010) 32(5):593–604. doi: 10.1016/j.immuni.2010.05.007
67. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol (2002) 23(11):549–55. doi: 10.1016/s1471-4906(02)02302-5
68. Abdelaziz MH, Abdelwahab SF, Wan J, Cai W, Huixuan W, Jianjun C, et al. Alternatively activated macrophages; a double-edged sword in allergic asthma. J Transl Med (2020) 18(1):58. doi: 10.1186/s12967-020-02251-w
69. Wang LX, Zhang SX, Wu HJ, Rong XL, Guo J. M2b macrophage polarization and its roles in diseases. J Leukoc Biol (2019) 106(2):345–58. doi: 10.1002/JLB.3RU1018-378RR
70. Arora S, Dev K, Agarwal B, Das P, Syed MA. Macrophages: Their role, activation and polarization in pulmonary diseases. Immunobiology. (2018) 223(4-5):383–96. doi: 10.1016/j.imbio.2017.11.001
71. Wang Q, Ni H, Lan L, Wei X, Xiang R, Wang Y. Fra-1 protooncogene regulates IL-6 expression in macrophages and promotes the generation of M2d macrophages. Cell Res (2010) 20(6):701–12. doi: 10.1038/cr.2010.52
72. Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol (2018) 233(9):6425–40. doi: 10.1002/jcp.26429
73. Tabas I, Bornfeldt KE. Macrophage phenotype and function in different stages of atherosclerosis. Circ Res (2016) 118(4):653–67. doi: 10.1161/CIRCRESAHA.115.306256
74. Biswas SK, Sica A, Lewis CE. Plasticity of macrophage function during tumor progression: regulation by distinct molecular mechanisms. J Immunol (2008) 180(4):2011–7. doi: 10.4049/jimmunol
75. Funes SC, Rios M, Escobar-Vera J, Kalergis AM. Implications of macrophage polarization in autoimmunity. Immunology. (2018) 154(2):186–95. doi: 10.1111/imm.12910
76. Kaul A, Gordon C, Crow MK, Touma Z, Urowitz MB, van Vollenhoven R, et al. Systemic lupus erythematosus. Nat Rev Dis Primers (2016) 2:16039. doi: 10.1038/nrdp.2016.39
77. Li Y, Lee PY, Reeves WH. Monocyte and macrophage abnormalities in systemic lupus erythematosus. Arch Immunol Ther Exp (Warsz). (2010) 58(5):355–64. doi: 10.1007/s00005-010-0093-y
78. Katsiari CG, Liossis SN, Sfikakis PP. The pathophysiologic role of monocytes and macrophages in systemic lupus erythematosus: a reappraisal. Semin Arthritis Rheumatol (2010) 39(6):491–503. doi: 10.1016/j.semarthrit.2008.11.002
79. Ma WT, Gao F, Gu K, Chen DK. The role of monocytes and macrophages in autoimmune diseases: A comprehensive review. Front Immunol (2019) 10:1140. doi: 10.3389/fimmu.2019.01140
80. Chalmers SA, Chitu V, Herlitz LC, Sahu R, Stanley ER, Putterman C. Macrophage depletion ameliorates nephritis induced by pathogenic antibodies. J Autoimmun (2015) 57:42–52. doi: 10.1016/j.jaut.2014.11.007
81. Kuriakose J, Redecke V, Guy C, Zhou J, Wu R, Ippagunta SK, et al. Patrolling monocytes promote the pathogenesis of early lupus-like glomerulonephritis. J Clin Invest. (2019) 129(6):2251–65. doi: 10.1172/JCI125116
82. Hill GS, Delahousse M, Nochy D, Rémy P, Mignon F, Méry JP, et al. Predictive power of the second renal biopsy in lupus nephritis: significance of macrophages. Kidney Int (2001) 59(1):304–16. doi: 10.1046/j.1523-1755.2001.00492.x
83. Yoshimoto S, Nakatani K, Iwano M, Asai O, Samejima K, Sakan H, et al. Elevated levels of fractalkine expression and accumulation of CD16+ monocytes in glomeruli of active lupus nephritis. Am J Kidney Dis (2007) 50(1):47–58. doi: 10.1053/j.ajkd.2007.04.012
84. Schiffer L, Bethunaickan R, Ramanujam M, Huang W, Schiffer M, Tao H, et al. Activated renal macrophages are markers of disease onset and disease remission in lupus nephritis. J Immunol (2008) 180(3):1938–47. doi: 10.4049/jimmunol.180.3.1938
85. Li H, Wu Q, Li J, Yang P, Zhu Z, Luo B, et al. Cutting edge: defective follicular exclusion of apoptotic antigens due to marginal zone macrophage defects in autoimmune BXD2 mice. J Immunol (2013) 190(9):4465–9. doi: 10.4049/jimmunol.1300041
86. Gaipl US, Voll RE, Sheriff A, Franz S, Kalden JR, Herrmann M. Impaired clearance of dying cells in systemic lupus erythematosus. Autoimmun Rev (2005) 4(4):189–94. doi: 10.1016/j.autrev.2004.10.007
87. Roszer T, Menéndez-Gutiérrez MP, Lefterova MI, Alameda D, Núñez V, Lazar MA, et al. Autoimmune kidney disease and impaired engulfment of apoptotic cells in mice with macrophage peroxisome proliferator-activated receptor gamma or retinoid X receptor alpha deficiency. J Immunol (2011) 186(1):621–31. doi: 10.4049/jimmunol.1002230
88. Roberts AW, Lee BL, Deguine J, John S, Shlomchik MJ, Barton GM. Tissue-resident macrophages are locally programmed for silent clearance of apoptotic cells. Immunity. (2017) 47(5):913–27. doi: 10.1016/j.immuni.2017.10.006
89. Denny MF, Chandaroy P, Killen PD, Caricchio R, Lewis EE, Richardson BC, et al. Accelerated macrophage apoptosis induces autoantibody formation and organ damage in systemic lupus erythematosus. J Immunol (2006) 176(4):2095–104. doi: 10.4049/jimmunol.176.4.2095
90. Li B, Yue Y, Dong C, Shi Y, Xiong S. Blockade of macrophage autophagy ameliorates activated lymphocytes-derived DNA induced murine lupus possibly via inhibition of proinflammatory cytokine production. Clin Exp Rheumatol (2014) 32(5):705–14.
91. Triantafyllopoulou A, Franzke CW, Seshan SV, Perino G, Kalliolias GD, Ramanujam M, et al. Proliferative lesions and metalloproteinase activity in murine lupus nephritis mediated by type I interferons and macrophages. Proc Natl Acad Sci U S A. (2010) 107(7):3012–7. doi: 10.1073/pnas.0914902107
92. Rönnblom L, Leonard D. Interferon pathway in SLE: one key to unlocking the mystery of the disease. Lupus Sci Med (2019) 6(1):e000270. doi: 10.1136/lupus-2018-000270
93. McNab F, Mayer-Barber K, Sher A, Wack A, O'Garra A. Type I interferons in infectious disease. Nat Rev Immunol (2015) 15(2):87–103. doi: 10.1038/nri3787
94. Bengtsson AA, Ronnblom L. Role of interferons in SLE. Best Pract Res Clin Rheumatol (2017) 31(3):415–28. doi: 10.1016/j.berh.2017.10.003
95. Tumurkhuu G, Montano E, Jefferies C. Innate immune dysregulation in the development of cardiovascular disease in lupus. Curr Rheumatol Rep (2019) 21(9):46. doi: 10.1007/s11926-019-0842-9
96. Boshuizen MC, de Winther MP. Interferons as essential modulators of atherosclerosis. Arterioscler Thromb Vasc Biol (2015) 35(7):1579–88. doi: 10.1161/ATVBAHA.115.305464
97. Crampton SP, Morawski PA, Bolland S. Linking susceptibility genes and pathogenesis mechanisms using mouse models of systemic lupus erythematosus. Dis Model Mech (2014) 7(9):1033–46. doi: 10.1242/dmm.016451
98. Li H, Fu YX, Wu Q, Zhou Y, Crossman DK, Yang P, et al. Interferon-induced mechanosensing defects impede apoptotic cell clearance in lupus. J Clin Invest. (2015) 125(7):2877–90. doi: 10.1172/JCI81059
99. Wang JH, Li J, Wu Q, Yang P, Pawar RD, Xie S, et al. Marginal zone precursor b cells as cellular agents for type I IFN-promoted antigen transport in autoimmunity. J Immunol (2010) 184(1):442–51. doi: 10.4049/jimmunol.0900870
100. Ramirez-Ortiz ZG, Prasad A, Griffith JW, Pendergraft WF 3rd, Cowley GS, Root DE, et al. The receptor TREML4 amplifies TLR7-mediated signaling during antiviral responses and autoimmunity. Nat Immunol (2015) 16(5):495–504. doi: 10.1038/ni.3143
101. Zhou J, Wu R, High AA, Slaughter CA, Finkelstein D, Rehg JE, et al. A20-binding inhibitor of NF-κB (ABIN1) controls toll-like receptor-mediated CCAAT/enhancer-binding protein β activation and protects from inflammatory disease. Proc Natl Acad Sci U S A. (2011) 108(44):E998–1006. doi: 10.1073/pnas.1106232108
102. Orme J, Mohan C. Macrophages and neutrophils in SLE-an online molecular catalog. Autoimmun Rev (2012) 11(5):365–72. doi: 10.1016/j.autrev.2011.10.010
103. Crispín JC, Hedrich CM, Tsokos GC. Gene-function studies in systemic lupus erythematosus. Nat Rev Rheumatol (2013) 9(8):476–84. doi: 10.1038/nrrheum.2013.78
104. Schaper F, de Leeuw K, Horst G, Bootsma H, Limburg PC, Heeringa P, et al. High mobility group box 1 skews macrophage polarization and negatively influences phagocytosis of apoptotic cells. Rheumatol (Oxford). (2016) 55(12):2260–70. doi: 10.1093/rheumatology/kew324
105. Hahn J, Euler M, Kilgus E, Kienhöfer D, Stoof J, Knopf J, et al. NOX2 mediates quiescent handling of dead cell remnants in phagocytes. Redox Biol (2019) 26:101279. doi: 10.1016/j.redox.2019.101279
106. Labonte AC, Kegerreis B, Geraci NS, Bachali P, Madamanchi S, Robl R, et al. Identification of alterations in macrophage activation associated with disease activity in systemic lupus erythematosus. PloS One (2018) 13(12):e0208132. doi: 10.1371/journal.pone.0208132
107. Iwata Y, Boström EA, Menke J, Rabacal WA, Morel L, Wada T, et al. Aberrant macrophages mediate defective kidney repair that triggers nephritis in lupus-susceptible mice. J Immunol (2012) 188(9):4568–80. doi: 10.4049/jimmunol.1102154
108. Vandivier RW, Henson PM, Douglas IS. Burying the dead: the impact of failed apoptotic cell removal (efferocytosis) on chronic inflammatory lung disease. Chest. (2006) 129(6):1673–82. doi: 10.1378/chest.129.6.1673
109. Poon IK, Lucas CD, Rossi AG, Ravichandran KS. Apoptotic cell clearance: basic biology and therapeutic potential. Nat Rev Immunol (2014) 14(3):166–80. doi: 10.1038/nri3607
110. Mohammadi S, Saghaeian-Jazi M, Sedighi S, Memarian A. Immunomodulation in systemic lupus erythematosus: induction of M2 population in monocyte-derived macrophages by pioglitazone. Lupus. (2017) 26(12):1318–27. doi: 10.1177/0961203317701842
111. Li F, Zhu X, Yang Y, Huang L, Xu J. TIPE2 alleviates systemic lupus erythematosus through regulating macrophage polarization. Cell Physiol Biochem (2016) 38(1):330–9. doi: 10.1159/000438633
112. Li F, Yang Y, Zhu X, Huang L, Xu J. Macrophage polarization modulates development of systemic lupus erythematosus. Cell Physiol Biochem (2015) 37(4):1279–88. doi: 10.1159/000430251
113. Horuluoglu B, Bayik D, Kayraklioglu N, Goguet E, Kaplan MJ, Klinman DM. PAM3 supports the generation of M2-like macrophages from lupus patient monocytes and improves disease outcome in murine lupus. J Autoimmun (2019) 99:24–32. doi: 10.1016/j.jaut.2019.01.004
114. Aparicio-Soto M, Montserrat-de la Paz S, Sanchez-Hidalgo M, Cardeno A, Bermudez B, Muriana FJG, et al. Virgin olive oil and its phenol fraction modulate monocyte/macrophage functionality: a potential therapeutic strategy in the treatment of systemic lupus erythematosus. Br J Nutr (2018) 120(6):681–92. doi: 10.1017/S0007114518001976
115. Aparicio-Soto M, Sánchez-Hidalgo M, Alarcón-de-la-Lastra C. An update on diet and nutritional factors in systemic lupus erythematosus management. Nutr Res Rev (2017) 30(1):118–37. doi: 10.1017/S0954422417000026
116. Allison AC. Mechanisms of action of mycophenolate mofetil. Lupus (2005) 14 Suppl 1:s2–8. doi: 10.1191/0961203305lu2109oa
117. Yoon BR, Yoo SJ, Choi Yh, Chung YH, Kim J, Yoo IS, et al. Functional phenotype of synovial monocytes modulating inflammatory T-cell responses in rheumatoid arthritis (RA). PloS One (2014) 9(10):e109775. doi: 10.1371/journal.pone.0109775
118. Yanni G, Whelan A, Feighery C, Bresnihan B. Synovial tissue macrophages and joint erosion in rheumatoid arthritis. Ann Rheum Dis (1994) 53(1):39–44. doi: 10.1136/ard.53.1.39
119. Patterson AM, Schmutz C, Davis S, Gardner L, Ashton BA, Middleton J. Differential binding of chemokines to macrophages and neutrophils in the human inflamed synovium. Arthritis Res (2002) 4(3):209–14. doi: 10.1186/ar408
120. Richards PJ, Williams AS, Goodfellow RM, Williams BD. Liposomal clodronate eliminates synovial macrophages, reduces inflammation and ameliorates joint destruction in antigen-induced arthritis. Rheumatol (Oxford). (1999) 38(9):818–25. doi: 10.1093/rheumatology/38.9.818
121. Hamilton JA, Tak PP. The dynamics of macrophage lineage populations in inflammatory and autoimmune diseases. Arthritis Rheumatol (2009) 60(5):1210–21. doi: 10.1002/art.24505
122. Huang QQ, Birkett R, Doyle R, Shi B, Roberts EL, Mao Q, et al. The role of macrophages in the response to TNF inhibition in experimental arthritis. J Immunol (2018) 200(1):130–8. doi: 10.4049/jimmunol.1700229
123. Boutet MA, Courties G, Nerviani A, Le Goff B, Apparailly F, Pitzalis C, et al. Novel insights into macrophage diversity in rheumatoid arthritis synovium. Autoimmun Rev (2021) 20(3):102758. doi: 10.1016/j.autrev.2021.102758
124. Corvaisier M, Delneste Y, Jeanvoine H, Preisser L, Blanchard S, Garo E, et al. IL-26 is overexpressed in rheumatoid arthritis and induces proinflammatory cytokine production and Th17 cell generation. PloS Biol (2012) 10(9):e1001395. doi: 10.1371/journal.pbio.1001395
125. Lee KA, Kim KW, Kim BM, Won JY, Min HK, Lee DW, et al. Promotion of osteoclastogenesis by IL-26 in rheumatoid arthritis. Arthritis Res Ther (2019) 21(1):283. doi: 10.1186/s13075-019-2070-0
126. Grabiec AM, Krausz S, de Jager W, Burakowski T, Groot D, Sanders ME, et al. Histone deacetylase inhibitors suppress inflammatory activation of rheumatoid arthritis patient synovial macrophages and tissue. J Immunol (2010) 184(5):2718–28. doi: 10.4049/jimmunol.0901467
127. Semerano L, Clavel G, Assier E, Denys A, Boissier MC. Blood vessels, a potential therapeutic target in rheumatoid arthritis? Joint Bone Spine (2011) 78(2):118–23. doi: 10.1016/j.jbspin.2010.06.004
128. Koch AE, Polverini PJ, Kunkel SL, Harlow LA, DiPietro LA, Elner VM, et al. Interleukin-8 as a macrophage-derived mediator of angiogenesis. Science. (1992) 258(5089):1798–801. doi: 10.1126/science.1281554
129. Burbano C, Villar-Vesga J, Vásquez G, Muñoz-Vahos C, Rojas M, Castaño D. Proinflammatory differentiation of macrophages through microparticles that form immune complexes leads to T- and b-cell activation in systemic autoimmune diseases. Front Immunol (2019) 10:2058. doi: 10.3389/fimmu.2019.02058
130. Smiljanovic B, Grützkau A, Sörensen T, Grün JR, Vogl T, Bonin M, et al. Synovial tissue transcriptomes of long-standing rheumatoid arthritis are dominated by activated macrophages that reflect microbial stimulation. Sci Rep (2020) 10(1):7907. doi: 10.1038/s41598-020-64431-4
131. Degboé Y, Rauwel B, Baron M, Boyer JF, Ruyssen-Witrand A, Constantin A, et al. Polarization of rheumatoid macrophages by TNF targeting through an IL-10/STAT3 mechanism. Front Immunol (2019) 10:3. doi: 10.3389/fimmu.2019.00003
132. Crotti C, Biggioggero M, Becciolini A, Agape E, Favalli EG. Mavrilimumab: a unique insight and update on the current status in the treatment of rheumatoid arthritis. Expert Opin Investig Drugs (2019) 28(7):573–81. doi: 10.1080/13543784.2019.1631795
133. Mortensen JH, Guo X, De Los Reyes M, Dziegiel MH, Karsdal MA, Bay-Jensen AC, et al. The VICM biomarker is released from activated macrophages and inhibited by anti-GM-CSFRα-mAb treatment in rheumatoid arthritis patients. Clin Exp Rheumatol (2019) 37(1):73–80.
134. Hu Y, Wang B, Shen J, Low SA, Putt KS, Niessen HWM, et al. Depletion of activated macrophages with a folate receptor-beta-specific antibody improves symptoms in mouse models of rheumatoid arthritis. Arthritis Res Ther (2019) 21(1):143. doi: 10.1186/s13075-019-1912-0
135. Paoletti A, Rohmer J, Ly B, Pascaud J, Rivière E, Seror R, et al. Monocyte/Macrophage abnormalities specific to rheumatoid arthritis are linked to miR-155 and are differentially modulated by different TNF inhibitors. J Immunol (2019) 203(7):1766–75. doi: 10.4049/jimmunol.1900386
136. Soler Palacios B, Estrada-Capetillo L, Izquierdo E, Criado G, Nieto C, Municio C, et al. Macrophages from the synovium of active rheumatoid arthritis exhibit an activin a-dependent pro-inflammatory profile. J Pathol (2015) 235(3):515–26. doi: 10.1002/path.4466
137. Orecchioni M, Ghosheh Y, Pramod AB, Ley K. Macrophage polarization: Different gene signatures in M1(LPS+) vs. Classically M2(LPS-) vs. Alternatively Activated Macrophages. Front Immunol (2019) 10:1084. doi: 10.3389/fimmu.2019.01084
138. Yang X, Chang Y, Wei W. Emerging role of targeting macrophages in rheumatoid arthritis: Focus on polarization, metabolism and apoptosis. Cell Prolif. (2020) 53(7):e12854. doi: 10.1111/cpr.12854
139. Song G, Lu Q, Fan H, Zhang X, Ge L, Tian R, et al. Inhibition of hexokinases holds potential as treatment strategy for rheumatoid arthritis. Arthritis Res Ther (2019) 21(1):87. doi: 10.1186/s13075-019-1865-3
140. Cao Y, Liu J, Huang C, Tao Y, Wang Y, Chen X, et al. Wilforlide a ameliorates the progression of rheumatoid arthritis by inhibiting M1 macrophage polarization. J Pharmacol Sci (2022) 148(1):116–24. doi: 10.1016/j.jphs.2021.10.005
141. Wang X, Tu J, Jiang J, Zhang Q, Liu Q, Körner H, et al. Angiotensin II type 2 receptor modulates synovial macrophage polarization by inhibiting GRK2 membrane translocation in a rat model of collagen-induced arthritis. J Immunol (2020) 205(11):3141–53. doi: 10.4049/jimmunol.2000561
142. Park JY, Kwon S, Kim SH, Kang YJ, Khang D. Triamcinolone-gold nanoparticles repolarize synoviocytes and macrophages in an inflamed synovium. ACS Appl Mater Interfaces. (2020) 12(35):38936–49. doi: 10.1021/acsami.0c09842
143. Li H, Feng Y, Zheng X, Jia M, Mei Z, Wang Y, et al. M2-type exosomes nanoparticles for rheumatoid arthritis therapy via macrophage re-polarization. J Control Release. (2022) 341:16–30. doi: 10.1016/j.jconrel.2021.11.019
144. Guo RB, Zhang XY, Yan DK, Yu YJ, Wang YJ, Geng HX, et al. Folate-modified triptolide liposomes target activated macrophages for safe rheumatoid arthritis therapy. Biomater Sci (2022) 10(2):499–513. doi: 10.1039/d1bm01520f
145. Yang Y, Guo L, Wang Z, Liu P, Liu X, Ding J, et al. Targeted silver nanoparticles for rheumatoid arthritis therapy via macrophage apoptosis and re-polarization. Biomaterials. (2021) 264:120390. doi: 10.1016/j.biomaterials.2020.120390
146. Pattanaik D, Brown M, Postlethwaite BC, Postlethwaite AE. Pathogenesis of systemic sclerosis. Front Immunol (2015) 6:272. doi: 10.3389/fimmu.2015.00272
147. Brown M, O'Reilly S. The immunopathogenesis of fibrosis in systemic sclerosis. Clin Exp Immunol (2019) 195(3):310–21. doi: 10.1111/cei.13238
148. LeRoy EC. Systemic sclerosis. a vascular perspective. Rheum Dis Clin North Am (1996) 22(4):675–94. doi: 10.1016/s0889-857x(05)70295-7
149. Guiducci S, Giacomelli R, Cerinic MM. Vascular complications of scleroderma. Autoimmun Rev (2007) 6(8):520–3. doi: 10.1016/j.autrev.2006.12.006
150. Kräling BM, Maul GG, Jimenez SA. Mononuclear cellular infiltrates in clinically involved skin from patients with systemic sclerosis of recent onset predominantly consist of monocytes/macrophages. Pathobiology. (1995) 63(1):48–56. doi: 10.1159/000163933
151. Misharin AV, Morales-Nebreda L, Reyfman PA, Cuda CM, Walter JM, McQuattie-Pimentel AC, et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J Exp Med (2017) 214(8):2387–404. doi: 10.1084/jem.20162152
152. Ballerie A, Lescoat A, Augagneur Y, Lelong M, Morzadec C, Cazalets C, et al. Efferocytosis capacities of blood monocyte-derived macrophages in systemic sclerosis. Immunol Cell Biol (2019) 97(3):340–7. doi: 10.1111/imcb.12217
153. Rudnik M, Hukara A, Kocherova I, Jordan S, Schniering J, Milleret V, et al. Elevated fibronectin levels in profibrotic CD14+ monocytes and CD14+ macrophages in systemic sclerosis. Front Immunol (2021) 12:642891. doi: 10.3389/fimmu.2021.642891
154. Liakouli V, Cipriani P, Marrelli A, Alvaro S, Ruscitti P, Giacomelli R. Angiogenic cytokines and growth factors in systemic sclerosis. Autoimmun Rev (2011) 10(10):590–4. doi: 10.1016/j.autrev.2011.04.019
155. Laurent P, Lapoirie J, Leleu D, Levionnois E, Grenier C, Jurado-Mestre B, et al. Fédération hospitalo-universitaire ACRONIM and the centre national de référence des maladies auto-immunes systémiques rares de l'Est et du sud-ouest (RESO). interleukin-1β-Activated microvascular endothelial cells promote DC-SIGN-Positive alternatively activated macrophages as a mechanism of skin fibrosis in systemic sclerosis. Arthritis Rheumatol (2022) 74(6):1013–26. doi: 10.1002/art.42061
156. Taniguchi T, Miyagawa T, Toyama S, Yamashita T, Nakamura K, Saigusa R, et al. CXCL13 produced by macrophages due to Fli1 deficiency may contribute to the development of tissue fibrosis, vasculopathy and immune activation in systemic sclerosis. Exp Dermatol (2018) 27(9):1030–7. doi: 10.1111/exd.13724
157. Distler O, Distler JH, Scheid A, Acker T, Hirth A, Rethage J, et al. Uncontrolled expression of vascular endothelial growth factor and its receptors leads to insufficient skin angiogenesis in patients with systemic sclerosis. Circ Res (2004) 95(1):109–16. doi: 10.1161/01.RES.0000134644.89917.96
158. Numajiri H, Kuzumi A, Fukasawa T, Ebata S, Yoshizaki-Ogawa A, Asano Y, et al. B cell depletion inhibits fibrosis via suppression of profibrotic macrophage differentiation in a mouse model of systemic sclerosis. Arthritis Rheumatol (2021) 73(11):2086–95. doi: 10.1002/art.41798
159. Toledo DM, Pioli PA. Macrophages in systemic sclerosis: Novel insights and therapeutic implications. Curr Rheumatol Rep (2019) 21(7):31. doi: 10.1007/s11926-019-0831-z
160. Lescoat A, Ballerie A, Lelong M, Augagneur Y, Morzadec C, Jouneau S, et al. Crystalline silica impairs efferocytosis abilities of human and mouse macrophages: Implication for silica-associated systemic sclerosis. Front Immunol (2020) 11:219. doi: 10.3389/fimmu.2020.00219
161. Sun C, Chen SY. RGC32 promotes bleomycin-induced systemic sclerosis in a murine disease model by modulating classically activated macrophage function. J Immunol (2018) 200(8):2777–85. doi: 10.4049/jimmunol.1701542
162. Taroni JN, Greene CS, Martyanov V, Wood TA, Christmann RB, Farber HW, et al. A novel multi-network approach reveals tissue-specific cellular modulators of fibrosis in systemic sclerosis. Genome Med (2017) 9(1):27. doi: 10.1186/s13073-017-0417-1
163. Sun W, Yan S, Yang C, Yang J, Wang H, Li C, et al. Mesenchymal stem cells-derived exosomes ameliorate lupus by inducing M2 macrophage polarization and regulatory T cell expansion in MRL/lpr mice. Immunol Invest. (2022) 51(6):1785–803. doi: 10.1080/08820139.2022.2055478
164. Bielecki M, Kowal K, Lapinska A, Chyczewski L, Kowal-Bielecka O. Increased release of soluble CD163 by the peripheral blood mononuclear cells is associated with worse prognosis in patients with systemic sclerosis. Adv Med Sci (2013) 58(1):126–33. doi: 10.2478/v10039-012-0076-9
165. Higashi-Kuwata N, Makino T, Inoue Y, Takeya M, Ihn H. Alternatively activated macrophages (M2 macrophages) in the skin of patient with localized scleroderma. Exp Dermatol (2009) 18(8):727–9. doi: 10.1111/j.1600-0625.2008.00828.x
166. Maier C, Ramming A, Bergmann C, Weinkam R, Kittan N, Schett G, et al. Inhibition of phosphodiesterase 4 (PDE4) reduces dermal fibrosis by interfering with the release of interleukin-6 from M2 macrophages. Ann Rheum Dis (2017) 76(6):1133–41. doi: 10.1136/annrheumdis-2016-210189
167. Yamashita T, Asano Y, Taniguchi T, Nakamura K, Saigusa R, Miura S, et al. Glycyrrhizin ameliorates fibrosis, vasculopathy, and inflammation in animal models of systemic sclerosis. J Invest Dermatol (2017) 137(3):631–40. doi: 10.1016/j.jid.2016.08.037
168. Huang J, Maier C, Zhang Y, Soare A, Dees C, Beyer C, et al. Nintedanib inhibits macrophage activation and ameliorates vascular and fibrotic manifestations in the Fra2 mouse model of systemic sclerosis. Ann Rheum Dis (2017) 76(11):1941–8. doi: 10.1136/annrheumdis-2016-210823
169. Skaug B, Khanna D, Swindell WR, Hinchcliff ME, Frech TM, Steen VD, et al. Global skin gene expression analysis of early diffuse cutaneous systemic sclerosis shows a prominent innate and adaptive inflammatory profile. Ann Rheum Dis (2020) 79(3):379–86. doi: 10.1136/annrheumdis-2019-215894
170. Laurent P, Sisirak V, Lazaro E, Richez C, Duffau P, Blanco P, et al. Innate immunity in systemic sclerosis fibrosis: Recent advances. Front Immunol (2018) 9:1702. doi: 10.3389/fimmu.2018.01702
171. Jeljeli M, Riccio LGC, Doridot L, Chêne C, Nicco C, Chouzenoux S, et al. Trained immunity modulates inflammation-induced fibrosis. Nat Commun (2019) 10(1):5670. doi: 10.1038/s41467-019-13636-x
172. Anderson MS, Bluestone JA. The NOD mouse: a model of immune dysregulation. Annu Rev Immunol (2005) 23:447–85. doi: 10.1146/annurev.immunol
173. Burg AR, Tse HM. Redox-sensitive innate immune pathways during macrophage activation in type 1 diabetes. Antioxid Redox Signal (2018) 29(14):1373–98. doi: 10.1089/ars.2017.7243
174. O'Reilly LA, Hutchings PR, Crocker PR, Simpson E, Lund T, Kioussis D, et al. Characterization of pancreatic islet cell infiltrates in NOD mice: effect of cell transfer and transgene expression. Eur J Immunol (1991) 21(5):1171–80. doi: 10.1002/eji.1830210512
175. Hanenberg H, Kolb-Bachofen V, Kantwerk-Funke G, Kolb H. Macrophage infiltration precedes and is a prerequisite for lymphocytic insulitis in pancreatic islets of pre-diabetic BB rats. Diabetologia. (1989) 32(2):126–34. doi: 10.1007/BF00505185
176. Willcox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG. Analysis of islet inflammation in human type 1 diabetes. Clin Exp Immunol (2009) 155(2):173–81. doi: 10.1111/j.1365-2249.2008.03860.x
177. Oschilewski U, Kiesel U, Kolb H. Administration of silica prevents diabetes in BB-rats. Diabetes (1985) 34(2):197–9. doi: 10.2337/diab.34.2.197
178. Kolb H, Burkart V, Appels B, Hanenberg H, Kantwerk-Funke G, Kiesel U, et al. Essential contribution of macrophages to islet cell destruction in vivo and in vitro. J Autoimmun (1990) 3 Suppl 1:117–20. doi: 10.1016/s0896-8411(09)90020-8
179. Jun HS, Santamaria P, Lim HW, Zhang ML, Yoon JW. Absolute requirement of macrophages for the development and activation of beta-cell cytotoxic CD8+ T-cells in T-cell receptor transgenic NOD mice. Diabetes. (1999) 48(1):34–42. doi: 10.2337/diabetes.48.1.34
180. Calderon B, Suri A, Unanue ER. In CD4+ T-cell-induced diabetes, macrophages are the final effector cells that mediate islet beta-cell killing: studies from an acute model. Am J Pathol (2006) 169(6):2137–47. doi: 10.2353/ajpath.2006.060539
181. Thornley TB, Agarwal KA, Kyriazis P, Ma L, Chipashvili V, Aker JE, et al. Contrasting roles of islet resident immunoregulatory macrophages and dendritic cells in experimental autoimmune type 1 diabetes. PloS One (2016) 11(3):e0150792. doi: 10.1371/journal.pone.0150792
182. Zhang J, Tan SB, Guo ZG. CD47 decline in pancreatic islet cells promotes macrophage-mediated phagocytosis in type I diabetes. World J Diabetes. (2020) 11(6):239–51. doi: 10.4239/wjd.v11.i6.239
183. Cantor J, Haskins K. Recruitment and activation of macrophages by pathogenic CD4 T cells in type 1 diabetes: evidence for involvement of CCR8 and CCL1. J Immunol (2007) 179(9):5760–7. doi: 10.4049/jimmunol.179.9.5760
184. Stoffels K, Overbergh L, Giulietti A, Kasran A, Bouillon R, Gysemans C, et al. NOD macrophages produce high levels of inflammatory cytokines upon encounter of apoptotic or necrotic cells. J Autoimmun (2004) 23(1):9–15. doi: 10.1016/j.jaut.2004.03.012
185. Marée AF, Kublik R, Finegood DT, Edelstein-Keshet L. Modelling the onset of type 1 diabetes: can impaired macrophage phagocytosis make the difference between health and disease? Philos Trans A Math Phys Eng Sci (2006) 364(1842). doi: 10.1098/rsta.2006.1769
186. Chung YH, Jun HS, Kang Y, Hirasawa K, Lee BR, Van Rooijen N, et al. Role of macrophages and macrophage-derived cytokines in the pathogenesis of kilham rat virus-induced autoimmune diabetes in diabetes-resistant BioBreeding rats. J Immunol (1997) 159(1):466–71. doi: 10.4049/jimmunol.159.1.466
187. Diana J, Lehuen A. Macrophages and β-cells are responsible for CXCR2-mediated neutrophil infiltration of the pancreas during autoimmune diabetes. EMBO Mol Med (2014) 6(8):1090–104. doi: 10.15252/emmm.201404144
188. Amrani A, Verdaguer J, Thiessen S, Bou S, Santamaria P. IL-1alpha, IL-1beta, and IFN-gamma mark beta cells for fas-dependent destruction by diabetogenic CD4(+) T lymphocytes. J Clin Invest. (2000) 105(4):459–68. doi: 10.1172/JCI8185
189. Jun HS, Yoon CS, Zbytnuik L, van Rooijen N, Yoon JW. The role of macrophages in T cell-mediated autoimmune diabetes in nonobese diabetic mice. J Exp Med (1999) 189(2):347–58. doi: 10.1084/jem.189.2.347
190. Marro BS, Legrain S, Ware BC, Oldstone MB. Macrophage IFN-I signaling promotes autoreactive T cell infiltration into islets in type 1 diabetes model. JCI Insight (2019) 4(2):e125067. doi: 10.1172/jci.insight.125067
191. Parsa R, Andresen P, Gillett A, Mia S, Zhang XM, Mayans S, et al. Adoptive transfer of immunomodulatory M2 macrophages prevents type 1 diabetes in NOD mice. Diabetes. (2012) 61(11):2881–92. doi: 10.2337/db11-1635
192. Li Y, Ding X, Tian X, Zheng J, Ding C, Li X, et al. Islet transplantation modulates macrophage to induce immune tolerance and angiogenesis of islet tissue in type I diabetes mice model. Aging (Albany NY). (2020) 12(23):24023–32. doi: 10.18632/aging.104085
193. Barra JM, Kozlovskaya V, Kharlampieva E, Tse HM. Localized immunosuppression with tannic acid encapsulation delays islet allograft and autoimmune-mediated rejection. Diabetes. (2020) 69(9):1948–60. doi: 10.2337/db20-0248
194. Chen Y, Nagy T, Guo TL. Glycated whey proteins protect NOD mice against type 1 diabetes by increasing anti-inflammatory responses and decreasing autoreactivity to self-antigens. J Funct Foods. (2019) 56:171–81. doi: 10.1016/j.jff.2019.03.015
195. Wang F, Sun F, Luo J, Yue T, Chen L, Zhou H, et al. Loss of ubiquitin-conjugating enzyme E2 (Ubc9) in macrophages exacerbates multiple low-dose streptozotocin-induced diabetes by attenuating M2 macrophage polarization. Cell Death Dis (2019) 10(12):892. doi: 10.1038/s41419-019-2130-z
196. Timper K, Hruz P, Beglinger C, Donath MY. Infliximab in the treatment of crohn disease and type 1 diabetes. Diabetes Care (2013) 36(7):e90–1. doi: 10.2337/dc13-0199
197. Green EA, Flavell RA. Tumor necrosis factor-alpha and the progression of diabetes in non-obese diabetic mice. Immunol Rev (1999) 169:11–22. doi: 10.1111/j.1600-065x.1999.tb01302.x
198. Von Scholten BJ, Kreiner FF, Gough SCL, von Herrath M. Current and future therapies for type 1 diabetes. Diabetologia. (2021) 64(5):1037–48. doi: 10.1007/s00125-021-05398-3
199. Citro A, Cantarelli E, Piemonti L. The CXCR1/2 pathway: Involvement in diabetes pathophysiology and potential target for T1D interventions. Curr Diabetes Rep (2015) 15(10):68. doi: 10.1007/s11892-015-0638-x
200. Li R, He Y, Zhu Y, Jiang L, Zhang S, Qin J, et al. Route to rheumatoid arthritis by macrophage-derived microvesicle-coated nanoparticles. Nano Lett (2019) 19(1):124–34. doi: 10.1021/acs.nanolett.8b03439
201. Tang TT, Lv LL, Wang B, Cao JY, Feng Y, Li ZL, et al. Employing macrophage-derived microvesicle for kidney-targeted delivery of dexamethasone: An efficient therapeutic strategy against renal inflammation and fibrosis. Theranostics. (2019) 9(16):4740–55. doi: 10.7150/thno.33520
Keywords: macrophage, systemic lupus erythematosus, rheumatic arthritis, systemic sclerosis, type 1 diabetes
Citation: Yang S, Zhao M and Jia S (2023) Macrophage: Key player in the pathogenesis of autoimmune diseases. Front. Immunol. 14:1080310. doi: 10.3389/fimmu.2023.1080310
Received: 26 October 2022; Accepted: 09 January 2023;
Published: 14 February 2023.
Edited by:
Yanan Ma, Memorial Sloan Kettering Cancer Center, United StatesReviewed by:
Mengmeng Jin, Rutgers, The State University of New Jersey, United StatesCopyright © 2023 Yang, Zhao and Jia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sujie Jia, c3VqaWVqaWFAMTI2LmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.