
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 14 February 2023
Sec. Molecular Innate Immunity
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1070779
This article is part of the Research Topic The Role of Innate Immune and Resident Cells in Allergic Diseases and their Potential in Therapeutic Intervention View all 9 articles
 Zahra Kanannejad1†
Zahra Kanannejad1† Saeede Soleimanian1†
Saeede Soleimanian1† Zahra Ghahramani2
Zahra Ghahramani2 Najmeh Sepahi1
Najmeh Sepahi1 Milad Mohkam1Soheila Alyasin1
Milad Mohkam1Soheila Alyasin1 Nasim Kheshtchin1,3*
Nasim Kheshtchin1,3*Allergic asthma is a respiratory disease initiated by type-2 immune responses characterized by secretion of alarmins, interleukin-4 (IL-4), IL-5, and IL-13, eosinophilic inflammation, and airway hyperresponsiveness (AHR). Immune checkpoints (ICPs) are inhibitory or stimulatory molecules expressed on different immune cells, tumor cells, or other cell types that regulate immune system activation and maintain immune homeostasis. Compelling evidence indicates a key role for ICPs in both the progression and prevention of asthma. There is also evidence of asthma development or exacerbation in some cancer patients receiving ICP therapy. The aim of this review is to provide an updated overview of ICPs and their roles in asthma pathogenesis, and to assess their implications as therapeutic targets in asthma.
Asthma is a respiratory disease initiated by crosstalk between the innate and adaptive immunity (1); however, T helper 2 (Th2) cells play a crucial role in driving airway inflammation and bronchial hyper-responsiveness, due to secretion of type-2 cytokines including interleukin-4 (IL-4), IL-5, and IL-13 (2, 3). Various effectors and signaling pathways contribute to the dissemination of allergic inflammatory responses that occur in epithelial, and stromal cells of the lung in allergic patients. Asthma is triggered by disruption of epithelial barrier integrity through environmental threats, such as airborne allergens. Consequently, epithelial cell (EC) injury results in the secretion of cytokines (TSLP, IL- 33, IL-25, and GM-CSF) and chemokines (TARC, CCL22, and CXCL10) from ECs. The release of alarmins promotes the stimulation of Th2 cells, type 2 innate lymphoid cells (ILC2s), and other immune cells such as macrophages, basophils and eosinophils (4). However, investigations reveal that other cytokines such as TGF-β1, which is involved in tissue remodeling during airway inflammation, also contributes to the pathogenesis of asthma through induction of Th2 and Th9 and suppression of Treg cells (5). In contrast, IL-37 down-regulates asthmatic airway inflammation by counterbalancing the disease-amplifying effects of IL-1β and IL-33 (6).
Asthma is a heterogeneous inflammatory disease characterized by complex pathophysiological pathways (7). Allergen- specific immunotherapy (AIT) through administration of increasing doses of relevant allergens to an allergic subject is the only disease- modifying treatment available for several common allergic conditions, such as asthma (8). Several biomarkers have been suggested by different studies as predictors of treatment efficacy, such as generation of allergen- specific IgG or IgG4 and IgA in serum (9, 10), suppression of local numbers of Th2/ILC2 cells or secretion of type 2 cytokines (11, 12) increased numbers of Th1 cells, Tregs, or Bregs (13–15), and induction of a recently introduced anti-inflammatory mediator, secretoglobin 1A1 (16), in the local environment. Various molecular effectors have been identified as therapeutic targets for severe asthma. For example, anti-asthma monoclonal antibodies against pro-allergic immunoglobulin E (IgE), IL-5, IL-4, and IL-13 have been developed. Since the regulation of T helper immune responses depends on the interaction between surface receptors and their ligands, inhibitory receptors could be considered as proper targets for asthma therapies (17, 18).
Immune checkpoints (ICPs) are inhibitory or stimulatory molecules expressed on the different immune cells, tumor cells, or other cell types that regulate immune system activation and maintain immune homeostasis. To date, many ICP molecules have been discovered including programmed cell death receptor-1 (PD-1), lymphocyte activation-gene-3 (LAG-3), T- cell membrane protein-3 (TIM-3), cytotoxic T- lymphocyte antigen-4 (CTLA-4), inducible co-stimulator (ICOS), and B7-H3. Stimulatory ICPs play important role in naïve T cell activation by providing the second signal (co-stimulatory signal), while signaling through inhibitory ICPs can direct activated T cells into a state called ‘exhaustion’ that is associated with reduced effector function and sustained expression of ICPs (19). Many pathogens and cancer cells can promote the inhibitory interactions to escape immune responses (20–26). Compelling evidence indicates a key role for ICPs in both progression and prevention of asthma (Table 1). Signals derived from costimulatory ICPs (for example CD28, ICOS, and OX40) together with IL-4, most likely derived from basophils, are required for initiating antigen- dependent polarization of naïve T cells toward Th2 cells and follicular helper T cells (TFH) (56). In contrast, antigen recognition in the presence of tolerogenic signals such as IL-10 and TGF-β leads to the development of telegenic dendritic cells (DCs) that promotes T regulatory cells (Tregs) development (57). Tolerogenic DCs may induce naïve T cell differentiation into Treg cells by providing inhibitory (PD-1 – PD-L1) and stimulatory (for example ICOS – ICOS-L) molecular interactions. Treg cells then inhibit sensitization and airway hyperresponsiveness through production of immunosuppressive cytokines (IL-10, TGF-β, and IL-35) and expression of inhibitory molecules (for example CTLA-4, PD-1, and LAG-3) and cytotoxic enzymes (granzyme B) (58). There is also evidence of asthma development or exacerbation in some cancer patients receiving ICP therapy. Based on the above considerations, the aim of this review is to provide an updated overview of ICPs and their roles in asthma pathogenesis (summarized in Figure 1), and to assess their implications as therapeutic targets in asthma.
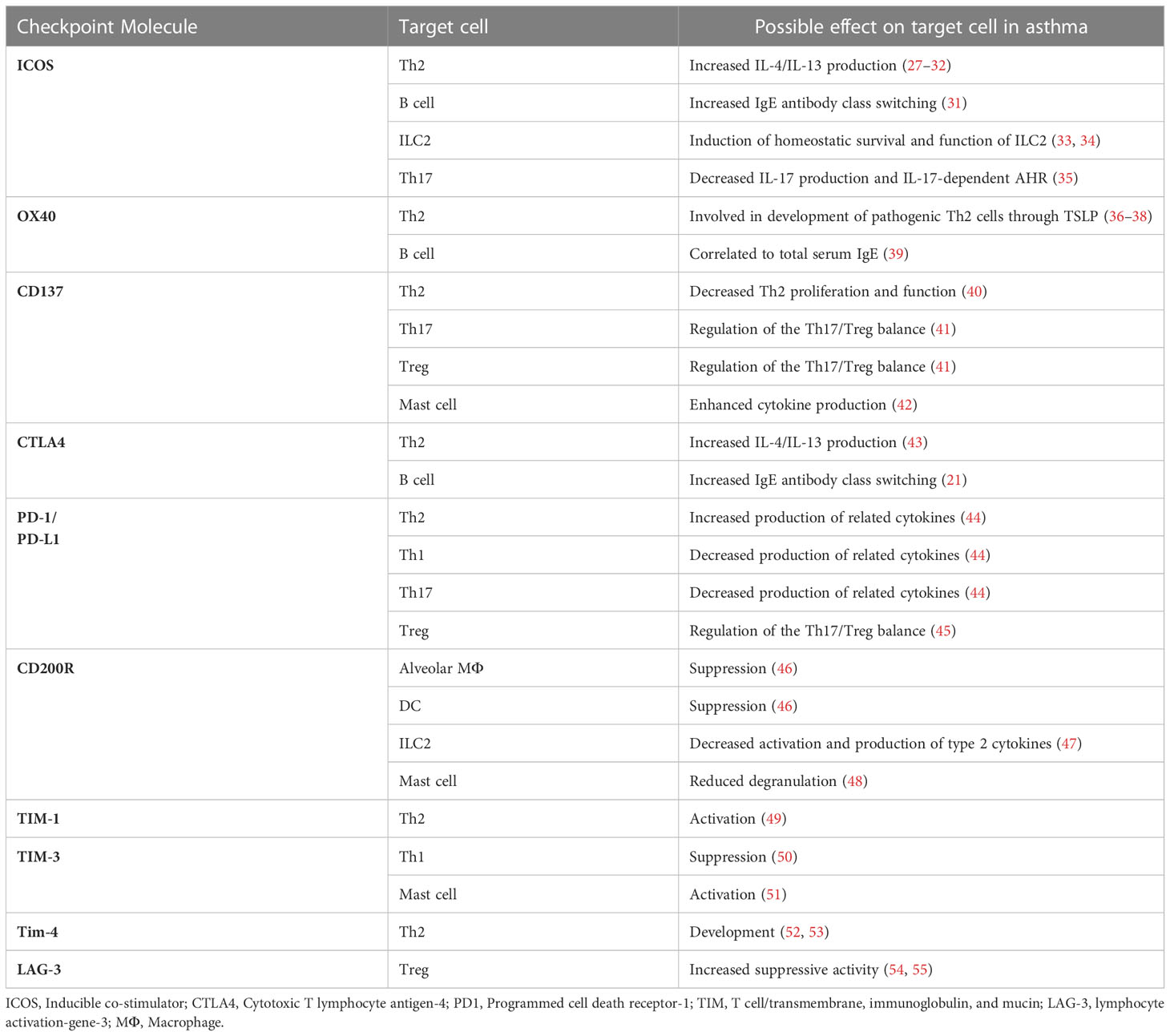
Table 1 Effects of co-inhibitory/co-stimulatory immune checkpoints in asthma.
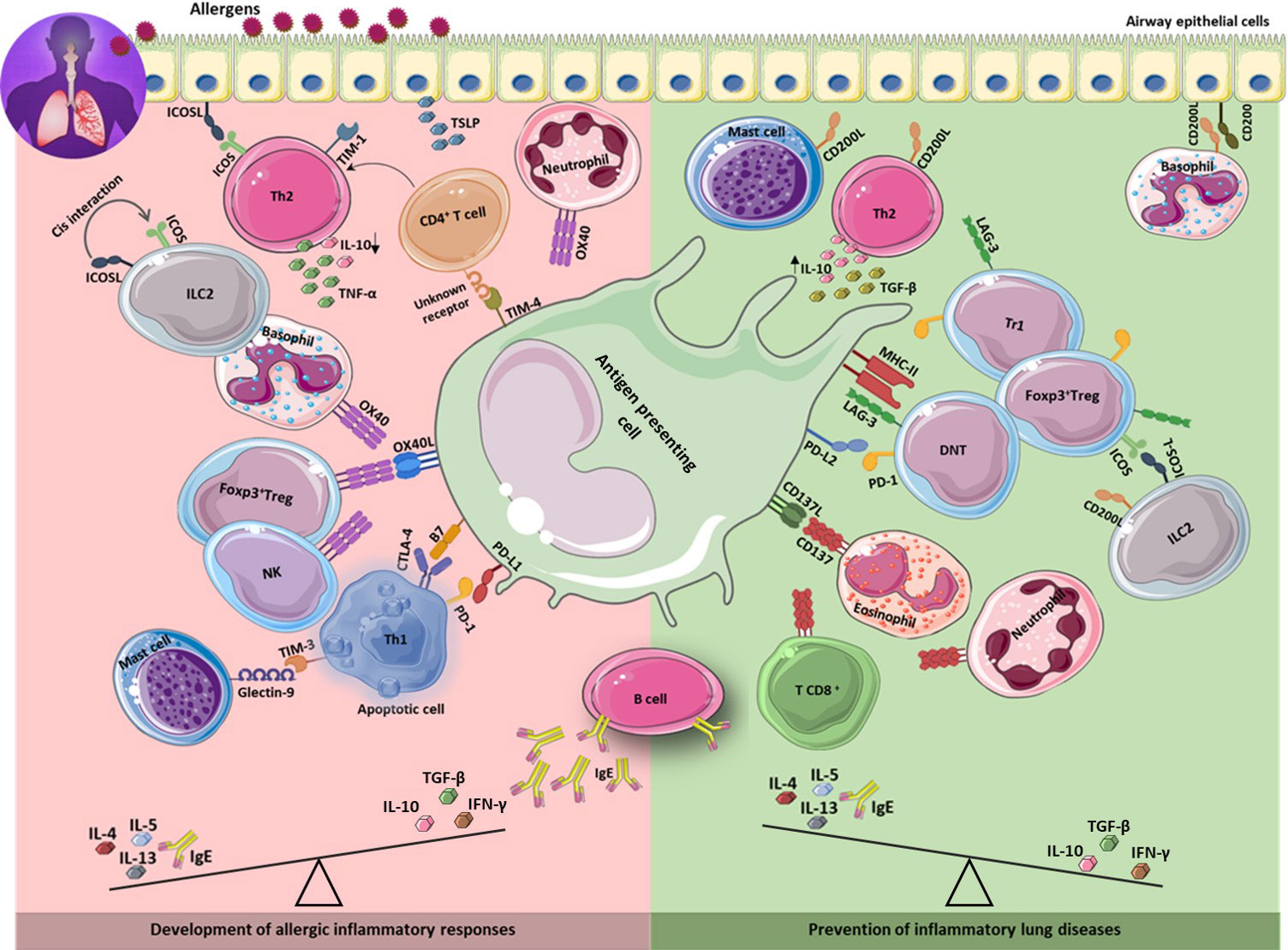
Figure 1 Immune checkpoints in the development and prevention of asthma. The development of asthma is associated with increased Th2 response results in secretion of IL-4, 5, 13, and IgE production. Inhibitory and stimulatory immune checkpoint molecules expressing on different immune cells as well as on lung epithelium, contribute to development (left) or prevention (right) of allergic inflammatory responses through interaction with their ligands on different cell types including APCs. Th2: T helper 2; IL: interleukin.
The inducible co-stimulator (ICOS), known as the third member of the CD28 family (59), is an activating co-stimulatory immune checkpoint expressed on activated T cells as well as Treg cells (60, 61). The ligand for ICOS, ICOSL (also known as B7-H2), is constitutively expressed on antigen- presenting cells (APCs), including B cells, DCs, and macrophages, and is downregulated upon binding to ICOS as an immunoregulatory mechanism (62). Its expression can be induced in non-lymphoid tissues such as lung epithelial cells and certain fibroblasts (63, 64). Interactions between ICOS and ICOSL trigger the activation of diverse signaling molecules that prevent apoptosis and play important roles in the development of memory and effector T cells and specific humoral immune responses. As discussed below, ICOS is critical for Th2 differentiation and function and for development of pulmonary inflammatory responses (27, 65).
Numerous studies using animal models with disrupted ICOS genes and antibody-blocking experiments have revealed the role of ICOS in tolerance induction. Preliminary studies in mouse models of Th1-mediated autoimmune diseases, including experimental autoimmune encephalomyelitis (EAE) and type 1 diabetes, using anti-ICOS mAbs have demonstrated disease exacerbation via disruption of the balance between effector and regulatory T cells (27, 66, 67). In allergy and asthma as Th2-mediated diseases, on the other hand, ICOS signaling regulates the magnitude of Th2-mediated pulmonary inflammation and clonal expansion (28–30). Functional studies have shown that ICOS-deficient mice exhibit profound defects in germinal center formation, antibody class switching and IL-4/IL-13 production (27, 31, 32). Similarly, in the context of allergic airway inflammation, ICOSL deletion leads to decreased serum IgE levels and inflammatory airway infiltration (68). Interestingly, a study using OVA-TCR transgenic mice showed that challenge with respiratory allergens leads to the induction of respiratory tolerance through development of Treg cells. This observation was at least partly, attributed to ICOS co-stimulation signals provided by pulmonary DCs suggesting a role for ICOS in respiratory tolerance (69).
Emerging evidence has revealed an interaction between ILC2 and Treg cells in allergic asthma, which involves ICOS: ICOSL ligation. The co-expression of ICOS and ICOSL on ILC2s has been demonstrated, and cis interaction of ICOS and ICOSL on ILC2s stimulates homeostatic survival and function of these cells (33, 34). Furthermore, induced Tregs (iTregs) have been shown to suppress ILC2-driven pro-inflammatory cytokines and pulmonary inflammation. This effect apparently involves Tran’s interaction of ICOS-L on ILC2 with ICOS on Tregs, which leads to decreased function of ILC2 via disruption of cis interaction of ICOS: ICOSL on ILCs. However, the authors also pointed out that iTreg-TGF-β signaling is the main mechanism of this suppression, and cell-cell contact through ICOS:ICOSL interaction is necessary for iTreg-mediated suppression (33, 34). Furthermore, suppression of IL-17 production by IL-35 producing ICOS+ Tregs and reversal of established IL-17-dependent AHR in mice is another example of ICOS involvement in controlling allergic asthma (35).
OX40 (CD134, TNFRSF4) belongs to the tumor necrosis factor receptor (TNFR) superfamily, which is predominantly expressed on activated CD4+ and CD8+ T cells (70–72). OX40 acts as a co-stimulatory receptor essential for regulating activation, survival, and expansion of conventional CD4+ and CD8+ T cells (73). It has been shown that the expression of OX40 is not restricted to activated conventional T cells but is also expressed on activated Treg cells, neutrophils and NK cells (74). The engagement of OX40 by its ligand OX40L (CD252) on antigen- presenting cells enhances T cell- mediated immune responses and contributes to the development of autoimmune diseases (75). Animal studies have revealed that OX40L is present in both asthmatic and non-asthmatic airway smooth muscle cells with higher levels of both OX40 and OX40L in asthma (76, 77). There is also an association between the expression level of OX40/OX40L and severity of asthma, since elevated expression was observed in bronchial submucosa of mild asthma, which was related to the accumulation of eosinophils and the level of IL-4 expression in the lamina propria (36). Another study investigated OX40L expression in atopic and non-atopic asthmatics, and found significantly higher levels of this molecule in atopic asthma. It was also shown that OX40L over expression in atopic patients was correlated with the specific hallmarks of the allergic asthma including total serum IgE suggesting a critical role for OX40L in the atopic mechanism (39). Indeed, involvement of OX40L in the induction phase of asthma has been suggested because the administration of a neutralizing antibody against OX40L prior to sensitization of mice prevented the induction of asthmatic responses with no effect on asthmatic responses during the challenge period. These observations provide evidence of the contribution of OX40 to the development of pathogenic Th2 cells, but not to the migration and activation of pathogenic Th2 cells in the lungs (78). OX40L has also been shown to be involved in development of Th2 responses to TSLP. TSLP activates human myeloid DCs to induce OX40L which triggers an inflammatory Th2 response characterized by high levels of TNF-α and low levels of IL-10 production, representing pathogenic Th2 cells involved in allergic inflammatory diseases such as asthma (36–38). Interestingly, exosomes derived from TSLP-activated DCs can induce differentiation of CD4+ T cells into Th2 cells in an OX40L-dependent manner (79). High expression of OX40L by activated basophils has also been demonstrated in a mouse model of allergic asthma. Blockade of OX40-OX40L interaction resulted in repressed differentiation of Th2 cells primed by basophils in vitro and attenuated OVA-induced eosinophilic inflammation in an experimental model (80). Such observations reveal essential modulatory functions of OX40–OX40L interactions in the development of allergic asthma, making them attractive candidates for clinical intervention.
CD137 (also known as 4-1BB) is a prototype co-stimulatory molecule for T cells (81). Treg cells are the only cell type with high levels of constitutive expression of CD137, while CD137 is expressed in lymphoid cells, including T cells of both CD4+ and CD8+ subsets, natural killer (NK) cells, NKT cells and myeloid lineage cells (82). Specific expression of CD137 has also been found in eosinophils derived from patients with allergic asthma or atopic dermatitis (83). Ligand for CD137 (CD137L) is a member of the TNF family and is expressed by APCs, which use it to co-stimulate T cell activity (84). The involvement of CD137/CD137L interactions in CD4+ T- cell-mediated inflammation has been demonstrated in various studies (81, 82). However, it is not clear whether this contribution involves activation of CD4+ T cells or other steps of inflammatory process (81). Moreover, these interactions have been suggested to be involved in the suppression of antigen-specific helper T cells and B cell-dependent humoral immune responses (83), and play an essential role in allergic asthma by regulating the Th17/Treg balance (41). It has been shown that administration of anti-CD137 in a mouse model of asthma not only prevented the development of asthma phenotype, but also completely reversed the established disease. Further investigations revealed the association of protection with reduced Th2 proliferation and function, as well as elevated secretion of the Th1-type cytokine IFN-γ (40). Confirming these observations, agonistic Abs to CD137 could directly co-inhibit Th2 responses and co-stimulate Th1 responses in vitro. However, CD137-mediated suppression of Th2 response is independent of IFN-γ (85). Cytokine production by mast cells following FcϵRI engagement was also enhanced in response to agonistic anti-CD137 antibodies, as demonstrated by another study (42). Interestingly, Stimulation of the CD137L using an agonistic antibody against CD137L instead of CD137 in an animal model of asthma prevented the development of asthma phenotype but failed to reverse the established disease. These results were partly attributed to IFN-γ-producing CD8+ T cells (86).
While multiple studies have indicated reduced airway hyper-reactivity, eosinophilic airway inflammation, mucus production, and IgE levels following administration of anti-CD137 mAb, indicating its potential to alleviate asthma (40, 87–90), a study using anti-CD137 antibody showed no reduction in airway hyper-reactivity and eosinophilic inflammation in a murine model of asthma, although abolished allergen-specific IgE was observed (91). Mice deficient in CD137 have also been shown to develop eosinophilic airway inflammation associated with Th2 cytokine production and allergen-specific serum IgE levels (92). In accordance with these observations, both soluble and membrane-bound CD137L levels in monocytes are reduced in patients with allergic asthma (41). Regarding the contribution of CD137 to the development of asthma, more experimental studies are required on the application of CD137 directed immunotherapies in the context of allergic diseases such as asthma.
CTLA-4, also known as CD152, is a member of the B7/CD28 family and has a high affinity for B7-1/2. CTLA-4 is constitutively expressed on Treg cells, but is inducible in conventional T cells (93). CTLA-4 competes with CD28 for B7 binding and produces inhibitory signals that counteract stimulatory signals originating from CD28:B7 ligation (94). CTLA-4 blockade leads to an augmented immune response owning to the potentiation of helper T cell function. Conversely, engagement of CTLA-4 in Tregs enhances their suppressive activity (95). Disruption of this delicate balance in immune regulation may lead to autoimmune or atopic diseases (96). Multiple polymorphism studies have investigated the association between CTLA-4 and asthma severity and AHR, providing evidence for involvement of these polymorphisms in controlling helper T cell- mediated responses and total serum IgE levels in patients with asthma (97, 98). The surface expression of T cell co-stimulatory molecules CTLA-4 and CD28 and their counter-ligands is differentially induced for T cell activation and expansion in atopic asthma (99). Recent studies have indicated an elevation in plasma soluble CTLA-4 and CS28 and their counter ligands B7-1 and B7-2 in asthmatic patients (100, 101). Generally, sCTLA-4 levels are higher in atopic and non-atopic asthma as severity increases (99, 100, 102–104) and its concentration is positively and significantly associated with total serum IgE and eosinophil count in atopic asthmatic patients (21, 43, 104, 105). The exact role of these soluble co-stimulatory molecules in the pathogenesis of allergic asthma is not clear; however, it may be a consequence of the dysregulation of T cell activation (101, 104).
In a study of children with recurrent wheezing with high- or low-risk factors for asthma, a reduced absolute number and percentage of CD4+CD25+CTLA-4+ T cells was shown compared to healthy children. Additionally, stimulation of PBMCs with house dust mite extract resulted in decreased expression of CTLA-4 mRNA in high-risk children, suggesting a potential association between defective expression of CTLA-4 and development of asthma (105). Cumulative evidence suggests a potential therapeutic capacity of CTLA-4 in diverse autoimmune diseases and atopy. In accordance with this evidence, a high dose allergen-CTLA-4-encoding DNA vaccine was shown to suppress Th2 immune responses induced by allergens in a mouse model of asthma (43).
Programmed cell death 1 protein (PD-1) is an inhibitory immune checkpoint molecule expressed by exhausted CD8+ T cells, activated T cells, naive and activated B cells (21). Signaling pathways triggered by the binding of PD-1 to its ligands, PD-L1 (B7-H1) and PD-L2 (B7-DC), are essential for regulating the balance between T-cell activation and tolerance, thereby contributing to the pathogenesis of various diseases such as autoimmunity, tumors, infectious diseases, and allergies (17, 106, 107). Primary role of PD-1 is the regulation of T cell responses via promotion of apoptosis in effector T cells and inhibition of apoptosis in Treg cells after antigen clearance (21). However, chronic engagement of TCR in the absence of costimulatory signals can cause PD-1 overexpression leading to anergy, exhaustion and tolerance of CD4+ and CD8+ T cells (108, 109). It has been revealed that ligation of PD-1 enhances production of Th2 cytokines by naïve T cell and decreases production of Th1 and Th17-related cytokines (44). Elevated expression of PD-1 and CTLA-4 in activated Th2 cells upon allergen exposure was observed in a mouse model of asthma. The long-term persistence of these exhausted pro-allergic Th2 cells during AIT with a reduction during up-dosing stage might explain the long-term treatment period in AIT to obtain clinical and immunological benefits (110). Involvement of PD-1 in pathogenesis of allergies has been investigated in several studies. It has been shown that altered PD-1/PD-L1 signaling might contribute to the balance between Th17 and Treg cells leading to development of asthma (45). In a mouse model of asthma sustained upregulation of PD-1 and PD-L2 has been observed in the lungs of asthmatic mice. Moreover, in vivo blockade of PD-L2 results in exacerbation of the disease due to elevated serum IgE levels, increased eosinophilic and lymphocytic inflammation, and elevated Th2 related cytokine production (111). These observations are in sharp contrast to the in vitro results with diminished proliferation of stimulated T cells and decreased production of Th2-related cytokines (111). Enhancement of AHR by PD-L2 blockade has also been illustrated by other investigations indicating a protective role of PD-L2 against the initiation and progression of airway inflammation (112, 113). In line with these observations, the results obtained from knock out animals showed opposing roles of PD-L1 and PD-L2 in asthma. While increased AHR, lung inflammation and Il-4 production by iNKT cells were observed in PD-L2-/-mice, PD-L1-/- mice showed decreased AHR and lung inflammation and increased secretion of IFN-Y by NK cells (114). Taken together, an opposing role for PD-L1 and PD-L2 is suggested since PD-L2 deficiency results in a more severe disease, while mice deficient in PD-L1 experience reduced AHR and lung inflammation.
CD200 receptor (CD200R) is an endogenous inhibitory signaling molecules that is mainly expressed in the lungs, predominantly by alveolar macrophages, neutrophils and mast cells (115). There are several isoforms of CD200R, among which CD200R1 is the best- characterized (116). CD200, a type-1 cell membrane glycoprotein, is the only known ligand of the CD200R family and is primarily expressed in pulmonary epithelial cells (115, 117). The CD200:CD200R1 inhibitory signaling pathway has been suggested as a mechanism to prevent an inflammatory response to harmless inhaled antigens. The activation of alveolar macrophages and DCs is tightly regulated by epithelial cells through the release of soluble mediators such as TGF-β, nitric oxide (NO), and IL-10, as well as their interactions with CD200 (118). Binding CD200 on airway epithelium to CD200R on alveolar macrophages and DCs initiates an inhibitory signal and thus represses activation of these cells preventing inflammatory lung diseases (46). It has been shown that the expression of CD200R1 is markedly up-regulated on alveolar macrophages, DCs and mast cells upon allergen exposure and/or in pulmonary infections (46, 119). In the context of influenza infection, previous reports have shown that alveolar macrophages in CD200-deficient mice are more active, leading to delayed resolution of inflammation and increased mortality (46). CD200R1 is also been found to be expressed in asthmatic inflammatory cells including basophils, Th2 cells, and ILC2 (115, 120). CD200R upon ligation prevents activation, proliferation and production of type 2 cytokines in activated ILC2s (47). Importantly, CD200R engagement on ILC2 using CD200-Fc chimeric protein in humanized mouse models of asthma dampened airway resistance, reduced eosinophilia and improved pulmonary dynamic compliance (47). Such observations provide evidence confirming regulatory role of CD200:CD200R as an important immunological checkpoint crucial for the maintenance of immune tolerance in the airway mucosa. The effect of CD200R engagement stimulation on AHR inhibition has also been attributed to local alterations in T cell responses and cytokine secretion (121). Other investigations have revealed that CD200:CD200R interaction diminishes allergic inflammation through reduced degranulation of mast cells and basophils (48, 122). Dysregulation of CD200 in asthma has been confirmed by several studies evaluating CD200 expression in PBMCs of asthmatic patients or serum levels of soluble CD200 which is probably produced by membrane shedding (123) or mRNA splicing (124) during asthma pathogenesis. However, the exact function of sCD200 in human diseases and its anti-inflammatory proprieties remain unclear. Further experimental investigations are needed to understand the cell specific functions of different isoforms of CD200R during homeostatic and inflammatory processes.
There has been a growing interest in the function of the T cell/transmembrane, immunoglobulin, and mucin (TIM) (gene family, which are the cell surface receptors for phosphatidylserine (PtdSer). These proteins with broad immune functions play essential roles in the regulation of innate and adaptive immune responses (125). Three members of TIM gene family (TIM-1, -3 and -4) are located on human chromosome 5q33 and have frequently been associated with asthma, allergy, and autoimmunity (126, 127). The molecules of TIM family are expressed on different lymphoid and myeloid immune cells, including T cells, B cells, NK cells, mast cells and macrophages (128, 129), and due to widespread manner of their function, the definition of TIM has been proposed to be transmembrane immunoglobulin and mucin” instead of “T cell/transmembrane”, immunoglobulin, and mucin (128). PtdSer on the surface of apoptotic cells is recognized specifically by immunoglobulin variable region domains of TIM-1, TIM-3, and TIM-4 which are expressed in mouse and human cells with different expression profiles, thereby playing distinct roles (126). TIM-1 is expressed on the surface of Th2 cells, generating a costimulatory signal that drives T cells activation in the context of asthma and allergy (49). TIM-3 is a co-inhibitory molecule that is preferentially expressed on Th1 and T-cytotoxic1 (Tc1) cells, and upon ligation to its ligand, promotes apoptosis of these cell populations (50). Therefore, there may be a reciprocal relationship between TIM 1 and TIM-3 in the regulation of Th1 and Th2 activity (126). TIM-4 is selectively expressed on APCs. Recent evidence indicates that the binding of TIM-4 to T cells via unknown receptors can promote the development of Th2 cells (52, 53). The immune response to infectious agents, tumor-specific antigens, and allergens results from a balance between Th1 and Th2 cells. The cross-regulation and differentiation of these subsets lead to two distinct scenarios; first, prevention of autoimmunity that results from the stimulation of Th2 cell cytokines and the second, regulation of asthma and allergic diseases that result from the stimulation of Th1 cytokines (130). Similarly, in the context of asthma and allergic rhinitis, the interaction between Tim-1 and Tim-4 causes the expansion of Th2 cells, mitigating asthmatic inflammation. In this scenario, Tim-3 is involved in the induction of Th2 cytokines (IL-4, IL-5, IL-13) through stimulation of mast cells (51). Consistent with this activity, TIM-3 by binding to its ligand, Galectin-9, stimulates apoptosis of Th1 cells thereby reducing the production of IFN-γ (128). Indeed, TIM-3, as a negative regulatory molecule, play a significant role in regulation of the balance between Th1 and Th2 which could affect the development of allergy and asthma. Thus, TIM3 upregulation in CD4+ T cells leads to a Th1/Th2 imbalance in allergic asthma patients. An in vitro study it was indicated that Tim-3 blocking dramatically reduced Th2-produced cytokines and promoted Th1-produced cytokines including TNF-α and IFN-γ in the supernatant of PBMCs. It has been shown that after co-culture of CD4+T cells and B cells, the production of IgG and IgE was decreased by blocking Tim-3 (51, 130). Therefore, Tim-3 blocking may be of therapeutic benefit in reducing airway inflammation by up regulating Th1 cytokines, dampening Th2 cytokines, and reducing IgG/IgE generation.
Lymphocyte activation gene-3 (LAG-3) is an inhibitory immune checkpoint expressed on various cell types, especially CD4+T cells, CD8+ T cells, NK cells, NKT cells, B cells, and Treg cells. Major histocompatibility complex-II (MHC-II) is a major ligand for LAG-3, and other ligands, including fibrinogen-like protein-1 (FGL-1), α-synuclein fibrils (a-syn), galectin-3 (Gal-3), and lymph node sinusoidal endothelial cell C-type lectin (LSECtin), have also been discovered. LAG-3 expression is induced upon TCR activation or by cytokines, especially IL-2, IL-12, IL-27, and IL-7. Once LAG-3 binds to its ligand, it inhibits T cell activation through its cytoplasmic inhibitory domains.
It has been shown that T regulatory type 1 (Tr1) cells express high level of LAG-3 for cell contact-mediated immune suppression during asthma (131). Activin-A has been reported to be the main cytokine for Tr1 generation and LAG-3 expression in allergic asthma (132). The adoptive transfer of LAG3+CD49b+CD4+ Tr1 cells improves atopic asthma (133). LAG3 is also essential for Foxp3+ Treg function, since LAG-3 deficient Tregs show reduced suppressive activity (54, 55). Li X et al., showed that IL-10 differentiated dendritic cells (DC10) of asthmatic individuals induced differentiation of CD25+Foxp3+LAG3+CTLA-4+ Tregs, which suppressed T effector response in a contact dependent manner (134). In addition to Tr1 and Foxp3+ Treg cells, ex vivo-generated double- negative T cells (DNT) exert an antigen-specific regulatory function through LAG-3 expression. Indeed, elevated expression of LAG-3 in DNT cells in a murine model of allergic asthma affected MHC-II antigen recognition and thus modulated allergen-specific immune regulation by these cells. Allergen-induced AHR, pulmonary inflammation, and allergen-specific IGE/IgG were inhibited (135). LAG-3 expression has also been reported in Treg-of-B cells, a subset of Treg cells which are induced from naïve T cells by follicular B cells in Peyer’s patches (136). LAG3 also has a crucial effect on the suppressive capacity of these cells. In a mouse model of asthma, the adoptive transfer of LAG3+ Treg-of B cells inhibited Th2 cytokine secretion and eosinophilic inflammation and ameliorated asthma symptoms (137). Elevated LAG-3 expression was also reported in lung B cells from sensitized mice in a murine model of asthma in a recent study by Habener et al. (138). This finding is in line with the reported expression of LAG-3 in immunosuppressive natural plasma cells in an animal model of infection (139), and the ability of LAG-3+ Tregs to suppress DC maturation and MHC-II-directed functions (140) indicates a crucial role for LAG-3 in the development and pathogenesis of asthma.
ICP molecules can be considered as predictive biomarkers for asthma susceptibility or severity. Numerous studies have reported a genetic association between susceptibility to asthma and ICPs. A meta-analysis study detected +49A/G polymorphism in CTLA-4 as an important risk factor for asthma susceptibility, especially in Asian individuals, children, and atopic patients (98). TIM-3 574T>G and TIM-1-416G>C single nucleotide polymorphisms are associated with an increased risk of asthma (128, 141). In addition to genetic associations, increased plasma concentration of CTLA-4 protein and cell expression of TIM-3 have been proposed as biomarkers for asthma susceptibility and severity in different studies (99, 102, 104, 142). Cellular expression level of some ICP can also be considered as biomarker of asthma. CD137 is increased in circulating eosinophils in IgE-mediated allergic asthma and is associated with increased IgE levels (143). In asthma, CD200 expression is reduced in peripheral blood cells of patients with asthma during exacerbation, and pharmacological modulation of this receptor has been shown to improve clinical and inflammatory outcomes in preclinical asthma models (144, 145).
Immune checkpoint inhibitors (ICIs) have been used for treatment of many different types of malignancies either as monotherapies or in combination with other treatments including chemotherapy and adjuvants showing remarkable clinical benefits (146). However, they are also linked to various immune-related adverse events (irAEs) caused by abnormal activation of autoreactive T cells with a prevalence of approximately 20–30% which can affect any organ in the body (147). Pulmonary toxicity is well documented with a wide range of toxicities reported and an overall incidence of 2.7–3.5% depending on the tumor type and treatment (148). Interestingly, lung toxicities appear to be more frequent with combination regimens than with monotherapies (149). The development of bronchial asthma or its exacerbations has also been reported in a few cases receiving ICI therapy (Table 2). However, there is a lack of knowledge regarding these types of irAEs, as only a limited number of cases have been reported. There are no established predictive markers for ICI-related toxicities. Peripheral blood eosinophilia, elevation of serum levels of total IgE, reversible airflow obstruction and elevated fractions of exhaled nitric oxide have been reported in these patients (150). Elevated eosinophil counts have been associated with an increased risk of toxicity and better clinical outcomes of ICI treatment in several types of advanced cancer (158–161). However, whether the proposed biomarkers have a specific predictive impact on ICI therapy outcomes is uncertain but, provides a foundation for future investigations in randomized controlled trials.

Table 2 Asthma as an IrAE in ICI-treated cancer patients reported in the literature.
Owing to their success in cancer therapy, ICIs have gained considerable interest as treatments for other immune-related diseases (162). However, it is controversial ICI therapy would be beneficial in treatment of such diseases due to a lack of knowledge about exact compensatory mechanisms in different immune checkpoint signaling pathways. In addition, it is unclear whether the reported alterations in the expression of immune checkpoint molecules in asthma are pathogenic or an outcome of the disease. Given the complex nature of immune responses, particularly in the context of immune-related diseases such as asthma, further investigations on agonistic or antagonistic targeting of one or more immune checkpoint axes are needed to evaluate the outcomes with a more comprehensive approach.
The microbiota can play various vital functions in the development of human immunity and the setup of immunological homeostasis (163). Lifestyle parameters comprising of hygiene, diet, encountering to bacteria or viruses, and medical intercessions with any medication, amend variability of phylogenetic composition and the cross- talk quality among adaptive and innate immune systems through skin and mucosal epithelia. Recently, several reports support the association of microbiota and their composition for offering protective impacts for healthiness (164). However, dysbiosis or alteration in microbial diversity has been associated with immune-related diseases including cancer and allergy, conditions that are accompanied by exaggerated or impaired immune tolerance, respectively (165). In this context, probiotics’ administration has been considered to modify this dysbiotic digression by reinstating the gap between the current and previous microbial communities.
Many functional mechanisms of probiotics or their derivatives (postbiotics) have been formerly described (166, 167). However, the exact mechanisms considering the function of the human microbiota remain unclear. The postulated mechanisms include reduction of mast cell infiltration, modulation of Th1/Th2 balance, and elevation of Treg population in peripheral lymph nodes (168, 169). In this section, we will discuss the possible interaction of microbiota/probiotics and immune checkpoints based on of new findings.
The formation of lung microbiota is crucial for the maturation of the immune system and for orchestrating respiratory function in health or disease (170). In this context, during the two first weeks of a neonatal mouse’s life, the drift of lung microbiota from Firmicutes and Gammaproteobacteria to Bacteroides was linked to the appearance of CD4+Helios−Foxp3+ Treg cells, which need the interaction with PD-L1 for progression in the lungs. Therefore, any dysbiosis or lack of microbiota as well as PD-L1 blocking leads to allergen responsiveness alongside adulthood (171). In another study, it was demonstrated that DCs of germ- free (GF) mice showed elevated levels of CD137, ICOSL, and OX40L than of controls in DCs. These differences in the activation status of the cells may direct the lung environment toward a Th2-biased immune response. Although, the precise function of DC populations or the associations of their activation status, in the course of allergic asthma stays unknown, it is obvious that the phenotype and number of DCs are affected by the commensal microbiota (172).
Overall, the knowledge of microbiota has provided new insights into pathogenesis of allergic diseases including asthma owing to the various immunomodulatory features of the mutualistic microorganisms. The microbiota effects can be indirect or direct via a cellular cross- talk with and between adaptive or innate immune cells, through the biomolecules exchanging which in turn could provoke inflammatory or regulatory cells. The consequent outcomes can be reconstitution or inflammation, with differing effects in allergy. However, the exact mechanisms of the possible role of microbiota in controlling allergic diseases, especially in the field of immune checkpoints are still unclear. Thus, additional investigations with a higher preference for identification of immune checkpoint functions in allergic diseases are of importance.
The balance between pro-/anti-inflammatory and the lung-resident immune cell network is of crucial importance in the maintenance of pulmonary homeostasis. The immune checkpoint molecules provide stimulatory/inhibitory signals during different stages of cellular and humoral immune responses. Ligation of CD28 by B7-1/-2 leads to a cascade of intracellular signaling that provides the second signal essential for naïve T cell activation and clonal expansion. CD28 costimulation is also required for B cell migration into the B cell follicles and provides proper signals for antibody production (173). Signals through an inhibitory immune checkpoint, ICOS, result in elevated IL-4/-13 secretion and thus increased IgE production by B cells in airways. In addition, CTLA-4 ligation by B7 molecules on DCs or activated lung epithelial cells, along with PD-1/PD-L1 interaction is involved in increased production of type-2 cytokines, IgE secretion, and formation of AHR. Other immune checkpoint molecules such as CD200 together with TNF receptor family members, CD137 and OX40, are also involved in the severity of pulmonary inflammation and AHR through signals that affect the balance of immune cells in the lungs. Indeed, the complex interactions between the signals through different immune checkpoints is necessary to limit the activation of type-2 immune responses and eosinophilic inflammation in pulmonary airways. Understanding these complex and comprehensive interactions not only provides insights into the pathogenesis of allergic asthma but also paves the way for setting up new strategies for therapeutic intervention in the context of allergic airway inflammation.
All authors took part in literature search and manuscript preparation and editing. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Zhu X, Cui J, Yi L, Qin J, Tulake W, Teng F, et al. The role of T cells and macrophages in asthma pathogenesis: a new perspective on mutual crosstalk. Mediators Inflammation (2020) 2020:7835284. doi: 10.1155/2020/7835284
2. Lloyd CM, Hessel EM. Functions of T cells in asthma: More than just TH2 cells. Nat Rev Immunol (2010) 10(12):838–48. doi: 10.1038/nri2870
3. Zissler UM, Chaker AM, Effner R, Ulrich M, Guerth F, Piontek G, et al. Interleukin-4 and interferon-γ orchestrate an epithelial polarization in the airways. Mucosal Immunol (2016) 9(4):917–26. doi: 10.1038/mi.2015.110
4. Komlósi ZI, van de Veen W, Kovács N, Szűcs G, Sokolowska M, O'Mahony L, et al. Cellular and molecular mechanisms of allergic asthma. Mol aspects Med (2021) 85:100995. doi: 10.1016/j.mam.2021.100995
5. Musiol S, Alessandrini F, Jakwerth CA, Chaker AM, Schneider E, Guerth F, et al. TGF-β1 drives inflammatory Th cell but not treg cell compartment upon allergen exposure. Front Immunol (2021) 12:763243. doi: 10.3389/fimmu.2021.763243
6. Schröder A, Lunding LP, Zissler UM, Vock C, Webering S, Ehlers JC, et al. IL-37 regulates allergic inflammation by counterbalancing pro-inflammatory IL-1 and IL-33. Allergy (2022) 77(3):856–69. doi: 10.1111/all.15072
7. Louhaichi S, Salhi M, Berraïes A, Hamdi B, Ammar J, Hamzaoui K, et al. Co-Inhibitory receptors in female asthmatic patients: Correlation with IL-17 and IL-26. AIMS Allergy Immunol (2018) 2(1):10–23. doi: 10.3934/Allergy.2018.1.10
8. Nagata M, Nakagome K. Allergen immunotherapy in asthma: current status and future perspectives. Allergol Int (2010) 59(1):15–9. doi: 10.2332/allergolint.09-RAI-0150
9. James LK, Shamji MH, Walker SM, Wilson DR, Wachholz PA, Francis JN, et al. Long-term tolerance after allergen immunotherapy is accompanied by selective persistence of blocking antibodies. J Allergy Clin Immunol (2011) 127(2):509–16.e5. doi: 10.1016/j.jaci.2010.12.1080
10. Pilette C, Nouri-Aria KT, Jacobson MR, Wilcock LK, Detry B, Walker SM, et al. Grass pollen immunotherapy induces an allergen-specific IgA2 antibody response associated with mucosal TGF-β expression. J Immunol (2007) 178(7):4658–66. doi: 10.4049/jimmunol.178.7.4658
11. Scadding GW, Eifan A, Lao-Araya M, Penagos M, Poon SY, Steveling E, et al. Effect of grass pollen immunotherapy on clinical and local immune response to nasal allergen challenge. Allergy (2015) 70(6):689–96. doi: 10.1111/all.12608
12. Lao-Araya M, Steveling E, Scadding GW, Durham SR, Shamji MH. Seasonal increases in peripheral innate lymphoid type 2 cells are inhibited by subcutaneous grass pollen immunotherapy. J Allergy Clin Immunol (2014) 134(5):1193–5.e4. doi: 10.1016/j.jaci.2014.07.029
13. Radulovic S, Jacobson MR, Durham SR, Nouri-Aria KT. Grass pollen immunotherapy induces Foxp3-expressing CD4+ CD25+ cells in the nasal mucosa. J Allergy Clin Immunol (2008) 121(6):1467–72.e1. doi: 10.1016/j.jaci.2008.03.013
14. Nouri-Aria KT, Wachholz PA, Francis JN, Jacobson MR, Walker SM, Wilcock LK, et al. Grass pollen immunotherapy induces mucosal and peripheral IL-10 responses and blocking IgG activity. J Immunol (2004) 172(5):3252–9. doi: 10.4049/jimmunol.172.5.3252
15. van de Veen W, Stanic B, Yaman G, Wawrzyniak M, Söllner S, Akdis DG, et al. IgG4 production is confined to human IL-10–producing regulatory b cells that suppress antigen-specific immune responses. J Allergy Clin Immunol (2013) 131(4):1204–12. doi: 10.1016/j.jaci.2013.01.014
16. Zissler UM, Jakwerth CA, Guerth F, Lewitan L, Rothkirch S, Davidovic M, et al. Allergen-specific immunotherapy induces the suppressive secretoglobin 1A1 in cells of the lower airways. Allergy (2021) 76(8):2461–74. doi: 10.1111/all.14756
17. Singh AK, Stock P, Akbari O. Role of PD-L1 and PD-L2 in allergic diseases and asthma. Allergy (2011) 66(2):155–62. doi: 10.1111/j.1398-9995.2010.02458.x
18. Bonamichi-Santos R, Aun MV, Kalil J, Castells MC, Giavina-Bianchi P. PD-L1 blockade during allergen sensitization inhibits the synthesis of specific antibodies and decreases mast cell activation in a murine model of active cutaneous anaphylaxis. Front Immunol (2021) 12:655958. doi: 10.3389/fimmu.2021.655958
20. Jansson A, Barnes E, Klenerman P, Harlén M, Sørensen P, Davis SJ, et al. A theoretical framework for quantitative analysis of the molecular basis of costimulation. J Immunol (Baltimore Md 1950) (2005) 175(3):1575–85. doi: 10.4049/jimmunol.175.3.1575
21. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol (2008) 26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331
22. Wykes MN, Lewin SR. Immune checkpoint blockade in infectious diseases. Nat Rev Immunol (2018) 18(2):91–104. doi: 10.1038/nri.2017.112
23. Illingworth J, Butler NS, Roetynck S, Mwacharo J, Pierce SK, Bejon P, et al. Chronic exposure to plasmodium falciparum is associated with phenotypic evidence of b and T cell exhaustion. J Immunol (Baltimore Md 1950) (2013) 190(3):1038–47. doi: 10.4049/jimmunol.1202438
24. Hou N, Zou Y, Piao X, Liu S, Wang L, Li S, et al. T-Cell immunoglobulin- and mucin-Domain-Containing molecule 3 signaling blockade improves cell-mediated immunity against malaria. J Infect Dis (2016) 214(10):1547–56. doi: 10.1093/infdis/jiw428
25. Chew GM, Fujita T, Webb GM, Burwitz BJ, Wu HL, Reed JS, et al. TIGIT marks exhausted T cells, correlates with disease progression, and serves as a target for immune restoration in HIV and SIV infection. PloS Pathog (2016) 12(1):e1005349. doi: 10.1371/journal.ppat.1005349
26. Pollock KM, Montamat-Sicotte DJ, Grass L, Cooke GS, Kapembwa MS, Kon OM, et al. PD-1 expression and cytokine secretion profiles of mycobacterium tuberculosis-specific CD4+ T-cell subsets; potential correlates of containment in HIV-TB Co-infection. PloS One (2016) 11(1):e0146905. doi: 10.1371/journal.pone.0146905
27. Dong C, Juedes AE, Temann U-A, Shresta S, Allison JP, Ruddle NH, et al. ICOS co-stimulatory receptor is essential for T-cell activation and function. Nature (2001) 409(6816):97–101. doi: 10.1038/35051100
28. Clay BS, Shilling RA, Bandukwala HS, Moore TV, Cannon JL, Welcher AA, et al. Inducible costimulator expression regulates the magnitude of Th2-mediated airway inflammation by regulating the number of Th2 cells. PloS One (2009) 4(11):e7525. doi: 10.1371/journal.pone.0007525
29. Tesciuba AG, Shilling RA, Agarwal MD, Bandukwala HS, Clay BS, Moore TV, et al. ICOS costimulation expands Th2 immunity by augmenting migration of lymphocytes to draining lymph nodes. J Immunol (2008) 181(2):1019–24. doi: 10.4049/jimmunol.181.2.1019
30. Vieira PL, Wassink L, Smith LM, Nam S, Kingsbury GA, Gutierrez-Ramos JC, et al. ICOS-mediated signaling regulates cytokine production by human T cells and provides a unique signal to selectively control the clonal expansion of Th2 helper cells. Eur J Immunol (2004) 34(5):1282–90. doi: 10.1002/eji.200324417
31. McAdam AJ, Greenwald RJ, Levin MA, Chernova T, Malenkovich N, Ling V, et al. ICOS is critical for CD40-mediated antibody class switching. Nature (2001) 409(6816):102–5. doi: 10.1038/35051107
32. Tafuri A, Shahinian A, Bladt F, Yoshinaga SK, Jordana M, Wakeham A, et al. ICOS is essential for effective T-helper-cell responses. Nature (2001) 409(6816):105–9. doi: 10.1038/35051113
33. Rigas D, Lewis G, Aron JL, Wang B, Banie H, Sankaranarayanan I, et al. Type 2 innate lymphoid cell suppression by regulatory T cells attenuates airway hyperreactivity and requires inducible T-cell costimulator–inducible T-cell costimulator ligand interaction. J Allergy Clin Immunol (2017) 139(5):1468–77.e2. doi: 10.1016/j.jaci.2016.08.034
34. Aron J, Akbari O. Regulatory T cells and type 2 innate lymphoid cell-dependent asthma. Allergy (2017) 72(8):1148–55. doi: 10.1111/all.13139
35. Whitehead GS, Wilson RH, Nakano K, Burch LH, Nakano H, Cook DN. IL-35 production by inducible costimulator (ICOS)–positive regulatory T cells reverses established IL-17–dependent allergic airways disease. J Allergy Clin Immunol (2012) 129(1):207–15.e5. doi: 10.1016/j.jaci.2011.08.009
36. Siddiqui S, Mistry V, Doe C, Stinson S, Foster M, Brightling C. Airway wall expression of OX40/OX40L and interleukin-4 in asthma. Chest (2010) 137(4):797–804. doi: 10.1378/chest.09-1839
37. Liu YJ. Thymic stromal lymphopoietin and OX40 ligand pathway in the initiation of dendritic cell-mediated allergic inflammation. J Allergy Clin Immunol (2007) 120(2):238–44; quiz 45-6. doi: 10.1016/j.jaci.2007.06.004
38. Seshasayee D, Lee WP, Zhou M, Shu J, Suto E, Zhang J, et al. In vivo blockade of OX40 ligand inhibits thymic stromal lymphopoietin driven atopic inflammation. J Clin Invest (2007) 117(12):3868–78. doi: 10.1172/JCI33559
39. Farres MN, Sabry MK, Ahmed EE, Elkady HM, Mohamed NA. OX40 ligand: A potential costimulatory molecule in atopic asthma. J Asthma (2014) 51(6):573–7. doi: 10.3109/02770903.2014.897729
40. Polte T, Foell J, Werner C, Hoymann HG, Braun A, Burdach S, et al. CD137-mediated immunotherapy for allergic asthma. J Clin Invest (2006) 116(4):1025–36. doi: 10.1172/JCI23792
41. Ai X, Shi G, Wan H, Shi Y, Hou X, Zhu H, et al. 4-1BB ligand-mediated imbalance of helper 17 T cells and regulatory T cells in patients with allergic asthma. J Int Med Res (2012) 40(3):1046–54. doi: 10.1177/147323001204000323
42. Nishimoto H, Lee SW, Hong H, Potter KG, Maeda-Yamamoto M, Kinoshita T, et al. Costimulation of mast cells by 4-1BB, a member of the tumor necrosis factor receptor superfamily, with the high-affinity IgE receptor. Blood (2005) 106(13):4241–8. doi: 10.1182/blood-2005-04-1358
43. Zhang F, Huang G, Hu B, Song Y, Shi Y. Induction of immune tolerance in asthmatic mice by vaccination with DNA encoding an allergen-cytotoxic T lymphocyte-associated antigen 4 combination. Clin Vaccine Immunol CVI (2011) 18(5):807–14. doi: 10.1128/CVI.00434-10
44. McAlees JW, Lajoie S, Dienger K, Sproles AA, Richgels PK, Yang Y, et al. Differential control of CD4(+) T-cell subsets by the PD-1/PD-L1 axis in a mouse model of allergic asthma. Eur J Immunol (2015) 45(4):1019–29. doi: 10.1002/eji.201444778
45. Xi X, Liu JM, Guo JY. Correlation of PD-1/PD-L1 signaling pathway with Treg/Th17 imbalance from asthmatic children. Int Arch Allergy Immunol (2018) 176(3-4):255–67. doi: 10.1159/000489338
46. Snelgrove RJ, Goulding J, Didierlaurent AM, Lyonga D, Vekaria S, Edwards L, et al. A critical function for CD200 in lung immune homeostasis and the severity of influenza infection. Nat Immunol (2008) 9(9):1074–83. doi: 10.1038/ni.1637
47. Shafiei-Jahani P, Helou DG, Hurrell BP, Howard E, Quach C, Painter JD, et al. CD200–CD200R immune checkpoint engagement regulates ILC2 effector function and ameliorates lung inflammation in asthma. Nat Commun (2021) 12(1):1–15. doi: 10.1038/s41467-021-22832-7
48. Zhang S, Cherwinski H, Sedgwick JD, Phillips JH. Molecular mechanisms of CD200 inhibition of mast cell activation. J Immunol (2004) 173(11):6786–93. doi: 10.4049/jimmunol.173.11.6786
49. McIntire JJ, Umetsu DT, DeKruyff RH. TIM-1, a novel allergy and asthma susceptibility gene. Springer Semin Immunopathol (2004) 25(3):335–48. doi: 10.1007/s00281-003-0141-3
50. Han G, Chen G, Shen B, Li Y. Tim-3: An activation marker and activation limiter of innate immune cells. Front Immunol (2013) 4:449. doi: 10.3389/fimmu.2013.00449
51. Kearley J, McMillan SJ, Lloyd CM. Th2-driven, allergen-induced airway inflammation is reduced after treatment with anti–Tim-3 antibody in vivo. J Exp Med (2007) 204(6):1289–94. doi: 10.1084/jem.20062093
52. Mizui M, Shikina T, Arase H, Suzuki K, Yasui T, Rennert PD, et al. Bimodal regulation of T cell-mediated immune responses by TIM-4. Int Immunol (2008) 20(5):695–708. doi: 10.1093/intimm/dxn029
53. Liu W, Xu L, Liang X, Liu X, Zhao Y, Ma C, et al. Tim-4 in health and disease: Friend or foe? Front Immunol (2020) 11:537. doi: 10.3389/fimmu.2020.00537
54. Huang CT, Workman CJ, Flies D, Pan X, Marson AL, Zhou G, et al. Role of LAG-3 in regulatory T cells. Immunity (2004) 21(4):503–13. doi: 10.1016/j.immuni.2004.08.010
55. Nguyen QT, Jang E, Le HT, Kim S, Kim D, Dvorina N, et al. IL-27 targets Foxp3+ tregs to mediate antiinflammatory functions during experimental allergic airway inflammation. JCI Insight (2019) 4(2):e123216. doi: 10.1172/jci.insight.123216
56. Komlósi ZI, van de Veen W, Kovács N, Szűcs G, Sokolowska M, O'Mahony L, et al. Cellular and molecular mechanisms of allergic asthma. Mol Aspects Med (2022) 85:100995. doi: 10.1016/j.mam.2021.100995
57. Huang H, Dawicki W, Lu M, Nayyar A, Zhang X, Gordon J. Regulatory dendritic cell expression of MHC II and IL-10 are jointly requisite for induction of tolerance in a murine model of OVA-asthma. Allergy (2013) 68(9):1126–35. doi: 10.1111/all.12203
58. Martín-Orozco E, Norte-Muñoz M, Martínez-García J. Regulatory T cells in allergy and asthma. Front Pediatr (2017) 5:117. doi: 10.3389/fped.2017.00117
59. Hutloff A, Dittrich AM, Beier KC, Eljaschewitsch B, Kraft R, Anagnostopoulos I, et al. ICOS is an inducible T-cell co-stimulator structurally and functionally related to CD28. Nature (1999) 397(6716):263–6. doi: 10.1038/16717
60. Burmeister Y, Lischke T, Dahler AC, Mages HW, Lam K-P, Coyle AJ, et al. ICOS controls the pool size of effector-memory and regulatory T cells. J Immunol (2008) 180(2):774–82. doi: 10.4049/jimmunol.180.2.774
61. McAdam AJ, Chang TT, Lumelsky AE, Greenfield EA, Boussiotis VA, Duke-Cohan JS, et al. Mouse inducible costimulatory molecule (ICOS) expression is enhanced by CD28 costimulation and regulates differentiation of CD4+ T cells. J Immunol (2000) 165(9):5035–40. doi: 10.4049/jimmunol.165.9.5035
62. Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu Rev Immunol (2005) 23:515. doi: 10.1146/annurev.immunol.23.021704.115611
63. Yoshinaga SK, Whoriskey JS, Khare SD, Sarmiento U, Guo J, Horan T, et al. T-Cell co-stimulation through B7RP-1 and ICOS. Nature (1999) 402(6763):827–32. doi: 10.1038/45582
64. Qian X, Agematsu K, Freeman GJ, Tagawa Yi, Sugane K, Hayashi T. The ICOS-ligand B7-H2, expressed on human type II alveolar epithelial cells, plays a role in the pulmonary host defense system. Eur J Immunol (2006) 36(4):906–18. doi: 10.1002/eji.200535253
65. Coyle AJ, Lehar S, Lloyd C, Tian J, Delaney T, Manning S, et al. The CD28-related molecule ICOS is required for effective T cell–dependent immune responses. Immunity (2000) 13(1):95–105. doi: 10.1016/S1074-7613(00)00011-X
66. Herman AE, Freeman GJ, Mathis D, Benoist C. CD4+ CD25+ T regulatory cells dependent on ICOS promote regulation of effector cells in the prediabetic lesion. J Exp Med (2004) 199(11):1479–89. doi: 10.1084/jem.20040179
67. Rottman JB, Smith T, Tonra JR, Ganley K, Bloom T, Silva R, et al. The costimulatory molecule ICOS plays an important role in the immunopathogenesis of EAE. Nat Immunol (2001) 2(7):605–11. doi: 10.1038/89750
68. Mak TW, Shahinian A, Yoshinaga SK, Wakeham A, Boucher L-M, Pintilie M, et al. Costimulation through the inducible costimulator ligand is essential for both T helper and b cell functions in T cell–dependent b cell responses. Nat Immunol (2003) 4(8):765–72. doi: 10.1038/ni947
69. Akbari O, Freeman GJ, Meyer EH, Greenfield EA, Chang TT, Sharpe AH, et al. Antigen-specific regulatory T cells develop via the ICOS–ICOS-ligand pathway and inhibit allergen-induced airway hyperreactivity. Nat Med (2002) 8(9):1024–32. doi: 10.1038/nm745
70. Paterson D, Jefferies W, Green J, Brandon M, Corthesy P, Puklavec M, et al. Antigens of activated rat T lymphocytes including a molecule of 50,000 Mr detected only on CD4 positive T blasts. Mol Immunol (1987) 24(12):1281–90. doi: 10.1016/0161-5890(87)90122-2
71. Mallett S, Fossum S, Barclay A. Characterization of the MRC OX40 antigen of activated CD4 positive T lymphocytes–a molecule related to nerve growth factor receptor. EMBO J (1990) 9(4):1063–8. doi: 10.1002/j.1460-2075.1990.tb08211.x
72. Calderhead DM, Buhlmann J, Van den Eertwegh A, Claassen E, Noelle R, Fell HP. Cloning of mouse Ox40: A T cell activation marker that may mediate TB cell interactions. J Immunol (1993) 151(10):5261–71. doi: 10.4049/jimmunol.151.10.5261
73. Sun G, Sun X, Li W, Liu K, Tian D, Dong Y, et al. Critical role of OX40 in the expansion and survival of CD4 T-cell-derived double-negative T cells. Cell Death Dis (2018) 9(6):616. doi: 10.1038/s41419-018-0659-x
74. Fu Y, Lin Q, Zhang Z, Zhang L. Therapeutic strategies for the costimulatory molecule OX40 in T-cell-mediated immunity. Acta Pharm Sin B (2020) 10(3):414–33. doi: 10.1016/j.apsb.2019.08.010
75. Fu N, Xie F, Sun Z, Wang Q. The OX40/OX40L axis regulates T follicular helper cell differentiation: Implications for autoimmune diseases. Front Immunol (2021) 12:670637. doi: 10.3389/fimmu.2021.670637
76. Burgess JK, Carlin S, Pack RA, Arndt GM, Au WW, Johnson PR, et al. Detection and characterization of OX40 ligand expression in human airway smooth muscle cells: A possible role in asthma? J Allergy Clin Immunol (2004) 113(4):683–9. doi: 10.1016/j.jaci.2003.12.311
77. Lei W, Zeng DX, Zhu CH, Liu GQ, Zhang XQ, Wang CG, et al. The upregulated expression of OX40/OX40L and their promotion of T cells proliferation in the murine model of asthma. J Thorac Dis (2014) 6(7):979–87. doi: 10.3978/j.issn.2072-1439.2014.06.34
78. Hoshino A, Tanaka Y, Akiba H, Asakura Y, Mita Y, Sakurai T, et al. Critical role for OX40 ligand in the development of pathogenic Th2 cells in a murine model of asthma. Eur J Immunol (2003) 33(4):861–9. doi: 10.1002/eji.200323455
79. Huang L, Zhang X, Wang M, Chen Z, Yan Y, Gu W, et al. Exosomes from thymic stromal lymphopoietin-activated dendritic cells promote Th2 differentiation through the OX40 ligand. Pathobiology (2019) 86(2-3):111–7. doi: 10.1159/000493013
80. Di C, Lin X, Zhang Y, Zhong W, Yuan Y, Zhou T, et al. Basophil-associated OX40 ligand participates in the initiation of Th2 responses during airway inflammation. J Biol Chem (2015) 290(20):12523–36. doi: 10.1074/jbc.M115.642637
81. Kwon B. CD137-CD137 ligand interactions in inflammation. Immune Network (2009) 9(3):84–9. doi: 10.4110/in.2009.9.3.84
82. Luu K, Shao Z, Schwarz H. The relevance of soluble CD137 in the regulation of immune responses and for immunotherapeutic intervention. J Leukocyte Biol (2020) 107(5):731–8. doi: 10.1002/JLB.2MR1119-224R
83. Lombardi V, Singh AK, Akbari O. The role of costimulatory molecules in allergic disease and asthma. Int Arch Allergy Immunol (2010) 151(3):179–89. doi: 10.1159/000242355
84. Dharmadhikari B, Wu M, Abdullah NS, Rajendran S, Ishak ND, Nickles E, et al. CD137 and CD137L signals are main drivers of type 1, cell-mediated immune responses. Oncoimmunology (2016) 5(4):e1113367. doi: 10.1080/2162402X.2015.1113367
85. Sun Y, Blink SE, Liu W, Lee Y, Chen B, Solway J, et al. Inhibition of Th2-mediated allergic airway inflammatory disease by CD137 costimulation. J Immunol (Baltimore Md 1950) (2006) 177(2):814–21. doi: 10.4049/jimmunol.177.2.814
86. Polte T, Jagemann A, Foell J, Mittler RS, Hansen G. CD137 ligand prevents the development of T-helper type 2 cell-mediated allergic asthma by interferon-gamma-producing CD8+ T cells. Clin Exp Allergy (2007) 37(9):1374–85. doi: 10.1111/j.1365-2222.2007.02785.x
87. Cho Y, Kwon B, Lee TH, Kim TB, Moon KA, La S, et al. 4-1BB stimulation inhibits allergen-specific immunoglobulin e production and airway hyper-reactivity but partially suppresses bronchial eosinophilic inflammation in a mouse asthma model. Clin Exp Allergy (2006) 36(3):377–85. doi: 10.1111/j.1365-2222.2006.02445.x
88. Behrendt A-K, Hansen G. CD27 costimulation is not critical for the development of asthma and respiratory tolerance in a murine model. Immunol Lett (2010) 133(1):19–27. doi: 10.1016/j.imlet.2010.06.004
89. Shreffler WG. CD137-mediated immunotherapy for allergic asthma. Pediatrics (2007) 120(Supplement_3):S153–S. doi: 10.1542/peds.2007-0846IIII
90. Kim B-J, Kwon J-W, Seo J-H, Choi W-A, Kim Y-J, Kang M-J, et al. Hu. 4-1BB-Fc fusion protein inhibits allergic inflammation and airway hyperresponsiveness in a murine model of asthma. Korean J Pediatr (2011) 54(9):373. doi: 10.3345/kjp.2011.54.9.373
91. Cho Y, Kwon B, Lee J, Oh Y, Yoo B, Moon H. Anti-4-1BB agonistic antibody abolishes allergen-specific IgE but does not reduce airway hyperreactivity and eosinophilic inflammation in a murine model of asthma. J Allergy Clin Immunol (2004) 113(2):S221. doi: 10.1016/j.jaci.2004.01.249
92. Behrendt AK, Meyer-Bahlburg A, Hansen G. CD137 deficiency does not affect development of airway inflammation or respiratory tolerance induction in murine models. Clin Exp Immunol (2012) 168(3):308–17. doi: 10.1111/j.1365-2249.2012.04572.x
93. Savage J, Sicherer S, Wood R. The natural history of food allergy. J Allergy Clin Immunol Pract (2016) 4(2):196–203. doi: 10.1016/j.jaip.2015.11.024
94. Sohn MH, Kim SH, Song TW, Kim KW, Kim ES, Park HS, et al. Cytotoxic T lymphocyte-associated antigen-4 gene polymorphisms confer susceptibility to atopic asthma in Korean children. Pediatr Pulmonol (2007) 42(6):542–7. doi: 10.1002/ppul.20622
95. Feng Q, Liu D, Lu Y, Liu Z. The interplay of renin-angiotensin system and toll-like receptor 4 in the inflammation of diabetic nephropathy. J Immunol Res (2020) 2020:6193407. doi: 10.1155/2020/6193407
96. Lee SY, Lee YH, Shin C, Shim JJ, Kang KH, Yoo SH, et al. Association of asthma severity and bronchial hyperresponsiveness with a polymorphism in the cytotoxic T-lymphocyte antigen-4 gene. Chest (2002) 122(1):171–6. doi: 10.1378/chest.122.1.171
97. Munthe-Kaas MC, Carlsen KH, Helms PJ, Gerritsen J, Whyte M, Feijen M, et al. CTLA-4 polymorphisms in allergy and asthma and the TH1/ TH2 paradigm. J Allergy Clin Immunol (2004) 114(2):280–7. doi: 10.1016/j.jaci.2004.03.050
98. Zheng Y, Wang H, Luo L, Liao L, You L, Wang J, et al. A meta-analysis of the association between CTLA-4 genetic polymorphism and susceptibility of asthma. Medicine (2018) 97(28):e11380. doi: 10.1097/MD.0000000000011380
99. Ip WK, Wong CK, Leung TF, Lam CW. Elevation of plasma soluble T cell costimulatory molecules CTLA-4, CD28 and CD80 in children with allergic asthma. Int Arch Allergy Immunol (2005) 137(1):45–52. doi: 10.1159/000084612
100. Ip WK, Wong CK, Leung TF, Lam CW. Plasma concentrations of soluble CTLA-4, CD28, CD80 and CD86 costimulatory molecules reflect disease severity of acute asthma in children. Pediatr Pulmonol. (2006) 41(7):674–82. doi: 10.1002/ppul.20432
101. Chen YQ, Shi HZ. CD28/CTLA-4–CD80/CD86 and ICOS–B7RP-1 costimulatory pathway in bronchial asthma. Allergy (2006) 61(1):15–26. doi: 10.1111/j.1398-9995.2006.01008.x
102. Shi H-Z, Mo X-Y, Zhong X-N. Soluble CTLA-4 in sera of patients with bronchial asthma. J Asthma (2004) 42(2):133–9. doi: 10.1081/JAS-200051331
103. Qin XJ, Shi HZ, Qin SM, Kang LF, Huang CP, Zhong XN. Effects of allergen inhalation and oral glucocorticoid on serum soluble CTLA-4 in allergic asthmatics. Allergy (2005) 60(6):774–9. doi: 10.1111/j.1398-9995.2005.00784.x
104. Wong CK, Lun SW, Ko FW, Ip WK, Hui DS, Lam CW. Increased expression of plasma and cell surface co-stimulatory molecules CTLA-4, CD28 and CD86 in adult patients with allergic asthma. Clin Exp Immunol (2005) 141(1):122–9. doi: 10.1111/j.1365-2249.2005.02815.x
105. Borrego LM, Arroz MJ, Videira P, Martins C, Guimaraes H, Nunes G, et al. Regulatory cells, cytokine pattern and clinical risk factors for asthma in infants and young children with recurrent wheeze. Clin Exp Allergy (2009) 39(8):1160–9. doi: 10.1111/j.1365-2222.2009.03253.x
106. Boussiotis VA. Molecular and biochemical aspects of the PD-1 checkpoint pathway. N Engl J Med (2016) 375(18):1767–78. doi: 10.1056/NEJMra1514296
107. Galvan Morales MA, Montero-Vargas JM, Vizuet-de-Rueda JC, Teran LM. New insights into the role of PD-1 and its ligands in allergic disease. Int J Mol Sci (2021) 22(21):11898. doi: 10.3390/ijms222111898
108. Wei F, Zhong S, Ma Z, Kong H, Medvec A, Ahmed R, et al. Strength of PD-1 signaling differentially affects T-cell effector functions. Proc Natl Acad Sci USA (2013) 110(27):E2480–9. doi: 10.1073/pnas.1305394110
109. Zuazo M, Arasanz H, Fernandez-Hinojal G, Garcia-Granda MJ, Gato M, Bocanegra A, et al. Functional systemic CD4 immunity is required for clinical responses to PD-L1/PD-1 blockade therapy. EMBO Mol Med (2019) 11(7):e10293. doi: 10.1093/annonc/mdz253.058
110. Wang SH, Zissler UM, Buettner M, Heine S, Heldner A, Kotz S, et al. An exhausted phenotype of TH2 cells is primed by allergen exposure, but not reinforced by allergen-specific immunotherapy. Allergy (2021) 76(9):2827–39. doi: 10.1111/all.14896
111. Oflazoglu E, Swart DA, Anders-Bartholo P, Jessup HK, Norment AM, Lawrence WA, et al. Paradoxical role of programmed death-1 ligand 2 in Th2 immune responses in vitro and in a mouse asthma model in vivo. Eur J Immunol (2004) 34(12):3326–36. doi: 10.1002/eji.200425197
112. Matsumoto K, Inoue H, Nakano T, Tsuda M, Yoshiura Y, Fukuyama S, et al. B7-DC regulates asthmatic response by an IFN-γ-dependent mechanism. J Immunol (2004) 172(4):2530–41. doi: 10.4049/jimmunol.172.4.2530
113. Lewkowich I, Lajoie S, Stoffers S, Suzuki Y, Richgels P, Dienger K, et al. PD-L2 modulates asthma severity by directly decreasing dendritic cell IL-12 production. Mucosal Immunol (2013) 6(4):728–39. doi: 10.1038/mi.2012.111
114. Akbari O, Stock P, Singh A, Lombardi V, Lee W, Freeman G, et al. PD-L1 and PD-L2 modulate airway inflammation and iNKT-cell-dependent airway hyperreactivity in opposing directions. Mucosal Immunol (2010) 3(1):81–91. doi: 10.1038/mi.2009.112
115. Wright GJ, Cherwinski H, Foster-Cuevas M, Brooke G, Puklavec MJ, Bigler M, et al. Characterization of the CD200 receptor family in mice and humans and their interactions with CD200. J Immunol (2003) 171(6):3034–46. doi: 10.4049/jimmunol.171.6.3034
116. Hatherley D, Barclay AN. The CD200 and CD200 receptor cell surface proteins interact through their n-terminal immunoglobulin-like domains. Eur J Immunol (2004) 34(6):1688–94. doi: 10.1002/eji.200425080
117. Hussell T, Bell TJ. Alveolar macrophages: Plasticity in a tissue-specific context. Nat Rev Immunol (2014) 14(2):81–93. doi: 10.1038/nri3600
118. Bissonnette EY, Lauzon-Joset J-F, Debley JS, Ziegler SF. Cross-talk between alveolar macrophages and lung epithelial cells is essential to maintain lung homeostasis. Front Immunol (2020) 11:583042. doi: 10.3389/fimmu.2020.583042
119. Rate A, Bosco A, McKenna KL, Holt PG, Upham JW. Airway epithelial cells condition dendritic cells to express multiple immune surveillance genes. PLoS One (2012) 7(9):e44941. doi: 10.1371/journal.pone.0044941
120. Blom LH, Martel BC, Larsen LF, Hansen CV, Christensen MP, Juel-Berg N, et al. The immunoglobulin superfamily member CD 200R identifies cells involved in type 2 immune responses. Allergy (2017) 72(7):1081–90. doi: 10.1111/all.13129
121. Lauzon-Joset J-F, Langlois A, Lai LJ, Santerre K, Lee-Gosselin A, Bossé Y, et al. Lung CD200 receptor activation abrogates airway hyperresponsiveness in experimental asthma. Am J Respir Cell Mol Biol (2015) 53(2):276–84. doi: 10.1165/rcmb.2014-0229OC
122. Holmannová D, Kolácková M, Kondélková K, Kunes P, Krejsek J, Andrys C. CD200/CD200R paired potent inhibitory molecules regulating immune and inflammatory responses; part I: CD200/CD200R structure, activation, and function. Acta Med (hradec Králové) (2012) 55(1):12–7. doi: 10.14712/18059694.2015.68
123. Twito T, Chen Z, Khatri I, Wong K, Spaner D, Gorczynski R. Ectodomain shedding of CD200 from the b-CLL cell surface is regulated by ADAM28 expression. Leukemia Res (2013) 37(7):816–21. doi: 10.1016/j.leukres.2013.04.014
124. Chen Z, Chen D-X, Kai Y, Khatri I, Lamptey B, Gorczynski RM. Identification of an expressed truncated form of CD200, CD200tr, which is a physiologic antagonist of CD200-induced suppression. Transplantation (2008) 86(8):1116–24. doi: 10.1097/TP.0b013e318186fec2
125. Su EW, Lin JY, Kane LP. TIM-1 and TIM-3 proteins in immune regulation. Cytokine (2008) 44(1):9–13. doi: 10.1016/j.cyto.2008.06.013
126. Freeman GJ, Casasnovas JM, Umetsu DT, DeKruyff RH. TIM genes: A family of cell surface phosphatidylserine receptors that regulate innate and adaptive immunity. Immunol Rev (2010) 235(1):172–89. doi: 10.1111/j.0105-2896.2010.00903.x
127. Lee J, Phong B, Egloff AM, Kane LP. TIM polymorphisms–genetics and function. Genes Immun (2011) 12(8):595–604. doi: 10.1038/gene.2011.75
128. Wei W, Huang J, Ma Y, Hao C. Association between gene polymorphisms of T cell immunoglobulin domain and mucin domain-3 and risk of asthma: A systematic review and meta-analysis. Iranian J Allergy Asthma Immunol (2021) 20(1):1. doi: 10.18502/ijaai.v20i1.5407
129. Liu Y, Chen H, Chen Z, Qiu J, Pang H, Zhou Z. Novel roles of the Tim family in immune regulation and autoimmune diseases. Front Immunol (2021) 12:748787. doi: 10.3389/fimmu.2021.748787
130. Chae S-C, Song J-H, Pounsambath P, Yuan H-Y, Lee J-H, Kim J-J, et al. Molecular variations in Th1-specific cell surface gene Tim-3. Exp Mol Med (2004) 36(3):274–8. doi: 10.1038/emm.2004.37
131. Thorburn AN, Hansbro PM. Harnessing regulatory T cells to suppress asthma: From potential to therapy. Am J Respir Cell Mol Biol (2010) 43(5):511–9. doi: 10.1165/rcmb.2009-0342TR
132. Tousa S, Semitekolou M, Morianos I, Banos A, Trochoutsou AI, Brodie TM, et al. Activin-a co-opts IRF4 and AhR signaling to induce human regulatory T cells that restrain asthmatic responses. Proc Natl Acad Sci USA (2017) 114(14):E2891–e900. doi: 10.1073/pnas.1616942114
133. Matsuda M, Doi K, Tsutsumi T, Fujii S, Kishima M, Nishimura K, et al. Regulation of allergic airway inflammation by adoptive transfer of CD4(+) T cells preferentially producing IL-10. Eur J Pharmacol (2017) 812:38–47. doi: 10.1016/j.ejphar.2017.06.037
134. Li X, Yang A, Huang H, Zhang X, Town J, Davis B, et al. Induction of type 2 T helper cell allergen tolerance by IL-10-differentiated regulatory dendritic cells. Am J Respir Cell Mol Biol (2010) 42(2):190–9. doi: 10.1165/rcmb.2009-0023OC
135. Tian D, Yang L, Wang S, Zhu Y, Shi W, Zhang C, et al. Double negative T cells mediate Lag3-dependent antigen-specific protection in allergic asthma. Nat Commun (2019) 10(1):4246. doi: 10.1038/s41467-019-12243-0
136. Okamura T, Fujio K, Shibuya M, Sumitomo S, Shoda H, Sakaguchi S, et al. CD4+CD25-LAG3+ regulatory T cells controlled by the transcription factor egr-2. Proc Natl Acad Sci USA (2009) 106(33):13974–9. doi: 10.1073/pnas.0906872106
137. Chu KH, Chiang BL. Characterization and functional studies of forkhead box protein 3(-) lymphocyte activation gene 3(+) CD4(+) regulatory T cells induced by mucosal b cells. Clin Exp Immunol (2015) 180(2):316–28. doi: 10.1111/cei.12583
138. Habener A, Happle C, Grychtol R, Skuljec J, Busse M, Dalüge K, et al. Regulatory b cells control airway hyperreactivity and lung remodeling in a murine asthma model. J Allergy Clin Immunol (2021) 147(6):2281–94.e7. doi: 10.1016/j.jaci.2020.09.041
139. Lino AC, Dang VD, Lampropoulou V, Welle A, Joedicke J, Pohar J, et al. LAG-3 inhibitory receptor expression identifies immunosuppressive natural regulatory plasma cells. Immunity (2018) 49(1):120–33.e9. doi: 10.1016/j.immuni.2018.06.007
140. Liang B, Workman C, Lee J, Chew C, Dale BM, Colonna L, et al. Regulatory T cells inhibit dendritic cells by lymphocyte activation gene-3 engagement of MHC class II. J Immunol (Baltimore Md 1950) (2008) 180(9):5916–26. doi: 10.4049/jimmunol.180.9.5916
141. Xie X, Shi X, Chen P, Rao L. Associations of TIM-1 genetic polymorphisms with asthma: A meta-analysis. Lung (2017) 195(3):353–60. doi: 10.1007/s00408-017-0006-5
142. Mosayebian A, Koohini Z, Hossein-Nataj H, Abediankenari S, Abedi S, Asgarian-Omran H. Elevated expression of Tim-3 and PD-1 immune checkpoint receptors on T-CD4+ lymphocytes of patients with asthma. Iranian J allergy asthma Immunol (2018) 17(6):517–25.
143. Heinisch IV, Bizer C, Volgger W, Simon HU. Functional CD137 receptors are expressed by eosinophils from patients with IgE-mediated allergic responses but not by eosinophils from patients with non-IgE-mediated eosinophilic disorders. J Allergy Clin Immunol (2001) 108(1):21–8. doi: 10.1067/mai.2001.116864
144. Aoki T, Matsumoto Y, Hirata K, Ochiai K, Okada M, Ichikawa K, et al. Expression profiling of genes related to asthma exacerbations. Clin Exp Allergy (2009) 39(2):213–21. doi: 10.1111/j.1365-2222.2008.03186.x
145. Dimeloe S, Richards DF, Urry ZL, Gupta A, Stratigou V, Farooque S, et al. 1α,25-dihydroxyvitamin D3 promotes CD200 expression by human peripheral and airway-resident T cells. Thorax (2012) 67(7):574–81. doi: 10.1136/thoraxjnl-2011-200651
146. Robert C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat Commun (2020) 11(1):3801. doi: 10.1038/s41467-020-17670-y
147. Yamagata A, Yokoyama T, Fukuda Y, Ishida T. Impact of interstitial lung disease associated with immune checkpoint inhibitors on prognosis in patients with non-small-cell lung cancer. Cancer Chemother Pharmacol (2021) 87(2):251–8. doi: 10.1007/s00280-020-04205-x
148. Delaunay M, Cadranel J, Lusque A, Meyer N, Gounant V, Moro-Sibilot D, et al. Immune-checkpoint inhibitors associated with interstitial lung disease in cancer patients. Eur Respir J (2017) 50(2):1700050. doi: 10.1183/13993003.00050-2017
149. Nishino M, Giobbie-Hurder A, Hatabu H, Ramaiya NH, Hodi FS. Incidence of programmed cell death 1 inhibitor-related pneumonitis in patients with advanced cancer: A systematic review and meta-analysis. JAMA Oncol (2016) 2(12):1607–16. doi: 10.1001/jamaoncol.2016.2453
150. Maeno K, Fukuda S, Oguri T, Niimi A. Nivolumab-induced asthma in a patient with non-small-cell lung cancer. Ann Oncol (2017) 28(11):2891. doi: 10.1093/annonc/mdx455
151. Roger A, Groh M, Lorillon G, Le Pendu C, Maillet J, Arangalage D, et al. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss) induced by immune checkpoint inhibitors. Ann Rheum Dis (2019) 78(8):e82. doi: 10.1136/annrheumdis-2018-213857
152. Sumi T, Nagahisa Y, Matsuura K, Sekikawa M, Yamada Y, Nakata H, et al. Successful management of severe bronchial asthma exacerbated by anti-PD-L1 treatment: A report of two cases. Respirol Case Rep (2021) 9(11):e0868. doi: 10.1002/rcr2.868
153. Harada M, Naoi H, Yasuda K, Ito Y, Kagoo N, Kubota T, et al. Programmed cell death-1 blockade in kidney carcinoma may induce eosinophilic granulomatosis with polyangiitis: a case report. BMC Pulm Med (2021) 21(1):6. doi: 10.1186/s12890-020-01375-5
154. Hamada K, Yoshimura K, Oshinomi K, Hirasawa Y, Ariizumi H, Ohkuma R, et al. A case of bronchial asthma as an immune-related adverse event of pembrolizumab treatment for bladder cancer: A case report. Medicine (2022) 101(2):e28339. doi: 10.1097/MD.0000000000028339
155. Xu W, Lian B, Cui C, Guo J. The combination therapy with the cytotoxic T lymphocyte-associated antigen-4 and programmed death 1 antibody-induced asthma in a patient with advanced melanoma. J Cancer Res Ther (2021) 17(3):808–10. doi: 10.4103/jcrt.jcrt_419_21
156. Kissoonsingh P, Sutton B, Iqbal SU, Pallan L, Steven N, Khoja L. Eosinophilic asthma secondary to adjuvant anti-PD-1 immune checkpoint inhibitor treatment in a melanoma patient. Case Rep oncological Med (2022) 2022:2658136. doi: 10.1155/2022/2658136
157. Uemura T, Fukumitsu K, Maeno K, Fukuda S, Onuki T, Kanemitsu Y, et al. Asthma caused by durvalumab after chemoradiotherapy in two patients with non-small cell lung cancer. Respirol Case Rep (2021) 9(9):e0835. doi: 10.1002/rcr2.835
158. Bernard-Tessier A, Jeanville P, Champiat S, Lazarovici J, Voisin AL, Mateus C, et al. Immune-related eosinophilia induced by anti-programmed death 1 or death-ligand 1 antibodies. Eur J Cancer (2017) 81:135–7. doi: 10.1016/j.ejca.2017.05.017
159. Simon SCS, Hu X, Panten J, Grees M, Renders S, Thomas D, et al. Eosinophil accumulation predicts response to melanoma treatment with immune checkpoint inhibitors. Oncoimmunology (2020) 9(1):1727116. doi: 10.1080/2162402X.2020.1727116
160. Weide B, Martens A, Hassel JC, Berking C, Postow MA, Bisschop K, et al. Baseline biomarkers for outcome of melanoma patients treated with pembrolizumab. Clin Cancer Res (2016) 22(22):5487–96. doi: 10.1158/1078-0432.CCR-16-0127
161. Kizawa R, Miura Y, Oda Y, Nagaoka Y, Ozaki Y, Kondoh C, et al. Eosinophilia during treatment of immune checkpoint inhibitors (ICIs) to predict succeeding onset of immune-related adverse events (irAEs). Am Soc Clin Oncol (2019). doi: 10.1093/annonc/mdz253.071
162. Wang Z, Huang J, Xie D, He D, Lu A, Liang C. Toward overcoming treatment failure in rheumatoid arthritis. Front Immunol (2021) 12:755844. doi: 10.3389/fimmu.2021.755844
163. Untersmayr E, Bax HJ, Bergmann C, Bianchini R, Cozen W, Gould HJ, et al. AllergoOncology: Microbiota in allergy and cancer–a European academy for allergy and clinical immunology position paper. Allergy (2019) 74(6):1037–51. doi: 10.1111/all.13718
164. Pascal M, Perez-Gordo M, Caballero T, Escribese MM, Lopez Longo MN, Luengo O, et al. Microbiome and allergic diseases. Front Immunol (2018) 9:1584. doi: 10.3389/fimmu.2018.01584
165. Wu J, Wang S, Zheng B, Qiu X, Wang H, Chen L. Modulation of gut microbiota to enhance effect of checkpoint inhibitor immunotherapy. Front Immunol (2021) 12:2554. doi: 10.3389/fimmu.2021.669150
166. Rangan KJ, Pedicord VA, Wang Y-C, Kim B, Lu Y, Shaham S, et al. A secreted bacterial peptidoglycan hydrolase enhances tolerance to enteric pathogens. Science (2016) 353(6306):1434–7. doi: 10.1126/science.aaf3552
167. Kawanabe-Matsuda H, Takeda K, Nakamura M, Makino S, Karasaki T, Kakimi K, et al. Dietary Lactobacillus-derived exopolysaccharide enhances immune-checkpoint blockade TherapyDietary exopolysaccharide enhances ICB therapy efficacy. Cancer Discovery (2022) 12:1336–55. doi: 10.1158/2159-8290.CD-21-0929
168. Lim SK, Kwon M-S, Lee J, Oh YJ, Jang J-Y, Lee J-H, et al. Weissella cibaria WIKIM28 ameliorates atopic dermatitis-like skin lesions by inducing tolerogenic dendritic cells and regulatory T cells in BALB/c mice. Sci Rep (2017) 7(1):1–9. doi: 10.1038/srep40040
169. Boonpiyathad T, Sözener ZC, Akdis M, Akdis CA. The role of treg cell subsets in allergic disease. Asian Pac J Allergy (2020) 38(3):139–49. doi: 10.12932/AP-030220-0754
170. Sharma G, Im S-H. Probiotics as a potential immunomodulating pharmabiotics in allergic diseases: current status and future prospects. Allergy Asthma Immunol Res (2018) 10(6):575–90. doi: 10.4168/aair.2018.10.6.575
171. Gollwitzer ES, Saglani S, Trompette A, Yadava K, Sherburn R, McCoy KD, et al. Lung microbiota promotes tolerance to allergens in neonates via PD-L1. Nat Med (2014) 20(6):642–7. doi: 10.1038/nm.3568
172. Herbst T, Sichelstiel A, Schär C, Yadava K, Bürki K, Cahenzli J, et al. Dysregulation of allergic airway inflammation in the absence of microbial colonization. Am J Resp Crit Care (2011) 184(2):198–205. doi: 10.1164/rccm.201010-1574OC
Keywords: immune checkpoint, asthma, co-stimulatory signals, co-inhibitory signals, autoimmunity
Citation: Kanannejad Z, Soleimanian S, Ghahramani Z, Sepahi N, Mohkam M, Alyasin S and Kheshtchin N (2023) Immune checkpoint molecules in prevention and development of asthma. Front. Immunol. 14:1070779. doi: 10.3389/fimmu.2023.1070779
Received: 15 October 2022; Accepted: 30 January 2023;
Published: 14 February 2023.
Edited by:
Milena Sokolowska, University of Zurich, SwitzerlandReviewed by:
Ulrich Matthias Zissler, Technical University of Munich, GermanyCopyright © 2023 Kanannejad, Soleimanian, Ghahramani, Sepahi, Mohkam, Alyasin and Kheshtchin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nasim Kheshtchin, bl9raGVzaHRjaGluQHN1bXMuYWMuaXI=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.