
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Immunol. , 11 August 2023
Sec. Autoimmune and Autoinflammatory Disorders: Autoinflammatory Disorders
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1061182
 Robin Jacquot1*†
Robin Jacquot1*† Maurine Jouret2†Mathieu Gerfaud Valentin1
Maurine Jouret2†Mathieu Gerfaud Valentin1 Maël Richard1
Maël Richard1 Yvan Jamilloux1Florent Rousset3Jean-François Emile4
Yvan Jamilloux1Florent Rousset3Jean-François Emile4 Julien Haroche5Lars Steinmüller2
Julien Haroche5Lars Steinmüller2 Franck Zekre2Alice Phan2
Franck Zekre2Alice Phan2 Alexandre Belot2
Alexandre Belot2 Pascal Seve1
Pascal Seve1H syndrome is a rare autosomal recessive genetic disorder characterized by the following clinical features: cutaneous hyperpigmentation, hypertrichosis, hepatosplenomegaly, heart anomalies, hearing loss, hypogonadism, short stature, hallux valgus, hyperglycemia, fixed flexion contractures of the toe joints, and the proximal interphalangeal joints. In rare cases, autoinflammatory and lymphoproliferative manifestations have also been reported. This disorder is due to loss-of-function mutations in SLC29A3 gene, which encode the equilibrative nucleoside transporter ENT3. This deficiency leads to abnormal function and proliferation of histiocytes. H syndrome is part of the R-group of histiocytosis. We report two different cases, one was diagnosed in adulthood and the other in childhood. The first case reported is a 37-year-old woman suffering from H syndrome with an autoinflammatory systemic disease that begins in adulthood (fever and diffuse organ’s infiltration) and with cutaneous, articular, auditory, and endocrinological manifestations since childhood. The second case reported is a 2-year-old girl with autoinflammatory, endocrine, and cutaneous symptoms (fever, lymphadenopathy, organomegaly, growth delay, and cutaneous hyperpigmentation). Homozygous mutations in SLC29A3 confirmed the diagnosis of H syndrome in both cases. Each patient was treated with Tocilizumab with a significant improvement for lymphoproliferative, autoinflammatory, and cutaneous manifestations. Both cases were reported to show the multiple characteristics of this rare syndrome, which can be diagnosed either in childhood or in adulthood. In addition, an overview of the literature suggested Tocilizumab efficiency.
H syndrome is an autosomal recessive disorder of abnormal histiocytic proliferation caused by mutations in SLC29A3 at chromosome 10q22.1 encoding the human equilibrative nucleoside transporter-3 (hENT3) protein. ENT3 is a member of the SLC29-equilibrating nucleoside transporter family, which has a widespread tissue distribution, and its cellular expression is particularly important in lysosomal and mitochondria membrane (1). Impaired phagocytosis by SLC29A3 mutations result in excessive inflammatory response and histiocytic proliferation underlying clinical features (2). By searching on PubMed Medline using the keywords, H syndrome and SLC29A3, approximately 130 patients suffering from H syndrome have been reported in the literature until October 2022.
The main clinical manifestations associate cutaneous and systemic features starting with the letter “H”: hyperpigmentation and hypertrichosis of the inner thighs, hepatosplenomegaly, heart anomalies, hearing loss, hypogonadism, and low height. Systemic features including recurrent fever, arthritis, joint deformity, diabetes, and symptoms associated with diffuse organ infiltration are also described (3).
Both the cases we report had inflammatory manifestations that positively responded with Tocilizumab. In addition, we provide an overview of the nine other cases of H syndrome treated with Tocilizumab in the literature.
A 37-year-old Moroccan woman was referred to our internal medicine department, for recurrent febrile attacks with abdominal pain and assessment of a fibro-inflammatory disease with mediastinal, pericardial, pleural, periaortic, perirenal, and retroperitoneum infiltration. She reported a sensorineural hearing loss and symmetrical hyperpigmentation with thickening of the legs 20 years prior to the current presentation.
When hospitalized, she complained of abdominal pain associated with nausea, vomiting, and fever. She reported tiredness, a weight loss of 8 kg, and night sweats for several weeks. On clinical examination, hyperpigmented and indurated cutaneous plaques inside both legs and sparing knees were found (Figures 1A, B). A camptodactyly in the fifth radii of both hands (Figure 1C) and hallux valgus and retraction of the toes were noticed. Laboratory test results revealed inflammation with C-reactive protein (CRP) at 97 mg/L (normal < 5 mg/L). There was no cytopenia. Two fasting blood glucose > 1.2 g/L led to diagnosis of diabetes with HbA1c concentration at 7.7%. An abdomino-pelvic computed tomography (CT) scan showed splenomegaly and retroperitoneal, periaortic, bilateral perirenal, pericardial, diffuse abdominal infiltration and right pleural effusion (Table 1).
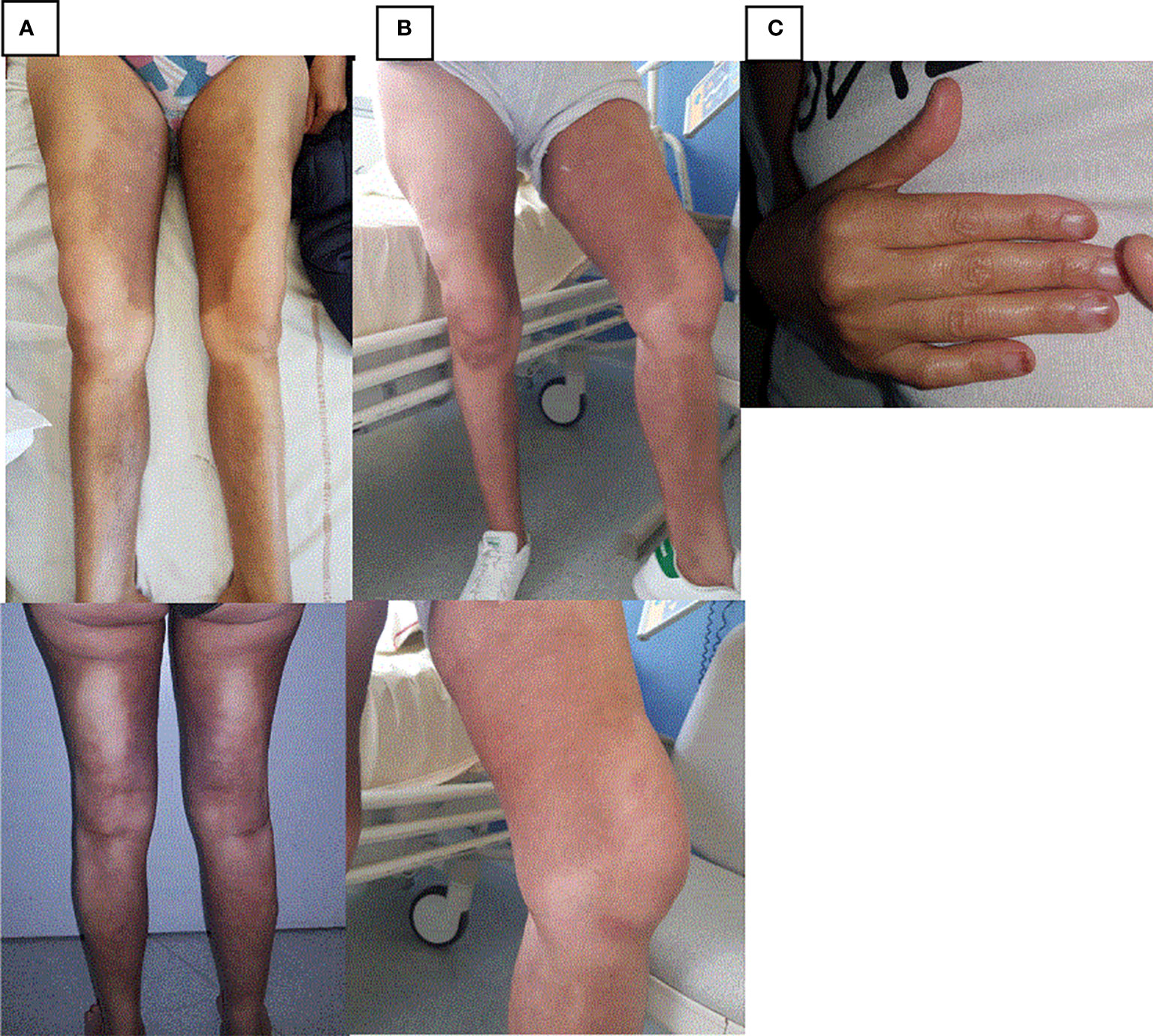
Figure 1 Skin lesions in case report 1. (A) Hyperpigmented and indurated cutaneous plaques inside both legs before treatment by Tocilizumab. (B) Hyperpigmented and indurated cutaneous plaques inside both legs 6 months after treatment by Tocilizumab. (C) Camptodactyly in the fifth radii.
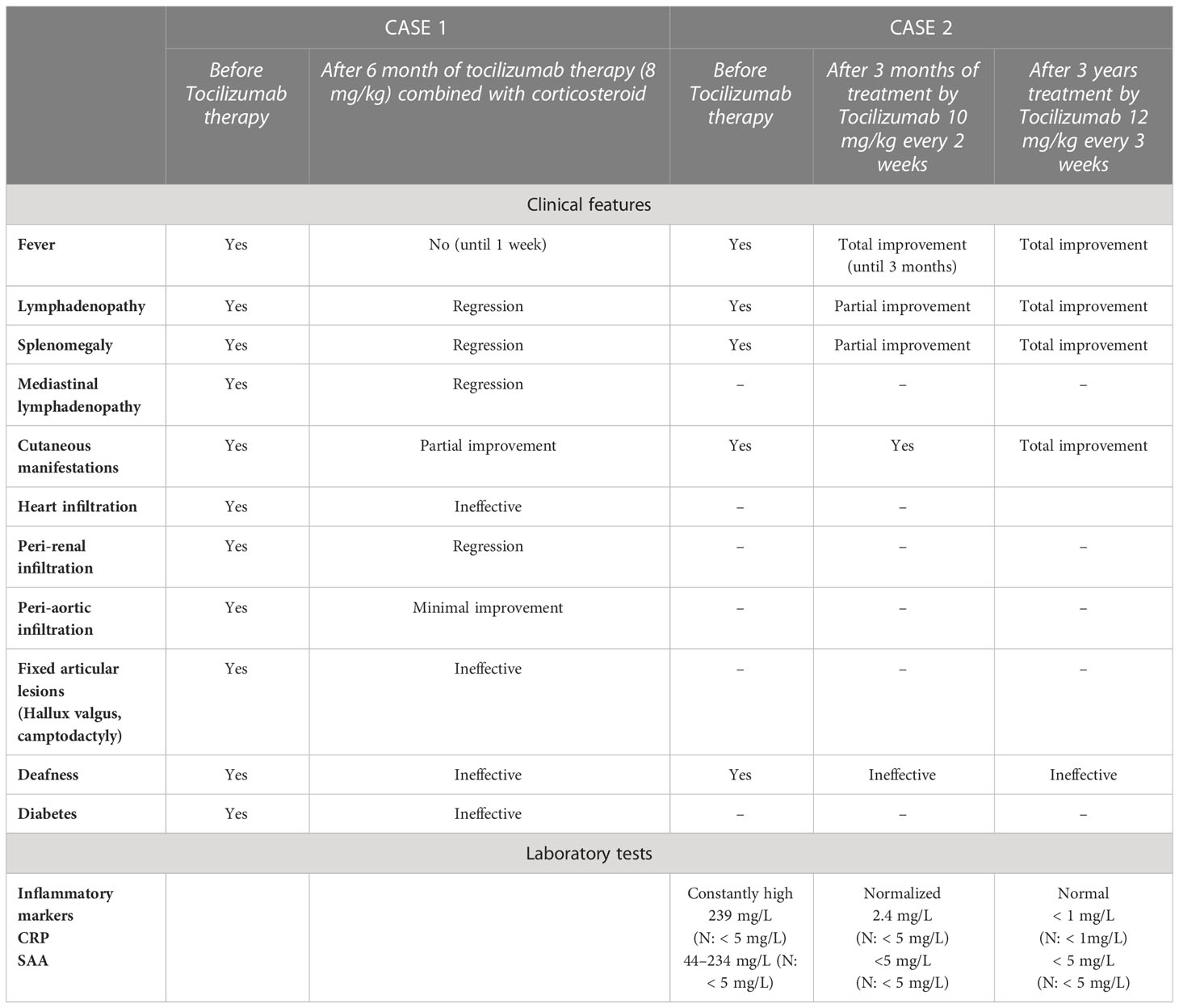
Table 1 Clinical and biological response to Tocilizumab therapy.
Investigations were conducted to exclude an infectious etiology (QuantiFERON® test, syphilis, Bartonella sp., Rickettsia sp., and Coxiella sp. serology). There was no evidence to support the thesis of an autoimmune disease (negative antinuclear and antineutrophil cytoplasmic antibodies). Due to diffuse organ infiltration with inflammatory syndrome and a negative initial etiological assessment, non-Langheransian histiocytosis, IgG4-related disease, and lymphoma were considered.
Further studies showed no bone infiltration on bone scintigraphy and PET scan. Periaortic fatty infiltration associated with posterior mediastinal adenomegaly, paracardiac tumor lesions, and pericardial effusion on pan aortic MRI were observed. No pituitary gland infiltration on brain MRI was described. Right pleural effusion and thickening of the right atrium were found on thoracic CT scan, confirmed by cardiac ultrasound with a 5-cm tumor of the right atrium wall. Muscle MRI was done because of the infiltration of the inside thighs and showed subcutaneous soft tissue infiltration with edema, predominantly on the medial side of thighs with no muscle infiltration.
Skin biopsy and right video thoracoscopy with pleural and pericardial biopsy were performed as well. Skin histology revealed fibrosis hypodermic infiltrate histiocytes, CD 163+, CD 68+, CD1a−, PS100−, Langherine−, CD20− (no B cells), weakly positive IgG4 marking, with no BRAF/V600E mutation, no The mitogen-activated extracellular signal-regulated kinase (MEK)/The extracellular signal-regulated kinase (ERK) pathway mutation, and no immunohistochemical staining of phosphorylated ERK. Pleural and pericardial biopsy revealed the same histology.
The systemic manifestations (mediastinal, pleural, cardiac, retroperitoneum including kidney infiltration) and the histopathological characteristics (histiocyte’s infiltration with CD68+ and CD163+) aroused suspicions of histiocytosis. There was no pathognomonic long-bone osteosclerosis at the metadiaphysis to support the diagnosis of Erdheim–Chester disease. According to the patient’s clinical signs (cutaneous hyperpigmentation, camptodactyly with retraction of toes, diabetes mellitus, and history of sensorineural hearing loss), we suspected histiocytosis–lymphadenopathy plus syndrome, i.e., the H syndrome. The diagnosis was confirmed by genetic analysis using next-generation sequencing, which showed a homozygous mutation in the SLCA29A3 gene c.1088G>A (p.Arg363Gln), which was performed thanks to Sanger sequencing.
Anti-IL6 therapy (Tocilizumab) was administered by intravenous injection (8 mg/kg/month) coupled with oral corticosteroid therapy (40 mg/day). Clinical features improved with a 8-kg gain, no more fever after 1 week, a decreased skin infiltration after 3 months, and the disappearance of splenomegaly. However, there was no result regarding deafness or joint damages. There was no inflammation on biological controls on the first month (Table 1). The reassessment CT scan on the 6 month showed a decrease in pericardial, mediastinal, perirenal, periaortic infiltration, and pleural effusion but no regression of atrium’s tumor.
The second case is a 2-year-old girl who was sent to our pediatric dermatology, nephrology, and rheumatology unit because of a hypertrichosis, hyperpigmentation, lymphoproliferative syndrome, and recurrent respiratory tract infections.
About her familial background, she was the offspring of consanguineous parents who comes from Algeria. She is the youngest daughter of a family with four children, two of whom already suffered from severe combined immune deficiency due to RAG2 mutations.
From the first months of her life, she suffered from numerous febrile attacks and recurrent respiratory tract infections that required several hospitalizations (Figure 2A). Sensorineural hearing loss was identified at the age of 3. During the early childhood, she developed mild asthma with reactive airway disease well controlled with long acting beta2-agonist and montelukast. Since the age of 6 months, she presented a growth delay. The endocrine assessment revealed a delayed bone maturation and a partial growth hormone (GH) deficiency. MRI of the pituitary gland was normal. She has been supplemented with GH since the age of 18 months with moderate growth improvement (Figure 2B).
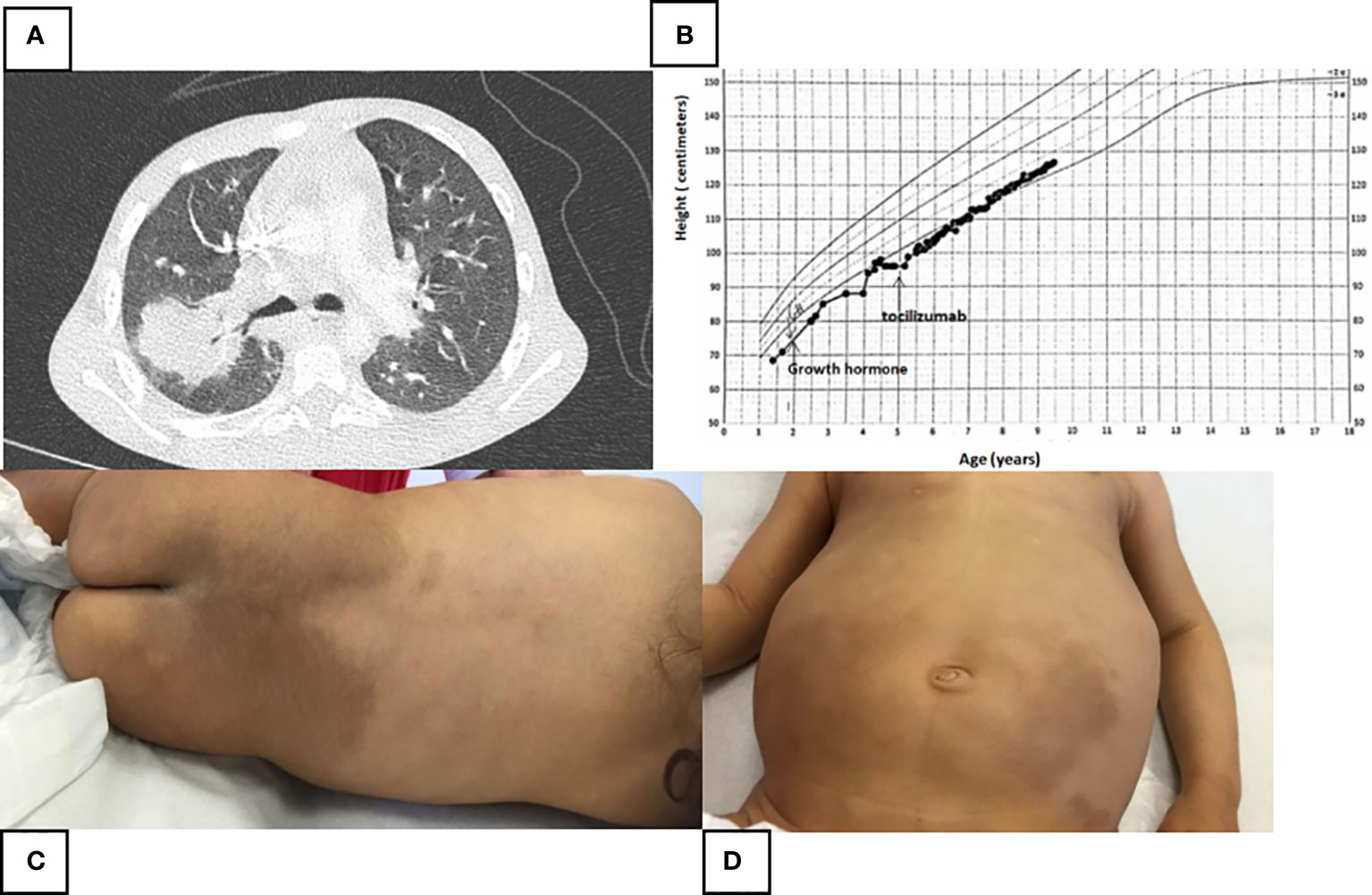
Figure 2 (A) Thoracic scanner (parenchymal window) performed at the age of 4 during a new feverish episode and respiratory difficulties. Acute postero-superior lobar pneumonia measuring 34 mm with air bronchogram, right hilar adenomegaly and sequelar mediobasal and posterobasal atelectasis. (B) Height growth chart. Moderate effect of growth hormone and Tocilizumab on the height. (C, D) Large areas of pigmentation of the lumbosacral region and both lower limbs with associated hypertrichosis.
Her clinical examination revealed a significant hepatosplenomegaly. The most striking symptom was diffuse lymphadenopathy with many bilaterally cervical, axillary, and inguinal lymph nodes. Large areas of pigmentation of the lumbosacral region and of both the lower limbs with associated hypertrichosis were observed. The buttocks, knees, and popliteal fossae were spared (Figures 2C, D). There was a discrete lichenified appearance at the back of the feet. A delayed tooth eruption was noted. There was no skeletal deformities or facial dysmorphia. No abnormality was detected on cardiac, neurological, and genitourinary examination. Her psychomotor development was normal.
At admission, laboratory tests showed elevation of inflammatory markers including C-reactive protein at 239 mg/L (normal, <5 mg/L), serum amyloid A at 234 mg/L (normal < 5 mg/L), and erythrocyte sedimentation rate at 84 mm/h (normal < 21 mm/h). Interferon signature was moderately high at 6.6 (normal, 2.3). During febrile attacks, a microcytic hypochromic anemia was found (lowest measured hemoglobin level, 83 g/L; lowest Mean corpuscular volume (MCV), 61.7 fL). There were no autoantibodies found that suggested autoimmune disease. The blood glucose level was normal. Bone marrow aspiration was performed due to severe anemia and revealed a cell-rich bone marrow with balanced hematopoiesis. Lymphadenopathy biopsy showed reactive lymphadenitis with no specific feature of histiocytosis or H syndrome. No histiocytes and no sign of emperipolesis were found.
First genetic screening excluded the contribution of RAG2; the patient did not carry the familial mutation. Genetic tests by Sanger sequencing were conducted and revealed a homozygous mutation p.Arg363Trp in exon 6 of the SLC29A3 gene. Hence, the diagnosis of H syndrome was confirmed. Regarding treatments, in the absence of an etiological diagnosis on the onset, Azathioprine (2 mg/kg/day) was introduced at the age of 3. Inflammatory markers remained elevated during treatment. No clinical improvement has been noted. We found the pathogenic mutation in SLC29A3 at the age of 5, and consequently, the Tocilizumab was introduced at the dose of 10 mg/kg every 2 weeks. Inflammatory markers normalized 3 months after the beginning of the treatment. The patient suffers from repeated bronchopneumopathy, requiring intravenous antibiotics during the first months of the treatment. To prevent this side effect, the intake of Tocilizumab was decreased to every 3 weeks with the same dose (10 mg/kg). This protocol helped in fighting the infections but caused a resurgence of the lymphoproliferative disease with elevated inflammatory factors and clinical relapse (splenomegaly). An new dosage of Tocilizumab (12 mg/kg every 3 weeks) and a preventive treatment with alternate antibiotics (amoxicillin, sulfamethoxazol/trimetroprim, and clarithromycin) was administrated.
After 2 years of treatment with this amount, when the patient turned 7, a remission without cutaneous, lymphoproliferative, and inflammatory signs was observed. However, it did not have any notable effect upon deafness (Table 1).
Immunological assessment was performed to search for immune deficiency that could explain repeated lung infections. There was no leukopenia and no thrombocytopenia. T- and B-cell immunophenotyping, vaccine responses, and T lymphocyte functional test (OKT3 and PHA) were normal before the Tocilizumab was started. Importantly, T-cell subsets were normal without abnormal naive T cells and no bias in the immune repertoire. Hypergammaglobulinemia with IgG levels at 12.9 g/L (normal between 4.82 and 8.96 g/L) and IgM levels at 1.72 g/L (normal between 0.54 and 1.53 g/L) was noted. IgA level was normal for the age. These biological results invalidated the diagnosis of primary immune deficiency. After 3 years of treatment with Tocilizumab, at the age of 8, B-cell, T-cell, and NK-cell lymphopenia was observed to be associated with low rate of IgG2 and IgG4.
In 2008, Molho-Pessach et al. first described H syndrome and reported mutation of SLC29A3 as the cause of this syndrome (3). The majority of patients with H syndrome are of Arab origin especially because the two most common mutations were found in individuals of Arab–Palestinian ancestry (1). Since the first description was made, reported H syndrome cases demonstrated considerable clinical variability, which leads to misdiagnosis. This heterogeneity in phenotype may be attributed to impaired tissue response to injury (4).
H syndrome is considered as a familial Rosai–Dorfman–Destombes (RDD) disease, classified as R-group in revised classification of histiocytosis (5, 6). It is an autosomal recessive disorder, which is mainly diagnosed among children thanks to early symptom developments and family history. Typical clinical manifestations are hyperpigmentation (91%) and hypertrichosis (68%) of the inner thighs, flexion contractures of proximal interphalangeal joints (56%), short stature (49%), diabetes, hepatosplenomegaly (40%), and hallux valgus (25%) (7). Diffuse lymphadenopathy may occur in 24% (7) of cases and can mimicking RDD like in our pediatric case (8). In addition, this syndrome is part of the spectrum of monogenic autoinflammatory diseases (9) with 25% of patients developing autoinflammatory complications like recurrent febrile attacks, persistently elevated acute phase reactants, or fibrosis. The quality of tissue repair can be affected, causing fibrosis and histiocytic invasion of organs (10). Symptoms may be related to this diffuse tissue infiltration (pericardial, perirenal, periaortic, and retroperitoneal) as demonstrated on our adult case. Depending on how the organs are affected, it may be confused with other types of histiocytosis. By perirenal infiltration and right atrial tumor, our adult case was initially presented as Erdheim–Chester disease (ECD) clinical phenotype, but the absence of bone infiltration has challenged the diagnosis.
H syndrome being held responsible for specific cutaneous symptoms, it should induce such a diagnosis, and it may be associated with autoinflammatory manifestations and lymphadenopathy. However, these aspects require particular attention not to miss differential diagnosis of lymphoproliferative syndromes [Castleman disease, autoimmune lymphoproliferative syndrome (ALPS) and non-Langerhans histiocytosis such as RDD and ECD] and genetic autoinflammatory diseases (mevalonate kinase deficiency) (11, 12).
The key element to diagnose this syndrome is a histological analysis of lymph nodes and/or infiltration tissues. Cutaneous and organ infiltration biopsy normally showed a dense dermal and subcutaneous infiltrate, composed of CD68+ histiocytes and later replaced by fibrosis (13). In our first case, the patient presented mediastinal, pericardial, pleural, periaortic, perirenal, and retroperitoneal diffuse infiltrations, which suggest clinical characteristics of ECD, but without bones involvement. Histopathological characteristics (CD 163+, CD 68+, CD1a−, PS100−, Langherine−) exclude the diagnosis of Langerhans histiocytosis and brings us to an ECD rather than an RDD because of lack of S-100 protein labeling. These clinical and histological immunophenotypes need to be considered because, traditionally, H syndrome phenotype is closer to RDD’s manifestations with S-100 protein positivity and no emperipolesis (7, 13). Therefore, histological analysis usually leads to a diagnosis of histiocytosis, with markers that rule our Langerhans histiocytosis but can be consistent with ECD or RDD histology. Histological analysis is essential to determine the proper diagnosis.
However, histological analyses have limits; as shown in our second case, reactive lymphadenitis were observed without specific microscopic features of histiocytosis. Biopsy of lymph node or infiltration tissues may help clinicians with histopathological characteristics (CD163, CD68, CD1a, PS100, and Langherine); however, there is no specific feature of syndrome H (13).
Genetic testing was performed after the informed consent provision revealed a homozygous mutation c.1088G>A p.Arg363Gln in exon 6 in senior patient report and c.1087C>T p.Arg363Trp in exon 6 of the SLC29A3 gene in pediatric patient report. The mutations highlighted within our studies have already been described among patients with H syndrome (14, 15). The residue 363 encodes for a highly conserved position in the protein. c.1087C>T p.Arg363Trp is predicted to be likely pathogenic by the browser Polyphen and is rare in the general population (allele frequency in gnomAD: q =0.00004773, <0.0001 is the threshold for recessive gene). c.1088G>A is predicted to be pathogenic by Polyphen and its low-frequency variant (q = 0.000007955).
In our pediatric case, it was found that both parents were heterozygous for this mutation. A highly conserved position in the protein, segregation of this mutation in the family members, and low frequency of the variant in the general population supported its pathogenicity.
Regarding the complexity of H syndrome, there is a lack of consensus about the treatment. Colchicine, anti-IL1, and Tumor necrosis factor (TNF)-alpha therapy seem to be ineffective (15). Systemic corticosteroids showed good results but with important harmful long-term effect, especially among diabetic patients diagnosed as H syndrome (16). Some of these patients partially responded to a methotrexate treatment followed by a notable improvement on hyperpigmentation, joint stiffness, and arthritis (17). Methotrexate can be combined with Tocilizumab or Ciclosporine to keep the disease under control (9, 18) (Table 2). Generally, Ciclosporine receded the symptoms, but arthritis and persistent inflammation did not improve with the mentioned treatment (9). The positive effect on mycophenolate mofetil is also described on hyperpigmentation and joint stiffness with promising results (24). What seems to be the advantage of Tocilizumab is the fact that this medication has higher success in the treatment of inflammatory manifestations such as arthritis and organ infiltration (16).
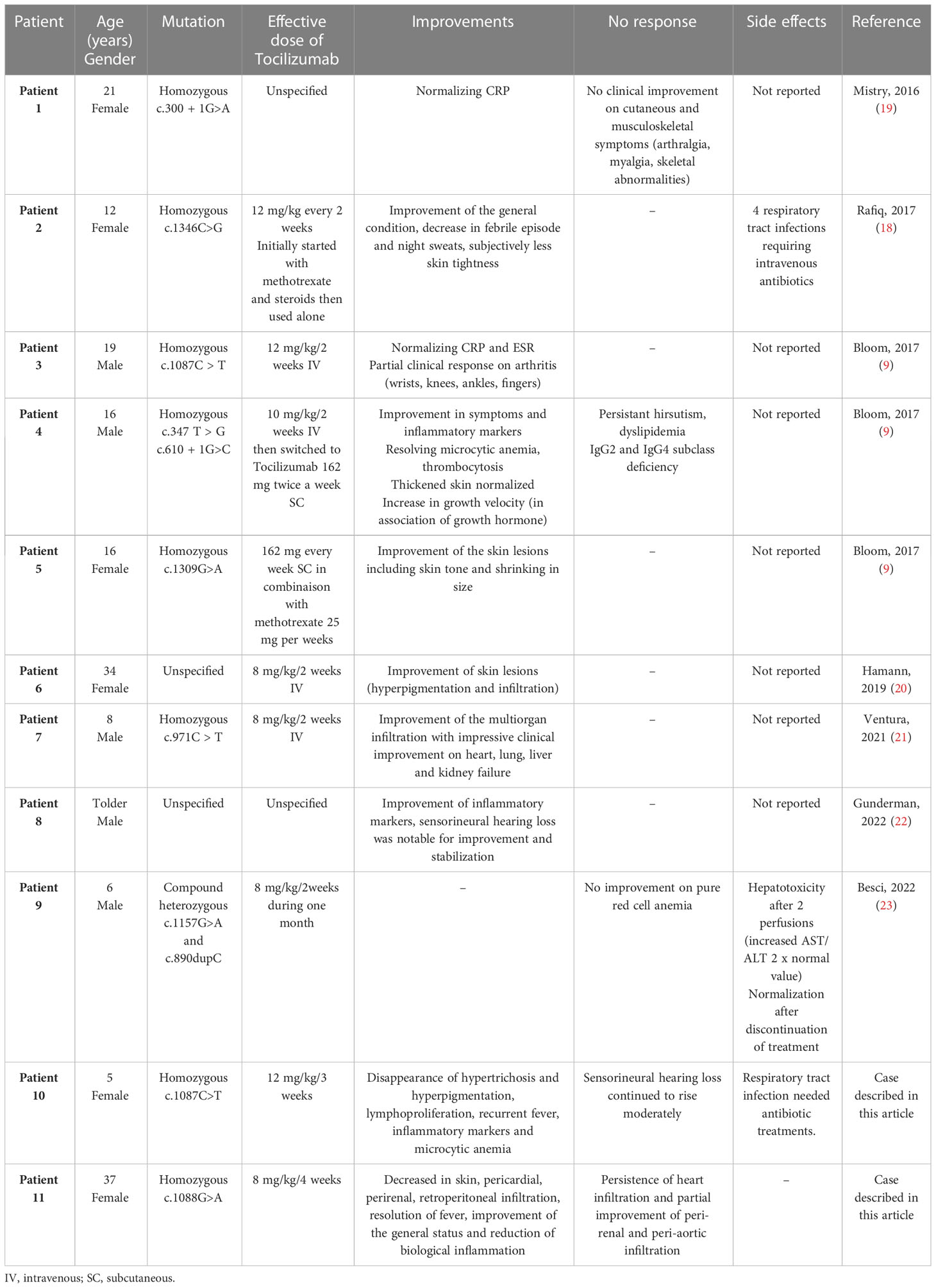
Table 2 Characteristics of H syndrome treated by Tocilizumab.
In the first case’s treatment, Tocilizumab (8 mg/kg/4 weeks) was the better option to start with. It improved the hyperpigmentation and pericardial, perirenal, and retroperitoneal infiltration; broke down the fever; improved the general status; and reduced the inflammation (Table 2). However, despite the use of Tocilizumab, a 5-cm right atrial infiltration remained as it was initially, which led to changing the treatment. No side effect was noticed.
In the second case, Tocilizumab happened to be effective when increased (10 mg/kg per 2 weeks). However, it needed to be balanced because of lower respiratory tract infections. A higher dose of 12 mg/kg/3 week, paired with preventive antibiotic treatment, proved to more effective. Typical posology for systemic juvenile idiopathic arthritis (S-JIA) approved by Food and Drug Administration (FDA) regarding children weighing <30 kg is 12 mg/kg every 2 weeks. The most common adverse effect is infections (mild gastroenteritis and nasopharyngeal and upper respiratory infections) along with biological abnormalities (neutropenia, thrombopenia, lymphopenia, and elevated transaminases) (25). In safety studies of Tocilizumab in patients with systemic-onset juvenile idiopathic arthritis, cases of bronchopneumonia are rare (n=4/112) and are treated with antibiotics (26).
To stress the efficiency of Tocilizumab, we can say that among the 134 H-syndrome-confirmed patients reviewed in the literature, there were 11 patients (including our cases) with pathogenic SCL29A3 mutations treated with Tocilizumab (Table 2).
The median age of the patients was 14 years (extreme values, 5–37 years). Doses used varied between 8 mg/kg/4 weeks and 12 mg/kg/2 weeks. Patients 1 and 9 did not respond to treatment with persistent cutaneous manifestations, arthralgia and skeletal abnormalities, and pure red cell aplasia. For one patient (our adult case), Tocilizumab was discontinued because of a partial response on tissue infiltration with Tocilizumab 8 mg/kg/4weeks. The other eight patients had notably responded to Tocilizumab. Improvement was described regarding cutaneous symptoms (5/6), arthritis (1 with partial response), microcytic anemia (2/2), recurrent fever (3/3), lymphoproliferation (1/1), growth velocity (2/2, adding to growth hormones), hearing loss (1/2), acute phase reactants (5/5), and multiorgan infiltration (2/2). Two patients benefited from subcutaneous Tocilizumab injections (162 mg every 2 weeks) with successful response. Regarding the side effects, two patients presented recurrent tract infections, which were checked, thanks to a lower dosage but no crucial need to discontinue the treatment. Only one patient presented a moderate hepatotoxicity (moderate elevation AST/ALT, 1/11), which required stopping Tocilizumab use.
Even though Tocilizumab seems to be effective for these patients, the data from this study must be interpreted according to the following limitations: retrospective data, small number of patients included, and lack of comparative information with other treatments.
In the first case, persistence of heart infiltration and partial improvement of peri-renal and peri-aortic infiltration, anti-MEK (COBIMETINIB) has been discussed in a multidisciplinary meeting, despite the absence of ERK hyperphosphorylation on biopsy.
Concerning our pediatric patient, she is currently 9 years old and has been treated with Tocilizumab for 4 years with a 12 mg/kg/3 weeks dose. Until now, no lymphoproliferation or clinical or biological inflammation was reported, and skin lesions have disappeared. Growth velocity has been improved thanks to the intake of growth hormone and the control of systemic inflammation with Tocilizumab, but sensorineural hearing loss continues to evolve moderately and slowly.
This report shows that H syndrome, due to mutation in SLC29A3, leads to complex clinical manifestations, which can be diagnosed in adulthood with a fibro-inflammatory disease-like presentation and in childhood with lymphoproliferative syndrome associated with inflammatory condition. We suggest that Tocilizumab should be considered as the first-choice treatment, mainly to control autoinflammatory component (skin lesions, lymphoproliferation, recurrent fever, arthritis, and systemic fibrosis). The treatment intensification with doses increased to 12 mg/kg/2 weeks may be considered when the clinical response is partial, prior to a change in treatment. Tocilizumab treatment may be taken as early as possible, ideally as a juvenile to avoid chronic inflammatory complications.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author AB declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Molho-Pessach V, Lerer I, Abeliovich D, Agha Z, Abu Libdeh A, Broshtilova V, et al. The H syndrome is caused by mutations in the nucleoside transporter hENT3. Am J Hum Genet (2008) 83(4):529–34. doi: 10.1016/j.ajhg.2008.09.013
2. Govindarajan R, Leung GPH, Zhou M, Tse CM, Wang J, Unadkat JD. Facilitated mitochondrial import of antiviral and anticancer nucleoside drugs by human equilibrative nucleoside transporter-3. Am J Physiology-Gastrointestinal Liver Physiol (2009) 296(4):G910–22. doi: 10.1152/ajpgi.90672.2008
3. Molho-Pessach V, Agha Z, Aamar S, Glaser B, Doviner V, Hiller N, et al. The H syndrome: A genodermatosis characterized by indurated, hyperpigmented, and hypertrichotic skin with systemic manifestations. J Am Acad Dermatol (2008) 59(1):79–85. doi: 10.1016/j.jaad.2008.03.021
4. Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity (2016) 44(3):450–62. doi: 10.1016/j.immuni.2016.02.015
5. Bruce-Brand C, Schneider JW, Schubert P. Rosai-Dorfman disease: an overview. J Clin Pathol (2020) 73(11):697–705. doi: 10.1136/jclinpath-2020-206733
6. Emile JF, Abla O, Fraitag S, Horne A, Haroche J, Donadieu J, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood (2016) 127(22):2672–81. doi: 10.1182/blood-2016-01-690636
7. Molho-Pessach V, Ramot Y, Camille F, Doviner V, Babay S, Luis SJ, et al. H syndrome: The first 79 patients. J Am Acad Dermatol (2014) 70(1):80–8. doi: 10.1016/j.jaad.2013.09.019
8. McClellan SF, Ainbinder DJ. Orbital rosai-dorfman disease: A literature review. Orbit (2013) 32(5):341–6. doi: 10.3109/01676830.2013.814689
9. Bloom JL, Lin C, Imundo L, Guthery S, Stepenaskie S, Galambos C, et al. H syndrome: 5 new cases from the United States with novel features and responses to therapy. Pediatr Rheumatol (2017) 15(1):76. doi: 10.1186/s12969-017-0204-y
10. Farooq M, Moustafa RM, Fujimoto A, Fujikawa H, Abbas O, Kibbi AG, et al. Identification of two novel mutations in SLC29A3 encoding an equilibrative nucleoside transporter (hENT3) in two distinct Syrian families with H syndrome: expression studies of SLC29A3 (hENT3) in human skin. Dermatology (2012) 224(3):277–84. doi: 10.1159/000338886
11. Dispenzieri A, Fajgenbaum DC. Overview of castleman disease. Blood (2020) 135(16):1353–64. doi: 10.1182/blood.2019000931
12. Bader-Meunier B, Florkin B, Sibilia J, Acquaviva C, Hachulla E, Grateau G, et al. Mevalonate kinase deficiency: A survey of 50 patients. Pediatrics (2011) 128(1):e152–9. doi: 10.1542/peds.2010-3639
13. Doviner V, Maly A, Ne’eman Z, Qawasmi R, Aamar S, Sultan M, et al. H syndrome: recently defined genodermatosis with distinct histologic features. A morphological, histochemical, immunohistochemical, and ultrastructural study of 10 cases. Am J Dermatopathol (2010) 32(2):118–28. doi: 10.1097/DAD.0b013e3181b28572
14. Molho-Pessach V, Suarez J, Perrin C, Chiaverini C, Doviner V, Tristan-Clavijo E, et al. The H syndrome: Two novel mutations affecting the same amino acid residue of hENT3. J Dermatol Sci (2010) 57(1):59–61. doi: 10.1016/j.jdermsci.2009.09.011
15. Melki I, Lambot K, Jonard L, Couloigner V, Quartier P, Neven B, et al. Mutation in the SLC29A3 gene: A new cause of a monogenic, autoinflammatory condition. PEDIATRICS (2013) 131(4):e1308–13. doi: 10.1542/peds.2012-2255
16. Nofal H, AlAkad R, Nofal A, Rabie E, Chaikul T, Chiu FP, et al. H syndrome: A review of treatment options and a hypothesis of phenotypic variability. Dermatol Ther (2021) 34(5). doi: 10.1111/dth.15082
17. di Dio F, Mariotti I, Coccolini E, Bruzzi P, Predieri B, Iughetti L. Unusual presentation of Rosai-Dorfman disease in a 14-month-old Italian child: a case report and review of the literature. BMC Pediatr (2016) 16:62. doi: 10.1186/s12887-016-0595-9
18. Rafiq NK, Hussain K, Brogan PA. Tocilizumab for the treatment of SLC29A3 mutation positive PHID syndrome. Pediatrics (2017) 140(5):e20163148. doi: 10.1542/peds.2016-3148
19. Mistry A, Parry D, Matthews B, Laws P, Goodfield M, Savic S. A case of SLC29A3 spectrum disorder—unresponsive to multiple immunomodulatory therapies. J Clin Immunol (2016) 36(5):429–433. doi: 10.1007/s10875-016-0301-6
20. Hamann P, de Risi-Pugliese T, Senet P, Barbaud A, Frances C. Efficacité du tocilizumab dans le syndrome H, un cas clinique. Ann Dermatol Venereol (2019) 146(12):A247–8. doi: 10.1016/j.annder.2019.09.389
21. Ventura-Espejo L, Gracia-Darder I, Escribá-Bori S, Amador-González ER, Martín-Santiago A, Ramakers J. Patient with H syndrome, cardiogenic shock, multiorgan infiltration, and digital ischemia. Pediatr Rheumatol (2021) 19(1):104. doi: 10.1186/s12969-021-00586-2
22. Gunderman LM, Valika T, Carol H, Khojah A. Improvement of SLC29A3 spectrum disorder-related sensorineural hearing loss after initiation of IL-6 inhibitor. BMJ Case Rep (2022) 15(6):e249191. doi: 10.1136/bcr-2022-24919
23. Besci Ö, Patel KA, Yıldız G, Tüfekçi Ö, Acinikli KY, Erbaş M, et al. Atypical comorbidities in a child considered to have type 1 diabetes led to the diagnosis of SLC29A3 spectrum disorder. Hormones (2022) 21:501–6. doi: 10.1007/s42000-022-00352-3
24. Behrangi E, Sadeghzadeh-Bazargan A, Khosravi S, Shemshadi M, Youssefian L, Vahidnezhad H, et al. Mycophenolate mofetil treatment of an H syndrome patient with a SLC29A3 mutation. Dermatol Ther (2020) 33(6):e14375. doi: 10.1111/dth.14375
25. Correll CK, Binstadt BA. Advances in the pathogenesis and treatment of systemic juvenile idiopathic arthritis. Pediatr Res (2014) 75(1–2):176–83. doi: 10.1038/pr.2013.187
26. Yokota S, Imagawa T, Mori M, Miyamae T, Aihara Y, Takei S, et al. Efficacy and safety of tocilizumab in patients with systemic-onset juvenile idiopathic arthritis: a randomised, double-blind, placebo-controlled, withdrawal phase III trial. Lancet (2008) 371(9617):998–1006. doi: 10.1016/S0140-6736(08)60454-7
Keywords: H syndrome, SLC29A3, tocilizumab, monogenic autoinflammatory disease, hyperpigmentation, recurrent febrile attacks, lymphoproliferative syndrome
Citation: Jacquot R, Jouret M, Valentin MG, Richard M, Jamilloux Y, Rousset F, Emile J-F, Haroche J, Steinmüller L, Zekre F, Phan A, Belot A and Seve P (2023) H syndrome treated with Tocilizumab: two case reports and literature review. Front. Immunol. 14:1061182. doi: 10.3389/fimmu.2023.1061182
Received: 04 October 2022; Accepted: 17 July 2023;
Published: 11 August 2023.
Edited by:
Enver Simsek, Eskişehir Osmangazi University, TürkiyeReviewed by:
Talha Munir, St James’s University Hospital, United KingdomCopyright © 2023 Jacquot, Jouret, Valentin, Richard, Jamilloux, Rousset, Emile, Haroche, Steinmüller, Zekre, Phan, Belot and Seve. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robin Jacquot, cm9iaW4uamFjcXVvdEBjaHUtbHlvbi5mcg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.