
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 06 February 2023
Sec. Inflammation
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1032469
This article is part of the Research Topic Women in Inflammation View all 8 articles
 Iwona Niemietz1,2
Iwona Niemietz1,2 Kelly L. Brown2,3*
Kelly L. Brown2,3*Background: Hyaluronan (HA) is an important structural component of the extracellular matrix and has well-described roles in maintaining tissue integrity and homeostasis. With inflammation, HA metabolism (synthesis and degradation) increases and results in higher concentrations of soluble HA. Previously, we demonstrated that (soluble) HA primed resting neutrophils for the oxidative burst in response to a secondary stimulus. Notably, HA-mediated priming was not dependent on degranulation, which is a hallmark of priming by classical agents such as TNFα. In this study, we queried the ability of HA to prime neutrophils to different stimuli and its capacity to modulate neutrophil function in the presence of TNFα.
Methods: Blood neutrophils from healthy donors were stimulated ex vivo with HA in the absence and presence of classic neutrophil agonists, inclusive of TNFα. Western blotting was used to assess the activation (phosphorylation) of p38 MAPK, and key neutrophil functions associated with priming and activation, such as intracellular and extracellular ROS production, degranulation, and apoptosis, were evaluated by standard chemiluminescence assays (ROS) and flow cytometry.
Results: Hyaluronan is capable of atypical priming and, with TNFα, co-priming neutrophils for an enhanced (rate and/or magnitude) oxidative burst to various secondary stimuli. In addition, HA can augment intracellular ROS production that is directly induced by TNFα in resting neutrophils, which coincided with the activation of p38 MAPK and apoptosis.
Conclusions: These data demonstrate that the extracellular matrix component HA is a key modulator of neutrophil function(s) in the presence of inflammatory agents such as TNFα. Moreover, it provides additional evidence for the diversity and complexity of neutrophil priming and activation during inflammation.
Hyaluronan (HA) is a naturally occurring glycosaminoglycan and a ubiquitous component of the extracellular matrix (ECM) (1). In homeostatic tissue, HA undergoes continuous synthesis and enzymatic degradation that is mediated by hyaluronan synthases and hyaluronidases, respectively. This basal rate of HA synthesis and corresponding degradation is increased during inflammation (2) and to an extent that circulating concentrations of HA are elevated in individuals with conditions characterized by systemic inflammation, such as rheumatoid arthritis (3, 4), myositis (5), and sepsis (6). High circulating concentrations of HA have also been observed in other diseases where damage-induced neutrophil infiltration is prominent, such as pulmonary disease (7–9), liver fibrosis (10), lupus nephritis (11) and vasculitis (12).
It is appreciated that the ECM, and components thereof that are liberated during inflammation, can actively participate in immune processes, often by direct engagement of immune cells (13), and ultimately shape the immune response (14). It has been speculated that HA that is degraded during inflammation (by hyaluronidases as per normal metabolism and/or by microbial enzymes and reactive oxygen species present in damaged tissues) may alert host immune cells. Early investigations to this effect provided evidence that HA may act as a damage-associated molecular pattern (DAMP) that induces cytokine production by engaging immune cells through CD44, the primary receptor for HA (15), or Toll-like receptors TLR2 and TLR4 (16–19). However, mounting evidence by our group (20) and others (21, 22) is to the contrary. We recently provided evidence that hyaluronan contributes to innate immune cell activation and inflammation not by the induction of cytokine production by human peripheral blood cells (23) but via priming neutrophils for a robust oxidative burst following secondary exposure to the G-protein coupled receptor agonist and bacterial peptide, N-formylmethionyl-leucyl-phenylalanine (fMLF).
Neutrophils are ‘first responders’ of the immune system and the first immune cells to migrate (from the blood stream) into injured and/or infected tissues. Extravasation is initiated by exogenous or host-derived environmental cues that, coincident with inducing a migratory phenotype, can prime and/or fully activate neutrophils to carry out key immune functions. Some stimuli, notably TNFα, put neutrophils in an alerted, or ‘primed’, state while others (e.g., fMLF) fully activate resting or primed neutrophils. Primed neutrophils have several key features including the ability to readily migrate to damaged tissues, possess heightened functional capacity in response to secondary stimuli, and have a prolonged lifespan (24). Primed neutrophils are commonly and conveniently identified by an altered abundance of adhesion molecules (CD11b and CD62L) on the neutrophil surface and enhanced production of reactive oxygen species (ROS) upon (secondary) activation. Classical priming agents, such as TNFα, impart these changes quickly, often within 10 - 20 minutes, and in large part via the mobilization and fusion of intracellular granules with the neutrophil plasma membrane (25), which allows for the deposition of granule-stored molecules (e.g., CD11b and the fMLF receptor FPR1) on the cell surface.
In a previous report by our group (23), we provided the first evidence that HA, in the form of both small (< 40kDa) and large polymers (1.01 – 1.8 MDa), is a neutrophil priming agent. However, and in contrast to TNFα and other classical priming agents that induce a primed neutrophil phenotype characterized by altered cell surface proteins (due to granule mobilization), enhanced ROS release to secondary stimuli, and delayed apoptosis, HA-mediated priming is limited to ROS release and is not dependent on granule mobilization. In this study, we strengthen and extend our previous observation of ‘atypical’ priming of neutrophils by hyaluronan, and we provide new evidence that hyaluronan can co-operate with TNFα to co-prime neutrophils for enhanced ROS release. Further, we demonstrate that HA is a modulator of direct effects (not dependent on secondary stimulation) that are induced in neutrophils by TNFα, specifically, the production of intracellular ROS and apoptosis. Altogether in this study, we demonstrate new, context-specific roles for HA in the inflammatory response, that are independent, but perhaps not unrelated to its role in maintaining tissue homeostasis.
Pharmaceutical grade hyaluronan (HA), 15M (1.01 - 1.8 MDa) was purchased from Lifecore Biomedical LLC (Chaska, MN, USA). Stocks were reconstituted in endotoxin-free water (GE Healthcare, Chicago, IL, USA) and confirmed to contain < 0.1 EU of endotoxin with a LAL assay (Pierce LAL Chromogenic Endotoxin Quantitation Kit, ThermoFisher Scientific, Waltham, MA, USA) as per manufacturer’s instructions.
Venous blood from healthy adult volunteers (Children’s and Women’s Research Ethics Board of the University of British Columbia certificate number H15-00351) was collected in sodium heparin vacutainers (BD Bioscience, San Jose, CA, USA). Polymorphonuclear cells (PMN) were isolated using dextran sedimentation (Sigma-Aldrich, Saint Louis, MO, USA) and Ficoll-Paque gradient centrifugation as described (26). Neutrophils consistently comprised no less than 95% of the PMN population (pocHi 100i™ Hematology Analyzer, Sysmex Corp., Kobe, Japan), thus PMN are referred to as neutrophils throughout this study. Isolated neutrophils were suspended in complete RPMI (RPMI 1640 with 10% fetal bovine serum (FBS), 2 mM L-glutamine and 1 mM sodium pyruvate; all from ThermoFisher Scientific, Waltham, MA, USA) and used immediately. Alternatively, neutrophils were kept on ice in Krebs-Ringer Glucose buffer (KRG, pH 7.3) containing 1 mM Ca2+ for a maximum of 6 h prior to use in downstream experiments. Unless otherwise stated, neutrophils (5 x 106/mL) were incubated at 37°C, 5% CO2 in the absence and presence of HA (200 μg/mL) and/or TNFα (10 ng/mL) for 20 min.
Following stimulation with TNFα in the absence and presence of HA, the neutrophils were washed, suspended in PBS/2% FBS/5 mM EDTA and stained 45 min on ice with PE-conjugated mouse anti-human CD11b mAb (1:50; Biolegend; #560914) and/or Alexa Fluor® 647-conjugated mouse anti-human CD35 mAb (1:40; Biolegend; #565329). A minimum of 20,000 events in the neutrophil gate, determined by FCS and SSC, were collected on an Attune NxT flow cytometer.
In this study, we specifically measured ROS generated by the NADPH oxidase using a luminol/isoluminol-amplified chemiluminescence assay as described (27). In brief, after incubation with TNFα in the absence and presence of HA, cells in suspension (1 x 105/well) were transferred to a 96-well white flat bottom plate (Nunc™, 1256602), equilibrated 5 min at 37°C in a 200 μL reaction mixture containing KRG with horseradish peroxidase (4 units/mL, Sigma-Aldrich, Saint Louis, MO, USA) and luminol or (membrane impermeable) isoluminol (10 μg/mL, Sigma, Saint Louis, MO, USA) for detection of total ROS and extracellular ROS (ecROS), respectively. To detect intracellular ROS (icROS), membrane impermeable antioxidants, superoxide dismutase (50 units/mL, Sigma, Saint Louis, MO, USA) and catalase (1,000 units/mL, Worthington Biochemical Corp., Lakewood, NJ, USA) were used in combination with luminol. Cells (1 x 105/well) suspended in the reaction mixture were exposed to N-Formylmethionyl-leucyl-phenylalanine (fMLF;100 nM; Sigma, Saint Louis, MO, USA), C5a (50 nM; PeproTech, Rocky Hill, NJ, USA; #300-70), wheat-germ agglutinin (WGA; 10 μg/mL; EY laboratories, San Mateo, CA, USA; #L-2101-25), galectin – 3 (20 μg/mL, courtesy of Dr. H. Leffler, Lund University, Sweden) or PMA (5 mM; Sigma-Aldrich) and chemiluminescence was recorded every 6 sec for a minimum of 10 min on a TECAN Infinite 200 Pro plate reader (TECAN Group, Männedorf, Switzerland). Alternatively, cells were stimulated with HA (200 μg/mL) and/or TNFα (10 ng/mL), and incubated directly in the Tecan at 37°C while chemiluminescence was recorded for a minimum of 60 min.
Neutrophils (5 x 106 cells/mL) were incubated in the absence and presence of LPS (anti-apoptotic; 100 ng/mL; In vivoGen, E.coli 0111:B4, San Diego, CA, USA), anti-CD95/FasL (pro-apoptotic; 10 μg/mL; Invitrogen by ThermoFisher Scientific, Waltham, MA, USA; #16-0958-81), HA (200 μg/mL) and/or TNFα (10 ng/mL; R&D Systems) for 6 or 20 h at 37°C, 5% CO2. Cells were stained sequentially with FITC-Annexin V (1:20, 10 min, BD Bioscience, San Jose, CA, USA) and 7-amino-actinomycin D (7-AAD; 1:100, 5 min, Biolegend, San Diego, CA, USA). A minimum of 50,000 events were acquired on an Attune NxT flow cytometer (ThermoFisher Scientific, Waltham, MA, USA).
Neutrophil lysates were prepared according to the method of Sarkar et al. (28). Briefly, following stimulation, neutrophils (2 x 106 cells) were spun at 350 x g for 10 min at 4°C. The cell pellet was lysed in 10% ice cold trichloroacetic acid (TCA) and kept on ice for 5 min. Precipitates were spun at 14, 000 x g for 5 min at 4°C. The cell pellet was washed twice in ice-cold acetone (14 000 x g/5min), left to dry for 1 minute at 100°C to ensure residual acetone evaporation, and suspended in 1 x Laemmli buffer. Samples were boiled for 5 min at 100°C and stored at -80°C until use. Lysates (an equivalent of 0.45 x 106 cells/lane) were separated by 10% SDS-PAGE and transferred onto polyvinylidene difluoride membranes (Immobilon-FL, MilliporeSigma). Membranes were blocked with 5% BSA (Sigma) in TBS-T (Tris-buffered saline containing 0.05% Tween-20) for 60 min at room temperature and incubated with primary antibodies overnight at 4°C. Membranes were washed in TBS-T (3 x 5 min) prior to staining with secondary antibodies. After staining with secondary antibodies for 60 min at room temperature, membranes were washed and imaged using a LI-COR Odyssey infrared imager (LI-COR Biosciences). The primary antibodies used were the following: rabbit anti-human p38 mAb (1:2,000; Cell Signalling Technology; #8690), mouse anti-human phospho-p38 mAb (Thr180/Tyr182; 1:2,000; Cell Signalling Technology; #9216). Secondary antibodies used: IRDye 800-conjugated goat anti-rabbit IgG monoclonal antibody (1:20,000; LI-COR; #926-32211; Lincoln, NE, USA) and IgG IRDye 680RD-conjugated goat anti-mouse monoclonal antibody (1:20,000; LI-COR; #926-68070).
All flow cytometry data were analyzed using FlowJo software (version 10.5, Tree Star Inc, OR, USA). Western blot data were analyzed using Image Studio Lite Quantification Software (LI-COR Biosciences). All data are expressed as mean + standard deviation (SD). Statistical analysis was performed using Graph Pad Prism (version 8.1.2 for Mac OS X, GraphPad Software, La Jolla, CA, USA) on raw data values, as described in each figure legend. Statistical significance is indicated by *p < 0.05, **p < 0.01, ***p< 0.001 and ****p< 0.0001.
We have demonstrated that hyaluronan can prime neutrophils towards secondary stimulation with the exogenous G-protein coupled receptor (GPCR) agonist fMLF (23), resulting in an increased release of extracellular ROS that, on average, was 50-60% greater than induced by fMLF alone (23). Although our prior investigations exclusively used fMLF as the ROS-inducing agent, heightened/amplified ROS production by primed neutrophils can be induced by a variety of both endogenous (e.g., C5a, galectin-3, GM-CSF) and exogenous (e.g., fMLF, LPS, PMA) stimuli (29). Therefore, we asked whether neutrophil responsiveness to ROS-inducing agonists other than fMLF can be primed by HA. Towards this, we evaluated a subset of well characterized agonists that induce ROS production in neutrophils through GPCR (C5a) and non-GPCR mediated signaling (lectins and PMA) and with known differences in the magnitude and kinetics of ROS release.
In line with the observed response of HA-primed neutrophils to fMLF, HA priming led to a heightened production of ROS by neutrophils subsequently exposed to the endogenous GPCR agonist, C5a. Relative to unprimed cells stimulated with C5a (100% peak ROS release), the average peak ROS release in HA-primed/C5a-stimulated cells was 136.6 ± 29.1% (Figures 1A, B).
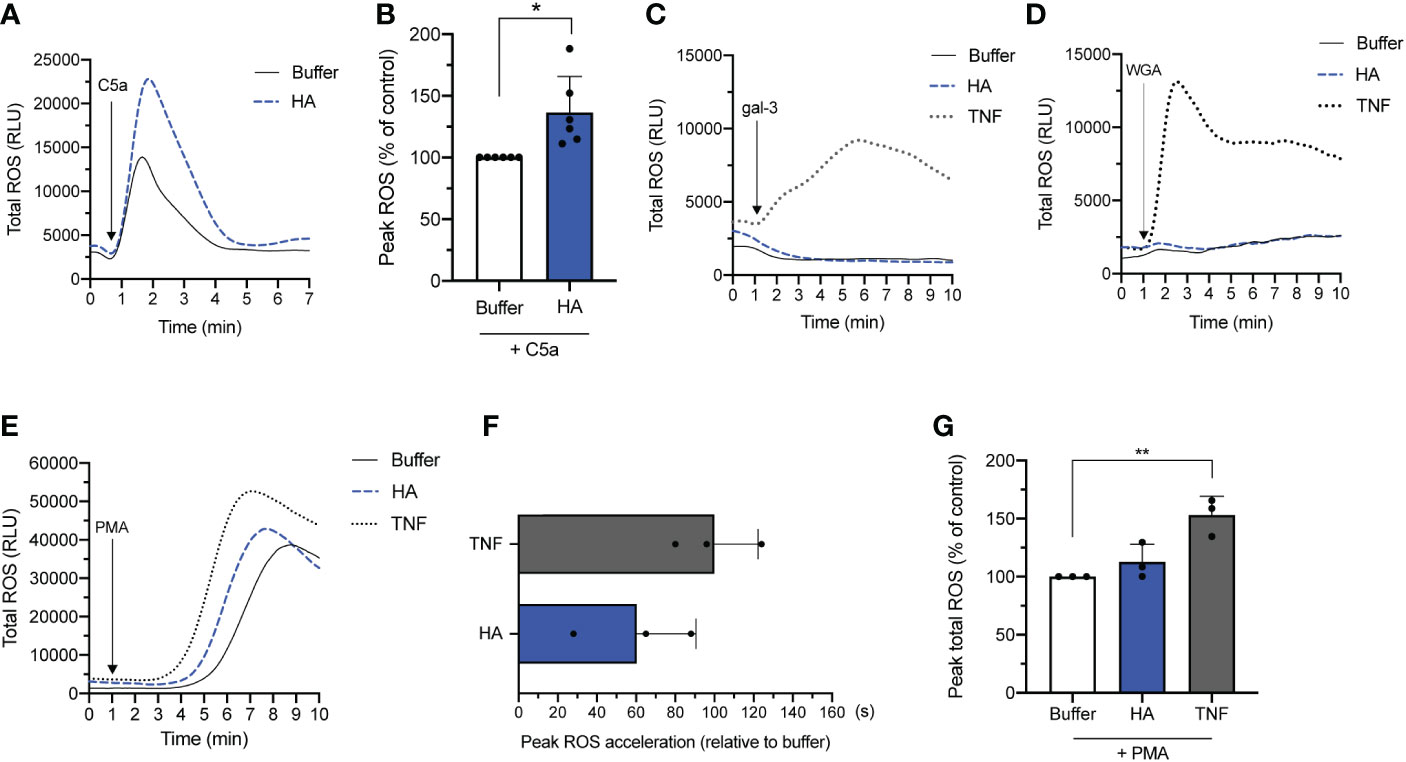
Figure 1 NADPH oxidase-derived ROS release by HA-primed neutrophils activated with various stimuli. Total ROS production (y-axis, relative light units (RLU)) by neutrophils over time (x-axis, min) after (A) priming without (buffer) or with HA (200 μg/mL) followed by addition of C5a (50 nM). Data is representative of 6 independent experiments. (B) Bar graphs show total ROS production (y-axis) of unstimulated (buffer) and HA-primed cells (HA) after stimulation with C5a (50 nM). The data is presented as mean percentage of peak total ROS (% ROS) + SD normalized to the response of unstimulated neutrophils to C5a, n = 6. (C). Total ROS release (y-axis, RLU) by neutrophils over time (x-axis, min) after priming in the absence (buffer) or presence of TNFα (10 ng/mL) or HA (200 μg/mL) followed by galectin-3 (20 μg/mL) or (D) WGA (wheat germ agglutinin; 10 μg/mL). Data is representative of 2 independent experiments. (E) Total ROS release (y-axis, RLU) by neutrophils over time (x-axis, min) after priming in the absence (buffer) or presence of TNFα (10 ng/mL) or HA (200 μg/mL) followed by PMA (5 nM). Data representative of 3 independent experiments. (F) Bar graphs show the acceleration in reaching peak ROS (x-axis, seconds; mean + SD) by cells primed with TNFα or HA relative to unprimed cells after PMA activation. (G) Bar graphs show total ROS production of unstimulated (buffer) and HA-primed cells (x-axis) after stimulation with PMA (5 nM). The data is presented as mean percentage of peak total ROS (% ROS) + SD normalized to the response of unstimulated neutrophils to PMA, n = 3. Statistical analysis was performed using two-tailed paired t-test (B) or repeated measures one-way ANOVA followed by Dunnett’s multiple comparisons test, (G); *p < 0.05, **p < 0.01.
Next, we evaluated the ability of HA to prime neutrophils for secondary stimulation with non-GPCR agonists. First, we tested the ability of HA to prime neutrophils towards galectin-3 and wheat germ agglutinin (WGA), which are lectins that crosslink cell surface glycoprotein receptors. Contrary to fMLF and C5a that can induce ROS production in resting and primed neutrophils, WGA and galectin-3 (30, 31) can only induce ROS release by classically primed neutrophils by virtue of granule mobilization that results in the deposition of cognate granule-stored receptors on the cell surface (32). In addition, lectin-induced release of ROS by primed neutrophils is lower in magnitude but longer in duration compared to fMLF and C5a; the kinetics and magnitude are shown in Figures 1C, D for TNF-α-primed neutrophils. Consistent with the inability of HA to induce neutrophil degranulation, neither galectin-3 nor WGA could provoke ROS release by neutrophils primed with HA.
Finally, we evaluated the responsiveness of HA-primed neutrophils to the diacylglycerol mimetic, PMA, which induces ROS production through a receptor-independent mechanism (33). PMA is a potent ROS-inducing stimulus that alone can induce an oxidative burst equivalent or greater in magnitude to primed/activated neutrophils. In this scenario, priming prior to secondary stimulation with PMA alters (accelerates) the kinetics of the oxidative burst more so than the magnitude; this heightened and hastened ROS production by TNFα-primed neutrophils in response to PMA is shown in Figure 1E (34). Like TNFα, HA-mediated priming also shortened the response time (i.e., time to peak ROS release) induced by PMA when compared to buffer. Neutrophils primed with HA reached peak ROS release considerably faster (60.0 ± 30.3 s) than unprimed cells (buffer) exposed to PMA (Figure 1F). By way of comparison, TNFα-primed/PMA-activated neutrophils reached peak ROS release 100.0 ± 22.2 sec before unprimed cells. The shortened ROS response time in HA-primed cells was accompanied by a (non-significant) increase in total ROS release (112.8 ± 15.1% of peak total ROS) that was substantially lower in magnitude than in TNFα-primed neutrophils (153.0 ± 16.3% of peak ROS) following PMA activation compared to unprimed neutrophils (buffer; 100% peak ROS release) (Figure 1G).
In summary, HA atypically primes neutrophils to respond to GPCR- and non-GPCR stimuli (C5a, PMA and fMLF) with the exception of stimuli (e.g., galectin-3, WGA) that are dependent on granule mobilization (of granule-stored receptors) to exert their ROS-inducing effect (32, 35). Notably, priming induced by HA could modify both the rate and/or magnitude of the oxidative burst.
Our previous data demonstrated that TNFα and HA elicited priming through overlapping but not identical mechanisms (23). One important difference is the induction of granule mobilization (and the potential deposition of ROS-inducing agonist receptors on the cell surface) by TNFα but not by HA. Our results show that TNFα was not able to induce an oxidative burst in unprimed or HA-primed neutrophils (Figure 2A). However, given the different mechanism(s) of priming (34, 36), we questioned if HA- and TNFα-mediated priming was synergistic. To do this, we measured fMLF-induced (total) ROS production by neutrophils primed (20 min) simultaneously with HA and TNFα. Results show that the kinetics of total ROS release was similar in HA-, TNFα-, and HA/TNFα-primed cells (Figure 2B). Yet, co-priming of neutrophils with TNFα and HA led to a heightened ROS response to subsequent fMLF activation (peak total ROS = 130.8 ± 16.9% of TNFα-primed/fMLF induced peak total ROS) than with TNFα priming alone (Figures 2B–D).
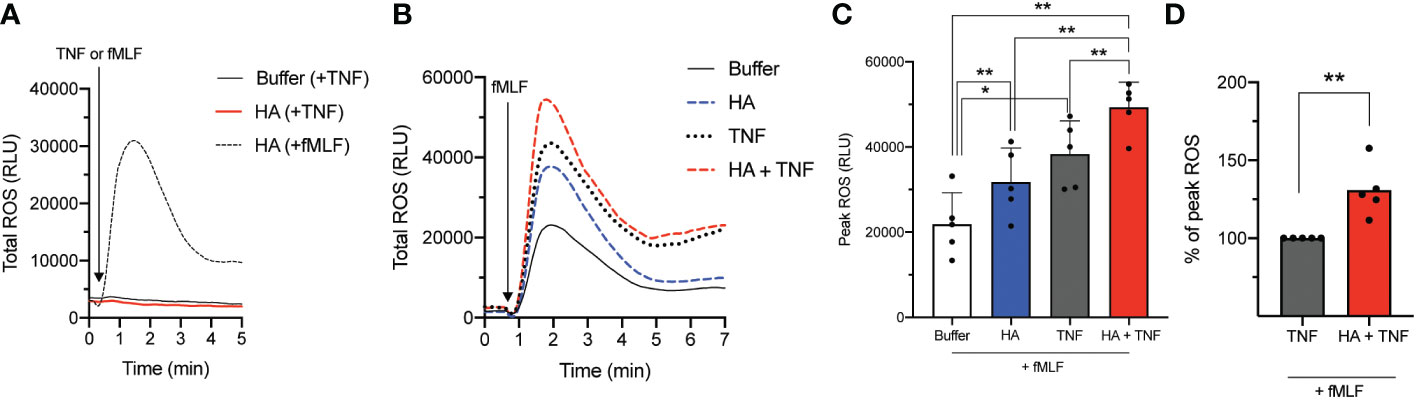
Figure 2 NADPH oxidase-derived reactive oxygen species (ROS) release by co-primed neutrophils. (A) Total ROS release (y-axis, relative light units (RLU)) by neutrophils over time (x-axis, min) after priming in the absence (buffer) or presence of HA (200 μg/mL) followed by fMLF (100 nM) or TNFα (10 ng/mL). (B) Total ROS production (y-axis, RLU) by neutrophils over time (x-axis, min) after priming without (buffer) and with TNFα (10 ng/mL), HA (200 μg/ml), or HA with TNFα followed by the addition of fMLF (100 nM). Histogram is representative of 5 independent experiments. (C) Bar graphs show mean peak total ROS (+SD) for HA-, TNFα−, or HA+TNFα-primed cells (x-axis, RLU) after stimulation with fMLF (D) Peak total ROS (mean + SD) in co-primed neutrophils (HA + TNFα) is presented as % of peak total ROS relative to the response of TNFα-primed neutrophils to fMLF, n = 5. Statistical analysis was performed using repeated measures one-way ANOVA with Sidak’s multiple comparison test (C, D); *p < 0.05, **p < 0.01.
Despite the inability of HA to induce granule exocytosis on its own (23), we questioned if granule mobilization was induced in neutrophils co-primed with TNFα and HA. Using flow cytometry, we measured CD35 and CD11b cell surface expression. Although changes in the abundance of CD11b is widely accepted and commonly utilized as a marker of neutrophil degranulation, inclusion of CD35 allows for the assessment of both gelatinase granule and secretory vesicle mobilization. This is important given that the storage of FPR1, the main receptor for fMLF, is predominantly in gelatinase vesicles (that contain CD11b and CD35), but it has also been detected in secretory vesicles (that contain CD35 mostly) (37) that are readily mobilized prior to the degranulation of gelatinase granules (37). In response to TNFα, there was a measured upregulation (mean fluorescence intensity, MFI) of cell surface CD35 and CD11b (Figures 3A, B for CD35 and Figures 3C, D for CD11b). The addition of HA to TNFα-stimulated neutrophils did not result in the enhanced expression of CD35 or CD11b, suggesting no additional (HA-mediated) mobilization of intracellular granules.
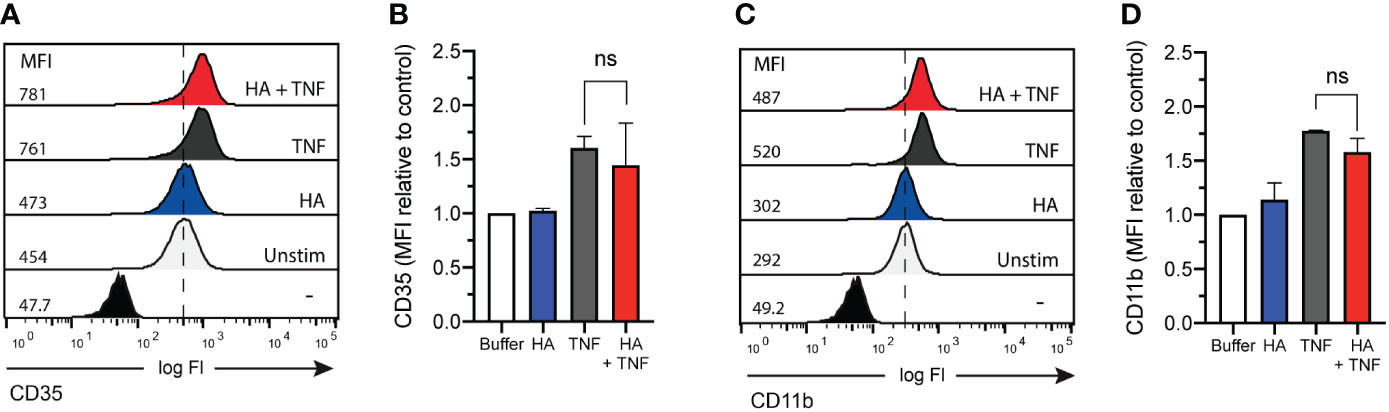
Figure 3 CD35 and CD11b surface expression on co-primed neutrophils. (A) CD35 and (C) CD11b surface expression (x-axis; log fluorescence intensity (log Fl)) on neutrophils (y-axis; cell counts) following incubation in the absence (unstimulated without (-) and with (Unstim) antibody staining) and presence of HA (200 μg/mL), TNFα (10 ng/mL), or TNFα and HA. Data is representative of 3 experiments. Bar graphs show mean (B) CD35 and (D) CD11b fluorescence (y-axis; + SD) for no stimulation, HA, TNFα, and HA + TNFα normalized to control (buffer, unstimulated). Statistical analysis was performed using ordinary one-way ANOVA followed by Sidak's multiple comparison test, (B, D); ns - not significant.
Although priming (by TNFα or HA) and co-priming (TNFα + HA) of neutrophils paired with fMLF activation led to an oxidative burst that was predominantly extracellular (23), some evidence suggests that TNFα can directly stimulate neutrophils to produce intracellular ROS (38, 39). Consistent with this, we observed substantially more intracellular ROS in neutrophils exposed to TNFα alone than cells incubated with HA or no stimulation at all (Figure 4A). The addition of HA significantly increased this intracellular, but not extracellular (data not shown), ROS production in TNFα-stimulated cells (Figures 4A–C).
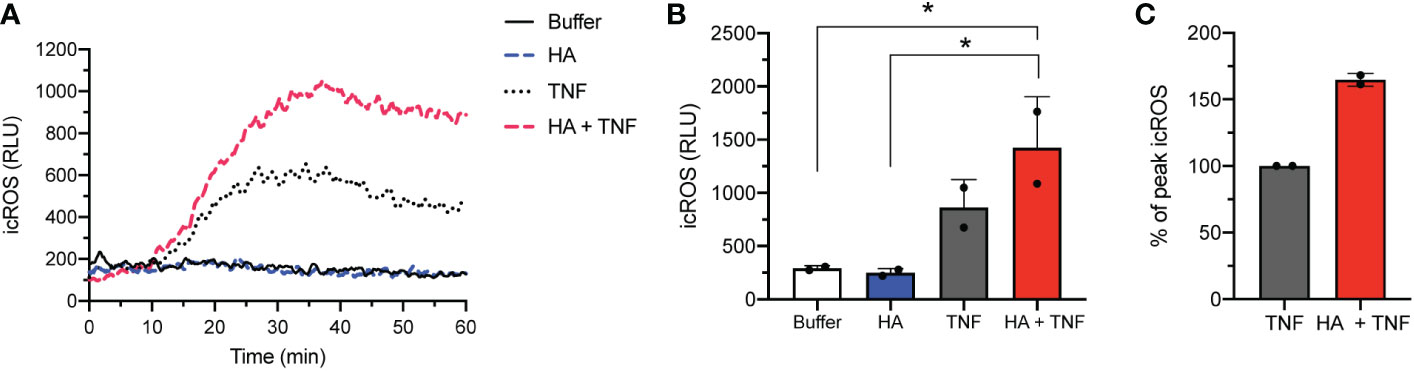
Figure 4 Intracellular ROS release in co-stimulated neutrophils. (A) Intracellular ROS production (y-axis, relative light units (RLU)) by neutrophils over time (x-axis, min) after stimulation without (buffer) and with TNFα (10 ng/mL), HA (200 μg/ml), or HA with TNFα. Histogram is representative of 2 independent experiments. (B) Bar graphs show mean peak intracellular ROS (icROS, y-axis, RLU) + SD for unstimulated (buffer) or HA-, TNFα−, and HA+TNFα-stimulated cells (x-axis). (C) Peak icROS (mean + SD) in co-stimulated (HA + TNFα) neutrophils is presented as % of peak icROS relative to the response of TNFα-stimulated neutrophils, n = 2. Statistical analysis was performed using ordinary one-way ANOVA with Tukey’s multiple comparison test (B); *p < 0.05.
Previous reports have demonstrated that modest levels of intracellular ROS that are induced by TNFα in resting neutrophils (38) can trigger apoptotic pathways and accelerate neutrophil apoptosis (40). Likewise, scavenging of intracellular oxygen species delayed apoptosis in neutrophils from healthy individuals and is consistent with delayed apoptosis of neutrophils in patients with chronic granulomatous disease (CGD) that are deficient in the production of NADPH oxidase-derived ROS (41–43). Our prior work shows that HA alone did not affect neutrophil apoptosis (23). However, based on our findings of enhanced production of intracellular ROS in TNFα−stimulated neutrophils in the presence of HA (Figures 4A–C), by extension, we questioned whether HA could modulate direct effects of TNFα on neutrophil longevity.
To evaluate a potential effect of HA on changes in neutrophil longevity induced by TNFα, the percent of viable, apoptotic, and necrotic neutrophils was determined by AnnexinV/7AAD staining after ex vivo culture of neutrophils with HA alone and in combination with TNFα for 6 h and 20 h. At these time points and by an as yet unknown mechanism, TNFα elicits an early (6 h) pro-apoptotic effect on neutrophils and a late (20 h) anti-apoptotic effect (44). After 6 h of neutrophil culture we did not observe a change in the rate of spontaneous apoptosis (sum of Annexin V+ 7-AAD– cells/Annexin V+ 7-AAD+ cells) in cells stimulated with HA (14.3 ± 5.2% apoptotic cells) compared to unstimulated cells (14.5 ± 6.0%) (Figures 5A, B). As expected, a greater percent of neutrophils, 21.9 ± 5.0%, were apoptotic after 6 h culture with TNFα alone. This pro-apoptotic effect of TNFα was enhanced (p = 0.07) by the presence of HA and resulted in 41.7 ± 8.4% apoptotic neutrophils by 6 h. The percent of apoptotic neutrophils exposed to HA/TNFα was comparable to cells stimulated with FasL, a potent pro-apoptotic stimulus (41.1 ± 12.3%), and significantly higher than measured in unstimulated cells (14.5 ± 6.0%). Expectedly, LPS delayed neutrophil apoptosis, with only 10.6 ± 6.6% apoptotic cells detected after 6 h (Figures 5A, B).
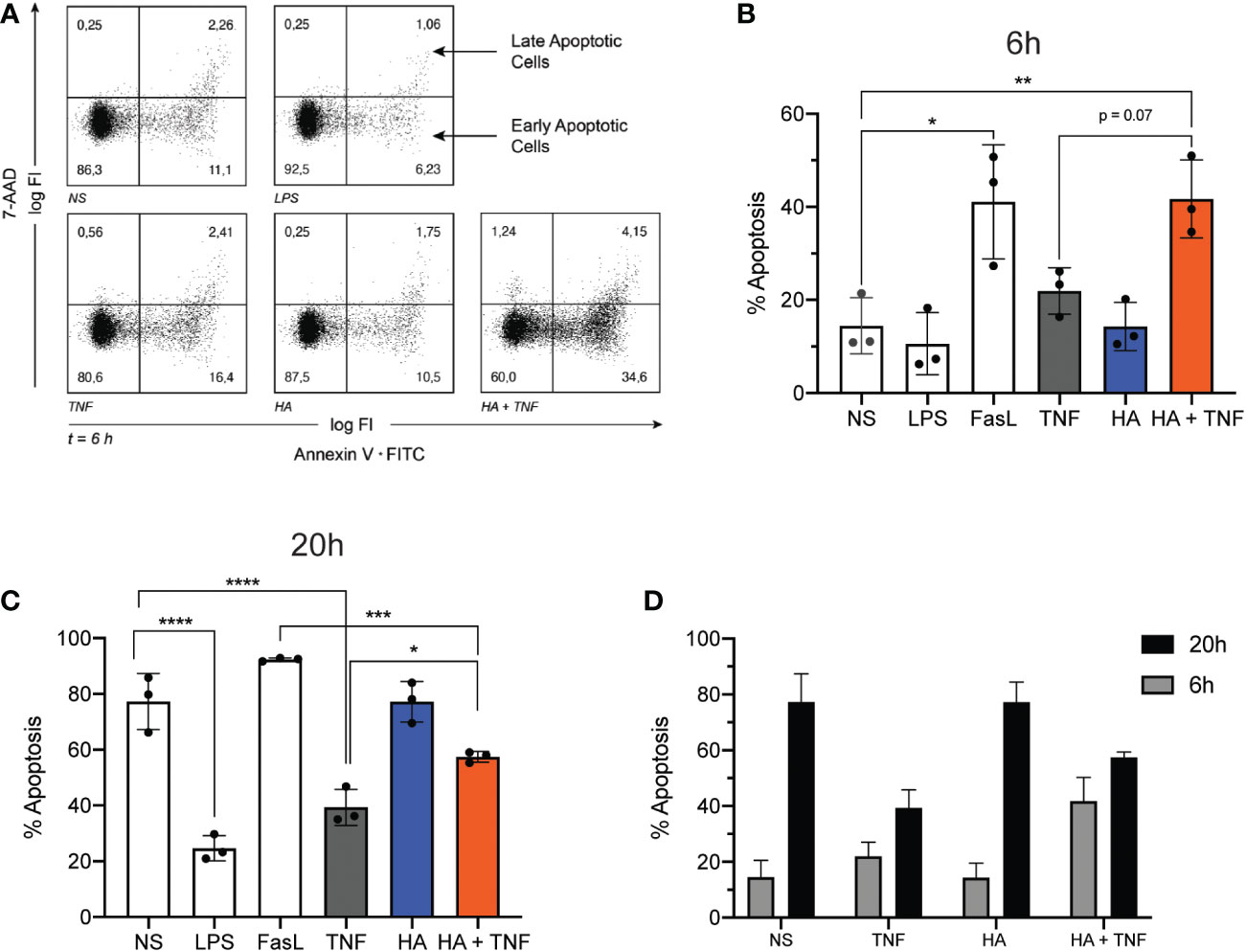
Figure 5 Neutrophil apoptosis in co-stimulated neutrophils. (A) Representative dot plots show Annexin V (x-axis; log fluorescence intensity (log FI)) and 7-AAD (y-axis; log FI) staining of neutrophils following ex vivo culture for 6 h (t = 6 h) in the absence of stimuli (NS) or presence of LPS (100 ng/mL), TNFα (10 ng/mL), HA (200 μg/ml), or HA + TNFα. Staining denotes viable (lower left quadrant, Annexin V– 7-AAD–), early apoptotic (lower right quadrant, Annexin V+ 7-AAD–) and late apoptotic (upper right quadrant, Annexin V+ 7-AAD+) neutrophils. Dot plot is representative of 3 independent experiments summarized in B. (B) Bar graphs show the mean percent of apoptotic cells (Annexin V+) + SD (y-axis) after 6 h of incubation for no stimulation (NS), LPS (100 ng/mL), FasL (10 μg/mL), TNFα 10 ng/mL, HA (200 μg/mL), and HA + TNFα, n = 3.(C) Bar graphs show the mean percent of apoptotic cells (Annexin V+) + SD (y-axis) after 20 h of incubation for FasL (10 μg/mL), LPS (100 ng/mL), TNFα (10 ng/mL), HA (200 μg/mL), and HA + TNFα, n = 3. (D) Bar graph shows side by side comparison of the mean percent of apoptotic cells after 6 h (black bars) and 20 h (grey bars) culture, n = 3. Statistical analysis was performed using ordinary one-way ANOVA followed by Tukey’s multiple comparison test, (B, C); *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Similar to the observed pro-apoptotic effect of HA at 6 h (Figures 5A, B), culture of neutrophils for 20 h with TNFα in the presence of HA reversed, at least in part, the anti-apoptotic effect exerted by TNFα alone. In the presence of TNFα, the percent of apoptotic cells was 39.4 ± 6.5% compared to 77.3 ± 10.1% in the absence of stimulation (spontaneous apoptosis). The addition of HA to TNFα-stimulated cells resulted in a significant increase in neutrophil apoptosis (57.5 ± 1.9% apoptotic cells, p = 0.03) (Figure 5C). Expectedly, neutrophil apoptosis was delayed in the presence of LPS (24.7 ± 4.5% apoptotic cells) and accelerated in the presence of FasL (92.4 ± 0.6%) (Figure 5C). HA alone had no effect on neutrophil apoptosis (Figure 5C). While confirming our previous results (23) showing that HA was unable to induce apoptosis on its own (summarized in Figure 5D), these data suggest a previously undescribed function of HA as a modulator of TNFα-mediated apoptosis.
The p38 MAPK is well-recognized for its role in cellular stress responses. In neutrophils, rapid phosphorylation of p38 MAPK has been observed during ROS production (45–47) and granule exocytosis (45, 48, 49). p38 MAPK has also been described as a key mediator of apoptosis (50, 51). To investigate the possibility of p38 MAPK activation in response to HA- and/or HA/TNFα stimulation, we assessed phosphorylation of p38 MAPK Thr180/Tyr182 residues over time using immunoblotting followed by densitometry analysis. Our results show that exposure of neutrophils to HA resulted in rapid phosphorylation of p38 MAPK by ~ 2 min post stimulation and is sustained over the observed 16 min time period (Figures 6A, B; 2.1- fold increase in phospho-p38 at 16 min, p = 0.0275). The addition of fMLF, which results in an oxidative burst in HA-primed neutrophils, was associated with a non-significant increase in p38 MAPK phosphorylation, compared to fMLF alone (Figures 6C, D).
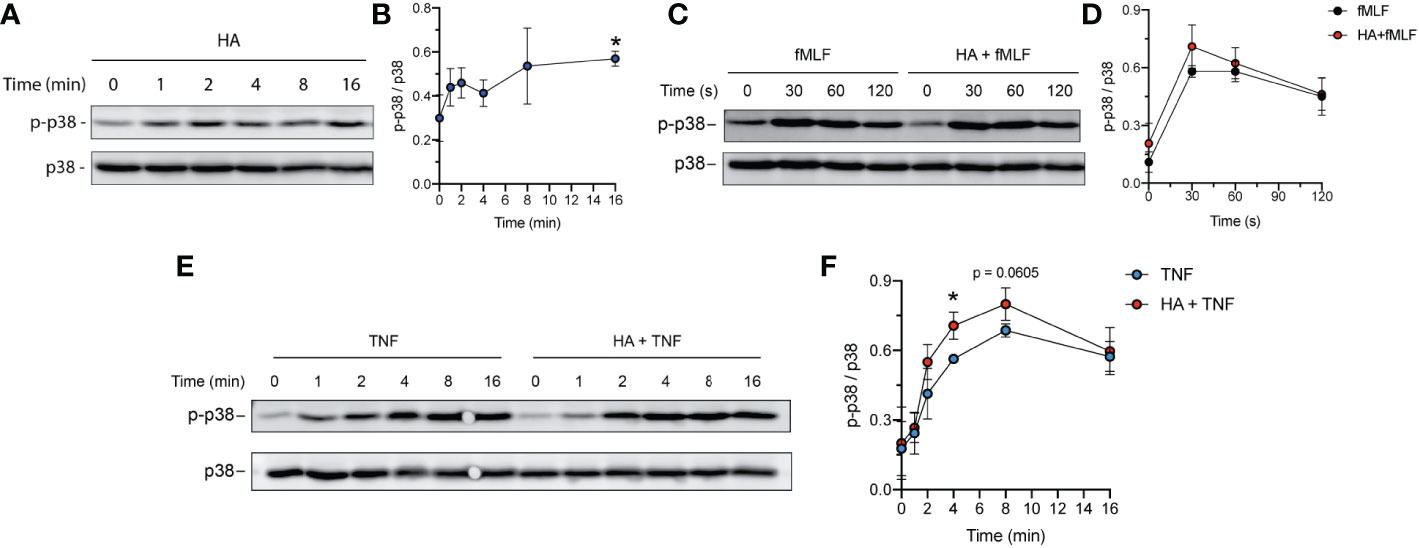
Figure 6 Total and phospho p38 MAPK in TNFα- and HA-stimulated neutrophils. (A) Western blot showing the phosphorylated and total p38 MAPK protein over time in neutrophils primed with HA (200 μg/mL). Data is representative of 3 independent experiments. (B) Graph shows quantification (mean ± SD) of phospho-p38 normalized to total p38 protein (y-axis) over time (x-axis), n = 3.(C) Western blot showing the phosphorylated and total p38 MAPK protein in neutrophils upon fMLF activation (100 nM, 0 – 2 min) at t = 16 min of priming in the presence (HA + fMLF) or absence (fMLF) of HA (200 μg/mL). Data is representative of 3 independent experiments. (D) Graph shows quantification (mean ± SD) of phospho-p38 normalized to total p38 protein (y-axis) over time (x-axis) during activation with fMLF at t = 16 min of priming with (HA + fMLF) and without (fMLF) HA, n = 3. (E) Abundance of phosphorylated and total p38 MAPK protein over time in neutrophils primed with TNFα (10 ng/mL) and HA + TNFα (200 μg/mL HA). Data is representative of 3 independent experiments. (F) Graph shows quantification (mean ± SD) of phospho-p38 normalized to total p38 (y-axis) over time (x-axis) in TNFα-primed and HA+TNFα co-primed neutrophils, n = 3. Statistical analysis for individual time points was performed using unpaired two-tailed t-test, (B, F); *p < 0.05.
In contrast to HA, TNFα induced phosphorylation of p38 MAPK had different kinetics, with steadily increasing phosphorylation over a 16-minute time period (Figure 6E). Immunoblotting and densitometry analysis of phosphorylated (Thr180/Tyr182) p38 and total p38 protein in neutrophils co-stimulated (from 0 – 16 min) with TNFα and HA demonstrated more pronounced phosphorylation of p38 (Thr180/Tyr182) in co-stimulated cells when compared to TNFα alone (Figure 6E). Phosphorylation of p38 MAPK in neutrophils exposed to HA/TNFα co-stimulation reached peak activation at ~ 4 min with a maximum fold increase in phosphorylation occurring between 2 - 8 min compared to cells stimulated with TNFα alone (Figure 6F). The observed phosphorylation of p38 MAPK in neutrophils exposed HA, as well as enhancement of p38 MAPK phosphorylation in the presence of TNFα, implies that HA-exerted effects are p38 MAPK-mediated.
Building on our previous report of HA as a novel and atypical priming agent for fMLF-induced ROS production by neutrophils (23), herein we demonstrate that HA can prime neutrophils for an augmented oxidative burst in response to classical endogenous and exogenous agonists (receptor and non-receptor mediated) other than fMLF, namely C5a and PMA. HA failed to prime cells for ROS production by lectins, which require an upregulation of granule-stored receptors to exert their ROS-inducing effect (32, 35), thus, providing additional confirmation that HA-mediated priming occurs in the absence of degranulation. Further, HA- and TNFα-induced priming are synergistic, leading to an oxidative burst of even greater magnitude that ‘singly-primed’ neutrophils. Co-priming for the neutrophil oxidative burst has been reported previously for TNFα and IFNα (52), but this is the first evidence of neutrophil co-priming with a component of the ECM. The observed synergy between TNFα and HA priming may produce a ‘primed phenotype’ that is found in neutrophils at sites of tissue damage/inflammation where they are in contact with both HA and TNFα simultaneously.
In addition to priming neutrophils, we provide evidence that HA can modulate direct effects of TNFα on neutrophils. Specifically, HA enhances TNFα-induced production of intracellular ROS and exerts a pro-apoptotic effect on neutrophils in the presence of TNFα. Generation of intracellular ROS has been reported to have a role in the transient, pro-apoptotic effect elicited by TNFα in neutrophils (40). Although select stimuli, e.g., GM-CSF, IFNγ, have been reported to enhance ROS production and apoptosis in neutrophils exposed to TNFα (39, 53), again, there is little, if any, prior evidence for a modulatory effect by extracellular matrix components. Few studies have focused on the influence of HA on cell apoptosis in activated cells and studies investigating the effect of HA alone show mixed results (54–57). In leukemic lymphocytes, CD44 stimulation exerted an anti-apoptotic rather than a pro-apoptotic effect (54). Similarly, as reported by Zhao et al., HA delayed apoptosis in murine neutrophils (58). It is important to note that endotoxin contamination of HA (58) was not tested in these studies and cannot be excluded as a driver of delayed apoptosis (59). In agreement with the data presented in this study, a pro-apoptotic effect of HA was observed in lymphoid cells (15), where HA induced cell death in activated but not resting T cells in a CD44-dependent manner.
Our study demonstrates that stimulation of neutrophils with HA is concomitant with the phosphorylation of p38 MAPK. Although the HA-induced activation of p38 was subtle and would benefit from further validation to determine its direct downstream consequences, our data are consistent with evidence that MAPKs, including p38, are active during priming and generation of the oxidative burst (38). It should be also noted that MAPKs have a key role in degranulation (48). However, the extent to which p38 activation contributes directly to NAPDH oxidase activation as opposed to granule mobilization is not known (34, 60). p38 MAPK activation may also regulate ROS generation by promoting cytoskeletal rearrangement that disrupts the interaction of actin with the cytosolic p40phox subunit of the NADPH oxidase (61, 62), and phosphorylation of the NADPH oxidase component p47phox. In the context of HA-mediated signaling via CD44, Lu et al. (63) demonstrated that medium-sized HA fragments (~ 500 kDa) stimulate p38 MAPK activation in neutrophils and suggest that this is via cytoskeletal rearrangements caused by HA binding. It should be noted that p38 MAPK is a mediator of cell apoptosis and at least one study showed that TNFα-induced apoptosis is dependent on the activation of p38 (40). However, as p38 controls ROS production in neutrophils (64) that, in turn, may induce apoptosis (40), it is plausible that the augmented cell death observed in HA/TNFα-stimulated cells is a result of HA-enhanced production of the NADPH-oxidase generated intracellular ROS. This is consistent with an absence of p38 activation in neutrophils stimulated with other pro-, as well as anti-apoptotic stimuli (50). Further analysis is needed to assess whether the enhanced activation of p38 observed in this study results in increased phosphorylation of the p47phox subunit of the NADPH oxidase leading to the observed amplification of ROS generation (65), and if so, whether this is due to independent signals induced separately by HA through CD44-mediated cytoskeletal rearrangement (66) and TNFα engagement and signaling through its cognate receptor (TNFR1).
It is recognized that p38 MAPK phosphorylation may result from TLR4 engagement (67) that may also lead to the activation of NF-κB (68). Demonstrating previously no difference in neutrophil functionality with HA size (< 40kDa up to 1.01 – 1.8kDa polymers) (23), in this study we used the high molecular mass HA polymers that are known to bind to CD44 rather than small HA fragments that may engage CD44 and/or TLR2/4 (summarized by Tavianatou et al. (69)). Accordingly, we found no evidence (based on immunoblot analysis of IκBα degradation) for the activation of the NF-κB pathway in neutrophils incubated with HA (data not shown). This is in agreement with our previous findings (23) showing that HA does not lead to neutrophil responses known to be induced by TLR4 engagement, namely IL-8 release, degranulation and prolonged longevity (70, 71)
Taken together, although HA does not have direct effects on neutrophil activation, our data suggests that is may be a key, context-specific modulator of neutrophil function, namely the production of reactive oxygen species and apoptosis. Considering the interplay between ROS and cell death (72) and in light of the observed modulating effect of HA on TNFα-induced intracellular ROS, the demonstrated pro-apoptotic role of HA is somewhat expected, yet intriguing. Given the potential toxicity of ROS, it is tempting to speculate that by enhancing ROS production, HA contributes to tissue damage. However, by virtue of the pleiotropic roles of both intra- and extra-cellular ROS (73), the consequences of HA-enhanced ROS production are likely more complex. For example, NADPH oxidase-generated ROS may act as a signaling molecule that, among other functions, can regulate cell apoptosis (74). When released into the extracellular space, it can also lead to gap formation between endothelial cells by disturbing junction proteins (75, 76) and inducing cytoskeletal reorganization (77, 78), thereby facilitating cell migration. When considering the pro-apoptotic effect HA had on TNFα stimulated neutrophils, which may be linked to the observed enhancement of TNFα-induced intracellular ROS production, it cannot be excluded that, paradoxically, HA contributes positively to the inflammatory process by facilitating a quick neutrophil response and ensuring a timely termination of inflammation (79); a role that may have pathological consequences when the balance of HA turnover is disturbed and HA is in excess. This could explain the contradicting results where HAS1 (HA synthesizing enzyme) deficiency resulted in chronic inflammation (80), while clearance of hyaluronan with hyaluronidase restored tissue homeostasis (81).
Given that HA is abundant throughout the body, it seems reasonable that HA-mediated effects on neutrophils (the most abundant immune cells) are not direct, but instead governed by the presence of inflammatory stimuli, such as fMLF or TNFα, to ensure that HA-enhanced immune responses are context specific (e.g., upon encountering danger). While an immunomodulatory effect of HA on activated cells has been described in other cells, e.g., T-lymphocytes (15), our findings provide the first report of such a role in neutrophils. HA modulation of TNFα-induced function, particularly the oxidative burst, is intriguing considering that enhanced ROS production at the inflammatory site is commonly thought to contribute to the progression of numerous inflammatory diseases (73), including rheumatic diseases driven by TNFα with concomitant ECM degradation, such as rheumatoid arthritis and vasculitis (82, 83).
In conclusion, our study advances the understanding of the contribution of HA to inflammatory processes that is independent of its previously described role as an inflammation-inducing damage-associated molecular pattern (DAMP) by providing evidence that HA is a modulator of neutrophil effector functions exerted by another stimulus present in the inflammatory milieu, namely TNFα (84).
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving human participants were reviewed and approved by Children’s and Women’s Research Ethics Board of the University of British Columbia. The patients/participants provided their written informed consent to participate in this study.
IN designed and performed all experiments, prepared figures, and wrote the manuscript. KB oversaw all aspects of experiment design, data analysis, interpretation of results and manuscript preparation. All authors contributed to the article and approved the submitted version.
Funding for this project was provided by The Arthritis Society – Ross Petty Chair in Pediatric Rheumatology (to KB) and the BC Children’s Hospital Foundation (IGAP Award to KB). Support was provided to IN by a UBC affiliated Andrew Nord Fellowship in Rheumatology and to KB from the Michael Smith Foundation for Health Research Scholar Award.
The authors wish to acknowledge Ms. Stephanie Hughes for exceptional technical support and Dr. Martina Sundqvist for technical assistance as well as valuable advice on experimental design and data interpretation. We thank Dr. Pauline Johnson for insightful comments on the experimental design and interpretation of data, as well as Dr. Hakon Leffler for providing galectin-3. The authors wish to thank the many blood donors that made this research possible.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Hascall V, Esko JD. Hyaluronan. In: Essentials of glycobiology. Cold Spring Harbor (NY (2015). p. 197–206.
2. Lee-Sayer SS DY, Arif AA, Olsson M, Brown KL, Johnson P. The where, when, how and why of hyaluronan binding by immune cells. Front Immunol (2015) 6(150). doi: 10.3389/fimmu.2015.00150
3. Goldberg RL, Huff JP, Lenz ME, Glickman P, Katz R, Thonar EJ. Elevated plasma levels of hyaluronate in patients with osteoarthritis and rheumatoid arthritis. Arthritis Rheumatol (1991) 34(7):799–807. doi: 10.1002/art.1780340704
4. Kida D, Yoneda M, Miyaura S, Ishimaru T, Yoshida Y, Ito T, et al. The SHAP-HA complex in sera from patients with rheumatoid arthritis and osteoarthritis. J Rheumatol (1999) 26(6):1230–8.
5. Silva MG, Shinjo SK. Hyaluronic acid in dermatomyositis and polymyositis: relationship with disease and cutaneous lesions. Bras Dermatol (2018) 93(1):72–5. doi: 10.1590/abd1806-4841.20186727
6. Anand D, Ray S, Srivastava LM, Bhargava S. Evolution of serum hyaluronan and syndecan levels in prognosis of sepsis patients. Clin Biochem (2016) 49(10-11):768–76. doi: 10.1016/j.clinbiochem.2016.02.014
7. Papakonstantinou E, Bonovolias I, Roth M, Tamm M, Schumann D, Baty F, et al. Serum levels of hyaluronic acid are associated with COPD severity and predict survival. Eur Respir J (2019) 53(3). doi: 10.1183/13993003.01183-2018
8. Wyatt HA, Dhawan A, Cheeseman P, Mieli-Vergani G, Price JF. Serum hyaluronic acid concentrations are increased in cystic fibrosis patients with liver disease. Arch Dis Child. (2002) 86(3):190–3. doi: 10.1136/adc.86.3.190
9. Yang S, Ling Y, Zhao F, Li W, Song Z, Wang L, et al. Hymecromone: a clinical prescription hyaluronan inhibitor for efficiently blocking COVID-19 progression. Signal Transduct Target Ther (2022) 7(1):91. doi: 10.1038/s41392-022-00952-w
10. McHutchison JG, Blatt LM, de Medina M, Craig JR, Conrad A, Schiff ER, et al. Measurement of serum hyaluronic acid in patients with chronic hepatitis c and its relationship to liver histology. consensus interferon study group. J Gastroenterol Hepatol (2000) 15(8):945–51. doi: 10.1046/j.1440-1746.2000.02233.x
11. Yung S, Tsang RC, Leung JK, Chan TM. Increased mesangial cell hyaluronan expression in lupus nephritis is mediated by anti-DNA antibody-induced IL-1beta. Kidney Int (2006) 69(2):272–80. doi: 10.1038/sj.ki.5000042
12. West DC, Yaqoob M. Serum hyaluronan levels follow disease activity in vasculitis. Clin Nephrol. (1997) 48(1):9–15.
13. Lu P, Takai K, Weaver VM, Werb Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb Perspect Biol (2011) 3(12). doi: 10.1101/cshperspect.a005058
14. Adair-Kirk TL, Senior RM. Fragments of extracellular matrix as mediators of inflammation. Int J Biochem Cell Biol (2008) 40(6-7):1101–10. doi: 10.1016/j.biocel.2007.12.005
15. Ruffell B, Johnson P. Hyaluronan induces cell death in activated T cells through CD44. J Immunol (2008) 181(10):7044–54. doi: 10.4049/jimmunol.181.10.7044
16. Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, et al. Regulation of lung injury and repair by toll-like receptors and hyaluronan. Nat Med (2005) 11(11):1173–9. doi: 10.1038/nm1315
17. Taylor KR, Yamasaki K, Radek KA, Di Nardo A, Goodarzi H, Golenbock D, et al. Recognition of hyaluronan released in sterile injury involves a unique receptor complex dependent on toll-like receptor 4, CD44, and MD-2. J Biol Chem (2007) 282(25):18265–75. doi: 10.1074/jbc.M606352200
18. Termeer C, Benedix F, Sleeman J, Fieber C, Voith U, Ahrens T, et al. Oligosaccharides of hyaluronan activate dendritic cells via toll-like receptor 4. J Exp Med (2002) 195(1):99–111. doi: 10.1084/jem.20001858
19. Sokolowska M, Chen LY, Eberlein M, Martinez-Anton A, Liu Y, Alsaaty S, et al. Low molecular weight hyaluronan activates cytosolic phospholipase A2alpha and eicosanoid production in monocytes and macrophages. J Biol Chem (2014) 289(7):4470–88. doi: 10.1074/jbc.M113.515106
20. Dong Y, Arif A, Olsson M, Cali V, Hardman B, Dosanjh M, et al. Endotoxin free hyaluronan and hyaluronan fragments do not stimulate TNF-alpha, interleukin-12 or upregulate co-stimulatory molecules in dendritic cells or macrophages. Sci Rep (2016) 6:36928. doi: 10.1038/srep36928
21. Olsson M, Bremer L, Aulin C, Harris HE. Fragmented hyaluronan has no alarmin function assessed in arthritis synovial fibroblast and chondrocyte cultures. Innate Immun (2018) 24(2):131–41. doi: 10.1177/1753425918759590
22. Safrankova B, Hermannova M, Nesporova K, Velebny V, Kubala L. Absence of differences among low, middle, and high molecular weight hyaluronan in activating murine immune cells in vitro. Int J Biol Macromol (2018) 107(Pt A):1–8. doi: 10.1016/j.ijbiomac.2017.08.131
23. Niemietz I, Moraes AT, Sundqvist M, Brown KL. Hyaluronan primes the oxidative burst in human neutrophils. J Leukoc Biol (2020) 108(2):705–13. doi: 10.1002/JLB.3MA0220-216RR
24. Vogt KL, Summers C, Chilvers ER, Condliffe AM. Priming and de-priming of neutrophil responses in vitro and in vivo. Eur J Clin Invest (2018) 48 Suppl 2:e12967. doi: 10.1111/eci.12967
25. Miralda I, Uriarte SM, McLeish KR. Multiple phenotypic changes define neutrophil priming. Front Cell Infect Microbiol (2017) 7:217. doi: 10.3389/fcimb.2017.00217
26. Sundqvist M, Wekell P, Osla V, Bylund J, Christenson K, Savman K, et al. Increased intracellular oxygen radical production in neutrophils during febrile episodes of periodic fever, aphthous stomatitis, pharyngitis, and cervical adenitis syndrome. Arthritis Rheumatol (2013) 65(11):2971–83. doi: 10.1002/art.38134
27. Dahlgren C, Karlsson A. Respiratory burst in human neutrophils. J Immunol Methods (1999) 232(1-2):3–14. doi: 10.1016/S0022-1759(99)00146-5
28. Sarkar A, Hellberg L, Bhattacharyya A, Behnen M, Wang K, Lord JM, et al. Infection with anaplasma phagocytophilum activates the phosphatidylinositol 3-Kinase/Akt and NF-kappaB survival pathways in neutrophil granulocytes. Infect Immun (2012) 80(4):1615–23. doi: 10.1128/IAI.05219-11
29. El-Benna J, Dang PM, Gougerot-Pocidalo MA. Priming of the neutrophil NADPH oxidase activation: role of p47phox phosphorylation and NOX2 mobilization to the plasma membrane. Semin Immunopathol (2008) 30(3):279–89. doi: 10.1007/s00281-008-0118-3
30. Karlsson A. Wheat germ agglutinin induces NADPH-oxidase activity in human neutrophils by interaction with mobilizable receptors. Infect Immun (1999) 67(7):3461–8. doi: 10.1128/IAI.67.7.3461-3468.1999
31. Sundqvist M, Welin A, Elmwall J, Osla V, Nilsson UJ, Leffler H, et al. Galectin-3 type-c self-association on neutrophil surfaces; the carbohydrate recognition domain regulates cell function. J Leukoc Biol (2018) 103(2):341–53. doi: 10.1002/JLB.3A0317-110R
32. Almkvist J, Faldt J, Dahlgren C, Leffler H, Karlsson A. Lipopolysaccharide-induced gelatinase granule mobilization primes neutrophils for activation by galectin-3 and formylmethionyl-Leu-Phe. Infect Immun (2001) 69(2):832–7. doi: 10.1128/IAI.69.2.832-837.2001
33. Bass DA, Gerard C, Olbrantz P, Wilson J, McCall CE, McPhail LC. Priming of the respiratory burst of neutrophils by diacylglycerol. independence from activation or translocation of protein kinase c. J Biol Chem (1987) 262(14):6643–9.
34. Brown GE, Stewart MQ, Bissonnette SA, Elia AE, Wilker E, Yaffe MB. Distinct ligand-dependent roles for p38 MAPK in priming and activation of the neutrophil NADPH oxidase. J Biol Chem (2004) 279(26):27059–68. doi: 10.1074/jbc.M314258200
35. Karlsson A, Follin P, Leffler H, Dahlgren C. Galectin-3 activates the NADPH-oxidase in exudated but not peripheral blood neutrophils. Blood (1998) 91(9):3430–8. doi: 10.1182/blood.V91.9.3430
36. Belambri SA, Rolas L, Raad H, Hurtado-Nedelec M, Dang PM, El-Benna J. NADPH oxidase activation in neutrophils: Role of the phosphorylation of its subunits. Eur J Clin Invest. (2018) 48 Suppl 2:e12951. doi: 10.1111/eci.12951
37. Borregaard N, Sorensen OE, Theilgaard-Monch K. Neutrophil granules: a library of innate immunity proteins. Trends Immunol (2007) 28(8):340–5. doi: 10.1016/j.it.2007.06.002
38. Suzuki K, Hino M, Kutsuna H, Hato F, Sakamoto C, Takahashi T, et al. Selective activation of p38 mitogen-activated protein kinase cascade in human neutrophils stimulated by IL-1beta. J Immunol (2001) 167(10):5940–7. doi: 10.4049/jimmunol.167.10.5940
39. Mol S, Hafkamp FMJ, Varela L, Simkhada N, Taanman-Kueter EW, Tas SW, et al. Efficient neutrophil activation requires two simultaneous activating stimuli. Int J Mol Sci (2021) 22(18). doi: 10.3390/ijms221810106
40. Geering B, Gurzeler U, Federzoni E, Kaufmann T, Simon HU. A novel TNFR1-triggered apoptosis pathway mediated by class IA PI3Ks in neutrophils. Blood (2011) 117(22):5953–62. doi: 10.1182/blood-2010-11-322206
41. Lundqvist-Gustafsson H, Bengtsson T. Activation of the granule pool of the NADPH oxidase accelerates apoptosis in human neutrophils. J Leukoc Biol (1999) 65(2):196–204. doi: 10.1002/jlb.65.2.196
42. Kasahara Y, Iwai K, Yachie A, Ohta K, Konno A, Seki H, et al. Involvement of reactive oxygen intermediates in spontaneous and CD95 (Fas/APO-1)-mediated apoptosis of neutrophils. Blood (1997) 89(5):1748–53. doi: 10.1182/blood.V89.5.1748
43. van den Berg JM, Weyer S, Weening JJ, Roos D, Kuijpers TW. Divergent effects of tumor necrosis factor alpha on apoptosis of human neutrophils. J Leukoc Biol (2001) 69(3):467–73. doi: 10.1189/jlb.69.3.467
44. Cross A, Moots RJ, Edwards SW. The dual effects of TNFalpha on neutrophil apoptosis are mediated via differential effects on expression of mcl-1 and bfl-1. Blood (2008) 111(2):878–84. doi: 10.1182/blood-2007-05-087833
45. Ward RA, Nakamura M, McLeish KR. Priming of the neutrophil respiratory burst involves p38 mitogen-activated protein kinase-dependent exocytosis of flavocytochrome b558-containing granules. J Biol Chem (2000) 275(47):36713–9. doi: 10.1074/jbc.M003017200
46. Nguyen GT, Green ER, Mecsas J. Neutrophils to the ROScue: Mechanisms of NADPH oxidase activation and bacterial resistance. Front Cell Infect Microbiol (2017) 7:373. doi: 10.3389/fcimb.2017.00373
47. DeLeo FR, Renee J, McCormick S, Nakamura M, Apicella M, Weiss JP, et al. Neutrophils exposed to bacterial lipopolysaccharide upregulate NADPH oxidase assembly. J Clin Invest. (1998) 101(2):455–63. doi: 10.1172/JCI949
48. Tandon R, Sha'afi RI, Thrall RS. Neutrophil beta2-integrin upregulation is blocked by a p38 MAP kinase inhibitor. Biochem Biophys Res Commun (2000) 270(3):858–62. doi: 10.1006/bbrc.2000.2540
49. Potera RM, Jensen MJ, Hilkin BM, South GK, Hook JS, Gross EA, et al. Neutrophil azurophilic granule exocytosis is primed by TNF-alpha and partially regulated by NADPH oxidase. Innate Immun (2016) 22(8):635–46. doi: 10.1177/1753425916668980
50. Aoshiba K, Yasui S, Hayashi M, Tamaoki J, Nagai A. Role of p38-mitogen-activated protein kinase in spontaneous apoptosis of human neutrophils. J Immunol (1999) 162(3):1692–700. doi: 10.4049/jimmunol.162.3.1692
51. Grab J, Rybniker J. The expanding role of p38 mitogen-activated protein kinase in programmed host cell death. Microbiol Insights (2019) 12:1178636119864594. doi: 10.1177/1178636119864594
52. Glennon-Alty L, Moots RJ, Edwards SW, Wright HL. Type I interferon regulates cytokine-delayed neutrophil apoptosis, reactive oxygen species production and chemokine expression. Clin Exp Immunol (2021) 203(2):151–9. doi: 10.1111/cei.13525
53. Salamone G, Giordano M, Trevani AS, Gamberale R, Vermeulen M, Schettinni J, et al. Promotion of neutrophil apoptosis by TNF-alpha. J Immunol (2001) 166(5):3476–83. doi: 10.4049/jimmunol.166.5.3476
54. Fedorchenko O, Stiefelhagen M, Peer-Zada AA, Barthel R, Mayer P, Eckei L, et al. CD44 regulates the apoptotic response and promotes disease development in chronic lymphocytic leukemia. Blood (2013) 121(20):4126–36. doi: 10.1182/blood-2012-11-466250
55. Leu SW, Shi L, Xu C, Zhao Y, Liu B, Li Y, et al. TLR4 through IFN-beta promotes low molecular mass hyaluronan-induced neutrophil apoptosis. J Immunol (2011) 186(1):556–62. doi: 10.4049/jimmunol.1001630
56. Sheehan KM, DeLott LB, West RA, Bonnema JD, DeHeer DH. Hyaluronic acid of high molecular weight inhibits proliferation and induces cell death in U937 macrophage cells. Life Sci (2004) 75(26):3087–102. doi: 10.1016/j.lfs.2004.02.038
57. Alaniz L, Garcia MG, Gallo-Rodriguez C, Agusti R, Sterin-Speziale N, Hajos SE, et al. Hyaluronan oligosaccharides induce cell death through PI3-K/Akt pathway independently of NF-kappaB transcription factor. Glycobiology (2006) 16(5):359–67. doi: 10.1093/glycob/cwj085
58. Zhao H, Ma Y, Zhang L. Low-molecular-mass hyaluronan induces pulmonary inflammation by up-regulation of mcl-1 to inhibit neutrophil apoptosis via PI3K/Akt1 pathway. Immunology (2018) 155(3):387–95. doi: 10.1111/imm.12981
59. Francois S, El Benna J, Dang PM, Pedruzzi E, Gougerot-Pocidalo MA, Elbim C. Inhibition of neutrophil apoptosis by TLR agonists in whole blood: involvement of the phosphoinositide 3-kinase/Akt and NF-kappaB signaling pathways, leading to increased levels of mcl-1, A1, and phosphorylated bad. J Immunol (2005) 174(6):3633–42. doi: 10.4049/jimmunol.174.6.3633
60. Dang PM, Stensballe A, Boussetta T, Raad H, Dewas C, Kroviarski Y, et al. A specific p47phox -serine phosphorylated by convergent MAPKs mediates neutrophil NADPH oxidase priming at inflammatory sites. J Clin Invest. (2006) 116(7):2033–43. doi: 10.1172/JCI27544
61. Canovas B, Nebreda AR. Diversity and versatility of p38 kinase signalling in health and disease. Nat Rev Mol Cell Biol (2021) 22(5):346–66. doi: 10.1038/s41580-020-00322-w
62. Hofmann M, Zaper J, Bernd A, Bereiter-Hahn J, Kaufmann R, Kippenberger S. Mechanical pressure-induced phosphorylation of p38 mitogen-activated protein kinase in epithelial cells via src and protein kinase c. Biochem Biophys Res Commun (2004) 316(3):673–9. doi: 10.1016/j.bbrc.2004.02.101
63. Lu CH, Lin CH, Li KJ, Shen CY, Wu CH, Kuo YM, et al. Intermediate molecular mass hyaluronan and CD44 receptor interactions enhance neutrophil phagocytosis and IL-8 production via p38- and ERK1/2-MAPK signalling pathways. Inflammation (2017) 40(5):1782–93. doi: 10.1007/s10753-017-0622-5
64. El-Benna J, Hurtado-Nedelec M, Marzaioli V, Marie JC, Gougerot-Pocidalo MA, Dang PM. Priming of the neutrophil respiratory burst: role in host defense and inflammation. Immunol Rev (2016) 273(1):180–93. doi: 10.1111/imr.12447
65. El Benna J, Han J, Park JW, Schmid E, Ulevitch RJ, Babior BM. Activation of p38 in stimulated human neutrophils: phosphorylation of the oxidase component p47phox by p38 and ERK but not by JNK. Arch Biochem Biophys (1996) 334(2):395–400. doi: 10.1006/abbi.1996.0470
66. Seveau S, Eddy RJ, Maxfield FR, Pierini LM. Cytoskeleton-dependent membrane domain segregation during neutrophil polarization. Mol Biol Cell (2001) 12(11):3550–62. doi: 10.1091/mbc.12.11.3550
67. Aomatsu K, Kato T, Fujita H, Hato F, Oshitani N, Kamata N, et al. Toll-like receptor agonists stimulate human neutrophil migration via activation of mitogen-activated protein kinases. Immunology (2008) 123(2):171–80. doi: 10.1111/j.1365-2567.2007.02684.x
68. McDonald PP, Bald A, Cassatella MA. Activation of the NF-kappaB pathway by inflammatory stimuli in human neutrophils. Blood (1997) 89(9):3421–33. doi: 10.1182/blood.V89.9.3421
69. Tavianatou AG, Caon I, Franchi M, Piperigkou Z, Galesso D, Karamanos NK. Hyaluronan: molecular size-dependent signaling and biological functions in inflammation and cancer. FEBS J (2019) 286(15):2883–908. doi: 10.1111/febs.14777
70. Drewniak A, Tool AT, Geissler J, van Bruggen R, van den Berg TK, Kuijpers TW. Toll-like receptor-induced reactivity and strongly potentiated IL-8 production in granulocytes mobilized for transfusion purposes. Blood (2010) 115(22):4588–96. doi: 10.1182/blood-2009-11-253245
71. Sabroe I, Prince LR, Jones EC, Horsburgh MJ, Foster SJ, Vogel SN, et al. Selective roles for toll-like receptor (TLR)2 and TLR4 in the regulation of neutrophil activation and life span. J Immunol (2003) 170(10):5268–75. doi: 10.4049/jimmunol.170.10.5268
72. Redza-Dutordoir M, Averill-Bates DA. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim Biophys Acta (2016) 1863(12):2977–92. doi: 10.1016/j.bbamcr.2016.09.012
73. Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB. Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal (2014) 20(7):1126–67. doi: 10.1089/ars.2012.5149
74. Sies H, Jones DP. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat Rev Mol Cell Biol (2020) 21(7):363–83. doi: 10.1038/s41580-020-0230-3
75. Krizbai IA, Bauer H, Bresgen N, Eckl PM, Farkas A, Szatmari E, et al. Effect of oxidative stress on the junctional proteins of cultured cerebral endothelial cells. Cell Mol Neurobiol (2005) 25(1):129–39. doi: 10.1007/s10571-004-1378-7
76. DeMaio L, Rouhanizadeh M, Reddy S, Sevanian A, Hwang J, Hsiai TK. Oxidized phospholipids mediate occludin expression and phosphorylation in vascular endothelial cells. Am J Physiol Heart Circ Physiol (2006) 290(2):H674–83. doi: 10.1152/ajpheart.00554.2005
77. Huot J, Houle F, Marceau F, Landry J. Oxidative stress-induced actin reorganization mediated by the p38 mitogen-activated protein kinase/heat shock protein 27 pathway in vascular endothelial cells. Circ Res (1997) 80(3):383–92. doi: 10.1161/01.RES.80.3.383
78. Moldovan L, Moldovan NI, Sohn RH, Parikh SA, Goldschmidt-Clermont PJ. Redox changes of cultured endothelial cells and actin dynamics. Circ Res (2000) 86(5):549–57. doi: 10.1161/01.RES.86.5.549
79. Greenlee-Wacker MC. Clearance of apoptotic neutrophils and resolution of inflammation. Immunol Rev (2016) 273(1):357–70. doi: 10.1111/imr.12453
80. Chan DD, Xiao WF, Li J, de la Motte CA, Sandy JD, Plaas A. Deficiency of hyaluronan synthase 1 (Has1) results in chronic joint inflammation and widespread intra-articular fibrosis in a murine model of knee joint cartilage damage. Osteoarthritis Cartilage. (2015) 23(11):1879–89. doi: 10.1016/j.joca.2015.06.021
81. Bell TJ, Brand OJ, Morgan DJ, Salek-Ardakani S, Jagger C, Fujimori T, et al. Defective lung function following influenza virus is due to prolonged, reversible hyaluronan synthesis. Matrix Biol (2019) 80:14–28. doi: 10.1016/j.matbio.2018.06.006
82. Feldmann M, Pusey CD. Is there a role for TNF-alpha in anti-neutrophil cytoplasmic antibody-associated vasculitis? lessons from other chronic inflammatory diseases. J Am Soc Nephrol. (2006) 17(5):1243–52. doi: 10.1681/ASN.2005121359
83. Feldmann M, Maini RN. Lasker clinical medical research award. TNF defined as a therapeutic target for rheumatoid arthritis and other autoimmune diseases. Nat Med (2003) 9(10):1245–50. doi: 10.1038/nm939
Keywords: neutrophils, priming, hyaluronan, tumor necrosis factor-alpha, reactive oxygen species
Citation: Niemietz I and Brown KL (2023) Hyaluronan promotes intracellular ROS production and apoptosis in TNFα-stimulated neutrophils. Front. Immunol. 14:1032469. doi: 10.3389/fimmu.2023.1032469
Received: 30 August 2022; Accepted: 17 January 2023;
Published: 06 February 2023.
Edited by:
Eva Reali, University of Milano-Bicocca, ItalyReviewed by:
Manuela Viola, University of Insubria, ItalyCopyright © 2023 Niemietz and Brown. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kelly L. Brown, a2Jyb3duQGJjY2hyLmNh
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.