
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 09 February 2023
Sec. Vaccines and Molecular Therapeutics
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1030879
This article is part of the Research TopicInsights in Vaccines and Molecular Therapeutics: 2022View all 13 articles
 Ravi Krishnan1*†
Ravi Krishnan1*† Catherine J. M. Stapledon1Helen Mostafavi2,3,4Joseph R. Freitas2,3,4Xiang Liu2,3,4
Catherine J. M. Stapledon1Helen Mostafavi2,3,4Joseph R. Freitas2,3,4Xiang Liu2,3,4 Suresh Mahalingam2,3,4*†
Suresh Mahalingam2,3,4*† Ali Zaid2,3,4†
Ali Zaid2,3,4†Introduction: There is an unmet medical need for effective anti-inflammatory agents for the treatment of acute and post-acute lung inflammation caused by respiratory viruses. The semi-synthetic polysaccharide, Pentosan polysulfate sodium (PPS), an inhibitor of NF-kB activation, was investigated for its systemic and local anti-inflammatory effects in a mouse model of influenza virus A/PR8/1934 (PR8 strain) mediated infection.
Methods: Immunocompetent C57BL/6J mice were infected intranasally with a sublethal dose of PR8 and treated subcutaneously with 3 or 6 mg/kg PPS or vehicle. Disease was monitored and tissues were collected at the acute (8 days post-infection; dpi) or post-acute (21 dpi) phase of disease to assess the effect of PPS on PR8-induced pathology.
Results: In the acute phase of PR8 infection, PPS treatment was associated with a reduction in weight loss and improvement in oxygen saturation when compared to vehicle-treated mice. Associated with these clinical improvements, PPS treatment showed a significant retention in the numbers of protective SiglecF+ resident alveolar macrophages, despite uneventful changes in pulmonary leukocyte infiltrates assessed by flow cytometry. PPS treatment in PR8- infected mice showed significant reductions systemically but not locally of the inflammatory molecules, IL-6, IFN-g, TNF-a, IL-12p70 and CCL2. In the post-acute phase of infection, PPS demonstrated a reduction in the pulmonary fibrotic biomarkers, sICAM-1 and complement factor C5b9.
Discussion: The systemic and local anti-inflammatory actions of PPS may regulate acute and post-acute pulmonary inflammation and tissue remodeling mediated by PR8 infection, which warrants further investigation.
Acute respiratory virus infections are a significant global disease burden, as evidenced by the challenges posed by the recent SARS-CoV-2 pandemic and both previous and ongoing influenza virus endemic cycles (1–3). Severe respiratory illness caused by these viruses represents a major challenge in the clinic, and there is an unmet need for therapeutics that limit acute lung inflammation in patients by helping to manage pathology (4, 5). Mouse models of respiratory viral infection such as influenza virus and other viral and bacterial illnesses have been utilized to study the development and progression of acute lung inflammation, and to better understand how to modulate inflammatory mechanisms that exacerbate disease and lead to defective healing (6–8). PR8 is the most pathogenic species of influenza virus and comprises of a number of highly pathogenic pandemic strains, such as H1N1 and H3N2, which can cause severe illness in the elderly, children and immunocompromised individuals (9). In general, infection of airway epithelial cells results in robust pro-inflammatory cytokine production which culminates in severe inflammation, and subsequent respiratory failure (10, 11). A number of therapeutic targets have been investigated including signaling pathway inhibitors, chemokine receptor antagonists or inhibitors of neutrophil recruitment (10). Polysaccharide-based compounds have been previously shown to inhibit viral entry in vitro, in Respiratory syncytial virus (RSV) and SARS-CoV-2 infection (12, 13) however, there is limited information on the potential ability of PPS to ameliorate acute and/or chronic lung inflammation. Currently the management of influenza-induced illness is limited to yearly vaccination or administration of short-lasting anti-viral drugs that must be administered within 48 hours of symptom onset (14). Early corticosteroid administration is often advised in conditions such as acute respiratory distress syndrome (ARDS) to reduce lung tissue damage and mortality risk, however there is uncertainty surrounding the beneficial effects of treatment with corticosteroids in patients with severe influenza (15). ARDS is a form of severe respiratory failure where acute inflammation of the lungs results in damage of the alveolar capillary barrier, ultimately leading to leakage of oedema fluid and airway obstruction (16). Therefore, polysaccharide-based compounds that have been shown to exert some antiviral and/or anti-inflammatory functions against respiratory viruses may offer an additional treatment strategy for viral-induced ARDS.
Pentosan polysulfate sodium (PPS) is a semi-synthetic, linear polysaccharide derived from beechwood (17) that is highly sulphated. The oral formulation of PPS is an approved treatment for interstitial cystitis and bladder pain syndrome in Europe and the United States for over twenty years (18). The multiple mechanisms of action of PPS have provided the scientific rationale for repurposing the drug in other indications with unmet medical needs such as osteoarthritis (19) and alphavirus-induced arthralgia (20, 21). As PPS was demonstrated to be effective in a guinea pig model of allergen-induced rhinitis via its actions as an inhibitor of the Th2 cytokines IL-4, IL-5 and IL-13 (22), we extended our investigations to explore the effects of PPS in acute inflammation mediated by viral infection of the lung. The actions of PPS reported in other studies provided the rationale for its use in ARDS, which involved anti-inflammatory actions mediated via inhibition of NF-κB activation (23–26); inhibition of complement (C3, C3Bb and C5b9) mediated tissue injury (27); inhibition of endothelial cell activation (28); and adverse tissue remodeling by inhibiting fibroblast growth factor-2 (FGF-2) (29) and matrix metalloproteinases (MMPs), ADAMTS-4 (30–32) and elastase (33).
Based on the reported mechanisms of action of PPS, this study aimed to investigate the specific effects of subcutaneous (SC) administration of PPS on the clinical symptoms of IAV infection including weight loss & oxygen saturation and lung pathological changes associated with inflammatory cellular infiltrates and fibrosis in a sublethal PR8 mouse model using the mouse adapted H1N1 pandemic A/PR8/1934 (termed PR8) strain. In addition, the effects of PPS on inflammatory biomarkers (cytokine, chemokine and complement profiles) locally within the lungs and systemically during the acute and post-acute disease phases of PR8 infection were also investigated. The aim of this study was to evaluate the anti-inflammatory and tissue preservation properties (tissue remodeling) of PPS in a mouse model of influenza infection, and to determine the use of PPS as a potential therapeutic in both acute and post-acute phase respiratory viral infections.
6–8-week-old C57BL/6J mice (purchased from Animal Resources Centre, Perth, WA) were infected intranasally with a sublethal dose of Influenza A virus (PR8) A/Puerto Rico/8/1934 (kindly donated by Prof. Carl Feng, The University of Sydney) inoculum at 300 plaque forming unit (pfu) equivalent, as determined by antibody-based endpoint dilution assay (EPDA). Mice were weighed and anesthetized with a mixture of Ketamine (80 mg/kg) and Xylazine (8 mg/kg), administered intraperitoneally in a volume of 200 μL sterile Phosphate Buffered Saline (PBS; Gibco). Mice were administered 50 μL viral inoculum (stock diluted in sterile PBS) evenly through both nostrils.
Mice were weighed and monitored daily for signs of disease and weight loss. Weight loss was calculated as a percentage of weight change from the starting weight for each individual mouse. In accordance with AEC guidelines (Griffith University AEC# MHIQ/06/20), mice that displayed early signs of disease, including ruffled fur and lethargy, in addition to weight loss greater than 10%, were provided with wet food in the cage and hydration gel packs.
Virus stock and tissue homogenates were titrated via EPDA. Vero E6 cells were seeded in tissue culture-treated 96-well plates until they reached 85% confluency, and serial dilutions (from neat/undiluted to 10-8) of virus inoculum were prepared in serum-free MEM. Virus dilutions were applied to the VeroE6 cell monolayer and cells were incubated for 3 days at 37°C (5% CO2). Following incubation, supernatant was removed and cells were fixed with 4% paraformaldehyde (PFA; v/v in PBS) for 5 minutes at room temperature (RT). PFA was washed off with PBS twice. Cells were permeabilized and blocked with 1% bovine serum albumin (BSA; w/v in PBS) with 0.05% Triton X-100 (v/v in PBS) for 30 minutes at RT. Blocking/permeabilising solution was removed and mouse anti-N nucleocapsid antibody (conjugated to FITC; Clone D67J; Cat#MA1-7322; Thermo Fisher Scientific) was added to the wells at a dilution of 1:2000 for 45 minutes. Cells were washed with PBS, and Hoechst 33258 (Cat#H1398; Thermo Fisher Scientific) (1:5000 of a 10 mg/mL stock in PBS) was added to the cells for 5 minutes at RT. Cells were washed and wells visualized on a Nikon Ti-E epifluorescence microscope (Nikon, Coherent Scientific, Australia) to determine the highest dilution where FITC-positive cells were present. Titer was determined as a tissue culture infectious dose titer (TCID), normalized to mL volume (and grams of tissue, where applicable).
PPS solution was prepared fresh for each daily injection. The powdered API (bene pharmaChem GmbH & Co. KG, Germany) was weighed and taken up in 1 mL sterile PBS (1X) and solution was mixed and filtered using a 0.22 µm PVDF syringe filter. PPS was prepared at the indicated concentration (3 mg/kg or 6 mg/kg) after weighing mice and administered individually in a 200 µL volume. PPS was administered daily as indicated (from Day 0, Day 2 or Day 5) either subcutaneously in the scruff of the neck, or intraperitoneally. Vehicle (1X PBS) was administered SC or IP in a 200 µL volume as indicated.
Tissues were collected and placed in 500 µL cold sterile PBS in pre-weighed 2 mL SafeLock homogenization tubes (Eppendorf, Hamburg, Germany) with 6 mm sterile steel beads. Tissues were weighed and frozen at -80°C. Shortly before homogenization, tubes were partially thawed at room temperature (RT) and homogenized using a QIAGen TissueLyzer II (QIAGen) for 30 seconds at 30 repetitions per second (rps). Tubes were centrifuged at 8,000 x g for 5 minutes at 4°C and supernatants collected to perform serial dilutions used for viral titrations or tissue cytokine quantification.
Lungs were collected from mice after euthanasia and placed 4% PFA (v/v in PBS) for 24 hours on a nutator at 4°C. Tissues were transferred to PBS for washing for 24 hours on a nutator at 4°C twice. Tissues were then transferred to histology cassettes and placed in 80% ethanol. Tissues were processed for paraffin embedding and non-consecutive 5 µm sections (25-30 µm apart starting at ~500 µm depth) were performed and stained with either haematoxylin and eosin (H&E) stain for cell infiltration or Masson Trichrome (MT) stain for detection of collagen. Slides were scanned using a Leica Aperio AT2 (Leica Biosystems) and images processed using QuPath software (21). For H&E-stained slides, nuclei were detected as individual objects using NucleiCount v9 algorithm on QuPath. Values generated from technical replicates of lung sections across three non-consecutive sections (25-30 µm apart) were pooled in the final analysis and graphed on GraphPad Prism (9.0). For the quantification of collagen positive fibers as an indication of tissue remodeling, a custom-designed script was devised to detect collagen fibers and quantified as a percentage of collagen-positive surface area relative to section surface area. Objects pertaining to lobes or sections of lobes were assigned using the “Simple Tissue Detection” macro, and small or torn/smeared sections were manually excluded. In addition, objects that included collagen-rich tissues such as airway, parts of trachea, bronchia and septum were manually excluded, or excised from pre-assigned objects. Using QuPath pixel classifier, specific annotations were assigned to collagen-rich structures within selected objects using the Wand tool. Non-collagen pixel objects were assigned specific annotations, and background was assigned as either “Negative” or “Ignore” annotation. Using the “Train Pixel Classifier” algorithm, the detection of blue, collagen-denoting pixels was verified on “Normal Resolution”. Pixel classifier algorithm was further trained to eliminate false or approximate collagen/blue pixel detection (5-7 iterations). A final validation was performed using “High” or “Very high” resolution, and classifier further applied to a random selection of images (2, 3). Upon final validation, the algorithm was input into a Script Editor to run multi-file batches.
Additional assessment of cellular inflammation (H&E-stained slides) and tissue remodeling-associated collagen (Masson Trichrome-stained slides) and fibrosis-associated morphological changes was performed by an expert histopathologist who was blinded to the identity of the sections. A validated pathology scoring system (22, 23) was used to assign scores according to a scale from 0 to 5 where: 0 = no change; 1 = minimal change (or possibly non-specific background or appearance compounded by lung collapse, i.e., not inflated); 2 = mild change; 3 = moderate change; 4 = severe change in <50% of lung lobe; 5 = severe change in >50% of lung lobe. Data was de-identified and transferred to GraphPad Prism (9.0) for plotting and statistical analysis. Technical replicates of lung sections across three non-consecutive sections (25-30 µm apart) were pooled in the final analysis and graphed on GraphPad Prism (9.0).
SpO2 measurements were performed using a StarrLife MouseOx sensor pulse oximetry measurement instrument. Briefly, mice were anesthetized using 1-2% isoflurane (in a nebulized air mixture) and hind legs shaved and depilated using a depilatory cream (Veet). Pulse oximetry was measured using a thigh sensor, and values were recorded for each mouse.
Lung tissues were harvested and fixed in 4% paraformaldehyde overnight. The tissues were subsequently washed in PBS and dehydrated in in 30% sucrose overnight. The tissues were embedded in optimal-cutting-temperature (OCT) compound (Sakura Finetek) and then frozen at -80°C. Tissue blocks were cut into 20 μm-thick sections and permeabilized in cold acetone. The sections were blocked with serum-free protein block (DAKO) and immunolabelled with antibodies against mouse CD169 (AbD Serotec), Gr-1 (BD Biosciences), and Collagen Type IV (Abcam). Nuclei were stained with 0.025 mg/ml Hoechst33258 (Thermo Fisher Scientific). The slides were mounted with ProLong Gold antifade (Thermo Fisher Scientific). Images were acquired using a confocal microscope (Olympus FV3000) using a 20x (0.95 NA) objective and processed using Imaris (v9.5; Bitplane).
Lungs were processed for flow cytometry analysis using a protocol adapted from Liu et al. (24), shown in Supplemental Figure 3. Briefly, mice were euthanized by ketamine/xylazine overdose, and perfused intracardially with ice-cold 1x Phosphate Buffered Saline (PBS; Gibco). Experimental procedures were performed under aseptic conditions and within a BSC-II cabinet. Lungs (left lobe) were collected in ice-cold PBS until processing for flow cytometry. Lungs were cut into small pieces using sterile scissors in 1 mL Digestion Solution (3mg/mL Type IV Collagenase [Worthington Co.] with 10 µg/mL DNAse I [Sigma Aldrich] in RPMI [Gibco]). Dissected lungs were then added to 2mL digestion solution and incubated for 1 hour at 37°C. Samples were added to 3 mL RPMI + 10% FCS and pipetted vigorously for 10 seconds using a Pasteur pipette, then filtered through a 70 µm cell strainer. The strainer was further rinsed with 5 mL RPMI, and samples centrifuged at 375 x g for 5 minutes at 4°C. Supernatant was decanted and pellets were resuspended in ACK lysis buffer (0.15M NH4Cl; 0.01M KHCO3; 0.1mM Na2EDTA; pH 7.2) for 5 minutes at RT to lyse red blood cells. Lysis was stopped with 10 mL PBS, followed by centrifugation at 375 x g for 5 minutes at 4°C. Single cells were resuspended in FACS buffer (PBS with 1% bovine serum albumin [BSA; Sigma Aldrich] and 0.5 mM EDTA) and placed on ice. For antibody staining, cells were first incubated with anti-mouse CD16/32 (Fc block; BD Biosciences) antibody for 15 minutes at 4°C before addition of fluorochrome-conjugated antibodies. Cells were immunolabelled with the following anti-mouse antibodies: CD45-BUV805 (BD Biosciences), CD11b-BV711 (BD Biosciences), CD64-BV650 (BD Biosciences), CD8-AlexaFluor594 (Biolegend), CD4-BV510 (BD Biosciences), CD3-APC (eBiosciences), SiglecF-PE (eBiosciences), Ly6C-PeCy7 (eBiosciences), Ly6G-BV786 (BD Biosciences), CCR2-BUV496 (BD Biosciences) and LIVE/DEAD NIR (Thermo Fisher Scientific) cell viability dye. Antibody staining was performed for 40 minutes in the dark at 4°C and cells were subsequently washed with ice-cold FACS buffer. Cells were fixed in 4% paraformaldehyde (v/v in PBS) for 5 min at 4°C, washed twice with FACS buffer, filtered through a 30 µm cell strainer and resuspended in 200 µL PBS. Counting beads (Sphero Calibration beads, BD Biosciences) were added to the samples before acquisition on a BD LSR Fortessa X20 flow cytometer, and data analyzed using FlowJo v10.7 (TreeStar Inc.).
Mice were euthanized by lethal overdose with ketamine/xylazine prior to cervical dislocation. The thoracic cage was dissected, and mice were exsanguinated via an excision of the right atrium. Lungs were collected in pre-weighed 2 mL homogenization tubes containing 500 µL ice-cold PBS and a 6 mm diameter stainless steel bead (QIAGEN; Cat#69989). Samples were kept on ice during the procedure, weighed and transferred to -80°C for storage. Prior to homogenization, samples were allowed to partially thaw at 4°C for 2 hours. Samples were loaded on a TissueLyzer II (QIAGEN) and homogenized for 30 seconds at 20 repetitions per second. Samples were then centrifuged at 5000 x g for 5 minutes at 4°C, and supernatant collected for either viral titration (as per plaque assay protocol) or cytokine/complement protein analysis. Cytokine analysis was performed using a multiplex cytokine bead array (CBA) Mouse Inflammation kit (BD Biosciences; Cat#552364) which enables simultaneous detection of pro-inflammatory cytokines CCL2, IL-6, IFN-γ, IL-12p70, IL-10 and TNF-α. Samples were processed as per manufacturer’s instructions and acquired on a BD LSR Fortessa flow cytometer. Data was exported and analyzed using the FCAP Array v3.0 software (BD Biosciences). Complement proteins were quantified using single ELISA kits (Novus Biologicals Mouse Complement C3a ELISA kit, Cat#NBP2-70037; Thermo Fisher Mouse Complement C5a ELISA kit, Cat#EHMC; Hycult Biotech Mouse Complement C3b ELISA kit, Cat#HK216-01; Biomatik mouse Terminal Complement Complex C5b9 (Cat#EKU10320); Cusabio mouse Factor B (Complement Factor Bb; CF-Bb) Cat#CSB-E04695m); R&D Systems mouse soluble ICAM-1 (sICAM-1) Quantikine Cat#MIC100) according to manufacturer’s instructions. Optical density values (450nm and 570nm) were read on a POLARStar Omega plate reader (BMG Labtech) and data exported to .csv files using the manufacturer’s proprietary software.
Statistical analyses were performed using GraphPad Prism v9. Comparisons between groups were performed using a one-way ANOVA with a Kruskal-Wallis test, and two-variable comparisons were performed using a two-way ANOVA. Datasets were assessed for normality (normal or non-normal distribution), and statistical tests were applied accordingly. Where applicable, an outlier test (ROUT; Q=5%) was performed to exclude outliers from the data set. Statistically significant differences are shown on plots as numerical p values (adjusted) or using asterisks (i.e., ns. Not significant; *p<0.05; **p<0.01; ***p<0.005; ****p<0.001).
Sub-lethal infection with the PR8 strain of influenza A virus causes ARDS in immunocompetent C57BL/6J mice, which is characterized by sharp weight loss generally observed between 5- and 7-days post-infection (dpi) (7, 8). To determine whether PPS was effective in reducing PR8-induced disease severity, mice were treated with either 3 or 6 mg/kg PPS subcutaneously (SC) at 2 hours post-PR8 infection with a sublethal dose [300 plaque forming units (pfu)] of intranasal PR8. For our acute phase study, vehicle (PBS), and PPS were administered every 24 hours from 0- 7 dpi. For our subsequent post-acute phase study, we monitored mice until 21 dpi. Mice infected with PR8 and administered vehicle (SC) displayed severe disease, as evidenced by the sharp weight loss starting from 6 dpi, reaching 10-15% weight loss from starting bodyweight (Figure 1A). Weight loss in mice treated with 3 mg/kg SC PPS was significantly reduced compared to that observed in vehicle-treated mice at 7 and 8 dpi (p<0.05), with the average weight loss approaching the 15% endpoint threshold at 8 dpi. In contrast, mice treated with 6 mg/kg SC PPS displayed less pronounced weight loss at 7 and 8 dpi compared to mice treated with 3 mg/kg PPS (Figure 1A). Based on previous reports of systemic effect of PPS (24), we also investigated the effect of PPS administered intraperitoneally (IP) in mice infected with PR8. Intraperitoneal administration of PPS led to a statistically significant reduction in weight loss at 6 (p<0.01) and 7 dpi, (p<0.005), but not 8 dpi (p=0.0809), in PR8-infected mice when compared to vehicle-treated mice (Supplemental Figure 1). As the reduction in weight loss in the IP group was not as pronounced as what was observed in the SC group, the subcutaneous route was chosen for all subsequent experiments.
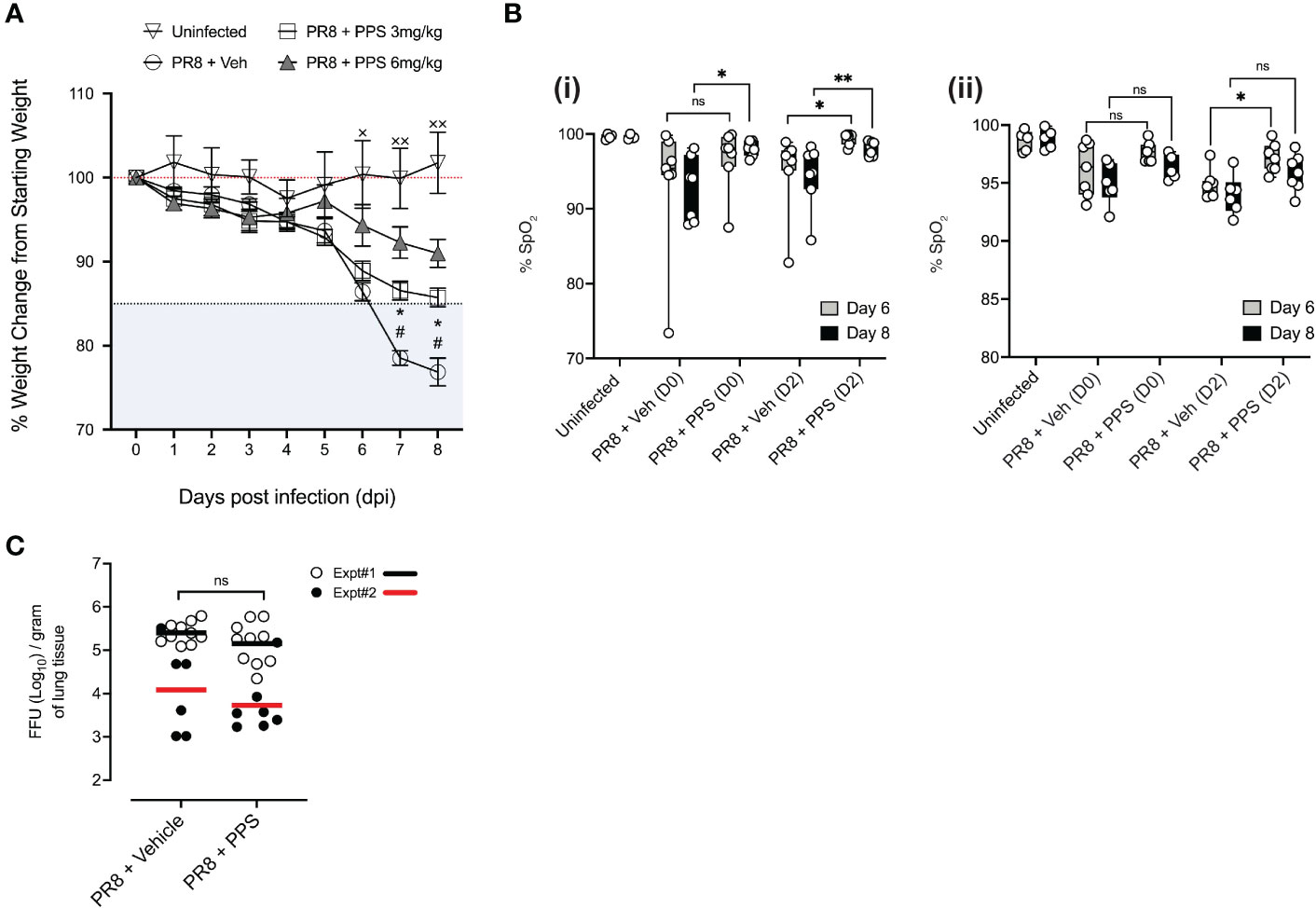
Figure 1 Effects of daily PPS treatment on sublethal PR8-induced disease assessments. Mice were treated daily with 3 mg/kg or 6 mg/kg PPS, or vehicle, starting from Day 0 to Day 7 post-infection. (A) Weight loss in uninfected mice, PR8-infected mice treated with 3 mg/kg or 6 mg/kg PPS or with vehicle SC #p<0.05 for PR8 + Veh compared with PR8 + PPS 3 mg/kg; *p<0.05 for PR8 + Veh compared with PR8 + PPS 6 mg/kg; ×p<0.05, ××p<0.01 for PR8 + Veh compared with Uninfected. Statistically significant differences were assessed by two-way ANOVA. (B) Pulse oximetry was measured at Day 6 and Day 8 post-infection across two independent experiments, shown in (i) and (ii). Mice were infected with PR8 and treated with PPS 3 mg/kg at Day 0 or Day 2 post-infection. Statistically significant differences were assessed by one-way ANOVA; p values shown on plots; *p<0.05; **p<0.01; ns, not significant. ((i) and (ii) n=7 mice per group; n=4 mice in the uninfected group). (C) Effect of PPS treatment on viral titres in the lungs of mice infected with PR8. PR8-infected mice treated with vehicle SC (PR8 + Vehicle), PR8-infected mice treated with PPS SC (PR8 + PPS); n=7-10 mice per group. Data shown from two independent experiments, denoted Expt#1 and Expt#2. Mean values for each experiment depicted by black (Expt#1) and red (Expt#2) lines on plots. Statistically significant differences were assessed on pooled data by student’s t-test; p values shown on plots. ns: not significant.
To better understand the physiological effect of PPS in the context of acute respiratory disease in PR8-infected mice, we measured blood oxygen saturation (SpO2) at 6 and 8 dpi in mice treated with 3 mg/kg PPS, across two independent experiments and tested whether the delayed administration of 3 mg/kg PPS, from 2 dpi, may exert a therapeutic effect in infected mice (Figure 1B; i and ii). In the first experiment (Figure 1B; i), infected mice treated with PPS from Day 0 demonstrated significantly improved oxygen saturation at 8 dpi (Figure 1B; i). Although the differences at 6 dpi were not statistically significant, SpO2 was higher in PPS-treated mice compared to vehicle-treated mice (Figure 1B; i). Mice treated with PPS from Day 2 showed a statistically significant increase in SpO2 at both 6 and 8 dpi, when compared with infected mice administered vehicle. In the repeat experiment (Figure 1B; ii), only mice treated with PPS from Day 2 demonstrated a significant increase in SpO2 when compared to vehicle-treated mice at 6 dpi. While not statistically significant, there was an improvement in SpO2 in mice treated with PPS from Day 2 compared with mice treated with vehicle (Figure 1B; ii).
Reducing tissue inflammation during virus-induced disease without enhancing viral replication is an important characteristic of any immune-modulatory compound in the context of pre-clinical assessment. To investigate the effects of PR8 infection and PPS as a treatment option, lung viral titers at 8 dpi were measured. Over the course of two independent experiments (Expt#1 and Expt#2), there were no statistically significant differences in viral titers between the lungs of mice treated with 3 mg/kg PPS and those that received vehicle (Figure 1C), indicating that reduced disease severity seen in PPS-treated mice at 8 dpi is not associated with a reduction in viral titers, nor does it confer an advantage for viral replication.
As the data suggested that PPS treatment could dampen severe PR8-induced disease without affecting lung viral titers, the impact of PPS on lung infiltration and tissue morphology was investigated histologically via haematoxylin & eosin (H&E) staining of lung sections (Figure 2A). Lung alveolar spacing appeared reduced in PR8-infected, vehicle-treated mice in both Day 0 and Day 2 treatment groups, and consolidation of the parenchyma - a pathological feature of respiratory infections that leads to an increase in density of the lung parenchyma (34) - was increased compared to PPS-treated mice. Upon further inspection, alveolar density appeared less pronounced in the Day 2 PPS cohort when compared to the Day 0 cohort. We did not observe any statistically significant reduction (p=0.4708) in leukocyte infiltrates in the lungs of mice treated from Day 0 with 3 mg/kg SC PPS compared to vehicle-treated mice, but PPS administered from Day 2 was associated with a significant reduction (p=0.0445) in infiltrates compared to infected control mice (Figure 2B). Quantitation of lung sections from uninfected control mice (uninfected + vehicle and uninfected + PPS) showed counts similar to those observed in uninfected, untreated mice (Figure 2B).
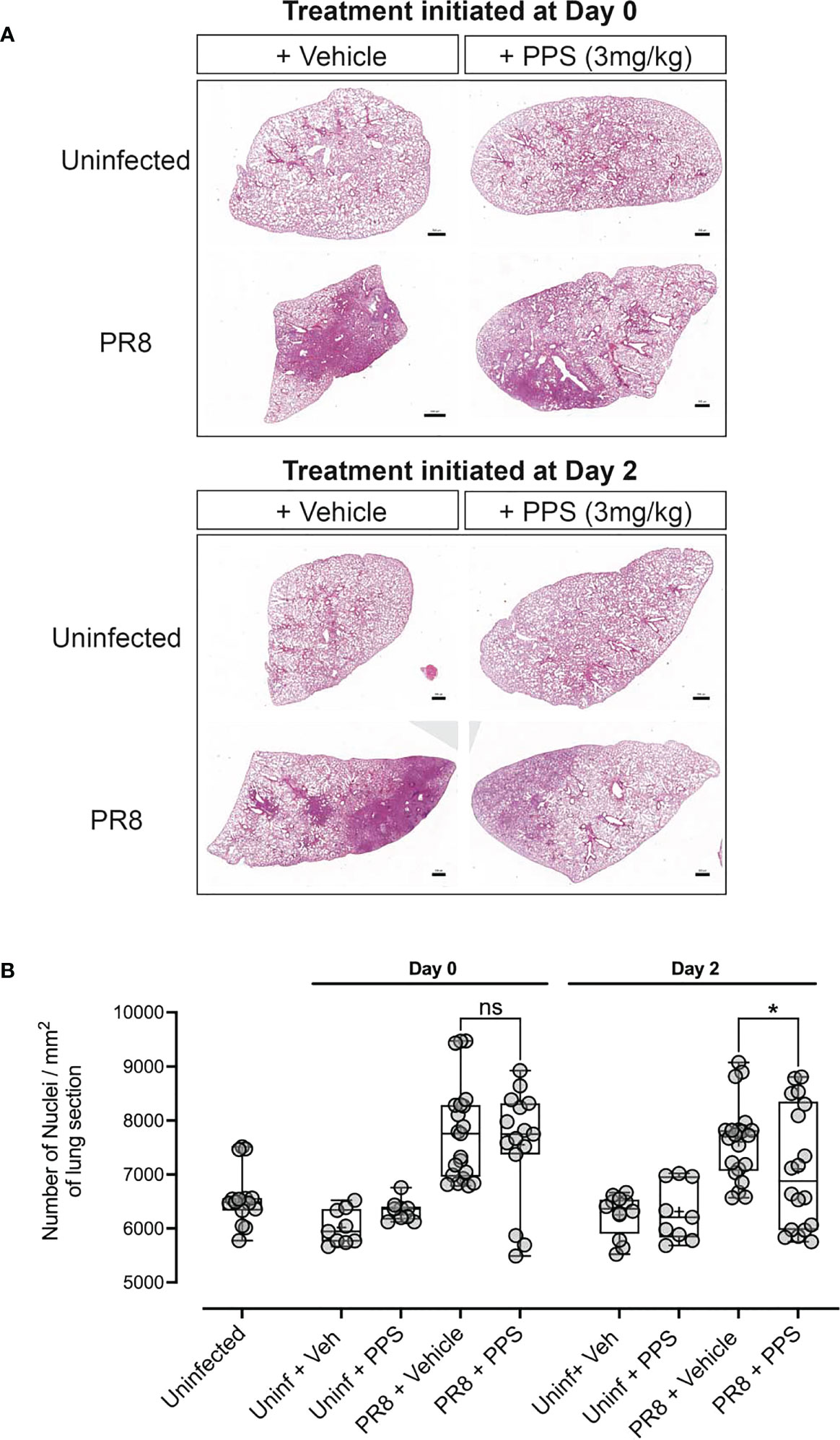
Figure 2 Histological analysis of lungs in PR8-infected mice treated with PPS. (A) H&E staining of lung sections and nuclei counts from uninfected mice and mice infected with PR8 and treated with 3 mg/kg PPS from Day 0 or Day 2 post-infection. (B) Quantification of cellular infiltrates in the lungs [from data shown in (A)] of PR8-infected mice treated with 3 mg/kg PPS. n=7 for all PR8 groups, n=3 for Uninfected + PPS, n=3 for Uninfected + Veh, n=5 for Uninfected). Each data point represents nuclei counts calculated on individually detected objects (using automated software object detection) in triplicate sections for each group. Statistically significant differences were assessed by one-way ANOVA with a Sidak post-test. *p<0.05 shown in plots. (ns, not significant). Box-whiskers plot show all data points (max/min), standard error, median (line in box) and mean (‘+’ in box). Each data point represents one pathological score assigned to a lobe assessed in triplicate sections for each group.
Next, the effect of PPS treatment on immune cell infiltration in the lungs of PR8-infected mice was assessed by flow cytometry. Data in Figure 1A indicated that while mice treated with 3 mg/kg PPS showed reduced weight loss compared to vehicle-treated mice, this effect was enhanced in mice treated with 6 mg/kg PPS, pointing towards a potential dose-dependent effect of PPS in the context of acute PR8-induced disease. To determine whether this effect could be explained by a difference in host immune responses, lungs were collected at 8 dpi and processed for immune cell analysis. The number of CD45+ cells was significantly higher in the lungs of PR8-infected mice compared to uninfected mice, but there were no statistically significant differences in the overall number of CD45+ cells between PR8-infected vehicle-treated mice and mice treated with SC PPS at either 3 or 6 mg/kg (Figure 3A). While infection with PR8 was associated with an increase in the number of CD4+ and CD8+ T cells in the lungs, there were no statistically significant differences between PR8-infected mice treated with vehicle and mice treated with PPS at either 3 or 6 mg/kg (Figure 3B). While absolute CD4+ T cell numbers in the lungs were not significantly different in the lungs of PPS-treated mice, we observed that the percentage of CD4+ T cells (of all CD45+ cells) was significantly higher in the lungs of mice treated with 3 mg/kg PPS compared with PR8-infected mice that received vehicle (Figure 3C). Despite not being statistically significant, PPS-treated mice (6 mg/kg) exhibited a higher percentage of CD4+ T cells than mice treated with vehicle. (Figure 3C).
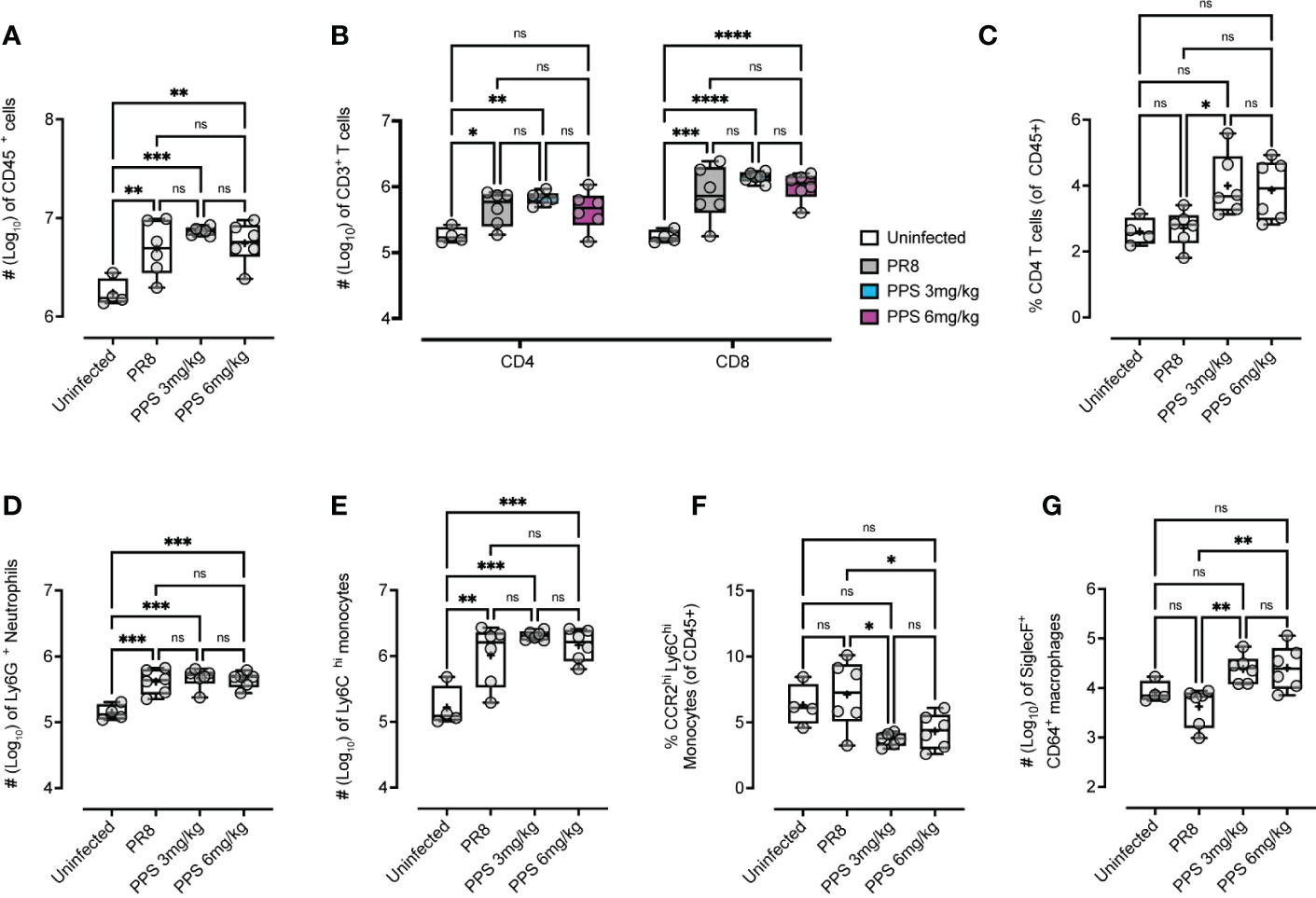
Figure 3 Flow cytometry analysis of changes in the number of cellular infiltrates in the lungs of mice infected with PR8 and treated with PPS (3 mg/kg and 6 mg/kg, SC). Enumeration of (A) total live CD45+ cells, (B) CD3+ CD4+ and CD8+ T cells, (C) the percentage of CD4+ T cells (of total CD45+ leukocytes), (D) Ly6G+ neutrophils, (E) Ly6ChiCD11b+ inflammatory monocytes (IM), (F) the percentage of CCR2hi inflammatory monocytes cells (of total CD45+ leukocytes) and (G) SiglecF+ CD64+ alveolar macrophages. Data shown as either Log10 number of cells (normalized to tissue weights) or % of cell subset of total CD45+ cells. Statistically significant differences were assessed by one-way ANOVA with a Tukey post-test (ns, not significant; *p<0.05; **p<0.01; ***p<0.005; ****p<0.001). Box-whiskers plot show all data points (max/min), standard error, median (line in box) and mean (‘+’ in box). Sample size: n=6 mice per group; n=4 mice for uninfected group.
In the myeloid compartment, there were no significant differences in the number of Ly6G+ neutrophils (Figure 3D), or Ly6Chi inflammatory monocytes (Figure 3E) between vehicle and PPS-treated groups, however, within CD45+ immune cells the subset of CCR2hi inflammatory monocytes was significantly reduced in the lungs of mice treated with PPS at both 3 and 6 mg/kg, when compared to vehicle-treated mice (Figure 3F). Interestingly, the number of SiglecF+ alveolar macrophages, a subset of lung-resident macrophages implicated in local immune responses against respiratory pathogens, and associated with protection against viral infection, was significantly higher in the lungs of mice treated with PPS at both 3 and 6 mg/kg, compared to vehicle-treated mice (Figure 3G). This was also observed in situ using confocal immunofluorescence microscopy of lung cryosections (Figure 4) collected at 8 dpi, which outlines the distribution of Gr-1-expressing monocytes and neutrophils, and CD169+ lung-resident alveolar macrophages in the lung tissue. Alveolar spacing in uninfected mice is characterized by low-density parenchyma, and sparse distribution of Gr-1+ and CD169+ cells. In contrast, PR8-infected lungs of mice treated with vehicle display areas of consolidation with dense clusters of immune cells, characteristic of large infiltration of circulating cells (neutrophils, monocytes) and alveolar macrophages. Interestingly, we observed a broader distribution of CD169+ alveolar macrophages - and conversely, a more restricted distribution of Gr-1+ cells across the parenchyma at 8 dpi in mice treated with PPS compared to vehicle-treated mice (Figure 4).
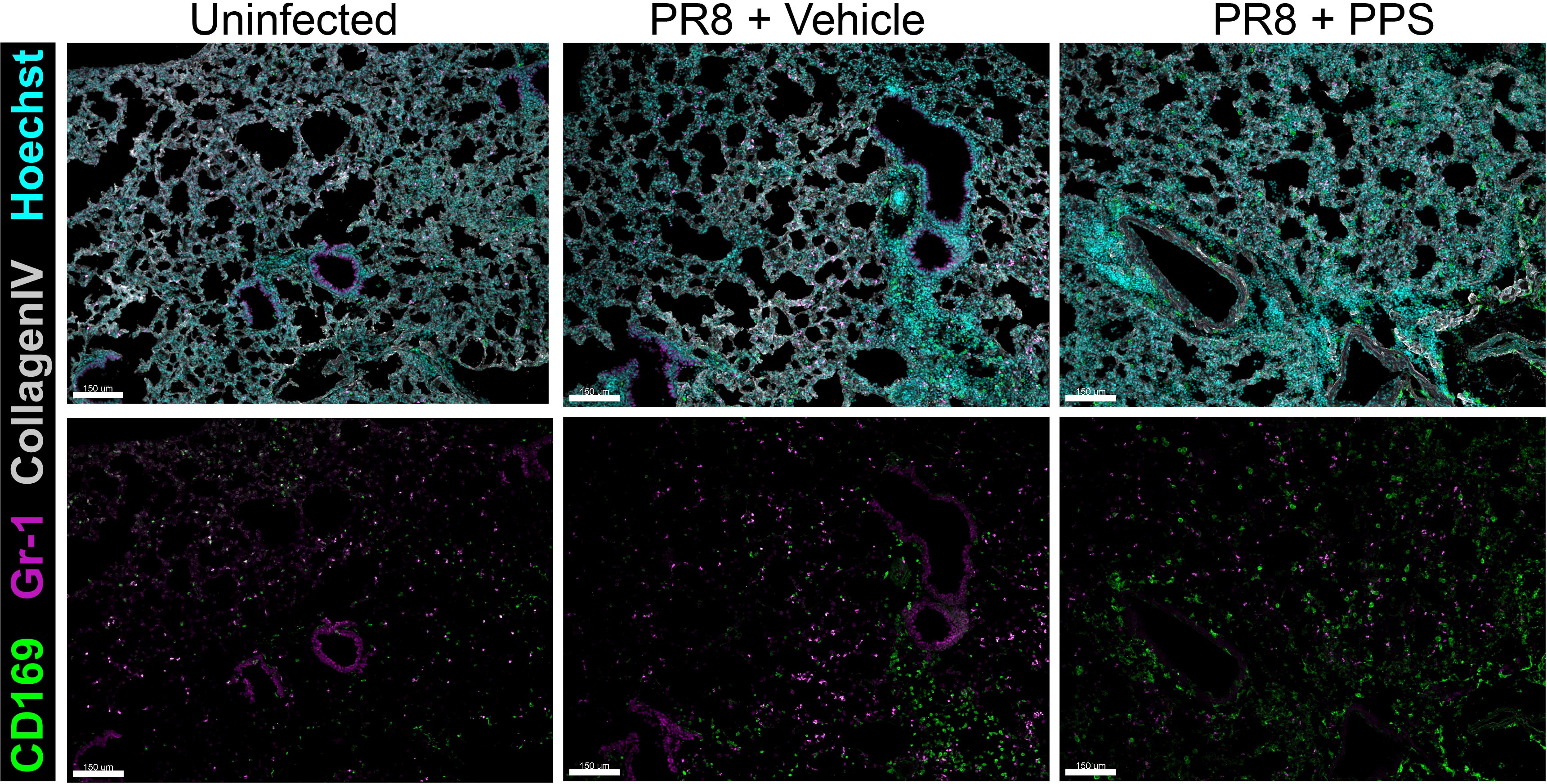
Figure 4 Immunofluorescence microscopy of lungs in PR8-infected mice treated with 6 mg/kg SC PPS. Representative confocal immunofluorescence micrographs of lung cryosections from uninfected mice, PR8-infected mice (vehicle-treated; PR8 + Vehicle) and PR8-infected mice treated with 6 mg/kg PPS (PR8 + PPS). Sections were immunolabelled with anti-mouse CD169, anti-mouse Gr-1 and anti-mouse Collagen Type IV. Nuclei were labelled using Hoechst (33258) and images acquired as z-stacks by confocal microscopy. Images shown as maximum intensity projections. Upper panels show merged channels. Scale bars = 150 μm.
PPS has been shown to interfere with components of the complement pathway, including C3a and C5a (27, 33), which are likely to contribute to airway inflammation in acute PR8 infection (29). Therefore, we determined lung and serum levels of C3a, C3b and C5a by ELISA at 8 dpi in PR8-infected mice treated with vehicle, 3 or 6 mg/kg PPS. Across three independent experiments, we observed variable outcomes in both serum and lungs. While one out of three experiments showed that lung C3a levels were significantly reduced in mice treated with 6 mg/kg PPS (Figure 5A), this difference was not reflected in the serum of mice from the same experiment (Figure 5B). Likewise, we did not observe any statistically significant differences in C3b in either the lungs (Figure 5C) or serum (Figure 5D), as was the case for lung (Figure 5E) and serum (Figure 5F) C5a levels between vehicle- and PPS-treated groups. Lung and serum levels of pro-inflammatory cytokines and chemokines (CCL2, IL-6, IL-10, IL-12p70, IFN-γ and TNF-α) were also measured in three independent experiments (Figure 6). In one experiments (Experiment 1, shown in Figure 6), serum levels of CCL2 were significantly reduced in PPS-treated mice (3 and 6 mg/kg), as were serum levels of IL-6 (6 mg/kg), IFN-γ (6 mg/kg) and IL-12p70 (6 mg/kg) compared with vehicle-treated mice (Figure 6). It is worth noting that Experiment 1 showed a more relevant baseline as cytokine levels in the control (uninfected) group were lower compared to those detected in infected groups, whereas this baseline cytokine readout in control mice was higher in Experiments 2 and 3 (Expt#2 and Expt#3; shown in Supplemental Figure 2). Overall, except for the significant reduction in IL-12p70 (Supplemental Figure 2; Expt#3) in the lungs of mice treated with 3 and 6 mg/kg PPS, we did not observe any significant differences in lung tissue cytokine/chemokine concentration following PPS treatment compared to PR8-infected mice treated with vehicle.
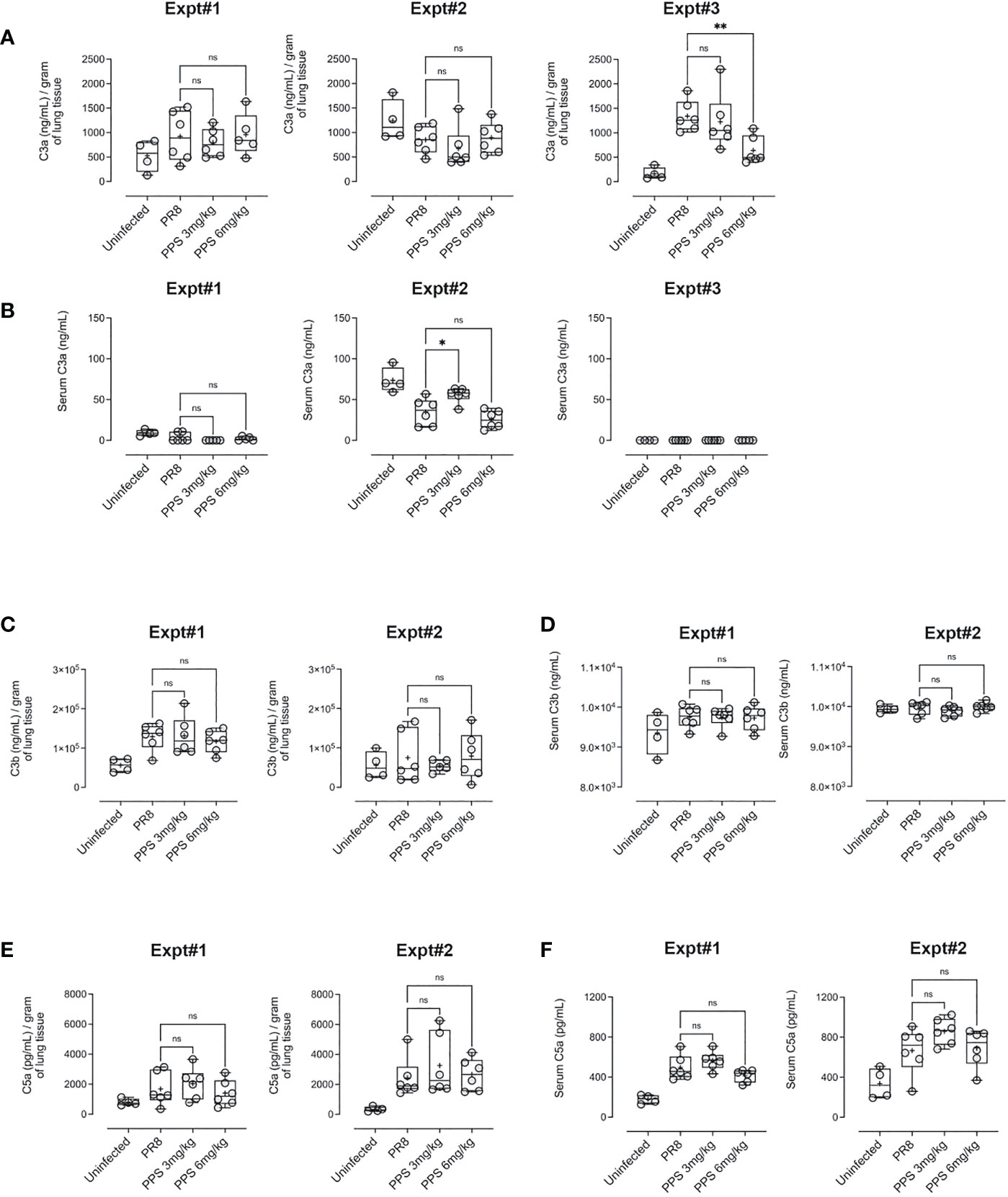
Figure 5 Complement protein measurement in the lungs and serum of PR8-infected mice treated with PPS. (A) Changes in lung levels of C3a, C3b and C5a measured by ELISA. Values normalised to tissue weights. (B) Changes in serum concentration of C3a, C3b and C5a measured by ELISA. Box-whiskers plot show all data points (max/min), standard error, median (line in box) and mean (‘+’ in box). Statistically significant differences were assessed by one-way ANOVA with a Sidak post-test (ns, not significant);. Data shown is from three individual, independent experiments denoted Expt#1, Expt#2 and Expt#3 (for C3a in 5a, 5b; total n=12-18 mice) and two independent experiments denoted Expt#1 and Expt#2 (for C3b in (C, D) and C5a in (E, F) total n= 7-12 mice).
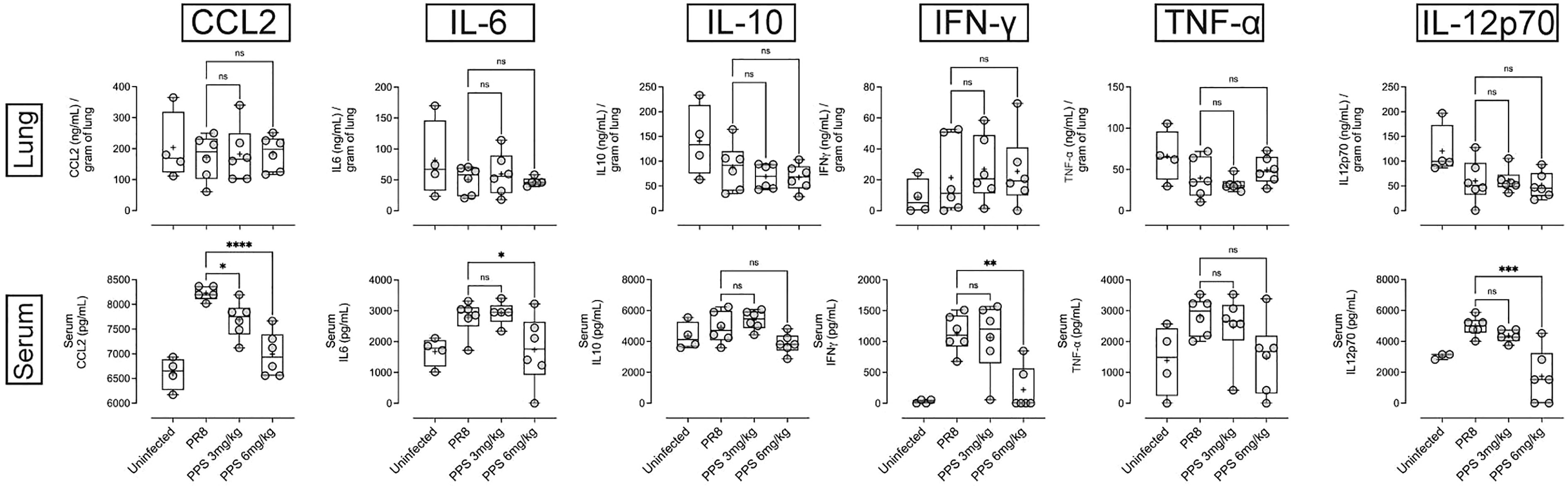
Figure 6 Pro-inflammatory cytokine/chemokine measurement in the lungs and serum of PR8-infected mice treated with PPS. Changes in lung and serum levels of CCL2, IL-6, IL-12p70, IL-10, IFN-γ and TNF-α measured by ELISA in Experiment 1. Values for lung concentrations were normalised to tissue weights. Values for serum concentrations are shown as per mL of serum. Box-whiskers plot show all data points (max/min), standard error, median (line in box) and mean (‘+’ in box). Statistically significant differences were assessed by one-way ANOVA with a Sidak post-test (*p<0.05; **p<0.01; ***p<0.001; ****p<0.0001; ns: not significant). (n=6 mice per group per experiment; Data shown is one of 3 independent experiments).
The development of chronic lung disease following severe PR8 infection, in particular lung consolidation and/or fibrosis that result from lung tissue damage and remodeling are clinically important, because they may give rise to chronic manifestations. As our data indicates that PPS treatment can reduce acute disease in PR8-infected mice, we asked whether this was associated with lung consolidation in the post-acute phase. In addition, we sought to determine whether SC PPS administered therapeutically during peak PR8 infection and disease (5 - 12 dpi) could result in reduced post-acute lung pathology, i.e., lung consolidation. PR8-infected mice were treated with vehicle or PPS, at 3 or 6 mg/kg, from 0 dpi to 7 dpi, or from 5 dpi to 12 dpi. Over 21 days of infection, we observed a significant reduction in weight loss in mice treated with PPS from Day 0 (Figure 7A), but this was confined to early (i.e., 5 and 6 dpi) time points for mice treated with 6 mg/kg PPS, and later (12, 14 and 16 dpi) time points for mice treated with 3 mg/kg PPS. Interestingly, mice treated therapeutically with PPS from Day 5 displayed more consistent reduction in weight loss (Figure 7B), as mice treated with 3 mg/kg PPS showed a significant reduction between 8 and 21 dpi, and mice treated with 6 mg/kg showing improved weight gain at 8, 9, 10 and 12 dpi.
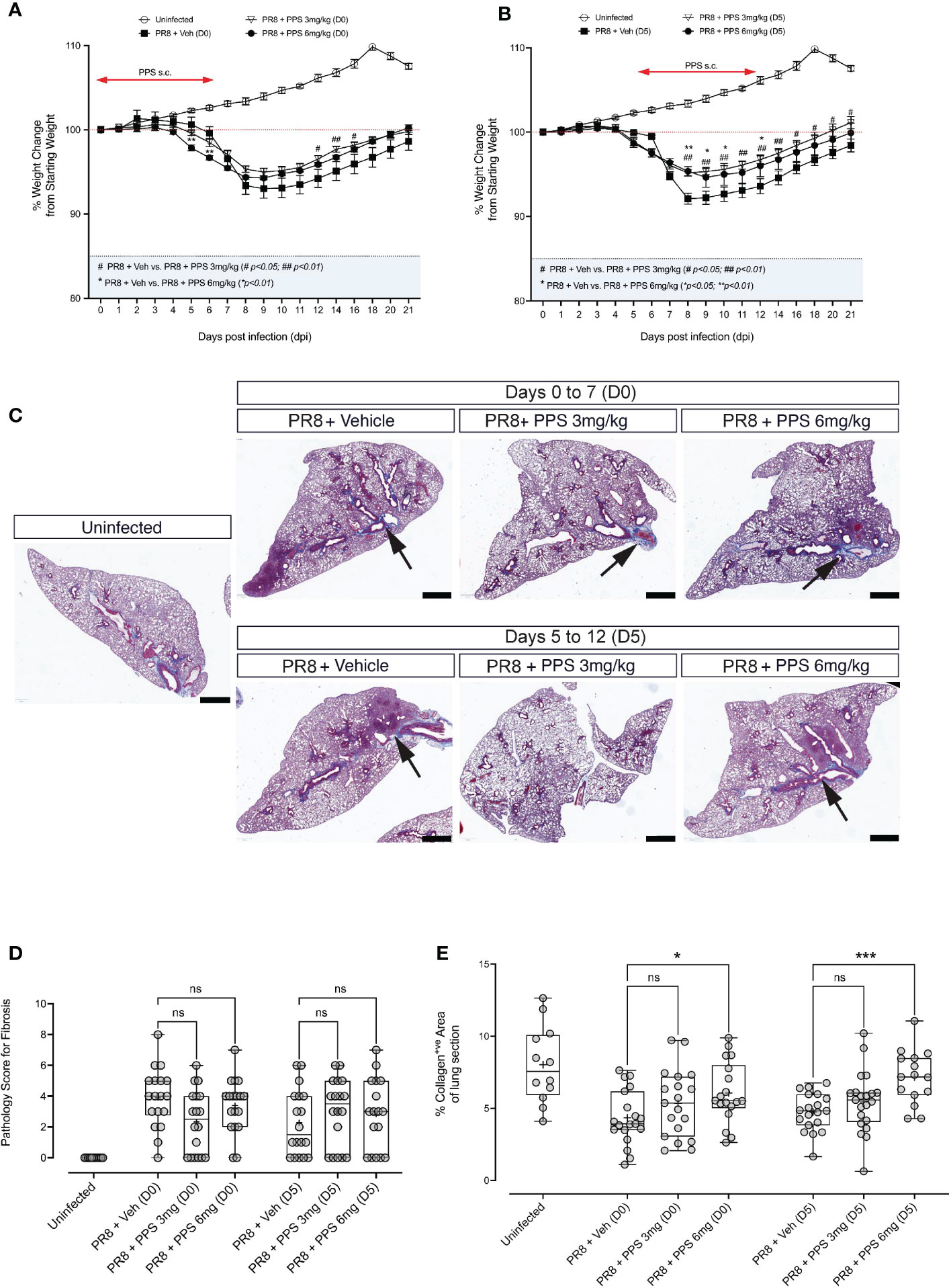
Figure 7 Histological analysis of post-acute (21 dpi) lung fibrosis and tissue remodeling. (A) Weight loss curves for PR8-infected mice administered 3 or 6 mg/kg PPS at Day 0 or (B) Day 5 post-infection. Mice were monitored and weighed daily until 12 dpi and every second day from 12 dpi until Day 21 dpi. Statistically significant differences in weight loss compared to vehicle-treated, PR8-infected mice shown as # (comparing PR8+Veh and PR8 + PPS 3mg/kg) and * (comparing PR8+Veh and PR8 + PPS 6mg/kg). Mean values for each dpi were analysed by two-way ANOVA with Dunnett’s post-test. */#p<0.05; **/##p<0.01. Sample size: n=6 mice per group; n=4 mice for uninfected group. (C) Representative micrographs of Masson-Trichrome (MT) stained lung sections from mice infected with PR8 and treated with SC PPS at 3 mg/kg or 6 mg/kg, with treatment starting at Day 0 or Day 5. Black arrows point to the blue MT staining which indicates areas of collagen deposition; red staining indicates cell nuclei. (D) Pathology scoring of lung sections shown in (A, B). Sections were assessed according to a pathology scoring matrix, where 0 = no change; 1 = minimal change (or possibly non-specific background or appearance compounded by lung collapse, i.e., not inflated); 2 = mild change; 3 = moderate change; 4 = severe change in <50% of lung lobe; 5 = severe change in >50% of lung lobe. n=6 per group; n=4 for uninfected group. Number of slides per group: n=3 per group. Box-whiskers plot show all data points (max/min), standard error, median (line in box) and mean (‘+’ in box). Statistically significant differences between groups were determined using a non-parametric one-way ANOVA (Kruskal-Wallis), and p values shown on the plots. (E) Automated quantification of collagen deposit and fibrosis in MT-stained lung sections in mice infected with PR8 and treated with SC PPS at 3 mg/kg or 6 mg/kg, with treatment starting at Day 0 or Day 5. Statistically significant differences between groups were determined using a non-parametric one-way ANOVA (Kruskal-Wallis). P values shown on plots: *p<0.05; **p<0.01; ***p<0.005; ns, not significant. Individual data points represent single value of % collagen signal coverage per lung section. Box-whiskers plot show all data points (max/min), standard error, median (line in box) and mean (‘+’ in box). (n=6 per group; n=4 for uninfected group. Number of slides per group: n=3 per group).
At 21 dpi, mice were euthanized and lung tissues were processed for histological analysis and stained with Masson’s Trichrome (MT) to determine the extent of collagen fibre deposition, a readout for lung tissue remodeling. Histological sections outline the deposition of collagen in tissues, which appears blue against a pink (eosin) counterstain (Figure 7C). Areas of lung alveolar space consolidation and more intense collagen stain was observed, however no clear visual differences between PPS- and vehicle-treated mice were observed. Therefore, lung tissue consolidation was assessed via two distinct approaches: 1) blinded, qualitative pathology scoring, and 2) automated/computer assisted quantification of tissue collagen. While some reduction in pathology score was observed in lungs of mice treated with PPS from Day 0 (D0; 3 and 6 mg/kg), these differences were not statistically significant (Figure 7D). Likewise, sections from mice treated from Day 5 (D5; 3 and 6 mg/kg) showed no statistically significant differences in pathology score compared to vehicle-treated mice (Figure 7D). Similarly, our analysis of collagen fiber deposition using a pixel classification algorithm did not reveal any significant reductions in collagen signal in MT-stained sections (Figure 7E).
We next asked whether cytokine and signaling pathways associated with fibrosis in the post-acute phase were impacted by PPS treatment during acute infection. As soluble ICAM-1 (sICAM-1), and the C5 complement pathway have been shown to be associated with lung fibrosis (35–38), we measured lung tissue concentrations of sICAM-1 and complement proteins, Complement Factor Bb (CF-Bb) and C5b9 by ELISA across two independent experiments (shown as Expt#4 and Expt#5 in Figure 8). In PR8-infected mice treated with 6 mg/kg PPS from both Day 0 and Day 5, sICAM-1 levels at 21 dpi were significantly reduced compared with vehicle-treated mice (Figure 8A). Levels of CF-Bb were not affected by PPS treatment (Figure 8B) at either concentration or dose timing. Finally, levels of C5b9 were significantly reduced (Figure 8C) in the lungs of mice treated with PPS at either 3 or 6 mg/kg doses, and in both Day 0 and Day 5 treatment groups.
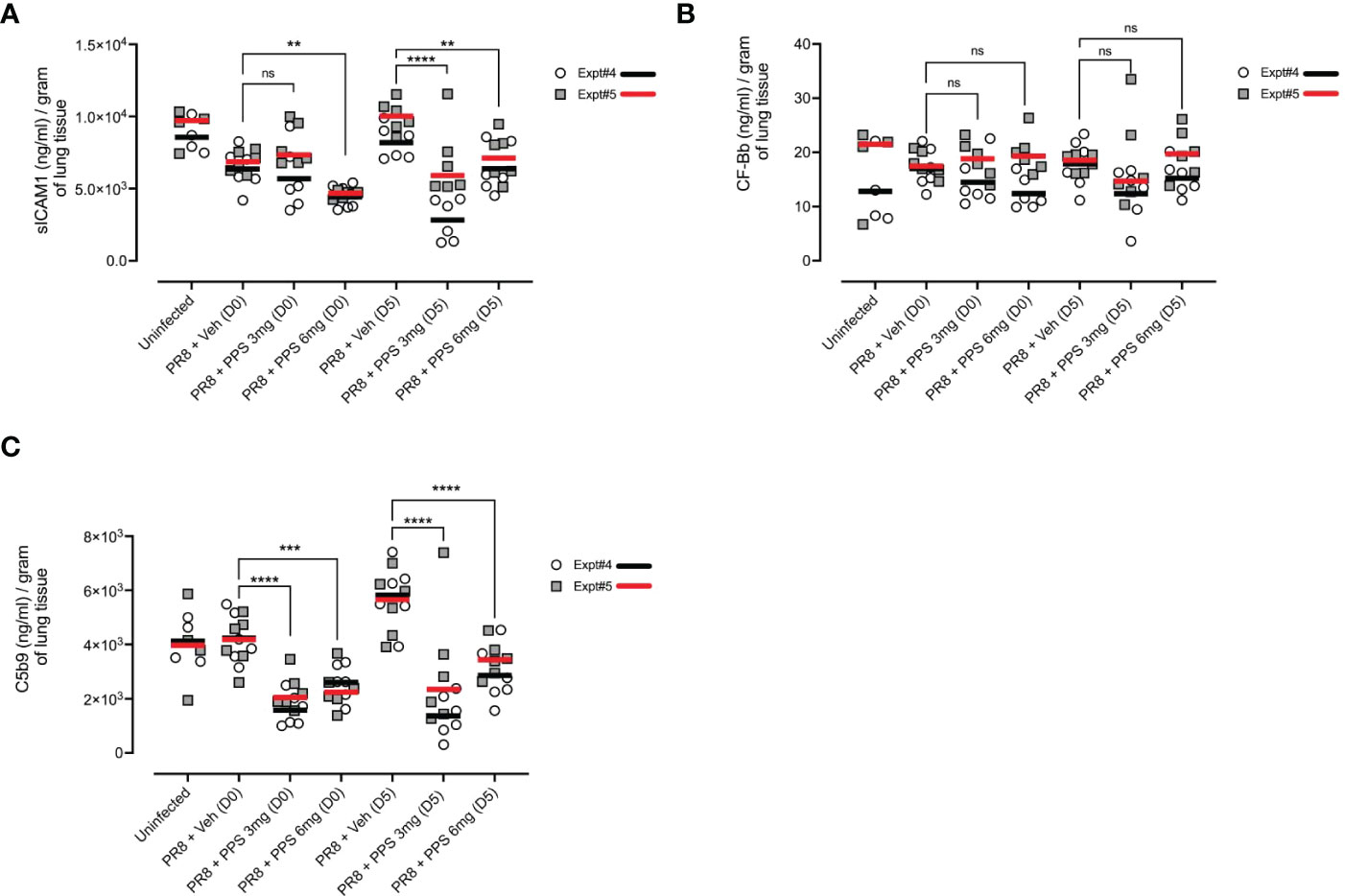
Figure 8 Measurement of pro-fibrotic and remodeling factors in the lungs of PR8-infected mice treated with PPS at 21 dpi. Mice were infected with PR8 and treated with 3 or 6 mg/kg PPS at Day 0 or Day 5 post-infection, daily for 7 days. Lung concentration of (A) sICAM-1 (B) Complement Factor Bb (CF-Bb) and (C) C5b9. All values normalised to tissue weights. Data shown from two independent experiments, denoted Expt#4 and Expt#5. Mean values for each experiment shown by black (Expt#4) and red (Expt#5) line on plot. Statistically significant differences were assessed on pooled data by a Student’s t-test. Statistically significant differences of pooled values were assessed by one-way ANOVA with a Sidak post-test (ns, not significant; **p<0.01; ***p<0.005; ****p<0.001). (sample size: n=4-6 mice/group).
In this study, a well-established experimental model of PR8 mediated pulmonary infection was examined to determine whether PPS, a semi-synthetic polysaccharide could favorably alter the outcome of PR8-induced disease in mice. PPS has been shown to interfere with NF-κB signaling, which consequently downregulates cytokines and complement proteins that contribute to potent inflammatory responses and tissue damage (26, 39, 40). In addition, the use of PPS in pre-clinical models of inflammatory viral disease, such as viral arthritis caused by arthritogenic alphaviruses, suggests that this compound may be suitable for treatment of virus-induced inflammatory pathologies that lead to tissue damage.
We found that PPS treatment protected PR8-infected mice from acute weight loss over the course of infection, and our PPS treatment may improve blood oxygen saturation (%SpO2) during acute PR8 infection. Interestingly, PPS-treated mice did exhibit disease signs (weight loss) compared to uninfected mice, albeit significantly less than vehicle-treated mice.
Early inhibition of viral attachment and entry represents a key advantage in acute disease management, and several antiviral drugs targeting these mechanisms have been developed and tested (10, 41, 42). PPS has been shown to inhibit human T-lymphotropic virus 1 (HTLV-1) replication in vitro (43), and recent studies indicate that PPS inhibits SARS-CoV-2 infection in vitro by modifying the receptor binding domain of S1 protein (44, 45). We found that at 8 dpi, viral titers in the lungs of mice infected with PR8 were not affected by PPS treatment, with no differences observed between infected mice treated with vehicle or with 3 mg/kg PPS. These findings suggest that the effect of PPS on weight loss and %SpO2 is independent of viral replication in the lungs.
Despite clear indications that PPS treatment helped reduce disease severity, histological analysis of lung sections did not show any significant changes in infiltration or overall pathology. However, flow cytometry analysis showed that rather than reducing overall inflammatory infiltrates in the lungs, PPS treatment was associated with the retention of alveolar macrophages, and a reduction in the proportion of CCR2+ inflammatory monocytes in the lungs of mice treated with 3 and 6 mg/kg PPS. Alveolar macrophages are a yolk sac-derived subset of tissue-resident macrophage which establishes early during development, and provides a first-line of defense against airway pathogens (46, 47). Alveolar macrophages act both by clearing airway pathogens via phagocytosis, or efferocytosis of apoptotic cells, and by limiting excessive inflammation through the release of anti-inflammatory factors, including transforming growth factor (TGFβ), Prostaglandin E2 (PGE2) and platelet-activating factor (PAF) into the tissue microenvironment (48, 49). These macrophages have been shown to be critical for protection against IAV infection (50, 51) and other respiratory pathogens such as Respiratory syncytial virus (RSV) and Streptococcus pneumoniae (49). Our results indicate that PPS may exert a protective effect in the lung that enables alveolar macrophages to persist and potentially assist in limiting tissue damage.
Although we did not observe a significant change in the number of T cells in the lungs, the proportion of CD4+ (but not CD8+) T cells was significantly higher in the lungs of mice treated with 3 mg/kg PPS, and higher (though not statistically significant) in the lungs of mice treated with 6 mg/kg PPS, suggesting that a reduced inflammatory milieu may facilitate the expansion of lung CD4+ T cells subsets, which may include helper type-1 CD4+ (Th1) T cells. Interestingly, while our observations indicated that alveolar macrophages were likely retained in the lungs of mice treated with PPS, other myeloid cell subsets were not significantly affected by PPS treatment.
The impact of pro-inflammatory chemokines and cytokines was investigated, be ELISA, and although we did not consistently observe decreased levels across all experimental repeats, some independent experiments showed clear evidence of reductions in CCL2, IL-6, IFN-γ and IL-12p70 in the serum of mice treated with PPS. Interestingly, only CCL2 serum levels appeared to be reduced in mice treated with both 3 and 6 mg/kg PPS, while IL-6, IFN-γ and IL-12p70 serum levels were only reduced in mice treated with 6 mg/kg PPS, compared to vehicle-treated mice. Of note, we did not observe any decreases in these markers in the lung tissue of mice treated with PPS, with the exception of IL-12p70, which was significantly reduced in the lungs of mice treated with both 3 and 6 mg/kg in one out of three experiments. High IL-6 expression is characteristic of PR8 infection, both in mice and humans (52, 53), and do contribute to the severity of systemic pro-inflammatory responses; although the IL6-IL-6R signaling pathway is required to enable neutrophil-mediated clearance of PR8 in the lungs (54), and a lack of IL-6 signaling in mice was shown to be detrimental in PR8 infection (55, 56). Our findings that IL-6 serum levels may be affected by higher PPS doses may account for the disease phenotype we observed in PPS-treated mice.
CCL2, which drives macrophage differentiation during inflammatory response (57) is also locally expressed in inflamed lungs and helps recruit CCR2+ monocytes from the circulation during PR8 infection (58). Although we only observed a decrease in serum CCL2 in mice treated with PPS, CCR2+ inflammatory monocytes were reduced in the lungs of mice treated with PPS. The systemic effect of PPS, administered subcutaneously, may affect serum chemokines (like CCL2, which act through a gradient) and in turn dampen recruitment of monocytes to the lungs, but our observations do not point toward direct interference with the CCL2/CCR2 axis.
The observation that CD4+ T cell expansion may be enhanced in the lungs of PPS-treated mice could be a result of an immunoregulatory effect of PPS, in particular in the context of IFN-γ. IFN-γ is a potent inflammatory and regulatory cytokine that is also elevated in the lungs following PR8 infection (59, 60), and is required for the activation of CD8+ cytotoxic and CD4+ helper T cells and also helps control neutrophilic infiltration in the lung, thereby limiting tissue damage (60). We observed a reduction in IFN-γ at 8 dpi in the serum, but not in the lungs. Therefore, it is possible that the marginal increase in CD4+ T cells we observed in the lungs of PPS-treated mice is independent of IFN-γ, as other cytokines including TGFβ, IL-12 or IL-6 – depending on the dominant helper T cell phenotype – may play a more defining role during PR8 infection, although studies have shown that at 8 dpi, Th1, TFH (follicular helper) and TREG (regulatory) T cell phenotypes, which are regulated by IFN-γ, IL-6/IL-21 and IL-10, respectively, are the main infiltrating CD4+ T cell subtypes found in the lungs of PR8-infected mice (61). Future studies should therefore dissect how PPS treatment impacts the phenotypic diversity of lung T cell subsets in PR8 infection.
One of the key outcomes of the effects of PPS was reduced weight loss in mice infected with PR8. Cachexia, mediated weight loss during disease, has been extensively described in a range of inflammatory illnesses, both acute and chronic (62). Cytokines that are most implicated with cachexia include IFN-γ, as well as IL-1β, TNF-α and IL-6 (63–65), yet only IL-6 and IFN-γ levels in the serum appeared to have been affected following PPS treatment. This indicates that, although TNF-α and IL-6 are well-accepted drivers of cachexia in disease (66, 67), our observations may be explained by the regulatory effect of PPS on NF-κB resulting in the downregulation of these cytokines, and further studies examining the transcriptional profile of cachexia-associated pathways may shed light on how PPS treatment ameliorates cachexia in PR8-infected mice.
We next asked whether the reduction in weight loss seen in PPS-treated mice during the acute phase would lead to improved post-acute pathology. Collagen production is increased after resolution of inflammation in most tissues, to aid with tissue repair – although in post-infectious pathology, this can develop into fibrosis (68). Earlier reports linked PPS with inhibition of matrix metalloproteinases (MMPs) and ADAMTS proteins (30–32) as well as FGF-2 and elastase (69, 70), all associated with the development of fibrosis. Expert histopathological scoring of lung structural changes determined that the hallmarks of fibrotic responses (e.g., consolidation, fibrosing alveolitis) in this model of PR8 infection were not clearly visible, suggesting that PR8-induced post-acute lung disease is distinct from well-characterized fibrosis models such as bleomycin-induced fibrosis models (71, 72). However, studies have reported that sICAM-1 levels in the lungs were associated with fibrosis (38, 68), and the complement system has also been shown to be relevant in fibrosis development in the lungs. C5 complement proteins were also shown to drive inflammation and neutrophil recruitment in PR8 infection in mice (73), and C5b9 and CF-Bb, which are integral components of the complement membrane attack complex, are recruited downstream of C5 activation (74, 75). PPS administered at a 6mg/kg dose either prophylactically or therapeutically showed significant reductions in lung sICAM-1 at 21 dpi in PR8-infected mice. Reduced levels of sICAM-1 in the post-acute phase suggests that PPS treatment both over early (Day 0 to Day 7) and late (Day 5 to Day 12) phases of PR8 infection may dampen post-acute lung consolidation, and this was reflected by our observations on the levels of complement proteins associated with fibrosis. Levels of C5b9 were significantly reduced in mice treated with PPS while levels of Complement Factor Bb (CF-Bb) were not affected by PPS treatment. C5b9, which triggers the complement’s membrane attack complex (MAC), was shown to be a feature of viral infection with SARS-CoV-2, as this complex is deposited in the alveolar septa of COVID-19 patients (76). While the exact role of C5b9 in PR8 infection remains poorly understood, a study showed that in a model bleomycin-induced fibrosis, mice lacking interleukin-17 (IL-17A) developed less severe fibrosis, and this was concomitant with a reduction in C5b9 deposition (35). CF-Bb is required for the induction of alternative complement pathways such as the mannan-binding lectin (MBL) pathway (77), which has been shown to exert a protective effect in PR8 infection in vitro (78), and this effect was dependent on alveolar macrophages. This suggests that the potential role of complement (and its associated pathways) in PR8 could be linked to the PPS-induced maintenance of alveolar macrophages we observed in this study.
In conclusion, this study investigated the potential therapeutic effects of the semi-synthetic polysaccharide, PPS in a model of lung inflammation mediated by PR8 (PR8 strain) infection. The key observations are that PPS demonstrated potential anti-inflammatory effects during the acute phase of pulmonary infection mediated by PR8 as observed by the reduction in weight loss and improvement in oxygen saturation following PPS treatment, as well as an enrichment in protective alveolar macrophages and CD4+ T cells in the lungs. In addition, the preceding anti-inflammatory actions of PPS during the acute phase may have had a role in modulating the post-acute pulmonary changes based on the observations of reduced fibrotic biomarkers.
The original contributions presented in the study are included in the article/Supplementary material. Further inquiries can be directed to the corresponding authors.
The animal study was reviewed and approved by Griffith University Animal Ethics Committee Griffith University, Parklands Dr, Southport QLD 4215.
Conceptualisation: RK, SM, AZ. Data curation: CS, AZ. Formal analysis: AZ. Funding acquisition: SM. Investigation: AZ, HM, JF, XL. Methodology: AZ, HM, JF, XL. Project administration: SM. Supervision: SM, AZ. Visualisation: AZ, SM. Writing – original draft: AZ, SM. Writing – review and editing: RK, CS, SM, AZ. All authors contributed to the article and approved the submitted version.
This study was conducted at the Menzies Health Institute Queensland, Griffith University, as a contract research project funded by Paradigm Biopharmaceuticals Ltd (https://paradigmbiopharma.com/). The funders did not have any role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript beyond those specified.
The authors would like to thank Griffith University’s Bioresource Facility staff for their help in animal husbandry and maintenance.
RK and CS are employees of Paradigm Biopharmaceuticals Ltd. As staff members of Paradigm Biopharmaceuticals Ltd, RK and CS are entitled to long-term and short-term incentives. The specific roles of these authors are articulated in the ‘author contributions’ section. This research may lead to the use of PPS towards the treatment of Influenza disease.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1030879/full#supplementary-material
Supplementary Figure 1 | Effect of subcutaneous and intraperitoneal administration of PPS in PR-8 infected mice. Weight loss in uninfected mice (uninfected; Uninf), uninfected mice treated with PPS (Uninf + PPS (sc)) or vehicle (Uninf + Veh (ip)) subcutaneously (SC) or intraperitoneally (IP), PR8-infected mice treated with PPS SC (PR8 + PPS (sc)) or IP (PR8 + PPS (ip)), and PR8-infected, vehicle-treated mice SC (PR8 + Veh (sc)) or IP (PR8 + Veh (ip)). **p<0.01, ***p<0.005 and ns: not significant for PR8 + Veh (ip) compared with PR8 + PPS (ip); ###p<0.005, ####p<0.0001 for PR8 + Veh (sc) compared with PR8 + PPS (sc). n=7 per group; n=4 for uninfected mice. Statistically significant differences were assessed by two-way ANOVA & p values shown on plots
Supplementary Figure 2 | Pro-inflammatory cytokine/chemokine measurement in the lungs and serum of PR8-infected mice treated with PPS. Changes in lung and serum levels of CCL2, IL-6, IL-12p70, IL-10, IFN-γ and TNF-α measured by ELISA in Experiments 2 (Expt#2) and Experiment 3 (Expt#3). Values for lung concentrations were normalised to tissue weights. Values for serum concentrations shown as per mL of serum. Box-whiskers plot show all data points (max/min), standard error, median (line in box) and mean (‘+’ in box). Statistically significant differences were assessed by one-way ANOVA with a Sidak post-test (*p<0.05; **p<0.01; ****p<0.0001; ns: not significant). (n=6 mice per group per experiment; Data shown from two of 3 independent experiments).
Supplementary Figure 3 | Gating strategy used for the identification of immune cells in the lungs of PR8-infected mice. Representative plots outlining sequential gating of CD45+ live cells, Ly6G+ neutrophils, CD11bhiLy6Chi inflammatory monocytes (IM; derived from green panel), CD64+SiglecF+ alveolar macrophages (AM; derived from blue panel) and CD4+ and CD8+ T cells (derived from pink square).
1. Flerlage T, Boyd DF, Meliopoulos V, Thomas PG, Schultz-Cherry S. Influenza virus and SARS-CoV-2: pathogenesis and host responses in the respiratory tract. Nat Rev Microbiol (2021) 19:425–41. doi: 10.1038/s41579-021-00542-7
2. Leung NHL. Transmissibility and transmission of respiratory viruses. Nat Rev Microbiol (2021) 19:528–45. doi: 10.1038/s41579-021-00535-6
3. Petersen E, Koopmans M, Go U, Hamer DH, Petrosillo N, Castelli F, et al. Comparing SARS-CoV-2 with SARS-CoV and influenza pandemics. Lancet Infect Dis (2020) 0:30484–9. doi: 10.1016/S1473-3099(20)30484-9
4. Moriyama M, Hugentobler WJ, Iwasaki A. Seasonality of respiratory viral infections. Annu Rev Virol (2020) 7:83–101. doi: 10.1146/annurev-virology-012420-022445
5. Behzadi MA, Leyva-Grado VH. Overview of current therapeutics and novel candidates against influenza, respiratory syncytial virus, and middle East respiratory syndrome coronavirus infections. Front Microbiol (2019) 10:1327. doi: 10.3389/fmicb.2019.01327
6. Matute-Bello G, Frevert CW, Martin TR. Animal models of acute lung injury. Am J Physiology-Lung Cell Mol Physiol (2008) 295:L379–99. doi: 10.1152/ajplung.00010.2008
7. Nguyen T-Q, Rollon R, Choi Y-K. Animal models for influenza research: Strengths and weaknesses. Viruses (2021) 13:1011. doi: 10.3390/v13061011
8. Rodriguez L, Nogales A, Martínez-Sobrido L. Influenza a virus studies in a mouse model of infection. JoVE (Journal Visualized Experiments) (2017) 127:e55898. doi: 10.3791/55898
9. Krammer F, Smith GJD, Fouchier RAM, Peiris M, Kedzierska K, Doherty PC, et al. Influenza. Nat Rev Dis Primers (2018) 4:1–21. doi: 10.1038/s41572-018-0002-y
10. Tavares LP, Teixeira MM, Garcia CC. The inflammatory response triggered by influenza virus: a two edged sword. Inflammation Res (2017) 66:283–302. doi: 10.1007/s00011-016-0996-0
11. Kalil AC, Thomas PG. Influenza virus-related critical illness: pathophysiology and epidemiology. Crit Care (2019) 23:258. doi: 10.1186/s13054-019-2539-x
12. Guimond SE, Mycroft-West CJ, Gandhi NS, Tree JA, Le TT, Spalluto CM, et al. Synthetic heparan sulfate mimetic pixatimod (PG545) potently inhibits SARS-CoV-2 by disrupting the spike–ACE2 interaction. ACS Cent Sci (2022) 8(5):527–45. doi: 10.1021/acscentsci.1c01293
13. Lundin A, Bergström T, Andrighetti-Fröhner CR, Bendrioua L, Ferro V, Trybala E. Potent anti-respiratory syncytial virus activity of a cholestanol-sulfated tetrasaccharide conjugate. Antiviral Res (2012) 93:101–9. doi: 10.1016/j.antiviral.2011.11.002
14. Research C for DE. Influenza (Flu) antiviral drugs and related information, in: FDA (2022). Available at: https://www.fda.gov/drugs/information-drug-class/influenza-flu-antiviral-drugs-and-related-information (Accessed April 19, 2022).
15. Zhou Y, Fu X, Liu X, Huang C, Tian G, Ding C, et al. Use of corticosteroids in influenza-associated acute respiratory distress syndrome and severe pneumonia: a systemic review and meta-analysis. Sci Rep (2020) 10:3044. doi: 10.1038/s41598-020-59732-7
16. Han S, Mallampalli RK. The acute respiratory distress syndrome: From mechanism to translation. J Immunol (2015) 194:855–60. doi: 10.4049/jimmunol.1402513
17. Degenhardt M, Ghosh P, W?tzig H. Studies on the structural variations of pentosan polysulfate sodium (NaPPS) from different sources by capillary electrophoresis. Archiv der Pharmazie (2001) 334:27–9. doi: 10.1002/1521-4184(200101)334:1<27::AID-ARDP27>3.0.CO;2-X
18. Parsons CL, Mulholland SG. Successful therapy of interstitial cystitis with pentosanpolysulfate. J Urol (1987) 138:513–6. doi: 10.1016/s0022-5347(17)43243-5
19. Sampson MJ, Kabbani M, Krishnan R, Nganga M, Theodoulou A, Krishnan J. Improved clinical outcome measures of knee pain and function with concurrent resolution of subchondral bone marrow edema lesion and joint effusion in an osteoarthritic patient following pentosan polysulphate sodium treatment: a case report. BMC Musculoskeletal Disord (2017) 18:396. doi: 10.1186/s12891-017-1754-3
20. Krishnan R, Duiker M, Rudd PA, Skerrett D, Pollard JGD, Siddel C, et al. Pentosan polysulfate sodium for Ross river virus-induced arthralgia: a phase 2a, randomized, double-blind, placebo-controlled study. BMC Musculoskeletal Disord (2021) 22:271. doi: 10.1186/s12891-021-04123-w
21. Rudd PA, Lim EXY, Stapledon CJM, Krishnan R, Herrero LJ. Pentosan polysulfate sodium prevents functional decline in chikungunya infected mice by modulating growth factor signalling and lymphocyte activation. PloS One (2021) 16:e0255125. doi: 10.1371/journal.pone.0255125
22. Sanden C, Mori M, Jogdand P, Jönsson J, Krishnan R, Wang X, et al. Broad Th2 neutralization and anti-inflammatory action of pentosan polysulfate sodium in experimental allergic rhinitis. Immunity Inflammation Dis (2017) 5:300–9. doi: 10.1002/iid3.164
23. Bwalya EC, Kim S, Fang J, Wijekoon HMS, Hosoya K, Okumura M. Pentosan polysulfate inhibits IL-1β-induced iNOS, c-jun and HIF-1α upregulation in canine articular chondrocytes. PloS One (2017) 12:e0177144. doi: 10.1371/journal.pone.0177144
24. Herrero LJ, Foo S-S, Sheng K-C, Chen W, Forwood MR, Bucala R, et al. Pentosan polysulfate: a novel glycosaminoglycan-like molecule for effective treatment of alphavirus-induced cartilage destruction and inflammatory disease. J Virol (2015) 89:8063–76. doi: 10.1128/JVI.00224-15
25. Sunaga T, Oh N, Hosoya K, Takagi S, Okumura M. Inhibitory effects of pentosan polysulfate sodium on MAP-kinase pathway and NF-κB nuclear translocation in canine chondrocytes in vitro. J Vet Med Sci (2012) 74:707–11. doi: 10.1292/jvms.11-0511
26. Wu J, Guan T, Zheng S, Grosjean F, Liu W, Xiong H, et al. Inhibition of inflammation by pentosan polysulfate impedes the development and progression of severe diabetic nephropathy in aging C57B6 mice. Lab Invest (2011) 91:1459–71. doi: 10.1038/labinvest.2011.93
27. Kilgore KS, Naylor KB, Tanhehco EJ, Park JL, Booth EA, Washington RA, et al. The semisynthetic polysaccharide pentosan polysulfate prevents complement-mediated myocardial injury in the rabbit perfused heart. J Pharmacol Exp Ther (1998) 285:987–94.
28. Kutlar A, Ataga KI, McMahon L, Howard J, Galacteros F, Hagar W, et al. A potent oral p-selectin blocking agent improves microcirculatory blood flow and a marker of endothelial cell injury in patients with sickle cell disease. Am J Hematol (2012) 87:536–9. doi: 10.1002/ajh.23147
29. Sun S, Zhao G, Liu C, Wu X, Guo Y, Yu H, et al. Inhibition of complement activation alleviates acute lung injury induced by highly pathogenic avian influenza H5N1 virus infection. Am J Respir Cell Mol Biol (2013) 49:221–30. doi: 10.1165/rcmb.2012-0428OC
30. Lupia E, Zheng F, Grosjean F, Tack I, Doublier S, Elliot SJ, et al. Pentosan polysulfate inhibits atherosclerosis in Watanabe heritable hyperlipidemic rabbits: differential modulation of metalloproteinase-2 and -9. Lab Invest (2012) 92:236–45. doi: 10.1038/labinvest.2011.154
31. Troeberg L, Mulloy B, Ghosh P, Lee M-H, Murphy G, Nagase H. Pentosan polysulfate increases affinity between ADAMTS-5 and TIMP-3 through formation of an electrostatically driven trimolecular complex. Biochem J (2012) 443:307–15. doi: 10.1042/BJ20112159
32. Vistnes M, Aronsen JM, Lunde IG, Sjaastad I, Carlson CR, Christensen G. Pentosan polysulfate decreases myocardial expression of the extracellular matrix enzyme ADAMTS4 and improves cardiac function in vivo in rats subjected to pressure overload by aortic banding. PloS One (2014) 9:e89621. doi: 10.1371/journal.pone.0089621
33. Klegeris A, Singh EA, McGeer PL. Effects of c-reactive protein and pentosan polysulphate on human complement activation. Immunology (2002) 106:381–8. doi: 10.1046/j.1365-2567.2002.01425.x
34. Tanaka N, Emoto T, Suda H, Kunihiro Y, Matsunaga N, Hasegawa S, et al. High-resolution computed tomography findings of influenza virus pneumonia: a comparative study between seasonal and novel (H1N1) influenza virus pneumonia. Jpn J Radiol (2012) 30:154–61. doi: 10.1007/s11604-011-0027-6
35. Cipolla E, Fisher AJ, Gu H, Mickler EA, Agarwal M, Wilke CA, et al. IL-17A deficiency mitigates bleomycin-induced complement activation during lung fibrosis. FASEB J (2017) 31:5543–56. doi: 10.1096/fj.201700289R
36. Fisher AJ, Cipolla E, Varre A, Gu H, Mickler EA, Vittal R. Potential mechanisms underlying TGF-β-mediated complement activation in lung fibrosis. Cell Mol Med Open Access (2017) 3:14. doi: 10.21767/2573-5365.100037
37. Okuda R, Matsushima H, Aoshiba K, Oba T, Kawabe R, Honda K, et al. Soluble intercellular adhesion molecule-1 for stable and acute phases of idiopathic pulmonary fibrosis. Springerplus (2015) 4:657. doi: 10.1186/s40064-015-1455-z
38. Tsoutsou PG, Gourgoulianis KI, Petinaki E, Mpaka M, Efremidou S, Maniatis A, et al. ICAM-1, ICAM-2 and ICAM-3 in the sera of patients with idiopathic pulmonary fibrosis. Inflammation (2004) 28:359–64. doi: 10.1007/s10753-004-6647-6
39. Berdal J-E, Mollnes TE, Wæhre T, Olstad OK, Halvorsen B, Ueland T, et al. Excessive innate immune response and mutant D222G/N in severe a (H1N1) pandemic influenza. J Infection (2011) 63:308–16. doi: 10.1016/j.jinf.2011.07.004
40. Chen P, Yuan Y, Zhang T, Xu B, Gao Q, Guan T. Pentosan polysulfate ameliorates apoptosis and inflammation by suppressing activation of the p38 MAPK pathway in high glucose-treated HK-2 cells. Int J Mol Med (2018) 41:908–14. doi: 10.3892/ijmm.2017.3290
41. Ison MG. Optimizing antiviral therapy for influenza: understanding the evidence. Expert Rev Anti Infect Ther (2015) 13:417–25. doi: 10.1586/14787210.2015.1018183
42. Liu J-W, Lin S-H, Wang L-C, Chiu H-Y, Lee J-A. Comparison of antiviral agents for seasonal influenza outcomes in healthy adults and children: A systematic review and network meta-analysis. JAMA Network Open (2021) 4:e2119151. doi: 10.1001/jamanetworkopen.2021.19151
43. Ma G, Yasunaga J, Ohshima K, Matsumoto T, Matsuoka M. Pentosan polysulfate demonstrates anti-human T-cell leukemia virus type 1 activities. In Vitro In Vivo. J Virol (2019) 93:e00413–19. doi: 10.1128/JVI.00413-19
44. Ennemoser M, Rieger J, Muttenthaler E, Gerlza T, Zatloukal K, Kungl AJ. Enoxaparin and pentosan polysulfate bind to the SARS-CoV-2 spike protein and human ACE2 receptor, inhibiting vero cell infection. Biomedicines (2021) 10:49. doi: 10.3390/biomedicines10010049
45. Bertini S, Alekseeva A, Elli S, Pagani I, Zanzoni S, Eisele G, et al. Pentosan polysulfate inhibits attachment and infection by SARS-CoV-2 in vitro: insights into structural requirements for binding. Thromb Haemost (2022) 0:984–97. doi: 10.1055/a-1807-0168
46. Perdiguero EG, Geissmann F. The development and maintenance of resident macrophages. Nat Immunol (2015) 17:2–8. doi: 10.1038/ni.3341
47. Perdiguero EG, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature (2014) 518:547–51. doi: 10.1038/nature13989
48. Allard B, Panariti A, Martin JG. Alveolar macrophages in the resolution of inflammation, tissue repair, and tolerance to infection. Front Immunol (2018) 9:1777. doi: 10.3389/fimmu.2018.01777
49. Clua P, Tomokiyo M, Raya Tonetti F, MdA I, García Castillo V, Marcial G, et al. The role of alveolar macrophages in the improved protection against respiratory syncytial virus and pneumococcal superinfection induced by the peptidoglycan of lactobacillus rhamnosus CRL1505. Cells (2020) 9:1653. doi: 10.3390/cells9071653
50. He W, Chen C-J, Mullarkey CE, Hamilton JR, Wong CK, Leon PE, et al. Alveolar macrophages are critical for broadly-reactive antibody-mediated protection against influenza a virus in mice. Nat Commun (2017) 8:846. doi: 10.1038/s41467-017-00928-3
51. Schneider C, Nobs SP, Heer AK, Kurrer M, Klinke G, van Rooijen N, et al. Alveolar macrophages are essential for protection from respiratory failure and associated morbidity following influenza virus infection. PloS Pathog (2014) 10:e1004053. doi: 10.1371/journal.ppat.1004053
52. Liu S, Yan R, Chen B, Pan Q, Chen Y, Hong J, et al. Influenza virus-induced robust expression of SOCS3 contributes to excessive production of IL-6. Front Immunol (2019) 10:1843. doi: 10.3389/fimmu.2019.01843
53. Wang J, Wang Q, Han T, Li Y-K, Zhu S-L, Ao F, et al. Soluble interleukin-6 receptor is elevated during influenza a virus infection and mediates the IL-6 and IL-32 inflammatory cytokine burst. Cell Mol Immunol (2015) 12:633–44. doi: 10.1038/cmi.2014.80
54. Dienz O, Rud JG, Eaton SM, Lanthier PA, Burg E, Drew A, et al. Essential role of IL-6 in protection against H1N1 influenza virus by promoting neutrophil survival in the lung. Mucosal Immunol (2012) 5:258–66. doi: 10.1038/mi.2012.2
55. Pyle CJ, Uwadiae FI, Swieboda DP, Harker JA. Early IL-6 signalling promotes IL-27 dependent maturation of regulatory T cells in the lungs and resolution of viral immunopathology. PloS Pathog (2017) 13:e1006640. doi: 10.1371/journal.ppat.1006640
56. Yang M-L, Wang C-T, Yang S-J, Leu C-H, Chen S-H, Wu C-L, et al. IL-6 ameliorates acute lung injury in influenza virus infection. Sci Rep (2017) 7:43829. doi: 10.1038/srep43829
57. Gschwandtner M, Derler R, Midwood KS. More than just attractive: How CCL2 influences myeloid cell behavior beyond chemotaxis. Front Immunol (2019) 10:2759. doi: 10.3389/fimmu.2019.02759
58. Seo S-U, Kwon H-J, Ko H-J, Byun Y-H, Seong BL, Uematsu S, et al. Type I interferon signaling regulates Ly6Chi monocytes and neutrophils during acute viral pneumonia in mice. PloS Pathog (2011) 7:e1001304. doi: 10.1371/journal.ppat.1001304
59. Califano D, Furuya Y, Roberts S, Avram D, McKenzie ANJ, Metzger DW. IFN-γ increases susceptibility to influenza a infection through suppression of group II innate lymphoid cells. Mucosal Immunol (2018) 11:209–19. doi: 10.1038/mi.2017.41
60. Stifter SA, Bhattacharyya N, Pillay R, Flórido M, Triccas JA, Britton WJ, et al. Functional interplay between type I and II interferons is essential to limit influenza a virus-induced tissue inflammation. PloS Pathog (2016) 12:e1005378. doi: 10.1371/journal.ppat.1005378
61. Hornick EE, Zacharias ZR, Legge KL. Kinetics and phenotype of the CD4 T cell response to influenza virus infections. Front Immunol (2019) 10:2351. doi: 10.3389/fimmu.2019.02351
64. Cahlin C, Körner A, Axelsson H, Wang W, Lundholm K, Svanberg E. Experimental cancer cachexia: the role of host-derived cytokines interleukin (IL)-6, IL-12, interferon-gamma, and tumor necrosis factor alpha evaluated in gene knockout, tumor-bearing mice on C57 bl background and eicosanoid-dependent cachexia. Cancer Res (2000) 60:5488–93.
65. Matthys P, Dijkmans R, Proost P, Van Damme J, Heremans H, Sobis H, et al. Severe cachexia in mice inoculated with interferon-gamma-producing tumor cells. Int J Cancer (1991) 49:77–82. doi: 10.1002/ijc.2910490115
66. Beutler B, Cerami A. Cachectin and tumour necrosis factor as two sides of the same biological coin. Nature (1986) 320:584–8. doi: 10.1038/320584a0
67. Radigan KA, Nicholson TT, Welch LC, Chi M, Amarelle L, Angulo M, et al. Influenza a virus infection induces muscle wasting via IL-6 regulation of the E3 ubiquitin ligase atrogin-1. J Immunol (2019) 202:484–93. doi: 10.4049/jimmunol.1701433
68. Fernandez IE, Amarie OV, Mutze K, Königshoff M, Yildirim AÖ, Eickelberg O. Systematic phenotyping and correlation of biomarkers with lung function and histology in lung fibrosis. Am J Physiol Lung Cell Mol Physiol (2016) 310:L919–927. doi: 10.1152/ajplung.00183.2015
69. Andrews JL, Ghosh P, Lentini A, Ternai B. The interaction of pentosan polysulphate (SP54) with human neutrophil elastase and connective tissue matrix components. Chem Biol Interact (1983) 47:157–73. doi: 10.1016/0009-2797(83)90155-2
70. Herbert JM, Cottineau M, Driot F, Pereillo JM, Maffrand JP. Activity of pentosan polysulphate and derived compounds on vascular endothelial cell proliferation and migration induced by acidic and basic FGF in vitro. Biochem Pharmacol (1988) 37:4281–8. doi: 10.1016/0006-2952(88)90608-9
71. Hay J, Shahzeidi S, Laurent G. Mechanisms of bleomycin-induced lung damage. Arch Toxicol (1991) 65:81–94. doi: 10.1007/BF02034932
72. Liu T, De Los Santos FG, Phan SH. The bleomycin model of pulmonary fibrosis. Methods Mol Biol (2017) 1627:27–42. doi: 10.1007/978-1-4939-7113-8_2
73. Garcia CC, Weston-Davies W, Russo RC, Tavares LP, Rachid MA, Alves-Filho JC, et al. Complement C5 activation during influenza a infection in mice contributes to neutrophil recruitment and lung injury. PloS One (2013) 8:e64443. doi: 10.1371/journal.pone.0064443
74. Bayly-Jones C, Bubeck D, Dunstone MA. The mystery behind membrane insertion: a review of the complement membrane attack complex. Philos Trans R Soc B: Biol Sci (2017) 372:20160221. doi: 10.1098/rstb.2016.0221
75. Xie CB, Jane-Wit D, Pober JS. Complement membrane attack complex: New roles, mechanisms of action, and therapeutic targets. Am J Pathol (2020) 190:1138–50. doi: 10.1016/j.ajpath.2020.02.006
76. Niederreiter J, Eck C, Ries T, Hartmann A, Märkl B, Büttner-Herold M, et al. Complement activation via the lectin and alternative pathway in patients with severe COVID-19. Front Immunol (2022) 13:835156. doi: 10.3389/fimmu.2022.835156
77. Petersen SV, Thiel S, Jensenius JC. The mannan-binding lectin pathway of complement activation: biology and disease association. Mol Immunol (2001) 38:133–49. doi: 10.1016/s0161-5890(01)00038-4
Keywords: influenza A virus, Pentosan polysulfate sodium, acute inflammation, cytokines and chemokines, lung consolidation, lung fibrosis, chronic lung inflammation, immune cell infiltrate
Citation: Krishnan R, Stapledon CJM, Mostafavi H, Freitas JR, Liu X, Mahalingam S and Zaid A (2023) Anti-inflammatory actions of Pentosan polysulfate sodium in a mouse model of influenza virus A/PR8/34-induced pulmonary inflammation. Front. Immunol. 14:1030879. doi: 10.3389/fimmu.2023.1030879
Received: 29 August 2022; Accepted: 30 January 2023;
Published: 09 February 2023.
Edited by:
Lee Mark Wetzler, Boston University, United StatesReviewed by:
Wayne Robert Thomas, University of Western Australia, AustraliaCopyright © 2023 Krishnan, Stapledon, Mostafavi, Freitas, Liu, Mahalingam and Zaid. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ravi Krishnan, ci5rcmlzaG5hbkBwYXJhZGlnbS5jb20uYXU=; Suresh Mahalingam, cy5tYWhhbGluZ2FtQGdyaWZmaXRoLmVkdS5hdQ==
†These authors share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.