
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 14 February 2023
Sec. B Cell Biology
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1030813
 Ophélie A. Martin1†
Ophélie A. Martin1† Morgane Thomas1‡Marie Marquet1‡Charlotte Bruzeau1Armand Garot1Mylène Brousse1
Morgane Thomas1‡Marie Marquet1‡Charlotte Bruzeau1Armand Garot1Mylène Brousse1 Sébastien Bender1,2Claire Carrion1Jee Eun Choi3
Sébastien Bender1,2Claire Carrion1Jee Eun Choi3 Bao Q. Vuong3Patricia J. Gearhart4
Bao Q. Vuong3Patricia J. Gearhart4 Robert W. Maul4
Robert W. Maul4 Sandrine Le Noir1*
Sandrine Le Noir1* Eric Pinaud1*
Eric Pinaud1*Intoduction: Two scaffold/matrix attachment regions (5’- and 3’-MARsEµ) flank the intronic core enhancer (cEµ) within the immunoglobulin heavy chain locus (IgH). Besides their conservation in mice and humans, the physiological role of MARsEµ is still unclear and their involvement in somatic hypermutation (SHM) has never been deeply evaluated.
Methods: Our study analyzed SHM and its transcriptional control in a mouse model devoid of MARsEµ, further combined to relevant models deficient for base excision repair and mismatch repair.
Results: We observed an inverted substitution pattern in of MARsEµ-deficient animals: SHM being decreased upstream from cEµ and increased downstream of it. Strikingly, the SHM defect induced by MARsEµ-deletion was accompanied by an increase of sense transcription of the IgH V region, excluding a direct transcription-coupled effect. Interestingly, by breeding to DNA repair-deficient backgrounds, we showed that the SHM defect, observed upstream from cEµ in this model, was not due to a decrease in AID deamination but rather the consequence of a defect in base excision repair-associated unfaithful repair process.
Discussion: Our study pointed out an unexpected “fence” function of MARsEµ regions in limiting the error-prone repair machinery to the variable region of Ig gene loci.
The IgH locus, encoding the immunoglobulin heavy chain, is among the most complex in mammals, with multiple cis-regulatory elements controlling stepwise DNA accessibility to recombination and mutation through mechanisms that mainly rely on transcription (1). Current studies of the dynamic processes that regulate chromatin conformation changes and subnuclear location have renewed interest in cis-regulatory regions that delimit differentially regulated chromosomal domains. Among such DNA regulatory regions, nuclear Scaffold/Matrix Attachment Regions (MARs) have been implicated in the structural and functional organization of these domains. The juxtaposition of MARs to intronic enhancer elements in both IgH and IgL loci and their conservation in humans, mice and rabbits (2) suggest that such regions serve physiological functions. They participate in the regulation of gene expression notably by increasing enhancer function and facilitating their action over large distances. Several proteins found to bind MARs are expressed ubiquitously or in a tissue-specific manner, respectively defining constitutive or facultative MARs (3). Once attached to the nuclear matrix in a tissue specific fashion, facultative MARs could form topological barriers that could isolate or fasten chromatin regions (3). Such barriers could induce DNA torsional strain with positive and negative DNA supercoiling, respectively, upstream and downstream from the RNA pol II-induced transcription bubble (4). The supercoils are then released by the action of dedicated topoisomerases (5).
The IgH Eµ enhancer region is a combination of both the core Eµ (cEµ) enhancer element (220 bp) and two 310–350-bp flanking MARs (MARsEµ) that were first defined by in vitro matrix-binding assays (6). This region, especially cEµ, controls early VDJ recombination events (7, 8) and is also involved in Ig µ chain expression in pre-B cells (9). However, its role in SHM remains unclear. An elegant model of deletion in the endogenous Eµ region of hybridoma cells, enforced for human AID expression, suggested the requirement of cEµ and a substantial function of MARsEµ for SHM (10). Similarly, when added to transgenes, cEµ and its flanking MARs contribute to Ig µ chain expression and high levels of SHM (11–16). In contrast, knock out (KO) models underlined the complexity of its physiological regulation. In a mouse model carrying the pre-rearranged VB1-8i region, Eµ deletion still resulted in a high level of SHM in Peyer’s patch germinal center (GC) B cells, arguing for a non-essential role of the enhancer (17). More clearly, deletion of cEµ in the mouse germline did not reduce SHM frequency but only slightly increased the proportion of unmutated alleles; this minor effect was likely due to the reduced inflow of peripheral and, consequently, GC B cells in this model (7). Strikingly, the role of MARsEµ was also elusive and somewhat controversial. Whereas their endogenous deletion, analysed in mouse chimeras by the RAG-2 complementation assay, demonstrated that MARsEµ are dispensable for VDJ recombination and IgH expression (18), the ambiguous function of MARsEµ was sustained by the discrepancy between their ability to either bind negative regulatory factors (19, 20), improve cEµ enhancer efficiency (21), or substitute for cEµ to maintain IgH expression (22). At the κ light chain locus (Igκ), the intronic enhancer Eiκ region also contains an upstream MAR. The implication of MAREiκ as an enhancer of SHM was first suggested in transgenic studies (23) and then tested in a KO mouse model that accumulated premature light chain rearrangements with a mild SHM defect (24), an effect comparable to one observed at IgH locus in hybridoma cells devoid of MARsEµ (10). At that time, while these studies instigated a variety of hypotheses accounting for MARs in modulating SHM (25), these were contradicted by a study comparing 3’Eκ-and MAREiκ- function in mouse KO models (26).
To address the controversy over the role of the scaffold in SHM, we generated a mouse model carrying a germline deletion of MARsEµ and bred it into DNA repair-deficient backgrounds. In our models devoid of MARsEµ and their wt counterparts, we proceeded to side by side comparison of total SHM, transcription patterns, AID targeting and error prone repair events leading to SHM, in regions located upstream and downstream from the intronic enhancer. Our study showed that the absence of MARsEµ allows some of the error-prone repair machinery to get access to the region downstream from the Eµ enhancer. We propose that MARsEµ act as physiological barriers for error-prone repair in activated B cells. As a rational hypothesis, our study suggests that the conservation of nuclear matrix attachment regions in Ig genes serves to optimize SHM events upstream from the intronic enhancer region.
We generated a mouse mutant line carrying an endogenous deletion of both the 5’ and 3’ IgH matrix attachment regions that flank the JH-CH intronic cEµ enhancer. Although generated with slightly different targeting vector backbone and homology arms, the resulting IgH allele, so-called MARsEµΔ (Figure 1A), is similar to that generated by Sakai et al. (18). Bone marrow subsets of B cell precursors were analysed in wt and homozygous MARsEµΔ/Δ deficient mice. When compared to age-matched wt animals, MARsEµΔ/Δ mice exhibited normal proportions and numbers of pre-proB, pro-B and pre-B cell precursors (Supplementary Table 1). Unlike endogenous deletion of the entire Eµ region (9), MARsEµ deletion did not modify Ig µ heavy chain expression in early B lineage cells since proportions of IgM-expressing bone marrow B cell populations (immature, transitional and mature recirculating B cell subsets) were comparable to those of wt (Supplementary Table S1 and Supplementary Figure S1A). Mature B cell subsets were also similar to wt in the spleen and peritoneal cavity of homozygous MARsEµΔ/Δ mutants (Supplementary Table S1 and Supplementary Figures S1B, C). In agreement with the normal inflow of mature B cells in MARsEµΔ/Δ animals, Peyer’s patches were efficiently colonized by naive and GC B cells. Numbers and proportions of GC B cells were even significantly increased in homozygous mutants (Figure 1B left panels and Supplementary Table S1). The similar proportion of proliferating KI67+ GC B cells in wt and MARsEµΔ/Δ mice implied that this increase was not due to over-proliferation of Peyer’s patch B cells (Figure 1B right panels). Finally, levels of serum Ig isotypes were unaffected in MARsEµΔ/Δ animals (Figure 1C). This normal B cell homeostasis in homozygous mutants confirmed that MARsEµ are dispensable for B cell ontogeny and antibody production. This statement is in agreement with previous studies of an analogous MAR region in the Igκ locus (24).
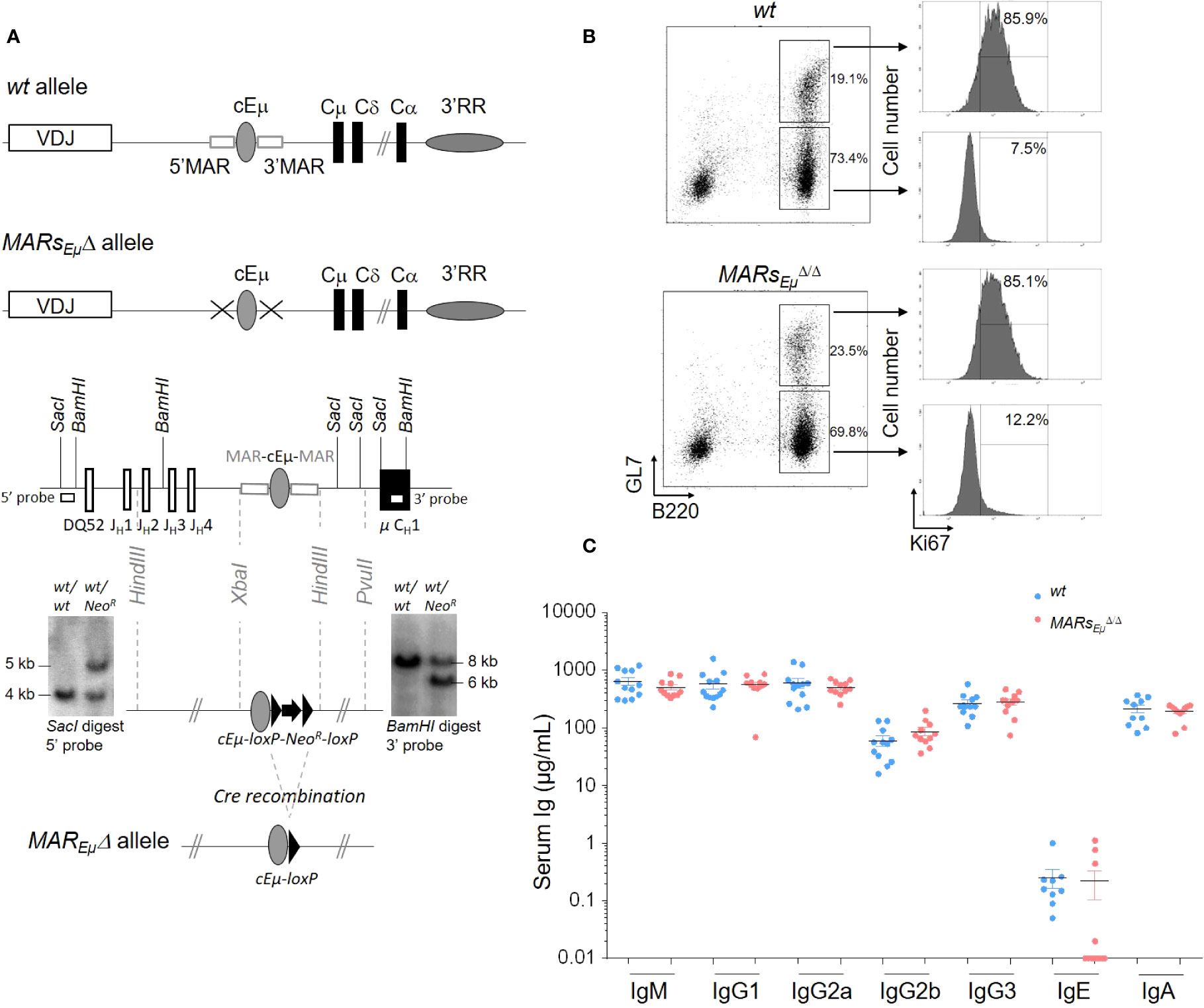
Figure 1 MARsEµ deletion supports efficient in vivo Ig isotype production and GC B cell development. (A) Schematic representation of wt and MARsEµΔ alleles (top). Targeting construct and Southern blot performed on recombinant ES cells with NeoR insertion. Hybridization with the 5′ probe detected 4 kbp and 5 kbp SacI genomic fragments respectively for wt and recombined alleles. Hybridization with the 3′ probe detected 8 kbp and 6 kbp BamHI genomic fragments respectively for wt and recombined alleles. MARsEµΔ allele preserved the cEµ enhancer after Cre-recombination. (B) Comparison of Peyer’s patch B cells subsets from wt and MARsEµΔ/Δ animals by flow cytometry: dot plots showed percentage of naïve (B220+/GL7-) and GC (B220+/GL7+) B cells (left panels) and, for each subset, the percentage of dividing cells (Ki67+) was indicated on cell count histogram plots (right panels). Experiments were performed twice with a minimum of 3 mice per group. (C) Immunoglobulin isotype secretion in sera from wt and MARsEµΔ/Δ mice determined by ELISA (n=9 to 12 mice, mean ± SEM).
To assess whether MARsEµ deletion could affect IgH somatic hypermutation, we first quantified mutations within the 500-bp regions downstream from the variable exons rearranged to JH3 and JH4 segments (Figure 2A) in Peyer’s patch GC B cells sorted from wt and MARsEµΔ/Δ (animals overall data reported in Figure 2 left, data from individual animals reported in Supplementary Figure S2 and Supplementary Tables S2A, B). For this we used two complementary sequencing methods: the first one, based on classical Sanger approach and GS junior technology, allowed to discriminate and exclude unmutated and clonally related sequences from the calculation of SHM frequency, as initially described (27). The second method used Ion proton deep sequencing coupled to DeMinEr filtering. Since using DNA templates including non-mutated alleles, this second approach underestimated the SHM frequency; but since including AID-deficient control samples as a reference, the method provided highly reproducible and reliable quantification of SHM in a DNA sample extracted from GC B cells (28). Interestingly, by using Sanger approach, MARsEµΔ/Δ GC B cells displayed significant differences in the distribution of mutations: an increased proportion of unmutated sequences (less than 10% in wt compared to 38% in MARsEµΔ/Δ) (from 30.6 to 45.5%, overall data collected from several mice, data from independent mice in Supplementary Figure S2A) Another effect of MARsEµ deletion on IgH SHM targeting was the strong decrease in highly mutated sequences (>10bp per sequence). In wt, the proportion of highly mutated alleles reached ~24% (from 18.9 to 28.3%) while in mutants they were barely present ( ∼2%) (Figure 2B left and Supplementary Fig S2A). When comparing only the mutated sequences, mutation frequency was decreased at least by two fold in MARsEµΔ/Δ mutants, with 7.6 mutations per 1000 bp (in average) compared to 14.9 in wt (in average) (Figure 2B left and Supplementary Figure S2A). By using next generation sequencing (NGS), the decreased SHM frequency was also highly significant (Figure 2C, 12.3‰ vs 8.3‰, p=0.008, individual mice in Supplementary Table S2A).
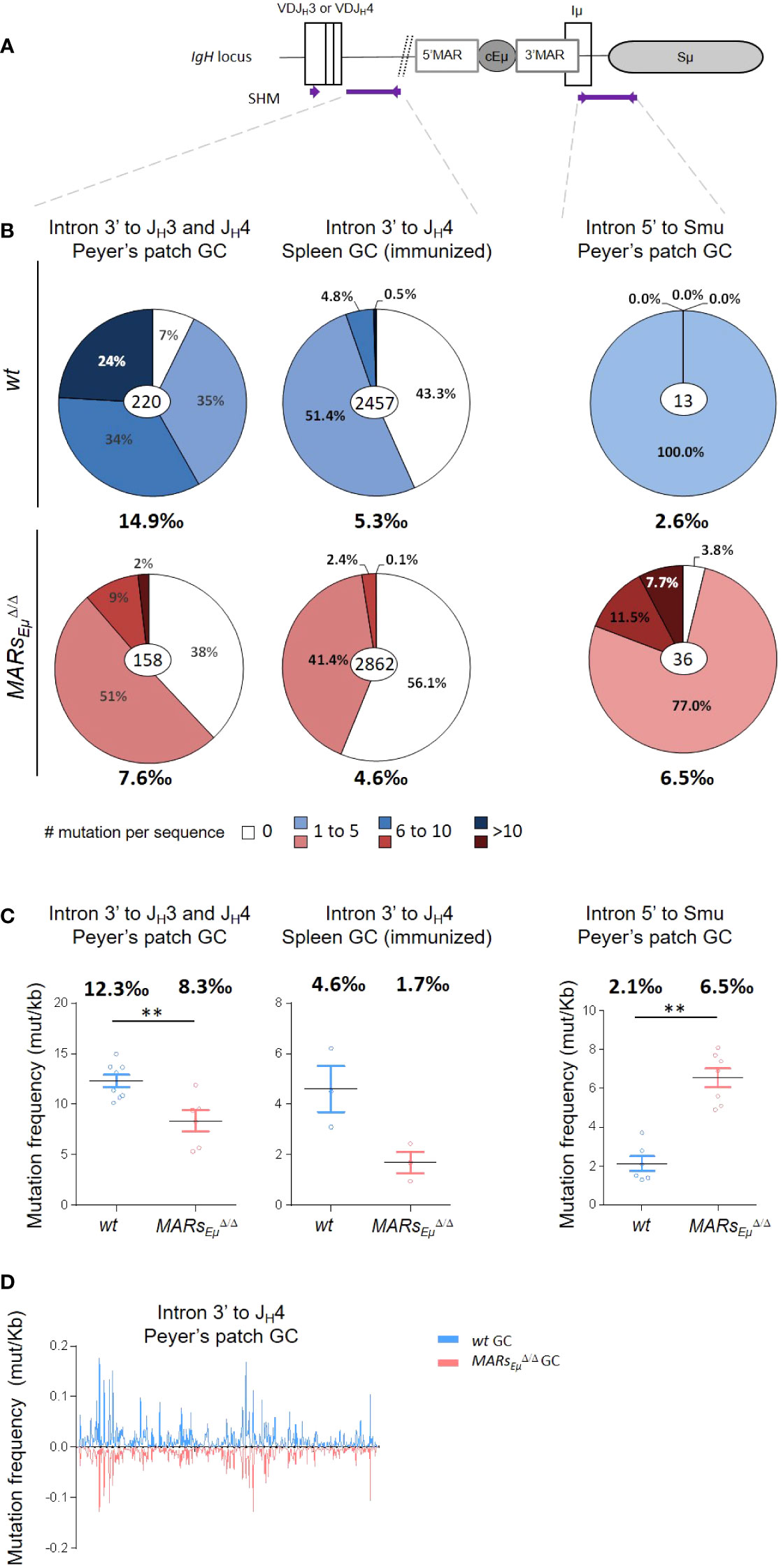
Figure 2 MARsEµ deletion impairs the overall SHM frequency and distribution within the IgH J-C intronic region. (A) Location of IgH regions (thick purple lines) tested for SHM, arrows represent primers used for PCR amplification. (B) Pie charts represent distribution of mutated sequences (proportional to the area in each slice, data obtained by Sanger and GS Junior sequencing method) quantified in wt and MARsEµΔ/Δ mice in individually recombined IgH alleles. For each genotype number of individual clones is indicated in the center (after removal of clonally related sequences based on VDJ junction) and overall mutation frequencies (mutation per 1000 bp in mutated clones) are indicated below. Left: SHM downstream from JH3 and JH4 segments in Peyer’s patch sorted GC B cells, data obtained after cloning and sequencing by classical Sanger method. Middle: SHM downstream from JH4 segments in spleen GC B cells sorted from SRBC-immunized mice, data obtained by NGS (GS Junior). Right: SHM downstream from cEµ region from Peyer’s patch GC sorted B cells, data obtained by classical Sanger method. (C) Graphical representation of SHM frequency in wt and MARsEµΔ/Δ mice, quantified by NGS (Ion Proton) submitted to DeMinEr filtering, a pipeline that identifies substitution frequency at each nucleotide based on an AicdaΔ/Δ control sample (28). Since no indication in sequence distribution is available using this method, data were represented as scattered plots, each point refers to a mutation frequency from one individual mice, mean mutation frequencies are indicated above. p-value was determined with two tailed Mann Whitney test; significant differences are indicated by: **P < 0.01 and error bars represent SEM of two independent experiments. (D) Mutation distribution along the JH4 intron in wt (top) and in MARsEµΔ/Δ (bottom).
To monitor SHM upon antigen challenge, we analyzed mutations by Sanger and NGS in a large number of GC B cells sorted from spleen of SRBC-immunized MARsEµΔ/Δ and wt mice. In parallel; we verified on spleen frozen section that efficient GC formation was indeed comparable between MARsEµΔ/Δ and wt mice (Supplementary Figure S2B). In the intronic region downstream from the JH4 segment, SHM frequency dropped from 5.3 (Sanger method, excluding unmutated clones) or 4.6 (NGS bulk method) mutations per 1000bp in wt cells to respectively 4.6 or 1.7 in MARsEµΔ/Δ cells (Figure 2B middle, and 2C, Supplementary FigureS2C, Supplementary Table S2B). Similarly to what was observed in spontaneous GC B cells from Peyer’s patches, NGS assays performed on few immunized animals also showed that SHM was decreased upon MARsEµ-deletion only eight days after antigen challenge (Figure 2C middle). Immunization assays also showed an increased proportion of unmutated or poorly mutated sequences in MARsEµ-deficient B cells (Figure 2B and Supplementary Figure S2C). This confirmed that the intronic region was less efficiently targeted by SHM in MARsEµ deficient mice. An identical SHM defect was also observed in mice harbouring deletion of the entire Eµ region (core enhancer and flanking MARs; (9)) (Supplementary Figure S2A top right); this data indicated that the SHM failure was the only consequence of MARs deletion. This hypothesis is completely consistent with a previous study showing that SHM efficiency was not affected by the endogenous deletion of the cEµ enhancer alone (7). The comparison between those three models is certainly relevant since all knock outs were created in murine germlines with similar mixed genetic backgrounds.
To reinforce the unbiased SHM evaluation within JH introns, we sought to evaluate the impact of MARsEµ deletion on acquired diversity of the VDJ exon by quantifying SHM events taking place in the out of frame (passenger) IgH alleles. This parallel unbiased analysis was made possible in a region physiologically highly targeted by SHM (overlapping parts of the FR3 and CDR3 domains) thanks to the IMGT/HighV-QUEST reference tool (29). Although focused on a restricted region of out of frame VDJ exons, this study performed on GC cells sorted from Peyer’s patches still highlighted a decreased SHM frequency in the MARsEµΔ/Δ model (∼27‰ in mutants compared to ∼32‰ in wt, Supplementary Table S3). This strongly suggests that the SHM defect observed within JH introns in the absence of MARsEµ extends to their respective upstream VDJ exons.
Since our MARsEµ deletion includes the 3’HinfI-XbaI genomic region that contains transcription start sites and part of the Iµ exon (6, 30), we also quantified SHM immediately downstream from this exon in a 629bp region described as mutated in GC B cells (Figure 2A) (31, 32). Unlike the intronic regions downstream from the rearranged VDJ exon, the overall mutation frequency downstream from Iµ was strongly increased in GC B cells devoid of MARsEµ region and reached 6.5 (in average) mutations per 1000 bp compared to 2.6 (in average) in wt cells (Figure 2B right). This suggests that the region downstream from the cEµ was more efficiently targeted in the absence of its MARs. This was supported by the very low proportion of unmutated sequences in wt (Figure 2B right and Supplementary Figure S3A) and the increased proportion of highly mutated sequences in MARsEµ-deficient GC B cells (Figure 2B right and Supplementary Figure S3A). This data was efficiently confirmed by NGS analysis that estimated that mutation frequency was increased by 3.5 fold in the MARsEµ-deficient GC B compared to wt mice (2.1‰ vs 6.5‰) (Figure 2C right and Supplementary Table S2C). In our mouse model, 3’MARs deletion brought Sµ region closer to the cEµ enhancer element. Given this, we also calculated SHM frequency in a 473bp region located at the same distance from the cEµ element in wt and MARsEµ-deficient mice. Similarly to what was reported in Figure 2, we also observed, at the same distance of cEµ, an increase in SHM frequency upon MARsEµ-deletion (Supplementary Figure S3B).
Analysis of mutation distribution in wt and in MARsEµΔ/Δ mice did not show any difference between models (Figure 2D), indicating that, while affecting SHM efficiency, the absence of MARsEµ region did not influence DNA sequence hotspot or preferences for SHM within the JH4 intron.
Importantly, our mouse model clearly assigns a specific function for endogenous MARsEµ on SHM at the IgH locus, in accord with the requirement of similar regions for efficient SHM previously pointed out in the endogenous Igκ Kappa light chain locus. This pioneer study, describing the specific deletion of a 420bp MAR region upstream from the intronic Kappa enhancer (Eiκ), highlighted a modest decrease in SHM by quantifying mutations downstream from the Jκ5 segment in GC B cells from Peyer’s patches (24). While our study suggests that MARsEµ optimizes SHM upstream from the cEµ enhancer; the presence of such regulatory regions does not prevent the SHM machinery to get access to downstream regions as reported in a recent study (33). This hypothesis is mostly supported by the increased SHM frequency downstream from the cEµ enhancer in the absence of MARsEµ, a finding consistent with previous works describing increased Sµ internal deletions in hybridomas devoid of MARs regions (34). We could speculate that one physiological function of MARsEµ regions in GC B cells is to tightly isolate the VDJ transcription unit by, at least temporarily, attaching the Eµ region to the nuclear matrix. Such a “locked” target conformation could provide an optimal environment for somatic mutations by trapping the transcription machinery and its co-factors including AID and error-prone repair factors. This topological barrier could, at the same time, partially protect downstream constant regions from SHM; although this configuration should be brief since regions downstream from Eµ are also efficiently targeted by AID in GC B cells (35).
It is well established that SHM in Ig V segments is coupled to transcription initiated at V promoters (36). To investigate transcription-related events in SHM-targeted regions upstream and downstream from Eµ, we precisely quantified the total amounts of steady state total IgH primary transcripts by using multiple q-PCR probes located respectively downstream from JH4 and JH3: the previously described probe A (37) (Figure 3A and Supplementary Figure S4) complemented by probes A’ and C (Supplementary Figures S4, S5). The use of cDNA templates conducted with random hexamers showed that the amount of total IgH primary transcripts running upstream from Eµ did not display significant variations between wt and MARsEµ-deficient cells in both GC and in vitro-stimulated samples by using probe A (Figure 3B) as well as with probes A’ and C, although an upward trend could be noticed in LPS-activated samples (Supplementary Figure S5A left). The intriguing discrepancy between the mutation phenotype observed in MARsEµ-deficient GC B cells and the silent effect on global transcription motivated a more complete study of transcription events occurring upstream from Eµ, particularly sense and antisense transcription since the latter has been found in cells undergoing SHM (38). To proceed, we generated cDNA templates with sense transcripts, initiated at the promoter of the rearranged VDJ segment, with three primers located downstream from the JH4 segment (S1 and S2) and within the cEµ enhancer (S3) (Figure 3C and Supplementary Figure S4, S5A). Reciprocally, we generated cDNA templates with antisense transcripts, initiated in the intronic regions upstream from Eµ as described by Perlot et al. (38), with four primers respectively located downstream from JH2 (AS0), JH3 (AS1) and JH4 (AS2 and AS3) (Figure 3E and Supplementary Figure S4 and S5B). For both sense and antisense, quantification of steady state transcripts was possible with the same probes A, A’ and C. For strand-specific quantification assays with a given probe, the baseline level was either provided by a control reaction (P-) measuring endogenous priming since devoid of primer or by one strand-specific template that cannot be detected by the probe (T-) as reported previously (39, 40). To note, strand-specific transcripts were optimally detected when primers and probes were closer (sense transcripts with primer S1/probe A or antisense transcripts with primer AS2/probe A). Side by side comparison of wt and MARsEµ-deficient activated B cells samples revealed several interesting differences. When quantified with optimal primer S1/probe A tandem, sense transcripts were significantly increased in the absence of MARsEµ (Figure 3C). In GC B cells, a two fold increase was noticed by using S1 template (Figure 3C, left bar graph, p=0.019). In in vitro-activated cells, an increase of sense transcription was also observed upon MARsEµ-deletion, this effect became significant for long transcripts that reach the cEµ (Figure 3C, right bar graphs, p=0.004 for S3/probe A). By using A’ and C probes, sense transcripts were hardly detectable in GC samples (Supplementary Figure S5A; middle bar graphs); although a significant increase was noticed with S3/probe C (p=0.043) and S2/probe C (p=0.02) tandems in LPS-activated samples upon MARsEµ-deletion (Supplementary Figure S5A; right bar graphs). As a potential consequence of the increased transcription of the VDJ unit in observed upon MARsEµ deletion, we measured by flow cytometry the level of intracellular Igµ chain in Peyer’s patch GC B cells (B220+/GL7+) of wt and MARsEµΔ/Δ mice. The significant increase of Igµ chain expression in the absence of MARsEµ region (Figure 3D, p=0.002) corroborate our sense-transcription data. This indicated that the absence of MARsEµ certainly did not hamper RNA pol II machinery to progress 3’ to the VDJ unit and might even facilitate this process in activated cells.
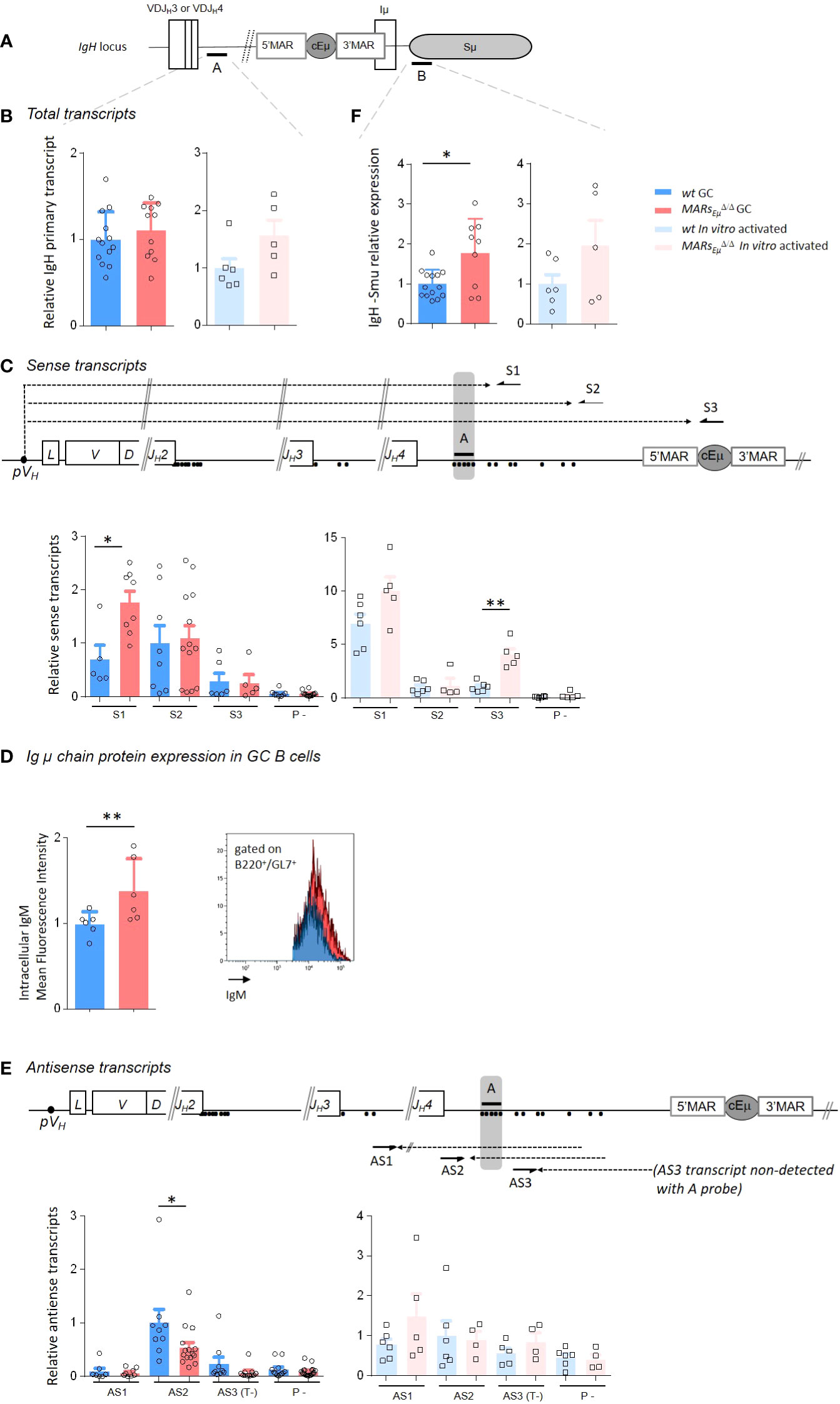
Figure 3 MARsEµ deletion impairs strand-specific transcription upstream from Eµ region. (A) IgH locus specifying location of q-PCR probes (A, B) used for transcripts quantification. (B) Total primary transcripts quantified downstream from the JH4 segment with probe A in Peyer’s patch GC B cells (dark colors) and in vitro-activated B cells (light colors) from wt and MARsEµΔ/Δ mice. (C) Detection of sense transcripts (dotted arrows) in murine IgH locus (not to scale). Arrows indicate primers (S1, S2, S3) downstream from JH3 and JH4 used for strand-specific reverse transcription. Primary sense transcripts were quantified with probe A (black bar) in Peyer’s patch GC B cells and in vitro-activated B cells from wt and MARsEµΔ/Δ mice. Dots indicate antisense transcript start sites according to Perlot et al. (38). Baseline levels were defined by using a RT template performed without primers (P-). Bar graphs show the relative quantity of sense transcripts obtained from template S1, S2 and S3 (mean ± SEM) from two to three independent experiments. (D) Intracellular IgM mean fluorescence intensity measured by flow cytometry in GC B cells from Peyer’s patches of wt and MARsEµΔ/Δ mice. Bar graphs indicate data from individual mice (n=6 mice in 2 independent experiments, mean ± SEM); a representative example of cell count overlay is shown. (E) Detection of antisense transcripts (dotted arrows) in murine IgH locus (not to scale). Arrows indicate primers (AS1, AS2, AS3) downstream from JH3 and JH4 used for strand-specific reverse transcription. Primary antisense transcripts were quantified with probe A (black bar) in Peyer’s patch GC B (dark colors) cells and in vitro-activated (light colors) B cells from wt and MARsEµΔ/Δ mice. Dots indicate antisense transcripts start sites according to the Alt study (38). Baseline levels were defined by using a RT template performed without primers (P-) or by using a strand-specific template that cannot be detected with A probe (T-). (F) Total primary transcripts quantified downstream from cEµ region with probe B in Peyer’s patch GC B cells (dark colors) and in vitro-activated B cells (light colors) from wt and MARsEµΔ/Δ mice. p-value was determined with two tailed Mann Whitney test; significant differences are indicated by: *P < 0.05; **P < 0.01 and error bars represent SEM of two to three independent experiments.
Globally less abundant than their sense counterparts, quantification of antisense transcripts running downstream from JH segments showed quite different patterns (Figure 3E and Supplementary Figure S5B). While quite similar levels were detected in LPS-activated samples (Figure 3E right and Supplementary Figure S5B), intronic antisense transcripts were about 2 fold less abundant in MARsEµ-deficient GC B cells when detection was allowed by optimal probe/primer combination (Figure 3E, left, p=0.025, AS2/probe A and Supplementary Figure S5B left, p=0.041 for AS3/probe A’). As a complementary experiment, sense and antisense transcription patterns were also determined in naive B cells in order to assess whether the influence of MARs was restricted to the activated stage. Data proved this was the case since, in naive cells sorted from Peyer’s patches, transcription patterns as well as intracellular IgM levels were unchanged in wt and MARsEµΔ/Δ mice (Supplementary Figures S6A, B); identical transcription patterns were also observed in splenic resting B cells (Supplementary Figure S6C).
The obvious unbalanced sense/antisense transcription ratio could result from either weak transcription efficiency or instability of antisense products. Nevertheless, Perlot et al. identified by RACE assays, in normal GC B cells, multiple antisense-transcript initiation start sites downstream from every JH region and raised the question of specific enhancers. Our current data refines this previous study by identifying MARsEµ as potential boosters of antisense transcripts that, given their proximity to the enhancer, could achieve some regulatory function like eRNA or PROMPT/uaRNA (41). Highlighting a correlation between mutation efficacy and strand-specific transcription pattern upstream from Eµ, our data support the idea that some level antisense transcription downstream from the VDJ exon could prepare to SHM (38). Seemingly transient, specific to cell subsets and occurring upstream from an enhancer, such antisense transcripts could be substrates for RNA exosome and lead to optimized SHM targeting as proposed by Basu and colleagues (42–44).
Since a strong increase of mutations was observed within the Sµ region in the absence of MARsEµ, we also sought to correlate SHM and transcription on the other side of cEµ by quantifying total transcripts (probe B) running in this region (Figure 3A). In this case and according to what could be expected, transcription was significantly increased in MARsEµ-deficient GC B cells from Peyer’s patches (Figure 3F left, p=0.04); a similar trend, although not significant, was observed in LPS activated B cells (Figure 3B right p=0.32). Accordingly, we also observed a modest but reproducible increase of CSR Cγ3 and Cγ1 in MARsEµ-deficient B cells stimulated in vitro respectively by LPS or by LPS + IL4 cocktail (Supplementary Figure S7). A similar modest CSR effect associated to Sµ internal deletions has been previously reported in hybridomas carrying the same MARsEµ-deletion (34). This indicated that the absence of MARsEµ leads to a global increase in transcription of the donor S region and consequently favours SHM targeting.
The significant changes in transcription patterns upstream and downstream from cEµ observed in our models put forward the hypothesis that MARsEµ act as physiological barriers in activated B cells, limiting sense transcription of the VDJ unit up to the intronic enhancer. For transcription running through the Sµ region, our data is in agreement with a repressive function of MARsEµ in activated B cells, in order to limit SHM targeting of this area. However, our data also suggest that MARsEµ act as transcriptional repressors of the VDJ unit in both naïve and activated cells; a statement in contradiction with our hypothesis that MARsEµ facilitates SHM upstream from cEµ. To settle such a discrepancy in our MARsEµ-deficient B cells, we first questioned AID deamination efficiency and second error-prone repair pathways processing in SHM targeted regions: upstream and downstream from cEµ.
One critical experiment needed to challenge the function of MARsEµ as physiological barrier for SHM machinery was to first assess whether IgH AID targeting could be impaired in the absence of MARsEµ. To proceed, we bred our MARsEµ-KO mice in a genetic background deficient for both base excision repair (UngΔ/Δ) and mismatch repair (Msh2Δ/Δ) in order to evaluate, on and unbiased manner, the DNA footprint of AID deamination upstream and downstream from cEµ (Figure 4A). As expected and according to the literature (45–47), models deficient for both BER and MMR displayed only transitions at C/G pairs reflecting cytidine deamination on respectively the template and non-template strands. By looking at deamination frequencies between control (UngΔ/Δ Msh2Δ/Δ) and mutant animals (UngΔ/Δ Msh2Δ/Δ MARsEµΔ/Δ), our data showed that AID activity upstream from cEµ was not impeded upon MARsEµ-deletion; while differences were not statistically significant (evaluated on n=3 to 4 mice of each genotype), cytidine deamination even tended to be increased in B cells devoid of MARsEµ, on both sides of cEµ (Figure 4B and Table S4A). When compared to control animals (UngΔ/Δ Msh2Δ/Δ), nucleotide substitution patterns were unchanged in the absence of MARsEµ (Figure 4B and Supplementary Figure S8A), proving identical strand-specific cytidine deamination: roughly 2/3 on the non-template strand (C to T substitutions) and 1/3 on the template strand (G to A substitutions). Besides imbalanced transcription upstream from Eµ, this data indicates that MARsEµ-deletion does not impact the choice of any DNA strand for AID targeting within intronic regions.
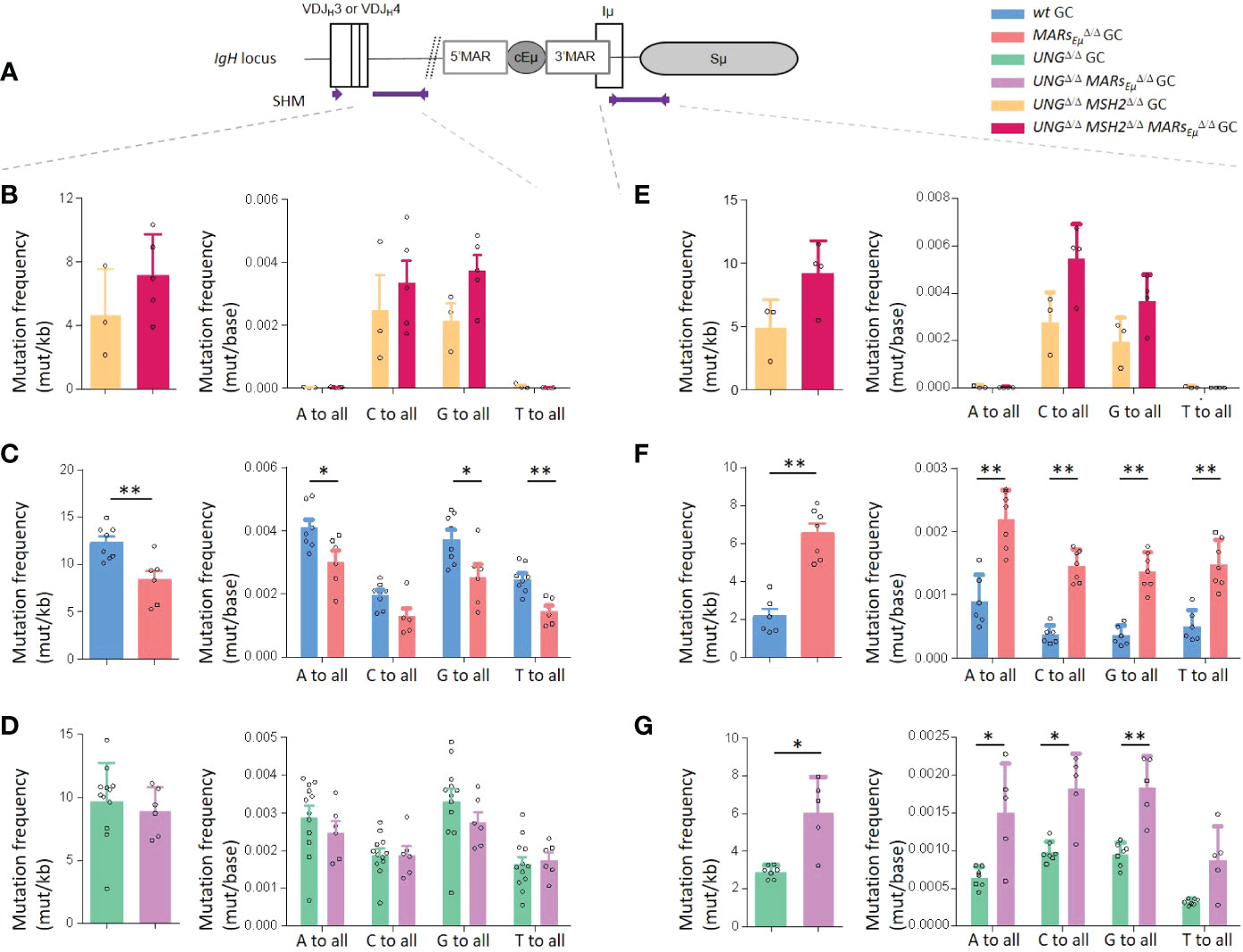
Figure 4 MARsEµ deletion impedes error-prone repair pathways upstream from Eµ region. Comparison of IgH SHM events occurring on both sides of cEµ in Peyer’s patch GC B cells sorted from wt and MARsEµΔ/Δ mice models, bred in genetic backgrounds deficient for base excision repair (Ung KO) and mismatch repair (Msh2 KO). Data were obtained by NGS (Ion Proton) combined to DeMinEr filtering (28). In each region, analyzed and represented as a panel, bar graphs report overall mutation frequencies (left) and detailed mutation frequencies at all bases (right). (A) Location of IgH regions (thick purple lines) tested for SHM, arrows represent primers used for PCR amplification. (B) SHM downstream from JH4 in double-deficient UngΔ/Δ Msh2Δ/Δ background. (C) SHM downstream from JH4 in DNA repair proficient (Ung+/+ Msh2+/+) background. (D) SHM downstream from JH4 in UngΔ/Δ background. (E) SHM downstream from cEµ in double-deficient UngΔ/Δ Msh2Δ/Δ background. (F) SHM downstream from cEµ in DNA repair proficient (Ung+/+ Msh2+/+) background. (G) SHM downstream from cEµ in UngΔ/Δ background. p-value was determined with two tailed Mann Whitney test; significant differences are indicated by: *P < 0.05; **P<0.01 and error bars represent SEM of two to three independent experiments.
This notable increased AID deamination footprint prompted by MARsEµ-deletion was in total agreement with the increased transcription observed in the corresponding regions of activated B cells. The obvious discrepancy between efficient C to U deamination events and the strong SHM targeting defect within the same JH intron region unravel the origin of the SHM defect in MARsEµ-deficient mice as a default of the mutagenic process occurring downstream from the normally-introduced U-G mismatches in DNA.
This prompted us to investigate whether MARsEµ-deletion could provoke skewed mutation patterns within SHM-targeted regions. In the intron region downstream from JH4, mutation frequency at each of the four bases in wt and MARsEµ-deficient backgrounds (Figure 4C and Table S2A) revealed a global significant decrease of mutations at all bases except for substitutions occurring at C in the absence of MARsEµ. Similarly, beyond significant differences for C>A, G>C, T>A and T>G events, individual mutation patterns unveiled a global decrease that did not offer any clear hypothesis regarding the mechanism impeding SHM upon MARsEµ-deletion. Factually, the unchanged relative proportion of transitions and transversions at C:G base pairs in MARsEµ deficient cells did not suggest any defect in UNG activity (45, 48, 49) (Supplementary Figure S8B).
To solve this paradox, we bred our MARsEµ-KO mice into base excision repair deficient background (UngΔ/Δ) and analysed SHM in the same region. Strikingly, in the absence of UNG, SHM frequency within the JH4 intron region was identical upon the presence (UngΔ/Δ control mice) or the absence of MARsEµ (UngΔ/Δ MARsEµΔ/Δ mice) (Figure 4D and Supplementary Table S4B). Beyond the expected increase of G/C transitions, a typical hallmark of UNG-deficient background, substitution frequencies at all four bases were also identical in UngΔ/Δ MARsEµΔ/Δ mice (Figure 4D) the same was true when looking at individual substitution events (Supplementary Figure S8C). The fact that the SHM deficiency induced by MARsEµ deletion was no more observed in UNG-deficient background (Figures 4C, D) strongly imply the involvement of BER pathway in the initial mutagenic defect. This same data also proved that SHM events occurring independently of UNG (e.g. altogether obtained by replication across U and/or processed by MMR pathway) took place normally within the JH intron in the absence of MARsEµ. Given this, a rational hypothesis to explain the origin of the SHM defect in our model was that abasic sites generated by UNG upstream from cEµ are processed differently upon the absence of MARsEµ. Our data suggests that U:G mismaches processed by UNG are accurately repaired in the absence of MARsEµ while these are normally subject to error-prone repair; sustaining for a specific function of MARsEµ in recruiting mutagenic factors associated to BER.
In contrast to what observed in the JH intron, substitution frequencies and mutation patterns downstream from cEµ evidenced a different function for such regulatory regions. In B cells capable of BER and MMR, the absence of MARsEµ significantly boosted mutations at all bases by at least two fold (Figure 4F), this was true for any kind of substitution (Supplementary Figure S8E). Substitution patterns collected in mutant animals devoid of BER and MMR highlighted a global “overtargeting” of the Sµ region induced by MARsEµ deletion (Figure 4E, Supplementary Figure S8D and Supplementary Table S4C). This was in line with the general increase in both Sµ germline transcription observed in this model. In models impaired for BER, our data showed that UNG-deficiency combined to deletion of MARs (UngΔ/Δ MARsEµΔ/Δ mice) maintained the SHM burden downstream from cEµ significantly higher than what observed for UNG alone (UngΔ/Δ control mice) (Figure 4G; Supplementary Figure S8F and Supplementary Table S4D). Such a comparison suggests that error-prone repair factors could more readily access to abasic sites generated in the S region when MARsEµ are missing. In this way, our data support the idea that MARsEµ act as physiological barrier that optimize SHM upstream from the Eµ region and rationalize the fact that MARs are evolutionary conserved downstream from Ig gene V regions (24); and moreover conserved structures in mammals (2).
One simplistic model would argue that the most important regulatory regions for IgH locus expression are conserved upon any reshaping event occurring in developing B lineage cells (VDJ recombination, CSR and SHM). Beyond the major enhancer regions, e.g. cEµ and the 3’RR, our current study identifies MARsEµ, also conserved upon any rearrangement, as part of these most critical IgH elements. Since flanking cEµ, MARsEµ could potentially interfere with the function of this regulatory element. Our current study showed that MARsEµ deletion impact the transcription pattern in this region but also points out, when compared to deletion of cEµ alone (7), that the core enhancer and MARsEµ act independently on SHM. Our data indicate that IgH MARsEµ delimit, upstream from the enhancer region, some error-prone repair processes coupled to BER.
Several studies indeed proposed that J-C intronic MARs help generate negative supercoiling and consequently increased ssDNA and potential other secondary structures that could promote accessibility to AID (50–52). The hypothesis that MARsEµ add again more DNA strain to the sense-transcribed VDJ transcription unit is relevant to the positive effect of topoisomerase depletion on AID targeting and SHM (51, 53, 54). In line with transcription dynamics, RNA pol II stalling (55, 56) and DIVAC regulatory regions are being proposed to facilitate AID targeting for SHM (57, 58). A model proposed by Alt and colleagues (59) would be that the optimal chromatin environment for AID-induced mutations would be provided by convergent transcription as the result of fine balance between sense and antisense events. As possible byproducts of RNA Pol II collision, antisense or regulatory transcripts in such regions remain transient and difficult to detect in a wt context; probably because processed by RNA exosome or other RNAse activities (60, 61). In line with our data, a recent study from the Basu group emphasize the critical function of the RNA exosome for the fine tuning of sense transcription of the VDJ exon, leading to modifications on strand-specific AID targeting and SHM (44). While also modifying sense and antisense transcription pattern, our study shows that MARsEµdeletion does not impede AID footprint but rather some of repair mechanisms acting downstream from the U-G mismatch. Since we hypothesize that UNG activity by itself is not necessarily impaired in our model, our data strongly suggest that only BER-dependent error-prone repair is impeded by MARsEµdeletion. This statement supposes that efficient MMR-dependent error-prone repair in this region does not require such a barrier. The question of MARs binding factors and their respective dynamic association to such regulatory regions needs to be further investigated. The literature already suggest that some of them, like the Special AT-rich binding factor 1 (SATB1), could act as accessory factors in BER (62). Recent findings, showing that UNG2-interacting protein FAM72A promotes error prone processing of U-G mismatch in Ig genes (48, 49), raise the question of its specific recruitment to AID-targeted regions; our current study suggests that MARsEµ could potentially interact with error-prone factors acting downstream from UNG2. Another future challenge remains to define whether some components of the nuclear matrix, nuclear filaments or proteins anchored in the envelope, could be involved in the anchorage of SHM targets.
To generate MARsEµ KO model, gene targeting for matrix attachment regions flanking the IgH Eµ enhancer element was performed by homologous recombination, in the murine E14 ES cell line, with a vector kindly provided by Dr. Frederick Alt that permitted replacement of the 995 pb region (including cEµ and its flanking MARs) by a 220 pb HinfI genomic fragment that reintroduced the cEµ enhancer, a 256 bp fragment containing plasmid sequence and a “loxP-pGK-NeoR-loxP” cassette (18). Once introduced in the mouse germline, the selection cassette was deleted in vivo by cre-loxP recombination as previously described (9) to obtain the MARsEµΔ/Δ IgH allele devoid of both 5’ and 3’ MARsEµ (respectively 344 pb XbaI-HinfI and 426bp HinfI-XbaI genomic fragments) (Figure 1A). Animal procedures were performed on 8 weeks old male and female mice. All models were created in mixed 129Sv;C57BL/6 background. Wt, MARsEµΔ/Δ, EµΔ/Δ (9), Msh2Δ/Δ, UngΔ/Δ (a kind gift of Dr S. Storck) and Aicda-/- (a kind gift of Pr. T. Honjo) homozygous mice were used for our experiments and maintained at 21–23°C with a 12-h light/dark cycle. All experiments comparing Wt, MARsEµΔ/Δ, EµΔ/Δ in DNA repair proficient background used systematically control mice carrying two IgH alleles of the same haplotype: IgHa (from 129/Ola strain). Procedures were reviewed and approved by the Ministère de l’Education Nationale de l’Enseignement Supérieur et Recherche autorisation APAFIS#16639-2018090612249522v2.
Genomic Southern blots were performed as follows: 20 µg genomic DNA were digested by SacI or BamHI and submitted to electrophoresis on a 0.7% agarose gel. DNA was transfered to nylon membranes (MP Biomedicals) by capillarity. Blots were hybridized with [32P]-labeled probes generated by random priming. Hybridization with 5’ probe (0.803 kpb SacI-SphI fragment) and 3’ probe (0.8 kpb XbaI-BamHI fragment) located outside the homology arms were used to identify ES cell clones in which MARsEµ were replaced by the loxP-pGK-NeoR-loxP cassette (Figure 1A).
Sera were collected at 8 weeks of age from non-immunized wt and MARsEµΔ/Δ mice and analyzed for the presence of different Ig classes and subclasses by ELISA as previously described (63).
Mice were challenged by intraperitoneal injection with 200µL 50% sheep red blood cell suspension and sacrificed 8 days later to collect GC B cells (B220+/GL7+) in the spleen. Efficient immunization was assessed by counting GC numbers on spleen frozen sections upon immunofluorescence labelling.
Spleens cryosections (10µm) were fixed and permeabilized for 20 minutes in cold acetone at -20°C. Sections were stained with the following primary Abs GL7-FITC (1/50), αB220 APC (1/1000), αIgD-AF594 (1/1000) diluted in PBS/FCS 2%/EDTA 2mM, overnight at 4°C. After 2 washes with PBS, DAPI was added. Images were acquired using an epifluorescent microscope (NIKON). GC structures were identified according to their content in B220+GL7+ cells.
Flow cytometry analysis was performed on LSR-Fortessa cell analyzer (BD Biosciences) on single-cell suspensions from fresh organs. Once washed with 2% fetal calf serum-PBS, lymphoid cells from bone marrow, spleen, peritoneal cavity and Peyer’s patches were labeled with various conjugated Abs: αB220-V450, αCD117-PE, αCD43-PE for bone marrow cells. αB220-V450, αCD21-PE, αCD23-FITC, αIgM total-PE, αIgD-FITC and αCD3e-FITC for splenocytes. αB220-V450, αIgM-PE, αCD5-FITC for peritoneal cavity. αB220-V450, αB220-APC, αIgA-FITC, αIgM-PE, αPNA-FITC, αFAS-PE, αKi67-FITC and GL7-FITC for Peyer’s Patches. (Southern Biotechnology Associates; eBioscience; Sigma and BD Biosciences). Flow cytometry cell sorting was performed on an ARIA 3 (BD Biosciences) apparatus on single-cell suspensions from spleens or Peyer’s patches. Once washed with 2% fetal calf serum-PBS, cells were labeled with PNA, GL7, αB220, and αFAS reagents and sorted based on distinct gates defined as germinal center B cells (B220+/GL7+ or B220+/PNAHigh/Fas+).
Splenocytes were collected, after red blood cells lysis, CD43+ cells were depleted using anti-CD43 MicroBeads (Miltenyi Biotec). CD43- splenic B cells were cultured for 3 days at a density of 1x106 cells per mL in RPMI 1640 supplemented in 10% serum calf fetal, sodium pyruvate (Lonza), amino acid (NEAA 100x Lonza) and Penicillin-Streptomycin (Gibco) with 1µg/ml LPS (In vivoGen) alone (for transcription assays) or plus 20ng/ml IL4 (PeproTech) (for CSR experiments).
SHM analysis within the JH4 intron was either performed by cloning followed by classical Sanger method as described (64) or performed directly on PCR products by next generation sequencing using GS Junior (Roche) or Ion Proton system (Applied Biosystem). SHM within Smu intron was performed by using the following primer SmuF: 5’-AAGGGCTTCTAAGCCAGTCC-3’and SmuR: 5’-TAGCCTGGGTCCCTCCTTAC-3’ and sequenced using GS junior sequencer. For GS Junior, sequencing libraries were prepared according to the manufacturer’s instructions, adaptor sequences were added to the previous amplification primer sequences in order to be compatible with the GS-Junior sequencing technology. Amplifications were performed with Phusion® High-Fidelity DNA Polymerase (New England Biolabs) according to the following program: DNA was denatured 40 s at 98°C and then submitted to 38 cycles consisting of 98°C for 10 s, 68°C for 30 s and 72°C for 30 s, and 1 cycle at 72°C for 10 min. PCR products were first purified using NucleoSpin kit (Macherey-Nagel) followed by Ampure bead purification (Beckman Coulter). PCR products were subjected to “PCR emulsion step” (GS Junior+ emPCR Kit (Lib-A), Roche) and sequenced using GS Junior sequencing kit XL+ (Roche) according to the manufacturer’s instructions. Raw sequences were aligned against reference sequences of IgHJ4-downstream intron or Smu and only full length sequences were kept for mutation analysis. For IgHJ4, clonally related sequences were removed based on the sequence of VDJ junction (i.e. CDR3) similarity. No further filtering steps were implemented in our analysis workflow. Mutations were called on each sequence using pairwise alignment algorithm (from biopython package) and only base substitutions were reported. Mutation frequencies were computed as the ratio between the sum of mutated bases in all complete sequences over the total number of aligned bases. For Ion Proton, sequencing libraries were prepared according to the user guide Ion Xpress™ Plus gDNA Fragment Library Preparation (Cat. no. 4471269, Life Technologies). Briefly, PCR products (100ng) were fragmented by enzymatic digestion (Ion Shear™ Plus Reagents Kit, Cat. no. 4471248) and ligated to Barcodes and Adapters (Ion Plus Fragment Library Kit, Cat. no. 4471252). After 200 bp size selection step on E-Gel precast agarose electrophoresis system, final amplification was performed. Raw data were processed using DeMinEr tool as described (28). In the case of studies performed in BER or MMR-deficient backgrounds of mixed 129Sv;C57BL/6 backgrounds, all SHM analysis were done by excluding nucleotides that differ between strains.
Total RNA was prepared by using TRIzol reagent (Ambion) procedures. RNA samples were first treated with DNase I (Invitrogen) for 15 min at 25°C. RT was performed on 200 ng of total RNA with random hexamers or with specific primer (10µM) (sequence available in Supplementary Figure S2.) using superscript III enzyme (Invitrogen). As control, we performed a reverse transcription without primer to determine the threshold (referred ad P- in bar graphs). Each real-time qPCR reaction was performed in duplicate on 10 ng of RNA equivalent, using TaqMan Universal (except for Sµ quantification we used SYBR green Mastermix (TAKARA)) on StepOnePlus system (Applied Biosystems). Primary transcription at IgH locus was quantified as previously described (37) and completed with both a set of primers and q-PCR probes close to JHsegments (listed in Supplementary Figure 2.) and a set of primers located 5’ to Sµ: Smu-Fw (5’-ACCCAGGCTAAGAAGGCAAT-3’), Smu-Rev (5’-CCTTCCTTCTGCGTATCCAT-3’). Relative mRNA levels were normalized to Gapdh transcripts with the appropriate TaqMan probe (Mm99999915_g1, Applied Biosystem). Data were analyzed by comparing threshold cycle (CT) values according to the 2-(δδCT) method. The wt mice templates used as calibrators were S2 for sense transcripts, AS0 or AS2 for antisense transcripts.
If not specified in the figure legend, Mann Whitney two-tailed tests were used for statistical analysis using GraphPad Prism software (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001).
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ebi.ac.uk/ena, PRJEB52221.
The animal study was reviewed and approved by Procedures were reviewed and approved by the Ministère de l’Education Nationale de l’Enseignement Supérieur et Recherche autorisation APAFIS#16639-2018090612249522v2.
OAM, MT, MM, AG, MB, SB, CC, CB, JC, EP and SN performed experiments. EP and SN conceived and supervised the study. MM developed the experimental model. OAM, BV, PG, RM, EP and SN wrote the manuscript. All authors contributed to the article and approved the submitted version.
OM and MT were supported by PhD fellowship of the french Ministère de l’Enseignement Supérieur, de la Recherche et de de l’Innovation. This work was supported by Région Nouvelle Aquitaine, La Ligue Contre le Cancer (comités 87, 23 to EP and SN); the Fondation ARC pour la recherche sur le cancer (PJA 20181207918 to EP and PhD continuation fellowship to MT), Institut CARNOT CALYM, INCa-Cancéropôle GSO Emergence (to EP).
The authors are grateful to BISCEm unit (Univ. Limoges, UAR 2015 CNRS, US 42 Inserm, CHU Limoges) for technical support regarding DNA-RNA sequencing, microscopy, histology, cytometry, cell sorting and animal core facility. We are grateful to Drs. Fred Alt for providing MARsEµ targeting construct, Zéliha Oruc-Ratinaud and Tiffany Marchiol regarding animal health status. We thank Dominik Schenten, Sébastien Storck, Christophe Sirac, Laurent Delpy, Brice Laffleur, Alexis Saintamand and Jeanne Moreau for discussions and helpful comments. We are grateful to Michel Cogné for critical review of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1030813/full#supplementary-material
Supplementary Figure 1 | Gating strategy to test developing B cells in mice. (A) Bone marrow B cell populations in wt and MARsEµΔ/Δ mice. Top row: B220+ CD117+ pre-pro B cells were stained with V450–anti-B220, PE–anti-CD117 Abs. Bottom row: B220+ CD43High pro-B and B220+CD43Low pre-B cell populations were stained with V450–anti-B220, FITC–anti-IgM, and PE–anti-CD43 Abs, gated on the IgM-negative population. (B) Splenic B cell subsets in wt and MARsEµΔ/Δ mice. Top row: CD21High CD23Low marginal zone and CD21Low CD23High follicular B cell populations were stained with V450-anti-B220, PE–anti-CD21 and FITC–anti-CD23 Abs, gated on B220+ population. Bottom row: IgM+IgD+ mature B cells were stained with V450-anti-B220, FITC–anti-IgD and PE–anti-IgM Abs. (C) Peritoneal cavity B cells in wt and MARsEµΔ/Δ mice. IgM+ CD5+ -B1a and IgM+ CD5- -B1b subsets were stained with V450-anti-B220, PE-anti-IgM, FITC-anti-CD5 Abs, gated on the B220+ population.
Supplementary Figure 2 | SHM frequency within JH4 intron. (A) SHM downstream from JH3 and JH4 segments in Peyer’s patch GC B cells sorted from wt and MARsEµΔ/Δ mice. For each genotype, pie charts represent distribution of mutated sequences (proportional to the area in each slice, data obtained by Sanger sequencing method) in individually recombined IgH alleles. Number of individual clones is reported in the center (after removal of clonally related sequences based on VDJ junction). Each pie chart represent SHM obtained from an individual experiment. Under each pie chart, SHM frequency, sequencing strategy and sample type (individual mice or pool) is indicated. Mean SHM frequency and p values are reported. (B) Bar graph displayed numbers of germinal center structures obtained in the spleen of wt and MARsEµΔ/Δ mice upon SRBC-immunization (left). Representative images of spleen section from immunized wt and MARsEµΔ/Δ animals (right) (C) Equivalent data representation than reported in A for SHM downstream from JH4 segments in splenic GC B cells sorted from SRBC-immunized wt and MARsEµΔ/Δ mice. Mean SHM frequency and p values are reported.
Supplementary Figure 3 | SHM frequency downstream from cEµ region. (A) SHM downstream from cEµ region in Peyer’s patch GC B cells sorted from wt and MARsEµΔ/Δ mice obtained by GS junior sequencing. For each genotype, pie charts represent distribution of mutated sequences (proportional to the area in each slice, data obtained by Sanger and GS Junior sequencing method) in individually recombined IgH alleles. Number of individual clones is reported in the center (after removal of clonally related sequences based on VDJ junction). Each pie chart represent SHM obtained from an individual experiment. Under each pie chart, SHM frequency, sequencing strategy and sample type (individual mice or pool) is indicated. Mean SHM frequency and p values are reported. (B) Left: schematic representation of both wt and MARsEµΔ alleles to illustrate the distance between cEµ and Sµ region. Blue and red bars indicate the 473bp-designed region to quantify SHM at the same distance from cEµ in the two models. Right: Bar graph showing SHM frequency in the corresponding regions of wt and MARsEµΔ/Δ mice, the analysis was performed on samples described in A.
Supplementary Figure 4 | Annotated nucleotide map of the IgH-JH1 to Eµ germline region from of 129 wt mice. All JH exons as well as coreEµ element are indicated by bold characters. Start sites for antisense transcripts are reported as (*) according to the Alt study (38). Location of primers used for strand-specific reverse transcription (S1, S2, S3, AS0, AS1, AS2, AS3) are indicated by underlines. TaqMan qPCR amplicons (C, A, A’) are highlighted in grey.
Supplementary Figure 5 | Sense and antisense transcripts quantified with IgH JH3 and JH 4 exons with additional TaqMan probes. (A) Murine IgH locus (not to scale) indicating location of primers (S1, S2, S3; black arrows) within introns downstream from JH3 and JH4 used for strand-specific reverse transcription to detect sense transcripts (dotted arrows). Black bars (A’ and C) indicate location of q-PCR probes. Total primary transcripts and primary sense transcripts were quantified with A’ and C probes in Peyer’s patch GC B cells (dark colors) and in vitro-activated B cells (light colors) from wt and MARsEµΔ/Δ mice. (B) Murine IgH locus (not to scale) indicating location of primers (AS0, AS1, AS2, AS3; black arrows) within introns downstream from JH2, JH3 and JH4 used for strand-specific reverse transcription to detect antisense transcripts (dotted arrows). Black bars (A’ and C) indicate location of q-PCR probes. Primary antisense transcripts quantified with A’ and C probes in Peyer’s patch GC B cells (dark colors) and in vitro-activated B cells (light colors) from wt and MARsEµΔ/Δ mice. Dots indicated antisense transcripts start sites according to the Alt study (38). Baseline level was either provided by using a RT template performed without primers (P-) or using one strand-specific template that cannot be detected with the current probe (T-). p-value was determined with two tailed Mann Whitney test; significant differences are indicated by: *P<0.05; **P<0.01 and error bars represent SEM of two to three independent experiments.
Supplementary Figure 6 | Sense and antisense transcription and expression in naive B cells. (A) IgH Sense (left bar graph) and antisense (right bar graph) transcripts quantified with A probe in naive B cells sorted from Peyer’s patch in wt and MARsEµΔ/Δ mice. (A) Intracellular IgM mean fluorescence intensities measured by flow cytometry in naive B cells from Peyer’s patches of wt and MARsEµΔ/Δ mice. (C) IgH sense (left bar graph) and antisense (right bar graph) transcripts quantified with A probe in resting splenic B cells sorted from wt and MARsEµΔ/Δ mice. Bar graphs show mean ± SEM of two independent experiments.
Supplementary Figure 7 | Comparison of in-vitro Ig class switching in wt and MARsEµΔ/Δ mice. Percentage of IgG3 and IgG1 positive cells measured by flow cytometry after respectively LPS or LPS + IL4 stimulation for 3 days of splenic B cells sorted from wt and MARsEµΔ/Δ mice. Bar graphs show mean ± SEM of one representative experiment for each condition, gating strategy is indicated below.
Supplementary Figure 8 | Base substitution patterns in BER- and MMR-deficient backgrounds. Comparison of SHM-related base substitution patterns, reported as frequencies, at IgH in Peyer’s patch GC B cells sorted from wt and MARsEµΔ/Δ mice models, bred in genetic backgrounds deficient for base excision repair (UngΔ/Δ) and mismatch repair (Msh2Δ/Δ). Data were obtained by NGS (Ion Proton) combined to DeMinEr filtering (28). (A) Substitution pattern downstream from JH4 in double-deficient UngΔ/Δ Msh2Δ/Δ background. (B) Substitution pattern downstream from JH4 in DNA repair proficient (Ung+/+ Msh2+/+) background. Relative proportions of transitions and transversions at dC/dG pairs are reported in an additional bar graph. (C) Substitution pattern downstream from JH4 in UngΔ/Δ background. (D) Substitution pattern downstream from Iµ in double-deficient UngΔ/Δ Msh2Δ/Δ background. (E) Substitution pattern downstream from Iµ in DNA repair proficient (Ung+/+ Msh2+/+) background. (F) Substitution pattern downstream from Iµ in UngΔ/Δ background. p-value was determined with two tailed Mann Whitney test; significant differences are indicated by: *P<0.05; **P<0.01 and error bars represent SEM of two to three independent experiments.
Supplementary Table 1 | MARsEµ deletion led to normal B-lineage cell development. Bone Marrow and peripheral B cell subsets counts in wt and MARsEµΔ/Δ mice. Absolute numbers are reported as mean± SEM. Significance was assessed with Student T test. P value is indicated when difference is significant.
Supplementary Table 2 | SHM data (NGS) from individual mice in DNA repair proficient background. Total number of mutations, total number of bp analyzed and mutation frequencies for wt and MARsEµΔ/Δ mice. (A) Data from intron 3’ to JH4 in Peyer’s patches GC B cells, (B) Data from spleen GC B cells from SRBC-immunized mice, (C) Data from intron 3’ to Iµ in Peyer’s patches GC B cells.
Supplementary Table 3 | Unselected SHM within out of frame VDJ exons in GC B cells sorted from Peyer’s patch. Unique VDJ junctions, rearranged to JH3 and JH4 segments, were obtained after amplification, cloning and sequencing by classical Sanger method of DNA extracted from from Peyer’s patch sorted GC B cells (n=2 to 3 independent samples corresponding to individual mice or pools of 7 to 12 mice of each genotype). SHM data were calculated after processing individually recombined IgH alleles by IMGT V-Quest software (https://www.imgt.org/IMGT_vquest); numbers of base substitutions identified as SHM by the software in the FR3 and CDR3 regions are reported in the table and were used to calculate SHM frequencies.
Supplementary Table 4 | SHM data (NGS) from individual mice in genetic backgrounds deficient for base excision repair (Ung-deficient) and mismatch repair (Msh2-deficient). Total number of mutations, total number of bp analyzed and mutation frequencies. (A) Data from intron 3’ to JH4 in Peyer’s patches GC B cells of UngΔ/Δ Msh2Δ/Δ and UngΔ/Δ Msh2Δ/Δ mice, (B) Data from UngΔ/Δ and UngΔ/Δ mice, (C) Data from intron 3’ to Iµ in Peyer’s patches GC B cells of UngΔ/Δ Msh2Δ/Δ and UngΔ/Δ Msh2Δ/Δ mice, (D) Data from UngΔ/Δ and UngΔ/Δ MARsEµΔ/Δ mice.
1. Perlot T, Alt FW. Cis-regulatory elements and epigenetic changes control genomic rearrangements of the IgH locus. Adv Immunol (2008) 99:1–32. doi: 10.1016/S0065-2776(08)00601-9
2. Scheuermann RH, Garrard WT. MARs of antigen receptor and co-receptor genes. Crit Rev Eukaryot Gene Expr (1999) 9:295–310. doi: 10.1615/CritRevEukarGeneExpr.v9.i3-4.140
3. Gluch A, Vidakovic M, Bode J. Scaffold/matrix attachment regions (S/MARs): relevance for disease and therapy. Handb Exp Pharmacol (2008), 186:67–103. doi: 10.1007/978-3-540-72843-6_4
4. Teves SS, Henikoff S. DNA Torsion as a feedback mediator of transcription and chromatin dynamics. Nucleus (2014) 5:211–8. doi: 10.4161/nucl.29086
5. Pommier Y, Sun Y, Huang S-YN, Nitiss JL. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat Rev Mol Cell Biol (2016) 17:703–21. doi: 10.1038/nrm.2016.111
6. Cockerill PN, Yuen MH, Garrard WT. The enhancer of the immunoglobulin heavy chain locus is flanked by presumptive chromosomal loop anchorage elements. J Biol Chem (1987) 262:5394–7. doi: 10.1016/S0021-9258(18)61200-1
7. Perlot T, Alt FW, Bassing CH, Suh H, Pinaud E. Elucidation of IgH intronic enhancer functions via germ-line deletion. Proc Natl Acad Sci USA (2005) 102:14362–7. doi: 10.1073/pnas.0507090102
8. Afshar R, Pierce S, Bolland DJ, Corcoran A, Oltz EM. Regulation of IgH gene assembly: role of the intronic enhancer and 5’DQ52 region in targeting DHJH recombination. J Immunol (2006) 176:2439–47. doi: 10.4049/jimmunol.176.4.2439
9. Marquet M, Garot A, Bender S, Carrion C, Rouaud P, Lecardeur S, et al. The eμ enhancer region influences h chain expression and b cell fate without impacting IgVH repertoire and immune response in vivo. J Immunol (2014) 193:1171–83. doi: 10.4049/jimmunol.1302868
10. Ronai D, Iglesias-Ussel MD, Fan M, Shulman MJ, Scharff MD. Complex regulation of somatic hypermutation by cis-acting sequences in the endogenous IgH gene in hybridoma cells. Proc Natl Acad Sci USA (2005) 102:11829–34. doi: 10.1073/pnas.0505449102
11. Azuma T, Motoyama N, Fields LE, Loh DY. Mutations of the chloramphenicol acetyl transferase transgene driven by the immunoglobulin promoter and intron enhancer. Int Immunol (1993) 5:121–30. doi: 10.1093/intimm/5.2.121
12. Giusti AM, Manser T. Hypermutation is observed only in antibody h chain V region transgenes that have recombined with endogenous immunoglobulin h DNA: implications for the location of cis-acting elements required for somatic mutation. J Exp Med (1993) 177:797–809. doi: 10.1084/jem.177.3.797
13. Motoyama N, Miwa T, Suzuki Y, Okada H, Azuma T. Comparison of somatic mutation frequency among immunoglobulin genes. J Exp Med (1994) 179:395–403. doi: 10.1084/jem.179.2.395
14. Bachl J, Wabl M. Enhancers of hypermutation. Immunogenetics (1996) 45:59–64. doi: 10.1007/s002510050167
15. Lin MM, Green NS, Zhang W, Scharff MD. The effects of e mu, 3’alpha (hs 1,2) and 3’kappa enhancers on mutation of an ig-VDJ-Cgamma2a ig heavy gene in cultured b cells. Int Immunol (1998) 10:1121–9. doi: 10.1093/intimm/10.8.1121
16. Ronai D, Berru M, Shulman MJ. Variegated expression of the endogenous immunoglobulin heavy-chain gene in the absence of the intronic locus control region. Mol Cell Biol (1999) 19:7031–40. doi: 10.1128/MCB.19.10.7031
17. Li F, Yan Y, Pieretti J, Feldman DA, Eckhardt LA. Comparison of identical and functional igh alleles reveals a nonessential role for eμ in somatic hypermutation and class-switch recombination. J Immunol (2010) 185:6049–57. doi: 10.4049/jimmunol.0902992
18. Sakai E, Bottaro A, Davidson L, Sleckman BP, Alt FW. Recombination and transcription of the endogenous ig heavy chain locus is effected by the ig heavy chain intronic enhancer core region in the absence of the matrix attachment regions. Proc Natl Acad Sci USA (1999) 96:1526–31. doi: 10.1073/pnas.96.4.1526
19. Kohwi-Shigematsu T, Maass K, Bode J. A thymocyte factor SATB1 suppresses transcription of stably integrated matrix-attachment region-linked reporter genes. Biochemistry (1997) 36:12005–10. doi: 10.1021/bi971444j
20. Wang Z, Goldstein A, Zong RT, Lin D, Neufeld EJ, Scheuermann RH, et al. Cux/CDP homeoprotein is a component of NF-muNR and represses the immunoglobulin heavy chain intronic enhancer by antagonizing the bright transcription activator. Mol Cell Biol (1999) 19:284–95. doi: 10.1128/MCB.19.1.284
21. Kaplan MH, Zong RT, Herrscher RF, Scheuermann RH, Tucker PW. Transcriptional activation by a matrix associating region-binding protein. contextual requirements for the function of bright. J Biol Chem (2001) 276:21325–30. doi: 10.1074/jbc.M100836200
22. Wiersma EJ, Ronai D, Berru M, Tsui FW, Shulman MJ. Role of the intronic elements in the endogenous immunoglobulin heavy chain locus. either the matrix attachment regions or the core enhancer is sufficient to maintain expression. J Biol Chem (1999) 274:4858–62. doi: 10.1074/jbc.274.8.4858
23. Goyenechea B, Klix N, Yélamos J, Williams GT, Riddell A, Neuberger MS, et al. Cells strongly expressing ig(kappa) transgenes show clonal recruitment of hypermutation: a role for both MAR and the enhancers. EMBO J (1997) 16:3987–94. doi: 10.1093/emboj/16.13.3987
24. Yi M, Wu P, Trevorrow KW, Claflin L, Garrard WT. Evidence that the igkappa gene MAR regulates the probability of premature V-J joining and somatic hypermutation. J Immunol (1999) 162:6029–39. doi: 10.4049/jimmunol.162.10.6029
25. Franklin A, Blanden RV. Hypothesis: biological role for J-c intronic matrix attachment regions in the molecular mechanism of antigen-driven somatic hypermutation. Immunol Cell Biol (2005) 83:383–91. doi: 10.1111/j.1440-1711.2005.01327.x
26. Inlay MA, Gao HH, Odegard VH, Lin T, Schatz DG, Xu Y. Roles of the ig kappa light chain intronic and 3’ enhancers in igk somatic hypermutation. J Immunol (2006) 177:1146–51. doi: 10.4049/jimmunol.177.2.1146
27. Rada C, Gupta SK, Gherardi E, Milstein C. Mutation and selection during the secondary response to 2-phenyloxazolone. Proc Natl Acad Sci U.S.A. (1991) 88:5508–12. doi: 10.1073/pnas.88.13.5508
28. Martin OA, Garot A, Le Noir S, Aldigier J-C, Cogné M, Pinaud E, et al. Detecting rare AID-induced mutations in b-lineage oncogenes from high-throughput sequencing data using the detection of minor variants by error correction method. J Immunol (2018) 201:950–6. doi: 10.4049/jimmunol.1800203
29. Alamyar E, Duroux P, Lefranc M-P, Giudicelli V. IMGT(®) tools for the nucleotide analysis of immunoglobulin (IG) and T cell receptor (TR) V-(D)-J repertoires, polymorphisms, and IG mutations: IMGT/V-QUEST and IMGT/HighV-QUEST for NGS. Methods Mol Biol (2012) 882:569–604. doi: 10.1007/978-1-61779-842-9_32
30. Lennon GG, Perry RP. C mu-containing transcripts initiate heterogeneously within the IgH enhancer region and contain a novel 5’-nontranslatable exon. Nature (1985) 318:475–8. doi: 10.1038/318475a0
31. Petersen S, Casellas R, Reina-San-Martin B, Chen HT, Difilippantonio MJ, Wilson PC, et al. AID is required to initiate Nbs1/gamma-H2AX focus formation and mutations at sites of class switching. Nature (2001) 414:660–5. doi: 10.1038/414660a
32. Nagaoka H, Muramatsu M, Yamamura N, Kinoshita K, Honjo T. Activation-induced deaminase (AID)-directed hypermutation in the immunoglobulin smu region: Implication of AID involvement in a common step of class switch recombination and somatic hypermutation. J Exp Med (2002) 195:529–34. doi: 10.1084/jem.20012144
33. Heltzel JHM, Maul RW, Yang W, Gearhart PJ. Promoter proximity defines mutation window for VH and VK genes rearranged to different J genes. J Immunol (2022), 208:2220–2226. doi: 10.4049/jimmunol.2101002.
34. Sakai E, Bottaro A, Alt FW. The ig heavy chain intronic enhancer core region is necessary and sufficient to promote efficient class switch recombination. Int Immunol (1999) 11:1709–13. doi: 10.1093/intimm/11.10.1709
35. Xue K, Rada C, Neuberger MS. The in vivo pattern of AID targeting to immunoglobulin switch regions deduced from mutation spectra in msh2-/- ung-/- mice. J Exp Med (2006) 203:2085–94. doi: 10.1084/jem.20061067
36. Fukita Y, Jacobs H, Rajewsky K. Somatic hypermutation in the heavy chain locus correlates with transcription. Immunity (1998) 9:105–14. doi: 10.1016/S1074-7613(00)80592-0
37. Tinguely A, Chemin G, Péron S, Sirac C, Reynaud S, Cogné M, et al. Cross talk between immunoglobulin heavy-chain transcription and RNA surveillance during b cell development. Mol Cell Biol (2012) 32:107–17. doi: 10.1128/MCB.06138-11
38. Perlot T, Li G, Alt FW. Antisense transcripts from immunoglobulin heavy-chain locus V(D)J and switch regions. Proc Natl Acad Sci USA (2008) 105:3843–8. doi: 10.1073/pnas.0712291105
39. Bolland DJ, Wood AL, Johnston CM, Bunting SF, Morgan G, Chakalova L, et al. Antisense intergenic transcription in V(D)J recombination. Nat Immunol (2004) 5:630–7. doi: 10.1038/ni1068
40. Zhao Y, Dunn-Walters DK, Barone F, Spencer J. Antisense transcripts of V(D)J rearrangements; artifacts caused by false priming? Mol Immunol (2009) 46:2357–62. doi: 10.1016/j.molimm.2009.03.020
41. Li W, Notani D, Rosenfeld MG. Enhancers as non-coding RNA transcription units: recent insights and future perspectives. Nat Rev Genet (2016) 17:207–23. doi: 10.1038/nrg.2016.4
42. Laffleur B, Basu U, Lim J. RNA Exosome and non-coding RNA-coupled mechanisms in AID-mediated genomic alterations. J Mol Biol (2017) 429:3230–3241. doi: 10.1016/j.jmb.2016.12.021
43. Lim J, Giri PK, Kazadi D, Laffleur B, Zhang W, Grinstein V, et al. Nuclear proximity of Mtr4 to RNA exosome restricts DNA mutational asymmetry. Cell (2017) 169:523–537.e15. doi: 10.1016/j.cell.2017.03.043
44. Laffleur B, Lim J, Zhang W, Chen Y, Pefanis E, Bizarro J, et al. Noncoding RNA processing by DIS3 regulates chromosomal architecture and somatic hypermutation in b cells. Nat Genet (2021) 53:230–42. doi: 10.1038/s41588-020-00772-0
45. Rada C, Di Noia JM, Neuberger MS. Mismatch recognition and uracil excision provide complementary paths to both ig switching and the A/T-focused phase of somatic mutation. Mol Cell (2004) 16:163–71. doi: 10.1016/j.molcel.2004.10.011
46. Shen HM, Tanaka A, Bozek G, Nicolae D, Storb U. Somatic hypermutation and class switch recombination in Msh6(-/-)Ung(-/-) double-knockout mice. J Immunol (2006) 177:5386–92. doi: 10.4049/jimmunol.177.8.5386
47. Liu M, Duke JL, Richter DJ, Vinuesa CG, Goodnow CC, Kleinstein SH, et al. Two levels of protection for the b cell genome during somatic hypermutation. Nature (2008) 451:841–5. doi: 10.1038/nature06547
48. Feng Y, Li C, Stewart JA, Barbulescu P, Seija Desivo N, Álvarez-Quilón A, et al. FAM72A antagonizes UNG2 to promote mutagenic repair during antibody maturation. Nature (2021) 600:324–8. doi: 10.1038/s41586-021-04144-4
49. Rogier M, Moritz J, Robert I, Lescale C, Heyer V, Abello A, et al. Fam72a enforces error-prone DNA repair during antibody diversification. Nature (2021) 600:329–33. doi: 10.1038/s41586-021-04093-y
50. Lebecque SG, Gearhart PJ. Boundaries of somatic mutation in rearranged immunoglobulin genes: 5’ boundary is near the promoter, and 3’ boundary is approximately 1 kb from V(D)J gene. J Exp Med (1990) 172:1717–27. doi: 10.1084/jem.172.6.1717
51. Shen HM, Storb U. Activation-induced cytidine deaminase (AID) can target both DNA strands when the DNA is supercoiled. Proc Natl Acad Sci USA (2004) 101:12997–3002. doi: 10.1073/pnas.0404974101
52. Wright BE, Schmidt KH, Davis N, Hunt AT, Minnick MF. II. correlations between secondary structure stability and mutation frequency during somatic hypermutation. Mol Immunol (2008) 45:3600–8. doi: 10.1016/j.molimm.2008.05.012
53. Kobayashi M, Sabouri Z, Sabouri S, Kitawaki Y, Pommier Y, Abe T, et al. Decrease in topoisomerase I is responsible for activation-induced cytidine deaminase (AID)-dependent somatic hypermutation. Proc Natl Acad Sci USA (2011) 108:19305–10. doi: 10.1073/pnas.1114522108
54. Maul RW, Saribasak H, Cao Z, Gearhart PJ. Topoisomerase I deficiency causes RNA polymerase II accumulation and increases AID abundance in immunoglobulin variable genes. DNA Repair (Amst) (2015) 30:46–52. doi: 10.1016/j.dnarep.2015.03.004
55. Kodgire P, Mukkawar P, Ratnam S, Martin TE, Storb U. Changes in RNA polymerase II progression influence somatic hypermutation of ig-related genes by AID. J Exp Med (2013) 210:1481–92. doi: 10.1084/jem.20121523
56. Maul RW, Cao Z, Venkataraman L, Giorgetti CA, Press JL, Denizot Y, et al. Spt5 accumulation at variable genes distinguishes somatic hypermutation in germinal center b cells from ex vivo-activated cells. J Exp Med (2014) 211:2297–306. doi: 10.1084/jem.20131512
57. Kohler KM, McDonald JJ, Duke JL, Arakawa H, Tan S, Kleinstein SH, et al. Identification of core DNA elements that target somatic hypermutation. J Immunol (2012) 189:5314–26. doi: 10.4049/jimmunol.1202082
58. Tarsalainen A, Maman Y, Meng F-L, Kyläniemi MK, Soikkeli A, Budzyńska P, et al. Ig enhancers increase RNA polymerase II stalling at somatic hypermutation target sequences. J Immunol (2022) 208:143–54. doi: 10.4049/jimmunol.2100923
59. Meng F-L, Du Z, Federation A, Hu J, Wang Q, Kieffer-Kwon K-R, et al. Convergent transcription at intragenic super-enhancers targets AID-initiated genomic instability. Cell (2014) 159:1538–48. doi: 10.1016/j.cell.2014.11.014
60. Basu U, Meng F-L, Keim C, Grinstein V, Pefanis E, Eccleston J, et al. The RNA exosome targets the AID cytidine deaminase to both strands of transcribed duplex DNA substrates. Cell (2011) 144:353–63. doi: 10.1016/j.cell.2011.01.001
61. Pefanis E, Wang J, Rothschild G, Lim J, Chao J, Rabadan R, et al. Noncoding RNA transcription targets AID to divergently transcribed loci in b cells. Nature (2014) 514:389–93. doi: 10.1038/nature13580
62. Kaur S, Coulombe Y, Ramdzan ZM, Leduy L, Masson J-Y, Nepveu A. Special AT-rich sequence-binding protein 1 (SATB1) functions as an accessory factor in base excision repair. J Biol Chem (2016) 291:22769–80. doi: 10.1074/jbc.M116.735696
63. Pinaud E, Khamlichi AA, Le Morvan C, Drouet M, Nalesso V, Le Bert M, et al. Localization of the 3’ IgH locus elements that effect long-distance regulation of class switch recombination. Immunity (2001) 15:187–99. doi: 10.1016/S1074-7613(01)00181-9
Keywords: B cell, immunoglobulin gene, MARs region, somatic hypermutation (SHM), UNG
Citation: Martin OA, Thomas M, Marquet M, Bruzeau C, Garot A, Brousse M, Bender S, Carrion C, Choi JE, Vuong BQ, Gearhart PJ, Maul RW, Le Noir S and Pinaud E (2023) The IgH Eµ-MAR regions promote UNG-dependent error-prone repair to optimize somatic hypermutation. Front. Immunol. 14:1030813. doi: 10.3389/fimmu.2023.1030813
Received: 29 August 2022; Accepted: 13 January 2023;
Published: 14 February 2023.
Edited by:
Paolo Casali, The University of Texas Health Science Center at San Antonio, United StatesReviewed by:
Uttiya Basu, Columbia University, United StatesCopyright © 2023 Martin, Thomas, Marquet, Bruzeau, Garot, Brousse, Bender, Carrion, Choi, Vuong, Gearhart, Maul, Le Noir and Pinaud. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eric Pinaud, ZXJpYy5waW5hdWRAdW5pbGltLmZy; Sandrine Le Noir, c2FuZHJpbmUubGUtbm9pckB1bmlsaW0uZnI=
†Present address: Ophélie A. Martin, Genome Damage and Stability Centre, School of Life Sciences, University of Sussex, Brighton, United Kingdom
‡These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.