 Xiaojuan Jiang
Xiaojuan Jiang Ruihao Zhou
Ruihao Zhou Yujun Zhang
Yujun Zhang Tao Zhu
Tao Zhu Qian Li
Qian Li Weiyi Zhang
Weiyi Zhang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 29 September 2022
Sec. Inflammation
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.999407
Chronic pain remains to be a clinical challenge and is recognized as a major health problem with varying impacts on quality of life. Currently, the first-line therapy for chronic pain is opioids, which are often accompanied by unwanted psychoactive side effects. Thus, new and effective treatments for chronic pain are urgently needed and eagerly pursued. Inflammatory cytokines, especially interleukin-17 (IL-17), are reportedly potential therapeutic targets owing to their pivotal role in chronic pain from the neuroinflammation perspective. Recently, substantial evidence confirmed that IL-17 and IL-17 receptors (IL-17Rs) were increased in neuropathic, inflammatory, and cancer pain models. Notably, IL-17/IL-17R antibodies also reportedly relieve or cure inflammatory- and pain-related diseases. However, existing studies have reported controversial results regarding IL-17/IL-17Rs as potential therapeutic targets in diverse animal models of chronic pain. In this review, we present a summary of published studies and discuss the evidence, from basic to clinical to research, regarding the role and mechanism of action between IL-17 and diverse kinds of chronic pain in animal models and clinical patients. Furthermore, we evaluated IL-17-based therapy as a potential therapeutic strategy for inflammatory- and pain-related disease. Importantly, we also discussed clinical trials of IL-17/IL-17R targeting monoclonal antibodies. Overall, we found that IL-17 is a potential therapeutic target for chronic pain from the perspective of neuroinflammation.
Chronic pain continues to be recognized as a major health problem with varying impacts on quality of life and increases health costs, which mainly includes neuropathic pain (NP), inflammatory pain, and cancer pain (1, 2). Chronic pain is projected to impact 37% of Americans by 2030, creating an additional economic burden of $635 billion (3, 4). The first-line therapy for chronic pain is opioids, which are often accompanied by unwanted psychoactive side effects. Thus, new and effective treatments for chronic pain are urgently needed and eagerly pursued. Recently, several research efforts have been devoted to treatment regimens for chronic pain, although there are still no effective analgesics with minimal side effects (5). Accordingly, the specific mechanisms underlying the pathogenesis of chronic pain are topics of continuous research. Neuroinflammation is an emerging target in pain relief (6). Many studies have proven that neuroinflammation is involved in multiple steps of chronic pain, such as promoting central (7) and peripheral sensitization (8).
IL-17 (also called IL-17A, CTLA8) cDNA was isolated and cloned from murine hybridomas and mainly secreted by CD4+ T cell and subtype T helper 17 cells. Th17 cells were the major source of IL-17, and other innate immune cells (e.g., eosinophils, monocytes, and neutrophils) could produce IL-17 in response to pathogens or tissue damage (9). Overall, IL-17 is produced from Th17 cells and other innate immune cells, inducing various products, including cytokines (IL-6, IL-1β, and tumor necrosis factor-alpha [TNF-α]), chemokines (CXCL1 and CCL20), and transforming growth factor-β (10).
The IL-17 family consists of six other family molecules (IL-17B, IL-17C, IL-17D, IL-17E (IL-25), and IL-17F) with structural identity (11). IL-17A and IL-17F are closely related linked genes that are usually coproduced by Type 17 cells. The IL-17 receptor family includes five members (IL-17RA to IL-17RE) that have such conserved structural characteristics as extracellular fibronectin III-like domains and Toll-IL-1R family (SEFIR) domains (12). Upon binding to the IL-17 receptor A/C heterodimeric complex, IL-17 can trigger multiple signal transduction pathways to stimulate gene transcription and increase messenger RNA stability (13). For example, the IL-17 signaling activates NF-κB, JNK, Erk1/2 and p38, CCAAT/enhancer-binding proteins (C/EBPs), Janus kinase (JAK) and phosphoinositol-3 kinase (PI3K) (12, 13).
Recently, numerous studies in humans and mice have shown that IL-17 plays a dominant role in many autoimmune diseases that affect nervous system structure and function, including multiple sclerosis, rheumatoid arthritis, psoriasis, asthma, and inflammatory bowel disease (14). Interestingly, at least some of these are attributed to the direct and indirect signaling effects of IL-17 on neurons and glial cells, such as microglia and astrocytes (15, 16). Besides, it was reported that IL-17 mainly expressed in DRG satellite glial cells expressing glutamine synthetase (GS). In contrast, IL-17Rs was detected in most neurons in DRG sections as well as in cultured DRG neurons. Recently, IL-17Rs were found to be widely expressed in small-sized mouse DRG IB4+ and CGRP+ neurons with a predominance in IB4+ neurons, and spinal cord neurons, including Somatostatin-positive (SOM)+ excitatory interneurons (expressed vesicular glutamate transporter VGLUT2) and PAX2+ inhibitory interneurons (17, 18). Thus, IL-17 and IL-17R in peripheral nervous systems may also mediate glial-neuron interactions.
IL-17 has been proven to play a critical role in neuroimmune interactions (19). For example, IL-17 released from activated microglia reportedly promotes oxygen-glucose deprivation reperfusion-induced neuronal apoptosis (20). Furthermore, IL-17 was found to have the ability to reduce the proliferation and differentiation of neural stem cells (NSCs) (21). NSCs reportedly have the potential for differentiation towards varying types of neural or glial cells (22). Notably, aside from producing IL-17, central nervous system astrocytes and microglia can also be affected by IL-17, which can lead to glial cell activation and damage, and promote the release of cytokines and chemokines, further damaging or promoting neuron growth and survival (23–25). These various effects of IL-17 may affect the cellular source and secondary release of other cytokines, and may be closely related to the target cell types.
Due to the effect of pro-inflammatory cytokines on communication between glia, immune cells, and neurons, pro-inflammatory cytokines have been implicated as integral to chronic pain development (26). In particular, IL-17 has been repeatedly verified to indirectly cause chronic pain by promoting immune cell infiltration to injured tissues (27) and releasing pro-nociceptive inflammatory factors (28), or by acting directly on neurons to regulate synaptic transmission (18). Moreover, many scientists have found that anti-IL-17 and anti-IL-17R antibodies can relieve pain (29). These studies all suggest that IL-17 plays a significant role in chronic pain initiation and progression. Recent studies also emphasize the tremendous potential of anti-IL-17/IL-17R antibodies as a potent drug for chronic pain. Hence, in this review we summarized the key evidence regarding the role of IL-17 in chronic pain.
NP originates from somatosensory nervous system damage, either in peripheral or central lesions, caused by various diseases (30). The main contributing factors in NP include drug-induced peripheral neuropathy (31) and nerve injury (32). To explore the mechanisms of NP, various animal models have been established, including peripheral nerve injury (PNI), spinal cord injury (SCI), and chemotherapy-induced peripheral neuropathy (CIPN) (33) models. Previous studies have found that IL-17 expression levels were increased in these animal models (29, 34, 35), indicating that IL-17 may play a critical role in the initiation and development of NP. Herein, we review the role of IL-17 in NP caused by PNI, SCI, and CIPN.
Many PNI animal models (including chronic constriction injury [CCI], partial sciatic nerve ligation [pSNL], complete sciatic transection [CST] and perineural inflammation [neuritis]) have been used to study the relationship between NP and IL-17 (36–38). Investigators explored IL-17 protein expression patterns by enzyme-linked immunoassay in the nerves of four PNI models (including CCI, pSNL, CST, and neuritis) (34) and found that on day three after modeling, only the pSNL model showed increased IL-17 expression in the sciatic nerve. In contrast, IL-17 levels increased in all four PNI models at day eight after modeling. Notably, IL-17 exerted a limited role in the acute nerve injury phase and associated acute pain, while mainly playing a vital role in the later stages of nerve injury, and is involved in the inflammatory response and development of NP. Future studies should explore the role of IL-17 in the maintenance and development of NP by blocking IL-17 activity or secretion in relation to pain behavior.
Mounting evidence suggests that IL-17 contributes to the dysregulation of immune cell infiltration and glial activation after PNI and the ensuing NP, especially in neuroinflammatory responses and pain hypersensitivity. IL-17 may also be involved in the cross-communication between IL-17 and other cytokine signaling systems in the pathogenesis of NP. IL-17 deficiency in a pSNL model significantly reduced T cell and macrophage infiltration in DRG neurons and spinal microglia as well as astrocyte activation (36). Notably, IL-17 knockout (KO) mice showed significantly reduced mechanical pain hypersensitivity, but not thermal pain hypersensitivity, suggesting that pSNL-induced NP is only partially regulated by IL-17 (36). This was also consistent with the finding that IL-17 mediated articular mechanical hypernociception in a model of antigen (methylated bovine serum albumin [mBSA])-induced arthritis in mice, suggesting that IL-17 exerts a key role in the development of mechanical abnormal pain, although to a lesser extent than that in thermal nociceptive sensitization (39). After long-term exposure to IL-17A, researchers observed that expression of transient receptor potential vanilloid (TRPV) 1 was not upregulated in DRG neurons, although TRPV4 was significantly upregulated (38). In fact, TRPV4 was considered a candidate transduction molecule for mechanical nociceptive sensitization, further indicating that the specific role of IL-17 in mechanical nociceptive sensitization is associated with TRPV4 activation and upregulation. Future studies should explore the unique role of IL-17 in the mechanical pain and thermal pain hypersensitivity with further mechanism.
Day et al. also reported similar results; IL-17 KO mice manifested obviously decreased mechanical pain hypersensitivity compared with wild-type (WT) mice, as well as decreased pro-inflammatory factors (TNF-α, IL-6, and interferon-c), anti-inflammatory factors (IL-10 and IL-13), enkephalin, β-endoglin, and porphinato (37). Moreover, the IL-17 cells related to lymphocyte activation in injured sciatic nerves and spinal cords (L4-6) were observed less in nude mice than in their heterozygous littermates, further indicating that IL-17 contributes to the nervous autoimmune reaction after nerve injury (40). Researchers have consistently proved that IL−17 promotes astrocyte and pro-inflammatory cytokine proliferation (IL−1β and IL−6) during the maintenance phase of pSNL-induced NP (41, 42). Overall, these results implicate that IL-17 can regulate the process of hypersensitivity (especially for mechanical pain hypersensitivity) by modulating inflammatory cell infiltration (T cells and macrophages), glial cell activation (microglia and astrocytes), and pro-/anti-inflammatory environments (see details above) within the injured nerve. The molecular and cellular characteristics of IL-17 in the nervous system after PNI and effects on pharmacological inhibition of IL-17 bioactivity in NP warrant further investigation.
IL-17 also reportedly responds or regulates some pain-related molecules in PNI. Furthermore, CCI-induced mechanical allodynia was reversed by inhibitory matrix metalloproteinase (MMP)-9/MMP-2, resulting in the concomitant inhibition of IL-17 and TNF-α expression in nerves (43). Moreover, acute connecting segment 1 (CS1)-containing fibronectin isoform (FN-CS1) therapy reduced mechanical allodynia and downregulated the expression of IL-17 in the injured nerve, indicating that FN-CS1 contributes to mechanical hyperalgesia via upregulation of Th17 cells after physical trauma (44). Interestingly, blocking IL-17 was found to ameliorate hyperalgesia 7 days after SNL and significantly reduce the expression of phosphorylated calcium/calmodulin-dependent protein kinase II (p-CaMKII) and phosphorylated cAMP-response element binding protein (p-CREB) in the spinal cord, whereas recombinant (r)IL-17A treatment resulted in opposing effects. Moreover, some experiments showed that CaMKII inhibitor KN93 not only reduced SNL- or rIL-17A-induced hyperalgesia, but also downregulated p-CREB expression levels. Results of in vitro data showed that KN93 can also suppress rIL-17A-induced CREB activation in primary cultured spinal neurons. IL-17A could play a major part in the maintenance of NP in the spinal cord via the CaMKII/CREB signaling pathway (42). Overall, IL-17 may respond to other pain-related factors such as the MMP2/MMP9 axis and FN-CS1, and may regulate the CaMKII/CREB signaling pathway to mediate NP.
Among patients with SCI, 70% have chronic pain, although no effective therapy has been reported, and the pathogenic mechanisms remain unclear (45). IL-17, IL-23, IL-6, IL-21, and phosphorylated (p)-signal transducer and activator of transcription (STAT) 3 expression were markedly upregulated and peaked at 24 h after SCI, indicating that IL-17 may contribute to the promotion of spinal cord neuroinflammation after SCI via transcription factor STAT3 activation (46). Furthermore, CCL20 aggravated neuroinflammation caused by SCI through Th17 cell recruitment and IL-17A upregulation. Furthermore, flow cytometry results demonstrated that CCL20 antibody reduced Th17 cell recruitment 14 days after SCI (47). More importantly, IL-17 activated astrocytes and increased vascular endothelial growth factor (VEGF) expression in an SCI model via Janus kinase (JAK)/STAT signaling pathway activation (48). IL-17 could activate the JAK/STAT pathway by mediating a rapid tyrosine phosphorylation of the JAK family (Tyk 2, JAK 1,2, and 3) and STAT 1, 2, 3, and 4. Future studies should focus on the role of IL-17 in spinal cord neuroinflammation process via multiple signaling pathways, such as IL-17/STAT3, IL-17/CCL20, and IL-17/VEGF/JAK/STAT.
CIPN is a dose-limiting side effect of common chemotherapy drugs (taxanes, vinca-alkaloids, platinum compounds, bortezomib, and thalidomide) (49). As previously reported, the CIPN prevalence rate was 68.1% after chemotherapy (50). Currently, we still do not know the role of IL-17 in CIPN.
Recently, researchers demonstrated the synaptic mechanisms of IL-17 in regulating pain transmission in a CIPN model (18). IL-17 released from DRG satellite glial cells influenced synaptic transmission in the spinal cord pain circuit thereby driving CIPN. IL-17 from exogenous administration or secreted by spinal cord astrocytes may enhance excitatory postsynaptic currents (EPSCs) and reduce inhibitory postsynaptic currents (IPSCs) and gamma-aminobutyric acid (GABA)-induced currents in SOM+ neurons with IL-17R detection in superficial spinal cord horn. Specifically, IL-17R-KO in SOM+ neurons reversed paclitaxel-induced excitability of SOM+ neurons and effectively relieved CIPN. Meanwhile, overexpression of IL-17 in astrocytes directly induced mechanical hyperalgesia, similar with the previously mentioned finding that PNI induces NP. IL-17 secreted by peripheral glial cells enhanced the excitability of primary sensory neurons through IL-17R, and participated in the peripheral sensitization of CIPN by the non-lymphocyte-derived IL-17 method. The specific role of IL-17 may be the direct mechanism by which it mediates NP. IL-17 is an important signal molecule in interactions between glial cells and neurons, and may become a new target for CIPN treatment. However, reports on the molecular mechanisms underlying the interaction between IL-17R and ion channels are lacking. Further studies are needed to investigate this mechanism.
As an innate immune response, inflammation functions as a defense barrier in the body. The most significant symptoms are redness, swelling, heat, and pain (51). Inflammatory pain is the most common clinical symptom in inflammatory disease, and is often caused by sensitizing nociceptive neurons (52). Currently, studies have found that cytokines play essential roles in inflammatory pain, including IL-6 and IL-1β (26, 53). A growing number of studies have suggested that IL-17 has several important implications in inflammatory pain.
Researchers found that IL-17A injected into the knee joints of rats promoted long-lasting sensitization of nociceptive C fibers of the joint to mechanical stimuli and rapid phosphorylation of extracellular signal-regulated kinase (ERK). Furthermore, protein kinase B was increased in the isolated culturing of DRG neuron with long-term exposure to IL-17A, which upregulated its excitability (54). Furthermore, IL-17A-KO mice had less mechanical hyperalgesia than WT mice in an inflammatory pain model, although thermal hyperalgesia was unaffected, possibly owing to regulation of TRPV4 receptors in DRG neurons (38). These data demonstrate the special role of IL-17 in mechanical hyperalgesia, which is associated with TRPV4 activation and upregulation, as well as the phosphorylation of ERK and protein kinase B.
Many studies report that IL-17 contributes to the sensation of pain in other inflammatory-related disorders in which pain is a symptom. The incidence of abnormal pain in an experimental autoimmune prostatitis rat model was higher in rat prostate tissue with upregulated IL-17 compared with that in normal control rat tissue (55). TNF-α, IL-1β, and CXCL1 expression increased after IL-17 injection in an arthritis animal model. Notably, treatment with MMP inhibitors, cyclooxygenase (COX) inhibitors, or sympathetic blockers alleviates IL-17-induced nociception. IL-17 injection also upregulated prostaglandin E2 (PGE-2) creation, MMP-9 activity, and mRNA expression levels of COX-2, MMP-9, and precursor preproendothelin-1 in the synovial membrane. These inflammatory mediators and chemokines play critical roles in the pathogenesis and progression of hyperalgesia response caused by innate and adaptive immunity (56). Taken together, IL-17 is a critical pro-nociceptive cytokine in mBSA-induced arthritis through the promotion of neutrophil migration and production of various pro-inflammatory mediators, such as TNF-α, IL-1β, CXCR1/2 chemokine ligands, MMPs, endothelins, prostaglandins, and sympathetic amines. Nevertheless, the detailed mechanisms remain unclear in these studies. IL-17 may bind to IL-17Rs in nucleus pulposus cells to enhance COX2 expression and PGE2 production via the p38/c-Fos and c-Jun N-terminal kinase (JNK/c-Jun) signaling pathways in nucleus pulposus cells to mediate intervertebral disc inflammation and low back pain (57). Based on these data, we can present a reasonable hypothesis: IL-17 activates key signaling pathways to promote the release of pro-nociceptive mediators, cytokines, and inflammatory factors, thereby activating the inflammatory cascade to change neuron sensitivity.
Among these inflammatory factors, TNF has been strongly associated with chronic pain (26) and a close relationship with IL-17 (58). IL-17 may induce hyperalgesia and the effect caused by neutrophil migration, which depend on TNF binding to TNF receptor 1 (TNFR1) (27). TNF has previously been reported to bind to TNFR 1 to mediate hyperalgesia in a CCI model of a mouse sciatic nerve (59). Because TNF-induced hyperalgesia has also been associated with neutrophil migration, IL-17 may cause acute hyperalgesia by promoting TNF release from fibroblasts that stimulates further keratinocyte chemoattractant production, which migrates neutrophil chemotaxis to the plantar tissue and produces analgesic mediators leading to nerve sensitivity. Furthermore, TNFR1 KO and TNF blocking reportedly reduced hyperalgesia caused by IL-17 in an mBAS mice model (39), validating the role of IL-17 and TNF in chronic pain from another perspective. From these results, we can present this hypothesis: IL-17 mediates inflammatory pain by promoting the production of cytokine and inflammatory factors and accumulation of neutrophils and lymphocytes in inflammatory sites.
In addition to promoting inflammatory factor production to mediate chronic pain, studies have also found that IL-17 has a direct effect by binding to IL-17Rs located in neurons. IL-17Rs have been widely expressed in DRG neurons of rats (17, 54). Moreover, IL-17 isoforms can upregulate tetrodotoxin-(TTX-) resistant sodium currents in isolated DRG neurons, implying that IL-17 may play a role in inflammation-evoked sensitization of sensory nociceptive neurons (17). However, the mechanism of the increased TTX- current is still unclear in this study. Another study provides new insight. IL-17 released by astrocytes and IL-17Rs is localized in NR1-immunoreactive neurons and upregulated in chronic pain models. This study also found that IL-17 antibody significantly subsided hyperalgesia and downregulated phosphorylated (p)-NR1 and IL-17RA compared with the control group (29). A body of evidence shows that N-methyl-D-aspartate (NMDA) receptor phosphorylation of neurons is strongly associated with hyperalgesia (60). These studies showed that IL-17 directly acted on nociceptive neurons with NMDA receptors to enhance NMDA receptor phosphorylation to change neuron sensitivity.
Among cancer patients with bone metastases, 75% experience chronic pain, which can seriously affect quality of life (61, 62). Thus, elucidating the mechanisms of cancer pain and early interventions are urgently needed. IL-17 has been proven important in the initiation and development of immune disorders and cancer (63, 64). Furthermore, a study found that IL-17A levels were negatively associated with clinical pain scores in cancer patients (65). Some studies have also begun to explore the role of IL-17 in the pain of cancer patients with bone metastases. Herein, we will review the potential mechanism of IL-17 in cancer pain.
T cells infiltrated the spinal cord in a rat model of bone cancer pain (BCP) and was accompanied by increased pain and a brief transient rise in regulatory T (Treg) cells, followed by an imbalance of Th17 cells (66). Researchers observed that IL-17 was elevated in both the blood and spinal cord, and IL-17R was upregulated in microglia. Increased production of IL-17/IL-17Rs may further promote microglia activation during BCP development. Furthermore, IL-17-neutralizing antibodies can relieve BCP, as well as suppress Th17/Treg infiltration and inhibit microglial activation. These results suggest that T cells infiltrate the spinal cord accompanied by an imbalance of Th17 cells during BCP progression, thereby promoting IL-17 production which can bind to IL-17R to activate microglial and further increase BCP (66). Thus, targeting infiltrated T cells and increased Th17/Treg ratios in the spinal cord may be a novel therapy for BCP. Further studies of are needed to investigate the role of IL-17 in therapeutic avenues for cancer neuroscience from translational frontiers and clinical opportunities aspects.
The above review respectively elaborates on the potential role of IL-17 in chronic pain from different diseases (Figure 1 and Table 1). However, reviews that comprehensively summarize the mechanistic role of IL-17 in chronic pain are lacking. Herein, we separate the mechanistic roles of IL-17 in chronic pain into direct (IL-17 directly binds to neuronal IL-17R to induce chronic pain) and indirect roles (IL-17 induces chronic pain by regulating infiltrating immune cells and pain mediator production).
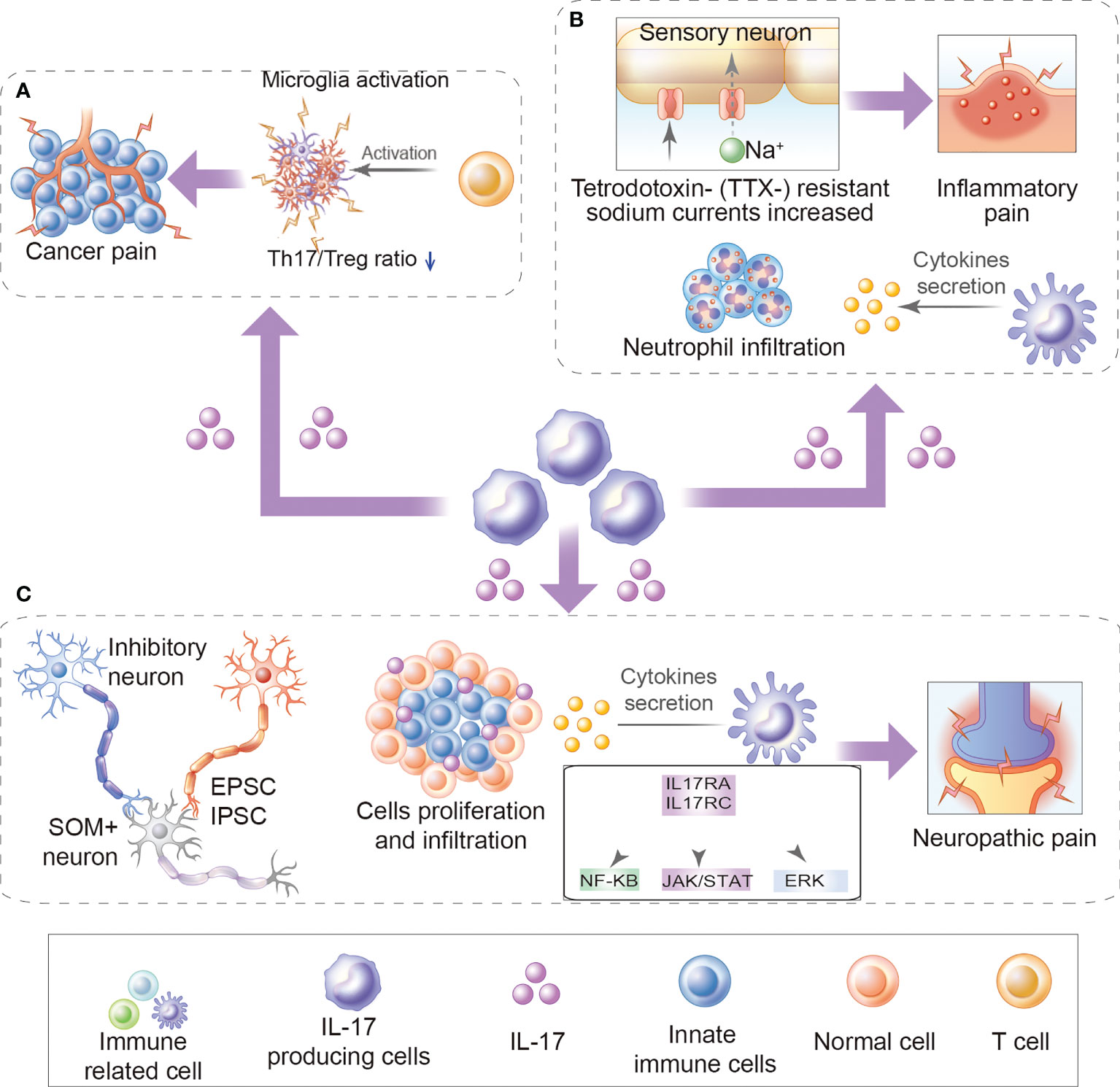
Figure 1 Role of IL-17 in chronic pain. (A) The role of IL-17 in cancer pain: For example, increased production of IL-17/IL-17Rs may further promote Th17/Treg infiltration and microglia activation during bone cancer pain development. (B) Inflammatory pain: For example, IL-17 mediates inflammatory pain by promoting the production of cytokine and inflammatory factors and accumulation of neutrophils and lymphocytes in inflammatory sites. Besides, IL-17 upregulates TTX- resistant sodium currents in isolated DRG neurons. (C) Neuropathic pain: For example, IL-17/IL17Rs regulate EPSCs and IPSCs, promote cytokine production, immune cell activation, and immune cell proliferation and infiltration, activates ERK and NF-κB, and JAK/STAT signal pathways.
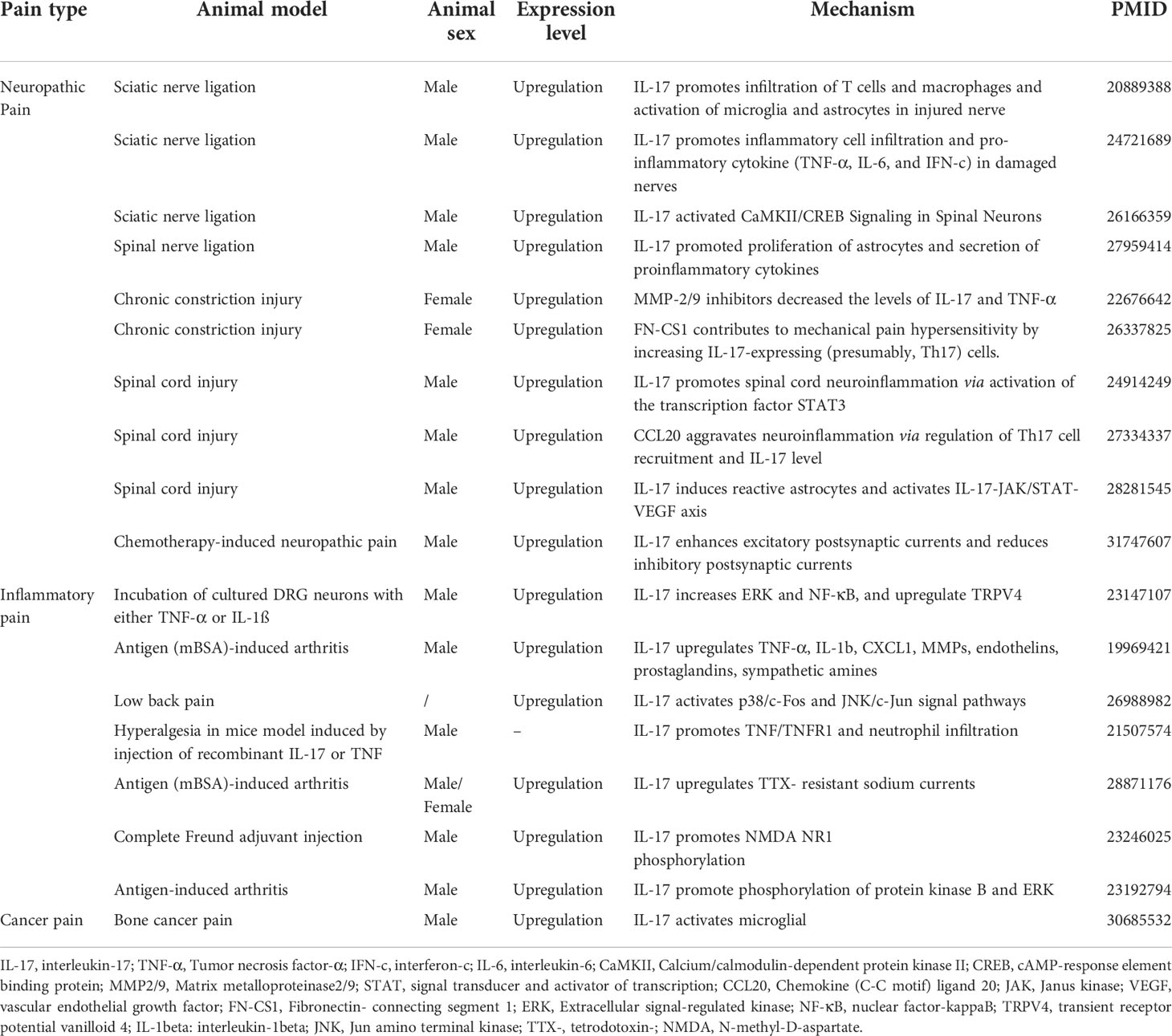
Table 1 Summary of characteristics of studies on the role of IL-17 in chronic pain.
IL-17 was mainly expressed in DRG satellite glial cells. In contrast, IL-17Rs were found to be widely expressed in small-sized mouse DRG IB4+ and CGRP+ neurons with a predominance in IB4+ neurons, although not in large-sized NF-200+ DRG neurons (17, 18). The respective localization of IL-17/IL-17R signaling offers a cellular basis for neuron-glial interaction and promotes both central and peripheral sensitization via the central and peripheral nervous systems. Here, we summarize the direct role of IL-17 in chronic pain (Figure 2).
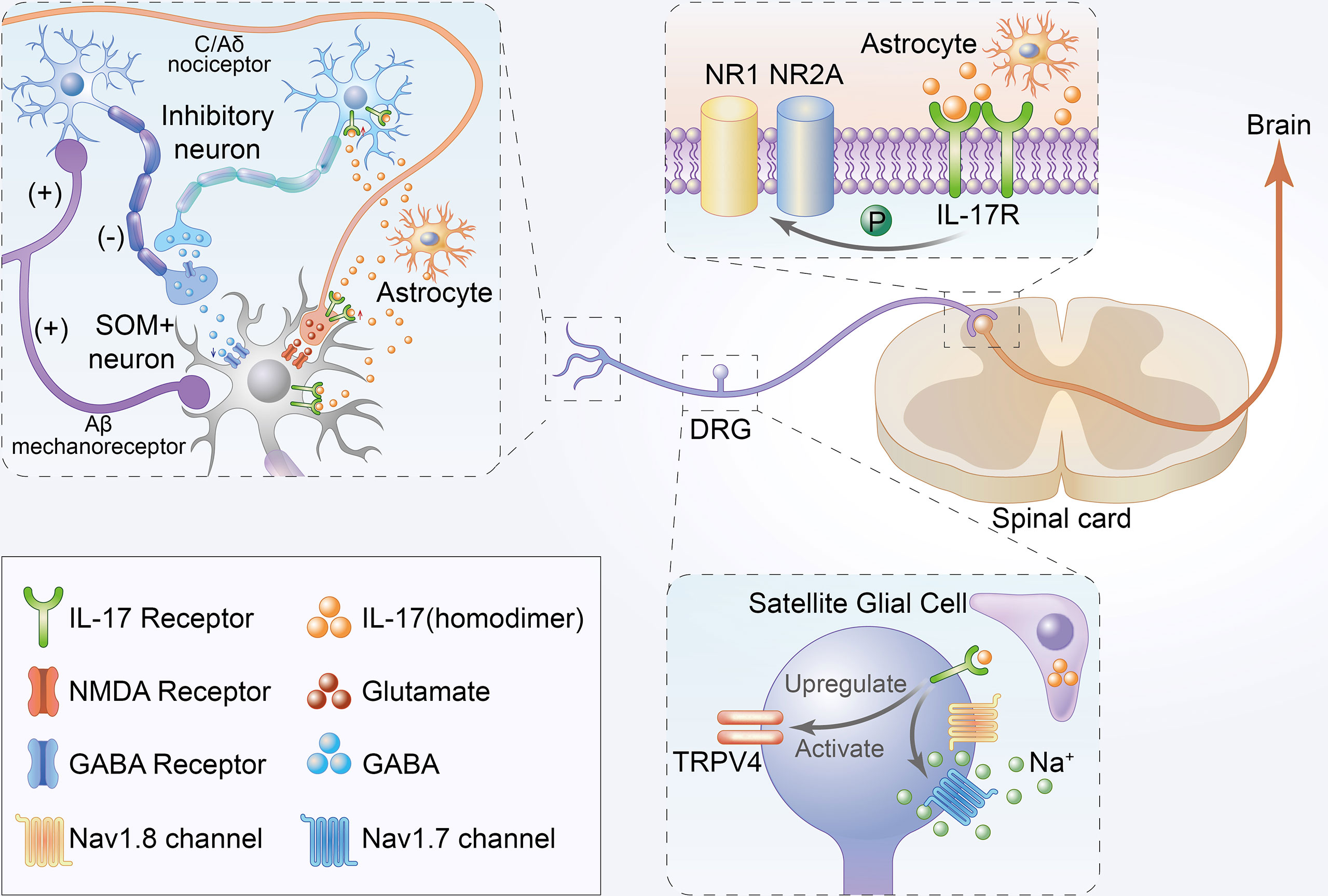
Figure 2 Schematic illustration of direct mechanisms of IL-17 in the development of chronic pain. Firstly, IL-17 produced by astrocytes not only enhances EPSCs but also suppresses inhibitory IPSCs and GABA-induced currents in the lamina II by interacting with neurons. Moreover, IL-17 from astrocytes can directly bind to IL-17Rs to mediate NAMD NR1 phosphorylation in the rat spinal cord. Thirdly, IL-17 from satellite glial cell can directly bind to IL-17R to upregulate and activate TRPV4 and increase tetrodotoxin-sensitive sodium currents in DRG neurons.
IL-17/IL-17Rs mediate neuron-glial interactions and neuronal hyperexcitability in chemotherapy-induced chronic pain (18). IL-17 produced by astrocytes not only enhances EPSCs but also suppresses inhibitory IPSCs and GABA-induced currents in the lamina II by interacting with neurons. Increased EPSCs and decreased IPSCs are reportedly associated with central sensitization and play key roles in the mechanisms of chronic pain (67, 68). Moreover, IL-17 is released by astrocytes, and IL-17R is localized in NR1-immunoreactive neurons with decreased p-NR1 and upregulated in chronic pain models (29). Some studies have established that hyperalgesia correlates with enhanced NR1 phosphorylation in the rat spinal cord (69).
IL-17 isoforms increased TTX- resistant sodium currents in isolated DRG neurons (17). TTX- resistant sodium currents were confirmed to be associated with chronic pain, and TTX- was also considered a potential therapeutic agent for pain (70). Remarkably, isolated and cultured rat DRG neurons showed significant TRPV4 upregulation upon long-term exposure to IL-17A (38), which is considered a candidate transduction molecule for mechanical hyperalgesia (71). TRPV4 activation was also associated with tetrodotoxin-sensitive sodium currents (72). In summary, the direct role of IL-17 in chronic pain may occur through binding with IL-17Rs located in neurons, thereby regulating the corresponding sodium currents in neurons.
Aside from the direct mechanisms, there are also important indirect mechanisms. IL-17 can mediate chronic pain by promoting the production of pain factors and activation of cells related to chronic pain (Figure 3). Microglia reportedly play an important role in chronic pain (73). IL-17 and IL-17R levels increased with microglia activation during BCP development (66). Furthermore, IL-17 KO decreased T cell and macrophage infiltration to injured sciatic nerves and microglia and astrocyte activation in the spinal cord (36). These cells were also strongly associated with chronic pain (74, 75).
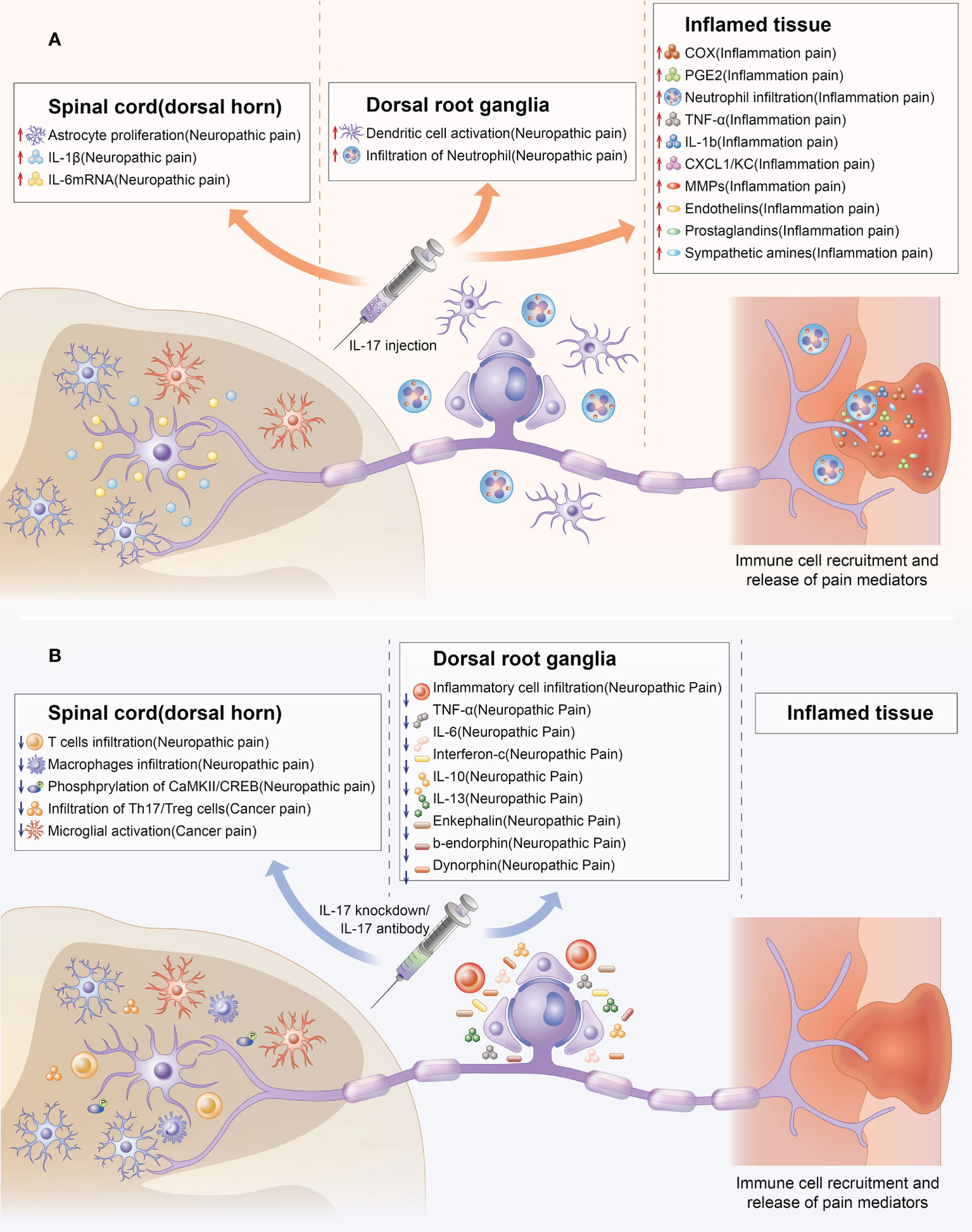
Figure 3 Indirect effect of IL-17 in chronic pain. Upregulation and downregulation of IL-17 can mediate chronic pain through chemical signals at several steps of nociceptive transmission. The colored boxes summarize the effects of IL-17 injection and IL-17 knockdown in the preclinical pain models in which they were described. IL-17 can mediate chronic pain by promoting the production of pain factors (such as IL-6,TNF-α, PGE2) and activation of cells (such as astrocyte, microglial, T cells, macrophage infiltration) related to chronic pain from spinal cord, DRG, and inflamed tissue levels. (A) Indirect effect of IL-17 upregulation in chronic pain. (B) Indirect effects of IL-17 knockdown in chronic pain.
Aside from activating immune cell-related chronic pain, IL-17 can also mediate chronic pain by inducing the production of other pain-related factors. IL-17 production was enhanced in a pSNL model and accompanied by significant upregulation of mRNA levels of IL−1β and IL−6. IL-1β and IL-6 are important in chronic pain (53, 76). Furthermore, IL-17 knockdown may decrease pro-inflammatory cytokines (TNF-α, IL-6, and interferon-γ) levels in damaged nerves (37). TNF-α and interferon-γ have also been confirmed to be closely related to chronic pain (77, 78). IL-17 injection into joints increased TNF-α, IL-1β, CXCL1, and PGE-2 production; MMP-9 and COX-2 activity; and MMP-9 mRNA expression in the synovial membrane (39). The inflammatory process has also been associated with chronic pain development (79–81). Furthermore, IL-17A enhanced COX2 expression and PGE-2 production via the p38/c-Fos and JNK/c-Jun signaling pathways. Moreover, IL-17A KO significantly reduced p-CaMKII and p-CREB levels, which have been associated with hyperalgesia and central sensitization in the spinal cord (42, 82, 83).
IL-17 has been associated with chronic pain in not only chronic pain animal models, but also in clinical patients. Th17 lymphocyte and IL-17 levels in lumbar disc herniation were positively correlated with visual analog scale scores recorded for preoperative pain intensity (84). The levels of IL-17A mRNA and protein expression in patients with bladder pain syndrome type 3C were higher than those in healthy volunteers (85). Furthermore, IL-17 levels had a positive correlation with pain severity in patients with osteonecrosis of the femoral head (86). In addition, IL-17 levels were negatively associated with clinical pain scores in patients with cancer (65). Interestingly, opioid requirements were increased in patients with the AA genotype of rs2275913 SNP of IL-17A during and after surgery (87).
IL-17/IL-17R upregulation was observed in rodent models of NP and patients with pain in many studies, and these data demonstrate that IL-17/IL-17R antibodies or IL-17-knockdown can effectively attenuate chronic pain (18, 42, 60). Hence, there are certainly reasons to believe that targeting IL-17/IL-17R signaling is a promising treatment for chronic pain. In addition, IL-17/IL-17R antibodies can relieve and cure autoimmune diseases (88) such as rheumatoid arthritis (89) and psoriatic disease (90).
Currently, IL-17/IL-17R blockers have been developed to treat inflammatory disease. Brodalumab (Kyntheum®) is a human anti-IL-17RA monoclonal antibody available for use in patients with moderate to severe psoriasis (91). Furthermore, brodalumab was associated with significant improvement in signs and symptoms of psoriatic arthritis versus placebo (92). Researchers found that mouse IL-17RA antibody was effective in suppressing neuronal hyperexcitability after paclitaxel, which might indicate brodalumab could be used to treat CIPN and neuropathic pain (18). While, Secukinumab (Cosentyx™) is the first fully human monoclonal immunoglobulin G1κ antibody targeting human IL-17A, with indications for psoriasis, psoriatic arthritis, and ankylosing spondylitis (93–95). Ixekizumab, a humanized immunoglobulin G4 variant IL-17A-neutralizing antibody can bind human and cynomolgus monkey IL-17A with high affinity to inhibit interactions between IL-17A and its receptors in binding assays and potently blocks IL-17A-induced GRO or KC secretion in cell-based assays (96). Ixekizumab has been widely applied to patients with psoriasis and psoriatic arthritis (97, 98). We summarized the stages and clinical trial results for drugs targeting IL-17/IL-17Rs in various diseases in Table 2. Thus, the proposal of drugs targeting IL-17/IL-17Rs is plausible for patients with pain. Although direct clinical trial evidence for these drugs in the treatment of chronic pain is lacking, some studies have demonstrated that IL-17/IL-17R blockers can relieve painful symptoms in patients (99). Secukinumab-treated patients had rapid improvement in pain and fatigue scores in an overall population after 1 and 4 weeks, respectively, indicating the potential of IL-17/IL-17R blockers for pain management.
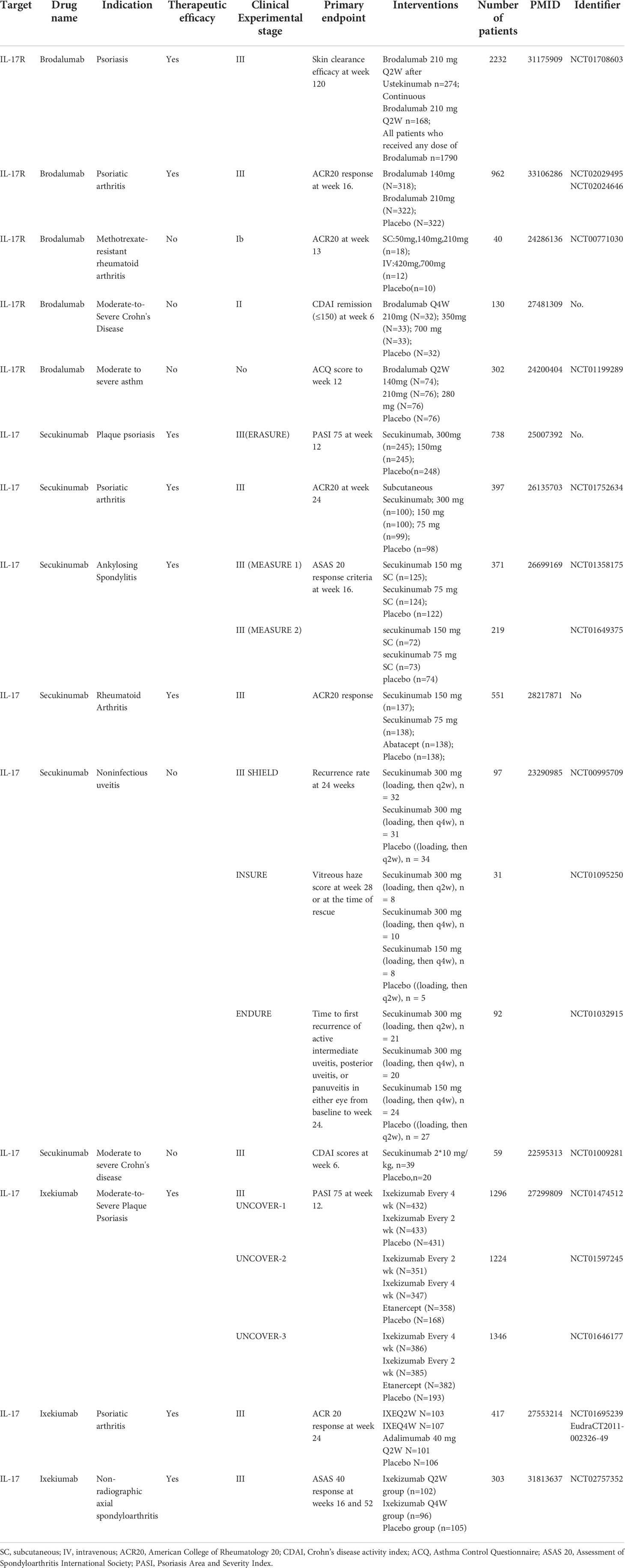
Table 2 Summary of IL-17/IL-17R drug targeting in various diseases.
Although great progress has been made in pain research over the past decade, there has been little translation of preclinical results into clinical practice. Currently, patients are frequently undertreated, and more effective analgesic drugs are urgently needed. Previous studies have shown that IL17 plays a critical role in the pathogenesis of neuropathic pain, inflammatory pain and cancer pain (Figure 1). Increased expression IL17 levels have been observed in various chronic pain models (Table 1). Notably, treatment with IL-17/IL-17R antibodies or IL-17-knockdown considerably attenuated chronic pain and relieve autoimmune diseases (Table 2). While reviewing the current evidence, we have discussed the direct and indirect relationship between IL17 and chronic pain (Figures 2, 3). IL-17/IL-17R blockers may be a novel and beneficial therapeutic tool for chronic pain management. However, these findings raise additional questions.
First, whether IL-17 blockers exert similar analgesic effects on other types of chronic pain (such as herpetic neuralgia and diabetes peripheral neuropathy) requires further investigation. Each type of chronic pain has unique characteristics. However, they may have the same pathogenesis. Therefore, it is important to reveal the beneficial effects of IL-17 inducers in other types of chronic pain.
Second, current studies have mostly focused on the analgesic effects of IL-17 blockers rather than the underlying mechanisms. For example, IL-17 exerted a limited role in the acute nerve injury phase and associated acute pain, while mainly playing a vital role in the later stages of nerve injury in the development of NP. Therefore, the detailed mechanisms underlying the analgesic effects of IL-17 blockers and the mechanisms by which so‐called IL-17 blockers repress IL-17 need to be elucidated. Recently, Secukinumab (IL-17 antibody)-treated patients showed rapid improvement in pain and fatigue scores in an overall population. The specific IL-17/IL-17R blockers in preclinical studies and clinical trials for autoimmune diseases and chronic pain area are further studies warranted.
Finally, current studies have primarily focused on the relationship between IL-17 (IL-17A) and chronic pain. However, other IL-17 family members (e.g., IL-17B, and IL17-F) are also involved in the mechanisms of chronic pain. Therefore, the involvement of additional members of the IL-17 family in chronic pain could be reported in the future. Large‐scale, multicenter, prospective clinical trials are needed. Given the analgesic effects of IL-17 blockers, efforts to discover more IL-17 blockers would have a large impact on clinical and public health.
XJJ, RHZ, and YJZ were the major contributors in writing and revising the manuscript. YJZ, QL and WYZ performed the literature search. XJJ, RHZ, and TZ discussed and revised the manuscript. QL and WYZ participated in the design of the review and helped to finalize the manuscript. All authors read and approved the final manuscript
We would like to thank Editage (www.editage.cn) for English language editing.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Treede RD, Rief W, Barke A, Aziz Q, Bennett MI, Benoliel R, et al. Chronic pain as a symptom or a disease: the IASP classification of chronic pain for the international classification of diseases (ICD-11). Pain (2019) 160(1):19–27. doi: 10.1097/j.pain.0000000000001384
2. Finnerup NB, Attal N, Haroutounian S, McNicol E, Baron R, Dworkin RH, et al. Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. Lancet Neurol (2015) 14(2):162–73. doi: 10.1016/s1474-4422(14)70251-0
3. Pizzo PA, Clark NM. Alleviating suffering 101–pain relief in the united states. N Engl J Med (2012) 366(3):197–9. doi: 10.1056/NEJMp1109084
4. Lorenz J, Casey KL. Imaging of acute versus pathological pain in humans. Eur J Pain (2005) 9(2):163–5. doi: 10.1016/j.ejpain.2004.07.009
5. Price TJ, Basbaum AI, Bresnahan J, Chambers JF, De Koninck Y, Edwards RR, et al. Transition to chronic pain: opportunities for novel therapeutics. Nat Rev Neurosci (2018) 19(7):383–4. doi: 10.1038/s41583-018-0012-5
6. Ji R-R, Xu Z-Z, Gao Y-J. Emerging targets in neuroinflammation-driven chronic pain. Nat Rev Drug Discovery (2014) 13(7):533–48. doi: 10.1038/nrd4334
7. Ji R-R, Nackley A, Huh Y, Terrando N, Maixner W. Neuroinflammation and central sensitization in chronic and widespread pain. Anesthesiology (2018) 129(2):343–66. doi: 10.1097/ALN.0000000000002130
8. Matsuda M, Huh Y, Ji R-R. Roles of inflammation, neurogenic inflammation, and neuroinflammation in pain. J Anesth (2019) 33(1):131–9. doi: 10.1007/s00540-018-2579-4
9. Pappu R, Ramirez-Carrozzi V, Ota N, Ouyang W, Hu Y. The IL-17 family cytokines in immunity and disease. J Clin Immunol (2010) 30(2):185–95. doi: 10.1007/s10875-010-9369-6
10. Lynde CW, Poulin Y, Vender R, Bourcier M, Khalil S. Interleukin 17A: toward a new understanding of psoriasis pathogenesis. J Am Acad Dermatol (2014) 71(1):141–50. doi: 10.1016/j.jaad.2013.12.036
11. Miossec P, Kolls JK. Targeting IL-17 and TH17 cells in chronic inflammation. Nat Rev Drug Discovery (2012) 11(10):763–76. doi: 10.1038/nrd3794
12. Gu C, Wu L, Li X. IL-17 family: cytokines, receptors and signaling. Cytokine (2013) 64(2):477–85. doi: 10.1016/j.cyto.2013.07.022
13. Suyama K, Sakai D, Watanabe M. The role of IL-17-Mediated inflammatory processes in the pathogenesis of intervertebral disc degeneration and herniation: A comprehensive review. Front Cell Dev Biol (2022) 10:857164. doi: 10.3389/fcell.2022.857164
14. Hu Y, Shen F, Crellin NK, Ouyang W. The IL-17 pathway as a major therapeutic target in autoimmune diseases. Ann NY Acad Sci (2011) 1217(1):60–76. doi: 10.1111/j.1749-6632.2010.05825.x
15. Waisman A, Hauptmann J, Regen T. The role of IL-17 in CNS diseases. Acta Neuropathol (2015) 129(5):625–37. doi: 10.1007/s00401-015-1402-7
16. Yang Q-Q, Zhou J-W. Neuroinflammation in the central nervous system: Symphony of glial cells. Glia (2019) 67(6):1017–35. doi: 10.1002/glia.23571
17. Ebbinghaus M, Natura G, Segond von Banchet G, Hensellek S, Böttcher M, Hoffmann B, et al. Interleukin-17A is involved in mechanical hyperalgesia but not in the severity of murine antigen-induced arthritis. Sci Rep (2017) 7(1):10334. doi: 10.1038/s41598-017-10509-5
18. Luo H, Liu H-Z, Zhang W-W, Matsuda M, Lv N, Chen G, et al. Interleukin-17 regulates neuron-glial communications, synaptic transmission, and neuropathic pain after chemotherapy. Cell Rep (2019) 29(8):2384-97.e5. doi: 10.1016/j.celrep.2019.10.085
19. Moynes DM, Vanner SJ. Lomax AE. participation of interleukin 17A in neuroimmune interactions. Brain Behav Immun (2014) 41:1–9. doi: 10.1016/j.bbi.2014.03.004
20. Lv M, Liu Y, Zhang J, Sun L, Liu Z, Zhang S, et al. Roles of inflammation response in microglia cell through toll-like receptors 2/interleukin-23/interleukin-17 pathway in cerebral ischemia/reperfusion injury. Neuroscience (2011) 176:162–72. doi: 10.1016/j.neuroscience.2010.11.066
21. Li Z, Li K, Zhu L, Kan Q, Yan Y, Kumar P, et al. Inhibitory effect of IL-17 on neural stem cell proliferation and neural cell differentiation. BMC Immunol (2013) 14:20. doi: 10.1186/1471-2172-14-20
22. Zahr SK, Kaplan DR, Miller FD. Translating neural stem cells to neurons in the mammalian brain. Cell Death Differ (2019) 26(12):2495–512. doi: 10.1038/s41418-019-0411-9
23. Das Sarma J, Ciric B, Marek R, Sadhukhan S, Caruso ML, Shafagh J, et al. Functional interleukin-17 receptor a is expressed in central nervous system glia and upregulated in experimental autoimmune encephalomyelitis. J Neuroinflamm (2009) 6:14. doi: 10.1186/1742-2094-6-14
24. Zimmermann J, Krauthausen M, Hofer MJ, Heneka MT, Campbell IL, Müller M. CNS-targeted production of IL-17A induces glial activation, microvascular pathology and enhances the neuroinflammatory response to systemic endotoxemia. PloS One (2013) 8(2):e57307. doi: 10.1371/journal.pone.0057307
25. Kawanokuchi J, Shimizu K, Nitta A, Yamada K, Mizuno T, Takeuchi H, et al. Production and functions of IL-17 in microglia. J Neuroimmunol (2008) 194(1-2):54–61. doi: 10.1016/j.jneuroim.2007.11.006
26. Vanderwall AG, Milligan ED. Cytokines in pain: Harnessing endogenous anti-inflammatory signaling for improved pain management. Front Immunol (2019) 10:3009. doi: 10.3389/fimmu.2019.03009
27. McNamee KE, Alzabin S, Hughes JP, Anand P, Feldmann M, Williams RO, et al. IL-17 induces hyperalgesia via TNF-dependent neutrophil infiltration. Pain (2011) 152(8):1838–45. doi: 10.1016/j.pain.2011.03.035
28. Ren K, Dubner R. Interactions between the immune and nervous systems in pain. Nat Med (2010) 16(11):1267–76. doi: 10.1038/nm.2234
29. Meng X, Zhang Y, Lao L, Saito R, Li A, Bäckman CM, et al. Spinal interleukin-17 promotes thermal hyperalgesia and NMDA NR1 phosphorylation in an inflammatory pain rat model. Pain (2013) 154(2):294–305. doi: 10.1016/j.pain.2012.10.022
30. Baron R, Binder A, Wasner G. Neuropathic pain: diagnosis, pathophysiological mechanisms, and treatment. Lancet Neurol (2010) 9(8):807–19. doi: 10.1016/S1474-4422(10)70143-5
31. Xu L, Zhang Y, Huang Y. Advances in the treatment of neuropathic pain. Adv Exp Med Biol (2016) 904:117–29. doi: 10.1007/978-94-017-7537-3_9
32. Wang X, Shen X, Xu Y, Xu S, Xia F, Zhu B, et al. The etiological changes of acetylation in peripheral nerve injury-induced neuropathic hypersensitivity. Mol Pain (2018) 14:1744806918798408. doi: 10.1177/1744806918798408
33. Finnerup NB, Kuner R, Jensen TS. Neuropathic pain: From mechanisms to treatment. Physiol Rev (2021) 101(1):259–301. doi: 10.1152/physrev.00045.2019
34. Noma N, Khan J, Chen IF, Markman S, Benoliel R, Hadlaq E, et al. Interleukin-17 levels in rat models of nerve damage and neuropathic pain. Neurosci Lett (2011) 493(3):86–91. doi: 10.1016/j.neulet.2011.01.079
35. Kleinschnitz C, Hofstetter HH, Meuth SG, Braeuninger S, Sommer C, Stoll G. T Cell infiltration after chronic constriction injury of mouse sciatic nerve is associated with interleukin-17 expression. Exp Neurol (2006) 200(2):480–5. doi: 10.1016/j.expneurol.2006.03.014
36. Kim CF, Moalem-Taylor G. Interleukin-17 contributes to neuroinflammation and neuropathic pain following peripheral nerve injury in mice. J Pain (2011) 12(3):370–83. doi: 10.1016/j.jpain.2010.08.003
37. Day Y-J, Liou J-T, Lee C-M, Lin Y-C, Mao C-C, Chou A-H, et al. Lack of interleukin-17 leads to a modulated micro-environment and amelioration of mechanical hypersensitivity after peripheral nerve injury in mice. Pain (2014) 155(7):1293–302. doi: 10.1016/j.pain.2014.04.004
38. Segond von Banchet G, Boettger MK, König C, Iwakura Y, Bräuer R, Schaible H-G. Neuronal IL-17 receptor upregulates TRPV4 but not TRPV1 receptors in DRG neurons and mediates mechanical but not thermal hyperalgesia. Mol Cell Neurosci (2013) 52:152–60. doi: 10.1016/j.mcn.2012.11.006
39. Pinto LG, Cunha TM, Vieira SM, Lemos HP, Verri WA, Cunha FQ, et al. IL-17 mediates articular hypernociception in antigen-induced arthritis in mice. Pain (2010) 148(2):247–56. doi: 10.1016/j.pain.2009.11.006
40. Li J, Wei G-H, Huang H, Lan Y-P, Liu B, Liu H, et al. Nerve injury-related autoimmunity activation leads to chronic inflammation and chronic neuropathic pain. Anesthesiology (2013) 118(2):416–29. doi: 10.1097/ALN.0b013e31827d4b82
41. Sun C, Zhang J, Chen L, Liu T, Xu G, Li C, et al. IL-17 contributed to the neuropathic pain following peripheral nerve injury by promoting astrocyte proliferation and secretion of proinflammatory cytokines. Mol Med Rep (2017) 15(1):89–96. doi: 10.3892/mmr.2016.6018
42. Yao C-Y, Weng Z-L, Zhang J-C, Feng T, Lin Y, Yao S. Interleukin-17A acts to maintain neuropathic pain through activation of CaMKII/CREB signaling in spinal neurons. Mol Neurobiol (2016) 53(6):3914–26. doi: 10.1007/s12035-015-9322-z
43. Liu H, Shiryaev SA, Chernov AV, Kim Y, Shubayev I, Remacle AG, et al. Immunodominant fragments of myelin basic protein initiate T cell-dependent pain. J Neuroinflamm (2012) 9:119. doi: 10.1186/1742-2094-9-119
44. Liu H, Dolkas J, Hoang K, Angert M, Chernov AV, Remacle AG, et al. The alternatively spliced fibronectin CS1 isoform regulates IL-17A levels and mechanical allodynia after peripheral nerve injury. J Neuroinflamm (2015) 12:158. doi: 10.1186/s12974-015-0377-6
45. Shiao R, Lee-Kubli CA. Neuropathic pain after spinal cord injury: Challenges and research perspectives. Neurotherapeutics (2018) 15(3):635–53. doi: 10.1007/s13311-018-0633-4
46. Zong S, Zeng G, Fang Y, Peng J, Tao Y, Li K, et al. The role of IL-17 promotes spinal cord neuroinflammation via activation of the transcription factor STAT3 after spinal cord injury in the rat. Mediators Inflammation (2014) 2014:786947. doi: 10.1155/2014/786947
47. Hu J, Yang Z, Li X, Lu H. C-c motif chemokine ligand 20 regulates neuroinflammation following spinal cord injury via Th17 cell recruitment. J Neuroinflamm (2016) 13(1):162. doi: 10.1186/s12974-016-0630-7
48. You T, Bi Y, Li J, Zhang M, Chen X, Zhang K, et al. IL-17 induces reactive astrocytes and up-regulation of vascular endothelial growth factor (VEGF) through JAK/STAT signaling. Sci Rep (2017) 7:41779. doi: 10.1038/srep41779
49. Miltenburg NC, Boogerd W. Chemotherapy-induced neuropathy: A comprehensive survey. Cancer Treat Rev (2014) 40(7):872–82. doi: 10.1016/j.ctrv.2014.04.004
50. Seretny M, Currie GL, Sena ES, Ramnarine S, Grant R, MacLeod MR, et al. Incidence, prevalence, and predictors of chemotherapy-induced peripheral neuropathy: A systematic review and meta-analysis. Pain (2014) 155(12):2461–70. doi: 10.1016/j.pain.2014.09.020
51. Medzhitov R. Inflammation 2010: new adventures of an old flame. Cell (2010) 140(6):771–6. doi: 10.1016/j.cell.2010.03.006
52. Millan MJ. The induction of pain: an integrative review. Prog Neurobiol (1999) 57(1):1-164. doi: 10.1016/s0301-0082(98)00048-3
53. Ren K, Torres R. Role of interleukin-1beta during pain and inflammation. Brain Res Rev (2009) 60(1):57–64. doi: 10.1016/j.brainresrev.2008.12.020
54. Richter F, Natura G, Ebbinghaus M, von Banchet GS, Hensellek S, König C, et al. Interleukin-17 sensitizes joint nociceptors to mechanical stimuli and contributes to arthritic pain through neuronal interleukin-17 receptors in rodents. Arthritis Rheum (2012) 64(12):4125–34. doi: 10.1002/art.37695
55. Liu X, Fan S, Zheng M, Chen J, Zhang J, Li H. The mediation of interleukin-17 and chemokine ligand 2 in pelvic pain of experimental autoimmune prostatitis. Exp Ther Med (2017) 14(1):51–8. doi: 10.3892/etm.2017.4448
56. Cunha TM, Verri WA, Valério DA, Guerrero AT, Nogueira LG, Vieira SM, et al. Role of cytokines in mediating mechanical hypernociception in a model of delayed-type hypersensitivity in mice. Eur J Pain (2008) 12(8):1059–68. doi: 10.1016/j.ejpain.2008.02.003
57. Li J-k, Nie L, Zhao Y-p, Zhang Y-q, Wang X, Wang S-s, et al. IL-17 mediates inflammatory reactions via p38/c-fos and JNK/c-jun activation in an AP-1-dependent manner in human nucleus pulposus cells. J Transl Med (2016) 14:77. doi: 10.1186/s12967-016-0833-9
58. Ten Bergen LL, Petrovic A, Krogh Aarebrot A, Appel S. The TNF/IL-23/IL-17 axis-head-to-head trials comparing different biologics in psoriasis treatment. Scand J Immunol (2020) 92(4):e12946. doi: 10.1111/sji.12946
59. Sommer C, Schmidt C, George A. Hyperalgesia in experimental neuropathy is dependent on the TNF receptor 1. Exp Neurol (1998) 151(1):138–42 doi: 10.1006/exnr.1998.6797.
60. Ultenius C, Linderoth B, Meyerson BA, Wallin J. Spinal NMDA receptor phosphorylation correlates with the presence of neuropathic signs following peripheral nerve injury in the rat. Neurosci Lett (2006) 399(1-2):85–90 doi: 10.1016/j.neulet.2006.01.018.
61. Coleman RE. Clinical features of metastatic bone disease and risk of skeletal morbidity. Clin Cancer Res (2006) 12(20 Pt 2):6243s–9s. doi: 10.1158/1078-0432.CCR-06-0931
62. Coleman RE, Croucher PI, Padhani AR, Clézardin P, Chow E, Fallon M, et al. Bone metastases. Nat Rev Dis Primers (2020) 6(1):83. doi: 10.1038/s41572-020-00216-3
63. Ruiz de Morales JMG, Puig L, Daudén E, Cañete JD, Pablos JL, Martín AO, et al. Critical role of interleukin (IL)-17 in inflammatory and immune disorders: An updated review of the evidence focusing in controversies. Autoimmun Rev (2020) 19(1):102429. doi: 10.1016/j.autrev.2019.102429
64. Zhao J, Chen X, Herjan T, Li X. The role of interleukin-17 in tumor development and progression. J Exp Med (2020) 217(1):jem.20190297. doi: 10.1084/jem.20190297
65. Fazzari J, Sidhu J, Motkur S, Inman M, Buckley N, Clemons M, et al. Applying serum cytokine levels to predict pain severity in cancer patients. J Pain Res (2020) 13:313–21. doi: 10.2147/JPR.S227175
66. Huo W, Liu Y, Lei Y, Zhang Y, Huang Y, Mao Y, et al. Imbalanced spinal infiltration of Th17/Treg cells contributes to bone cancer pain via promoting microglial activation. Brain Behav Immun (2019) 79:139–51. doi: 10.1016/j.bbi.2019.01.024
67. Xu Z-Z, Liu X-J, Berta T, Park C-K, Lü N, Serhan CN, et al. Neuroprotectin/protectin D1 protects against neuropathic pain in mice after nerve trauma. Ann Neurol (2013) 74(3):490–5. doi: 10.1002/ana.23928
68. Zeilhofer HU, Benke D, Yevenes GE. Chronic pain states: pharmacological strategies to restore diminished inhibitory spinal pain control. Annu Rev Pharmacol Toxicol (2012) 52:111–33. doi: 10.1146/annurev-pharmtox-010611-134636
69. Daulhac L, Maffre V, Mallet C, Etienne M, Privat A-M, Kowalski-Chauvel A, et al. Phosphorylation of spinal n-methyl-d-aspartate receptor NR1 subunits by extracellular signal-regulated kinase in dorsal horn neurons and microglia contributes to diabetes-induced painful neuropathy. Eur J Pain (2011) 15(2):169.e1-12. doi: 10.1016/j.ejpain.2010.06.003
70. Nieto FR, Cobos EJ, Tejada MÁ, Sánchez-Fernández C, González-Cano R, Cendán CM. Tetrodotoxin (TTX) as a therapeutic agent for pain. Mar Drugs (2012) 10(2):281–305. doi: 10.3390/md10020281
71. Levine JD, Alessandri-Haber N. TRP channels: targets for the relief of pain. Biochim Biophys Acta (2007) 1772(8):989-1003. doi: 10.1016/j.bbadis.2007.01.008
72. Li L, Liu C, Chen L, Chen L. Hypotonicity modulates tetrodotoxin-sensitive sodium current in trigeminal ganglion neurons. Mol Pain (2011) 7:27. doi: 10.1186/1744-8069-7-27
73. Chen G, Zhang Y-Q, Qadri YJ, Serhan CN, Ji R-R. Microglia in pain: Detrimental and protective roles in pathogenesis and resolution of pain. Neuron (2018) 100(6):1292–311. doi: 10.1016/j.neuron.2018.11.009
74. Ji R-R, Donnelly CR, Nedergaard M. Astrocytes in chronic pain and itch. Nat Rev Neurosci (2019) 20(11):667–85. doi: 10.1038/s41583-019-0218-1
75. Chen O, Donnelly CR, Ji R-R. Regulation of pain by neuro-immune interactions between macrophages and nociceptor sensory neurons. Curr Opin Neurobiol (2020) 62:17–25. doi: 10.1016/j.conb.2019.11.006
76. De Jongh RF, Vissers KC, Meert TF, Booij LHDJ, De Deyne CS, Heylen RJ. The role of interleukin-6 in nociception and pain. Anesth Analg (2003) 96(4):1096-103. doi: 10.1213/01.ANE.0000055362.56604.78
77. Li Q-Y, Xu H-Y, Yang H-J. [Effect of proinflammatory factors TNF-α,IL-1β, IL-6 on neuropathic pain]. Zhongguo Zhong Yao Za Zhi (2017) 42(19):3709–12. doi: 10.19540/j.cnki.cjcmm.20170907.004
78. Racz I, Nadal X, Alferink J, Baños JE, Rehnelt J, Martín M, et al. Interferon-gamma is a critical modulator of CB(2) cannabinoid receptor signaling during neuropathic pain. J Neurosci (2008) 28(46):12136–45. doi: 10.1523/JNEUROSCI.3402-08.2008
79. Silva RL, Lopes AH, Guimarães RM, Cunha TM. CXCL1/CXCR2 signaling in pathological pain: Role in peripheral and central sensitization. Neurobiol Dis (2017) 105:109–16. doi: 10.1016/j.nbd.2017.06.001
80. Ma W, Quirion R. Does COX2-dependent PGE2 play a role in neuropathic pain? Neurosci Lett (2008) 437(3):165–9. doi: 10.1016/j.neulet.2008.02.072
81. Escolano-Lozano F, Gries E, Schlereth T, Dimova V, Baka P, Vlckova E, et al. Local and systemic expression pattern of MMP-2 and MMP-9 in complex regional pain syndrome. J Pain (2021) 22(10):1294–302. doi: 10.1016/j.jpain.2021.04.002
82. Hoeger-Bement MK, Sluka KA. Phosphorylation of CREB and mechanical hyperalgesia is reversed by blockade of the cAMP pathway in a time-dependent manner after repeated intramuscular acid injections. J Neurosci (2003) 23(13):5437–45. doi: 10.1523/JNEUROSCI.23-13-05437.2003
83. Gu Y, Chen S, Mo Y, Tu Y, Chen N, Zhao X, et al. Electroacupuncture attenuates CFA-induced inflammatory pain by regulating CaMKII. Neural Plast (2020) 2020:8861994. doi: 10.1155/2020/8861994
84. Cheng L, Fan W, Liu B, Wang X, Nie L. Th17 lymphocyte levels are higher in patients with ruptured than non-ruptured lumbar discs, and are correlated with pain intensity. Injury (2013) 44(12):1805–10. doi: 10.1016/j.injury.2013.04.010
85. Logadottir Y, Delbro D, Fall M, Gjertsson I, Jirholt P, Lindholm C, et al. Cytokine expression in patients with bladder pain syndrome/interstitial cystitis ESSIC type 3C. J Urol (2014) 192(5):1564–8. doi: 10.1016/j.juro.2014.04.099
86. Zou D, Zhang K, Yang Y, Ren Y, Zhang L, Xiao X, et al. Th17 and IL-17 exhibit higher levels in osteonecrosis of the femoral head and have a positive correlation with severity of pain. Endokrynol Pol (2018) 69(3):283–90. doi: 10.5603/EP.a2018.0031
87. Ohka S, Nishizawa D, Hasegawa J, Takahashi K, Nakayama K, Ebata Y, et al. Association between rs2275913 single-nucleotide polymorphism of the interleukin-17A gene and perioperative analgesic use in cosmetic orthognathic surgery. Neuropsychopharmacol Rep (2018) 38(2):67–74. doi: 10.1002/npr2.12010
88. Zhu S, Qian Y. IL-17/IL-17 receptor system in autoimmune disease: mechanisms and therapeutic potential. Clin Sci (Lond) (2012) 122(11):487–511. doi: 10.1042/CS20110496
89. Martin DA, Churchill M, Flores-Suarez L, Cardiel MH, Wallace D, Martin R, et al. A phase ib multiple ascending dose study evaluating safety, pharmacokinetics, and early clinical response of brodalumab, a human anti-IL-17R antibody, in methotrexate-resistant rheumatoid arthritis. Arthritis Res Ther (2013) 15(5):R164. doi: 10.1186/ar4347
90. Mitra A, Raychaudhuri SK, Raychaudhuri SP. IL-17 and IL-17R: an auspicious therapeutic target for psoriatic disease. Actas Dermosifiliogr (2014) 105 Suppl 1:21–33. doi: 10.1016/S0001-7310(14)70015-8
91. Papp KA, Leonardi C, Menter A, Ortonne J-P, Krueger JG, Kricorian G, et al. Brodalumab, an anti-interleukin-17-receptor antibody for psoriasis. N Engl J Med (2012) 366(13):1181–9. doi: 10.1056/NEJMoa1109017
92. Mease PJ, Helliwell PS, Hjuler KF, Raymond K, McInnes I. Brodalumab in psoriatic arthritis: results from the randomised phase III AMVISION-1 and AMVISION-2 trials. Ann Rheum Dis (2021) 80(2):185–93. doi: 10.1136/annrheumdis-2019-216835
93. Garnock-Jones KP. Secukinumab: a review in moderate to severe plaque psoriasis. Am J Clin Dermatol (2015) 16(4):323–30. doi: 10.1007/s40257-015-0143-7
94. Kerschbaumer A, Smolen JS, Dougados M, de Wit M, Primdahl J, McInnes I, et al. Pharmacological treatment of psoriatic arthritis: a systematic literature research for the 2019 update of the EULAR recommendations for the management of psoriatic arthritis. Ann Rheum Dis (2020) 79(6):778–86. doi: 10.1136/annrheumdis-2020-217163
95. Blair HA. Secukinumab: A review in ankylosing spondylitis. Drugs (2019) 79(4):433–43. doi: 10.1007/s40265-019-01075-3
96. Markham A. Ixekizumab: First global approval. Drugs (2016) 76(8):901–5. doi: 10.1007/s40265-016-0579-y
97. Toussirot E. Ixekizumab: an anti- IL-17A monoclonal antibody for the treatment of psoriatic arthritis. Expert Opin Biol Ther (2018) 18(1):101–7. doi: 10.1080/14712598.2018.1410133
98. Blegvad C, Skov L, Zachariae C. Ixekizumab for the treatment of psoriasis: an update on new data since first approval. Expert Rev Clin Immunol (2019) 15(2):111–21. doi: 10.1080/1744666X.2019.1559730
99. Deodhar A, Conaghan PG, Kvien TK, Strand V, Sherif B, Porter B, et al. Secukinumab provides rapid and persistent relief in pain and fatigue symptoms in patients with ankylosing spondylitis irrespective of baseline c-reactive protein levels or prior tumour necrosis factor inhibitor therapy: 2-year data from the MEASURE 2 study. Clin Exp Rheumatol (2019) 37(2):260–9.
Keywords: interleukin-17, inflammatory cytokines, neuroinflammation, neuropathic pain, inflammatory pain, cancer, chronic pain
Citation: Jiang X, Zhou R, Zhang Y, Zhu T, Li Q and Zhang W (2022) Interleukin-17 as a potential therapeutic target for chronic pain. Front. Immunol. 13:999407. doi: 10.3389/fimmu.2022.999407
Received: 21 July 2022; Accepted: 20 September 2022;
Published: 29 September 2022.
Edited by:
Eva Reali, University of Milano-Bicocca, ItalyReviewed by:
Wuping Sun, Affiliated Nanshan Hospital of Shenzhen University Health Science Center, ChinaCopyright © 2022 Jiang, Zhou, Zhang, Zhu, Li and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qian Li, aHhsaXFpYW5AZm94bWFpbC5jb20=; Weiyi Zhang, emhhbmd3ZWl5aUB3Y2hzY3UuY24=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.