 Zhangyan Jing1†
Zhangyan Jing1† Xudong Zhang
Xudong Zhang- 1Department of Pharmacology, School of Medicine, Shenzhen Campus of Sun Yat-sen University, Sun Yat-sen University, Shenzhen, Guangdong, China
- 2Guangdong Provincial Key Laboratory of Medical Molecular Diagnostics, Key Laboratory of Stem Cell and Regenerative Tissue Engineering, School of Basic Medical Sciences, Guangdong Medical University, Dongguan, China
The pathogeny of type 1 diabetes (T1D) is mainly provoked by the β-cell loss due to the autoimmune attack. Critically, autoreactive T cells firsthand attack β-cell in islet, that results in the deficiency of insulin in bloodstream and ultimately leads to hyperglycemia. Hence, modulating immunity to conserve residual β-cell is a desirable way to treat new-onset T1D. However, systemic immunosuppression makes patients at risk of organ damage, infection, even cancers. Biomaterials can be leveraged to achieve targeted immunomodulation, which can reduce the toxic side effects of immunosuppressants. In this review, we discuss the recent advances in harness of biomaterials to immunomodulate immunity for T1D. We investigate nanotechnology in targeting delivery of immunosuppressant, biological macromolecule for β-cell specific autoreactive T cell regulation. We also explore the biomaterials for developing vaccines and facilitate immunosuppressive cells to restore immune tolerance in pancreas.
1 Introduction
Type 1 diabetes (T1D) is a metabolic syndrome caused by the loss of autoimmune tolerance to insulin-producing islet β-cells (1). Pancreatic infiltrated islet-autoreactive T cells destroy the β-cells, resulting in insufficient insulin secretion, that elicits hyperglycemia (2). T1D is commonly occurred in children and adolescents with age from 1 to 15 years old, and around 1.2 million cases worldwide are diagnosed with T1D by 2021 (3). Moreover, the incidence of T1D is rising ever year. There are annually almost 30,000 new cases in United States and 13,000 new cases in China (3).
There have been many applications of immunotherapy for T1D. Currently, insulin replacement therapy is the main form of treatment for T1D since the discovery of insulin in 1921 by Frederick Banting and his colleague. Hereafter, pharmaceutical companies have developed a variety of insulin analogues such as fast-acting insulin and long-acting insulin analogues for the treatment of T1D (4). However, exogenous insulin injection cannot accurately mimic the physiological insulin secretion by the β-cells through sensing the high level of blood glucose (5). Thus, the careless of management of blood glucose could cause hyperglycemia, and long-term hyperglycemia may lead to many diabetic complications such as diabetic heart disease, diabetic neuropathy, diabetic nephropathy, etc. (4). On the other hand, excess insulin administration may also cause hypoglycemia followed by coma and even the risk of death (6). Therefore, glucose-sensing and responsive insulin delivery system represents an ideal treatment of T1D. Numerous strategies were proposed, such as glucose oxidase-based systems, glucose-binding molecule (particle)-based systems and phenylboronic acid-based systems, which were reviewed in detail before (7).
Islet transplantation is another alternatively treatment for T1D. Shapiro and colleagues successfully performed islet transplant on a patient firstly in 2000, and this standard procedure was known as “Edmon protocol” (8). Although islet transplantation can be technically achieved, it still faces many problems such as the lack of donors, cumbersome isolation of islets, immune rejection of transplantation, long-term need for immunosuppressants, and islet death caused by immune rejection and hypoxia (3). To conquer these issues, utilizing biomaterial to encapsulate islets, β-cells or induced pluripotent stem cells enhances our insight into better transplantation in remission of autoimmune responses, amelioration of hypoxia and protection against fibrosis. Various strategies, including modification of grafts, encapsulation of functional materials (e.g., can produce oxygen), co-transplantation with other cells or molecules, offer an attractive modality.
T1D is considered as a T cell-mediated autoimmune disease, and involves in both of innate immune and acquired immune cells (9). Critically, the abnormality of peripheral immune tolerance system such as the loss of regulatory T cells (Tregs) and decrease of programmed cell death-ligand 1 (PD-L1) also leads to T1D (10). Thus, inhibiting the immune activity and restoring self-tolerance to pancreatic β-cells are novel treatment strategies for T1D. Immunosuppressive agent cyclosporine A were firstly used in the study of treatment for T1D (11). Although cyclosporine A could protect β-cells, its nephrotoxicity led to the termination of the studies (11). Next, anti-CD3 monoclonal antibody was utilized to deplete T cells to avoid autoimmune attack the residual β-cells, that could permanently reverse the diabetes in the NOD mice (12). Regardless of anti-CD3 monoclonal antibody may also cause the risk of infection, it is still under the study of phase III clinical trial, and may become an potent antibody drug for T1D treatment (13). T cell immune checkpoints are a class of receptors present on the surface of T cells, or intracellular metabolic enzymes that regulate T cell activity, such as programmed death-1 (PD-1), T cell immunoglobulin and mucin domain-containing protein 3 (TIM-3), and lymphocyte activation gene-3 (LAG-3) (14). Many types of cancer cells abnormally express immune checkpoint inhibitory ligands such as PD-L1, enabling cancer cells to escape from immune elimination (14). Therefore, monoclonal antibodies of anti-PD-1/PD-L1 have been developed to treat cancer and have achieved significant efficacy in the clinic therapy (15). However, some patients developed autoimmune diabetes after receiving antibody therapy (16). Thus, immunosuppressive checkpoint ligands on β-cells may play an important role in maintaining immune tolerance and protecting cells from the development of T1D (17). However, these immunosuppressive checkpoint ligands such as PD-L1 have been demonstrated as novel and potent therapeutic candidates to treat T1D. Additionally, Tregs can directly inhibit the activity of effector T cells. Therefore, Tregs play a critical role in peripheral immune tolerance and the prevention of autoimmune diseases (10). To restore the immune tolerance of the pancreatic microenvironment, it is essential to induce the expansion of Tregs (10). Autoantigen-based vaccines such as insulin and GAD 65 peptides could induce autoantigen-specific Tregs to treat T1D (18).
Although the systemic immunosuppression could protect residual β-cells from autoimmune attack and reverse the diabetes, it still makes patients under the risk of organ damage. We discuss the recent advances in biomaterial used to immunomodulate immunity for T1D, including the controlled release of chemical immunosuppressive agent, targeted delivery of biological macromolecule for autoreactive T cell regulation by nanotechnology, exploring the biomaterials to develop diabetic vaccine and facilitating immunosuppressive cells to restore immune tolerance in pancreas (Figure 1).
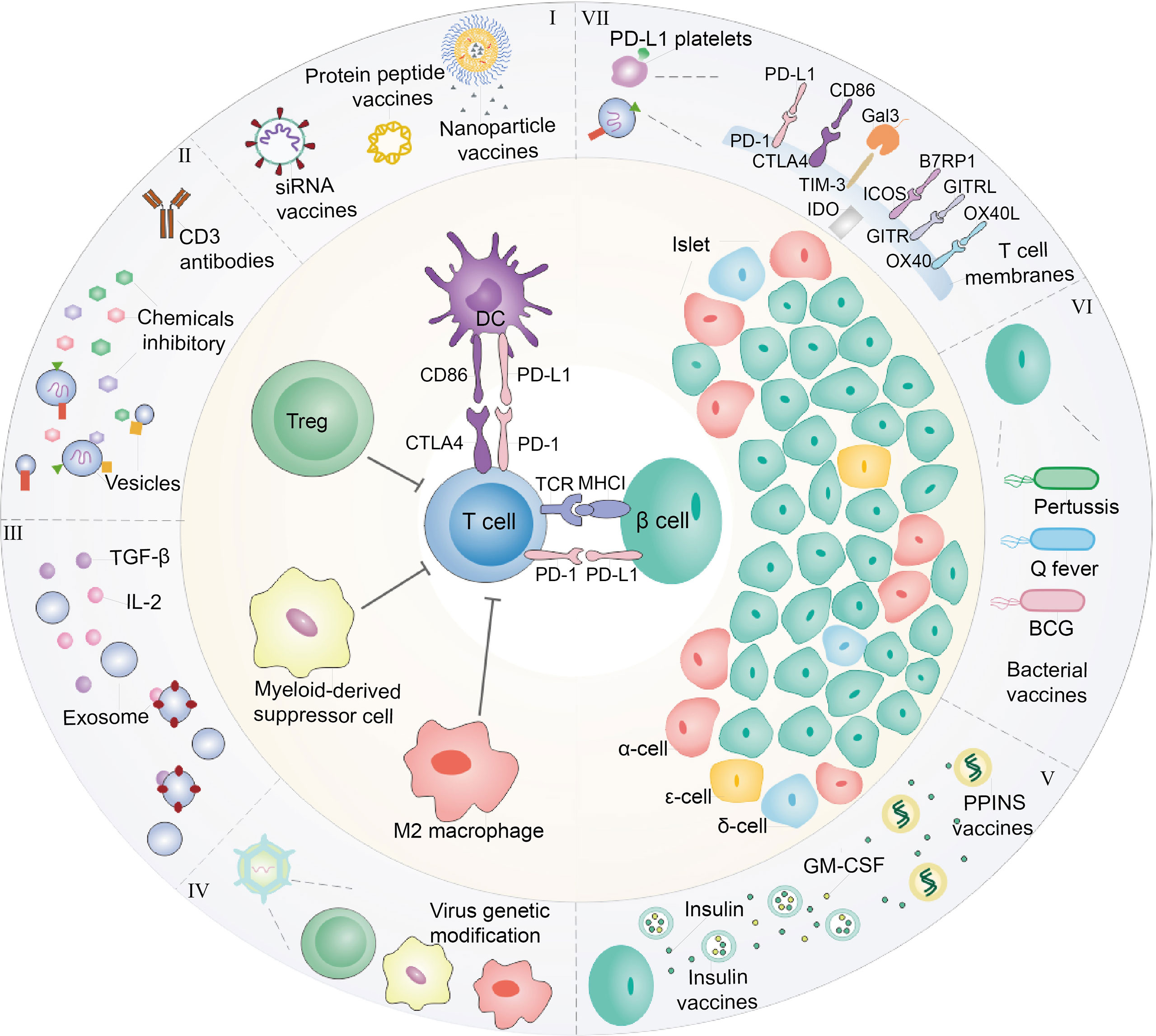
Figure 1 Diagram of interaction between immune cells and β-cells. At present, immunotherapy is commonly explored to treat T1D. (I) In the perspective of DCs, nanoparticles, or liposomes encapsulated with GAD56 peptides, siRNA molecules in pancreatic associated APCs Sema3E, Protein gene as T1D vaccines are often used. (II) For Tregs, the recovery of immune tolerance and immune homeostasis can be achieved by the use of CD3 monoclonal antibody and the combination with chemical inhibitors (prednisone azathioprine cyclosporin rapamycin et al.) to treat T1D. In order to enhance the therapeutic effect and reduce the toxic and side effects, it is considered to encapsulate the drug within nanoparticles or vesicles. (III) In addition, immunosuppressive factors such as transforming growth factor-β (TGF-β) and Interleukin-2 (IL-2) also have been employed for treating T1D. (IV) Immunosuppressive cells including Tregs, MDSC and M2 macrophage were cultured or engineered to modulate immunity of T cells for T1D therapy, which also could secrete exosomes with overexpression of TGF-β and IL-2. These immunosuppressive cells are often stimulated in vivo or re-infused into the body after the selective amplification or virus genetic modification in vitro to enhance their function of inhibiting T cells. (V) In addition, the insulin gene is modified and edited to produce insulin peptide vaccine, which can continuously secrete insulin in the body with only one injection. In cases where the β-cells have been damaged finally cause hyperglycemia, hence, foreign insulin is often utilized to maintain blood sugar levels. In addition to direct insulin injection, insulin and GM-CSF wrapped in cell membrane vesicles are currently considered to be injected into the body to achieve the purpose of sustained release. (VI) Moreover, β-cells are susceptible to bacterial infection and cause to damage. Therefore, it is common to detoxify pathogenic bacteria and make vaccines to achieve the purpose of prevention or treatment of T1D. Q fever vaccines, BCG vaccines and Coxsackie virus B1 vaccines are common bacteria that are used as T1D vaccines. (VII) On the other hand, T1D is caused by the direct β-cells attack by T cells. Thus, blocking T cells can be used to protect β-cells. For example, platelets expressing PD-L1 can accurately identify pathological T cells, and bind to PD-1 on T cell to abate its attack. Beside PD-1/PD-L1 immune checkpoint, CTLA4/CD86, TIM3/Gal3, ICOS/B7RP1, etc., also could be considered as T1D therapy targets.
2 Immune suppressive agents
As previously described, T1D is the result of deficiency of endogenous insulin due to the cell-mediated autoimmune destruction of pancreatic β-cells. Thus, how to preserve β-cells for sufficient production and release of insulin would provide a platform. To date, there have been several methods to enable the strategy of immune intervention: by either blocking autoimmune response against β-cells or achieving the replication of preexisting β-cells.
2.1 Prednisone
Prednisone is a synthetic glucocorticoid (GC) prodrug that is converted by the liver into prednisolone (a β-hydroxy group instead of the oxo group at position 11), which is a active steroid. In current study, prednisone is a synthetic corticosteroid, was administered to patients with T1D to roughly mimic the natural hormonal response to acute stress and to induce insulin resistance. Genomic effects are mediated by glucocorticoids, which bind to specific receptor proteins in target tissues and direct their production or inhibition. Therefore, most of the effects of glucocorticoids may not be seen for several hours.
Insulin requirements to maintain normoglycemia during glucocorticoid therapy and stress are often difficult to estimate. To simulate insulin resistance during stress, adults with T1D were given a three-day course of prednisone. Studies by Wendy C. Bevier et al. suggest that in adults with T1D, insulin requirements during prednisone-induced insulin resistance may need to increase by 70% or more to normalize blood sugar levels (19). Moreover, A. Secchi. et al. observed the curative effect of prednisone on the remission of newly diagnosed T1D patients and found that prednisone can also induce the remission of insulin dependent diabetes mellitus (IDDM). Remission may occur early in the onset of adult T1D (20). Prednisone broadly inhibits inflammatory cytokines caused by RTK bypass signaling and inhibition of EGFR or other RTK targets and thus may be a useful drug in combination with RTK pathway inhibition. Ke Gong et al. found similar results with dexamethasone, suggesting this could be GC. In particular, prednisone induces bypass RTK signaling, shutting down and repressing key resistance signals (21).While prednisone has a number of side effects and can induce significant problems with sustained use, it continues to be widely used in multiple inflammatory and rheumatologic conditions because it is highly effective.
2.2 Azathioprine
Azathioprine (AZA) and thiopurine are widely used for induction and maintenance of remission in steroid dependent patients with inflammatory bowel disease (IBD) (22). As the antilymphocyte adaptive agent, azathioprine and prednisone could induce immunosuppression and reduce insulin requirements at the onset of diabetes. However, the remission of autoimmunity was limited by the duration of the treatment, demonstrating that the tolerance to pancreatic β-cell autoantigens was not re-established (23).
Geliebter et al. delivered azathioprine after thymectomy for the treatment of T1D, which exhibited a good therapeutic effect on T1D. And Sun-275 azathioprine for slow release for the tertiary prevention of diabetes in newly diagnosed T1D patients, showing more effective results (24). Insulin dosage in patients with T1D can be effectively reduced by the delivery of Prednisone (19).
2.3 Cyclosporin A
Cyclosporine A (CsA) is an immunosuppressant agent that is widely used in clinical practice, predominantly for the prevention of rejection in various types of post-allogenic organ transplantation. In particular, cyclic peptides combine several properties such as high affinity, target selectivity and stability (enzymatic and chemical), which are important properties for therapeutics and make them ideal as orally delivered candidate drugs. The current interim analysis of the prospective and randomized reverse study showed that switching from tacrolimus to cyclosporine improved glucose metabolism and reversed new-onset diabetes after transplantation (NODAT) in majority of patients. The change of drug used was safe and did not result in an increased incidence of acute rejection episodes. Stiller C.R. et al. showed that the utilization of cyclosporine reduced insulin requirement and improved glycemic control in patients with recent T1D (25). However, cyclosporine could not fully establish immune tolerance and had other toxic side effects (25, 26). Furthermore, CsA induced nephrotoxicity, and azathioprine was ineffective as a single agent (23). Although some patients could achieve short-term control of diabetes, more researches are required to determine whether cyclosporine can be used safely to maintain glycemic control and prevent long-term consequences of the disease.
Short-term control of diabetes can be achieved by delivering Cyclosporin. Persistent remission appears to be dependent on continued administration of cyclosporine. Drug delivery decreases not only the leucocyte migration inhibition as previously observed (25), but also the lymphocyte-mediated cytotoxicity, which represents the late stage of cellular immune reactions against pancreatic tissue. Hence, it is promising to target the pancreas to control release of CsA in vivo through nanoparticles, such as cell membrane vesicles, polymer material PEG wrapping could achieve targeting therapy and reduce the toxicity of CsA for T1D.
2.4 Rapamycin
Rapamycin (RAPA) is a macrolide anti-immune antibiotic produced by Streptococcus. RAPA cannot directly block the transcription, translation and the secretion of gene products (IL-2, IL-3, IL-4, CM-CSF, TNF-α and IFN-γ, etc.) RAPA is different from nucleotide biosynthesis, it inhibits immune activation pathways that are insensitive to CsA and FK506. through blocking Ca2+-independent T cell activation and inhibiting IL-2-dependent entry of activated T cells into S phase.
Further, RAPA has been used in clinical transplantation for many years (27), including islet transplantation in T1D patients safely (28). In mice, RAPA monotherapy prevented from developing T1D but failed to induce disease reversal (29). Studies have shown that RAPA, as a targeted inhibitor of the mTOR pathway, exerts anti-inflammatory and anti-tumor effects in a variety of diseases by inhibiting the activation of the mToR pathway (30, 31). Interestingly, RAPA/GABA combination therapy effectively suppressed autoimmune responses to islet cells and improved islet function in patients with new-onset diabetes. In particular, after a hyperglycemic episode, patients treated with the RAPA/GABA combine significantly achieved diabetes improvement compared with patients treated with RAPA or GABA alone. This protective effect of RAPA/GABA combination treatment in NOD mice works through two distinct mechanisms (32). RAPA induces Tregs and thus suppresses targeted autoimmune responses to islet cells, which may be related to the reduction in insulitis observed in RAPA-treated NOD mice simultaneously. Manirarora and Wei treated NOD mice with a combination of IL-2/anti-IL-2 Ab complex, islet Ag peptide, and RAPA. Following combination treatment, CD4+CD25+Foxp3+ Tregs significantly expanded in vivo, expressed canonical Tregs markers, enhanced suppressive function in vitro, and spontaneously inhibited T1D in NOD mice. This novel approach to peripheral immune regulation will facilitate the progress of new immunotherapeutic strategies to prevent the development of T1D or promote resistance to islet transplantation without the long-term use of immunosuppressive drugs (33).
Lee Jung Seok et al. show that polymerized ursodeoxycholic acid, selected from a panel of bile acid polymers and formulated into nanoparticles for oral administration of RAPA, delayed diabetes in mice with chemically induced pancreatic inflammation attack (34). Nanoparticles acted as protective insulin carriers and high-affinity bile acid receptor agonists, increased intestinal absorption of insulin, polarized intestinal macrophages to an M2 phenotype, and preferentially accumulated in mouse pancreas, where they interacted with pancreatic islets associated with cellular bile. These nanoparticles could bind to acidic membrane receptor TGR5 with high affinity and activate glucagon-like peptide and endogenous insulin secretion. In mice, the nanoparticles also reversed inflammation, restored metabolic function and prolonged the animals’ survival. The metabolic and immunomodulatory functions of ingestible bile acid polymer nanocarriers may provide translational opportunities for the prevention and treatment of T1D. Moreover, RAPA can be modified. For example, drug delivery in cell membranes or cell vesicles, or in combination with other biological materials.
2.5 Anti-CD3 antibody
CD3 has five peptide chains, namely γ chain, δ chain, ϵ chain, ζ chain and η chain, all of which are transmembrane proteins. The transmembrane region of the CD3 molecule is connected to the transmembrane region of the two TCR peptide chains through a salt bridge to form a TCR-CD3 complex, which together participate in the recognition of antigens by T cells. The activation signal generated by TCR recognition of antigen is transduced into T cells by CD3. Preclinical studies suggested that mAbs against CD3 could reverse hyperglycemia at presentation and induce tolerance to recurrent disease. Based on previous study, the anti-CD3 monoclonal antibody could induce immune-suppressive Tregs and restoration of self-tolerance (35). Moreover, anti-CD3 specific antibody was used with a human proinsulin II to achieve remission even reversion of diabetes by synergistic effect (36). Interestingly, transgenic Lactococcus lactis combined with oral insulin and IL-10 combined with low-dose anti-CD3 in T1D mice can restore autoimmune tolerance and block the killing of β-cells by T cells (37). In addition, Stewart et al. combined a subcutaneously administered dual-size biodegradable microparticle (MP) platform with anti-CD3 to treated NOD mice. According to MP platform, DCs recruitment can be increased and Tregs expression can be induced to achieve immune tolerance. In theory, the combination of MP platform and anti-CD3 in the treatment of T1D mice seems to be a good direction. Sadly, there was no therapeutic benefit when considering the combination of biomaterials with CD3 antibodies (38). In this way, biomaterial combination therapy for T1D can be better utilized.
3 Immune suppressive factor
Immunosuppressive cytokines mainly include immune checkpoint molecules such as TGF-β, IL-2 and CD3. TGF-β is an important factor in inducing macrophages towards M2-type polarization and can promote IL-10 expression (39).IL-10 reduces the expression of MHC class II molecules and co-stimulatory molecules CD80 and CD86 on the surface of antigen presenting cells (APCs) such as dendritic cells (DCs) and macrophages, and mediates the phosphorylation of STAT3, and interferes with IFN-γ-induced monocyte activation and expression, leading to the function inhibition of APCs.
3.1 TGF-β
TGF-β inhibits effector cells directly or indirectly by disrupting DCs differentiation, migration, and antigen presentation. That is, TGF-β has two effects, both directly inhibiting T cells and inducing Tregs to establish a tolerance mechanism (40).
TGF-β is an immunoregulatory cytokine that enhances the development of an immune tolerance microenvironment by inhibiting antigen-specific inflammatory processes such as those found in autoimmune diseases and cancer. Li et al. used in vitro CD4+ T cell flow cytometry to verify forkhead transcription factor positive (Foxp3+) expression and TGF-β release (Figure 2) (41). In a TGF-β-rich environment, DCs, which uptake antigens, become tolerogenic and mediate the differentiation of antigen-specific CD4+ T cells into anti-inflammatory Tregs (42), which inhibits both the formation of antigen-specific effector CD4+ T cells and the activation and proliferation of cytotoxic CD8+ T cells (43). In order to avoid unavoidable inflammatory response induced by biomaterial implantation itself, TGF-β was designed to release from layered PLG scaffolds and showed great potential to enhance the function of encapsulated cells by decreasing the production of inflammatory cytokine (44). Considering immunomodulatory effect of MSC, in this study, researchers engineered MSCs with TGF-β gene to increase MSC potency for T1D therapy in mouse model. Engineered TGF-β/MSCs could restore several T1D functions, including modulating harmful immune responses, and could be a powerful tool for cell therapy of T1D compared to MSCs alone (45). To achieve sustained stimulation of regulatory immune cells, Ivan Koprivica et al. orally introduced microparticles (MPs) loaded with all-trans retinoic acid (ATRA) and transforming growth factor-β (TGF-β) to C57BL/6 mice treated with multiple low doses of streptozotocin (MLDS) for T1D induction. Mice treated with ATRA/TGF-β MP had significantly reduced disease incidence and immune cell infiltration into islets. In conclusion, oral administration of ATRA/TGF-β MPs ameliorated T1D by enhancing tolerogenic dendritic cells (tolDCs) and Tregs, suppressing Th1 responses, and preventing immune cells from entering islets (46). Current clinical practice relies on long-term systemic immunosuppression, leading to serious adverse events. To circumvent these adverse effects, polylactic-co-glycolic acid (PLGA) microparticles (MPs) were designed for localized and controlled release of immunomodulatory TGF-β1. In vitro co-incubation of TGF-β-releasing PLGA MPs with naive CD4+ T cells efficiently generated polyclonal and antigen-specific inducible regulatory T cells (iTregs) with potent immunosuppressive capabilities. Co-implantation of TGF-β1-releasing PLGA MPs with Balb/c mouse pancreatic islets into the extrahepatic epididymal fat pad (EFP) of diabetic C57BL/6J mice resulted in rapid allograft engraftment and local compatibility with PLGA MPs. TGF-β1 release is supported. However, the presence of TGF-β1-PLGA MPs did not confer significant graft protection compared to untreated controls, although insulin expression did not change and intra-islet CD3+ T cell infiltration was reduced and peripheral CD4+ T cells were measured elevated in - long-term graft site functional grafts. Examination of the broader effects of TGF-β1/PLGA MPs on the host immune system suggests a local nature of the immunomodulation, with no systemic effects observed. In conclusion, this approach established the feasibility of local and modular microparticle delivery systems for immunomodulation at liver explant sites. This approach can be readily adapted to deliver larger doses or other agents in the local graft microenvironment, similar to multi-drug approaches to prevent graft rejection (41).
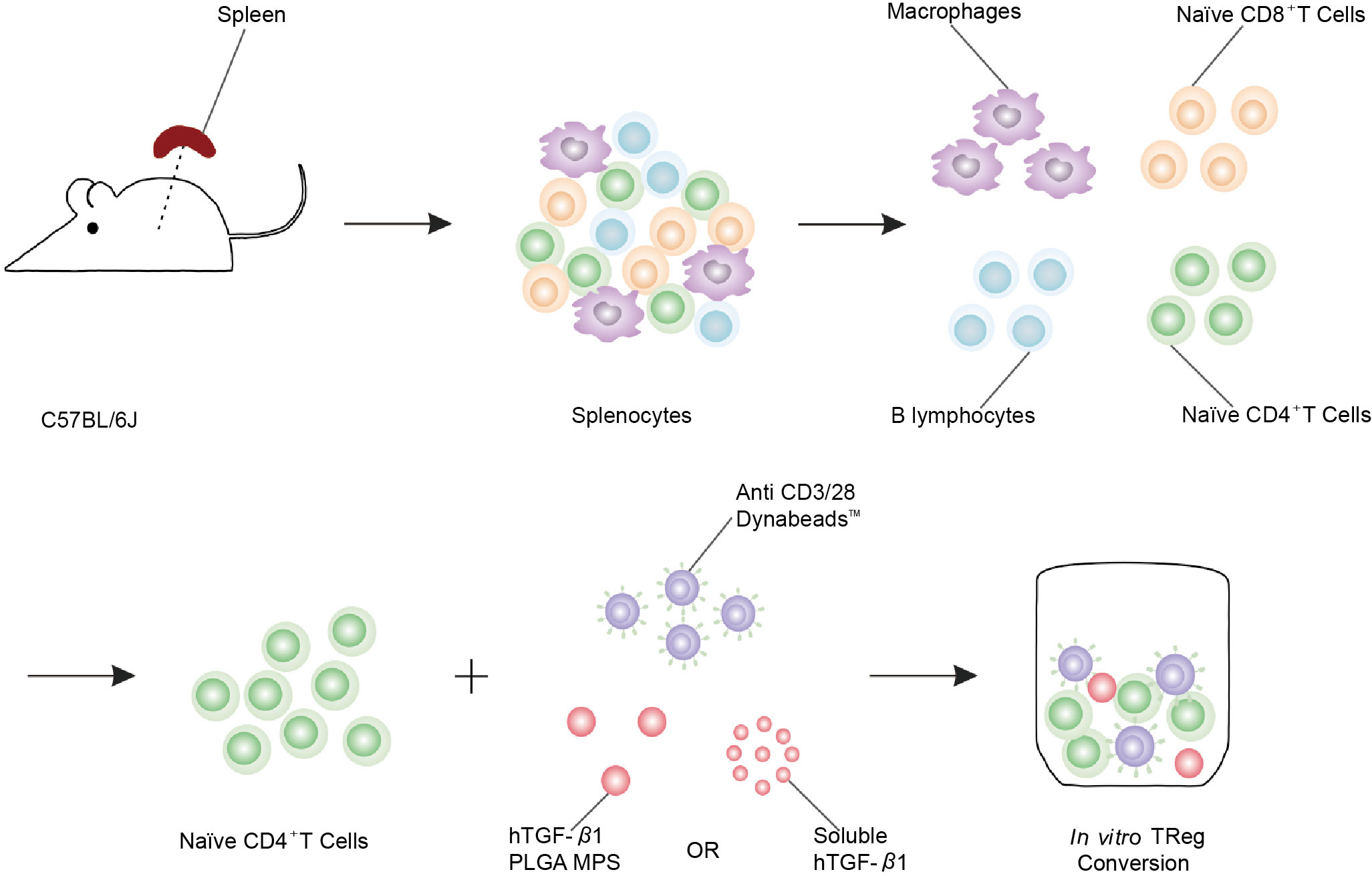
Figure 2 Schematic of polyclonal induced Treg (iTreg) conversion assay. Poly (lactic-co-glycolic acid) (PLGA) microparticles (MPs) were designed for the local and controlled release of immunomodulatory TGF-β1. PLGA MPs release TGF-β1, which can efficiently promote the conversion of naïve CD4+ T cells isolated from spleen to iTreg (41).
Recent studies have demonstrated the unique potency of cell surface-bound TGF-β1 on Tregs to promote infection tolerance, i.e., confer suppressive capacity from one cell to another. To mimic this property, TGF-β1 was selectively chemically bound to an inert and viable polymer grafting platform using staudinger ligation. Inert beads tethered with TGF-β 1 were capable of efficiently converting naïve CD4+ CD62Lhi T cells to functional Treg. These results illustrated the unique function of the tethered TGF-β1 biomaterial platform to function as “infectious” Tregs and provide a compelling approach to generating a tolerant microenvironment for allogeneic transplantation.
3.2 IL-2
As IL-2/IL-2R might be the activation pathway relative to T1D (47), the safe and effective dose range of IL-2 for T1D therapy was investigated, showing that appropriate dosage of IL-2 facilitated β-cells persistence (47–49).What’s more, a recent clinical trial verified previous conclusion that IL-2 is a major role both in the development of T1D and the function of Tregs (50).
However, it is necessary to optimize the specificity of IL-2 since it can both activate Tregs cells and pathogenic T cells. Two major strategies have showed to be feasible in this area. One is antibody-cytokine conjugates. It was mentioned that different IL-2 and IL-2 monoclonal antibodies (mAbs) complexes could be used to achieve opposite effects. For example, IL-2 could enhance the proliferation of CD8+ T cells by binding to JES6-1 mAbs, as well as stimulating CD4+ Tregs with the combination of S4B6 mAbs, according to its selectivity resulting from conformational change (51, 52). And the other is chemical modifications of IL-2. A human IL-2, whose conformation was modified, could specifically expand Tregs in vivo to avoid off-target undesired effects (53). UFKA-20, an anti-human IL-2 antibody, was also identified and demonstrated the preference of stimulating CD4+ Tregs cells through binding to IL-2 (54). H9-RETR was designed as an engineered IL-2 variants and specific for IL-2Rb, holding the promise of a novel approach for treating T1D (55).
In addition, IL-2 conjugates with polymers, such as PEGylated IL-2, exert promising progress and are under further study. PEGylated IL-2 molecules that preferentially bind to different IL-2R conformations are being explored in oncology to controllably activate the IL-2 system and shift the balance in the tumor microenvironment in favor of effector T cells (Teffs). Overwijk et al. induced tumor regression by increasing intratumorally proliferation, activation and effector function of CD8+ T and NK cells (56). Cristina Izquierdo et al. demonstrated that combined treatment of Ag7/2.5 mi tetramer and IL-2: mAb complex is effective in preventing T1D. The treatment led to a large expansion of Ag-specific Foxp3+ Treg that acquire markers of activation, suppression and homing, and is accompanied by the proliferation of an antigen-specific Foxp3− population that produced anti-inflammatory IL-10 (57). Jonathan T. Sockolosky et al. engineered IL-2 cytokine-receptor orthogonal (ortho) pairs that interact with one another, transmitting native IL-2 signals, but do not interact with their natural cytokine and receptor counterparts. Introduction of ortho IL-2Rb into T cells enabled the selective cellular targeting of orthoIL-2 to engineered CD4+ and CD8+ T cells in vitro and in vivo, with limited off-target effects and negligible toxicity. OrthoIL-2 pairs were efficacious in a preclinical mouse cancer model of adoptive cell therapy and may therefore represent a synthetic approach to achieving selective potentiation of engineered cells. OrthoIL-2 may be a synthetic approach to achieve selective enhancement in engineered cells (58). By modifying IL-2, the half-life of the drug can be prolonged, the cell specificity can be improved, and it can act more specifically on effector T cells or regulatory T cells for the treatment of tumors or autoimmune diseases. The sustained-release inhibitors used in the treatment of tumors can be used in the treatment of diabetes to improve the targeting of drugs.
4 Immune checkpoint
Immune checkpoints are immune homeostasis negative regulatory mechanisms that maintains autoimmune tolerance (59). Currently known immune checkpoints include programmed death-1/PD-L1(PD-1/PD-L1), cytotoxic T lymphocyte antigen 4 (CTLA4), Galactose lectin 3 (Gal3), indoleamine 2,3 dioxygenase (IDO) and so on. Of these, PD-1 and CTLA4 are the most well-known (60), although their modes of action and signaling pathways are distinct, they both contribute to maintain autoimmune tolerance. PD-1 is absent in immature T cells and memory T cells at rest, but it is expressed when the TCR is activated. Meanwhile, transcriptional activation is required for PD-1 expression on activated T cells. A classical immunoreceptor tyrosine inhibitory motif (ITIM) and an immunoreceptor tyrosine switch motif (ITSM) are also found in PD-1. On the other hand, CTLA4 is involved in down-regulating the magnitude of T cell response, mainly by competing with CD28 to share ligands CD80 (B 7.1) and CD86 (B 7.2) (59, 61). Nivolumab (PD-1 antibody), lambrolizumab (PD-1 antibody), and ipilimumab (CTLA4 antibody) are the most common immune checkpoint mAbs on the market today. These medications, rather than targeting tumor cells. As a result, immune checkpoint inhibition is frequently employed during tumor treatment to break tumor immunological tolerance. T1D, on the other hand, can move in the reverse direction of treating tumors, reverting over immunity to immunological homeostasis.
4.1 PD-1/PD-L1 targeting therapy
The immune checkpoint signaling axis PD-1/PD-L1 can effectively control T cell activity and prevent autoimmune attacks. To generate a self-amplifying signal loop, IFN-γ and IL-2 may increase PD-L1 expression, reconstruct the PD-1/PD-L1 inhibitory axis, suppressing CD4+ T cell activation and restoring immunological tolerance to over-activated T cells and over-produced pro-inflammatory cytokines (60, 61). It has been consistently proven that PD-1/PD-L1 interaction plays a central role in the induction and regulation of autoimmune diabetes progression in NOD mice (62). In addition, to gain more about how CD4+ T cells affect diabetes, Pauken transferred a small number of naive CD4+ T cells from BDC2.5 mice into prediabetic NOD mice, allowing the cells to be activated by endogenous autoantigens. The transferred BDC2.5 Tcells were activated and differentiated into T-BET + IFN-γ producing cells, which infiltrated the pancreas. The absence of PD-1 on CD4+ T cells leads to an increase in the number of cells in the spleen, pancreatic draining lymph nodes, and pancreas. At the same time, PD-1 deletion also increased the expression of chemokine receptor CXCR3. That is, PD-1 regulates islet responsive CD4+ T cells in a cellular manner by inhibiting proliferation, restraining pancreatic invasion and limiting diabetes mellitus (63). However, tumor patients developed insulin-dependent diabetes after taking the anti-PD-1 antibody (nivolumab) (64). Thus, the safety of systemic administration of anti-PD-1 antibody remains challenging.
In NOD mouse model, β-cells and hematopoietic stem and progenitor cell (HSPC) lack PD-L1, making them vulnerable to CD4+ T cell attack. Ben Naser et al. established a PD-L1 deficient HSPC mouse model to explore PD-L1 immunotherapy. They engineered mouse Lineage−c-kit+ (KL) cells in vitro to produce PD-L1 and transplanted the modified KL cells into a mouse pancreas. PD-L1 expression defects identified in T1D patients can be regulated by drugs encapsulated in human HSPC that could also inhibit in vitro autoimmune responses (65). Xudong Zhang et al. used genetically engineered megakaryocytes to overexpress PD-L1 and produced platelets with immunosuppressive function. In neo-hyperglycemic NOD mice, platelets overexpressing PD-L1 accumulated in the inflammatory pancreas, inhibited the activity of autoreactive T cells, protected insulin-producing β-cells from destruction, and maintained immune tolerance in the pancreas (Figure 3) (66). Yoshihara et al. generated human islet-like organs (HILOs) from induced pluripotent stem cells, which were driven by atypical WNT4 signaling for metabolic maturation. The cells were cultured into 3D multicellular spheres (MCSs) through gel, and the MCSs was coated with sodium alginate. It was transplanted into the renal sac of NOD/SCID mice with streptozotocin (STZ) induced diabetes. For allografts, they protected HILO xenografts with overexpression of PD-L1. The blood glucose of STZ mice returned to normal 50 days after allograft (67). Au et al. uses pretargeting and glycochemistry ways. The β-cells were first being delivered with nanoparticles encapsulated with targeted Ac4ManNAz, enabling β-cells to have high levels of surface-active azide groups. The PD-L1 immunoglobulin fusion protein (PD-L1-IG) was then given dibenzylcycloctane (DBCO) function. PD-L1 can easily bind to the surface of natural β-cells. In NOD mice, it was demonstrated that bioorthogonal staining promoted azide-alkyne cycloaddition effectively and selectively conjugated PD-L1 to β-cells. Moreover, in vivo functionalized β-cells present both islet specific antigen and PD-L1 to participating T cells, reversing early-onset T1D by reducing IFN-γ-expressing cytotoxic T cells and inducing antigen-specific tolerance (68).
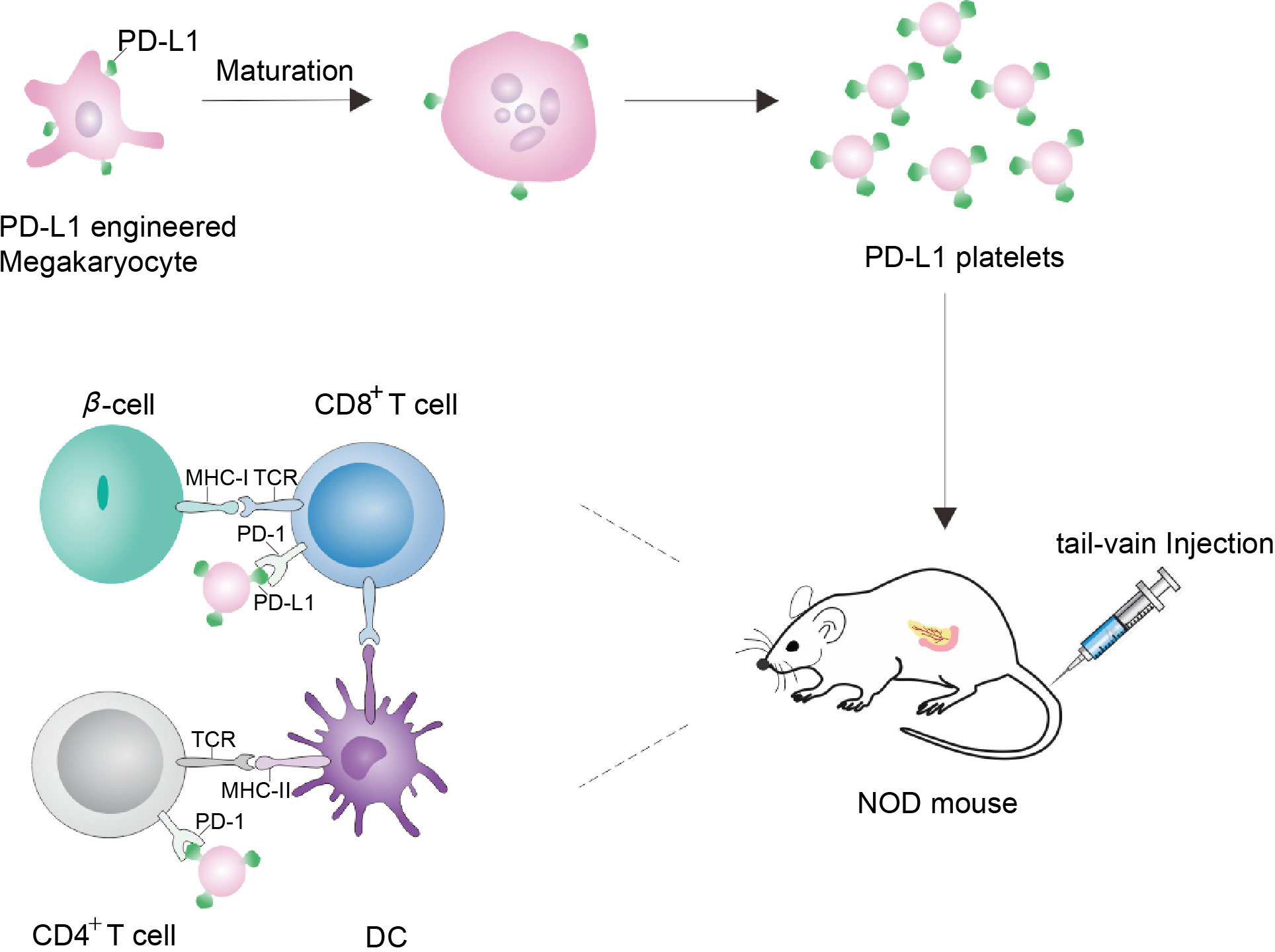
Figure 3 Schematic diagram of NOD mice treated by genetically engineering megakaryocytes to secrete platelets expressing PD-L1. Megakaryocytes were genetically engineered to express PD-L1. As PD-L1-megakaryocytes grow mature, the fragmentation of the proplatelets released platelets. Via tail-vein injection, PD-L1 platelets tend to accumulate in the inflamed pancreas, where PD-L1 can bind to PD-1 on the surface of CD4+/CD8+ T cells to protect β-cells, thereby protecting the pancreas and realizing the treatment of NOD mice (66).
However, immune checkpoint inhibitors also have adverse reactions in the treatment of insulin-dependent diabetes. Through 6 years of cases in two academic institutions, Stamatouli et al. found that a minority number of patients who received anti-PD-1 or anti-PD-L1 checkpoint inhibited developed ketoacidosis, pancreatitis and other autoimmune diseases (69). As a result, a new requirement for the development of PD-L1 for the treatment of diabetes is how to prevent the development of autoimmune diseases in a minority percentage of individuals.
4.2 Engineering CTLA4 for targeting therapy
CTLA4 is a T cell negative regulator that regulates T cell activation by competing with the costimulatory protein CD28 and binding to the shared ligands CD80 (B7.1) and CD86 (B7.2) (70). Meanwhile, the long-term survival of islet allografts produced by the soluble fusion protein CTLA4-immunoglobulin (CTLA4-Ig) is dependent on the host’s effective tryptophan catabolism. CTLA4 function in vivo by regulating tryptophan catabolism, but it also acts as a ligand of B7 receptor molecules, transducing intracellular signals (71). In order to treat diabetes mellitus, El Khatib et al. overexpressed synthetic PD-L1-CTLA4-Ig polyprotein using a cell-targeted AAV8 vector. Sadly, in NOD mice injected with proteins during early hyperglycemia, PD-L1-CTLA4-Ig alone was unable to normalize the state. However, the PD-L1-CTLA4-Ig expression vector prevented the islets from rejection for at least 4 months when drug-induced diabetic mice received MHC-matched transplanted islets. While transplanting PD-L1-CTLA4-Ig-expressing MHC-matched islets into T1D animals with established disease prevented immediate excretion of the transplanted islets, it eventually failed to sustain long-term immunological tolerance. Therefore, it is useful to design PD-L1 and CTLA4 gene-based strategies for the treatment of diabetes (72). Intriopirocrine green (ICG) is a photothermal agent to encapsulate imiquimod (Toll-like receptor 7 agonist) with polylactic acid coglycololic acid (PLGA). The formed PLGA-ICG-R837 nanoparticles, which trigger photothermal ablation by a near-infrared laser, can produce tumor-associated antigens in vivo, while combined with CTLA4 antibody therapy, which can prevent recurrence after tumor elimination. Can similar biomaterials be used for the treatment of T1D? In addition to the current treatment for diabetes with CTLA4-Ig, utilizing CTLA4 to prevent the onslaught of autoimmune cells to the islets is a suitable place to start (73). In reality, a “CAR-T” that expresses CTLA4-Ig was created in a method akin to CAR-T to continuous production of CTLA4-Ig in vivo, which can lessen the long-term immunosuppressive effectiveness and autoimmune rejection brought by islet transplantation.
4.3 Galactose lectin
Galactose lectin family plays an important role in cell proliferation and activation, but its function is easily reversed by the influence of the microenvironment. Galactose lectin has been continuously explored since 1975 when abnormal expression of galactose lectin was found in cancer. At present, Gal1 and Gal3 have been identified as the main molecules that mainly promote the growth of tumor cells and evade immune surveillance. At the same time, studies have suggested that the overexpression of Gal1 is related to the poor prognosis of patients with tumors, and the cure of hepatocellular carcinoma is often based on the expression of Gal1 (74, 75). Overexpression of Gal1 enables DCs to acquire interleukin-27 (IL-27) dependent regulation, promoting IL-10-mediated T cell tolerance and inhibiting autoimmune inflammation (76). It has also been found that Gal1 is related to the activation of MAPK signaling pathway and phosphatidylinositol-3 kinase (PI3K) signaling pathway. Gal9 in the galactose lectin family has shown great potential in the treatment of various cancers, and it was discovered that TIM-3 is a set of signaling pathways with Gal9 (77). The TIM-3/Gal9 pathway does not play a direct immunosuppressive role. Instead, TIM-3+ T cells were used to interact with CD4+CD25+ Tregs expressing Gal9 to activate T help 1 response and achieve immunosuppression (78).
In addition, Gal3 can also control the release of inflammatory cytokines into the body and the uptake of glucose by adipose tissue. Among some of the lectins, Gal3 is mostly secreted by macrophages. When fed a typical diet, without any accompanying inflammatory disease, galactose agglutinin-3 deficient (LGALS3 -/-) mice had smaller bodies and less epididymal white adipose tissue (eWAT). Further, metformin therapy could decrease these galactoagglutinin-3 levels (79). Mice receiving Gal3 injections developed insulin resistance and glucose intolerance, but obese mice receiving Gal3 treatment had enhanced insulin sensitivity due to pharmacological or gene loss. Gal3 was demonstrated to impair the insulin-mediated regulation of glucose output in primary mouse hepatocytes, lower the insulin-stimulated glucose uptake in muscle cells and 3T3-L1 adipocytes, and directly increase the chemotaxis of macrophages in in vitro tests. Gal3 can bind directly to the insulin receptor (IR) and suppress the following signals. So Gal3 inhibition is a novel target for the treatment of insulin sensitivity (80). Can pancreatitis be decreased or perhaps reversed by reducing or removing Gal3 since it is recognized to be the primary cause of pancreatitis? Iacobini et al. fed both wild-type (LGALS3+/+) and galactin-3 defective (LGALS3-/-) mice an atherogenic diet more than a longer length of time before the levels of fibrosis, inflammation, and steatosis in wild-type mice were much lower than those in wild-type mice. Gal3 was also discovered to be the receptor for advanced lipid oxidation end products in the liver. Gal3 ablation protected against diet-induced nonalcoholic steatohepatitis and reduced inflammation, hepatocyte damage, and fibrosis in terms of reducing the formation of lipoxidation end products in the liver (81). The application of the galactose lectin family is very promising, which can be considered as combined with other immunosuppressive agents, immunosuppressive factors, etc., to detreat T1D.
4.4 IDO
Initially, it was thought that the indoleamine 2,3 dioxygenase (IDO) enzyme directly affected how the immune system responded to illnesses and infectious agents. But as the years progressed, our knowledge of IDO has grown more and more comprehensive. In addition to limiting microbial growth and increasing immune response as an antibacterial, IDO may also have a role in modulating immune response activation and the development of immunological tolerance (82). The upregulation of plasma nephropathy (Kyn) by indoxyamide2,3-dioxygenase 1 (IDO1) in adipocytes has also been discovered. Mice were protected from obesity by IDO1 consumption in fat cells, which diminished Kyn buildup. For diabetic, obese mice, this offers a fresh perspective (83). Additional research on the location and expression of IDO in pancreatic tissues showed that diabetes patients with new-onset or other autoimmune diseases had significantly lower levels of IDO expression in insulin-secreting islets (84).
In C57BL/6 mice deficient in the Toll-like receptors9 (TLR9) gene, the lack of IDO induction in the pancreatic lymph nodes was likewise associated with the lack of STZ induction. It caused STZ mice to display some symptoms of a more serious illness. In order to prevent and treat T1D, attention should be paid to the TLR9-IDO axis (85). Due to decreased expression and catalytic activity of IDO1 in plasmacytoid dendritic cells (pDCs), TGF-β is unable to activate the IDO1 signaling pathway in NOD mice. Pallotta et al. pretransfected NOD pDCs with IDO1 to activate the IDO1 promoter, boost atypical NF-κB and TGF-β, and inhibit the production of pro-inflammatory cytokines, interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α), in such autoimmune diabetic animals. The presentation of pancreatic cell self-antigens in vivo could be inhibited by pDCs after TGF-β was applied to NOD pDCs with IDO1 overexpression (86). Moreover, the proteasome inhibitor bortezomib (BTZ) was utilized to restore IDO1 protein expression in vitro in pDCs. In autoimmune diabetic mice with prediabetes, treatment with BTZ can reverse the disease. But in hyperglycemic mice, there was no therapeutic efficacy (87). Dermal fibroblasts may also be used in addition to pDCs. dermal fibroblasts expressing IDO were given to newly unwell NOD mice by Zhang et al. in different doses. Only a small percentage of mice receiving low dosage therapy had their blood sugar levels reversed, but most animals receiving high dose treatment with dermal fibroblasts had their blood sugar levels returned to normal. It is amusing to note that high doses of IDO produced by fibroblasts reduced immune cell infiltration significantly while restoring islet cell function in NOD mice. Additionally, it lowered CD8+ T cells and T helper cell 17 in interactive NOD mice and increased Tregs in a number of organs (88).
There are different methods to accomplish IDO expression besides cell regulation. In inflammatory bodies, Dolpady et al. noticed that probiotics VSL#3, which were rich in lactobacilli, raised the release of IDO and IL-33 while actually lowering the expression of IL-1. By promoting CD103+ DCs differentiation tolerance and lessening Th1 and Th17 cells differentiation/expansion in intestinal mucosa and autoimmune disease, oral administration of lactobacteria-rich probiotics VSL#3 to NOD mice, either alone or in combination with retinoic acid, can modify their own intestinal microflora (89). Therefore, it makes sense to control IDO expression in a variety of methods in purpose of treating T1D.
5 Immunosuppressive cell therapy
As an autoimmune disease, T1D is induced by immune dysregulation, subsequently leading to the attack of pathogenic T cells to autologous β-cells. In view of this, several immunomodulatory therapies, such as delivering immunosuppressive agents and immune inhibitory biomacromolecule, have been proposed as described above. Most of them are focus on Treg, an immunosuppressive cell contributing to suppress overly pathogenic T cells and restoring autoimmune tolerance. However, more effective treatments are desired for complete remission of T1D due to the limited immune tolerance of the above therapies. Herein, adoptive suppressive immune cell therapy would be introduced to provide a broad overview of overcoming the above challenges.
5.1 Treg
In a research based on mice, CD4+CD25+ Treg was found to play an important role in relieving immune response and preventing the process of various autoimmune diseases (90). The Foxp3 was then identified as a special marker of CD4+CD25+ Treg, regulating their development and function (91). How Treg exerts immunosuppressive effects was discussed (92), and the further mechanism is still under study. Herein we demonstrate Treg as a kind of cell-based biomaterial applied in T1D therapy (Figure 4).
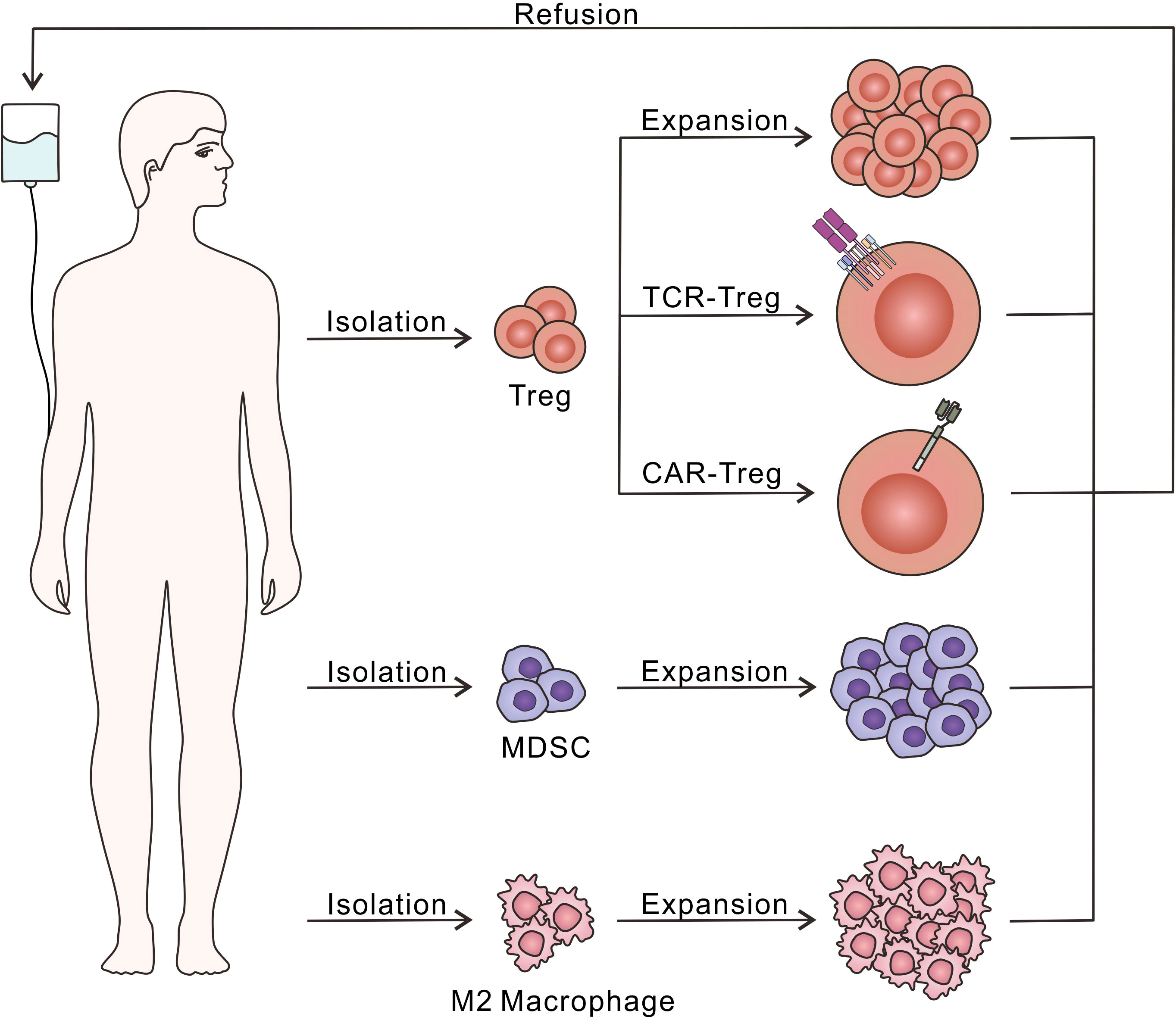
Figure 4 Diagram of adoptive immunosuppressive cell therapy. Treg, MDSC and M2 macrophage could be isolated and expand in vitro through different culture conditions, followed by the refusion into the host to relieve excessive autoimmune response. Moreover, Treg have been further modified with TCR or CAR to recognize T1D-related autoantigens, which is beneficial to enhance the immunosuppressive effects.
5.1.1 Expansion of Treg in vitro
Since natural CD4+CD25+ T cells generally keeps a small amount in vivo, the expansion ex vivo is considerable. In a research of Tang et al., decreased CD25 expression of Treg was observed in the islets of T1D mice. Moreover, low-dose IL-2 was found to be conducive to the survival of Treg and remission of T1D (48). Later, TGF-β/TGF-β receptor signals were demonstrated to have key roles in CD4+CD25+ Treg regulation (42). Therefore, IL-2 and TGF-β were used to co-cultured with naïve T cells, which would be stimulated to differentiate into CD4+CD25+ T cells (93, 94). Other in vitro factors, such as anti-CD3 and anti-CD28, acted as a way of indirect antigen presentation, were combined with fluorescence-activated cell sorting technology for better therapy (95). As for direct antigen presentation, DCs, function as APCs, were employed to expand Treg for stronger alloantigen-specific capacity, benefiting to ease autoimmune disease in vivo (96–98). With the low-dose antigen, DCs could stimulate CD4+ T cells to generate a group of antigen-specific Treg, and without additional purification, it could ameliorate T1D.
However, the marker of CD4, CD25, and Foxp3 could not define the whole Treg subset, since some of CD4+CD25-CD127- T cells also express Foxp3. Further research demonstrated that CD127 could be an excellent Treg marker, especially for those suppressor cells without antigen presentation (99). CD4+CD25+highCD127- Treg have been initially confirmed the prolonged survival of β-cells, insulin dependence and safety within one year of single or double Treg infusion (100)CD4+CD25+CD127lo/- polyclonal Treg was isolated from patients, expanded in vitro, showing its enhanced suppressive activity in vivo after being re-infused in a phase 1 trial (101). Ex vivo expanded autologous CD3+CD4+CD25highCD127− Treg was infused and contributed to the remission of T1D in children (102).
In some researches, stem cell-derived autoantigen-specific Treg was also applied in T1D treatment, showing autoantigen-specific and inhibition autoantigen-specific accumulation (103). A novel idea was validated to make Treg as a protective coating of transplanted pancreatic islets. Specifically, Treg was bound to target cells through streptavidin-biotin system or biotin-polyethylene glycol-succinimidyl valeric acid ester molecule, maintaining the activity and function of islets and preventing them from being immune attack (104, 105). However, in vivo experiments were required to further demonstrate the coating strategy.
Despite extensive research effort, it seemed that there were some of antigen-specific Treg playing a significant role to reverse T1D in the large number of in vitro expanded Treg, though the former represents only a very small fraction of the latter. In a T-cell exhausted mice T1D model, autoantigen-specific Treg showed superior capacity in T1D remission than polyclonal Tregs (106). Therefore, compared to selectively isolate and expand antigen-specific Treg, it may be a more effective pathway to engineer T cells to be antigen-specific Treg.
5.1.2 Engineered Treg
As for the uncertainty of in vitro amplification, direct gene-edited Treg contributed an attractive route. T cell receptor (TCR), which recognizes T1D-related autoantigens, was constructed on Treg via lentiviral gene transfer and enabled its feasibility on Treg. Specifically, Treg was endowed with ectopic expression of TCRs, which was identified and isolated from T cells of human origin, leading to antigen dependent inhibitory capacity and protecting islets from β-cell destruction (107, 108).
Compared to TCR-Treg, Chimeric antigen receptors (CARs)-Treg owns a unique advantage of being non major histocompatibility complex (MHC)-restricted, which means CAR transfer would be applicable to most patients, instead of varying from individuals.
HLA-A2 specific CAR Treg maybe the first concept of proof for therapy. Herein, extracellular single-chain Fragment Variable (scFv) was fused to extracellular signaling domain, enhancing our insight into creating Treg of various antigen specificity via using different CARs (109). A modular system based on CAR-Treg was developed. Usually, CAR was designed to interact with specific target antigen. Herein, the CAR was engineered to bind to Fluorescein Isothiocyanate (FITC), a fluorophore frequently conjugated to antibodies, which endowed it with enormous scalability to act with desired antigen in prolonging allograft survival and maintaining immune tolerance (110). To increase Treg yield and further ensure its security, Foxp3 was transduced to CD4+ T cell to generate converted Treg (cTreg), exhibiting the similar phenotype and function as natural Treg. Moreover, insulin-specific CAR construction of cTreg could make it activated and proliferative. It fulfilled a concept-on-proof of redirecting T effector cells to Treg through transduce Foxp3 (111).
Despite promising results, a number of issues remain to be solved, especially in the long-term safety and validity, no matter the large-scale expansion technology, or gene-editing method like TCR Treg and CAR Treg, the lineage stability of Treg in manual operation remains unknown. Recent data indicated that no Foxp3 loss in expression during over a year follow-up period (101). However, to prove the inevitability between lineage stability and Foxp3+ of Treg, more robust evidence is needed. As for gene-editing, it is particularly worrying about Good Manufacturing Practice, including the construction and optimization of TCR/CAR, purification of TCR/CAR Treg. Considering the widely existing neurotoxicity CAR-T used in oncology research, CAR Treg may also face the similar challenging hurdles.
5.2 MDSC
A population cell of myeloid origin with immunoregulatory activity named myeloid-derived suppressor cells (MDSC) is of interest, allowing for great potency in the fields of transplantation and autoimmune diseases (112). In T1D, MDSC exhibited immunosuppressive ability to some extent, its characteristic and mechanism had been discussed in the pre-existing review (113). Here, we focus on the studies related to MDSC adoptive therapy.
Alessia Zoso et al. proposed a novel human MDSC subset named fibrocytic MDSC to induce Treg expansion and normoglycemia in a xenogeneic mouse model of T1D (114). However, there was another research pointed that the adoptive transfer of MDSC could not always effectively prevent T1D autoimmunity in a stringent model (115). Combined therapy with adjunct immunosuppressive agents or addition factors would be needed (115, 116). Whitfield-Larry F et al. utilized hemagglutinin (HA) peptides to assist MDSC, making it capable of downregulating autoimmune responses and preventing diabetes onset (116).
In addition to direct action in assisting the establishment of immune tolerance, MDSC also protected transplanted islets from being disrupted. It reported single dose of MDSC was given to the treated mice and prolonged the survival of transplanted islets, indicating MDSC owned the capacity of immunosuppression. Moreover, the expression of C-C chemokine receptor type 2 (CCR2) contributed to MDSC migration, which means much more supervision would be required (117).
However, one obvious drawback limits the development of MDSC, that is the small number of them within the whole body. It usually means that it would be difficult to precisely isolate the “appropriate” MDSC, especially in autologous patients. Generic MDSC from healthy donors may suffer from mismatch genetic background and further Graft Versus Host Disease (GvHD). Exploring extra in vitro inducers for better MDSC production and in vivo combinations for effective function has still the long way to go.
5.3 M2 macrophages
It has been reported that early macrophage infiltration plays an important role in the process of T1D (118). There were two major activation states of macrophage, including proinflammatory (M1) and anti-inflammatory (M2, also named alternatively activated macrophages). A strategy of alternatively activated macrophages was proposed and generated keen interest. Similar to adoptive transfer therapy with other cells, M2 macrophages was isolated from macrophages, monocytes, or bone marrow–derived myeloid precursors, followed by in vitro induction using cytokine such as IL-4 and colony-stimulating factor-1 (CSF-1) (119, 120). The mechanism of M2 macrophage protection has been illustrated in other review (121). Here we focus on the application of M2 macrophage therapy.
In STZ induced diabetic mice, adoptive transfer of M2 macrophage expanding in vitro showed mitigated injury of islets and kidneys, mainly due to the protection of M2 macrophage from the destruction of β-cells (120, 122). Roham Parsa et al. developed an optimized in vitro culture protocol, that was IL-4/IL-10/TGF-β. Most of NOD mice exhibited good therapeutic effects of T1D for more than three months after being injected the induced M2 macrophage. Surprisingly, it was observed that the transferred M2 macrophage was specifically accumulated in inflamed pancreas and promoted β-cell survival (123). A study revealed that M2 phenotypical cell line could be stabilized by neutrophil gelatinase‐associated lipocalin transduction, which overexpressed IL-10 and low secreted TGF‐β, relieving diabetic kidney disease much better than non-modified bone marrow‐derived M2 (124). A small-scale clinical trial indicated that monocyte of newly diagnosed T1D patients could differentiate M2 macrophages in vitro while that of long-standing T1D patients could not. The differentiated M2 macrophages could use for adoptive therapy (125). In our opinion, M2 plasticity make it a potent candidate for cell therapy through genome editing and reprogramming. However, the limited proliferation halted the progress, indicating more advanced co-cultured and modified methods are still needed to developed.
6 Vaccine
There is a lot of data to prove the point that viruses can induce T1D in animals. The mouse encephalitis myocarditis (EMC-D) virus D variant is the most prevalent. Due mostly to the rapid death of β-cells by virus replication in cells, high titer EMC-D virus infection progresses to diabetes within 3 days. Macrophages are drawn to the islets by low titer EMC-D virus infection. An important element in the elimination of remaining β-cells is the production of soluble mediators by activated macrophages (126–128). In addition to the above mentioned, autoimmune responses can be modified from immune checkpoints. It can also be done by antigen-specific pathways. In addition to the above mentioned, autoimmune responses can be modified from immune checkpoints. It can also be done by antigen-specific pathways. Vaccines have been suggested as a means of treating and preventing T1D at beginning with its development. By controlling the autoantigen immune response and halting further degeneration of pancreatic β-cells, vaccines have been demonstrated clear advantages in immunotherapy. It can not only significantly lessen the pain brought on by diabetes complications but even help reverse diabetes (18). Inflammation is a major contributor to T1D, hence, strategies for reducing or eliminating inflammation are frequently used in the vaccine selection and formulation process.
6.1 Bacteria
From the first pertussis vaccine found to stimulate the production of immunoglobulin by lymphocytes in rats to the later treatment of gas-sensitive inflammation in mice (129–131). In 1984, to reduce inflammation, a study explored the protective effect of pertussigen on insulin-dependent diabetic mice. The pertussis vaccine was divided into two parts, one was boiled and the other was left intact, and injected into STZ mice. T1D was not observed in mice treated with either regimen (132). It has been suggested to use Tregs to inhibit or remove pathogenic T cells in order to prevent or safeguard the function of β-cells. Trials have started in children with new-onset diabetes and adults with autoimmune diabetes (LADA) based on this notion (133). Traditionally Q fever vaccines have traditionally been used to treat acute febrile illness after human contact with livestock. After professor Kevin Lafferty first demonstrated that Q fever vaccine protects NOD mice against diabetes, Bonn et al. explored whether Q fever vaccine could prevent diabetes in humans. They vaccinated more than 40 newly diagnosed T1D patients with Q fever, patients did not develop a second autoimmune disease from vaccination to 6 months later. These results, making vaccine against hepatitis virus colonies be a new way to protect insulin-producing β-cells (134). On the other hand, the soluble chimeric protein CTLA4-Ig can treat psoriasis when injected into the body. No T cell proliferation was observed in the patient’s lesions, but there were abnormal antibody responses to T cell-dependent neoantigens (135). Bacille Calmette-Guerin (BCG), a derivative of Mycobacterium bovis, induces upregulation of the c-Mcy genes that controls glucose homeostasis and is most commonly used to trigger long-term adjustment of blood sugar levels to near-normal levels in patients with T1D (136, 137). In addition, patients with long-term T1D received two doses of BCG for 8 years of follow-up. The BCG vaccine returned blood sugar levels to normal for three years after administration and remained stable for the next five years (138). Therefore, it is promising to employ BCG to treat T1D. From the perspective of adjuvant, peptide nanofibers have been used to assist BCG vaccine expansion of CD4+ T cells (137). It may be possible to extract microvesicles of Mycobacterium bovis and load them with immunosuppressive drugs as a strategy to treat T1D. Rotavirus mainly infects intestinal cells and causes dehydrated gastroenteritis (139). Hence, children must be given rotavirus at an early age. Notably, between 2006 and 2017, the incidence of diabetes in children from aged 0 to 4 declined by 3.4% per year after infants were vaccinated against rotavirus in the United States (140). Rotavirus spike protein is considered as a novel vehicle protein, which can greatly improve antigen immunogenicity when combined with vaccine (36, 141). Thus, whether combination of rotavirus spike protein or MEC-D to make a preventive vaccine could prevent T1D caused by MEC-D.
Moreover, Miller et al. synthesized an oral live attenuated salmonella vaccine that delivers its own antigens and TGF-β expression vectors to immune cells in the intestinal mucosa. DCs are present throughout the secondary lymphoid tissue after animal vaccination, and CD103+ DCs induce tolerance effects and intestinal homing. TGF-β significantly increased the expression of programmed death ligand-1 (PD-L-1 or CD274) in dendritic cells in MLN and PP of treated mice. TGF-β also increased the level of CTLA-4 in CD4+ T cells in MLN and PP (142). Based on this, Harrison et al. made a T1D vaccines using live attenuated Salmonella MvP728 (DhtrA/DpurD), cytokines (IL10 and TGF-β) and proinsulin (PPI) antigen combined with a subtherapeutic dose of anti-CD3 monoclonal antibody. The vaccine was able to reduce insulitis and prevent and reverse diabetes in NOD mice. It also reduced the risk of inflammatory response and sepsis by detoxifying lipopolysaccharide (LPS) with lipid A against live salmonella strains (143). Considering that the vaccine itself may trigger autoimmunity and the selection of the enterovirus serotype that the vaccine targets, Larsson et al. used formalin inactivated Coxsackie virus B1 (CVB1) as a novel non-adjuvant vaccine to explore its efficacy and autoimmune safety in the treatment of diabetes. Prediabetic NOD mice were inoculated with CVB1 vaccine and then infected with CVB1. The vaccinated mice produced high titers of CVB1 neutralizing antibodies, but showed no sign of vaccine-related side effects and no increase in insulin autoantibodies. At the same time, the onset of diabetes was accelerated in NOD pre-diabetic mice infected with CVB1 (144). Bacterial vaccines have great potential for the treatment of diabetes, either by using bacterial proteins as adjuvants to enhance antigen immunogenicity or by combining nanoparticles with bacteria.
6.2 Genetic engineering
Genetic engineering has been a hot topic in recent decades. It is possible to use gene editing and other methods to give organisms functions that they do not have (Figure 5). For patients with T1D, it is of great significance to re-enshrine the function of producing and secreting insulin.
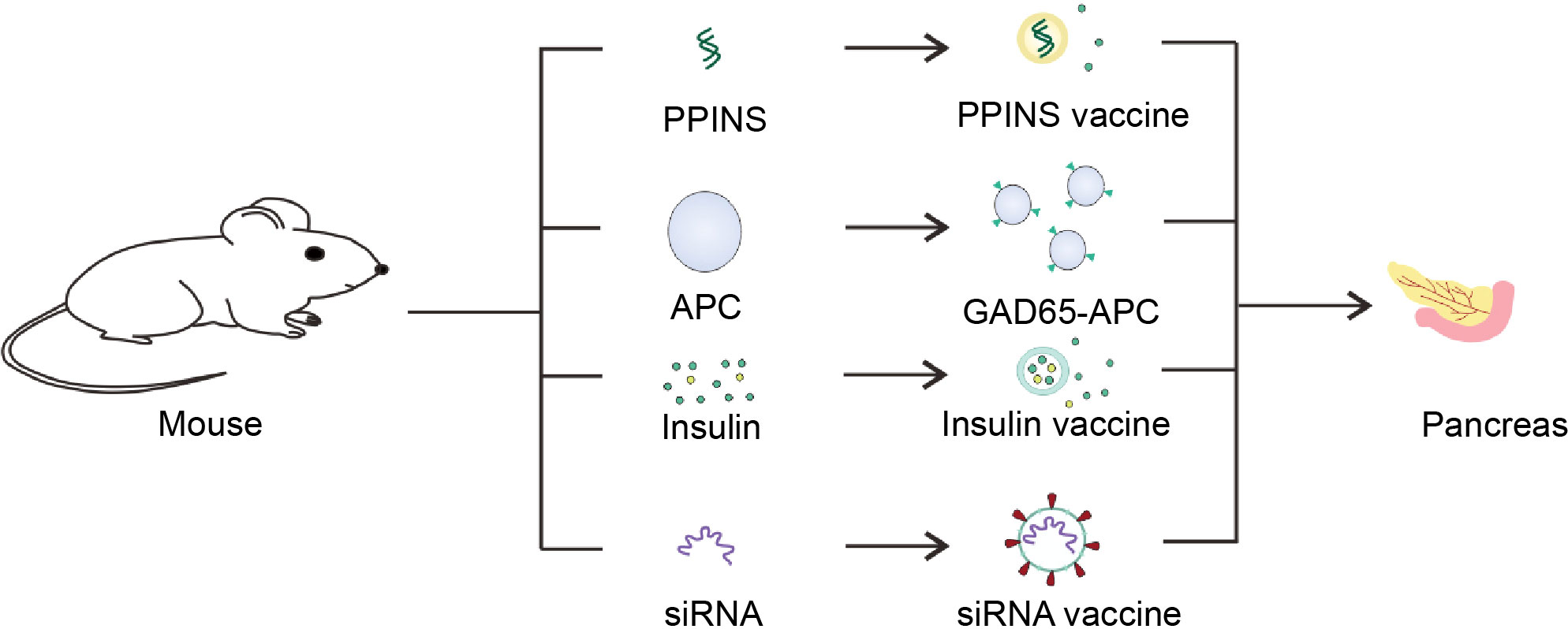
Figure 5 Schematic diagram of the T1D vaccine. Insulin gene was isolated from mice and utilized to made PPINS vaccine. Liposomes and other nanomaterials are used to coat insulin and GM-CSF to provide exogenous insulin, which are mainly used to make sustained release insulin vaccines. On the other hand, siRNA vaccines derived from siRNA molecules of APC associated with the pancreas and cell vaccines genetically engineered to express GAD65 antigen could protect the pancreas from T cell killing.
6.2.1 Proinsulin
For patients with T1D, insulin injections are commonly used. However, considering long-term insulin injection, it will cause body edema, hypoglycemia and other adverse reactions. There are many ways to get insulin into the body. Apart from the common intraperitoneal injection, there are nasal inhalation, oral and so on. Under continuous stimulation of high glucose, insulin nasal mucosal injection can maintain the stability of blood glucose and autoimmune tolerance (143). Meanwhile, blood glucose levels can be reduced with oral insulin, however the first-pass effect can lower the quantity of insulin that is actually useful. Therefore, there are new difficulties in increasing the amount of insulin that is effective (145).
For daily insulin injections, a way to produce a continuous supply of insulin in the body with a single injection is urgently needed. Sifter et al. analyzed the structural characteristics of the variation in the proinsulin proteins (PPINS) and designed PPINS as antigenic subcellular targeted vaccines. The potential of PPINS variants to inhibit the development of spontaneous diabetes was then tested in female NOD mice expressing h-2G7 haplotype susceptible to diabetes with CD8+ T cell-reactive antigens. They found that PPINS antigens excluded from endoplasmic reticulum (ER) expression did not induce CD8+ T cells or autoimmune diabetes in RIP-B7.1 TG mice, but effectively inhibited the development of spontaneous diabetes in NOD mice and CD8+ T cell-mediated autoimmune diabetes mice (146). Glinka et al. co-inoculated PPINS with mutated B7-1 molecule (B7-1WA) to protect NOD mice prone to autoimmune diabetes. The mutated B7-1 molecule binds to the negative T cell regulator CTLA4, but not CD28. Co-delivery of the plasmid encoding the PPINS-GAD65 fusion structure and B7-1WA has a protective effect on insulitis and diabetes. In adoptive transfer experiments, DNA vaccination produced protective CD4+ Treg with a CD25+ or CD25 phenotype. In addition, the number of T cells with TR-related markers, such as CTLA4, Foxp3, and membrane-bound TGF-β, increased in the vaccinated mice. At the same time, CD4+ regulatory T cells (Tr) can inhibit the response of T cells to islet antigen (147). Yoshida et al. constructed DNA plasmid vectors that encode membrane binding to self-antigen proinsulin (MB-PPI) and PPI/GAD65w (GAD65 total protein) respectively. He injected mB-PPI and PPI/GAD65wDNA plasmid vectors together with MB7.1-IGG1/CD40L into female NOD mice. Five injections of DNA were given every three weeks. A significant delay and reduction in the incidence of autoimmune diabetes was observed. In vitro experiments showed that the spleen cells of protected mice did not produce increased IFN-γ when stimulated by insulin and GAD65 peptide (148). Monoclonal antibodies using CTLA4 usually have a masking effect, replacing a single amino acid in B7-1 (W88>A; B7-1wa) does not bind to CD28 or CTLA4. A plasmid encoding B7-1 or B7-1wa was constructed as a cell surface or Ig fusion protein. In the binding state, B7-1-IG enhanced CD3-mediated T cell activation, but B7-1wa-Ig had an inhibitory effect consistent with CTLA4 ligand. The B7-1wa (membrane-bound form) plasmid was combined with the PPINS plasmid, and the gene transfer was amplified by electroporation, and co-injected into non-obese autoimmune diabetic mice. Co-delivery of B7-1wa and PPINS cDNA eliminates insulin responsiveness and improves diabetes. Injection of either plasmid alone could not suppress the immune cell response to itself (149). In addition, Chang et al. designed a DNA vaccine combined with membrane-bound preproinsulin (mbPPI) and mB7.1/CD40L. The vaccine is able to promote the effective transmission of the autoantigen PPI, and its mutation B7.1 binds to CTLA4 but not CD28, well preventing cell recognition and killing by T cells (150). Patients with chronic T1D and the HLA-DRB1-0401 genotype received subcutaneous injections of proinsulin peptide epitopes (C19-A3) limited by the human leukocyte antigen DR4 (HLA-DR4). At various doses, IL-10-secreting peptide-specific T cells were seen, but proinsulin-specific pro-inflammatory T cells and systemic hypersensitivity were not observed (151).
6.2.2 Polypeptide
In obese diabetic mice, Sema3E induces plexinD1-positive inflammatory macrophages into visceral white adipose tissue to regulate their biological defects. Yoshida et al. selected two antigenic peptides to produce neutralizing antibodies against amino acids 385-394(KVNGGKYGTT) or 359-368(HKEGPEYHWS) of Sema3E. The Ten terminus or lysine of each candidate peptide was conjugated by glutaraldehyde to the keyhole limpet hemocyanin (KLH) and given to obese wild-type male mice with Freund’s adjuvant. Vaccination with KLH-conjugated HKEGPEYHWS (Sema3E vaccine) significantly increased Sema3E antibody titer by screening. Sema3E vaccine inhibited plexind1-positive cell swelling, improved chronic inflammation in visceral white adipose tissue, and improved systemic glucose tolerance in obese mice (148). Consider that small interfering RNA (siRNA) was hindered by low transfection efficiency in vivo, Leconet et al. found that lipids or polyethyleneimine delivery agents can effectively treat siRNA molecules in pancreatic associated APCs. In terms of treatment, he found that short-term treatment with the lipid/ALOX15-specific siRNA complex promoted long-term protection against T1D in young wild-type (WT) NOD mice. In pancreatic associated CD11b+ cells, the PD-L1 pathway is upregulated and Treg are increased (152). β2 macroglobulin (β2m) was a universal signal component of MHC-I molecules when fused with cd3-Z chain. Therefore, the design of connecting the H-2KD binding insulin B chain peptide insulin B chain, amino acid 15-23 (IbsB15-23) to the N-terminal of β2m/CD3-ζ, made the mRNA encoding chimeric MHC-I receptor could target effector CD8 to diabetic CD8+ T cells, which reduced pancreatitis in NOD mice (153). Meanwhile, M2-polarized bone marrow derived macrophages (BMDMs) can also be used to secrete exosomes (Exos) containing miRNA to inhibit inflammation and maintain immune tolerance. After injection of M2 BMDM Exos in obese mice, miRNA consumption in exosomes blocked the ability of M2 BMDM Exos to enhance insulin sensitivity. Further analysis of miRNA showed that miR-690 was highly expressed in exosomes, and both in vivo and in vitro experiments proved that miR-690 targets NAD kinase (Nadk) and plays an important role in regulating macrophage inflammation and insulin signaling. Therefore, miR-690, as an insulin-sensitized miRNA, can be further explored (154). HSP60 peptide DiaPep277 is recognized to prevent adult humans and NOD mice from developing diabetes-related cell function decline. However, when the insulin dose and the level of glycosylated hemoglobin were treated in children with early-onset T1DM, there was no distinction between the patients in the control group and those in the blank control group. DiaPep277 thus does not maintain cell function (155, 156). Therefore, designing vaccinations to shield β-cells from damage now presents a new challenge.
6.2.3 GAD65
Young NOD mice exhibited spontaneous Th1 response to glutamic acid decarboxylase (GAD65), which resulted in T cell autoimmunity. In order to prevent the autoimmunity before it happens naturally, Tian et al. introduced the notion of inducing active tolerance through the involvement of a Th2 immune response on GAD65 and testing if the T1D caused by series of Thl reactions can be prevented. As a result, young NOD mice were given a single intranasal injection of the GAD65 polypeptide, allowing for the detection of the high level of GAD65 IgGl antibody expression in the mice. GAD65 peptide administered intravenously reduced IFN-γ elevated IL-5 response, and promoted Th1 response with Th2 phenotypic transfer, decreased the prevalence of long-term T1D and insulitis (157). Agardh et al. evaluated the safety of alum formula for human recombinant GAD65 after suggesting an enhancement to its GAD65 polypeptide. Subcutaneous injections of various dosages of the modified vaccination did not significantly alter c-peptide levels or HbA1c in persons with underlying autoimmune diabetes mellitus (LADA), whether fasting or hyperglycemic stimulation. It has been established that adding alum is a good way to enhance the GAD65 polypeptide vaccination (158). Moreover, GAD65 could also be packaged into vesicles to regulate hBA1c levels through nerve stimulation (159). Fengchun Li et al. modified the syngeneic splenocytes, and used retrovirus particles to transfer the gene encoding GAD65 into the spleen cells of NOD mice to make a lymphoid vaccine. A few weeks after the vaccine was injected into four-week-old NOD mice, high doses of the vaccine were found to reduce both insulitis and blood sugar levels (149). In addition, anti-CD3/GAD65 combination to treat T1D. The results showed that the amplification of GAD65-specific Treg was related to the mouse genotype. This provides further insights into the use of immunotherapy in the treatment of T1D. To improve the therapeutic effect, targeted individual immune monitoring is needed (160).
6.3 Cytokines
Liposomes, nanoparticles and other inorganic materials show certain advantages and are widely used in the treatment of tumor diseases (161). Treatment of tumors requires breaking immune tolerance, while for T1D, it is to restore immune homeostasis and protect β-cells. Therefore, inorganic materials can also be used as adjuvants to enhance the therapeutic effect of vaccines.
Yoon et al. has developed a vaccine-based approach using two synthetic controlled-release biomaterials, poly (lactate-co-polycerids; PLGA) microparticles (MPs) encapsulate denature-resistant insulin and PuraMatrix™ peptide hydrogel containing granulocyte macrophage colony stimulating factor (GM-CSF) and CpGODN1826 (CpG). Interestingly, three subcutaneous injections of this hydrogel (GM-CSF/CpG)/insulin-MP vaccine protected most NOD mice against T1D infection. At the same time, the stimulating effect of CpG increased IL-10 production. Multiple injections of insulin-containing agents under the skin can form granulomas, creating a kind of microenvironment that recruits immune cells. Accordingly, this injectable hydrogel/MP based vaccine system make it possible to prevent T1D with (162). Based on inhibition of autoreactive T cells by Foxp3+ Treg, impaired tolerance promotes the destruction of insulin-producing β-cells in autoimmune T1D. So Hyoty and Knip treated human hematopoietic stem cell transplanted NG-HLA-DQ8 transgenic mice with upon subimmunogenic vaccine. It was observed that the hypoimmunogenicity boosted the levels of insulin-specific Foxp3+ Treg in the human immune system. Moreover, the expression of Foxp3, CTLA4, IL-2RA and TIGIT increased, which effectively inhibited effector T cells. Therefore, inducing the expression of highly efficient human insulin-specific Foxp3+ Treg in vivo can be the direction of new human insulin mimics (163). Interleukin-1β (IL-1β) is a key cytokine involved in inflammatory diseases, which can be treated from the perspective of decreased IL-1β activity. Cavellti-Weder et al. synthesized a novel vaccine against IL-1β. The vaccine hIL1bQb consists of full-length, recombinant IL-1β coupled virus-like particles. IL-1β-specific antibodies were induced immediately after vaccination in preclinical apes, while neutralizing antibodies were delayed. In a clinical study of 48 patients with type 2 diabetes, neutralizing IL-1β specific antibody responses were detected after multiple injections of anti-IL-1β vaccine. The patient’s blood glucose and body weight were improved without obvious adverse reactions (164). In addition, Spohn et al. chemically cross-conjugated IL-1β with virus-like particles of phage Qβ to form a detoxifying version of the IL-1β vaccine. The vaccine was well tolerated in mice and neutralized the biological activity of IL-1β. Nor did infecting mice with Listeria monocytogenes or Mycobacterium tuberculosis induce an immune response. And the IL-1β vaccine also improved glucose tolerance in diet-induced type 2 diabetes (165). In addition, DCs expressing interleukin 4 (IL-4) were induced by lentiviral transduction. To avoid the dual risk of carcinogenicity or immune-heredity of cell modification with viral vectors, DCs were used electrocuted to “translation-enhanced” of IL-4mRNA (eDC/IL-4) in NOD mice, and demonstrated that electroporation did not affect the therapeutic effect. After a single injection of eDC/IL-4 shortly after the onset of hyperglycemia in NOD mice, some mice could maintain blood glucose stability for several months. The eDC/IL-4 treatment enhanced the function of Treg and modulated T-assisted responses to reduce pathogenicity (166). Microcarriers are known to direct DCs to the site of administration and, once phagocytic, the contents can form a DC phenotype. Therefore, promaxtm microspheres were proposed to form antisense oligonucleotides and enable autologous dendritic cells to inhibit diabetes. When subcutaneously injected into NOD mice, the vaccine enhanced the expression of CD25+Foxp3+ Treg. In vitro experiments also demonstrated that the vaccine could inhibit NOD-derived pancreatic cell antigen activity (167). Au et al. synthesized β-cells with high expression of PD-L1, CD86 and GAL-9 and subcutaneously injected β-cells with high express co-inhibitory immune checkpoint ligands. It can induce islet antigen specific immune tolerance to autoreactive T cells and reverse early onset hyperglycemia (168).
The vaccination has demonstrated some benefits in the management of diabetes, starting with bacteria, insulin, β-cells, and GAD65, though diabetes patients could not be fully treated for a variety of reasons. Therefore, in order to maintain autoimmune tolerance, it is important to decide whether to combine a vaccine with immunological checkpoint or anti-inflammatory agents, as well as to manufacture insulin to maintain normal blood glucose levels.
7 Conclusion and perspective
In summary, as we have review above, there are three aspects for the treatment of new-onset diabetes by immune modulation. Firstly, for immunosuppressive factors, the mostly employed ones are TGF-β, IL-2, etc. Currently, organic synthetic materials, such as PLGA and PEGylated, have been used to encapsulate immunosuppressive factors to achieve sustained drug release to control blood glucose. Alternatively, nanoparticles or device reduce the toxic side effects and enhance efficacy of the immunosuppressive drugs. In addition to the current approaches, it can use cell membrane vesicles to encapsulate the epidemic suppressor. Secondly, the most common and promising treatment for T1D is the vaccine. To prevent T1D, the vaccines based on virus or bacteria that elicit T1D can be injected into the body in advance. There are many therapeutic vaccines available even the patients have with T1D. It is common to use proinsulin as a vaccine, or the insulin peptide also could be wrapped in liposomes or other biological materials as nanoparticles vaccines, that facilitate the capture by DC. However, for patients whose islets are not completely damaged, how to restore immune tolerance should be considered. GAD56 is another common vaccine candidates, which can robustly induce Treg cells to play the immunosuppressive role. Mixing a variety of immunosuppressive factors together with vaccines is a promising therapeutic strategy for T1D therapy. For T1D vaccine, enhance of the immunogenicity of the antigen is a promising way. As for adjuvants, adjuvant vaccines are extremely important. Bacterial membrane derived vesicles had been intensively exploring as cancer vaccine adjuvant that can also are be considered for enhancing immunogenicity of the T1D antigens. For example, proinsulin peptide vaccines can be coated with bacterial membranes to promote the robust immune response. Thirdly, engineered cell-based therapy such as Tregs, MDSC, M2 macrophages that could be employed to restore the immune tolerance. Cells could be isolated from patients and proliferated in vitro based on specific amplification or engineering approaches to maintain immune tolerance, then transfuse to individual to treat T1D. We envision that the future applications of immunosuppressive cells could be in combination with immunosuppressive agents’ target delivery or immune checkpoint blockade of islet-autoreactive T cell to prevent its pathological killing of β-cells. Alternatively, inflammatory suppressive cytokines could be overexpressed in immunosuppressive cells, whose exosomes could be extracted to treat T1D together with insulin.
Together, combining biomaterials to form drug delivery nanoparticles or device holds great potential for treating T1D, and further research and exploration in the delicate design of biomaterials and unknown mechanistic signaling pathways would make a huge contribution.
8 Future and challenges
The particularity of biomaterials can achieve some purposes of T1D treatment. Treating T1D from an immunological perspective has many merits. Firstly, immune checkpoint modulation is one of the most promising ways in protecting β-cells from T cell attack. At present, PD-1/PD-L1, CTLA4, GAL3, IDO and other checkpoints have been paid more and more attention in cancer immunotherapy. Except these above, the ICOS/ICOSL signaling deserves attention. It has shown potent antitumor activity in combination with CTLA4 blockers in tumor immunotherapy (69). These immune suppressive legends such as PD-L1, Gal-9 and CTLA4 could be engineered to treat T1D through reactively inhibit the T cell activity. In addition, these negative immune checkpoint proteins also be wrapped exosome, platelets, hydrogels and other nanoparticles to inhibit T cells to protecting β-cells. However, how to target delivery these native immune checkpoint proteins to pancreas and reduce their off-target immune suppression is still a challenge.
Secondly, for vaccines, the preventive and therapeutic functions cannot be ignored, though the induced immune response rate is relatively low by current antigen candidates. The components of a vaccine are mainly divided into two parts, antigen and adjuvant. To improve the effect, it can not only improve the immunogenicity of autoantigen, but also adjust the adjuvant to cause autoimmunity. Since EMC-D can rapidly destroy β-cells by replicating in cells, it is considerable to modify the structure of EMC-D to remove pathogenic toxicity and act as an antigen to cause immune response at the same time, so as to prevent T1D. In addition, whether bacterial membrane vesicle can be engineered to contain multiple surface proteins to inhibit inflammation is worth considering. For example, how about inhibiting the expression of IL-1β, IL-6, IL-10 simultaneously?
Thirdly, referred to immunosuppressive cells, in vitro expansion has achieved impressive success and numerous clinical trials were ongoing, especially in Treg. During this process, more efficient cell types were detected, such as antigen-specific Treg, which encourages researchers to explore new ways to achieve it. So far, major challenges are summarized as follows: 1) Optimized separation and amplification process of specific cells; 2) limited expandability based on the patient’s condition; 3) Further confirmation of efficacy and mechanism of engineered immunosuppressive cells.
Hence, challenges to biomaterials applied in T1D still persist today. However, we envisage that employing biomaterials to facilitate the treatment of T1D based on immunotherapy is promising.
Author contributions
ZJ, YL, YM contributed equally to this work. XiZ, ZJ, YL, YM wrote the manuscript. XuZ, ZJ, XiZ helped to revised the manuscript. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (31971268, 32201084), grants from Science, Technology & Innovation Commission of Shenzhen Municipality (JCYJ20200109142610136, JCYJ20180507181654186), Guangdong Basic and Applied Basic Research Foundation (2019A1515010855), Guangdong Basic and Applied Basic Research Foundation (2020A1515110166), the Natural Science Foundation of Guangdong Province (No. 2020A1515010802, No. 2022A1515012289), Shenzhen Science and Technology Program (Grant No. RCYX20200714114643121), University of Chinese Academy of Sciences-Shenzhen Hospital Research Funding (HRF-2020004), the Health system scientific research project of Shenzhen Guangming District Science and innovation Bureau (2020R01073, 2020R01061), Special fund for economic development of ShenZhen Guangming District (2021R01128), Doctoral personnel scientific research start-up Fund project of Guangdong Medical University (GDMUB2022037), Fundamental Research Funds for the Central Universities (19lgzd45), and the Health system scientific research project of Shenzhen Guangming District Science and innovation Bureau (2020R01073, 2020R01061).
Acknowledgments
Xudong Zhang, Pingping Li, Wen-I Yeh, Alessia Zoso, Roham Parsa, Kin Man Au, Katja Stifter, Wilhem Leconet at al. for improvement.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Roep BO, Thomaidou S, Van Tienhoven R, Zaldumbide A. Type 1 diabetes mellitus as a disease of the beta-cell (do not blame the immune system?). Nat Rev Endocrinol (2021) 17:150–61. doi: 10.1038/s41574-020-00443-4
2. Lehuen A, Diana J, Zaccone P, Cooke A. Immune cell crosstalk in type 1 diabetes. Nat Rev Immunol (2010) 10:501–13. doi: 10.1038/nri2787
3. Ghasemi A, Akbari E, Imani R. An overview of engineered hydrogel-based biomaterials for improved beta-cell survival and insulin secretion. Front bioeng Biotechnol (2021) 9:662084. doi: 10.3389/fbioe.2021.662084
4. Waldron-Lynch F, Herold KC. Continuous glucose monitoring: Long live the revolution! Nat Clin Pract Endocrinol Metab (2009) 5:82–3. doi: 10.1038/ncpendmet1044
5. Espona-Noguera A, Ciriza J, Canibano-Hernandez A, Orive G, Hernandez RMM, Saenz Del Burgo L, et al. Review of advanced hydrogel-based cell encapsulation systems for insulin delivery in type 1 diabetes mellitus. Pharmaceutics (2019) 11(11):594. doi: 10.3390/pharmaceutics11110597
6. Battelino T, Bode BW. Continuous glucose monitoring in 2015. Diabetes Technol Ther (2016) 18 suppl 1:s10–21. doi: 10.1089/dia.2016.2502
7. Wang J, Wang Z, Yu J, Kahkoska AR, Buse JB, Gu Z. Glucose-responsive insulin and delivery systems: Innovation and translation. Adv mat (2020) 32:e1902004. doi: 10.1002/adma.201902004
8. Shapiro AM, Lakey JR, Ryan EA, Korbutt GS, Toth E, Warnock GL, et al. Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N Engl J Med (2000) 343:230–8. doi: 10.1056/NEJM200007273430401
9. Boldison J, Wong FS. Immune and pancreatic beta cell interactions in type 1 diabetes. Trends Endocrinol Metab (2016) 27:856–67. doi: 10.1016/j.tem.2016.08.007
10. Raffin C, Vo LT, Bluestone JA. Treg cell-based therapies: challenges and perspectives. Nat Rev Immunol (2020) 20:158–72. doi: 10.1038/s41577-019-0232-6
11. Bougneres PF, Carel JC, Castano L, Boitard C, Gardin JP, Landais P, et al. Factors associated with early remission of type i diabetes in children treated with cyclosporine. N Engl J Med (1988) 318:663–70. doi: 10.1056/NEJM198803173181103
12. Keymeulen B, Vandemeulebroucke E, Ziegler AG, Mathieu C, Kaufman L, Hale G, et al. Insulin needs after cd3-antibody therapy in new-onset type 1 diabetes. N Engl J Med (2005) 352:2598–608. doi: 10.1056/NEJMoa043980
13. Gaglia J, Kissler S. Anti-cd3 antibody for the prevention of type 1 diabetes: a story of perseverance. Biochemistry (2019) 58:4107–11. doi: 10.1021/acs.biochem.9b00707
14. Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: Toward combination strategies with curative potential. Cell (2015) 161:205–14. doi: 10.1016/j.cell.2015.03.030
15. Sharma P, Allison JP. The future of immune checkpoint therapy. Science (2015) 348:56–61. doi: 10.1126/science.aaa8172
16. Farina KA, Kane MP. Programmed cell death-1 monoclonal antibody therapy and type 1 diabetes mellitus: A review of the literature. J Pharm Pract (2021) 34:133–40. doi: 10.1177/0897190019850929
17. Colli ML, Hill JLE, Marroqui L, Chaffey J, Dos Santos RS, Leete P, et al. Pdl1 is expressed in the islets of people with type 1 diabetes and is up-regulated by interferons-alpha and-gamma via irf1 induction. Ebiomedicine (2018) 36:367–75. doi: 10.1016/j.ebiom.2018.09.040
18. Desai S, Buchade S, Chitlange S, Sharma H, Bhombe D, Shewale S, et al. Vaccines for type 1 diabetes: Prevention or reversal? Curr Diabetes Rev (2021) 17:30–6. doi: 10.2174/1573399816666200330145501
19. Secchi A, Pontiroli AE, Falqui L, Pastore RM, Scorza R, Meroni PL, et al. Efficacy of prednisone to induce remission in recent onset type i (insulin dependent) diabetic patients. Klin wochenschr (1987) 65:244. doi: 10.1007/BF01715857
20. Bevier WC, Zisser HC, Jovanovic L, Finan DA, Palerm CC, Seborg DE, et al. Use of continuous glucose monitoring to estimate insulin requirements in patients with type 1 diabetes mellitus during a short course of prednisone. J Diabetes Sci Technol (2008) 2:578–83. doi: 10.1177/193229680800200408
21. Gong K, Guo G, Beckley NA, Yang X, Zhang Y, Gerber DE, et al. Comprehensive targeting of resistance to inhibition of rtk signaling pathways by using glucocorticoids. Nat Commun (2021) 12(1):7014. doi: 10.1038/s41467-021-27276-7
22. Cassieri C, Mastromatteo AM, Pica R, Zippi M, Corazziari ES, Paoluzi P, et al. Azathioprine in the maintainance remission in inflammatory bowel disease patients: 7-year follow up. Eur J Public Health (2020) 30. doi: 10.1093/eurpub/ckaa166.591
23. Silverstein J, Maclaren N, Riley W, Spillar R, Radjenovic D, Johnson S. Immunosuppression with azathioprine and prednisone in recent-onset insulin-dependent diabetes mellitus. N Engl J Med (1988) 319:599–604. doi: 10.1056/NEJM198809083191002
24. Geliebter R, Chia D, Derrick K. Sun-275 azathioprine for tertiary prevention of diabetes in patients with newly diagnosed type 1 diabetes. J Endocrine Soc (2019) 3:SUN–275. doi: 10.1210/js.2019-SUN-275
25. Stiller CR, Dupre J, Gent M, Heinrichs D, Jenner MR, Keown PA, et al. Effects of cyclosporine in recent-onset juvenile type 1 diabetes: impact of age and duration of disease. J Pediatr (1987) 111:1069–72. doi: 10.1016/S0022-3476(87)80058-6
26. Wilkin TJ. Insulin resistance and progression to type 1 diabetes in the european nicotinamide diabetes intervention trial (endit): Response to bingley et al. Diabetes Care (2008) 31:e29. doi: 10.2337/dc07-2202
27. Abraham RT, Wiederrecht GJ. Immunopharmacology of rapamycin. Annu Rev Immunol (1996) 14:483–510. doi: 10.1146/annurev.immunol.14.1.483
28. Piemonti L, Maffi P, Monti L, Lampasona V, Perseghin G, Magistretti P, et al. Beta cell function during rapamycin monotherapy in long-term type 1 diabetes. Diabetologia (2011) 54:433–9. doi: 10.1007/s00125-010-1959-6
29. Valle A, Jofra T, Stabilini A, Atkinson M, Roncarolo MG, Battaglia M. Rapamycin prevents and breaks the anti-cd3-induced tolerance in nod mice. Diabetes (2009) 58:875–81. doi: 10.2337/db08-1432
30. Kon N, Ou Y, Wang SJ, Li H, Rustgi AK, Gu W. Mtor inhibition acts as an unexpected checkpoint in p53-mediated tumor suppression. Genes Dev (2021) 35:59–64. doi: 10.1101/gad.340919.120
31. Zhao X-F, Liao Y, Alam MM, Mathur R, Feustel P, Mazurkiewicz JE, et al. Microglial mtor is neuronal protective and antiepileptogenic in the pilocarpine model of temporal lobe epilepsy. J Neurosc (2020) 40:7593–608. doi: 10.1523/JNEUROSCI.2754-19.2020
32. He S, Zhang Y, Wang D, Tao K, Zhang S, Wei L, et al. Rapamycin/gaba combination treatment ameliorates diabetes in nod mice. Mol Immunol (2016) 73:130–7. doi: 10.1016/j.molimm.2016.01.008
33. Manirarora JN, Wei CH. Combination therapy using il-2/il-2 monoclonal antibody complexes, rapamycin, and islet autoantigen peptides increases regulatory t cell frequency and protects against spontaneous and induced type 1 diabetes in nonobese diabetic mice. J Immunol (2015) 195:5203–14. doi: 10.4049/jimmunol.1402540
34. Lee JS, Han P, Chaudhury R, Khan S, Bickerton S, Mchugh MD, et al. Metabolic and immunomodulatory control of type 1 diabetes via orally delivered bile-acid-polymer nanocarriers of insulin or rapamycin. Nat BioMed Eng (2021) 5:983–97. doi: 10.1038/s41551-021-00791-0
35. Herold KC, Hagopian W, Auger JA, Poumian-Ruiz E, Taylor L, Donaldson D, et al. Anti-cd3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med (2002) 346:1692–8. doi: 10.1056/NEJMoa012864
36. Bresson D, Togher L, Rodrigo E, Chen Y, Bluestone JA, Herold KC, et al. Anti-cd3 and nasal proinsulin combination therapy enhances remission from recent-onset autoimmune diabetes by inducing tregs. J Clin Invest (2006) 116:1371–81. doi: 10.1172/JCI27191
37. Cook DP, Cunha J, Martens PJ, Sassi G, Mancarella F, Ventriglia G, et al. Intestinal delivery of proinsulin and il-10 via lactococcus lactis combined with low-dose anti-cd3 restores tolerance outside the window of acute type 1 diabetes diagnosis. Front Immunol (2020) 11 1103. doi: 10.3389/fimmu.2020.01103
38. Stewart JM, Posgai AL, Leon JJ, Haller MJ, Keselowsky BG. Combination treatment with antigen-specific dual-sized microparticle system plus anti-cd3 immunotherapy fails to synergize to improve late-stage type 1 diabetes prevention in nonobese diabetic mice. ACS Biomat Sci Eng (2020) 6:5941–58. doi: 10.1021/acsbiomaterials.0c01075
39. Liu VC, Wong LY, Jang T, Shah AH, Park I, Yang X, et al. Tumor evasion of the immune system by converting cd4+cd25- t cells into cd4+cd25+ t regulatory cells: role of tumor-derived tgf-beta. J Immunol (2007) 178:2883–92. doi: 10.4049/jimmunol.178.5.2883
40. Waldron-Lynch F, Herold KC. Immunomodulatory therapy to preserve pancreatic beta-cell function in type 1 diabetes. Nat Rev Drug Discovery (2011) 10:439–52. doi: 10.1038/nrd3402
41. Li Y, Frei AW, Labrada IM, Rong Y, Liang JP, Samojlik MM, et al. Immunosuppressive plga tgf-beta1 microparticles induce polyclonal and antigen-specific regulatory t cells for local immunomodulation of allogeneic islet transplants. Front Immunol (2021) 12:653088. doi: 10.3389/fimmu.2021.653088
42. Green EA, Gorelik L, Mcgregor CM, Tran EH, Flavell RA. Cd4+cd25+ t regulatory cells control anti-islet cd8+ t cells through tgf-beta-tgf-beta receptor interactions in type 1 diabetes. Proc Natl Acad Sci u s(2003) 100:10878–83. doi: 10.1073/pnas.1834400100
43. Perol L, Lindner JM, Caudana P, Nunez NG, Baeyens A, Valle A, et al. Loss of immune tolerance to il-2 in type 1 diabetes. Nat Commun (2016) 7:13027. doi: 10.1038/ncomms13027
44. Liu JMH, Zhang J, Zhang X, Hlavaty KA, Ricci CF, Leonard JN, et al. Transforming growth factor-beta 1 delivery from microporous scaffolds decreases inflammation post-implant and enhances function of transplanted islets. Biomaterials (2016) 80:11–9. doi: 10.1016/j.biomaterials.2015.11.065
45. Daneshmandi S, Karimi MH, Pourfathollah AA. Tgf-beta engineered mesenchymal stem cells (tgf-beta/mscs) for treatment of type 1 diabetes (t1d) mice model. Int immunopharmacol (2017) 44:191–6. doi: 10.1016/j.intimp.2017.01.019
46. Koprivica I, Gajic D, Saksida T, Cavalli E, Auci D, Despotovic S, et al. Orally delivered all-trans-retinoic acid- and transforming growth factor-beta-loaded microparticles ameliorate type 1 diabetes in mice. Eur J Pharmacol (2019) 864:172721. doi: 10.1016/j.ejphar.2019.172721
47. Hartemann A, Bensimon G, Payan CA, Jacqueminet S, Bourron O, Nicolas N, et al. Low-dose interleukin 2 in patients with type 1 diabetes: A phase 1/2 randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol (2013) 1:295–305. doi: 10.1016/S2213-8587(13)70113-X
48. Tang Q, Adams JY, Penaranda C, Melli K, Piaggio E, Sgouroudis E, et al. Central role of defective interleukin-2 production in the triggering of islet autoimmune destruction. Immunity (2008) 28:687–97. doi: 10.1016/j.immuni.2008.03.016
49. Rosenzwajg M, Churlaud G, Mallone R, Six A, Derian N, Chaara W, et al. Low-dose interleukin-2 fosters a dose-dependent regulatory t cell tuned milieu in t1d patients. J autoimmun (2015) 58:48–58. doi: 10.1016/j.jaut.2015.01.001
50. TODD JA, EVANGELOU M, CUTLER AJ, PEKALSKI ML, WALKER NM, STEVENS HE, et al. Regulatory t cell responses in participants with type 1 diabetes after a single dose of interleukin-2: A non-randomised, open label, adaptive dose-finding trial. PLoS Med (2016) 13:e1002139. doi: 10.1371/journal.pmed.1002139
51. Boyman O, Kovar M, Rubinstein MP, Surh CD, Sprent J. Selective stimulation of t cell subsets with antibody-cytokine immune complexes. Science (2006) 311:1924–7. doi: 10.1126/science.1122927
52. Spangler JB, Tomala J, Luca VC, Jude KM, Dong S, Ring AM, et al. Antibodies to interleukin-2 elicit selective t cell subset potentiation through distinct conformational mechanisms. Immunity (2015) 42:815–25. doi: 10.1016/j.immuni.2015.04.015
53. Trotta E, Bessette PH, Silveria SL, Ely LK, Jude KM, Le DT, et al. A human anti-il-2 antibody that potentiates regulatory t cells by a structure-based mechanism. Nat Med (2018) 24:1005–14. doi: 10.1038/s41591-018-0070-2
54. Karakus U, Sahin D, Mittl PRE, Mooij P, Koopman G, Boyman O. Receptor-gated il-2 delivery by an anti-human il-2 antibody activates regulatory t cells in three different species. Sci Transl Med (2020) 12(574):eabb9283. doi: 10.1126/scitranslmed.abb9283
55. Mitra S, Ring AM, Amarnath S, Spangler JB, Li P, Ju W, et al. Interleukin-2 activity can be fine tuned with engineered receptor signaling clamps. Immunity (2015) 42:826–38. doi: 10.1016/j.immuni.2015.04.018
56. Overwijk WW, Tagliaferri MA, Zalevsky J. Engineering il-2 to give new life to t cell immunotherapy. Annu Rev Med (2021) 72:281–311. doi: 10.1146/annurev-med-073118-011031
57. Izquierdo C, Ortiz AZ, Presa M, Malo S, Montoya A, Garabatos N, et al. Treatment of t1d via optimized expansion of antigen-specific tregs induced by il-2/anti-il-2 monoclonal antibody complexes and peptide/mhc tetramers. Sci Rep (2018) 8(1):8106. doi: 10.1038/s41598-018-26161-6
58. Sockolosky JT, Trotta E, Parisi G, Picton L, Su LL, Le AC, et al. Selective targeting of engineered t cells using orthogonal il-2 cytokine-receptor complexes. Science (2018) 359:1037–42. doi: 10.1126/science.aar3246
59. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated b7-h1 promotes t-cell apoptosis: A potential mechanism of immune evasion. Nat Med (2002) 8:793–800. doi: 10.1038/nm730
60. De La Rosa M, Rutz S, Dorninger H, Scheffold A. Interleukin-2 is essential for cd4+cd25+ regulatory t cell function. Eur J Immunol (2004) 34:2480–8. doi: 10.1002/eji.200425274
61. Gauci ML, Laly P, Vidal-Trecan T, Baroudjian B, Gottlieb J, Madjlessi-Ezra N, et al. Autoimmune diabetes induced by pd-1 inhibitor-retrospective analysis and pathogenesis: A case report and literature review. Cancer Immunol Immunother (2017) 66:1399–410. doi: 10.1007/s00262-017-2033-8
62. Ansari MJ, Salama AD, Chitnis T, Smith RN, Yagita H, Akiba H, et al. The programmed death-1 (pd-1) pathway regulates autoimmune diabetes in nonobese diabetic (nod) mice. J Exp Med (2003) 198:63–9. doi: 10.1084/jem.20022125
63. Pauken KE, Jenkins MK, Azuma M, Fife BT. Pd-1, but not pd-l1, expressed by islet-reactive cd4+ t cells suppresses infiltration of the pancreas during type 1 diabetes. Diabetes (2013) 62:2859–69. doi: 10.2337/db12-1475
64. Hughes J, Vudattu N, Sznol M, Gettinger S, Kluger H, Lupsa B, et al. Precipitation of autoimmune diabetes with anti-pd-1 immunotherapy. Diabetes Care (2015) 38:e55–7. doi: 10.2337/dc14-2349
65. Ben Nasr M, Tezza S, D'Addio F, Mameli C, Usuelli V, Maestroni A, et al. PD-L1 genetic overexpression or pharmacological restoration in hematopoietic stem and progenitor cells reverses autoimmune diabetes. Sci Transl Med (2017) 9(416):caam75. doi: 10.1126/scitranslmed.aam7543
66. Zhang X, Kang Y, Wang J, Yan J, Chen Q, Cheng H, et al. Engineered pd-l1-expressing platelets reverse new-onset type 1 diabetes. Adv mat (2020) 32:e1907692. doi: 10.1002/adma.201907692
67. Yoshihara E, O'connor C, Gasser E, Wei Z, Oh TG, Tseng TW, et al. Immune-evasive human islet-like organoids ameliorate diabetes. Nature (2020) 586:606–11. doi: 10.1038/s41586-020-2631-z
68. Au KM, Tisch R, Wang AZ. In vivo bioengineering of beta cells with immune checkpoint ligand as a treatment for early-onset type 1 diabetes mellitus. ACS nano (2021) 15:19990–20002. doi: 10.1021/acsnano.1c07538
69. Stamatouli AM, Quandt Z, Perdigoto AL, Clark PL, Kluger H, Weiss SA, et al. Collateral damage: Insulin-dependent diabetes induced with checkpoint inhibitors. Diabetes (2018) 67:1471–80. doi: 10.2337/dbi18-0002
70. Zappasodi R, Merghoub T, Wolchok JD. Emerging concepts for immune checkpoint blockade-based combination therapies. Cancer Cell (2018) 34:690. doi: 10.1016/j.ccell.2018.09.008
71. Grohmann U, Orabona C, Fallarino F, Vacca C, Calcinaro F, Falorni A, et al. Ctla-4-ig regulates tryptophan catabolism in vivo. Nat Immunol (2002) 3:1097–101. doi: 10.1038/ni846
72. El Khatib MM, Sakuma T, Tonne JM, Mohamed MS, Holditch SJ, Lu B, et al. Beta-cell-targeted blockage of pd1 and ctla4 pathways prevents development of autoimmune diabetes and acute allogeneic islets rejection. Gene Ther (2015) 22:430–8. doi: 10.1038/gt.2015.18
73. Chen Q, Xu L, Liang C, Wang C, Peng R, Liu Z. Photothermal therapy with immune-adjuvant nanoparticles together with checkpoint blockade for effective cancer immunotherapy. Nat Commun (2016) 7:13193. doi: 10.1038/ncomms13193
74. Carabias P, Espelt MV, Bacigalupo ML, Rojas P, Sarrias L, Rubin A, et al. Galectin-1 confers resistance to doxorubicin in hepatocellular carcinoma cells through modulation of p-glycoprotein expression. Cell Death Dis (2022) 13:79. doi: 10.1038/s41419-022-04520-6
75. Bacigalupo ML, Carabias P, Troncoso MF. Contribution of galectin-1, a glycan-binding protein, to gastrointestinal tumor progression. World J Gastroenterol (2017) 23:5266–81. doi: 10.3748/wjg.v23.i29.5266
76. Sundblad V, Morosi LG, Geffner JR, Rabinovich GA. Galectin-1: A jack-of-all-trades in the resolution of acute and chronic inflammation. J Immunol (2017) 199:3721–30. doi: 10.4049/jimmunol.1701172
77. Kandel S, Adhikary P, Li G, Cheng K. The tim3/gal9 signaling pathway: An emerging target for cancer immunotherapy. Cancer Lett (2021) 510:67–78. doi: 10.1016/j.canlet.2021.04.011
78. Li X, Chen Y, Liu X, Zhang J, He X, Teng G, et al. Tim3/gal9 interactions between t cells and monocytes result in an immunosuppressive feedback loop that inhibits th1 responses in osteosarcoma patients. Int immunopharmacol (2017) 44:153–9. doi: 10.1016/j.intimp.2017.01.006
79. Baek JH, Kim SJ, Kang HG, Lee HW, Kim JH, Hwang KA, et al. Galectin-3 activates ppargamma and supports white adipose tissue formation and high-fat diet-induced obesity. Endocrinology (2015) 156:147–56. doi: 10.1210/en.2014-1374
80. Li P, Liu S, Lu M, Bandyopadhyay G, Oh D, Imamura T, et al. Hematopoietic-derived galectin-3 causes cellular and systemic insulin resistance. Cell (2016) 167 973–84.e12. doi: 10.1016/j.cell.2016.10.025
81. Iacobini C, Menini S, Ricci C, Blasetti Fantauzzi C, Scipioni A, Salvi L, et al. Galectin-3 ablation protects mice from diet-induced nash: a major scavenging role for galectin-3 in liver. J Hepatol (2011) 54:975–83. doi: 10.1016/j.jhep.2010.09.020
82. Baban B, Penberthy WT, Mozaffari MS. The potential role of indoleamine 2,3 dioxygenase (ido) as a predictive and therapeutic target for diabetes treatment: A mythical truth. Epma J (2010) 1:46–55. doi: 10.1007/s13167-010-0009-2
83. Huang T, Song J, Gao J, Cheng J, Xie H, Zhang L, et al. Adipocyte-derived kynurenine promotes obesity and insulin resistance by activating the ahr/stat3/il-6 signaling. Nat Commun (2022) 13(1):3489. doi: 10.1038/s41467-022-31126-5
84. Anquetil F, Mondanelli G, Gonzalez N, Rodriguez Calvo T, Zapardiel Gonzalo J, Krogvold L, et al. Loss of ido1 expression from human pancreatic beta-cells precedes their destruction during the development of type 1 diabetes. Diabetes (2018) 67:1858–66. doi: 10.2337/db17-1281
85. Fallarino F, Volpi C, Zelante T, Vacca C, Calvitti M, Fioretti MC, et al. Ido mediates tlr9-driven protection from experimental autoimmune diabetes. J Immunol (2009) 183:6303–12. doi: 10.4049/jimmunol.0901577
86. Pallotta MT, Orabona C, Bianchi R, Vacca C, Fallarino F, Belladonna ML, et al. Forced ido1 expression in dendritic cells restores immunoregulatory signalling in autoimmune diabetes. J Cell Mol Med (2014) 18:2082–91. doi: 10.1111/jcmm.12360
87. Mondanelli G, Albini E, Pallotta MT, Volpi C, Chatenoud L, Kuhn C, et al. The proteasome inhibitor bortezomib controls indoleamine 2,3-dioxygenase 1 breakdown and restores immune regulation in autoimmune diabetes. Front Immunol (2017) 8:428. doi: 10.3389/fimmu.2017.00428
88. Zhang Y, Jalili RB, Kilani RT, Elizei SS, Farrokhi A, Khosravi-Maharlooei M, et al. Ido-expressing fibroblasts protect islet beta cells from immunological attack and reverse hyperglycemia in non-obese diabetic mice. J Cell Physiol (2016) 231:1964–73. doi: 10.1002/jcp.25301
89. Dolpady J, Sorini C, Di Pietro C, Cosorich I, Ferrarese R, Saita D, et al. Oral probiotic VSL#3 prevents autoimmune diabetes by modulating microbiota and promoting indoleamine 2,3-dioxygenase-enriched tolerogenic intestinal environment. J Diabetes Res (2016) 2016:7569431. doi: 10.1155/2016/7569431
90. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated t cells expressing il-2 receptor alpha-chains (cd25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol (1995) 155:1151–64.
91. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of cd4+cd25+ regulatory t cells. Nat Immunol (2003) 4:330–6. doi: 10.1038/ni904
92. Waldmann H, Hilbrands R, Howie D, Cobbold S. Harnessing foxp3+ regulatory t cells for transplantation tolerance. J Clin Invest (2014) 124:1439–45. doi: 10.1172/JCI67226
93. Horwitz DA, Zheng SG, Gray JD, Wang JH, Ohtsuka K, Yamagiwa S. Regulatory t cells generated ex vivo as an approach for the therapy of autoimmune disease. Semin Immunol (2004) 16:135–43. doi: 10.1016/j.smim.2003.12.009
94. Nishimura E, Sakihama T, Setoguchi R, Tanaka K, Sakaguchi S. Induction of antigen-specific immunologic tolerance by in vivo and in vitro antigen-specific expansion of naturally arising foxp3+cd25+cd4+ regulatory t cells. Int Immunol (2004) 16:1189–201. doi: 10.1093/intimm/dxh122
95. Putnam AL, Brusko TM, Lee MR, Liu W, Szot GL, Ghosh T, et al. Expansion of human regulatory t-cells from patients with type 1 diabetes. Diabetes (2009) 58:652–62. doi: 10.2337/db08-1168
96. Yamazaki S, Inaba K, Tarbell KV, Steinman RM. Dendritic cells expand antigen-specific foxp3+ cd25+ cd4+ regulatory t cells including suppressors of alloreactivity. Immunol Rev (2006) 212:314–29. doi: 10.1111/j.0105-2896.2006.00422.x
97. Tarbell KV, Yamazaki S, Olson K, Toy P, Steinman RM. Cd25+ cd4+ t cells, expanded with dendritic cells presenting a single autoantigenic peptide, suppress autoimmune diabetes. J Exp Med (2004) 199:1467–77. doi: 10.1084/jem.20040180
98. Tarbell KV, Petit L, Zuo X, Toy P, Luo X, Mqadmi A, et al. Dendritic cell-expanded, islet-specific cd4+ cd25+ cd62l+ regulatory t cells restore normoglycemia in diabetic nod mice. J Exp Med (2007) 204:191–201. doi: 10.1084/jem.20061631
99. Liu W, Putnam AL, Xu-Yu Z, Szot GL, Lee MR, Zhu S, et al. Cd127 expression inversely correlates with foxp3 and suppressive function of human cd4+ t reg cells. J Exp Med (2006) 203:1701–11. doi: 10.1084/jem.20060772
100. Marek-Trzonkowska N, Mysliwiec M, Dobyszuk A, Grabowska M, Derkowska I, Juscinska J, et al. Therapy of type 1 diabetes with cd4(+)cd25(high)cd127-regulatory t cells prolongs survival of pancreatic islets - results of one year follow-up. Clin Immunol (2014) 153:23–30. doi: 10.1016/j.clim.2014.03.016
101. Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, et al. Type 1 diabetes immunotherapy using polyclonal regulatory t cells. Sci Transl Med (2015) 7:315ra189. doi: 10.1126/scitranslmed.aad4134
102. Marek-Trzonkowska N, Mysliwiec M, Dobyszuk A, Grabowska M, Techmanska I, Juscinska J, et al. Administration of cd4+cd25highcd127- regulatory t cells preserves beta-cell function in type 1 diabetes in children. Diabetes Care (2012) 35:1817–20. doi: 10.2337/dc12-0038
103. Haque M, Lei F, Xiong X, Das JK, Ren X, Fang D, et al. Stem cell-derived tissue-associated regulatory t cells suppress the activity of pathogenic cells in autoimmune diabetes. JCI Insight (2019) 4(7):e126471. doi: 10.1172/jci.insight.126471
104. Marek N, Krzystyniak A, Ergenc I, Cochet O, Misawa R, Wang LJ, et al. Coating human pancreatic islets with cd4(+)cd25(high)cd127(-) regulatory t cells as a novel approach for the local immunoprotection. Ann Surg (2011) 254:512–8; discussion 518-9. doi: 10.1097/SLA.0b013e31822c9ca7
105. Golab K, Kizilel S, Bal T, Hara M, Zielinski M, Grose R, et al. Improved coating of pancreatic islets with regulatory t cells to create local immunosuppression by using the biotin-polyethylene glycol-succinimidyl valeric acid ester molecule. Transplant Proc (2014) 46:1967–71. doi: 10.1016/j.transproceed.2014.05.075
106. Cabello-Kindelan C, Mackey S, Sands A, Rodriguez J, Vazquez C, Pugliese A, et al. Immunomodulation followed by antigen-specific treg infusion controls islet autoimmunity. Diabetes (2020) 69:215–27. doi: 10.2337/db19-0061
107. Yeh WI, Seay HR, Newby B, Posgai AL, Moniz FB, Michels A, et al. Avidity and bystander suppressive capacity of human regulatory t cells expressing de novo autoreactive t-cell receptors in type 1 diabetes. Front Immunol (2017) 8 1313. doi: 10.3389/fimmu.2017.01313
108. Hull CM, Nickolay LE, Estorninho M, Richardson MW, Riley JL, Peakman M, et al. Generation of human islet-specific regulatory t cells by tcr gene transfer. J autoimmun (2017) 79:63–73. doi: 10.1016/j.jaut.2017.01.001
109. Macdonald KG, Hoeppli RE, Huang Q, Gillies J, Luciani DS, Orban PC, et al. Alloantigen-specific regulatory t cells generated with a chimeric antigen receptor. J Clin Invest (2016) 126:1413–24. doi: 10.1172/JCI82771
110. Pierini A, Iliopoulou BP, Peiris H, Perez-Cruz M, Baker J, Hsu K, et al. T Cells expressing chimeric antigen receptor promote immune tolerance. JCI Insight (2017) 2(20):e92865. doi: 10.1172/jci.insight.92865
111. Tenspolde M, Zimmermann K, Weber LC, Hapke M, Lieber M, Dywicki J, et al. Regulatory t cells engineered with a novel insulin-specific chimeric antigen receptor as a candidate immunotherapy for type 1 diabetes. J autoimmun (2019) 103:102289. doi: 10.1016/j.jaut.2019.05.017
112. Jordan KR, Kapoor P, Spongberg E, Tobin RP, Gao D, Borges VF, et al. Immunosuppressive myeloid-derived suppressor cells are increased in splenocytes from cancer patients. Cancer Immunol Immunother (2017) 66:503–13. doi: 10.1007/s00262-016-1953-z
113. Wang S, Tan Q, Hou Y, Dou H. Emerging roles of myeloid-derived suppressor cells in diabetes. Front Pharmacol (2021) 12:798320. doi: 10.3389/fphar.2021.798320
114. Zoso A, Mazza EM, Bicciato S, Mandruzzato S, Bronte V, Serafini P, et al. Human fibrocytic myeloid-derived suppressor cells express ido and promote tolerance via treg-cell expansion. Eur J Immunol (2014) 44:3307–19. doi: 10.1002/eji.201444522
115. Drujont L, Carretero-Iglesia L, Bouchet-Delbos L, Beriou G, Merieau E, Hill M, et al. Evaluation of the therapeutic potential of bone marrow-derived myeloid suppressor cell (mdsc) adoptive transfer in mouse models of autoimmunity and allograft rejection. PLoS One (2014) 9:e100013. doi: 10.1371/journal.pone.0100013
116. Whitfield-Larry F, Felton J, Buse J, Su MA. Myeloid-derived suppressor cells are increased in frequency but not maximally suppressive in peripheral blood of type 1 diabetes mellitus patients. Clin Immunol (2014) 153:156–64. doi: 10.1016/j.clim.2014.04.006
117. Qin J, Arakawa Y, Morita M, Fung JJ, Qian S, Lu L. C-c chemokine receptor type 2-dependent migration of myeloid-derived suppressor cells in protection of islet transplants. Transplantation (2017) 101:1793–800. doi: 10.1097/TP.0000000000001529
118. Dahlen E, Dawe K, Ohlsson L, Hedlund G. Dendritic cells and macrophages are the first and major producers of tnf-alpha in pancreatic islets in the nonobese diabetic mouse. J Immunol (1998) 160:3585–93. doi: 10.1097/TP.0000000000001529
119. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest (2012) 122:787–95. doi: 10.1172/JCI59643
120. Ricardo SD, Goor V, Eddy AA. Macrophage diversity in renal injury and repair. J Clin Invest (2008) 118:3522–30. doi: 10.1172/JCI36150
121. Jensen DM, Hendricks KV, Mason AT, Tessem JS. Good cop, bad cop: The opposing effects of macrophage activation state on maintaining or damaging functional beta-cell mass. Metabolites (2020) 10(12):485. doi: 10.3390/metabo10120485
122. Zheng D, Wang Y, Cao Q, Lee VW, Zheng G, Sun Y, et al. Transfused macrophages ameliorate pancreatic and renal injury in murine diabetes mellitus. Nephron Exp Nephrol (2011) 118:e87–99. doi: 10.1159/000321034
123. Parsa R, Andresen P, Gillett A, Mia S, Zhang XM, Mayans S, et al. Adoptive transfer of immunomodulatory m2 macrophages prevents type 1 diabetes in nod mice. Diabetes (2012) 61:2881–92. doi: 10.2337/db11-1635
124. Guiteras R, Sola A, Flaquer M, Manonelles A, Hotter G, Cruzado JM. Exploring macrophage cell therapy on diabetic kidney disease. J Cell Mol Med (2019) 23:841–51. doi: 10.1111/jcmm.13983
125. Juhas U, Ryba-Stanislawowska M, Brandt-Varma A, Mysliwiec M, Mysliwska J. Monocytes of newly diagnosed juvenile dm1 patients are prone to differentiate into regulatory il-10(+) m2 macrophages. Immunol Res (2019) 67:58–69. doi: 10.1007/s12026-019-09072-0
126. Yoon JW, Jun HS. Viruses cause type 1 diabetes in animals. Ann n y Acad Sci (2006) 1079:138–46. doi: 10.1196/annals.1375.021
127. Hober D, Alidjinou EK. Enteroviral pathogenesis of type 1 diabetes: queries and answers. Curr Opin Infect Dis (2013) 26:263–9. doi: 10.1097/QCO.0b013e3283608300
128. Racaniello VR. One hundred years of poliovirus pathogenesis. Virology (2006) 344:9–16. doi: 10.1016/j.virol.2005.09.015
129. Schvartzman JM, Thompson CB, Finley LWS. Metabolic regulation of chromatin modifications and gene expression. J Cell Biol (2018) 217:2247–59. doi: 10.1083/jcb.201803061
130. Kroes MM, Miranda-Bedate A, Jacobi RHJ, Van Woudenbergh E, Den Hartog G, Van Putten JPM, et al. Bordetella pertussis-infected innate immune cells drive the anti-pertussis response of human airway epithelium. Sci Rep (2022) 12(1):3622. doi: 10.1038/s41598-022-07603-8
131. Huang SW, Taylor G, Basid A. The effect of pertussis vaccine on the insulin-dependent diabetes induced by streptozotocin in mice. Pediatr Res (1984) 18:221–6. doi: 10.1203/00006450-198402000-00021
132. Shpilsky GF, Takahashi H, Aristarkhova A, Weil M, Ng N, Nelson KJ, et al. Bacillus calmette-guerin 's beneficial impact on glucose metabolism: Evidence for broad based applications. Iscience (2021) 24:103150. doi: 10.1016/j.isci.2021.103150
133. Tian J, Kaufman DL. Antigen-based therapy for the treatment of type 1 diabetes. Diabetes (2009) 58:1939–46. doi: 10.2337/db09-0451
134. Bonn D. Q fever vaccine on trial for type i diabetes. Mol Med Today (1999) 5:143. doi: 10.1016/S1357-4310(99)01450-1
135. Abrams JR, Lebwohl MG, Guzzo CA, Jegasothy BV, Goldfarb MT, Goffe BS, et al. Ctla4ig-mediated blockade of t-cell costimulation in patients with psoriasis vulgaris. J Clin Invest (1999) 103:1243–52. doi: 10.1172/JCI5857
136. Kuhtreiber WM, Takahashi H, Keefe RC, Song Y, Tran L, Luck TG, et al. Bcg vaccinations upregulate myc, a central switch for improved glucose metabolism in diabetes. Iscience (2020) 23:101085. doi: 10.1016/j.isci.2020.101085
137. Files MA, Naqvi KF, Saito TB, Clover TM, Rudra JS, Endsley JJ. Self-adjuvanting nanovaccines boost lung-resident cd4(+) t cell immune responses in bcg-primed mice. NPJ Vaccines (2022) 7:48. doi: 10.1038/s41541-022-00466-0
138. Kuhtreiber WM, Tran L, Kim T, Dybala M, Nguyen B, Plager S, et al. Long-term reduction in hyperglycemia in advanced type 1 diabetes: The value of induced aerobic glycolysis with bcg vaccinations. NPJ Vaccines (2018) 3:23. doi: 10.1038/s41541-018-0062-8
139. Park WJ, Yoon YK, Park JS, Pansuriya R, Seok YJ, Ganapathy R. Rotavirus spike protein deltavp8* as a novel carrier protein for conjugate vaccine platform with demonstrated antigenic potential for use as bivalent vaccine. Sci Rep (2021) 11:22037. doi: 10.1038/s41598-021-01549-z
140. Rogers MAM, Basu T, Kim C. Lower incidence rate of type 1 diabetes after receipt of the rotavirus vaccine in the united states 2001-2017. Sci Rep (2019) 9(1):7727. doi: 10.1038/s41598-019-44193-4
141. Bresson D, Fradkin M, Manenkova Y, Rottembourg D, Von Herrath M. Genetic-induced variations in the gad65 t-cell repertoire governs efficacy of anti-cd3/gad65 combination therapy in new-onset type 1 diabetes. Mol Ther (2010) 18:307–16. doi: 10.1038/mt.2009.197
142. Miller TW, Shirley TL, Wolfgang WJ, Kang X, Messer A. Dna vaccination against mutant huntingtin ameliorates the hdr6/2 diabetic phenotype. Mol Ther (2003) 7:572–9. doi: 10.1016/S1525-0016(03)00063-7
143. Harrison LC, Honeyman MC, Steele CE, Stone NL, Sarugeri E, Bonifacio E, et al. Pancreatic beta-cell function and immune responses to insulin after administration of intranasal insulin to humans at risk for type 1 diabetes. Diabetes Care (2004) 27:2348–55. doi: 10.2337/diacare.27.10.2348
144. Larsson PG, Lakshmikanth T, Laitinen OH, Utorova R, Jacobson S, Oikarinen M, et al. A preclinical study on the efficacy and safety of a new vaccine against coxsackievirus b1 reveals no risk for accelerated diabetes development in mouse models. Diabetologia (2015) 58:346–54. doi: 10.1007/s00125-014-3436-0
145. Bergerot I, Fabien N, Mayer A, Thivolet C. Active suppression of diabetes after oral administration of insulin is determined by antigen dosage. Ann n y Acad Sci (1996) 778:362–7. doi: 10.1111/j.1749-6632.1996.tb21144.x
146. Stifter K, Schuster C, Krieger J, Spyrantis A, Boehm BO, Schirmbeck R. Preproinsulin designer antigens excluded from endoplasmic reticulum suppressed diabetes development in nod mice by dna vaccination. Mol Ther Methods Clin Dev (2019) 12:123–33. doi: 10.1016/j.omtm.2018.12.002
147. Glinka Y, Chang Y, Prud'homme GJ. Protective regulatory t cell generation in autoimmune diabetes by dna covaccination with islet antigens and a selective ctla-4 ligand. Mol Ther (2006) 14:578–87. doi: 10.1016/j.ymthe.2006.03.021
148. Yoshida Y, Shimizu I, Hayashi Y, Ikegami R, Suda M, Katsuumi G, et al. Peptide vaccine for semaphorin3e ameliorates systemic glucose intolerance in mice with dietary obesity. Sci Rep (2019) 9(1):3858. doi: 10.1038/s41598-019-40325-y
149. Prud'homme GJ, Chang Y, Li X. Immunoinhibitory dna vaccine protects against autoimmune diabetes through cdna encoding a selective ctla-4 (cd152) ligand. Hum Gene Ther (2002) 13:395–406. doi: 10.1089/10430340252792521
150. Chang Y, Yap S, Ge X, Piganelli J, Bertera S, Giannokakis N, et al. Dna vaccination with an insulin construct and a chimeric protein binding to both ctla4 and cd40 ameliorates type 1 diabetes in nod mice. Gene Ther (2005) 12:1679–85. doi: 10.1038/sj.gt.3302578
151. Thrower SL, James L, Hall W, Green KM, Arif S, Allen JS, et al. Proinsulin peptide immunotherapy in type 1 diabetes: Report of a first-in-man phase i safety study. Clin Exp Immunol (2009) 155:156–65. doi: 10.1111/j.1365-2249.2008.03814.x
152. Leconet W, Petit P, Peraldi-Roux S, Bresson D. Nonviral delivery of small interfering rna into pancreas-associated immune cells prevents autoimmune diabetes. Mol Ther (2012) 20:2315–25. doi: 10.1038/mt.2012.190
153. Fishman S, Lewis MD, Siew LK, De Leenheer E, Kakabadse D, Davies J, et al. Adoptive transfer of mrna-transfected t cells redirected against diabetogenic cd8 t cells can prevent diabetes. Mol Ther (2017) 25:456–64. doi: 10.1016/j.ymthe.2016.12.007
154. Ying W, Gao H, Dos Reis FCG, Bandyopadhyay G, Ofrecio JM, Luo Z, et al. Mir-690, an exosomal-derived mirna from m2-polarized macrophages, improves insulin sensitivity in obese mice. Cell Metab (2021) 33781–90.e5. doi: 10.1016/j.cmet.2020.12.019
155. Lazar L, Ofan R, Weintrob N, Avron A, Tamir M, Elias D, et al. Heat-shock protein peptide diapep277 treatment in children with newly diagnosed type 1 diabetes: a randomised, double-blind phase ii study. Diabetes Metab Res Rev (2007) 23:286–91. doi: 10.1002/dmrr.711
156. Raz I, Avron A, Tamir M, Metzger M, Symer L, Eldor R, et al. Treatment of new-onset type 1 diabetes with peptide diapep277 is safe and associated with preserved beta-cell function: extension of a randomized, double-blind, phase ii trial. Diabetes Metab Res Rev (2007) 23:292–8. doi: 10.1002/dmrr.712
157. Tian J, Atkinson MA, Clare-Salzler M, Herschenfeld A, Forsthuber T, Lehmann PV, et al. Nasal administration of glutamate decarboxylase (gad65) peptides induces th2 responses and prevents murine insulin-dependent diabetes. J Exp Med (1996) 183:1561–7. doi: 10.1084/jem.183.4.1561
158. Agardh CD, Cilio CM, Lethagen A, Lynch K, Leslie RD, Palmer M, et al. Clinical evidence for the safety of gad65 immunomodulation in adult-onset autoimmune diabetes. J Diabetes Complicat (2005) 19:238–46. doi: 10.1016/j.jdiacomp.2004.12.003
159. Buddhala C, Hsu CC, Wu JY. A novel mechanism for gaba synthesis and packaging into synaptic vesicles. Neurochem Int (2009) 55:9–12. doi: 10.1016/j.neuint.2009.01.020
160. Stabler CL, Li Y, Stewart JM, Keselowsky BG. Engineering immunomodulatory biomaterials for type 1 diabetes. Nat Rev mat (2019) 4:429–50. doi: 10.1038/s41578-019-0112-5
161. Soldevilla MM, Villanueva H, Meraviglia-Crivelli D, Menon AP, Ruiz M, Cebollero J, et al. Icos costimulation at the tumor site in combination with ctla-4 blockade therapy elicits strong tumor immunity. Mol Ther (2019) 27:1878–91. doi: 10.1016/j.ymthe.2019.07.013
162. Yoon YM, Lewis JS, Carstens MR, Campbell-Thompson M, Wasserfall CH, Atkinson MA, et al. A combination hydrogel microparticle-based vaccine prevents type 1 diabetes in non-obese diabetic mice. Sci Rep (2015) 5:13155. doi: 10.1038/srep13155
163. Hyoty H, Knip M. Developing a vaccine for type 1 diabetes through targeting enteroviral infections. Expert Rev Vaccines (2014) 13:989–99. doi: 10.1586/14760584.2014.933078
164. Cavelti-Weder C, Timper K, Seelig E, Keller C, Osranek M, Lassing U, et al. Development of an interleukin-1beta vaccine in patients with type 2 diabetes. Mol Ther (2016) 24:1003–12. doi: 10.1038/mt.2015.227
165. Spohn G, Schori C, Keller I, Sladko K, Sina C, Guler R, et al. Preclinical efficacy and safety of an anti-il-1beta vaccine for the treatment of type 2 diabetes. Mol Ther Methods Clin Dev (2014) 1:14048. doi: 10.1038/mtm.2014.48
166. Creusot RJ, Chang P, Healey DG, Tcherepanova IY, Nicolette CA, Fathman CG. A short pulse of il-4 delivered by dcs electroporated with modified mrna can both prevent and treat autoimmune diabetes in nod mice. Mol Ther (2010) 18:2112–20. doi: 10.1038/mt.2010.146
167. Santoni De Sio FR, Cascio P, Gritti A, Ncri M, Sampaolesi M, Colleoni S, et al. 1106. High proteasome activity is a common feature among stem cells of different lineages and restricts lentiviral gene transfer. Mol Ther (2007) 15:s422–s42. doi: 10.1016/s1525-0016(16)45312-8
Keywords: biomaterial, type 1 diabetes, immune modulation, immune checkpoint, vaccine
Citation: Jing Z, Li Y, Ma Y, Zhang X, Liang X and Zhang X (2022) Leverage biomaterials to modulate immunity for type 1 diabetes. Front. Immunol. 13:997287. doi: 10.3389/fimmu.2022.997287
Received: 18 July 2022; Accepted: 20 September 2022;
Published: 02 November 2022.
Edited by:
Yuanzeng Min, University of Science and Technology of China, ChinaReviewed by:
Juan Mi, University of Science and Technology of China, ChinaXueying Zheng, University of Science and Technology of China, China
Copyright © 2022 Jing, Li, Ma, Zhang, Liang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xudong Zhang, emhhbmd4ZDU2QG1haWwuc3lzdS5lZHUuY24=; Xin Liang, bGlhbmd4aW5naWJoQGdkbXUuZWR1LmNu
†These authors have contributed equally to this work