
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 23 September 2022
Sec. Multiple Sclerosis and Neuroimmunology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.996469
This article is part of the Research Topic Community series in Reassessing the Immune System Contribution in Multiple Sclerosis: Therapeutic Target, Biomarkers of Disease and Immune Pathogenesis - Volume II View all 5 articles
 Rongzeng Liu1
Rongzeng Liu1 Shushu Du1
Shushu Du1 Lili Zhao1
Lili Zhao1 Sahil Jain2Kritika Sahay3
Sahil Jain2Kritika Sahay3 Albert Rizvanov4
Albert Rizvanov4 Vera Lezhnyova4
Vera Lezhnyova4 Timur Khaibullin5
Timur Khaibullin5 Ekaterina Martynova4
Ekaterina Martynova4 Svetlana Khaiboullina4*
Svetlana Khaiboullina4* Manoj Baranwal3*
Manoj Baranwal3*Multiple sclerosis (MS) is a chronic inflammatory disease of the central nervous system (CNS) characterized by destruction of the myelin sheath structure. The loss of myelin leads to damage of a neuron’s axon and cell body, which is identified as brain lesions on magnetic resonance image (MRI). The pathogenesis of MS remains largely unknown. However, immune mechanisms, especially those linked to the aberrant lymphocyte activity, are mainly responsible for neuronal damage. Th1 and Th17 populations of lymphocytes were primarily associated with MS pathogenesis. These lymphocytes are essential for differentiation of encephalitogenic CD8+ T cell and Th17 lymphocyte crossing the blood brain barrier and targeting myelin sheath in the CNS. B-lymphocytes could also contribute to MS pathogenesis by producing anti-myelin basic protein antibodies. In later studies, aberrant function of Treg and Th9 cells was identified as contributing to MS. This review summarizes the aberrant function and count of lymphocyte, and the contributions of these cell to the mechanisms of MS. Additionally, we have outlined the novel MS therapeutics aimed to amend the aberrant function or counts of these lymphocytes.
Multiple sclerosis (MS) is a chronic inflammatory disease of the central nervous system (CNS) characterized by disruption of myelin sheath integrity and damage to axons and bodies of neurons (1). Initially, this destruction is microscopic in size, which accumulates over the years, presenting with CNS lesions, which could appear disseminated temporary and spatially. Clinical manifestation of MS has a wide range of neurological symptoms ranging from a loss of sensation to impaired muscle function, visual loss, cognitive dysfunction, etc. (2). The MS diagnostic criteria reflect a pattern of CNS lesion development by incorporating the signs of disseminated CNS damage identified with magnetic resonance imaging (MRI) and clinical symptoms (3, 4). Although diagnosed in all age groups, most MS patients are between 20 and 40 years old (5–8). Among them, there is a higher prevalence of MS in women as compared to men (8).
Pathogenesis of autoimmune disease is explained by a loss of immune tolerance to self-proteins due to a combination of genetic susceptibility and environmental provocation. This could result in the generation of autoreactive T and B cells. This review summarizes the latest advances in our understanding of the autoreactive lymphocyte populations that are identified as playing role in the pathogenesis of MS. The cells recently found to be affected in MS include GM-CSF-secreting effector T cells (9), DR2a and DR2b-derived self-peptides specific CD4+ T cell clones (10), glutamate producing Th17 cells (11), GM-CSF+ CXCR4+T cells (12), CD4+CD25hiCD127lowFOXP3+ Tregs (13), MBP-specific CD8+ T cells (14), circulating CD8+ CD20+ T memory cells (specific to myelin) (15) and regulatory B cells (Bregs) producing IL-10 and IL-35 (16). The autoreactive CD4+ T cells cross-reacting with DR2a and DR2b-derived self-peptides shown as a contributing to MS pathogenesis (10). The glutamate producing Th17 cells could cause damage to oligodendrocytes, exposing myelin antigens as a target to autoreactive lymphocytes (11). GM-CSF+ CXCR4+T cells express significantly higher amounts of VLA-4, a CXCR4 ligand, associated with an increase in lymphocyte trafficking to the CNS (17). Studies have shown that the MBP-specific CD8+ T cells could exacerbate brain inflammation (18). Circulating CD8+ CD20+ T memory cells were found with up-regulation of activation markers, pro-inflammatory cytokines, and adhesion molecules, indicating that they could have a high pathogenic potential (19).
About 2.8 million cases of MS were diagnosed worldwide in 2020 (7). Interestingly, a greater burden of MS was reported in countries with high socio-economic status (20). This observation is supported by the high-risk of MS in Northern Europe and North America, where ≥100 cases are diagnosed per 100,000 population (20, 21). A lower risk of MS is found in Africa and Asia, where the prevalence is <30 cases per 100,000 population (22–24). An analysis of MS frequency led to identification of three zones of global MS prevalence: high (30-80 cases per 100,000 population), medium (5-25 cases per 100,000 population) and low (<5 cases per 100,000 population) (25). An updated five-zone scale of MS prevalence was proposed by Wade: very high (170–350 cases per 100,000 population), high (70–170 cases per 100,000 population), medium (38–70 cases per 100,000 population), low (13–38 cases per 100,000 population), and very low (0–13 cases per 100,000 population) (26). The very high and high prevalence zones of MS were reported in North America and Northern Europe, while Asia and Africa were in the lower zone (27). It appears that populations with European ancestry (28) are at a higher risk, due to higher MS prevalence reported in Australia and New Zealand (27). This assumption is also supported by finding higher MS prevalence in Russia (30-<60 cases per 100,000 population) as compared to that in China (0-<30 cases per 100,000 population) (27). Within Russia, the incidence rate of MS is also higher in the North-West regions, where the population is predominantly of European ancestry (29). In China, MS prevalence remains low, although an increasing trend was recently reported (30, 31). It also appears that residence sites are one of the risk factors of MS, as more MS cases were diagnosed at high latitude and high altitude locations (31).
The pathogenesis of MS remains largely unknown mainly due to the limited understanding of the disease ethology. Multiple risk factors of MS were identified representing unrelated elements such as latitude, serum vitamin D (vitD) levels, genetics and virus infection (32). However, neither one of these factors was recognized as the single cause of the disease suggesting that the pathogenesis of the disease is multifactorial.
The role of the genetics in MS pathogenesis is widely recognized and supported by a large body of evidence. This includes a higher frequency of MS found in siblings (33–35) as well as in individuals closely related to MS probands (36). Also finding the link between Human leukocyte Antigen (HLA) DRB1*1501 haplotype and MS in Northern European populations supported this assumption (37). A separate subset of haplotypes, the HLA-DRB1*0301, HLA-DRB1*0405 and HLA-DRB1*1303 haplotypes, is found in MS from Mediterranean region (38). Similarly, the association between HLA-DRB1 was demonstrated in Russian and Chinese MS patients (39, 40).
Epigenetic modifications, which includes histones and DNA post-translational modification, could represent the environment induced modification of gene expression (41). These epigenetic modifications were also implemented to play role in pathogenesis of MS (42). Supporting this assumption was the study demonstrating differentially methylated region (DMR) composed of eight hypomethylated CPGs in HLA-DRB1 in MS (43). This data emphasising the contribution of hypermethylation of ring finger protein 39 (rnf39) at an independent MHC site to MS pathogenesis. In a recent study, the DMR hypomethylation in the HLA-DRB1*15:01 locus was found in the blood cells of MS patients. This study demonstrated that the hypomethylation of this MS susceptibility gene locus can induce the high expression of HLA-DRB1, found in these patients (44). Additionally, a significant hypermethylation of Foxp3 promoter was reported in EAE mice (45). It was demonstrated that expression of FOXP3 gene in EAE is significantly reduced by methylation of conserved non-coding sequence 2 (CNS2).
Other extensively documented risk factors are latitude and serum VitD levels. A latitudinal gradient with an increased south to north prevalence of MS was documented in Northern America and Northern Europe (46). Similar patterns of higher number of MS cases in the northern as compared to southern regions within the same country is also reported (47, 48). Latitudinal gradient with lower number of cases in the south regions is found in Russia and China (29, 31). The latitude dependent risk of MS could be explained by the lesser sunlight exposure in the north as compared to south regions (32). This lesser sun exposure could also explain the commonly found lower VitD serum levels in MS patients (49). The effect of VitD levels was explained by decreased intensity of ultraviolet B (UVB) radiation in northern latitudes (50). In addition to regulation of VitD synthesis, UVB could ameliorate symptoms of MS by inhibiting production of pro-inflammatory cytokines (51).
The role of viral infection in pathogenesis of MS was suggested in multiple studies (52–54). It is based on the epidemiological evidence of higher risk of MS in individuals infected with Epstein-Barr virus (EBV) and diagnosed with infectious mononucleosis (52). In a recent study, Bjornevik et al. presented strong evidence of the association between EBV infection and MS (53). This study also identified EBV as a potential infectious agent causing MS. The role of this virus in MS pathogenesis is also supported by elevated anti-EBV serum antibody titers (55) and by detection of EBV in MS lesions (56, 57). The mechanisms of EBV causing MS remains largely unknown. It is believed that EBV could inhibit the production of antiviral cytokines and proteins, and interfere with the antigen processing and presentation (58). Production of the autoreactive immune cells responsible for an aberrant self-targeting immune response is also suggested (59). This assumption is supported by findings of antibodies cross reacting with myelin basic protein (MBP) and EBV latent membrane protein 1 (LMP1) (60). This cross reactivity was explained by the homology between MBP and LMP1. The role of LMP1 in MS pathogenesis was supported by the demonstration that the exposure to this virus protein could induce myelin-reactive antibodies in vivo (61).
The gut microbiome contributes to the activation and development of innate and adaptive immune responses (62). The imbalance in the interaction between microbiota and immune cells could contribute to the pathogenesis of immune-mediated disorders, such as MS. Supporting this assumption are multiple studies demonstrating changes in the microbiome in MS (63–66). These changes in the microbiome were linked to MS activity, such as the risk of relapse and detection of new lesions (67). Alterations in the gut microbiome were demonstrated in the different forms of MS, where the short-chain fatty acids producing bacteria reduced in RRMS, while bacteria associated with excessive DNA oxidation were found in SPMS (65).
Studies have demonstrated that changes in the gut microbiome could affect lymphocyte activation and differentiation. It was demonstrated that MS gut microbiota could inhibit the interaction between T-cell C-C chemokine receptor type 9 (CCR9) and its ligand chemokine (CCL25) leading to a reduction of CCR9+ CD4+ T cell counts in circulation (68). A correlation has been found between a high frequency of Th17 and increased Firmicutes/Bacteroidetes ratio (69) which was evident by an increased Streptococcus and decreased Prevotella species number. The authors state that these patients had higher MS activity. In another study, a reduced number of Clostridia species, producing short-chain fatty acids was found in PPMS (70). Using EAE model of MS, the therapeutic potency of short-chain fatty acids was demonstrated which was linked to the development of Tregs (71). Dysbacteriosis characterized by increased Methanobrevibacter and Akkermansia species combined with decreased Butyricimonas species was reported in MS (70). Supporting the role of Methanobrevibacter in MS pathogenesis was finding the reduced duration of relapse in patients with higher of this species (65). Also, Akkermansia species could contribute to MS pathogenesis by promoting inflammatory response by blood leukocytes (72).
These data suggest that MS is characterized by a change in gut microbiome, which could contribute to a reduction of Tregs, while promoting the development of pathogenic Th17 lymphocytes. Also, dysbacteriosis could support inflammation and facilitate the differentiation of lymphocytes with proinflammatory activity.
Smoking was identified as an MS risk factor in many studies (73–75). Manouchehrinia et al’s study confirmed smoking as a risk factor by demonstrating that “ever-smokers” had a 41% higher chance of MS diagnosis as compared to “never smokers” (75). Smoking could affect the immune system function by inducing oxidative stress, the release of pro-inflammatory cytokines, and increasing nitric oxide (76–78). Cigarette smoking could contribute to MS pathogenesis by affecting the differentiation and activation of the different lymphocyte populations. It was demonstrated that smoke could trigger the development of auto-aggressive T cells, cross-reactive with CNS antigens in MS (79). Proinflammatory T cells producing IL-17 were increased in MS smokers (80). A study by Hedstrom et al. had shown a 41% risk of having MS in subjects having HLA-DRB1*15 without HLA-A*02 (81). Smoking could also have epigenetic mechanisms contributing to MS pathogenesis by affecting DNA methylation (82).
These data indicate multiple mechanisms of cigarette smoking contributing to MS pathogenesis. It could be suggested that smoking contributes to MS pathogenesis by the combined effect of activation of pro-inflammatory leukocytes, epigenetic modification, and genetic markers of predisposition to the disease.
The sex of the patient contributes to the risk of MS, due to a reported 3:1 female to male ratio (83). It appears that puberty, early onset of menarche in particular contributes to the risk of MS diagnosis (84). It remains unclear whether this is a result of the rising level of sex hormones. However, evidence suggests that female sex hormones could contribute to the progression of the disease. This assumption is supported by reports showing amelioration of MS symptoms during pregnancy (85). Also, a reduction of disability levels during pregnancy in MS was reported (86), although, lack of long-term effect on disability scores was also documented in other studies (85, 87).
Although the sex of the patient appears to be a risk factor, there is still a lack of strong evidence establishing sex hormones as contributing to onset of MS pathogenesis. Therefore, it could be suggested that sex could contribute to MS when combined with other risk factors. Supporting this assumption is the study by Irizar et al. demonstrating the association between female sex and HLA-DRB1*15:01 haplotype (88). A combination of HLA-DRB1*15:01, an established MS risk factor, with female sex was also demonstrated by Chao et al. (89). The authors also found that this haplotype is more likely to be transmitted from a mother to a girl rather than to a boy.
Female sex hormones were shown to modulate the immune response by promoting the development of Tregs, reducing Th1 and Th17 cells activity, as well as supporting Th2 lymphocytes differentiation (90–92). The anti-inflammatory effect of estrogens was also confirmed using EAE, an MS model (93, 94).
Childhood obesity was reported as a risk factor for MS (95, 96). It appears that having a high body mass index (BMI) in childhood could increase the probability of MS diagnosis (96). The odds ratio for MS diagnosis increased as BMI increased reaching 3.76 in extremely obese individuals (65). Interestingly, obesity was significantly associated with MS diagnosis in girls, while it was not found in boys (95). In another study, obesity was demonstrated as a risk factor during adolescence (97), while it had a limited contribution in children. These differences could be attributed to using different approaches, BMI or body silhouettes, to assess obesity (96, 98). Still, childhood obesity is believed to contribute to MS diagnosis, as Ascherio et al. suggested reduction of childhood obesity could reduce MS diagnosis by15% (99).
Chronic inflammation is commonly found in obese patients (100). Activation of inflammatory cytokines such as IFN-γ and TNF-α was demonstrated in obese individuals (101). This inflammation could be promoted by a disturbed gut microbiome, which is often found in this group of patients (102). This gut dysbiosis could shift the Treg/Th17 ratio toward pathogenic Th17 population (103), which has been linked to pathogenesis of MS (104). Association between obesity and low levels of vitD, another risk factor of MS was found (46, 105, 106). These data suggest that the higher risk of MS could be the combined result of many risk factors, such as inflammation, Th17 activation, and low vitD levels induced by obesity.
Several hypotheses were developed to explain the pathogenesis of MS, including the role of viral infection and genetic predisposition, which are studied more extensively. Although the trigger of MS remains unknown, the consensus is that pathogenesis is based on the activation of auto-aggression against myelin proteins, causing defects in the CNS structure. These myelin proteins form a multilamellar myelin sheath around the axons and neuron cell bodies. The sheath is composed of several proteins such as MBP, myelin-associated glycoprotein (MAG) and proteolipid protein (107). Also, a myelin oligodendrocyte glycoprotein (MOG) is located on the surface of the myelin sheath (108) and functions as an adhesive receptor as well as connects neighbouring myelinated fibers (109).
Abnormalities in immune mechanisms were suggested as protagonists in pathogenies of MS. This assumption is based on finding of the reduced number and activity of circulating T-regulatory (Tregs) cells, which correlated with an exacerbation of the disease symptoms (110, 111). Tregs counts were shown to be reduced during relapse, and restored during remission in MS patients (111). The role of immune mechanisms in the pathogenesis of MS was also confirmed by histological studies, indicating the presence of leukocyte infiltrate in the plaques within the CNS (112, 113). One of the most substantial pieces of evidence supporting the immune mechanisms of the disease is the detection of antibodies and leukocyte clones specific to brain antigens, among which the MBP appears the most immunogenic (114, 115). However, the mechanism of the autoimmune response activation in MS remains unclear.
There is substantial clinical and experimental data identifying MOG as a target for auto-reactive leukocytes in MS. This assumption is supported by detection of anti-MOG antibodies in some MS patients (116, 117), indicating the development of auto-reactive B cells. An increased count of MOG-reactive T cells in blood and cerebrospinal fluid (CSF) of MS patients was reported (117). The role of MOG in MS pathogenesis was also confirmed in the experimental autoimmune encephalitis (EAE) mouse model, where this glycoprotein was shown as inducing an encephalitogenic T cell response and antibody-mediated demyelination (118). Augmentation of demyelination in EAE by monoclonal antibodies directed against MOG further supports the pathogenic role of this protein in brain demyelination (119). Some of the strongest evidence for MOG contribution to MS pathogenesis comes from the clinical study by Berger et al. with detection of anti-MOG and anti-MBP IgM, predicting the conversion of a clinically isolated syndrome, a fits episode of neurological symptoms prior the disease, to MS (116).
In addition to MOG, studies have demonstrated the presence of circulating antibodies against MBP in MS patients (114, 115). Accumulation of T- lymphocytes specific for MBP was also demonstrated in the CNS (120) and CSF (18). Additionally, studies showed prolonged circulation of MBP specific lymphocytes in the blood of MS patients (18). The role of MBP-specific lymphocytes in the pathogenesis of MS was demonstrated in the EAE model, where the transfer of lymphocytes specific for this myelin protein induced symptoms similar to those of MS (121). Phenotype studies have identified a population of myelin-specific lymphocytes as effector cells or memory cells (122, 123).
These auto-aggressive T lymphocytes could cross the blood-brain barrier (BBB) and initiate myelin destruction in the CNS. BBB is formed by tightly connected endothelial cells located at the luminal surface of the brain blood vessels (124). In addition to the endothelial cells, astrocytes, a neuroglia cells, maintain the integrity of BBB (125) by locating between the endothelial cells and neurons. Astrocytes form a perivascular end-feet at the BBB controlling the movement of molecules between brain and blood (126). This barrier function of BBB was shown to be altered in many neuroinflammatory diseases, including MS (127). It is believed that the disruption of the BBB integrity is one of the critical initial steps in MS pathogenesis, which is required for auto-reactive T cells infiltration of CNS (125). The mechanism of T lymphocyte infiltration could be explained by an increased expression of adhesion molecules on BBB endothelial cells in MS (128). The role of adhesion molecules in leucocyte trans-BBB migration was also supported by their expression on inflammatory cells such as macrophages and lymphocytes in MS lesions (129, 130).
Once the BBB integrity is disrupted in MS, it could lead to CNS infiltration with activated lymphocytes, which is facilitated by an increased expression of adhesion molecules on endothelial cells, and inflammatory cytokines (131). CD4+T lymphocytes play a key role in initiating and maintaining the autoimmune response in MS (131). Studies have also shown the essential role of CD8+ T cells in maintaining the myelin damage and inflammation. It is generally accepted that cytokines secreted by Th1 cells, such as IFN-γ and TNF-β, can activate macrophages to damage oligodendrocytes, resulting in pathological myelination (132). HLA-E-restricted CD8+ Tregs induced by IFN-γ, could reduce CD4+T lymphocyte counts and induce secretion TGF-β by local cells (133). Studies have shown that inhibiting Th1 and/or Th17 cells and increasing the number of circulating Tregs could suppress the progression of MS (134). CD8+T cells rather that CD4+ lymphocytes were found more characteristic in MS plaques (135).
MS pathogenesis is explained by leukocytes, mainly T and B lymphocytes, infiltration of the brain and spinal cord (136). It was suggested that the inflammation in the CNS selectively recruits autoreactive T cells which could target the autoantigens in brain tissue (26). It is believed that the autologous myelin reactive T cells are initially primed to CNS autoantigens in the periphery and then cross the BBB entering the brain (137). Within the brain tissue, these leukocytes could activate microglia and macrophages, promoting local inflammation (138) (Figure 1). It appears that the lymphocyte distribution is population specific where CD8+ T cells are mostly found at the edge, while CD4+ T cells are located deep within the lesions (139). Also, their role in the diseases’ stage was demonstrated as CD8+ T cells were frequently found in acute lesions (140). In addition to the autoreactive lymphocyte’s migration across the BBB, the reduced regulatory T cell function was shown to promote autoimmune response in MS (141). The hyperactivation of Th1 cells could lead to tissue damage and chronic inflammation (142), which is frequently found in autoimmune diseases (143). Th17 cells are known for secreting high levels of interleukin (IL) 17A, IL-17F, IL-21 and for low production of interferon γ (IFN-γ) (144). Cytokines, such as transforming growth factor β (TGF-β), IL-6, IL-1β and IL-23, are implicated in Th17 development and maintenance (145). In another study, the high frequency of Th1, Th17 and granulocyte-macrophage colony-stimulating factor (GM-CSF)-secreting effector T cells have been reported in MS (146). Together, these cells could cause the death of myelin-producing oligodendrocytes, directly damage the myelin sheath of the nerve fibers, and impact brain tissue integrity forming lesions in the CNS.
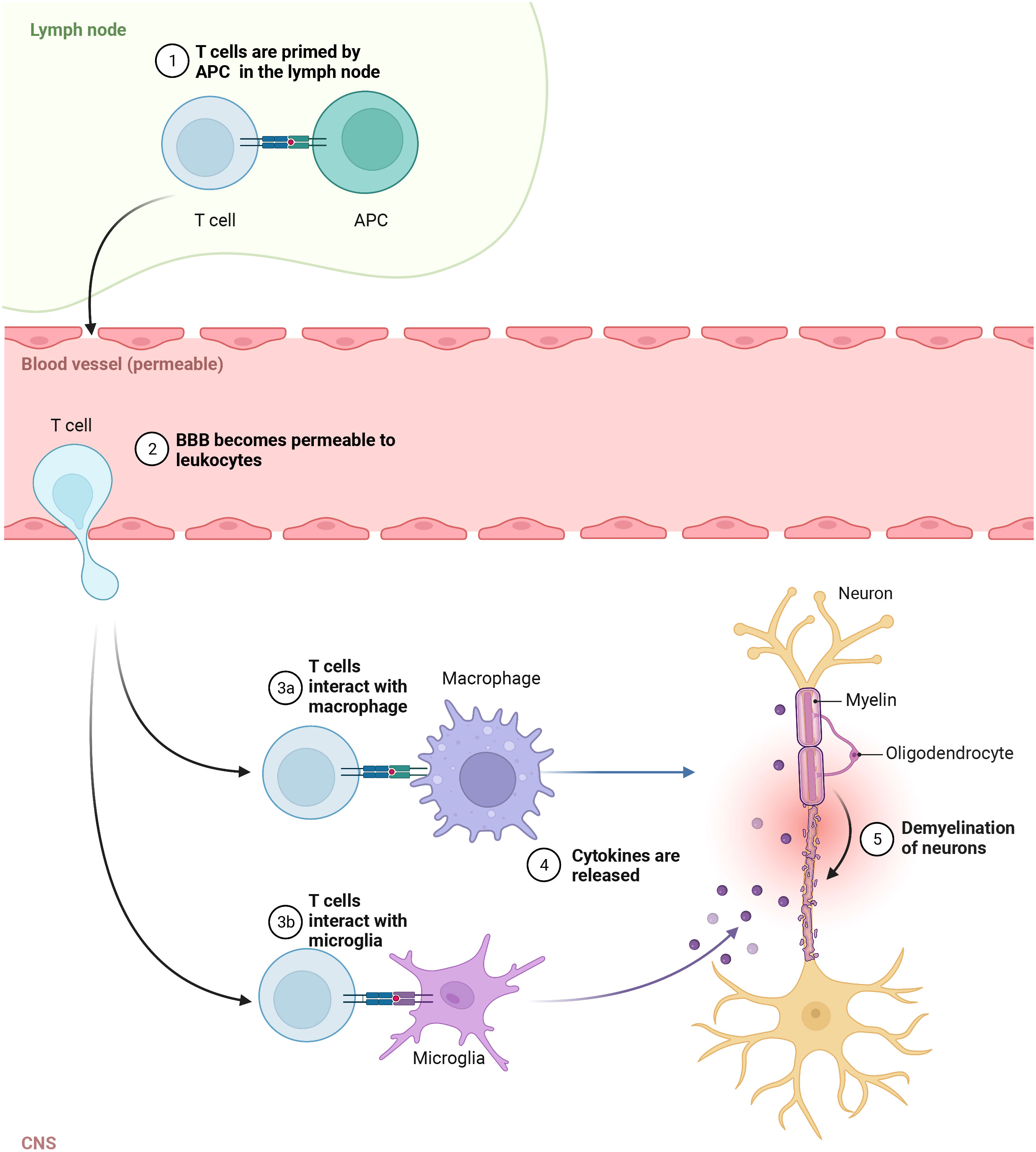
Figure 1 Autoreactive T cells in pathogenesis of MS. In the lymph node, T cells are primed to CNS autoantigens presented by APC, then cross the Blood brain barrier (BBB) and enter the brain. In CNS, they activate macrophages and microglia, which release cytokines promoting local inflammation and tissue injury.
The key role of CD4+ T cells was demonstrated when activated myelin-reactive CD4+ T cells were found in the blood and CSF of MS patients (12). In contrast, only non-activated myelin-reactive T cells were detected in controls. Study of CD4+ cells contribution to MS pathogenesis identified the HLA-DR15 allele as an antigen-presenting structure and epitope source to autoreactive CD4+ T cells (147). The authors identified the autoreactive CD4+ T cell clones that can cross-react with DR2a and DR2b-derived self-peptides, as well as peptides from foreign agents shown as potential cause of MS (147). CD4+ T cells could coordinate the adaptive immune response by releasing cytokines and chemokines supporting the activation of myelin-specific CD4+ T cells and their infiltration of the brain (10) (Figure 2).
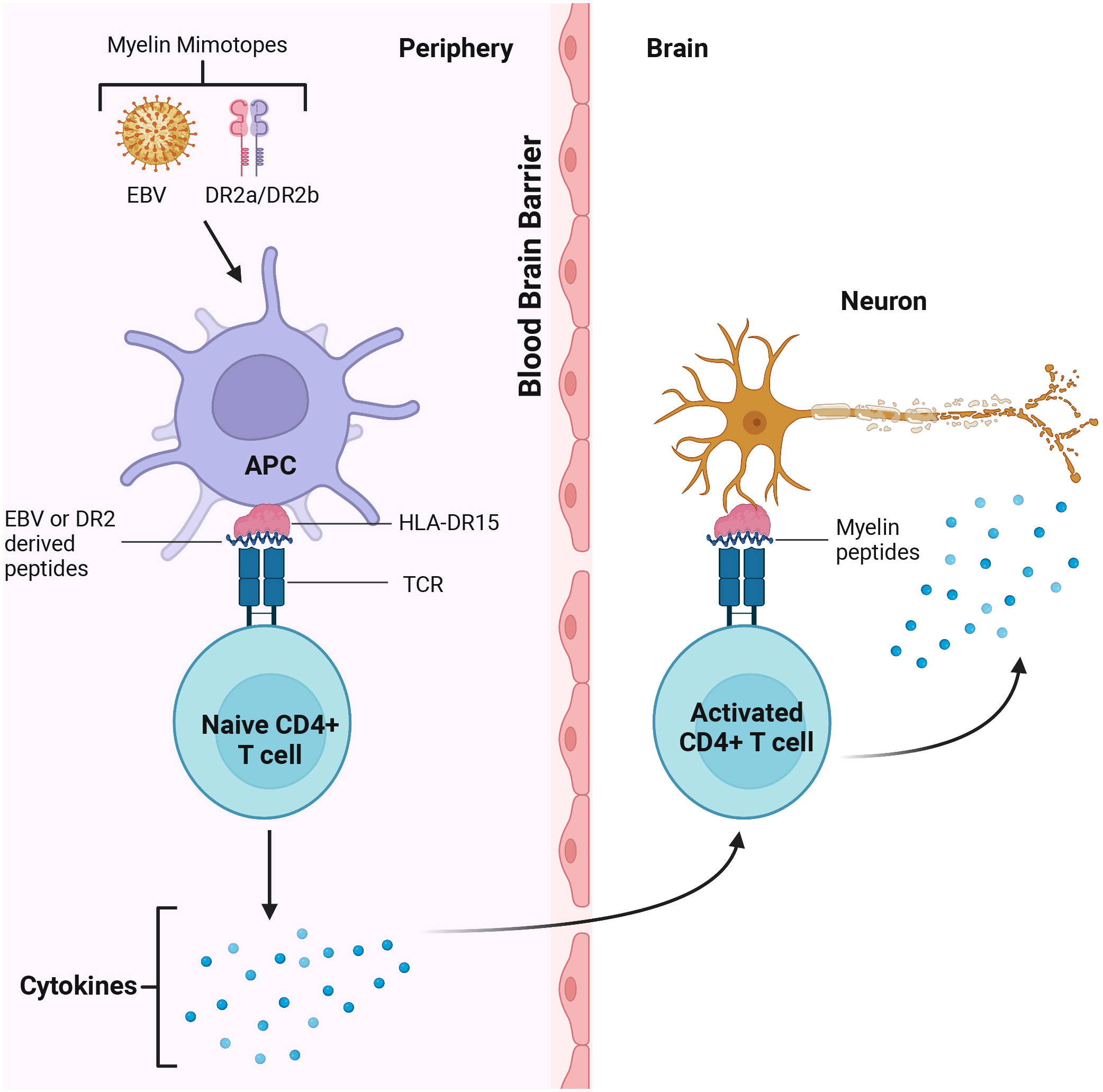
Figure 2 CD4+T cells cross react with Epstein Barr Virus (EBV) and DR2 derived peptides in the periphery and myelin in the brain. Myelin mimotopes could induce autoreactive CD4+ T cells by presenting EBV and DR2a/DR2b derived peptides sharing similarities with CNS self-antigens (i.e. myelin). These T cells could recognize the target antigen on microglia, leading to activation of CD4+ T cells. Activated T cells release cytokines which could trigger demyelination and axon loss, CNS inflammation and tissue injury.
CD4+ T cells could differentiate into distinct Th cell populations, identified by the repertoire of the surface receptors and the cytokines secreted (148). To date, several Th cell populations are recognized and characterized by secretion of the lineage-defining cytokines (149). Among these, Th1 and Th17 cell counts were shown to be increased at the onset of MS (150). Secretion of IFN-γ is characteristic for Th1 cells, which are essential to mediate T-cell immune response (151). Th17 cells are characterized by expression of retinoic acid-related orphan receptor gamma t (RORγt) as well as a signal transducer and activator of transcription 3 (STAT3) factor regulating the production of pro-inflammatory cytokines IL-17, IL-6, IL-21, IL-22, IL-23 and tumor necrosis factor α (TNF-α) (152). Another subset of lymphocytes, named Th9 producing IL-9, was shown as involved in regulation of the balance between Th17 and Tregs (153, 154). An additional population of Th lymphocytes was identified by Galli et al. which was characterized by secretion of GM-CSF and expression of CXCR4 in relapsing-remitting MS (RRMS) (146, 151).
High Th17 cell counts and elevated IL-17 mRNA transcription in the blood of MS patients were found correlated with the severity of the disease (155). Large numbers of Th17 cells were found in CSF at the early stage of MS (156). These cells were characterized by high-level expression of very late antigen-4 (VLA-4), a surface adhesion molecule for CD4+T cells, directing their migration across the BBB (156). Therefore, it was suggested that the inhibition of VLA-4 expression can reduce the CD4+ T cells migration to CNS and limit CNS inflammation (156). Th17 cells have multifactorial contribution to MS pathogenesis. These cells could produce glutamate upon contacting with oligodendrocytes in CNS (11, 157). This high glutamate level could cause damage to oligodendrocytes, exposing the myelin antigens as a target to autoreactive lymphocytes (157). Th17 could contribute to chronic CNS inflammation (151). It was shown that Th17.1 subpopulation, expressing the highest level of IL-23 receptor and granzyme B, has a greater capacity to migrate through the BBB (156). Upon stimulation with IL-23, these IL-23R+ Th17 lymphocytes could produce GM-CSF, which could change their phenotype to be more encephalitogenic and support CNS inflammation (158). This is consistent with findings in EAE model, where Th17 cells, maintained in the presence of IL-23, became cytotoxic to oligodendrocytes and neurons (156). Similarly, upregulation of GM-CSF and more encephalitogenic phenotype of Th17 was demonstrated upon exposure these cells to IL-1β (158), a proinflammatory cytokine product of inflammasome (159).
IL-9-producing CD4+ Th cells, Th9, are the most recently discovered lymphocyte subset (154). These cells are characterized by a substantial production of IL-9, as well as IL-10 and IL-21 (160). Initially identified as a sub-lineage of Th2 cells, they require IL-4 for differentiation (161). However, unlike Th2, TGF-β was shown critical for Th9 differentiation (162). The role of Th9 in the pathogenesis of MS was suggested as the IL-9 levels in CSF correlated inversely with indexes of inflammatory activity, neurodegeneration, and progression disability (163). These data suggest that Th9 product, IL-9, could have anti-inflammatory effect in MS, consequently promoting remission. Supporting this statement were findings using the EAE model. It was demonstrated that mice with EAE receiving Th9 cell had fewer lymphocyte infiltration in the meninges as compared to those receiving Th1 and Th17 cells (164). Protective role of IL-9 was also shown by using IL-9R knockout mice, which had a severe course of EAE (165). Interestingly, it appears that TGF-β could support the differentiation of Th9 as well as non-pathogenic Th17 (166). These, non-pathogenic Th17 were also shown to produce IL-9 (166). These data suggest that IL-9 produced by Th9 and non-pathogenic Th17 could be a potential therapeutic target for treatment of MS.
An expanded Th cell subset was identified in CSF of RRMS patients, which was characterized by expression of GM-CSF and CXCR4 (146). CXCR4 could promote migration of lymphocyte subsets with encephalitogenic capacity across BBB in MS. Supporting the role of these cells in pathogenesis of MS was a finding in twins expressing significantly higher amounts of VLA-4, a CXCR4 ligand, associated with an increased lymphocyte trafficking to the CNS (167). GM-CSF and CXCR4 expressing cells were increased in MS as compared to other inflammatory and non-inflammatory conditions (146). The number of these cells was also reduced under the effective disease-modifying therapy by using dimethyl fumarate (DMF). These data indicate that these cells could represent a specific therapeutic target in MS. This statement was supported by the fact that knocking out GM-CSF substantially decreased the disease severity, which was associated with lower lymphocyte infiltration and inflammation in CNS (168).
Tregs have an immune-modulating function by the release of IL-10 and TGF-β (169). Multiple types of Tregs were shown to contribute the maintenance of the immune homoeostasis, by keeping autoreactive T and B cells from developing into the auto-aggressive type (Figure 3). In MS, the aberrant activity of effector T cells could be a result of reduced Treg function (170, 171). This assumption of insufficient Tregs function is supported by decreased counts of resting population of this lymphocytes in MS as compared to controls (172). Also, T cells appear resistant to regulation by Tregs in MS. This deficiency could lead to differentiation of pathogenic lymphocytes in MS (110, 173).Therefore, these population of T cells could be a potential target for MS (174). Support for this is found in approved disease-modifying drug, such as IFN-β for MS increased Treg cell count, which could explain the mechanism of their therapeutic efficacy (174, 175).
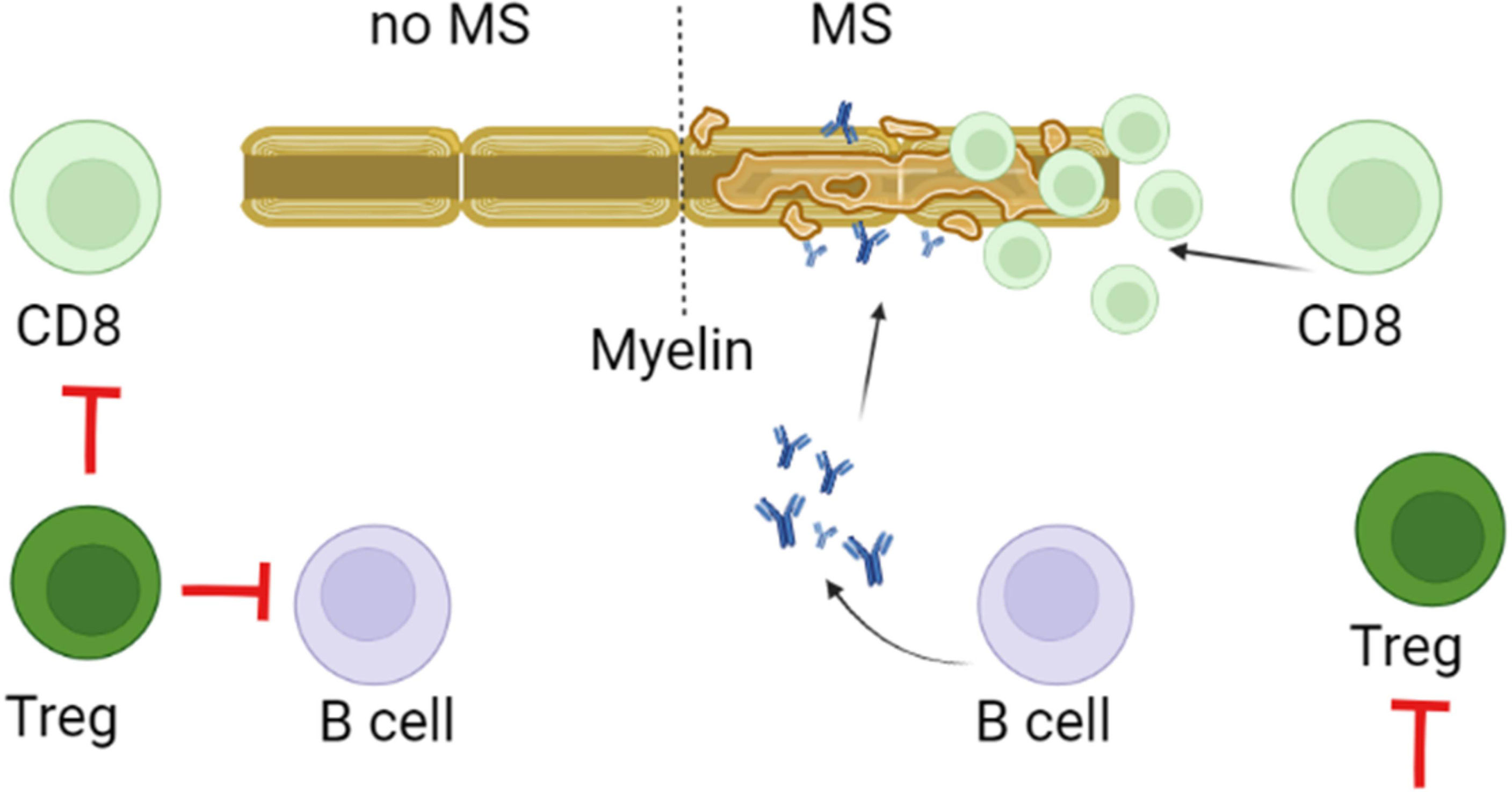
Figure 3 Role of Tregs in pathogenesis of MS. Tregs control of the immune homeostasis focused on preventing differentiation, proliferation and survival of autoreactive myelin targeting CD8+, Th17 and B cells. This control depends on the direct contact as well as the release of IL-10 and TGF-β by Tregs. It is believed that in MS, decreased activity and lower counts could reduce the regulatory capacity of Tregs. This could promote survival, proliferation and differentiation of myelin reactive CD8, Th17 and B cells.
Since myelin-reactive T cells are found in healthy individuals, it was suggested that MS pathogenesis could be associated with insufficient immune regulation by Tregs (176). Tregs could suppress the activity of Th1 and Th17 effector cells (Teff), cytotoxic CD8+ T cells (Tc), and antigen-presenting cells (APCs) by direct contact and secretion of suppressive cytokines (177). By presenting myelin, APCs could trigger activation and differentiation of pathogenic Th1 and Th17 cells, releasing pro-inflammatory cytokines such as IFN-γ, TNF-α and IL-17 (178). The encephalitic lymphocyte activation could be suppressed by Tregs which inhibits the inflammatory response by reducing pro-inflammatory cytokine release and by promoting T cell anergy or apoptosis (177). However, the functionally defective Tregs could contribute to the pathogenesis of MS (176). There are two Tregs subpopulations, that express either CD4 or CD8 markers (179, 180). CD4+Tregs express the cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) which contributes to inhibition of B and T cells proliferation and APCs function (181–183). The CD8+Tregs are characterized by the ability to suppress activation and proliferation of autoreactive CD4+ effector cells by cell-to-cell contact (184). Several immunosuppressive markers such as CD25, CTLA-4, PD-1, GITR, and transcription factors FoxP3 and Helios were found on both CD8+ and CD4+ Tregs (185). It was shown that Helios could stabilize the suppressive phenotype of both CD4+ Tregs and CD8+ Tregs, by activating the STAT5 signalling pathway (186). Interestingly, the expression of Helios in RRMS was shown to be lower as compared to controls (186). These data suggest that these Tregs in MS could have an unstable phenotype and, when migrated to CNS lesions having inflammatory milieu, they fail to continue maintaining immunoregulatory function (187).
Studies have shown that the MBP-specific CD8+ T cells could exacerbate brain inflammation (18, 188). The encephalitogenic effect of myelin-specific CD8+ T cells in MS could involve the FasL dependent mechanisms, especially those promoting the formation of intracerebellar lesions (188). CD8+ T cells are the primary lymphocytes found in the CNS brain lesions of MS patients and mice with EAE (18). Supporting the role of CD8+ T cells in MS pathogenesis, was a finding that the adoptive transfer of CD8+ enriched MOG-specific T cells could induce EAE in mice (189). In another study, an increased number of circulating CD8+ CD20+ T memory cells, specific to myelin antigens was found in MS as compared to control subjects. The expression of CD20 on these T cells correlated with up-regulation of activation markers, pro-inflammatory cytokines and adhesion molecules, indicating that they could have high pathogenic potential. This assumption was supported by experiments with an adoptive transfer of CD20+ T cells in EAE mice, which caused the deterioration of the brain tissue integrity and aggravation of the disease severity (190). The proportion of memory and CD20+CD8+ T cells, specific for myelin antigens, was significantly reduced following anti-CD20 treatment, indicating that the depletion of CD20+ T cells has therapeutic potential in MS (190).
Originally, MS was believed to be a primarily T-cell mediated disease with an imbalance of pro- and anti-inflammatory cells in CNS inflammation. However, more evidence suggests that B cells are also involved in the pathogenesis of disease. Though not completely clear, but both, antibody-dependent and independent mechanisms, are now proposed as mechanisms of B-cell mediated CNS injury in MS (188). Aberrant stimulation of B cells and plasma cells are suggested in MS (191). These cells could also inflict damage by producing autoantibodies against specific myelin antigens. They could produce autoantibodies affecting the course of MS (192). In the EAE model, B cells were shown to interact with CD4+ T cells and initiate an adaptive immune response to myelin antigens. Reduced inflammation and alleviated clinical scores were shown in rats with EAE after inhibiting B cell immunity (193, 194). These data suggest that B cells could be used as therapeutic targets for MS.
The direct evidence of the role of B cells in immunity comes from histology analysis of the CNS. Lymphatic follicle-like aggregates containing B cells were shown in meninges of MS patients, similar to the secondary lymphatic tissue developed at sites of chronic inflammation (195). The role of these aggregates in MS pathogenesis is supported by finding their association with more severe neuropathology and clinical disease. These data identified the CNS location of auto-reactive B cells in MS, which is the important for our understanding of the disease pathogenesis as well as it provides the site for the potential therapeutic targeting. Significant B cell infiltration is often found along the small blood vessels at the site of myelin destruction, suggesting the continuous production of mature B cells in CNS causing local humoral immune response (196). This sustained proliferation of B cells producing anti-myelin antibodies could cause damage to the cerebral cortex and exacerbate the progression of MS. Recently, B-cell depletion with anti-CD20 monoclonal antibodies was demonstrated as highly effective for treatment of all forms of MS (197–200). The depletion of B cells suppressed the inflammation and reduced the MS relapses, suggesting that B cells play a central role in the pathogenesis of MS.
Additionally, B cells could produce cytokines contributing to MS pathogenesis. By producing IL-6, B cells could bias towards differentiation of pathogenic Th cells capable of triggering EAE (201, 202). This IL-6 derived pathogenic Th lymphocytes were linked to expression of Fc fragment of IgM receptor (FCMR) gene in Th17.1 population (203). The surface receptor FCMR was identified as unique regulator of inflammatory autoimmune responses and an attractive target for therapeutic intervention. Inhibition of FCMR activity in vivo at an early or late stages of EAE could prevent the disease progression (204). Additionally, B cells could produce GM-CSF, a cytokine linked to the pathogenesis of MS (145, 205).
The role of regulatory Bregs in pathogenesis of MS is extensively explored in the last two decades. Bregs can mediate immune tolerance and inhibit inflammation by producing IL-10 and IL-35 (206). IL-10 is anti-inflammatory cytokine which can suppress the development of Th1 and enhance polarization of Th2 lymphocytes (207, 208). Similar to IL-10, IL-35 has strong anti-inflammatory activity by inhibiting differentiation of pathogenic Th1 and Th17 as well as promoting development of Bregs and Tregs (209, 210). In addition to IL-10 and IL-35, Bregs could secrete TGF-β, thus, suppressing Th1 proliferation while promoting Treg cells differentiation (211). The role of Bregs in pathogenesis of MS was demonstrated in a study, where the reduction of the naïve/memory Bregs was found during a relapse (212). Further, Kim et al. demonstrated reduced counts of immature and highly regulatory Bregs in MS (213). These data support the notion that Bregs could be a novel target for MS therapy. The study has shown that treatment with alemtuzumab, an anti-CD52 monoclonal antibody, increased these lymphocyte counts, which correlated with the efficacy of MS therapy (214). Similar results were presented by Zhang et al. confirming the efficacy of alemtuzumab treatment which was linked to reconstitution of these lymphocyte subset population (215).
Studies suggest that EBV could be one of the triggers of MS (215). EBV infection also has a connection to B cells as it could establish latent infection in memory B cells (216). Infection of naïve and mature B cells with EBV could happen via CD21, CD35 and HLA-DR, which contribute to the viral load and the expansion of B cells (217). EBV produces latent member protein 1 (LMP1), which can promote the survival and immortalization of B cells (56). These LMP1 expressing B cells are often found in the brain of MS (56). Therefore, it was suggested that the increased prevalence and the association between EBV and MS could be a consequence of chronic B cell activation in these patients (218).
In conclusion, it appears that B cells are involved in the MS pathogenesis through (I) presenting antigenic peptides to T cells and driving the self-proliferation of brain homing T cells (possibly through memory B cells) (219), (II) producing regulatory cytokines and chemokines and contributing to differentiation of lymphocyte subpopulations and (III) serving as a site for EBV infection.
Cytokines are small molecules with pleiotropic function enabling cell communication in health and in pathology. Cytokines could define the resolution of the pathological process by contributing to the recovery and development of the immune response. However, in some cases, they can support the destruction of the host tissue, promoting the aberrant immune response. The role of cytokines in pathogenesis of MS was demonstrated in multiple studies identifying molecules contributing to the development of inflammation, auto-aggressive T lymphocytes and nervous tissue damage (220–224). Cytokines could contribute to the pathogenesis of chronic inflammation in MS as well as orchestrating leukocytes trans-BBB migration.
The current paradigm of MS pathogenesis is explained by activation of Th1 and Th17 lymphocytes (225). Activation of these lymphocyte populations is managed by a specific set of cytokines. The Th1 cells differentiation is regulated by IFN-γ, IL-2 and TNF-α (150, 226). Also, activated Th1 can secrete IFN-γ and TNF-α, which maintain the inflammatory milieu and provide positive feedback to support differentiation of naïve T cells to Th1 (227). These Th1 lymphocytes assist differentiation of cytotoxic T lymphocytes (CTLs), playing a central role in myelin disintegration (228). There are vast numbers of data supporting the role of IFN-γ and TNF-α in pathogenesis of MS. These data demonstrated that IFN-γ is expressed in MS lesions (222, 223, 226, 229). Also, it appears that the overexpression of IFN-γ in the CNS could contribute to demyelination by opposing the remyelination (230). Similar to IFN-γ, increased serum and CSF levels of TNF-α in MS patients correlated with the disease severity and progression (231–233). Additionally, the exacerbation of the disease symptoms was shown to correlate with an increased serum level of this cytokine (234). Demyelination appears to depend on TNF-α expression as it correlates with the loss of myelin in grey matter at the time of diagnosis and postmortem (235).
Th17 lymphocytes differentiation is tightly regulated and requires TGF-β and IL-6 (236). Cytokines IL-21 and IL-23 support the proliferation and stabilization of Th17 lymphocyte population (145). In the absence of TGF-β, a cocktail of IL-6, IL-23 and IL-1β could control differentiation of Th17 (166). It was demonstrated that the exposure of Th17 lymphocytes to IL-23 results in conversion of non-pathogenic to pathogenic phenotype (145, 166). Increased serum level of IL-6, IL-23 and IL-1β was demonstrated in MS (145, 223, 237). Interestingly, reduced in serum, while increased in CFS was the level of TGF-β, a key regulator of Th17 differentiation (238, 239). This elevated TGF-β in CSF correlated with the disease relapse (240).
Studies have shown the Th17 lymphocyte plasticity, where they could shift to Th1 cells (238, 241). This was confirmed by finding Th17 phenotype lymphocyte population producing IL-17 as well as IFN-γ, a cytokine characteristic for Th1 cells (242). Similarly, Th17/Th1 phenotype of Th17 lymphocytes was reported by Cosmi et al. in patients with juvenile idiopathic arthritis (242). This ability of Th17 to convert to pathogenic Th1 lymphocytes was demonstrated using EAE model of MS (243). It appears that, depending on the cytokine microenvironment (243), Th17 could obtain the pathogenic features of Th1 lymphocytes, producing IFN-γ (244). The leading role in this switch is played by IL-12, IL-23 and IFN-γ (243).
Cytokines could also contribute to migration of autoreactive T and B cells across the BBB. Supporting this, TNF-α was shown to affect the distribution of tight junction and adherents junction molecules, holding the endothelial cells together and reducing BBB permeability (244). Also, a combination of TNF-α and IFN-γ could alter the architecture of the junction proteins. Additionally, these cytokines could increase the expression of adhesion molecules such as intercellular adhesion molecule 1 (ICAM), vascular cell adhesion molecule 1 (VCAM-1) and selectins, which are required for leukocytes trans-endothelial migration (245, 246). Similarly, IL-6 was shown to increase the VCAM-1 expression and leukocyte recruitment to the spinal cord. Cytokines, associated with Th17 lymphocyte activation also contribute to lymphocyte migration across the BBB. It was demonstrated that endothelial cells express IL-17R and IL-22R, promoting disruption of BBB integrity in MS patients (247). It appears that IL-17 promotes lymphocyte migration by increased expression of chemokine (C-C motif) ligand 2 (CCL2), IL-6 and chemokine (C-X-C motif) ligand 8 (CXCL8) by endothelial cells of BBB. Increased serum level of these chemokines was also found in MS patients (222).
These data suggest that cytokines play a role in pathogenesis of MS by: I. directing differentiation of T lymphocytes towards the auto-reactive Th1 and Th17 phenotype and II. disrupting the integrity of BBB and facilitating migration of these reactive leukocytes. Therefore, cytokines as well as autoreactive lymphocytes could be therapeutic targets for treatment of MS (Figure 4).
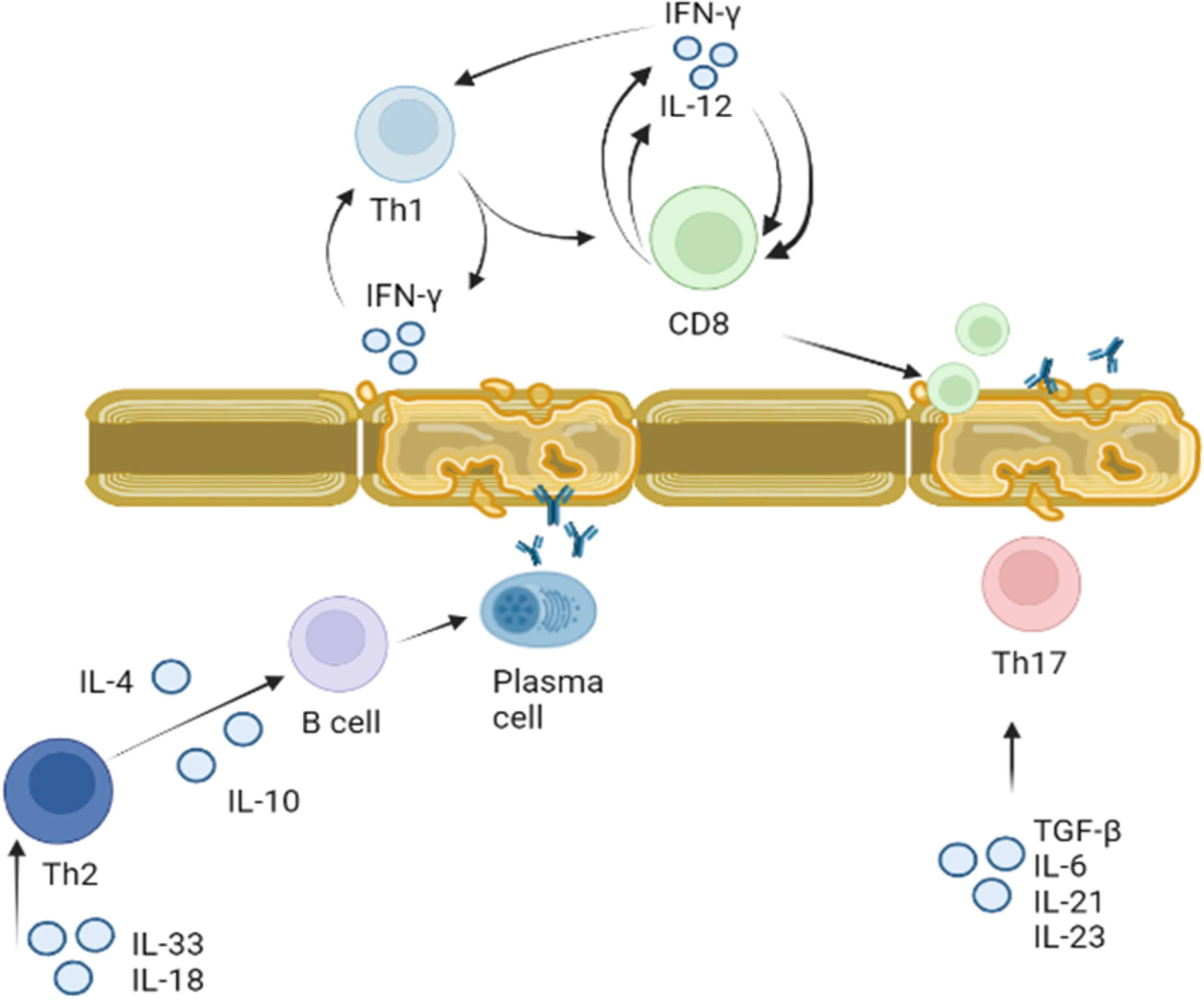
Figure 4 Role of cytokines in activation of T and B lymphocytes in MS. Activation of Th1 and Th2 lymphocytes was demonstrated in MS. Th1 lymphocyte differentiation is regulated by IFN-γ. Once Th1 cells are activated they produce more IFN-γ stimulating differentiation and proliferation of CD8 cells. CD8 cells also release IFN-γ, which together with Il-12 supports CD8 and Th1 proliferation. Activated myelin primed activated CD8 lymphocytes could damage myelin sheets. Activated by IL-18 and IL-33, Th2 lymphocytes secrete IL-4 and IL-10 which support proliferation and differentiation of B cells. Myelin antigen primed B cells could terminally differentiate to plasma cells producing anti-BMP antibodies targeting myelin sheets. Th17 lymphocytes proliferation and differentiation is supported by IL-6, IL-21, IL-23 and TGF-β. These activated Th17 lymphocytes could also target axon myelin sheets and contribute to the local inflammation.
MS remains life-lasting disease where its management is aimed to facilitate the recovery after the relapse, delay the disease progression and alleviation of symptoms (222, 248). Here, we summarize the treatments strategies with a focus on T-cell targeting. A schematic representation of the FDA approval for MS therapeutics is summarized in Figure 5.
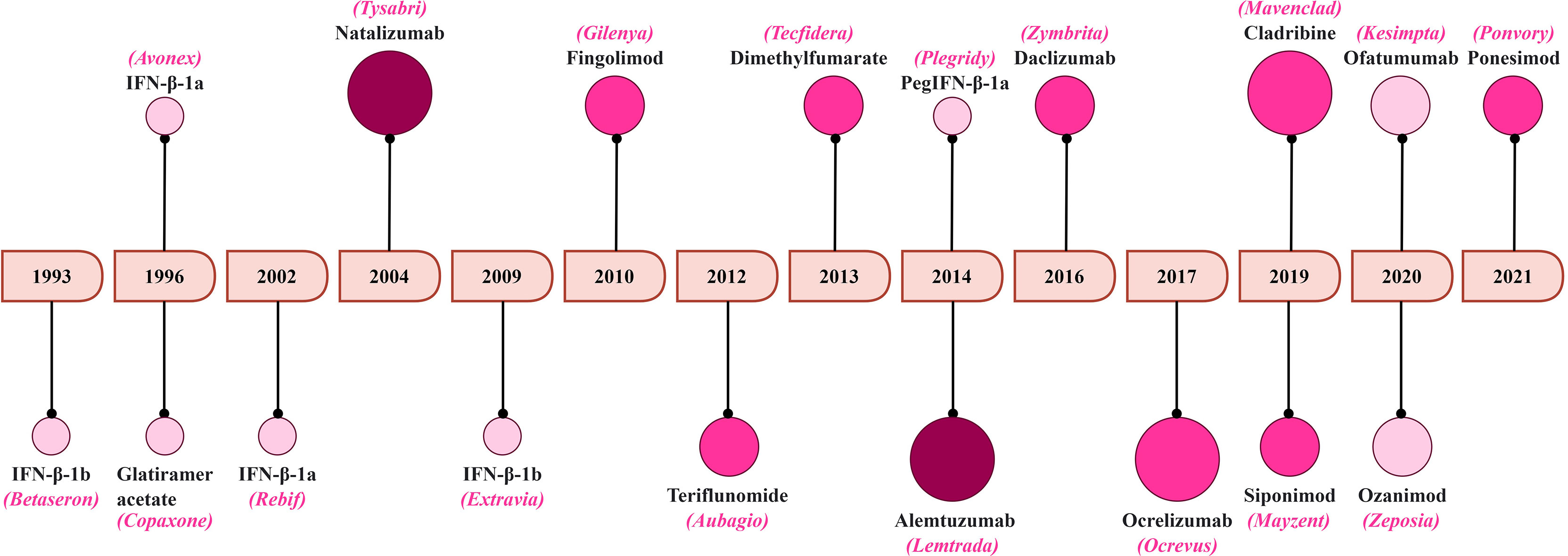
Figure 5 A timeline of FDA approval for MS therapeutics. The increasing color gradient in the circles indicates potential for higher toxicity, while the increasing size of the circles indicates higher efficacy.
DMTs such as IFN-β and glatiramer acetate, were amongst the first agents commonly prescribed to MS patients (248). Their therapeutic effects were explained by decreasing pro-inflammatory cytokine production, reducing formation of MRI lesions, inhibiting immune cell activation, and lowering matrix metalloproteinase activity (249–251). Three major IFN-β products were approved as the first line MS treatment based on the phase III clinical trial results (252). It was demonstrated that the mechanism of IFN-β therapeutic effect is based on the enhancement of the forkhead box P3 (FoxP3) expression in CD4+ and CD25+ Treg cells (253) and downregulation of HLA expression on antigen presenting cells (254). The side-effects, such as flu-like symptoms, elevated level of hepatic enzymes and injection-site reaction, are mild and could be managed by symptomatic treatment (255). Glatiramer acetate is an alternative to IFN-β, with similar therapeutic effects; however, it has aberrant effects such as dyspnea, palpitation, chest tightness and flushing (256, 257).
With our advances in the understanding the lymphocyte role in the pathogenesis of MS, the novel approaches were developed to correct the aberrant immune response and leukocyte infiltration in CNS. The research now focused on the identification of immune-modulating agents and monoclonal antibodies, with the ability to restrain lymphocyte diapedesis into the CNS through the BBB. Addressing this pressing need, the therapeutic potentials of S1P inhibitors in MS were explored. S1P is a sphingolipid that regulates lymphocyte migration and enhances T cell survival (258–260). Fingolimod was the first oral S1P1 modulator approved by the FDA in 2010 for the treatment of MS (248, 251). It is an anti-inflammatory drug with the mechanism of action based on degrading the sphingosine-1-phosphate (S1P) receptor on lymphocytes (250, 251), which is required for their migration to the CNS (248). Phase III clinical trial data indicate higher efficacy of fingolimod in reduction of the relapse rate as compared to IFN-β (261). This drug has multiple side effects including gastroenteritis, back pain, lymphopenia, macular edema and, possibly, bradycardia and atrioventricular blockage. Therefore, it is recommended only for treatment of patients with no prior infection history. Siponimod (also known as BAF312) and ozanimod are also S1P1 modulating agents that displayed therapeutic effects by selectively inhibiting S1P in phase II clinical trials (262, 263). Ponesimod, a selective S1P1 modulator, was tested in double-blind, placebo-controlled trial demonstrating a significant reduction of T1 brain lesions and delayed relapses (264). Later, a phase III clinical trial showed a higher efficacy of ponesimod as compared to teriflunomide on MS activity (265). Ponesimod was well tolerated without safety concerns for MS patients.
A group of monoclonal antibody-based therapeutics were shown to be effective in treatment of MS. One of them is natalizumab, a humanized monoclonal antibody, demonstrated efficacy in Phase III trial by reducing annualized relapse rate, preventing lesion accumulation and decreasing progression of disability (266). The mechanism of therapeutic potency could be explained by prevention of lymphocyte adhesion to VCAM on endothelial cells and α4-integrin receptor on lymphocytes (267). This could block the migration of lymphocytes across the BBB and entering the CNS. Natalizumab also demonstrated better relapse reduction rates and longer-lasting effects as compared to IFN-β (268, 269). It should be noted that natalizumab has limitations such as induction of progressive multifocal leukoencephalopathy, high infusion-related adverse responses and ‘rebounding’ of the diseased-state upon treatment cessation (248, 270, 271).
Since B-cells play a central role in MS pathogenesis, the therapeutic efficacy of monoclonal antibodies targeting this lymphocyte population, such as rituximab, ocrelizumab and ofatumumab, was also assessed in clinical trials. The objective of targeting B-cells is to prevent their diapedesis across the BBB, decrease interaction with T cells as well as restrict pro-inflammatory cytokine production (272). Rituximab and ocrelizumab are widely used monoclonal antibodies, which target B-cell surface antigen CD20 (207, 230). Ocrelizumab selectively depletes B-cells, while preserving the ability for B-cell reconstitution (248). Ofatumumab also inhibits B cell activation and has a similar efficacy as that of ocrelizumab to control this lymphocytes (248, 251). The effectiveness of rituximab and ocrelizumab was demonstrated in phase II trials where decreased number of MRI lesions was found in MS patients (251, 273). Also, the therapeutic potency of ocrelizumab was demonstrated in a phase III trial where ~50% reduction in annual relapse rate was documented in MS (232). Studies have shown that rituximab could be used in patients suffering simultaneously with other autoimmune disorders (274).
Another monoclonal antibody-based therapeutic targeting T and B lymphocytes is alemtuzumab, which binds to CD52 (275). Expression of CD52 was shown on activated T lymphocytes, Tregs and B cells (276–278). It is believed that alemtuzumab depletes CD52 expressing lymphocytes by antibody-dependent cell-mediated cytolysis (ADCC) and complement-dependent cytolysis (CDC) (275). It should be noted that the lymphocyte population gradually becomes reconstituted where B cell counts reach the baseline within 6 months, while T cell counts reach a normal range within 12 months (279, 280). In clinical trials, alemtuzumab has demonstrated higher efficacy in reducing relapses as compared with IFNβ-1α and decreasing confirmed disability worsening (CDW) (281, 282).
Additionally, multiple immune-modulating agents are recommended for treatment of MS such as anti-inflammatory agents (283–285), diapedesis inhibitors (286), inhibitors of myelin degradation (249, 253), inhibitors of pro-inflammatory cytokines production (287, 288). Each of these therapeutics, however, have limitations or side effects. It was demonstrated that laquinimod could cause cardiovascular complications (289). Also, mitoxantrone was linked to the development of acute leukemia and hematologic side-effects (268, 290). Hepatotoxicity and teratogenic effects are shown as complications for teriflunomide therapy (248, 290), while hepatotoxicity and progressive multifocal leukoencephalopathy were described in patients treated with DMF (291).
A novel approach focused on targeting the autologous myelin-reactive T-cells vaccine (292) and administration of autologous, myelin-reactive thymic and Tr1 Tregs was tested in MS (293). Early TCV trials reported decreased counts of the pathogenic, MBP-specific T-cells, which was combined with low toxicity (294). However, failure to meet the primary and secondary study endpoints was reported in phase II clinical study using TCV in MS (295). In 2002, an open label study pointed at a need for multiple TCV doses to prevent relapse post first vaccination. Encouraging results were reported in 2004 during a TCV trial using MBP and MOG activated T-cells in 20 patients, having low therapeutic efficacy of the standard treatment options (294). In this study, the therapeutic effect was determined by the reduction of lesions count, relapse rate and neurological disability. Similar results were reported in phase I/II clinical trial with TCV in 19 subjects where reduced frequency of the relapse without side-effects was reported (296).
Some studying conducted in the last decade also confirmed safety and good tolerability of TCV. In 2012, Karussis et al. investigated the TCV vaccination efficacy in a blinded clinical trial where RPMS patients received multiple injections of attenuated T-cells reactive to 9 different myelin peptides (175). This study demonstrated TCV safety without serious adverse events. In 2016, Seledtsova et al. conducted a study by immunizing 39 patients with PPMS using multiple autologous polyclonal TCV (297). None of the vaccine-treated patients experienced any significant side effect during the entire follow-up period. The results suggest that polyclonal TCV is safe to use, able to induce measurable, long-lasting, anti-inflammatory immune effects in patients with advanced MS. Although TCV has demonstrated safety and limited side effects, still this information is coming from studying using small, selected groups of patients. Only single phase I/II clinical trial was conducted so far, while there was no phase III clinical trial completed. Therefore, the therapeutic efficacy of TCV remains under the investigation.
One of the novel approaches is based on targeting the lymphocyte populations shown as contributing to MS pathogenesis. One of these lymphocyte population is Tregs, known to control the expansion of auto-aggressive lymphocytes (298). Supporting the potential of these cells are therapeutic target was the effective relapse prevention, even when Tregs were administered post the onset of EAE (299). In another study, nitric oxide-induced CD4+CD25+FoxP3- Tregs were shown to suppress the migration of immune cells through BBB, decreased proliferation of Th17 and lowered secretion of IL-17 in EAE mice model (300). Recently, adoptive cell transfer therapy using engineered Tregs, expressing MOG specific-TCR, was used for treatment of EAE mice (301). Authors reported a positive outcome at both, the initial and the peak stages, in EAE. In another recent study, a phase I trial using CD4+CD25highCD127-FoxP3+ Tregs reported no adverse effects such as relapse of MS, enlarging T2 lesions or visual impairment progression, upon intrathecal administration (110). These reports support the potential of Tregs based cell-therapy in MS, though, further validation in large size cohorts MS patients is required.
There were several reports of the therapeutic potency of engineering antigen-specific Tregs such as a chimeric antigen receptor T cells or CAR-T cells, in MS. Fransson et al. used CD4+ T cells, expressing MOG-targeting CAR and FoxP3, to start treating EAE mice at the height of the clinical symptoms (302). Authors have reported an effective suppression of EAE symptoms which was coupled with reduced IL-12 and IFN-γ production. Also, repeated attempts to induce EAE failed to produce symptoms in treated mice, indicating development of a prolonged protection by Tregs (302). In another study, human Tregs were transduced with MBP-specific TCR and used for treatment of EAE (303). The transduced cells ameliorated EAE symptoms and upregulated the expression of several Tregs markers such as FoxP3, latency-associated peptide and Helios (303). The limiting factor of Treg treatment is that the engineering target-specific Tregs is challenging as there is little known about MS-specific autoantigens (304). Also, individual epitope variation could lead to changes in the target antigens, further compounding the engineering process (305).
Our understanding of the therapeutic mechanisms of drugs used for MS treatments remains largely unknown.
It was suggested that IFN-β could increase the production of anti-inflammatory cytokines, inhibit the secretion of pro-inflammatory IL-17 as well as reduce the migration of leukocytes across BBB (306). Following IFN-β administration, increased expression of Th2 (IL-10 and IL-4) and reduced Th1 (IFN-γ and TNF-α) cytokines in CD4+ T cells of MS patients were reported (307). Also, direct interaction between IFN-β and CD4+ T cells may result in regulation of IL-17 expression via type I IFN receptor-mediated activation of (STAT)-1 signaling pathway and suppression of STAT3 activity (308) Further, IFN-β could suppress IL-17 secretion by T cells via IFN-α/β receptor signaling (309). Additionally, IFN-β was shown to reduce the level of ICAM-1, which is essential for T cells binding to endothelial cells and crossing BBB (310).
The mechanism of Glatiramer acetate, another DMT drug, the therapeutic effect could also target the Th17 cell population by inhibiting the production of IL-17, IFN-γ and IL-6, cytokines required for differentiation of this subset of lymphocytes (311). Glatiramer acetate could promote the development of anti-inflammatory T cells, T-helper 2, and regulatory T cells (312, 313). It was suggested that these Th2 cells could be accumulated in the CNS, producing anti-inflammatory cytokines and reducing the encephalitic effect of pathogenic T cells (313). Glatiramer acetate has been shown to activate Foxp3 which promotes the development of CD4+CD25+ Tregs (311).
The therapeutic effect of phosphorylated fingolimod is based on its ability to bind to the S1P receptor (subtypes 1,3,4 and 5) on astrocytes, oligodendrocytes, neurons, microglia, and BBB (314). This interaction induces irreversible internalization and degradation of the receptor, which results in lymphocyte sequestration to the secondary lymphoid organs (315). This results in the reduction of peripheral blood lymphocyte count, especially of T cells with naive and memory phenotypes (315). The therapeutic effect of siponimod is similar to that of phosphorylated fingolimod (316). The main differences are that siponimod activity does not require phosphorylation and it mainly targets the SIP receptor subtype 5 (317). Siponimod has neuroprotective activity by increasing oligodendrocyte precursors maturation, limiting astrogliosis, and promoting remyelination (316). Another drug, ozanimod, has similar mechanisms as phosphorylated fingolimod and siponimod by targeting S1P receptor (318).Ozanimod can bind to both, S1P-1 and SIP-5 subtype receptors, reducing peripheral T cell counts and their migration across the BBB (318).
Natalizumab administration decreases leukocyte migration into the CNS (319) by blocking the interaction between VCAM-1 on endothelial cells and α4β1-integrin on lymphocytes (320).Another monoclonal antibody-based drug, rituximab, could induce the apoptotic death of B and T cells, especially those with pro-inflammatory CD3+CD20+ phenotype (321). Rituximab could also interact with a core epitope on the extracellular CD20 loop (321) inducing apoptosis via ADCC and complement-dependent cytotoxicity mechanisms (322). Natalizumab’s and rituximab’s ability to decrease B cell counts could also contribute to their therapeutic effect on MS (323). As compared to rituximab, ocrelizumab appears to have lower ability to produce an immune response combined with higher therapeutic efficacy (199, 324) This monoclonal antibody-based drug produces a lower level of neutralizing autoantibodies combined with more ADCC complement-dependent cytotoxicity, as compared to rituximab (324).On the downside, ocrelizumab administration could have higher infusion-related reactions (324). Reduction of B cells and CD20+ T combined with increased counts of Tregs was demonstrated when ofatumumab is used for the treatment of MS (325). Additionally, ofatumumab was shown to limit CD4+ T cells secretion of IFN-γ, TNFα, and GM-CSF, as well as a decrease in Th17.1 cell counts (325).
The active metabolite of DMF, monomethyl fumarate (MMF) (326), was shown to decrease peripheral blood mononuclear cell counts by inducing apoptosis. The reduced Th1 (IFN-γ and TNF-α) and increased Th2 (IL-4 and IL-5) cytokines production were reported in MS patients treated with DMF (327). MMF could disrupt the expression of genes responsible for the production of adhesion molecules by interfering with the expression of the nuclear factor kappa B transcription factor (328). Additionally, MMF could protect glial cells against oxidative stress leading to significantly reduced axonal damage in MS (327). Intrathecal methotrexate (ITMTX) is hypothesized to reduce astroglial proliferation and scarring (329).
Laquinimod administration impedes Th17 proinflammatory response and reduces the production of pro-inflammatory IL-12 and TNF-α, while promoting secretion of anti-inflammatory IL-4 and IL-10 cytokines (330). These effects were explained by the ability of laquinimod to inhibit NF-κB pathway (331). The migration of macrophages, CD4+, and CD8+ T cells into the CNS was shown to be reduced by laquinimod, by downregulation of the VLA-4 mediated lymphocytes adhesiveness (332). Additionally, decreased axonal damage, synaptic loss, and inflammatory demyelination have been reported in laquinimod’ treated MS (333). Increased serum level of the brain-derived neuroprotective factor was also demonstrated in MS receiving laquinimod, which could contribute to the drug’s neuroprotective efficacy (334).
It was suggested that mitoxantrone could induce apoptosis in B cells, reduce the secretion of pro-inflammatory cytokines, and suppress T cells (335, 336).The mechanism of the therapeutic effect of teriflunomide could include the inhibition of the dihydro-orotate dehydrogenase enzyme required for de novo pyrimidine synthesis in lymphocytes (337). This reduced intracellular level of pyrimidine could have a cytostatic effect on B and T cells, consequently reducing their counts in circulation (337). Cladribine is also suggested to affect the B and T cells, as the absolute lymphocyte counts decreased rapidly upon treatment (288). The phosphorylated cladribine was shown to disrupt DNA synthesis by inhibiting enzymes involved in the cell cycle, which was proposed as a mechanism of B and T cell depletion (338)
TCV therapeutic efficacy is based on the activation of anti-idiotypic and anti-ergotypic immune mechanisms, by administration of attenuated, self-activated, myelin-reactive T cells (339). This could lead to a direct depletion of pathogenic Th1 cells, as well as activate CD4+ Th2 cells that produce anti-inflammatory cytokines IL-4 and IL-10. Elevated levels of FoxP3+ Tregs, γδT and NK cells were reported when using TCV (340–343).
Disruption of myelin sheath integrity causes the neuron damage commonly found in CNS tissues from MS. Damage of the laminated structure of myelin sheath covering neuron axon and cells body is believed to be primarily done by Th1 and Th17 lymphocytes. These lymphocytes could produce inflammatory cytokines as well activate pathogenic CTL and Th17 damaging myelin. The reduced activity and counts of Th9 and Tregs was also demonstrated in MS. This data suggests the limited control over lymphocytes differentiation could lead to activation of pathogenic Th1 and Th17 cells. In addition to T lymphocytes, B cells producing anti-myelin proteins antibodies were shown in CNS of MS. These B cells could produce pro-inflammatory cytokines sustaining pathogenic inflammatory milieu in CNS. Also, B cells could facilitate antigen presentation required to activate pathogenic Th1 and Th17 lymphocytes.
These data support the notion of targeting T and B lymphocytes for treatment of MS. Monoclonal antibodies inhibiting lymphocyte migration to CNS were shown effective in MS. Also, anti-CD20 antibodies demonstrated therapeutic efficacy which was explained by reduction of B lymphocyte counts in MS. CAR-T approach targeting Tregs has shown the therapeutic potency in EAE model of MS. Additionally, TCV reducing the myelin-reactive T-cell counts was demonstrated as having therapeutic efficacy in MS.
MB, SK, and AR contributed to conception and design of the study. SD and LZ organized the database. RL wrote the first draft of the manuscript. SD, LZ, SJ, KS, VL, TK, and EM wrote sections of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
This work was funded by grant of NSFC (81901663). Also, this work was supported by the Kazan Federal University Strategic Academic Leadership Program (PRIORITY-2030).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Lassmann H. Multiple sclerosis pathology. Cold Spring Harb Perspect Med (2018) 8(3):a028936. doi: 10.1101/cshperspect.a028936
2. Ford H. Clinical presentation and diagnosis of multiple sclerosis. Clin Med (Northfield Il) (2020) 20(4):380–3. doi: 10.7861/clinmed.2020-0292
3. McDonald WI, Compston A, Edan G, Goodkin D, Hartung H-P, Lublin FD, et al. Recommended diagnostic criteria for multiple sclerosis: Guidelines from the international panel on the diagnosis of multiple sclerosis. Ann Neurol (2001) 50(1):121–7. doi: 10.1002/ana.1032
4. Thompson AJ, Banwell BL, Barkhof F, Carroll WM, Coetzee T, Comi G, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol (2018) 17(2):162–73. doi: 10.1016/S1474-4422(17)30470-2
5. National Institute of Neurological Disorders S (US). Multiple sclerosis: Hope through research (2022). Available at: https://www.ninds.nih.gov/health-information/patient-caregiver-education/hope-through-research/multiple-sclerosis-hope-through-research.
6. Gbaguidi B, Guillemin F, Soudant M, Debouverie M, Mathey G, Epstein J. Age-period-cohort analysis of the incidence of multiple sclerosis over twenty years in Lorraine, France. Sci Rep (2022) 12(1):1001. doi: 10.1038/s41598-022-04836-5
7. Reich DS, Lucchinetti CF, Calabresi PA. Multiple sclerosis. N Engl J Med (2018) 378(2):169–80. doi: 10.1056/NEJMra1401483
8. Freiin von Hövel F, Kefalakes E, Grothe C. What can we learn from FGF-2 isoform-specific mouse mutants? differential insights into FGF-2 physiology In vivo. Int J Mol Sci (2020) 22(1):390. doi: 10.3390/ijms22010390
9. Rasouli J, Ciric B, Imitola J, Gonnella P, Hwang D, Mahajan K, et al. Expression of GM-CSF in T cells is increased in multiple sclerosis and suppressed by IFN-β therapy. J Immunol (2015) 194(11):5085–93. doi: 10.4049/jimmunol.1403243
10. Wang J, Jelcic I, Mühlenbruch L, Haunerdinger V, Toussaint NC, Zhao Y, et al. HLA-DR15 molecules jointly shape an autoreactive T cell repertoire in multiple sclerosis. Cell (2020) 183(5):1264–81.e20. doi: 10.1016/j.cell.2020.09.054
11. Larochelle C, Wasser B, Jamann H, Löffel JT, Cui Q-L, Tastet O, et al. Pro-inflammatory T helper 17 directly harms oligodendrocytes in neuroinflammation. Proc Natl Acad Sci (2021) 118(34):223–43. doi: 10.1073/pnas.2025813118
12. Galli E, Hartmann FJ, Schreiner B, Ingelfinger F, Arvaniti E, Diebold M, et al. GM-CSF and CXCR4 define a T helper cell signature in multiple sclerosis. Nat Med (2019) 25(8):1290–300. doi: 10.1038/s41591-019-0521-4
13. Khosravi M, Majdinasab N, Amari A, Ghadiri AA. Increased frequency of CD4+CD25high CD127low regulatory T cells in patients with multiple sclerosis. Gene Rep (2019) 17:100456. doi: 10.1016/j.genrep.2019.100456
14. Salou M, Nicol B, Garcia A, Laplaud DA. Involvement of CD8+ T cells in multiple sclerosis. Front Immunol (2015) Vol. 6. doi: 10.3389/fimmu.2015.00604
15. Vlaming M, Bilemjian V, Freile JÁ, Lourens HJ, van Rooij N, Huls G, et al. CD20 positive CD8 T cells are a unique and transcriptionally-distinct subset of T cells with distinct transmigration properties. Sci Rep (2021) 11(1):20499. doi: 10.1038/s41598-021-00007-0
16. Radomir L, Kramer MP, Perpinial M, Schottlender N, Rabani S, David K, et al. The survival and function of IL-10-producing regulatory b cells are negatively controlled by SLAMF5. Nat Commun (2021) 12(1):1893. doi: 10.1038/s41467-021-22230-z
17. Aram J, Frakich N, Morandi E, Alrouji M, Samaraweera A, Onion D, et al. Increased IL-2 and reduced TGF-β upon T-cell stimulation are associated with GM-CSF upregulation in multiple immune cell types in multiple sclerosis. Biomedicines. (2020) 8(7):226. doi: 10.3390/biomedicines8070226
18. Wagner CA, Roqué PJ, Mileur TR, Liggitt D, Goverman JM. Myelin-specific CD8+ T cells exacerbate brain inflammation in CNS autoimmunity. J Clin Invest. (2019) 130(1):203–13. doi: 10.1172/JCI132531
19. Gingele S, Skripuletz T, Jacobs R. Role of CD20 + T cells in multiple sclerosis: implications for treatment with ocrelizumab. Neural Regener Res (2020) 15(4):663. doi: 10.4103/1673-5374.266913
20. Moghaddam VK, Dickerson AS, Bazrafshan E, Seyedhasani SN, Najafi F, Hadei M, et al. Socioeconomic determinants of global distribution of multiple sclerosis: an ecological investigation based on global burden of disease data. BMC Neurol (2021) 21(1):145. doi: 10.1186/s12883-021-02170-3
21. Willumsen JS, Aarseth JH, Myhr K-M, Midgard R. High incidence and prevalence of MS in møre and romsdal county, Norway, 1950–2018. Neurol - Neuroimmunol Neuroinflammation. (2020) 7(3):e713. doi: 10.1212/NXI.0000000000000713
22. Cheong WL, Mohan D, Warren N, Reidpath DD. Multiple sclerosis in the Asia pacific region: A systematic review of a neglected neurological disease. Front Neurol (2018) 9. doi: 10.3389/fneur.2018.00432
23. Badry R, Farghaly W, Rageh T, Shehata G, Metwally N, El Tallawy H, et al. Prevalence of multiple sclerosis in Al quseir city, red Sea governorate, Egypt. Neuropsychiatr Dis Treat (2016) 155:1–15. doi: 10.2147/NDT.S87348
24. Nelson LM, Wallin MT, Marrie RA, Culpepper WJ, Langer-Gould A, Campbell J, et al. A new way to estimate neurologic disease prevalence in the united states. Neurology. (2019) 92(10):469–80. doi: 10.1212/WNL.0000000000007044
25. Kurtzke JF. A reassessment of the distribution of multiple sclerosis. Acta Neurol Scand (2009) 51(2):110–36. doi: 10.1111/j.1600-0404.1975.tb01364.x
26. Wade BJ. Spatial analysis of global prevalence of multiple sclerosis suggests need for an updated prevalence scale. Mult Scler Int (2014) 2014:1–7. doi: 10.1155/2014/124578
27. Wallin MT, Culpepper WJ, Nichols E, Bhutta ZA, Gebrehiwot TT, Hay SI, et al. Global, regional, and national burden of multiple sclerosis 1990–2016: a systematic analysis for the global burden of disease study 2016. Lancet Neurol (2019) 18(3):269–85. doi: 10.1016/S1474-4422(18)30443-5
28. Chi C, Shao X, Rhead B, Gonzales E, Smith JB, Xiang AH, et al. Admixture mapping reveals evidence of differential multiple sclerosis risk by genetic ancestry. PloS Genet (2019) 15(1):e1007808. doi: 10.1371/journal.pgen.1007808
29. Boyko A, Melnikov M. Prevalence and incidence of multiple sclerosis in Russian federation: 30 years of studies. Brain Sci (2020) 10(5):305. doi: 10.3390/brainsci10050305
30. Liu X, Cui Y, Han J. Estimating epidemiological data of multiple sclerosis using hospitalized data in Shandong province, China. Orphanet J Rare Dis (2016) 11(1):73. doi: 10.1186/s13023-016-0457-4
31. Tian D-C, Zhang C, Yuan M, Yang X, Gu H, Li Z, et al. Incidence of multiple sclerosis in China: A nationwide hospital-based study. Lancet Reg Heal - West Pacific. (2020) 1:100010. doi: 10.1016/j.lanwpc.2020.100010
32. Tarlinton R, Khaibullin T, Granatov E, Martynova E, Rizvanov A, Khaiboullina S. The interaction between viral and environmental risk factors in the pathogenesis of multiple sclerosis. Int J Mol Sci (2019) 20(2):303. doi: 10.3390/ijms20020303
33. O’Gorman C, Lin R, Stankovich J, Broadley SA. Modelling genetic susceptibility to multiple sclerosis with family data. Neuroepidemiology. (2013) 40(1):1–12. doi: 10.1159/000341902
34. Nielsen NM, Westergaard T, Rostgaard K, Frisch M, Hjalgrim H, Wohlfahrt J, et al. Familial risk of multiple sclerosis: A nationwide cohort study. Am J Epidemiol. (2005) 162(8):774–8. doi: 10.1093/aje/kwi280
35. Willer CJ, Dyment DA, Risch NJ, Sadovnick AD, Ebers GC. Twin concordance and sibling recurrence rates in multiple sclerosis. Proc Natl Acad Sci (2003) 100(22):12877–82. doi: 10.1073/pnas.1932604100
36. Compston A, Coles A. Multiple sclerosis. Lancet. (2002) 359(9313):1221–31. doi: 10.1016/S0140-6736(02)08220-X
37. Schmidt H, Williamson D, Ashley-Koch A. HLA-DR15 haplotype and multiple sclerosis: A HuGE review. Am J Epidemiol. (2007) 165(10):1097–109. doi: 10.1093/aje/kwk118
38. Marrosu MG. Dissection of the HLA association with multiple sclerosis in the founder isolated population of Sardinia. Hum Mol Genet (2001) 10(25):2907–16. doi: 10.1093/hmg/10.25.2907
39. Gusev EI, Sudomoina MA, Boĭko AN, Turetskaia RL, Demina TL, Alekseev LP, et al. [Genetic predisposition factors for multiple sclerosis (based on the data from genotyping patients of the Russian ethnic group)]. Zhurnal Nevrol i psikhiatrii Im SS Korsakova. (1997) 97(5):39–46.
40. Wu X-M, Wang C, Zhang K-N, Lin A-Y, Kira J, Hu G-Z, et al. Association of susceptibility to multiple sclerosis in southern han Chinese with HLA-DRB1, -DPB1 alleles and DRB1-DPB1 haplotypes: distinct from other populations. Mult Scler J (2009) 15(12):1422–30. doi: 10.1177/1352458509345905
41. Liu L, Li Y, Tollefsbol TO. Gene-environment interactions and epigenetic basis of human diseases. Curr Issues Mol Biol (2008) 10(1–2):25–36. doi: 10.1038/s41598-019-52894-z
42. Castro K, Casaccia P. Epigenetic modifications in brain and immune cells of multiple sclerosis patients. Mult Scler J (2018) 24(1):69–74. doi: 10.1177/1352458517737389
43. Maltby VE, Lea RA, Sanders KA, White N, Benton MC, Scott RJ, et al. Differential methylation at MHC in CD4+ T cells is associated with multiple sclerosis independently of HLA-DRB1. Clin Epigenetics. (2017) 9(1):71. doi: 10.1186/s13148-017-0371-1
44. Kular L, Liu Y, Ruhrmann S, Zheleznyakova G, Marabita F, Gomez-Cabrero D, et al. DNA Methylation as a mediator of HLA-DRB1*15:01 and a protective variant in multiple sclerosis. Nat Commun (2018) 9(1):2397. doi: 10.1038/s41467-018-04732-5
45. Noori-Zadeh A, Mesbah-Namin SA, Saboor-Yaraghi AA. Epigenetic and gene expression alterations of FOXP3 in the T cells of EAE mouse model of multiple sclerosis. J Neurol Sci (2017) 375:203–8. doi: 10.1016/j.jns.2017.01.060
46. Sintzel MB, Rametta M, Reder AT. Vitamin d and multiple sclerosis: A comprehensive review. Neurol Ther (2018) 7(1):59–85. doi: 10.1007/s40120-017-0086-4
47. Benjaminsen E, Olavsen J, Karlberg M, Alstadhaug KB. Multiple sclerosis in the far north - incidence and prevalence in nordland county, Norway, 1970–2010. BMC Neurol (2014) 14(1):226. doi: 10.1186/s12883-014-0226-8
48. Fromont A, Binquet C, Sauleau EA, Fournel I, Bellisario A, Adnet J, et al. Geographic variations of multiple sclerosis in France. Brain. (2010) 133(7):1889–99. doi: 10.1093/brain/awq134
49. Tao C, Simpson S, van der Mei I, Blizzard L, Havrdova E, Horakova D, et al. Higher latitude is significantly associated with an earlier age of disease onset in multiple sclerosis. J Neurol Neurosurg Psychiatry (2016) 87(12):1343–9. doi: 10.1136/jnnp-2016-314013
50. Mendes MM, Hart KH, Lanham-New SA, Botelho PB. Exploring the impact of individual UVB radiation levels on serum 25-hydroxyvitamin d in women living in high versus low latitudes: A cross-sectional analysis from the d-SOL study. Nutrients. (2020) 12(12):3805. doi: 10.3390/nu12123805
51. Wang Y, Marling SJ, Beaver EF, Severson KS, Deluca HF, light selectively inhibits spinal cord inflammation UV. And demyelination in experimental autoimmune encephalomyelitis. Arch Biochem Biophys (2015) 567:75–82. doi: 10.1016/j.abb.2014.12.017
52. Loosen SH, Doege C, Meuth SG, Luedde T, Kostev K, Roderburg C. Infectious mononucleosis is associated with an increased incidence of multiple sclerosis: Results from a cohort study of 32,116 outpatients in Germany. Front Immunol (2022) 2:13. doi: 10.3389/fimmu.2022.937583
53. Bjornevik K, Cortese M, Healy BC, Kuhle J, Mina MJ, Leng Y, et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science (80-) (2022) 375(6578):296–301. doi: 10.1126/science.abj8222
54. Langer-Gould A, Wu J, Lucas R, Smith J, Gonzales E, Amezcua L, et al. Epstein-Barr Virus, cytomegalovirus, and multiple sclerosis susceptibility. Neurology. (2017) 89(13):1330–7. doi: 10.1212/WNL.0000000000004412
55. Munger K, Levin L, O’Reilly E, Falk K, Ascherio A. Anti-Epstein–Barr virus antibodies as serological markers of multiple sclerosis: A prospective study among united states military personnel. Mult Scler J (2011) 17(10):1185–93. doi: 10.1177/1352458511408991
56. Moreno MA, Or-Geva N, Aftab BT, Khanna R, Croze E, Steinman L, et al. Molecular signature of Epstein-Barr virus infection in MS brain lesions. Neurol - Neuroimmunol Neuroinflammation. (2018) 5(4):e466. doi: 10.1212/NXI.0000000000000466
57. Serafini B, Rosicarelli B, Franciotta D, Magliozzi R, Reynolds R, Cinque P, et al. Dysregulated Epstein-Barr virus infection in the multiple sclerosis brain. J Exp Med (2007) 204(12):2899–912. doi: 10.1084/jem.20071030
58. Hatton OL, Harris-Arnold A, Schaffert S, Krams SM, Martinez OM. The interplay between Epstein–Barr virus and b lymphocytes: implications for infection, immunity, and disease. Immunol Res (2014) 58(2–3):268–76. doi: 10.1007/s12026-014-8496-1
59. Pender MP. The essential role of Epstein-Barr virus in the pathogenesis of multiple sclerosis. Neurosci. (2011) 17(4):351–67. doi: 10.1177/1073858410381531
60. Gabibov AG, Belogurov AA, Lomakin YA, Zakharova MY, Avakyan ME, Dubrovskaya VV, et al. Combinatorial antibody library from multiple sclerosis patients reveals antibodies that cross-react with myelin basic protein and EBV antigen. FASEB J (2011) 25(12):4211–21. doi: 10.1096/fj.11-190769
61. Lomakin Y, Arapidi GP, Chernov A, Ziganshin R, Tcyganov E, Lyadova I, et al. Exposure to the Epstein–Barr viral antigen latent membrane protein 1 induces myelin-reactive antibodies in vivo. Front Immunol (2017) 8. doi: 10.3389/fimmu.2017.00777
62. Zheng D, Liwinski T, Elinav E. Interaction between microbiota and immunity in health and disease. Cell Res (2020) 30(6):492–506. doi: 10.1038/s41422-020-0332-7
63. Cantoni C, Lin Q, Dorsett Y, Ghezzi L, Liu Z, Pan Y, et al. Alterations of host-gut microbiome interactions in multiple sclerosis. eBioMedicine. (2022) 76:103798. doi: 10.1016/j.ebiom.2021.103798
64. Ventura RE, Iizumi T, Battaglia T, Liu M, Perez-Perez GI, Herbert J, et al. Gut microbiome of treatment-naïve MS patients of different ethnicities early in disease course. Sci Rep (2019) 9(1):16396. doi: 10.3390/ijms17020234
65. Jangi S, Gandhi R, Cox LM, Li N, von Glehn F, Yan R, et al. Alterations of the human gut microbiome in multiple sclerosis. Nat Commun (2016) 28;7(1):12015. doi: 10.1038/ncomms12015
66. Wekerle H. Brain autoimmunity and intestinal microbiota: 100 trillion game changers. Trends Immunol (2017) 38(7):483–97. doi: 10.1016/j.it.2017.03.008
67. Horton MK, McCauley K, Fadrosh D, Fujimura K, Graves J, Ness J, et al. Gut microbiome is associated with multiple sclerosis activity in children. Ann Clin Transl Neurol (2021) 8(9):1867–83. doi: 10.1002/acn3.51441
68. Kadowaki A, Saga R, Lin Y, Sato W, Yamamura T. Gut microbiota-dependent CCR9+CD4+ T cells are altered in secondary progressive multiple sclerosis. Brain. (2019) 142(4):916–31. doi: 10.1093/brain/awz012
69. Cosorich I, Dalla-Costa G, Sorini C, Ferrarese R, Messina MJ, Dolpady J, et al. High frequency of intestinal T h 17 cells correlates with microbiota alterations and disease activity in multiple sclerosis. Sci Adv (2017) 3(7):1–4. doi: 10.1126/sciadv.1700492
70. Schepici G, Silvestro S, Bramanti P, Mazzon E. The gut microbiota in multiple sclerosis: An overview of clinical trials. Cell Transplant. (2019) 28(12):1507–27. doi: 10.1177/0963689719873890
71. Haghikia A, Jörg S, Duscha A, Berg J, Manzel A, Waschbisch A, et al. Dietary fatty acids directly impact central nervous system autoimmunity via the small intestine. Immunity. (2015) 43(4):817–29. doi: 10.1016/j.immuni.2015.09.007
72. Cekanaviciute E, Yoo BB, Runia TF, Debelius JW, Singh S, Nelson CA, et al. Gut bacteria from multiple sclerosis patients modulate human T cells and exacerbate symptoms in mouse models. Proc Natl Acad Sci (2017) 114(40):10713–8. doi: 10.1073/pnas.1711235114
73. Poorolajal J, Bahrami M, Karami M, Hooshmand E. Effect of smoking on multiple sclerosis: A meta-analysis. J Public Health (Bangkok) (2016) 3:fdw030. doi: 10.1016/j.puhe.2016.05.004
74. Wingerchuk DM. Smoking: effects on multiple sclerosis susceptibility and disease progression. Ther Adv Neurol Disord (2012) 5(1):13–22. doi: 10.1177/1756285611425694
75. Manouchehrinia A, Huang J, Hillert J, Alfredsson L, Olsson T, Kockum I, et al. Smoking attributable risk in multiple sclerosis. Front Immunol (2022) 3:13. doi: 10.3389/fimmu.2022.840158
76. Caliri AW, Tommasi S, Besaratinia A. Relationships among smoking, oxidative stress, inflammation, macromolecular damage, and cancer. Mutat Res Mutat Res (2021) 787:108365. doi: 10.1016/j.mrrev.2021.108365
77. Baskara I, Kerbrat S, Dagouassat M, Nguyen HQ, Guillot-Delost M, Surenaud M, et al. Cigarette smoking induces human CCR6+Th17 lymphocytes senescence and VEGF-a secretion. Sci Rep (2020) 10(1):6488. doi: 10.1038/s41598-020-63613-4
78. Tawa M, Kinoshita T, Masuoka T, Yamashita Y, Nakano K, Nishio M, et al. Impact of cigarette smoking on nitric oxide-sensitive and nitric oxide-insensitive soluble guanylate cyclase-mediated vascular tone regulation. Hypertens Res (2020) 43(3):178–85. doi: 10.1038/s41440-019-0363-y
79. Hedström AK, Katsoulis M, Hössjer O, Bomfim IL, Oturai A, Sondergaard HB, et al. The interaction between smoking and HLA genes in multiple sclerosis: replication and refinement. Eur J Epidemiol (2017) 32(10):909–19. doi: 10.1007/s10654-017-0250-2
80. Ammitzbøll C, von Essen MR, Börnsen L, Petersen ER, McWilliam O, Ratzer R, et al. GPR15+ T cells are Th17 like, increased in smokers and associated with multiple sclerosis. J Autoimmun (2019) 97:114–21. doi: 10.1016/j.jaut.2018.09.005
81. Hedström A, Olsson T, Alfredsson L. Smoking is a major preventable risk factor for multiple sclerosis. Mult Scler J (2016) 22(8):1021–6. doi: 10.1177/1352458515609794
82. Marabita F, Almgren M, Sjöholm LK, Kular L, Liu Y, James T, et al. Smoking induces DNA methylation changes in multiple sclerosis patients with exposure-response relationship. Sci Rep (2017) 7(1):14589. doi: 10.1038/s41598-017-14788-w
83. Coyle PK. What can we learn from sex differences in MS? J Pers Med (2021) 11(10):1006. doi: 10.3390/jpm11101006
84. Lulu S, Graves J, Waubant E. Menarche increases relapse risk in pediatric multiple sclerosis. Mult Scler J (2016) 22(2):193–200. doi: 10.1177/1352458515581873
85. Ysrraelit MC, Correale J. Impact of sex hormones on immune function and multiple sclerosis development. Immunology (2019) 156(1):9–22. doi: 10.1111/imm.13004
86. Jokubaitis VG, Spelman T, Kalincik T, Lorscheider J, Havrdova E, Horakova D, et al. Predictors of long-term disability accrual in relapse-onset multiple sclerosis. Ann Neurol (2016) 80(1):89–100. doi: 10.1002/ana.24682
87. D’Amico E, Leone C, Patti F. Offspring number does not influence reaching the disability’s milestones in multiple sclerosis: A seven-year follow-up study. Int J Mol Sci (2016) 17(2):234. doi: 10.1055/s-0036-1579693
88. Irizar H, Muñoz-Culla M, Zuriarrain O, Goyenechea E, Castillo-Triviño T, Prada A, et al. HLA-DRB1*15:01 and multiple sclerosis: A female association? Mult Scler J (2012) 18(5):569–77. doi: 10.1177/1352458511426813
89. Chao MJ, Herrera BM, Ramagopalan SV, Deluca G, Handunetthi L, Orton SM, et al. Parent-of-origin effects at the major histocompatibility complex in multiple sclerosis. Hum Mol Genet (2010) 19(18):3679–89. doi: 10.1093/hmg/ddq282
90. Rodenas MC, Cabas I, Gómez-González NE, Arizcun M, Meseguer J, Mulero V, et al. Estrogens promote the production of natural neutralizing antibodies in fish through G protein-coupled estrogen receptor 1. Front Immunol (2017) 8. doi: 10.3389/fimmu.2017.00736
91. AbdulHussain G, Azizieh F, Makhseed M, Raghupathy R. Effects of progesterone, dydrogesterone and estrogen on the production of Th1/Th2/Th17 cytokines by lymphocytes from women with recurrent spontaneous miscarriage. J Reprod Immunol (2020) 140:103132. doi: 10.1016/j.jri.2020.103132
92. Xiong Y, Zhong Q, Palmer T, Benner A, Wang L, Suresh K, et al. Estradiol resolves pneumonia via ERβ in regulatory T cells. JCI Insight (2021) 6(3):324–33. doi: 10.1172/jci.insight.133251
93. Wang C, Dehghani B, Li Y, Kaler LJ, Proctor T, Vandenbark AA, et al. Membrane estrogen receptor regulates experimental autoimmune encephalomyelitis through up-regulation of programmed death 1. J Immunol (2009) 182(5):3294–303. doi: 10.4049/jimmunol.0803205
94. Subramanian S, Matejuk A, Zamora A, Vandenbark AA, Offner H. Oral feeding with ethinyl estradiol suppresses and treats experimental autoimmune encephalomyelitis in SJL mice and inhibits the recruitment of inflammatory cells into the central nervous system. J Immunol (2003) 170(3):1548–55. doi: 10.4049/jimmunol.170.3.1548
95. Langer-Gould A, Brara SM, Beaber BE, Koebnick C. Childhood obesity and risk of pediatric multiple sclerosis and clinically isolated syndrome. Neurology. (2013) 80(6):548–52. doi: 10.1212/WNL.0b013e31828154f3
96. Munger KL, Bentzen J, Laursen B, Stenager E, Koch-Henriksen N, Sørensen TI, et al. Childhood body mass index and multiple sclerosis risk: A long-term cohort study. Mult Scler J (2013) 19(10):1323–9. doi: 10.1177/1352458513483889
97. Hedström A, Olsson T, Alfredsson L. Body mass index during adolescence, rather than childhood, is critical in determining MS risk. Mult Scler J (2016) 22(7):878–83. doi: 10.1177/1352458515603798
98. Tehard B, Mjv L, Cc Nougué, Clavel-Chapelon F. Anthropometric measurements and body silhouette of women. J Am Diet Assoc (2002) 102(12):1779–84. doi: 10.1016/S0002-8223(02)90381-0
99. Ascherio A, Munger K. Epidemiology of multiple sclerosis: From risk factors to prevention–an update. Semin Neurol (2016) 36(02):103–14. doi: 10.1038/cdd.2015.176
100. Ellulu MS, Patimah I, Khaza’ai H, Rahmat A, Abed Y. Obesity and inflammation: The linking mechanism and the complications. Arch Med Sci (2017) 4:851–63. doi: 10.5114/aoms.2016.58928
101. Schmidt FM, Weschenfelder J, Sander C, Minkwitz J, Thormann J, Chittka T, et al. Inflammatory cytokines in general and central obesity and modulating effects of physical activity. PloS One (2015) 10(3):e0121971. doi: 10.1371/journal.pone.0121971
102. Michalovich D, Rodriguez-Perez N, Smolinska S, Pirozynski M, Mayhew D, Uddin S, et al. Obesity and disease severity magnify disturbed microbiome-immune interactions in asthma patients. Nat Commun (2019) 10(1):5711. doi: 10.1038/s41467-019-13751-9
103. Croce S, Avanzini MA, Regalbuto C, Cordaro E, Vinci F, Zuccotti G, et al. Adipose tissue immunomodulation and Treg/Th17 imbalance in the impaired glucose metabolism of children with obesity. Children (2021) 8(7):554. doi: 10.3390/children8070554
104. Moser T, Akgün K, Proschmann U, Sellner J, Ziemssen T. The role of TH17 cells in multiple sclerosis: Therapeutic implications. Autoimmun Rev (2020) 19(10):102647. doi: 10.1016/j.autrev.2020.102647
105. Vranić L, Mikolašević I, Milić S. Vitamin d deficiency: Consequence or cause of obesity? Medicina (B Aires) (2019) 55(9):541. doi: 10.3390/medicina55090541
106. Walsh JS, Bowles S, Evans AL. Vitamin d in obesity. Curr Opin Endocrinol Diabetes Obes (2017) 24(6):389–94. doi: 10.1097/MED.0000000000000371
107. Brady S, Siegel G, Albers RW, Price DL. Basic neurochemistry: molecular, cellular and medical aspects. 7th ed. Price DL, editor. (National Institutes of Health, USA: Elsevier) (2005).
108. Peschl P, Bradl M, Höftberger R, Berger T, Reindl M. Myelin oligodendrocyte glycoprotein: Deciphering a target in inflammatory demyelinating diseases. Front Immunol (2017) 8. doi: 10.3389/fimmu.2017.00529
109. Stadelmann C, Timmler S, Barrantes-Freer A, Simons M. Myelin in the central nervous system: Structure, function, and pathology. Physiol Rev (2019) 99(3):1381–431. doi: 10.1152/physrev.00031.2018
110. Verma ND, Lam AD, Chiu C, Tran GT, Hall BM, Hodgkinson SJ. Multiple sclerosis patients have reduced resting and increased activated CD4+CD25+FOXP3+T regulatory cells. Sci Rep (2021) 11(1):10476. doi: 10.1038/s41598-021-88448-5
111. Rodi M, Dimisianos N, de Lastic A-L, Sakellaraki P, Deraos G, Matsoukas J, et al. Regulatory cell populations in relapsing-remitting multiple sclerosis (RRMS) patients: Effect of disease activity and treatment regimens. Int J Mol Sci (2016) 17(9):1398. doi: 10.3390/ijms17091398
112. Ramaglia V, Sheikh-Mohamed S, Legg K, Park C, Rojas OL, Zandee S, et al. Multiplexed imaging of immune cells in staged multiple sclerosis lesions by mass cytometry. Elife (2019) 8:43–6. doi: 10.7554/eLife.48051
113. Ahmed SM, Fransen NL, Touil H, Michailidou I, Huitinga I, Gommerman JL, et al. Accumulation of meningeal lymphocytes correlates with white matter lesion activity in progressive multiple sclerosis. JCI Insight (2022) 7(5):1–3. doi: 10.1172/jci.insight.151683
114. Hedegaard CJ, Chen N, Sellebjerg F, Sørensen PS, Leslie RGQ, Bendtzen K, et al. Autoantibodies to myelin basic protein (MBP) in healthy individuals and in patients with multiple sclerosis: A role in regulating cytokine responses to MBP. Immunology (2009) 128(1pt2):e451–61. doi: 10.1111/j.1365-2567.2008.02999.x
115. Martinsen V, Kursula P. Multiple sclerosis and myelin basic protein: Insights into protein disorder and disease. Amino Acids (2022) 54(1):99–109. doi: 10.1007/s00726-021-03111-7
116. Cobo-Calvo Á, d’Indy H, Ruiz A, Collongues N, Kremer L, Durand-Dubief F, et al. Frequency of myelin oligodendrocyte glycoprotein antibody in multiple sclerosis. Neurol - Neuroimmunol Neuroinflammation (2020) 7(2):e649. doi: 10.1212/NXI.0000000000000649
117. Cross H, Sabiq F, Ackermans N, Mattar A, Au S, Woodhall M, et al. Myelin oligodendrocyte glycoprotein (MOG) antibody positive patients in a multi-ethnic Canadian cohort. Front Neurol (2021) 11. doi: 10.3389/fneur.2020.525933
118. Hellings N, Barée M, Verhoeven C, MB D’hooghe, Medaer R, Bernard CCA, et al. T-Cell reactivity to multiple myelin antigens in multiple sclerosis patients and healthy controls. J Neurosci Res (2001) 63(3):290–302. doi: 10.1002/1097-4547(20010201)63:3<290::AID-JNR1023>3.0.CO;2-4
119. Kinzel S, Lehmann-Horn K, Torke S, Häusler D, Winkler A, Stadelmann C, et al. Myelin-reactive antibodies initiate T cell-mediated CNS autoimmune disease by opsonization of endogenous antigen. Acta Neuropathol (2016) 132(1):43–58. doi: 10.1007/s00401-016-1559-8
120. Goverman J. Autoimmune T cell responses in the central nervous system. Nat Rev Immunol (2009) 9(6):393–407. doi: 10.1038/nri2550
121. Doronin VB, Parkhomenko TA, Castellazzi M, Padroni M, Pastore M, Buneva VN, et al. Comparison of antibodies hydrolyzing myelin basic protein from the cerebrospinal fluid and serum of patients with multiple sclerosis. PloS One (2014) 9(9):7:e10780. doi: 10.1371/journal.pone.0107807
122. Hohlfeld R, Steinman L. T Cell–transfer experimental autoimmune encephalomyelitis: Pillar of multiple sclerosis and autoimmunity. J Immunol (2017) 198(9):3381–3. doi: 10.4049/jimmunol.1700346
123. Arneth B. Early activation of CD4+ and CD8+ T lymphocytes by myelin basic protein in subjects with MS. J Transl Med (2015) 13(1):341. doi: 10.1186/s12967-015-0715-6
124. Mockus TE, Munie A, Atkinson JR, Segal BM. Encephalitogenic and regulatory CD8 T cells in multiple sclerosis and its animal models. J Immunol (2021) 206(1):3–10. doi: 10.4049/jimmunol.2000797
125. Sweeney MD, Zhao Z, Montagne A, Nelson AR, Zlokovic BV. Blood-brain barrier: From physiology to disease and back. Physiol Rev (2019) 99(1):21–78. doi: 10.1152/physrev.00050.2017
126. Kadry H, Noorani B, Cucullo L. A blood–brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS. (2020) 17(1):69. doi: 10.1186/s12987-020-00230-3
127. Abbott NJ, Rönnbäck L, Hansson E. Astrocyte–endothelial interactions at the blood–brain barrier. Nat Rev Neurosci (2006) 7(1):41–53. doi: 10.1038/nrn1824
128. Correale J, Villa A. The blood–brain-barrier in multiple sclerosis: Functional roles and therapeutic targeting. Autoimmunity (2007) 40(2):148–60. doi: 10.1080/08916930601183522
129. Ikeguchi R, Shimizu Y, Kondo A, Kanda N, So H, Kojima H, et al. Melanoma cell adhesion molecule expressing helper T cells in CNS inflammatory demyelinating diseases. Neurol - Neuroimmunol Neuroinflammation (2021) 8(6):e1069. doi: 10.1212/NXI.0000000000001069
130. Ziliotto N, Zivadinov R, Jakimovski D, Baroni M, Bergsland N, Ramasamy DP, et al. Relationships among circulating levels of hemostasis inhibitors, chemokines, adhesion molecules, and MRI characteristics in multiple sclerosis. Front Neurol (2020) 11. doi: 10.3389/fneur.2020.553616
131. Yan X-B, Zhao Y-F, Yang Y-M, Wang N, He B-Z, Qiu X-T. Impact of astrocyte and lymphocyte interactions on the blood-brain barrier in multiple sclerosis. Rev Neurol (Paris) (2019) 175(6):396–402. doi: 10.1016/j.neurol.2018.12.006
132. Næss LM, Oftung F, Aase A, Michaelsen TE, Pollard AJ. T-Cell responses against meningococcal antigens. In: Meningococcal vaccines. (New Jersey: Humana Press). (2001) p. 339–48.
133. Frisullo G, Nociti V, Iorio R, Plantone D, Patanella AK, Tonali PA, et al. CD8+Foxp3+ T cells in peripheral blood of relapsing-remitting multiple sclerosis patients. Hum Immunol (2010) 71(5):437–41. doi: 10.1016/j.humimm.2010.01.024
134. Wang L, Liang Y. MicroRNAs as T lymphocyte regulators in multiple sclerosis. Front Mol Neurosci (2022) 15, 865529. doi: 10.3389/fnmol.2022.865529
135. Lassmann H. Pathogenic mechanisms associated with different clinical courses of multiple sclerosis. Front Immunol (2019) 10:9. doi: 10.3389/fimmu.2018.03116
136. Cayrol R, Wosik K, Berard JL, Dodelet-Devillers A, Ifergan I, Kebir H, et al. Activated leukocyte cell adhesion molecule promotes leukocyte trafficking into the central nervous system. Nat Immunol (2008) 9(2):137–45. doi: 10.1038/ni1551
137. Hohlfeld R, Dornmair K, Meinl E, Wekerle H. The search for the target antigens of multiple sclerosis, part 1: Autoreactive CD4+ T lymphocytes as pathogenic effectors and therapeutic targets. Lancet Neurol (2016) 15(2):198–209. doi: 10.1016/S1474-4422(15)00334-8
138. Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol (2005) 23(1):683–747. doi: 10.1146/annurev.immunol.23.021704.115707
139. Van Kaer L, Postoak JL, Wang C, Yang G, Wu L. Innate, innate-like and adaptive lymphocytes in the pathogenesis of MS and EAE. Cell Mol Immunol (2019) 16(6):531–9. doi: 10.1038/s41423-019-0221-5
140. Denic A, Wootla B, Rodriguez M. CD8 + T cells in multiple sclerosis. Expert Opin Ther Targets (2013) 17(9):1053–66. doi: 10.1517/14728222.2013.815726
141. Smolders J, Fransen NL, Hsiao C-C, Hamann J, Huitinga I. Perivascular tissue resident memory T cells as therapeutic target in multiple sclerosis. Expert Rev Neurother (2020) 20(8):835–48. doi: 10.1080/14737175.2020.1776609
142. Nyirenda MH, Morandi E, Vinkemeier U, Constantin-Teodosiu D, Drinkwater S, Mee M, et al. TLR2 stimulation regulates the balance between regulatory T cell and Th17 function: A novel mechanism of reduced regulatory T cell function in multiple sclerosis. J Immunol (2015) 194(12):5761–74. doi: 10.4049/jimmunol.1400472
143. Penberthy W, Tsunoda I. The importance of NAD in multiple sclerosis. Curr Pharm Des (2009) 15(1):64–99. doi: 10.2174/138161209787185751
144. Dargahi N, Katsara M, Tselios T, Androutsou M-E, de Courten M, Matsoukas J, et al. Multiple sclerosis: Immunopathology and treatment update. Brain Sci (2017) 7(12):78. doi: 10.3390/brainsci7070078
145. Wu X, Tian J, Wang S. Insight into non-pathogenic Th17 cells in autoimmune diseases. Front Immunol (2018) 9. doi: 10.3389/fimmu.2018.01112
146. Midgley A, Barakat D, Braitch M, Nichols C, Nebozhyn M, Edwards LJ, et al. PAF-r on activated T cells: Role in the IL-23/Th17 pathway and relevance to multiple sclerosis. Immunobiology (2021) 226(1):152023. doi: 10.1016/j.imbio.2020.152023
147. Chitnis T. The role of CD4 T cells in the pathogenesis of multiple sclerosis. In (2007) p:43–72. doi: 10.1016/S0074-7742(07)79003-7
148. Freedman SN, Shahi SK, Mangalam AK. The “Gut feeling”: Breaking down the role of gut microbiome in multiple sclerosis. Neurotherapeutics. (2018) 15(1):109–25. doi: 10.1007/s13311-017-0588-x
149. Kumar BV, Connors TJ, Farber DL. Human T cell development, localization, and function throughout life. Immunity. (2018) 48(2):202–13. doi: 10.1016/j.immuni.2018.01.007
150. Kunkl M, Frascolla S, Amormino C, Volpe E, Tuosto L. T Helper cells: The modulators of inflammation in multiple sclerosis. Cells. (2020) 9(2):482. doi: 10.3390/cells9020482
151. Arneth B. Contributions of T cells in multiple sclerosis: What do we currently know? J Neurol (2021) 268(12):4587–93. doi: 10.1007/s00415-020-10275-x
152. Krishnarajah S, Becher B, Cells TH. Cytokines in encephalitogenic disorders. Front Immunol (2022) 13:822919. doi: 10.3390/ijms23063185
153. Jadidi-Niaragh F, Mirshafiey A. Th17 cell, the new player of neuroinflammatory process in multiple sclerosis. Scand J Immunol (2011) 74(1):1–13. doi: 10.1111/j.1365-3083.2011.02536.x
154. Perumal NB, Kaplan MH. Regulating Il9 transcription in T helper cells. Trends Immunol (2011) 32(4):146–50. doi: 10.1016/j.it.2011.01.006
155. Elyaman W, Khoury SJ. Th9 cells in the pathogenesis of EAE and multiple sclerosis. Semin Immunopathol (2017) 39(1):79–87. doi: 10.1007/s00281-016-0604-y
156. Baharlou R, Khezri A, Razmkhah M, Habibagahi M, Hosseini A, Ghaderi A, et al. Increased interleukin-17 transcripts in PeripheralBlood mononuclear cells, a link between T-helper 17 and proinflammatory responses in bladder cancer. Iran Red Crescent Med J (2015) 17(2):33–9. doi: 10.5812/ircmj.9244
157. Schwab N, Schneider-Hohendorf T, Wiendl H. Therapeutic uses of anti-α4-integrin (anti-VLA-4) antibodies in multiple sclerosis. Int Immunol (2015) 27(1):47–53. doi: 10.1093/intimm/dxu096
158. Starost L, Lindner M, Herold M, Xu YKT, Drexler HCA, Heß K, et al. Extrinsic immune cell-derived, but not intrinsic oligodendroglial factors contribute to oligodendroglial differentiation block in multiple sclerosis. Acta Neuropathol. (2020) 140(5):715–36. doi: 10.1007/s00401-020-02217-8
159. El-Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, et al. The encephalitogenicity of TH17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol (2011) 12(6):568–75. doi: 10.1038/ni.2031
160. Martin-Sanchez F, Diamond C, Zeitler M, Gomez AI, Baroja-Mazo A, Bagnall J, et al. Inflammasome-dependent IL-1β release depends upon membrane permeabilisation. Cell Death Differ 2016 237. (2016) 23(7):1219–31. doi: 10.1371/journal.pone.0199694
161. Kaplan MH, Hufford MM, Olson MR. The development and in vivo function of T helper 9 cells. Nat Rev Immunol (2015) 15(5):295–307. doi: 10.1038/nri3824
162. Goswami R, Jabeen R, Yagi R, Pham D, Zhu J, Goenka S, et al. STAT6-dependent regulation of Th9 development. J Immunol (2012) 188(3):968–75. doi: 10.4049/jimmunol.1102840
163. Veldhoen M, Uyttenhove C, van Snick J, Helmby H, Westendorf A, Buer J, et al. Transforming growth factor-β ‘reprograms’ the differentiation of T helper 2 cells and promotes an interleukin 9–producing subset. Nat Immunol (2008) 9(12):1341–6. doi: 10.1038/ni.1659
164. Matsushita T, Tateishi T, Isobe N, Yonekawa T, Yamasaki R, Matsuse D, et al. Characteristic cerebrospinal fluid Cytokine/Chemokine profiles in neuromyelitis optica, relapsing remitting or primary progressive multiple sclerosis. PloS One (2013) 8(4):e61835. doi: 10.1371/journal.pone.0061835
165. Jäger A, Dardalhon V, Sobel RA, Bettelli E, Kuchroo VK. Th1, Th17, and Th9 effector cells induce experimental autoimmune encephalomyelitis with different pathological phenotypes. J Immunol (2009) 183(11):7169–77. doi: 10.4049/jimmunol.0901906
166. Elyaman W, Bradshaw EM, Uyttenhove C, Dardalhon V, Awasthi A, Imitola J, et al. IL-9 induces differentiation of T h 17 cells and enhances function of FoxP3 + natural regulatory T cells. Proc Natl Acad Sci (2009) 106(31):12885–90. doi: 10.1073/pnas.0812530106
167. Ghoreschi K, Laurence A, Yang X-P, Tato CM, McGeachy MJ, Konkel JE, et al. Generation of pathogenic TH17 cells in the absence of TGF-β signalling. Nature. (2010) 467(7318):967–71. doi: 10.1038/nature09447
168. Ingelfinger F, Gerdes LA, Kavaka V, Krishnarajah S, Friebel E, Galli E, et al. Twin study reveals non-heritable immune perturbations in multiple sclerosis. Nature (2022) 603(7899):152–8. doi: 10.1038/s41586-022-04419-4
169. Bar-Or A, Li R. Cellular immunology of relapsing multiple sclerosis: Interactions, checks, and balances. Lancet Neurol (2021) 20(6):470–83. doi: 10.1016/S1474-4422(21)00063-6
170. Danikowski KM, Jayaraman S, Prabhakar BS. Regulatory T cells in multiple sclerosis and myasthenia gravis. J Neuroinflammation (2017) 14(1):117. doi: 10.1186/s12974-017-0892-8
171. Baecher-Allan C, Kaskow BJ, Weiner HL. Multiple sclerosis: Mechanisms and immunotherapy. Neuron (2018) 97(4):742–68. doi: 10.1016/j.neuron.2018.01.021
172. Mexhitaj I, Nyirenda MH, Li R, O’Mahony J, Rezk A, Rozenberg A, et al. Abnormal effector and regulatory T cell subsets in paediatric-onset multiple sclerosis. Brain (2019) 142(3):617–32. doi: 10.1093/brain/awz017
173. Chwojnicki K, Iwaszkiewicz-Grześ D, Jankowska A, Zieliński M, Łowiec P, Gliwiński M, et al. Administration of CD4+CD25highCD127–FoxP3+ regulatory T cells for relapsing-remitting multiple sclerosis: A phase 1 study. BioDrugs (2021) 35(1):47–60. doi: 10.1007/s40259-020-00462-7
174. Kitz A, Singer E, Hafler D. Regulatory T cells: From discovery to autoimmunity. Cold Spring Harb Perspect Med (2018) 8(12):a029041. doi: 10.1101/cshperspect.a029041
175. Duffy SS, Keating BA, Moalem-Taylor G. Adoptive transfer of regulatory T cells as a promising immunotherapy for the treatment of multiple sclerosis. Front Neurosci (2019) 13. doi: 10.3389/fnins.2019.01107
176. Tapia-Maltos M, Treviño-Frenk I, García-González H, Rosetti M, Barriga-Maldonado V, Morales-Ramírez F, et al. Identification of regulatory T cell molecules associated with severity of multiple sclerosis. Mult Scler J (2021) 27(11):1695–705. doi: 10.1177/1352458520977045
177. Calahorra L, Camacho-Toledano C, Serrano-Regal MP, Ortega MC, Clemente D. Regulatory cells in multiple sclerosis: From blood to brain. Biomedicines (2022) 10(2):335. doi: 10.3390/biomedicines10020335
178. Fletcher JM, Lalor SJ, Sweeney CM, Tubridy N, Mills KHG. T Cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin Exp Immunol (2010) 162(1):1–11. doi: 10.1111/j.1365-2249.2010.04143.x
179. Mishra S, Srinivasan S, Ma C, Zhang N. CD8+ regulatory T cell – a mystery to be revealed. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.708874
180. Fehérvari Z, Sakaguchi S. CD4+ tregs and immune control. J Clin Invest. (2004) 114(9):1209–17. doi: 10.1172/JCI200423395
181. Zhao H, Liao X, Kang Y. Tregs: Where we are and what comes next? Front Immunol (2017) 8. doi: 10.3389/fimmu.2017.01578
182. Ha D, Tanaka A, Kibayashi T, Tanemura A, Sugiyama D, Wing JB, et al. Differential control of human treg and effector T cells in tumor immunity by fc-engineered anti–CTLA-4 antibody. Proc Natl Acad Sci (2019) 116(2):609–18. doi: 10.1073/pnas.1812186116
183. Sobhani N, Tardiel-Cyril DR, Davtyan A, Generali D, Roudi R, Li Y. CTLA-4 in regulatory T cells for cancer immunotherapy. Cancers (Basel) (2021) 13(6):1440. doi: 10.3390/cancers13061440
184. Dummer CD, Carpio VN, Gonçalves LFS, Manfro RC, Veronese FV. FOXP3+ regulatory T cells: From suppression of rejection to induction of renal allograft tolerance. Transpl Immunol (2012) 26(1):1–10. doi: 10.1016/j.trim.2011.08.009
185. Murphy KA, Bhamidipati K, Rubin SJS, Kipp L, Robinson WH, Lanz TV. Immunomodulatory receptors are differentially expressed in b and T cell subsets relevant to autoimmune disease. Clin Immunol (2019) 209:108276. doi: 10.1016/j.clim.2019.108276
186. Kim H-J, Barnitz RA, Kreslavsky T, Brown FD, Moffett H, Lemieux ME, et al. Stable inhibitory activity of regulatory T cells requires the transcription factor Helios. Sci (80-) (2015) 350(6258):334–9. doi: 10.1126/science.aad0616
187. Machcińska M, Kierasińska M, Michniowska M, Maruszewska-Cheruiyot M, Szewczak L, Rola R, et al. Reduced expression of PD-1 in circulating CD4+ and CD8+ tregs is an early feature of RRMS. Int J Mol Sci (2022) 23(6):3185. doi: 10.3390/ijms23063185
188. Goldschmidt CH, Hua LH. Re-evaluating the use of IFN-β and relapsing multiple sclerosis: Safety, efficacy and place in therapy. Degener Neurol Neuromuscul Dis (2020) Volume 10:29–38. doi: 10.2147/DNND.S224912
189. Kaskow BJ, Baecher-Allan C. Effector T cells in multiple sclerosis. Cold Spring Harb Perspect Med (2018) 8(4):a029025. doi: 10.1101/cshperspect.a029025
190. Sabatino JJ, Wilson MR, Calabresi PA, Hauser SL, Schneck JP, Zamvil SS. Anti-CD20 therapy depletes activated myelin-specific CD8 + T cells in multiple sclerosis. Proc Natl Acad Sci (2019) 116(51):25800–7. doi: 10.1073/pnas.1915309116
191. Ochs J, Nissimov N, Torke S, Freier M, Grondey K, Koch J, et al. Proinflammatory CD20 + T cells contribute to CNS-directed autoimmunity. Sci Transl Med (2022) 14(638):1244–50. doi: 10.1126/scitranslmed.abi4632
192. Kinzel S, Weber MS. B cell-directed therapeutics in multiple sclerosis: Rationale and clinical evidence. CNS Drugs (2016) 30(12):1137–48. doi: 10.1007/s40263-016-0396-6
193. Comi G, Bar-Or A, Lassmann H, Uccelli A, Hartung H, Montalban X, et al. Role of b cells in multiple sclerosis and related disorders. Ann Neurol (2021) 89(1):13–23. doi: 10.1002/ana.25927
194. Myhr K-M, Torkildsen Ø, Lossius A, Bø L, Holmøy T. B cell depletion in the treatment of multiple sclerosis. Expert Opin Biol Ther (2019) 19(3):261–71. doi: 10.1080/14712598.2019.1568407
195. Parker Harp CR, Archambault AS, Sim J, Shlomchik MJ, Russell JH, Wu GF. B cells are capable of independently eliciting rapid reactivation of encephalitogenic CD4 T cells in a murine model of multiple sclerosis. PloS One (2018) 13(6):e0199694. doi: 10.3390/ijms34063133
196. Batoulis H, Wunsch M, Birkenheier J, Rottlaender A, Gorboulev V, Kuerten S. Central nervous system infiltrates are characterized by features of ongoing b cell-related immune activity in MP4-induced experimental autoimmune encephalomyelitis. Clin Immunol (201) 158(1):47–58. doi: 10.1016/j.clim.2015.03.009
197. Chacko JA, Strati P, Stout PW, Archer RL, Baltz BP, Chacko JG. Primary mediastinal large b-cell lymphoma in a patient on fingolimod for relapsing-remitting multiple sclerosis. Mult Scler Relat Disord (2021) 49:102776. doi: 10.1016/j.msard.2021.102776
198. Bar-Or A, O’Brien SM, Sweeney ML, Fox EJ, Cohen JA. Clinical perspectives on the molecular and pharmacological attributes of anti-CD20 therapies for multiple sclerosis. CNS Drugs (2021) 35(9):985–97. doi: 10.1007/s40263-021-00843-8
199. Gingele S, Jacobus T, Konen F, Hümmert M, Sühs K-W, Schwenkenbecher P, et al. Ocrelizumab depletes CD20+ T cells in multiple sclerosis patients. Cells (2018) 8(1):12. doi: 10.3390/cells8010012
200. Hauser SL, Bar-Or A, Comi G, Giovannoni G, Hartung H-P, Hemmer B, et al. Ocrelizumab versus interferon beta-1a in relapsing multiple sclerosis. N Engl J Med (2017) 376(3):221–34. doi: 10.1056/NEJMoa1601277
201. Sellebjerg F, Blinkenberg M, Sorensen PS. Anti-CD20 monoclonal antibodies for relapsing and progressive multiple sclerosis. CNS Drugs (2020) 34(3):269–80. doi: 10.1007/s40263-020-00704-w
202. Yang C, Lai W, Zhou J, Zheng X, Cai Y, Yang W, et al. Betaine ameliorates experimental autoimmune encephalomyelitis by inhibiting dendritic cell–derived IL-6 production and Th17 differentiation. J Immunol (2018) 200(4):1316–24. doi: 10.4049/jimmunol.1700920
203. Barr TA, Shen P, Brown S, Lampropoulou V, Roch T, Lawrie S, et al. B cell depletion therapy ameliorates autoimmune disease through ablation of IL-6–producing b cells. J Exp Med (2012) 209(5):1001–10. doi: 10.1084/jem.20111675
204. van Langelaar J, van der Vuurst de Vries RM, Janssen M, Wierenga-Wolf AF, Spilt IM, Siepman TA, et al. T Helper 17.1 cells associate with multiple sclerosis disease activity: perspectives for early intervention. Brain (2018) 141(5):1334–49. doi: 10.1093/brain/awy069
205. Brenner D, Brüstle A, Lin GHY, Lang PA, Duncan GS, Knobbe-Thomsen CB, et al. Toso controls encephalitogenic immune responses by dendritic cells and regulatory T cells. Proc Natl Acad Sci (2014) 111(3):1060–5. doi: 10.1073/pnas.1323166111
206. Li R, Rezk A, Miyazaki Y, Hilgenberg E, Touil H, Shen P, et al. Proinflammatory GM-CSF–producing b cells in multiple sclerosis and b cell depletion therapy. Sci Transl Med (2015) 7(310):1–14. doi: 10.1126/scitranslmed.aab4176
207. Lundqvist S, Modvig S, Fischer EA, Frederiksen JL, Degn M. Frequency and immunophenotype of IL10-producing regulatory b cells in optic neuritis. Immunology (2019) 156(3):259–69. doi: 10.1111/imm.13024
208. Carter NA, Vasconcellos R, Rosser EC, Tulone C, Muñoz-Suano A, Kamanaka M, et al. Mice lacking endogenous IL-10–producing regulatory b cells develop exacerbated disease and present with an increased frequency of Th1/Th17 but a decrease in regulatory T cells. J Immunol (2011) 186(10):5569–79. doi: 10.4049/jimmunol.1100284
209. Ran Z, Yue-Bei L, Qiu-Ming Z, Huan Y. Regulatory b cells and its role in central nervous system inflammatory demyelinating diseases. Front Immunol (2020) 11. doi: 10.3389/fimmu.2020.01884
210. Zhang Y, Wei S, Wu Q, Shen X, Dai W, Zhang Z, et al. Interleukin-35 promotes breg expansion and interleukin-10 production in CD19+ b cells in patients with ankylosing spondylitis. Clin Rheumatol (2022) 41(8):2403–16. doi: 10.1007/s10067-022-06137-8
211. Guo K, Zhang X. Cytokines that modulate the differentiation of Th17 cells in autoimmune uveitis. J Immunol Res (2021) 2021:1–19. doi: 10.1155/2021/6693542
212. Huai G, Markmann JF, Deng S, Rickert CG. TGF-β-secreting regulatory b cells: unsung players in immune regulation. Clin Transl Immunol (2021) 10(4):12–9. doi: 10.1002/cti2.1270
213. Knippenberg S, Peelen E, Smolders J, Thewissen M, Menheere P, Cohen Tervaert JW, et al. Reduction in IL-10 producing b cells (Breg) in multiple sclerosis is accompanied by a reduced naïve/memory breg ratio during a relapse but not in remission. J Neuroimmunol (2011) 239(1–2):80–6. doi: 10.1016/j.jneuroim.2011.08.019
214. Kim Y, Kim G, Shin H-J, Hyun J-W, Kim S-H, Lee E, et al. Restoration of regulatory b cell deficiency following alemtuzumab therapy in patients with relapsing multiple sclerosis. J Neuroinflammation (2018) 15(1):300. doi: 10.1186/s12974-018-1334-y
215. Zhang X, Tao Y, Chopra M, Ahn M, Marcus KL, Choudhary N, et al. Differential reconstitution of T cell subsets following immunodepleting treatment with alemtuzumab (Anti-CD52 monoclonal antibody) in patients with relapsing–remitting multiple sclerosis. J Immunol (2013) 191(12):5867–74. doi: 10.4049/jimmunol.1301926
216. Mrozek-Gorska P, Buschle A, Pich D, Schwarzmayr T, Fechtner R, Scialdone A, et al. Epstein–Barr Virus reprograms human b lymphocytes immediately in the prelatent phase of infection. Proc Natl Acad Sci (2019) 116(32):16046–55. doi: 10.1073/pnas.1901314116
217. Ressing ME, van Gent M, Gram AM, Hooykaas MJG, Piersma SJ, Wiertz EJHJ. Immune evasion by Epstein-Barr virus. Curr Top Microbiol Immunol (2015) 1:355–81. doi: 10.1007/978-3-319-22834-1_12
218. Agostini S, Mancuso R, Guerini FR, D’Alfonso S, Agliardi C, Hernis A, et al. HLA alleles modulate EBV viral load in multiple sclerosis. J Transl Med (2018) 16(1):80. doi: 10.1186/s12967-018-1450-6
219. Bar-Or A, Pender MP, Khanna R, Steinman L, Hartung H-P, Maniar T, et al. Epstein–Barr Virus in multiple sclerosis: Theory and emerging immunotherapies. Trends Mol Med (2020) 26(3):296–310. doi: 10.1016/j.molmed.2019.11.003
220. Jelcic I, Al Nimer F, Wang J, Lentsch V, Planas R, Jelcic I, et al. Memory b cells activate brain-homing, autoreactive CD4+ T cells in multiple sclerosis. Cell. (2018) 175(1):85–100.e23. doi: 10.1016/j.cell.2018.08.011
221. Palle P, Monaghan KL, Milne SM, Wan ECK. Cytokine signaling in multiple sclerosis and its therapeutic applications. Med Sci (2017) 5(4):23. doi: 10.3390/medsci5040023
222. Bai Z, Chen D, Wang L, Zhao Y, Liu T, Yu Y, et al. Cerebrospinal fluid and blood cytokines as biomarkers for multiple sclerosis: A systematic review and meta-analysis of 226 studies with 13,526 multiple sclerosis patients. Front Neurosci (2019) 13. doi: 10.3389/fnins.2019.01026
223. Khaibullin T, Ivanova V, Martynova E, Cherepnev G, Khabirov F, Granatov E, et al. Elevated levels of proinflammatory cytokines in cerebrospinal fluid of multiple sclerosis patients. Front Immunol (2017) 8. doi: 10.3389/fimmu.2017.00531
224. Goyal M, Khanna D, Rana PS, Khaibullin T, Martynova E, Rizvanov AA, et al. Computational intelligence technique for prediction of multiple sclerosis based on serum cytokines. Front Neurol (2019) 10. doi: 10.3389/fneur.2019.00781
225. Wagner CA, Roqué PJ, Goverman JM. Pathogenic T cell cytokines in multiple sclerosis. J Exp Med (2020) 217(1):21–33. doi: 10.1084/jem.20190460
226. Kisuya J, Chemtai A, Raballah E, Keter A, Ouma C. The diagnostic accuracy of Th1 (IFN-γ, TNF-α, and IL-2) and Th2 (IL-4, IL-6 and IL-10) cytokines response in AFB microscopy smear negative PTB- HIV co-infected patients. Sci Rep (2019) 9(1):2966. doi: 10.1038/s41598-019-39048-x
227. Frade-Barros AF, Ianni BM, Cabantous S, Pissetti CW, Saba B, Lin-Wang HT, et al. Polymorphisms in genes affecting interferon-γ production and Th1 T cell differentiation are associated with progression to chagas disease cardiomyopathy. Front Immunol (2020) 11. doi: 10.3389/fimmu.2020.01386
228. Peschl P, Schanda K, Zeka B, Given K, Böhm D, Ruprecht K, et al. Human antibodies against the myelin oligodendrocyte glycoprotein can cause complement-dependent demyelination. J Neuroinflammation (2017) 14(1):208. doi: 10.1186/s12974-017-0984-5
229. Martynova E, Goyal M, Johri S, Kumar V, Khaibullin T, Rizvanov AA, et al. Serum and cerebrospinal fluid cytokine biomarkers for diagnosis of multiple sclerosis. Mediators Inflamm (2020) 2020:1–10. doi: 10.1155/2020/2727042
230. Ottum PA, Arellano G, Reyes LI, Iruretagoyena M, Naves R. Opposing roles of interferon-gamma on cells of the central nervous system in autoimmune neuroinflammation. Front Immunol (2015) 6. doi: 10.3389/fimmu.2015.00539
231. Lin W, Lin Y. Interferon-γ inhibits central nervous system myelination through both STAT1-dependent and STAT1-independent pathways. J Neurosci Res (2010) 12:2569–77. doi: 10.1002/jnr.22425
232. Ap K, Sr O, Anc Simão, Almeida Erd De, Hk M, Lopes J, et al. Cytokine profile in relapsing-remitting multiple sclerosis patients and the association between progression and activity of the disease. Mol Med Rep (2013) 7(3):1010–20. doi: 10.3892/mmr.2013.1256
233. Fresegna D, Bullitta S, Musella A, Rizzo FR, De Vito F, Guadalupi L, et al. Re-examining the role of TNF in MS pathogenesis and therapy. Cells (2020) 9(10):2290. doi: 10.3390/cells9102290
234. Magliozzi R, Pezzini F, Pucci M, Rossi S, Facchiano F, Marastoni D, et al. Changes in cerebrospinal fluid balance of TNF and TNF receptors in naïve multiple sclerosis patients: Early involvement in compartmentalised intrathecal inflammation. Cells (202) 10(7):1712. doi: 10.3390/cells10071712
235. Comabella M, Romera C, Camiña M, Perkal H, Moro MA, Leza JC, et al. TNF–α converting enzyme (TACE) protein expression in different clinical subtypes of multiple sclerosis. J Neurol (2006) 253(6):701–6. doi: 10.1007/s00415-006-0090-6
236. Magliozzi R, Howell OW, Nicholas R, Cruciani C, Castellaro M, Romualdi C, et al. Inflammatory intrathecal profiles and cortical damage in multiple sclerosis. Ann Neurol (2018) 83(4):739–55. doi: 10.1002/ana.25197
237. Shajarian M, Alsahebfosoul F, Etemadifar M, Sedaghat N, Shahbazi M, Firouzabadi FP, et al. IL-23 plasma level measurement in relapsing remitting multiple sclerosis (RRMS) patients compared to healthy subjects. Immunol Invest (2015) 44(1):36–44. doi: 10.3109/08820139.2014.930477
238. Moharami S, Nourazarian A, Nikanfar M, Laghousi D, Shademan B, Joodi Khanghah O, et al. Investigation of serum levels of orexin-a, transforming growth factor β, and leptin in patients with multiple sclerosis. J Clin Lab Anal (2022) 36(1):344–49. doi: 10.1002/jcla.24170
239. Lee WK, Kim Y, Jang H, Sim JH, Choi HJ, Shin Y, et al. Exogenous transforming growth factor-β in brain-induced symptoms of central fatigue and suppressed dopamine production in mice. Int J Mol Sci (2021) 22(5):2580. doi: 10.3390/ijms22052580
240. Donninelli G, Studer V, Brambilla L, Zecca C, Peluso D, Laroni A, et al. Immune soluble factors in the cerebrospinal fluid of progressive multiple sclerosis patients segregate into two groups. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.633167
241. Hirahara K, Poholek A, Vahedi G, Laurence A, Kanno Y, Milner JD, et al. Mechanisms underlying helper T-cell plasticity: Implications for immune-mediated disease. J Allergy Clin Immunol (2013) 131(5):1276–87. doi: 10.1016/j.jaci.2013.03.015
242. Kato H, Endres J, Fox DA. The roles of IFN-γ versus IL-17 in pathogenic effects of human Th17 cells on synovial fibroblasts. Mod Rheumatol (2013) 3:49–51. doi: 10.3109/s10165-012-0811-x
243. Loos J, Schmaul S, Noll TM, Paterka M, Schillner M, Löffel JT, et al. Functional characteristics of Th1, Th17, and ex-Th17 cells in EAE revealed by intravital two-photon microscopy. J Neuroinflammation (2020) 17(1):357. doi: 10.1186/s12974-020-02021-x
244. Harbour SN, Maynard CL, Zindl CL, Schoeb TR, Weaver CT. Th17 cells give rise to Th1 cells that are required for the pathogenesis of colitis. Proc Natl Acad Sci (2015) 112(22):7061–6. doi: 10.1073/pnas.1415675112
245. Sonar SA, Shaikh S, Joshi N, Atre AN, Lal G. IFN-γ promotes transendothelial migration of CD4 + T cells across the blood–brain barrier. Immunol Cell Biol (2017) 95(9):843–53. doi: 10.1038/icb.2017.56
246. Qiu Y, Zhang C, Chen A, Wang H, Zhou Y, Li Y, et al. Immune cells in the BBB disruption after acute ischemic stroke: Targets for immune therapy? Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.678744
247. Linker RA, Lühder F, Kallen K-J, Lee D-H, Engelhardt B, Rose-John S, et al. IL-6 transsignalling modulates the early effector phase of EAE and targets the blood-brain barrier. J Neuroimmunol. (2008) 205(1–2):64–72. doi: 10.1016/j.jneuroim.2008.09.007
248. Engel S, Zipp F. Preventing disease progression in multiple sclerosis–insights from large real-world cohorts. Genome Med (2022) 14(1):41. doi: 10.1186/s13073-022-01044-8
249. Hauser SL, Cree BAC. Treatment of multiple sclerosis: A review. Am J Med (2020) 133(12):1380–1390.e2. doi: 10.1016/j.amjmed.2020.05.049
250. Dhib-Jalbut S, Marks S. Interferon- mechanisms of action in multiple sclerosis. Neurology (2010) 74(Issue 1, Supplement 1):S17–24. doi: 10.1212/WNL.0b013e3181c97d99
251. Constantinescu C, Tanasescu R, Ionete C, Chou I-J. Advances in the treatment of relapsing - remitting multiple sclerosis. BioMed J (2014) 37(2):41. doi: 10.4103/2319-4170.130440
252. Gholamzad M, Ebtekar M, Ardestani MS, Azimi M, Mahmodi Z, Mousavi MJ, et al. A comprehensive review on the treatment approaches of multiple sclerosis: Currently and in the future. Inflammation Res (2019) 68(1):25–38. doi: 10.1007/s00011-018-1185-0
253. Ali R, Nicholas RSJ, Muraro PA. Drugs in development for relapsing multiple sclerosis. Drugs (2013) 73(7):625–50. doi: 10.1007/s40265-013-0030-6
254. Weber MS, Hohlfeld R, Zamvil SS. Mechanism of action of glatiramer acetate in treatment of multiple sclerosis. Neurotherapeutics (2007) 4(4):647–53. doi: 10.1016/j.nurt.2007.08.002
255. Kieseier BC. The mechanism of action of interferon-β in relapsing multiple sclerosis. CNS Drugs (2011) 25(6):491–502. doi: 10.2165/11591110-000000000-00000
256. Ford C, Goodman A, Johnson K, Kachuck N, Lindsey J, Lisak R, et al. Continuous long-term immunomodulatory therapy in relapsing multiple sclerosis: results from the 15-year analysis of the US prospective open-label study of glatiramer acetate. Mult Scler J (2010) 16(3):342–50. doi: 10.1177/1352458509358088
257. Melendez-Torres GJ, Armoiry X, Court R, Patterson J, Kan A, Auguste P, et al. Comparative effectiveness of beta-interferons and glatiramer acetate for relapsing-remitting multiple sclerosis: systematic review and network meta-analysis of trials including recommended dosages. BMC Neurol (2018) 18(1):162. doi: 10.1186/s12883-018-1162-9
258. La Mantia L, Di Pietrantonj C, Rovaris M, Rigon G, Frau S, Berardo F, et al. Interferons-beta versus glatiramer acetate for relapsing-remitting multiple sclerosis. Cochrane Database Syst Rev (2016) 2016(11):22–34. doi: 10.1002/14651858.CD009333.pub3
259. Baeyens A, Fang V, Chen C, Schwab SR. Exit strategies: S1P signaling and T cell migration. Trends Immunol (2015) 36(12):778–87. doi: 10.1016/j.it.2015.10.005
260. Graeler M, Goetzl EJ. Activation-regulated expression and chemotactic function of sphingosine 1-phosphate receptors in mouse splenic T cells. FASEB J (2002) 16(14):1874–8. doi: 10.1096/fj.02-0548com
261. Behrangi N, Fischbach F, Kipp M. Mechanism of siponimod: Anti-inflammatory and neuroprotective mode of action. Cells (2019) 8(1):24. doi: 10.3390/cells8010024
262. Derfuss T, Ontaneda D, Nicholas J, Meng X, Hawker K. Relapse rates in patients with multiple sclerosis treated with fingolimod: Subgroup analyses of pooled data from three phase 3 trials. Mult Scler Relat Disord (2016) 8:124–30. doi: 10.1016/j.msard.2016.05.015
263. Kleiter I, Ayzenberg I, Hoepner R. Fingolimod for multiple sclerosis and emerging indications: Appropriate patient selection, safety precautions, and special considerations. Ther Clin Risk Manag (2016) 261:412–42. doi: 10.2147/TCRM.S65558
264. Goodman AD, Anadani N, Gerwitz L. Siponimod in the treatment of multiple sclerosis. Expert Opin Investig Drugs (2019) 28(12):1051–7. doi: 10.1080/13543784.2019.1676725
265. Ruggieri S, Quartuccio ME, Prosperini L. Ponesimod in the treatment of relapsing forms of multiple sclerosis: An update on the emerging clinical data. Degener Neurol Neuromuscul Dis (2022) Volume 12:61–73. doi: 10.2147/DNND.S313825
266. Kappos L, Fox RJ, Burcklen M, Freedman MS, Havrdová EK, Hennessy B, et al. Ponesimod compared with teriflunomide in patients with relapsing multiple sclerosis in the active-comparator phase 3 OPTIMUM study. JAMA Neurol (2021) 78(5):558. doi: 10.1001/jamaneurol.2021.0405
267. Cohen JA, Arnold DL, Comi G, Bar-Or A, Gujrathi S, Hartung JP, et al. Safety and efficacy of the selective sphingosine 1-phosphate receptor modulator ozanimod in relapsing multiple sclerosis (RADIANCE): A randomised, placebo-controlled, phase 2 trial. Lancet Neurol (2016) 15(4):373–81. doi: 10.1016/S1474-4422(16)00018-1
268. Brandstadter R, Katz Sand I. The use of natalizumab for multiple sclerosis. Neuropsychiatr Dis Treat (2017) Volume 13:1691–702. doi: 10.2147/NDT.S114636
269. Craddock J, Markovic-Plese S. Immunomodulatory therapies for relapsing-remitting multiple sclerosis: Monoclonal antibodies, currently approved and in testing. Expert Rev Clin Pharmacol (2015) 8(3):283–96. doi: 10.1586/17512433.2015.1036030
270. Spelman T, Kalincik T, Jokubaitis V, Zhang A, Pellegrini F, Wiendl H, et al. Comparative efficacy of first-line natalizumab vs IFN-β or glatiramer acetate in relapsing MS. Neurol Clin Pract (2016) 6(2):102–15. doi: 10.1212/CPJ.0000000000000227
271. Li H, Shi F-H, Huang S-Y, Zhang S-G, Gu Z-C, Wei J-F. Clinical adverse effects of natalizumab. Med (Baltimore) (2018) 97(28):e11507. doi: 10.1097/MD.0000000000011507
272. Ryerson LZ, Foley J, Chang I, Kister I, Cutter G, Metzger RR, et al. Risk of natalizumab-associated PML in patients with MS is reduced with extended interval dosing. Neurology (2019) 93(15):e1452–e1462. doi: 10.1212/WNL.0000000000008243
273. Li R, Patterson KR, Bar-Or A. Reassessing b cell contributions in multiple sclerosis. Nat Immunol (2018) 19(7):696–707. doi: 10.1038/s41590-018-0135-x
274. Hauser SL, Kappos L, Montalban X, Craveiro L, Chognot C, Hughes R, et al. Safety of ocrelizumab in patients with relapsing and primary progressive multiple sclerosis. Neurology (2021) 97(16):e1546–59. doi: 10.1212/WNL.0000000000012700
276. Ruck T, Bittner S, Wiendl H, Meuth S. Alemtuzumab in multiple sclerosis: Mechanism of action and beyond. Int J Mol Sci (2015) 16(7):16414–39. doi: 10.3390/ijms160716414
277. Bhamidipati K, Silberstein JL, Chaichian Y, Baker MC, Lanz TV, Zia A, et al. CD52 is elevated on b cells of SLE patients and regulates b cell function. Front Immunol (2021) 11. doi: 10.3389/fimmu.2020.626820
278. Watanabe T, Masuyama J, Sohma Y, Inazawa H, Horie K, Kojima K, et al. CD52 is a novel costimulatory molecule for induction of CD4+ regulatory T cells. Clin Immunol (2006) 120(3):247–59. doi: 10.1016/j.clim.2006.05.006
279. Hu Y, Turner MJ, Shields J, Gale MS, Hutto E, Roberts BL, et al. Investigation of the mechanism of action of alemtuzumab in a human CD52 transgenic mouse model. Immunology (2009) 128(2):260–70. doi: 10.1111/j.1365-2567.2009.03115.x
280. Havrdova E, Horakova D, Kovarova I. Alemtuzumab in the treatment of multiple sclerosis: key clinical trial results and considerations for use. Ther Adv Neurol Disord (2015) 8(1):31–45. doi: 10.1177/1756285614563522
281. Zhang J, Shi S, Zhang Y, Luo J, Xiao Y, Meng L, et al. Alemtuzumab versus interferon beta 1a for relapsing-remitting multiple sclerosis. Cochrane Database Syst Rev (2017) 2018(5):1522–32. doi: 10.1002/14651858.CD010968.pub2
282. Coles AJ, Twyman CL, Arnold DL, Cohen JA, Confavreux C, Fox EJ, et al. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: A randomised controlled phase 3 trial. Lancet (2012) 380(9856):1829–39. doi: 10.1016/S0140-6736(12)61768-1
283. Fox EJ, Alroughani R, Brassat D, Broadley S, Cohen JA, Hartung HP, et al. Efficacy of alemtuzumab is durable over 6 years in patients with active relapsing-remitting multiple sclerosis and an inadequate response to prior therapy in the absence of continuous treatment (CARE-MS II). InMULTIPLE Scler J (2016) 22:596–7. doi: 10.1177/1352458519881759
284. Berenguer-Ruiz L, Sempere AP, Gimenez-Martinez J, Gabaldon-Torres L, Tahoces L, Sanchez-Perez R, et al. Rescue therapy using rituximab for multiple sclerosis. Clin Neuropharmacol (2016) 39(4):178–81. doi: 10.1097/WNF.0000000000000156
285. Gray O, McDonnell GV, Forbes RB. Methotrexate for multiple sclerosis. Cochrane Database Syst Rev (2004) 2010(10):12–43. doi: 10.1002/14651858.CD003208.pub2
286. Kaye J, Piryatinsky V, Birnberg T, Hingaly T, Raymond E, Kashi R, et al. Laquinimod arrests experimental autoimmune encephalomyelitis by activating the aryl hydrocarbon receptor. Proc Natl Acad Sci (2016) 113(41):54–8. doi: 10.1073/pnas.1607843113
287. Scannevin RH, Chollate S, Jung M, Shackett M, Patel H, Bista P, et al. Fumarates promote cytoprotection of central nervous system cells against oxidative stress via the nuclear factor (Erythroid-derived 2)-like 2 pathway. J Pharmacol Exp Ther (2012) 341(1):274–84. doi: 10.1124/jpet.111.190132
288. Rammohan K, Coyle PK, Sylvester E, Galazka A, Dangond F, Grosso M, et al. The development of cladribine tablets for the treatment of multiple sclerosis: A comprehensive review. Drugs (2020) 80(18):1901–28. doi: 10.1007/s40265-020-01422-9
289. Fischer HJ, Finck TLK, Pellkofer HL, Reichardt HM, Lühder F. Glucocorticoid therapy of multiple sclerosis patients induces anti-inflammatory polarization and increased chemotaxis of monocytes. Front Immunol (2019) 10. doi: 10.3389/fimmu.2019.01200
290. Sørensen PS, Comi G, Vollmer TL, Montalban X, Kappos L, Dadon Y, et al. Laquinimod safety profile. Int J MS Care (2017) 19(1):16–24. doi: 10.7224/1537-2073.2015-024
291. Ferreira CM, de V-PEF, de OVM, PR S, JA D, Monreal MTFD, et al. Hepatotoxicity associated with the use of teriflunomide in a patient with multiple sclerosis. Med (Baltimore) (2021) 100(51):e28246. doi: 10.1097/MD.0000000000028246
292. Comi G, Freedman MS, Kappos L, Olsson TP, Miller AE, Wolinsky JS, et al. Pooled safety and tolerability data from four placebo-controlled teriflunomide studies and extensions. Mult Scler Relat Disord (2016) 5:97–104. doi: 10.1016/j.msard.2015.11.006
293. Mills EA, Mao-Draayer Y. Aging and lymphocyte changes by immunomodulatory therapies impact PML risk in multiple sclerosis patients. Mult Scler J (2018) 24(8):1014–22. doi: 10.1177/1352458518775550
294. Zhang JZ, Rivera VM, Tejada-Simon MV, Yang D, Hong J, Li S, et al. T Cell vaccination in multiple sclerosis: results of a preliminary study. J Neurol (2002) 249(2):212–8. doi: 10.1007/PL00007867
295. Rolfes L, Pawlitzki M, Pfeuffer S, Huntemann N, Wiendl H, Ruck T, et al. Failed, interrupted, or inconclusive trials on immunomodulatory treatment strategies in multiple sclerosis: Update 2015–2020. BioDrugs (2020) 34(5):587–610. doi: 10.1007/s40259-020-00435-w
296. Achiron A, Lavie G, Kishner I, Stern Y, Sarova-Pinhas I, Ben-Aharon T, et al. T Cell vaccination in multiple sclerosis relapsing–remitting nonresponders patients. Clin Immunol (2004) 113(2):155–60. doi: 10.1016/j.clim.2004.06.004
297. Seledtsova GV, Ivanova IP, Shishkov AA, Seledtsov VI. Immune responses to polyclonal T-cell vaccination in patients with progressive multiple sclerosis. J Immunotoxicol (2016) 13(6):879–84. doi: 10.1080/1547691X.2016.1223767
298. Karussis D, Shor H, Yachnin J, Lanxner N, Amiel M, Baruch K, et al. T Cell vaccination benefits relapsing progressive multiple sclerosis patients: A randomized, double-blind clinical trial. PloS One (2012) 7(12):e50478. doi: 10.1371/journal.pone.0050478
299. Afzali B, Lombardi G, Lechler RI, Lord GM. The role of T helper 17 (Th17) and regulatory T cells (Treg) in human organ transplantation and autoimmune disease. Clin Exp Immunol (2007) 148(1):32–46. doi: 10.1111/j.1365-2249.2007.03356.x
300. Stephens LA, Malpass KH, Anderton SM. Curing CNS autoimmune disease with myelin-reactive Foxp3 + treg. Eur J Immunol (2009) 39(4):1108–17. doi: 10.1002/eji.200839073
301. Niedbala W, Besnard A-G, Jiang HR, Alves-Filho JC, Fukada SY, Nascimento D, et al. Nitric oxide–induced regulatory T cells inhibit Th17 but not Th1 cell differentiation and function. J Immunol (2013) 191(1):164–70. doi: 10.4049/jimmunol.1202580
302. Malviya M, Saoudi A, Bauer J, Fillatreau S, Liblau R. Treatment of experimental autoimmune encephalomyelitis with engineered bi-specific Foxp3+ regulatory CD4+ T cells. J Autoimmun (2020) 108:102401. doi: 10.1016/j.jaut.2020.102401
303. Fransson M, Piras E, Burman J, Nilsson B, Essand M, Lu B, et al. CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J Neuroinflammation (2012) 9(1):576. doi: 10.1186/1742-2094-9-112
304. Kim YC, Zhang A-H, Yoon J, Culp WE, Lees JR, Wucherpfennig KW, et al. Engineered MBP-specific human tregs ameliorate MOG-induced EAE through IL-2-triggered inhibition of effector T cells. J Autoimmun (2018) 92:77–86. doi: 10.1016/j.jaut.2018.05.003
305. Trzonkowski P, Bacchetta R, Battaglia M, Berglund D, Bohnenkamp HR, ten Brinke A, et al. Hurdles in therapy with regulatory T cells. Sci Transl Med (2015) 7(304):87–97. doi: 10.1126/scitranslmed.aaa7721
306. Jakimovski D, Kolb C, Ramanathan M, Zivadinov R, Weinstock-Guttman B. Interferon β for multiple sclerosis. Cold Spring Harb Perspect Med (2018) 8(11):a032003. doi: 10.1101/cshperspect.a032003
307. Karimi L, Eskandari N, Shaygannejad V. The effect of interferon-beta therapy on T-helper 17/miR-326 and T-helper 1/miR-29b-3p axis in relapsing-remitting multiple sclerosis patients. Neuroimmunomodulation (2022) 29(3):177–85. doi: 10.1159/000519777
308. Adamczyk B, Morawiec N, Arendarczyk M, Baran M, Wierzbicki K, Sowa P, et al. Multiple sclerosis immunomodulatory therapies tested for effectiveness in COVID-19. Neurol Neurochir Pol (2021) 55(4):357–68. doi: 10.5603/PJNNS.a2021.0051
309. Wang D, Ghosh D, Islam SMT, Moorman CD, Thomason AE, Wilkinson DS, et al. IFN-β facilitates neuroantigen-dependent induction of CD25 + FOXP3 + regulatory T cells that suppress experimental autoimmune encephalomyelitis. J Immunol (2016) 197(8):2992–3007. doi: 10.4049/jimmunol.1500411
310. Bolívar S, Anfossi R, Humeres C, Vivar R, Boza P, Muñoz C, et al. Plays both pro- and anti-inflammatory roles in the rat cardiac fibroblast through differential STAT protein activation. Front Pharmacol (2018) 9. doi: 10.3389/fphar.2018.01368
311. Melnikov M, Sharanova S, Sviridova A, Rogovskii V, Murugina N, Nikolaeva A, et al. The influence of glatiramer acetate on Th17-immune response in multiple sclerosis. PloS One (2020) 15(10):e0240305. doi: 10.1371/journal.pone.0240305
312. Perumal J, Filippi M, Ford C, Johnson K, Lisak R, Metz L.Glatiramer acetate as therapy for multiple sclerosis. In: Multiple sclerosis therapeutics p. (Detroit, USA: CRC Press) 446–77.
313. Prod’homme T, Zamvil SS. The evolving mechanisms of action of glatiramer acetate. Cold Spring Harb Perspect Med (2019) 9(2):a029249. doi: 10.1101/cshperspect.a029249
314. Chun J, Kihara Y, Jonnalagadda D, Blaho VA. Fingolimod: Lessons learned and new opportunities for treating multiple sclerosis and other disorders. Annu Rev Pharmacol Toxicol (2019) 59(1):149–70. doi: 10.1146/annurev-pharmtox-010818-021358
315. Bordet R, Camu W, De Seze J, Laplaud D-A, Ouallet J-C, Thouvenot E. Mechanism of action of s1p receptor modulators in multiple sclerosis: The double requirement. Rev Neurol (Paris) (2020) 176(1–2):100–12. doi: 10.1016/j.neurol.2019.02.007
316. Cohan SL, Benedict RHB, Cree BAC, DeLuca J, Hua LH, Chun J. The two sides of siponimod: Evidence for brain and immune mechanisms in multiple sclerosis. CNS Drugs (2022) 36(7):703–19. doi: 10.1007/s40263-022-00927-z
317. Chun J, Giovannoni G, Hunter SF. Sphingosine 1-phosphate receptor modulator therapy for multiple sclerosis: Differential downstream receptor signalling and clinical profile effects. Drugs (2021) 81(2):207–31. doi: 10.1007/s40265-020-01431-8
318. Lassiter G, Melancon C, Rooney T, Murat A-M, Kaye JS, Kaye AM, et al. Ozanimod to treat relapsing forms of multiple sclerosis: A comprehensive review of disease, drug efficacy and side effects. Neurol Int (2020) 12(3):89–108. doi: 10.3390/neurolint12030016
319. Khoy K, Mariotte D, Defer G, Petit G, Toutirais O, Le Mauff B. Natalizumab in multiple sclerosis treatment: From biological effects to immune monitoring. Front Immunol (2020) 11:549842. doi: 10.3389/fimmu.2020.549842
320. Sellebjerg F, Cadavid D, Steiner D, Villar LM, Reynolds R, Mikol D. Exploring potential mechanisms of action of natalizumab in secondary progressive multiple sclerosis. Ther Adv Neurol Disord (2016) 9(1):31–43. doi: 10.1177/1756285615615257
321. Chisari CG, Sgarlata E, Arena S, Toscano S, Luca M, Patti F. Rituximab for the treatment of multiple sclerosis: A review. J Neurol (2022) 269(1):159–83. doi: 10.1007/s00415-020-10362-z
322. Zhong M, van der Walt A, Campagna MP, Stankovich J, Butzkueven H, Jokubaitis V. The pharmacogenetics of rituximab: Potential implications for anti-CD20 therapies in multiple sclerosis. Neurotherapeutics (2020) 17(4):1768–84. doi: 10.1007/s13311-020-00950-2
323. Cencioni MT, Mattoscio M, Magliozzi R, Bar-Or A, Muraro PA. B cells in multiple sclerosis — from targeted depletion to immune reconstitution therapies. Nat Rev Neurol (2021) 17(7):399–414. doi: 10.1038/s41582-021-00498-5
324. Cotchett KR, Dittel BN, Obeidat AZ. Comparison of the efficacy and safety of anti-CD20 b cells depleting drugs in multiple sclerosis. Mult Scler Relat Disord (2021) 49:102787. doi: 10.1016/j.msard.2021.102787
325. von Essen MR, Hansen RH, Højgaard C, Ammitzbøll C, Wiendl H, Sellebjerg F. Ofatumumab modulates inflammatory T cell responses and migratory potential in patients with multiple sclerosis. Neurol - Neuroimmunol Neuroinflammation (2022) 9(4):e200004. doi: 10.1212/NXI.0000000000200004
326. Hosseini A, Masjedi A, Baradaran B, Hojjat-Farsangi M, Ghalamfarsa G, Anvari E, et al. Dimethyl fumarate: Regulatory effects on the immune system in the treatment of multiple sclerosis. J Cell Physiol (2019) 234(7):9943–55. doi: 10.1002/jcp.27930
327. Garcia-Mesa Y, Xu HN, Vance P, Gruenewald AL, Garza R, Midkiff C, et al. Dimethyl fumarate, an approved multiple sclerosis treatment, reduces brain oxidative stress in SIV-infected rhesus macaques: Potential therapeutic repurposing for HIV neuroprotection. Antioxidants (2021) 10(3):416. doi: 10.3390/antiox10030416
328. Breuer J, Herich S, Schneider-Hohendorf T, Chasan AI, Wettschureck N, Gross CC, et al. Dual action by fumaric acid esters synergistically reduces adhesion to human endothelium. Mult Scler J (2018) 24(14):1871–82. doi: 10.1177/1352458517735189
329. Zufferey R, Dull T, Mandel RJ, Bukovsky A, Quiroz D, Naldini L, et al. Self-inactivating lentivirus vector for safe and efficient In vivo gene delivery. J Virol (1998) 72(12):9873–80. doi: 10.1128/JVI.72.12.9873-9880.1998
330. Thöne J, Linker R. Laquinimod in the treatment of multiple sclerosis: A review of the data so far. Drug Des Devel Ther (2016) 10:1111–8. doi: 10.2147/DDDT.S55308
331. Zhou Y, Cui C, Ma X, Luo W, Zheng SG, Qiu W. Nuclear factor κB (NF-κB)–mediated inflammation in multiple sclerosis. Front Immunol (2020) 11. doi: 10.3389/fimmu.2020.00391
332. Rouhi F, Mohammadpour Z, Noureini SK, Abbastabar H, Harirchian MH, Bitarafan S. The effects and side effects of laquinimod for the treatment of multiple sclerosis patients: A systematic review and meta-analysis of clinical trials. Eur J Clin Pharmacol (2020) 76(5):611–22. doi: 10.1007/s00228-019-02827-6
333. Wheeler MA, Quintana FJ. Regulation of astrocyte functions in multiple sclerosis. Cold Spring Harb Perspect Med (2019) 9(1):a029009. doi: 10.1101/cshperspect.a029009
334. Ponath G, Park C, Pitt D. The role of astrocytes in multiple sclerosis. Front Immunol (2018) 9. doi: 10.3389/fimmu.2018.00217
335. Bross M, Hackett M, Bernitsas E. Approved and emerging disease modifying therapies on neurodegeneration in multiple sclerosis. Int J Mol Sci (2020) 21(12):4312. doi: 10.3390/ijms21124312
336. Dias-Carvalho A, Ferreira M, Reis-Mendes A, Ferreira R, Bastos ML, Fernandes E, et al. Chemobrain: Mitoxantrone-induced oxidative stress, apoptotic and autophagic neuronal death in adult CD-1 mice. Arch Toxicol (2022) 96(6):1767–82. doi: 10.1007/s00204-022-03261-x
337. Klotz L, Eschborn M, Lindner M, Liebmann M, Herold M, Janoschka C, et al. Teriflunomide treatment for multiple sclerosis modulates T cell mitochondrial respiration with affinity-dependent effects. Sci Transl Med (2019) 11(490):14–20. doi: 10.1126/scitranslmed.aao5563
338. Jacobs BM, Ammoscato F, Giovannoni G, Baker D, Schmierer K. Cladribine: Mechanisms and mysteries in multiple sclerosis. J Neurol Neurosurg Psychiatry (2018) 89(12):1266–71. doi: 10.1136/jnnp-2017-317411
339. Wang Z, Liu X, Cao F, Bellanti JA, Zhou J, Zheng SG. Prospects of the use of cell therapy to induce immune tolerance. Front Immunol (2020) 11. doi: 10.3389/fimmu.2020.00792
340. Ramagopalan SV, Handel AE, Giovannoni G, Rutherford Siegel S, Ebers GC, Chaplin G. Relationship of UV exposure to prevalence of multiple sclerosis in England. Neurology (2011) 76(16):1410–4. doi: 10.1212/WNL.0b013e318216715e
341. Hong J, Zang YCQ, Nie H, Zhang JZ. CD4 + regulatory T cell responses induced by T cell vaccination in patients with multiple sclerosis. Proc Natl Acad Sci (2006) 103(13):5024–9. doi: 10.1073/pnas.0508784103
342. Hermans G, Medaer R, Raus J, Stinissen P. Myelin reactive T cells after T cell vaccination in multiple sclerosis: Cytokine profile and depletion by additional immunizations. J Neuroimmunol (2000) 102(1):79–84. doi: 10.1016/S0165-5728(99)00157-5
Keywords: T lymphocytes, B lymphocytes, biomarkers, immune pathogenesis, CNS, multiple sclerosis, therapy
Citation: Liu R, Du S, Zhao L, Jain S, Sahay K, Rizvanov A, Lezhnyova V, Khaibullin T, Martynova E, Khaiboullina S and Baranwal M (2022) Autoreactive lymphocytes in multiple sclerosis: Pathogenesis and treatment target. Front. Immunol. 13:996469. doi: 10.3389/fimmu.2022.996469
Received: 17 July 2022; Accepted: 08 September 2022;
Published: 23 September 2022.
Edited by:
Luisa María Villar, Ramón y Cajal University Hospital, SpainReviewed by:
Victor Rivera, Baylor College of Medicine, United StatesCopyright © 2022 Liu, Du, Zhao, Jain, Sahay, Rizvanov, Lezhnyova, Khaibullin, Martynova, Khaiboullina and Baranwal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Svetlana Khaiboullina, c3Yua2hhaWJvdWxsaW5hQGdtYWlsLmNvbQ==; Manoj Baranwal, bWFub2ouYmFyYW53YWxAdGhhcGFyLmVkdQ==;YmFyYW53YWwubWFub2pAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.