 Yan Zhao1
Yan Zhao1 Yuansong Bai
Yuansong Bai
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 23 September 2022
Sec. Cancer Immunity and Immunotherapy
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.992762
This article is part of the Research Topic Plasticity of Immune Cells in Tumor Microenvironment View all 7 articles
Gastric cancer (GC) is a malignancy with a high incidence and mortality, and the emergence of immunotherapy has brought survival benefits to GC patients. Compared with traditional therapy, immunotherapy has the advantages of durable response, long-term survival benefits, and lower toxicity. Therefore, targeted immune cells are the most promising therapeutic strategy in the field of oncology. In this review, we introduce the role and significance of each immune cell in the tumor microenvironment of GC and summarize the current landscape of immunotherapy in GC, which includes immune checkpoint inhibitors, adoptive cell therapy (ACT), dendritic cell (DC) vaccines, reduction of M2 tumor-associated macrophages (M2 TAMs), N2 tumor-associated neutrophils (N2 TANs), myeloid-derived suppressor cells (MDSCs), effector regulatory T cells (eTregs), and regulatory B cells (Bregs) in the tumor microenvironment and reprogram TAMs and TANs into tumor killer cells. The most widely used immunotherapy strategies are the immune checkpoint inhibitor programmed cell death 1/programmed death-ligand 1 (PD-1/PD-L1) antibody, cytotoxic T lymphocyte–associated protein 4 (CTLA-4) antibody, and chimeric antigen receptor T (CAR-T) in ACT, and these therapeutic strategies have significant anti-tumor efficacy in solid tumors and hematological tumors. Targeting other immune cells provides a new direction for the immunotherapy of GC despite the relatively weak clinical data, which have been confirmed to restore or enhance anti-tumor immune function in preclinical studies and some treatment strategies have entered the clinical trial stage, and it is expected that more and more effective immune cell–based therapeutic methods will be developed and applied.
Cancer is the leading cause of death, and the incidence of gastric cancer (GC) remains high (1), most of which are advanced or metastatic at the time of diagnosis since a lack of reliable global screening strategies and specific symptoms (1); the chemotherapy-based comprehensive treatment is mainly the treatment method for this part of GC (2). In recent years, immunotherapy has received extensive attention from scholars, and some GC patients can benefit from this treatment. Compared with traditional chemotherapy, immunotherapy has the advantages of durable response, long-term survival benefits, lower toxicity, and active against brain metastases (3–6). At present, the widely used immunotherapy is immune checkpoint inhibitors (ICI) including programmed cell death 1/programmed death-ligand 1 (PD-1/PD-L1) antibody, cytotoxic T lymphocyte–associated protein 4 (CTLA-4) antibody, and chimeric antigen receptor T (CAR-T) in adoptive cell therapy (ACT). In addition, therapies targeting other immune cells have also demonstrated effectiveness in preclinical anti-tumor studies. Therefore, therapy targeting immune cells will become the most promising therapeutic strategy for GC.
Immune cells are the target cells of immunotherapy which as part of the tumor microenvironment (TME) include dendritic cells (DCs), natural killer (NK) cells, tumor-associated macrophages (TAMs), tumor-associated neutrophils (TANs), myeloid-derived suppressor cells (MDSCs), T cells, and B cells (7). Initiating immune cells to kill tumors needs to go through 3 steps: 1. Neoantigens produced by the tumor are released and captured by DCs for processing. 2. DCs present the captured antigens on major histocompatibility complex (MHC) molecules to T cells, resulting in the priming and activation of effector T cells against the cancer-specific antigens. 3. Activated T cells migrate and infiltrate tumor tissue to destroy their targeted cancer cells (8). Tumor cells can evade recognition and clearance by the immune system through various mechanisms in the above three steps. Among them, M2 TAMs, N2 TANs, MDSCs, effector regulatory T cells (eTregs), regulatory B cells (Bregs), and inhibitory targets on various immune cells play an important role in tumor immune escape, whereas the tumor-infiltrating DCs, NK, M1 TAMs, and N1 TANs are beneficial to anti-tumor immunity. In addition, anti-tumor immunity can be restored by increasing the number or enhancing the function of anti-tumor immune cells or by reducing the number of weakening the function of immunosuppressive cells (Figure 1). Based on this, a lot of trials have been carried out for targeting immune cells to treat tumors. This review introduces the impact of immune cells on the occurrence and development of GC from the perspective of immune cells in the tumor microenvironment of GC and summarizes the current strategies for targeting immune cells to treat GC.
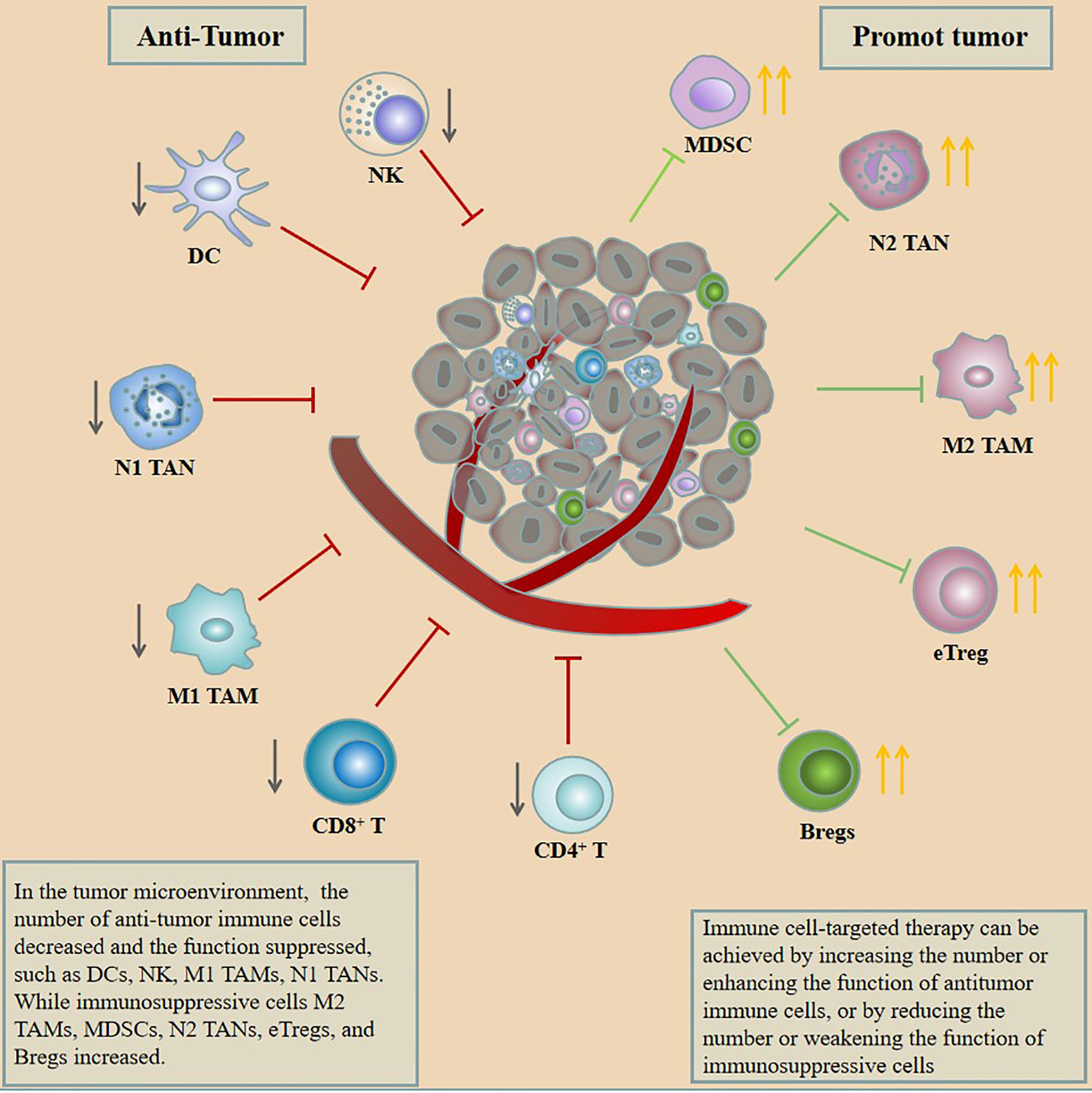
Figure 1 In the tumor microenvironment, the number of anti-tumor immune cells decreased and the function suppressed, such as DCs, NK, M1 TAMs, and N1 TANs, while immunosuppressive cells M2 TAMs, MDSCs, N2 TANs, eTregs, and Bregs increased. Immune cell–targeted therapy can be achieved by increasing the number or enhancing the function of anti-tumor immune cells or by reducing the number or weakening the function of immunosuppressive cells.
Targeted T-cell immunotherapy is the most studied and promising therapeutic approach in the field of GC. Numerous clinical trials have been conducted and some of these treatments have achieved remarkable results. T cells are part of the adaptive immune system, and cellular components are phagocytosed after tumor cell lysis and expressed on antigen-presenting cells (APCs) then exposing them to mature lymphocytes and leading to tumor suppression (9). Different subsets of T cells determine different tumor prognoses (10). Among which T-cell subsets that are beneficial to the prognosis and survival of GC include high levels of CD8+ and CD4+ T-cell infiltration (11), high CD45RO memory T cells (12, 13), and high T helper (TH)1/TH2 ratio (14). While the T-cell subsets associated with GC occurrence and progression are mainly tumor infiltrative forkhead box P3-positive (FOXP3+) Tregs (15, 16), FOXP3 is a transcription factor that is specifically expressed by natural Tregs (17). It is well known that HP is the main pathogenic factor of GC and studies have shown that the expression level of FOXP3+ Tregs in HP-infected patients is significantly higher than that in non-infected patients (18), and the number of FOXP3+ Tregs is significantly elevated in gastritis and GC in HP-infected patients (19). In addition, the expression level of FOXP3 Tregs is closely related to the occurrence and development of GC (20, 21). A significant increase in the proportion of Tregs has been reported in tumor-draining lymph nodes in patients with GC (22). In addition, highly expressed FOXP3+ Tregs in sentinel lymph nodes in GC could predict the metastasis to downstream lymph nodes (23). However, the correlation between Tregs infiltration and GC prognosis remains unclear, and there have been many contradictory results because of different Tregs markers, location distances, and intracellular interactions. As reported, a high FOXP3/CD4 ratio has been associated with poor prognosis in gastric cardia cancer (24). The intratumoral infiltration of FOXP3+ Tregs suggests a poor prognosis in patients after radical gastrectomy (21). Moreover, an increase of FOXP3+ Tregs infiltration in GC tissues and a high proportion of Tregs/CD8+ T cells both predict a poor prognosis (25). However, there are some reports to the contrary, for example, patients with high FOXP3 Tregs cells have a good prognosis in the microsatellite unstable GC population (26). In gastric cardia cancer, stromal infiltration of Tregs may inhibit GC progression caused by chronic inflammation (27). The high expressions of epithelial-infiltrating CD8+ T cells and FOXP3+ Tregs in the GC microenvironment also predict a good prognosis; the authors suggested that the spatial distance between Tregs and CD8+ T cells determines their effect on prognosis, with an intervening distance of 30–110 μm predicting a favorable prognosis (28). In addition, the reason for these two contradictory reports may also be related to different Tregs subsets; some subsets may specifically mediate immunosuppression, such as ICOS Tregs cells that play a major suppressive role in the GC tumor microenvironment and which are mainly found in advanced GC patients (29). Therefore, distinguishing Tregs subsets is also valuable for predicting the prognosis of GC patients, and immunotherapies targeting certain Tregs subsets may lead to better efficacy. T cell–based immunotherapies include ICIs [PD-1/PD-L1, CTLA-4, lymphocyte activation gene 3 (LAG-3), T-cell immunoglobulin and mucin domain-3 (TIM-3), T-cell immunoreceptor with immunoglobulin, and ITIM domain (TIGIT)], ACTs [CAR-T, tumor-infiltrating lymphocyte (TIL), and cytokine-induced killer (CIK)], and Tregs-targeted therapy strategy.
The anticancer activity of T cells depends on the detection of antigenic peptides present on the MHC by the T-cell receptor (TCR) (30) and the interaction between co-stimulatory signals and CD80/CD86 (B7 molecule) expressed on APCs and with cytokine interaction (31). Tumor cells can escape the killing of tumor cells by T cells through downregulating MHC and upregulating the expression of inhibitory receptors such as PD-L1, CTLA-4, LAG-3, TIM-3 and TIGIT (32–34), and blocking the binding of these inhibitory receptors to ligands can restore T-cell anti-tumor activity (35). PD-1/PD-L1 monoclonal antibodies (mAbs) demonstrate significant anti-tumor effect and prolong overall survival (OS) in various malignancies (36–38). PD-1 is an immunosuppressive receptor that is highly expressed on TILs (35), whereas PD-L1 as the main ligand of PD-1 is highly expressed on the surface of tumor cells (39). Tumor cells transmit negative regulatory signals to T cells through the interaction of PD-1 and PD-L1 thus inhibiting T cells activation and reducing cytokine secretion and promoting lymphocyte apoptosis, which was considered to be the main factors in promoting tumor immune escape (40), so antibodies that block PD-1 or PD-L1 can restore anti-tumor activity (41). More than 40% of GC patients express PD-L1 in gastric carcinoma tissues, and the expression of PD-L1 is significantly correlated to tumor size, invasion, lymph node metastasis (LNM), and survival time of patients (42).
Anti–PD-1/PD-L1 mAbs have demonstrated good efficacy in the treatment of GC. In the first-line treatment of GC, pembrolizumab combined with chemotherapy has a higher objective response rate (ORR) in people with a combined positive score (CPS) of ≥10, and the survival benefit of pembrolizumab monotherapy for GC was greater than that of chemotherapy in patients with microsatellite instability-high (MSI-H) (43). In the second-line treatment of gastric and esophagogastric junction adenocarcinoma, pembrolizumab improved OS compared with paclitaxel in patients with a CPS of ≥ 10, and, in the MSI-H subgroup, the OS significantly benefit; moreover, pembrolizumab had a better safety profile than paclitaxel (44). In the third-line treatment of GC, the ORR of pembrolizumab in the treatment of PD-L1–positive GC (CPS ≥1) patients was 15.5% (45), and, based on this study, the pembrolizumab was approved in the treatment of PD-L1–positive advanced GC by FDA. Nivolumab is another PD-1 inhibitor, which improved ORR, disease control rate (DCR), progression-free survival (PFS), and OS when compared with placebo in the third-line or later-line treatment of GC (46). Currently, nivolumab has been approved in China (47), Japan, South Korea, and other countries as a third- or later-line therapeutic option in heavily pretreated patients with unresectable advanced or recurrent gastric/gastroesophageal junction cancer (G/GEJ) cancer (48). In addition, nivolumab combined with chemotherapy [capecitabine plus oxaliplatin (CapeOx) or leucovorin calcium plus fluorouracil plus oxaliplatin (FOLFOX)] in the first-line treatment of metastatic G/GEJ cancer was compared with chemotherapy alone, combination therapy demonstrated PFS benefit in patients with CPS ≥ 1 and in all randomized patients, and statistical significance in patients with CPS ≥5 and the median OS (mOS) of combination therapy was longer than chemotherapy alone in this population. Thus, nivolumab combined with FOLFOX/CapeOx is recommended for late-stage GC with PD-L1 CPS of ≥5 (49). Several phase II studies have shown that combined therapy using anti-human epidermal growth factor receptor 2 (HER2) drugs combined with PD-1 antibody or antiangiogenic inhibitor combined with PD-1 antibody could be a potential treatment strategy in HER2-positive GC patients (50, 51). Such regimens are currently being investigated in stage III clinical trials. Sintilimab (anti–PD-1 mAb) plus CapeOx showed promising efficacy with encouraging pCR rate, significant downstaging effect, and good safety profile in neoadjuvant immunotherapy for GC. This combination regimen might present a new option for patients with locally advanced, resectable G/GEJ adenocarcinoma (52). In addition, the KEYNOTE-585 study is an ongoing phase III study conducted to evaluate the efficacy and safety of pembrolizumab plus chemotherapy compared with placebo plus chemotherapy as neoadjuvant/adjuvant treatment for localized G/GEJ cancer (53). A phase III study investigating the efficacy and safety of neoadjuvant-adjuvant anti–PD-L1 antibody durvalumab and 5-fluorouracil-leucovorin-oxaliplatin-docetaxel (FLOT) chemotherapy followed by adjuvant durvalumab monotherapy in patients with resectable G/GEJ cancer is also underway recruiting (54). Data from these clinical trials may lay the foundation for the use of immunologic drugs in neoadjuvant treatment of GC.
PD-1 and PD-L1 mAbs have an encouraging survival advantage in GC. Unfortunately, PD-1 and PD-L1 antibodies failed to improve OS and PFS in some trials. For example, the KEYNOTE-061 study showed that second-line treatment with pembrolizumab compared with paclitaxel did not significantly prolong OS in patients with PD-L1 CPS ≥ 1 (44). In KEYNOTE-062, pembrolizumab or pembrolizumab in combination with chemotherapy in first-line treatment of advanced GC, ICI monotherapy, or in combination with chemotherapy did not demonstrate significantly improved survival rates compared to chemotherapy (43). Although PD-1/PD-L1 mAbs have obtained positive results in some clinical trials, most of the benefited groups are only those with high PD-L1 CPS score, MSI-H, or high tumor mutational burden (TMB) (44), and it has also been reported that patients with Epstein–Barr virus (EBV)–positive tumors can respond significantly to anti–PD-1 antibody (55). Unfortunately, the abovementioned benefit population does not account for much of advanced GC (the clinical trial results mentioned above are summarized in Table 1).
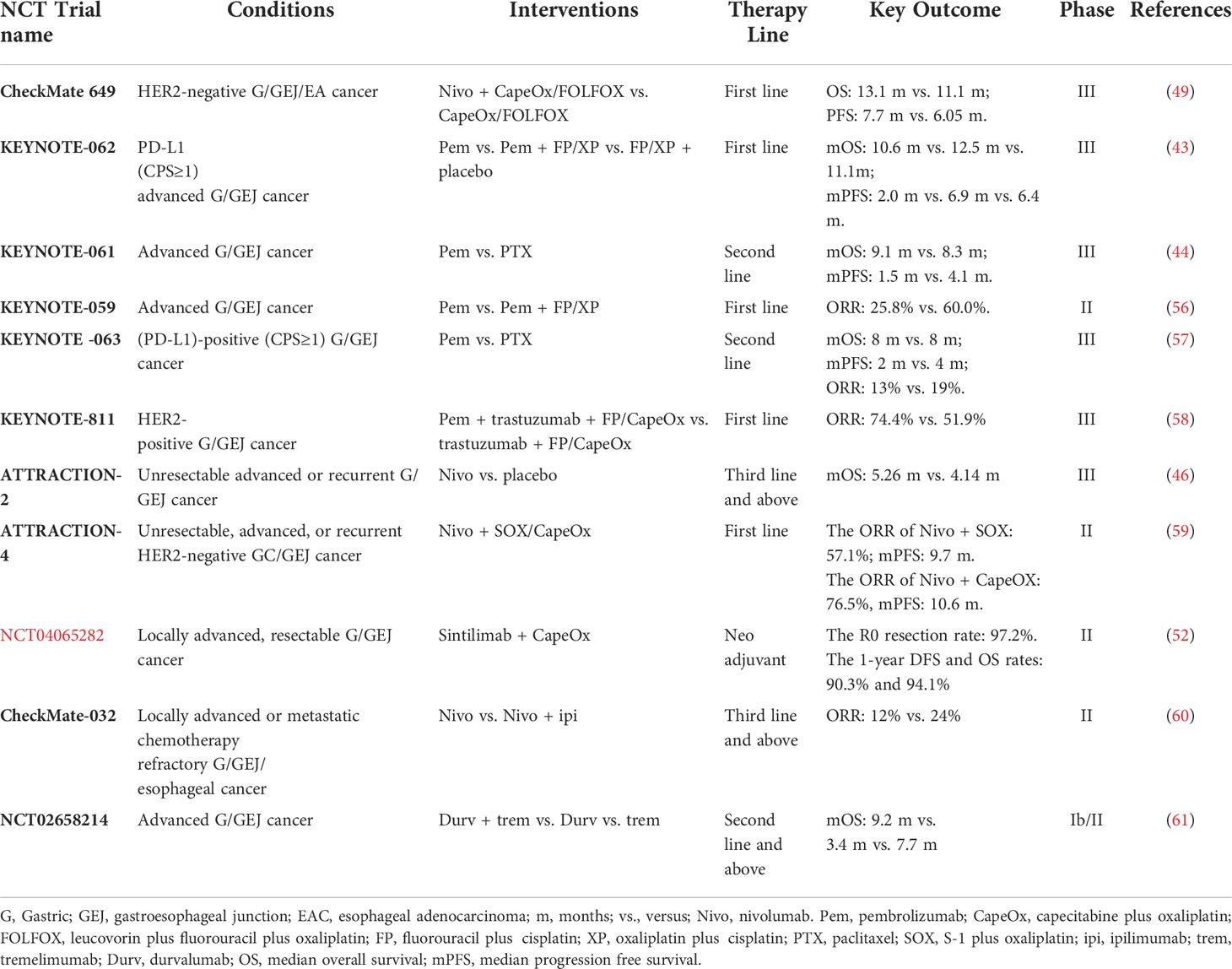
Table 1 Results of major clinical trials of PD-1/PD-L1 and CTLA-4 immune checkpoint inhibitors in the treatment of gastric cancer.
CTLA-4 inhibits T-cell activity by binding to B7 on the surface of APCs (62), whereas anti–CTLA-4 antibodies can relieve the inhibition of T cells by specifically binding to CTLA-4. These antibodies mainly include ipilimumab and tremelimumab. Ipilimumab monotherapy showed 14% ORR in chemotherapy-progressed advanced GC in a phase I/II clinical trial (CheckMate-032) (63). However, ipilimumab did not have significant survival benefits as maintenance therapy after first-line chemotherapy in phase II clinical trial (64). Another anti–CTLA-4 antibody drug tremetimumab acquired 1.7-month PFS and 7.7-month OS in second-line treatment of G/GEJ cancer in IB/II clinical trial (61). Although no exciting anti-tumor activity was shown in all patients, some patients have observed durable anti-tumor activity, ipilimumab suggests that some patients may benefit from tremelimumab therapy (65). In addition, in the treatment regimen of anti–PD-1 combined with anti–CTLA-4, CheckMate-032 showed that the ORR of the combination therapy group (nivolumab combined with ipilimumab) in the treatment of esophagogastric cancer was higher than that of the nivolumab monotherapy group, and the benefit of combination therapy was more obvious in the PD-L1–positive and MSI-H subgroups (60) (summary in Table 1).
Antagonizing LAG-3 can restore the anti-tumor activity of T cells (66). LAG-3 as an inhibitory receptor expressed on activated T cells can downregulate T-cell function and participate in T-cell exhaustion and tumor immune escape thus promoting tumor progression (67, 68), and this immunoglobulin superfamily gene was also expressed on NK, DCs, and B cells (66, 69). Tregs cells expressing LAG-3 can suppress tumor-specific T cells and produce high levels of the immunoregulatory cytokines interleukin (IL)-10 and transforming growth factor-beta (TGF-β) (70, 71). The LAG-3 inhibitor relatlimab has been shown to deplete leukemia cells and restore NK and T cell–mediated anti-tumor activity in chronic lymphocytic leukemia (72). So far, more than 10 LAG-3 blockers such as relatlimab (73), ieramilimab (74), and HLX26 (68) have entered clinical trials. In addition, multi-targeted ICI combination therapy targeting LAG-3 can improve anti-tumor efficacy because LAG-3 is frequently co-expressed with PD-1 on TILs (73). Anti–LAG-3 plus anti–PD-1 therapy prolonged PFS in patients with advanced melanoma compared with anti–LAG-3 and anti–PD-1 alone in a phase III melanoma trial (68). At present, several clinical trials targeting LAG-3 in the treatment of GC are being carried out (Table 2), which is expected to become the next immunotherapy target for the treatment of GC.
Table 2 Clinical trials of targeting LAG-3 in gastric cancer.
Targeting TIM-3 is another strategy based on T-cell immunotherapy. TIM-3 is an important tumor immune checkpoint expressed on a variety of immune cells including effector T cells, monocytes, NK, and DCs (75, 76), which can inhibit innate and T-cell immune response (77, 78), participate in immune escape (79), and promote immune tolerance (80). Blocking TIM-3 pathway can positively regulate innate and adaptive immunity, alleviate T-cell depletion, and increase the secretion of interferon-γ (IFN-γ) by NK and T cells (75, 81). TIM-3 and its ligands are highly expressed in various solid tumors such as GC, and its overexpression was associated with the aggressiveness, late stage, and poor prognosis of malignant tumors (75, 82–84). Therefore, TIM-3 can be used as a prognostic biomarker for various solid tumors. Three polymorphisms (−574G/T, −882C/T, and −1516G/T) within the TIM-3 gene were found to be significantly associated with GC in evaluating the association between TIM-3 gene variants and GC development, among which the −1516 polymorphism genotype was associated with distant metastasis of GC (85). The limited clinical efficacy of anti–TIM-3 antibody (LY3321367) led to the early termination of the study despite which has good pharmacokinetics/pharmacodynamics in the treatment of advanced solid tumors (86). However, it did not block the exploration of this target. Currently, there are two clinical studies on anti–TIM-3 treatment of GC, and we look forward to the announcement of positive clinical data. At the same time, the strategy of multi-target–combined blockade was also being explored in therapy. Dual blockade of TIM-3 and PD-1 seems to improve the anti-tumor ability of T cells because blocking PD-1 can upregulate the expression of TIM-3 (87). Facts also proved this point of view, a clinical trial of TIM-3 antibody (sabatolimab) combined with PD-1 antibody (spartalizumab) showed anti-tumor activity and well tolerated in the treatment of advanced solid tumors (88). However, there are also studies that reported a bispecific antibody (bsAb) against TIM-3 and PD-L1 resulting in early study termination in the treatment of advanced solid tumors because all patients developed anti-drug antibodies (89). Therefore, strategies for targeting TIM-3 and in combination with other treatments require further research and improvement.
TIGIT is an emerging immune checkpoint expressed on tumor-infiltrating CD8+ T cells, TH, Tregs, and NK cells in various solid tumors including GC (90), which is associated with poor prognosis in various malignancies (91). Binding of TIGIT to CD155 ligand overexpressed on tumor cells led to immune escape while also suppressing innate and adaptive immunity, including inhibition of NK-mediated cytotoxicity and IFN-γ production, inhibition of DCs produces IL-12, inhibition of CD4+ T, CD8+ T cells proliferation, and induction of IL-10 production (92). Based on the negative regulatory effect of TIGIT on tumor immunity, currently, there are a variety of anti-TIGIT mAbs as monotherapy or combined with PD-1/PD-L1 antibody or combined with chemotherapy to treat tumors. Clinical data showed that anti-TIGIT antibody tiragolumab plus anti–PD-L1 antibody atezolizumab improved overall response and PFS and was well tolerated compared with anti–PD-L1 monotherapy in patients with PD-L1–positive metastatic NSCLC (93, 94). A phase II clinical trial (NCT04933227) in GC is underway to explore the efficacy and safety of atezolizumab in combination with tiragolumab and chemotherapy in the first-line treatment of HER2-negative recurrent or metastatic G/GEJ cancer.
The ACT is T lymphocytes in peripheral blood or tumor-infiltrating is isolated and activated, expanded or genetically modified in vitro, and then infused back into the patient’s body. These treated T lymphocytes have potent anticancer activity (95). A meta-analysis showed that ACT treatment of GC could significantly improve PFS and prolong OS (96). ACT includes CAR-T (97), TIL ACT (98), and CIK ACT (97). CAR-T therapy is a promising therapeutic strategy in the field of cancer because it can cause target cell death and does not require activation of APCs (99, 100), which has an obvious curative effect in the treatment of hematological tumors (101). In the strategy of CAR-T treatment of GC, multiple targets have been proved to be effective in the treatment of xenograft mouse GC model, such as mesothelin (MSLN), prostate stem cell antigen (PSCA), B7 homology 3 (B7H3) protein, and NK group 2 member D (NKG2D) (102–105). In addition, CAR-T targeting carcinoembryonic antigen (CEA), HER2, CLDN18.2, FOLR1, c-Met, CD133, and CDH17 had a significant anti-tumor effect in the corresponding target-positive GC mouse model and the tumor appeared partially or even complete regression (106–112). CAR-T cells targeting ICAM-1 can effectively eliminate tumors developing in the lungs in animal models of GC (113). Modified CAR-T on the basis of the original target can play a role in increasing the efficacy, such as EGFR-CAR-T that secretes PD-1 scFv, c-Met CAR-T that adds PD-1/CD28 chimeric switch receptor (CSR), and bispecific human trophoblast cell surface antigen 2/PD-L1 CAR-T, which all have been proven to increase the efficacy of the original CAR-T therapy in mouse GC models (114–116). Up to now, there are as many as 30 clinical trials of CAR-T in the treatment of GC. Among them, the interim results of the phase I clinical trial of CLDN18.2-targeted CAR-T cells (CT041) in GC showed that CT041 treatment of CLDN18.2-positive GC patients had 57.1% ORR and 75.0% DCR; the 6-month OS rate was 81.2% and the safety was acceptable (117).
TIL ACT is usually isolated from the fresh tumor tissue of the patient and then returned to the patient after activation and expansion in vitro. GC TILs can be obtained from primary tumors (PTs), metastatic lymph nodes, and malignant ascites in GC patients, and TILs from different sites can recognize tumor cells in corresponding sites (118). TIL ACT has seen good efficacy in the treatment of GC. A clinical study showed that the survival time of the TIL treatment group was 50% longer than that of the chemotherapy group (119). Another report showed that intravenous injection of autologous TIL combined with recombinant IL -2 resulted in 13.0% tumor elimination (complete remission) and 21.7% tumor growth inhibition (partial response) of advanced GC patients (120). It has seen significant anti-tumor effects and good safety not only in the treatment of GC but also in various solid tumors (98). Therefore, it is a promising immunotherapy for the treatment of solid tumors. However, the wide clinical application of TIL ACT may be limited due to the high preparation cost, long production time, and the need for specialized facilities and personnel to produce TIL.
CIK has strong anti-tumor activity, MHC-independent, and antibody-dependent cytotoxicity (121). CIK can be produced by peripheral blood mononuclear cells (PBMCs) under the action of IFN-γ, IL-2, and anti-CD3 antibodies (122) and finally differentiate into CD3 CD56 CD8 NK-T cells. The cytotoxicity of CIK depends on the interaction of NKG2D receptors with NKG2D ligands and is mediated by perforin and can also regulate and enhance host cellular immune function by secreting cytokines and chemokines (123). In addition, CIK therapy can improve the survival rate of GC patients and can significantly improve the OS and disease-free survival (DFS) of patients with stage III GC when combined with chemotherapy (124). The 5-year OS and DFS were significantly prolonged after CIK treatment compared with the control group in patients with locally advanced GC (125). In addition, chemotherapy combined with CIK/DC-cytokine induced killer cell (DC-CIK) can significantly improve the OS rate, DFS rate, and T lymphocyte response in the treatment of postoperative patients with GC (105), and the survival was beneficial compared to chemotherapy alone (126).
Tregs in TME can be classified into three types according to the expression of FOXP3 and CD45RA: non-Tregs (FOXP3lowCD45RA−), naive Tregs (FOXP3lowCD45RA+), and effector Tregs (eTregs) (FOXP3highCD45RA−). Non-Tregs cannot exert an inhibitory effect but can secrete pro-inflammatory cytokines. Naive Tregs are only weakly suppressive, whereas eTregs, which differentiate from naive Tregs after antigenic stimulation, possess the strong suppressive activity and stable function (127). Tregs express CTLA-4, PD-1, inducible T-cell costimulator (ICOS), glucocorticoid-induced TNFR-related protein (GITR), tumor necrosis factor receptor 4 (OX40), vascular endothelial growth factor receptor-2 (VEGFR2), chemokine receptor 4 (CCR4) and CCR8 receptors, which can mediate tumor immunosuppression (128); and participate in co-stimulatory receptors on the surface of APCs to modulate APC activity, leading to weaken or abrogated signals from APC to naive/effector cells. It can secrete immunosuppressive cytokines (IL-10, TGF-β, and IL-35) and immunosuppressive metabolites (tryptophan and adenosine), deplete the cytokine IL-2, and inhibit APC maturation (such as DCs) and tumor antigen-specific T-cell responses (128). eTregs may also cause metabolic disruption to prevent naive/effector cell proliferation. Under certain circumstances, eTregs could have a direct cytotoxic effect through the production of perforin/granzyme and induce apoptosis in effector cells. Dead Tregs can also rapidly convert ATP into adenosine in tumors, which then binds to receptors on the T-cell surface and affects T-cell function (129, 130), which in turn leads to the occurrence and progression of cancer (Figure 2).
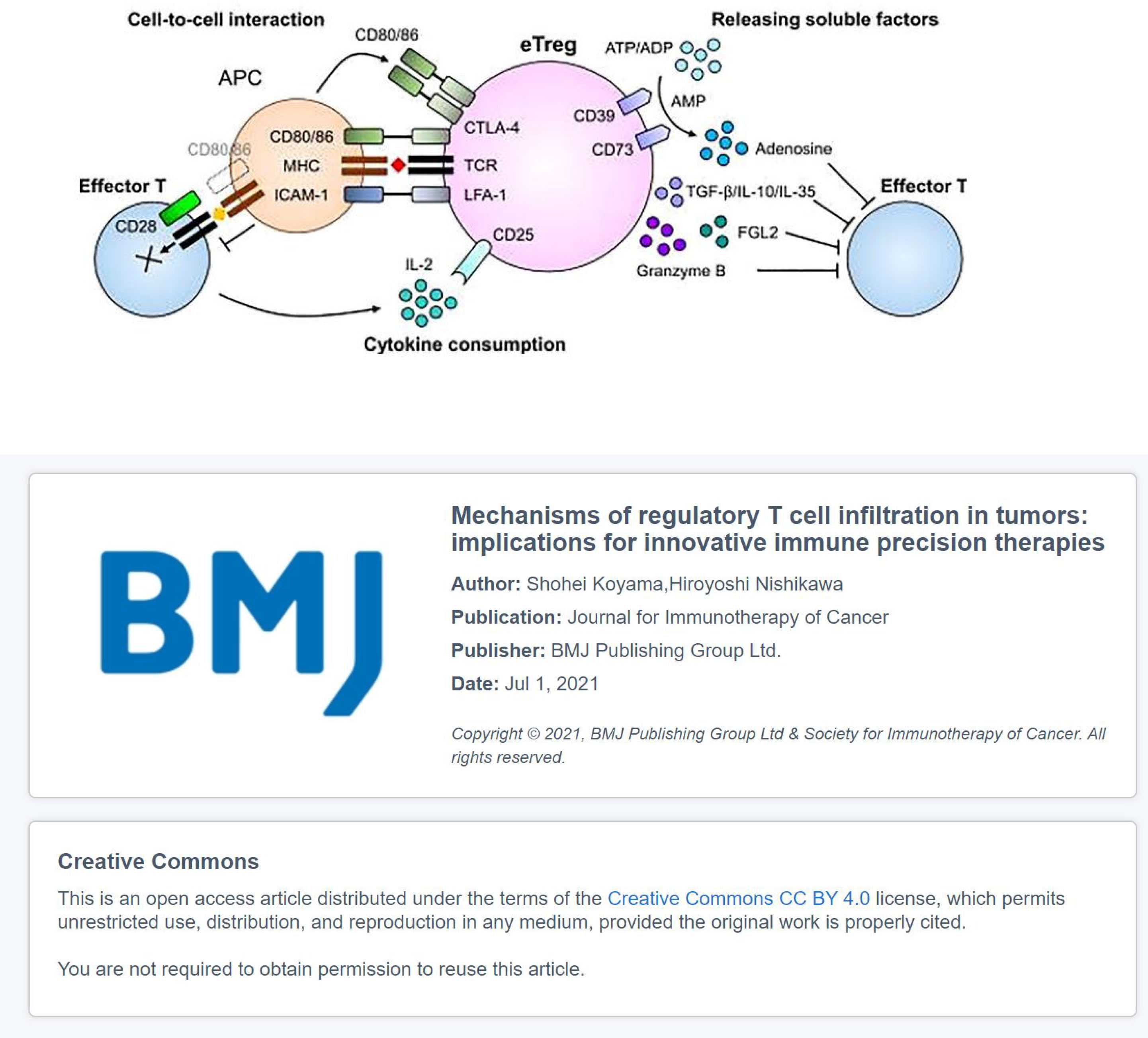
Figure 2 The immunosuppressive mechanisms of eTregs: The co-inhibitory receptor CTLA-4 in Tregs binds to CD80 and inhibits co-stimulatory signaling from APCs; Tregs can secrete inhibitory cytokines, including IL-10, TGF-β, and IL-35; Tregs can kill effector cells by granzyme and perforin and bind to the Fc fragment of IgG receptor IIB (FcγRIIB) in CD8+ T cells by secreting Fgl2, leading to their apoptosis. Tregs influence effector cell function: Tregs contain higher affinity receptor CD25 of IL-2, which compete with effector T cells to deplete IL-2, thereby inhibiting the growth of effector T cells; CD39 and CD73 expressed on the cell surface of Tregs act as ectonucleotidases that hydrolyze ATP or ADP to AMP and AMP to adenosine, respectively, thereby inhibiting effector T cells (131).
Eliminating the immunosuppressive effect of Tregs is beneficial to restoring the tumor immune response, and the strategies can be achieved by reducing Tregs, including direct strategies acting on Tregs surface molecules and indirect strategies to reduce Tregs through other ways. Direct strategies can be achieved by blocking surface molecules such as CD25, CTLA-4, PD-1, ICOS, GITR, OX40, and VEGFR2 on Tregs. The CD25-blocking mAb daclizumab can result in the Tregs of patients significantly and a long-term decrease in the treatment of metastatic breast cancer patients (132). Loss or inhibition of CTLA-4 resulted in decreased Tregs function, and anti–CTLA-4 antibodies promoted anti-tumor activity by selectively reducing intratumoral Tregs (133). In addition, ICOS antibody KY1044, anti-GITR antibody TRX518, anti-VEGFR2 antibody ramucirumab (RAM), and chemotherapy drugs, such as low-dose cyclophosphamide, cyclosporine A, and tacrolimus, all can reduce Tregs in tumor patients (131, 134–136), while anti-OX40 antibody produced anti-tumor activity by blocking the inhibitory effect of Tregs (137). The strategies to indirectly reduce Tregs can be achieved by blocking the chemokine and/or cytokine axis, intracellular signaling pathways, and metabolites of Tregs. Tregs can migrate to TME under the action of chemokines such as CCR4-CCL17/22, CCR8-CCL1, CCR10-CCL28, and CXCR3-CCL9/10/11 (128), so blocking chemokines and chemokine receptors can inhibit Tregs migration thus indirectly reduce Tregs in the TME. Anti-CCR4 mAb and anti-CCR8 mAb have been shown to selectively deplete tumor-infiltrating Tregs (138, 139). In addition, the tyrosine kinase inhibitor imatinib can selectively induce Tregs apoptosis by reducing the intensity of TCR signaling through the inhibition of lymphocyte-specific protein tyrosine kinase (LCK) (140). Specific inhibitors of phosphoinositide 3-kinase (PI3K) δ can also improve cancer immunotherapy by reducing the number of Tregs in the tumor microenvironment (141). At the same time, adjusting metabolites in the TME also affects Tregs numbers, such as targeting fatty acid uptake (e.g., inhibition of fatty acid transporter CD36 and lactate transporter monocarboxylate transporter 1), blocking fatty acid oxidation (carnitine palmitoyltransferase 1a inhibitor), and blocking fatty acid synthase (acetyl-CoA carboxylase inhibitor (5-(tetradecyloxy)-2-furoic acid)) all can inhibit Tregs proliferation and reduce Tregs in TME (131).
Strategies to reduce Tregs either directly or indirectly both can weaken the immunosuppressive effect of Tregs. However, reducing Tregs in the TME may also reduce systemic Tregs and thereby increase the risk of immune-related adverse events (irAEs), such as autoimmune-related toxicities. Therefore, strategies should be used that selectively deplete eTregs in the TME with little effect on systemic Tregs and other Tregs subtypes in order to ensure the safety and efficacy of Tregs cell–targeted therapy. Therefore, the Tregs-targeted therapy strategy needs to be further improved.
The most widely used and the most complete data are PD-1/PD-L1 mAbs and CTLA-4 mAbs in ICIs despite having various strategies for T cell–based immunotherapy in the treatment of GC. Particularly, PD-1 mAbs have clinical data on the first, second, and third lines. Many studies have shown that PD-1 and PD-L1 mAbs have an encouraging survival advantage in GC, which have been approved in several countries for the treatment of advanced GC. Unfortunately, PD-1 and PD-L1 antibodies failed to improve OS and PFS in some trials, and the benefit population in positive clinical trial results is only those with high PD-L1 CPS score, MSH-H, or high TMB, but this group of people is not many. Multi-target blockade therapy may be the future treatment direction as the discovery of successive multiple immune checkpoint receptors. In addition to ICIs recommended by guidelines, CAR-T therapy in ACT has also seen high ORR and DCR in clinical trials and other ACTs also have good prospects. However, the wide clinical application of ACT is limited because the preparation process is cumbersome and its high cost of ACT. At the same time, selective removal of eTregs cells in the TME is also the future direction because most current Tregs-targeted therapies lack selectivity.
The application of DCs in anti-tumor therapy mainly includes cancer vaccines and DC-CIK. Studies have been conducted combining DCs vaccination with chemotherapy (142), radiotherapy (143), targeted therapy (144), and other immunotherapies (145). DCs are used in cancer vaccines related to the anti-tumor mechanism of DCs. DCs are professional APCs that capture antigens released by tumor cells and present them to T cells in tumor-draining lymph nodes thus resulting in the generation of tumor-specific cytotoxic T lymphocytes (CTLs) (146). DCs can also stimulate NK and B cells to activate humoral immunity (146, 147). Studies showed that tumor-infiltrating DCs were associated with clinical stage, invasion, metastasis, and better prognosis in GC patients (148, 149). GC patients with many DCs invasion had lower LNM and lymphatic invasion, and also 5-year survival (78%) and OS higher than patients with little DC invasion (150, 151).
DC vaccines have shown OS benefits in solid tumors such as prostate cancer (152), melanoma (153), glioblastoma (154), and ovarian cancer (155). The studies showed that inactivated tumor cells (156), tumor lysates (153), tumor vesicles (157), synthetic tumor peptides (158), or synthetic tumor antigen mRNA (159) all can be used as DC vaccine–loaded antigens. Antigens that can be used as GC vaccines include melanoma-associated antigen (MAGE) A3, HER2 (p369) peptide, gastin-17 diphtheria toxoid (g17DT), URLC10 or VEGFR1 epitope, and heat shock protein GP96 (160). Up to now, there are five clinical trials of DC vaccines in the treatment of GC (Table 3).
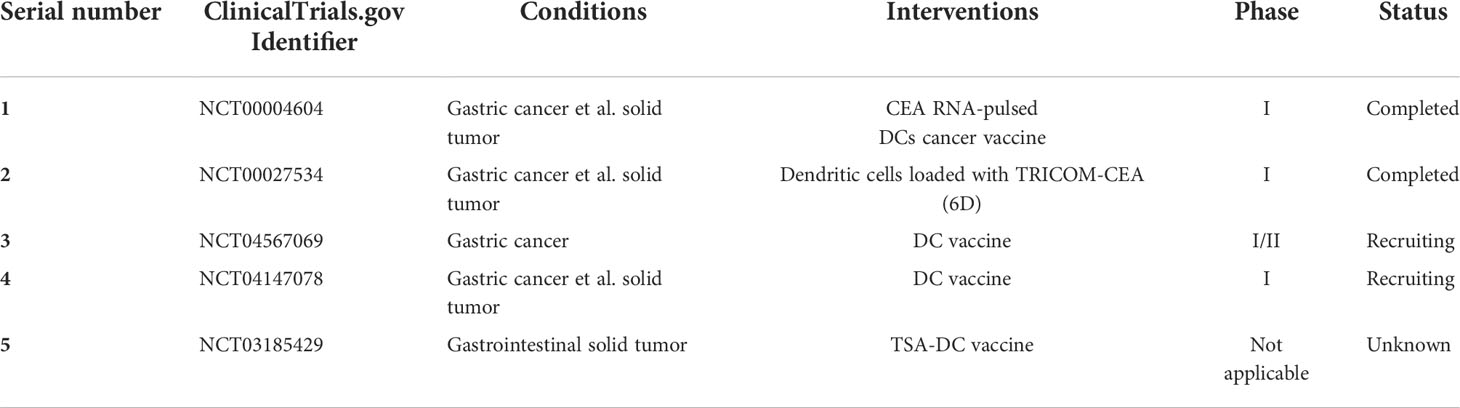
Table 3 Clinical trials of DC vaccine in the treatment of gastric cancer.
Unfortunately, up to now, there are not many positive results of DC vaccines in the treatment of GC. Although phase I/II clinical trial has shown that Wilms tumor 1 (WT1)–targeted DC vaccine was a potential treatment in advanced cancer including GC, only three GC patients were included in the enrolled patients and only one GC patient was effective (161). Therefore, strategies to target multiple antigens have been explored in order to improve the efficacy of GC vaccines. For example, OTSGC-A24 achieved an impressive OS (5.7 months) in the treatment of advanced GC, which is a HLA-A*24:02 peptide-conjugated vaccine targeting FOXM1, DEPDC1, KIF20A, URLC10, and VEGFR1 (162). In addition, whole GC cells can also be fused with DCs to generate DC tumor hybrids, which have the advantage of combining the powerful antigen-presenting capacity of DCs with all antigens expressed by tumor cells (163, 164). At the same time, DC vaccines can also be combined with chemotherapy, radiotherapy, and ICIs to increase efficacy. Moreover, DC vaccines combined with neoadjuvant chemotherapy (NAC) showed that the combined treatment was safe and increased pathological complete remission (tpCR) in the treatment of HER2-negative breast cancer (165). DC vaccines combined with chemotherapy (carboplatin/pemetrexed) also had good efficacy and tolerability as the first-line drug therapy for patients with advanced non-squamous non–small cell lung cancer without oncogenic drivers (166). In addition, DC vaccines combined with radiotherapy can significantly inhibit tumor growth and improve survival rate, which has been confirmed in many tumor types such as melanoma and esophageal cancer (156, 167). DC vaccines combined with ICIs are also an effective treatment strategy, and preclinical studies had shown that DC vaccines combined with PD-1 inhibitors led to smaller tumor volume and better OS in the treatment of hepatocellular carcinoma (HCC) (145). In terms of toxic and side effects, the side effects of DC vaccines mainly include influenza-like symptoms, fever, and local reactions at the injection site, so it is safe for cancer patients. At present, more than 200 clinical trials have shown high immunogenicity and safety of DC vaccines (158).
Another method of DCs treatment of tumors is DC-CIK. Studies had shown that DC-CIK combined with chemotherapy was effective and tolerable in the treatment of non–small cell lung cancer, breast cancer, colorectal cancer, GC, and other solid tumors. DC-CIK combined with chemotherapy can enhance cellular immune function and inhibit tumor invasion and metastasis in the treatment of advanced non–small cell lung cancer (168). DC-CIK combined with capecitabine prolonged PFS in the treatment of patients with recurrent and metastatic triple-negative breast cancer (169). In addition, DC-CIK combined with adjuvant chemotherapy can significantly prolong the DFS of patients with postoperative colorectal cancer (170). In the treatment of GC, a meta-analysis showed that DC-CIK combined with chemotherapy can significantly improve the OS rate, DFS rate, and T lymphocyte reaction in patients after GC surgery (171). In addition, DC-CIK combined with S-1 and cisplatin had good PFS and OS in the treatment of advanced GC, and the combination therapy was safe and the toxicity was tolerable (172).
Tumor-infiltrating DCs are associated with a better prognosis in GC, but only a few mature DCs in the tumor microenvironment. DC-based immunotherapies such as cancer vaccines and DC-CIK have limited efficacy as a single treatment for cancer. Therefore, in order to increase the anti-tumor effect, it is very necessary to find the reasons for the low efficacy or combine it with other treatments to treat tumors.
NK cells are responsible for destroying tumor cells and preventing tumor initiation and progression. Activated NK cells can exert direct cytotoxicity through death receptor signaling, perforin, or release granzymes, which can also modulate other parts of the immune response by producing cytokines and chemokines (173). However, tumor cells can escape NK cell destruction by binding to inhibitory receptors expressed on the surface of NK cells. At the same time, the overproduction of TGF-β and other anti-inflammatory cytokines and chemokines in the tumor microenvironment can inhibit NK cell activation (174, 175); downregulate NK cell–activating receptors NKp30, NKp44, NKG2D, and CD16 and co-receptors NKp80 and DNAM-1; upregulate checkpoint receptors TIGIT, TIM-3, LAG-3, and PD-1; impair the expression and secretion of CD107; and secrete a variety of immunosuppressive factors (175, 176). Studies have shown that NK cell inhibitory receptor antibodies can restore NK cell activity. The widely used anti–PD-1 and anti–PD-L1 can enhance NK cell–mediated anti-tumor effects. The expression of PD-1 on NK cells interacting with PD-L1 on cancer cells can decrease the responses of NK cells, whereas blocking PD-1 and PD-L1 can increase NK cells in vivo and trigger strong NK cell responses and cytotoxicity in mouse tumor models (177, 178).
The treatment strategies based on NK cells for GC include NK cell adoptive therapy (such as autologous NK cell infusion, allogeneic NK cell infusion, and CAR-NK), blocking the inhibitory receptors expressed on NK cells, and increasing the activity of NK cells (such as increased activating receptors expression on NK cells, activation of NK cells by cytokines, and increased immune clearance of tumors by NK cells).
At present, NK cell adoptive therapies for GC have been carried out in more than 20 clinical trials. Five of 19 patients achieved complete hematologic remission in the clinical trial of haploidentical NK cell therapy for acute myeloid leukemia (AML) (179). However, no objective clinical response was observed in patients with melanoma treated with autologous NK cells activated in vitro (180), which suggests that it is more effective in donors with mismatched killer immunoglobulin receptor (KIR) ligand in NK cell adoptive therapy. In addition, NK cells can also be modified into CAR-NK, which is similar to CAR-T cell activity in vivo and safer than CAR-T cells (181, 182), and no strict HLA-matching requirements (183). Preclinical studies in the treatment of GC have shown that mesothelin-targeted CAR-NK cells can effectively eliminate GC cells in both subcutaneous and intraperitoneal tumors and significantly prolong the survival time of the mouse (184). In addition, studies on CAR-NK therapy targeting HER2, Mucin-1, EpCAM, or PMSA for GC are being carried out (184). CAR-NK therapy is becoming a promising treatment strategy in cancer immunotherapy based on the existing research data and less toxic side effects.
Increasing the anti-tumor activity of NK cells can be achieved by increasing the expression of activating receptors on the surface of NK cells, activating NK cells with cytokines, and increasing the immune clearance of tumors by NK cells. Retroviral transduction with DTCR (PD1-DAP10/NKG2D) can significantly increase the expression of NKG2D (an activating receptor on the surface of NK cells) on the surface of NK92 cells, thus enhancing cytotoxicity against GC cells SGC-7901, and DTCR-NK92 cells showed strong anti-tumor activity in the GC mouse model (185). In addition, the cytokines can activate NK cells and promote their proliferation to enhance anti-tumor activity, such as IL-2, IL-12, IL-15, and IL-18 (186–188). NK cells activated by IL-2 which combined with anti–PD-1 can inhibit tumor growth in xenograft GC models (189). In addition, IL-15 can increase the infiltration of NK cells in the tumor (190) and improve the survival rate in the treatment of GC liver metastasis–bearing mice (191). Primer of blood NK cells with recombinant human (rh)IL-12, rhIL-15, and rhIL-18 (12/15/18) can lead to memory-like NK cell differentiation and enhance tumor response (192). At the same time, increasing the immune clearance of NK cells against tumors can also play an anti-tumor effect. NK cells carry out immune clearance of tumors by releasing cytotoxic particles, antibody-dependent cell-mediated cytotoxicity (ADCC), and protein-activated target cell apoptotic systems synthesized on the cell surface [FasL and tumor necrosis factor-α (TNF-α)]. The mAbs trastuzumab (193), pertuzumab (194), cetuximab (195), rituximab (196), and anti-CD3 × anticancer bsAbs (197) all can play anti-tumor effect by enhancing ADCC ability and NK cell activity.
Blocking the expression of inhibitory receptors on NK cells can restore NK cell function, reverse NK cell depletion, increase NK cell cytotoxicity against tumors, and inhibit tumor growth. The efficacy increased when combined with other targeted drugs or ICIs (195, 198). The inhibitory receptors expressed on NK cells include KIR, leukocyte immunoglobulin-like receptor (LILR), killer lectin-like receptor (KLR) (173), inhibitory receptor composed of NKG2A and CD94, B7H3 protein receptor, sialic acid–binding immunoglobulin-like lectin (Siglecs), TIM-3, LAG-3, TIGIT, CD-47, etc. (199–207). Blocking these inhibitory receptors can restore the activity of NK cells. In addition, immunoglobulin-like transcript 2 (also known as LILRB1) can also inhibit the proliferation and cytotoxic activity of infiltrating NK cells in GC tumor tissues (208), so blocking this receptor can enhance the activation and proliferation of NK cells. The elimination of leukemia cells increased by activating NK cell cytotoxicity when combined with lenalidomide in chronic lymphocytic leukemia patients (209). Currently, several clinical trials are underway to block NK cell inhibitory receptors (210). In addition, blocking TGF-β1 signaling can prevent dysfunction of NK cells and thus restore their activity (211), and gene silencing of the PI3K catalytic subunit PI3KCB can enhance the lytic activity of NK cells against tumors (212).
NK cells are more cytotoxic to tumors, less immunogenic, have faster response, and do not need additional connections to activate receptors when compared with effector T cells. For example, the current hot spot treatment strategy CAR-NK is a promising treatment strategy for GC due to its unique recognition mechanism, strong cytotoxicity, clinical safety, and the ability to reduce the risk of allogeneic reactions. However, the application of CAR-NK cells is limited because the complex preparation and expensive and solid tumors have no immune-specific target antigen, a loss of tumor antigens, low persistence, and other factors. Another method of targeting NK cells to treat tumors is to restore and increase the anti-tumor activity of NK cells, and some effects have been seen in related studies but the efficacy is limited when used alone; combination therapy may be a strategy to solve this problem.
TAMs are one of the most important components of the tumor microenvironment and are potential targets for cancer therapy. At present, the most studied is the “reprogramming” of TAMs from tumor support cells to tumor killer cells, that is, the reconversion of M2 TAMs to M1 TAMs. In addition, treatment strategies based on TAMs also include limiting monocyte recruitment and localization and CAR macrophage (CAR-M) therapy. Macrophages are divided into two subtypes: M1 or classically activated macrophages and M2 or alternatively activated macrophages. M1 TAMs have the inhibition effects of tumor and anti-angiogenic (213), whereas M2 TAMs can promote the occurrence and metastasis of tumor cells, inhibit the anti-tumor response mediated by T cells, and promote tumor progression and tumor angiogenesis (214, 215). In tumors, M2 macrophages are dominating in the tumor microenvironment as the tumor progresses (216). Chronic inflammation is a feature in GC tumor microenvironment, which is derived from infections such as Helicobacter pylori. These pathogens can impair the response of M1 TAMs, induce the state of M2-like, increase macrophage apoptosis, and promote disease progression (217). In addition, TAMs are related to the occurrence, development, and prognosis of GC, and CD204-positive (an M2-polarized macrophage receptor) TAMs are an important risk factor for a gastric adenoma to develop into adenocarcinoma (218). At the same time, the number of TAMs can predict the size and stage of GC in the GC tumor microenvironment (219) and involved in tumor invasion and metastasis (220). Furthermore, M2 TAMs were associated with poor prognosis and were an independent prognostic factor for GC (221).
The effect of phagocytosing tumor cells can increase by “Reprogramming” TAMs from tumor-supporting cells to tumor-killer cells, including the use of targeted antibodies (for example, targeting MФ surface receptors involved in immune response regulation and targeting the circulation cytokines/growth factors), gene therapy, small-molecule inhibitors, episomal vector delivery of nucleic acids, etc. Among them, the most studied therapeutic strategy is targeted antibodies, and the targets include colony-stimulating factor 1 (CSF-1)/CSF 1 receptor (CSF-1R), Toll-like receptors (TLR7, TLR8, and TLR9), histone deacetylase (HDAC), PI3Kγ, CD40, and CD47 (222). Neutralizing antibodies or small-molecule inhibitors of CSF-1/CSF-1R, CD40 antibodies, and TGF-β blockers all have been shown to reprogram M2 TAMs to M1 TAMs (222–224). Furthermore, TLR agonists can induce M1 polarization by increasing the release of pro-inflammatory mediators (225), and this therapy has shown promise in preclinical solid tumor models and in the clinic (226–228). The inhibition of HDAC or PI3Kγ exerted anti-tumor effects by downregulating M2 and upregulating M1 molecules (222). CD47 also affects TAMs polarization, and the anti-CD47 antibody increased the ability of macrophages to phagocytose tumor cells by blocking the interaction of CD47 with SIRPα on macrophages, which has been demonstrated in various preclinical models of solid tumors (229, 230). In addition, the suppression effect of macrophages can be abolished by inhibiting monocyte recruitment and localization to tumor tissue by targeting macrophage chemokines or their receptors (e.g., chemokine 2, chemokine 5, and CSF-1R). For example, blocking the CCL2/CCR2 axis can inhibit monocyte recruitment, TAM infiltration, and M2 polarization. Knocking down CCR2 or blocking CCL2/CCR2 signaling with CCR2 antagonists can inhibit tumor growth and metastasis, reduce postoperative recurrence, and improve survival (231). CCL5-CCR5 and CXCL12/CXCR4 also mediate TAMs recruitment and polarization, so blocking their mediated signaling was also a potential therapeutic strategy (232, 233). Macrophage recruitment was also promoted when CSF-1R binds to its ligand CSF-1 (234, 235), whereas the CSF-1R antagonists PLX 3397 or pexidartinib prevented the recruitment of monocytes from the circulation to cancerous tissues (236). In addition, drugs such as bisphosphonates also affect TAM infiltration and polarization. For example, zoledronic acid has been shown to modulate the tumor microenvironment by reducing TAM infiltration and polarization state (237, 238), which can also reduce angiogenesis by macrophage (239). At the same time, the efficacy of some chemotherapy drugs (such as trabetidine) may be related to the ability to kill TAMs (240). In addition, radiation therapy also affects TAMs as low-dose radiation therapy can reprogram TAMs to an anti-tumor phenotype (241).
CAR-M has also been developed and applied based on the good efficacy of CAR-T cells in hematological tumors (242). The CAR based on CD3ζ was highly active in human macrophages, which can drive phagocytosis and kill target tumor cells in a Syk-dependent manner (243). Studies have shown that CAR-M can transform M2 TAMs into M1 TAMs, upregulate the antigen presentation mechanism, recruit antigens and present them to T cells, express pro-inflammatory cytokines and chemokines, and resist the effects of immunosuppressive cytokines. CAR-M exhibited the effects of antigen-specific phagocytosis and tumor clearance in vitro. The tumor burden is reduced and OS is prolonged after infusion of CAR-M of human in mouse xenograft tumor models (243). At present, the treatment of CAR-M has entered the clinical trial stage and one of them is a study of CAR-M in the treatment of HER2-overexpressing solid tumors, including HER2-positive GC and other solid tumors. Another is a study of CAR-M in breast cancer. In addition, studies on the combination of CAR-M and T-cell checkpoint inhibitors are also ongoing based on the interaction between CAR-M and the adaptive immune system (243).
TAMs are the most abundant immune cells in the tumor microenvironment. Many strategies targeting TAMs to treat tumors have been carried out in preclinical studies and were proved effective, looking forward to the data of these therapeutic methods in clinical studies.
MDSCs are a highly heterogeneous group of myeloid-derived cells whose most important function is immunosuppression (244). Inhibiting DC function and anti-tumor T-cell response, inducing NK cell apoptosis, promoting M2 TAMs differentiation, and increasing the number of Tregs (244, 245) (Figure 3) can promote the growth and metastasis of PTs (246, 247). Expansion of MDSCs was associated with resistance to treatment and poor prognosis in malignant tumors (246). In GC patients, the levels of MDSCs was associated with cancer stage and survival (248), such that higher levels of MDSCs were associated with later tumor stage and poor prognosis (249–251), as well as with higher mortality and risk of tumor recurrence and progression. In addition, patients with high MDSCs levels had significantly shorter OS than patients with low MDSC levels in patients with stage IV gastrointestinal cancer (252).
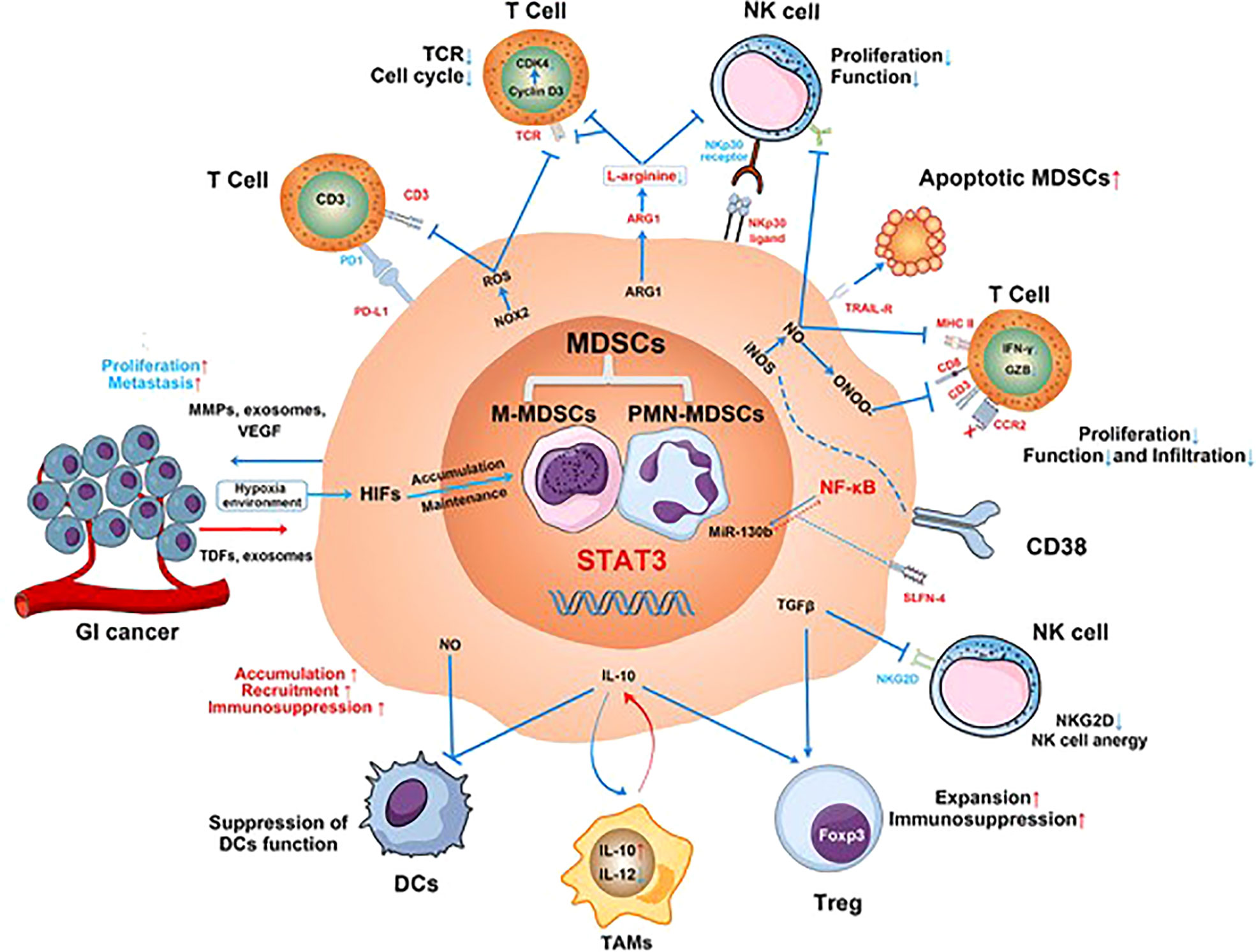
Figure 3 The mechanisms involved in MDSC-mediated immunosuppression in gastrointestinal (GI) cancer. MDSCs suppress proliferation and function of T cells and NK cells; reduce CD8+ T-cell infiltration; inhibit the function of DCs; inhibit the antigen presentation of DCs to CD4+ T cells; promote M2 macrophage differentiation; and promote Tregs expansion and immunosuppression. Additionally, the effect of ADCC function and anergy of NK cells is induced by the production of nitric oxide (NO) and the inhibition of NKG2D by TGF-β, respectively. MDSCs secrete matrix metalloproteinases (MMPs), exosomes, and vascular endothelial growth factors (VEGF) to promote GI cancer cell proliferation and metastasis (245).
Eliminating the immunosuppression of MDSCs is beneficial for restoring anti-tumor immunity, and strategies include reducing circulating and tumor-infiltrating MDSCs and eliminating the immunosuppressive functions of MDSCs (244). The reduction of circulating and tumor-infiltrating MDSCs can be achieved with some chemotherapy drugs, targeted drugs, all-trans retinoic acid (ATRA), or by blocking the chemokine receptor on MDSCs. Low-dose chemotherapy drugs such as 5-fluorouracil (253), paclitaxel (254), gemcitabine (255), platinum (256), and Adriamycin (257) all have been shown to reduce MDSCs in cancer patients. 5-Fluorouracil combined with oxaliplatin reduced the number of MDSCs in the mouse model of GC (258). Epirubicin or docetaxel can reduce MDSCs and inhibit MDSCs function in GC patients and can also induce MDSCs apoptosis through mitogen-activated protein kinase (MAPK) and NF-kappa B signaling pathway (259). In addition, the targeted drugs apatinib and ATRA both can downregulate the proportion of MDSCs and reduce the number of MDSCs in blood circulation (260, 261). At the same time, ATRA can restore the accumulation of intratumoral MDSCs induced by anti-VEGFR2 (262). The number of MDSCs can be reduced by blocking the chemokine or cytokine receptors (e.g., CCR5, CSF-1R, CXCR1, and CXCR2) targeting MDSCs, which prevented myeloid marrow cells from trafficking into peripheral lymphoid organs or the tumor microenvironment (263–266). In addition, anti-CCR5 therapy also can reduce granulocyte-like MDSCs (G-MDSCs) and monocyte MDSCs (M-MDSCs) in peripheral and tumors of GC patients (267). At the same time, anti-CSF-1R can significantly reduce the ratio of MDSCs in tumor-infiltrating immune cells (268) and resulted in greater inhibition of tumor angiogenesis and tumor growth when combined with anti-VEGFR-2 antibodies (269). In addition, SX-682 (a small-molecule inhibitor of CXCR1 and CXCR2) can also inhibit the migration of MDSCs and eliminate the accumulation of MDSCs in tumor (270). The targeted drug Bruton’s tyrosine kinase ibrutinib had been confirmed to play an anti-tumor effect by reducing the recruitment and the number of MDSCs and inducing the maturation of MDSCs (271).
In addition, the growth of tumors can be driven by the interaction between immune cells and cancer stem cells (CSCs) in the tumor microenvironment. CSCs are the main cause of tumor metastasis, drug resistance, and recurrence (272). A study showed that GC tissue–derived mesenchymal stem cells (MSCs) can impair the anti-tumor immune response of PBMCs through disruption of the Treg/TH17 balance (273). MSCs can induce MDSCs in the tumor microenvironment (274), induce the generation of G-MDSCs, and modulate their activation, and can also coordinate MDSCs to transform the bone marrow (BM) microenvironment into an immunosuppressive environment (275). In addition, the metastasis of GC can be promoted through the inhibition of serine/threonine protein kinase 24 (STK24) expressed in normal and GC tissues, and this promotion was achieved by inducing overexpression of the GC stem cell marker CD44, enhancing CD11b+Ly6C+ MDSCs in the mouse spleen, and inducing their expansion (276). Infection with H. pylori [classified as a group 1 carcinogen by the World Health Organization (WHO)] can promote gastric stem cell–like properties by altering the microenvironment of the gastric mucosa (277), such as promoting the migration of myeloid cell differentiation factor Schlafen 4+ (SLFN4+) MDSCs into gastric metaplasia (278), enhancing the infiltration of MDSCs in the tumor microenvironment, and increasing their number (279), which are beneficial to the generation, proliferation, and survival of GC CSCs (280). Therefore, reducing the number of MDSCs will attenuate the generation of GC stem cells, and targeting CSCs can also play an anti-tumor effect by indirectly reducing MDSCs.
Treatments to inhibit the immunosuppressive function of MDSCs include phosphodiesterase-5 inhibitors (e.g., sildenafil and tadalafil) (281, 282), cyclooxygenase-2 inhibitors (COX-2), triterpenoids, and some targeted drugs (283). Phosphodiesterase-5 inhibitors can reduce the function of MDSCs by downregulating the expression of ARG1, IL4Ra, and ROS (282), which have shown positive results in patients with head and neck squamous cell carcinoma and melanoma in clinical trials (284, 285). The function of MDSCs has been suppressed by celecoxib as an immunomodulator of targeting COX-2 (286). In addition, triterpenoids can inhibit the suppression of effector T cells by MDSCs-mediated and have shown promising anticancer results in phase I clinical trials (287). The targeted drug tyrosine kinase inhibitor sunitinib can modulate anti-tumor immunity by reversing the immunosuppression mediated by MDSCs (288). In addition to mediating MDSCs migration, the CCR1 and CCR5 silenced in vivo can also lead to repolarization of MDSCs into tumor-killing neutrophils thus playing an anti-tumor effect (256).
MDSCs play a crucial role in promoting tumor progression and metastasis and generating immunosuppressive TME. The efficacy is limited in targeting MDSC treatments as monotherapy although they have seen efficacy in preclinical studies, whereas, combining with ICIs, they have seen synergistic effects in animal tumor models and have entered the clinical trials stage (244). Therefore, the anti-tumor efficacy may be increased by combining anti-MDSCs therapy with other anti-tumor means or combining multiple anti-MDSCs therapies, which is a promising therapeutic strategy.
Neutrophils can also be targets for anticancer therapy and strategies mainly include suppressing neutrophil immunosuppression by altering neutrophil recruitment and migration, depleting neutrophils at tumor sites, increasing neutrophil anti-tumor activity, and altering neutrophil polarity.
Neutrophils in the BM are released and migrated to the tumor microenvironment under the stimulation of mediators such as granulocyte CSF (G-CSF), granulocyte-macrophage CSF (GM-CSF), and chemokines such as CXC and CCL3 (289). TANs exhibits two subtypes under the action of cytokines in the tumor microenvironment: N1 TANs with anti-tumor effect and N2 TANs with tumor support activity (290). N1 TANs can directly kill tumor cells by releasing ROS and reactive nitrogen species (RNS), which can also promote the activation of T cells and the recruitment of M1 TAMs, whereas N2 TANs inhibits the function of NK cells and recruits M2 TAMs and Tregs. It can also release matrix metalloproteinase 9 (MMP9) to promote angiogenesis and the spread of tumor cells (291). The N2 TANs phenotype increased in the TME since the high expression of TGF-β in the tumor microenvironment (292). Neutrophils are highly enriched and can enhance GC cell migration, invasion, and epithelial-mesenchymal transition (EMT) by secreting IL-17a in GC (293). The infiltration of neutrophils is closely related to the development of metachronous GC after endoscopic submucosal dissection (ESD) (294). Furthermore, the high levels of TANs are associated with disease progression and poor prognosis in GC (295). TANs are an independent risk factor for LNM in patients with early GC (EGC) (296). In addition, one study established a method to measure N2 TANs (cN2: CD15 minus CD66) and the results showed that cN2 TANs were closely associated with clinicopathological factors such as T stage, lymphatic, and perineural invasion in GC and were an independent marker of poor prognosis in DFS and OS (290). In addition, treatments can affect TANs, and the density of TANs decreased in tumor tissues of GC after neoadjuvant therapy compared with untreated GC (297).
The number of TANs can be reduced by decreasing neutrophil migration and recruitment to tumor sites in the tumor microenvironment. G-CSF can support tumor progression by mobilizing TANs (298), and neutralizing G-CSF or neutralizing IL-17, the upstream regulator of G-CSF can prevent neutrophil accumulation and downregulate the T-cell inhibitory phenotype of neutrophils (299). Furthermore, the CXCL/CXCR1/2 signaling axis is critical for neutrophil recruitment (300) and inhibition of CXCR1/2 signaling can reduce neutrophil recruitment (301); in addition, CXCR2 blockade as a single drug can prevent TAN accumulation and reduce tumor burden in tumor-bearing mice and can also enhance the efficacy of chemotherapy and immunotherapy (302, 303). The chemokine CXCL8 and the chemokine receptor CXCR4 are also involved in the recruitment of neutrophils in the tumor microenvironment. Inhibiting CXCL8 or blocking CXCR4 can inhibit the infiltration of TANs in TME (304–307). IL-6 can also attract TANs to the tumor environment and can attenuate and reverse the pro-inflammatory effects of neutrophils in the tumor microenvironment thus leading to immune-killing resistance. Therefore, immunotherapy targeting IL-6 is a potential target for tumor treatment via TANs (308). In addition, some targeted drugs such as ALK inhibitor lorlatinib and c-Met inhibitor capmatinib can also reduce TANs by inhibiting the entry of neutrophils in the BM into circulation (309, 310).
Depletion of TANs at tumor sites or neutrophil subsets with tumor-promoting functions is also a therapeutic strategy. The morphology and functions of polymorphonuclear MDSCs (PMN-MDSCs) are very similar to N2 TANs. The research showed that targeting TRAIL-R2 resulted in the elimination of different populations of MDSCs such as PMN-MDSCs and eMDSCs, without affecting mature myeloid or lymphoid cells (311). Splenectomy or the tyrosine kinase inhibitor sorafenib (low dose) can also attenuate or inhibit the inhibitory effect of tumor PMN-MDSCs on T-cell proliferation and cytotoxic activity (312).
In addition, it can also treat tumors by increasing the anti-tumor ability of neutrophils, and targeting Fc receptors on neutrophils can play an anti-tumor effect through antigen-dependent cytotoxicity (ADCC). Neutrophil-dependent ability induced by different tumor-associated antigens to kill tumor cells had been confirmed in extensive preclinical experiments in CD89 transgenic mice (including breast, colon, renal cell carcinoma, and T- and B-cell lymphoma) (313). Blockade of the Fas ligands that are upregulated by PMN-MDSCs can improve the anti-tumor efficacy of adoptive T-cell therapy in the TiRP melanoma model and improve the efficacy of checkpoint blockade in transplanted tumors (314).
Altering neutrophil polarization is also a therapeutic strategy, such that the immunosuppressive cytokine TGF-β can differentiate neutrophils into the N2 phenotype, and blocking TGF-β using the TGF-β inhibitor SM16 resulted in the accumulation of N1 TANs (312). Furthermore, type I IFN also can polarize TANs into the N1 phenotype in mouse tumor models, and similar changes were observed in melanoma patients with treated IFN-β (315).
A variety of therapeutic strategies targeting neutrophils are being carried out, but most of them are in the preclinical research stage; therefore, further validation of data from clinical trials is required.
B cells can not only participate in the humoral immune response by producing antibodies and cytokines but also have a role in antigen presentation and immune regulation. In tumors, B cells are mainly concentrated in the tumor margin and the lymph nodes close to the tumor (316). In addition, B cells infiltrating the tumor margin differentiate into Bregs under the action of growth factors and different signaling pathways, which can support tumor growth by suppressing anti-tumor responses through producing anti-inflammatory cytokines and expressing inhibitory molecules (317) (Figure 4). For example, it can inhibit TH17, TH1, and CD8+ T-cell responses; inhibit CD4+ T-cell proliferation and induce their death (319); inhibit the production of IFN-γ and TNF-α (320); promote Tregs expansion (320) and increase the expression of CTLA4 on Tregs (321); secrete TGF-β, IL-10, and IL-35 (322); and affect the balance of TH1/TH2 (323). In addition, Bregs also express PD-1 and PD-L1 (322). In GC, Bregs are significantly increased in tumor tissues compared with surrounding tissues (324, 325), and the frequency of which in peripheral blood is significantly higher than that in the healthy control group (326). Studies have shown that Bregs were significantly associated with poor prognosis in GC patients. The 5-year OS rate in BregLow GC patients was significantly better than that in BregHigh patients (326). Therefore, reducing Bregs can help increase the anti-tumor response based on Bregs-mediated immune escape. The treatments can take measures such as reducing the number of Bregs and reversing B cell–mediated immunosuppression. The number of Bregs can be reduced by proteasome inhibitor bortezomib, MEK inhibitor cobimetinib, and CD22 antibodies (327–329). Anti–IL-10 antibodies can inhibit the secretion of IFN-γ and TNF-α by Bregs (324). In addition, reversing B cell–mediated immunosuppression is also a therapeutic strategy. For example, the widely used ICI PD-1 antibody (nivolumab or pidilizumab) has been shown to reverse B cell–mediated immunosuppression (318). Above all, therapeutic approaches targeting Bregs have achieved some success but they are mostly based on preclinical studies with limited data and needed more research to support.
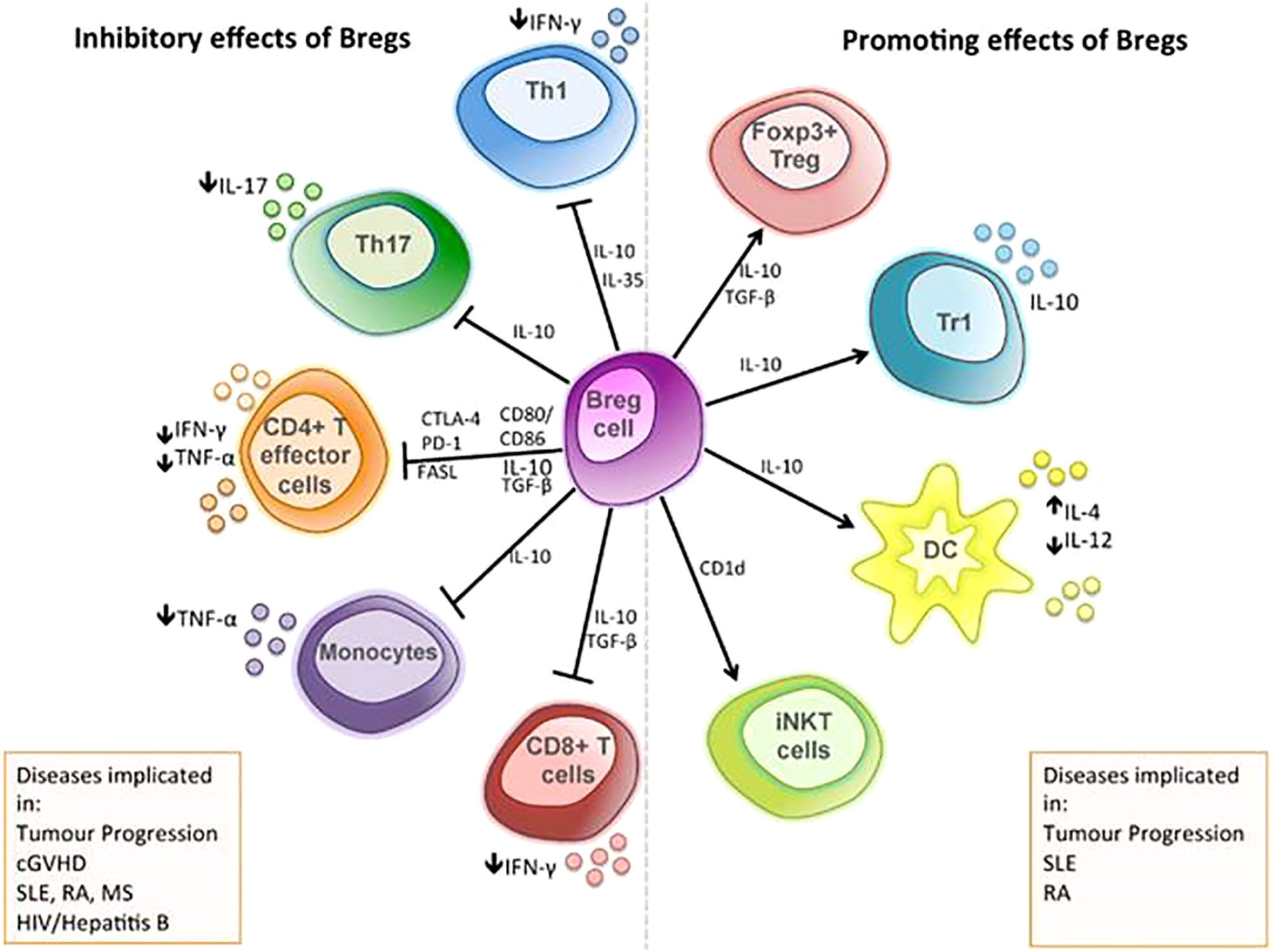
Figure 4 The immunosuppressive mechanisms of Bregs: the functional mechanisms of Bregs are mediated through the release of soluble factors, such as IL-10, TGF-β, and IL-35, and through direct cell-cell contact via co-stimulatory molecules, including the inhibition of T-cell differentiation into type 1 T helper (TH1) cell and type 17 T helper (TH17) cell; inhibit the production of pro-inflammatory cytokine by CD4+ effector T cells; inhibit the production of TNF-α by monocytes; and inhibit the responses of cytotoxic CD8+ T cell. Bregs can initiate apoptosis of effector T cells through the expression of FASL and can also promote the differentiation of Foxp3+ T cells and type 1 regulatory T (Tr1) cell, alter cytokine production by dendritic cells, and support the maintenance of iNKT cells, which may have regulatory functions (318).
The incidence of GC remains high in China, South Korea, Japan, and other Asian countries, and most patients are diagnosed in the middle or late stages because early cancer screening has not been popularized in some areas and the symptoms are not typical. The OS has not been significantly improved in the treatment of advanced GC after years of effort. However, the emergence of immunotherapy has brought hope to these patients. This review introduces the effects of various immune cells on the occurrence, development and prognosis of GC in the tumor microenvironment. Among them, tumor-infiltrating DCs, NK, M1 TAMs, and N1 TANs are beneficial to anti-tumor immunity and are associated with better prognosis in GC, whereas M2 TAMs, MDSCs, N2 TANs, eTregs, and Bregs and the expressions of PD-1, CTLA-4, LAG-3, TIM-3, and TIGIT on various immune cells can promote immune escape and are associated with poor prognosis in GC. Based on the importance of tumor-infiltrating immune cells to patient survival, the database Tumor Immunoassay Resource (TIMER) conducts a comprehensive analysis of tumor immunology and clinical and genomics data, which is used to estimate the abundance of six tumor-infiltrating immune cell (TIIC) subsets (B cells, CD4 T cells, CD8 T cells, macrophages, neutrophils, and DCs) to comprehensively study the molecular characterization of tumor-immune interactions. The TIMER database consists of six functional modules, including the association of TIIC abundance with gene expression (Gene), OS (Survival), somatic mutations (Mutation), and DNA somatic copy number alterations (SCNA), as well as analysis of differential gene expression (DiffExp) and gene-gene correlations (Correlation) (330). For example, using the TIMER database found that TGF-β2 had the ability to regulate immune cell recruitment and activation in gastric adenocarcinoma (STAD), and it might be an important regulator of immune cell infiltration and a valuable prognostic biomarker in GC patients (331). The expression of collagen family members positively correlated with infiltration of macrophages and expression of M2 macrophage markers, and with significant effects on tumor immunology (332). In addition, the expression of long non-coding RNA (lncRNA) target genes NOX4, COL8A1, and CHST1 was positively correlated with the degree of infiltration of CD8+ T cells, CD4+ T cells, macrophages, neutrophils, and DCs in the immune microenvironment, and these lncRNA target genes may be involved in the formation of the tumor immune microenvironment (333). Applying the TIMER tool analysis also showed that the expression of proteasome 26S subunit and ATPase gene (PSMC) family members was correlated with tumor purity, immune infiltration profile, and markers of different types of immune cells, which may become a new and important prognostic biomarker for tumor development (334). Therefore, the application of the TIMER database can help to discover new therapeutic targets and new immune evasion mechanisms.
The number of anti-tumor immune cells is reduced and their functions are suppressed in the tumor microenvironment, whereas the number of immunosuppressive cells is increased. Increasing the number or enhancing the function of anti-tumor immune cells, or reducing the number or weakening the function of immunosuppressive cells can restore anti-tumor immunity. Based on this, many studies have been carried out to target immune cells as a strategy for treating tumors, such as the ICIs PD-1/PD-L1 antibody and CTLA-4 antibody, which are widely used in clinical practice. The ICIs expressed on a variety of immune cells but mainly restore the anti-tumor activity of T cells. In addition, T cells also express LAG-3, TIM-3, TIGIT, and other receptors, and these immunosuppressive receptors are potential therapeutic targets and have seen anti-tumor efficacy in preclinical studies. ACTs are also promising treatment methods based on T-cell immunotherapy, such as CAR-T, TIL, and CIK. Among them, CAR-T has achieved amazing efficacy in hematological tumors, and CAR-T targeting CLDN18.2 has seen better ORR and DCR in the treatment of GC. However, the wide clinical application is limited due to the complicated preparation process and high cost of ACT, such as TIL and CIK. DC-CIK also belongs to ACT in DC-based therapy. Another DC-based treatment is the cancer vaccine, and these two treatment strategies can see anti-tumor effects when combined with other treatments such as chemotherapy. The anti-tumor activity of immune cells can be restored by reducing immunosuppressive cells or inhibiting the activity of immunosuppressive cells, such as M2 TAMs, MDSCs, N2 TANs, eTregs, Bregs, and the reduction of immunosuppressive cells can be achieved by blocking the migration of immunosuppressive cells into the TME. For example, CSF-1/CSF-1R mediates the chemotaxis of TAMs, TANs, and MDSCs; CXCR1/2 mediates the chemotaxis of TANs and MDSCs; CXCR4 participates in the chemotaxis of TAMs, TANs, and Tregs; and CCL5-CCR5 participates in the chemotaxis of TAMs and MDSCs. Blocking the interaction of these chemokines with their receptors can reduce the migration of immunosuppressive cells to the TME. In addition, “reprogramming” immune cells from tumor support cells to tumor killer cells can also increase anti-tumor activity. For example, M1 TAMs and N1 TANs can inhibit and kill tumors, whereas M2 TAMs and N2 TANs promote tumor progression and tumor angiogenesis. Using TGF-β inhibitors or type I IFN can polarize TANs to N1 phenotype. Meanwhile, TLR agonists, CD40 and CD47 antibodies, CSF-1/CSF-1R inhibitors or neutralizing antibodies, and TGF-β, HDAC, and PI3Kγ inhibitors all can reprogram TAMs to M1 TAMs. These reprogramming therapeutic strategies have seen anti-tumor efficacy in preclinical tumor models. In addition, CAR-NK and CAR-M have been developed and applied for anti-tumor therapy based on the success of CAR-T in hematological tumors, and both have entered the clinical trial stage. Among them, the activity of CAR-NK is similar to CAR-T cells and safer than that and does not have strict HLA-matching requirements, so it is a potential strategy for the treatment of GC. The treatment strategies to increase anti-tumor immune cells or their functions are ACTs which was mentioned before, such as CAR-T, CAR-NK, CAR-M, TIL, CIK, DC-CIK, etc.
There are many strategies for targeting immune cells to treat tumors, among which the treatment strategies targeting T cells have the most significant effect and the fastest development and have shown good clinical efficacy in hematological tumors and various solid tumors, such as ICIs PD-1/PD-L1, CTLA-4, and CAR-T in adoptive immune cell therapy. However, these immunotherapies also have limitations in the treatment of GC. For example, the aforementioned anti–PD-1/PD-L1 antibodies and anti–CTLA-4 antibodies have failed to improve OS and PFS in patients compared with chemotherapy in some trials. Although positive results have been obtained in some clinical trials, most of the benefited populations are only those with high PD-L1 CPS score, MSH-H, or high TMB, but unfortunately these populations do not account for much of advanced GC. ICI can also cause the occurrence of fatal toxic events. According to statistics, the toxicity-related mortality rates were 0.36% (anti–PD-1), 0.38% (anti–PD-L1), and 1.08% (anti–CTLA-4). A total of 613 fatal ICI toxicity events were reported in the WHO pharmacovigilance database (Vigilyze) from 2009 to January 2018, of which anti–PD-1/PD-L1-related deaths were usually from pneumonia (35%), followed by hepatitis (22%) and neurotoxicity (15%), whereas anti–CTLA-4–related deaths were mostly due to colitis (70%) (335). The CAR-T cell therapy also has limitations in the treatment of GC, including life-threatening–related toxicity such as cytokine release syndrome (CRS), hemophagocytic lymphohistiocytosis and/or macrophage activation syndrome (MAS), and immune effector cell-associated neurotoxicity syndrome (ICANS). Moreover, the efficacy of CAR-T in the treatment of GC is still limited. In addition, there is still antigen escape and the immunosuppressive tumor microenvironment and physical tumor barriers such as the tumor stroma limit the penetration and mobility of CAR-T cells (336). Other strategies for targeting immune cells to treat tumors have shown efficacy in preclinical animal models but have not been widely used in the clinic, which is partly because some treatment strategies are in clinical trials, and results have not yet been published. Another part of the reason may be related to the insignificant efficacy. For example, the two cancer treatment strategies based on DCs (vaccine and DC-CIK) are only effective when they are combined with other treatment options such as chemotherapy. The widely used ICIs are only effective for some tumor patients, whereas other patients will develop primary or secondary drug resistance. The reason for the poor efficacy of immunotherapy may be related to the complex microenvironment in which the tumor is located. The interaction of tumor cells, immune cells, and stromal cells in the tumor microenvironment constitutes a huge immune suppression network that leads to tumor immune escape, but the current immunotherapy only targets one type of cells or a certain target on a type of cells, whereas the immunosuppressive environment is composed of multiple cells and multiple targets. For example, more than 10 kinds of immunosuppressive receptors expressed on T cells have been confirmed, and there are many unexplored inhibitory receptors. The development of multi-cell and multi-target therapeutic strategies may be the future development direction. Based on this, multi-target combination strategies have been used for tumor treatment, such as PD-1/PD-L1 inhibitor combined with CTLA-4 inhibitor and PD-1 inhibitor combined with anti–LAG-3 (74, 337). At the same time, the PD-1/LAG-3 dual antibody has been developed and has entered the clinical trial stage (68). In addition, therapeutic strategies targeting immune cells have also achieved good results in combination with other treatments, such as combination with chemotherapy drugs. It has been confirmed that drug therapy plays a role in remodeling the tumor immune microenvironment, such as decreased density of CD8+ cells and increased density of FoxP3+ cells and B cells (CD20+) in PTs after NAC, but PD-L1 expression did not change (338). In addition, PD-L1 expression and CD8+ T-cell infiltration were increased in the tumor microenvironment after treatment with the targeted drug anti-VEGFR2 antibody RAM in GC, and PD-1 expression in eTregs cells and CD8+ T cells was significantly reduced in TILs (136). However, this multi-target or targeting multi-cell or combined with other therapies to treat tumors may bring more toxic side effects while increasing the anti-tumor efficacy. Therefore, there is a long way to go for anti-tumor therapy targeting immune cells to achieve synergy and detoxification.
YZ planned, designed, and wrote the majority of the manuscript. MS guided the project and wrote the manuscript. YB and YL planned and guided the project and wrote the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: Globcan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2021) 71(3):209–49. doi: 10.3322/caac.21660
2. Olnes MJ, Martinson HA. Recent advances in immune therapies for gastric cancer. Cancer Gene Ther (2021) 28(9):924–34. doi: 10.1038/s41417-021-00310-y
3. Sheng J, Li H, Yu X, Yu S, Chen K, Pan G, et al. Efficacy of PD-1/PD-L1 inhibitors in patients with non-small cell lung cancer and brain metastases: A real-world retrospective study in China. Thorac Cancer. (2021) 12(22):3019–31. doi: 10.1111/1759-7714.14171
4. Ma L, Jin G, Yao K, Yang Y, Chang R, Wang W, et al. Safety and efficacy of anti-PD-1/PD-L1 inhibitors compared with docetaxel for NSCLC: A systematic review and meta-analysis. Front Pharmacol (2021) 12:699892. doi: 10.3389/fphar.2021.699892
5. Tan S, Li D, Zhu X. Cancer immunotherapy: Pros, cons and beyond. BioMed Pharmacother. (2020) 124:109821. doi: 10.1016/j.biopha.2020.109821
6. Li S, Gao J, Xu Q, Zhang X, Huang M, Dai X, et al. A signature-based classification of gastric cancer that stratifies tumor immunity and predicts responses to PD-1 inhibitors. Front Immunol (2021) 12:693314. doi: 10.3389/fimmu.2021.693314
7. Khalaf K, Hana D, Chou JT, Singh C, Mackiewicz A, Kaczmarek M. Aspects of the tumor microenvironment involved in immune resistance and drug resistance. Front Immunol (2021) 12:656364. doi: 10.3389/fimmu.2021.656364
8. Chen DS, Mellman I. Oncology meets immunology: The cancer-immunity cycle. Immunity. (2013) 39(1):1–10. doi: 10.1016/j.immuni.2013.07.012
9. Chiang CL, Balint K, Coukos G, Kandalaft LE. Potential approaches for more successful dendritic cell-based immunotherapy. Expert Opin Biol Ther (2015) 15(4):569–82. doi: 10.1517/14712598.2015.1000298
10. Basu A, Ramamoorthi G, Albert G, Gallen C, Beyer A, Snyder C, et al. Differentiation and regulation of TH cells: A balancing act for cancer immunotherapy. Front Immunol (2021) 12:669474. doi: 10.3389/fimmu.2021.669474
11. Zhang N, Cao M, Duan Y, Bai H, Li X, Wang Y. Prognostic role of tumor-infiltrating lymphocytes in gastric cancer: A meta-analysis and experimental validation. Arch Med Sci (2019) 16(5):1092–103. doi: 10.5114/aoms.2019.86101
12. Liu T, Peng L, Yu P, Zhao Y, Shi Y, Mao X, et al. Increased circulating Th22 and Th17 cells are associated with tumor progression and patient survival in human gastric cancer. J Clin Immunol (2012) 32(6):1332–9. doi: 10.1007/s10875-012-9718-8
13. Wakatsuki K, Sho M, Yamato I, Takayama T, Matsumoto S, Tanaka T, et al. Clinical impact of tumor-infiltrating CD45RO⁺ memory T cells on human gastric cancer. Oncol Rep (2013) 29(5):1756–62. doi: 10.3892/or.2013.2302
14. Ubukata H, Motohashi G, Tabuchi T, Nagata H, Konishi S, Tabuchi T. Evaluations of interferon-γ/Interleukin-4 ratio and Neutrophil/Lymphocyte ratio as prognostic indicators in gastric cancer patients. J Surg Oncol (2010) 102(7):742–7. doi: 10.1002/jso.21725
15. Kindlund B, Sjöling A, Yakkala C, Adamsson J, Janzon A, Hansson LE, et al. CD4+ regulatory T cells in gastric cancer mucosa are proliferating and express high levels of IL-10 but little TGF-β. Gastric Cancer. (2017) 20(1):116–25. doi: 10.1007/s10120-015-0591-z
16. Yuan XL, Chen L, Li MX, Dong P, Xue J, Wang J, et al. Elevated expression of Foxp3 in tumor-infiltrating treg cells suppresses T-cell proliferation and contributes to gastric cancer progression in a COX-2-Dependent manner. Clin Immunol (2010) 134(3):277–88. doi: 10.1016/j.clim.2009.10.005
17. Liu X, Zhang Z, Zhao G. Recent advances in the study of regulatory T cells in gastric cancer. Int Immunopharmacol. (2019) 73:560–7. doi: 10.1016/j.intimp.2019.05.009
18. Bagheri N, Shirzad H, Elahi S, Azadegan-Dehkordi F, Rahimian G, Shafigh M, et al. Downregulated regulatory T cell function is associated with increased peptic ulcer in helicobacter pylori-infection. Microb Pathog (2017) 110:165–75. doi: 10.1016/j.micpath.2017.06.040
19. Jang TJ. The number of Foxp3-positive regulatory T cells is increased in helicobacter pylori gastritis and gastric cancer. Pathol Res Pract (2010) 206(1):34–8. doi: 10.1016/j.prp.2009.07.019
20. Chen SL, Cai SR, Zhang XH, Peng JJ, Li WF, Zhai ET, et al. Expression of CD4+CD25+ regulatory T cells and Foxp3 in peripheral blood of patients with gastric carcinoma. J Biol Reg Homeost Ag. (2016) 30(1):197–204.
21. Perrone G, Ruffini PA, Catalano V, Spino C, Santini D, Muretto P, et al. Intratumoural FOXP3-positive regulatory T cellsAre associated with adverse prognosis in radically resected gastric cancer. Eur J Cancer. (2008) 44(13):1875–82. doi: 10.1016/j.ejca.2008.05.017
22. Kawaida H, Kono K, Takahashi A, Sugai H, Mimura K, Miyagawa N, et al. Distribution of CD4+ CD25high regulatory T-cells in tumor-draining lymph nodes in patients with gastric cancer. J Surg Res (2005) 124(1):151–7. doi: 10.1016/j.jss.2004.10.004
23. Lee HE, Park DJ, Kim WH, Kim HH, Lee HS. High FOXP3+ regulatory T-cell density in the sentinel lymph node is associated with downstream non-sentinel lymph-node metastasis in gastric cancer. Br J Cancer. (2011) 105(3):413–9. doi: 10.1038/bjc.2011.248
24. Kim HI, Kim H, Cho HW, Kim SY, Song KJ, Hyung WJ, et al. The ratio of intra-tumoral regulatory T cells (Foxp3+)/Helper T cells (CD4+) is a prognostic factor and associated with recurrence pattern in gastric cardia cancer. J Surg Oncol (2011) 104(7):728–33. doi: 10.1002/jso.22038
25. Shen Z, Zhou S, Wang Y, Li RL, Zhong C, Liang C, et al. Higher intratumoral infiltrated Foxp3+ treg numbers and Foxp3+/CD8+ ratio are associated with adverse prognosis in resectable gastric cancer. J Cancer Res Clin Oncol (2010) 136(10):1585–95. doi: 10.1007/s00432-010-0816-9
26. Kim KJ, Lee KS, Cho HJ, Kim YH, Yang HK, Kim WH, et al. Prognostic implications of tumor-infiltrating FoxP3+ regulatory T cells and CD8+ cytotoxic T cells in microsatellite-unstable gastric cancers. Hum Pathol (2014) 45(2):285–93. doi: 10.1016/j.humpath.2013.09.004
27. Haas M, Dimmler A, Hohenberger W, Grabenbauer GG, Niedobitek G, Distel LV. Stromal regulatory T-cells are associated with a favourable prognosis in gastric cancer of the cardia. BMC Gastroenterol (2009) 9:65. doi: 10.1186/1471-230X-9-65
28. Feichtenbeiner A, Haas M, Büttner M, Grabenbauer GG, Fietkau R, Distel LV. Critical role of spatial interaction between CD8⁺ and Foxp3⁺ cells in human gastric cancer: The distance matters. Cancer Immunol Immun (2014) 63(2):111–9. doi: 10.1007/s00262-013-1491-x
29. Nagase H, Takeoka T, Urakawa S, Morimoto-Okazawa A, Kawashima A, Iwahori K, et al. ICOS+ Foxp3+ TILs in gastric cancer are prognostic markers and effector regulatory T cells associated with helicobacter pylori. Int J Cancer. (2017) 140(3):686–95. doi: 10.1002/ijc.30475
30. Venetis K, Invernizzi M, Sajjadi E, Curigliano G, Fusco N. Cellular immunotherapy in breast cancer: The quest for consistent biomarkers. Cancer Treat Rev (2020) 90:102089. doi: 10.1016/j.ctrv.2020.102089
31. Finn OJ. A believer’s overview of cancer immunosurveillance and immunotherapy. J Immunol (2018) 200(2):385–91. doi: 10.4049/jimmunol.1701302
32. Gu SS, Zhang W, Wang X, Jiang P, Traugh N, Li Z, et al. Therapeutically increasing MHC-I expression potentiates immune checkpoint blockade. Cancer Discovery (2021) 11(6):1524–41. doi: 10.1158/2159-8290.CD-20-0812
33. Zhu D, Xu R, Huang X, Tang Z, Tian Y, Zhang J, et al. Deubiquitinating enzyme OTUB1 promotes cancer cell immunosuppression via preventing ER-associated degradation of immune checkpoint protein PD-L1. Cell Death Differ (2021) 28(6):1773–89. doi: 10.1038/s41418-020-00700-z
34. Kamal AM, Wasfey EF, Elghamry WR, Sabry OM, Elghobary HA, Radwan SM. Genetic signature of CTLA-4, BTLA, TIM-3 and LAG-3 molecular expression in colorectal cancer patients: Implications in diagnosis and survival outcomes. Clin Biochem (2021) 96:13–8. doi: 10.1016/j.clinbiochem.2021.06.007
35. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. (2012) 12(4):252–64. doi: 10.1038/nrc3239
36. Oh S, Kim E, Lee H. Comparative impact of PD-1 and PD-L1 inhibitors on advanced esophageal or Gastric/Gastroesophageal junction cancer treatment: A systematic review and meta-analysis. J Clin Med (2021) 10(16):3612. doi: 10.3390/jcm10163612
37. Clarke E, Eriksen JG, Barrett S. The effects of PD-1/PD-L1 checkpoint inhibitors on Recurrent/Metastatic head and neck squamous cell carcinoma: A critical review of the literature and meta-analysis. Acta Oncol (2021) 60(11):1534–42. doi: 10.1080/0284186X.2021.1964699
38. Behrouzieh S, Sheida F, Rezaei N. Review of the tecent clinical trials for PD-1/PD-L1 based lung cancer immunotherapy. Expert Rev Anticanc. (2021) 21(12):1355–70. doi: 10.1080/14737140.2021.1996230
39. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat Med (2002) 8(8):793–800. doi: 10.1038/nm730
40. Jiang X, Wang J, Deng X, Xiong F, Ge J, Xiang B, et al. Role of the tumor microenvironment in PD-L1/PD-1-Mediated tumor immune escape. Mol Cancer. (2019) 18(1):10. doi: 10.1186/s12943-018-0928-4
41. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. (2014) 515(7528):568–71. doi: 10.1038/nature13954
42. Wu C, Zhu Y, Jiang J, Zhao J, Zhang XG, Xu N. Immunohistochemical localization of programmed death-1 ligand-1 (PD-L1) in gastric carcinoma and its clinical significance. Acta Histochem (2006) 108(1):19–24. doi: 10.1016/j.acthis.2006.01.003
43. Shitara K, Van Cutsem E, Bang YJ, Fuchs C, Wyrwicz L, Lee KW, et al. Efficacy and safety of pembrolizumab or pembrolizumab plus chemotherapy vs chemotherapy alone for patients with first-line, advanced gastric cancer: The KEYNOTE-062 phase 3 randomized clinical trial. JAMA Oncol (2020) 6(10):1571–80. doi: 10.1001/jamaoncol.2020.3370
44. Shitara K, Özgüroğlu M, Bang YJ, Di Bartolomeo M, Mandalà M, Ryu MH, et al. Pembrolizumab versus paclitaxel for previously treated, advanced gastric or gastro-oesophageal junction cancer (KEYNOTE-061): A randomised, open-label, controlled, phase trial. Lancet. (2018) 392(10142):123–33. doi: 10.1016/S0140-6736(18)31257-1
45. Fuchs CS, Doi T, Jang RW, Muro K, Satoh T, Machado M, et al. Safety and efficacy of pembrolizumab monotherapy in patients with previously treated advanced gastric and gastroesophageal junction cancer: Phase 2 clinical KEYNOTE-059 trial. JAMA Oncol (2018) 4(5):e180013. doi: 10.1001/jamaoncol.2018.0013
46. Kang YK, Boku N, Satoh T, Ryu MH, Chao Y, Kato K, et al. Nivolumab in patients with advanced gastric or gastro-oesophageal junction cancer refractory to, or intolerant of, At least two previous chemotherapy regimens (ONO-4538-12, ATTRACTION-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. (2017) 390(10111):2461–71. doi: 10.1016/S0140-6736(17)31827-5
47. Wang FH, Zhang XT, Li YF, Tang L, Qu XJ, Ying JE, et al. The Chinese society of clinical oncology (CSCO): Clinical guidelines for the diagnosis and treatment of gastric cancer, 2021. Cancer Commun (Lond). (2021) 41(8):747–95. doi: 10.1002/cac2.12193
48. Chen LT, Satoh T, Ryu MH, Chao Y, Kato K, Chung HC, et al. A phase 3 study of nivolumab in previously treated advanced gastric or gastroesophageal junction cancer (ATTRACTION-2): 2-year update data. Gastric Cancer. (2020) 23(3):510–19. doi: 10.1007/s10120-019-01034-7
49. Janjigian YY, Shitara K, Moehler M, Garrido M, Salman P, Shen L, et al. First-line nivolumab plus chemotherapy versus chemotherapy alone for advanced gastric, gastro-oesophageal junction, and oesophageal adenocarcinoma (CheckMate 649): A randomised, open-label, phase 3 trial. Lancet. (2021) 398(10294):27–40. doi: 10.1016/S0140-6736(21)00797-2
50. Janjigian YY, Maron SB, Chatila WK, Millang B, Chavan SS, Alterman C, et al. First-line pembrolizumab and trastuzumab in HER2-positive oesophageal, gastric, or gastro-oesophageal junction cancer: An open-label, single-arm, phase 2 trial. Lancet Oncol (2020) 21(6):821–31. doi: 10.1016/S1470-2045(20)30169-8
51. Peng Z, Wei J, Wang F, Ying J, Deng Y, Gu K, et al. Camrelizumab combined with chemotherapy followed by camrelizumab apatinib as first-line therapy for advanced gastric or gastroesophageal junction adenocarcinoma. Clin Cancer Res (2021) 27(11):3069–78. doi: 10.1158/1078-0432.CCR-20-4691
52. Jiang H, Yu X, Li N, Kong M, Ma Z, Zhou D, et al. Efficacy and safety of neoadjuvant sintilimab, oxaliplatin and capecitabine in patients with locally advanced, resectable gastric or gastroesophageal junction adenocarcinoma: Early results of a phase 2 study. J Immunother Cancer. (2022) 10(3):e003635. doi: 10.1136/jitc-2021-003635
53. Bang YJ, Van Cutsem E, Fuchs CS, Ohtsu A, Tabernero J, Ilson DH, et al. KEYNOTE-585: Phase III study of perioperative chemo-therapy with or without pembrolizumab for gastric cancer. Future Oncol (2019) 15(9):943–52. doi: 10.2217/fon-2018-0581
54. Janjigian YY, Van Cutsem E, Muro K, Wainberg Z, Al-Batran SE, Hyung WJ, et al. MATTERHORN: Phase III study of durvalumab plus FLOT chemotherapy in resectable Gastric/Gastroesophageal junction cancer. Future Oncol (2022) 18(20):2465–73. doi: 10.2217/fon-2022-0093
55. Kim ST, Cristescu R, Bass AJ, Kim KM, Odegaard JI, Kim K, et al. Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat Med (2018) 24(9):1449–58. doi: 10.1038/s41591-018-0101-z
56. Bang YJ, Kang YK, Catenacci DV, Muro K, Fuchs CS, Geva R, et al. Pembrolizumab alone or in combination with chemotherapy as first-line therapy for patients with advanced gastric or gastroesophageal junction adenocarcinoma: Results from the phase II nonrandomized KEYNOTE-059 study. Gastric Cancer. (2019) 22(4):828–37. doi: 10.1007/s10120-018-00909-5
57. Chung HC, Kang YK, Chen Z, Bai Y, Wan Ishak WZ, Shim BY, et al. Pembrolizumab versus paclitaxel for previously treated advanced gastric or gastroesophageal junction cancer (KEYNOTE-063): A randomized, open-label, phase 3 trial in Asian patients. Cancer. (2022) 128(5):995–1003. doi: 10.1002/cncr.34019
58. Janjigian YY, Kawazoe A, Yañez P, Li N, Lonardi S, Kolesnik O, et al. The KEYNOTE-811 trial of dual PD-1 and HER2 blockade in HER2-positive gastric cancer. Nature. (2021) 600(7890):727–30. doi: 10.1038/s41586-021-04161-3
59. Boku N, Ryu MH, Kato K, Chung HC, Minashi K, Lee KW, et al. Safety and efficacy of nivolumab in combination with s-1/Capecita bine plus oxaliplatin in patients with previously untreated, unresectable, advanced, or recurrent Gastric/Gastroesophageal junction cancer: Interim results of a randomized, phase II trial (ATTRACTION-4). Ann Oncol (2019) 30(2):250–8. doi: 10.1093/annonc/mdy540
60. Janjigian YY, Bendell J, Calvo E, Kim JW, Ascierto PA, Sharma P, et al. CheckMate-032 study: Efficacy and safety of nivolumab and nivolumab plus ipilimumab in patients with metastatic esophagogastric cancer. J Clin Oncol (2018) 36(28):2836–44. doi: 10.1200/JCO.2017.76.6212
61. Kelly RJ, Lee J, Bang YJ, Almhanna K, Blum-Murphy M, Catenacci DVT, et al. Safety and efficacy of durvalumab and tremelimumab alone or in combination in patients with advanced gastric and gastroesophageal junction adenocarcinoma. Clin Cancer Res (2020) 26(4):846–54. doi: 10.1158/1078-0432.CCR-19-2443
62. Sznol M, Melero I. Revisiting anti-CTLA-4 antibodies in combination with PD-1 blockade for cancer immunotherapy. Ann Oncol (2021) 32(3):295–97. doi: 10.1016/j.annonc.2020.11.018
63. Janjigian YY, Bendell JC, Calvo E, Kim JW, Ascierto PA, Sharma P, et al. CheckMate-032: Phase I/II, open-label study of safety and activity of nivolumab (Nivo) alone or with ipilimumab (Ipi) in advanced and metastatic (A/M) gastric cancer (GC). J Clin Oncol (2016) 34:4010. doi: 10.1200/JCO.2016.34.15_suppl.4010
64. Moehler MH, Cho JY, Kim YH, Kim JW, Di Bartolomeo M, Ajani JA, et al. A randomized, open-label, two-arm phase II trial comparing the efficacy of sequential ipilimumab (Ipi) versus best supportive care (BSC) following first-line (1L) chemotherapyin patients with unresectable, locally Advanced/Metastatic (A/M) gastric or gastro-esophageal junction (G/GEJ) cancer. J Clin Oncol (2016) 34:4011. doi: 10.1200/JCO.2016.34.15_suppl.4011
65. Li K, Zhang A, Li X, Zhang H, Zhao L. Advances in clinical immunotherapy for gastric cancer. BBA-Reviochim Cancer. (2021) 1876(2):188615. doi: 10.1016/j.bbcan.2021.188615
66. Lythgoe MP, Liu DSK, Annels NE, Krell J, Frampton AE. Gene of the month: Lymphocyte-activation gene 3 (LAG-3). J Clin Pathol (2021) 74(9):543–7. doi: 10.1136/jclinpath-2021-207517
67. Sung E, Ko M, Won JY, Jo Y, Park E, Kim H, et al. LAG-3xPD-L1 bispecific antibody potentiates antitumor responses of T cells through dendritic cell activation. Mol Ther (2022) 30(8):2800–16. doi: 10.1016/j.ymthe.2022.05.003
68. Tian J, Liu Y, Zhang T, Yue L, Xiao Y, Guo C. LAG-3 is a promising inhibitory immune checkpoint for antitumor immunotherapy. Expert Rev Anticancer. (2022) 22(3):289–96. doi: 10.1080/14737140.2022.2039124
69. Chocarro L, Blanco E, Zuazo M, Arasanz H, Bocanegra A, Fernández-Rubio L, et al. Understanding LAG-3 signaling. Int J Mol Sci (2021) 22(10):5282. doi: 10.3390/ijms22105282
70. Camisaschi C, Casati C, Rini F, Perego M, De Filippo A, Triebel F, et al. LAG-3 expression defines a subset of CD4(+)CD25(high) Foxp3(+) regulatory T cells that are expanded at tumor sites. J Immunol (2010) 184(11):6545–51. doi: 10.4049/jimmunol.0903879
71. Wei T, Zhang J, Qin Y, Wu Y, Zhu L, Lu L, et al. Increased expression of immunosuppressive molecules on intratumoral and circulating regulatory T cells in non-Small-Cell lung cancer patients. Am J Cancer Res (2015) 5(7):2190–201.
72. Sordo-Bahamonde C, Lorenzo-Herrero S, González-Rodríguez AP, Payer ÁR, González-García E, López-Soto A, et al. LAG-3 block-ade with relatlimab (BMS-986016) restores anti-leukemic responses in chronic lymphocytic leukemia. Cancers (Basel). (2021) 13(9):2112. doi: 10.3390/cancers13092112
73. Sun HW, Chen J, Wu WC, Yang YY, Xu YT, Yu XJ, et al. Retinoic acid synthesis deficiency fosters the generation of polymorphonuclear myeloid-derived suppressor cells in colorectal cancer. Cancer Immunol Res (2021) 9(1):20–33. doi: 10.1158/2326-6066.CIR-20-0389
74. Schöffski P, Tan DSW, Martín M, Ochoa-de-Olza M, Sarantopoulos J, Carvajal RD, et al. Phase I/II study of the LAG-3 inhibitor ieramilimab (LAG525) ± anti-PD-1 spartalizumab (PDR001) in patients with advanced malignancies. J Immunother Cancer. (2022) 10(2):e003776. doi: 10.1136/jitc-2021-003776
75. Zeidan AM, Komrokji RS, Brunner AM. TIM-3 pathway dysregulation and targeting in cancer. Expert Rev Anticancer. (2021) 21(5):523–34. doi: 10.1080/14737140.2021.1865814
76. Lu X. Structure and functions of T-cell immunoglobulin-domain and mucin-domain protein 3 in cancer. Curr Med Chem (2022) 29(11):1851–65. doi: 10.2174/0929867328666210806120904
77. Dixon KO, Tabaka M, Schramm MA, Xiao S, Tang R, Dionne D, et al. TIM-3 restrains anti-tumour immunity by regulating inflammasome activation. Nature. (2021) 595(7865):101–6. doi: 10.1038/s41586-021-03626-9
78. Pagliano O, Morrison RM, Chauvin JM, Banerjee H, Davar D, Ding Q, et al. Tim-3 mediates T cell trogocytosis to limit antitumor immunity. J Clin Invest. (2022) 132(9):e152864. doi: 10.1172/JCI152864
79. Granier C, Gey A, Dariane C, Mejean A, Timsit MO, Blanc C, et al. Tim-3: A novel biomarker and therapeutic target in oncology. Med Sci (Paris). (2018) 34(3):231–7. doi: 10.1051/medsci/20183403011
80. Gomes de Morais AL, Cerdá S, de Miguel M. New checkpoint inhibitors on the road: Targeting TIM-3 in solid tumors. Curr Oncol Rep (2022) 24(5):651–8. doi: 10.1007/s11912-022-01218-y
81. Saleh R, Toor SM, Elkord E. Targeting TIM-3 in solid tumors: Innovations in the preclinical and translational realm and therapeutic potential. Expert Opin Ther Tar. (2020) 24(12):1251–62. doi: 10.1080/14728222.2020.1841750
82. Yang F, Zeng Z, Li J, Ren X, Wei F. TIM-3 and CEACAM1 are prognostic factors in head and neck squamous cell carcinoma. Front Mol Biosci (2021) 8:619765. doi: 10.3389/fmolb.2021.619765
83. Chen H, Wang M, Weng T, Wei Y, Liu C, Yang L, et al. The prognostic and clinicopathological significance of Tim-3 and PD-1 expression in the prognosis of upper urinary tract urothelial carcinoma. Urol Oncol (2021) 39(11):743–53. doi: 10.1016/j.urolonc.2021.05.039
84. Cheng S, Han F, Xu Y, Qu T, Ju Y. Expression of Tim-3 in breast cancer tissue promotes tumor progression. Int J Clin Exp Patho. (2018) 11(3):1157–66.
85. Cao B, Zhu L, Zhu S, Li D, Zhang C, Xu C, et al. Genetic variations and haplotypes in TIM-3 gene and the risk of gastric cancer. Cancer Immunol Immun (2010) 59(12):1851–7. doi: 10.1007/s00262-010-0910-5
86. Harding JJ, Moreno V, Bang YJ, Hong MH, Patnaik A, Trigo J, et al. Blocking TIM-3 in treatment-refractory advanced solid tumors: A phase IA/B study of LY3321367 with or without an anti-PD-L1 antibody. Clin Cancer Res (2021) 27(8):2168–78. doi: 10.1158/1078-0432.CCR-20-4405
87. Tian T, Li Z. Targeting Tim-3 in cancer with resistance to PD-1/PD-L1 blockade. Front Oncol (2021) 11:731175. doi: 10.3389/fonc.2021.731175
88. Curigliano G, Gelderblom H, Mach N, Doi T, Tai D, Forde PM, et al. Phase I/Ib clinical trial of sabatolimab, an anti-TIM-3 antibody, alone and in combination with spartalizumab, an anti-PD-1 antibody, in advanced solid tumors. Clin Cancer Res (2021) 27(13):3620–9. doi: 10.1158/1078-0432.CCR-20-4746
89. Hellmann MD, Bivi N, Calderon B, Shimizu T, Delafontaine B, Liu ZT, et al. Safety and immunogenicity of LY3415244, a bispecific antibody against TIM-3 and PD-L1, in patients with advanced solid tumors. Clin Cancer Res (2021) 27(10):2773–81. doi: 10.1158/1078-0432.CCR-20-3716
90. Xu D, Zhao E, Zhu C, Zhao W, Wang C, Zhang Z, et al. TIGIT and PD-1 may serve as potential prognostic biomarkers for gastric cancer. Immunobiology. (2020) 225(3):151915. doi: 10.1016/j.imbio.2020.151915
91. Ge Z, Peppelenbosch MP, Sprengers D, Kwekkeboom J. TIGIT, the next step towards successful combination immune checkpoint therapy in cancer. Front Immunol (2021) 12:699895. doi: 10.3389/fimmu.2021.699895
92. Chauvin JM, Zarour HM. TIGIT in cancer immunotherapy. J Immunother Cancer. (2020) 8(2):e000957. doi: 10.1136/jitc-2020-000957
93. Rodriguez-Abreu D, Johnson ML, Hussein MA, Cobo M, Patel AJ, Secen NM, et al. Primary analysis of a randomized, double-blind, phase II study of the anti-TIGIT antibody tiragolumab (Tira) plus atezolizumab (Atezo) versus placebo plus atezo as first-line (1L) treatment in patients with PD-L1-Selected NSCLC (CITYSCAPE). J Clin Oncol (2020) 38. doi: 10.1200/JCO.2020.38.15_suppl.9503
94. Ahn MJ, Niu J, Kim DW, Rasco D, Mileham KF, Chung HC, et al. Vibostolimab, an anti-TIGIT antibody, as monotherapy and in combination with pembrolizumab in anti-PD-1/PD-L1-Refractory NSCLC. Ann Oncol (2020) 31:S887. doi: 10.1016/j.annonc.2020.08.1714
95. Rohaan MW, Wilgenhof S, Haanen JBAG. Adoptive celular therapies: the current landscape. Virchows Arch (2019) 474(4):449–61. doi: 10.1007/s00428-018-2484-0
96. Shen D, Liu ZH, Xu JN, Xu F, Lin QF, Lin F, et al. Efficacy of adoptive cellular therapy in patients with gastric cancer: A meta-analysis. Immunotherapy. (2016) 8(8):971–81. doi: 10.2217/imt.16.10
97. Rodríguez Pérez Á, Campillo-Davo D, Van Tendeloo VFI, Benítez-Ribas D. Cellular immunotherapy: A clinical state-of-the-Art of a new paradigm for cancer treatment. Clin Transl Oncol (2020) 22(11):1923–37. doi: 10.1007/s12094-020-02344-4
98. Kumar A, Watkins R, Vilgelm AE. Cell therapy with TILs: Training and taming T cells to fight cancer. Front Immunol (2021) 12:690499. doi: 10.3389/fimmu.2021.690499
99. Miao L, Zhang Z, Ren Z, Tang F, Li Y. Obstacles and coping strategies of CAR-T cell immunotherapy in solid tumors. Front Immunol (2021) 12:687822. doi: 10.3389/fimmu.2021.687822
100. Dana H, Chalbatani GM, Jalali SA, Mirzaei HR, Grupp SA, Suarez ER, et al. CAR-T cells: Early successes in blood cancer and challenges in solid tumors. Acta Pharm Sin B (2021) 11(5):1129–47. doi: 10.1016/j.apsb.2020.10.020
101. Haslauer T, Greil R, Zaborsky N, Geisberger R. CAR T-cell therapy in hematological malignancies. Int J Mol Sci (2021) 22(16):8996. doi: 10.3390/ijms22168996
102. Lv J, Zhao R, Wu D, Zheng D, Wu Z, Shi J, et al. Mesothelin is a target of chimeric antigen receptor T cells for treating gastric cancer. J Hematol Oncol (2019) 12(1):18. doi: 10.1186/s13045-019-0704-y
103. Tao K, He M, Tao F, Xu G, Ye M, Zheng Y, et al. Development of NKG2D-based chimeric antigen receptor-T cells for gastric cancer treatment. Cancer Chemoth Pharm (2018) 82(5):815–27. doi: 10.1007/s00280-018-3670-0
104. Sun F, Yu X, Ju R, Wang Z, Wang Y. Antitumor responses in gastric cancer by targeting B7H3 via chimeric antigen receptor T cells. Cancer Cell Int (2022) 22(1):50. doi: 10.1186/s12935-022-02471-8
105. Wu D, Lv J, Zhao R, Wu Z, Zheng D, Shi J, et al. PSCA is a target of chimeric antigen receptor T cells in gastric cancer. biomark Res (2020) 8:3. doi: 10.1186/s40364-020-0183-x
106. Jiang H, Shi Z, Wang P, Wang C, Yang L, Du G, et al. Claudin18.2-specific chimeric antigen receptor engineered T cells for the treatment of gastric cancer. J Natl Cancer Inst (2019) 111(4):409–18. doi: 10.1093/jnci/djy134
107. Kim M, Pyo S, Kang CH, Lee CO, Lee HK, Choi SU, et al. Folate receptor 1 (FOLR1) targeted chimeric antigen receptor (CAR) T cells for the treatment of gastric cancer. PLoS One (2018) 13(6):e0198347. doi: 10.1371/journal.pone.0198347
108. Han Y, Liu C, Li G, Li J, Lv X, Shi H, et al. Antitumor effects and persistence of a novel HER2 CAR T cells directed to gastric cancer in preclinical models. Am J Cancer Res (2018) 8(1):106–19.
109. Kang CH, Kim Y, Lee DY, Choi SU, Lee HK, Park CH. C-Met-Specific chimeric antigen receptor T cells demonstrate anti-tumor effect in c-met positive gastric cancer. Cancers (Basel). (2021) 13(22):5738. doi: 10.3390/cancers13225738
110. Chi X, Yang P, Zhang E, Gu J, Xu H, Li M, et al. Significantly increased anti-tumor activity of carcinoembryonic antigen-specific chimeric antigen receptor T cells in combination with recombinant human IL-12. Cancer Med (2019) 8(10):4753–65. doi: 10.1002/cam4.2361
111. Feng Z, He X, Zhang X, Wu Y, Xing B, Knowles A, et al. Potent suppression of neuroendocrine tumors and gastrointestinal cancers by CDH17CAR T cells without toxicity to normal tissues. Nat Cancer. (2022) 3(5):581–94. doi: 10.1038/s43018-022-00344-7
112. Han Y, Sun B, Cai H, Xuan Y. Simultaneously target of normal and stem cells-like gastric cancer cells via cisplatin and anti-CD133 CAR-T combination therapy. Cancer Immunol Immun (2021) 70(10):2795–803. doi: 10.1007/s00262-021-02891-x
113. Jung M, Yang Y, McCloskey JE, Zaman M, Vedvyas Y, Zhang X, et al. Chimeric antigen receptor T cell therapy targeting ICAM-1 in gastric cancer. Mol Ther Oncolytics. (2020) 18:587–601. doi: 10.1016/j.omto.2020.08.009
114. Zhou JT, Liu JH, Song TT, Ma B, Amidula N, Bai C. EGLIF-CAR-T cells secreting PD-1 blocking antibodies significantly mediate the elimination of gastric cancer. Cancer Manag Res (2020) 12:8893–902. doi: 10.2147/CMAR.S260915
115. Chen C, Gu YM, Zhang F, Zhang ZC, Zhang YT, He YD, et al. Construction of PD1/CD28 chimeric-switch receptor enhances anti-tumor ability of c-met CAR-T in gastric cancer. Oncoimmunology. (2021) 10(1):1901434. doi: 10.1080/2162402X.2021.1901434
116. Zhao W, Jia L, Zhang M, Huang X, Qian P, Tang Q, et al. The killing effect of novel bi-specific Trop2/PD-L1 CAR-T cell targeted gastric cancer. Am J Cancer Res (2019) 9(8):1846–56.
117. Qi C, Gong J, Li J, Liu D, Qin Y, Ge S, et al. Claudin18.2-specific CAR T cells in gastrointestinal cancers: Phase 1 trial interim results. Nat Med (2022) 28(6):1189–98. doi: 10.1038/s41591-022-01800-8
118. Kono K, Ichihara F, Iizuka H, Sekikawa T, Matsumoto Y. Differences in the recognition of tumor-specific CD8+ T cells derived from solid tumor, metastatic lymph nodes and ascites in patients with gastric cancer. Int J Cancer. (1997) 71(6):978–81. doi: 10.1002/(sici)1097-0215(19970611)71:6<978::aid-ijc12>3.0.co;2-2
119. Kono K, Takahashi A, Ichihara F, Amemiya H, Iizuka H, Fujii H, et al. Prognostic significance of adoptive immunotherapy with tumor-associated lymphocytes in patients with advanced gastric cancer: A randomized trial. Clin Cancer Res (2002) 8(6):1767–71. doi: 10.1093/carcin/23.6.1089
120. Xu X, Xu L, Ding S, Wu M, Tang Z, Fu W, et al. Treatment of 23 patients with advanced gastric cancer by intravenously transfer of autologous tumor-infiltrating lymphocytes combined with rIL-2. Chin Med Sci J (1995) 10(3):185–7.
121. Faghfuri E, Shadbad MA, Faghfouri AH, Soozangar N. Cellular immunotherapy in gastric cancer: Adoptive cell therapy and dendritic cell-based vaccination. Immunotherapy. (2022) 14(6):475–88. doi: 10.2217/imt-2021-0285
122. Guo Y, Han W. Cytokine-induced killer (CIK) cells: From basic research to clinical translation. Chin J Cancer. (2015) 34(3):99–107. doi: 10.1186/s40880-015-0002-1
123. Introna M, Correnti F. Innovative clinical perspectives for CIK cells in cancer patients. Int J Mol Sci (2018) 19(2):358. doi: 10.3390/ijms19020358
124. Hu G, Zhong K, Wang S, Wang S, Ding Q, Xu F, et al. Cellular immunotherapy plus chemotherapy ameliorates survival in gastric cancer patients: A meta-analysis. Int J Clin Oncol (2020) 25(10):1747–56. doi: 10.1007/s10147-020-01750-6
125. Shi L, Zhou Q, Wu J, Ji M, Li G, Jiang J, et al. Efficacy of adjuvant immunotherapy with cytokine-induced kller cells in patients with locally dvanced gastric cancer. Cancer Immunol Immun (2012) 61(12):2251–9. doi: 10.1007/s00262-012-1289-2
126. Jiang JT, Shen YP, Wu CP, Zhu YB, Wei WX, Chen LJ, et al. Increasing the frequency of CIK cells adoptive immunotherapy may decrease risk of death in gastric cancer patients. World J Gastroentero. (2010) 16(48):6155–62. doi: 10.3748/wjg.v16.i48.6155
127. Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity. (2009) 30(6):899–911. doi: 10.1016/j.immuni.2009.03.019
128. Ohue Y, Nishikawa H. Regulatory T (Treg) cell in cancer: Can treg cells be a new therapeutic target? Cancer Sci (2019) 110(7):2080–9. doi: 10.1111/cas.14069
129. Caridade M, Graca L, Ribeiro RM. Mechanisms underlying CD4+ treg immune regulation in the adult: From experiments to models. Front Immunol (2013) 4:378. doi: 10.3389/fimmu.2013.00378
130. Maj T, Wang W, Crespo J, Zhang H, Wang W, Wei S, et al. Oxidative stress controls regulatory T cell apoptosis and suppress or activity and PD-L1-Blockade resistance in tumor. Nat Immunol (2017) 18(12):1332–41. doi: 10.1038/ni.3868
131. Nishikawa H, Koyama S. Mechanisms of regulatory T cell infiltration in tumors: Implications for innovative immune precision therapies. J Immunother Cancer. (2021) 9(7):e002591. doi: 10.1136/jitc-2021-002591
132. Rech AJ, Mick R, Martin S, Recio A, Aqui NA, Powell DJ Jr, et al. CD25 blockade depletes and selectively reprograms regulatory T cells in concert with immunotherapy in cancer patients. Sci Transl Med (2012) 4(134):134ra62. doi: 10.1126/scitranslmed.3003330
133. Selby MJ, Engelhardt JJ, Quigley M, Henning KA, Chen T, Srinivasan M, et al. Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol Res (2013) 1(1):32–42. doi: 10.1158/2326-6066.CIR-13-0013
134. Sainson RCA, Thotakura AK, Kosmac M, Borhis G, Parveen N, Kimber R, et al. An antibody targeting ICOS increases intratumoral cytotoxic to regulatory T-cell ratio and induces tumor regression. Cancer Immunol Res (2020) 8(12):1568–82. doi: 10.1158/2326-6066.CIR-20-0034
135. Zappasodi R, Sirard C, Li Y, Budhu S, Abu-Akeel M, Liu C, et al. Rational design of anti-GITR-Based combination immunotherapy. Nat Med (2019) 25(5):759–66. doi: 10.1038/s41591-019-0420-8
136. Tada Y, Togashi Y, Kotani D, Kuwata T, Sato E, Kawazoe A, et al. Targeting VEGFR2 with ramucirumab strongly impacts Effector/Activated regulatory T cells and CD8+ T cells in the tumor microenvironment. J Immunother Cancer. (2018) 6(1):106. doi: 10.1186/s40425-018-0403-1
137. Piconese S, Valzasina B, Colombo MP. OX40 triggering blocks suppression by regulatory T cells and facilitates tumor rejection. J Exp Med (2008) 205(4):825–39. doi: 10.1084/jem.20071341
138. Kidani Y, Nogami W, Yasumizu Y, Kawashima A, Tanaka A, Sonoda Y, et al. CCR8-targeted specific depletion of clonally expanded treg cells in tumor tissues evokes potent tumor immunity with long-lasting memory. Proc Natl Acad Sci USA (2022) 119(7):e2114282119. doi: 10.1073/pnas.2114282119
139. Saito T, Kurose K, Kojima T, Funakoshi T, Sato E, Nishikawa H, et al. Phase ib study on the humanized anti-CCR4 antibody, KW-0761, in advanced solid tumors. Nagoya J Med Sci (2021) 83(4):827–40. doi: 10.18999/nagjms.83.4.827
140. Tanaka A, Nishikawa H, Noguchi S, Sugiyama D, Morikawa H, Takeuchi Y, et al. Tyrosine kinase inhibitor imatinib augments tumor immunity by depleting effector regulatory T cells. J Exp Med (2020) 217(2):e20191009. doi: 10.1084/jem.20191009
141. Ahmad S, Abu-Eid R, Shrimali R, Webb M, Verma V, Doroodchi A, et al. Differential PI3Kδ signaling in CD4+ T-cell subsets enables selective targeting of T regulatory cells to enhance cancer immunotherapy. Cancer Res (2017) 77(8):1892–904. doi: 10.1158/0008-5472.CAN-16-1839
142. Bernal-Estévez DA, Ortíz Barbosa MA, Ortíz-Montero P, Cifuentes C, Sánchez R, Parra-López CA. Autologous dendritic cells in combination with chemotherapy restore responsiveness of T cells in breast cancer patients: A single-arm phase I/II trial. Front Immunol (2021) 12:669965. doi: 10.3389/fimmu.2021.669965
143. Choi CW, Jeong MH, Park YS, Son CH, Lee HR, Koh EK. Combination treatment of stereotactic body radiation therapy and immature dendritic cell vaccination for augmentation of local and systemic effects. Cancer Res Treat (2019) 51(2):464–73. doi: 10.4143/crt.2018.186
144. Ogasawara M, Miyashita M, Ota S. Vaccination of urological cancer patients with WT1 peptide-pulsed dendritic cells in combination with molecular targeted therapy or conventional chemotherapy induces immunological and clinical responses. Ther Apher Dial. (2018) 22(3):266–77. doi: 10.1111/1744-9987.12694
145. Teng CF, Wang T, Shih FY, Shyu WC, Jeng LB. Therapeutic efficacy of dendritic cell vaccine combined with programmed death 1 inhibitor for hepatocellular carcinoma. J Gastroen Hepatol (2021) 36(7):1988–96. doi: 10.1111/jgh.15398
146. Zanna MY, Yasmin AR, Omar AR, Arshad SS, Mariatulqabtiah AR, Nur-Fazila SH, et al. Review of dendritic cells, their role in clinical immunology, and distribution in various animal species. Int J Mol Sci (2021) 22(15):8044. doi: 10.3390/ijms22158044
147. Russo E, Laffranchi M, Tomaipitinca L, Del Prete A, Santoni A, Sozzani S, et al. NK cell anti-tumor surveillance in a myeloid cell-shaped environment. Front Immunol (2021) 12:787116. doi: 10.3389/fimmu.2021.787116
148. Ananiev J, Gulubova MV, Manolova IM. Prognostic significance of CD83 positive tumor-infiltrating dendritic cells and expression of TGF-beta 1 in human gastric cancer. Hepatogastroenterology. (2011) 58(110-111):1834–40. doi: 10.5754/hge10320
149. Ishigami S, Natsugoe S, Uenosono Y, Hata Y, Nakajo A, Miyazono F, et al. Infiltration of antitumor immunocytes into the sentinel node in gastric cancer. J Gastrointest Surg (2003) 7(6):735–9. doi: 10.1016/s1091-255x(03)00076-3
150. Ishigami S, Natsugoe S, Tokuda K, Nakajo A, Xiangming C, Iwashige H, et al. Clinical impact of intratumoral natural killer cell and dendritic cell infiltration in gastric cancer. Cancer Lett (2000) 159(1):103–8. doi: 10.1016/s0304-3835(00)00542-5
151. Niccolai E, Taddei A, Prisco D, Amedei A. Gastric cancer and the epoch of immunotherapy approaches. World J Gastroentero. (2015) 21(19):5778–93. doi: 10.3748/wjg.v21.i19.5778
152. Hafron JM, Wilfehrt HM, Ferro C, Harmon M, Flanders SC, McKay RR. Real-world effectiveness of sipuleucel-T on overall survival in men with advanced prostate cancer treated with androgen receptor-targeting agents. Adv Ther (2022) 39(6):2515–32. doi: 10.1007/s12325-022-02085-6
153. Vreeland TJ, Clifton GT, Hale DF, Chick RC, Hickerson AT, Cindass JL, et al. A phase IIb randomized controlled trial of the TLPLDC vaccine as adjuvant therapy after surgical resection of stage III/IV melanoma: A primary analysis. Ann Surg Oncol (2021) 28(11):6126–37. doi: 10.1245/s10434-021-09709-1
154. Bota D, Piccioni D, Duma C, LaRocca R, Kesari S, Abedi M, et al. Phase II trial of AV-GBM-1: Dendritic cell vaccine pulsed with lysate enriched for autologous tumor-initiating cell antigens in the treatment of patients with newly diagnosed glioblastoma. J Immunother Cancer. (2021) . 9:A1001. doi: 10.1136/jitc-2021-SITC2021.952
155. Cibula D, Rob L, Mallmann P, Knapp P, Klat J, Chovanec J, et al. Dendritic cell-based immunotherapy (DCVAC/OvCa) combined with second-line chemotherapy in platinum-sensitive ovarian cancer (SOV02): A randomized, open-label, phase 2 trial. Gynecol Oncol (2021) 162(3):652–60. doi: 10.1016/j.ygyno.2021.07.003
156. Seitz C, Rückert M, Deloch L, Weiss EM, Utz S, Izydor M, et al. Tumor cell-based vaccine generated with high hydrostatic pressure synergizes with radiotherapy by generating a favorable anti-tumor immune microenvironment. Front Oncol (2019) 9:805. doi: 10.3389/fonc.2019.00805
157. Chulpanova DS, Kitaeva KV, James V, Rizvanov AA, Solovyeva VV. Therapeutic prospects of extracellular vesicles in cancer treatment. Front Immunol (2018) 9:1534. doi: 10.3389/fimmu.2018.01534
158. Nava S, Lisini D, Frigerio S, Bersano A. Dendritic cells and cancer immunotherapy: The adjuvant effect. Int J Mol Sci (2021) 22(22):12339. doi: 10.3390/ijms222212339
159. Miao L, Zhang Y, Huang L. mRNA vaccine for cancer immunotherapy. Mol Cancer. (2021) 20(1):41. doi: 10.1186/s12943-021-01335-5
160. Dolcetti R, De Re V, Canzonieri V. Immunotherapy for gastric cancer: Time for a personalized approach? Int J Mol Sci (2018) 19(6):1602. doi: 10.3390/ijms19061602
161. Zhang W, Lu X, Cui P, Piao C, Xiao M, Liu X, et al. Phase I/II clinical trial of a wilms’ tumor 1-targeted dendriti cell vaccination-based immunotherapy in patients with advanced cancer. Cancer Immunol Immun (2019) 68(1):121–30. doi: 10.1007/s00262-018-2257-2
162. Sundar R, Rha SY, Yamaue H, Katsuda M, Kono K, Kim HS, et al. A phase I/Ib study of OTSGC-A24 combined peptide vaccine in advanced gastric cancer. BMC Cancer. (2018) 18(1):332. doi: 10.1186/s12885-018-4234-8
163. Koido S. Dendritic-tumor fusion cell-based cancer vaccines. Int J Mol Sci (2016) 17(6):828. doi: 10.3390/ijms17060828
164. Takakura K, Kajihara M, Ito Z, Ohkusa T, Gong J, Koido S. Dendritic-tumor fusion cells in cancer immunotherapy. Discovery Med (2015) 19(104):169–74.
165. Santisteban M, Solans BP, Hato L, Urrizola A, Mejías LD, Salgado E, et al. Final results regarding the addition of dendritic cell vaccines to neoadjuvant chemotherapy in early HER2-negative breast cancer patients: Clinical and translational analysis. Ther Adv Med Oncol (2021) 13:17588359211064653. doi: 10.1177/17588359211064653
166. Zhong R, Ling X, Cao S, Xu J, Zhang B, Zhang X, et al. Safety and efficacy of dendritic cell-based immunotherapy (DCVAC/LuCa) combined with Carboplatin/Pemetrexed for patients with advanced non-squamous non-Small-Cell lung cancer without oncogenic drivers. ESMO Open (2022) 7(1):100334. doi: 10.1016/j.esmoop.2021.100334
167. Wang C, Pu J, Yu H, Liu Y, Yan H, He Z, et al. A dendritic cell vaccine combined with radiotherapy activates the specific immune response in patients with esophageal cancer. J Immunother. (2017) 40(2):71–6. doi: 10.1097/CJI.0000000000000155
168. Wang L, Dai Y, Zhu F, Qiu Z, Wang Y, Hu Y. Efficacy of DC-CIK-Based immunotherapy combined with chemotherapy in the treatment of intermediate to advanced non-small cell lung cancer. Am J Transl Res (2021) 13(11):13076–83.
169. Chen S, Yang Y, Jiao Y, Sun H, Yan Z. Capecitabine metronomic chemotherapy combined with autologous CIK cell immunotherapy in the treatment of recurrent and metastatic triple-negative breast cancer. J BUON. (2021) 26(3):734–40.
170. Xu H, Qin W, Feng H, Song D, Yang X, Zhang J. Analysis of the clinical efficacy of dendritic cell-cytokine induced killer cell-based adoptive immunotherapy for colorectal cancer. Immunol Invest. (2021) 50(6):622–33. doi: 10.1080/08820139.2020.1781881
171. Wang X, Tang S, Cui X, Yang J, Geng C, Chen C, et al. Cytokine-induced killer Cell/Dendritic cell-Cytokine-Induced killer cell immunotherapy for the postoperative treatment of gastric cancer: A systematic review and meta-analysis. Med (Baltimore). (2018) 97(36):e12230. doi: 10.1097/MD.0000000000012230
172. Qiao G, Wang X, Zhou L, Zhou X, Song Y, Wang S, et al. Autologous dendritic cell-cytokine induced killer cell immunotherapy combined with s-1 plus cisplatin in patients with advanced gastric cancer: A prospective study. Clin Cancer Res (2019) 25(5):1494–504. doi: 10.1158/1078-0432.CCR-18-2360
173. Wu SY, Fu T, Jiang YZ, Shao ZM. Natural killer cells in cancer biology and therapy. Mol Cancer. (2020) 19(1):120. doi: 10.1186/s12943-020-01238-x
174. Devillier R, Chrétien AS, Pagliardini T, Salem N, Blaise D, Olive D. Mechanisms of NK cell dysfunction in the tumor microenvironment and current clinical approaches to harness NK cell potential for immunotherapy. J Leukoc Biol (2021) 109(6):1071–88. doi: 10.1002/JLB.5MR0920-198RR
175. Hu Z, Xu X, Wei H. The adverse impact of tumor microenvironment on NK-cell. Front Immunol (2021) 12:633361. doi: 10.3389/fimmu.2021.633361
176. van Vliet AA, Georgoudaki AM, Raimo M, de Gruijl TD, Spanholtz J. Adoptive NK cell therapy: A promising treatment prospect for metastatic melanoma. Cancers (Basel). (2021) 13(18):4722. doi: 10.3390/cancers13184722
177. Hsu J, Hodgins JJ, Marathe M, Nicolai CJ, Bourgeois-Daigneault MC, Trevino TN, et al. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J Clin Invest. (2018) 128(10):4654–68. doi: 10.1172/JCI99317
178. Oyer JL, Gitto SB, Altomare DA, Copik AJ. PD-L1 blockade enhances anti-tumor efficacy of NK cells. Oncoimmunology. (2018) 7(11):e1509819. doi: 10.1080/2162402X.2018.1509819
179. Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood. (2005) 105(8):3051–7. doi: 10.1182/blood-2004-07-2974
180. Parkhurst MR, Riley JP, Dudley ME, Rosenberg SA. Adoptive transfer of autologous natural killer cells leads to high levels of circulating natural killer cells but does not mediate tumor regression. Clin Cancer Res (2011) 17(19):6287–97. doi: 10.1158/1078-0432.CCR-11-1347
181. Li Y, Hermanson DL, Moriarity BS, Kaufman DS. Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell (2018) 23(2):181–92.e5. doi: 10.1016/j.stem.2018.06.002
182. Quintarelli C, Sivori S, Caruso S, Carlomagno S, Falco M, Boffa I, et al. Efficacy of third-party chimeric antigen receptor modified peripheral blood natural killer cells for adoptive cell therapy of b-cell precursor acute lymphoblastic leukemia. Leukemia. (2020) 34(4):1102–15. doi: 10.1038/s41375-019-0613-7
183. Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med (2020) 382(6):545–53. doi: 10.1056/NEJMoa1910607
184. Gong Y, Klein Wolterink RGJ, Wang J, Bos GMJ, Germeraad WTV. Chimeric antigen receptor natural killer (CAR-NK) cell design and engineering for cancer therapy. J Hematol Oncol (2021) 14(1):73. doi: 10.1186/s13045-021-01083-5
185. Li M, Zhi L, Yin M, Guo C, Zhang H, Lu C, et al. A novel bispecific chimeric PD1-DAP10/NKG2D receptor augments NK92-cell therapy efficacy for human gastric cancer SGC-7901 cell. Biochem Bioph Res Co. (2020) 523(3):745–52. doi: 10.1016/j.bbrc.2020.01.005
186. Hu W, Wang G, Huang D, Sui M, Xu Y. Cancer immunotherapy based on natural killer cells: Current progress and new opportunities. Front Immunol (2019) 10:1205. doi: 10.3389/fimmu.2019.01205
187. Fang F, Xiao W, Tian Z. NK cell-based immunotherapy for cancer. Semin Immunol (2017) 31:37–54. doi: 10.1016/j.smim.2017.07.009
188. Hernandez R, Põder J, LaPorte KM, Malek TR. Engineering IL-2 for immunotherapy of autoimmunity and cancer. Nat Rev Immunol (2022) 25. doi: 10.1038/s41577-022-00680-w
189. Abdolahi S, Ghazvinian Z, Muhammadnejad S, Ahmadvand M, Aghdaei HA, Ebrahimi-Barough S, et al. Adaptive NK cell therapy modulated by anti-PD-1 antibody in gastric cancer model. Front Pharmacol (2021) 12:733075. doi: 10.3389/fphar.2021.733075
190. Bergamaschi C, Pandit H, Nagy BA, Stellas D, Jensen SM, Bear J, et al. Heterodimeric IL-15 delays tumor growth and promotes intratumoral CTL and dendritic cell accumulation by a cytokine network involving XCL1, IFN-γ, CXCL9 and CXCL10. J Immunother Cancer. (2020) 8(1):e000599. doi: 10.1136/jitc-2020-000599
191. Wang W, Jin J, Dai F, Long Z, Liu X, Cai H, et al. Interleukin-15 suppresses gastric cancer liver metastases by enhancing natural killer cell activity in a murine model. Oncol Lett (2018) 16(4):4839–46. doi: 10.3892/ol.2018.9303
192. Becker-Hapak MK, Shrestha N, McClain E, Dee MJ, Chaturvedi P, Leclerc GM, et al. A fusion protein complex that combines IL-12, IL-15, and IL-18 signaling to induce memory-like NK cells for cancer immunotherapy. Cancer Immunol Res (2021) 9(9):1071–87. doi: 10.1158/2326-6066.CIR-20-1002
193. Ehlers FAI, Beelen NA, van Gelder M, Evers TMJ, Smidt ML, Kooreman LFS, et al. ADCC-inducing antibody trastuzumab and selection of KIR-HLA ligand mismatched donors enhance the NK cell anti-breast cancer response. Cancers (Basel). (2021) 13(13):3232. doi: 10.3390/cancers13133232
194. Zhang C, Hu Y, Shi C. Targeting natural killer cells for tumor immunotherapy. Front Immunol (2020) 11:60. doi: 10.3389/fimmu.2020.00060
195. André P, Denis C, Soulas C, Bourbon-Caillet C, Lopez J, Arnoux T, et al. Anti-NKG2A mAb is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK cells. Cell. (2018) 175(7):1731–43.e13. doi: 10.1016/j.cell.2018.10.014
196. Li Y, Huang K, Liu L, Qu Y, Huang Y, Wu Y, et al. Effects of complement and serum IgG on rituximab-dependent natural killer cell-mediated cytotoxicity against raji cells. Oncol Lett (2019) 17(1):339–47. doi: 10.3892/ol.2018.9630
197. Wang Z, Yin C, Lum LG, Simons A, Weiner GJ. Bispecific antibody-activated T cells enhance NK cell-mediated antibody-dependent cellular cytotoxicity. J Hematol Oncol (2021) 14(1):204. doi: 10.1186/s13045-021-01216-w
198. Lian S, Xie R, Ye Y, Lu Y, Cheng Y, Xie X, et al. Dual blockage of both PD-L1 and CD47 enhances immunotherapy against circulating tumor cells. Sci Rep (2019) 9(1):4532. doi: 10.1038/s41598-019-40241-1
199. González-Ochoa S, Tellez-Bañuelos MC, Méndez-Clemente AS, Bravo-Cuellar A, Hernández Flores G, Palafox-Mariscal LA, et al. Combination blockade of the IL6R/STAT-3 axis with TIGIT and its impact on the functional activity of NK cells against prostate cancer cells. J Immunol Res (2022) 2022:1810804. doi: 10.1155/2022/1810804
200. Jiang W, Li F, Jiang Y, Li S, Liu X, Xu Y, et al. Tim-3 blockade elicits potent anti-multiple myeloma immunity of natural killer cells. Front Oncol (2022) 12:739976. doi: 10.3389/fonc.2022.739976
201. Deng M, Wu D, Zhang Y, Jin Z, Miao J. MiR-29c downregulates tumor-expressed B7-H3 to mediate the antitumor NK-cell functions in ovarian cancer. Gynecol Oncol (2021) 162(1):190–9. doi: 10.1016/j.ygyno.2021.04.013
202. Yu X, Huang X, Chen X, Liu J, Wu C, Pu Q, et al. Characterization of a novel anti-human lymphocyte activation gene 3 (LAG-3) antibody for cancer immunotherapy. MAbs. (2019) 11(6):1139–48. doi: 10.1080/19420862.2019.1629239
203. Grabenstein S, Barnard KN, Anim M, Armoo A, Weichert WS, Bertozzi CR, et al. Deacetylated sialic acids modulates immune mediated cytotoxicity via the sialic acid-siglec pathway. Glycobiology. (2021) 31(10):1279–94. doi: 10.1093/glycob/cwab068
204. Georgiev H, Ravens I, Papadogianni G, Bernhardt G. Coming of age: CD96 emerges as modulator of immune responses. Front Immunol (2018) 9. doi: 10.3389/fimmu
205. Yalniz FF, Daver N, Rezvani K, Kornblau S, Ohanian M, Borthakur G, et al. A pilot trial of lirilumab with or without azacitidine for patients with myelodysplastic syndrome. Cl Lymph Myelom Leuk. (2018) 18(10):658–63.e2. doi: 10.1016/j.clml.2018.06.011
206. Bagot M, Porcu P, Marie-Cardine A, Battistella M, William BM, Vermeer M, et al. IPH4102, a first-in-Class anti-KIR3DL2 monoclonal antibody, in patients with relapsed or refractory cutaneous T-cell lymphoma: An international, first-in-Human, open-label, phase 1 trial. Lancet Oncol (2019) 20(8):1160–70. doi: 10.1016/S1470-2045(19)30320-1
207. Tinker AV, Hirte HW, Provencher D, Butler M, Ritter H, Tu D, et al. Dose-ranging and cohort-expansion study of monalizumab (IPH2201) in patients with advanced gynecologic malignancies: A trial of the Canadian cancer trials group (CCTG): IND221. Clin Cancer Res (2019) 25(20):6052–60. doi: 10.1158/1078-0432.CCR-19-0298
208. Wan R, Wang ZW, Li H, Peng XD, Liu GY, Ou JM, et al. Human leukocyte antigen-G inhibits the anti-tumor effect of natural killer cells via immunoglobulin-like transcript 2 in gastric cancer. Cell Physiol Biochem (2017) 44(5):1828–41. doi: 10.1159/000485819
209. Villa-Álvarez M, Sordo-Bahamonde C, Lorenzo-Herrero S, Gonzalez-Rodriguez AP, Payer AR, Gonzalez-Garcia E, et al. Ig-like transcript 2 (ILT2) blockade and lenalidomide restore NK cell function in chronic lymphocytic leukemia. Front Immunol (2018) 9:2917. doi: 10.3389/fimmu.2018.02917
210. Khan M, Arooj S, Wang H. NK cell-based immune checkpoint inhibition. Front Immunol (2020) 11:167. doi: 10.3389/fimmu.2020.00167
211. Shaim H, Shanley M, Basar R, Daher M, Gumin J, Zamler DB, et al. Targeting the αv Integrin/TGF-β axis improves natural killer cell function against glioblastoma stem cells. J Clin Invest. (2021) 131(14):e142116. doi: 10.1172/JCI142116
212. Bommarito D, Martin A, Forcade E, Nastke MD, Ritz J, Bellucci R. Enhancement of tumor cell susceptibility to natural killer cell activity through inhibition of the PI3K signaling pathway. Cancer Immunol Immun (2016) 65(3):355–66. doi: 10.1007/s00262-016-1804-y
213. Raskov H, Orhan A, Gaggar S, Gögenur I. Cancer-associated fibroblasts and tumor-associated macrophages in cancer and cancer immunotherapy. Front Oncol (2021) 11:668731. doi: 10.3389/fonc.2021.668731
214. Ngambenjawong C, Gustafson HH, Pun SH. Progress in tumor-associated macrophage (TAM)-targeted therapeutics. Adv Drug Delivery Rev (2017) 114:206–21. doi: 10.1016/j.addr.2017.04.010
215. Hao S, Meng J, Zhang Y, Liu J, Nie X, Wu F, et al. Macrophage phenotypic mechanomodulation of enhancing bone regeneration by superparamagnetic scaffold upon magnetization. Biomaterials. (2017) 140:16–25. doi: 10.1016/j.biomaterials.2017.06.013
216. Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol (2018) 233(9):6425–40. doi: 10.1002/jcp.26429
217. Hardbower DM, Asim M, Murray-Stewart T, Casero RA Jr, Verriere T, Lewis ND, et al. Arginase 2 deletion leads to enhanced M1 macrophage activation and upregulated polyamine metabolism in response to helicobacter pylori infection. Amino Acids (2016) 48(10):2375–88. doi: 10.1007/s00726-016-2231-2
218. Taniyama D, Taniyama K, Kuraoka K, Zaitsu J, Saito A, Nakatsuka H, et al. Long-term follow-up study of gastric adenoma; tumor-associated macrophages are associated to carcinoma development in gastric adenoma. Gastric Cancer. (2017) 20(6):929–39. doi: 10.1007/s10120-017-0713-x
219. Zhou Z, Yang Z, Wang J, Wang Q, Zhang H, Ju X. Research progress on Tumour-Associated macrophages in gastric cancer (Review). Oncol Rep (2021) 45(4):35. doi: 10.3892/or.2021.7986
220. Wang Z, Yang Y, Cui Y, Wang C, Lai Z, Li Y, et al. Tumor-associated macrophages regulate gastric cancer cell invasion and metastasis through TGFβ2/NF-κB/Kindlin-2 axis. Chin J Cancer Res (2020) 32(1):72–88. doi: 10.21147/j.issn.1000-9604.2020.01.09
221. Zhang AZ, Yuan X, Liang WH, Zhang HJ, Li Y, Xie YF, et al. Immune infiltration in gastric cancer microenvironment and its clinical significance. Front Cell Dev Biol (2022) 9:762029. doi: 10.3389/fcell.2021.762029
222. Bart VMT, Pickering RJ, Taylor PR, Ipseiz N. Macrophage reprogramming for therapy. Immunology. (2021) 163(2):128–44. doi: 10.1111/imm.13300
223. Anderson NR, Minutolo NG, Gill S, Klichinsky M. Macrophage-based approaches for cancer immunotherapy. Cancer Res (2021) 81(5):1201–8. doi: 10.1158/0008-5472.CAN-20-2990
224. Fujiwara T, Yakoub MA, Chandler A, Christ AB, Yang G, Ouerfelli O, et al. CSF1/CSF1R signaling inhibitor pexidartinib (PLX3397) reprograms tumor-associated macrophages and stimulates T-cell infiltration in the sarcoma microenvironment. Mol Cancer Ther (2021) 20(8):1388–99. doi: 10.1158/1535-7163.MCT-20-0591
225. Cai H, Zhang Y, Wang J, Gu J. Defects in macrophage reprogramming in cancer therapy: The negative impact of PD-L1/PD-1. Front Immunol (2021) 12:690869. doi: 10.3389/fimmu.2021.690869
226. Adams S, Kozhaya L, Martiniuk F, Meng TC, Chiriboga L, Liebes L, et al. Topical TLR7 agonist imiquimod can induce immune-mediated rejection of skin metastases in patients with breast cancer. Clin Cancer Res (2012) 18(24):6748–57. doi: 10.1158/1078-0432.CCR-12-1149
227. Feng Y, Mu R, Wang Z, Xing P, Zhang J, Dong L, et al. A toll-like receptor agonist mimicking microbial signal to generate tumor-suppressive macrophages. Nat Commun (2019) 10(1):2272. doi: 10.1038/s41467-019-10354-2
228. Rodell CB, Ahmed MS, Garris CS, Pittet MJ, Weissleder R. Development of adamantane-conjugated TLR7/8 agonists for supramolecular delivery and cancer immunotherapy. Theranostics. (2019) 9(26):8426–36. doi: 10.7150/thno.35434
229. Wang J, Zhang H, Yin X, Bian Y. Anti-CD47 antibody synergizes with cisplatin against laryngeal cancer by enhancing phagocytic ability of macrophages. Clin Exp Immunol (2021) 205(3):333–42. doi: 10.1111/cei.13618
230. Wu H, Liu J, Wang Z, Yuan W, Chen L. Prospects of antibodies targeting CD47 or CD24 in the treatment of glioblastoma. CNS Neurosci Ther (2021) 27(10):1105–17. doi: 10.1111/cns.13714
231. Li X, Yao W, Yuan Y, Chen P, Li B, Li J, et al. Targeting of tumour-infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut. (2017) 66(1):157–67. doi: 10.1136/gutjnl-2015-310514
232. Nie Y, Huang H, Guo M, Chen J, Wu W, Li W, et al. Breast phyllodes tumors recruit and repolarize tumor-associated macrophages via secreting CCL5 to promote malignant progression, which can be inhibited by CCR5 inhibition therapy. Clin Cancer Res (2019) 25(13):3873–86. doi: 10.1158/1078-0432.CCR-18-3421
233. Li X, Bu W, Meng L, Liu X, Wang S, Jiang L, et al. CXCL12/CXCR4 pathway orchestrates CSC-like properties by CAF recruited tumor associated macrophage in OSCC. Exp Cell Res (2019) 378(2):131–8. doi: 10.1016/j.yexcr.2019.03.013
234. Gambardella V, Castillo J, Tarazona N, Gimeno-Valiente F, Martínez-Ciarpaglini C, Cabeza-Segura M, et al. The role of tumor associated macrophages in gastric cancer development and their potential as a therapeutic target. Cancer Treat Rev (2020) 86:102015. doi: 10.1016/j.ctrv.2020.102015
235. Cannarile MA, Weisser M, Jacob W, Jegg AM, Ries CH, Rüttinger D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J Immunother Cancer. (2017) 5(1):53. doi: 10.1186/s40425-017-0257-y
236. Guan W, Li F, Zhao Z, Zhang Z, Hu J, Zhang Y. Tumor-associated macrophage promotes the survival of cancer cells upon docetaxel chemotherapy via the CSF1/CSF1R-CXCL12/CXCR4 axis in castration-resistant prostate cancer. Genes (Basel). (2021) 12(5):773. doi: 10.3390/genes12050773
237. Lv J, Chen FK, Liu C, Liu PJ, Feng ZP, Jia L, et al. Zoledronic acid inhibits thyroid cancer stemness and metastasis by repressing M2-like tumor-associated macrophages induced wnt/β-catenin pathway. Life Sci (2020) 256:117925. doi: 10.1016/j.lfs.2020.117925
238. Colon-Echevarria CB, Ortiz T, Maldonado L, Hidalgo-Vargas MJ, Pérez-Morales J, Aquino-Acevedo AN, et al. Zoledronic acid abrogates restraint stress-induced macrophage infiltration, PDGF-AA expression, and ovarian cancer growth. Cancers (Basel). (2020) 12(9):2671. doi: 10.3390/cancers12092671
239. Comito G, Pons Segura C, Taddei ML, Lanciotti M, Serni S, Morandi A, et al. Zoledronic acid impairs stromal reactivity by inhibiting M2-macrophages polarization and prostate cancer-associated fibroblasts. Oncotarget. (2017) 8(1):118–32. doi: 10.18632/oncotarget.9497
240. Germano G, Frapolli R, Belgiovine C, Anselmo A, Pesce S, Liguori M, et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell (2013) 23(2):249–62. doi: 10.1016/j.ccr.2013.01.008
241. Shree T, Olson OC, Elie BT, Kester JC, Garfall AL, Simpson K, et al. Macrophages and cathepsin proteases blunt chemotherapeutic response in breast cancer. Gene Dev (2011) 25(23):2465–79. doi: 10.1101/gad.180331.111
242. Sloas C, Gill S, Klichinsky M. Engineered CAR-macrophages as adoptive immunotherapies for solid tumors. Front Immunol (2021) 12:783305. doi: 10.3389/fimmu.2021.783305
243. Klichinsky M, Ruella M, Shestova O, Lu XM, Best A, Zeeman M, et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat Biotechnol (2020) 38(8):947–53. doi: 10.1038/s41587-020-0462-y
244. Law AMK, Valdes-Mora F, Gallego-Ortega D. Myeloid-derived suppressor cells as a therapeutic target for cancer. Cells. (2020) 9(3):561. doi: 10.3390/cells9030561
245. Cui C, Lan P, Fu L. The role of myeloid-derived suppressor cells in gastrointestinal cancer. Cancer Commun (Lond). (2021) 41(6):442–71. doi: 10.1002/cac2.12156
246. Bayik D, Lee J, Lathia JD. The role of myeloid-derived suppressor cells in tumor growth and metastasis. Exp Suppl. (2022) 113:189–217. doi: 10.1007/978-3-030-91311-3_7
247. Wang Y, Ding Y, Guo N, Wang S. MDSCs: Key criminals of tumor pre-metastatic niche formation. Front Immunol (2019) 10:172. doi: 10.3389/fimmu.2019.00172
248. Wang L, Chang EW, Wong SC, Ong SM, Chong DQ, Ling KL. Increased myeloid-derived suppressor cells in gastric cancer correlate with cancer stage and plasma S100A8/A9 proinflammatory proteins. J Immunol (2013) 190(2):794–804. doi: 10.4049/jimmunol.1202088
249. Wang PF, Song SY, Wang TJ, Ji WJ, Li SW, Liu N, et al. Prognostic role of pretreatment circulating MDSCs in patients with solid malignancies: A meta-analysis of 40 studies. Oncoimmunology. (2018) 7(10):e1494113. doi: 10.1080/2162402X.2018.1494113
250. Moaaz M, Lotfy H, Elsherbini B, Motawea MA, Fadali G. TGF-β enhances the anti-inflammatory effect of tumor- infiltrating CD33+11b+HLA-DR myeloid-derived suppressor cells in gastric cancer: A possible relation to MicroRNA-494. Asian Pac J Cancer P. (2020) 21(11):3393–403. doi: 10.31557/APJCP.2020.21.11.3393
251. Chen X, Wang Y, Wang J, Wen J, Jia X, Wang X, et al. Accumulation of T-helper 22 cells, interleukin-22 and myeloid-derived suppressor cells promotes gastric cancer progression in elderly patients. Oncol Lett (2018) 16(1):253–61. doi: 10.3892/ol.2018.8612
252. Shibata M, Gonda K, Nakajima T, Matsumoto Y, Nakamura I, Ohki S, et al. Pretreatment serum levels of circulating myeloid derived suppressor cells (MDSC) as a prognostic indicator in patients with gastrointestinal cancer. J Clin Oncol (2014) 32:3059. doi: 10.1200/jco.2014.32.15_suppl.3059
253. Lin F, Chen H, Jiang T, Zheng J, Liu Q, Yang B, et al. The effect of low-dose chemotherapy on the tumor microenvironment and its antitumor activity combined with anti-PD-1 antibody. Immunotherapy. (2022) 9. doi: 10.2217/imt-2021-0018
254. Burkert SC, Shurin GV, White DL, He X, Kapralov AA, Kagan VE, et al. Targeting myeloid regulators by paclitaxel-loaded enzymatically degradable nanocups. Nanoscale. (2018) 10(37):17990–8000. doi: 10.1039/c8nr04437f
255. Wang H, He X, Fang D, Wang X, Guan J, Shi ZW, et al. Gemcitabine-facilitated modulation of the tumor microenvironment and PD-1/PD-L1 blockade generate a synergistic antitumor effect in a murine hepatocellular carcinoma model. Clin Res Hepatol Gas. (2022) 46(4):101853. doi: 10.1016/j.clinre.2021.101853
256. Van Wigcheren GF, De Haas N, Mulder TA, Horrevorts SK, Bloemendal M, Hins-Debree S, et al. Cisplatin inhibits frequency and suppressive activity of monocytic myeloid-derived suppressor cells in cancer patients. Oncoimmunology. (2021) 10(1):1935557. doi: 10.1080/2162402X.2021.1935557
257. Xu W, Li S, Li M, Zhou H, Yang X. Upregulation of CD3ζ and l-selectin in antigen-specific cytotoxic T lymphocytes by eliminating myeloid-derived suppressor cells with doxorubicin to improve killing efficacy of neuroblastoma cells in vitro. J Clin Lab Anal (2022) 36(1):e24158. doi: 10.1002/jcla.24158
258. Kim W, Chu TH, Nienhüser H, Jiang Z, Del Portillo A, Remotti HE, et al. PD-1 signaling promotes tumor-infiltrating myeloid derived suppressor cells and gastric tumorigenesis in mice. Gastroenterology. (2021) 160(3):781–96. doi: 10.1053/j.gastro.2020.10.036
259. Li N, Han D, Sun J, Li Y, Zhang J, Zhang Y, et al. Subtypes of MDSCs in mechanisms and prognosis of gastric cancer and are inhibited by epirubicin and paclitaxel. Discovery Med (2018) 25(137):99–112.
260. Chen H, Jiang T, Lin F, Guan H, Zheng J, Liu Q, et al. PD-1 inhibitor combined with apatinib modulate the tumor microenvironment and potentiate anti-tumor effect in mice bearing gastric cancer. Int Immunopharmacol. (2021) 99:107929. doi: 10.1016/j.intimp.2021.107929
261. Tobin RP, Jordan KR, Robinson WA, Davis D, Borges VF, Gonzalez R, et al. Targeting myeloid-derived suppressor cells using all trans retinoic acid in melanoma patients treated with ipilimumab. Int Immunopharmacol. (2018) 63:282–91. doi: 10.1016/j.intimp.2018.08.007
262. Bauer R, Udonta F, Wroblewski M, Ben-Batalla I, Santos IM, Taverna F, et al. Blockade of myeloid-derived suppressor cell expansion with all-trans retinoic acid increases the efficacy of antiangiogenic therapy. Cancer Res (2018) 78(12):3220–32. doi: 10.1158/0008-5472.CAN-17-3415
263. Bullock K, Richmond A. Suppressing MDSC recruitment to the tumor microenvironment by antagonizing CXCR2 to enhance the efficacy of immunotherapy. Cancers (Basel). (2021) 13(24):6293. doi: 10.3390/cancers13246293
264. Hemmatazad H, Berger MD. CCR5 is a potential therapeutic target for cancer. Expert Opin Ther Tar. (2021) 25(4):311–27. doi: 10.1080/14728222.2021.1902505
265. Sun L, Clavijo PE, Robbins Y, Patel P, Friedman J, Greene S, et al. Inhibiting myeloid-derived suppressor cell trafficking enhances T cell immunotherapy. JCI Insight (2019) 4(7):e126853. doi: 10.1172/jci.insight.126853
266. Li M, Li M, Yang Y, Liu Y, Xie H, Yu Q, et al. Remodeling tumor immune microenvironment via targeted blockade of PI3K-γ and CSF-1/CSF-1R pathways in tumor associated macrophages for pancreatic cancer therapy. J Control Release. (2020) 321:23–35. doi: 10.1016/j.jconrel.2020.02.011
267. Yang L, Wang B, Qin J, Zhou H, Majumdar APN, Peng F. Blockade of CCR5-mediated myeloid derived suppressor cell accumulation enhances anti-PD1 efficacy in gastric cancer. Immunopharm Immunot. (2018) 40(1):91–7. doi: 10.1080/08923973.2017.1417997
268. Saung MT, Zheng L. Adding combination immunotherapy consisting of cancer vaccine, anti-PD-1 and anti-CSF1R antibodies to gemcitabine improves anti-tumor efficacy in murine model of pancreatic ductal adenocarcinoma. Ann Pancreat Cancer. (2019) 2:21. doi: 10.21037/apc.2019.11.01
269. Priceman SJ, Sung JL, Shaposhnik Z, Burton JB, Torres-Collado AX, Moughon DL, et al. Targeting distinct tumor-infiltrating myeloid cells by inhibiting CSF-1 receptor: Combating tumor evasion of antiangiogenic therapy. Blood. (2010) 115(7):1461–71. doi: 10.1182/blood-2009-08-237412
270. Greene S, Robbins Y, Mydlarz WK, Huynh AP, Schmitt NC, Friedman J, et al. Inhibition of MDSC trafficking with SX-682, a CXCR1/2 inhibitor, enhances NK-cell immunotherapy in head and neck cancer models. Clin Cancer Res (2020) 26(6):1420–31. doi: 10.1158/1078-0432.CCR-19-2625
271. Good L, Benner B, Carson WE. Bruton’s tyrosine kinase: An emerging targeted therapy in myeloid cells within the tumor microenvironment. Cancer Immunol Immun (2021) 70(9):2439–51. doi: 10.1007/s00262-021-02908-5
272. Zhu Z, Xu J, Li L, Ye W, Xu G, Chen B, et al. Effect of gastric cancer stem cell on gastric cancer invasion, migration and angiogenesis. Int J Med Sci (2020) 17(13):2040–51. doi: 10.7150/ijms.46774
273. Wang M, Chen B, Sun XX, Zhao XD, Zhao YY, Sun L, et al. Gastric cancer tissue-derived mesenchymal stem cells impact peripheral blood mononuclear cells via disruption of Treg/Th17 balance to promote gastric cancer progression. Exp Cell Res (2017) 361(1):19–29. doi: 10.1016/j.yexcr.2017.09.036
274. Giallongo C, Tibullo D, Parrinello NL, La Cava P, Di Rosa M, Bramanti V, et al. Granulocyte-like myeloid derived suppressor cells (G-MDSC) are increased in multiple myeloma and are driven by dysfunctional mesenchymal stem cells (MSC). Oncotarget. (2016) 7(52):85764–75. doi: 10.18632/oncotarget.7969
275. Giallongo C, Romano A, Parrinello NL, La Cava P, Brundo MV, Bramanti V, et al. Mesenchymal stem cells (MSC) regulate activation of granulocyte-like myeloid derived suppressor cells (G-MDSC) in chronic myeloid leukemia patients. PLoS One (2016) 11(7):e0158392. doi: 10.1371/journal.pone.0158392
276. Chen YL, Wang CY, Fang JH, Hsu HP. Serine/Threonine-protein kinase 24 is an inhibitor of gastric cancer metastasis through suppressing CDH1 gene and enhancing stemness. Am J Cancer Res (2021) 11(9):4277–93.
277. Wizenty J, Tacke F, Sigal M. Responses of gastric epithelial stem cells and their niche to helicobacter pylori infection. Ann Transl Med (2020) 8(8):568. doi: 10.21037/atm.2020.02.178
278. Ding L, Hayes MM, Photenhauer A, Eaton KA, Li Q, Ocadiz-Ruiz R, et al. Schlafen 4-expressing myeloid-derived suppressor cells are induced during murine gastric metaplasia. J Clin Invest. (2016) 126(8):2867–80. doi: 10.1172/JCI82529
279. Mesali H, Ajami A, Hussein-Nattaj H, Rafiei A, Rajabian Z, Asgarian-Omran H, et al. Regulatory T cells and myeloid-derived suppressor cells in patients with peptic ulcer and gastric cancer. Iran J Immunol (2016) 13(3):167–77.
280. He J, Hu W, Ouyang Q, Zhang S, He L, Chen W, et al. Helicobacter pylori infection induces stem cell-like properties in correa cascade of gastric cancer. Cancer Lett (2022) 542:215764. doi: 10.1016/j.canlet.2022.215764
281. Zhang T, Xiong H, Ma X, Gao Y, Xue P, Kang Y, et al. Supramolecular tadalafil nanovaccine for cancer immunotherapy by alleviating myeloid-derived suppressor cells and heightening immunogenicity. Small Methods (2021) 5(6):e2100115. doi: 10.1002/smtd.202100115
282. Tai LH, Alkayyal AA, Leslie AL, Sahi S, Bennett S, Tanese de Souza C, et al. Phosphodiesterase-5 inhibition reduces postoperative metastatic disease by targeting surgery-induced myeloid derived suppressor cell-dependent inhibition of natural killer cell cytotoxicity. Oncoimmunology. (2018) 7(6):e1431082. doi: 10.1080/2162402X.2018.1431082
283. Tang F, Tie Y, Hong W, Wei Y, Tu C, Wei X. Targeting myeloid-derived suppressor cells for premetastatic niche disruption after tumor resection. Ann Surg Oncol (2021) 28(7):4030–48. doi: 10.1245/s10434-020-09371-z
284. Weed DT, Zilio S, Reis IM, Sargi Z, Abouyared M, Gomez-Fernandez CR, et al. The reversal of immune exclusion mediated by tadalafil and an anti-tumor vaccine also induces PDL1 upregulation in recurrent head and neck squamous cell carcinoma: Interim analysis of a phase I clinical trial. Front Immunol (2019) 10:1206. doi: 10.3389/fimmu.2019.01206
285. Hassel JC, Jiang H, Bender C, Winkler J, Sevko A, Shevchenko I, et al. Results of a pilot trial with tadalafil in patients with metastatic melanoma (TaMe). Oncoimmunology. (2017) 6(9):e1326440. doi: 10.1080/2162402X.2017.1326440
286. Veltman JD, Lambers ME, van Nimwegen M, Hendriks RW, Hoogsteden HC, Aerts JG, et al. COX-2 inhibition improves immunotherapy and is associated with decreased numbers of myeloid-derived suppressor cells in mesothelioma. celecoxib influences MDSC function. BMC Cancer. (2010) 10:464. doi: 10.1186/1471-2407-10-464
287. Creelan BC, Gabrilovich DI, Gray JE, Williams CC, Tanvetyanon T, Haura EB, et al. Safety, pharmacokinetics, and pharmacodynamics of oral omaveloxolone (RTA 408), a synthetic triterpenoid, in a first-in-Human trial of patients with advanced solid tumors. Onco Targets Ther (2017) 10:4239–50. doi: 10.2147/OTT.S136992
288. Ko JS, Zea AH, Rini BI, Ireland JL, Elson P, Cohen P, et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res (2009) 15(6):2148–57. doi: 10.1158/1078-0432.CCR-08-1332
289. Mishalian I, Granot Z, Fridlender ZG. The diversity of circulating neutrophils in cancer. Immunobiology. (2017) 222(1):82–8. doi: 10.1016/j.imbio.2016.02.001
290. Kim EY, Abdul-Ghafar J, Chong Y, Yim K. Calculated tumor-associated neutrophils are associated with the tumor-stroma ratio and predict a poor prognosis in advanced gastric cancer. Biomedicines. (2022) 10(3):708. doi: 10.3390/biomedicines10030708
291. Giese MA, Hind LE, Huttenlocher A. Neutrophil plasticity in the tumor microenvironment. Blood. (2019) 133(20):2159–67. doi: 10.1182/blood-2018-11-844548
292. Shaul ME, Fridlender ZG. Cancer-related circulating and tumor-associated neutrophils -subtypes, sources and function. FEBS J (2018) 285(23):4316–42. doi: 10.1111/febs.14524
293. Li S, Cong X, Gao H, Lan X, Li Z, Wang W, et al. Tumor-associated neutrophils induce EMT by IL-17a to promote migration and invasion in gastric cancer cells. J Exp Clin Canc Res (2019) 38(1):6. doi: 10.1186/s13046-018-1003-0
294. Sugimoto T, Yamaji Y, Sakitani K, Isomura Y, Yoshida S, Yamada A, et al. Neutrophil infiltration and the distribution of intestinal metaplasia is associated with metachronous gastric cancer following endoscopic submucosal dissection. Can J Gastroenterol (2015) 29(6):321–5. doi: 10.1155/2015/950734
295. Mollinedo F. Neutrophil degranulation, plasticity, and cancer metastasis. Trends Immunol (2019) 40(3):228–42. doi: 10.1016/j.it.2019.01.006
296. Wang Y, Zhai J, Zhang T, Han S, Zhang Y, Yao X, et al. Tumor-associated neutrophils can predict lymph node metastasis in early gastric cancer. Front Oncol (2020) 10:570113. doi: 10.3389/fonc.2020.570113
297. Hoffmann A, Behrens HM, Heckl S, Krüger S, Becker T, Röcken C. Neoadjuvant/Perioperative treatment affects spatial distribution and densities of tumor associated neutrophils and CD8+ lymphocytes in gastric cancer. J Pers Med (2021) 11(11):1184. doi: 10.3390/jpm11111184
298. Yeo B, Redfern AD, Mouchemore KA, Hamilton JA, Anderson RL. The dark side of granulocyte-colony stimulating factor: A supportive therapy with potential to promote tumour progression. Clin Exp Metastasis. (2018) 35(4):255–67. doi: 10.1007/s10585-018-9917-7
299. Coffelt SB, Kersten K, Doornebal CW, Weiden J, Vrijland K, Hau CS, et al. IL-17-Producing γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nature. (2015) 522(7556):345–8. doi: 10.1038/nature14282
300. Teijeira Á, Garasa S, Gato M, Alfaro C, Migueliz I, Cirella A, et al. CXCR1 and CXCR2 chemokine receptor agonists produced by tumors induce neutrophil extracellular traps that interfere with immune cytotoxicity. Immunity. (2020) 52(5):856–71.e8. doi: 10.1016/j.immuni.2020.03.001
301. Powell D, Lou M, Barros Becker F, Huttenlocher A. Cxcr1 mediates recruitment of neutrophils and supports proliferation of tumor initiating astrocytes in vivo. Sci Rep (2018) 8(1):13285. doi: 10.1038/s41598-018-31675-0
302. Nywening TM, Belt BA, Cullinan DR, Panni RZ, Han BJ, Sanford DE, et al. Targeting both tumour-associated CXCR2+ neutrophils and CCR2+ macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut. (2018) 67(6):1112–23. doi: 10.1136/gutjnl-2017-313738
303. Steele CW, Karim SA, Leach JDG, Bailey P, Upstill-Goddard R, Rishi L, et al. CXCR2 inhibition profoundly suppresses metastases and augments immunotherapy in pancreatic ductal adenocarcinoma. Cancer Cell (2016) 29(6):832–45. doi: 10.1016/j.ccell.2016.04.014
304. Li A, Zhang N, Zhao Z, Chen Y, Zhang L. Overexpression of B7-H4 promotes renal cell carcinoma progression by recruiting tumor-associated neutrophils via upregulation of CXCL8. Oncol Lett (2020) 20(2):1535–44. doi: 10.3892/ol.2020.11701
305. Eash KJ, Means JM, White DW, Link DC. CXCR4 is a key regulator of neutrophil release from the bone marrow under basal and stress granulopoiesis conditions. Blood. (2009) 113(19):4711–9. doi: 10.1182/blood-2008-09-177287
306. Eash KJ, Greenbaum AM, Gopalan PK, Link DC. CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. J Clin Invest. (2010) 120(7):2423–31. doi: 10.1172/JCI41649
307. Jung K, Heishi T, Incio J, Huang Y, Beech EY, Pinter M, et al. Targeting CXCR4-dependent immunosuppressive Ly6Clow monocytes improves antiangiogenic therapy in colorectal cancer. P Natl Acad Sci USA (2017) 114(39):10455–60. doi: 10.1073/pnas.1710754114
308. Liu QUS, Li A, Xu H, Han X, Wu K. Targeting interlukin-6 to relieve immunosuppression in tumor microenvironment. Tumour Biol (2017) 39(6):1010428317712445. doi: 10.1177/1010428317712445
309. Nielsen SR, Strøbech JE, Horton ER, Jackstadt R, Laitala A, Bravo MC, et al. Suppression of tumor-associated neutrophils by lorlatinib attenuates pancreatic cancer growth and improves treatment with immune checkpoint blockade. Nat Commun (2021) 12(1):3414. doi: 10.1038/s41467-021-23731-7
310. Glodde N, Bald T, van den Boorn-Konijnenberg D, Nakamura K, O’Donnell JS, Szczepanski S, et al. Reactive neutrophil responses dependent on the receptor tyrosine kinase c-MET limit cancer immunotherapy. Immunity. (2017) 47(4):789–802 e9. doi: 10.1016/j.immuni.2017.09.012
311. Domiuez GA, Condamine T, Mony S, Hashimoto A, Wang F, Liu Q, et al. Selective targeting of myeloid-derived suppressor cells in cancer patients using DS-8273a, an agonistic TRAIL-R2 antibody. Clin Cancer Res (2017) 23(12):2942–50. doi: 10.1158/1078-0432.CCR-16-1784
312. Wu C, Ning H, Liu M, Lin J, Luo S, Zhu W, et al. Spleen mediates a distinct hematopoietic progenitor response supporting tumor-promoting myelopoiesis. J Clin Invest. (2018) 128(8):3425–38. doi: 10.1172/JCI97973
313. Aleyd Heineke MH, van Egmond M. The era of the immunoglobulin a fc receptor FcαRI; its function and potential as target in disease. Immunol Rev (2015) 268(1):123–38. doi: 10.1111/imr.12337
314. Zhu J, Powis de Tenbossche CG, Cané S, Colau D, van Baren N, Lurquin C, et al. Resistance to cancer immunotherapy mediated by apoptosis of tumor-infiltrating lymphocytes. Nat Commun (2017) 8(1):1404. doi: 10.1038/s41467-017-00784-1
315. Andziki L, Kasnitz N, Stahnke S, Wu CF, Gereke M, von Köckritz-Blickwede M, et al. Type I IFNs induce anti-tumor polarization of tumor associated neutrophils in mice and human. Int J Cancer. (2016) 138(8):1982–93. doi: 10.1002/ijc.29945
316. Anderson NM, Simon MC. The tumor microenvironment. Curr Biol (2020) 30(16):R921–5. doi: 10.1016/j.cub.2020.06.081
317. Jing Y, Xu F, Liang W, Liu J, Zhang L. Role of regulatory b cells in gastric cancer: Latest evidence and therapeutics strategies. Int Immunopharmacol. (2021) 96:107581. doi: 10.1016/j.intimp.2021.107581
318. Sarvaria A, Madrigal JA, Saudemont A. B cell regulation in cancer and anti-tumor immunity. Cell Mol Immunol (2017) 14(8):662–74. doi: 10.1038/cmi
319. Tian J, Zekzer D, Hanssen L, Lu Y, Olcott A, Kaufman DL. Lipopolysaccharide-activated b cells down-regulate Th1 immunity and rrevent autoimmune diabetes in nonobese diabetic mice. J Immunol (2001) 167(2):1081–9. doi: 10.4049/jimmunol.167.2.1081
320. Matsushita T. Regulatory and effector b cells: Friends or foes? J Dermatol Sci (2019) 93(1):2–7. doi: 10.1016/j.jdermsci.2018.11.008
321. Alhabbab RY, Nova-Lamperti E, Aravena O, Burton HM, Lechler RI, Dorling A, et al. Regulatory b cells: Development, phenotypes, functions, and role in transplantation. Immunol Rev (2019) 292(1):164–79. doi: 10.1111/imr.12800
322. Catalán D, Mansilla MA, Ferrier A, Soto L, Oleinika K, Aguillón JC, et al. Immunosuppressive mechanisms of regulatory b cells. Front Immunol (2021) 12:611795. doi: 10.3389/fimmu.2021.611795
323. Moulin V, Andris F, Thielemans K, Maliszewski C, Urbain J, Moser M. B lymphocytes regulate dendritic cell (DC) function in vivo: Increased interleukin 12 production by DCs from b cell-deficient mice results in T helper cell type 1 deviation. J Exp Med (2000) 192(4):475–82. doi: 10.1084/jem.192.4.475
324. Li W, Song D, Li H, Liang L, Zhao N, Liu T. Reduction in peripheral CD19+CD24hCD27+ b cell frequency predicts favourable clinical course in XELOX-treated patients with advanced gastric cancer. Cell Physiol Biochem (2017) 41(5):2045–52. doi: 10.1159/000475435
325. Wang K, Liu J, Li J. IL-35-Producing b cells in gastric cancer patients. Med (Baltimore). (2018) 97(19):e0710. doi: 10.1097/MD.0000000000010710
326. Murakami Y, Saito H, Shimizu S, Kono Y, Shishido Y, Miyatani K, et al. Increased regulatory b cells are involved in immune evasion in patients with gastric cancer. Sci Rep (2019) 9(1):13083. doi: 10.1038/s41598-019-49581-4
327. Zou ZQ, Guo TT, Cui J, Zhang L, Pan L. Onset of regulatory b cells occurs at initial stage of b cell dysfunction in multiple myeloma. Blood. (2019) 134. doi: 10.1182/blood-2019-128703
328. Yarchoan M, Mohan AA, Dennison L, Vithayathil T, Ruggieri A, Lesinski GB, et al. MEK inhibition suppresses b regulatory cells and augments anti-tumor immunity. PLoS One (2019) 14(10):e0224600. doi: 10.1371/journal.pone.0224600
329. Matsushita T, Horikawa M, Iwata Y, Tedder TF. Regulatory b cells (B10 cells) and regulatory T cells have independent roles in controlling experimental autoimmune encephalomyelitis initiation and late-phase immunopathogenesis. J Immunol (2010) 185(4):2240–52. doi: 10.4049/jimmunol.1001307
330. Li T, Fan J, Wang B, Traugh N, Chen Q, Liu JS, et al. TIMER: A web server for comprehensive analysis of tumor-infiltrating immune cells. Cancer Res (2017) 77(21):e108–10. doi: 10.1158/0008-5472.CAN-17-0307
331. Xiao Z, Hu L, Yang L, Wang S, Gao Y, Zhu Q, et al. TGFβ2 is a prognostic-related biomarker and correlated with immune infiltrates in gastric cancer. J Cell Mol Med (2020) 24(13):7151–62. doi: 10.1111/jcmm.15164
332. Chen Y, Chen W, Dai X, Zhang C, Zhang Q, Lu J. Identification of the collagen family as prognostic biomarkers and immune-associated targets in gastric cancer. Int Immunopharmacol. (2020) 87:106798. doi: 10.1016/j.intimp.2020.106798
333. Chen Q, Hu L, Chen K. Construction of a nomogram based on a hypoxia-related lncRNA signature to improve the prediction of gastric cancer prognosis. Front Genet (2020) 11:570325. doi: 10.3389/fgene.2020.570325
334. Kao TJ, Wu CC, Phan NN, Liu YH, Ta HDK, Anuraga G, et al. Prognoses and genomic analyses of proteasome 26S subunit, ATPase (PSMC) family genes in clinical breast cancer. Aging (Albany NY). (2021) 13(14):17970. doi: 10.18632/aging.203345
335. Christina Svensson M, Lindén A, Nygaard J, Borg D, Hedner C, Nodin B, et al. T Cells, b cells, and PD-L1 expression in esophageal and gastric adenocarcinoma before and after neoadjuvant chemotherapy: Relationship with histopathological response and survival. Oncoimmunology (2021) 10(1):1921443. doi: 10.1080/2162402X.2021.1921443
336. Wang DY, Salem JE, Cohen JV, Chandra S, Menzer C, Ye F, et al. Fatal toxic effects associated with immune checkpoint inhibitors: A systematic review and meta-analysis. JAMA Oncol (2018) 4(12):1721–8. doi: 10.1001/jamaoncol.2018.3923
337. Sterner RC, Sterner RM. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J (2021) 11(4):69. doi: 10.1038/s41408-021-00459-7
338. Zhang PP, Wang J, Ding DZ, Zhang L, Cheng C, Chen DK. Efficacy and safety of PD-1/PD-L1 inhibitors combined with CTLA-4 inhibitor versus chemotherapy for advanced lung cancer: A meta-analysis. Med (Baltimore). (2021) 100(35):e27121. doi: 10.1097/MD.0000000000027121

Keywords: gastric cancer, immunotherapy, immune cells, tumor microenvironment, treatment strategy
Citation: Zhao Y, Bai Y, Shen M and Li Y (2022) Therapeutic strategies for gastric cancer targeting immune cells: Future directions. Front. Immunol. 13:992762. doi: 10.3389/fimmu.2022.992762
Received: 13 July 2022; Accepted: 29 August 2022;
Published: 23 September 2022.
Edited by:
Valentyn Oksenych, University of Oslo, NorwayReviewed by:
Chih-Yang Wang, Taipei Medical University, TaiwanCopyright © 2022 Zhao, Bai, Shen and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yapeng Li, bGl5YXBlbmdAamx1LmVkdS5jbg==; Meili Shen, c2hlbm1laWxpQGpsdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.