 Benjy J. Y. Tan
Benjy J. Y. Tan Kenji Sugata1
Kenji Sugata1 Masahiro Ono
Masahiro Ono Yorifumi Satou
Yorifumi Satou- 1Division of Genomics and Transcriptomics, Joint Research Center for Human Retrovirus Infection, Kumamoto University, Kumamoto, Japan
- 2Department of Life Sciences, Imperial College London, London, United Kingdom
Human T-cell leukemia virus type 1 (HTLV-1), a retrovirus which mainly infects CD4+ T cells and causes adult T-cell leukemia/lymphoma (ATL), is primarily transmitted via direct cell-to-cell transmission. This feature generates a wide variety of infected clones in hosts, which are maintained via clonal proliferation, resulting in the persistence and survival of the virus. The maintenance of the pool of infected cells is achieved by sculpting the immunophenotype of infected cells and modulating host immune responses to avoid immune surveillance. Here, we review the processes undertaken by HTLV-1 to modulate and subvert host immune responses which contributes to viral persistence and development of ATL.
Introduction
Human T-cell leukemia virus type 1 (HTLV-1), the first human retrovirus discovered (1–3), was identified as the etiological agent of adult T cell leukemia/lymphoma (ATL) (4, 5) and to date, one of the only seven human viruses with strong epidemiological links to human cancers (6). The virus primarily infects CD4+ T cells and induces a lifelong infection in infected individuals as asymptomatic carriers (ACs) (Figure 1). Nevertheless, approximately 3-5% of infected individuals eventually develop ATL, a malignant CD4+ T cell neoplasm (7) (Figure 1). HTLV-1 is also associated with various other inflammatory diseases including uveitis, dermatitis, arthropathy (8) and most notably HTLV-1-associated myelopathy/tropical spastic paraparesis (HAM/TSP) which affects 1-4% of infected individuals (7, 9, 10) (Figure 1).
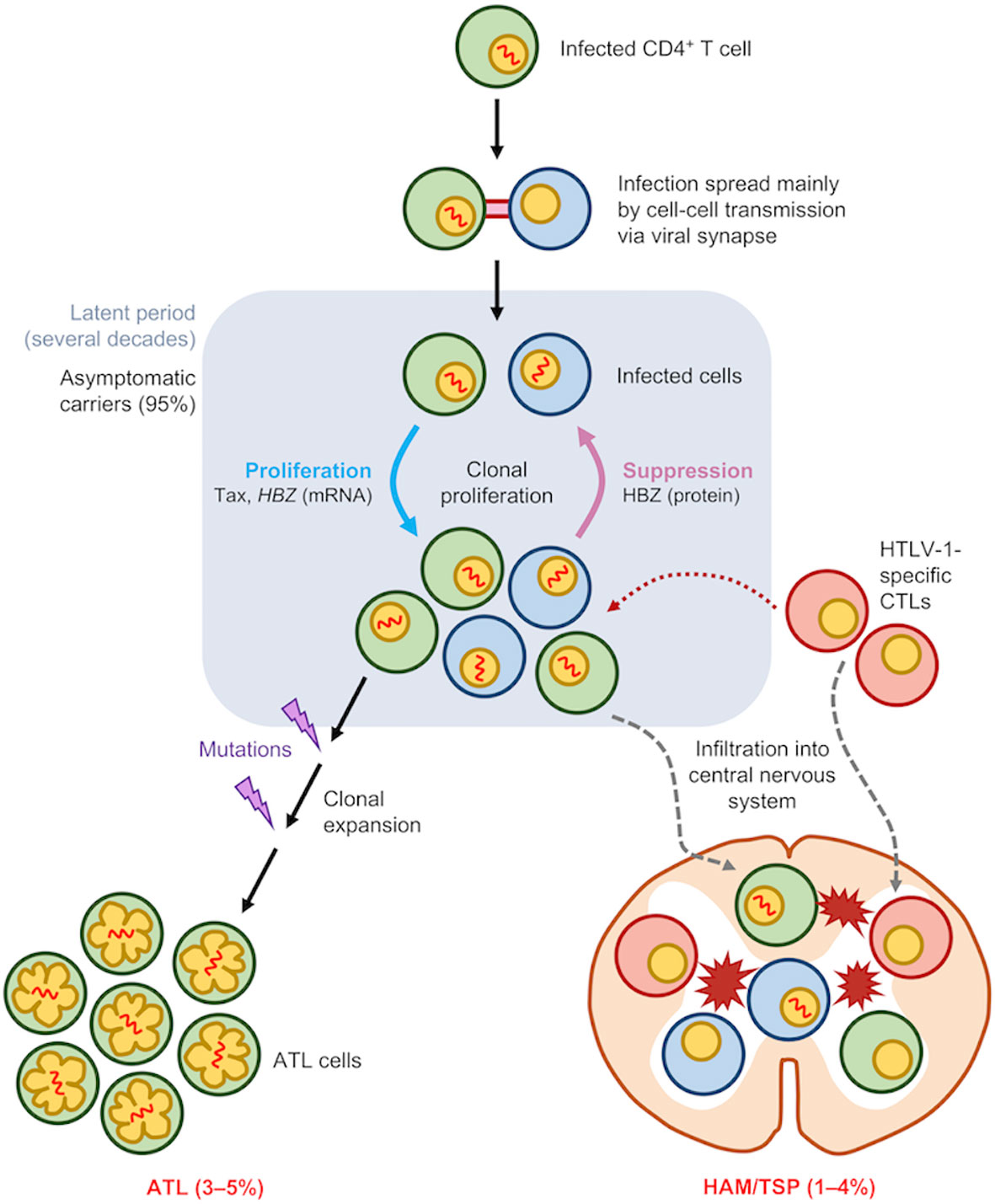
Figure 1 Natural history of HTLV-1 infection. HTLV-1 primarily infects CD4+ T cells and spread mainly by cell-to-cell transmission via viral synapse. The pool of infected cells are maintained by clonal proliferation which is promoted by Tax, HBZ mRNA and other viral accessory proteins while HBZ protein and host CTLs act to suppress them. Most HTLV-1-infected individuals remain life-long asymptomatic carriers. However, in approximately 5% of infected individuals, acquisition and accumulation of certain mutations leads to malignant transformation of infected cells into adult T-cell leukemia (ATL) cells. Additionally, about 4% of infected individuals develop HTLV-1-associated myelopathy/tropical spastic paraparesis (HAM/TSP), which is caused by infiltration of infected cells and CTLs into the central nervous system.
HTLV-1 is mainly transmitted through three routes: (1) sexual intercourse, (2) breastfeeding, and (3) blood transfusion and needle sharing (11). The peculiar thing about HTLV-1 transmission is that cell-free infection of HTLV-1 is extremely inefficient (12). Instead, HTLV-1 primarily relies on direct cell-to-cell transmission via virological synapse (13), viral biofilm (14) or cellular conduits such as tunneling nanotubes (15). Within an infected individual, HTLV-1 presence is then maintained largely via mitotic division of infected cells (16) (Figure 1) with a minor contribution by infectious spread (17).
In fact, immune cells are constantly surveilling the body to look for and eliminate foreign pathogens or pre-cancerous and cancerous cells (18). This process is called immune surveillance (18) and is carried out by both the innate and acquired immune system. Immune response to HTLV-1 has been extensively reviewed elsewhere (19–22) with most of the recent work focusing on the innate immune responses, including retroviral restriction factors (23, 24) and chemokines (25). HTLV-1 mainly infects CD4+ T cells, which are one of the major players of the cell-mediated immune response primarily carried out by T cells (26). Cell-mediated immune response is activated upon antigen presentation by professional antigen-presenting cells (APCs) such as dendritic cells (DCs) and B cells to T cells (26). Elimination of the foreign pathogen or cancerous cells is then carried out through the production of inflammatory cytokines and the killing action of effector cells including cytotoxic T lymphocytes (CTLs), macrophages and natural killer (NK) cells (26). To ensure long-term survival and circumvent this immune surveillance, HTLV-1 would have developed certain traits to modulate host cell’s immunophenotype, immune response and environment so that the infected cells have a survival advantage.
In this review, we will discuss on what is known so far on the virus–host interplay which renders infected cells invisible from immune surveillance and thus contributing to HTLV-1 persistence in infected individuals. The article is divided into 3 major sections which chiefly describes: (1) immune escape mechanisms utilized by HTLV-1-infected cells for persistence, (2) malignant transformation of infected cells plus additional mechanisms employed by leukemic cells, and (3) new findings on the hijack of physiological T-cell activation mechanisms for immune evasion.
Immune escape mechanisms used by HTLV-1-infected cells for persistence
During the infectious phase, thousands of infected-cell clones are established before plateauing and only those which escaped initial CTL detection continue to live on for decades in infected individuals (27, 28). Here, we will review some of the host and virus factors which sculps the clonal landscape of HTLV-1-infected cells.
Regulation of HTLV-1 provirus transcription
The HTLV-1 provirus is around 9 kb in length and encodes several structural (gag, pol and env), regulatory (tax and rex) and accessory proteins [p12, p13, p30 and HTLV-1 bZIP factor (HBZ)]. Among them, Tax and HBZ are important for the maintenance and proliferation of the pool of infected cells. Tax, which is encoded in the sense strand, is a highly immunogenic protein; while HBZ, encoded in the anti-sense strand, has a lower immunogenicity (29, 30). As such, there is a need for different expression pattern between tax and hbz to minimize the activity of Tax-specific CTLs.
Transcription from the sense and anti-sense strand of the provirus is controlled by different mechanisms. Sense strand transcription is strongly enhanced by Tax protein, which recruits cAMP response element binding protein (CREB) and the transcriptional coactivators CBP/p300 to the Tax response elements in the 5’ long terminal repeat (LTR) (31). This transcriptional burst is then suppressed by the viral proteins p30, which binds and retains Tax mRNA in the nucleus (32), and HBZ, which interacts with the KIX domain of p300 (33). Interestingly, HBZ acts in a negative feedback loop to control provirus expression and re-establish provirus latency as a small surge of HBZ transcription is observed during the late stage of the Tax burst (34).
Epigenetically, the 5’ LTR is heavily methylated which silences sense strand transcription. However, this only extends up to roughly three quarters of the provirus with the HBZ and 3’ LTR remaining free of DNA methylation, allowing continued expression of HBZ (35, 36). This unique pattern of epigenetic modification is achieved through the insulator-binding protein CCCTC-binding factor (CTCF) that acts to define boundaries between transcriptionally active and inactive regions of the genome by restricting the spread of epigenetic modifications (37). Binding of CTCF to the proviral DNA at a defined epigenetic border helps to regulate and modify provirus transcription. Additionally, a novel enhancer region was recently identified near the 3’ LTR which acts to enhance transcription from the 3’ LTR as well as acting as another barrier to prevent spreading of epigenetic modifications towards the 3’ LTR (38).
Differences in promoter activity, epigenetic modifications as well as regulatory elements as described above contribute to the contrasting transcriptional kinetics of Tax and HBZ whereby the tax gene is being expressed transiently in rare, self-limiting bursts (34, 39) while the hbz gene is continuously expressed at low levels (40, 41) (Figure 2).
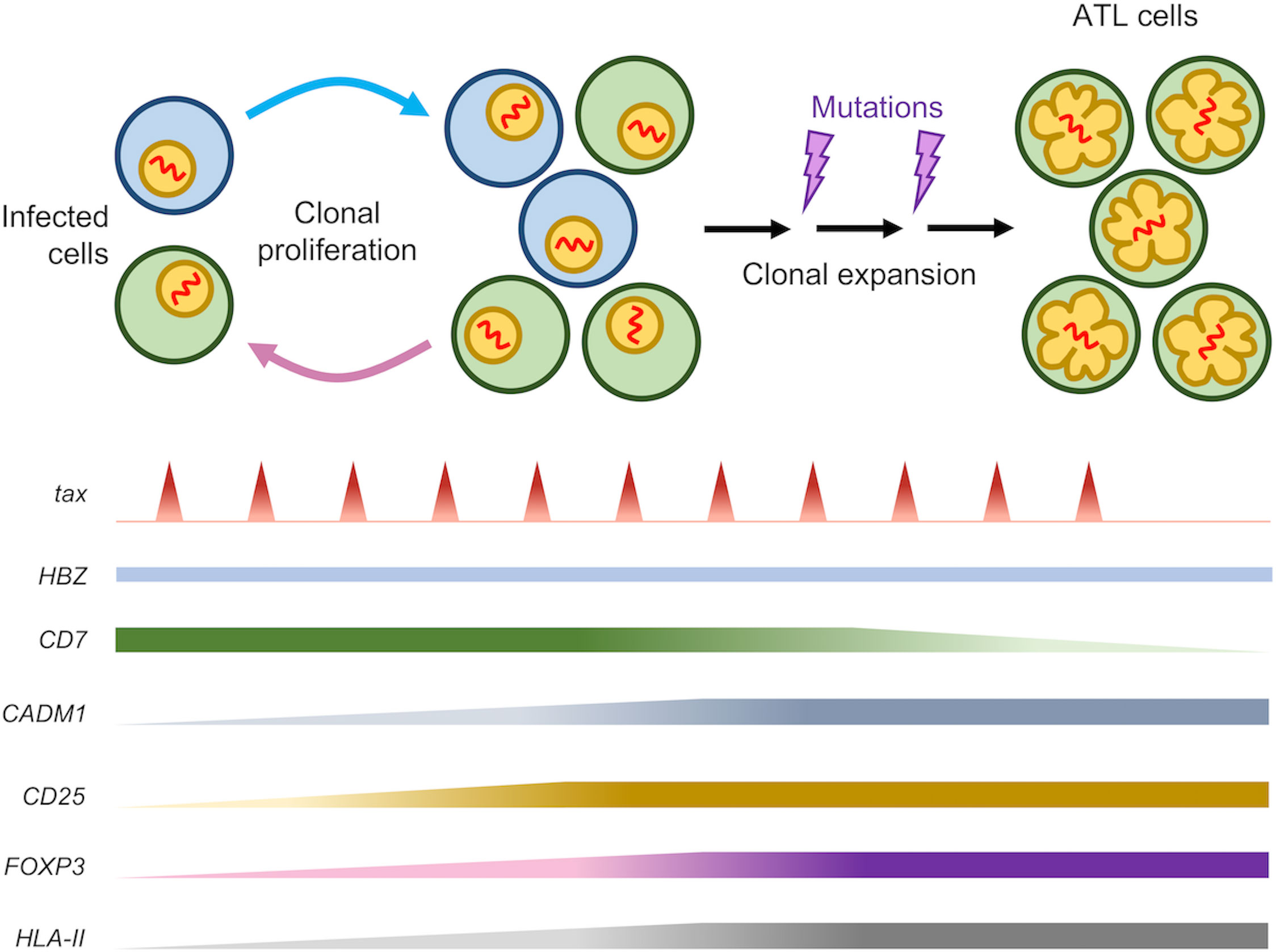
Figure 2 Expression pattern of several related genes during HTLV-1 persistence and leukemogenesis. After a long latency period, about 5% of HTLV-1-infected individuals develop ATL. Both Tax and HBZ are critical for leukemogenesis with the HBZ gene being constantly expressed while tax is transcribed in rare, short, self-limiting bursts. Infected cells exhibit increased expression of genes related to T-cell activation (CD25) as well as Treg (FOXP3). Subsequent accumulation of certain mutations during the lifetime of infected clone potentiates leukemic transformation which is accompanied by a loss of CD7 expression and an increase in CADM1 expression. Expression of CD25 and FOXP3, as well as HLA-II, is maintained throughout the latency phase as well as in ATL cells after malignant transformation.
CTLs response against HTLV-1-infected cells
Among the various viral proteins, Tax is the major viral antigen targeted by CTL (42, 43). The antigen specificity and quality of the CTL response is determined by the human leukocyte antigen (HLA) class I alleles of infected individuals. Several studies reported that individuals with HLA-A*02 had stronger affinities towards various Tax epitopes, especially Tax11-19, which confers a lower proviral load due to the selective pressure against Tax-expressing cells. The same studies also revealed that individuals with HLA-Cw*08 are associated with lower proviral load while HLA-B*54 is associated with higher proviral load (44–48).
Notably, HBZ-specific CTLs have a significantly lower frequency than Tax-specific CTLs in infected individuals (30, 49) and are unable to lyse infected cells (29). It has been suggested that the lower frequency and weak immune response of HBZ-specific CTLs is a consequence of the low expression and antigenicity as an immunogen. This is not surprising, given the indispensable roles of HBZ in maintaining the survival and proliferation of infected cells (50, 51) in vivo which necessitates the need for persistent expression (Figure 3). Such mechanism is not specific to HTLV-1 as it was observed in other oncogenic viruses as well, such as EBNA-1 in Epstein-Barr virus (EBV) (52) and E7 in human papilloma virus (HPV) (53). Furthermore, HBZ in its RNA form elicits additional functions including anti-apoptotic activity and suppression of Tax transcription to evade Tax-specific CTLs (54) (Figure 3).
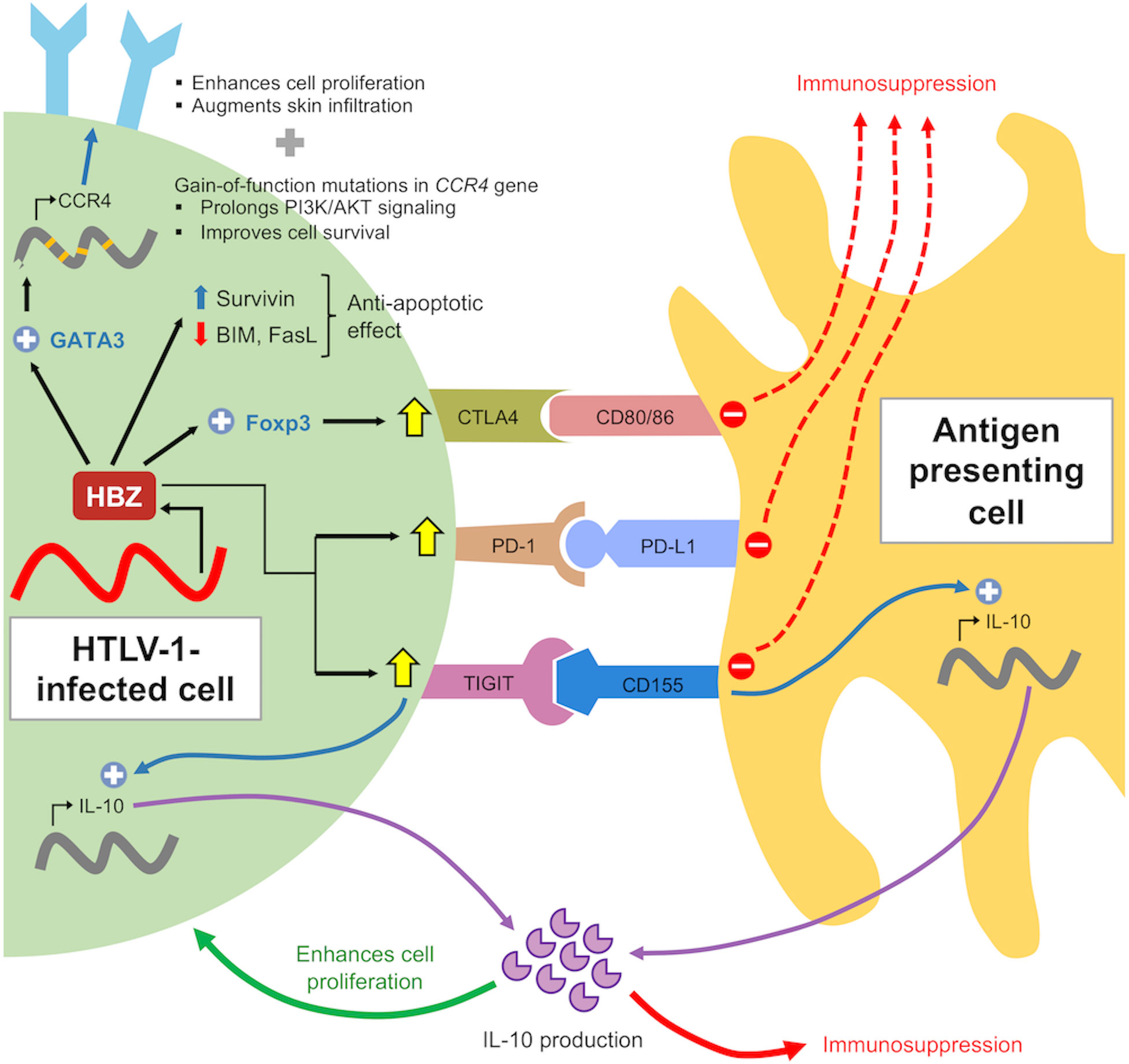
Figure 3 Role of HBZ in immune evasion of infected cells. HBZ induces expression of co-inhibitory molecules to suppress immune activation and confers anti-apoptotic properties by altering expression pattern of pro- and anti-apoptotic proteins. HBZ also induces IL-10 expression in infected cells and antigen presenting cells through TIGIT. IL-10 acts to suppress host immune response as well as enhancing cellular proliferation of infected cells through modulation of STAT signaling by HBZ (not shown).
In terms of clonal selection during viral persistence, the expression of Tax confers a disadvantage towards the survival of infected cells whereas HBZ might not. This selective advantage is not specific for ATL cells, but rather a general aspect of HTLV-1 infection, which was demonstrated in a comprehensive DNA-capture-seq analysis whereby in all HTLV-1-infected individuals, the proportion of 5’ defective provirus is around 15% but 3’ defective ones are extremely rare (~2.5%) (55). Analysis of viral genes expression also showed that Tax is not expressed in approximately 60% of ACs and ATL cases but HBZ expression can be seen in all of them (40, 56, 57), which is consistent with the notion that Tax expression is unfavorable for survival in vivo.
Integration preference and provirus structure
HTLV-1 prefers to integrate into transcriptionally active, accessible regions in the host genome with a slight bias to transcriptional start sites (TSS). However, as these proviruses are transcriptionally active, viral genes are expressed resulting in a majority of these cells being eliminated. Most of the infected cells that survived this initial selection are those with integration sites in transcriptionally quiescent regions (58–60). Nevertheless, some of these clones with provirus integrated in transcriptionally active regions do survive and remain in circulation at least partially due to the spatial organization of the chromosome harboring the integrated provirus (61). Although HTLV-1 shows no preference for any given chromosome during initial integration, the clones that survived and persisted in vivo are frequently found in the long arm of acrocentric chromosomes (chromosomes 13, 14, 15, 21 and 22) and close to the centromere (61, 62). The centromere-proximal regions of these chromosomes are situated in transcriptionally repressive regions in the nucleus – the nucleolar periphery or nuclear lamina – hence these regions are transcriptionally quiescent which favors the survival of these clones (61).
Besides integration site preference, the proviral structure also determines clonal survival and longevity. Defective proviruses are preferentially selected as these may lack the 5’ LTR and flanking regions coding immunogenic proteins, Tax in particular, which confers survival advantage by evading Tax-specific CTLs (63–65). The loss of the 5’ LTR has been well characterized in ATL patients where there are about 30% defective proviruses (55, 66, 67) while in ACs and HAM/TSP patients, about 15–20% of provirus are defective (55), suggesting that deletion of certain regions from the provirus genome for immune evasion is a general feature of HTLV-1-infected clones (55). Several studies have demonstrated that oncogenesis can still occur despite the absence of Tax, indicating that Tax is important for de novo infection but may be dispensable for leukemic transformation and maintenance of the clonal population of infected cells (64). Thus, both proviral integration site and structure plays a role in determining the fate of infected cells in vivo.
Immunophenotype remodeling through Foxp3 expression
Tregs, defined as CD4+Foxp3+ T cells, play a role in suppressing excessive immune activity and function as ‘guardians’ of immune homeostasis. Interestingly, however, 60–70% of ATL cells express Foxp3 (68–71). Hence, ATL was previously considered as a tumor of HTLV-1-infected Tregs (72). However, recent findings have demonstrated the immunophenotypic modifying properties of HBZ induces some features of Treg in conventional CD4+ T cells, although it does not confer suppressive function. HBZ induces Foxp3 expression via Smad3-dependent TGF-β signaling (73). However, HBZ then interferes with the DNA binding activity and function of Foxp3 by direct physical interaction (74). Thus, Foxp3+ HTLV-1-infected and Foxp3+ ATL cells may not always possess the suppressive function of Tregs (75–77). The key question here is why does HTLV-1 alter the immunophenotype of its host cell to mimic the ‘guardians’ of immune homeostasis? One plausible explanation is that Foxp3+ infected T cells can evade immune surveillance via the expression of immune checkpoint molecules such as CTLA-4, PD-1 and TIGIT and immunoregulatory cytokines including IL-35 and IL-10. The similarities and differences of immunosuppressive mechanism between Treg and HTLV-1-infected cells still require further understanding and further studies utilizing single-cell methods for precise immunophenotyping will provide more evidence on how HTLV-1 evade host immunity (78, 79).
Host restriction factors
Restriction factors, the first line of antiviral defense, are host cellular proteins that recognize and interfere with specific steps of the virus replication cycle, thus blocking infection. Restriction factors were first described as counteractors of HIV-1 infection and some well-characterized ones include APOBEC3G (apolipoprotein B mRNA-editing enzyme, catalytic subunit-like 3G), SAMHD1 (sterile alpha motif and histidine aspartate domain-containing protein 1), Tetherin/BST-2 (bone marrow stromal antigen 2) and TRIM5α (tripartite motif 5α) (80).
However, as HTLV-1 rarely produce free viral particles and spread primarily via cell-to-cell transmission, CTL response against immunogenic viral proteins, particularly Tax, is the predominant immune response against HTLV-1, especially during the initial stages of infection with the role of restriction factors remain poorly understood (23, 81). Nevertheless, in view of the complexity of clonal persistence and the wide spectrum of associated diseases caused by HTLV-1, mechanisms of intrinsic immunity may yet have a role to play. Studies pertaining to HTLV-1 restriction factors are rather limited with the majority of them discovered through studies in HIV-1 infection. The role of restriction factors in HTLV-1 infection has been extensively reviewed elsewhere (23, 24) and is summarized briefly in Table 1.

Table 1 Restriction factors and their impact on anti-HTLV-1 immunity.
Malignant transformation of infected cells and additional mechanisms employed by leukemic cells
Among the many infected clones, only a selected cell clone will eventually undergo leukemic transformation. The long latent period before ATL onset suggests that malignant transformation occurs when HTLV-1-infected cells acquire a certain set of genetic and epigenetic alterations (90–92) while escaping host’s immune surveillance. Once HTLV-1-infected cells become malignant, they will have to escape not only HTLV-1-specific immune surveillance but also cancer-specific ones (93).
Modulation of immune checkpoint expression
HTLV-1 is also known to modulate the expression of immune checkpoint molecules on the cell surface of infected cells for immune evasion. Several studies have shown that HTLV-1 infection upregulates both the expression of stimulatory and inhibitory immune checkpoint molecules and is further augmented by leukemic transformation and ATL progression (78, 79, 94). For instance, the viral protein Tax is involved in the overexpression of OX40 (95, 96), a costimulatory molecule which promotes cellular proliferation, enhances cell survival and suppresses Treg activity (97). Interestingly, HTLV-1 selectively enhances expression of particular coinhibitory receptors while suppressing the others. For example, Kinosada et al. (98) showed the involvement of the viral protein HBZ in this process whereby HBZ enhances the expression of TIGIT and PD-1 but suppresses the expression of BTLA and LAIR-1 (98).
TIGIT is a competitive inhibitor of the costimulatory receptor CD226 for binding with the CD155 ligand, resulting in poor T cell activation. Physiologically, TIGIT-mediated signaling inhibits T cell proliferation through the interaction of its cytoplasmic ITIM domains with the THEMIS : SHP complex (99). However, HTLV-1-infected and ATL cells can proliferate in vivo despite the upregulation of TIGIT, indicating that downstream TIGIT signaling is impaired. This impairment thus enhances cellular proliferation and is mediated by HBZ which interacts with THEMIS and weakens its interaction with the phosphatases SHP1 and SHP2 (100). Additionally, HBZ also suppresses CD226 expression (100).
An increase in TIGIT-mediated signaling was correlated with increased IL-10 production as shown in TIGIT+CD4+ T cells of HBZ-transgenic mice (100). Likewise, an elevated IL-10 levels was observed in the serum of HTLV-1-infected and ATL patients (101, 102). This increased IL-10 production not only occurs from DCs, but from infected T-cells as well (100, 102). IL-10 is an immunoregulatory cytokine with the functions of suppressing inflammation and Th1 responses (103). Therefore, this elevated expression leads to the generation of an immunosuppressive microenvironment which enhances survival of HTLV-1-infected and ATL cells. Furthermore, IL-10 was reported to support proliferation of ATL cells despite not known to promote T cell proliferation normally (104). This stark contrast of IL-10 function is attributed to the viral protein HBZ as it can modulate IL-10 signaling by interacting with STAT3 (105). This indigenous strategy by HTLV-1 enables the virus to modulate IL-10-mediated signaling pathways for suppressing host immune response in a paracrine manner, while supporting proliferation of infected cells in an autocrine manner.
The role of PD-1 upregulation in HTLV-1 infection is much less established. Expression of PD-1, as well as its ligand PD-L1, is shown to be augmented in HTLV-1-infected individuals and ATL patients (106, 107). Several studies showed that upon PD-1/PD-L1 blockade, CTL function was restored indicating operating PD-1/PD-L1 axis during HTLV-1 infection which diminishes CTL function (106–108). The PD-1 ligands, PD-L1 and PD-L2, are expressed on the surface of dendritic cells and reverse signaling from these ligands stimulates IL-10 expression and confer dendritic cells with an immunosuppressive phenotype (109). Thus, PD-1 upregulation by HTLV-1 in infected cells is a possible mechanism for escaping immune surveillance. Another study by Koya et al. (78) reported that upregulated PD-L1 in malignant cells can be transferred to the microenvironment and alter the anti-tumor immune response (78). However, contrary to the reports above, Takeuchi et al. (110) reported a significant association between PD-L1 expression and improved survival of ATL patients (110). Additionally, similar to TIGIT, the suppressive signaling from PD-1 is impaired via HBZ interaction with the THEMIS : SHP complex, thus avoiding growth suppression (98, 100). Furthermore, a clinical trial using the anti-PD-1 antibody, nivolumab, showed that all three participants showed dramatic progression of ATL with a rapid expansion of predominant ATL clones, which was a result of an unanticipated loss of ATL suppression rather than a selective advantage for a specific clone after PD-1 blockade (111, 112). These contrasting findings indicate that further studies are warranted to understand the role and importance of PD-1/PD-L1 upregulation by HTLV-1 and whether PD-1/PD-L1 blockade can be used efficiently as immunotherapy for ATL patients.
CCR4 expression
CCR4 expression is a well-known hallmark of ATL cells (113). CCR4 upregulation is reported to be induced by HBZ-mediated GATA3 expression which then activates the promoter of the CCR4 gene. Upregulated CCR4 is associated with enhanced cellular proliferation and chemotactic properties of ATL cells (114). Additionally, HTLV-1-infected cells produces IFN-γ (115), which induces production of the CCR4 ligands CCL17 and CCL22 in keratinocytes and endothelial cells. This chemoattracts HTLV-1-infected cells and augments infiltration into the skin (114). CCL22 production in infected cells is also enhanced by the Tax protein, which in turn chemoattracts and maintains a high frequency of CD4+Foxp3+CCR4+ regulatory T cells (Treg) in circulation. These Treg in turn suppresses and reduced the efficiency of HTLV-1-specific CTL response which promotes survival of ATL cells (70, 116). Furthermore, frequent gain-of-function mutations are observed in the CCR4 gene in ATL patients. This improves cellular metabolism and survival by prolonging the PI3K/AKT signaling (90, 117). These findings spurred the way for the development of mogamulizumab, a monoclonal antibody against CCR4, which was currently being adopted for treatment of relapsed ATL (118–121).
New findings on the hijack of physiological T-cell activation mechanisms for immune evasion
We have thus far discussed on the immunomodulation mechanisms of HTLV-1 and the role of HBZ which is summarized in Figure 3. It is well established that HTLV-1-infected and ATL cells exhibit a highly activated phenotype. Physiologically, activated T cells are not long-lived and will undergo apoptosis to restore T cell homeostasis, which is largely achieved by cytokine deprivation and negative regulators such as Foxp3 and CTLA4 (122). HTLV-1-infected and ATL cells, which are highly activated, also exhibits a sustained expression of Foxp3 and CTLA4 (Figure 2) but these cells are not targeted for cell killing. This implies that HTLV-1 must have developed ways to avoid these negative regulatory mechanisms while maintaining a highly activated phenotype.
HLA class II (HLA-II) are important molecules for the regulation of immune responses by CD4+ T cells and normally found only on professional APCs and in T cells after activation. We have recently shown that HTLV-1-infected cells also upregulate HLA-II to present antigen to T cells. However, HTLV-1-infected cells are not efficient APCs as they lack the CD28 ligands CD80/CD86 and cannot provide the key costimulatory signaling in T cells (79). This in turn may contribute to the immune escape of infected cells as the lack of costimulatory signals make responder T cells anergic, in a manner similar to tolerogenic DCs (123). The suppression of T cell activation by tolerogenic DCs is mediated by IL-10 (see above) and a similar mechanism may be operating here as well. Additionally, our study showed that T cells express anergy-related genes upon stimulation by HLA-II on HTLV-1-infected cells, which suggests that HLA-II induction in HTLV-1-infected cells may induce other mechanisms for T cell anergy to inhibit anti-HTLV-1 T cell response.
Additionally, HTLV-1-infected cells also upregulate the expression of CIITA, a master regulator of HLA-II expression. We showed that promoter III of CIITA was more open in infected cells and its activity increases proportionally to levels of Tax (79) which suggests that Tax contributes to the upregulation of HLA-II during HTLV-1 infection.
In addition to the role in inducing stable HLA-II complexes, previous studies showed that CIITA is a potent transcriptional repressor for Tax-mediated HTLV-1 expression and a potential host restriction factor (23). Tax-mediated HTLV-1 expression is induced by the assembly of a multiprotein promoter complex containing CREB, CBP and PCAF on the viral LTR promoter (31). Tosi et al. (124) showed that CIITA disrupts the assembly of this promoter complex by physically interacting with Tax, thus inhibiting viral replication (124). Furthermore, it was also reported that CIITA can inhibit Tax-mediated NF-κB activation (125). This implies that CIITA may be crucial to counteract HTLV-1 infection and oncogenic transformation. Therefore, it is unexpected that HTLV-1-infected cells, which rarely express Tax, exhibits a high CIITA activity with increased HLA-II expression.
To reconcile the evidence above, here we propose the model that HTLV-1 hijacks the CIITA-HLA-II axis to enhance its survival and persistence in vivo (Figure 4). Firstly, irrespective of when Tax expression occurs (either during initial infection or a Tax burst in the latency phase), induction of Tax leads to increased CIITA expression. This then enhances presentation of HLA-II molecules on the cell surface of infected cells which confers infected cells with an immunosuppressive phenotype. CIITA can also bind and sequester Tax to work in a negative feedback loop, thus suppressing Tax-mediated expression. This hijack of host’s cellular factors allows HTLV-1 to halt Tax expression or burst, thus silencing the provirus. Additionally, this also helps to reduce the amount of Tax available for antigen presentation, thus minimizing CTL response against the virus. However, there are still several ambiguous points, chiefly: (1) how Tax interacts with the promoter of CIITA, and (2) how the expression of CIITA and HLA-II is maintained throughout the latent period. We speculated that it is highly possible that similar to CCR4 expression in ATL cells (90, 113, 117), infected cells acquire genetic or epigenetic alterations associated with constitutive expression of the CIITA gene. Another possible explanation is that infected cells modulates expression of certain microRNAs which sustains CIITA expression, similar to how epigenetic downregulation of miR31 expression leads to NFκB activation in the absence of Tax (126). Secondly, by inducing HLA-II expression in host cells, HTLV-1 modulates the immune system to be less responsive and effective against Tax-expressing cells. This dual role hypothesis certainly warrants further investigation to elucidate the role of CIITA in HTLV-1 infection.
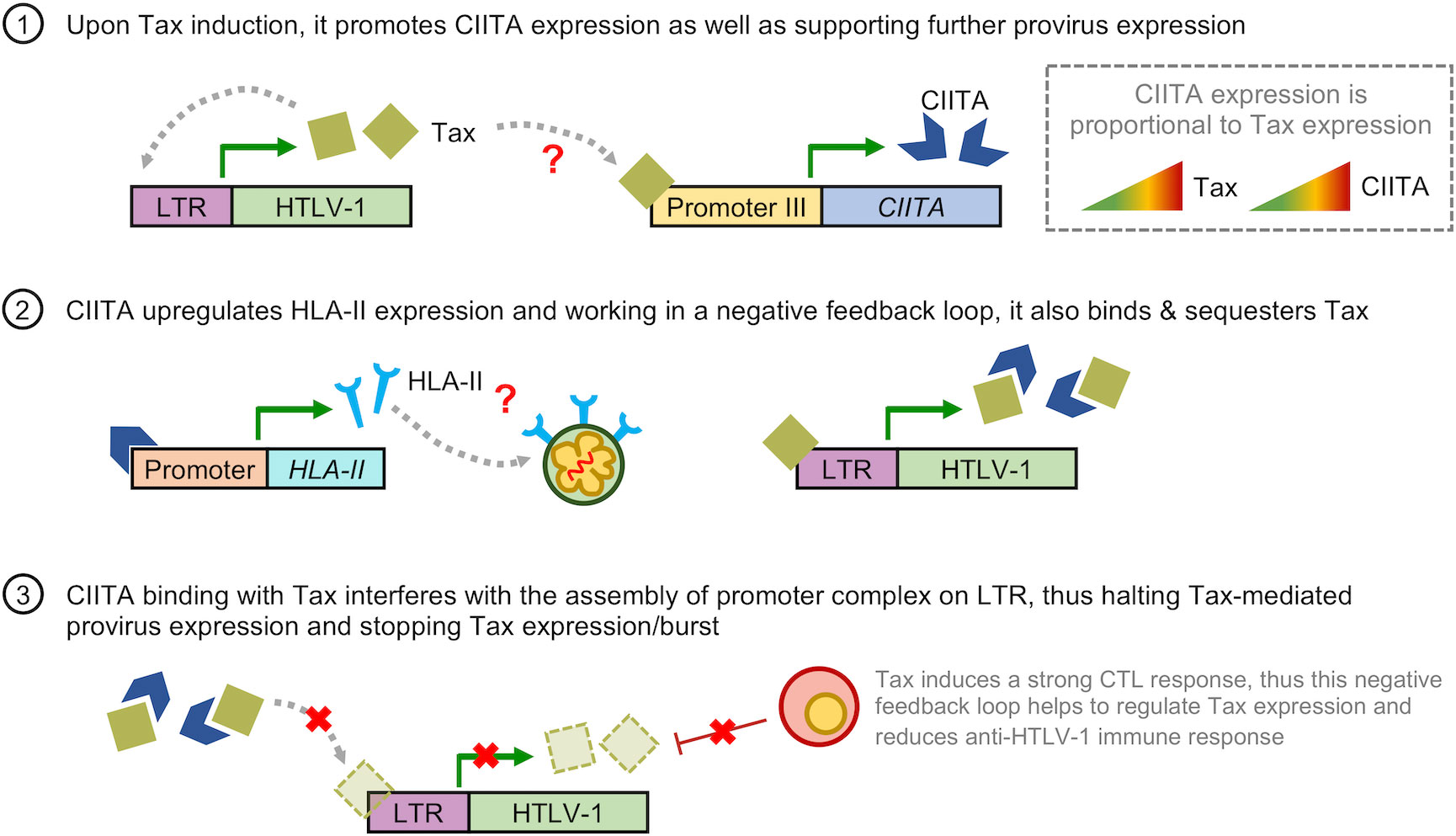
Figure 4 Hypothetical model on how HTLV-1 hijacks the CIITA-HLA-II axis for immune evasion. (1) Upon induction of Tax either during initial infection or a Tax burst, it promotes CIITA expression as well as further supporting HTLV-1 provirus expression in a positive feedback loop. As the level of Tax increases, so does the levels of CIITA. (2) CIITA induces expression of HLA-II-related genes, leading to upregulation of HLA-II molecules on the cell surface of HTLV-1-infected and ATL cells. This confers an immunosuppressive phenotype to these cells. Additionally, CIITA also binds and sequesters Tax. (3) Working in a negative feedback loop, binding of CIITA and Tax reduces the amount of Tax available for the assembly of promoter complex on the LTR, leading to a reduction in Tax-mediated expression of the HTLV-1 provirus, thus halting Tax expression or burst. Additionally, this also reduces the amount of Tax available for antigen presentation to Tax-specific CTLs, thus reducing CTL activity against HTLV-1. The red question marks in (1) and (2) indicates the points in which the regulation of CIITA in HTLV-1 infection is still unclear; namely (1) how Tax interacts with the promoter of CIITA, and (2) how CIITA and HLA-II expression is maintained throughout the latent period.
Concluding remarks
HTLV-1 has developed different and unique strategies in order to escape immune surveillance and induce disease in hosts. This remarkable ability of HTLV-1 to outmaneuver host immune surveillance while maintaining a pool of viral reservoir is the major obstacle in drafting effective treatment and prevention strategies. We expect that further uncovering of the immune escape mechanisms of HTLV-1 will lead to the development of innovative methods to reconstitute and restore normal immune homeostasis.
Author contributions
BT and YS designed the article concept and scope. BJYT wrote the manuscript and conceptualized the figures. KS, MO, and YS provided significant inputs and critically revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by JSPS KAKENHI Grant Number JP20240141 and AMED Grant Numbers JP22wm0325015 and JP22jm0210074 to YS.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Poiesz BJ, Ruscetti FW, Gazdar AF, Bunn PA, Minna JD, Gallo RC. Detection and isolation of type c retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc Natl Acad Sci USA (1980) 77(12 II):7415–9. doi: 10.1073/pnas.77.12.7415
2. Gallo RC. The discovery of the first human retrovirus: HTLV-1 and HTLV-2. Retrovirology (2005) 2:1–7. doi: 10.1186/1742-4690-2-17
3. Coffin JM. The discovery of HTLV-1, the first pathogenic human retrovirus. Proc Natl Acad Sci USA (2015) 112(51):15525–9. doi: 10.1073/pnas.1521629112
4. Uchiyama T, Yodoi J, Sagawa K, Takatsuki K, Uchino H. Adult T-cell leukemia: clinical and hematologic features of 16 cases. Blood (1977) 50(3):481–92. doi: 10.1182/blood.V50.3.481.481
5. Takatsuki K. Discovery of adult T-cell leukemia. Retrovirology (2005) 2:1–3. doi: 10.1186/1742-4690-2-16
6. White MK, Pagano JS, Khalili K. Viruses and human cancers: A long road of discovery of molecular paradigms. Clin Microbiol Rev (2014) 27(3):463–81. doi: 10.1128/CMR.00124-13
7. Tagaya Y, Matsuoka M, Gallo R. 40 years of the human T-cell leukemia virus: past, present, and future. F1000Research (2019) 8:228. doi: 10.12688/f1000research.17479.1
8. Bangham CRM. Human T cell leukemia virus type 1: Persistence and pathogenesis. Annu Rev Immunol (2018) 36:43–71. doi: 10.1146/annurev-immunol-042617-053222
9. Gessain A, Vernant JC, Maurs L, Barin F, Gout O, Calender A, et al. Antibodies to human T-lymphotropic virus type-I in patients with tropical spastic paraparesis. Lancet (1985) 326(8452):407–10. doi: 10.1016/S0140-6736(85)92734-5
10. Osame M, Usuku K, Izumo S, Ijichi N, Amitani H, Igata A, et al. HTLV-I associated myelopathy, a new clinical entity. Lancet (1986) 327(8488):1031–2. doi: 10.1016/S0140-6736(86)91298-5
11. Matsuoka M, Jeang KT. Human T-cell leukaemia virus type 1 (HTLV-1) infectivity and cellular transformation. Nat Rev Cancer (2007) 7(4):270–80. doi: 10.1038/nrc2111
12. Derse D, Hill SA, Lloyd PA, Chung H, Morse BA. Examining human T-lymphotropic virus type 1 infection and replication by cell-free infection with recombinant virus vectors. J Virol (2001) 75(18):8461–8. doi: 10.1128/JVI.75.18.8461-8468.2001
13. Igakura T, Stinchcombe JC, Goon PKC, Taylor GP, Weber JN, Griffiths GM, et al. Spread of HTLV-I between lymphocytes by virus-induced polarization of the cytoskeleton. Sci (80- ) (2003) 299(5613):1713–6. doi: 10.1126/science.1080115
14. Pais-Correia AM, Sachse M, Guadagnini S, Robbiati V, Lasserre R, Gessain A, et al. Biofilm-like extracellular viral assemblies mediate HTLV-1 cell-to-cell transmission at virological synapses. Nat Med (2010) 16(1):83–9. doi: 10.1038/nm.2065
15. Hiyoshi M, Takahashi N, Eltalkhawy YM, Noyori O, Lotfi S, Panaampon J, et al. M-sec induced by HTLV-1 mediates an efficient viral transmission. PloS Pathog (2021) 17(11):1–23. doi: 10.1371/journal.ppat.1010126
16. Wattel E, Cavrois M, Gessain A, Wain-Hobson S. Clonal expansion of infected cells: A way of life for HTLV-I. J Acquir Immune Defic Syndr Hum Retrovirol (1996) 13(SUPPL. 1):S92–9. doi: 10.1097/00042560-199600001-00016
17. Laydon DJ, Sunkara V, Boelen L, Bangham CRM, Asquith B. The relative contributions of infectious and mitotic spread to HTLV-1 persistence. PloS Comput Biol (2020) 16(9):1–25. doi: 10.1371/journal.pcbi.1007470
18. Swann JB, Smyth MJ. Immune surveillance of tumors. J Clin Invest (2007) 117(5):1137–46. doi: 10.1172/JCI31405
19. Bangham CRM, Osame M. Cellular immune response to HTLV-1. Oncogene (2005) 24(39):6035–46. doi: 10.1038/sj.onc.1208970
20. Bangham CRM. CTL quality and the control of human retroviral infections. Eur J Immunol (2009) 39(7):1700–12. doi: 10.1002/eji.200939451
21. Journo C, Mahieux R. HTLV-1 and innate immunity. Viruses (2011) 3(8):1374–94. doi: 10.3390/v3081374
22. Kannagi M, Hasegawa A, Takamori A, Kinpara S, Utsunomiya A. The roles of acquired and innate immunity in human T-cell leukemia virus type 1-mediated diseases. Front Microbiol (2012) 3:1–10. doi: 10.3389/fmicb.2012.00323
23. Forlani G, Shallak M, Ramia E, Tedeschi A, Accolla RS. Restriction factors in human retrovirus infections and the unprecedented case of CIITA as link of intrinsic and adaptive immunity against HTLV-1. Retrovirology (2019) 16(1):1–12. doi: 10.1186/s12977-019-0498-6
24. Carcone A, Journo C, Dutartre H. Is the HTLV-1 retrovirus targeted by host restriction factors? Viruses (2022) 14(8):1611. doi: 10.3390/v14081611
25. Zargari R, Mahdifar M, Mohammadi A, Vahidi Z, Hassanshahi G, Rafatpanah H. The role of chemokines in the pathogenesis of HTLV-1. Front Microbiol (2020) 11:1–16. doi: 10.3389/fmicb.2020.00421
26. Marshall JS, Warrington R, Watson W, Kim HL. An introduction to immunology and immunopathology. Allergy Asthma Clin Immunol (2018) 14(s2):1–10. doi: 10.1186/s13223-018-0278-1
27. Cook LBM, Melamed A, Demontis MA, Laydon DJ, Fox JM, Tosswill JHC, et al. Rapid dissemination of human T-lymphotropic virus type 1 during primary infection in transplant recipients. Retrovirology (2016) 13(1):1–9. doi: 10.1186/s12977-015-0236-7
28. Bangham CRM, Matsuoka M. Human T-cell leukaemia virus type 1: Parasitism and pathogenesis. Philos Trans R Soc B Biol Sci (2017) 372(1732):20160272. doi: 10.1098/rstb.2016.0272
29. Suemori K, Fujiwara H, Ochi T, Ogawa T, Matsuoka M, Matsumoto T, et al. HBZ is an immunogenic protein, but not a target antigen for human T-cell leukemia virus type 1-specific cytotoxic T lymphocytes. J Gen Virol (2009) 90(8):1806–11. doi: 10.1099/vir.0.010199-0
30. MacNamara A, Rowan A, Hilburn S, Kadolsky U, Fujiwara H, Suemori K, et al. HLA class I binding of HBZ determines outcome in HTLV-1 infection. PloS Pathog (2010) 6(9):e1001117. doi: 10.1371/journal.ppat.1001117
31. Kashanchi F, Brady JN. Transcriptional and post-transcriptional gene regulation of HTLV-1. Oncogene (2005) 24(39):5938–51. doi: 10.1038/sj.onc.1208973
32. Nicot C, Dundr M, Johnson JM, Fullen JR, Alonzo N, Fukumoto R, et al. HTLV-1-encoded p30II is a post-transcriptional negative regulator of viral replication. Nat Med (2004) 10(2):197–201. doi: 10.1038/nm984
33. Clerc I, Polakowski N, André-Arpin C, Cook P, Barbeau B, Mesnard JM, et al. An interaction between the human T cell leukemia virus type 1 basic leucine zipper factor (HBZ) and the KIX domain of p300/CBP contributes to the down-regulation of tax-dependent viral transcription by HBZ. J Biol Chem (2008) 283(35):23903–13. doi: 10.1074/jbc.M803116200
34. Billman MR, Rueda D, Bangham CRM. Single-cell heterogeneity and cell-cycle-related viral gene bursts in the human leukaemia virus HTLV-1. Wellcome Open Res (2017) 2(0):87. doi: 10.12688/wellcomeopenres.12469.2
35. Taniguchi Y, Nosaka K, Yasunaga JI, Maeda M, Mueller N, Okayama A, et al. Silencing of human T-cell leukemia virus type I gene transcription by epigenetic mechanisms. Retrovirology (2005) 2:1–16. doi: 10.1186/1742-4690-2-64
36. Miura M, Miyazato P, Satou Y, Tanaka Y, Bangham CRM. Epigenetic changes around the pX region and spontaneous HTLV-1 transcription are CTCF-independent [version 2; referees: 2 approved]. Wellcome Open Res (2018) 3(0):1–24. doi: 10.12688/wellcomeopenres.14741.2
37. Satou Y, Miyazato P, Ishihara K, Yaguchi H, Melamed A, Miura M, et al. The retrovirus HTLV-1 inserts an ectopic CTCF-binding site into the human genome. Proc Natl Acad Sci USA (2016) 113(11):3054–9. doi: 10.1073/pnas.1423199113
38. Matsuo M, Ueno T, Monde K, Sugata K, Tan BJY, Rahman A, et al. Identification and characterization of a novel enhancer in the HTLV-1 proviral genome. Nat Commun (2022) 13(1):1–16. doi: 10.1038/s41467-022-30029-9
39. Mahgoub M, Yasunaga JI, Iwami S, Nakaoka S, Koizumi Y, Shimura K, et al. Sporadic on/off switching of HTLV-1 tax expression is crucial to maintain the whole population of virus-induced leukemic cells. Proc Natl Acad Sci USA (2018) 115(6):E1269–78. doi: 10.1073/pnas.1715724115
40. Satou Y, Yasunaga JI, Yoshida M, Matsuoka M. HTLV-I basic leucine zipper factor gene mRNA supports proliferation of adult T cell leukemia cells. Proc Natl Acad Sci USA (2006) 103(3):720–5. doi: 10.1073/pnas.0507631103
41. Saito M, Matsuzaki T, Satou Y, Yasunaga JI, Saito K, Arimura K, et al. In vivo expression of the HBZ gene of HTLV-1 correlates with proviral load, inflammatory markers and disease severity in HTLV-1 associated myelopathy/tropical spastic paraparesis (HAM/TSP). Retrovirology (2009) 6:1–11. doi: 10.1186/1742-4690-6-19
42. Jacobson S, Shida H, McFarlin DE, Fauci AS, Koenig S. Circulating CD8+ cytotoxic T lymphocytes specific for HTLV-I pX in patients with HTLV-I associated neurological disease. Nature (1990) 348(6298):245–8. doi: 10.1038/348245a0
43. Kannagi M, Harada S, Maruyama I, Inoko H, Igarashi H, Kuwashima G, et al. Predominant recognition of human t cell leukemia virus type i (Htlv-i) px gene products by human cd8 + cytotoxic t cells directed against htlv-l-infected cells. Int Immunol (1991) 3(8):761–7. doi: 10.1093/intimm/3.8.761
44. Kannagi M, Shida H, Igarashi H, Kuruma K, Murai H, Aono Y, et al. Target epitope in the tax protein of human T-cell leukemia virus type I recognized by class I major histocompatibility complex-restricted cytotoxic T cells. J Virol (1992) 66(5):2928–33. doi: 10.1128/jvi.66.5.2928-2933.1992
45. Parker CE, Nightingale S, Taylor GP, Weber J, Bangham CR. Circulating anti-tax cytotoxic T lymphocytes from human T-cell leukemia virus type I-infected people, with and without tropical spastic paraparesis, recognize multiple epitopes simultaneously. J Virol (1994) 68(5):2860–8. doi: 10.1128/jvi.68.5.2860-2868.1994
46. Hausmann S, Biddison WE, Smith KJ, Ding YH, Garboczi DN, Utz U, et al. Peptide recognition by two HLA-A2/Tax11-19-specific T cell clones in relationship to their MHC/peptide/TCR crystal structures. J Immunol (1999) 162(9):5389–97.
47. Jeffery KJM, Usuku K, Hall SE, Matsumoto W, Taylor GP, Procter J, et al. HLA alleles determine human T-lymphotropic virus-I (HTLV-I) proviral load and the risk of HTLV-i-associated myelopathy. Proc Natl Acad Sci USA (1999) 96(7):3848–53. doi: 10.1073/pnas.96.7.3848
48. Jeffery KJM, Siddiqui AA, Bunce M, Lloyd AL, Vine AM, Witkover AD, et al. The influence of HLA class I alleles and heterozygosity on the outcome of human T cell lymphotropic virus type I infection. J Immunol (2000) 165(12):7278–84. doi: 10.4049/jimmunol.165.12.7278
49. Rowan AG, Suemori K, Fujiwara H, Yasukawa M, Tanaka Y, Taylor GP, et al. Cytotoxic T lymphocyte lysis of HTLV-1 infected cells is limited by weak HBZ protein expression, but non-specifically enhanced on induction of tax expression. Retrovirology (2014) 11(1):1–12. doi: 10.1186/s12977-014-0116-6
50. Gazon H, Lemasson I, Polakowski N, Césaire R, Matsuoka M, Barbeau B, et al. Human T-cell leukemia virus type 1 (HTLV-1) bZIP factor requires cellular transcription factor JunD to upregulate HTLV-1 antisense transcription from the 3′ long terminal repeat. J Virol (2012) 86(17):9070–8. doi: 10.1128/JVI.00661-12
51. Terol M, Gazon H, Lemasson I, Duc-Dodon M, Barbeau B, Césaire R, et al. HBZ-mediated shift of JunD from growth suppressor to tumor promoter in leukemic cells by inhibition of ribosomal protein S25 expression. Leukemia (2017) 31(10):2235–43. doi: 10.1038/leu.2017.74
52. Stuber G, Dillner J, Modrow S, Wolf H, Székely L, Klein G, et al. HLA-A0201 and HLA-B7 binding peptides in the EBV-encoded EBNA-1, EBNA-2 and BZLF-1 proteins detected in the MHC class I stabilization assay. low proportion of binding motifs for several HLA class I alleles in EBNA-1. Int Immunol (1995) 7(4):653–63. doi: 10.1093/intimm/7.4.653
53. Shi W, Bu P, Liu J, Polack A, Fisher S, Qiao L. Human papillomavirus type 16 E7 DNA vaccine: Mutation in the open reading frame of E7 enhances specific cytotoxic T-lymphocyte induction and antitumor activity. J Virol (1999) 73(9):7877–81. doi: 10.1128/JVI.73.9.7877-7881.1999
54. Matsuoka M, Mesnard JM. HTLV-1 bZIP factor: The key viral gene for pathogenesis. Retrovirology (2020) 17(1):4–11. doi: 10.1186/s12977-020-0511-0
55. Katsuya H, Islam S, Tan BJY, Ito J, Miyazato P, Matsuo M, et al. The nature of the HTLV-1 provirus in naturally infected individuals analyzed by the viral DNA-Capture-Seq approach. Cell Rep (2019) 29(3):724–35.e4. doi: 10.1016/j.celrep.2019.09.016
56. Takeda S, Maeda M, Morikawa S, Taniguchi Y, Yasunaga JI, Nosaka K, et al. Genetic and epigenetic inactivation of TAX gene in adult t-cell leukemia cells. Int J Cancer (2004) 109(4):559–67. doi: 10.1002/ijc.20007
57. Andrade RG, Gonçalves P de C, Ribeiro MA, Romanelli LCF, Ribas JG, Torres EB, et al. Strong correlation between tax and HBZ mRNA expression in HAM/TSP patients: Distinct markers for the neurologic disease. J Clin Virol (2013) 56(2):135–40. doi: 10.1016/j.jcv.2012.10.003
58. Doi K, Wu X, Taniguchi Y, Yasunaga JI, Satou Y, Okayama A, et al. Preferential selection of human T-cell leukemia virus type I provirus integration sites in leukemic versus carrier states. Blood (2005) 106(3):1048–53. doi: 10.1182/blood-2004-11-4350
59. Gillet NA, Malani N, Melamed A, Gormley N, Carter R, Bentley D, et al. The host genomic environment of the provirus determines the abundance of HTLV-1-infected T-cell clones. Blood (2011) 117(11):3113–22. doi: 10.1182/blood-2010-10-312926
60. Melamed A, Laydon DJ, Gillet NA, Tanaka Y, Taylor GP, Bangham CRM. Genome-wide determinants of proviral targeting, clonal abundance and expression in natural HTLV-1 infection. PloS Pathog (2013) 9(3):1–13. doi: 10.1371/journal.ppat.1003271
61. Melamed A, Fitzgerald TW, Wang Y, Ma J, Birney E, Bangham CRM. Selective clonal persistence of human retroviruses in vivo: Radial chromatin organization, integration site, and host transcription. Sci Adv (2022) 8(17):eabm6210. doi: 10.1126/sciadv.abm6210
62. Cook LB, Melamed A, Niederer H, Valganon M, Laydon D, Foroni L, et al. The role of HTLV-1 clonality, proviral structure, and genomic integration site in adult T-cell leukemia/lymphoma. Blood (2014) 123(25):3925–31. doi: 10.1182/blood-2014-02-553602
63. Tamiya S, Matsuoka M, Etoh KI, Watanabe T, Kamihira S, Yamaguchi K, et al. Two types of defective human T-lymphotropic virus type I provirus in adult T-cell leukemia. Blood (1996) 88(8):3065–73. doi: 10.1182/blood.V88.8.3065.bloodjournal8883065
64. Miyazaki M, Yasunaga J-I, Taniguchi Y, Tamiya S, Nakahata T, Matsuoka M. Preferential selection of human T-cell leukemia virus type 1 provirus lacking the 5′ long terminal repeat during oncogenesis. J Virol (2007) 81(11):5714–23. doi: 10.1128/JVI.02511-06
65. Takenouchi H, Umeki K, Sasaki D, Yamamoto I, Nomura H, Takajo I, et al. Defective human T-lymphotropic virus type 1 provirus in asymptomatic carriers. Int J Cancer (2011) 128(6):1335–43. doi: 10.1002/ijc.25450
66. Kimura N, Kikuchi M, Masuda Y, Kobari S, Sumiyoshi Y, Eguchi F, et al. Defective provirus form of human T-cell leukemia virus type I in adult T-cell Leukemia/Lymphoma: Clinicopathological features. Cancer Res (1991) 51(17):4639–42.
67. Korber B, Okayama A, Donnelly R, Tachibana N, Essex M. Polymerase chain reaction analysis of defective human T-cell leukemia virus type I proviral genomes in leukemic cells of patients with adult T-cell leukemia. J Virol (1991) 65(10):5471–6. doi: 10.1128/jvi.65.10.5471-5476.1991
68. Karube K, Ohshima K, Tsuchiya T, Yamaguchi T, Kawano R, Suzumiya J, et al. Expression of FoxP3, a key molecule in CD4+CD25+ regulatory T cells, in adult T-cell leukaemia/lymphoma cells. Br J Haematol (2004) 126(1):81–4. doi: 10.1111/j.1365-2141.2004.04999.x
69. Karube K, Aoki R, Sugita Y, Yoshida S, Nomura Y, Shimizu K, et al. The relationship of FOXP3 expression and clinicopathological characteristics in adult T-cell leukemia/lymphoma. Mod Pathol (2008) 21(5):617–25. doi: 10.1038/modpathol.2008.25
70. Toulza F, Heaps A, Tanaka Y, Taylor GP, Bangham CRM. High frequency of CD4+FoxP3+ cells in HTLV-1 infection: Inverse correlation with HTLV-l-specific CTL response. Blood (2008) 111(10):5047–53. doi: 10.1182/blood-2007-10-118539
71. Satou Y, Utsunomiya A, Tanabe J, Nakagawa M, Nosaka K, Matsuoka M. HTLV-1 modulates the frequency and phenotype of FoxP3+CD4+ T cells in virus-infected individuals. Retrovirology (2012) 9:1–12. doi: 10.1186/1742-4690-9-46
72. Kohno T, Yamada Y, Akamatsu N, Kamihira S, Imaizumi Y, Tomonaga M, et al. Possible origin of adult T-cell leukemia/lymphoma cells from human T lymphotropic virus type-1-infected regulatory T cells. Cancer Sci (2005) 96(8):527–33. doi: 10.1111/j.1349-7006.2005.00080.x
73. Zhao T, Satou Y, Sugata K, Miyazato P, Green PL, Imamura T, et al. HTLV-1 bZIP factor enhances TGF-β signaling through p300 coactivator. Blood (2011) 118(7):1865–76. doi: 10.1182/blood-2010-12-326199
74. Satou Y, Yasunaga Ji, Zhao T, Yoshida M, Miyazato P, Takai K, et al. HTLV-1 bZIP factor induces T-cell lymphoma and systemic inflammation in vivo. PloS Pathog (2011) 7(2):e1001274. doi: 10.1371/journal.ppat.1001274
75. Chen S, Ishii N, Ine S, Ikeda S, Fujimura T, Ndhlovu LC, et al. Regulatory T cell-like activity of Foxp3+ adult T cell leukemia cells. Int Immunol (2006) 18(2):269–77. doi: 10.1093/intimm/dxh366
76. Shimauchi T, Kabashima K, Tokura Y. Adult T-cell leukemia/lymphoma cells from blood and skin tumors express cytotoxic T lymphocyte-associated antigen-4 and Foxp3 but lack suppressor activity toward autologous CD8+ T cells. Cancer Sci (2008) 99(1):98–106. doi: 10.1111/j.1349-7006.2007.00646.x
77. Toulza F, Nosaka K, Takiguchi M, Pagliuca T, Mitsuya H, Tanaka Y, et al. FoxP3+ regulatory T cells are distinct from leukemia cells in HTLV-1-associated adult T-cell leukemia. Int J Cancer (2009) 125(10):2375–82. doi: 10.1002/ijc.24664
78. Koya J, Saito Y, Kameda T, Kogure Y, Yuasa M, Nagasaki J, et al. Single-cell analysis of the multicellular ecosystem in viral carcinogenesis by HTLV-1. Blood Cancer Discov (2021) 2(5):450–67. doi: 10.1158/2643-3230.BCD-21-0044
79. Tan BJY, Sugata K, Reda O, Matsuo M, Uchiyama K, Miyazato P, et al. HTLV-1 infection promotes excessive T cell activation and transformation into adult T cell leukemia/ lymphoma. J Clin Invest (2021) 131(24):e150472. doi: 10.1172/JCI150472
80. Colomer-Lluch M, Ruiz A, Moris A, Prado JG. Restriction factors: From intrinsic viral restriction to shaping cellular immunity against HIV-1. Front Immunol (2018) 9:1–18. doi: 10.3389/fimmu.2018.02876
81. Asquith B, Bangham CRM. How does HTLV-I persist despite a strong cell-mediated immune response? Trends Immunol (2008) 29(1):4–11. doi: 10.1016/j.it.2007.09.006
82. Navarro F, Bollman B, Chen H, König R, Yu Q, Chiles K, et al. Complementary function of the two catalytic domains of APOBEC3G. Virology (2005) 333(2):374–86. doi: 10.1016/j.virol.2005.01.011
83. Derse D, Hill SA, Princler G, Lloyd P, Heidecker G. Resistance of human T cell leukemia virus type 1 to APOBEC3G restriction is mediated by elements in nucleocapsid. Proc Natl Acad Sci USA (2007) 104(8):2915–20. doi: 10.1073/pnas.0609444104
84. Fan J, Ma G, Nosaka K, Tanabe J, Satou Y, Koito A, et al. APOBEC3G generates nonsense mutations in human T-cell leukemia virus type 1 proviral genomes In vivo. J Virol (2010) 84(14):7278–87. doi: 10.1128/JVI.02239-09
85. Ilinskaya A, Derse D, Hill S, Princler G, Heidecker G. Cell-cell transmission allows human T-lymphotropic virus 1 to circumvent tetherin restriction. Virology (2013) 436(1):201–9. doi: 10.1016/j.virol.2012.11.012
86. Sze A, Belgnaoui SM, Olagnier D, Lin R, Hiscott J, Van Grevenynghe J. Host restriction factor SAMHD1 limits human T cell leukemia virus type 1 infection of monocytes via STING-mediated apoptosis. Cell Host Microbe (2013) 14(4):422–34. doi: 10.1016/j.chom.2013.09.009
87. Nozuma S, Matsuura E, Kodama D, Tashiro Y, Matsuzaki T, Kubota R, et al. Effects of host restriction factors and the HTLV-1 subtype on susceptibility to HTLV-1-associated myelopathy/tropical spastic paraparesis. Retrovirology (2017) 14(1):1–11. doi: 10.1186/s12977-017-0350-9
88. Leal FE, Menezes SM, Costa EAS, Brailey PM, Gama L, Segurado AC, et al. Comprehensive antiretroviral restriction factor profiling reveals the evolutionary imprint of the ex vivo and in vivo IFN-β response in HTLV-1-associated neuroinflammation. Front Microbiol (2018) 9:1–12. doi: 10.3389/fmicb.2018.00985
89. Bai XT, Nicot C. MiR-28-3p is a cellular restriction factor that inhibits human T cell leukemia virus, type 1 (HTLV-1) replication and virus infection. J Biol Chem (2015) 290(9):5381–90. doi: 10.1074/jbc.M114.626325
90. Kataoka K, Nagata Y, Kitanaka A, Shiraishi Y, Shimamura T, Yasunaga JI, et al. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat Genet (2015) 47(11):1304–15. doi: 10.1038/ng.3415
91. Rowan AG, Dillon R, Witkover A, Melamed A, Demontis MA, Gillet NA, et al. Evolution of retrovirus-infected premalignant T-cell clones prior to adult T-cell leukemia/lymphoma diagnosis. Blood (2020) 135(23):2023–32. doi: 10.1182/blood.2019002665
92. Yamagishi M, Kubokawa M, Kuze Y, Suzuki A, Yokomizo A, Kobayashi S, et al. Chronological genome and single-cell transcriptome integration characterizes the evolutionary process of adult T cell leukemia-lymphoma. Nat Commun (2021) 12(1):1–16. doi: 10.1038/s41467-021-25101-9
93. Zitvogel L, Tesniere A, Kroemer G. Cancer despite immunosurveillance: Immunoselection and immunosubversion. Nat Rev Immunol (2006) 6(10):715–27. doi: 10.1038/nri1936
94. Clements DM, Crumley B, Chew GM, Davis E, Bruhn R, Murphy EL, et al. Phenotypic and functional analyses guiding combination immune checkpoint immunotherapeutic strategies in HTLV-1 infection. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.608890
95. Miura S, Ohtani K, Numata N, Niki M, Ohbo K, Ina Y, et al. Molecular cloning and characterization of a novel glycoprotein, gp34, that is specifically induced by the human T-cell leukemia virus type I transactivator p40tax. Mol Cell Biol (1991) 11(3):1313–25. doi: 10.1128/mcb.11.3.1313-1325.1991
96. Baum PR, Gayle RB, Ramsdell F, Srinivasan S, Sorensen RA, Watson ML, et al. Molecular characterization of murine and human OX40/OX40 ligand systems: Identification of a human OX40 ligand as the HTLV-1-regulated protein gp34. EMBO J (1994) 13(17):3992–4001. doi: 10.1002/j.1460-2075.1994.tb06715.x
97. Croft M, So T, Duan W, Soroosh P. The significance of OX40 and OX40L to T-cell biology and immune disease. Immunol Rev (2009) 229(1):173–91. doi: 10.1111/j.1600-065X.2009.00766.x
98. Kinosada H, Yasunaga JI, Shimura K, Miyazato P, Onishi C, Iyoda T, et al. HTLV-1 bZIP factor enhances T-cell proliferation by impeding the suppressive signaling of Co-inhibitory receptors. PloS Pathog (2017) 13(1):1–24. doi: 10.1371/journal.ppat.1006120
99. Paster W, Bruger AM, Katsch K, Grégoire C, Roncagalli R, Fu G, et al. A THEMIS : SHP 1 complex promotes T-cell survival. EMBO J (2015) 34(3):393–409. doi: 10.15252/embj.201387725
100. Yasuma K, Yasunaga JI, Takemoto K, Sugata K, Mitobe Y, Takenouchi N, et al. HTLV-1 bZIP factor impairs anti-viral immunity by inducing Co-inhibitory molecule, T cell immunoglobulin and ITIM domain (TIGIT). PloS Pathog (2016) 12(1):1–22. doi: 10.1371/journal.ppat.1005372
101. Inagaki A, Ishida T, Ishii T, Komatsu H, Iida S, Ding J, et al. Clinical significance of serum Th1-, Th2- and regulatory T cells-associated cytokines in adult T-cell leukemia/lymphoma: High interleukin-5 and -10 levels are significant unfavorable prognostic factors. Int J Cancer (2006) 118(12):3054–61. doi: 10.1002/ijc.21688
102. Kagdi H, Demontis MA, Ramos JC, Taylor GP. Switching and loss of cellular cytokine producing capacity characterize in vivo viral infection and malignant transformation in human T- lymphotropic virus type 1 infection. PloS Pathog (2018) 14(2):1–25. doi: 10.1371/journal.ppat.1006861
103. Ng THS, Britton GJ, Hill EV, Verhagen J, Burton BR, Wraith DC. Regulation of adaptive immunity; the role of interleukin-10. Front Immunol (2013) 4:1–13. doi: 10.3389/fimmu.2013.00129
104. Sawada L, Nagano Y, Hasegawa A, Kanai H, Nogami K, Ito S, et al. IL-10-mediated signals act as a switch for lymphoproliferation in human T-cell leukemia virus type-1 infection by activating the STAT3 and IRF4 pathways. PloS Pathog (2017) 13(9):1–23. doi: 10.1371/journal.ppat.1006597
105. Higuchi Y, Yasunaga JI, Mitagami Y, Tsukamoto H, Nakashima K, Ohshima K, et al. HTLV-1 induces T cell malignancy and inflammation by viral antisense factor-mediated modulation of the cytokine signaling. Proc Natl Acad Sci USA (2020) 117(24):13740–9. doi: 10.1073/pnas.1922884117
106. Shimauchi T, Kabashima K, Nakashima D, Sugita K, Yamada Y, Hino R, et al. Augmented expression of programmed death-1 in both neoplastic and non-neoplastic CD4+ T-cells in adult T-cell leukemia/lymphoma. Int J Cancer (2007) 121(12):2585–90. doi: 10.1002/ijc.23042
107. Kozako T, Yoshimitsu M, Fujiwara H, Masamoto I, Horai S, White Y, et al. PD-1/PD-L1 expression in human T-cell leukemia virus type 1 carriers and adult T-cell leukemia/lymphoma patients. Leukemia (2009) 23(2):375–82. doi: 10.1038/leu.2008.272
108. Kozako T, Yoshimitsu M, Fujiwara H, Masamoto I, Horai S, Akimoto M, et al. CTL exhaustion in persistent HTLV-1 infection and ATLL is restored through PD-1/PD-L1 pathway. Blood (2007) 110(11):511–1. doi: 10.1182/blood.V110.11.511.511
109. Kuipers H, Muskens F, Willart M, Hijdra D, van Assema FBJ, Coyle AJ, et al. Contribution of the PD-1 ligands/PD-1 signaling pathway to dendritic cell-mediated CD4+ cell activation. Eur J Immunol (2006) 36(9):2472–82. doi: 10.1002/eji.200635978
110. Takeuchi M, Miyoshi H, Nakashima K, Kawamoto K, Yamada K, Yanagida E, et al. Comprehensive immunohistochemical analysis of immune checkpoint molecules in adult T cell leukemia/lymphoma. Ann Hematol (2020) 99(5):1093–8. doi: 10.1007/s00277-020-03967-x
111. Ratner L, Waldmann TA, Janakiram M, Brammer JE. Rapid progression of adult T-cell leukemia–lymphoma after PD-1 inhibitor therapy. N Engl J Med (2018) 378(20):1947–8. doi: 10.1056/NEJMc1803181
112. Rauch DA, Conlon KC, Janakiram M, Brammer JE, Harding JC, Ye BH, et al. Rapid progression of adult T-cell leukemia/lymphoma as tumor-infiltrating tregs after PD-1 blockade. Blood (2019) 134(17):1406–14. doi: 10.1182/blood.2019002038
113. Yoshie O, Fujisawa R, Nakayama T, Harasawa H, Tago H, Izawa D, et al. Frequent expression of CCR4 in adult T-cell leukemia and human T-cell leukemia virus type 1-transformed T cells. Blood (2002) 99(5):1505–11. doi: 10.1182/blood.V99.5.1505
114. Sugata K, Yasunaga JI, Kinosada H, Mitobe Y, Furuta R, Mahgoub M, et al. HTLV-1 viral factor HBZ induces CCR4 to promote T-cell migration and proliferation. Cancer Res (2016) 76(17):5068–79. doi: 10.1158/0008-5472.CAN-16-0361
115. Yamamoto-Taguchi N, Satou Y, Miyazato P, Ohshima K, Nakagawa M, Katagiri K, et al. HTLV-1 bZIP factor induces inflammation through labile Foxp3 expression. PloS Pathog (2013) 9(9):1–12. doi: 10.1371/journal.ppat.1003630
116. Toulza F, Nosaka K, Tanaka Y, Schioppa T, Balkwill F, Taylor GP, et al. Human T-lymphotropic virus type 1-induced CC chemokine ligand 22 maintains a high frequency of functional FoxP3 + regulatory T cells. J Immunol (2010) 185(1):183–9. doi: 10.4049/jimmunol.0903846
117. Nakagawa M, Schmitz R, Xiao W, Goldman CK, Xu W, Yang Y, et al. Gain-of-function CCR4 mutations in adult T cell leukemia/lymphoma. J Exp Med (2014) 211(13):2497–505. doi: 10.1084/jem.20140987
118. Ishii T, Ishida T, Utsunomiya A, Inagaki A, Yano H, Komatsu H, et al. Defucosylated humanized aanti-CCR4 monoclonal antibody KW-0761 as a novel immunotherapeutic agent for adult T-cell leukemia/lymphoma. Clin Cancer Res (2010) 16(5):1520–31. doi: 10.1158/1078-0432.CCR-09-2697
119. Ishida T, Joh T, Uike N, Yamamoto K, Utsunomiya A, Yoshida S, et al. Defucosylated anti-CCR4 monoclonal antibody (KW-0761) for relapsed adult T-cell leukemia-lymphoma: A multicenter phase II study. J Clin Oncol (2012) 30(8):837–42. doi: 10.1200/JCO.2011.37.3472
120. Ishida T, Jo T, Takemoto S, Suzushima H, Uozumi K, Yamamoto K, et al. Dose-intensified chemotherapy alone or in combination with mogamulizumab in newly diagnosed aggressive adult T-cell leukaemia-lymphoma: A randomized phase II study. Br J Haematol (2015) 169(5):672–82. doi: 10.1111/bjh.13338
121. Yonekura K, Kusumoto S, Choi I, Nakano N, Ito A, Suehiro Y, et al. Mogamulizumab for adult T-cell leukemia-lymphoma: a multicenter prospective observational study. Blood Adv (2020) 4(20):5133–54. doi: 10.1182/bloodadvances.2020003053
122. Bending D, Ono M. From stability to dynamics: understanding molecular mechanisms of regulatory T cells through Foxp3 transcriptional dynamics. Clin Exp Immunol (2019) 197(1):14–23. doi: 10.1111/cei.13194
123. Domogalla MP, Rostan PV, Raker VK, Steinbrink K. Tolerance through education: How tolerogenic dendritic cells shape immunity. Front Immunol (2017) 8:1–14. doi: 10.3389/fimmu.2017.01764
124. Tosi G, Forlani G, Andresen V, Turci M, Bertazzoni U, Franchini G, et al. Major histocompatibility complex class II transactivator CIITA is a viral restriction factor that targets human T-cell lymphotropic virus type 1 tax-1 function and inhibits viral replication. J Virol (2011) 85(20):10719–29. doi: 10.1128/JVI.00813-11
125. Forlani G, Abdallah R, Accolla RS, Tosi G. The major histocompatibility complex class II transactivator CIITA inhibits the persistent activation of NF-κB by the human T cell lymphotropic virus type 1 tax-1 oncoprotein. J Virol (2016) 90(7):3708–21. doi: 10.1128/JVI.03000-15
Keywords: human T-cell leukemia virus type 1 (HTLV-1), adult T cell leukemia/lymphoma (ATL), viral immune response, cancer immune response, immune escape, clonal persistence, HLA-II
Citation: Tan BJY, Sugata K, Ono M and Satou Y (2022) HTLV-1 persistence and leukemogenesis: A game of hide-and-seek with the host immune system. Front. Immunol. 13:991928. doi: 10.3389/fimmu.2022.991928
Received: 12 July 2022; Accepted: 27 September 2022;
Published: 10 October 2022.
Edited by:
Toshiki Watanabe, St. Marianna University School of Medicine, JapanReviewed by:
Kazumi Nakano, The University of Tokyo, JapanAnne Van Den Broeke, Université libre de Bruxelles, Belgium
Helene Dutartre, UMR5308 Centre International de Recherche en Infectiologie (CIRI), France
Copyright © 2022 Tan, Sugata, Ono and Satou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Benjy J. Y. Tan, YmVuanl0YW5Aa3VtYW1vdG8tdS5hYy5qcA==; Yorifumi Satou, eS1zYXRvdUBrdW1hbW90by11LmFjLmpw