
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 16 September 2022
Sec. Inflammation
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.991044
This article is part of the Research Topic Gasdermins in the Defense Against Pathogens View all 8 articles
 Yuhong Pan1,2,3†
Yuhong Pan1,2,3† Wenjun Cai1,2,3†
Wenjun Cai1,2,3† Juan Huang1,2,3†
Juan Huang1,2,3† Anchun Cheng1,2,3*
Anchun Cheng1,2,3* Mingshu Wang1,2,3
Mingshu Wang1,2,3 Zhongqiong Yin3
Zhongqiong Yin3 Renyong Jia1,2,3*
Renyong Jia1,2,3*In the early 2000s, caspase-1, an important molecule that has been shown to be involved in the regulation of inflammation, cell survival and diseases, was given a new function: regulating a new mode of cell death that was later defined as pyroptosis. Since then, the inflammasome, the inflammatory caspases (caspase-4/5/11) and their substrate gasdermins (gasdermin A, B, C, D, E and DFNB59) has also been reported to be involved in the pyroptotic pathway, and this pathway is closely related to the development of various diseases. In addition, important apoptotic effectors caspase-3/8 and granzymes have also been reported to b involved in the induction of pyroptosis. In our article, we summarize findings that help define the roles of inflammasomes, inflammatory caspases, gasdermins, and other mediators of pyroptosis, and how they determine cell fate and regulate disease progression.
Cells can die through different pathways with unique morphological changes and physiological outcomes. Programmed cell death, such as apoptosis, necroptosis, and pyroptosis, are mediated by distinct sets of host proteins that coordinate various biological outcomes (1–3). Apoptosis is characterized by the activation of the caspase family, initiating caspases receiving intrinsic/extrinsic apoptosis signals and ultimately activating executioner caspases to trigger the cell death. During apoptosis, cell shrinks and divides into membrane-encapsulated apoptotic bodies that are normally phagocytosed by macrophages, resulting in non-inflammatory cell death.
Compared with apoptosis, pyroptosis is a necroptotic and inflammatory programmed cell death mode mediated by inflammatory caspases (3). Yet another type of necroptotic and inflammatory programmed cell death that executes independently of caspases is called necroptosis (4). Pyroptosis is a recently discovered cell death pathway that is regulated by various microbial infections (eg, Legionella, Salmonella and Francisella) (5). Surprisingly, caspase-1, known to play a role in inflammation and cell survival, was identified as an important player in this novel cell death pathway. Caspase-1 was first known as IL-1β-converting enzyme (ICE), because of its ability to process inactive precursors of interleukin 1β (IL-1β) and IL-18 into mature and active cytokines (6). Furthermore, activation of caspase-1 not only produces inflammatory cytokines, but also leads to cell death characterized by rupture of the plasma membrane and release of pro-inflammatory cellular contents (7, 8). The term pyroptosis, derived from the Greek roots pyro (fire/fever) and ptosis (to-sis, falling), was first proposed by D’Souza et al. in 2001 to define this inflammatory cell death (9).
In this timeline article, we discuss the seminal findings revealing the mechanistic of this newly discovered cell death pathway, including the molecules involved, such as inflammasomes, inflammatory caspases and gasdermins (Figure 1), and how inflammatory caspases determine cell fate in the context of microbial infection.
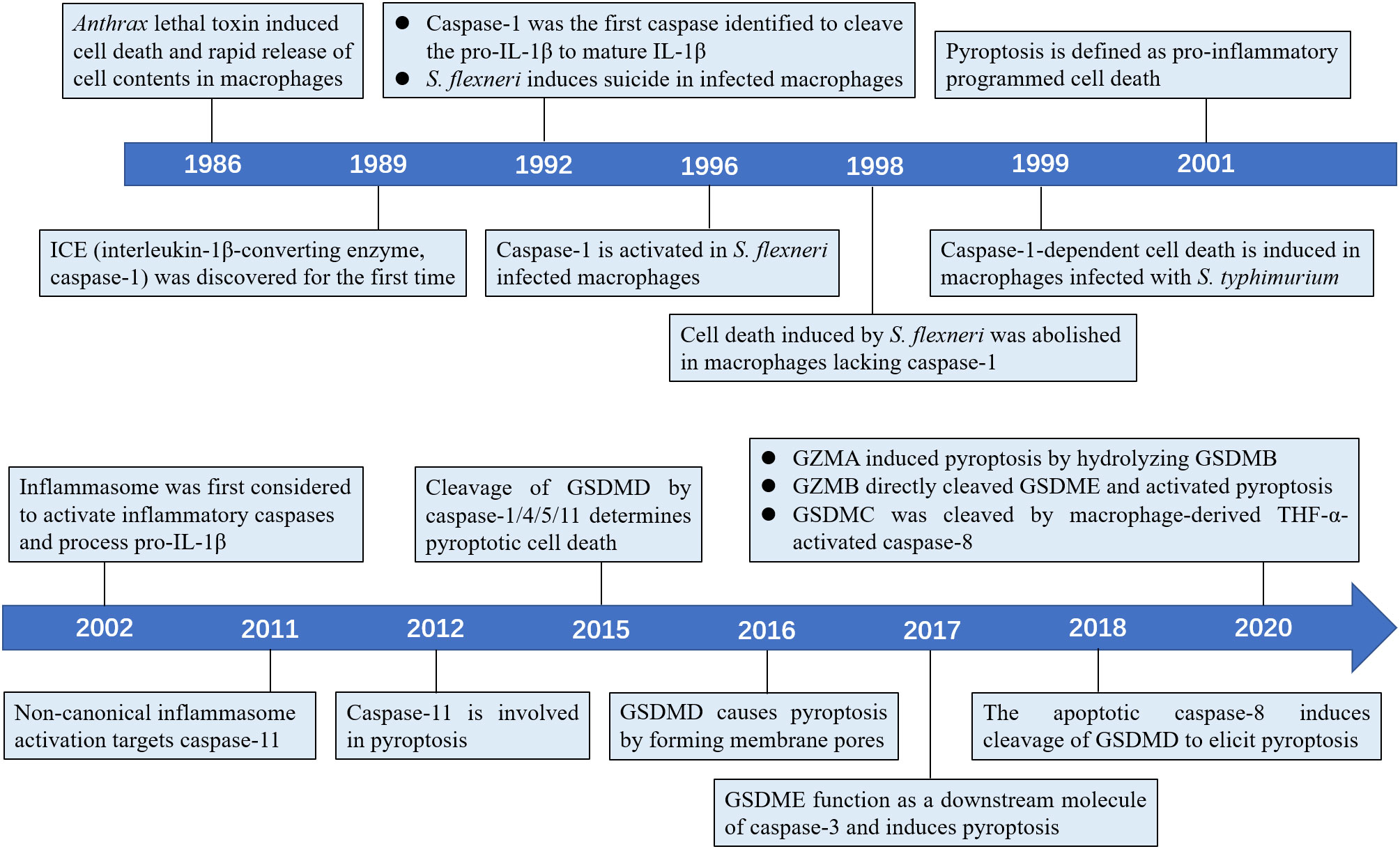
Figure 1 Timeline of the key discoveries about the pyroptosis.
The earliest study of pyroptosis dates back to 1986, when Friedlander found that treatment of mouse macrophages with Bacillus anthrax lethal toxin (LeTx) triggers cell death and release of cellular contents (10). Caspase-1, an inflammatory caspase responsible for processing pro-IL-1β to mature IL-1β, was first discovered in 1989 by Cerretti et al. and Thornberry et al. (11–13). In 1992, Zychlinsky et al. observed suicide in macrophages infected with Gram-negative bacterial pathogen Shigella flexneri, the first time discovery of pyroptosis (14). In 1996, Chen et al. found that the invasion plasmid antigen B (ipaB) of S. flexneri can directly bind to caspase-1 and cause caspase-1 activation in macrophages (15). A new research in 1998 found S. flexneri inability to induce cell death in macrophages lacking caspase-1 (16). In addition, a study in 1999 found that macrophages infected with Salmonella typhimurium also induced caspase-1-dependent cell death (17). As previous studies have shown that caspase-1 can cleave the pro-inflammatory cytokines pro-IL-1β and pro-IL-18 (12, 18), the term pyroptosis was therefore coined in 2001 to indicate inflammatory caspase-1-dependent cell death (19). In 2002, the inflammasome was first identified to activate inflammatory caspases and process pro-IL-1β (20). Later, Petr et al. reported that non-canonical caspase-11 induces cell death in a caspase-1-independent manner following host infection with Salmonella (21). Today, the concept of pyroptosis has been expanded to include cell death performed by most inflammatory caspases, such as human caspase-4/5, and mouse caspase-11 (2). Further, several studies have revealed that gasdermins serve as specific substrates of caspase-1/4/5/11 and as an effector molecule for pyroptosis (22, 23). In 2017, new research showed that GSDME can convert caspase-3-mediated apoptosis induced by TNF or chemotherapy drugs to pyroptosis, providing new insights into cancer chemotherapy (24, 25).
Pyroptosis, or caspase-1-dependent cell death is mediated by inflammasome assembly with cleavage of GSDMD and release of IL-1β/18 (26–28). Caspase-1 is initiated by multiple canonical inflammasome ligands and triggers pyroptosis (29). Inflammasomes are multi-protein signaling platforms that regulate inflammatory responses and coordinate host antimicrobial defense (30). In addition, inflammasomes have also been implicated in non-microbial diseases. Substantial evidence suggests that the inflammasome and its associated cytokines perform critical roles in tumorigenesis such as proliferation, invasion, and metastasis (31–33). Inflammasome assembly starts with cytoplasmic pattern recognition receptors (PRRs) that recognize pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs) (34). Most inflammasomes are composed of three components: (i) the nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), (ii) an adapter apoptosis-associated speck-like protein (ASC), (iii) pro-caspase-1. All NLRs have a central NOD domain, most members have a C-terminal leucine-rich repeats (LRRs) domain and a variable N-terminal protein-protein interaction domain. Depending on whether N-terminal has a pyrin domain (PYD) or a caspase activation and recruitment domain (CARD), NLRs can be subdivided into NLRPs or NLRCs (35, 36). Certain members of the NLR family, including NLRP1, NLRP3, NLRC4, already possess the ability to form inflammasomes, while others, like NLRP2, NLRP6, NLRP12, are considered putative inflammasome sensors (37). The adaptor protein ASC contains two death-fold domains: an N-terminal PYD and a C-terminal CARD. ASC uses its PYD and CARD to bind to the PYD of NLR and CARD of pro-caspase-1, respectively, bridging upstream inflammasome sensor molecules to pro-caspase-1 (38). Notably, some PRRs directly recruit pro-caspase-1 due to the inclusion of CARD (30). After the inflammasome is successfully assembled, caspase-1 is hydrolyzed into two fragments, which then dimerize into active cleaved caspase-1 (39). On one side, caspase-1 cleaves pro-IL-1β/18 to generate mature IL-1β/18 (40). On the other side, caspase-1 specifically cleaves the linker between N-terminal gasdermin D (GSDMD-N) and C-terminal GSDMD (GSDMD-C), which is necessary for pyroptosis (23). Finally, GSDMD-N oligomerizes to form transmembrane and non-selective pores of ~10-14 nm in diameters, releasing cellular contents and inflammatory cytokines IL-1β and IL-18 and leading to pyroptosis (41, 42).
To date, several receptor proteins of the NLRs have been shown to assemble the inflammasome, including NLRP1, NLRP3, NLRC4 (NLRP2/6/12), and two cytoplasmic PRRs: absent in melanoma 2 (AIM2) and pyrin (30) (Figure 2). Compared to NLRP3, NLRP1 has additional function-to-find domain (FIIND) and CARD domains at C-terminal (43). Thus, NLRP1 does not require ASC to activate pro-caspase-1, resulting in IL-β secretion (44). Human NLRP1 has three paralogs in mice: NLRP1a/b/c, but these NLRP1s in mice lack PYD. Using an in vitro system consisting of recombinant proteins, it was confirmed that NLRP1 in humans can be activated by ATP and muramyl dipeptide (45). Additionally, inhibitors of dipeptidyl peptidases 8 and 9 (DPP8/9) initiate the NLRP1 inflammasome, triggering pyroptosis of resting lymphocytes in humans and rodents (46). Importantly, NLRP1 acts as an innate immune sensor to produce IL-18 in response to metabolic stress, thereby preventing obesity and metabolic syndrome (47), and NLRP1b is also an innate immune sensor of Toxoplasma infection (48). Notably, NLRP1b activates caspase-1 to induce macrophage pyroptosis in response to B. anthracis lethal toxin (LeTx) (49), and the protease component of LeTx is required for inflammasome activation, whereas anthrax lethal factor (LF) was reported to cleave NLRP1b (50).
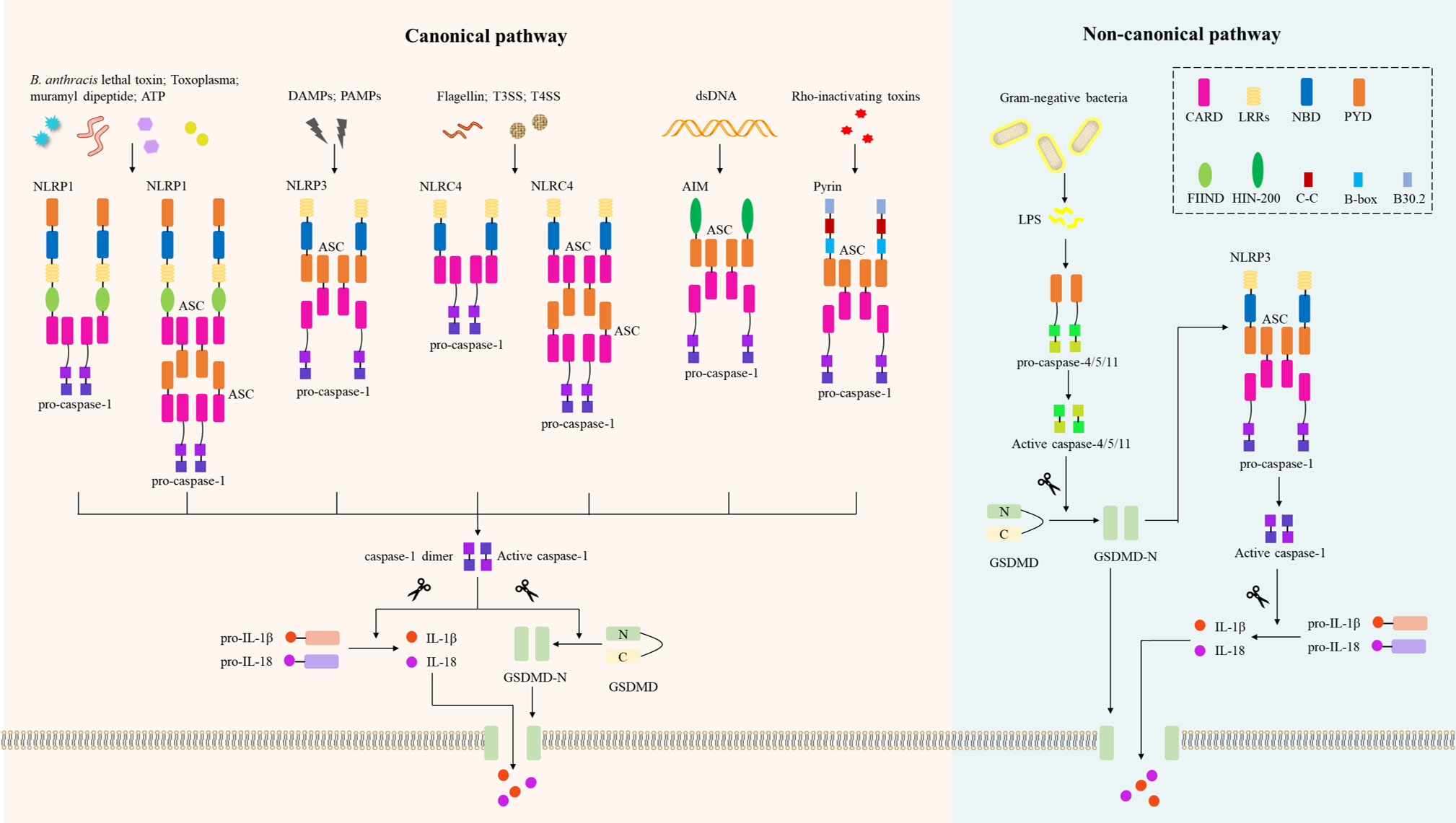
Figure 2 Inflammasomes in the canonical and non-canonical pyroptotic pathway. In the canonical pathway, danger-associated molecular patterns (DAMPs) or pathogen-associated molecular patterns (PAMPs) activate their respective inflammasome sensors, including NLRP1, NLRP3, NLRC4, AIM2, or Pyrin. B. Anthrax toxins, Toxoplasma, ATP, etc. activate the NLRP1 inflammasome, and NLRP1 recruits pro-caspase-1 via ASC or via CARD-CARD interaction. Multiple DAMPs and PAMPs activate the NLRP3 inflammasome, which subsequently recruits ASCs and pro-caspase-1. NLRC4 senses bacterial flagellin and T3SS/T4SS components and activates caspase-1 in an ASC-dependent or ASC-independent manner. The AIM2 inflammasome assembles when AIM2 senses host- or pathogen-derived dsDNA; the pyrin inflammasome is activated by Rho-inactivating toxins, and both AIM2 and pyrin activate caspase-1 in an ASC-dependent manner. Activated caspase-1 induces proteolytic cleavage of GSDMD to generate N-GSDMD, which then forms the GSDMD-N pore and induces pyroptosis. Activated caspase-1 also induces proteolytic maturation of IL-1β and IL-18 to their active forms, which are secreted through the GSDMD-N pore. In the non-canonical pathway, LPS from Gram-negative bacteria are recognized by caspase-4 or caspase-5 in human cells or caspase-11 in mouse cells. These activated inflammatory caspases directly cleave GSDMD and form GSDMD-N pores to trigger pyroptosis. The GSDMD-N-terminal fragment also activates the NLRP3 inflammasome and induces caspase-1-dependent maturation of IL-1β and IL-18. CARD, caspase recruitment domain; LRRs, leucine-rich repeats; BD, nucleotide-binding and oligomerization domain; PYD, pyrin domain; N FIIND, a functional-to-find domain; HIN-200, hematopoietic interferon-inducible nuclear protein 200; CC, coiled-coil; B-box, B-box-type zinc finger; AIM2, absent in melanoma 2; ASC, apoptosis-associated speck-like protein containing a caspase recruitment domain; GSDMD, gasdermin D; LPS, lipopolysaccharide.
Several molecular and cellular events have been proposed as triggers for NLRP3 inflammasome, such as K+ efflux, Ca2+ signaling, the production of reactive oxygen species (ROS), mitochondrial dysfunction, and lysosomal disruption (31). NIMA-related kinase 7 (NEK7) is an essential protein that mediates NLRP3 inflammasome assembly and activation downstream of K+ efflux (51), but prevents inflammasome formation during the mitotic phase of the cell cycle (52). New data demonstrate that NEK7 mediates NLRP3 inflammasome activation by bridging adjacent NLRP3 subunits (53). Notably, different NLRP3 stimuli all cause disassembly of trans-Golgi network (TGN), and NLRP3 is recruited to dispersed TGN (dTGN) via phosphatidylinositol 4-phosphate (PtdIns4P) on dTGN to activate downstream signaling cascades (54). Importantly, Szekanecz et al. (55) found that NLRP3 inflammasome-IL-1 pathway could serve as therapeutic targets for gout; another study found that cardamonin from a herbal medicinal prevents LPS-induced septic shock by inhibiting the NLRP3 inflammasome (56). Therefore, NLRP3 is emerging as a potential drug target for the treatment of inflammatory diseases (57).
NAIPs (NLR family apoptosis inhibitory proteins) were identified as immunosensors required for NLRC4 inflammasome activation (58). Human NAIP2 and NAIP5 recognize bacterial flagellin and type III/IV secretion system (T3SS/T4SS) apparatus components, respectively, to activate NLRC4 inflammasome-mediated innate immunity (59), resulting in cleavage of pro-caspase-1, and GSDMD-N pores formation and pyroptosis (60). Compared with human NAIPs, mouse NAIP1, NAIP2, NAIP5 and NAIP6 recognize the needle of T3SS, the inner rod of the T3SS, flagellin and flagellin, respectively (61). Of note, several gain-of-function mutations in NLRC4 have been implicated in the development of autoinflammatory disease (62).
AIM2 has been identified as a DNA receptor that induces inflammasome formation (63). AIM2 contains an N-terminal PYD and a C-terminal DNA-binding HIN-200 domain, senses cytoplasmic negatively charged double-strand DNA (dsDNA) through its positively charged HIN-200 domain, and interacts with ASC through its PYD (64). The ASC speck recruits pro-caspase-1 via CARD-CARD interaction, driving pro-IL-1β/18 proteolytic cleavage and pyroptosis (65). The release of microbial DNA into the cytoplasm leads to activation of the AIM2 inflammasome during infection by Listeria, Francisella, Mycobacterium, vaccinia virus, mouse cytomegalovirus, Plasmodium and Aspergillus (66). Similar to other inflammasomes, AIM2 inflammasome is precisely regulated in cells and has been reported to be negatively regulated by interferon-inducible protein p202 in mBMDMs (67). Notably, IFN-I boosts AIM2-dependent caspase-1 activation by promoting AIM2 protein expression (68). However, improper recognition of self-DNA by AIM2 can lead to the development of autoimmune and inflammatory diseases such as psoriasis, arthritis and dermatitis (65).
Pyrin is a candidate PRR that binds to ASC adaptor to form a caspase-1-activating complex by sensing pathogen modifications and inactivation of Rho GTPase (69). Interestingly, pyrin does not directly recognize molecular patterns (PAMPs and DAMPs), but responds to infection-induced disturbances in cytoplasmic homeostasis, these perturbations are called ‘homeostasis-altering molecular processes’ (HAMPs) (70). Rho-inactivating toxins including Vibrio parahaemolyticus VopS, Clostridium difficile glycosyltransferase TcdB, and Clostridium botulinum ADP-ribosylating C3 toxin could trigger the assemble of pyrin inflammasome. Inactivation of RhoA by Yersinia effectors YopE and YopT (Yersinia ectoprotein) triggers the pyrin inflammasome (71), while YopM was identified as a microbial inhibitor of the pyrin inflammasome (72). Furthermore, mutations in B30.2/SPRY domain of pyrin lead to pathogen-independent activation of pyrin and trigger autoinflammatory disease familial Mediterranean fever (FMF). Another dominantly inherited autoinflammatory disorder distinct from FMF, characterized by recurrent childhood-onset neutrophilic dermatosis, is induced by mutations in the gene encoding pyrin, MEFV (S242R) (73).
The astrocytic NLRP2 inflammasome is a vital component in CNS inflammatory responses, suggesting NLRP2 inflammasome could become a therapeutic target for suppressing CNS injury-induced inflammation (74). Growing evidence suggests that microbial signals (eg. type I IFNs), metabolic signals (eg. PPARγ activators), and microbial components (metabolites, RNA, lipoteichoic acid (LTA), LPS) can activate the NLRP6 inflammasome (75). Importantly, microbiota-related metabolites taurine, histamine and spermine shape the host-microbiota interface by co-regulating NLRP6 inflammasome signaling and epithelial IL-18 secretion (76). NLRP12 induces the production of inflammasome-dependent cytokines IL-1β/18 following Yersinia or Plasmodium salina infection, thereby preventing severe infections by these pathogens. Of note, NLRP12 is also a core component in maintaining intestinal inflammation and intestinal homeostasis. In contrast, NLRP12 exhibits as a negative mediator of NF-κB and MAPK signaling during infection with S. typhimurium, Klebsiella pneumoniae, Mycobacterium tuberculosis, or vesicular stomatitis virus, as well as during colon tumorigenesis. NLRP12 also negatively regulates canonical and non-canonical pathways in T cells and contributes to the exacerbation of autoimmune diseases. We speculate that the functional shift of NLRP12 as an inflammasome or negative regulator depends on the environment (77).
Interestingly, caspase-1-driven, pore-induced pyroptosis induces multifaceted defense against intracellular bacteria that is facilitated by pathogens entrapment in cellular debris, termed pore-induced intracellular traps (PIT) (78). Another study found that during inflammasomes activation, cells can undergo pyroptosis and release cytokines without triggering cell death, and Toll-like receptor adapter protein SARM may be participated (79). It is worth noting that thymocytes and macrophages from caspase-1-deficient animals typically undergo apoptosis (40). Furthermore, Francisella tularensis activates AIM2/ASC-dependent caspase-3-mediated apoptosis in macrophages lacking caspase-1 (80). Analysis of macrophages and primary cells treated with LPS plus nigericin or S.typhimurium indicated apoptosis became evident in GSDMD(-/-) cells, showing a inhibition of apoptosis by pyroptosis, and caspase-1 may devote to the apoptotic caspases activation in GSDMD(-/-) cells (81). These findings highlight the crosstalk between pyroptosis and apoptosis pathways and underscore the versatility of the inflammasome.
In the non-canonical pyroptotic pathway, human caspase-4/5 and the mouse homolog caspase-11 bind directly to lipopolysaccharide (LPS) of Gram-negative bacteria with high specificity and affinity (82). Binding to LPS is mediated by the CARD of the caspase, resulting in autoproteolysis and oligomerization of caspase (83). Subsequently, GSDMD is cleaved by inflammatory caspase-4/5/11 to generate N-terminal fragments, and GSDMD-N forms pores that impair cell membrane integrity (84), leading to K+ efflux (23), induction of NLRP3 inflammasome assembly, and ultimately pyroptosis (Figure 2) (85, 86). Although caspase-4/5/11 can’t cleave pro-IL-1β/18, they could regulate IL-1β/18 maturation and secretion by activating NLRP3/caspase-1 signaling (22, 85, 87). During non-canonical inflammasome signaling, caspase-11 generates active caspase-11 protease by self-cleaving at D285, which mediates GSDMD cleavage and NLRP3-dependent IL-1β production (88). Importantly, caspase-11 dimerization is required to initiate its auto-cleavage ability. Like caspase-1-deficient mice (89), caspase-11 mutant mice protect against LPS-induced endotoxic shock (40), besides, IL-1α and IL-1β production following LPS stimulation is blocked in caspase-11 mutant mice. These results indicate that caspase-11 is necessary for the activation of caspase-1 (90). Additionally, mice lacking caspase-11 had reduced numbers of apoptotic cells and lacked caspase-3 activation, suggesting that caspase-11 is also responsible for activating caspase-3 under certain pathological conditions (91).
In the face of cytoplasmic invading bacteria, caspase-4 activation via guanylate-binding proteins (GBPs) platform is critical for inducing GSDMD-dependent pyroptosis and processing of IL-18 (92). Yang et. al (93) reported that cytosolic LPS triggers caspase-11-dependent pannexin-1 channel cleavage, resulting in ATP release and subsequent activation of purinergic P2X7 receptors to mediate cytotoxicity. Cytosolic LPS-induced pyroptosis was abolished in the absence of P2X7 or pannexin-1. These data show the indispensable roles of pannexin-1 and P2X7 downstream of caspase-11 in pyroptosis. Of note, the oxidized phospholipid 1-palmitoyl-2-ara-chidonoyl-sn-glycero-3-phosphorylcholine (oxPAPC) competes with LPS to bind caspase-4 and caspase-11, thereby inhibiting LPS-induced macrophages pyroptosis, but not in dendritic cells (94). In addition, an epithelial cell-intrinsic non-canonical inflammasome has an important role in antimicrobial defense of intestinal mucosal surface, driving infected cells excretion to limit the intraepithelial proliferation of pathogens (95, 96). These studies suggest that non-canonical activation of inflammatory caspases by LPS initiates a novel paradigm for innate immunity.
GSDMD is required for pyroptosis induction and IL-1β/18 secretion in both canonical and non-canonical inflammasome responses (81). GSDMD contains two domains, 31 kDa N-terminal (GSDMD-N) and 22 kDa C-terminal (GSDMD-C), which are linked with a long loop (23). Inflammasome-associated caspases (caspase-1/4/5/11) cleaves the linker between the GSDMD-N and GSDMD-C, which is necessary to induce pyroptosis, while its C-terminal domain binds the N-terminal domain to suppress pyroptosis (97). The cleavage releases intramolecular suppression of GSDMD-N, showing pyroptosis-inducing ability. Studies have also found that apoptotic caspase-8 is able to cleave GSDMD to trigger pyroptosis (98, 99). Since GSDMD-N exhibits strong and specific binding to phosphoinositide and cardiolipin, which are known to exist in mammalian cytoplasmic membranes and bacterial inner membranes, respectively, suggesting that GSDMD-N could directly target the membrane and lyse it (100). GSDMD-N migrates to plasma membrane to form functional pores that disrupt the integrity of the cell membrane and release cytokines including IL-1β and IL-18. The gasdermin pores have an diameter of 10~14 nm and contain 16 symmetrical protomers (101). GSDMD-N can also destroy bacteria, including Escherichia coli, Bacillus megaterium protoplasts, and Staphylococcus aureus (41). Additionally, the N-terminal of GSDMD also initiates activation of NLRP3-dependent inflammasome (22, 23, 102), most likely requiring K+ efflux induced by GSDMD-N-induced membrane pores (86). It is worth noting that GSDMD-deficient cells were resistant to pyroptosis induced by LPS and known canonical inflammasome ligands, with reduced IL-1β release despite intact caspase-1 processing (23). But GSDMD-deficient cells may eventually die, possibly because of caspase-3/7 cleavage by caspase-1 (103, 104). In addition to its central role in pyroptosis, GSDMD also plays an essential function in NETosis (105). Caspase activation and specific recognition are the complete molecular mechanisms of GSDMD-mediated pyroptosis. Different caspases have different selective preferences for peptide sequences and thus have different substrate recognition profiles. Unlike caspases in the apoptotic pathway, GSDMD is the only physiological substrate known to caspase-4/5/11. The specific self-cleavage of caspase-4 and caspase-11 occurs at different subunits, which is critical for their activation and GSDMD cleavage (106). GSDMD is cleaved at the Asp275 site, which can release self-inhibition and initiate pyroptosis. Interestingly, caspase-3/7 cleaved GSDMD at Asp87 site, causing inactivation of the pyroptotic activity of GSDMD (107, 108). Succination of GSDMD on Cys192 prevents its processing and oligomerization, which limits pore formation, cytokine release, and cell death (109).
Other members of gasdermin (GSDM) family are becoming regulators of programmed cell death in various processes regulating cell proliferation and differentiation. The GSDM protein family is conservative in vertebrates and includes six homologs in human, termed GSDMA, GSDMB, GSDMC, GSDMD, GSDME/DFNA5, and DFNB59 (Table 1). Although mouse lacks GSDMB, it has three GSDMA homologs and four GSDMC homologs besides GSDMD. In addition to DFNB59, all GSDMs have two domains with a variable linker, and share similar auto-inhibitory structures. DFNB59 retains GSDM N-terminal sequence homology, while is biased at its C-terminal. Furthermore, GSDME and DFNB59 are expressed in heart, brain, kidney and inner ear; on the contrary, the remaining GSDMs are mainly expressed in the skin system and the gastrointestinal tract (epithelium) (110). The GSDM-N domains of GSDMA/GSDMA3, GSDMB, GSDMC, and GSDME, but not full-length proteins, all could trigger pyroptosis in mammalian cells and eliminate bacteria (23), the pore-forming activity of GSDM-N domains are necessary for pyroptosis (100).
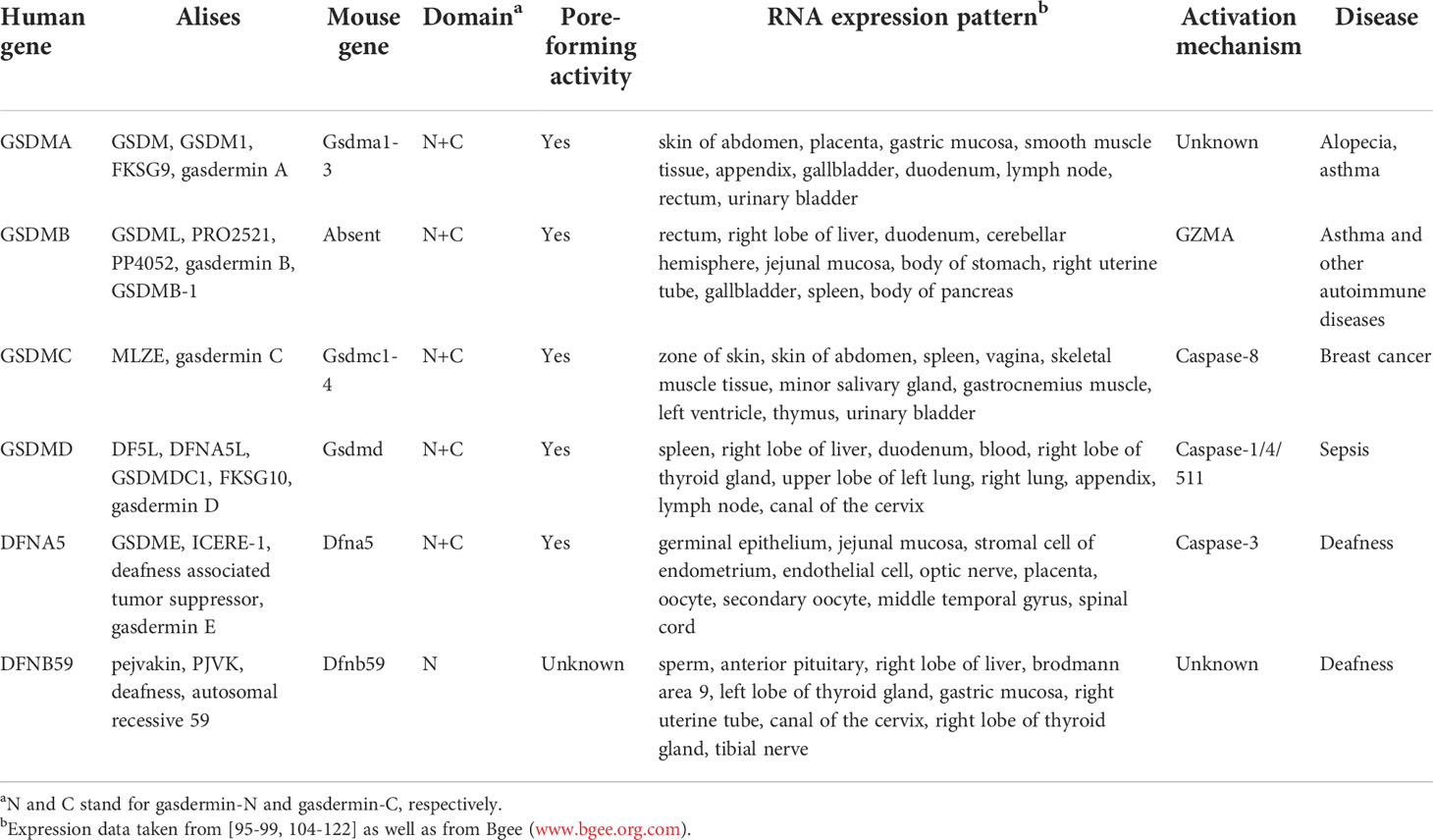
Table 1 Properties and functions of gasdermin family proteins.
Notably, the expression of GSDMA was decreased in gastric and esophageal cancers, suggesting its tumor suppressor effect (111). Further, gain-of-function mutations of GSDMA3 that lead to skin defects and alopecia disrupt autoinhibition, resulting in GSDMA3-N domain-induced pyroptosis (23). GSDMA3-N also regulates mitochondrial oxidative stress by targeting mitochondrial (112). Human GSDMB polymorphisms are strongly associated with autoimmune diseases including inflammatory bowel disease (IBD) and asthma (113, 114). Among them, GSDMB cleavage by lymphocyte-derived granzyme A (GZMA) exerts its pore-forming ability and promotes pyroptosis, a killing mechanism that enhances antitumor immunity (115). Interestingly, GSDMB is regarded as an oncogene because it is overexpressed in several cancers including gastric cancer (116), breast cancer (117) and cervical cancer (118). Therefore, a more in-depth study on the role of GSDMB in pyroptosis and cancers are required. Although artificial mutated GADMC can cause cytotoxicity in cell lines, the understanding of GSDMC is limited (100). In addition, the expression of GSDMC is related to metastatic melanoma, whereas GSDMD expression is detected in gastric cancers (GCs) (111). More recently, PD-L1 converts TNF-mediated apoptosis into pyroptosis in breast cancer cells. Under the stimulation of TNF-α, caspase-8 specifically lyses GSDMC to produce N-GSDMC, and forms pores on the cell membrane to induce pyroptosis. In addition, antibiotic chemotherapy drugs can also trigger caspase-8/GSDMC-mediated pyroptotic death in breast cancer cells (119). GSDME, also called deafness autosomal dominant 5 (DFNA5), genetic mutations in GSDME induce nonsyndromic hearing loss in humans (120, 121). In line with gain-of-function of the deafness mutation, GSDME knockout mice show no hearing impairment (122). Of note, GSDME was identified as a key molecule regulating apoptotic cell disassembly and progression to pyroptosis (24). GSDME-N permeabilizes mitochondrial membranes, releases cytochrome c (Cyt C) and activates apoptosomes. Like GSDME-N, GSDMD-N also permeates mitochondria, linking activation of apoptotic bodies to downstream inflammasome activation (123). Importantly, GSDME is emerging as an important tumor suppressor gene and a valuable molecular marker of human cancers, since GSDME regulated pyroptosis conduces to the cytotoxicity of various chemotherapeutic agents (124, 125). Among them, GSDME is specifically cleaved by caspase-3 in its linker, resulting in a GSDME-N fragment that penetrates the membrane and induces pyroptosis, providing new insights into cancer chemotherapy (25). Additionally, the cell-killer granzyme B (GZMB) is released to tumor cells by cytotoxic lymphocytes, followed by cleavage of GSDME by GZMB, inducing tumor cell pyroptosis (126). DFNB59 is also associated with deafness and is first human gene involved in nonsyndromic deafness caused by neuronal defects (120). It is worth noting that DFNB59 is also peroxisomes-associated and participated in its oxidative stress-induced proliferation (127).
Caspase-1-meidated innate immunity plays a role in the control of various bacterial infections, including S. typhimurium (128), S. flexneri (129), Listeria (130), Legionella pneumophila (131, 132), etc (Table 2). Caspase-1 substrates IL-1β and IL-18 are associated with restriction of S. typhimurium invasion, with IL-18 being important for resistance to systemic infection, while IL-1β is involved in the intestinal stage of the disease. Thus, caspase-1 is essential for the host’s innate immune defense S. typhimurium (146). Furthermore, casp1(-/-) mice were more susceptible to S. typhimurium invasion compared to casp1(-/-) casp11(-/-) mice, while both strains had elevated bacterial loads compared to wild-type mice, while bacterial quantities were similar between wild-type and casp11(-/-) mice, suggesting that caspsse-1, but not caspase-11, is protective against S. typhimurium, and caspase-11-dependent cell death is detrimental to host in the absence of caspase-1-mediated innate immunity (21). Although S. typhimurium normally resides in the vacuole, a specific mutant (sifA) enters the cytoplasm. The mutants trigger caspase-11 with elevated clearance of S. typhimurium sifA, suggesting caspase-11 is essential for innate immunity against cytosolic but not vacuolar bacteria (135). Another study reported caspase-11 is needed for intestinal tissue secretion of IL-18 in response to S. typhimurium infection and to control cecal bacterial populations (95). Importantly, S. typhimurium lacking the flagellin genes fliC and fljB exhibit a weakened ability to cause inflammation in vivo, probably due to its lack of motility and/or impaired activation of flagellin-sensing NLRC4-caspases-1 inflammasome and TLR5 (147–150). In contrast, S. typhimurium strains that overexpress flagellin because of lack of YdiV, a transcriptional suppressor of flagellin-encoding gene fliC, cause macrophage pyroptosis and induce elevated levels of IL-1β and TNF, ultimately fail in mouse tissue colonization (133).
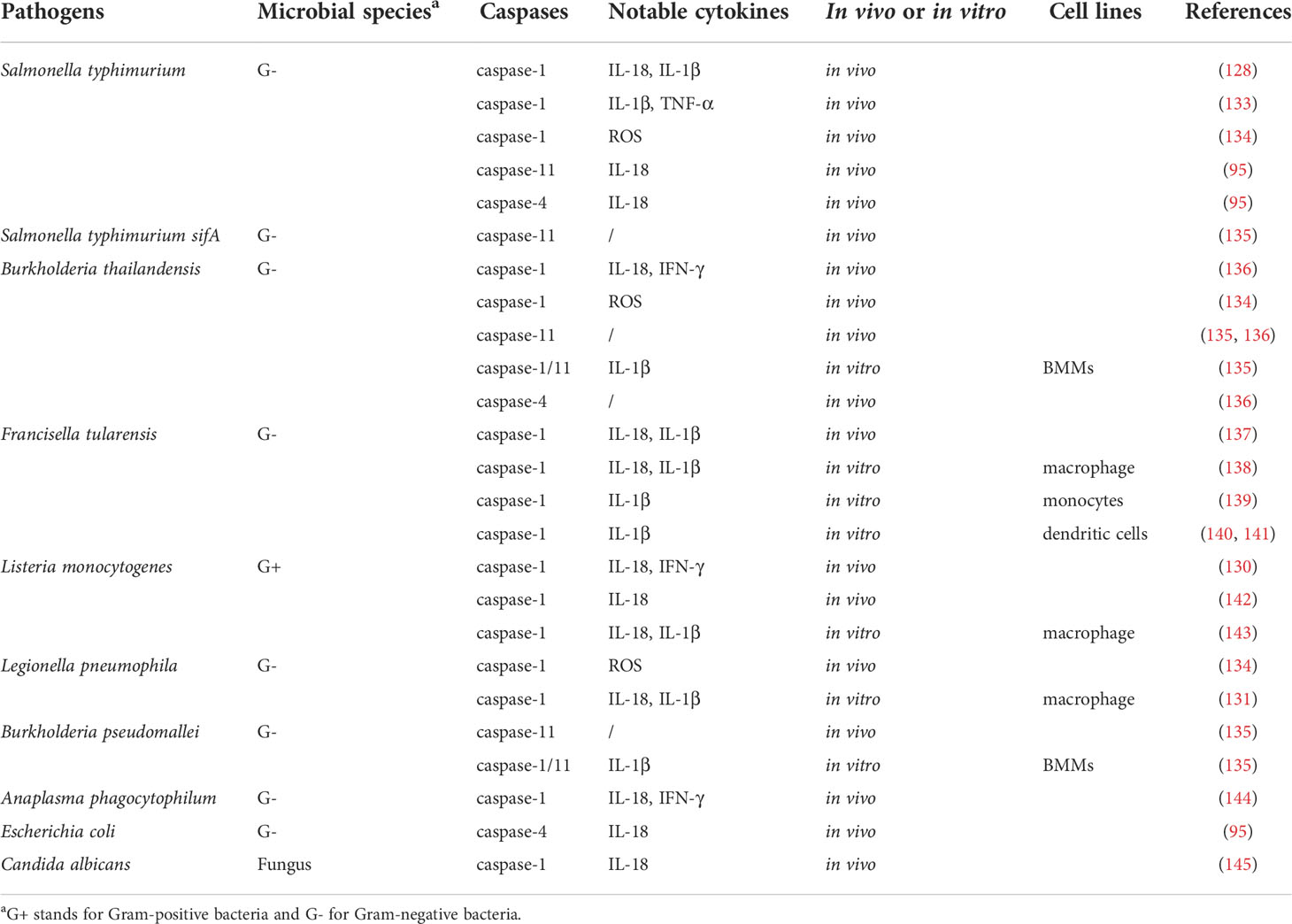
Table 2 Inflammatory caspases are involved in defense against multiple pathogen infections.
Caspase-1 is also involved in the cytoplasmic recognition of L. pneumophila flagellin by mouse macrophages, thereby limiting their infection (131). The NLRC4-caspase-1 inflammasome can trigger an IL-18-driven natural killer (NK) cell response that directs sterilizing immunity to intracellular bacterial pathogens such as Listeria (142, 143). Furthermore, activation of IL-18 by caspase-1 promotes IFN-γ to initiate caspase-11 and clearance of the cytoplasmic invasive bacterium Burkholderia thailandensis (136), whereas innate immunity to F. tularensis depends on the ASC/caspase-1 axis (137). New research finds that mice lacking caspase-1 are more susceptible to Francisella compared to mice lacking IL-1β and IL-18, suggesting cell death itself and other caspase-1-dependent pathways help restrict infection (151). Additionally, caspase-1 and ASC are indispensable for IFN-γ to control Anaplasma phagocytophilum infection (144). Monosodium urate (MSU) or calcium pyrophosphate dihydrate (CPPD) crystals are associated with inflammatory responses in gout and pseudogout. MSU and CPPD bind to the NLRP3 inflammasome that activate caspase-1, producing active IL-1β/18 (152). Other findings suggest that pyroptosis protects against bacterial infections even in the absence of IL-1α/β and IL-18 (96). For example, caspase-1-mediated pyroptosis releases bacteria in macrophages, such as B. thailandensis, L. pneumophila, and S. typhimurium, which are taken up by neutrophils and absorbed by intracellular ROS, independent of IL-1β/18 (134). Another mechanism by which pyroptosis clears bacteria is to crowd out infected cells from tissues. Enterocytes infected with S. typhimurium induce caspase-1/11 inflammasome activation, leading to the physical extrusion of infected enterocytes from intestine (95, 96).
Activation of caspase-1 also regulates the progression of adaptive immune response, and IL-18 can promote the differentiation of CD4+ T cells and the production of IFN-γ (153). Among them, caspase-1-dependent IL-18 production was used to express sustained Th1 responsiveness to Candida albicans (145). Whereas, lack of caspase-1 down-regulates CD4+ T cell-induced IFN-γ after stimulation with A. phagocytophilum in vitro (144). In contrast, Listeria-specific acquired immunity was normal in casp1(-/-) mice, while the cytokine profile is skewed towards T(h)2 compared to wild-type mice (130). In addition, aluminum-containing adjuvants can stimulate caspase-1 to activate a robust humoral immune response (154), and can also induce the release of IL-1β/18 of macrophages and dendritic cells to establish adaptive cellular immunity (154, 155), among which, the NLRP3 inflammasome is indispensable for alum-induced pro-caspase-1 processing (156, 157). Nonetheless, the regulation of antibody production by caspase-1 remains controversial (156, 157).
While pyroptosis protects the host against infections, inappropriate or excessive caspase-1 activity and pyroptosis may be detrimental. Unexamined inflammasome activation inhibits both IL-17-mediated antimicrobial responses against Pseudomonas aeruginosa and CD8+ T cell-mediated immune responses against Listeria (158–160). Additionally, overactivation of caspase-1 has been associated with myocardial infarction (5), ischemic brain injury (161), chronic colitis (162), and acute renal failure (163). It is also involved in triggering endotoxic shock (164) and exacerbating neurodegenerative diseases such as Huntington’s disease (165). Since caspase-1 deficiency or pharmacological inhibition prevents inflammation, cell death, and organ dysfunction related to these diseases, caspase-1 is a potential therapeutic target. Of note, neutralization of caspase-1-activated cytokines IL-1β/18 failed to achieve the protective effect of caspase-1 deficiency against sepsis and renal failure, suggesting that caspase-1 has other roles in disease besides activating cytokines effect (163, 166, 167).
In LPS-transfected human monocyte THP-1 cell line, caspase-4, but not caspase-5, has been found to promote IL-1β production and cell death. However, caspase-4 and caspase-5 exhibited non-redundant functions in THP-1 cell against S. typhimurium infection (85, 168). Likewise, casp11(-/-) mice expression human caspase-4 were observed to support caspase-1 activation and IL-1β/18 secretion in face of endotoxin LPS in bone marrow-derived macrophages, whereas casp11(-/-) mice are generally resistant to LPS-induced lethality. Unlike casp11(-/-) mice, casp11(-/-) mice expressing caspase-4 are protected from lethal B. thailandensis invasion (136). It is worth noting that the enteropathogenic E. coli (EPEC) virulence factor NleF binds caspase-4 and restricts the processing of caspase-4 and IL-18 in intestinal epithelial cells (IECs) (169). Further studies showed that caspase-4 is required for IL-1α secretion and pyroptosis in human macrophages infected with Yersinia, L. pneumophila, or S. Typhimurium; while caspase-4 was not necessary for IL-1β secretion (170). Thus, modulation of caspase-4 expression and activation represents a therapeutic target for systemic inflammatory response syndromes, sepsis, septic shock and related diseases (171).
Interestingly, intracellular Salmonella inhibits flagellin expression below the T-cell activation threshold to avoid triggering pyroptosis early in infection, thereby limiting inflammation and allowing bacteria to replicate intracellularly (172). Pathogens generate factors that directly suppress caspase-1 activation as well. Capase-1 has an important role in innate immune response to P. aeruginosa, virulent P. aeruginosa strains expressing ExoU can circumvent this innate immune response (173, 174). Johnston et al. discovered a poxvirus-encoded protein that inhibits caspase-1 activation by disrupting inflammasome formation (175). The IAV NS1 protein repress the cleavage of pro-IL-1β/18 and caspase-1-dependent apoptosis in primary human macrophages by controlling caspase-1 activation (176). M. tuberculosis-encoded Zn2+ metalloprotease prevents inflammasome activation and IL-1β processing (177). Yersinia translocates type III secretion proteins, counteracting the caspase-1 activating ability of type III secretion system, and Yersinia strains lacking all type III secretion proteins have an increased capacity to trigger caspase-1 (178, 179). Additionally, targeting of Rac1 by Yersinia effector YopE suppress caspase-1-mediated cleavage and secretion of IL-1β (180). Actinobacillus actinomycetemcomitans is an oral bacterium that produces a leukotoxin that selectively lyses primate neutrophils and monocytes by activating caspase-1 (181).
Atherosclerosis is an inflammatory disease associated with endothelial dysfunction in vascular wall. The disease is characterized by the accumulation of plaques rich in fat and calcium, where cell death is triggered. Although the complete mechanism of plaque formation has not been elucidated, direct activation of the AIM2 pyroptotic pathway was found to lead to GSDMD activation and vascular smooth muscle cell pyroptosis (182). NLRP3 inflammasome activation also contributes to the vascular inflammatory response driving atherosclerosis development and progression (183). Specific inhibition of the NLRP3 inflammasome using MCC950 may be a promising therapeutic approach to inhibit the development of atherosclerotic lesions (184). MCC950 also improved cardiac function in a pig myocardial infarction model (185). IL-1β is a crucial cytokine promoting the inflammatory reaction in heart failure, Tet2 mutation in blood cells can increase atherosclerotic plaque size due to elevated IL-1β signaling in mice (186, 187). Emerging evidence suggests IL-1β as a target for atherosclerosis therapy (188, 189). In addition, enhanced cardiomyocyte (CM) NLRP3 inflammasome signaling promotes atrial fibrillation (AF) (190). The work of Takeshi et al. showed that targeting the early inflammatory response induced by CM CaMKIIδ (Ca2+/calmodulin-dependent protein kinase II δ) signaling could prevent progression to heart failure (191). Another study showed that aspirin exerts a protective effect against cardiovascular disease by inhibiting the activation of NLRP3 in vascular endothelial cells (192).
Substantial evidence confirms that the NLRP3 inflammasome is associated with liver failure and liver disease (193, 194). Hepatocellular carcinoma (HCC) is a primary malignancy of hepatocytes, Wei et al. showed that the expression of NLRP3 inflammasome was significantly downregulated in human HCC (195). The team further found that estrogen inhibits HCC cells through NLRP3 inflammasome-mediated pyroptosis (196, 197). Moreover, the expression of DFNA5 in HCC cells is significantly reduced, and overexpression of DFNA5 can inhibit cell proliferation (198). Hepatic steatosis or cirrhosis is closely related to the occurrence of HCC (199). Emerging evidence suggests an important role for the NLRP3 inflammasome in nonalcoholic fatty liver disease (200). As an executor of pyroptosis, GSDMD plays a key role in the pathogenesis of steatohepatitis, by controlling cytokine secretion, NF-ĸB activation, and lipogenesis (201). Interestingly, GSDMD can prevent non-infectious liver injury by regulating apoptosis and necroptosis (202).
Lung cancer is one of the leading causes of death and the most common cancer in the world (203). GSDMD was found to be upregulated in non-small cell lung cancer (NSCLC), knockdown of GSDMD attenuated tumor proliferation by promoting apoptosis and inhibiting EGFR/Akt signaling in NSCLC (204). Another study found that polyphyllin VI induced caspase-1-mediated pyroptosis by inducing the ROS/NF-κB/NLRP3/GSDMD signal axis in NSCLC (205). In addition, polydatin inhibits the proliferation and metastasis of NSCLC cells by inhibiting NLRP3 inflammasome activation through the NF-κB pathway (206). And AIM2 promotes NSCLC cell growth through an inflammasome-dependent pathway (207). LncRNA-XIST is an oncogene in various tumors and plays a regulatory role in the proliferation, invasion and metastasis of NSCLC cells (208–210). Generally, downregulation of lncRNA-XIST inhibits the development of NSCLC by activating miR-335/SOD2/ROS cascade-associated pyroptosis (211–213). Chemotherapeutic of paclitaxel and cisplatin induces pyroptosis in A549 lung cancer cells via activation of caspase-3/GSDME (214).
Diabetic kidney disease (DKD) is a common clinical complication in diabetic patients and the leading cause of chronic kidney disease (CKD) in many developed and developing regions. Mitochondrial ROS promote tubular damage in diabetic nephropathy via the TXNIP/NLRP3/IL-1β axis (215). In turn, inhibition of NLRP3 inflammasome activation ameliorates renal injury and fibrosis in diabetic nephropathy (DN) (216), suggesting that targeting the NLRP3 inflammasome is a promising approach for the treatment of DN (217). In addition, TLR4/NF-κB signaling induces GSDMD-mediated pyroptosis in tubular cells in DKD (218), while global TLR4 knockout results in decreased renal inflammation, fibrosis and podocytopathy (219). Geniposide (GE) and Catalpol (Cat) can effectively block the oxidative stress and inflammatory response accompanying pyroptosis, thereby inhibiting the development of DN, and the mechanism may be related to the APMK/SIRT1/NF-κB pathway (220, 221). Overexpression of long non-coding RNAs MALAT1 and KCNQ1OT1 promoted high glucose (HG)-induced inflammation, oxidative stress and pyroptosis; whereas lncRNA GAS5 had the opposite effect (222–224). The findings of Pang et al. demonstrated that Irisin protects against vascular calcification (VC) by inducing autophagy and inhibiting vascular smooth muscle cell (VSMC) pyroptosis in CKD, and Irisin may serve as an effective therapeutic agent for CKD-related VC (225). Renal ischemia/reperfusion (I/R) injury is closely related to the activation of oxidative stress and pyroptosis, and the methyltransferase EZH2 may be a potential therapeutic target for renal I/R injury (226).
Alzheimer’s disease (AD) is a neurodegenerative disorder caused by the accumulation of misfolded amyloid-β (Aβ) peptide in the brain. Only recently has neuroinflammation emerged as an important component of AD pathology (227). It is now clear that Aβ-induced activation of NLRP3 inflammasome leads to the synthesis of neurotoxic factors in microglia, which ultimately cause the development of AD (228). The connection between the NLRP3 inflammasome and the pathogenesis of AD was further confirmed using the gene-knockout approach in mice. NLRP3(-/-) or casp11(-/-) mice carrying AD mutations did not show loss of spatial memory (229). Clinical studies have shown that active caspase-1 is elevated in AD patients’s brains. Thus, both animal and clinical studies have demonstrated the vital role of NLRP3 inflammasome in the pathogenesis of AD, and strategies targeting the NLRP3-caspase-1 pathway are currently being explored to find treatments for AD (230–233). Parkinson’s disease (PD) is a neurodegenerative disease affecting dopaminergic motor neurons in the midbrain, and misfolding and abnormal aggregation of the neuronal protein α-synuclein are implicated in the pathogenesis of PD (234). Intracellular misfolded α-Syn can also be transferred extracellularly by exocytosis, and extracellular α-Syn aggregates induce microglial activation and astrocytes to secrete IL-1β (235). Chronic expression of IL-1β induces dopaminergic cell death in the substantia nigra (SN), accompanied by microglial activation and inflammatory cell infiltration (236). Furthermore, Gaia et al. observed that IL-1β secretion involves NLRP3 inflammasome activation (237). NLRP3(-/-) mice are resistant to neurotoxin-induced death of dopaminergic neurons and display reduced serum levels of IL-1β and IL-18 compared to the control mice (238). The above studies show that the dopamine signaling pathway and the NLRP3 inflammasome are mutually regulated, and inhibiting the inflammatory process of neurons will be helpful for the treatment of PD (239, 240).
More than 20 years have passed since pyroptosis was rst discovered, during which time, by exploring its underlying mechanisms and its role in disease development. We understand that pyroptosis is an inflammatory and programmed form of cell death that can be triggered by a variety of host or microbial factors, such as DNA damage and infection. As vital players in the pyroptosis pathway, NLRP3, caspase-1/4/5/11 and GSDMD are potential therapeutic targets. Although pyroptosis is becoming the focus of a variety of studies, many aspects remain to be explored, including its triggers, initiation mechanisms, and physiological functions. We have not determined what other elements are involved in the regulation of pyroptosis and the physiological roles of other members of gasdermin family during pyroptosis. Also, we lack authentic evidence to sustain a major role of pyroptosis in human pathology, and this crucial hurdle could be crossed when we witness the first clinical trial that leverage comprehension of pyroptotic pathway to favorably treat diseases.
Conceptualization, YP and WC. Formal analysis, YP and JH. Funding acquisition, AC, MW and RJ. Investigation, YP. Methodology, YP. Project administration, AC and RJ. Supervision, RJ and ZY. Validation, RJ. Writing original draft, YP. All the authors have read and agreed to the published version of the manuscript.
This work was supported by the National Natural Science Foundation of China (32172833/31902267), Sichuan Veterinary Medicine and Drug Innovation Group of China Agricultural Research System (CARS-SVDIP), and Sichuan Province Research Program (2022NSFSC0078).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
IL-1β, interleukin 1β; ICE, IL-1β-converting enzyme; PRRs, pattern recognition receptors; PAMPs, pathogen-associated molecular patterns; DAMPs, danger-associated molecular patterns; NLRs, nucleotide-binding oligomerization domain (NOD)-like receptors; ASC, apoptosis-associated speck-like protein; LRRs, leucine-rich repeats; PYD, pyrin domain; CARD, caspase activation and recruitment domain; GSDMD, gasdermin D; AIM2, absent in melanoma 2; LPS, lipopolysaccharide.
1. Weinlich R, Oberst A, Beere HM, Green DR. Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol (2017) 18(2):127–36. doi: 10.1038/nrm.2016.149
2. Man SM, Kanneganti T-DJNRI. Converging roles of caspases in inflammasome activation, cell death and innate immunity. Nat Rev Immunol (2016) 16(1):7–21. doi: 10.1038/nri.2015.7
3. Walle LV, Lamkanfi MJCB. Pyroptosis. Curr Biol (2016) 26(13):R568–R72. doi: 10.1016/j.cub.2016.02.019
4. Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, et al. Fas triggers an alternative, Caspase-8–independent cell death pathway using the kinase rip as effector molecule. Nat Immunol (2000) 1(6):489–95. doi: 10.1038/82732
5. Frantz S, Ducharme A, Sawyer D, Rohde LE, Kobzik L, Fukazawa R, et al. Targeted deletion of caspase-1 reduces early mortality and left ventricular dilatation following myocardial infarction. J Mol Cell Cardiol (2003) 35(6):685–94. doi: 10.1016/s0022-2828(03)00113-5
6. Fantuzzi G, Dinarello CA. Interleukin-18 and interleukin-1 beta: Two cytokine substrates for ice (Caspase-1). J Clin Immunol (1999) 19(1):1–11. doi: 10.1023/a:1020506300324
7. Brennan MA, Cookson BT. Salmonella induces macrophage death by caspase-1-Dependent necrosis. Mol Microbiol (2000) 38(1):31–40. doi: 10.1046/j.1365-2958.2000.02103.x
8. Fink SL, Cookson BT. Caspase-1-Dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol (2006) 8(11):1812–25. doi: 10.1111/j.1462-5822.2006.00751.x
9. D’Souza CA, Heitman J. Dismantling the cryptococcus coat. Trends Microbiol (2001) 9(3):112–3. doi: 10.1016/s0966-842x(00)01945-4
10. Friedlander AM. Macrophages are sensitive to anthrax lethal toxin through an acid-dependent process. J Biol Chem (1986) 261(16):7123–6. doi: 10.1016/S0021-9258(17)38364-3
11. Black RA, Kronheim SR, Merriam JE, March CJ, Hopp TP. A pre-Aspartate-Specific protease from human leukocytes that cleaves pro-Interleukin-1 B. J Biol Chem (1989) 264(10):5323–6. doi: 10.1016/S0021-9258(18)83546-3
12. Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, et al. A novel heterodimeric cysteine protease is required for interleukin-1βprocessing in monocytes. Nature (1992) 356(6372):768–74. doi: 10.1038/356768a0
13. Cerretti DP, Kozlosky CJ, Mosley B, Nelson N, Van Ness K, Greenstreet TA, et al. Molecular cloning of the interleukin-1β converting enzyme. Science (1992) 256(5053):97–100. doi: 10.1126/science.1373520
14. Zychlinsky A, Prevost MC, Sansonetti PJJN. Shigella flexneri induces apoptosis in infected macrophages. Nature (1992) 358(6382):167–9. doi: 10.1038/358167a0
15. Chen Y, Smith MR, Thirumalai K, Zychlinsky A. A bacterial invasin induces macrophage apoptosis by binding directly to ice. EMBO J (1996) 15(15):3853–60. doi: 10.1002/j.1460-2075.1996.tb00759.x
16. Hilbi H, Moss JE, Hersh D, Chen Y, Arondel J, Banerjee S, et al. Shigella-induced apoptosis is dependent on caspase-1 which binds to ipab. J Biol Chem (1998) 273(49):32895–900. doi: 10.1074/jbc.273.49.32895
17. Hersh D, Monack DM, Smith MR, Ghori N, Falkow S, Zychlinsky A. The salmonella invasin sipb induces macrophage apoptosis by binding to caspase-1. Proc Natl Acad Sci USA (1999) 96(5):2396–401. doi: 10.1073/pnas.96.5.2396
18. Kostura MJ, Tocci MJ, Limjuco G, Chin J, Cameron P, Hillman AG, et al. Identification of a monocyte specific pre-interleukin 1 beta convertase activity. Proc Natl Acad Sci USA (1989) 86(14):5227–31. doi: 10.1073/pnas.86.14.5227
19. Cookson BT, Brennan MA. Pro-inflammatory programmed cell death. Trends Microbiol (2001) 3(9):113–4. doi: 10.1016/s0966-842x(00)01936-3
20. Martinon F, Burns K, Tschopp J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proil-B. Mol Cell (2002) 10(2):417–26. doi: 10.1016/s1097-2765(02)00599-3
21. Broz P, Ruby T, Belhocine K, Bouley DM, Kayagaki N, Dixit VM, et al. Caspase-11 increases susceptibility to salmonella infection in the absence of caspase-1. Nature (2012) 490(7419):288–91. doi: 10.1038/nature11419
22. Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin d for non-canonical inflammasome signalling. Nature (2015) 526(7575):666–71. doi: 10.1038/nature15541
23. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of gsdmd by inflammatory caspases determines pyroptotic cell death. Nature (2015) 526(7575):660–5. doi: 10.1038/nature15514
24. Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. Cleavage of Dfna5 by caspase-3 during apoptosis mediates progression to secondary Necrotic/Pyroptotic cell death. Nat Commun (2017) 8:14128. doi: 10.1038/ncomms14128
25. Wang Y, Gao W, Shi X, Ding J, Liu W, He H, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature (2017) 547(7661):99–103. doi: 10.1038/nature22393
26. Xia X, Wang X, Cheng Z, Qin W, Lei L, Jiang J, et al. The role of pyroptosis in cancer: Pro-cancer or pro-”Host”? Cell Death Dis (2019) 10(9):650. doi: 10.1038/s41419-019-1883-8
27. Frank D, Vince JE. Pyroptosis versus necroptosis: Similarities, differences, and crosstalk. Cell Death Differ (2019) 26(1):99–114. doi: 10.1038/s41418-018-0212-6
28. Jackson DN, Theiss AL. Gut bacteria signaling to mitochondria in intestinal inflammation and cancer. Gut Microbes (2020) 11(3):285–304. doi: 10.1080/19490976.2019.1592421
29. Ghayur T, Banerjee S, Hugunin M, Butler D, Herzog L, Carter A, et al. Caspase-1 processes ifn-Gamma-Inducing factor and regulates lps-induced ifn-gamma production. Nature (1997) 386(6625):619–23. doi: 10.1038/386619a0
30. Broz P, Dixit VM. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat Rev Immunol (2016) 16(7):407–20. doi: 10.1038/nri.2016.58
31. He Y, Hara H, Núñez G. Mechanism and regulation of Nlrp3 inflammasome activation. Trends Biochem Sci (2016) 41(12):1012–21. doi: 10.1016/j.tibs.2016.09.002
32. Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
33. Massagué J, Obenauf AC. Metastatic colonization by circulating tumour cells. Nature (2016) 529(7586):298–306. doi: 10.1038/nature17038
34. Place DE, Kanneganti T-D. Recent advances in inflammasome biology. Curr Opin Immunol (2018) 50:32–8. doi: 10.1016/j.coi.2017.10.011
35. Zitvogel L, Kepp O, Galluzzi L, Kroemer G. Inflammasomes in carcinogenesis and anticancer immune responses. Nat Immunol (2012) 13(4):343–51. doi: 10.1038/ni.2224
36. Lamkanfi M. Emerging inflammasome effector mechanisms. Nat Rev Immunol (2011) 11(3):213–20. doi: 10.1038/nri2936
37. Sharma D, Kanneganti T-D. The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation. J Cell Biol (2016) 213(6):617–29. doi: 10.1083/jcb.201602089
38. Fernandes-Alnemri T, Wu J, Yu JW, Datta P, Miller B, Jankowski W, et al. The pyroptosome: A supramolecular assembly of asc dimers mediating inflammatory cell death Via caspase-1 activation. Cell Death Differ (2007) 14(9):1590–604. doi: 10.1038/sj.cdd.4402194
39. Sollberger G, Strittmatter GE, Garstkiewicz M, Sand J, Beer H-D. Caspase-1: The inflammasome and beyond. Innate Immun (2014) 20(2):115–25. doi: 10.1177/1753425913484374
40. Li P, Allen H, Banerjee S, Franklin S, Herzog L, Johnston C, et al. Mice deficient in il-1 beta-converting enzyme are defective in production of mature il-1 beta and resistant to endotoxic shock. Cell (1995) 80(3):401–11. doi: 10.1016/0092-8674(95)90490-5
41. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin d causes pyroptosis by forming membrane pores. Nature (2016) 535(7610):153–8. doi: 10.1038/nature18629
42. Sborgi L, Rühl S, Mulvihill E, Pipercevic J, Heilig R, Stahlberg H, et al. Gsdmd membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J (2016) 35(16):1766–78. doi: 10.15252/embj.201694696
43. Chavarría-Smith J, Mitchell PS, Ho AM, Daugherty MD, Vance RE. Functional and evolutionary analyses identify proteolysis as a general mechanism for Nlrp1 inflammasome activation. PloS Pathog (2016) 12(12):e1006052. doi: 10.1371/journal.ppat.1006052
44. Schroder K, Tschopp J. The inflammasomes. Cell (2010) 140(6):821–32. doi: 10.1016/j.cell.2010.01.040
45. Faustin B, Lartigue L, Bruey J-M, Luciano F, Sergienko E, Bailly-Maitre B, et al. Reconstituted Nalp1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol Cell (2007) 25(5):713–24. doi: 10.1016/j.molcel.2007.01.032
46. Johnson DC, Okondo MC, Orth EL, Rao SD, Huang H-C, Ball DP, et al. Dpp8/9 inhibitors activate the Card8 inflammasome in resting lymphocytes. Cell Death Dis (2020) 11(8):628. doi: 10.1038/s41419-020-02865-4
47. Murphy AJ, Kraakman MJ, Kammoun HL, Dragoljevic D, Lee MKS, Lawlor KE, et al. Il-18 production from the Nlrp1 inflammasome prevents obesity and metabolic syndrome. Cell Metab (2016) 23(1):155–64. doi: 10.1016/j.cmet.2015.09.024
48. Ewald SE, Chavarria-Smith J, Boothroyd JC. Nlrp1 is an inflammasome sensor for toxoplasma gondii. Infect Immun (2014) 82(1):460–8. doi: 10.1128/IAI.01170-13
49. Boyden ED, Dietrich WF. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat Genet (2006) 38(2):240–4. doi: 10.1038/ng1724
50. Levinsohn JL, Newman ZL, Hellmich KA, Fattah R, Getz MA, Liu S, et al. Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. PloS Pathog (2012) 8(3):e1002638. doi: 10.1371/journal.ppat.1002638
51. He Y, Zeng MY, Yang D, Motro B, Núñez G. Nek7 is an essential mediator of Nlrp3 activation downstream of potassium efflux. Nature (2016) 530(7590):354–7. doi: 10.1038/nature16959
52. Shi H, Wang Y, Li X, Zhan X, Tang M, Fina M, et al. Nlrp3 activation and mitosis are mutually exclusive events coordinated by Nek7, a new inflammasome component. Nat Immunol (2016) 17(3):250–8. doi: 10.1038/ni.3333
53. Sharif H, Wang L, Wang WL, Magupalli VG, Andreeva L, Qiao Q, et al. Structural mechanism for Nek7-licensed activation of Nlrp3 inflammasome. Nature (2019) 570(7761):338–43. doi: 10.1038/s41586-019-1295-z
54. Chen J, Chen ZJ. Ptdins4p on dispersed trans-golgi network mediates Nlrp3 inflammasome activation. Nature (2018) 564(7734):71–6. doi: 10.1038/s41586-018-0761-3
55. Szekanecz Z, Szamosi S, Kovács GE, Kocsis E, Benkő S. The Nlrp3 inflammasome - interleukin 1 pathway as a therapeutic target in gout. Arch Biochem Biophys (2019) 670:82–93. doi: 10.1016/j.abb.2019.01.031
56. Wang Z, Xu G, Gao Y, Zhan X, Qin N, Fu S, et al. Cardamonin from a medicinal herb protects against lps-induced septic shock by suppressing Nlrp3 inflammasome. Acta Pharm Sin B (2019) 9(4):734–44. doi: 10.1016/j.apsb.2019.02.003
57. Yang Y, Wang H, Kouadir M, Song H, Shi F. Recent advances in the mechanisms of Nlrp3 inflammasome activation and its inhibitors. Cell Death Dis (2019) 10(2):128. doi: 10.1038/s41419-019-1413-8
58. Kofoed EM, Vance RE. Innate immune recognition of bacterial ligands by naips determines inflammasome specificity. Nature (2011) 477(7366):592–5. doi: 10.1038/nature10394
59. Zhao Y, Yang J, Shi J, Gong Y-N, Lu Q, Xu H, et al. The Nlrc4 inflammasome receptors for bacterial flagellin and type iii secretion apparatus. Nature (2011) 477(7366):596–600. doi: 10.1038/nature10510
60. Duncan JA, Canna SW. The Nlrc4 inflammasome. Immunol Rev (2018) 281(1):115–23. doi: 10.1111/imr.12607
61. Lightfield KL, Persson J, Brubaker SW, Witte CE, von Moltke J, Dunipace EA, et al. Critical function for Naip5 in inflammasome activation by a conserved carboxy-terminal domain of flagellin. Nat Immunol (2008) 9(10):1171–8. doi: 10.1038/ni.1646
62. Romberg N, Vogel TP, Canna SW. Nlrc4 inflammasomopathies. Curr Opin Allergy Clin Immunol (2017) 17(6):398–404. doi: 10.1097/ACI.0000000000000396
63. Jin T, Perry A, Jiang J, Smith P, Curry JA, Unterholzner L, et al. Structures of the hin domain : DNA complexes reveal ligand binding and activation mechanisms of the Aim2 inflammasome and Ifi16 receptor. Immunity (2012) 36(4):561–71. doi: 10.1016/j.immuni.2012.02.014
64. Fernandes-Alnemri T, Yu J-W, Datta P, Wu J, Alnemri ES. Aim2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature (2009) 458(7237):509–13. doi: 10.1038/nature07710
65. Man SM, Karki R, Kanneganti T-D. Aim2 inflammasome in infection, cancer, and autoimmunity: Role in DNA sensing, inflammation, and innate immunity. Eur J Immunol (2016) 46(2):269–80. doi: 10.1002/eji.201545839
66. Fernandes-Alnemri T, Yu J-W, Juliana C, Solorzano L, Kang S, Wu J, et al. The Aim2 inflammasome is critical for innate immunity to francisella tularensis. Nat Immunol (2010) 11(5):385–93. doi: 10.1038/ni.1859
67. Roberts TL, Idris A, Dunn JA, Kelly GM, Burnton CM, Hodgson S, et al. Hin-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science (2009) 323(5917):1057–60. doi: 10.1126/science.1169841
68. Jones JW, Kayagaki N, Broz P, Henry T, Newton K, O’Rourke K, et al. Absent in melanoma 2 is required for innate immune recognition of francisella tularensis. Proc Natl Acad Sci USA (2010) 107(21):9771–6. doi: 10.1073/pnas.1003738107
69. Xu H, Yang J, Gao W, Li L, Li P, Zhang L, et al. Innate immune sensing of bacterial modifications of rho gtpases by the pyrin inflammasome. Nature (2014) 513(7517):237–41. doi: 10.1038/nature13449
70. Heilig R, Broz P. Function and mechanism of the pyrin inflammasome. Eur J Immunol (2018) 48(2):230–8. doi: 10.1002/eji.201746947
71. Chung LK, Park YH, Zheng Y, Brodsky IE, Hearing P, Kastner DL, et al. The yersinia virulence factor yopm hijacks host kinases to inhibit type iii effector-triggered activation of the pyrin inflammasome. Cell Host Microbe (2016) 20(3):296–306. doi: 10.1016/j.chom.2016.07.018
72. Ratner D, Orning MPA, Proulx MK, Wang D, Gavrilin MA, Wewers MD, et al. The yersinia pestis effector yopm inhibits pyrin inflammasome activation. PloS Pathog (2016) 12(12):e1006035. doi: 10.1371/journal.ppat.1006035
73. Masters SL, Lagou V, Jéru I, Baker PJ, Van Eyck L, Parry DA, et al. Familial autoinflammation with neutrophilic dermatosis reveals a regulatory mechanism of pyrin activation. Sci Transl Med (2016) 8(332):332ra45. doi: 10.1126/scitranslmed.aaf1471
74. Minkiewicz J, de Rivero Vaccari JP, Keane RW. Human astrocytes express a novel Nlrp2 inflammasome. Glia (2013) 61(7):1113–21. doi: 10.1002/glia.22499
75. Li R, Zhu S. Nlrp6 inflammasome. Mol Aspects Med (2020) 76:100859. doi: 10.1016/j.mam.2020.100859
76. Levy M, Thaiss CA, Zeevi D, Dohnalová L, Zilberman-Schapira G, Mahdi JA, et al. Microbiota-modulated metabolites shape the intestinal microenvironment by regulating Nlrp6 inflammasome signaling. Cell (2015) 163(6):1428–43. doi: 10.1016/j.cell.2015.10.048
77. Tuladhar S, Kanneganti T-D. Nlrp12 in innate immunity and inflammation. Mol Aspects Med (2020) 76:100887. doi: 10.1016/j.mam.2020.100887
78. Jorgensen I, Zhang Y, Krantz BA, Miao EA. Pyroptosis triggers pore-induced intracellular traps (Pits) that capture bacteria and lead to their clearance by efferocytosis. J Exp Med (2016) 213(10):2113–28. doi: 10.1084/jem.20151613
79. Carty M, Kearney J, Shanahan KA, Hams E, Sugisawa R, Connolly D, et al. Cell survival and cytokine release after inflammasome activation is regulated by the toll-Il-1r protein sarm. Immunity (2019) 50(6):1412–24. doi: 10.1016/j.immuni.2019.04.005
80. Pierini R, Juruj C, Perret M, Jones CL, Mangeot P, Weiss DS, et al. Aim2/Asc triggers caspase-8-Dependent apoptosis in francisella-infected caspase-1-Deficient macrophages. Cell Death Differ (2012) 19(10):1709–21. doi: 10.1038/cdd.2012.51
81. He W-t, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin d is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res (2015) 25(12):1285–98. doi: 10.1038/cr.2015.139
82. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature (2011) 479(7371):117–21. doi: 10.1038/nature10558
83. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular lps. Nature (2014) 514(7521):187–92. doi: 10.1038/nature13683
84. Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N, et al. Gsdmd P30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci USA (2016) 113(28):7858–63. doi: 10.1073/pnas.1607769113
85. Baker PJ, Boucher D, Bierschenk D, Tebartz C, Whitney PG, D’Silva DB, et al. Nlrp3 inflammasome activation downstream of cytoplasmic lps recognition by both caspase-4 and caspase-5. Eur J Immunol (2015) 45(10):2918–26. doi: 10.1002/eji.201545655
86. Rühl S, Broz P. Caspase-11 activates a canonical Nlrp3 inflammasome by promoting k(+) efflux. Eur J Immunol (2015) 45(10):2927–36. doi: 10.1002/eji.201545772
87. Shi J, Gao W, Shao F. Pyroptosis: Gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci (2017) 42(4):245–54. doi: 10.1016/j.tibs.2016.10.004
88. Ross C, Chan AH, Von Pein J, Boucher D, Schroder K. Dimerization and auto-processing induce caspase-11 protease activation within the non-canonical inflammasome. Life Sci Alliance (2018) 1(6):e201800237. doi: 10.26508/lsa.201800237
89. Kuida K, Lippke JA, Ku G, Harding MW, Livingston DJ, Su MS, et al. Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science (1995) 267(5206):2000–3. doi: 10.1126/science.7535475
90. Wang S, Miura M, Jung YK, Zhu H, Li E, Yuan J. Murine caspase-11, an ice-interacting protease, is essential for the activation of ice. Cell (1998) 92(4):501–9. doi: 10.1016/s0092-8674(00)80943-5
91. Kang SJ, Wang S, Hara H, Peterson EP, Namura S, Amin-Hanjani S, et al. Dual role of caspase-11 in mediating activation of caspase-1 and caspase-3 under pathological conditions. J Cell Biol (2000) 149(3):613–22. doi: 10.1083/jcb.149.3.613
92. Wandel MP, Kim B-H, Park E-S, Boyle KB, Nayak K, Lagrange B, et al. Guanylate-binding proteins convert cytosolic bacteria into caspase-4 signaling platforms. Nat Immunol (2020) 21(8):880–91. doi: 10.1038/s41590-020-0697-2
93. Yang D, He Y, Muñoz-Planillo R, Liu Q, Núñez G. Caspase-11 requires the pannexin-1 channel and the purinergic P2x7 pore to mediate pyroptosis and endotoxic shock. Immunity (2015) 43(5):923–32. doi: 10.1016/j.immuni.2015.10.009
94. Chu LH, Indramohan M, Ratsimandresy RA, Gangopadhyay A, Morris EP, Monack DM, et al. The oxidized phospholipid oxpapc protects from septic shock by targeting the non-canonical inflammasome in macrophages. Nat Commun (2018) 9(1):996. doi: 10.1038/s41467-018-03409-3
95. Knodler LA, Crowley SM, Sham HP, Yang H, Wrande M, Ma C, et al. Noncanonical inflammasome activation of caspase-4/Caspase-11 mediates epithelial defenses against enteric bacterial pathogens. Cell Host Microbe (2014) 16(2):249–56. doi: 10.1016/j.chom.2014.07.002
96. Sellin ME, Müller AA, Felmy B, Dolowschiak T, Diard M, Tardivel A, et al. Epithelium-intrinsic Naip/Nlrc4 inflammasome drives infected enterocyte expulsion to restrict salmonella replication in the intestinal mucosa. Cell Host Microbe (2014) 16(2):237–48. doi: 10.1016/j.chom.2014.07.001
97. Liu Z, Wang C, Yang J, Zhou B, Yang R, Ramachandran R, et al. Crystal structures of the full-length murine and human gasdermin d reveal mechanisms of autoinhibition, lipid binding, and oligomerization. Immunity (2019) 51(1):43–9. doi: 10.1016/j.immuni.2019.04.017
98. Chen KW, Demarco B, Heilig R, Shkarina K, Boettcher A, Farady CJ, et al. Extrinsic and intrinsic apoptosis activate pannexin-1 to drive Nlrp3 inflammasome assembly. EMBO J (2019) 38(10):e101638. doi: 10.15252/embj.2019101638
99. Sarhan J, Liu BC, Muendlein HI, Li P, Nilson R, Tang AY, et al. Caspase-8 induces cleavage of gasdermin d to elicit pyroptosis during infection. Proc Natl Acad Sci USA (2018) 115(46):E10888–E97. doi: 10.1073/pnas.1809548115
100. Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature (2016) 535(7610):111–6. doi: 10.1038/nature18590
101. Evavold CL, Ruan J, Tan Y, Xia S, Wu H, Kagan JC. The pore-forming protein gasdermin d regulates interleukin-1 secretion from living macrophages. Immunity (2018) 48(1):35–44. doi: 10.1016/j.immuni.2017.11.013
102. Case CL, Kohler LJ, Lima JB, Strowig T, de Zoete MR, Flavell RA, et al. Caspase-11 stimulates rapid flagellin-independent pyroptosis in response to legionella pneumophila. Proc Natl Acad Sci USA (2013) 110(5):1851–6. doi: 10.1073/pnas.1211521110
103. Akhter A, Gavrilin MA, Frantz L, Washington S, Ditty C, Limoli D, et al. Caspase-7 activation by the Nlrc4/Ipaf inflammasome restricts legionella pneumophila infection. PloS Pathog (2009) 5(4):e1000361. doi: 10.1371/journal.ppat.1000361
104. Lamkanfi M, Kanneganti T-D, Van Damme P, Vanden Berghe T, Vanoverberghe I, Vandekerckhove J, et al. Targeted peptidecentric proteomics reveals caspase-7 as a substrate of the caspase-1 inflammasomes. Mol Cell Proteomics (2008) 7(12):2350–63. doi: 10.1074/mcp.M800132-MCP200
105. Sollberger G, Choidas A, Burn GL, Habenberger P, Di Lucrezia R, Kordes S, et al. Gasdermin d plays a vital role in the generation of neutrophil extracellular traps. Sci Immunol (2018) 3(26):eaar6689. doi: 10.1126/sciimmunol.aar6689
106. Kesavardhana S, Malireddi RKS, Kanneganti TD. Caspases in cell death, inflammation, and pyroptosis. Annu Rev Immunol (2020) 38:567–95. doi: 10.1146/annurev-immunol-073119-095439
107. Liu X, Xia S, Zhang Z, Wu H, Lieberman J. Channelling inflammation: Gasdermins in physiology and disease. Nat Rev Drug Discovery (2021) 20(5):384–405. doi: 10.1038/s41573-021-00154-z
108. Yu P, Zhang X, Liu N, Tang L, Peng C, Chen X. Pyroptosis: Mechanisms and diseases. Signal Transduct Target Ther (2021) 6(1):128. doi: 10.1038/s41392-021-00507-5
109. Humphries F, Shmuel-Galia L, Ketelut-Carneiro N, Li S, Wang B, Nemmara VV, et al. Succination inactivates gasdermin d and blocks pyroptosis. Science (2020) 369(6511):1633–7. doi: 10.1126/science.abb9818
110. Zheng Z, Deng W, Lou X, Bai Y, Wang J, Zeng H, et al. Gasdermins: Pore-forming activities and beyond. Acta Biochim Biophys Sin (Shanghai) (2020) 52(5):467–74. doi: 10.1093/abbs/gmaa016
111. Saeki N, Usui T, Aoyagi K, Kim DH, Sato M, Mabuchi T, et al. Distinctive expression and function of four gsdm family genes (Gsdma-d) in normal and malignant upper gastrointestinal epithelium. Genes Chromosomes Cancer (2009) 48(3):261–71. doi: 10.1002/gcc.20636
112. Lin P-H, Lin H-Y, Kuo C-C, Yang L-T. N-terminal functional domain of gasdermin A3 regulates mitochondrial homeostasis Via mitochondrial targeting. J BioMed Sci (2015) 22:44. doi: 10.1186/s12929-015-0152-0
113. Chao KL, Kulakova L, Herzberg O. Gene polymorphism linked to increased asthma and ibd risk alters gasdermin-b structure, a sulfatide and phosphoinositide binding protein. Proc Natl Acad Sci U.S.A. (2017) 114(7):E1128–E37. doi: 10.1073/pnas.1616783114
114. Zhao C-N, Fan Y, Huang J-J, Zhang H-X, Gao T, Wang C, et al. The association of gsdmb and Ormdl3 gene polymorphisms with asthma: A meta-analysis. Allergy Asthma Immunol Res (2015) 7(2):175–85. doi: 10.4168/aair.2015.7.2.175
115. Zhou Z, He H, Wang K, Shi X, Wang Y, Su Y, et al. Granzyme a from cytotoxic lymphocytes cleaves gsdmb to trigger pyroptosis in target cells. Science (2020) 368(6494):eaaz7548. doi: 10.1126/science.aaz7548
116. Komiyama H, Aoki A, Tanaka S, Maekawa H, Kato Y, Wada R, et al. Alu-derived cis-element regulates tumorigenesis-dependent gastric expression of gasdermin b (Gsdmb). Genes Genet Syst (2010) 85(1):75–83. doi: 10.1266/ggs.85.75
117. Hergueta-Redondo M, Sarrio D, Molina-Crespo Á, Vicario R, Bernadó-Morales C, Martínez L, et al. Gasdermin b expression predicts poor clinical outcome in Her2-positive breast cancer. Oncotarget (2016) 7(35):56295–308. doi: 10.18632/oncotarget.10787
118. Sun Q, Yang J, Xing G, Sun Q, Zhang L, He F. Expression of gsdml associates with tumor progression in uterine cervix cancer. Transl Oncol (2008) 1(2):73–83. doi: 10.1593/tlo.08112
119. Hou J, Zhao R, Xia W, Chang C-W, You Y, Hsu J-M, et al. Pd-L1-Mediated gasdermin c expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis. Nat Cell Biol (2020) 22(10):1264–75. doi: 10.1038/s41556-020-0575-z
120. Delmaghani S, del Castillo FJ, Michel V, Leibovici M, Aghaie A, Ron U, et al. Mutations in the gene encoding pejvakin, a newly identified protein of the afferent auditory pathway, cause Dfnb59 auditory neuropathy. Nat Genet (2006) 38(7):770–8. doi: 10.1038/ng1829
121. Van Laer L, Huizing EH, Verstreken M, van Zuijlen D, Wauters JG, Bossuyt PJ, et al. Nonsyndromic hearing impairment is associated with a mutation in Dfna5. Nat Genet (1998) 20(2):194–7. doi: 10.1038/2503
122. Van Laer L, Pfister M, Thys S, Vrijens K, Mueller M, Umans L, et al. Mice lacking Dfna5 show a diverging number of cochlear fourth row outer hair cells. Neurobiol Dis (2005) 19(3):386–99. doi: 10.1016/j.nbd.2005.01.019
123. Rogers C, Erkes DA, Nardone A, Aplin AE, Fernandes-Alnemri T, Alnemri ES. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat Commun (2019) 10(1):1689. doi: 10.1038/s41467-019-09397-2
124. Kim MS, Chang X, Yamashita K, Nagpal JK, Baek JH, Wu G, et al. Aberrant promoter methylation and tumor suppressive activity of the Dfna5 gene in colorectal carcinoma. Oncogene (2008) 27(25):3624–34. doi: 10.1038/sj.onc.1211021
125. Akino K, Toyota M, Suzuki H, Imai T, Maruyama R, Kusano M, et al. Identification of Dfna5 as a target of epigenetic inactivation in gastric cancer. Cancer Sci (2007) 98(1):88–95. doi: 10.1111/j.1349-7006.2006.00351.x
126. Zhang Z, Zhang Y, Xia S, Kong Q, Li S, Liu X, et al. Gasdermin e suppresses tumour growth by activating anti-tumour immunity. Nature (2020) 579(7799):415–20. doi: 10.1038/s41586-020-2071-9
127. Delmaghani S, Defourny J, Aghaie A, Beurg M, Dulon D, Thelen N, et al. Hypervulnerability to sound exposure through impaired adaptive proliferation of peroxisomes. Cell (2015) 163(4):894–906. doi: 10.1016/j.cell.2015.10.023
128. Lara-Tejero M, Sutterwala FS, Ogura Y, Grant EP, Bertin J, Coyle AJ, et al. Role of the caspase-1 inflammasome in salmonella typhimurium pathogenesis. J Exp Med (2006) 203(6):1407–12. doi: 10.1084/jem.20060206
129. Sansonetti PJ, Phalipon A, Arondel J, Thirumalai K, Banerjee S, Akira S, et al. Caspase-1 activation of il-1beta and il-18 are essential for shigella flexneri-induced inflammation. Immunity (2000) 12(5):581–90. doi: 10.1016/s1074-7613(00)80209-5
130. Tsuji NM, Tsutsui H, Seki E, Kuida K, Okamura H, Nakanishi K, et al. Roles of caspase-1 in listeria infection in mice. Int Immunol (2004) 16(2):335–43. doi: 10.1093/intimm/dxh041
131. Molofsky AB, Byrne BG, Whitfield NN, Madigan CA, Fuse ET, Tateda K, et al. Cytosolic recognition of flagellin by mouse macrophages restricts legionella pneumophila infection. J Exp Med (2006) 203(4):1093–104. doi: 10.1084/jem.20051659
132. Zamboni DS, Kobayashi KS, Kohlsdorf T, Ogura Y, Long EM, Vance RE, et al. The Birc1e cytosolic pattern-recognition receptor contributes to the detection and control of legionella pneumophila infection. Nat Immunol (2006) 7(3):318–25. doi: 10.1038/ni1305
133. Stewart MK, Cummings LA, Johnson ML, Berezow AB, Cookson BT. Regulation of phenotypic heterogeneity permits salmonella evasion of the host caspase-1 inflammatory response. Proc Natl Acad Sci USA (2011) 108(51):20742–7. doi: 10.1073/pnas.1108963108
134. Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, et al. Caspase-1-Induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol (2010) 11(12):1136–42. doi: 10.1038/ni.1960
135. Aachoui Y, Leaf IA, Hagar JA, Fontana MF, Campos CG, Zak DE, et al. Caspase-11 protects against bacteria that escape the vacuole. Science (2013) 339(6122):975–8. doi: 10.1126/science.1230751
136. Aachoui Y, Kajiwara Y, Leaf IA, Mao D, Ting JPY, Coers J, et al. Canonical inflammasomes drive ifn-Γ to prime caspase-11 in defense against a cytosol-invasive bacterium. Cell Host Microbe (2015) 18(3):320–32. doi: 10.1016/j.chom.2015.07.016
137. Mariathasan S, Weiss DS, Dixit VM, Monack DM. Innate immunity against francisella tularensis is dependent on the Asc/Caspase-1 axis. J Exp Med (2005) 202(8):1043–9. doi: 10.1084/jem.20050977
138. Henry T, Brotcke A, Weiss DS, Thompson LJ, Monack DM. Type I interferon signaling is required for activation of the inflammasome during francisella infection. J Exp Med (2007) 204(5):987–94. doi: 10.1084/jem.20062665
139. Gavrilin MA, Bouakl IJ, Knatz NL, Duncan MD, Hall MW, Gunn JS, et al. Internalization and phagosome escape required for francisella to induce human monocyte il-1beta processing and release. Proc Natl Acad Sci USA (2006) 103(1):141–6. doi: 10.1073/pnas.0504271103
140. Ben Nasr A, Haithcoat J, Masterson JE, Gunn JS, Eaves-Pyles T, Klimpel GR. Critical role for serum opsonins and complement receptors Cr3 (Cd11b/Cd18) and Cr4 (Cd11c/Cd18) in phagocytosis of francisella tularensis by human dendritic cells (Dc): Uptake of francisella leads to activation of immature dc and intracellular survival of the bacteria. J Leukoc Biol (2006) 80(4):774–86. doi: 10.1189/jlb.1205755
141. Li H, Nookala S, Bina XR, Bina JE, Re F. Innate immune response to francisella tularensis is mediated by Tlr2 and caspase-1 activation. J Leukoc Biol (2006) 80(4):766–73. doi: 10.1189/jlb.0406294
142. Maltez VI, Tubbs AL, Cook KD, Aachoui Y, Falcone EL, Holland SM, et al. Inflammasomes coordinate pyroptosis and natural killer cell cytotoxicity to clear infection by a ubiquitous environmental bacterium. Immunity (2015) 43(5):987–97. doi: 10.1016/j.immuni.2015.10.010
143. Cervantes J, Nagata T, Uchijima M, Shibata K, Koide Y. Intracytosolic listeria monocytogenes induces cell death through caspase-1 activation in murine macrophages. Cell Microbiol (2008) 10(1):41–52. doi: 10.1111/j.1462-5822.2007.01012.x
144. Pedra JHF, Sutterwala FS, Sukumaran B, Ogura Y, Qian F, Montgomery RR, et al. Asc/Pycard and caspase-1 regulate the il-18/Ifn-Gamma axis during anaplasma phagocytophilum infection. J Immunol (2007) 179(7):4783–91. doi: 10.4049/jimmunol.179.7.4783
145. Mencacci A, Bacci A, Cenci E, Montagnoli C, Fiorucci S, Casagrande A, et al. Interleukin 18 restores defective Th1 immunity to candida albicans in caspase 1-deficient mice. Infect Immun (2000) 68(9):5126–31. doi: 10.1128/IAI.68.9.5126-5131.2000
146. Raupach B, Peuschel S-K, Monack DM, Zychlinsky A. Caspase-1-Mediated activation of interleukin-1beta (Il-1beta) and il-18 contributes to innate immune defenses against salmonella enterica serovar typhimurium infection. Infect Immun (2006) 74(8):4922–6. doi: 10.1128/IAI.00417-06
147. Iqbal M, Philbin VJ, Withanage GSK, Wigley P, Beal RK, Goodchild MJ, et al. Identification and functional characterization of chicken toll-like receptor 5 reveals a fundamental role in the biology of infection with salmonella enterica serovar typhimurium. Infect Immun (2005) 73(4):2344–50. doi: 10.1128/IAI.73.4.2344-2350.2005
148. Schmitt CK, Ikeda JS, Darnell SC, Watson PR, Bispham J, Wallis TS, et al. Absence of all components of the flagellar export and synthesis machinery differentially alters virulence of salmonella enterica serovar typhimurium in models of typhoid fever, survival in macrophages, tissue culture invasiveness, and calf enterocolitis. Infect Immun (2001) 69(9):5619–25. doi: 10.1128/IAI.69.9.5619-5625.2001
149. Stecher B, Hapfelmeier S, Müller C, Kremer M, Stallmach T, Hardt W-D. Flagella and chemotaxis are required for efficient induction of salmonella enterica serovar typhimurium colitis in streptomycin-pretreated mice. Infect Immun (2004) 72(7):4138–50. doi: 10.1128/IAI.72.7.4138-4150.2004
150. Vijay-Kumar M, Wu H, Jones R, Grant G, Babbin B, King TP, et al. Flagellin suppresses epithelial apoptosis and limits disease during enteric infection. Am J Pathol (2006) 169(5):1686–700. doi: 10.2353/ajpath.2006.060345
151. Henry T, Monack DM. Activation of the inflammasome upon francisella tularensis infection: Interplay of innate immune pathways and virulence factors. Cell Microbiol (2007) 9(11):2543–51. doi: 10.1111/j.1462-5822.2007.01022.x
152. Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the Nalp3 inflammasome. Nature (2006) 440(7081):237–41. doi: 10.1038/nature04516
153. Nakanishi K, Yoshimoto T, Tsutsui H, Okamura H. Interleukin-18 regulates both Th1 and Th2 responses. Annu Rev Immunol (2001) 19:423–74. doi: 10.1146/annurev.immunol.19.1.423
154. Eisenbarth SC, Colegio OR, O’Connor W, Sutterwala FS, Flavell RA. Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature (2008) 453(7198):1122–6. doi: 10.1038/nature06939
155. Kool M, Pétrilli V, De Smedt T, Rolaz A, Hammad H, van Nimwegen M, et al. Cutting edge: Alum adjuvant stimulates inflammatory dendritic cells through activation of the Nalp3 inflammasome. J Immunol (2008) 181(6):3755–9. doi: 10.4049/jimmunol.181.6.3755
156. Franchi L, Núñez G. The Nlrp3 inflammasome is critical for aluminium hydroxide-mediated il-1beta secretion but dispensable for adjuvant activity. Eur J Immunol (2008) 38(8):2085–9. doi: 10.1002/eji.200838549
157. Li H, Willingham SB, Ting JP-Y, Re F. Cutting edge: Inflammasome activation by alum and alum’s adjuvant effect are mediated by Nlrp3. J Immunol (2008) 181(1):17–21. doi: 10.4049/jimmunol.181.1.17
158. Cohen TS, Prince AS. Activation of inflammasome signaling mediates pathology of acute p. aeruginosa pneumonia. J Clin Invest (2013) 123(4):1630–7. doi: 10.1172/JCI66142
159. Faure E, Mear J-B, Faure K, Normand S, Couturier-Maillard A, Grandjean T, et al. Pseudomonas aeruginosa type-3 secretion system dampens host defense by exploiting the Nlrc4-coupled inflammasome. Am J Respir Crit Care Med (2014) 189(7):799–811. doi: 10.1164/rccm.201307-1358OC
160. Theisen E, Sauer J-D. Listeria monocytogenes-induced cell death inhibits the generation of cell-mediated immunity. Infect Immun (2017) 85(1):e00733-16. doi: 10.1128/IAI.00733-16
161. Schielke GP, Yang GY, Shivers BD, Betz AL. Reduced ischemic brain injury in interleukin-1 beta converting enzyme-deficient mice. J Cereb Blood Flow Metab (1998) 18(2):180–5. doi: 10.1097/00004647-199802000-00009
162. Siegmund B, Lehr H-A, Fantuzzi G, Dinarello CA. Il-1β-Converting enzyme (Caspase-1) in intestinal inflammation. Proc Natl Acad Sci U.S.A. (2001) 98(23):13249–54. doi: 10.1073/pnas.231473998
163. Wang W, Faubel S, Ljubanovic D, Mitra A, Falk SA, Kim J, et al. Endotoxemic acute renal failure is attenuated in caspase-1-Deficient mice. Am J Physiol Renal Physiol (2005) 288(5):F997–1004. doi: 10.1152/ajprenal.00130.2004
164. Li P, Allen H, Banerjee S, Franklin S, Herzog L, Johnston C, et al. Mice deficient in il-1β-Converting enzyme are defective in production of mature il-1β and resistant to endotoxic shock. Cell (1995) 80(3):401–11. doi: 10.1016/0092-8674(95)90490-5
165. Ona VO, Li M, Vonsattel JP, Andrews LJ, Khan SQ, Chung WM, et al. Inhibition of caspase-1 slows disease progression in a mouse model of huntington’s disease. Nature (1999) 399(6733):263–7. doi: 10.1038/20446
166. Faubel S, Lewis EC, Reznikov L, Ljubanovic D, Hoke TS, Somerset H, et al. Cisplatin-induced acute renal failure is associated with an increase in the cytokines interleukin (Il)-1beta, il-18, il-6, and neutrophil infiltration in the kidney. J Pharmacol Exp Ther (2007) 322(1):8–15. doi: 10.1124/jpet.107.119792
167. Sarkar A, Hall MW, Exline M, Hart J, Knatz N, Gatson NT, et al. Caspase-1 regulates escherichia coli sepsis and splenic b cell apoptosis independently of interleukin-1beta and interleukin-18. Am J Respir Crit Care Med (2006) 174(9):1003–10. doi: 10.1164/rccm.200604-546OC
168. Schmid-Burgk JL, Gaidt MM, Schmidt T, Ebert TS, Bartok E, Hornung V. Caspase-4 mediates non-canonical activation of the Nlrp3 inflammasome in human myeloid cells. Eur J Immunol (2015) 45(10):2911–7. doi: 10.1002/eji.201545523
169. Pallett MA, Crepin VF, Serafini N, Habibzay M, Kotik O, Sanchez-Garrido J, et al. Bacterial virulence factor inhibits caspase-4/11 activation in intestinal epithelial cells. Mucosal Immunol (2017) 10(3):602–12. doi: 10.1038/mi.2016.77
170. Casson CN, Yu J, Reyes VM, Taschuk FO, Yadav A, Copenhaver AM, et al. Human caspase-4 mediates noncanonical inflammasome activation against gram-negative bacterial pathogens. Proc Natl Acad Sci USA (2015) 112(21):6688–93. doi: 10.1073/pnas.1421699112
171. Kajiwara Y, Schiff T, Voloudakis G, Gama Sosa MA, Elder G, Bozdagi O, et al. A critical role for human caspase-4 in endotoxin sensitivity. J Immunol (2014) 193(1):335–43. doi: 10.4049/jimmunol.1303424
172. Cummings LA, Barrett SLR, Wilkerson WD, Fellnerova I, Cookson BT. Flic-specific Cd4+ T cell responses are restricted by bacterial regulation of antigen expression. J Immunol (2005) 174(12):7929–38. doi: 10.4049/jimmunol.174.12.7929
173. Sutterwala FS, Mijares LA, Li L, Ogura Y, Kazmierczak BI, Flavell RA. Immune recognition of pseudomonas aeruginosa mediated by the Ipaf/Nlrc4 inflammasome. J Exp Med (2007) 204(13):3235–45. doi: 10.1084/jem.20071239
174. Miao EA, Ernst RK, Dors M, Mao DP, Aderem A. Pseudomonas aeruginosa activates caspase 1 through ipaf. Proc Natl Acad Sci USA (2008) 105(7):2562–7. doi: 10.1073/pnas.0712183105
175. Johnston JB, Barrett JW, Nazarian SH, Goodwin M, Ricciuto D, Ricuttio D, et al. A poxvirus-encoded pyrin domain protein interacts with asc-1 to inhibit host inflammatory and apoptotic responses to infection. Immunity (2005) 23(6):587–98. doi: 10.1016/j.immuni.2005.10.003
176. Stasakova J, Ferko B, Kittel C, Sereinig S, Romanova J, Katinger H, et al. Influenza a mutant viruses with altered Ns1 protein function provoke caspase-1 activation in primary human macrophages, resulting in fast apoptosis and release of high levels of interleukins 1beta and 18. J Gen Virol (2005) 86(Pt 1):185–95. doi: 10.1099/vir.0.80422-0
177. Master SS, Rampini SK, Davis AS, Keller C, Ehlers S, Springer B, et al. Mycobacterium tuberculosis prevents inflammasome activation. Cell Host Microbe (2008) 3(4):224–32. doi: 10.1016/j.chom.2008.03.003
178. Shin H, Cornelis GR. Type iii secretion translocation pores of yersinia enterocolitica trigger maturation and release of pro-inflammatory il-1beta. Cell Microbiol (2007) 9(12):2893–902. doi: 10.1111/j.1462-5822.2007.01004.x
179. Bergsbaken T, Cookson BT. Macrophage activation redirects yersinia-infected host cell death from apoptosis to caspase-1-Dependent pyroptosis. PloS Pathog (2007) 3(11):e161. doi: 10.1371/journal.ppat.0030161
180. Schotte P, Denecker G, Van Den Broeke A, Vandenabeele P, Cornelis GR, Beyaert R. Targeting Rac1 by the yersinia effector protein yope inhibits caspase-1-Mediated maturation and release of interleukin-1beta. J Biol Chem (2004) 279(24):25134–42. doi: 10.1074/jbc.M401245200
181. Kelk P, Johansson A, Claesson R, Hänström L, Kalfas S. Caspase 1 involvement in human monocyte lysis induced by actinobacillus actinomycetemcomitans leukotoxin. Infect Immun (2003) 71(8):4448–55. doi: 10.1128/IAI.71.8.4448-4455.2003
182. Pan J, Han L, Guo J, Wang X, Liu D, Tian J, et al. Aim2 accelerates the atherosclerotic plaque progressions in apoe-/- mice. Biochem Biophys Res Commun (2018) 498(3):487–94. doi: 10.1016/j.bbrc.2018.03.005
183. Grebe A, Hoss F, Latz E. Nlrp3 inflammasome and the il-1 pathway in atherosclerosis. Circ Res (2018) 122(12):1722–40. doi: 10.1161/CIRCRESAHA.118.311362
184. van der Heijden T, Kritikou E, Venema W, van Duijn J, van Santbrink PJ, Slütter B, et al. Nlrp3 inflammasome inhibition by Mcc950 reduces atherosclerotic lesion development in apolipoprotein e-deficient mice-brief report. Arterioscler Thromb Vasc Biol (2017) 37(8):1457–61. doi: 10.1161/ATVBAHA.117.309575
185. van Hout GPJ, Bosch L, Ellenbroek GHJM, de Haan JJ, van Solinge WW, Cooper MA, et al. The selective Nlrp3-inflammasome inhibitor Mcc950 reduces infarct size and preserves cardiac function in a pig model of myocardial infarction. Eur Heart J (2017) 38(11):828–36. doi: 10.1093/eurheartj/ehw247
186. Sano S, Oshima K, Wang Y, MacLauchlan S, Katanasaka Y, Sano M, et al. Tet2-mediated clonal hematopoiesis accelerates heart failure through a mechanism involving the il-1β/Nlrp3 inflammasome. J Am Coll Cardiol (2018) 71(8):875–86. doi: 10.1016/j.jacc.2017.12.037
187. Swirski FK. Inflammation and cvd in 2017: From clonal haematopoiesis to the cantos trial. Nat Rev Cardiol (2018) 15(2):79–80. doi: 10.1038/nrcardio.2017.208
188. Libby P. Interleukin-1 beta as a target for atherosclerosis therapy: Biological basis of cantos and beyond. J Am Coll Cardiol (2017) 70(18):2278–89. doi: 10.1016/j.jacc.2017.09.028
189. Abbate A, Toldo S, Marchetti C, Kron J, Van Tassell BW, Dinarello CA. Interleukin-1 and the inflammasome as therapeutic targets in cardiovascular disease. Circ Res (2020) 126(9):1260–80. doi: 10.1161/CIRCRESAHA.120.315937
190. Yao C, Veleva T, Scott L, Cao S, Li L, Chen G, et al. Enhanced cardiomyocyte Nlrp3 inflammasome signaling promotes atrial fibrillation. Circulation (2018) 138(20):2227–42. doi: 10.1161/CIRCULATIONAHA.118.035202
191. Suetomi T, Willeford A, Brand CS, Cho Y, Ross RS, Miyamoto S, et al. Inflammation and Nlrp3 inflammasome activation initiated in response to pressure overload by Ca/Calmodulin-dependent protein kinase ii Δ signaling in cardiomyocytes are essential for adverse cardiac remodeling. Circulation (2018) 138(22):2530–44. doi: 10.1161/CIRCULATIONAHA.118.034621
192. Zhou X, Wu Y, Ye L, Wang Y, Zhang K, Wang L, et al. Aspirin alleviates endothelial gap junction dysfunction through inhibition of Nlrp3 inflammasome activation in lps-induced vascular injury. Acta Pharm Sin B (2019) 9(4):711–23. doi: 10.1016/j.apsb.2019.02.008
193. Kim S-J, Lee S-M. Nlrp3 inflammasome activation in d-galactosamine and lipopolysaccharide-induced acute liver failure: Role of heme oxygenase-1. Free Radic Biol Med (2013) 65:997–1004. doi: 10.1016/j.freeradbiomed.2013.08.178
194. Ganz M, Csak T, Nath B, Szabo G. Lipopolysaccharide induces and activates the Nalp3 inflammasome in the liver. World J Gastroenterol (2011) 17(43):4772–8. doi: 10.3748/wjg.v17.i43.4772
195. Wei Q, Mu K, Li T, Zhang Y, Yang Z, Jia X, et al. Deregulation of the Nlrp3 inflammasome in hepatic parenchymal cells during liver cancer progression. Lab Invest (2014) 94(1):52–62. doi: 10.1038/labinvest.2013.126
196. Wei Q, Guo P, Mu K, Zhang Y, Zhao W, Huai W, et al. Estrogen suppresses hepatocellular carcinoma cells through erβ-mediated upregulation of the Nlrp3 inflammasome. Lab Invest (2015) 95(7):804–16. doi: 10.1038/labinvest.2015.63
197. Wei Q, Zhu R, Zhu J, Zhao R, Li M. E2-induced activation of the Nlrp3 inflammasome triggers pyroptosis and inhibits autophagy in hcc cells. Oncol Res (2019) 27(7):827–34. doi: 10.3727/096504018X15462920753012
198. Wang C-J, Tang L, Shen D-W, Wang C, Yuan Q-Y, Gao W, et al. The expression and regulation of Dfna5 in human hepatocellular carcinoma Dfna5 in hepatocellular carcinoma. Mol Biol Rep (2013) 40(12):6525–31. doi: 10.1007/s11033-013-2581-8
199. Musso G, Cassader M, Gambino R. Non-alcoholic steatohepatitis: Emerging molecular targets and therapeutic strategies. Nat Rev Drug Discovery (2016) 15(4):249–74. doi: 10.1038/nrd.2015.3
200. Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, et al. Inflammasome-mediated dysbiosis regulates progression of nafld and obesity. Nature (2012) 482(7384):179–85. doi: 10.1038/nature10809
201. Xu B, Jiang M, Chu Y, Wang W, Chen D, Li X, et al. Gasdermin d plays a key role as a pyroptosis executor of non-alcoholic steatohepatitis in humans and mice. J Hepatol (2018) 68(4):773–82. doi: 10.1016/j.jhep.2017.11.040
202. Yang C, Sun P, Deng M, Loughran P, Li W, Yi Z, et al. Gasdermin d protects against noninfectious liver injury by regulating apoptosis and necroptosis. Cell Death Dis (2019) 10(7):481. doi: 10.1038/s41419-019-1719-6
204. Gao J, Qiu X, Xi G, Liu H, Zhang F, Lv T, et al. Downregulation of gsdmd attenuates tumor proliferation Via the intrinsic mitochondrial apoptotic pathway and inhibition of Egfr/Akt signaling and predicts a good prognosis in Non−Small cell lung cancer. Oncol Rep (2018) 40(4):1971–84. doi: 10.3892/or.2018.6634
205. Teng J-F, Mei Q-B, Zhou X-G, Tang Y, Xiong R, Qiu W-Q, et al. Polyphyllin vi induces caspase-1-Mediated pyroptosis Via the induction of Ros/Nf-Kb/Nlrp3/Gsdmd signal axis in non-small cell lung cancer. Cancers (Basel) (2020) 12(1):193. doi: 10.3390/cancers12010193
206. Zou J, Yang Y, Yang Y, Liu X. Polydatin suppresses proliferation and metastasis of non-small cell lung cancer cells by inhibiting Nlrp3 inflammasome activation Via nf-Kb pathway. BioMed Pharmacother (2018) 108:130–6. doi: 10.1016/j.biopha.2018.09.051
207. Zhang M, Jin C, Yang Y, Wang K, Zhou Y, Zhou Y, et al. Aim2 promotes non-Small-Cell lung cancer cell growth through inflammasome-dependent pathway. J Cell Physiol (2019) 234(11):20161–73. doi: 10.1002/jcp.28617
208. Liu A, Liu L, Lu H. Lncrna xist facilitates proliferation and epithelial-mesenchymal transition of colorectal cancer cells through targeting mir-486-5p and promoting neuropilin-2. J Cell Physiol (2019) 234(8):13747–61. doi: 10.1002/jcp.28054
209. Chen Z, Hu X, Wu Y, Cong L, He X, Lu J, et al. Long non-coding rna xist promotes the development of esophageal cancer by sponging mir-494 to regulate Cdk6 expression. BioMed Pharmacother (2019) 109:2228–36. doi: 10.1016/j.biopha.2018.11.049
210. Zhang J, Li W-Y, Yang Y, Yan L-Z, Zhang S-Y, He J, et al. Lncrna xist facilitates cell growth, migration and invasion Via modulating H3 histone methylation of Dkk1 in neuroblastoma. Cell Cycle (2019) 18(16):1882–92. doi: 10.1080/15384101.2019.1632134
211. Sun W, Zu Y, Fu X, Deng Y. Knockdown of lncrna-xist enhances the chemosensitivity of nsclc cells Via suppression of autophagy. Oncol Rep (2017) 38(6):3347–54. doi: 10.3892/or.2017.6056
212. Tang H, Zhu J, Du W, Liu S, Zeng Y, Ding Z, et al. Cpne1 is a target of mir-335-5p and plays an important role in the pathogenesis of non-small cell lung cancer. J Exp Clin Cancer Res (2018) 37(1):131. doi: 10.1186/s13046-018-0811-6
213. Wang Y, Shi P, Chen Q, Huang Z, Zou D, Zhang J, et al. Mitochondrial ros promote macrophage pyroptosis by inducing gsdmd oxidation. J Mol Cell Biol (2019) 11(12):1069–82. doi: 10.1093/jmcb/mjz020
214. Zhang C-C, Li C-G, Wang Y-F, Xu L-H, He X-H, Zeng Q-Z, et al. Chemotherapeutic paclitaxel and cisplatin differentially induce pyroptosis in A549 lung cancer cells via caspase-3/Gsdme activation. Apoptosis (2019) 24(3-4):312–25. doi: 10.1007/s10495-019-01515-1
215. Han Y, Xu X, Tang C, Gao P, Chen X, Xiong X, et al. Reactive oxygen species promote tubular injury in diabetic nephropathy: The role of the mitochondrial ros-Txnip-Nlrp3 biological axis. Redox Biol (2018) 16:32–46. doi: 10.1016/j.redox.2018.02.013
216. Wang S, Li Y, Fan J, Zhang X, Luan J, Bian Q, et al. Interleukin-22 ameliorated renal injury and fibrosis in diabetic nephropathy through inhibition of Nlrp3 inflammasome activation. Cell Death Dis (2017) 8(7):e2937. doi: 10.1038/cddis.2017.292
217. Wang M-Z, Wang J, Cao D-W, Tu Y, Liu B-H, Yuan C-C, et al. Fucoidan alleviates renal fibrosis in diabetic kidney disease inhibition of Nlrp3 inflammasome-mediated podocyte pyroptosis. Front Pharmacol (2022) 13:790937. doi: 10.3389/fphar.2022.790937
218. Wang Y, Zhu X, Yuan S, Wen S, Liu X, Wang C, et al. Tlr4/Nf-Kb signaling induces gsdmd-related pyroptosis in tubular cells in diabetic kidney disease. Front Endocrinol (Lausanne) (2019) 10:603. doi: 10.3389/fendo.2019.00603
219. Jialal I, Major AM, Devaraj S. Global toll-like receptor 4 knockout results in decreased renal inflammation, fibrosis and podocytopathy. J Diabetes Complications (2014) 28(6):755–61. doi: 10.1016/j.jdiacomp.2014.07.003
220. Li F, Chen Y, Li Y, Huang M, Zhao W. Geniposide alleviates diabetic nephropathy of mice through Ampk/Sirt1/Nf-Kb pathway. Eur J Pharmacol (2020) 886:173449. doi: 10.1016/j.ejphar.2020.173449
221. Chen J, Yang Y, Lv Z, Shu A, Du Q, Wang W, et al. Study on the inhibitive effect of catalpol on diabetic nephropathy. Life Sci (2020) 257:118120. doi: 10.1016/j.lfs.2020.118120
222. Li X, Zeng L, Cao C, Lu C, Lian W, Han J, et al. Long noncoding rna Malat1 regulates renal tubular epithelial pyroptosis by modulated mir-23c targeting of Elavl1 in diabetic nephropathy. Exp Cell Res (2017) 350(2):327–35. doi: 10.1016/j.yexcr.2016.12.006
223. Zhu B, Cheng X, Jiang Y, Cheng M, Chen L, Bao J, et al. Silencing of Kcnq1ot1 decreases oxidative stress and pyroptosis of renal tubular epithelial cells. Diabetes Metab Syndr Obes (2020) 13:365–75. doi: 10.2147/DMSO.S225791
224. Xie C, Wu W, Tang A, Luo N, Tan Y. Lncrna Gas5/Mir-452-5p reduces oxidative stress and pyroptosis of high-Glucose-Stimulated renal tubular cells. Diabetes Metab Syndr Obes (2019) 12:2609–17. doi: 10.2147/DMSO.S228654
225. Pang Q, Wang P, Pan Y, Dong X, Zhou T, Song X, et al. Irisin protects against vascular calcification by activating autophagy and inhibiting Nlrp3-mediated vascular smooth muscle cell pyroptosis in chronic kidney disease. Cell Death Dis (2022) 13(3):283. doi: 10.1038/s41419-022-04735-7
226. Liu H, Chen Z, Weng X, Chen H, Du Y, Diao C, et al. Enhancer of zeste homolog 2 modulates oxidative stress-mediated pyroptosis in vitro and in a mouse kidney ischemia-reperfusion injury model. FASEB J (2020) 34(1):835–52. doi: 10.1096/fj.201901816R
227. Heneka MT, Golenbock DT, Latz E. Innate immunity in alzheimer’s disease. Nat Immunol (2015) 16(3):229–36. doi: 10.1038/ni.3102
228. Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, et al. The Nalp3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol (2008) 9(8):857–65. doi: 10.1038/ni.1636
229. Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, et al. Nlrp3 is activated in alzheimer’s disease and contributes to pathology in App/Ps1 mice. Nature (2013) 493(7434):674–8. doi: 10.1038/nature11729
230. Zhang X, Wang R, Hu D, Sun X, Fujioka H, Lundberg K, et al. Oligodendroglial glycolytic stress triggers inflammasome activation and neuropathology in alzheimer’s disease. Sci Adv (2020) 6(49):eabb8680. doi: 10.1126/sciadv.abb8680
231. Lonnemann N, Hosseini S, Marchetti C, Skouras DB, Stefanoni D, D’Alessandro A, et al. The Nlrp3 inflammasome inhibitor Olt1177 rescues cognitive impairment in a mouse model of alzheimer’s disease. Proc Natl Acad Sci U.S.A. (2020) 117(50):32145–54. doi: 10.1073/pnas.2009680117
232. Zhang Y, Dong Z, Song W. Nlrp3 inflammasome as a novel therapeutic target for alzheimer’s disease. Signal Transduct Target Ther (2020) 5(1):37. doi: 10.1038/s41392-020-0145-7
233. Wu A-G, Zhou X-G, Qiao G, Yu L, Tang Y, Yan L, et al. Targeting microglial autophagic degradation in Nlrp3 inflammasome-mediated neurodegenerative diseases. Ageing Res Rev (2021) 65:101202. doi: 10.1016/j.arr.2020.101202
234. Poewe W, Seppi K, Tanner CM, Halliday GM, Brundin P, Volkmann J, et al. Parkinson Disease. Nat Rev Dis Primers (2017) 3:17013. doi: 10.1038/nrdp.2017.13
235. Béraud D, Maguire-Zeiss KA. Misfolded A-synuclein and toll-like receptors: Therapeutic targets for parkinson’s disease. Parkinsonism Relat Disord (2012) 18 Suppl 1:S17–20. doi: 10.1016/S1353-8020(11)70008-6
236. Ferrari CC, Pott Godoy MC, Tarelli R, Chertoff M, Depino AM, Pitossi FJ. Progressive neurodegeneration and motor disabilities induced by chronic expression of il-1beta in the substantia nigra. Neurobiol Dis (2006) 24(1):183–93. doi: 10.1016/j.nbd.2006.06.013
237. Codolo G, Plotegher N, Pozzobon T, Brucale M, Tessari I, Bubacco L, et al. Triggering of inflammasome by aggregated A-synuclein, an inflammatory response in synucleinopathies. PloS One (2013) 8(1):e55375. doi: 10.1371/journal.pone.0055375
238. Yan Y, Jiang W, Liu L, Wang X, Ding C, Tian Z, et al. Dopamine controls systemic inflammation through inhibition of Nlrp3 inflammasome. Cell (2015) 160(1-2):62–73. doi: 10.1016/j.cell.2014.11.047
239. Tan E-K, Chao Y-X, West A, Chan L-L, Poewe W, Jankovic J. Parkinson Disease and the immune system - associations, mechanisms and therapeutics. Nat Rev Neurol (2020) 16(6):303–18. doi: 10.1038/s41582-020-0344-4
Keywords: pyroptosis, caspase-1, gasdermins, inflammation, disease
Citation: Pan Y, Cai W, Huang J, Cheng A, Wang M, Yin Z and Jia R (2022) Pyroptosis in development, inflammation and disease. Front. Immunol. 13:991044. doi: 10.3389/fimmu.2022.991044
Received: 11 July 2022; Accepted: 30 August 2022;
Published: 16 September 2022.
Edited by:
Kuo-Feng Hua, National Ilan University, TaiwanReviewed by:
Jingwen Zhao, Tianjin Medical University, ChinaCopyright © 2022 Pan, Cai, Huang, Cheng, Wang, Yin and Jia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anchun Cheng, Y2hlbmdhbmNodW5AdmlwLjE2My5jb20=; Renyong Jia, amlhcnlAc2ljYXUuZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.