 Dengpan Song
Dengpan Song Chi-Tai Yeh2*
Chi-Tai Yeh2* Jian Wang
Jian Wang Fuyou Guo
Fuyou Guo
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 05 September 2022
Sec. Inflammation
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.989503
Intracerebral hemorrhage (ICH) is a highly harmful neurological disorder with high rates of mortality, disability, and recurrence. However, effective therapies are not currently available. Secondary immune injury and cell death are the leading causes of brain injury and a poor prognosis. Pyroptosis is a recently discovered form of programmed cell death that differs from apoptosis and necrosis and is mediated by gasdermin proteins. Pyroptosis is caused by multiple pathways that eventually form pores in the cell membrane, facilitating the release of inflammatory substances and causing the cell to rupture and die. Pyroptosis occurs in neurons, glial cells, and endothelial cells after ICH. Furthermore, pyroptosis causes cell death and releases inflammatory factors such as interleukin (IL)-1β and IL-18, leading to a secondary immune-inflammatory response and further brain damage. The NOD-like receptor protein 3 (NLRP3)/caspase-1/gasdermin D (GSDMD) pathway plays the most critical role in pyroptosis after ICH. Pyroptosis can be inhibited by directly targeting NLRP3 or its upstream molecules, or directly interfering with caspase-1 expression and GSDMD formation, thus significantly improving the prognosis of ICH. The present review discusses key pathological pathways and regulatory mechanisms of pyroptosis after ICH and suggests possible intervention strategies to mitigate pyroptosis and brain dysfunction after ICH.
Intracerebral hemorrhage (ICH) is a common and devastating cerebrovascular disease with high morbidity and mortality rates, accounting for approximately 15% of all strokes and 50% of stroke-related mortality, and approximately 2.8 million deaths occur worldwide each year (1, 2). Following initial insult, secondary brain injury in patients with ICH contributes to poor outcomes, and effective therapy is still lacking (3, 4). Neuroinflammation plays a critical role in the pathogenesis of secondary brain injury (4, 5).
Pyroptosis is a recently discovered process that can lead to an inflammatory response. In contrast to common cell death pathways, such as necrosis, apoptosis, and autophagy, pyroptosis is characterized by unique expression patterns and mechanisms. Morphologically, pyroptosis exhibits features of both necrosis and apoptosis. Morphological changes include rupture of the necrosis-like cell membrane, pore formation, cell swelling, release of proinflammatory intracellular contents, nuclear condensation, and deoxyribonucleic acid (DNA) fragmentation. In contrast to apoptosis, pyroptosis is characterized by intact mitochondria and ‘balloon-shaped’ vesicle formation but not cytochrome c release (6, 7). Pore formation is a key factor in pyroptosis, and these pores allow the release of inflammatory cytokines such as interleukin (IL)-1β and IL-18. These inflammatory factors exacerbate the local inflammatory response, leading to neuronal death, destruction of the blood-brain barrier (BBB), and other types of secondary inflammatory damage in ICH (8).
Molecularly, pyroptosis is characterized by gasdermin-mediated cell death (9). The gasdermin protein family is made up of a pore-forming aminoterminal domain (N-terminal domain), which is automatically suppressed by the carboxyterminal domain (C-terminal domain) (9). Recent research suggests that gasdermin D (GSDMD) is one of the most important members. Activated caspase-1/4/5/11 effectively cleaves GSDMD at the aspartic acid site in the linker loop. Cleavage generates an N-terminal cleavage product (GSDMD-N) that binds to the plasma membrane and forms pores that trigger pyroptosis and release of the inflammatory cytokines IL-1β and IL-18 (10–13).
Normally, gasdermin is activated by two caspase-dependent pathways, including the canonical caspase-1 pathway and the noncanonical caspase-4/5/11 pathway (7). Caspase-1/4/5/11 are usually activated by inflammasomes or lipopolysaccharide (LPS). Inflammasomes are polyprotein complexes that are assembled from particular pattern recognition receptors (PRR) that recognize damage-associated molecular patterns (DAMP) and pathogen-associated molecular patterns (PAMP) in aberrant cellular settings. Toll-like receptors (TLR) and C-type lectin receptors (CLR) are transmembrane receptors, while retinoic acid-inducible gene (RIG) I-like receptors (RLR) and NOD-like receptors are cytosolic receptors (NLR) (14–16). Currently, the most widely studied inflammasome is the NOD-like receptor protein 3 (NLRP3) inflammasome, which has been shown to mediate the appearance of pyroptosis in a variety of diseases, including dilated cardiomyopathy, ischemic stroke, myocardial ischemia, ICH, inflammation and neoplastic diseases (17–24).
The classical pyroptosis pathway is mediated by caspase-1 (Figure 1). Pyroptosis signals trigger different cytoplasmic sensor proteins and promote the activation and release of different inflammasomes, as represented by NLRP3. Inflammasomes recruit pro-caspase-1 through an apoptosis-associated speck-like protein that contains a caspase recruitment domain (ASC), and the three factors bind and oligomerize to form inflammasomes (25). The activated inflammasome cleaves inactive pro-caspase-1 into mature caspase-1, which then converts pro-IL-1β and pro-IL-18 to mature IL-1β and IL-18, respectively (26). GSDMD is also cleaved by caspase-1, which separates its N-terminal pore-forming domain from the C-terminal repressor domain. The N-terminal fragment forms extensive gas-soluble pores on the cell membrane by dissolving phospholipids or cardiolipins, subsequently promoting the release of IL-1β and IL-18 and recruiting more extracellular inflammatory cytokines to cause inflammation (27, 28).
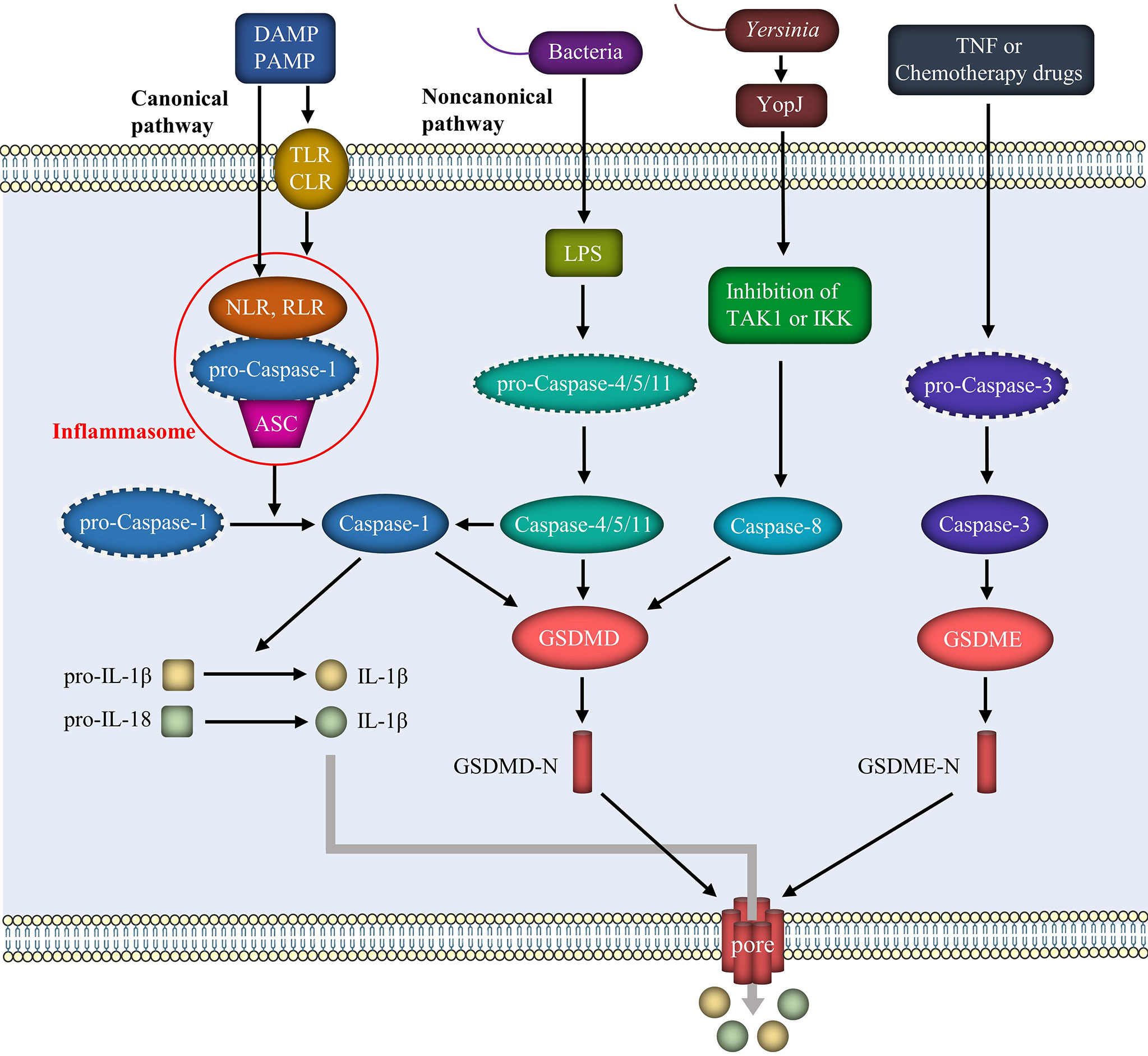
Figure 1 Mechanism of pyroptosis. The pathogenesis of pyroptosis includes mainly the canonical pathway mediated by the inflammasome and caspase-1, the noncanonical pathway mediated by LPS and caspase-4/5/11, and some other pathways involving caspase-8 and caspase-3. These pathways result in the cleavage of GSDMD or GSDME and the separation of the N-terminal fragments. GSDMD-N or GSDME-N forms pores in the cell membrane, leading to the release of inflammatory factors such as IL-1β and IL-18.
LPS released from Gram-negative bacteria, such as E. coli, binds directly to and activates the host immune receptors caspase-4/5 (in humans) and caspase-11 (in mice) through endocytosis. On the one hand, activated caspase-4/5/11 directly cleaves GSDMD, exerting the same effect as caspase-1 and leading to perforation of the cell membrane. On the other hand, in the presence of NLRP3 and the adapter protein ASC, activated caspase-4/5/11 interacts with caspase-1 to activate it, thus activating pyroptosis through the classic pathway. Furthermore, GSDMD-N oligomerization produces pores in the membrane after cleavage of GSDMD, as in the classical route, mediating the release of IL-1β and IL-18 into the extracellular space and causing an inflammatory cascade (29).
Tumor necrosis factor (TNF) or chemotherapy drugs activate caspase-3 to cleave gasdermin E (GSDME). The N-terminal fragment of GSDME perforates the cell membrane and causes pyroptosis (30). Furthermore, GSDMD-induced pyroptosis is regulated by caspase-8. Inhibition of TGF-β activated kinase-1 (TAK1) or inhibitor of κB kinase (IKK) by the Yersinia effector protein YopJ results in RIPK1- and caspase-8-dependent cleavage of GSDMD, resulting in pyroptosis (31). Pyroptosis is characterized by the release of IL-1β and IL-18. Caspase-8-induced pyroptosis occurs simultaneously with NLRP3 activation and IL-1β secretion (32). Furthermore, as a typical activator, ATP activates the NLRP3 inflammasome in macrophages, leading to caspase-1/GDSDMD-mediated pyroptosis (33). In addition to caspase-mediated pyroptosis, neutrophil serine endopeptidase activates GSDMD through the cleavage of L-cysteine and activates neutrophils via a mechanism independent of caspase-1/11 (34).
Caspase-1 activation is the key factor in the classical pathway of pyroptosis (Figure 2). The important roles of caspase-1 in pyroptosis and secondary immune inflammation after ICH have been documented. Several animal studies have confirmed that an increase in caspase-1 levels is observed at least 6 h after ICH in mice, and these levels generally peak at 24-72 h (35–37). Furthermore, by inhibiting caspase-1, IL-1β expression and matrix metalloproteinase-9 (MMP-9) activity are reduced, subsequently reducing BBB damage, improving nerve function, and attenuating brain edema. These effects might be mediated by the JNK pathway (38).
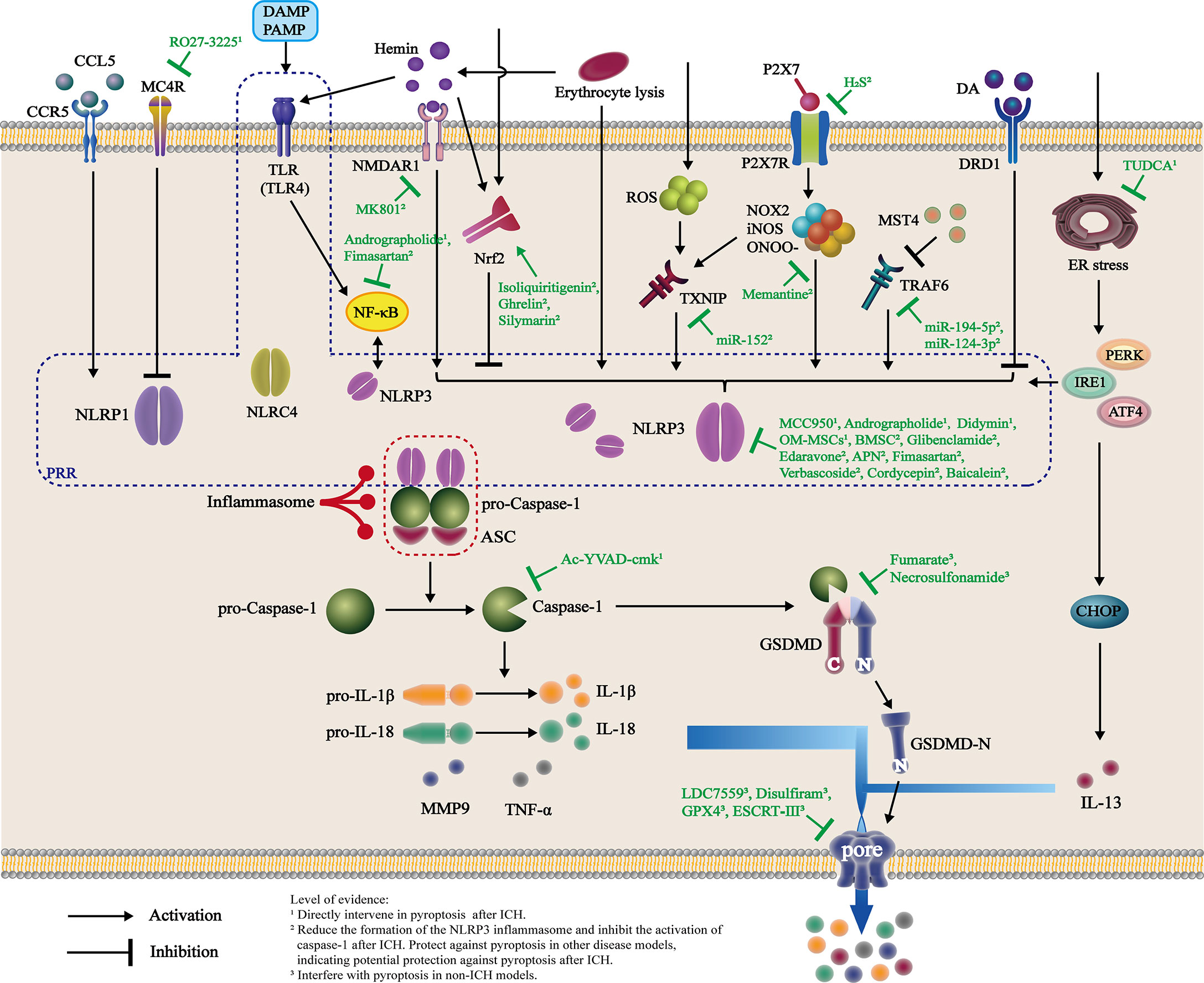
Figure 2 Activation and intervention of the pyroptosis pathway after ICH. Changes in the internal environment of the brain after ICH directly or indirectly activate PRR, as represented by NLRP3, through multiple pathways. These molecules recruit ASC and pro-caspase-1 and interact to form inflammasomes, which cleave pro-caspase-1 into active caspase-1. Caspase-1 cleaves both pro-IL-1β and pro-IL-18 into mature IL-1β and IL-18, respectively, and GSDMD to form GSDMD-N fragments. GSDMD-N binds to the plasma membrane and forms pores in the membrane, inducing the release of mature inflammatory factors such as IL-1β, IL-18, and TNF-α and subsequently recruiting more extracellular inflammatory cytokines and causing inflammation. Currently, animal- and cell-based experiments have shown that various drugs intervene in the formation of inflammasomes after ICH through different targets. Additionally, some candidate drugs are believed to directly interfere with pyroptosis after ICH, reducing the release of inflammatory factors and inhibiting neuroinflammation.
Activation of inflammasomes initiates the classical pyroptosis signaling pathway. To date, the best characterized inflammasome is assembled by NLRP3, which has been shown to be involved in the pathogenesis of atherosclerosis and brain injury caused by ischemia and ischemia/reperfusion (35, 39). NLRP3 is expressed in microglia and other types of brain cells, but this inflammasome component is not expressed after ICH. The current view is that microglial activation and polarization are important causes of secondary brain injury after ICH. ICH activates the NLRP3 inflammasome, and NLRP3 production increases biphasically, reaching a peak at 24 and 72 h after ICH (36). NLRP3 knockdown reduces brain edema and decreases myeloperoxidase (MPO) levels at 24 h and improves neurological function between 24 and 72 h after ICH (35). The expression of caspase-1 increases with increasing expression of NLRP3. Attenuation of ICH-induced NLRP3 overproduction results in decreases in caspase-1 and IL-1β expression, indicating that the NLRP3/caspase-1 signaling axis may mediate IL-1β overproduction after ICH (36). Similarly, the NLRP3 inflammasome amplifies the inflammatory response by releasing IL-1β and promoting neutrophil infiltration after ICH (35).
Direct evidence of changes in the morphology of pyroptotic cells is the formation of pores in the cell membrane. Li et al. directly confirmed the presence of neuronal pyroptosis after ICH using electron microscopy (40). Protecting neurons as much as possible is critical in the treatment of ICH and directly affects the prognosis. Although several studies have attempted to investigate the expression of inflammasome components and upstream regulators of canonical inflammasomes after ICH, few have explored the key protein GSDMD, which is downstream of canonical and noncanonical inflammasome activation. Furthermore, we propose to study the degree and changes in neuronal pyroptosis at different time points after ICH to verify whether pyroptosis plays a major role in secondary immune injury after ICH.
Another interesting question is the possible mechanism underlying the formation of NLRP3 and the activation of caspase-1. Researchers have shown that the ATP-gated transmembrane cation channel purinergic 2X7 receptor (P2X7R) and the NLRP3 inflammasomes are activated after ICH. Silencing of the P2X7R gene substantially reduces brain edema and neurological impairment by suppressing the activation of the NLRP3 inflammasome and IL-1β/IL-18 production. Furthermore, elevated levels of NADPH oxidase 2 (NOX2) and inducible nitric oxide synthase (iNOS), as well as increased levels of their cytotoxic product peroxynitrite (ONOO−), which are important molecules associated with secondary immune injury after ICH, are markedly attenuated by treatment with a selective P2X7R inhibitor. This effect is accompanied by the downregulation of inflammasome components and IL-1β/IL-18 levels and decreased neutrophil activation. In particular, the degradation of ONOO− substantially reduces the activation of the inflammasome and the release of IL-1β/IL-18 (36, 41–43). These studies indicated that P2X7R is upstream of NLRP3 activation. P2X7R increases inflammatory progression and brain injury in ICH mice, potentially increasing IL-1β/IL-18 release and neutrophil infiltration through the NLRP3 inflammasome. ONOO−, a putative downstream signaling molecule of P2X7R, could play a key role in the activation of the NLRP3 inflammasome. Although P2X7 activation clearly peaks 24 h after ICH, ONOO−, NLRP3 and caspase-1 activation exhibit biphasic trends, with peaks occurring at 24 and 72 h. Therefore, we propose that the P2X7 pathway is only one of the causes of pyroptosis and manifests itself primarily as pyroptosis 24 h after ICH.
Thioredoxin binding protein (TXNIP), a member of the α-arrestin protein superfamily, is involved in inflammation and participates in the response of the reactive oxygen species (ROS)/TXNIP signaling pathway to oxidative stress together with ROS (44). TXNIP is involved in the induction of pyroptosis after diabetic nephropathy, alcoholic liver disease, and intestinal ischemia (45–47). TXNIP activates the NLRP3 inflammasome and facilitates the release of inflammatory cytokines after binding to NLRP3 (48). Zuo et al. found that thrombin release after ICH activates ROS/TXNIP/NLRP3 signaling in BV2 cells, thus activating the pyroptotic signaling pathway NLRP3/caspase-1/ASC, promoting the expression of IL-1β, IL-18 and TNF-α, and increasing the inflammatory response and cell death (49).
TRAF6 is a vital binding protein of the TNF and the TLR superfamily and is notably involved in innate immunity and acquired immunity. TRAF6 is involved in the pyroptosis process in cerebral ischemia, sepsis, acute pancreatitis, and other diseases and is associated with the manifestation of ICH (50–53). Mammalian sterile-20-like kinase 4 (MST4) is classified as a member of the GCK subfamily and is expressed at high levels in the thymus and brain (54, 55). MST4 also mediates cell growth and apoptosis, affecting cell migration, differentiation, and proliferation (54). As a negative regulator of inflammation, the kinase MST4 limits inflammatory responses through direct phosphorylation of the adapter TRAF6 (56–58). TRAF6 interacts with NLRP3 to promote activation of the NLRP3 inflammasome. TRAF6 overexpression after ICH increases NLRP3 inflammasome levels and other inflammatory factors, increasing cell death rate and reducing cell viability (56). However, since relevant studies are limited to activation of the NLRP3 inflammasome, it remains to study whether it causes pyroptosis through caspase-1 in ICH.
Nrf2, an important transcription factor and the main regulator of cellular oxidative stress, induces antioxidant responses and increases the expression of downstream proteins, such as NAD(P)H:quinone oxidoreductase 1 (NQO1) and heme oxygenase 1 (HO-1) (59–63). Many studies have found that Nrf2 activation plays an essential role in protecting the brain from oxidative damage caused by ICH. Nrf2 activation in mice with ICH increases the expression of a series of antioxidant enzymes, detoxifying enzymes, and proteins, thus alleviating early brain injury after ICH and exerting a neuroprotective effect by activating Nrf2 (64, 65). These neuroprotective effects include improving neurological dysfunction, alleviating brain edema, reducing inflammatory cell infiltration, and accelerating hematoma clearance. Nrf2 knockout mice show more brain damage than control mice (66–72). Nrf2 inhibits pyroptosis by suppressing NLRP3 activation in microglia and vascular endothelial cells (73–74). Furthermore, Nrf2 activation inhibits expression of NLRP3/caspase-1/IL-1β and alleviates early brain damage after ICH by reducing neuronal death and the inflammatory response (75–77). Therefore, although direct evidence is not available, we postulate that Nrf2 participates in the mechanism that mediates pyroptosis after ICH.
A previous study reported that the neurotransmitter dopamine inhibits the activation of the NLRP3 inflammasome via the DRD1 (78). Mechanisms involved in pyroptosis in Parkinson’s disease and spinal cord injury have been extensively investigated (79–82). Wang et al. reported that DRD1 activation decreases brain edema and improves neurobehaviors 24 and 72 h after ICH, respectively. Further mechanistic studies showed that DPD1 activation decreases IL-1β release and inhibits microglial activation and neutrophil infiltration by inhibiting pyroptosis and inflammation mediated by the NLRP3/caspase-1 pathway (83). However, there is no direct evidence that DRD1 is involved in mediating pyroptosis in the ICH model.
In addition to NLRP3, which has been widely studied, NLRP1 is another important component of the inflammasome in focal cell death after ICH. Jun et al. showed that the expression of the C-C chemokine ligand 5 (CCL5), the C-C chemokine receptor type 5 (CCR5), and NLRP1 in the brain is upregulated and reached a peak 24 h after ICH. CCR5 activation, which is mediated in part by CCR5/PKA/CREB/NLR1, enhances neuronal pyroptosis and neurological impairment in mice after ICH (4). Melocortin 4 receptor (MC4R), a seven-transmembrane G-protein coupled receptor, has previously been shown to exert anti-inflammatory and neuroprotective effects after traumatic brain injury and cerebral ischemia. Using the MC4R agonist RO27-3225, Chen et al. found that the MC4R/ASK1/JNK/p38 MAPK signaling pathway is also involved in NLRP1-dependent neuronal pyroptosis after ICH (37). The component of the NLRC4 inflammasome, another member of the NLR family, has recently been shown to play a critical role in pyroptosis and the resulting inflammation after ICH. Similarly to NLRP3 and NLRP1, NLRC4 amplifies inflammation by facilitating the processing of caspase-1, IL-1β, IL-18, and TNF-α. Furthermore, elimination of NLRC4 attenuates neuronal death, BBB damage, brain edema, and neurological deficiency 3 days after ICH (84). Therefore, it is essential to explore the role of the two pathways in pyroptosis after ICH and to see if they are related to the NLRP3/caspase-1 pathway.
Erythrocyte lysis is also involved in pyroptosis after ICH. The volume of the hematoma peaks and stabilizes from 24 to 72 h due to injury caused by primary rupture of the blood vessels and secondary destruction of the BBB and then begins to decrease (85–88). Furthermore, erythrocyte lysis and infiltration of neutrophils, macrophages, and microglia were observed at 24 h, and their levels peaked at 72 h, consistent with the increase in NLRP3 and caspase-1 levels described in other studies (89). The severity of perihematomal neuronal loss is proportional to the degree of erythrolysis. Furthermore, the addition of lysates to microglia increased the expression of NLRP3 and caspase-1 in vitro. On the one hand, lysis of red blood cells can induce oxidative stress and pyroptosis by causing ferroptosis (36, 90–92). Hemin, on the other hand, is a hemoglobin degradation product that is postulated to be a DAMP involved in the classical pyroptosis pathway (93). The results reported by Wen et al. showed that heme induces NLRP3-mediated inflammatory injury. Furthermore, the authors determined that the effect of heme was mediated by the N-methyl-D-aspartic acid receptor 1 (NMDAR1). NMDAR1 plays a pivotal role in hemin-induced NLRP3-mediated inflammatory damage through synergistic activation (94). Similarly, Lin et al. performed animal and cell studies and showed that hemin enhances microglial activation through TLR4, which in turn induces nuclear transcription factor-κB (NF-κB) activation through the MyD88/TRIF signaling pathway (95). Finally, the expression of IL-1β, IL-6, and TNF-α and the resulting inflammatory injury increases after ICH (95). When NF-κB, a downstream TLR molecule, is activated, it translocates to the nucleus and binds to DNA, ensuring full expression of NLRP3, pro-IL-1β, and pro-IL-18 mRNAs and proteins (96).
Another study provided that microRNA (miR)-223 is a potential negative regulator of NLRP3 formation using TargetScan (a program used to look for potential regulatory microRNAs). Inhibition of NLRP3 expression by miR-223 reduces microglial inflammation induced by erythrocyte lysis induced by erythrocyte lysis and neuronal injury after ICH in mice. Cell-based experiments also showed that miR-223 levels in microglia and ICH mice decrease after erythrocyte lysis (97). These studies indicate that erythrocyte lysis may be involved in the induction of cell death after ICH. Although the study proposes a possible role for pyroptosis, other groups have not replicated the results.
Furthermore, Nrf2 is also strongly associated with erythrocyte rupture products after ICH. Both free hemoglobin (Hb) released by rupture of the erythrocyte and free heme released by hemoglobin decomposition are cytotoxic and cause neuronal death and secondary inflammation (98–100). Haptoglobin (Hp) and hemopexin (Hx) interact with Hb and heme, respectively, to reduce their toxicity (101, 102). According to Zhao et al., Nrf2 activation increases Hp and Hx expression after ICH, exerting neuroprotective effects (71, 103). However, more studies are needed to determine whether this protective effect is related to pyroptosis.
After ICH, other molecules or signaling pathways are activated and involved in pyroptosis. ICH leads to ER stress, which can induce neurological impairment through crosstalk with programmed cell death (PCD). Specifically, ER stress is caused by hypoxia and altered calcium homeostasis after ICH, mainly through the NLRP3/caspase-1 pathway. The ER stress-mediated NLRP3/caspase-1 pathway requires inositol-requiring protein-1 (IRE1) or protein kinase RNA-like endoplasmic reticulum kinase (PERK). ER stress induces pyroptosis (104). Furthermore, Chen et al. showed that ER stress through IL-13 causes neuronal pyroptosis after ICH. The AMP-dependent transcription factor-4 (ATF-4) and inositol-requiring kinase 1α (pIRE1α)/CCAAT-enhancer-binding protein homologous protein (CHOP) pathways, which are downstream targets of the PERK pathway, are also involved (85).
Most studies on pyroptosis after ICH have focused on neuronal and glial cell death. The two significant pathological changes observed after ICH are neuronal death and BBB destruction. Xu et al. found that NLRP3 is activated in hemin-exposed cerebral microvascular endothelial bEnd3 cells and induces cell death. Furthermore, inhibition of NLRP3 after ICH alleviates brain edema and damage to the BBB. Based on this finding, the primary hematoma caused by the rupture of the blood vessel is likely to destroy the BBB by causing pyroptosis in the endothelium, forming a positive feedback loop that further exacerbates hemorrhage and secondary brain injury (7, 105). Suppression of caspase-1 improves angiogenesis and the prognosis of ischemia (106). Furthermore, Wu et al. found that caspase-1 activation and upregulation of IL-1β, JNK, and MMP-9 expression are also closely related to the disruption of the BBB (38). Pericytes and astrocytes, two other cells components of the BBB, should be investigated in future research on post-ICH pyroptosis.
Microglia respond to acute brain injury by activating and developing classic M1-like (proinflammatory) or alternative M2-like (anti-inflammatory) phenotypes, a process termed polarization (107–109). Xia et al. performed cell-based experiments examining a mouse model of hyperlipidemic pancreatitis and found that macrophage M1 polarization is accompanied by activation of the caspase-1-mediated canonical pyroptosis pathway, leading to inflammation and damage to pancreatic tissue (110). Activation of the NLRP3 inflammasome has been shown in a variety of diseases to promote polarization of microglia/macrophages to the M1 phenotype (111–114). In individuals with ICH, selective inhibitors of the NLRP3 inflammasome reduce the polarization of M1 microglia around the hematoma, increase the number of M2 cells, and reduce neuroinflammation after ICH (61, 115). Selective inhibitors of caspase-1 exert a similar effect (116). Microglial polarization after ICH may be closely related to pyroptosis. Therefore, we hypothesized that M1 polarization of microglia after ICH is accompanied by activation of the pyroptosis pathway and that inflammatory factors released by the pyroptosis pathway likely induce subsequent microglial polarization, thus aggravating the secondary immune-inflammatory response. However, since no direct evidence exists, more experimental studies are needed to verify this hypothesis.
Chemotherapeutic drugs activate caspase-3 and induce pyroptosis by disrupting GSDME signaling in the treatment of lung, stomach, and colon cancer (30, 117–120). Caspase-3 is a well-recognized key molecule that causes cell apoptosis (121, 122). A conversion between apoptosis and pyroptosis occurs through a mechanism mediated by caspase-3/GSDME (121). Many studies have documented caspase-3 activation leading to apoptosis after ICH, and many signaling pathways have been shown to be involved (122–124). However, studies are needed to determine whether caspase-3/GSDME mediates pyroptosis after ICH in the future.
Recently, complement-mediated inflammation has also been reported to play a crucial role in ICH, including the secretion of IL-1β. Complement-induced ICH neuroinflammation depends on activation of NLRP3 (125). Although previous studies have shown that complement is widely involved in the pyroptosis observed in individuals with rheumatoid arthritis, membranous nephropathy, and infection (126–129), its role in pyroptosis after ICH and the specific underlying mechanism remain to be studied.
Inflammasome activation, such as NLRP3 activation, is associated with the appearance of pyroptosis, which results in the release of inflammatory mediators. Therefore, approaches targeting inflammasome activation are effective therapeutic strategies to alleviate pyroptosis and inflammatory injury after ICH (Figure 2, Table 1).
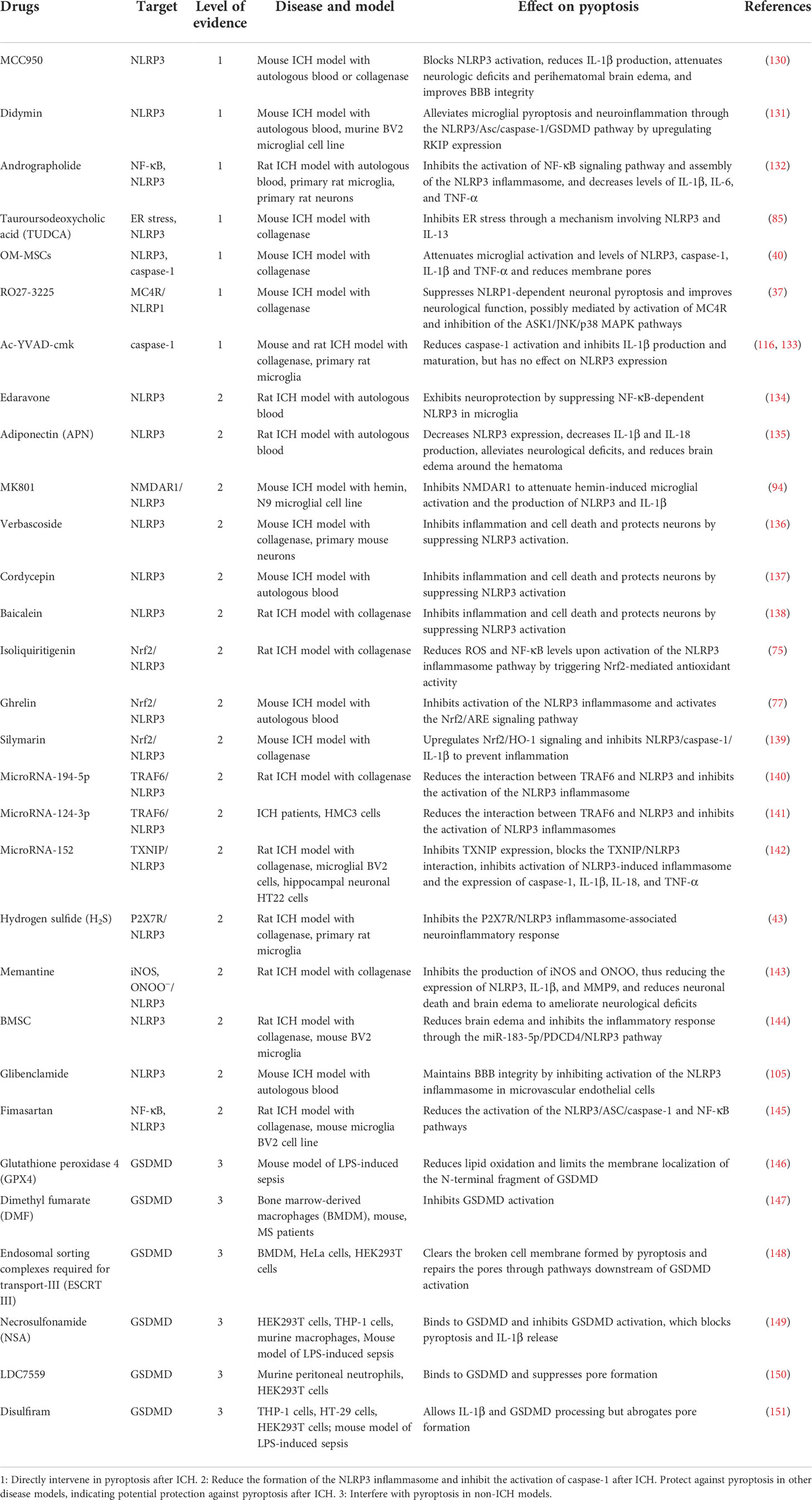
Table 1 Potential therapeutic drugs and targets for pyroptosis after ICH.
MCC950 is a potent, selective, small molecule NLRP3 inhibitor that blocks NLRP3 activation at nanomolar concentrations. MCC950 reduces IL-1β production, attenuates neurologic deficits and perihematomal brain edema, improves BBB integrity, and decreases pyroptosis after induction of ICH induction by injection of autologous blood or collagenase (130). Due to the high efficiency and specificity of MCC950, it is now widely used as a positive control to study the effect of NLRP3 inhibition. Edaravone and adiponectin (APN) exert similar effects to MCC950 in alleviating neurodegeneration and reducing the expression of mRNAs and proteins caspase-1, IL-1β and NF-κB (56, 130, 134, 135).
Didymin, a citrus dietary flavonoid, attenuates microglial activation and neutrophil infiltration by upregulating the expression of Raf kinase inhibitor protein (RKIP) and downregulating the expression of molecules related to pyroptosis, such as NLRP3, caspase-1, and GSDMD, and inflammatory cytokines, such as TNF-α and MPO, after ICH. Binding of RKIP to ASC interrupts the activation and assembly of the inflammasome (131). Andrographolide is one of the main active diterpenoid constituents of traditional Chinese medicine. It has been reported to exert anti-inflammatory effects mediated by the TLR4/NF-κB signaling pathway in treating liver inflammation and fibrosis (152, 153). Li et al. found that andrographolide improves neurobehavioral deficits and brain edema by inhibiting activation of the NF-κB signaling pathway and NLRP3 inflammasome assembly and reducing levels of IL-1β; andrographolide also improves ICH-induced microglial and neuronal pyroptosis. Furthermore, andrographolide significantly reduces the TNF-α and IL-6 levels by inhibiting the NF-κB signaling pathway (132). Other active ingredients in herbal medicine, such as verbascoside, cordycepin, and baicalein, inhibit inflammation and cell death and protect neurons following ICH by suppressing NLRP3 expression (136–138). Because of the common pharmacological actions, these drugs are likely to lessen pyroptosis after ICH.
Although the correlation between endothelial pyroptosis and BBB destruction after ICH has rarely been studied, Xu et al. have shown that glibenclamide maintains BBB integrity in an experimental ICH model by inhibiting the activation of the NLRP3 inflammasome in microvascular endothelial cells (105). Controlling and maintaining blood pressure after ICH are essential treatment effects. Controlling hypertension is vital to prevent the recurrence of ICH and to prevent ischemia and hypoxia, or even ischemic stroke caused by excessive decreases in blood pressure. Treatment with low doses of fimasartan, an angiotensin II receptor blocker, alleviates edema, improves neurological function, and alleviates secondary brain injury by significantly reducing activation of the NLRP3/ASC/caspase-1 and NF-κB pathways after ICH without changing mean blood pressure (145).
Cell-based therapy is a promising avenue for ICH treatment. Treatments with mesenchymal stem cells (MSCs) of various sources have produced favorable neuroprotective and neurorestorative effects on ICH (154–156). Hypoxia-preconditioned ORs (OM-MSCs) attenuate microglial activation and reduce levels of IL-1β and TNF-α. Furthermore, hypoxia-preconditioned OM-MSCs mitigate pyroptosis by reducing the levels of proteins associated with pyroptosis in perihematomal brain tissues, decreasing the expression of microglial NLRP3 and caspase-1, and reducing membrane pores in microglia after ICH (40). Similarly, bone marrow mesenchymal stem cell (BMSC) transplantation contributes to attenuating neurological deficits after ICH (157). BMSCs improve neurological function, reduce brain edema, and inhibit the inflammatory response after diabetes-induced ICH through the miR-183-5p/PDCD4/NLRP3 pathway (144).
Nrf2 regulation is closely related to NLRP3 expression. Intraperitoneal administration of isoliquiritigenin after ICH reduces early brain impairment and neurological deficits; The mechanisms underlying these effects involve negative regulation of ROS production and activation of NF-κB through the NLRP3 inflammasome pathway and activation of the Nrf2-mediated antioxidant system (75). Ghrelin, a peptide that participates in the brain-intestinal axis, has been shown to exert neuroprotective effects in many different neurological diseases, including subarachnoid hemorrhage (SAH), ischemic stroke, and traumatic brain injury (TBI) (158–161). Recent studies have shown that ghrelin protects against ICH-induced SBI by inhibiting activation of the NLRP3 inflammasome and activating the Nrf2/antioxidant response element (ARE) signaling pathway (77). Silymarin acts as a neuroprotective compound after ICH injury by activating Nrf2/HO-1 signaling and inhibiting NLRP3/caspase-1/IL-1β signaling to prevent inflammation (139).
Studies of drugs targeting TRAF6 have also shown that these drugs regulate the expression of NLRP3. Wu et al. found that selective inhibition of MST4 expression by hesperidin increases the expression of NLRP3 inflammasome-related proteins, thus exacerbating neurological deficits and brain edema. Therefore, MST4 alleviates inflammatory progression and brain injury in mice with ICH, perhaps by reducing the activation of the NLRP3 inflammasome (56). Both microRNA-194-5p and microRNA-124-3p target TRAF6. Both molecules exert the same effect on TRAF6 through the same mechanism. Overexpression of miR-194-5p or miR-124-3p reduces the interaction between TRAF6 and NLRP3, thus inhibiting the activation of the NLRP3 inflammasome and reducing neuroinflammation during ICH (140, 141).
MicroRNA-152 has been reported to possess anti-inflammatory properties, and its expression is downregulated after ICH. Overexpression of miR-152 in microglial BV2 cells reduces the hemin-induced inflammatory response and ROS generation, thus reducing the death of cocultured neuronal HT22 cells. Furthermore, overexpression of miR-152 by intracerebroventricular injection of a lentivirus in rats with ICH significantly alleviates brain edema, hematoma, and neurological deficits. Mechanistically, miR-152 significantly inhibits the expression of TXNIP induced by ICH, and its overexpression blocks the TXNIP/NLRP3 interaction, thus inhibiting the activation of the NLRP3 inflammasome and the expression of caspase-1, IL-1β, IL-18, and TNF-α to reduce neuroinflammation in vivo and in vitro (142).
Emerging studies have revealed the important physiological and pathophysiological roles of hydrogen sulfide (H2S) as a transmitter of NLRP3 inflammasome-associated neuroinflammation in the central nervous system. Exogenous administration of H2S reduces brain edema, microglial accumulation, and neurological deficits after ICH by inhibiting the neuroinflammatory response associated with the P2X7R/NLRP3 inflammasome, suggesting that it may be related to the pyroptosis pathway (43). Furthermore, Chen et al. also found that memantine inhibits the production of iNOS and ONOO−, thus reducing the expression of NLRP3, IL-1β and MMP9, reducing neuronal death and brain edema, and improving neurological deficits (143).
NMDAR1 is an important receptor that regulates NLRP3 after ICH. The NMDAR1 inhibitor MK801 has also been shown to attenuate hemin-induced microglial activation, which subsequently leads to a decrease in the production of NLRP3 and IL-1β in microglia (87). Tauroursodeoxycholic acid (TUDCA) alleviates neuronal pyroptosis by inhibiting ER stress after ICH through a mechanism involving NLRP3 and IL-13 (83). However, it remains to be explored whether they directly affect the pyroptosis process downstream of NLRP3.
Few studies have been conducted on non-NLRP3 inflammasomes. As mentioned above, NLRP1, NLRC4, and some TLRs may also be involved in the process of pyroptosis after ICH. RO27-3225, an MC4R agonist, was found to suppress NLRP1-dependent neuronal pyroptosis and improved neurological function, possibly mediated by activation of the MC4R and inhibition of the ASK1/JNK/p38 MAPK signaling pathway after ICH (37).
Bypassing inflammasomes and directly interfering with molecules downstream of the focal death pathway have also been shown to be effective treatments for ICH. Ac-YVAD-cmk, a well-known selective inhibitor of caspase-1, reduces caspase-1 activation and inhibits IL-1β production and maturation, but it does not have an effect on NLRP3 expression. Ac-YVAD-cmk inhibits pyroptosis, decreases inflammatory factor secretion or activation, and affects microglial polarization and aggregation, resulting in improvements in neurological function and behavioral performance and amelioration of brain edema after ICH. Neuroprotection is associated with decreased expression of IL-1β, IL-18, and MMP-9 and inhibition of ZO-1 degradation (38, 116, 133).
The cleavage and activation of gasdermin is the most direct cause of membrane pores and the release of inflammatory factors that induce the inflammatory response. Therefore, direct inhibition of gasdermin proteins or recombining C-terminal and N-terminal fragments could also inhibit pyroptosis. Kang et al. found that glutathione peroxidase 4 (GPX4) decreases lipid oxidation and thus limits membrane localization of the N-terminal fragment of GSDMD and negatively regulates GSDMD-mediated pyroptosis in LPS-induced sepsis (146). According to Fitzgerald et al., fumarate, an intermediate in the tricarboxylic acid cycle, succinylates cysteine in GSDMD, inhibits its interaction with and activation of cysteine proteases, and ultimately inhibits the occurrence of pyroptosis. Related therapeutics, such as tecfidera, have been used to treat multiple sclerosis (MS) (147). Furthermore, the endosomal sorting complexes required for transport-III (ESCRT III)-dependent exocytosis can clear the broken cell membrane caused by pyroptosis and repair the pores in the cell membrane after GSDMD activation, thus strongly inhibiting pyroptosis and IL-1β release (148). Necrosulfonamide (NSA) was originally identified as a cysteine-reactive drug that targets the mixed-lineage kinase domain-like protein (MLKL) to inhibit necroptosis (162, 163). Furthermore, NSA binds directly to GSDMD and inhibits its activation. NSA blocks pyroptosis and IL-1β release in human and murine monocytes/macrophages without interfering with inflammasome formation and GSDME-mediated cell death (149, 162). LDC7559 inhibits the formation of neutrophil extracellular traps (NET), presumably by binding to GSDMD and suppressing pore formation (150). Disulfiram also covalently modifies human/mouse Cys191/Cys192 in GSDMD to block pore formation. Interestingly, disulfiram still allows IL-1β and GSDMD processing, but abrogates pore formation, thus preventing IL-1β release and pyroptosis in LPS-induced sepsis (151). Various methods have been developed to curb pyroptosis by regulating GSDMD and repairing pores. It should be emphasized that current studies mainly focus on the upstream molecular regulatory mechanisms of pyroptosis after ICH. No studies have explored the role and mechanism of related drugs targeting GSDMD regulation and cell membrane repair in pyroptosis in ICH. Due to their similar mechanisms of action, these treatments are likely to play equally important roles in the pyroptosis process after ICH and therefore should be explored in detail.
ICH still threatens life due to high morbidity and mortality. Currently, many original studies have focused on the research hotspot of pyroptosis. The mechanism of pyroptosis in various diseases, such as sepsis and tumors, has gradually become known due to recent studies. However, basic research on the role of pyroptosis in ICH and related interventions for ICH is lacking. Although there is increasing evidence to support the link between pyroptosis and ICH, there is no detailed summary of the available knowledge. This review focuses on ICH and comprehensively and systematically introduces recent research findings, mechanisms of pyroptosis after ICH, and potential intervention strategies. Pyroptosis in neurons, microglia, and endothelial cells after ICH is closely related to impairments in neural function, activation of immune inflammation, and destruction of the BBB. The NLRP3/caspase-1/GSDMD pathway induces pyroptosis after ICH. Specifically, changes in the brain microenvironment after ICH activate various NLRP3 upstream molecules, including NMDAR1, TXNIP, and so on. It induces NLRP3 and contributes to the assembly of inflammasomes, thus activating caspase-1. In addition to increasing the release of IL-1β, IL-18, TNF-α and other inflammatory factors, caspase-1 also cleaved GSDMD to release GSDMD-N, which formed pores in the cell membrane and promoted the release of inflammatory factors. On the one hand, released inflammatory factors damage normal tissues through the immune response; on the other hand, they can also reactivate the above pyroptosis reaction, forming a positive feedback effect, and further aggravating brain injury. It would also be interesting to investigate whether pyroptosis plays a role in mediating the interaction between the immune system and the autonomic nervous system after ICH (164, 165).
Evidence indicates that pyroptosis could play an essential role in ICH injury and that the targeted intervention has achieved positive results in preclinical research. However, it is unclear whether pyroptosis is the leading cause of secondary brain injury after ICH. Although we have proposed the above assumptions based on preclinical studies, most studies focused on the NLRP3 inflammasome. Therapies targeting downstream caspase-1 and GSDMD of pyroptosis have not been tested. Therefore, the specific mechanism of pyroptosis after ICH needs further exploration. We also suggest that future studies on pyroptosis after ICH should be more complete. In addition to NLRP3, it is unclear whether NLRP3 and NLRC4 play the same critical role in the pyroptosis process of pyroptosis after ICH.
Meanwhile, caspase-1/11 and GSDMD are specific or universal markers of pyroptosis, and their intervention effect after ICH needs clarification. We propose that caspase-1 might have a connecting role in the pyroptosis process after ICH and suggest exploring and developing potential drugs targeting it. Erythrocyte breakdown after ICH might be one of the causes of pyroptosis. We hypothesize that controlled erythrocyte lysis is critical against pyroptosis. Investigating this possibility is worthwhile. Currently, relevant compounds may have multiple targets, but they do not target pyroptosis-related molecules. It is worth investigating whether the main target of these drugs is primarily pyroptosis.
Like other modes of cell death, pyroptosis ultimately leads to brain cell death and the resulting histopathology and functional dysfunction (Figure 3). Pyroptosis of cerebral microvascular endothelial cells after ICH leads to disruption of the BBB. It further aggravates the formation of hematoma and brain edema. The hematoma mechanically compresses the surrounding normal brain tissue. Exudation and lysis of the erythrocyte can further enhance pyroptosis, as described above. It remains unclear whether pericytes and astrocytes undergo pyroptosis and participate in the disruption of the BBB after ICH. Primary brain damage may be due to pyroptosis in many neurons, leading to neurological deficits and a poor prognosis. The most crucial characteristic of pyroptosis, which is different from other modes of cell death, is that many inflammatory factors produced in the pyroptosis process permeate brain tissue by forming cell membrane pores. Pyroptosis of microglia after ICH may be the most critical initiator. The release of inflammatory factors, such as IL-1β, IL-18 and TNF-α, leads to secondary immune injury. The excessive immune response will lead to more neuronal death, increased neurological deficits, and a worse prognosis. Therefore, we should focus on the vital role of microglia in pyroptosis after ICH and further explore whether there is a causal relationship between microglia activation and polarization and pyroptosis.
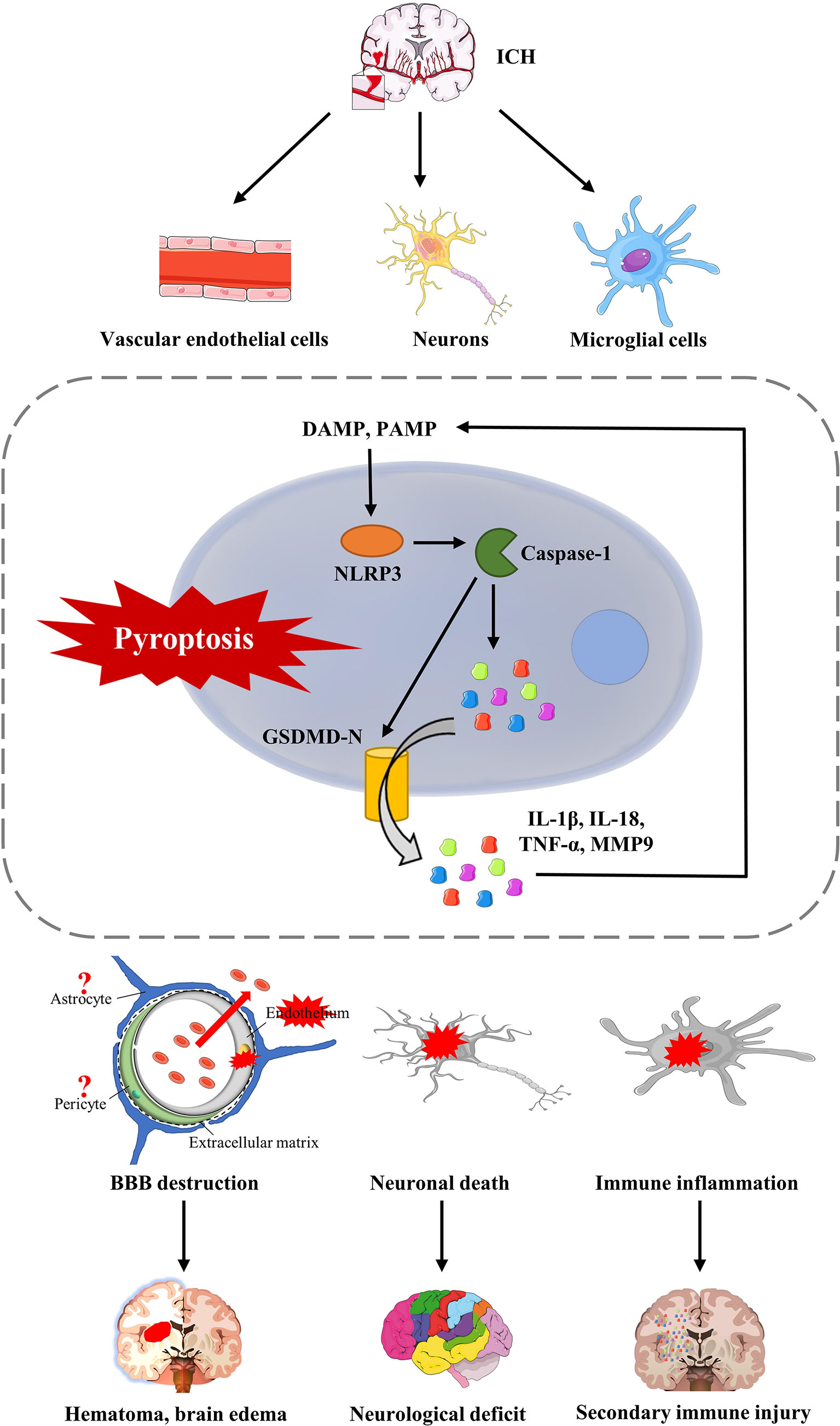
Figure 3 The consequences of pyrotosis after ICH. Pyroptosis of cerebral microvascular endothelial cells after ICH can lead to disruption of the BBB, further aggravated by the formation of hematoma and brain edema. Furthermore, pyroptosis, which occurs in neurons, leads to the death of functional neurons, which is a fundamental cause of neurological impairment and complications after ICH. Furthermore, pyroptosis of microglia after ICH will release various inflammatory factors, such as IL-1β, IL-18, and TNF-α, and recruit more extracellular inflammatory factors, leading to secondary immune injury.
In conclusion, current research shows that inhibition of pyroptosis can alleviate tissue damage after ICH. A breakthrough in the treatment of ICH could be achieved by targeting pyroptosis, which could eventually be an effective strategy to improve the prognosis of patients. However, most current studies focus only on the underlying mechanism; in addition, most studies use cell and animal models rather than clinical trials. Currently, no relevant clinical research has been completed. Therefore, basic scientists and clinicians must carefully design translational research and clinical trials on pyroptosis to test novel therapeutic strategies to treat ICH.
FG, JW, and DS conceptualized the manuscript outline. DS performed the literature search, wrote the manuscript, and designed the figures. FG, JW, and C-TY provided comments and proof-reading. All authors read and approved the final manuscript.
This work was supported by the National Key Research and Development Program of China (2021YFE0204700).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.

1. Ren H, Han R, Chen X, Liu X, Wan J, Wang L, et al. Potential therapeutic targets for intracerebral hemorrhage-associated inflammation: an update. J Cereb Blood Flow Metab (2020) 40:1752–68. doi: 10.1177/0271678X20923551
2. Schrag M, Kirshner H. Management of intracerebral hemorrhage: jacc focus seminar. J Am Coll Cardiol (2020) 75:1819–31. doi: 10.1016/j.jacc.2019.10.066
3. Hua W, Chen X, Wang J, Zang W, Jiang C, Ren H, et al. Mechanisms and potential therapeutic targets for spontaneous intracerebral hemorrhage. Brain Hemorrhages (2020) 1:99–104. doi: 10.1016/j.hest.2020.02.002
4. Yan J, Xu W, Lenahan C, Huang L, Wen J, Li G, et al. Ccr5 activation promotes nlrp1-dependent neuronal pyroptosis via ccr5/pka/creb pathway after intracerebral hemorrhage. Stroke (2021) 52:4021–32. doi: 10.1161/STROKEAHA.120.033285
5. Boltze J, Aronowski JA, Badaut J, Buckwalter MS, Caleo M, Chopp M, et al. New mechanistic insights, novel treatment paradigms, and clinical progress in cerebrovascular diseases. Front Aging Neurosci (2021) 13:623751. doi: 10.3389/fnagi.2021.623751
6. Herr DR, Yam T, Tan W, Koh SS, Wong W, Ong WY, et al. Ultrastructural characteristics of dha-induced pyroptosis. Neuromolecular Med (2020) 22:293–303. doi: 10.1007/s12017-019-08586-y
7. Fang Y, Gao S, Wang X, Cao Y, Lu J, Chen S, et al. Programmed cell deaths and potential crosstalk with blood-brain barrier dysfunction after hemorrhagic stroke. Front Cell Neurosci (2020) 14:68. doi: 10.3389/fncel.2020.00068
8. Zhu H, Wang Z, Yu J, Yang X, He F, Liu Z, et al. Role and mechanisms of cytokines in the secondary brain injury after intracerebral hemorrhage. Prog Neurobiol (2019) 178:101610. doi: 10.1016/j.pneurobio.2019.03.003
9. Shi J, Gao W, Shao F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci (2017) 42:245–54. doi: 10.1016/j.tibs.2016.10.004
10. Gou X, Xu D, Li F, Hou K, Fang W, Li Y. Pyroptosis in stroke-new insights into disease mechanisms and therapeutic strategies. J Physiol Biochem (2021) 77:511–29. doi: 10.1007/s13105-021-00817-w
11. Aglietti RA, Dueber EC. Recent insights into the molecular mechanisms underlying pyroptosis and gasdermin family functions. Trends Immunol (2017) 38:261–71. doi: 10.1016/j.it.2017.01.003
12. Qi X. Formation of membrane pores by gasdermin-n causes pyroptosis. Sci China Life Sci (2016) 59:1071–3. doi: 10.1007/s11427-016-5109-3
13. Chen X, He WT, Hu L, Li J, Fang Y, Wang X, et al. Pyroptosis is driven by non-selective gasdermin-d pore and its morphology is different from mlkl channel-mediated necroptosis. Cell Res (2016) 26:1007–20. doi: 10.1038/cr.2016.100
15. Liang F, Zhang F, Zhang L, Wei W. The advances in pyroptosis initiated by inflammasome in inflammatory and immune diseases. Inflammation Res (2020) 69:159–66. doi: 10.1007/s00011-020-01315-3
16. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol (2016) 16:407–20. doi: 10.1038/nri.2016.58
17. Alsaadi M, Tezcan G, Garanina EE, Hamza S, McIntyre A, Rizvanov AA, et al. Doxycycline attenuates cancer cell growth by suppressing nlrp3-mediated inflammation. Pharm (Basel) (2021) 14:852. doi: 10.3390/ph14090852
18. Bellut M, Papp L, Bieber M, Kraft P, Stoll G, Schuhmann MK. Nlpr3 inflammasome inhibition alleviates hypoxic endothelial cell death in vitro and protects blood-brain barrier integrity in murine stroke. Cell Death Dis (2021) 13:20. doi: 10.1038/s41419-021-04379-z
19. Li Z, Hu J, Bao C, Gao C, Zhang N, Cardona CJ, et al. Activation of the nlrp3 inflammasome and elevation of interleukin-1beta secretion in infection by sever fever with thrombocytopenia syndrome virus. Sci Rep (2022) 12:2573. doi: 10.1038/s41598-022-06229-0
20. Song S, Guo R, Mehmood A, Zhang L, Yin B, Yuan C, et al. Liraglutide attenuate central nervous inflammation and demyelination through ampk and pyroptosis-related nlrp3 pathway. CNS Neurosci Ther (2022) 28:422–34. doi: 10.1111/cns.13791
21. Wang X, Li X, Liu S, Brickell AN, Zhang J, Wu Z, et al. Pcsk9 regulates pyroptosis via mtdna damage in chronic myocardial ischemia. Basic Res Cardiol (2020) 115:66. doi: 10.1007/s00395-020-00832-w
22. Ye A, Li W, Zhou L, Ao L, Fang W, Li Y. Targeting pyroptosis to regulate ischemic stroke injury: molecular mechanisms and preclinical evidences. Brain Res Bull (2020) 165:146–60. doi: 10.1016/j.brainresbull.2020.10.009
23. Zeng C, Duan F, Hu J, Luo B, Huang B, Lou X, et al. Nlrp3 inflammasome-mediated pyroptosis contributes to the pathogenesis of non-ischemic dilated cardiomyopathy. Redox Biol (2020) 34:101523. doi: 10.1016/j.redox.2020.101523
24. Zhu W, Cao FS, Feng J, Chen HW, Wan JR, Lu Q, et al. Nlrp3 inflammasome activation contributes to long-term behavioral alterations in mice injected with lipopolysaccharide. Neuroscience (2017) 343:77–84. doi: 10.1016/j.neuroscience.2016.11.037
25. Broz P. Immunology: caspase target drives pyroptosis. Nature (2015) 526:642–3. doi: 10.1038/nature15632
26. Mandal P, Feng Y, Lyons JD, Berger SB, Otani S, DeLaney A, et al. Caspase-8 collaborates with caspase-11 to drive tissue damage and execution of endotoxic shock. Immunity (2018) 49:42–55. doi: 10.1016/j.immuni.2018.06.011
27. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of gsdmd by inflammatory caspases determines pyroptotic cell death. Nature (2015) 526:660–5. doi: 10.1038/nature15514
28. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin d causes pyroptosis by forming membrane pores. Nature (2016) 535:153–8. doi: 10.1038/nature18629
29. Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin d for non-canonical inflammasome signalling. Nature (2015) 526:666–71. doi: 10.1038/nature15541
30. Wang Y, Gao W, Shi X, Ding J, Liu W, He H, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature (2017) 547:99–103. doi: 10.1038/nature22393
31. Orning P, Weng D, Starheim K, Ratner D, Best Z, Lee B, et al. Pathogen blockade of tak1 triggers caspase-8-dependent cleavage of gasdermin d and cell death. Science (2018) 362:1064–9. doi: 10.1126/science.aau2818
32. Gram AM, Booty LM, Bryant CE. Chopping gsdmd: caspase-8 has joined the team of pyroptosis-mediating caspases. EMBO J (2019) 38:e102065. doi: 10.15252/embj.2019102065
33. Kost A, Kasprowska D, Labuzek K, Wiaderkiewicz R, Gabryel B. [Autophagy in brain ischemia]. Postepy Hig Med Dosw (Online) (2011) 65:524–33. doi: 10.5604/17322693.955650
34. Kambara H, Liu F, Zhang X, Liu P, Bajrami B, Teng Y, et al. Gasdermin d exerts anti-inflammatory effects by promoting neutrophil death. Cell Rep (2018) 22:2924–36. doi: 10.1016/j.celrep.2018.02.067
35. Ma Q, Chen S, Hu Q, Feng H, Zhang JH, Tang J. Nlrp3 inflammasome contributes to inflammation after intracerebral hemorrhage. Ann Neurol (2014) 75:209–19. doi: 10.1002/ana.24070
36. Feng L, Chen Y, Ding R, Fu Z, Yang S, Deng X, et al. P2x7r blockade prevents nlrp3 inflammasome activation and brain injury in a rat model of intracerebral hemorrhage: involvement of peroxynitrite. J Neuroinflamm (2015) 12:190. doi: 10.1186/s12974-015-0409-2
37. Chen S, Zuo Y, Huang L, Sherchan P, Zhang J, Yu Z, et al. The mc4 receptor agonist ro27-3225 inhibits nlrp1-dependent neuronal pyroptosis via the ask1/jnk/p38 mapk pathway in a mouse model of intracerebral haemorrhage. Br J Pharmacol (2019) 176:1341–56. doi: 10.1111/bph.14639
38. Wu B, Ma Q, Khatibi N, Chen W, Sozen T, Cheng O, et al. Ac-Yvad-cmk decreases blood-brain barrier degradation by inhibiting caspase-1 activation of interleukin-1beta in intracerebral hemorrhage mouse model. Transl Stroke Res (2010) 1:57–64. doi: 10.1007/s12975-009-0002-z
39. Yuan B, Shen H, Lin L, Su T, Zhong S, Yang Z. Recombinant adenovirus encoding nlrp3 rnai attenuate inflammation and brain injury after intracerebral hemorrhage. J Neuroimmunol (2015) 287:71–5. doi: 10.1016/j.jneuroim.2015.08.002
40. Liu J, He J, Huang Y, Ge L, Xiao H, Zeng L, et al. Hypoxia-preconditioned mesenchymal stem cells attenuate microglial pyroptosis after intracerebral hemorrhage. Ann Transl Med (2021) 9:1362. doi: 10.21037/atm-21-2590
41. Ahsan H. 3-nitrotyrosine: a biomarker of nitrogen free radical species modified proteins in systemic autoimmunogenic conditions. Hum Immunol (2013) 74:1392–9. doi: 10.1016/j.humimm.2013.06.009
42. Wanyong Y, Zefeng T, Xiufeng X, Dawei D, Xiaoyan L, Ying Z, et al. Tempol alleviates intracerebral hemorrhage-induced brain injury possibly by attenuating nitrative stress. Neuroreport (2015) 26:842–9. doi: 10.1097/WNR.0000000000000434
43. Zhao H, Pan P, Yang Y, Ge H, Chen W, Qu J, et al. Endogenous hydrogen sulphide attenuates nlrp3 inflammasome-mediated neuroinflammation by suppressing the p2x7 receptor after intracerebral haemorrhage in rats. J Neuroinflamm (2017) 14:163. doi: 10.1186/s12974-017-0940-4
44. Zhang X, Zhang JH, Chen XY, Hu QH, Wang MX, Jin R, et al. Reactive oxygen species-induced txnip drives fructose-mediated hepatic inflammation and lipid accumulation through nlrp3 inflammasome activation. Antioxid Redox Signal (2015) 22:848–70. doi: 10.1089/ars.2014.5868
45. Heo MJ, Kim TH, You JS, Blaya D, Sancho-Bru P, Kim SG. Alcohol dysregulates mir-148a in hepatocytes through foxo1, facilitating pyroptosis via txnip overexpression. Gut (2019) 68:708–20. doi: 10.1136/gutjnl-2017-315123
46. An X, Zhang Y, Cao Y, Chen J, Qin H, Yang L. Punicalagin protects diabetic nephropathy by inhibiting pyroptosis based on txnip/nlrp3 pathway. Nutrients (2020) 12:1516. doi: 10.3390/nu12051516
47. Jia Y, Cui R, Wang C, Feng Y, Li Z, Tong Y, et al. Metformin protects against intestinal ischemia-reperfusion injury and cell pyroptosis via txnip-nlrp3-gsdmd pathway. Redox Biol (2020) 32:101534. doi: 10.1016/j.redox.2020.101534
48. Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol (2010) 11:136–40. doi: 10.1038/ni.1831
49. Ye X, Zuo D, Yu L, Zhang L, Tang J, Cui C, et al. Ros/txnip pathway contributes to thrombin induced nlrp3 inflammasome activation and cell apoptosis in microglia. Biochem Biophys Res Commun (2017) 485:499–505. doi: 10.1016/j.bbrc.2017.02.019
50. Dou Y, Tian X, Zhang J, Wang Z, Chen G. Roles of traf6 in central nervous system. Curr Neuropharmacol (2018) 16:1306–13. doi: 10.2174/1570159X16666180412094655
51. Yan W, Sun W, Fan J, Wang H, Han S, Li J, et al. Sirt1-ros-traf6 signaling-induced pyroptosis contributes to early injury in ischemic mice. Neurosci Bull (2020) 36:845–59. doi: 10.1007/s12264-020-00489-4
52. Li LL, Dai B, Sun YH, Zhang TT. The activation of il-17 signaling pathway promotes pyroptosis in pneumonia-induced sepsis. Ann Transl Med (2020) 8:674. doi: 10.21037/atm-19-1739
53. Wei B, Gong Y, Yang H, Zhou J, Su Z, Liang Z. Role of tumor necrosis factor receptorassociated factor 6 in pyroptosis during acute pancreatitis. Mol Med Rep (2021) 24:848. doi: 10.3892/mmr.2021.12488
54. Lin JL, Chen HC, Fang HI, Robinson D, Kung HJ, Shih HM. Mst4, a new ste20-related kinase that mediates cell growth and transformation via modulating erk pathway. Oncogene (2001) 20:6559–69. doi: 10.1038/sj.onc.1204818
55. Qian Z, Lin C, Espinosa R, LeBeau M, Rosner MR. Cloning and characterization of mst4, a novel ste20-like kinase. J Biol Chem (2001) 276:22439–45. doi: 10.1074/jbc.M009323200
56. Wu X, Zhang Y, Zhang Y, Xia L, Yang Y, Wang P, et al. Mst4 attenuates nlrp3 inflammasome-mediated neuroinflammation and affects the prognosis after intracerebral hemorrhage in mice. Brain Res Bull (2021) 177:31–8. doi: 10.1016/j.brainresbull.2021.09.006
57. Jiao S, Zhang Z, Li C, Huang M, Shi Z, Wang Y, et al. The kinase mst4 limits inflammatory responses through direct phosphorylation of the adaptor traf6. Nat Immunol (2015) 16:246–57. doi: 10.1038/ni.3097
58. Liu X, You J, Peng X, Wang Q, Li C, Jiang N, et al. Mammalian ste20-like kinase 4 inhibits the inflammatory response in aspergillus fumigatus keratitis. Int Immunopharmacol (2020) 88:107021. doi: 10.1016/j.intimp.2020.107021
59. Calkins MJ, Johnson DA, Townsend JA, Vargas MR, Dowell JA, Williamson TP, et al. The nrf2/are pathway as a potential therapeutic target in neurodegenerative disease. Antioxid Redox Signal (2009) 11:497–508. doi: 10.1089/ars.2008.2242
60. Denzer I, Munch G, Friedland K. Modulation of mitochondrial dysfunction in neurodegenerative diseases via activation of nuclear factor erythroid-2-related factor 2 by food-derived compounds. Pharmacol Res (2016) 103:80–94. doi: 10.1016/j.phrs.2015.11.019
61. Xiao L, Zheng H, Li J, Wang Q, Sun H. Neuroinflammation mediated by nlrp3 inflammasome after intracerebral hemorrhage and potential therapeutic targets. Mol Neurobiol (2020) 57:5130–49. doi: 10.1007/s12035-020-02082-2
62. Zhang X, Wu Q, Lu Y, Wan J, Dai H, Zhou X, et al. Cerebroprotection by salvianolic acid b after experimental subarachnoid hemorrhage occurs via nrf2- and sirt1-dependent pathways. Free Radic Biol Med (2018) 124:504–16. doi: 10.1016/j.freeradbiomed.2018.06.035
63. Zhang Z, Zhang Z, Lu H, Yang Q, Wu H, Wang J. Microglial polarization and inflammatory mediators after intracerebral hemorrhage. Mol Neurobiol (2017) 54:1874–86. doi: 10.1007/s12035-016-9785-6
64. Lan X, Han X, Li Q, Wang J. (-)-epicatechin, a natural flavonoid compound, protects astrocytes against hemoglobin toxicity via nrf2 and ap-1 signaling pathways. Mol Neurobiol (2017) 54:7898–907. doi: 10.1007/s12035-016-0271-y
65. Wang J, Jiang C, Zhang K, Lan X, Chen X, Zang W, et al. Melatonin receptor activation provides cerebral protection after traumatic brain injury by mitigating oxidative stress and inflammation via the nrf2 signaling pathway. Free Radic Biol Med (2019) 131:345–55. doi: 10.1016/j.freeradbiomed.2018.12.014
66. Chang CF, Cho S, Wang J. (-)-epicatechin protects hemorrhagic brain via synergistic nrf2 pathways. Ann Clin Transl Neurol (2014) 1:258–71. doi: 10.1002/acn3.54
67. Iniaghe LO, Krafft PR, Klebe DW, Omogbai E, Zhang JH, Tang J. Dimethyl fumarate confers neuroprotection by casein kinase 2 phosphorylation of nrf2 in murine intracerebral hemorrhage. Neurobiol Dis (2015) 82:349–58. doi: 10.1016/j.nbd.2015.07.001
68. Ren H, Han R, Liu X, Wang L, Koehler RC, Wang J. Nrf2-bdnf-trkb pathway contributes to cortical hemorrhage-induced depression, but not sex differences. J Cereb Blood Flow Metab (2021) 41:3288–301. doi: 10.1177/0271678X211029060
69. Shang H, Yang D, Zhang W, Li T, Ren X, Wang X, et al. Time course of keap1-nrf2 pathway expression after experimental intracerebral haemorrhage: correlation with brain oedema and neurological deficit. Free Radic Res (2013) 47:368–75. doi: 10.3109/10715762.2013.778403
70. Wang J, Fields J, Zhao C, Langer J, Thimmulappa RK, Kensler TW, et al. Role of nrf2 in protection against intracerebral hemorrhage injury in mice. Free Radic Biol Med (2007) 43:408–14. doi: 10.1016/j.freeradbiomed.2007.04.020
71. Zhao X, Sun G, Zhang J, Strong R, Dash PK, Kan YW, et al. Transcription factor nrf2 protects the brain from damage produced by intracerebral hemorrhage. Stroke (2007) 38:3280–6. doi: 10.1161/STROKEAHA.107.486506
72. Zhao X, Sun G, Zhang J, Ting SM, Gonzales N, Aronowski J. Dimethyl fumarate protects brain from damage produced by intracerebral hemorrhage by mechanism involving nrf2. Stroke (2015) 46:1923–8. doi: 10.1161/STROKEAHA.115.009398
73. Hu Q, Zhang T, Yi L, Zhou X, Mi M. Dihydromyricetin inhibits nlrp3 inflammasome-dependent pyroptosis by activating the nrf2 signaling pathway in vascular endothelial cells. Biofactors (2018) 44:123–36. doi: 10.1002/biof.1395
74. Arioz BI, Tastan B, Tarakcioglu E, Tufekci KU, Olcum M, Ersoy N, et al. Melatonin attenuates lps-induced acute depressive-like behaviors and microglial nlrp3 inflammasome activation through the sirt1/nrf2 pathway. Front Immunol (2019) 10:1511. doi: 10.3389/fimmu.2019.01511
75. Zeng J, Chen Y, Ding R, Feng L, Fu Z, Yang S, et al. Isoliquiritigenin alleviates early brain injury after experimental intracerebral hemorrhage via suppressing ros- and/or nf-kappab-mediated nlrp3 inflammasome activation by promoting nrf2 antioxidant pathway. J Neuroinflamm (2017) 14:119. doi: 10.1186/s12974-017-0895-5
76. Cheng Y, Liu M, Tang H, Chen B, Yang G, Zhao W, et al. Itraq-based quantitative proteomics indicated nrf2/optn-mediated mitophagy inhibits nlrp3 inflammasome activation after intracerebral hemorrhage. Oxid Med Cell Longev (2021) 2021:6630281. doi: 10.1155/2021/6630281
77. Cheng Y, Chen B, Xie W, Chen Z, Yang G, Cai Y, et al. Ghrelin attenuates secondary brain injury following intracerebral hemorrhage by inhibiting nlrp3 inflammasome activation and promoting nrf2/are signaling pathway in mice. Int Immunopharmacol (2020) 79:106180. doi: 10.1016/j.intimp.2019.106180
78. Yan Y, Jiang W, Liu L, Wang X, Ding C, Tian Z, et al. Dopamine controls systemic inflammation through inhibition of nlrp3 inflammasome. Cell (2015) 160:62–73. doi: 10.1016/j.cell.2014.11.047
79. Al MA, Wu Y, Monalisa I, Jia C, Zhou K, Munir F, et al. Role of pyroptosis in spinal cord injury and its therapeutic implications. J Adv Res (2021) 28:97–109. doi: 10.1016/j.jare.2020.08.004
80. Wang S, Yuan YH, Chen NH, Wang HB. The mechanisms of nlrp3 inflammasome/pyroptosis activation and their role in parkinson's disease. Int Immunopharmacol (2019) 67:458–64. doi: 10.1016/j.intimp.2018.12.019
81. Li Q, Wang Z, Xing H, Wang Y, Guo Y. Exosomes derived from mir-188-3p-modified adipose-derived mesenchymal stem cells protect parkinson's disease. Mol Ther Nucleic Acids (2021) 23:1334–44. doi: 10.1016/j.omtn.2021.01.022
82. Cai M, Zhuang W, Lv E, Liu Z, Wang Y, Zhang W, et al. Kaemperfol alleviates pyroptosis and microglia-mediated neuroinflammation in parkinson's disease via inhibiting p38mapk/nf-kappab signaling pathway. Neurochem Int (2022) 152:105221. doi: 10.1016/j.neuint.2021.105221
83. Wang T, Nowrangi D, Yu L, Lu T, Tang J, Han B, et al. Activation of dopamine d1 receptor decreased nlrp3-mediated inflammation in intracerebral hemorrhage mice. J Neuroinflamm (2018) 15:2. doi: 10.1186/s12974-017-1039-7
84. Gan H, Zhang L, Chen H, Xiao H, Wang L, Zhai X, et al. The pivotal role of the nlrc4 inflammasome in neuroinflammation after intracerebral hemorrhage in rats. Exp Mol Med (2021) 53:1807–18. doi: 10.1038/s12276-021-00702-y
85. Chen G, Gao C, Yan Y, Wang T, Luo C, Zhang M, et al. Inhibiting er stress weakens neuronal pyroptosis in a mouse acute hemorrhagic stroke model. Mol Neurobiol (2020) 57:5324–35. doi: 10.1007/s12035-020-02097-9
86. Jia P, He J, Li Z, Wang J, Jia L, Hao R, et al. Profiling of blood-brain barrier disruption in mouse intracerebral hemorrhage models: collagenase injection vs. Autologous arterial whole Blood infusion. Front Cell Neurosci (2021) 15:699736. doi: 10.3389/fncel.2021.699736
87. Yang H, Ni W, Jiang H, Lei Y, Su J, Gu Y, et al. Histone deacetylase inhibitor scriptaid alleviated neurological dysfunction after experimental intracerebral hemorrhage in mice. Behav Neurol (2018) 2018:6583267. doi: 10.1155/2018/6583267
88. Yang J, Li Q, Wang Z, Qi C, Han X, Lan X, et al. Multimodality mri assessment of grey and white matter injury and blood-brain barrier disruption after intracerebral haemorrhage in mice. Sci Rep (2017) 7:40358. doi: 10.1038/srep40358
89. Dang G, Yang Y, Wu G, Hua Y, Keep RF, Xi G. Early erythrolysis in the hematoma after experimental intracerebral hemorrhage. Transl Stroke Res (2017) 8:174–82. doi: 10.1007/s12975-016-0505-3
90. Li Q, Weiland A, Chen X, Lan X, Han X, Durham F, et al. Ultrastructural characteristics of neuronal death and white matter injury in mouse brain tissues after intracerebral hemorrhage: coexistence of ferroptosis, autophagy, and necrosis. Front Neurol (2018) 9:581. doi: 10.3389/fneur.2018.00581
91. Qin D, Wang J, Le A, Wang TJ, Chen X, Wang J. Traumatic brain injury: ultrastructural features in neuronal ferroptosis, glial cell activation and polarization, and blood-brain barrier breakdown. Cells-Basel (2021) 10:1009. doi: 10.3390/cells10051009
92. Wan J, Ren H, Wang J. Iron toxicity, lipid peroxidation and ferroptosis after intracerebral haemorrhage. Stroke Vasc Neurol (2019) 4:93–5. doi: 10.1136/svn-2018-000205
93. Grace PM, Strand KA, Galer EL, Urban DJ, Wang X, Baratta MV, et al. Morphine paradoxically prolongs neuropathic pain in rats by amplifying spinal nlrp3 inflammasome activation. Proc Natl Acad Sci USA (2016) 113:E3441–50. doi: 10.1073/pnas.1602070113
94. Weng X, Tan Y, Chu X, Wu XF, Liu R, Tian Y, et al. N-methyl-d-aspartic acid receptor 1 (nmdar1) aggravates secondary inflammatory damage induced by hemin-nlrp3 pathway after intracerebral hemorrhage. Chin J Traumatol (2015) 18:254–8. doi: 10.1016/j.cjtee.2015.11.010
95. Lin S, Yin Q, Zhong Q, Lv FL, Zhou Y, Li JQ, et al. Heme activates tlr4-mediated inflammatory injury via myd88/trif signaling pathway in intracerebral hemorrhage. J Neuroinflamm (2012) 9:46. doi: 10.1186/1742-2094-9-46
96. Segovia J, Sabbah A, Mgbemena V, Tsai SY, Chang TH, Berton MT, et al. Tlr2/myd88/nf-kappab pathway, reactive oxygen species, potassium efflux activates nlrp3/asc inflammasome during respiratory syncytial virus infection. PloS One (2012) 7:e29695. doi: 10.1371/journal.pone.0029695
97. Yang Z, Zhong L, Xian R, Yuan B. Microrna-223 regulates inflammation and brain injury via feedback to nlrp3 inflammasome after intracerebral hemorrhage. Mol Immunol (2015) 65:267–76. doi: 10.1016/j.molimm.2014.12.018
98. Camejo G, Halberg C, Manschik-Lundin A, Hurt-Camejo E, Rosengren B, Olsson H, et al. Hemin binding and oxidation of lipoproteins in serum: mechanisms and effect on the interaction of ldl with human macrophages. J Lipid Res (1998) 39:755–66.
99. Wang J, Dore S. Heme oxygenase-1 exacerbates early brain injury after intracerebral haemorrhage. Brain (2007) 130:1643–52. doi: 10.1093/brain/awm095
100. Wang J, Dore S. Heme oxygenase 2 deficiency increases brain swelling and inflammation after intracerebral hemorrhage. Neuroscience (2008) 155:1133–41. doi: 10.1016/j.neuroscience.2008.07.004
101. Lee MY, Kim SY, Choi JS, Lee IH, Choi YS, Jin JY, et al. Upregulation of haptoglobin in reactive astrocytes after transient forebrain ischemia in rats. J Cereb Blood Flow Metab (2002) 22:1176–80. doi: 10.1097/01.wcb.0000037989.07114.d1
102. Grinberg LN, O'Brien PJ, Hrkal Z. The effects of heme-binding proteins on the peroxidative and catalatic activities of hemin. Free Radic Biol Med (1999) 27:214–9. doi: 10.1016/s0891-5849(99)00082-9
103. Zhao X, Aronowski J. Nrf2 to pre-condition the brain against injury caused by products of hemolysis after ich. Transl Stroke Res (2013) 4:71–5. doi: 10.1007/s12975-012-0245-y
104. Mohammed TS, Tsai ST, Hung HY, Hu WF, Pang CY, Chen SY, et al. A role for endoplasmic reticulum stress in intracerebral hemorrhage. Cells-Basel (2020) 9:750. doi: 10.3390/cells9030750
105. Xu F, Shen G, Su Z, He Z, Yuan L. Glibenclamide ameliorates the disrupted blood-brain barrier in experimental intracerebral hemorrhage by inhibiting the activation of nlrp3 inflammasome. Brain Behav (2019) 9:e1254. doi: 10.1002/brb3.1254
106. Deroide N, Li X, Lerouet D, Van Vre E, Baker L, Harrison J, et al. Mfge8 inhibits inflammasome-induced il-1beta production and limits postischemic cerebral injury. J Clin Invest (2013) 123:1176–81. doi: 10.1172/JCI65167
107. Lan X, Han X, Li Q, Yang QW, Wang J. Modulators of microglial activation and polarization after intracerebral haemorrhage. Nat Rev Neurol (2017) 13:420–33. doi: 10.1038/nrneurol.2017.69
108. Lan X, Han X, Liu X, Wang J. Inflammatory responses after intracerebral hemorrhage: from cellular function to therapeutic targets. J Cereb Blood Flow Metab (2019) 39:184–6. doi: 10.1177/0271678X18805675
109. Xiong XY, Liu L, Yang QW. Functions and mechanisms of microglia/macrophages in neuroinflammation and neurogenesis after stroke. Prog Neurobiol (2016) 142:23–44. doi: 10.1016/j.pneurobio.2016.05.001
110. Xia W, Lu Z, Chen W, Zhou J, Zhao Y. Excess fatty acids induce pancreatic acinar cell pyroptosis through macrophage m1 polarization. BMC Gastroenterol (2022) 22:72. doi: 10.1186/s12876-022-02146-8
111. Ye Y, Jin T, Zhang X, Zeng Z, Ye B, Wang J, et al. Meisoindigo protects against focal cerebral ischemia-reperfusion injury by inhibiting nlrp3 inflammasome activation and regulating microglia/macrophage polarization via tlr4/nf-kappab signaling pathway. Front Cell Neurosci (2019) 13:553. doi: 10.3389/fncel.2019.00553
112. Jiao Y, Zhang T, Zhang C, Ji H, Tong X, Xia R, et al. Exosomal mir-30d-5p of neutrophils induces m1 macrophage polarization and primes macrophage pyroptosis in sepsis-related acute lung injury. Crit Care (2021) 25:356. doi: 10.1186/s13054-021-03775-3
113. Ran Y, Su W, Gao F, Ding Z, Yang S, Ye L, et al. Curcumin ameliorates white matter injury after ischemic stroke by inhibiting microglia/macrophage pyroptosis through nf-kappab suppression and nlrp3 inflammasome inhibition. Oxid Med Cell Longev (2021) 2021:1552127. doi: 10.1155/2021/1552127
114. Zhang J, Liu X, Wan C, Liu Y, Wang Y, Meng C, et al. Nlrp3 inflammasome mediates m1 macrophage polarization and il-1beta production in inflammatory root resorption. J Clin Periodontol (2020) 47:451–60. doi: 10.1111/jcpe.13258
115. Chen W, Guo C, Huang S, Jia Z, Wang J, Zhong J, et al. Mitoq attenuates brain damage by polarizing microglia towards the m2 phenotype through inhibition of the nlrp3 inflammasome after ich. Pharmacol Res (2020) 161:105122. doi: 10.1016/j.phrs.2020.105122
116. Lin X, Ye H, Siaw-Debrah F, Pan S, He Z, Ni H, et al. Ac-Yvad-cmk inhibits pyroptosis and improves functional outcome after intracerebral hemorrhage. BioMed Res Int (2018) 2018:3706047. doi: 10.1155/2018/3706047
117. Yu J, Li S, Qi J, Chen Z, Wu Y, Guo J, et al. Cleavage of gsdme by caspase-3 determines lobaplatin-induced pyroptosis in colon cancer cells. Cell Death Dis (2019) 10:193. doi: 10.1038/s41419-019-1441-4
118. Zheng X, Zhong T, Ma Y, Wan X, Qin A, Yao B, et al. Bnip3 mediates doxorubicin-induced cardiomyocyte pyroptosis via caspase-3/gsdme. Life Sci (2020) 242:117186. doi: 10.1016/j.lfs.2019.117186
119. Wang Y, Yin B, Li D, Wang G, Han X, Sun X. Gsdme mediates caspase-3-dependent pyroptosis in gastric cancer. Biochem Biophys Res Commun (2018) 495:1418–25. doi: 10.1016/j.bbrc.2017.11.156
120. Zhang CC, Li CG, Wang YF, Xu LH, He XH, Zeng QZ, et al. Chemotherapeutic paclitaxel and cisplatin differentially induce pyroptosis in a549 lung cancer cells via caspase-3/gsdme activation. Apoptosis (2019) 24:312–25. doi: 10.1007/s10495-019-01515-1
121. Porter AG, Janicke RU. Emerging roles of caspase-3 in apoptosis. Cell Death Differ (1999) 6:99–104. doi: 10.1038/sj.cdd.4400476
122. Sun DB, Xu MJ, Chen QM, Hu HT. Significant elevation of serum caspase-3 levels in patients with intracerebral hemorrhage. Clin Chim Acta (2017) 471:62–7. doi: 10.1016/j.cca.2017.05.021
123. Xu H, Cao J, Xu J, Li H, Shen H, Li X, et al. Gata-4 regulates neuronal apoptosis after intracerebral hemorrhage via the nf-kappab/bax/caspase-3 pathway both in vivo and in vitro. Exp Neurol (2019) 315:21–31. doi: 10.1016/j.expneurol.2019.01.018
124. Liu X, Wu G, Tang N, Li L, Liu C, Wang F, et al. Glymphatic drainage blocking aggravates brain edema, neuroinflammation via modulating tnf-alpha, il-10, and aqp4 after intracerebral hemorrhage in rats. Front Cell Neurosci (2021) 15:784154. doi: 10.3389/fncel.2021.784154
125. Yao ST, Cao F, Chen JL, Chen W, Fan RM, Li G, et al. Nlrp3 is required for complement-mediated caspase-1 and il-1beta activation in ich. J Mol Neurosci (2017) 61:385–95. doi: 10.1007/s12031-016-0874-9
126. Wu XY, Li KT, Yang HX, Yang B, Lu X, Zhao LD, et al. Complement c1q synergizes with ptx3 in promoting nlrp3 inflammasome over-activation and pyroptosis in rheumatoid arthritis. J Autoimmun (2020) 106:102336. doi: 10.1016/j.jaut.2019.102336
127. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular lps. Nature (2014) 514:187–92. doi: 10.1038/nature13683
128. Wang H, Lv D, Jiang S, Hou Q, Zhang L, Li S, et al. Complement induces podocyte pyroptosis in membranous nephropathy by mediating mitochondrial dysfunction. Cell Death Dis (2022) 13:281. doi: 10.1038/s41419-022-04737-5
129. Jiang Y, Li J, Teng Y, Sun H, Tian G, He L, et al. Complement receptor c5ar1 inhibition reduces pyroptosis in hdpp4-transgenic mice infected with mers-cov. Viruses (2019) 11:39. doi: 10.3390/v11010039
130. Ren H, Kong Y, Liu Z, Zang D, Yang X, Wood K, et al. Selective nlrp3 (pyrin domain-containing protein 3) inflammasome inhibitor reduces brain injury after intracerebral hemorrhage. Stroke (2018) 49:184–92. doi: 10.1161/STROKEAHA.117.018904
131. Gu L, Sun M, Li R, Zhang X, Tao Y, Yuan Y, et al. Didymin suppresses microglia pyroptosis and neuroinflammation through the asc/caspase-1/gsdmd pathway following experimental intracerebral hemorrhage. Front Immunol (2022) 13:810582. doi: 10.3389/fimmu.2022.810582
132. Li X, Wang T, Zhang D, Li H, Shen H, Ding X, et al. Andrographolide ameliorates intracerebral hemorrhage induced secondary brain injury by inhibiting neuroinflammation induction. Neuropharmacology (2018) 141:305–15. doi: 10.1016/j.neuropharm.2018.09.015
133. Liang H, Sun Y, Gao A, Zhang N, Jia Y, Yang S, et al. Ac-Yvad-cmk improves neurological function by inhibiting caspase-1-mediated inflammatory response in the intracerebral hemorrhage of rats. Int Immunopharmacol (2019) 75:105771. doi: 10.1016/j.intimp.2019.105771
134. Miao H, Jiang Y, Geng J, Zhang B, Zhu G, Tang J. Edaravone administration confers neuroprotection after experimental intracerebral hemorrhage in rats via nlrp3 suppression. J Stroke Cerebrovasc Dis (2020) 29:104468. doi: 10.1016/j.jstrokecerebrovasdis.2019.104468
135. Wang S, Yao Q, Wan Y, Wang J, Huang C, Li D, et al. Adiponectin reduces brain injury after intracerebral hemorrhage by reducing nlrp3 inflammasome expression. Int J Neurosci (2020) 130:301–8. doi: 10.1080/00207454.2019.1679810
136. Zhou H, Zhang C, Huang C. Verbascoside attenuates acute inflammatory injury caused by an intracerebral hemorrhage through the suppression of nlrp3. Neurochem Res (2021) 46:770–7. doi: 10.1007/s11064-020-03206-9
137. Cheng Y, Wei Y, Yang W, Song Y, Shang H, Cai Y, et al. Cordycepin confers neuroprotection in mice models of intracerebral hemorrhage via suppressing nlrp3 inflammasome activation. Metab Brain Dis (2017) 32:1133–45. doi: 10.1007/s11011-017-0003-7
138. Chen X, Zhou Y, Wang S, Wang W. Mechanism of baicalein in brain injury after intracerebral hemorrhage by inhibiting the ros/nlrp3 inflammasome pathway. Inflammation (2022) 45:590–602. doi: 10.1007/s10753-021-01569-x
139. Yuan R, Fan H, Cheng S, Gao W, Xu X, Lv S, et al. Silymarin prevents nlrp3 inflammasome activation and protects against intracerebral hemorrhage. BioMed Pharmacother (2017) 93:308–15. doi: 10.1016/j.biopha.2017.06.018
140. Wan SY, Li GS, Tu C, Chen WL, Wang XW, Wang YN, et al. Micronar-194-5p hinders the activation of nlrp3 inflammasomes and alleviates neuroinflammation during intracerebral hemorrhage by blocking the interaction between traf6 and nlrp3. Brain Res (2021) 1752:147228. doi: 10.1016/j.brainres.2020.147228
141. Fang Y, Hong X. Mir-124-3p inhibits microglial secondary inflammation after basal ganglia hemorrhage by targeting traf6 and repressing the activation of nlrp3 inflammasome. Front Neurol (2021) 12:653321. doi: 10.3389/fneur.2021.653321
142. Hu L, Zhang H, Wang B, Ao Q, He Z. Microrna-152 attenuates neuroinflammation in intracerebral hemorrhage by inhibiting thioredoxin interacting protein (txnip)-mediated nlrp3 inflammasome activation. Int Immunopharmacol (2020) 80:106141. doi: 10.1016/j.intimp.2019.106141
143. Chen X, Xiang X, Xie T, Chen Z, Mou Y, Gao Z, et al. Memantine protects blood-brain barrier integrity and attenuates neurological deficits through inhibiting nitric oxide synthase ser1412 phosphorylation in intracerebral hemorrhage rats: involvement of peroxynitrite-related matrix metalloproteinase-9/nlrp3 inflammasome activation. Neuroreport (2021) 32:228–37. doi: 10.1097/WNR.0000000000001577
144. Ding H, Jia Y, Lv H, Chang W, Liu F, Wang D. Extracellular vesicles derived from bone marrow mesenchymal stem cells alleviate neuroinflammation after diabetic intracerebral hemorrhage via the mir-183-5p/pdcd4/nlrp3 pathway. J Endocrinol Invest (2021) 44:2685–98. doi: 10.1007/s40618-021-01583-8
145. Yang X, Sun J, Kim TJ, Kim YJ, Ko SB, Kim CK, et al. Pretreatment with low-dose fimasartan ameliorates nlrp3 inflammasome-mediated neuroinflammation and brain injury after intracerebral hemorrhage. Exp Neurol (2018) 310:22–32. doi: 10.1016/j.expneurol.2018.08.013
146. Kang R, Zeng L, Zhu S, Xie Y, Liu J, Wen Q, et al. Lipid peroxidation drives gasdermin d-mediated pyroptosis in lethal polymicrobial sepsis. Cell Host Microbe (2018) 24:97–108. doi: 10.1016/j.chom.2018.05.009
147. Humphries F, Shmuel-Galia L, Ketelut-Carneiro N, Li S, Wang B, Nemmara VV, et al. Succination inactivates gasdermin d and blocks pyroptosis. Science (2020) 369:1633–7. doi: 10.1126/science.abb9818
148. Ruhl S, Shkarina K, Demarco B, Heilig R, Santos JC, Broz P. Escrt-dependent membrane repair negatively regulates pyroptosis downstream of gsdmd activation. Science (2018) 362:956–60. doi: 10.1126/science.aar7607
149. Rathkey JK, Zhao J, Liu Z, Chen Y, Yang J, Kondolf HC, et al. Chemical disruption of the pyroptotic pore-forming protein gasdermin d inhibits inflammatory cell death and sepsis. Sci Immunol (2018) 3:eaat6689. doi: 10.1126/sciimmunol.aat2738
150. Sollberger G, Choidas A, Burn GL, Habenberger P, Di Lucrezia R, Kordes S, et al. Gasdermin d plays a vital role in the generation of neutrophil extracellular traps. Sci Immunol (2018) 3. doi: 10.1126/sciimmunol.aar6689
151. Hu JJ, Liu X, Xia S, Zhang Z, Zhang Y, Zhao J, et al. Fda-approved disulfiram inhibits pyroptosis by blocking gasdermin d pore formation. Nat Immunol (2020) 21:736–45. doi: 10.1038/s41590-020-0669-6
152. Lin L, Li R, Cai M, Huang J, Huang W, Guo Y, et al. Andrographolide ameliorates liver fibrosis in mice: involvement of tlr4/nf-kappab and tgf-beta1/smad2 signaling pathways. Oxid Med Cell Longev (2018) 2018:7808656. doi: 10.1155/2018/7808656
153. Lee TY, Chang HH, Wen CK, Huang TH, Chang YS. Modulation of thioacetamide-induced hepatic inflammations, angiogenesis and fibrosis by andrographolide in mice. J Ethnopharmacol (2014) 158 Pt A:423–30. doi: 10.1016/j.jep.2014.10.056
154. Vinukonda G, Liao Y, Hu F, Ivanova L, Purohit D, Finkel DA, et al. Human cord blood-derived unrestricted somatic stem cell infusion improves neurobehavioral outcome in a rabbit model of intraventricular hemorrhage. Stem Cells Transl Med (2019) 8:1157–69. doi: 10.1002/sctm.19-0082
155. Feng M, Zhu H, Zhu Z, Wei J, Lu S, Li Q, et al. Serial 18f-fdg pet demonstrates benefit of human mesenchymal stem cells in treatment of intracerebral hematoma: a translational study in a primate model. J Nucl Med (2011) 52:90–7. doi: 10.2967/jnumed.110.080325
156. Gao L, Xu W, Li T, Chen J, Shao A, Yan F, et al. Stem cell therapy: a promising therapeutic method for intracerebral hemorrhage. Cell Transplant (2018) 27:1809–24. doi: 10.1177/0963689718773363
157. Vaquero J, Otero L, Bonilla C, Aguayo C, Rico MA, Rodriguez A, et al. Cell therapy with bone marrow stromal cells after intracerebral hemorrhage: impact of platelet-rich plasma scaffolds. Cytotherapy (2013) 15:33–43. doi: 10.1016/j.jcyt.2012.10.005
158. Cheyuo C, Wu R, Zhou M, Jacob A, Coppa G, Wang P. Ghrelin suppresses inflammation and neuronal nitric oxide synthase in focal cerebral ischemia via the vagus nerve. Shock (2011) 35:258–65. doi: 10.1097/SHK.0b013e3181f48a37
159. Ersahin M, Toklu HZ, Erzik C, Cetinel S, Akakin D, Velioglu-Ogunc A, et al. The anti-inflammatory and neuroprotective effects of ghrelin in subarachnoid hemorrhage-induced oxidative brain damage in rats. J Neurotrauma (2010) 27:1143–55. doi: 10.1089/neu.2009.1210
160. Hao XK, Wu W, Wang CX, Xie GB, Li T, Wu HM, et al. Ghrelin alleviates early brain injury after subarachnoid hemorrhage via the pi3k/akt signaling pathway. Brain Res (2014) 1587:15–22. doi: 10.1016/j.brainres.2014.08.069
161. Lee J, Costantini TW, D'Mello R, Eliceiri BP, Coimbra R, Bansal V. Altering leukocyte recruitment following traumatic brain injury with ghrelin therapy. J Trauma Acute Care Surg (2014) 77:709–15. doi: 10.1097/TA.0000000000000445
162. Xia S, Hollingsworth LT, Wu H. Mechanism and regulation of gasdermin-mediated cell death. Cold Spring Harb Perspect Biol (2020) 12:a036400. doi: 10.1101/cshperspect.a036400
163. Sun L, Wang H, Wang Z, He S, Chen S, Liao D, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of rip3 kinase. Cell (2012) 148:213–27. doi: 10.1016/j.cell.2011.11.031
164. Jiang C, Wang Y, Hu Q, Shou J, Zhu L, Tian N, et al. Immune changes in peripheral blood and hematoma of patients with intracerebral hemorrhage. FASEB J (2020) 34:2774–91. doi: 10.1096/fj.201902478R
Keywords: intracerebral hemorrhage, pyroptosis, inflammasome, secondary immune-inflammatory response, caspase-1, NLRP3
Citation: Song D, Yeh C-T, Wang J and Guo F (2022) Perspectives on the mechanism of pyroptosis after intracerebral hemorrhage. Front. Immunol. 13:989503. doi: 10.3389/fimmu.2022.989503
Received: 11 July 2022; Accepted: 17 August 2022;
Published: 05 September 2022.
Edited by:
Sermin Genc, Dokuz Eylul University, TurkeyReviewed by:
Prativa Sherchan, Loma Linda University, United StatesCopyright © 2022 Song, Yeh, Wang and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fuyou Guo, Y2h5b3U2NjZAaG90bWFpbC5jb20=; Jian Wang, amlhbndhbmcyMDIwQG91dGxvb2suY29t; Chi-Tai Yeh, Y3R5ZWh0bXVAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.