
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 25 August 2022
Sec. Vaccines and Molecular Therapeutics
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.985450
This article is part of the Research Topic Precision Vaccinology for Infectious Diseases View all 26 articles
 Sara Aiman1
Sara Aiman1 Yahya Alhamhoom2
Yahya Alhamhoom2 Fawad Ali3
Fawad Ali3 Noor Rahman4*
Noor Rahman4* Luca Rastrelli5
Luca Rastrelli5 Asifullah Khan4Qurat ul Ain Farooq1Abbas Ahmed6Asif Khan7
Asifullah Khan4Qurat ul Ain Farooq1Abbas Ahmed6Asif Khan7 Chunhua Li1*
Chunhua Li1*The emerging monkeypox virus (MPXV) is a zoonotic orthopoxvirus that causes infections in humans similar to smallpox. Since May 2022, cases of monkeypox (MPX) have been increasingly reported by the World Health Organization (WHO) worldwide. Currently, there are no clinically validated treatments for MPX infections. In this study, an immunoinformatics approach was used to identify potential vaccine targets against MPXV. A total of 190 MPXV-2022 proteins were retrieved from the ViPR database and subjected to various analyses including antigenicity, allergenicity, toxicity, solubility, IFN-γ, and virulence. Three outer membrane and extracellular proteins were selected based on their respective parameters to predict B-cell and T-cell epitopes. The epitopes are conserved among different strains of MPXV and the population coverage is 100% worldwide, which will provide broader protection against various strains of the virus globally. Nine overlapping MHC-I, MHC-II, and B-cell epitopes were selected to design multi-epitope vaccine constructs linked with suitable linkers in combination with different adjuvants to enhance the immune responses of the vaccine constructs. Molecular modeling and structural validation ensured high-quality 3D structures of vaccine constructs. Based on various immunological and physiochemical properties and docking scores, MPXV-V2 was selected for further investigation. In silico cloning revealed a high level of gene expression for the MPXV-V2 vaccine within the bacterial expression system. Immune and MD simulations confirmed the molecular stability of the MPXV-V2 construct, with high immune responses within the host cell. These results may aid in the development of experimental vaccines against MPXV with increased potency and improved safety.
The monkeypox virus (MPXV) is an orthopoxvirus belonging to the Poxviridae family that causes diseases in humans and animals. Monkeypox (MPX) is a zoonotic disease in which the virus is usually transmitted through animal-human contact, with symptoms similar to smallpox but with reduced mortality (1). MPXV is rather large (200-250 nanometers), brick-shaped with a lipoprotein envelope, and a linear double-stranded DNA genome (2). Currently, MPXVs are classified into two clades. The Central African Congo Basin clade has been reported more frequently than the West African clade. The Congo Basin clade has recorded occurrences of human-to-human transmission, whereas the West African clade has not (3). In May 2022, MPXV cases were reported by the World Health Organization (WHO). Several cases of MPX have been identified in geographically diverse countries. Human-to-human transmission of MPXV occurs because of close contact with lesions, bodily fluids, respiratory droplets, and infected objects such as bedding. Consumption of undercooked meat and other diseased animal products is a potential risk factor (4). The early symptoms of MPX include fatigue, headache, fever, myalgia, and lymphadenopathy- a key feature that differentiates MPX from smallpox. After 1-2 days, mucosal lesions appear in the mouth followed by centrifugally concentrated skin lesions on the face, hands, and feet. The rash can spread to the rest of the body and the number of lesions can vary from a few to thousands. MPX usually takes 6-13 days to incubate, but it can take up to 21 days (5). MPX is often self-limiting, although it can be severe in some people, including children, pregnant women, and individuals with immunosuppression due to other medical conditions. According to reports by the Centers for Disease Control and Prevention (CDC) and World Health Organization (WHO) 2022, homosexual men comprise a large number of cases. However, anyone in close contact with an infected individual is at high risk of infection.
The first isolate of MPXV was identified in 1958 when monkeys shipped from Singapore fell sick in a research facility in Denmark (6). However, the first human case of MPX was confirmed in 1970 when the virus was isolated from a child suspected of having smallpox in the Democratic Republic of the Congo (7). Human MPX cases and sporadic clusters have also been reported outside of Africa. The first MPX outbreak in humans outside Africa was reported in 2003 in the midwest United States. This outbreak was linked to contact with prairie dogs housed in giant Gambian rats and dormice imported from Ghana. This has resulted in over 50 human cases of MPX in the U.S (8). A case of MPX was reported in a traveler from Nigeria to Israel in October 2018 (9). One case occurred in May 2019 when a man traveled from Nigeria to Singapore (10). Three members of a family traveling from Nigeria to the United Kingdom were reported to be infected with MPXV in May 2021 (11), one case was reported in July 2021 (Nigeria to Texas) (12), and one was reported in November 2021 (Nigeria to Maryland) (13). As of May 2022, clusters of human MPX have been reported in several non-endemic countries across the world (CDC and WHO, 2022) (Figure 1). This outbreak has been linked to a virus from the West African clade, which is commonly associated with milder symptoms, and in this case, human-to-human transmission has been suspected. Further investigations are underway to understand the epidemiology, sources of disease, and viral transmission patterns.
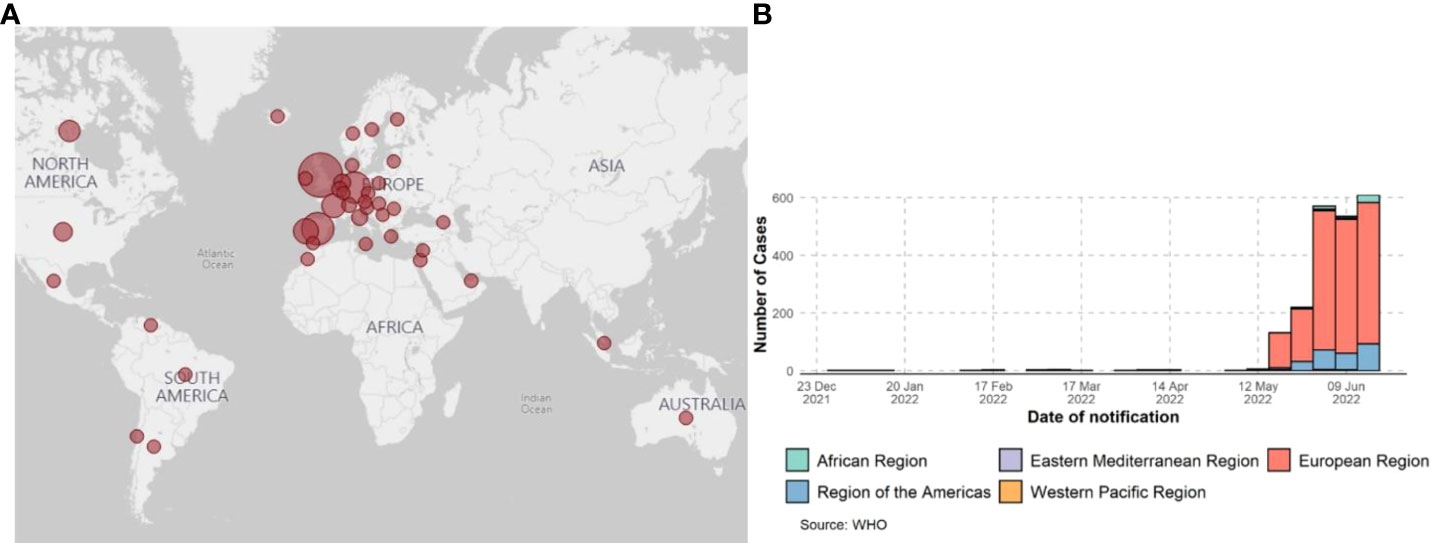
Figure 1 Global surveillance of MPX confirmed cases from January 2022 to 15 June 2022, data as of 15 June 2022. (A) CDC report; 2022 MPXV and Orthopoxvirus Outbreak Global Map - https://www.cdc.gov/. (B) Confirmed cases of MPXV by WHO report.
Vaccinia vaccination provided coincident immunity against MPXVs in the past; however, multiple observational studies have indicated approximately 85% efficiency in avoiding MPX. The eradication of smallpox following a lack of vaccine efforts has allowed MPX to gain clinical relevance. Currently, there are no clinically validated treatments for MPX infection (7). Therefore, new therapeutic strategies against emerging strains of the MPXV are urgently needed. The immune system plays a crucial role in pathogenesis as well as in the fight against viral infections and cancers. Advancements in immuno-informatics and bioinformatics techniques have facilitated the identification of novel therapeutic targets against a variety of pathogenic strains. Multi-epitope-based vaccination is an emerging strategy for the prevention of pathogenic diseases (14–17). The identification of immunogenic antigens is crucial for developing effective vaccines (18). Potent multi-epitope vaccine constructs have overlapping B- and T-cell epitopes in each antigenic peptide sequence, which induces cellular or humoral responses against target viral infections (14). In this study, we used reverse vaccinology and biophysical techniques to design a multi-epitope vaccine against MPXV infection. The entire protein sequence data of the latest 2022 strain of MPXV were used to identify lead B- and T-cell epitopes among the selected antigenic peptides. Overlapping lead epitopes have been used in the construction of chimeric vaccine structures. The efficacy of the designed vaccine constructs was evaluated using immuno-informatics, binding potential with immune receptor proteins, and in silico cloning into a host-vector expression system.
The systematic workflow followed in the current study to design a multi-epitope vaccine construct against monkeypox is shown in Figure 2.
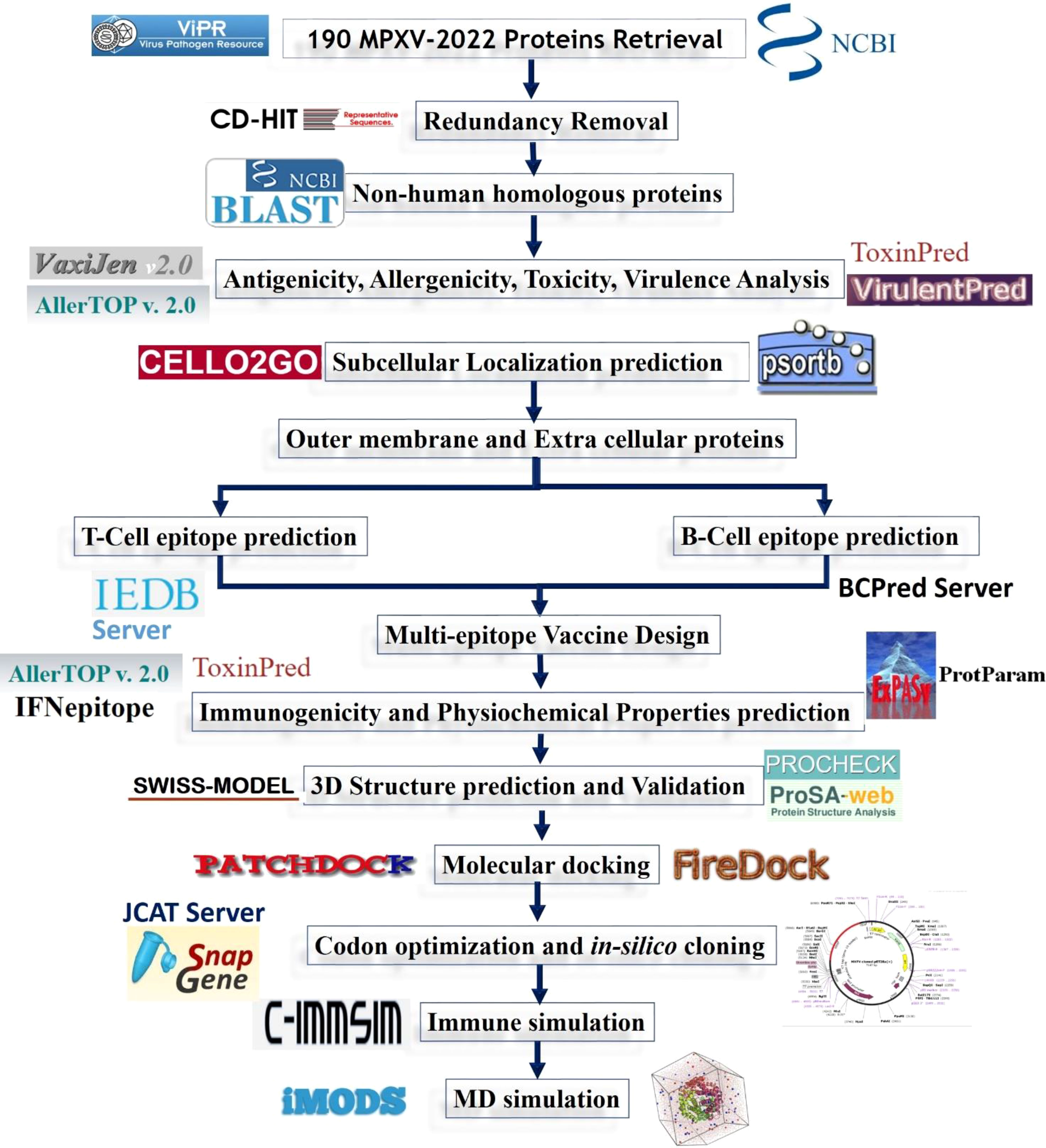
Figure 2 Systematic workflow of the designed study.
All proteins of the MPXV_USA_2022_FL001 strain were retrieved from the virus pathogen resource (VIPR) database (19) and confirmed with NCBI data entry. The entire amino acid sequences of the proteins were obtained in FASTA format based on data submitted to the NCBI in May 2022 with humans as the host of the virus. The CD-hit suite was used to remove redundancy and obtain non-paralogous protein sequences with a threshold of 80% sequence similarity (20). Sequences homologous to human proteins were removed using the NCBI-BLASTp program (21) with a threshold of setting an e-value cut-off of 10−4, percent identity ≤ 35, query coverage ≤ 70, bit score ≤ 100, and the rest of the parameters were set as default. AllerTOP2.0 web tool was used to determine the allergenicity of non-human homologous viral proteins (22). The antigenicity of the selected proteins was evaluated using the VaxiJen v 2.0 web server with a threshold of above 0.4 (23). The ToxinPred web server was used to assess the toxicity of shortlisted viral proteins (24). The virulence potentials of antigenic, non-toxic, and non-allergenic proteins were examined using the Virulentpred web tool (25). Non-human homologous viral proteins were subjected to subcellular localization using PSORTb version 3.0.2 (26) and CELLO2GO V.2.5 (27) web servers. Outer-membrane and extracellular proteins were selected as suitable vaccine candidates (17).
Proteins located in the outer membrane and extracellular region were selected as suitable vaccine candidates. The surface topology of these proteins was chosen to identify the immunogenic determinants (epitopes) for chimeric vaccine design. T cell epitopes are represented by major histocompatibility complex (MHC) molecules as class I (MHC-I) and II (MHC-II), which are recognized by two separate subsets of T-cells, CD8, and CD4, respectively (28). T-cell epitopes of the selected protein sequences were predicted using the Immune Epitope Database (IEDB) (29). The stabilized matrix method (SMM) scoring method and a neutral network-based tool (net MHC-1.1) were utilized for MHC-I and MHC-II prediction. The top binding overlapping epitopes with a calling criterion of IC50 200 nM and a length of 12-20 residues were prioritized (21, 30). The BCpred server was used to predict B-cell epitopes with the cut-off value set at > 0.8 and other default parameters. BCpred identifies linear B-cell epitopes that are critical for inducing a humoral immune response that stimulates B lymphocytes to produce antibodies (31). The IFNepitope web server was used to identify interferon-inducing epitopes from MHC-II binding epitopes. The IFNepitope server predicts the region of antigenic protein sequences that causes interferon-gamma (IFN-γ) induction. IFN-γ was predicted using the SVM model, and IFN-gamma vs. non-IFN-gamma model was selected for prediction (32). Epitopes positive for IFN-γ inducers were selected for further analysis.
Peptide vaccines are typically weak immunogens; however, integrating immunodominant epitopes to form a multi-epitope peptide vaccine can improve immunogenicity. B- and T-cell epitopes are immunodominant and crucial for inducing strong immunogenic responses to toxins and contagions (33). Overlapping B-cell, MHC-I, MHC-II, and IFN epitopes were selected based on cut-off values and manual comparisons. The rationale behind selecting overlapping B- and T-cell epitopes was to ensure that the designed vaccine could generate both humoral and cytotoxic immune responses (30). Effective synthesis of vaccine constructs is affected by variations in the expression and distribution of HLA alleles across different regions and ethnicities worldwide (34). The IEDB population coverage tool (http://tools.iedb.org/population/) was used to calculate the population coverage of the selected MHC-I and MHC-II epitopes and the associated HLA-binding alleles. Based on the distribution of HLA binding alleles, this tool calculates the population coverage of each epitope in various regions around the world (35). The IEDB epitope conservancy tool (http://tools.iedb.org/conservancy/) was used to determine the conservation of selected epitopes among various strains of MPXV.
A multi-epitope vaccine construct is also comprised of a strong immunostimulatory adjuvant to improve immunogenicity and activate long-term innate and adaptive immune responses (36). Four different adjuvants (HBHA protein, β-defensin, 50S ribosomal protein L7/L12 adjuvants, and HBHA conserved peptide sequences) were incorporated into the design of the multi-epitope vaccine. EAAAK linkers were used to join adjuvant sequences at the N-terminus of the vaccine constructs. HEYGAEALERAG and GGGS linkers were used to join the final epitopes (17). The EAAAK linker was used as a stiff spacer to bind the N terminus of the adjuvant to the epitope. HEYGAEALERAG and GGGS linkers are based on the method of Solanki & Tiwari (2018) as flexible linkers. All of these linkers are used for the best expression, bioactivity improvement, and to generate high immunogenic responses by the designed vaccine (37).
Various physicochemical properties of the vaccine constructs were evaluated using the ProtParam tool on the ExPASy server (38). The multi-epitope vaccine constructs were subjected to immunogenic analysis. The allergenicity of the vaccine constructs was predicted using the AllerTOP2.0 web tool (22). The antigenicity of the vaccine constructs was determined using the VaxiJen v2.0 web server with a threshold of above 0.4 (23). The SOLpro web server was used to predict the solubility of vaccine constructs (39). The toxicity of the vaccine constructs was evaluated using the ToxinPred server (40).
The three-dimensional structures of the multi-epitope vaccine constructs were predicted using the Swiss Model server (41). The predicted vaccine structures were refined using the DeepRefiner web server (42). The refined 3D structures of vaccine constructs were further validated using the ERRAT tool, PROCHECK suite of programs (43) of structure validation server SAVES v6.0 (https://saves.mbi.ucla.edu/) and ProSA-Web (https://prosa.services.came.sbg.ac.at/prosa.php). Understanding the binding pattern between designed vaccines and the TLR4 immune cell receptor is crucial to generate successful immune responses (44). The vaccine constructs were docked into the human TLR4 receptor (PDB ID: 4G8A) (45) using the PatchDock web server to evaluate immune receptor-vaccine interactions (46). All heteroatoms and cocrystallized ligands were removed from the TLR4 3D structure using Molecular Operating Environment (MOE) software for receptor preparation and preprocessing (47). The Fire Dock (Fast Interaction Refinement in Molecular Docking) server was used to refine the results of the molecular docking (48).
Codon optimization and in silico cloning were performed for the final vaccine construct (V2). The Java Codon Adaptation (JCat) tool was used for reverse translation and codon optimization of the vaccine construct to obtain higher expression of the cloned sequence in the E. coli expression system (49). JCat provides the percentage of GC content and codon adaptation index (CAI) to evaluate the expression potential of the cloned vaccine sequence. For favorable transcriptional and translational efficacy, the optimum value reported for CAI is 1 and the GC content is 30%-70% (17, 45). Finally, the Snapgene tool (https://www.snapgene.com/) was used for restriction cloning of the optimized vaccine construct in the E. coli expression system. pET28a_TIAL1 (E. coli plasmid) was retrieved from the Addgene server (https://www.addgene.org/).
Computational immune simulation of the finalized multi-epitope vaccine construct was performed using the C-ImmSim server to evaluate the immunogenic potential of the vaccine (50). The server uses a position-specific score matrix (PSSM) along with various machine learning approaches to predict multi-epitope vaccines and immunological interactions. It is a cellular-level agent-based model that obtains information about humoral and cellular responses of the mammalian immune system in response to antigens (51). Immune simulation performed for the vaccine construct was based on a protocol previously described by Bibi et al. (15). The simulation parameters were set as default for a period of 1 h, 84 h, and 168 h along with human host leukocyte antigens selection (HLA-A*0101 & A*0201, HLA-B*0702 & B*3901, and HLA-DRB1*0101 & DRB1*0401). The volume of the simulation was set at 10 and random seed at 12345 with vaccine injection not containing LPS. An immune simulation was conducted for 1000 simulation steps.
A molecular dynamics (MD) simulation study was conducted for the MPXV-V2-TLR4 complex with the best docking analysis results. The iMODS web server was used for MD simulations, energy minimization, and calculation of protein flexibility (52). iMODS is based on normal mode analysis (NMA) in the internal (dihedral) coordinates of macromolecules that naturally reproduce the collective functional movements of biological macromolecules. iMODS generates practical transition pathways between two homologous structures of the macromolecules based on these modes. The server defines potential conformational changes, detects elastic network potentials, models resolution with numerous coarse-grained atomic representations, and provides an improved affine-model-based arrow representation of the complex domain dynamics of macromolecules. The server investigates the structural dynamics of proteins and docked protein complexes with other proteins and ligands to provide values of deformability, eigenvalues, variance, B-factor (mobility profiles), covariance map, and elastic network data based on NMA (53). The docked PDB file for the MPXV-V2-TLR4 complex was submitted to the iMODS server and the results were obtained based on the default settings for all parameters.
All the proteins available for the latest strain of MPXV retrieved from the VIPR database yielded 190 protein sequences with NCBI and GenBank information (Supplementary file 1). Duplicate sequences were removed, and 186 non-paralogous sequences were obtained using CD-Hit recourse with a threshold of 80% sequence similarity (Supplementary file 2). A total of 43 sequences remained after the removal of human-homologous sequences. These proteins were subjected to further analysis based on antigenicity, allergenicity, toxicity, and virulence (Table S1). The subcellular localization identified three proteins, URP85109, URP84966, and URP85049, in the outer membrane and extracellular region to be prioritized as vaccine candidates (Figure 3; Table 1). Based on VaxiJen (23) prediction, the finalized proteins were highly antigenic with prediction scores of 0.5734, 0.5992, and 0.5316 respectively, at a threshold of >0.4. Moreover, the prioritized vaccine candidate proteins are non-allergenic, non-toxic, and involved in virulence, implying that host cell-induced immunogenic responses target only the virus rather than the host (40).
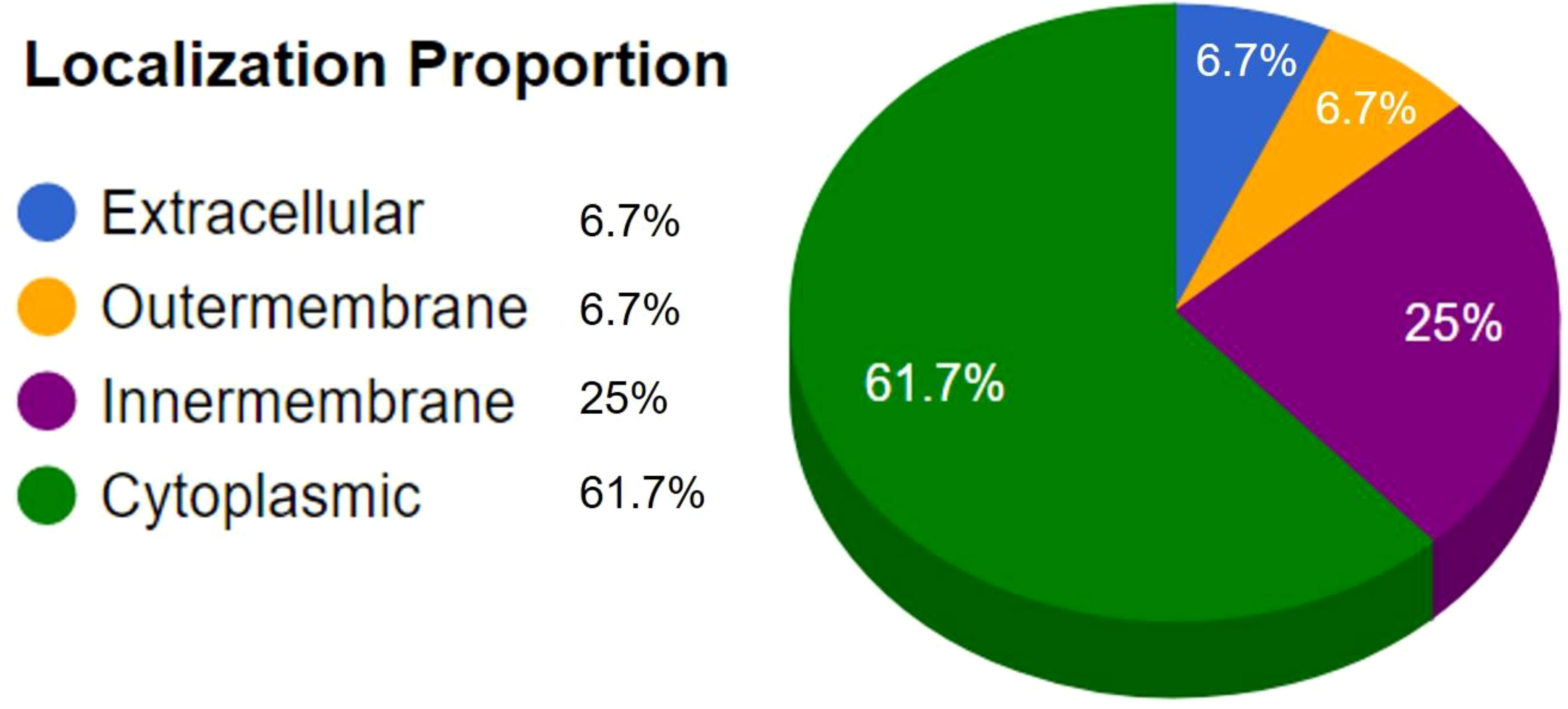
Figure 3 Subcellular localization results predicted by CELLO2GO.

Table 1 Subcellular localization results predicted by PSORTb web server.
The three proteins prioritized in this study were subjected to further analysis to identify lead epitopes for designing chimeric vaccine constructs against MPXV. T-cell (MHC-I and MHC-II) epitopes for the selected proteins were predicted based on an IC50 threshold of < 200 nM using the IEDB server. Overlapping B-cell epitopes were predicted with BCpred scores > 0.8 and 75% specificity. Three overlapping lead epitopes were predicted for each prioritized protein which was used to design vaccine constructs. Nine epitopes were prioritized based on their high antigenicity, IFN-positivity, low toxicity, and low allergenic reactions (Table 2). The ultimate objective was to identify lead epitopes with the capability to induce humoral and cell-mediated immunogenic responses and host interferons. The conservation of the selected epitopes was confirmed in various strains of MPXV. The use of conserved epitopes in the multi-epitope vaccine construct would provide broader protection against different strains of MPXV (54). The conservation of the selected epitopes is presented in Table 3. The selected epitopes demonstrated a 100% coverage of the global population (Table 4). The IEDB results revealed that the population coverage of the predicted epitopes was high in countries most affected by MPXV, including European countries, Israel, Canada, and the United States (Figure 4).
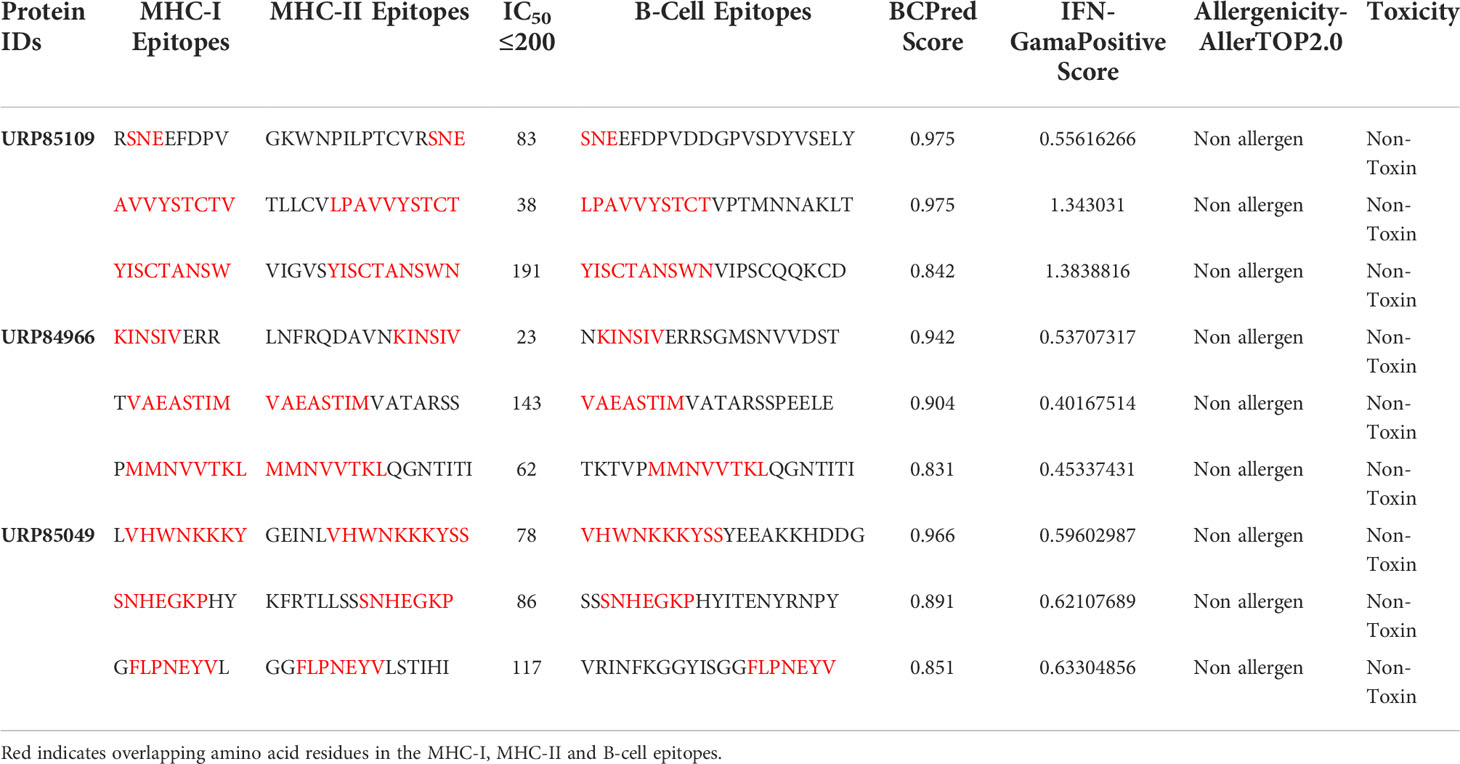
Table 2 T-cell and B-cell epitope prediction showing overlapping epitopes, IFN-γ epitope identification, allergenicity, and toxicity analysis.
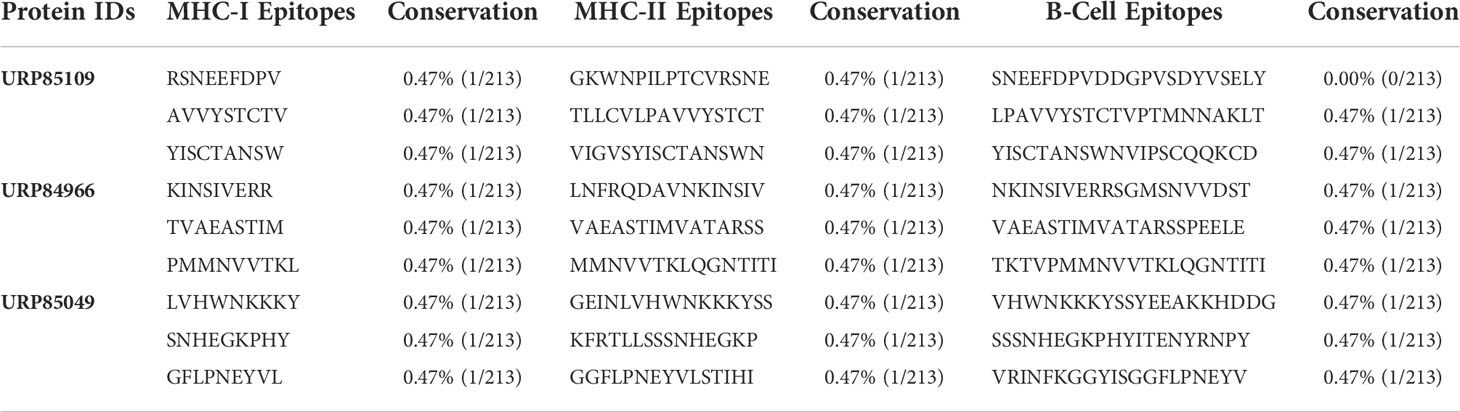
Table 3 Epitope conservancy of the selected B- and T- cell epitopes calculated by IEDB.
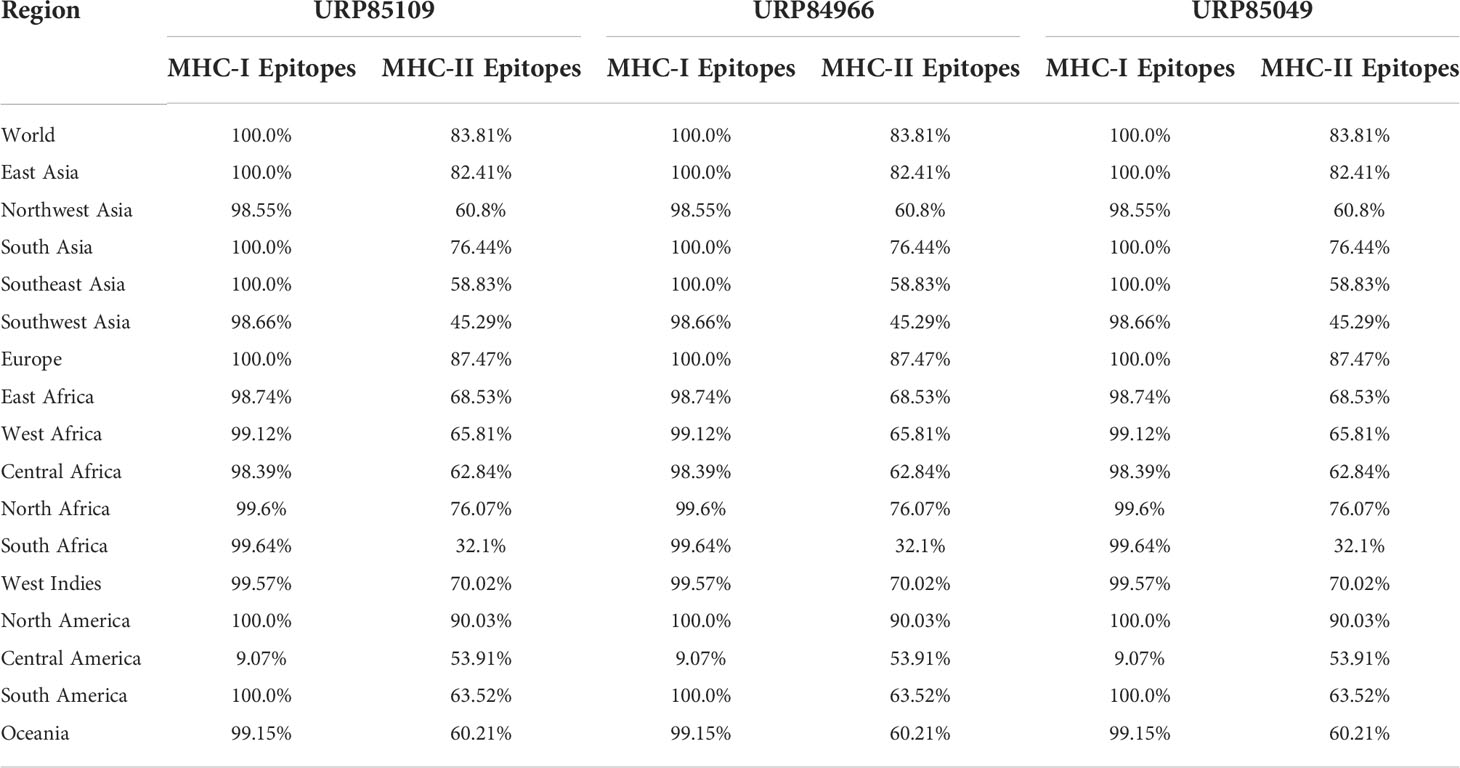
Table 4 Population coverage of MHC epitopes in different regions across the world calculated by IEDB.
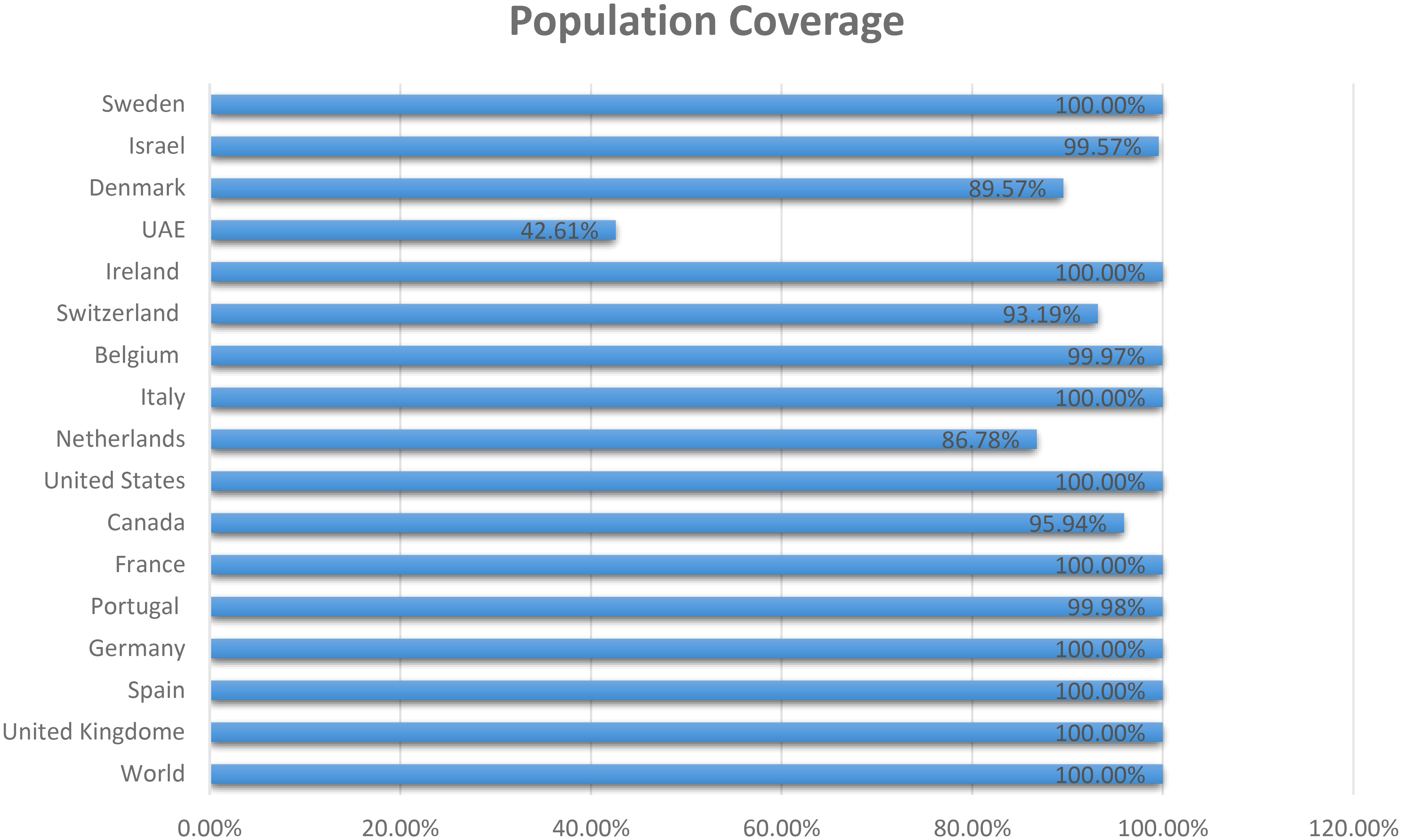
Figure 4 Population coverage of MHC epitopes in highly infected regions of the world calculated by IEDB.
Several combinations of selected lead epitopes were used to design a chimeric vaccine construct. The epitopes were linked using GGGS and HEYGAEALERAG linkers. Linkers provide stability to vaccine constructs and functional prevention of each epitope, allowing them to function independently after injection into the human host (55). The epitopes were linked to four different adjuvants: HBHA protein, beta-defensin, 50S ribosomal protein L7/L12 adjuvants, and HBHA conserved peptide sequences at the N-terminal with the help of EAAAK linkers to boost immunogenic responses. Moreover, PADRE peptide sequences have been incorporated into the designed vaccine constructs to avoid complications caused by HLA-DR variation in different populations worldwide. Previous studies have reported that vaccine constructs incorporating the PADRE peptide exhibit enhanced immune protection and high cytotoxic T lymphocyte (CTL) responses (56). The immunological properties of the vaccine constructs showed that all four constructs were non-allergenic and non-toxic. Antigenicity scores > 0.9, calculated by the ANTIGENpro server indicate the substantial antigenic nature of the multi-epitope vaccine constructs. ANTIGENpro evaluates antigenicity by 10-fold cross-validation of peptide sequences based on known datasets and identification of protective aspects of the antigenic sequences (57). VaxiJen 2.0 scores for all constructs ranged from 0.44 to 0.47, which is equal to the default threshold typically used for viruses. SOLpred scores > 0.97 indicated high solubility of the vaccine constructs upon expression (Table S2). The physiochemical properties of the vaccine constructs calculated by the ProtParam server indicated that the molecular weights of all these constructs ranged from ~41 kDa to ~54 kDa. GRAVY scores from -0.189 to -0.378 indicate the hydrophilic nature of the designed vaccine constructs. The theoretical pI values were in the range of 5.25 to 8.74. Aliphatic index scores ranged from 64.81 to 75.10, which indicates the thermostability of these constructs. The instability index scores were predicted to range from 30 to 40, indicating the stability of these constructs at various temperatures (Table 5). No significant changes were observed in the physicochemical properties of the constructs, as the amino acid content for all these constructs was similar, with the only difference being the adjuvant. Immunogenic and physiochemical property analysis signified the capability of vaccine constructs to initiate a significant immunogenic response within the human host. However, further experimental research is required to verify the accuracy of these results.
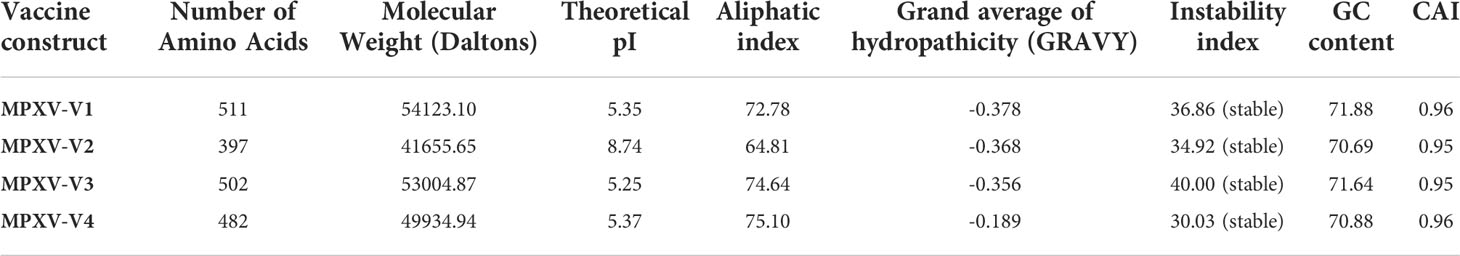
Table 5 Physiochemical properties of the vaccine constructs using ProtParam server and JCAT server.
A stable and functional three-dimensional structure of a vaccine is crucial for studying its molecular interactions with immune receptor proteins (58). Prediction of the vaccine constructs was anticipated by homology modeling approaches using the Swiss Model server (Figure 5A), refined by the DeepRefiner web server, and subjected to structural validation analysis (Figure 5B). The Ramachandran plots of the vaccine constructs revealed that 99.0% residues of V1 (HBHA), 84.5% residues of V2 (beta-defensin construct), 84.5% residues of V3 (HBHA conserved), and 89.4% residues of V4 (50S ribosomal) appeared in the favored region of the plots (Figure 5C). With the ERRAT, the overall quality factor of the refined vaccine constructs ranged from 85% to 100%. Similarly, the ProSA-web server evaluated the Z score for the vaccine constructs to be -5.94 to -1.66 (Figure 5D). The overall results from RAMPAGE, the ERRAT web tool, and the ProSa-web server determined the excellent quality of the designed vaccine 3D structures (Table 6). Molecular docking is an efficient method for identifying the optimal binding between designed vaccine constructs and receptor molecules. The PatchDock server (a blind docking technique) was used to dock the vaccine constructs with the surface human TLR4 immune receptor. The top ten results were further subjected to the FireDock web server to refine the docked complexes. FireDock resolves the protein flexibility issues that may occur during protein-peptide docking, enabling high-throughput complex refinement. The docking scores of all the vaccine-receptor complexes were similar. However, the V2 construct (i.e. Beta defensin adjuvant) showed the lowest binding energy (-12.08 kcal/mol) with the TLR4 receptor during this study (Figure 6). The docking analysis revealed strong binding affinities between the vaccine constructs and the TLR4 receptor (Table 7).
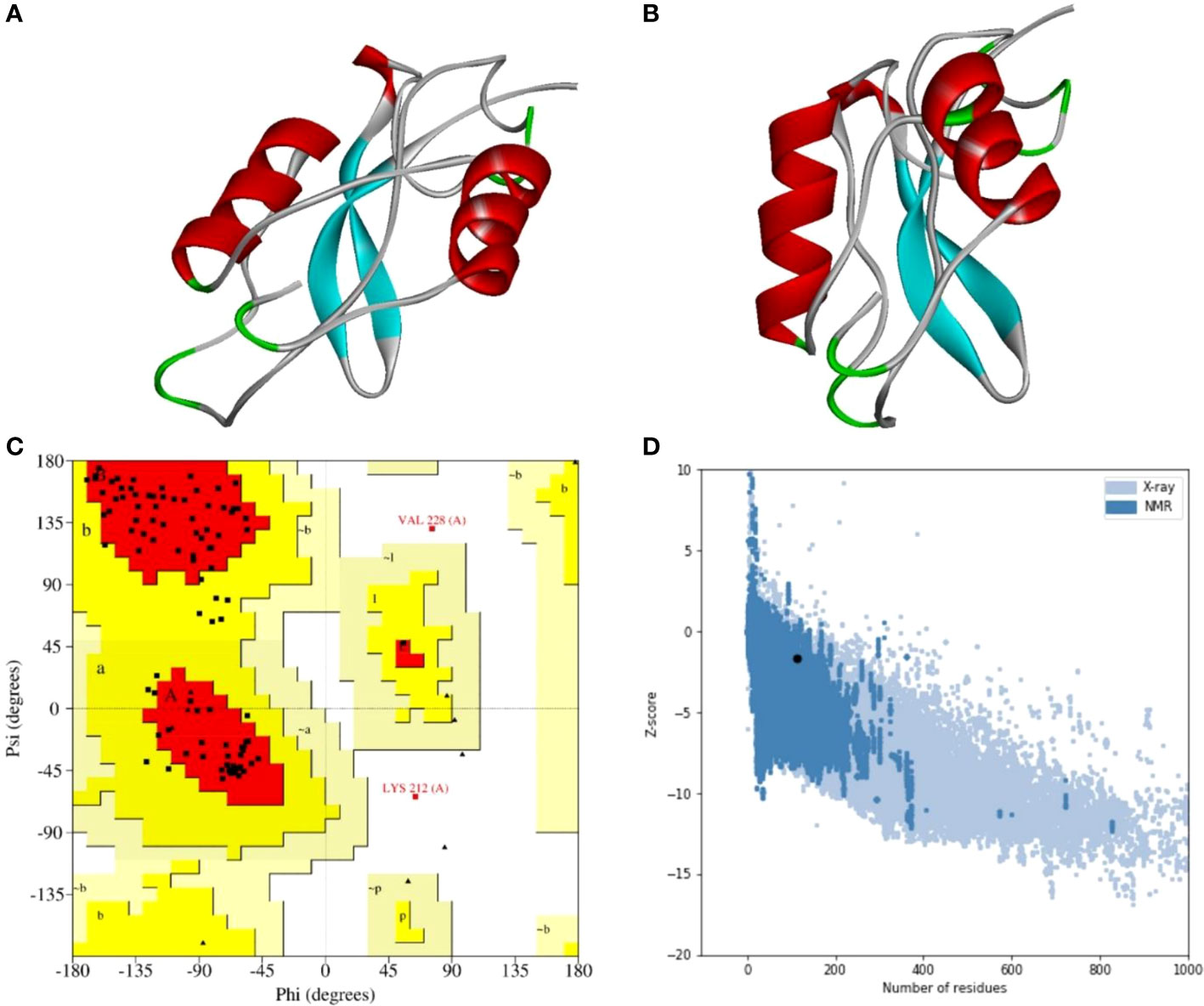
Figure 5 Three-dimensional structure analysis, structure refinement, and structure validation of MPXV-V2. (A) The 3D model of the multi-epitope vaccine was built by the Swiss Model using a homology modeling approach. (B) Refine 3D structure of MPXV-V2 obtained from DeepRefiner web-server. (C) Ramachandran plot analysis shows 84.5% residues in the favored region, 13.4% residues in the allowed region, and 2.1% residues in the disallowed region of the plot. (D) ProSA-web results with a Z-score of -1.66.

Table 6 3D structural validation of vaccine constructs via ERRAT, PROCHECK (Ramachandran plot favoured region), and ProSA-Web Server.
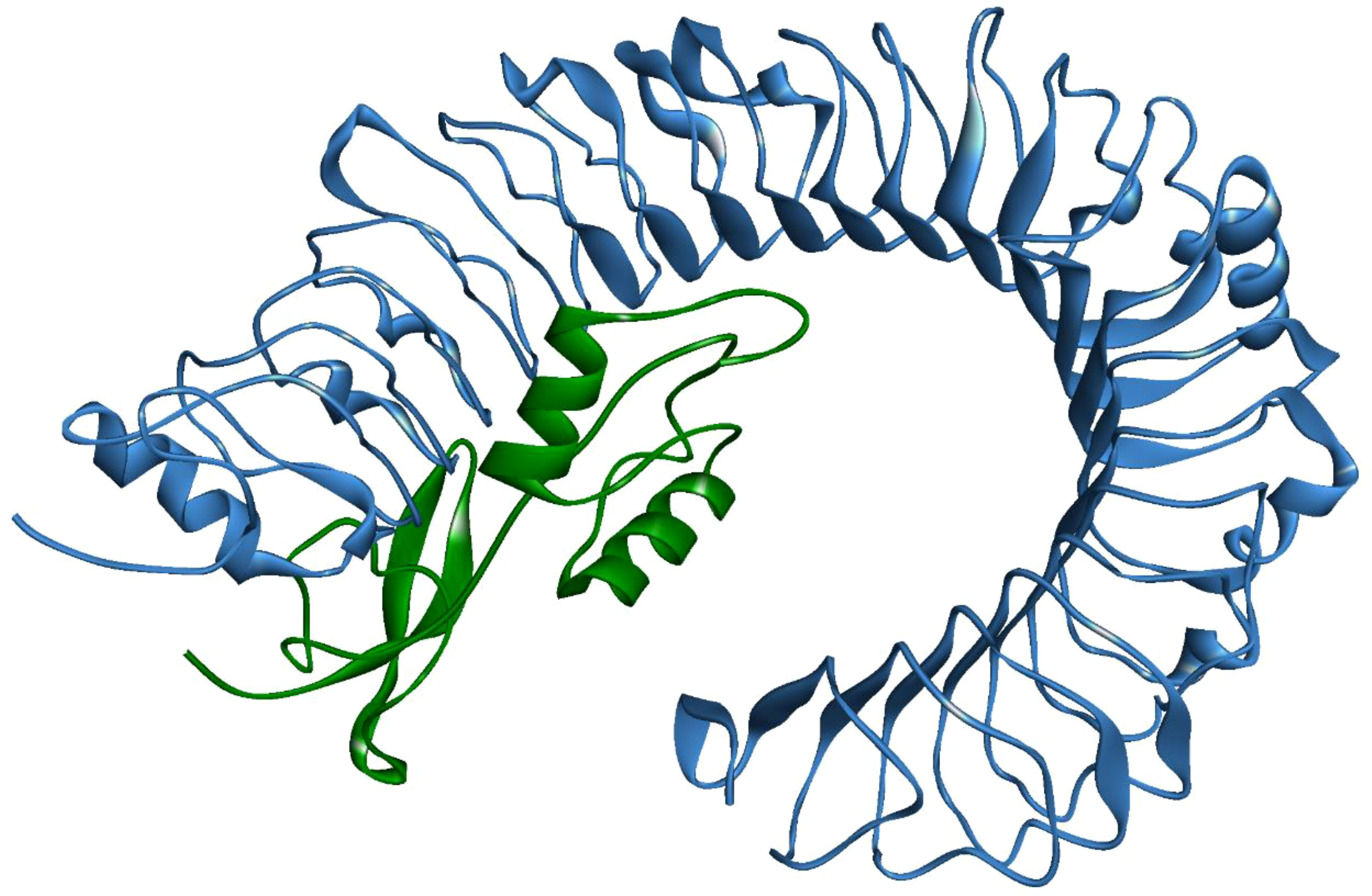
Figure 6 MPXV-V2 docked complex with TLR4 receptor. Blue indicates TLR4 and Green indicates the vaccine construct.
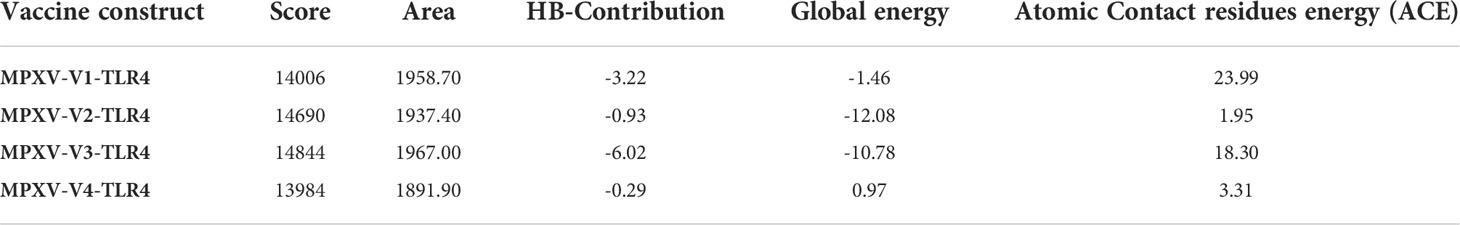
Table 7 Docking score, the interface area, the contribution of the hydrogen bonds, global energy, and atomic contact residues energy of vaccine constructs in HLA and TLR4 molecules.
The sequences of the designed vaccine constructs were subjected to codon optimization using the JCat web server. The peptide sequences were reverse-translated into DNA sequences to achieve higher expression levels of the vaccine constructs in the E. coli expression system. The CAI values for all constructs were predicted to be~0.95, and the average GC content of the adapted sequences was ~70% indicating an acceptable range for higher expression of the designed vaccine in the E. coli host (Tables 4; Table S3). Finally, SnapGene software was used to construct a recombinant plasmid sequence by introducing the adapted codon sequence of the final vaccine construct V2 into the plasmid vector pET28a(+), thereby ensuring heterologous cloning and expression in the E. coli system (Figure 7).
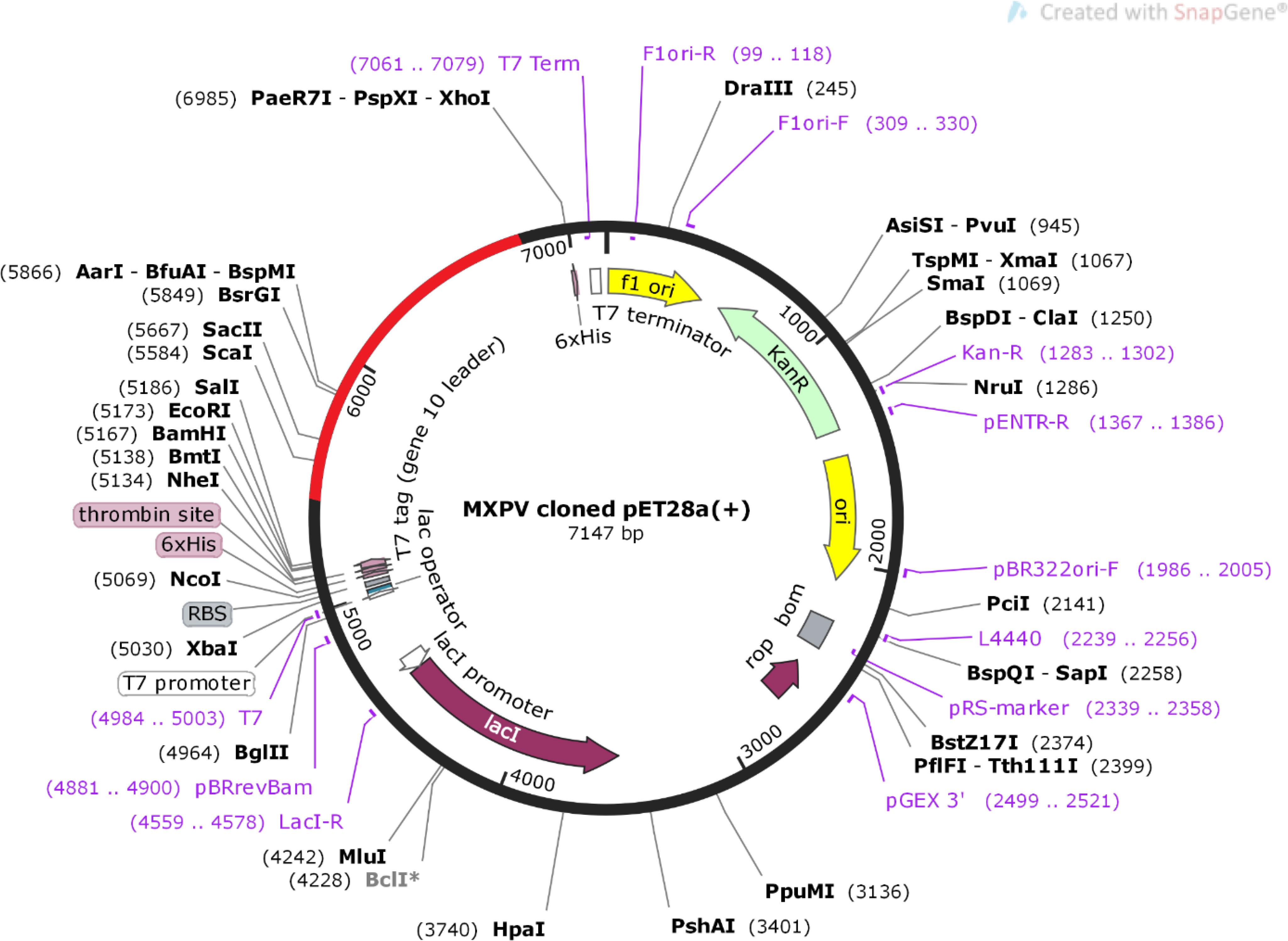
Figure 7 In silico restricted cloning of MPXV-V2 vaccine into E.coli expression vector pET28a (+).
Immune simulation predictions resulted in a significant increase in the secondary responses induced by the prioritized vaccine construct. In principle, this trend is consistent with the development of real-time immune responses. Elevated levels of IgM were the primary simulated response. The secondary and tertiary simulated responses exhibited significant increases in B-cell populations, as well as high levels of IgG1 + IgG2, IgM, and IgM + IgG antibodies. However, a decrease in the antigen levels was observed (Figures 8A, B). This suggests the generation of immunological memory, as evidenced by the increased level of memory B-cell population along with isotype switching. This led to a rapid antigen decrease following the subsequent chimeric antigen exposure (Figure 8C). The cytotoxic (TC) and helper (TH) T-cell populations were predicted to have a similar higher response, with the development of corresponding memory upon subsequent antigen exposure (Figures 8D, E). Additionally, during the immunization period, macrophage, dendritic cell, and natural killer cell populations were triggered and maintained at elevated levels (Figures 8F–H). Increased concentrations of cytokines, such as IFN-y, and interleukins, such as IL-2, were also observed (Figure 8I). These findings suggest that the predicted vaccine construct elicited encouraging immune responses against MPXV.
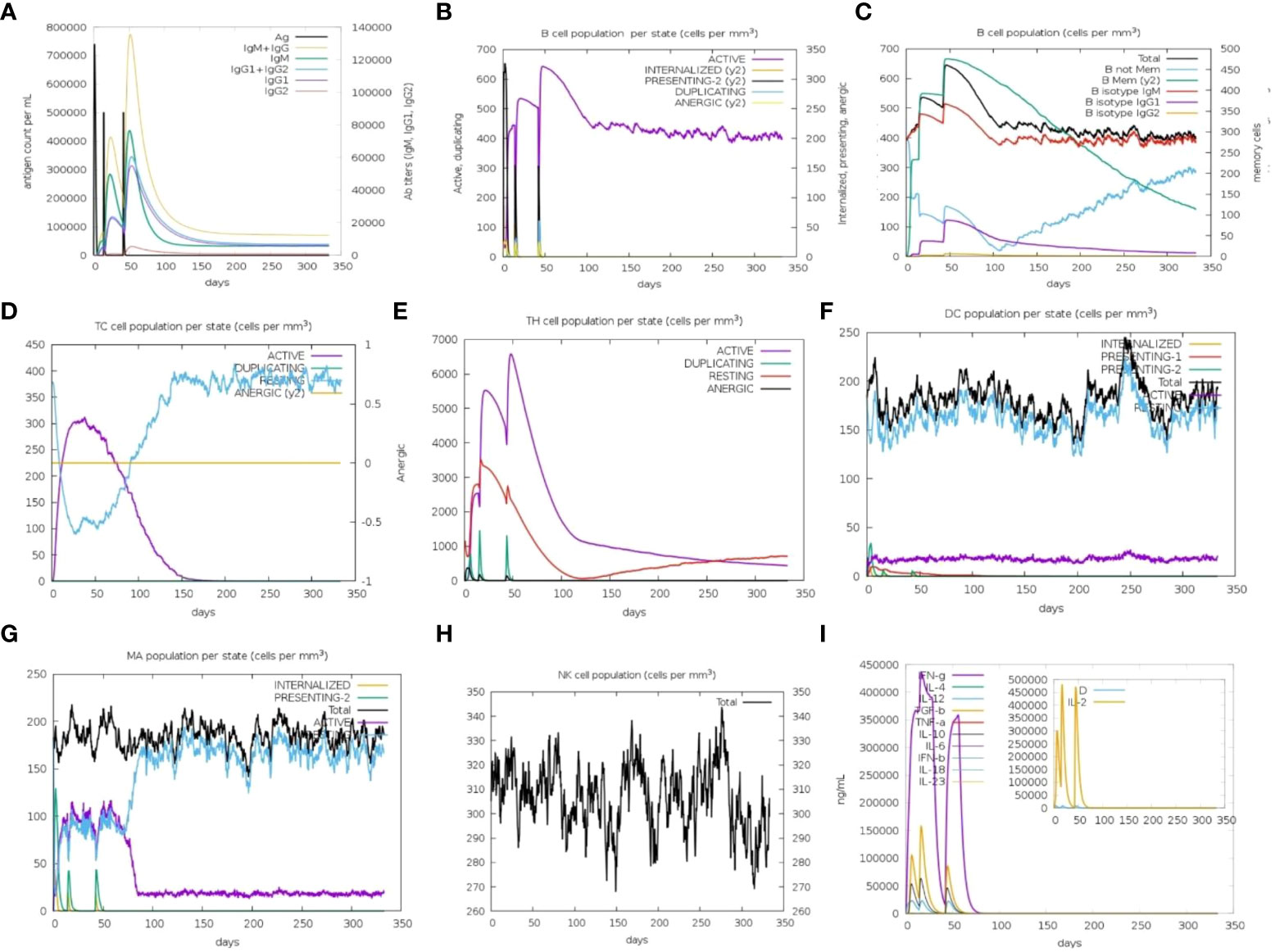
Figure 8 In silico immune simulation to predict the immunological potential of designed vaccine MPXV-V2 chimeric peptide predicted by C-ImmSim Server. (A) Increased level of immunoglobin antibodies with a decrease in antigen levels upon vaccine injections. (B) Increased levels of B-cell populations with a decrease in antigen levels upon vaccine injections. (C) Rising B-cell populations after repeated exposure to antigen. (D, E) The increase in the population of T-cytotoxic and T-helper cells upon repeated antigen exposure. (F–H) The population increase of dendritic cells, macrophages, and natural killer cells during the immunization period. (I) Increased concentrations of cytokine and interleukin levels after the repeated antigen exposure. The inset plot shows the danger signal together with leukocyte growth factor IL-2.
The MPXV-V2 construct was finalized based on the lowest global energy upon docking with the TLR4 receptor. MPXV-V2-TLR4 complex was subjected to MD simulation for energy minimization and protein stability analysis. A simulation analysis was performed to determine the movement of atoms and molecules in the designed vaccine within a biological system using the iMODS webserver. iMODS uses a normal mode analysis approach to describe the collective functional motion of macromolecules. Each normal mode comprises a frequency that is correlated with the relative motion amplitude, and a deformation vector that specifies the direction of an atomic displacement of the macromolecules to determine the molecular flexibility of these molecules within the cellular environment (59). The results of the MD simulation and normal mode analysis (NMA) of the vaccine MPXV-V2 and TLR4 docked complex are shown in Figure 9A. To simulate possible transitions, the input structure was deformed iteratively along the lowest modes, and the RMSD of the target structure was minimized based on the local and global superposition of the structures. Main-chain deformability is a measure of the atomic displacements summed over all modes of residues at every atomic position. The deformability graph of the complex depicts the peaks that indicate deformable regions of the protein. The flexible regions (hinges/linkers) of the chain have high values, and the lower values are usually in the rigid regions of the main chain residues. The deformability graph of the complex shows the peaks in the graphs that correlate to the deformed regions of the protein (Figure 9B). The NMA-derived B-factor determines the relative amplitude of atomic displacements of the molecular complex around equilibrium confirmation. The B-factor graph illustrates the relationship between the mobility of the docked complex NMA and the PDB scores (represents the average RMSD) (Figure 9C). The eigenvalue associated with each normal mode represents the motion stiffness. This value is directly related to the energy required to deform a structure. The lower the eigenvalue, the easier the deformation of the carbon alpha atoms. The eigenvalue of the MPXV-V2 and TLR4 complex was 1.030787e-05, which represented the stability of the complex (Figure 9D). The variance graph is associated with each normal mode of the complex representing individual (purple) and cumulative (green) variances and is inversely related to the eigenvalue (Figure 9E). The covariance map of the complex indicates coupling between pairs of residues in the system. Covariance analysis is used to describe the correlated (red), uncorrelated (white), or anti-correlated (blue) atomic movements in the dynamical regions of the complex molecule (Figure 9F). The correlation matrix was computed using the Cα Cartesian coordinates and Equation 2 by Ichiye and Karplus 1991 (60). The elastic network model of the complex defines the relationship between the atoms. Each dot in the graph represents a spring that connects the corresponding pair of atoms. Dots are colored according to their stiffness, and the darker greys imply stiffer regions, whereas the lighter dots indicate flexible regions (Figure 9G).
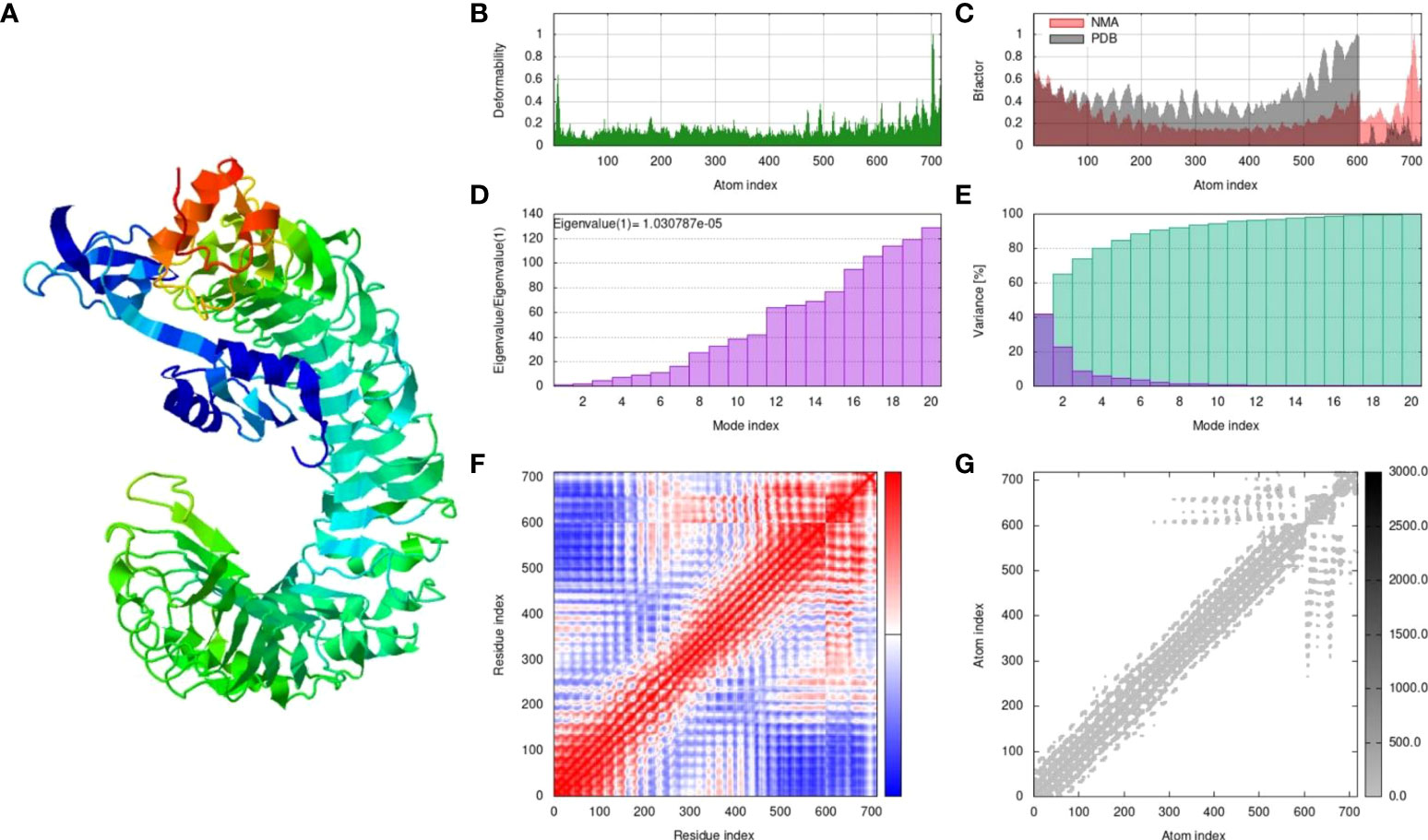
Figure 9 The results of molecular dynamics simulation of vaccine MPXV-V2 and TLR4 complex achieved by iMODS server. (A) NMA mobility, (B) deformability, (C) B-factor which indicates an averaged RMS, (D) eigenvalues (E) colored bars show the individual (purple) and cumulative (green) variances (F) Covariance matrix indicates correlated (red), uncorrelated (white), and anti-correlated (blue) motions of paired residues and (G) The elastic network model (grey regions indicates stiffer regions).
Prevention of epidemic MPXV is challenging because of the reported increase in cases of human MPX and sporadic clusters worldwide. The available vaccines offer limited protection against MPX, especially in children and adults with underlying health conditions (7). Therefore, novel therapeutic strategies are required for emerging MPXV infections. Advancements in reverse vaccinology, as well as the availability of genomic and proteomic data, have assisted in vaccine design. Furthermore, the implementation of advanced bioinformatics tools is more beneficial than traditional approaches (61). Epitope-based vaccines are a novel therapeutic approach for the design and development of suitable vaccines with high potency, logistic viability, and improved safety. Multi-epitope vaccines have the potential to generate specific immunogenic responses based on conserved epitopes in complete antigenic sequences, thus avoiding responses against unfavorable epitopes that might induce immunopathogenic or immune-modulating responses against the host (62, 63). To date, there is no specific treatment for MPX, and vaccination against MPX infection is the primary preventive measure. The goal of this study was to use immunoinformatic techniques to design novel multi-epitope MPX vaccine constructs capable of inducing immunogenic responses in infected individuals.
Three outer membrane and extracellular MPXV proteins were retrieved based on parameters such as antigenic behavior, non-allergenic and non-toxic nature, and virulence capabilities, to identify T-cell and B-cell epitopes. This method determines the suitability of vaccine candidates for experimental validation during vaccine development (64). T-cell epitopes (MHC-I and MHC-II) are crucial for adaptive immunity. MHC-I epitopes induce durable immunity to eliminate viruses and infected cells from the host, whereas MHC-II epitopes are responsible for generating both cellular and humoral immune responses (65). These epitopes induce a CD4+ helper T-cell response, leading to CD8+ T-cell memory generation and B-cell activation (66, 67). MHC-I and MHC-II epitopes overlapping B-cell epitopes were selected and linked using multiple combinations of immune enhancers and four different adjuvant peptide sequences to design vaccine constructs. ANTIGENpro and Vaxijen v2.0 determined high antigenicity scores for the designed multi-epitope constructs. All designed vaccine constructs were non-allergens and non-toxins. These immunological properties strengthen its potential as a vaccine candidate. physicochemical properties of the predicted vaccine constructs were also investigated.
Structural information is important for vaccine development to study interactions between antigens and receptor molecules. The 3D structures of the vaccine constructs were predicted and further improved after refinement. The refined 3D structural analysis confirmed the structural stability of the designed vaccine constructs and showed the maximum number of residues in the favorable region of the Ramachandran plot. The high-quality predicted structures of the multi-epitope vaccine constructs were validated using ERRAT and ProSA-web predictions. The molecular weights of the vaccine constructs were within the desired range (> 30 kDa). The physicochemical properties of these constructs revealed that they are highly soluble and extremely stable upon expression. The solubility of an overexpressed recombinant peptide in an E. coli expression system is a crucial prerequisite for most functional and biochemical studies (68). Additionally, the estimated instability scores infer that the designed vaccine constructs will be extremely stable upon expression, thus enhancing its potential as a vaccine. Confirmation of immunoreactivity based on serological analysis is a key step in validating a constructed vaccine (69) that must be expressed in a suitable expression system. The E. coli expression system is considered the most suitable for the cloning and development of recombinant peptides (70, 71).
Molecular docking analysis was performed to examine the ability of the designed vaccines to bind to the TLR4 immune cell receptor. TLR receptors are important for immune cells activation to generate adaptive immune responses and hence play a significant role in innate immunity. Previous studies have reported the involvement of TLR4 in the recognition of viral peptide structures, which trigger the production of inflammatory cytokines (72, 73). Molecular docking analysis confirmed strong binding affinities between the vaccine constructs and the active site of the receptor protein. This determines the ability of the designed vaccine to generate stable immunogenic responses. The best, stable, and most effective vaccine candidate was selected based on docking poses, interacting atoms, and binding free energies. MPXV-V2 was selected as the best vaccine construct based on its lowest global energy and was considered for immune simulation and molecular dynamic simulation studies.
The predictions of the immune simulation results were similar to those of the natural cellular immunogenic responses. Repeated exposure to antigens induced a strong immune response. The development of memory in B- and T-cells was observed with a long-lasting adaptive immunity as memory in B-cells lasts for several months. High levels of T-cytotoxic, T-helper cells, and Ig production were observed, along with an increase in IFN-γ and IL-2 levels after the first injection. The consistent increase in levels upon repeated antigen exposure indicated humoral responses to the vaccine. Simpson index D for the investigation of clonal specificity suggests the possibility of diverse immunogenic responses. The stability of the lead vaccine (MPXV-V2-TLR4) docked complex was validated using MD simulations. This analysis confirmed the strong molecular interactions of the vaccine with the immune receptor, thus ensuring the molecular stability of the multi-epitope vaccine complex in a cellular environment. This implies that the vaccine construct designed in this study is capable of generating strong immune responses with high gene expression. In silico investigations utilizing immunoinformatics, techniques are helpful and can contribute to direct laboratory assays, thereby saving time and money. The next step is in vitro immunological assays to validate the designed vaccine, evaluate the immunogenicity of the multi-epitope vaccine construct, and design a challenge-protection preclinical trial to validate the results of this study.
This study highlights a multi-epitope-based vaccine design of the MPXV protein component, which is an alternative approach to tackle antigenic complexity. The current study has some limitations. The design of vaccine constructs based on immunoinformatics relies on many predictions. The accuracy of these prediction methods is not perfect, and the degree of protection against MPXV infection is uncertain. Moreover, the immunoinformatics approach has several challenges, including standard benchmarking, limited prediction approaches, and a lack of exact datasets for various computational analyses. Several successful cases have been reported in recent years (74). Therefore, the prediction results, i.e. the proposed vaccine construct awaits investigation using in vitro and in -vivo bioassays for experimental validation to prove its safety.
In the present study, reverse vaccinology and immuno-informatics approaches were used to identify potential therapeutic vaccine candidate proteins against the emerging MPXV, based on antigenicity, allergenicity, toxicity, and virulence. Overlapping B- and T-cell epitopes from the selected proteins were used to design multi-epitope-based vaccine constructs. The population coverage of the selected epitopes was 100% worldwide and were conserved among different strains of MPXV, hence ensuring broader protection against different strains of the virus globally. These epitopes were joined using suitable linker and adjuvant sequences to enhance the immunogenicity of the vaccines. Immunological and physiochemical analysis identified MPXV-V2 as an ideal vaccine construct with the lowest global energy and binding affinity for the TLR4 immune receptor. MPXV-V2 has the capability of effective gene expression in the E. coli expression system. Moreover, immune stimulation, revealed that the MPXV-V2 vaccine has the ability to generate humoral and cellular immune responses against the MPXV. The MD simulation analysis confirmed the stability of the vaccine in the cellular environment. Additional experimental and clinical assays are required to validate the results of the present study.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
CL, and SA conceived the basic idea. SA, FA, NR, and AA performed the analysis and prepared the initial draft. QF, YA, LR, AK, and AK reviewed the critical analysis and helped in draft preparation. NR, CL finalized the draft and supervised the overall study. All authors read and approved the final manuscript.
This work was supported by the National Natural Science Foundation of China [31971180].
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.985450/full#supplementary-material
MPXV, Monkeypox Virus; CDC, Centers for Disease Control and Prevention; WHO, World Health Organization; NCBI, National Center for Biotechnology Information; BLAST, Basic Local Alignment Search Tool; CTL, Cytotoxic T Lymphocyte; MHC-I, Major Histocompatibility Complex Class I; MHC-II, Major Histocompatibility Complex Class II; IEDB, Immune Epitope Database; SMM, Stabilize Matrix Method; LPS, Lipopolysaccharide; JCAT, Java Codon Adaptation Tool; CAI, Codon Adaptation Index; NMA, Normal Mode Analysis; MD, Molecular Dynamic; RMSD, Root-Mean-Square Deviation.
2. Alakunle E, Moens U, Nchinda G, Okeke MI. Monkeypox virus in Nigeria: Infection biology, epidemiology, and evolution. Viruses (2020) 12(11):1257. doi: 10.3390/v12111257
3. Sklenovská N, Van Ranst M. Emergence of monkeypox as the most important orthopoxvirus infection in humans. Front Public Health (2018) 6:241. doi: 10.3389/fpubh.2018.00241
4. Grant R, Nguyen LL, Breban R. Modelling human-to-human transmission of monkeypox. Bull World Health Organ (2020) 98(9):638–40. doi: 10.2471/BLT.19.242347
5. McCollum AM, Damon IK. Human monkeypox. Clin Infect Dis (2014) 58(2):260–7. doi: 10.1093/cid/cit703
6. Cho CT, Wenner HA. Monkeypox virus. Bacteriol Rev (1973) 37(1):1–18. doi: 10.1128/br.37.1.1-18.1973
7. Ladnyj ID, Ziegler P, Kima E. A human infection caused by monkeypox virus in basankusu territory, democratic republic of the Congo. Bull World Health Organ (1972) 46(5):593–7.
8. Multistate outbreak of monkeypox–Illinois, Indiana, and Wisconsin, 2003. MMWR Morb Mortal Wkly Rep (2003) 52(23):537–40.
9. Erez N, Achdout H, Milrot E, Schwartz Y, Wiener-Well Y, Paran N, et al. Diagnosis of imported monkeypox, Israel, 2018. Emerg Infect Dis (2019) 25(5):980–3. doi: 10.3201/eid2505.190076
10. Yong SEF, Ng OT, Ho ZJM, Mak TM, Marimuthu K, Vasoo S, et al. Imported monkeypox, Singapore. Emerg Infect Dis (2020) 26(8):1826–30. doi: 10.3201/eid2608.191387
11. Hobson G, Adamson J, Adler H, Firth R, Gould S, Houlihan C, et al. Family cluster of three cases of monkeypox imported from Nigeria to the United Kingdom, may 2021. Euro Surveill (2021) 26(32):2100745. doi: 10.2807/1560-7917.ES.2021.26.32.2100745
12. Rao AK, Schulte J, Chen TH, Hughes CM, Davidson W, Neff JM, et al. Monkeypox in a traveler returning from Nigeria - Dallas, Texas, July 2021. MMWR Morb Mortal Wkly Rep (2022) 71(14):509–16. doi: 10.15585/mmwr.mm7114a1
13. Costello V, Sowash M, Gaur A, Cardis M, Pasieka H, Wortmann G, et al. Imported monkeypox from international traveler, Maryland, USA, 2021. Emerg Infect Dis (2022) 28(5):1002–5. doi: 10.3201/eid2805.220292
14. Zhang L. Multi-epitope vaccines: A promising strategy against tumors and viral infections. Cell Mol Immunol (2018) 15(2):182–4. doi: 10.1038/cmi.2017.92
15. Bibi S, Ullah I, Zhu B, Adnan M, Liaqat R, Kong W-B, et al. In silico analysis of epitope-based vaccine candidate against tuberculosis using reverse vaccinology. Sci Rep (2021) 11(1):1249. doi: 10.1038/s41598-020-80899-6
16. Sibuyi NRS, Martin DR, Goboza M, Klein A, Madiehe AM, et al. Immunoinformatics design of a novel epitope-based vaccine candidate against dengue virus. Sci Rep (2021) 11(1):19707. doi: 10.1038/s41598-021-99227-7
17. Aiman S, Ali F., Zia A., Aslam M., Han Z., Shams S, et al. Core genome mediated potential vaccine targets prioritization against clostridium difficile via reverse vaccinology-an immuno-informatics approach. J Biol Res (2022) 29. doi: 10.26262/jbrt.v29i0.8481
18. Sette A, Rappuoli R. Reverse vaccinology: Developing vaccines in the era of genomics. Immunity (2010) 33(4):530–41. doi: 10.1016/j.immuni.2010.09.017
19. Pickett BE, Sadat EL, Zhang Y, Noronha JM, Squires RB, Hunt V, et al. ViPR: An open bioinformatics database and analysis resource for virology research. Nucleic Acids Res (2012) 40(Database issue):D593–8. doi: 10.1093/nar/gkr859
20. Huang Y, Niu B, Gao Y, Fu L, Li W. CD-HIT suite: A web server for clustering and comparing biological sequences. Bioinformatics (2010) 26(5):680–2. doi: 10.1093/bioinformatics/btq003
21. Aslam M, Shehroz M., Hizbullah Shah M., Khan MA., Afridi SG, et al. Potential druggable proteins and chimeric vaccine construct prioritization against brucella melitensis from species core genome data. Genomics (2020) 112(2):1734–45. doi: 10.1016/j.ygeno.2019.10.009
22. Dimitrov I, Flower DR, Doytchinova I. AllerTOP–a server for in silico prediction of allergens. BMC Bioinf (2013) 14 Suppl 6(Suppl 6):S4. doi: 10.1186/1471-2105-14-S6-S4
23. Doytchinova IA, Flower DR. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinf (2007) 8(1):4. doi: 10.1186/1471-2105-8-4
24. Gupta S, Kapoor P., Chaudhary K., Gautam A., Kumar R., Raghava GP, et al. Peptide toxicity prediction. Methods Mol Biol (2015) 1268:143–57. doi: 10.1007/978-1-4939-2285-7_7
25. Garg A, Gupta D. VirulentPred: A SVM based prediction method for virulent proteins in bacterial pathogens. BMC Bioinf (2008) 9:62. doi: 10.1186/1471-2105-9-62
26. Yu NY, Wagner JR., Laird MR., Melli G., Rey S., Lo R, et al. PSORTb 3.0: Improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics (2010) 26(13):1608–15. doi: 10.1093/bioinformatics/btq249
27. Yu C-S, Cheng CW., Su WC., Chang KC., Huang SW., Hwang JK, et al. CELLO2GO: A web server for protein subCELlular LOcalization prediction with functional gene ontology annotation. PloS One (2014) 9(6):e99368. doi: 10.1371/journal.pone.0099368
28. Ahmed RKS, Maeurer MJ. T-Cell epitope mapping. In: Schutkowski M, Reineke U, editors, Epitope Mapping Protocols. 2nd ed. (Totowa, NJ: Humana Press) (2009). p. 427–38.
29. Fleri W, Paul S., Dhanda SK., Mahajan S., Xu X., Peters B, et al. The immune epitope database and analysis resource in epitope discovery and synthetic vaccine design. Front Immunol (2017) 8(278). doi: 10.3389/fimmu.2017.00278
30. Rahman N, Ali F., Basharat Z., Shehroz M., Khan MK., Jeandet P, et al. Vaccine design from the ensemble of surface glycoprotein epitopes of SARS-CoV-2: An immunoinformatics approach. Vaccines (Basel) (2020) 8(3):423. doi: 10.3390/vaccines8030423
31. Sanchez-Trincado JL, Gomez-Perosanz M, Reche PA. Fundamentals and methods for T- and b-cell epitope prediction. J Immunol Res (2017) 2017:2680160. doi: 10.1155/2017/2680160
32. Dhanda SK, Vir P, Raghava GP. Designing of interferon-gamma inducing MHC class-II binders. Biol Direct (2013) 8:30. doi: 10.1186/1745-6150-8-30
33. Li W, Joshi MD., Singhania S., Ramsey KH., Murthy AK. Peptide vaccine: Progress and challenges. Vaccines (Basel) (2014) 2(3):515–36. doi: 10.3390/vaccines2030515
34. Adhikari UK, Tayebi M, Rahman MM. Immunoinformatics approach for epitope-based peptide vaccine design and active site prediction against polyprotein of emerging oropouche virus. J Immunol Res (2018) 2018:6718083. doi: 10.1155/2018/6718083
35. Bui HH, Sidney J., Dinh K., Southwood S., Newman MJ., Sette A., et al. Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinf (2006) 7:153. doi: 10.1186/1471-2105-7-153
36. Nezafat N, Karimi Z., Eslami M., Mohkam M., Zandian S., Ghasemi Y. Designing an efficient multi-epitope peptide vaccine against vibrio cholerae via combined immunoinformatics and protein interaction based approaches. Comput Biol Chem (2016) 62:82–95. doi: 10.1016/j.compbiolchem.2016.04.006
37. Solanki V, Tiwari V. Subtractive proteomics to identify novel drug targets and reverse vaccinology for the development of chimeric vaccine against acinetobacter baumannii. Sci Rep (2018) 8(1):9044. doi: 10.1038/s41598-018-26689-7
38. Gasteiger E., Hoogland C., Gattiker A., Wilkins MR., Appel RD, Bairoch A, et al. Protein identification and analysis tools in the ExPASy server. The proteomics protocols handbook (2005), 571–607.
39. Magnan CN, Randall A, Baldi P. SOLpro: Accurate sequence-based prediction of protein solubility. Bioinformatics (2009) 25(17):2200–7. doi: 10.1093/bioinformatics/btp386
40. Gupta S, Kapoor P., Chaudhary K., Gautam A., Kumar R., Open Source Drug Discovery C., Raghava GPS., et al. In silico approach for predicting toxicity of peptides and proteins. PloS One (2013) 8(9):e73957. doi: 10.1371/journal.pone.0073957
41. Biasini M, Bienert S., Waterhouse A., Arnold K., Studer G., Schmidt T, et al. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res (2014) 42(W1):W252–8. doi: 10.1093/nar/gku340
42. Shuvo MH, Gulfam M, Bhattacharya D. DeepRefiner: High-accuracy protein structure refinement by deep network calibration. Nucleic Acids Res (2021) 49(W1):W147–52. doi: 10.1093/nar/gkab361
43. Lovell SC, Davis IW., Arendall WB. III, De Bakker PI., Word JM., Prisant MG, et al. Structure validation by cα geometry: ϕ,ψ and cβ deviation. Proteins: Structure Function Bioinf (2003) 50(3):437–50. doi: 10.1002/prot.10286
44. Ismail S, Ahmad S, Azam SS. Vaccinomics to design a novel single chimeric subunit vaccine for broad-spectrum immunological applications targeting nosocomial enterobacteriaceae pathogens. Eur J Pharm Sci (2020) 146:105258. doi: 10.1016/j.ejps.2020.105258
45. Ismail S, Abbasi SW., Yousaf M., Ahmad S., Muhammad K., Waheed Y, et al. Design of a multi-epitopes vaccine against hantaviruses: An immunoinformatics and molecular modelling approach. Vaccines (Basel) (2022) 10(3). doi: 10.3390/vaccines10030378
46. Schneidman-Duhovny D, Inbar Y., Nussinov R., Wolfson HJ. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucleic Acids Res (2005) 33(suppl_2):W363–7. doi: 10.1093/nar/gki481
47. Rasheed MA, Raza S., Zohaib A., Riaz MI., Amin A., Awais M, et al. Immunoinformatics based prediction of recombinant multi-epitope vaccine for the control and prevention of SARS-CoV-2. Alexandria Eng J (2021) 60(3):3087–97. doi: 10.1016/j.aej.2021.01.046
48. Andrusier N, Nussinov R, Wolfson HJ. FireDock: Fast interaction refinement in molecular docking. Proteins: Structure Function Bioinf (2007) 69(1):139–59. doi: 10.1002/prot.21495
49. Grote A, Hiller K., Scheer M., Münch R., Nörtemann B., Hempel DC, et al. JCat: A novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res (2005) 33(Web Server issue):W526–31. doi: 10.1093/nar/gki376
50. Rapin N, Lund O., Bernaschi M, Castiglione F. Computational immunology meets bioinformatics: The use of prediction tools for molecular binding in the simulation of the immune system. PloS One (2010) 5(4):e9862. doi: 10.1371/journal.pone.0009862
51. Nain Z, Abdulla F., Rahman MM., Karim MM., Khan MSA., Sayed SB, et al. Proteome-wide screening for designing a multi-epitope vaccine against emerging pathogen elizabethkingia anophelis using immunoinformatic approaches. J Biomol Structure Dynamics (2020) 38(16):4850–67. doi: 10.1080/07391102.2019.1692072
52. López-Blanco JR, Aliaga JI., Quintana-Ortí ES., Chacón P. iMODS: Internal coordinates normal mode analysis server. Nucleic Acids Res (2014) 42(Web Server issue):W271–6. doi: 10.1093/nar/gku339
53. Sarkar B, Ullah MA. Designing novel subunit vaccines against herpes simplex virus-1 using reverse vaccinology approach. BioRxiv (2020), 01.10.901678. doi: 10.1101/2020.01.10.901678
54. Bui HH, Sidney J., Li W., Fusseder N., Sette A. Development of an epitope conservancy analysis tool to facilitate the design of epitope-based diagnostics and vaccines. BMC Bioinf (2007) 8:361. doi: 10.1186/1471-2105-8-361
55. Chen H, Wu B., Zhang T., Jia J., Lu J., Chen Z, et al. Effect of linker length and flexibility on the clostridium thermocellum esterase displayed on bacillus subtilis spores. Appl Biochem Biotechnol (2017) 182(1):168–80. doi: 10.1007/s12010-016-2318-y
56. Wu C-Y, Monie A., Pang X., Hung C-F., Wu TC. Improving therapeutic HPV peptide-based vaccine potency by enhancing CD4+ T help and dendritic cell activation. J Biomed Sci (2010) 17(1):88. doi: 10.1186/1423-0127-17-88
57. Cheng J, Randall AZ., Sweredoski MJ., Baldi P. SCRATCH: A protein structure and structural feature prediction server. Nucleic Acids Res (2005) 33(suppl_2):W72–6. doi: 10.1093/nar/gki396
58. Mehmood A, Kaushik AC, Wei DQ. Prediction and validation of potent peptides against herpes simplex virus type 1 via immunoinformatic and systems biology approach. Chem Biol Drug Des (2019) 94(5):1868–83. doi: 10.1111/cbdd.13602
59. Dykeman EC, Sankey OF. Normal mode analysis and applications in biological physics. J Phys Condens Matter (2010) 22(42):423202. doi: 10.1088/0953-8984/22/42/423202
60. Ichiye T, Karplus M. Collective motions in proteins: A covariance analysis of atomic fluctuations in molecular dynamics and normal mode simulations. Proteins (1991) 11(3):205–17. doi: 10.1002/prot.340110305
61. Lim HX, Lim J., Jazayeri SD., Poppema S., Poh CL.. Development of multi-epitope peptide-based vaccines against SARS-CoV-2. BioMed J (2021) 44(1):18–30. doi: 10.1016/j.bj.2020.09.005
62. Zhou WY, Shi Y., Wu C., Zhang WJ., Mao XH., Guo G, et al. Therapeutic efficacy of a multi-epitope vaccine against helicobacter pylori infection in BALB/c mice model. Vaccine (2009) 27(36):5013–9. doi: 10.1016/j.vaccine.2009.05.009
63. Vartak A, Sucheck SJ. Recent advances in subunit vaccine carriers. Vaccines (Basel) (2016) 4(2):12. doi: 10.3390/vaccines4020012
64. Naz K, Naz A., Ashraf ST., Rizwan M., Ahmad J., Baumbach J, et al. PanRV: Pangenome-reverse vaccinology approach for identifications of potential vaccine candidates in microbial pangenome. BMC Bioinf (2019) 20(1):123. doi: 10.1186/s12859-019-2713-9
65. Singhvi N, Singh Y., Shukla P. Computational approaches in epitope design using DNA binding proteins as vaccine candidate in mycobacterium tuberculosis. Infection, Genetics and Evolution (2020) 83:104357. doi: 10.1016/j.meegid.2020.104357
66. Kar T, Narsaria U., Basak S., Deb D., Castiglione F., Mueller DM, et al. A candidate multi-epitope vaccine against SARS-CoV-2. Sci Rep (2020) 10(1):1–24. doi: 10.1038/s41598-020-67749-1
67. Sauer K, Harris T. An effective COVID-19 vaccine needs to engage T cells. Front Immunol (2020) 11:581807. doi: 10.3389/fimmu.2020.581807
68. Khatoon N, Pandey RK, Prajapati VK. Exploring leishmania secretory proteins to design b and T cell multi-epitope subunit vaccine using immunoinformatics approach. Sci Rep (2017) 7(1):8285. doi: 10.1038/s41598-017-08842-w
69. Gori A, Longhi R., Peri C., Colombo G. Peptides for immunological purposes: design, strategies and applications. Amino Acids (2013) 45(2):257–68. doi: 10.1007/s00726-013-1526-9
70. Chen R. Bacterial expression systems for recombinant protein production: E. coli and beyond. Biotechnol Adv (2012) 30(5):1102–7. doi: 10.1016/j.biotechadv.2011.09.013
71. Rosano GL, Ceccarelli EA. Recombinant protein expression in escherichia coli: Advances and challenges. Front Microbiol (2014) 5(172). doi: 10.3389/fmicb.2014.00172
72. Compton T, Kurt-Jones EA., Boehme KW., Belko J., Latz E., Golenbock DT, et al. Human cytomegalovirus activates inflammatory cytokine responses via CD14 and toll-like receptor 2. J Virol (2003) 77(8):4588–96. doi: 10.1128/JVI.77.8.4588-4596.2003
73. Vaure C, Liu Y. A comparative review of toll-like receptor 4 expression and functionality in different animal species. Front Immunol (2014) 5(316):. doi: 10.3389/fimmu.2014.00316
Keywords: reverse vaccinology, monkeypox virus, multi-epitope vaccine construct, vaccine candidates, immune simulation, molecular dynamic simulation
Citation: Aiman S, Alhamhoom Y, Ali F, Rahman N, Rastrelli L, Khan A, Farooq QuA, Ahmed A, Khan A and Li C (2022) Multi-epitope chimeric vaccine design against emerging Monkeypox virus via reverse vaccinology techniques- a bioinformatics and immunoinformatics approach. Front. Immunol. 13:985450. doi: 10.3389/fimmu.2022.985450
Received: 03 July 2022; Accepted: 29 July 2022;
Published: 25 August 2022.
Edited by:
Puthen Veettil Jithesh, Hamad bin Khalifa University, QatarReviewed by:
Prakash Ghosh, International Centre for Diarrhoeal Disease Research (ICDDR), BangladeshCopyright © 2022 Aiman, Alhamhoom, Ali, Rahman, Rastrelli, Khan, Farooq, Ahmed, Khan and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Noor Rahman, bm9vcmJpb2NoZW1pc3RAZ21haWwuY29t; Chunhua Li, Y2h1bmh1YWxpQGJqdXQuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.