
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 15 September 2022
Sec. Cancer Immunity and Immunotherapy
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.984728
This article is part of the Research Topic Identification and Characterization of Molecular Targets in Hepatocellular Carcinoma View all 22 articles
 Zelin Tian1†
Zelin Tian1† Chen Xu1†Peijun Yang1Zhibin Lin1
Chen Xu1†Peijun Yang1Zhibin Lin1 Wenlong Wu1
Wenlong Wu1 Wenjie Zhang1,2Jian Ding1
Wenjie Zhang1,2Jian Ding1 Rui Ding1
Rui Ding1 Xuan Zhang1*Kefeng Dou1*
Xuan Zhang1*Kefeng Dou1*Hepatocellular carcinoma(HCC) is the sixth most common cancer in the world and is usually caused by viral hepatitis (HBV and HCV), alcoholic, and non-alcoholic fatty liver disease(NAFLD). Viral hepatitis accounts for 80% of HCC cases worldwide. In addition, With the increasing incidence of metabolic diseases, NAFLD is now the most common liver disease and a major risk factor for HCC in most developed countries. This review mainly described the specificity and similarity between the pathogenesis of viral hepatitis(HBV and HCV)-induced HCC and NAFLD-induced HCC. In general, viral hepatitis promotes HCC development mainly through specific encoded viral proteins. HBV can also exert its tumor-promoting mechanism by integrating into the host chromosome, while HCV cannot. Viral hepatitis-related HCC and NASH-related HCC differ in terms of genetic factors, and epigenetic modifications (DNA methylation, histone modifications, and microRNA effects). In addition, both of them can lead to HCC progression through abnormal lipid metabolism, persistent inflammatory response, immune and intestinal microbiome dysregulation.
Hepatocellular carcinoma, accounting for 75-85% of primary liver cancer cases, is the sixth most common cancer and the third leading cause of cancer death globally in 2020, with approximately 906,000 new cases and 830,000 deaths annually (1). Patients with HCC are usually asymptomatic in the early stage and are often in the advanced stage when they have typical symptoms, such as liver pain, jaundice, ascites, and liver failure (2). Common treatments for HCC include radiofrequency ablation(RFA), hepatic resection, liver transplantation, transcatheter arterial chemoembolization(TACE), tyrosine-kinase inhibitors (sorafenib), radiotherapy, and immune oncology (2, 3). Hepatitis B virus (HBV) or hepatitis C virus (HCV) infection, alcohol abuse, and NAFLD are the most common risk factors for HCC (2, 4–6).
Although the burden of nonalcoholic steatohepatitis (NASH)-induced HCC is increasing, chronic viral hepatitis (HBV and HCV) remains the leading cause of HCC, causing 80% of cases worldwide (5, 6). Liver cirrhosis caused by chronic HBV or HCV infection is an important factor in the development of HCC. It is worth noting that HCC can also appear in patients with chronic viral hepatitis infection without cirrhosis (7). Patients with viral hepatitis often have co-infections. Chronic hepatitis B (CHB) infection affected approximately 257 million people worldwide, of which 48-60 million were co-infected with HDV and 2.6 million were co-infected with HCV (8). Individuals co-infected with HBV/HCV have an increased incidence of HCC and a poorer prognosis compared with HBV or HCV mono-infection (2, 9). In the progression of CHB to HCC, synergistic risk factors include male sex, alcohol abuse, high viral load(HBV DNA > 106 U/mL), HBV genotype C, presence of cirrhosis, and hepatitis B e-antigen positivity (10, 11). The main treatment for CHB is the use of nucleotide analogues (NA) to inhibit HBV replication (1, 12). HCV infection is another major cause of chronic liver disease. Additional risk factors that may increase the risk of HCC in patients with HCV infection include male sex, diabetes and obesity, alcohol abuse, and HCV genotype 3 (6, 13). The development of direct-acting antiviral therapy (DAA) has improved the prognosis of HCV-induced HCC, and achieving sustained virologic response (i.e., virological cure, SVR) is associated with a significant reduction in HCC risk (14, 15).
With the increasing incidence of metabolic diseases such as diabetes and obesity, NAFLD has become an increasingly serious health problem (16). NAFLD can develop into NASH, liver fibrosis, liver cirrhosis, and eventually HCC. In recent years, with the widespread vaccination of the hepatitis B vaccine and the popularization of anti-HBV and HCV treatment, the incidence of virus-induced HCC has steadily decreased. NAFLD/NASH has gradually developed into one of the main causes of HCC in developed countries (16, 17). Patients with NAFLD have a very low risk of progressing to cirrhosis, but patients with NASH have a significantly increased risk of progressing to cirrhosis and even HCC. Compared with viral hepatitis-related HCC, NASH-related HCC patients tend to be older, have a better liver function, larger tumor size, and longer overall survival(OS) (18). In addition, some patients can directly progress to HCC without cirrhosis, and these patients always have a worse prognosis (19).
In this review, we discuss the similarities and differences in the molecular mechanisms of viral hepatitis and NASH-induced HCC. We accept that this review will help clinicians in the diagnosis and treatment of HCC patients, and provide guidance for the development of new molecular therapeutic targets and therapeutic drugs.
HBV is a para-retrovirus that was discovered by American geneticist Baruch Blumberg in 1965. Its genome is a 3.2-kb double-stranded loop of DNA (20). HBV has 10 genotypes (A to J), with genotypes C, B, F, D, and A are associated with the development of HCC (6). The HBV genome consists of 4 overlapping open reading frames (ORFs): Pre-S/S, X, P, and pre-C/C, which are transcribed to produce 5 messenger RNAs (mRNAs) (21). Viral protein products include 3 surface proteins (also known as large/pre-S1 (L-HBsAg), medium/pre-S2 (M-HBsAg), and small/major (S-HBsAg)), the excreted “e” antigen (HBeAg), the core antigen (HBcAg), the X protein (HBx), and the viral polymerase (DNA polymerase, reverse transcriptase, and RNaseH activity) (Figure 1). HBx is required for HBV replication and plays an important role in both HBV and HBV-induced HCC progression (21).
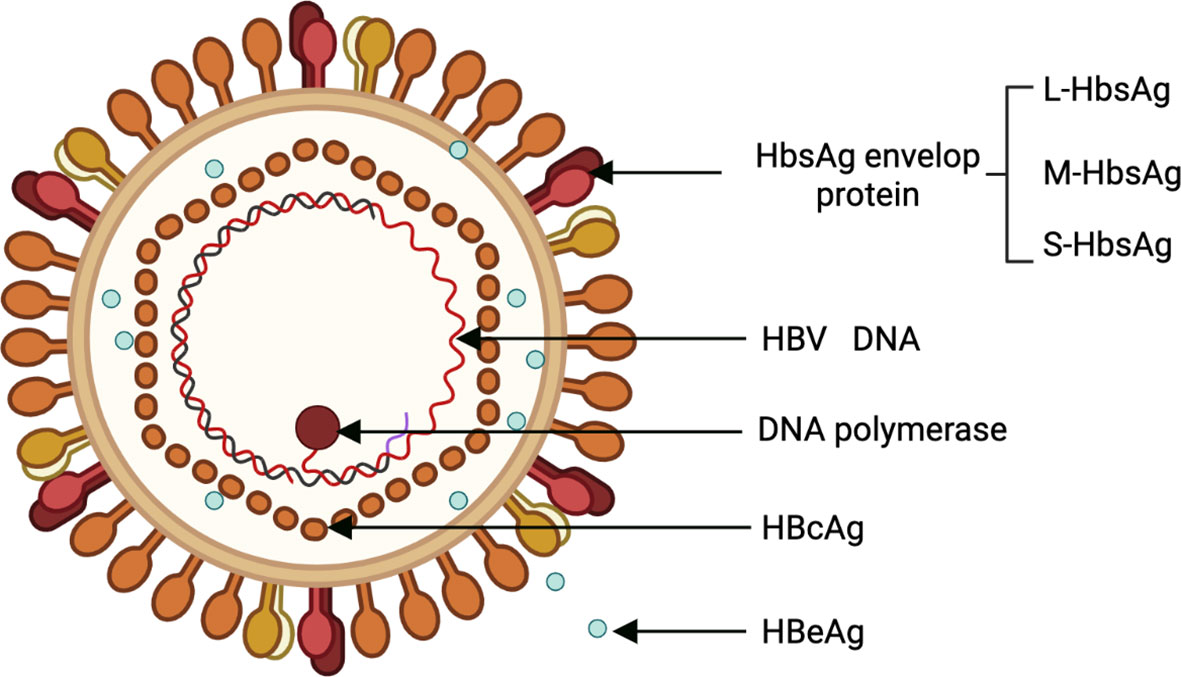
Figure 1 Structural of HBV. HBV, Hepatitis B virus.
HBV DNA integration into the host chromosome is not an essential step in the HBV life cycle (22). However, this phenomenon can lead to the instability of the host genome, insertional mutations in proto-oncogenes and tumor suppressor genes, which in turn promote the occurrence of HCC (23). HBV DNA integration was observed in approximately 80% of HBV-induced HCC patients, and the frequency of integration was significantly higher in tumor tissues than in adjacent tissues (24). Integration sites tend to be located near repeat regions, CpG islands, and telomeres, leading to chromosomal instability (24). There are many target genes affected by HBV genome integration, such as TERT, MLL4, CCNE1, MLL2, ARID1A, ARID1B, ARID2, MLL3, et al. (25–27). Integration of HBV DNA can also induce the persistent expression of mutated and truncated HBsAg, HBcAg, and HBx proteins. High expression rates of these proteins can promote HCC development through endoplasmic reticulum and mitochondrial stress responses (28, 29).
Epigenetic changes include all chromatin changes, while DNA sequence does not change, and can be divided into three types: DNA methylation, histone modification, and RNA-related silencing.
In HBV-related HCC, DNA hypermethylation occurs at CpG islands in the promoter regions of specific tumor suppressor genes, resulting in the silencing of tumor suppressor genes, which in turn promotes the occurrence of HCC (30, 31). Persistent HBV infection can cause hypermethylation of p16INK4A, and HBx may play a key role in this process (32). RASSF1A (a cell cycle-related tumor suppressor protein) methylation occurs in more than 50% of HBV-infected livers, and its methylation level is also considerably increased early in the pathogenesis of HCC (33). CDH1 encoded a protein named epithelial cadherin (E-cadherin), which plays a crucial role in the epithelial-mesenchymal transition process. HBx can downregulate E-cadherin protein levels via promoting CDH1 hypermethylation (34). In contrast to hypermethylation, DNA hypomethylation is assumed to be a genome-wide event in HCC, which can lead to genomic instability. HBx can selectively promote regional hypermethylation of specific tumor suppressor genes by upregulating DNMT1, DNMT3A1, and DNMT3A2, and can also induce global hypomethylation of HSATII by downregulating DNMT3B (35).
Histones can be reversibly modified by acetylation, methylation, phosphorylation, or ubiquitination. These modifications have implications for gene activation, gene repression, DNA repair, and cancer development (36). Experiments by Liu et al. demonstrated that HBx can promote the expression of IGF-II by inducing the hypomethylation of the P3 and P4 promoters in HCC cells and HCC specimens. HBx can bind to MBD2 and CBP/p300 to promote MBD2-HBx-CBP/p300 complex formation, which in turn promotes the acetylation of the corresponding histones H3 and H4, providing new insights into the pathogenesis of HBx-mediated HCC (37). Arzumanyan’s study revealed that HBx protein can promote epigenetic modulation of E-cadherin transcriptional activity through histone deacetylation and miR-373 (34). Histone H3 lysine 4 methyltransferase SMYD3 has been shown to promote the transcriptional activation of genes involved in the development of HCC, such as C-MYC, JAK/STAT3, CDK2, and MMP2 (38–40).
MicroRNAs are small non-coding RNA with 19-25 nucleotides in length, which lead to gene silencing through translation inhibition or targeted degradation of mRNA. In recent years, more and more studies have shown that some MicroRNAs can be regulated by HBV infection and play a key role in hepatocarcinogenesis (41, 42). For example, miR-18a, miR-21, miR-221, miR-222, and miR-224 were upregulated in HBV-related HCC tissues, while miR-26a, miR-101, miR-122, miR-125b, miR-145, miR-199a, miR-199b, miR-200a, and miR-223 were upregulated in HBV-related HCC tissues. These miRNAs have been proved to affect HCC progression via targeting JAK/STAT, PI3K/MAPK, TP53, WNT/β-catenin pathways (41).
HBx protein plays an indispensable role in the life cycle of HBV and the progression of HCC. HBx plays its role mainly through the following four mechanisms: 1) HBx gene can be integrated into hepatocyte genome and affect genomic stability; 2) Induced epigenetic modifications such as DNA methylation, histone acetylation, and MicroRNA expression; 3) Oxidative stress induced by interaction with mitochondria and other proteins; 4) Participate in the regulation of proto-oncogene activation and tumor suppressor gene inactivation (8). The first two of these mechanisms have been described above. HBx is the most common open reading frame(ORF) integrated into the host genome in HBV-induced HCC specimens and the integrated HBx is frequently mutated (43, 44). And both of them appear to be important steps in HCC tumorigenesis (45, 46). HBx can trans-activate cellular promoters and enhancers and participate in the regulation of inflammatory proliferation-related signaling pathways, such as NF-κB, Ras/Raf mitogen-activated protein kinase (MAPK), c-Jun N-terminal kinase, Jak1/STAT, protein kinase C (PKC) and Src kinase, etc (47, 48). HBx in the cytoplasm can also bind to p53, prevent p53 nuclear localization, lead to dysregulation of cell cycle checkpoints, and inhibit p53-dependent apoptosis and DNA repair (49, 50).
HBx can also induce upregulation of vascular endothelial growth factor (VEGF) and angiogenic factor ang2(ANG2), stabilize HIF1α, and promote the angiogenesis of HCC (51, 52).
In addition, HBsAg can enhance the malignant potential of HBV-induced HCC by enhancing the IL-6-STAT3 pathway (53). HBV core protein can increase the production of cytokines and is related to the host immune response, both of which play a role in HBV-related HCC (54, 55).
Due to the lack of proofreading function of HBV reverse transcriptase, the replication error rate of HBV DNA is much higher than that of other DNA viruses. Among the three envelope protein forms of HBsAg (L-HBsAg, M-HBsAg, and S-HBsAg), the amino acid position between 99-169 of S-HBsAg is called the main hydrophilic region (MHR), and the antigenic cluster “A” is located in it. Mutations in MHR will affect HBsAg antigenicity and lead to vaccine-induced immune escape (56). Pres/S region mutations may give rise to endoplasmic reticulum stress, oxidative DNA damage, and genomic instability in hepatocytes (57). In addition, K130M/V131I double mutations in the X gene, A1762T/G1764A double mutations in the basic core promoter (BCP), and mutations in the reverse transcriptase region will increase the risk of HCC (58–61).
HCV genome is a 9.6 kb positive-strand single-stranded RNA virus with highly conserved 5’ and 3’ untranslated regions, encoding 3 structural proteins (core, E1, E2) and 7 nonstructural proteins (p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B) (Figure 2) (62). Unlike HBV viruses, HCV cannot stably integrate into the host genome and requires continuous replication to gain viability (63). There are 6 major genotypes of HCV, and genotypes 3 and 6 infections have been reported to have a higher risk in hepatocarcinogenesis (13, 64).
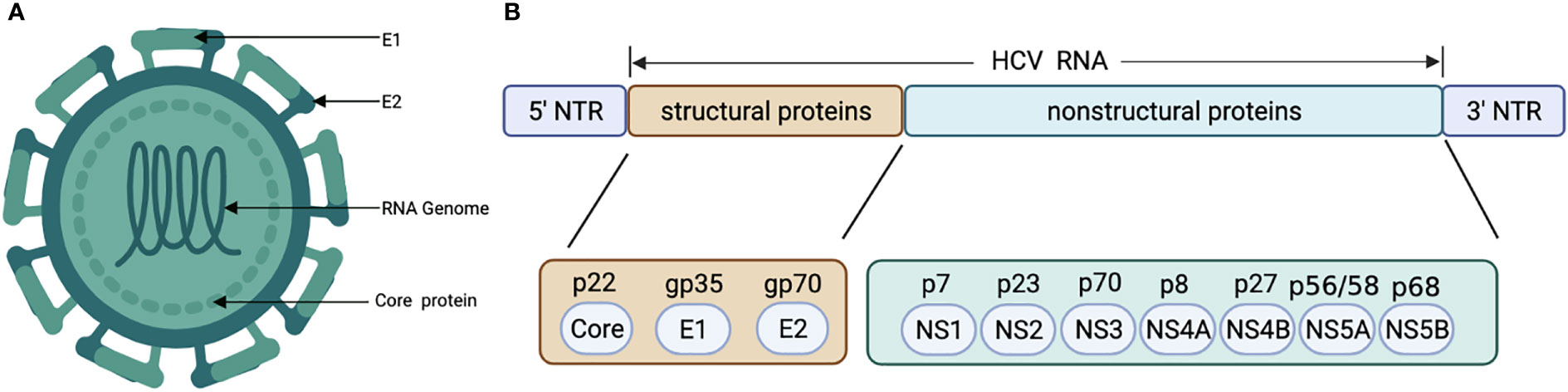
Figure 2 Structural (A) and genetic organization (B) of HCV. HCV, Hepatitis C virus.
Unlike HBV, HCV is not integrated into the host genome, but can promote HCC progression through epigenetic dysregulation.
As with HBV-positive HCC, DNA methylation also plays an important role in HCV-positive HCC. The clinical study of Zekri showed that the progression of HCV-induced HCC was associated with increased DNA promoter methylation (65). SOCS-1, as a negative regulator of the JAK/STST pathway, often acts as a tumor suppressor gene (66). The methylation of SOCS-1 is more common in HCV-positive HCC (compared to HCV-negative HCC) (67). HCV protein can down-regulate Gadd45β expression by promoting hypermethylation of Gadd45β promoter, resulting in defective cell cycle arrest and leading to hepatocarcinogenesis (68). Duong’s study revealed that HCV could reduce the transcriptional activation of interferon- alpha(IFN-α) by promoting STAT1 and PP2Ac hypomethylation (69). In addition, Hypermethylation of APCαp15αp14αp73αp16αO6MGMT, and IGF2 can also affect the progression of HCV-induced HCC (30, 70, 71).
Hamdane’s research exposed a paradigm that chronic HCV infection induces 27 histone 3 (H3K27Ac) acetylation modifications can promote hepatocarcinogenesis (72). In addition, acetylation of histone H3 lysine 9 (H3K9Ac) plays a similar role in HCV-mediated carcinogenesis (73). Histone demethylase member KDM5B/JARID1B can enhance HCV-induced HCC cell proliferation via regulating its downstream genes E2F1 and E2F2 (74). HCV core protein can induce dysregulation of HOX gene by impairing histone H2A mono-ubiquitination, which will promote HCC development (75).
Studies have reported that specific liver and serum MicroRNAs are involved in the pathogenesis of HCV-induced HCC, including miR-193b, miR-155, miR-122, etc (76). In general, the mechanism of MicroRNAs in HCV-related HCC is not in-depth enough, and the number of related reports is smaller than that of HBV-related HCC reports.
There are 10 kinds of HCV gene products, among which the core, NS3, NS5A, and NS5B proteins potentiate carcinogenic pathways. They play an important role in promoting cell proliferation, regulating cytokines, oxidative stress, apoptosis, and HCV-related metabolic disorders and liver disease progression (77). HCV NS5B can form cytoplasmic complexes with Rb, leading to activation of E2F-dependent transcription and increased cell proliferation (78). HCV NS5A and NS3 can bind to P53 and down-regulate the expression of cell cycle regulation gene P21 (79, 80). P73 can interact with HCV core protein, leading to nuclear translocation of the core protein and promotes cell proliferation in a p53-dependent manner (81). HCV core protein can also induce oxidative DNA damage, enhance ROS production, and inhibit apoptosis (82).
There are relatively few studies on the correlation between HCV characteristic mutations and HCC development (compared with HBV), among which HCV core gene mutations are the most studied. Studies have shown that HCV core A028C, G209A, C219U/A, U264C, A271C/U, C378U/A, G435A/C, and G481A mutations were significantly associated with increased HCC risk, while U303C/A mutation predicted reduced HCC risk (83).
The progression of NAFLD to NASH is a complex multi-factor process, whose detailed mechanism has not been fully elucidated. It is currently mainly accepted by the public as the “two-hit hypothesis” (84). The core idea is that liver steatosis and insulin resistance are the “first hit”, which leads to riglycerides accumulate in liver cells (85). Then, under the joint action of inflammatory factors, oxidative stress and endoplasmic reticulum stress, liver dysfunction such as hepatocyte inflammation, liver fibrosis and cirrhosis is developed, namely the “second hit” (86).
In recent years, there is a new consensus on the “multiple parallel hit hypothesis” to replace the “two-hit hypothesis”. The multiple parallel hit hypothesis suggested that NASH is the result of a combination of genetic differences, insulin resistance, lipid metabolism abnormalities, endoplasmic reticulum stress, mitochondrial dysfunction, and intestinal microbiota (87, 88).
Genetic mutations in the protein-like phospholipase domain-containing protein 3 PNPLA3 gene is the most well-known mutations associated with NASH-related HCC progression (89, 90). PNPLA3 rs738409 c.444C>G minor allele (encoding the I148M variant) is associated with increased lipid accumulation and fibrosis in the liver. It also predisposes individuals to fatty liver-related diseases ranging from simple steatosis to steatohepatitis, NASH, and HCC (91). Overexpression of I148M PNPLA3 protein in mouse liver promoted steatosis by triggering metabolic reprogramming and driving inflammatory pathways (92).
17β-HSD13 is thought to be the pathogenic protein of NAFLD development (93). Chen et al. demonstrated that hydroxysteroid 17-Beta Dehydrogenase 13 (HSD17B13) was low expression in HCC and was associated with poor prognosis (94). HSD17B13 rs72613567 (a splice variant with an adenine insertion) reduced the risk of NASH and progressive liver injury (95).
In addition, TM6SF2 rs58542926 variant and MBOAT7 rs641738 variant have also been proved to be genetic variants susceptible to NAFLD-related HCC, and their effects are not necessarily mediated by the development of liver fibrosis (96–98).
Epigenetic changes such as abnormal DNA methylation are considered to be an important mechanism for NASH progression. It induced gene silencing associated with DNA damage and repair, lipid and glucose metabolism, and fibrosis progression via enzyme methyltransferase (DNMT) (99). Kuramoto’s study further confirmed that NASH-specific DNA methylation change may be involved in the development of NASH-associated multistage HCC (100, 101).
The histone deacetylase 8 (HDAC8) has been defined as a modifier of chromatin tissue in NASH-associated HCC (102, 103). HDAC8 can inhibit p53/P21 mediated apoptosis and stimulate β-catenin dependent cell proliferation. Knockdown of HDAC8 can reverse insulin resistance and reduce NAFLD-related tumorigenicity (103).
Accumulating evidence has demonstrated the role of microRNAs in epigenetic dysregulation of metabolic processes in NAFLD, NASH, and HCC. Takaki’s study showed that silencing of miR-122 is an early event in NASH and may be a novel molecular marker for assessing HCC risk in NASH patients (104). In addition, miR-21, miR-29, miR-23, miR-155, miR-221, miR-222, miR-106, miR-93, and miR-519 have also been confirmed to be associated with carcinogenic effects associated with NASH (105).
Viral hepatitis-induced and NASH-induced HCC have been described in detail in a large number of previous reviews, but there is no article to summarize and discuss them together. In the previous section, we have discussed the specific pathways of viral hepatitis-induced and NASH-induced HCC, respectively. In this section, we will describe the common mechanism of them, and we will select the most important research hotspots for description, including metabolic pathways, inflammatory pathways, and intestinal microbiota dysfunction.
Metabolic disorders, including dyslipidemia, insulin resistance and impaired blood glucose control have been identified as contributing factors to the progression of NASH (17). Insulin resistance and hyperinsulinemia can enhance the expression of IGF-1, trigger IRS-1/2 signal cascades, activate downstream PI3K-Akt and MAPK pathways, induce cell proliferation and inhibit apoptosis (105, 106) (Figure 3). In addition, the accumulation of excess lipids can also lead to the over-production of free fatty acids (FFA), which produce specific lipid toxicity and influence liver cell metabolism through a cascade of signals (107). It is worth mentioning that elevated iron levels have been observed in NASH patients and are considered a risk factor for HCC development (108).
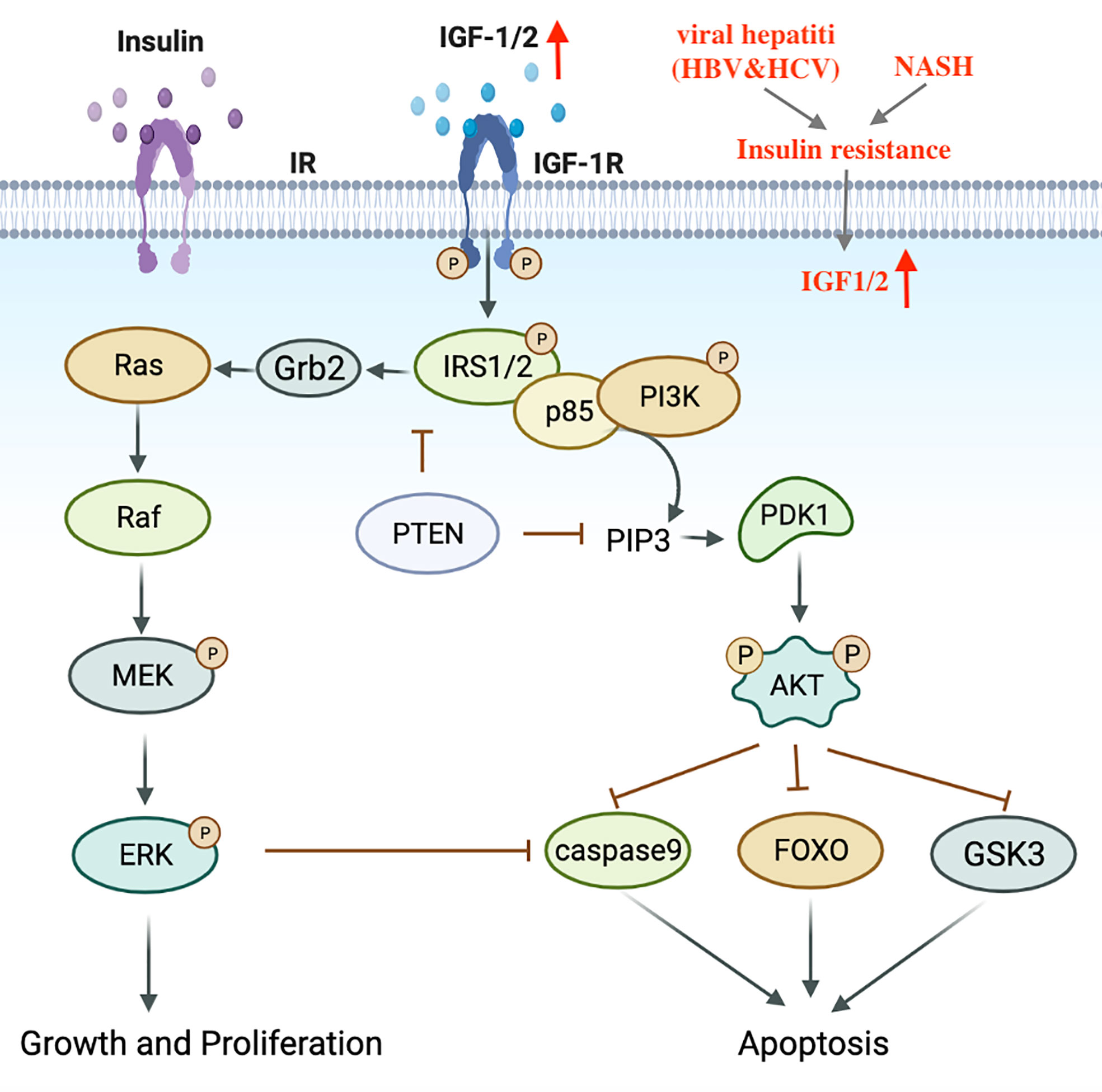
Figure 3 Transduction mechanism of the IGF1 signaling pathway in HCV, NASH-induced HCC. IGF, Insulin-like growth factor; IRS, Insulin receptor substrate.
In contrast to HBV viral hepatitis, HCV is commonly associated with hepatic steatosis (109). HCV core protein plays an important role in regulating lipid metabolism. Transgenic mice that express HCV core protein can develop insulin resistance, lipid accumulation in the liver, and eventually progresses to HCC (110, 111). Koike et al. revealed that HCV core protein can bind to retinol-like X receptor (RXR)-α and continuously activate peroxisome proliferator-activated receptor-α (PPAR-α), continuously activate peroxisome proliferator-activated receptor-α (PPAR-α), promote steatosis, and induce oxidative stress, eventually leading to the occurrence of liver cancer (112, 113).
Overall, hepatitis C and NASH have similar metabolic dysregulation, including hepatic steatosis, dyslipidemia, insulin resistance, and oxidative stress. But at the same time, there are differences. The metabolic dysfunction of HCV is mainly induced by the core protein, while the metabolic dysfunction of NASH is more complicated and the specific mechanisms remain to be explored. Compared with NASH, the incidence of HCV-induced HCC is higher (114).
More than 90% of HCC occurs in the context of liver inflammation. Chronic inflammation induces immune cells to secrete a variety of cytokines, including TNFα, IL-6, leptin, adiponectin, chemokines, and so on (115) (Figure 4). In NASH, chronic HBV, and HCV, common mechanisms driving HCC development include: the persistence of liver inflammation, immune-mediated liver injury, and ultimately up-regulated release of pro-inflammatory factors TNF-α and IL-6. TNF-α is one of the most clearly characterized pro-tumor cytokines in HCC. It can simultaneously activate NF-κB and JNK signaling pathways, promote cell survival, inhibit cell apoptosis (116–118). IL-6-mediated STAT3 activation is a major driver of hepatocyte repair and replication, which promotes HCC development (117). Studies have also shown that IL-6 expression is upregulated and STAT3 is over-activated in HCC patients (119). The chemokine CKLF1 is overexpressed in HCC and is associated with tumor stage, vascular invasion, and prognosis. It can promote the progression of HCC by activating the IL6/STAT3 pathway (120).

Figure 4 Schematic diagram of IL6 and TNF-α inflammatory pathways in HBV, HCV, NASH-induced HCC. IL-6, Interleukin-6; TNF-αα Tumor necrosis factor-alpha; HCC, Hepatocellular carcinoma.
Specifically, PNPLA3 polymorphism can also enhance inflammatory signaling through the IL-6/STAT3 and CCL5 pathways. PNPLA3-I148M mutant mice have been reported to spontaneously develop hepatic steatosis (121). PNPLA3-I148M HCC cells can promote proliferation via IL6/STAT3 and enhance the activation of hepatic stellate cells by upregulating the expression of chemokine ligand 5 and collagen 1α1 (120, 122).
HBx protein has carcinogenic activity and can regulate a variety of inflammatory pathways in hepatocarcinogenesis, including MAPK, NF-κB, IL-6/STAT3, and PI3K signaling pathways (48). HBsAg can inhibit the activation of STAT3 in NK cells, leading to HBV clearance disorder and accelerate the progression of HBV hepatitis to HCC (123, 124). Meanwhile, immunosuppressive microenvironment also plays an important role in promoting tumor progression. In HBV-related HCC patients, increased peripheral blood neutrophil/lymphocyte ratio (NLR) and increased Foxp3 + Treg cell number are positively correlated with disease progression (125, 126).
As for hepatitis C, the pro-inflammatory status is maintained mainly by affecting STAT3 and NF-κB pathways (127). HCV core proteins, NS4B, and NS5B can enhance TNF-α-induced cell death by inhibiting NF-κB activation (128). HCV can also promote the transcription of STAT3 by upregulating miR-135A-5p and inhibiting its regulatory factor PTPRD, driving the progression of HCC (129).
Due to the tight anatomical functional crosstalk between the gut and liver, the gut microbiota and its metabolites can influence liver disease progression through the “gut-liver axis” (130). Animal models and human studies have demonstrated that increased intestinal permeability can lead to dysbiosis, resulting in the influx of pathogen-associated molecular patterns(PAMPs) and gut microbiota-derived metabolites into the liver, which further triggers hepatic inflammation and hepatocarcinogenesis (131). At the molecular level, PAMPs, such as lipopolysaccharide (LPS), enter the liver through the portal vein and are recognized by TLRs (TLR4 and TLR9) in immune cells, resulting in the production of a series of cytokines (IL, TNF, IFN) that cause liver cell damage (132). It has been reported that changes in intestinal flora and dysbiosis exist in both NASH and chronic viral hepatitis (131, 133–135).
In general, the progression from HBV, HCV, and NASH infection to HCC is the result of the accumulation of multiple factors and the interaction of multiple mechanisms. For HBV-induced HCC, HBV DNA integration was observed in nearly 80% of tumor tissues. HBV-induced epigenetic dysregulation (DNA methylation, Histone modification, MicroRNAs), HBV-encoded proteins (HBx, HbsAg, and core protein), and genetic variations play a central role in HCC development. For HCV-induced HCC, HCV-induced epigenetic dysregulation, HCV-encoded proteins (NS3, NS5A, NS5B, and core protein), and genetic variations can lead to hepatocarcinogenesis. For NASH-induced HCC, it is more dependent on genetic factors (PNPLA3, 17β-HSD13, TM6SF2 variant) and epigenetic dysregulation.
In addition, some of the same mechanisms also exist in the process of viral hepatitis and NASH-induced HCC, lipid metabolism disorders, persistent pro-inflammatory, immune responses, and intestinal microbiota dysbiosis are all involved and play a crucial role. Abnormal lipid accumulation, insulin resistance, oxidative stress, and other metabolic disorders are present in both hepatitis C and NASH-induced HCC. Persistent liver inflammation, immune-mediated liver damage, and upregulation release of proinflammatory cytokines TNF-α and IL-6 are present in NASH, chronic HBV, and HCV-induced HCC. Due to the presence of the gut-liver axis, intestinal microbiological changes and dysbiosis caused by viral hepatitis and NASH can further trigger hepatic inflammation and hepatocarcinogenesis through PAMPs.
In early stage HCC, the most effective treatment options are surgical resection, liver transplantation, or percutaneous local ablation. Systemic therapy with various drugs targeting the tumor microenvironment (TME) for unresectable HCC has been shown to be effective. Multi tyrosine kinase inhibitors(TKIs), such as sorafenib, lenvatinib, regorafenib, cabozantinib, and the vascular endothelial growth factor inhibitor (ramucirumab), have been widely used in clinical. Sorafenib exerts anti-tumor effects via inhibiting vascular endothelial growth factor receptor (VEGFR), Raf-1, B-Raf, and platelet-derived growth factor receptor (PDGFR) (136, 137). In addition to TKIs, new therapeutic strategies such as immune checkpoint inhibitors (ICIs) have also progressed in recent years (138). Two anti-PD-1 drugs, nivolumab and pembrolizumab, have been approved as second-line treatment for patients with sorafenib-refractory advanced HCC in the United States (139, 140). Even so, the prognosis of patients with advanced HCC is still not optimistic, and the prognosis of unresectable HCC remains poor. Exploring the pathogenesis of viral hepatitis and NASH-induced HCC may provide guidance for the development of new molecular therapeutic targets and therapeutic drugs. At the same time, it may also play an important role in judging the prognosis of patients and providing individualized treatment.
In general, the innovation of this paper is that we discuss viral hepatitis-induced and NASH-induced HCC together for the first time, and analyze differences and connections of them, which have not been seen in the published reviews. However, the description of the potential molecular pathogenesis in the article is not in-depth. We will focus on a certain direction to further investigate the underlying molecular mechanism in our future studies.
Writing—original draft preparation: ZT, CX, PY, WW, ZL, WZ, and JD; writing—review and editing: XZ and KD. All authors contributed to the article and approved the submitted version.
This work was supported by the Academician expert workstation of Shaanxi Province, and the National Natural Science Foundation of China (No.81870446, 82070671, 81900571, 82070681).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2021) 71:209–49. doi: 10.3322/caac.21660
2. Kulik L, El-Serag HB. Epidemiology and management of hepatocellular carcinoma. Gastroenterology (2019) 156:477–491.e471. doi: 10.1053/j.gastro.2018.08.065
3. Hartke J, Johnson M, Ghabril M. The diagnosis and treatment of hepatocellular carcinoma. Semin Diagn Pathol (2017) 34:153–9. doi: 10.1053/j.semdp.2016.12.011
4. Golabi P, Rhea L, Henry L, Younossi ZM. Hepatocellular carcinoma and non-alcoholic fatty liver disease. Hepatol Int (2019) 13:688–94. doi: 10.1007/s12072-019-09995-8
5. Yang JD, Hainaut P, Gores GJ, Amadou A, Plymoth A, Roberts LR. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat Rev Gastroenterol Hepatol (2019) 16:589–604. doi: 10.1038/s41575-019-0186-y
6. El-Serag HB. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology (2012) 142:1264–1273.e1261. doi: 10.1053/j.gastro.2011.12.061
7. Russo FP, Zanetto A, Pinto E, Battistella S, Penzo B, Burra P, et al. Hepatocellular carcinoma in chronic viral hepatitis: Where do we stand? Int J Mol Sci (2022) 23:500–16. doi: 10.3390/ijms23010500
8. D’Souza S, Lau KC, Coffin CS, Patel TR. Molecular mechanisms of viral hepatitis induced hepatocellular carcinoma. World J Gastroenterol (2020) 26:5759–83. doi: 10.3748/wjg.v26.i38.5759
9. Jia HD, Liang L, Li C, Wu H, Wang H, Liang YJ, et al. Long-term surgical outcomes of liver resection for hepatocellular carcinoma in patients with HBV and HCV Co-infection: A multicenter observational study. Front Oncol (2021) 11:700228. doi: 10.3389/fonc.2021.700228
10. EASL. 2017 Clinical practice guidelines on the management of hepatitis b virus infection. J Hepatol (2017) 67:370–98. doi: 10.1016/j.jhep.2017.03.021
11. Frager SZ, Schwartz JM. Hepatocellular carcinoma: Epidemiology, screening, and assessment of hepatic reserve. Curr Oncol (2020) 27:S138–s143. doi: 10.3747/co.27.7181
12. Baran B. Nucleos(t)ide analogs in the prevention of hepatitis b virus related hepatocellular carcinoma. World J Hepatol (2015) 7:1742–54. doi: 10.4254/wjh.v7.i13.1742
13. Kanwal F, Kramer JR, Ilyas J, Duan Z, El-Serag HB. HCV genotype 3 is associated with an increased risk of cirrhosis and hepatocellular cancer in a national sample of US Veterans HCV. Hepatol (2014) 60:98–105. doi: 10.1002/hep.27095
14. Ioannou GN, Green PK, Berry K. HCV eradication induced by direct-acting antiviral agents reduces the risk of hepatocellular carcinoma. J Hepatol (2017) 68:25–32. doi: 10.1016/j.jhep.2017.08.030
15. Morgan RL, Baack B, Smith BD, Yartel A, Pitasi M, Falck-Ytter Y. Eradication of hepatitis c virus infection and the development of hepatocellular carcinoma: A meta-analysis of observational studies. Ann Intern Med (2013) 158:329–37. doi: 10.7326/0003-4819-158-5-201303050-00005
16. Huang DQ, El-Serag HB, Loomba R. Global epidemiology of NAFLD-related HCC: Trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol (2021) 18:223–38. doi: 10.1038/s41575-020-00381-6
17. Anstee QM, Reeves HL, Kotsiliti E, Govaere O, Heikenwalder M. From NASH to HCC: Current concepts and future challenges. Nat Rev Gastroenterol Hepatol (2019) 16:411–28. doi: 10.1038/s41575-019-0145-7
18. Reddy SK, Steel JL, Chen HW, DeMateo DJ, Cardinal J, Behari J, et al. Outcomes of curative treatment for hepatocellular cancer in nonalcoholic steatohepatitis versus hepatitis c and alcoholic liver disease. Hepatology (2012) 55:1809–19. doi: 10.1002/hep.25536
19. Leung C, Yeoh SW, Patrick D, Ket S, Marion K, Gow P, et al. Characteristics of hepatocellular carcinoma in cirrhotic and non-cirrhotic non-alcoholic fatty liver disease. World J Gastroenterol (2015) 21:1189–96. doi: 10.3748/wjg.v21.i4.1189
20. Blumberg BS. Australia Antigen and the biology of hepatitis b. Science (1977) 197:17–25. doi: 10.1126/science.325649
21. Seeger C, Mason WS. Hepatitis b virus biology. Microbiol Mol Biol Rev (2000) 64:51–68. doi: 10.1128/MMBR.64.1.51-68.2000
22. Ringelhan M, McKeating JA, Protzer U. Viral hepatitis and liver cancer. Philos Trans R Soc Lond B Biol Sci (2017) 372:372. doi: 10.1098/rstb.2016.0274
23. Tu T, Budzinska MA, Shackel NA, Urban S. HBV DNA integration: Molecular mechanisms and clinical implications. Viruses (2017) 9:75–92. doi: 10.3390/v9040075
24. Zhao LH, Liu X, Yan HX, Li WY, Zeng X, Yang Y, et al. Genomic and oncogenic preference of HBV integration in hepatocellular carcinoma. Nat Commun (2016) 7:12992. doi: 10.1038/ncomms12992
25. Sung WK, Zheng H, Li S, Chen R, Liu X, Li Y, et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat Genet (2012) 44:765–9. doi: 10.1038/ng.2295
26. Tamori A, Yamanishi Y, Kawashima S, Kanehisa M, Enomoto M, Tanaka H, et al. Alteration of gene expression in human hepatocellular carcinoma with integrated hepatitis b virus DNA. Clin Cancer Res (2005) 11:5821–6. doi: 10.1158/1078-0432.CCR-04-2055
27. Fujimoto A, Totoki Y, Abe T, Boroevich KA, Hosoda F, Nguyen HH, et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat Genet (2012) 44:760–4. doi: 10.1038/ng.2291
28. Wang HC, Huang W, Lai MD, Su IJ. Hepatitis b virus pre-s mutants, endoplasmic reticulum stress and hepatocarcinogenesis. Cancer Sci (2006) 97:683–8. doi: 10.1111/j.1349-7006.2006.00235.x
29. Wang HC, Wu HC, Chen CF, Fausto N, Lei HY, Su IJ. Different types of ground glass hepatocytes in chronic hepatitis b virus infection contain specific pre-s mutants that may induce endoplasmic reticulum stress. Am J Pathol (2003) 163:2441–9. doi: 10.1016/S0002-9440(10)63599-7
30. Rongrui L, Na H, Zongfang L, Fanpu J, Shiwen J. Epigenetic mechanism involved in the HBV/HCV-related hepatocellular carcinoma tumorigenesis. Curr Pharm Des (2014) 20:1715–25. doi: 10.2174/13816128113199990533
31. Zhang D, Guo S, Schrodi SJ. Mechanisms of DNA methylation in virus-host interaction in hepatitis b infection: Pathogenesis and oncogenetic properties. Int J Mol Sci (2021) 22:9858–70. doi: 10.3390/ijms22189858
32. Zhu R, Li BZ, Li H, Ling YQ, Hu XQ, Zhai WR, et al. Association of p16INK4A hypermethylation with hepatitis b virus X protein expression in the early stage of HBV-associated hepatocarcinogenesis. Pathol Int (2007) 57:328–36. doi: 10.1111/j.1440-1827.2007.02104.x
33. Um TH, Kim H, Oh BK, Kim MS, Kim KS, Jung G, et al. Aberrant CpG island hypermethylation in dysplastic nodules and early HCC of hepatitis b virus-related human multistep hepatocarcinogenesis. J Hepatol (2011) 54:939–47. doi: 10.1016/j.jhep.2010.08.021
34. Arzumanyan A, Friedman T, Kotei E, Ng IO, Lian Z, Feitelson MA. Epigenetic repression of e-cadherin expression by hepatitis b virus x antigen in liver cancer. Oncogene (2012) 31:563–72. doi: 10.1038/onc.2011.255
35. Park IY, Sohn BH, Yu E, Suh DJ, Chung YH, Lee JH, et al. Aberrant epigenetic modifications in hepatocarcinogenesis induced by hepatitis b virus X protein. Gastroenterology (2007) 132:1476–94. doi: 10.1053/j.gastro.2007.01.034
36. Berger SL. The complex language of chromatin regulation during transcription. Nature (2007) 447:407–12. doi: 10.1038/nature05915
37. Liu XY, Tang SH, Wu SL, Luo YH, Cao MR, Zhou HK, et al. Epigenetic modulation of insulin-like growth factor-II overexpression by hepatitis b virus X protein in hepatocellular carcinoma. Am J Cancer Res (2015) 5:956–78.
38. Sarris ME, Moulos P, Haroniti A, Giakountis A, Talianidis I. Smyd3 is a transcriptional potentiator of multiple cancer-promoting genes and required for liver and colon cancer development. Cancer Cell (2016) 29:354–66. doi: 10.1016/j.ccell.2016.01.013
39. Wang Y, Xie BH, Lin WH, Huang YH, Ni JY, Hu J, et al. Amplification of SMYD3 promotes tumorigenicity and intrahepatic metastasis of hepatocellular carcinoma via upregulation of CDK2 and MMP2. Oncogene (2019) 38:4948–61. doi: 10.1038/s41388-019-0766-x
40. Yang L, He J, Chen L, Wang G. Hepatitis b virus X protein upregulates expression of SMYD3 and c-MYC in HepG2 cells. Med Oncol (2009) 26:445–51. doi: 10.1007/s12032-008-9144-1
41. Xu J, An P, Winkler CA, Yu Y. Dysregulated microRNAs in hepatitis b virus-related hepatocellular carcinoma: Potential as biomarkers and therapeutic targets. Front Oncol (2020) 10:1271. doi: 10.3389/fonc.2020.01271
42. Zhu HT, Liu RB, Liang YY, Hasan AME, Wang HY, Shao Q, et al. Serum microRNA profiles as diagnostic biomarkers for HBV-positive hepatocellular carcinoma. Liver Int (2017) 37:888–96. doi: 10.1111/liv.13356
43. Unsal H, Yakicier C, Marçais C, Kew M, Volkmann M, Zentgraf H, et al. Genetic heterogeneity of hepatocellular carcinoma. Proc Natl Acad Sci USA (1994) 91:822–6. doi: 10.1073/pnas.91.2.822
44. Yeh CT, Shen CH, Tai DI, Chu CM, Liaw YF. Identification and characterization of a prevalent hepatitis b virus X protein mutant in Taiwanese patients with hepatocellular carcinoma. Oncogene (2000) 19:5213–20. doi: 10.1038/sj.onc.1203903
45. Wollersheim M, Debelka U, Hofschneider PH. A transactivating function encoded in the hepatitis b virus X gene is conserved in the integrated state. Oncogene (1988) 3:545–52.
46. Paterlini P, Poussin K, Kew M, Franco D, Brechot C. Selective accumulation of the X transcript of hepatitis b virus in patients negative for hepatitis b surface antigen with hepatocellular carcinoma. Hepatology (1995) 21:313–21.
47. Nguyen DH, Ludgate L, Hu J. Hepatitis b virus-cell interactions and pathogenesis. J Cell Physiol (2008) 216:289–94. doi: 10.1002/jcp.21416
48. Murakami S. Hepatitis b virus X protein: A multifunctional viral regulator. J Gastroenterol (2001) 36:651–60. doi: 10.1007/s005350170027
49. Knoll S, Fürst K, Thomas S, Villanueva Baselga S, Stoll A, Schaefer S, et al. Dissection of cell context-dependent interactions between HBx and p53 family members in regulation of apoptosis: a role for HBV-induced HCC. Cell Cycle (2011) 10:3554–65. doi: 10.4161/cc.10.20.17856
50. Geng M, Xin X, Bi LQ, Zhou LT, Liu XH. Molecular mechanism of hepatitis b virus X protein function in hepatocarcinogenesis. World J Gastroenterol (2015) 21:10732–8. doi: 10.3748/wjg.v21.i38.10732
51. Yoo YG, Na TY, Seo HW, Seong JK, Park CK, Shin YK, et al. Hepatitis b virus X protein induces the expression of MTA1 and HDAC1, which enhances hypoxia signaling in hepatocellular carcinoma cells. Oncogene (2008) 27:3405–13. doi: 10.1038/sj.onc.1211000
52. Sanz-Cameno P, Martín-Vílchez S, Lara-Pezzi E, Borque MJ, Salmerón J, Muñoz de Rueda P, et al. Hepatitis b virus promotes angiopoietin-2 expression in liver tissue: Role of HBV x protein. Am J Pathol (2006) 169:1215–22. doi: 10.2353/ajpath.2006.051246
53. Song J, Zhang X, Ge Q, Yuan C, Chu L, Liang HF, et al. CRISPR/Cas9-mediated knockout of HBsAg inhibits proliferation and tumorigenicity of HBV-positive hepatocellular carcinoma cells. J Cell Biochem (2018) 119:8419–31. doi: 10.1002/jcb.27050
54. Chen M, Sällberg M, Hughes J, Jones J, Guidotti LG, Chisari FV, et al. Immune tolerance split between hepatitis b virus precore and core proteins. J Virol (2005) 79:3016–27. doi: 10.1128/JVI.79.5.3016-3027.2005
55. Kanda T, Wu S, Sasaki R, Nakamura M, Haga Y, Jiang X, et al. HBV core protein enhances cytokine production. Diseases (2015) 3:213–20. doi: 10.3390/diseases3030213
56. Caligiuri P, Cerruti R, Icardi G, Bruzzone B. Overview of hepatitis b virus mutations and their implications in the management of infection. World J Gastroenterol (2016) 22:145–54. doi: 10.3748/wjg.v22.i1.145
57. Pollicino T, Cacciola I, Saffioti F, Raimondo G. Hepatitis b virus PreS/S gene variants: Pathobiology and clinical implications. J Hepatol (2014) 61:408–17. doi: 10.1016/j.jhep.2014.04.041
58. Yang Y, Sun JW, Zhao LG, Bray F, Xiang YB. Quantitative evaluation of hepatitis b virus mutations and hepatocellular carcinoma risk: a meta-analysis of prospective studies. Chin J Cancer Res (2015) 27:497–508. doi: 10.3978/j.issn.1000-9604.2015.10.05
59. Yeh CT, Chen T, Hsu CW, Chen YC, Lai MW, Liang KH, et al. Emergence of the rtA181T/sW172* mutant increased the risk of hepatoma occurrence in patients with lamivudine-resistant chronic hepatitis b. BMC Cancer (2011) 11:398. doi: 10.1186/1471-2407-11-398
60. Baptista M, Kramvis A, Kew MC. High prevalence of 1762(T) 1764(A) mutations in the basic core promoter of hepatitis b virus isolated from black africans with hepatocellular carcinoma compared with asymptomatic carriers. Hepatology (1999) 29:946–53. doi: 10.1002/hep.510290336
61. An P, Xu J, Yu Y, Winkler CA. Host and viral genetic variation in HBV-related hepatocellular carcinoma. Front Genet (2018) 9:261. doi: 10.3389/fgene.2018.00261
62. Bartenschlager R, Lohmann V, Penin F. The molecular and structural basis of advanced antiviral therapy for hepatitis c virus infection. Nat Rev Microbiol (2013) 11:482–96. doi: 10.1038/nrmicro3046
63. Hoshida Y, Fuchs BC, Bardeesy N, Baumert TF, Chung RT. Pathogenesis and prevention of hepatitis c virus-induced hepatocellular carcinoma. J Hepatol (2014) 61:S79–90. doi: 10.1016/j.jhep.2014.07.010
64. Lee MH, Hsiao TI, Subramaniam SR, Le AK, Vu VD, Trinh HN, et al. HCV genotype 6 increased the risk for hepatocellular carcinoma among Asian patients with liver cirrhosis. Am J Gastroenterol (2017) 112:1111–9. doi: 10.1038/ajg.2017.123
65. Zekri Ael R, Nassar AA, El-Din El-Rouby MN, Shousha HI, Barakat AB, El-Desouky ED, et al. Disease progression from chronic hepatitis c to cirrhosis and hepatocellular carcinoma is associated with increasing DNA promoter methylation. Asian Pac J Cancer Prev (2014) 14:6721–6. doi: 10.7314/apjcp.2013.14.11.6721
66. Zhang J, Li H, Yu JP, Wang SE, Ren XB. Role of SOCS1 in tumor progression and therapeutic application. Int J Cancer (2012) 130:1971–80. doi: 10.1002/ijc.27318
67. Yang B, Guo M, Herman JG, Clark DP. Aberrant promoter methylation profiles of tumor suppressor genes in hepatocellular carcinoma. Am J Pathol (2003) 163:1101–7. doi: 10.1016/S0002-9440(10)63469-4
68. Higgs MR, Lerat H, Pawlotsky JM. Downregulation of Gadd45beta expression by hepatitis c virus leads to defective cell cycle arrest. Cancer Res (2010) 70:4901–11. doi: 10.1158/0008-5472.CAN-09-4554
69. Duong FH, Filipowicz M, Tripodi M, La Monica N, Heim MH. Hepatitis c virus inhibits interferon signaling through up-regulation of protein phosphatase 2A. Gastroenterology (2004) 126:263–77. doi: 10.1053/j.gastro.2003.10.076
70. Zekri AR, Bahnasy AA, Shoeab FE, Mohamed WS, El-Dahshan DH, Ali FT, et al. Methylation of multiple genes in hepatitis c virus associated hepatocellular carcinoma. J Adv Res (2014) 5:27–40. doi: 10.1016/j.jare.2012.11.002
71. Couvert P, Carrié A, Pariès J, Vaysse J, Miroglio A, Kerjean A, et al. Liver insulin-like growth factor 2 methylation in hepatitis c virus cirrhosis and further occurrence of hepatocellular carcinoma. World J Gastroenterol (2008) 14:5419–27. doi: 10.3748/wjg.14.5419
72. Hamdane N, Jühling F, Crouchet E, El Saghire H, Thumann C, Oudot MA, et al. HCV-induced epigenetic changes associated with liver cancer risk persist after sustained virologic response. Gastroenterology (2019) 156:2313–2329.e2317. doi: 10.1053/j.gastro.2019.02.038
73. Perez S, Kaspi A, Domovitz T, Davidovich A, Lavi-Itzkovitz A, Meirson T, et al. Hepatitis c virus leaves an epigenetic signature post cure of infection by direct-acting antivirals. PloS Genet (2019) 15:e1008181. doi: 10.1371/journal.pgen.1008181
74. Shigekawa Y, Hayami S, Ueno M, Miyamoto A, Suzaki N, Kawai M, et al. Overexpression of KDM5B/JARID1B is associated with poor prognosis in hepatocellular carcinoma. Oncotarget (2018) 9:34320–35. doi: 10.18632/oncotarget.26144
75. Kasai H, Mochizuki K, Tanaka T, Yamashita A, Matsuura Y, Moriishi K. Induction of HOX genes by hepatitis c virus infection via Impairment Histone H2A Monoubiquitination. J Virol (2021) 95:e01784–20.
76. Kanda T, Goto T, Hirotsu Y, Moriyama M, Omata M. Molecular mechanisms driving progression of liver cirrhosis towards hepatocellular carcinoma in chronic hepatitis b and c infections: A review. Int J Mol Sci (2019) 20:3057–73. doi: 10.3390/ijms20061358
77. Goto K, Roca Suarez AA, Wrensch F, Baumert TF, Lupberger J. Hepatitis c virus and hepatocellular carcinoma: When the host loses its grip. Int J Mol Sci (2020) 21:1358–81. doi: 10.3390/ijms21093057
78. Machida K, Liu JC, McNamara G, Levine A, Duan L, Lai MM. Hepatitis c virus causes uncoupling of mitotic checkpoint and chromosomal polyploidy through the Rb pathway. J Virol (2009) 83:12590–600. doi: 10.1128/JVI.02643-08
79. Majumder M, Ghosh AK, Steele R, Ray R, Ray RB. Hepatitis c virus NS5A physically associates with p53 and regulates p21/waf1 gene expression in a p53-dependent manner. J Virol (2001) 75:1401–7. doi: 10.1128/JVI.75.3.1401-1407.2001
80. He QQ, Cheng RX, Sun Y, Feng DY, Chen ZC, Zheng H. Hepatocyte transformation and tumor development induced by hepatitis c virus NS3 c-terminal deleted protein. World J Gastroenterol (2003) 9:474–8. doi: 10.3748/wjg.v9.i3.474
81. Alisi A, Giambartolomei S, Cupelli F, Merlo P, Fontemaggi G, Spaziani A, et al. Physical and functional interaction between HCV core protein and the different p73 isoforms. Oncogene (2003) 22:2573–80. doi: 10.1038/sj.onc.1206333
82. Tardif KD, Waris G, Siddiqui A. Hepatitis c virus, ER stress, and oxidative stress. Trends Microbiol (2005) 13:159–63. doi: 10.1016/j.tim.2005.02.004
83. Hu Z, Muroyama R, Kowatari N, Chang J, Omata M, Kato N. Characteristic mutations in hepatitis c virus core gene related to the occurrence of hepatocellular carcinoma. Cancer Sci (2009) 100:2465–8. doi: 10.1111/j.1349-7006.2009.01338.x
84. Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology (1998) 114:842–5. doi: 10.1016/S0016-5085(98)70599-2
85. Sumida Y, Niki E, Naito Y, Yoshikawa T. Involvement of free radicals and oxidative stress in NAFLD/NASH. Free Radic Res (2013) 47:869–80. doi: 10.3109/10715762.2013.837577
86. Kawano Y, Cohen DE. Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. J Gastroenterol (2013) 48:434–41. doi: 10.1007/s00535-013-0758-5
87. Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology (2010) 52:1836–46. doi: 10.1002/hep.24001
88. Takaki A, Kawai D, Yamamoto K. Multiple hits, including oxidative stress, as pathogenesis and treatment target in non-alcoholic steatohepatitis (NASH). Int J Mol Sci (2013) 14:20704–28. doi: 10.3390/ijms141020704
89. Dongiovanni P, Meroni M, Longo M, Fargion S, Fracanzani AL. Genetics, immunity and nutrition boost the switching from NASH to HCC. Biomedicines (2021) 9:1524–50. doi: 10.3390/biomedicines9111524
90. Valenti L, Dongiovanni P, Ginanni Corradini S, Burza MA, Romeo S. PNPLA3 I148M variant and hepatocellular carcinoma: A common genetic variant for a rare disease. Dig Liver Dis (2013) 45:619–24. doi: 10.1016/j.dld.2012.12.006
91. Dongiovanni P, Donati B, Fares R, Lombardi R, Mancina RM, Romeo S, et al. PNPLA3 I148M polymorphism and progressive liver disease. World J Gastroenterol (2013) 19:6969–78. doi: 10.3748/wjg.v19.i41.6969
92. Banini BA, Kumar DP, Cazanave S, Seneshaw M, Mirshahi F, Santhekadur PK, et al. Identification of a metabolic, transcriptomic, and molecular signature of patatin-like phospholipase domain containing 3-mediated acceleration of steatohepatitis. Hepatology (2021) 73:1290–306. doi: 10.1002/hep.31609
93. Su W, Wang Y, Jia X, Wu W, Li L, Tian X, et al. Comparative proteomic study reveals 17β-HSD13 as a pathogenic protein in nonalcoholic fatty liver disease. Proc Natl Acad Sci USA (2014) 111:11437–42. doi: 10.1073/pnas.1410741111
94. Chen J, Zhuo JY, Yang F, Liu ZK, Zhou L, Xie HY, et al. 17-beta-hydroxysteroid dehydrogenase 13 inhibits the progression and recurrence of hepatocellular carcinoma. Hepatobiliary Pancreat Dis Int (2018) 17:220–6. doi: 10.1016/j.hbpd.2018.04.006
95. Pirola CJ, Garaycoechea M, Flichman D, Arrese M, San Martino J, Gazzi C, et al. Splice variant rs72613567 prevents worst histologic outcomes in patients with nonalcoholic fatty liver disease. J Lipid Res (2019) 60:176–85. doi: 10.1194/jlr.P089953
96. Donati B, Dongiovanni P, Romeo S, Meroni M, McCain M, Miele L, et al. MBOAT7 rs641738 variant and hepatocellular carcinoma in non-cirrhotic individuals. Sci Rep (2017) 7:4492. doi: 10.1038/s41598-017-04991-0
97. Stickel F, Buch S, Nischalke HD, Weiss KH, Gotthardt D, Fischer J, et al. Genetic variants in PNPLA3 and TM6SF2 predispose to the development of hepatocellular carcinoma in individuals with alcohol-related cirrhosis. Am J Gastroenterol (2018) 113:1475–83. doi: 10.1038/s41395-018-0041-8
98. Yang J, Trépo E, Nahon P, Cao Q, Moreno C, Letouzé E, et al. PNPLA3 and TM6SF2 variants as risk factors of hepatocellular carcinoma across various etiologies and severity of underlying liver diseases. Int J Cancer (2019) 144:533–44. doi: 10.1002/ijc.31910
99. Tryndyak VP, Han T, Muskhelishvili L, Fuscoe JC, Ross SA, Beland FA, et al. Coupling global methylation and gene expression profiles reveal key pathophysiological events in liver injury induced by a methyl-deficient diet. Mol Nutr Food Res (2011) 55:411–8. doi: 10.1002/mnfr.201000300
100. Kuramoto J, Arai E, Tian Y, Funahashi N, Hiramoto M, Nammo T, et al. Genome-wide DNA methylation analysis during non-alcoholic steatohepatitis-related multistage hepatocarcinogenesis: Comparison with hepatitis virus-related carcinogenesis. Carcinogenesis (2017) 38:261–70. doi: 10.1093/carcin/bgx005
101. Tian Y, Arai E, Makiuchi S, Tsuda N, Kuramoto J, Ohara K, et al. Aberrant DNA methylation results in altered gene expression in non-alcoholic steatohepatitis-related hepatocellular carcinomas. J Cancer Res Clin Oncol (2020) 146:2461–77. doi: 10.1007/s00432-020-03298-4
102. de Conti A, Dreval K, Tryndyak V, Orisakwe OE, Ross SA, Beland FA, et al. Inhibition of the cell death pathway in nonalcoholic steatohepatitis (NASH)-related hepatocarcinogenesis is associated with histone H4 lysine 16 deacetylation. Mol Cancer Res (2017) 15:1163–72. doi: 10.1158/1541-7786.MCR-17-0109
103. Tian Y, Wong VW, Wong GL, Yang W, Sun H, Shen J, et al. Histone deacetylase HDAC8 promotes insulin resistance and β-catenin activation in NAFLD-associated hepatocellular carcinoma. Cancer Res (2015) 75:4803–16. doi: 10.1158/0008-5472.CAN-14-3786
104. Takaki Y, Saito Y, Takasugi A, Toshimitsu K, Yamada S, Muramatsu T, et al. Silencing of microRNA-122 is an early event during hepatocarcinogenesis from non-alcoholic steatohepatitis. Cancer Sci (2014) 105:1254–60. doi: 10.1111/cas.12498
105. Kutlu O, Kaleli HN, Ozer E. Molecular pathogenesis of nonalcoholic steatohepatitis- (NASH-) related hepatocellular carcinoma. Can J Gastroenterol Hepatol (2018) 2018:8543763. doi: 10.1155/2018/8543763
106. Chettouh H, Lequoy M, Fartoux L, Vigouroux C, Desbois-Mouthon C. Hyperinsulinaemia and insulin signalling in the pathogenesis and the clinical course of hepatocellular carcinoma. Liver Int (2015) 35:2203–17. doi: 10.1111/liv.12903
107. Hirsova P, Ibrabim SH, Gores GJ, Malhi H. Lipotoxic lethal and sublethal stress signaling in hepatocytes: relevance to NASH pathogenesis. J Lipid Res (2016) 57:1758–70. doi: 10.1194/jlr.R066357
108. Nelson JE, Wilson L, Brunt EM, Yeh MM, Kleiner DE, Unalp-Arida A, et al. Relationship between the pattern of hepatic iron deposition and histological severity in nonalcoholic fatty liver disease. Hepatology (2011) 53:448–57. doi: 10.1002/hep.24038
109. Hwang SJ, Lee SD. Hepatic steatosis and hepatitis c: Still unhappy bedfellows? J Gastroenterol Hepatol (2011) 26 Suppl 1:96–101. doi: 10.1111/j.1440-1746.2010.06542.x
110. Moriya K, Fujie H, Shintani Y, Yotsuyanagi H, Tsutsumi T, Ishibashi K, et al. The core protein of hepatitis c virus induces hepatocellular carcinoma in transgenic mice. Nat Med (1998) 4:1065–7. doi: 10.1038/2053
111. Leslie J, Geh D, Elsharkawy AM, Mann DA, Vacca M. Metabolic dysfunction and cancer in HCV: shared pathways and mutual interactions. J Hepatol (2022) 77:219–36. doi: 10.1016/j.jhep.2022.01.029
112. Koike K. Steatosis, liver injury, and hepatocarcinogenesis in hepatitis c viral infection. J Gastroenterol (2009) 44 Suppl 19:82–8. doi: 10.1007/s00535-008-2276-4
113. Tanaka N, Moriya K, Kiyosawa K, Koike K, Gonzalez FJ, Aoyama T. PPARalpha activation is essential for HCV core protein-induced hepatic steatosis and hepatocellular carcinoma in mice. J Clin Invest (2008) 118:683–94. doi: 10.1172/JCI33594
114. Koike K, Moriya K. Metabolic aspects of hepatitis c viral infection: Steatohepatitis resembling but distinct from NASH. J Gastroenterol (2005) 40:329–36. doi: 10.1007/s00535-005-1586-z
115. Yang YM, Kim SY, Seki E. Inflammation and liver cancer: Molecular mechanisms and therapeutic targets. Semin Liver Dis (2019) 39:26–42. doi: 10.1055/s-0038-1676806
116. Taniguchi K, Karin M. NF-κB, inflammation, immunity and cancer: Coming of age. Nat Rev Immunol (2018) 18:309–24. doi: 10.1038/nri.2017.142
117. Ringelhan M, Pfister D, O’Connor T, Pikarsky E, Heikenwalder M. The immunology of hepatocellular carcinoma. Nat Immunol (2018) 19:222–32. doi: 10.1038/s41590-018-0044-z
118. Das M, Garlick DS, Greiner DL, Davis RJ. The role of JNK in the development of hepatocellular carcinoma. Genes Dev (2011) 25:634–45. doi: 10.1101/gad.1989311
119. Aleksandrova K, Boeing H, Nöthlings U, Jenab M, Fedirko V, Kaaks R, et al. Inflammatory and metabolic biomarkers and risk of liver and biliary tract cancer. Hepatology (2014) 60:858–71. doi: 10.1002/hep.27016
120. Liu Y, Liu L, Zhou Y, Zhou P, Yan Q, Chen X, et al. CKLF1 enhances inflammation-mediated carcinogenesis and prevents doxorubicin-induced apoptosis via IL6/STAT3 signaling in HCC. Clin Cancer Res (2019) 25:4141–54. doi: 10.1158/1078-0432.CCR-18-3510
121. Smagris E, BasuRay S, Li J, Huang Y, Lai KM, Gromada J, et al. Pnpla3I148M knockin mice accumulate PNPLA3 on lipid droplets and develop hepatic steatosis. Hepatology (2015) 61:108–18. doi: 10.1002/hep.27242
122. Bruschi FV, Claudel T, Tardelli M, Caligiuri A, Stulnig TM, Marra F, et al. The PNPLA3 I148M variant modulates the fibrogenic phenotype of human hepatic stellate cells. Hepatology (2017) 65:1875–90. doi: 10.1002/hep.29041
123. Zheng B, Yang Y, Han Q, Yin C, Pan Z, Zhang J. STAT3 directly regulates NKp46 transcription in NK cells of HBeAg-negative CHB patients. J Leukoc Biol (2019) 106:987–96. doi: 10.1002/JLB.2A1118-421R
124. Sun C, Sun H, Zhang C, Tian Z. NK cell receptor imbalance and NK cell dysfunction in HBV infection and hepatocellular carcinoma. Cell Mol Immunol (2015) 12:292–302. doi: 10.1038/cmi.2014.91
125. Liu X, He L, Han J, Wang L, Li M, Jiang Y, et al. Association of neutrophil-lymphocyte ratio and T lymphocytes with the pathogenesis and progression of HBV-associated primary liver cancer. PloS One (2017) 12:e0170605. doi: 10.1371/journal.pone.0170605
126. Tajiri K, Baba H, Kawai K, Minemura M, Yasumura S, Takahara T, et al. Neutrophil-to-lymphocyte ratio predicts recurrence after radiofrequency ablation in hepatitis b virus infection. J Gastroenterol Hepatol (2016) 31:1291–9. doi: 10.1111/jgh.13287
127. Virzì A, Roca Suarez AA, Baumert TF, Lupberger J. Oncogenic signaling induced by HCV infection. Viruses (2018) 10:10. doi: 10.3390/v10100538
128. Park J, Kang W, Ryu SW, Kim WI, Chang DY, Lee DH, et al. Hepatitis c virus infection enhances TNFα-induced cell death via suppression of NF-κB. Hepatology (2012) 56:831–40. doi: 10.1002/hep.25726
129. Van Renne N, Roca Suarez AA, Duong FHT, Gondeau C, Calabrese D, Fontaine N, et al. miR-135a-5p-mediated downregulation of protein tyrosine phosphatase receptor delta is a candidate driver of HCV-associated hepatocarcinogenesis. Gut (2018) 67:953–62. doi: 10.1136/gutjnl-2016-312270
130. Albillos A, de Gottardi A, Rescigno M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J Hepatol (2020) 72:558–77. doi: 10.1016/j.jhep.2019.10.003
131. Chen YH, Wu WK, Wu MS. Microbiota-associated therapy for non-alcoholic steatohepatitis-induced liver cancer: A review. Int J Mol Sci (2020) 21:21. doi: 10.3390/ijms21175999
132. Rattan P, Minacapelli CD, Rustgi V. The microbiome and hepatocellular carcinoma. Liver Transpl (2020) 26:1316–27. doi: 10.1002/lt.25828
133. Levrero M, Zucman-Rossi J. Mechanisms of HBV-induced hepatocellular carcinoma. J Hepatol (2016) 64:S84–s101. doi: 10.1016/j.jhep.2016.02.021
134. Yang R, Xu Y, Dai Z, Lin X, Wang H. The immunologic role of gut microbiota in patients with chronic HBV infection. J Immunol Res (2018) 2018:2361963. doi: 10.1155/2018/2361963
135. Preveden T, Scarpellini E, Milić N, Luzza F, Abenavoli L. Gut microbiota changes and chronic hepatitis c virus infection. Expert Rev Gastroenterol Hepatol (2017) 11:813–9. doi: 10.1080/17474124.2017.1343663
136. Huang A, Yang XR, Chung WY, Dennison AR, Zhou J. Targeted therapy for hepatocellular carcinoma. Signal Transduct Target Ther (2020) 5:146. doi: 10.1038/s41392-020-00264-x
137. Llovet JM, Castet F, Heikenwalder M, Maini MK, Mazzaferro V, Pinato DJ, et al. Immunotherapies for hepatocellular carcinoma. Nat Rev Clin Oncol (2022) 19:151–72. doi: 10.1038/s41571-021-00573-2
138. Liu Z, Lin Y, Zhang J, Zhang Y, Li Y, Liu Z, et al. Molecular targeted and immune checkpoint therapy for advanced hepatocellular carcinoma. J Exp Clin Cancer Res (2019) 38:447. doi: 10.1186/s13046-019-1412-8
139. El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet (2017) 389:2492–502. doi: 10.1016/S0140-6736(17)31046-2
Keywords: hepatocellular carcinoma, viral hepatitis, non-alcoholic steatohepatitis, molecular pathogenesis, HBV - hepatitis B virus, HCV (hepatitis C)
Citation: Tian Z, Xu C, Yang P, Lin Z, Wu W, Zhang W, Ding J, Ding R, Zhang X and Dou K (2022) Molecular pathogenesis: Connections between viral hepatitis-induced and non-alcoholic steatohepatitis-induced hepatocellular carcinoma. Front. Immunol. 13:984728. doi: 10.3389/fimmu.2022.984728
Received: 02 July 2022; Accepted: 31 August 2022;
Published: 15 September 2022.
Edited by:
Surendra Kumar Shukla, University of Oklahoma, United StatesReviewed by:
Aneesha Dasgupta, Purdue University Indianapolis, United StatesCopyright © 2022 Tian, Xu, Yang, Lin, Wu, Zhang, Ding, Ding, Zhang and Dou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xuan Zhang, emhhbmd4dWFudGpAMTYzLmNvbQ==; Kefeng Dou, ZG91a2VmQGZtbXUuZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.