 Eric Jou
Eric Jou Noe Rodriguez-Rodriguez
Noe Rodriguez-Rodriguez Andrew N. J. McKenzie
Andrew N. J. McKenzie
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 03 October 2022
Sec. Cytokines and Soluble Mediators in Immunity
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.981479
This article is part of the Research TopicModulating Cytokines as Treatment for Autoimmune Diseases and Cancer: Volume IIView all 8 articles
Colorectal cancer (CRC) is the second leading cause of cancer-related death worldwide, and is largely refractory to current immunotherapeutic interventions. The lack of efficacy of existing cancer immunotherapies in CRC reflects the complex nature of the unique intestinal immune environment, which serves to maintain barrier integrity against pathogens and harmful environmental stimuli while sustaining host-microbe symbiosis during homeostasis. With their expression by barrier epithelial cells, the cytokines interleukin-25 (IL-25) and IL-33 play key roles in intestinal immune responses, and have been associated with inappropriate allergic reactions, autoimmune diseases and cancer pathology. Studies in the past decade have begun to uncover the important roles of IL-25 and IL-33 in shaping the CRC tumour immune microenvironment, where they may promote or inhibit tumorigenesis depending on the specific CRC subtype. Notably, both IL-25 and IL-33 have been shown to act on group 2 innate lymphoid cells (ILC2s), but can also stimulate an array of other innate and adaptive immune cell types. Though sometimes their functions can overlap they can also produce distinct phenotypes dependent on the differential distribution of their receptor expression. Furthermore, both IL-25 and IL-33 modulate pathways previously known to contribute to CRC tumorigenesis, including angiogenesis, tumour stemness, invasion and metastasis. Here, we review our current understanding of IL-25 and IL-33 in CRC tumorigenesis, with specific focus on dissecting their individual function in the context of distinct subtypes of CRC, and the potential prospects for targeting these pathways in CRC immunotherapy.
CRC affects the colon or rectum, and is third on the list of most common cancers globally (1). Despite improvements in early detection and diagnosis, CRC remains the second leading cause of cancer-related mortality worldwide, underlying ~10% of total cancer deaths. It is estimated that this incidence may increase by a further 60% by 2030 (2). CRC is a heterogenous disease encompassing different subtypes. The majority (~80%) of sporadic human CRCs have causative loss-of-function mutations in the adenomatous polyposis coli (APC) tumour suppressor gene, and progress over several years from benign polyps to metastatic disease (3). Other rarer subtypes of CRC, such as microsatellite-instability-high (MSI-high) CRC (~15% of CRC) primarily driven by mismatch-repair-deficiency (4), and colitis-associated colorectal cancer (CAC) secondary to inflammatory bowel disease (IBD) (1-2% of CRC) (5, 6), have distinct driver mutations and disease etiologies.
The intestinal tract is inhabited and visited by huge numbers of immune cells, and harbors the largest proportion of the host immune system (7). With its continuous exposure to the external environment during the processing of dietary intake, and its hosting of an enormous and diverse microbiome, the intestinal immune compartment must protect the host from harmful pathogens, while maintaining tolerance to self-tissues, dietary antigens, and hosting commensal microorganisms. Through embryonic development (Stras et al., 2019) and subsequent exposure to external environmental stimuli after birth (8) the intestinal immune system adapts continuously during its education by host and environmental factors (9–12), and develops a highly specialised compartmentalisation required for its proper function (13, 14). A complex array of immune interactions between secreted molecules and cell surface receptors across a diverse collection of epithelial, stromal and immune cells help maintain immune homeostasis and tolerance during the steady state, but can also react rapidly to infection to orchestrate protective immunity (15, 16). Dysregulation of this intricate system has been implicated in a wide range of pathologies including autoimmunity and cancer (17–19).
IL-25 and IL-33 are epithelial cell-derived cytokines that play important roles in intestinal immunity. Both IL-25 and IL-33 are potent activators of type-2 immunity, which is critical in parasite helminth expulsion, and promoting tissue repair and regeneration (20–22). Nevertheless, recent studies suggest that there are important differences in their tissue-specific functions (23). For example, in the intestines, IL-33 and IL-25 are produced by non-overlapping cellular sources, and have distinct regulatory mechanisms behind their storage and release (24). Furthermore, IL-33 and IL-25 mediate effector functions through distinct receptors, with differential expression of these receptors on shared and distinct cell types. In the past decade, emerging studies have begun to uncover the previously unappreciated role of IL-25 and IL-33 in CRC disease pathogenesis and in shaping the tumour niche, with distinct effects specific to the different subtypes of CRC.
In this review, we outline how the dynamics of intestinal immune responses driven by IL-25 and IL-33 impact CRC development in the context of CRC subtypes, with reference to how different mouse models provide insight (sometimes imperfect) into the immune reactions in the tissues in different subtypes of CRC. Consequently, we examine how targeting the inhibition of IL-25, IL-33 and type 2 intestinal immunity, to release the brakes on type 1 anti-cancer immunity, may be beneficial in certain CRC subtypes, but detrimental in others.
IL-25, also known as IL-17E, is part of the IL-17 family of cytokines. IL-25 binds a heterodimeric receptor, composed of interleukin-17 receptor A (IL-17RA) and interleukin-17 receptor B (IL-17BR) (25), which is expressed by Th2 cells, Tregs, ILC2s, NKT cells, and signals through Act1, TRAF6, NF-κB, MAPK and STAT5-mediated pathways (Figure 1) (26–29). In the intestines, IL-25 is secreted by specialised epithelial chemosensory cells named tuft cells that express the marker doublecortin-like kinase 1 (DCLK1) (30), which are estimated to constitute ~1 - 7% of the intestinal epithelial compartment in mice, with variations possibly due to the effects of the microbiota on the mucosal immune system (31). Functionally and phenotypically similar cells exist in other tissues but they have received different names, such as brush cells in the airways (32).
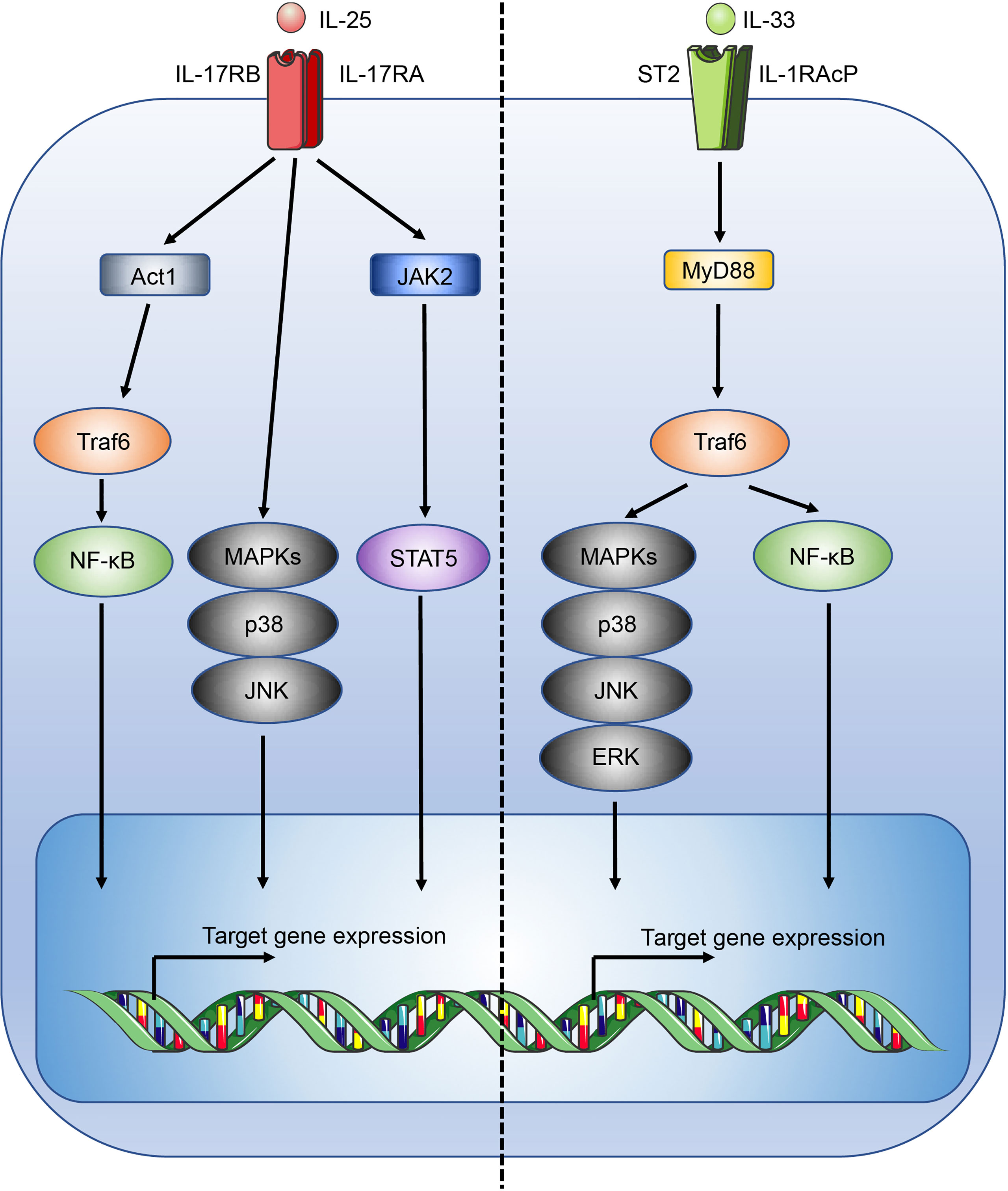
Figure 1 Overview of the IL-25 and IL-33 signalling pathways. Figure depicts the IL-25 (left) and IL-33 (right) signalling pathways. Act1, CIKS; c-Jun N-terminal kinase, JNK.
Conversely, IL-33 is constitutively expressed at high levels by a wide range of cell types including epithelial cells at mucosal barriers, endothelial cells, fibroblasts, and stromal cells (33, 34). While IL-33 expression within the intestinal epithelial compartment is widespread, it is not, unlike IL-25, produced by tuft cells (30, 35). The distinct expression pattern of IL-25 and IL-33 is reflective of the structural and functional differences of the two cytokines. IL-33 is a member of the IL-1 family of cytokines and lacks a secretory signal peptide (36) and instead contains a nuclear localisation sequence at its N-terminus, allowing storage in the nucleus where it is thought to act as a transcriptional repressor (37, 38). Without a secretory signal sequence IL-33 is not secreted like conventional cytokines such as IL-25, and is instead released upon cell damage (39), in line with its postulated role as an “alarmin”. To avoid inappropriate inflammation, apoptotic turnover of epithelial cells activates caspases that inactivate IL-33 (39). Notably, a recent report has identified a role for the pore-forming protein gasdermin C in the atypical secretion of IL-33 by intestinal epithelial cells (40). During inflammation, IL-33 expression is further upregulated, and can be processed extracellularly by mast cell chymases and tryptases into mature forms, increasing IL-33 activity by 10 - 30 fold (41). Subsequently, inactivation of IL-33 in the extracellular space occurs through oxidation (42).
IL-33 mediates its immune functions through a heterodimeric receptor complex comprising the IL-33 specific receptor ST2 (interleukin 1 receptor-like 1, Il1rl1), coupled with the IL-1 receptor accessory protein (IL-1RAcP) coreceptor (43, 44). ST2 is typically expressed by a wide range of immune cells involved with type 2 immunity, including ILC2s, mast cells, basophils, dendritic cells, eosinophils, M2-like macrophages, and Th2 cells (45). Upon IL-33 binding to ST2, signalling occurs through the recruitment of the adaptors MyD88 and TNF receptor associated factor 6 (Traf6) leading to the activation of mitogen-activated protein kinases (MAPKs) and NF-κB (Figure 1) (46). In the lungs IL-33 potently stimulates airway contraction, mucus production, and goblet cell hyperplasia through early induction of IL-13 in ILC2s, and plays important roles in allergic diseases and asthma (47, 48). While immune cells associated with type 2 immunity may co-express IL-17BR and ST2 allowing a degree of potentiation in the induction of a type 2 immune response, IL-33 and IL-25 are shown to induce distinct activation phenotypes, for example in ILC2s, indicating functional divergence (47, 49).
While best characterised for their role in type 2 immunity, IL-25 and IL-33 have additional functions in modulating other arms of immunity. IL-33 has been shown to also support Th1, Th17 and Treg responses (50–52). Activation of innate NK cells, and adaptive Th1 and CD8+ T cells by the classical type 1 immunity polarising cytokine IL-12, induces the upregulation of the IL-33 receptor ST2, and IL-33 exposure greatly increases their ability to produce IFNγ, the signature effector cytokine of classical type 1 responses. It is postulated that IL-33 supports Th17 immunity through mast cells, and activated Th17 cells have recently been shown to express ST2 in the gut (53). Conversely, IL-25 has been shown to suppress both Th1 and Th17 immunity in inflammatory conditions (54, 55). IL-33 also plays essential roles in the differentiation and maintenance of ST2+ Tregs in the intestines, supporting their function during inflammation (52). In line with the wide range of functions, both IL-25 and IL-33 have been implicated in a broad range of diseases. IL-25 signaling is proposed to be involved in infection, asthma and allergy, psoriasis, autoimmunity including inflammatory bowel disease (IBD), rheumatoid arthritis, multiple sclerosis, Sjögren’s Syndrome and cancer (56–59). Similarly, IL-33 has been implicated in the pathogenesis of chronic obstructive pulmonary disease (COPD), asthma, IBD, obesity, diabetes, cardiovascular and musculoskeletal diseases, and cancer (60, 61).
Despite recent advances, immunotherapy in CRC patients is largely disappointing (62), in particular for APC gene-mutation initiated, microsatellite-stable (MSS) CRC (63, 64) which do not respond to checkpoint inhibitors (4, 65). Tumour vaccines and adoptive T cell transfers have also shown little efficacy in these patients (17). APC inhibits the Wnt signaling pathway (Figure 2), and loss-of-function mutations in APC lead to accumulation and nuclear translocation of β-catenin, resulting in aberrant upregulation of Wnt signaling and colorectal tumour initiation (3, 63, 68, 69). Currently, immunotherapy in CRC is largely limited to MSI-high CRC where checkpoint inhibitors show efficacy (4, 70). The differential efficacy of checkpoint inhibitors against MSI-high and MSS CRC reflects the distinct immune microenvironment in different subtypes of CRC (71). MSI-high tumours have defects in the DNA mismatch-repair pathway, most commonly due to epigenetic silencing of the MutL homolog 1 (MLH1) gene or inactivation of MSH2, and consequently accrue extensive genomic mutations that lead to the expression of neoantigens by the tumours, and their immune recognition by neo-antigen specific cytotoxic T cells (72, 73). This immunogenicity underlies the striking efficacy of immunotherapeutic checkpoint inhibitors in these patients (KEYNOTE-177) (74). In addition, CAC secondary to IBD is a rarer subtype of CRC that is mutationally distinct from other human CRC subtypes, with whole-exome sequence analysis revealing only 13% incidence of APC mutation in CAC (6, 63). Importantly, immuno-profiling of CAC and sporadic CRC samples showed different immune patterns consistent with distinct disease phenotypes, with decreased immune cells that resemble CD8+ T cells and regulatory T cells (Tregs) in CAC compared to sporadic CRC (75). Studies investigating the role of IL-25 and IL-33 have found conflicting results across the various mouse models that resemble distinct human CRC subtypes, where they may either promote or inhibit tumorigenesis depending on the CRC subtype through different downstream immune mechanisms. As such, it is important to recognize that findings in a specific CRC subtype may not extend to encompass all CRC. A range of mouse models have been developed to model different subtypes of CRC, and these have been discussed in depth by recent reviews (76, 77).
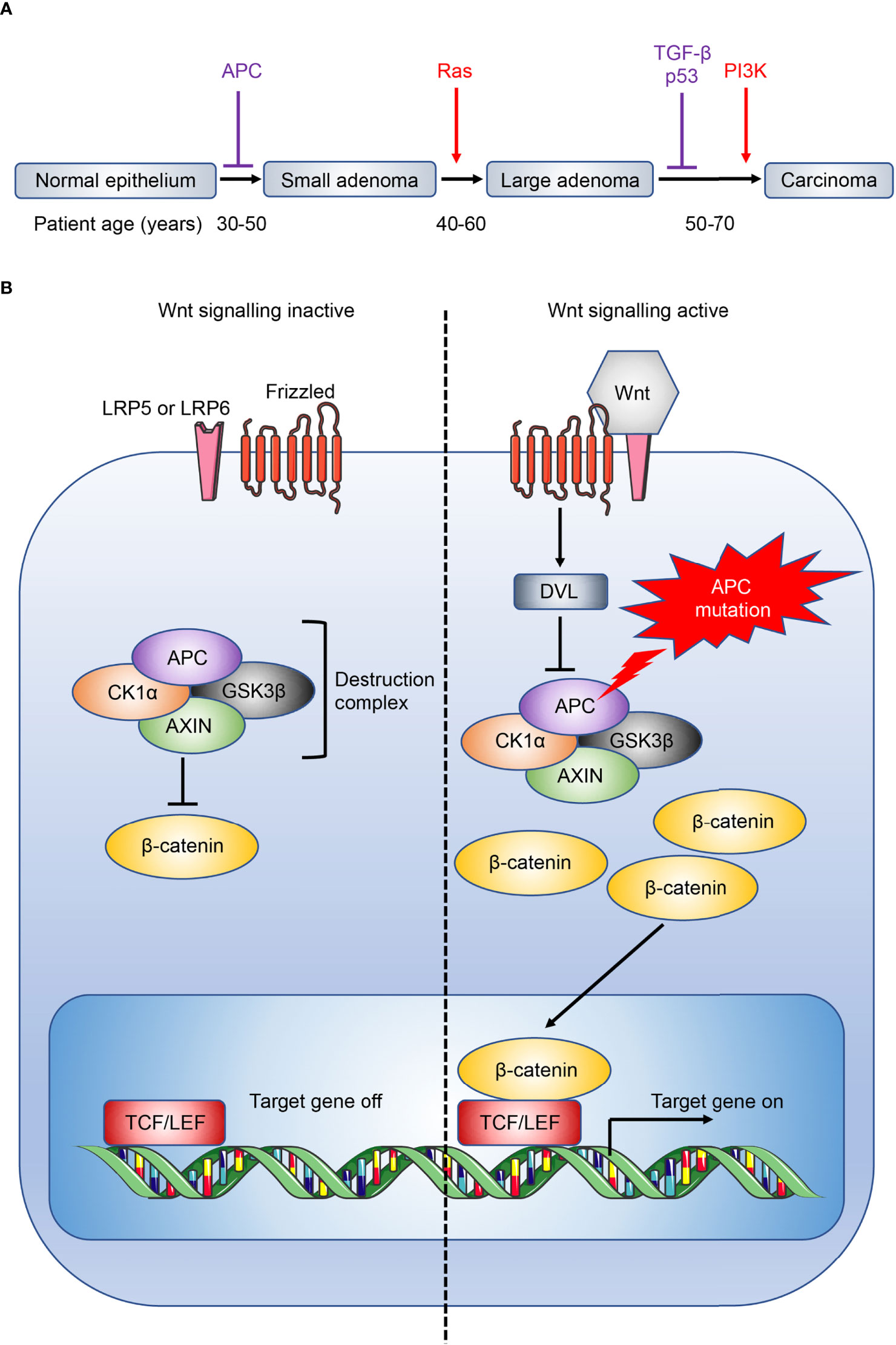
Figure 2 Overview of CRC development and Wnt signaling pathway. (A) CRC development is associated with a gradual accumulation of genetic alterations involving the loss of tumour suppressors (purple) or gain of oncogenic function (red). Loss of the tumour suppressor adenomatous polyposis coli (APC) facilitates the initial transition from normal epithelium to adenomas. Patient age when the driver genes are commonly mutated are also shown (66). Ras, rat sarcoma virus; TGF-β, transforming growth factor-β; p53, tumour protein p53; PI3K, phosphatidylinositol 3-kinase. (B) The destruction complex, consisting of APC, AXIN, casein kinase 1α (CK1α) and glycogen synthase kinase 3β (GSK3β), induces the continuous degradation of β-catenin in the absence of Wnt signaling. The destruction complex is disassembled when Wnt ligand binds to its receptor frizzled and coreceptor lipoprotein receptor-related protein 5 or 6 (LRP5 or LRP6 respectively), or when APC is mutated, leading to the accumulation and nuclear translocation of β-catenin, and downstream Wnt signaling (67). DVL, dishevelled; TCF/LEF, T cell factor/lymphoid enhancer factor.
The prominent roles of IL-33 and IL-25 in modulating the intestinal immune response made them attractive candidates for investigation in CRC and as potential targets for immunotherapy. In the past decade many studies have shown that IL-33 modulates CRC pathogenesis, while in more recent years the roles of IL-25 in CRC are increasingly being recognised. IL-25 has been found to be highly expressed in human CRC by two histopathological studies (78, 79). Analysis of IL-25 expression in healthy colons and CRC showed comparable levels in one report (78), while a more recent study found IL-25 expression to be elevated in human CRC tumours compared to the adjacent normal gut (80). Similar to the homeostatic intestine and mirroring IL-25 expression during parasitic infection (30), DCLK1+ tuft cells were found to be the main source of IL-25 in tumours in mouse models of APC-mutation-mediated CRC (Apc1322T/+) (81) and azoxymethane (AOM)/dextran sodium sulphate (DSS)-mediated model of colitis associated cancer (CAC) (80), indicating that IL-25 expression by tuft cells is conserved across different subtypes of CRC. Recently, analysis of the publicly available gene expression databases found that high IL25 expression by CRC tumours was associated with reduced CRC patient disease-free survival (81). Others have shown that human patients with high CRC IL-25 expression have worse 5-year overall survival than stage IV CRC patients with low IL-25 expression (80), indicating that IL-25 may contribute to CRC disease pathogenesis. As unselected CRC patients were assessed in these studies, the majority of patients would have sporadic APC-mutation-mediated CRC (85% of total CRC). In Apc1322T/+ mice, genetic deficiency of IL-25 reduced intestinal tumour burden and virtually doubled survival, and antibody-mediated blockade of IL-25-signalling similarly reduced CRC (81). These studies indicate a pro-tumoral role of IL-25 in autochthonous APC-mutation-mediated CRC. Conversely, IL-25 has conflicting roles in CAC. Genetic deficiency of IL-25 reduced CAC tumour burden and prolonged survival in one study (80), however others reported that antibody-mediated blockade of IL-25 instead enhanced CAC (82). As both studies employed the AOM/DSS-model of CAC, this suggests that congenital deficiency of IL-25 may have distinct effects compared to acute blockade in CAC. Finally, IL-25 injections inhibited heterotypic subcutaneous tumour growth across a range of human cancer cell lines in immunocompromised T cell-deficient nude mice, including a HT-29 CRC cell line (83). Therefore, the role of IL-25 in CRC is likely dependent on the disease stage and CRC subtype (Figure 3), and hence may be reflected differently in the mouse model investigated.
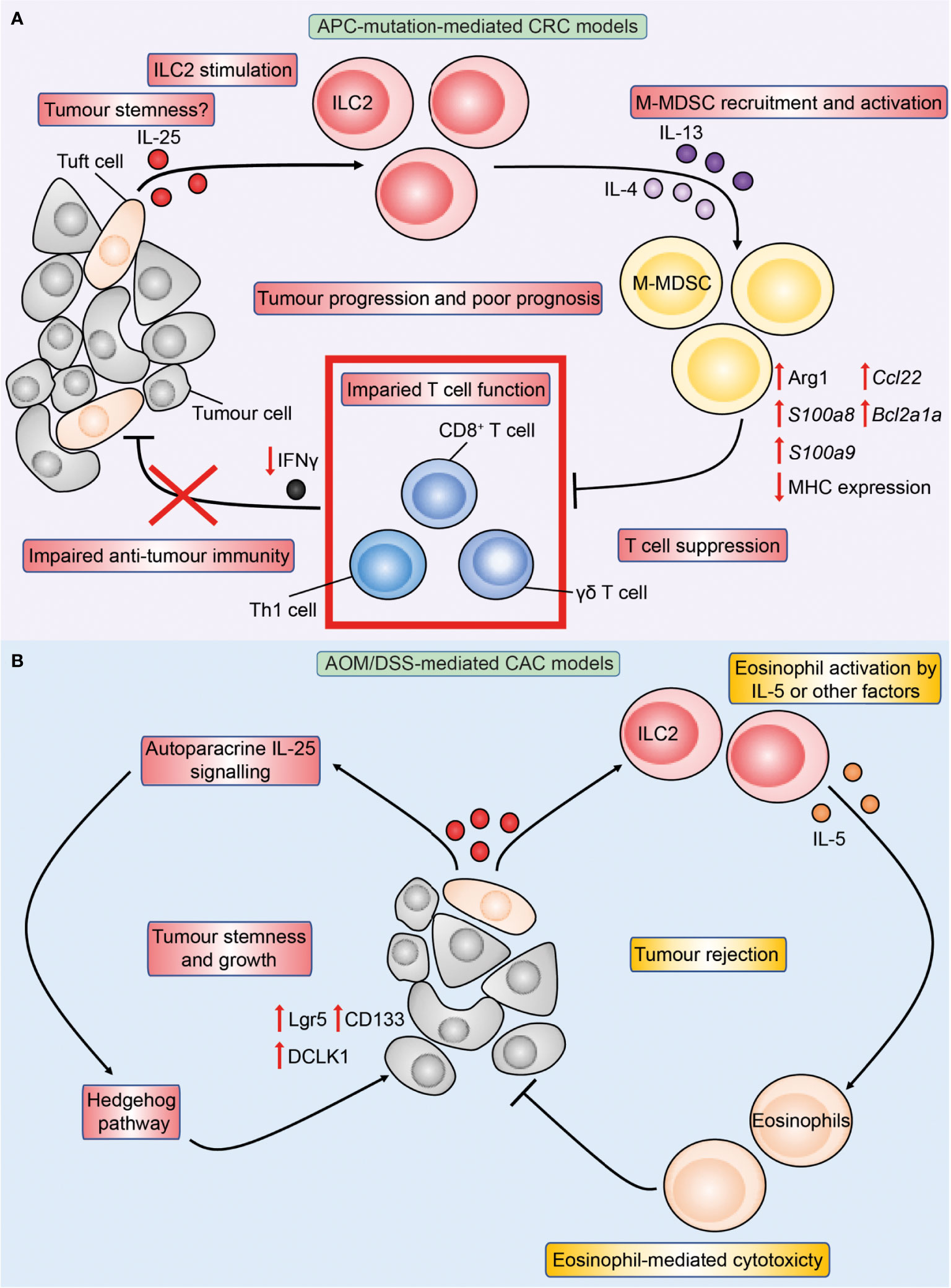
Figure 3 Overview of IL-25 in APC-mutation-mediated CRC and colitis-associated cancer (CAC) models. Figure depicts the immune-regulatory role of IL-25 characterised in preclinical models of APC-mutation-mediated CRC (A) and CAC (B). (A) In the intestinal tumour microenvironment, DCLK1+ tuft-like cells are the main source of IL-25, and the latter may in turn promote tumour stemness. IL-25 activates tumour IL-25R+ ILC2s to produce IL-4 and IL-13, which increases arginase 1 (Arg1) expression in tumour M-MDSCs leading to enhanced M-MDSC suppressive capacity. M-MDSC-mediated T cell suppression reduces T cell proliferation and IFNγ production, leading to impaired anti-tumour immunity and tumour progression. (B) In the AOM/DSS model of CAC, IL-25 reduces tumour burden through eosinophils. This may be via ILC2-derived IL-5 or other yet-to-be identified factors promoting eosinophil activation and cytotoxicity against tumours in a CD8+ T cell-independent manner. Conversely, IL-25 may act in an autoparacrine manner to promote tumour stemness characterised by upregulation of stem markers Lgr5, CD133 and DCLK1 downstream IL-25 signaling in a Hedgehog pathway-dependent manner.
In CRC, IL-33 is predominantly expressed by tumour epithelial cells and is upregulated in both human and mouse (Apcmin/+) intestinal tumours compared to the adjacent normal gut (84). IL-33 expression has similarly been detected in the AOM/DSS model of CAC (85). Unlike tumour IL25 gene expression which is associated with poor survival in CRC patients, IL33 expression was not found to correlate with differential CRC patient survival (81, 86). While an earlier study found no association of tumour ST2 expression with CRC patient survival (86), two recent studies showed that CRC patients who have increased tumour IL1RL1 expression and high densities of ST2-positive cells showed reduced overall survival (87, 88), indicating that ST2+ cells may contribute to CRC pathogenesis. Conversely, others have found that decreased IL33 and ST2 expression is associated with advanced human CRC and poor survival respectively, and ST2+ CRC showed reduced vascular invasion and lymph node metastasis (89, 90), indicating that the role of IL-33 and ST2 in CRC is likely complex and may depend on the CRC subtype and disease stage.
Studies of IL-33 in mice have similarly demonstrated conflicting results where IL-33 may either promote or inhibit CRC (Figure 4) (84, 90–92). In Apc-mutant mice, reports are consistent with IL-33 exerting a pro-tumoral role (81, 84, 91). Genetic deficiency of IL-33 or antibody-mediated blockade of ST2 led to fewer tumours (81, 84) while transgenic overexpression of IL-33 specifically in intestinal epithelial cells via the villin promoter enhanced tumorigenesis in Apcmin/+ mice (91). In a separate model, through IL-33-mediated upregulation of COX-2 in tumour cells, IL-33 transgenic mice with subcutaneous implants of MC38 tumours showed increased growth and proliferation markers compared to wild type mice, which was abrogated upon treatment with a selective COX2 inhibitor (93). Meanwhile, the role of IL-33 in CAC is more contentious. Genetic deficiency of ST2 (St2-/-) reduced tumour burden in the AOM/DSS mouse model of CAC, and was associated with improved intestinal barrier integrity reducing bacterial translocation and inflammation (90). Accordingly, inhibition of the epidermal growth factor receptor (EGFR) via gefitinib in AOM/DSS mice led to decreased tumour burden, and this correlated with a reduction in intestinal epithelial cell Il33 expression which the authors attributed to for the pro-tumoral effect of EGFR signalling (85). However, further investigation is required to determine how much of the reduction in tumour burden can be attributed to a reduction in Il33 expression, rather than to a direct effect of EGFR inhibition. Conversely, in another study genetic deficiency of IL-33 significantly enhanced tumour burden in AOM/DSS-treated mice (92). Therefore regulation of IL-33 in vivo is multi-layered and highly complex, and its impact on CRC tumorigenesis may be affected by the overall tumour microenvironment and the presence of different immune cell populations, which may underlie the conflicting results.
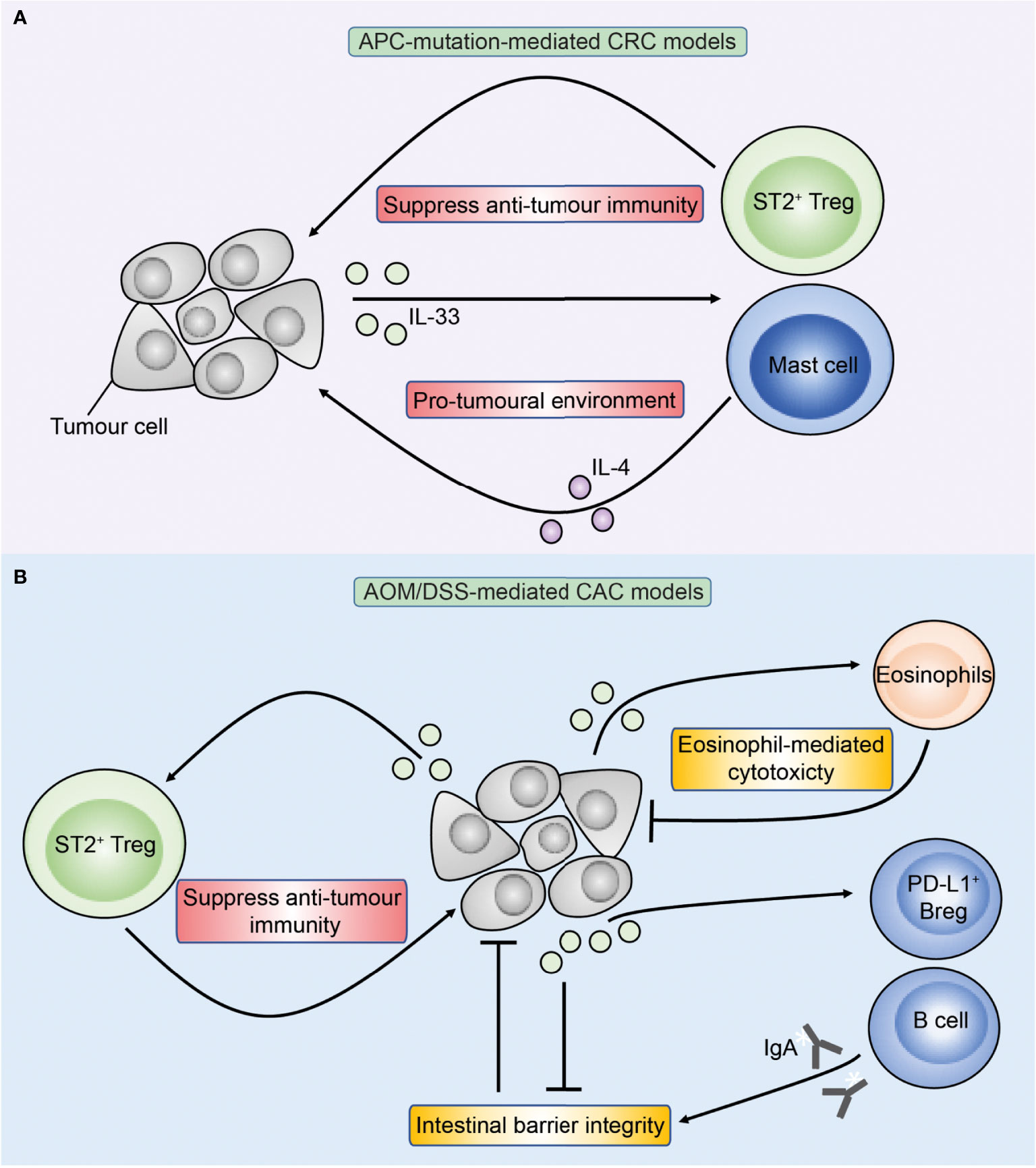
Figure 4 Overview of IL-33 in APC-mutation-mediated CRC and colitis-associated cancer (CAC) models. Figure depicts the role of IL-33 in preclinical models of APC-mutation-mediated CRC (A) and CAC (B). (A) In APC-mutation-mediated CRC models, IL-33 is proposed to promote tumorigenesis through activating ST2+ Tregs and mast cells which may have pro-tumoral properties through suppressing anti-tumour T cells and production of IL-4 respectively. (B) In AOM/DSS-mediated models of CAC, IL-33 can similarly promote tumorigenesis through ST2+ Tregs, but also via direct action on ST2+ epithelial tumour cells compromising the intestinal barrier resulting in colitis and CAC. Conversely, IL-33 may inhibit CAC through sustaining intestinal barrier integrity via stimulating B cells to produce IgA, and also via direct activation of anti-tumoral eosinophils.
Intriguingly, while both IL-25 and IL-33 expression is elevated in human CRC compared to the adjacent normal gut, in some studies expression of both cytokines was found to be highest in early colorectal adenomas compared to adenocarcinoma (79, 86). Therefore, both IL-25 and IL-33 and downstream effector functions may be affected by temporal factors during CRC disease progression providing additional complexity. Various mechanisms have been proposed on how IL-25 and IL-33 may either promote and inhibit CRC pathogenesis, involving many different pathways associated with CRC development. These will be discussed in turn below.
As potent orchestrators of intestinal immunity, IL-25 and IL-33 can modulate CRC pathogenesis through various downstream immune cells. Despite their overlapping function in asthma and inflammatory diseases associated with type 2 immunity, existing studies suggest that IL-25 and IL-33 elicit largely distinct immune pathways in CRC. Specifically, IL-25 has been shown to preferentially affect ILC2s and myeloid-derived suppressor cells (MDSCs) in CRC (81), while IL-33 exerts a more dominant effect on Th2 cells, Tregs, and mast cells (84, 87, 88, 91, 92, 94–96), although both can modulate eosinophils (82, 97). Adding to the complexity, the downstream immune response elicited by IL-25 and IL-33 is also dependent on the CRC subtypes, in line with the distinct immune profiles in the latter.
Until recently, the role of IL-25 in cancer has been largely speculative and few studies to date investigated the downstream immune mechanisms of IL-25 in CRC. Our group recently reported that IL-25 can promote CRC through ILC2s. ILC2s are innate immune cells that orchestrate type-2 immunity by providing an early innate source of the type-2 cytokines IL-4, IL-5, IL-9, and IL-13 (16, 21, 98, 99) to induce intestinal parasitic helminth worm expulsion (21, 99) and sustain tissue repair and regeneration (20–22). ILC2s can express the receptors for IL-25 and IL-33, but expression may vary dependent on the tissue immune microenvironment and inflammatory mediators. Intestinal ILC2s express higher levels of IL-17BR compared to those found in other tissues such as the bone marrow, skin, fat and lung, while expression of the IL-33 receptor ST2 is minimal (100). This suggests that IL-25 may play a more dominant role in stimulating ILC2s in the intestinal environment.
Importantly, recent studies have reported an abundance of ILC2s in human CRC (101–103), and that tumour-infiltrating ILC2s in human CRC express IL-17BR, which is mirrored in APC-mutant mice (81). Ablation of IL-25 signaling reduced tumour load, ILC2s and extended survival of IL-25-deficient Apc1322T/+ mice. ILC2-deficient mice (Roraf/fIl7rCre/+Apc1322T/+ mice) also developed fewer tumours, which correlated with increased frequencies of Th1 cells and CD8+ T cells, and a reduction in MDSCs. Th1 and CD8+ T cells are potent producers of IFNγ, which have been shown to be anti-tumoral by several groups in APC-mutant mouse models (104, 105). Critically, rIL-25 treatment increased tumour burden in ILC2-replete but not ILC2-deplete Apc1322T/+ mice, indicating an essential role for ILC2 in driving IL-25-mediated CRC tumorigenesis (81). Importantly, rIL-33 treatment was able to increase tumour burden independently of ILC2s in the same model. Furthermore, IL-25-signalling blockade via anti-IL17BR antibody administration in Apc1322T/+ mice reduced ILC2s and pro-tumoral MDSCs while increasing IFNγ+ CD4+ and CD8+ T cells in colonic adenocarcinomas, indicating a potential therapeutic intervention to block the pro-tumoral role of the IL-25-ILC2-monocytic MDSC (M-MDSC) axis in CRC. MDSCs mediate suppression of anti-tumour adaptive T cells through a plethora of mechanisms including expression of checkpoint ligands such as programmed death-ligand 1 (PD-L1) (Figure 5) (106–114). Mechanistically, ILC2-derived IL-4 and IL-13 in response to IL-25 signaling activated tumour M-MDSCs derived from Apc1322T/+ mice to express Arginase 1 (Arg1) and suppress CD8+ T cells (81) . In human CRC samples, ILC2s positively correlated with M-MDSCs, while negatively correlating with Th1 cells and CD8+ T cells. Many independent studies have confirmed Th1 and CD8+ T cell infiltration to be amongst the strongest positive prognostic factors for improved CRC patient survival across all stages of disease (115–121). These results indicate that an IL-25-ILC2-M-MDSC pathway acts independent of IL-33-mediated pathways to produce a cancer-permissive immune niche in APC-mutation-mediated CRC (Figure 3), and may potentially be targeted in patients. By contrast, ILC2-deficiency increased tumour burden in an AOM/DSS model of CAC indicating an anti-tumoral role of ILC2s. Thus, differential mechanisms or interactions between the IL-25-ILC2 axis and pro-tumoral MDSCs may exist in CAC (122).
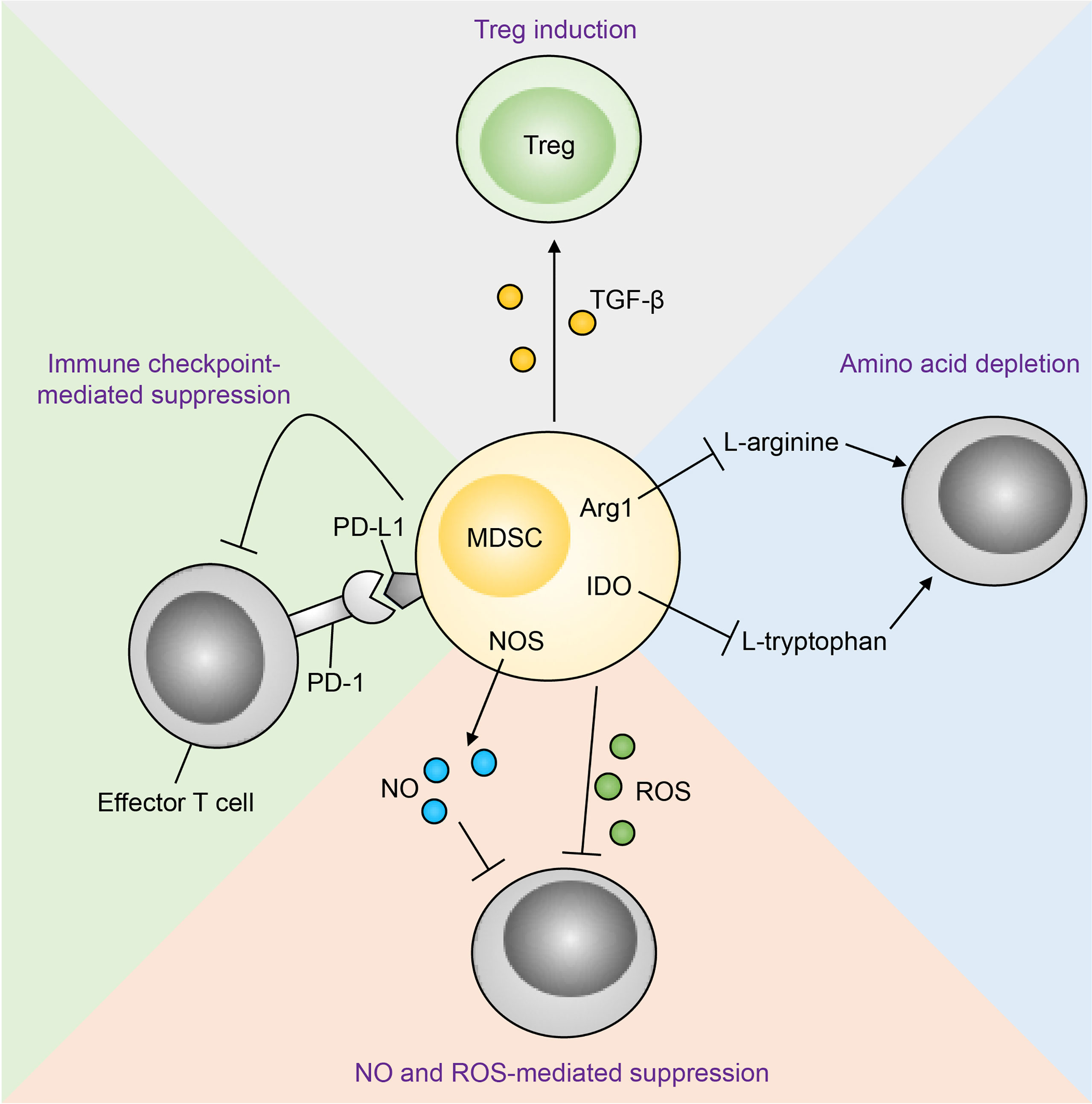
Figure 5 Mechanisms of MDSC-mediated T cell suppression. Immunosuppression of effector T cells by MDSCs occurs through multiple mechanisms. MDSC-derived nitric oxide (NO) and reactive oxygen species (ROS) inhibit T cell effector function and promote apoptosis. MDSCs can also suppress effector T cells through amino acid depletion MDSCs express arginase 1 (Arg1) and indoleamine-pyrrole 2,3-dioxygenase (IDO), which deplete the amino acids L-arginine and L-tryptophan respectively in the tumour microenvironment. Metabolic suppression of T cells ensues as these amino acids are essential for T cell effector function. In addition, MDSCs express programmed death-ligand 1 (PD-L1), which interacts with its receptor programmed cell death protein 1 (PD-1) on T cells, resulting in T cell functional exhaustion. Finally, MDSC-derived transforming growth factor-β (TGF-β) converts T cells to a regulatory phenotype (Tregs). NOS, nitric oxide synthase.
IL-33 has been shown to sustain MDSCs in human and mouse breast cancer (123), and there is some evidence that IL-33 may also affect MDSC recruitment in CRC. In an orthotopic caecal implant model of CRC using MC38 cells, overexpression of IL-33 in tumour cells led to increased liver metastasis that was associated with an increased recruitment of CD11b+Gr1+ MDSCs and tumour angiogenesis (124). However, a more recent study found no effect of IL-33 treatment on tumour MDSC frequency in a subcutaneous CT26 implant model of CRC (97). These discrepancies may be due to the different site of implant or CRC cell line used. MC38 cells have a higher accumulation of mutations than CT26 cells (125) and mutational load is known to affect tumour immunogenicity and downstream immune mechanisms (126). Equally the differing immune response tendencies between the two mouse strains BALB/c (CT26) and C57BL/6 (MC38) may also contribute to the differences observed. Critically, cell line implant models do not retain the original tumour architecture and cellular complexity, and CT26 and MC38 models do not harbor mutations in Apc (125, 127). Whether IL-33 modulates MDSCs in autochthonous APC-mutation-mediated CRC or CAC, allowing potential functional interaction with the IL-25-ILC2 axis in CRC through MDSCs, remains to be established.
Studies in APC-mutant mice found that decreased tumour burden upon IL-33-deficiency (but not IL-25-deficiency) is associated with reduced tumour mast cells (81, 84), suggesting a potential pro-tumoral role downstream IL-33 signalling. Indeed, mast cells were found to be increased in intestinal tumours from ApcΔ468 mice, and depletion of mast cells through the generation of chimeric mice reduced tumour development (128). However, another study found that genetic deficiency of the atypical chemokine receptor 2 (Ackr2-/-) in Apcmin/+ mice led to reduced tumour burden, which the authors attributed to an increase in mast cells that can recruit CD8+ T cells through leukotriene B4 (LTB4) (129). Mast cells may exert anti-tumoral properties in CAC. In an AOM/DSS model of CAC, deficiency in hematopoietic prostaglandin D synthase (H-PGDS) exacerbated tumorigenesis, and adoptive transfer of mast cells deficient in H-PGDS, but not wild type mast cells, into mast cell-deficient (Kitw-sh/w-sh) mice exacerbated tumorigenesis (130). It is well-established that IL-33 promotes mast cell activation (131), and as discussed above both IL-33 and mast cells can either promote and inhibit CRC. In human intestinal tumours, mast cells are increased in adenomas and CRC compared to the adjacent normal gut (128, 132, 133), and high tumour mast cell infiltration has been associated with reduced (133) or improved (134) survival in CRC patients. It is recognised that mast cell function in cancer can be tumour stage dependent, for example in promoting early but not late stage prostate cancer growth through MMP9 production (135). Similarly, human CRC IL33 and ST2 expression varies according to tumour stage (89, 90). Whether differential IL-33 expression during the stages of tumour progression affect downstream mast cell function, and whether mast cells directly contribute to the tumour-modulatory effects of IL-33 in CRC remains to be established. Mechanistic studies that directly assess the role of mast cells downstream of IL-33 in the context of CRC are required.
Genetic overexpression of IL-33 led to increased tumour burden and expression of Il4, Il5 and Il13 in Th2 cells, and this correlated with increased tumorigenesis in the Apcmin/+ mice (91), while genetic deficiency of IL-33 reduced overall tumour Il4 and Il13 expression (84). On the other hand, IL-25-deficiency had no effect on Th2 cell frequency in Apc1322T/+ mice, and instead acts via modulating ILC2 numbers and their production of IL-4 and IL-13 (81). Collectively, these studies suggest that IL-25-mediated activation of ILC2s, and IL-33-mediated stimulation of Th2 cells, may act as independent parallel pathways to induce the production of IL-4 and IL-13 leading to the activation of MDSCs to suppress anti-tumour T cells and promote CRC.
Studies suggest that IL-33 may modulate CRC tumorigenesis through Tregs. Like MDSCs, Tregs are immunosuppressive cells that can counteract an anti-tumoral immune response, and hence are traditionally associated with pro-tumoral functions (136). In a CT26 subcutaneous model of CRC, exogenous IL-33 increased tumour size and correlated with ST2+ Treg tumour infiltration (95). Overexpression of IL-33 in Apcmin/+ mice increased tumour burden and correlated with an increase in colonic ST2+ Tregs (91), while our group showed that IL-33-deficiency reduced tumour Tregs in Apc1322T/+ mice (81), although these studies did not directly assess the functional importance of Tregs. On the other hand, genetic deficiency of IL-25 or rIL-25 treatment did not alter tumour Treg frequency in Apc1322T/+ mice (81), suggesting that tumour Tregs preferentially respond to IL-33 and not IL-25 in CRC.
However, whilst Tregs have been implicated in IL-33-mediated CRC tumorigenesis, studies are largely associative and functional depletion studies in the setting of IL-33 perturbation are required. Furthermore, whilst Tregs are often assumed to be pro-tumoral due to their immunosuppressive nature across many cancers (137), the role of Tregs in CRC is more opaque. Certain microbiota are associated with promoting CRC (138, 139), and Tregs may prevent CRC through repressing microbe-induced inflammation (140). In human prognostic studies, a recent systematic review and meta-analysis compiling eight studies found Tregs to be associated with improved survival in human CRC (Odds ratio 0.71, 95% CI 0.62 to 0.82) while correlating with poor survival in most other cancer type assessed (137). In animal models, whilst Treg ablation (DEREG mice) in the AOM/DSS model of CAC reduced tumorigenesis (141), Treg depletion in Apcmin/+DEREG mice instead increased tumorigenesis (142), supporting the proposal that Tregs may have a protective role in sporadic APC-mutation-mediated CRC. In CAC, the pro-tumoral role of Tregs has been attributed to thymic stromal lymphopoietin (TSLP) rather than IL-33 in one study, as Treg-specific deletion of the TSLP receptor (TSLPR) (Foxp3IRES-creTslprf/f) but not ST2 (Foxp3IRES-creIl1rl1f/f) reduced AOM/DSS-mediated CAC (143). While others have found through bone marrow chimera experiments that Treg-specific deletion of ST2 delayed AOM/DSS-mediated CAC disease progression suggesting that Tregs contribute to the pro-tumoral role of IL-33 in CAC (96). A recent immunoprofiling study in human CRC found relative enrichment of Tregs in sporadic CRC compared to CAC (75), consistent with the distinct disease phenotype and immune etiology which may explain the opposite function of Tregs observed. However, as Tregs are heterogeneous with many functionally distinct subsets characterized (144), IL-33-responsive ST2+ Treg subsets may potentially promote APC-mutation-mediated CRC and these should be specifically assessed in future studies (87). For now, care must be taken before designating Tregs to be responsible for IL-33-mediated CRC (in particular APC-mutation-mediated CRC) in settings where there is a concomitant rise in tumour Treg infiltration and increased cancer burden, in the absence of functional assessments.
Studies have found B cells to inhibit intestinal tumorigenesis in CAC downstream of IL-33. Recently, mice with defects in circadian rhythm (Bmal1-/-) were reported to show increased susceptibility to DSS-induced colitis and heightened tumour burden in AOM/DSS-mediated CAC (145). Mechanistically, Bmal1 induces IL-33 expression and IL-33 was found to be necessary to sustain protective intraepithelial PD-L1+ regulatory B cells (Bregs). Bregs inhibited pathogenic Th17 cells in an IL-33 and PD-L1-dependent manner thereby suppressing colitis and subsequent CAC. Others have similarly found that genetic IL-33-deficiency results in increased susceptibility to AOM/DSS-mediated CAC (92). In that study, IL-33 promoted B cell production of IgA, which was necessary for microbial homeostasis in the intestines, and defects in the IL-33-B cell-IgA pathway led to dysbiosis characterised by increased mucolytic and colitogenic bacteria, increased IL-1α response, and increased CAC. Cohousing IL-33-deficient and IL-33-replete mice before DSS treatment allowed equilibration of the gut microbiota and abrogated the increased colitis in IL-33-deficient mice seen upon DSS treatment. B cells have also been shown to suppress tumorigenesis in Apcmin/+ mice similar to in CAC (146). However, currently there are no studies that assessed the role of B cells downstream IL-25 or IL-33 outside CAC.
In the lungs, both IL-25 and IL-33 have been shown to contribute to eosinophil-mediated airway inflammation (147). Currently, studies on eosinophils downstream of IL-25 or IL-33 in CRC mainly focussed on animal models of CAC. Exogenous IL-33 treatment reduced tumour burden in the AOM/DSS model of CAC and correlated with increased eosinophil infiltration (97). Mechanistically, IL-33 treatment enhanced IL-5 and eotaxin-2 expression contributing to eosinophil recruitment. The same study also assessed the role of IL-33 and eosinophils in a heterotypic subcutaneous model of CRC, and found that the anti-tumoral effect of IL-33 was abrogated in mice genetically deficient in eosinophils (ΔdblGATA-1), and adoptive transfer of eosinophils restored the anti-tumour efficacy of IL-33 treatment. Others have found IL-25 to similarly suppress CAC through eosinophils. Antibody-mediated neutralisation of IL-25 increased tumour burden in the AOM/DSS model of CAC and correlated with reduced eosinophils (82), although functional studies were not performed. Therefore, eosinophils may act as a converging node that integrate signals from both IL-25 and IL-33 during intestinal tumorigenesis, particularly CAC. Whether IL-25 and IL-33-mediated eosinophilia occurs in other subtypes of CRC inhibiting tumorigenesis remains to be established, however unlike CAC, most studies are concordant with IL-25 and IL-33 promoting tumorigenesis in APC-mutation-mediated CRC (81, 84, 91). Given that eosinophils are predominantly anti-tumoral in CRC including in Apcmin/+ mice (148), this could suggest that the IL-25 and IL-33-eosinophil axis may be less prominent in APC-mutation-mediated CRC relative to other pro-tumorigenic pathways compared to in CAC.
Due to their broad range of functions IL-25 and IL-33 and can also modulate CRC through additional mechanisms.
CSCs play important roles in conferring cancer resistance against treatment due to their quiescent nature, and yet are able to regenerate tumours upon insult, and have therefore become attractive therapeutic targets in recent years (149). Both IL-25 and IL-33 have been shown to promote CRC tumour cell stemness. In vitro treatment with IL-33 enhanced sphere formation by primary human CRC cells and HT29 cell line, with a concomitant increase in expression of stem cell markers NANOG, NOTCH3, OCT3, OCT4 and LGR5 resulting in treatment resistance against 5-fluorouracil in a ST2-dependent manner (94). Others have identified that a small proportion of DCLK1+ tuft cells are Lgr5+ (stem cell marker) and may act as tumour stem cells during intestinal tumorigenesis, and that DCLK1 expression readily differentiates intestinal tumour stem cells from normal stem cells (80, 150). DCLK1+Lgr5+ cells can be identified in intestinal tumours that arose following activation of Wnt signaling in Lgr5+ stem cells (150), and a recent study confirmed DCLK1 as a downstream target of Wnt signaling (151). DCLK1+Lgr5+ tumour stem cells resemble tuft cells in their gene expression profile and upregulate other tuft cell markers such as COX-1 (150). IL-25 expression in CRC was reported to be predominantly by DCLK1+ tuft-like cells in Apc1322T/+ mice (81). Whether it is the acquisition of DCLK1+ expression by tumour stem cells due to aberrant Wnt signaling (for example due to loss of APC) which leads to their ability to produce IL-25 remains to be explored. Furthermore, CRC CSCs also express the IL-17BR suggesting potential autocrine signaling (152). Indeed IL-25 may reciprocally promote tumour stemness, as in vitro treatment of HT-29 and SW620 human CRC cell lines with IL-25 induced expression of stem markers CD133, Lgr5 and OCT4 through hedgehog signaling, and similarly in the AOM/DSS model of CAC genetic IL-25-deficiency reduced tumour burden and expression of Lgr5, CD133 and DCLK1 (80). Importantly, depletion of DCLK1+ tuft cells reduced tumour burden in models of CRC both in vitro and in vivo (152, 153), indicating that these IL-25-expressing tuft cells can support intestinal tumorigenesis. IL-25 and IL-33 may synergistically promote CRC stemness, indicating that simultaneous blockade of both molecules may be required for maximal therapeutic benefit in attempts to target CSCs.
Both IL-25 and IL-33 have been implicated in promoting angiogenesis, and angiogenesis mediated by the VEGF family of proteins plays an important and non-redundant role in CRC development and progression, in both patients and mouse models of CRC (154–157). Currently, several VEGF or VEGFR inhibitors (e.g. bevacizumab) are approved by the FDA for treatment of CRC patients (158). In a mouse CRC cell line implant model, transgenic overexpression of IL-33 in MC38 cells (MC38-IL-33) resulted in 3-fold higher microvessel density and increased cancer metastasis to the liver compared to vector control (124). Interestingly, IL-33-mediated angiogenesis in this setting was thought to be VEGF independent, as tumour VEGF expression was lower in MC38-IL-33 tumours compared to control. This suggests that combined blockade of IL-33 and VEGF may potentially further reduce angiogenesis in CRC. Evidence suggests that IL-25 may also promote angiogenesis in CRC. Colonic Vegfa expression was found to be reduced upon neutralisation of IL-25, or genetic IL-25-deficiency, in AOM/DSS-treated mice with CAC (82), suggesting that IL-25 may promote tumour VEGF-A expression. Indeed, IL-25 has been reported to induce VEGF expression and promote angiogenesis by human vascular endothelial cells in vitro (159), and in vivo in a mouse model of asthma where intranasal administration of IL-25 increased VEGF expression and airway vascularity (160). Angiogenesis blockers have proved beneficial clinically against CRC, and combined treatment through the inhibition of IL-25 or IL-33 may provide additional benefits to further reduce tumour angiogenesis.
IL-25 and IL-33 have been shown to modulate CRC invasion. MMPs are zinc-dependent proteolytic metalloenzymes with roles in degrading extracellular matrix (ECM) proteins. MMP9 is part of the gelatinase family that breakdowns the ECM proteins gelatin and type IV collagen, thereby promoting tumour invasion and metastasis (161, 162). In CRC, increased tumour MMP-9 expression is associated with advanced cancer stages, lymph node metastasis and reduced patient survival (163, 164). However, a study found that genetic deficiency of MMP9 enhanced CAC development and reduced survival in AOM/DSS-treated mice (165) suggesting an anti-tumoral role in CAC in contrast to APC-mutation-driven CRC, where genetic deficiency of MMP9 decreased tumour number by 40% in Apcmin/+ mice (166). Genetic IL-25 deficiency led to a reduction in tumour Mmp9 expression in AOM/DSS mice (82), indicating that IL-25 may induce MMP9 expression in CRC. The differential effect of MMP9 in different CRC subtypes may contribute to the opposite effect of IL-25 seen in APC-mutant mice (Apc1322T/+) where it promotes tumorigenesis (81), while inhibiting CAC in some studies (82). Interestingly, genetic IL-25-deficiency also led to a reduction in tumour Mmp2 in AOM/DSS-mediated CAC (82). Similar to MMP9, MMP2 is part of the gelatinase family and is associated with poor overall survival in CRC patients (167). Mechanistically, how IL-25 modulates MMP expression in CRC remains to be established by future studies, but their co-expression may provide additional correlative markers for targeting treatments to specific CRC subtypes.
IL-33 may promote CRC tumour invasion and metastasis through desmoplastic reactions. Desmoplasia during CRC progression is largely mediated by cancer-associated fibroblasts (CAFs) (168), and CRC patients with abundant desmoplasia are more likely to have lymphatic metastases (169). CAFs in human CRC express IL-33, and IL-33 expression levels in CAFs correlated with the degree of human CRC cell line migration when treated with CAF conditioned media (169). In the metastatic lymph node from human CRC patients, IL-33 expression is detected in CAF-like cells at areas of high desmoplasia at the tumour invasive front. In addition, CRC epithelial IL-33 expression was also found to correlate with increased desmoplasia, indicating that both epithelial and CAF-derived IL-33 can promote CRC desmoplasia, invasion and metastasis. Mechanistically, IL-33 promotes epithelial mesenchymal transition (EMT) characterised by decreased E-cadherin and increased vimentin and N-cadherin expression in HT29 human CRC cells, and induced migration in a wound healing assay in vitro. In another study, IL-33 stimulation of primary human CRC cells increased expression of MMP2 and MMP9, and shRNA-mediated knockdown of ST2 or inhibition of MMP2 and MMP9 reduced invasion in vitro (170). Therefore, both IL-25 and IL-33 may promote CRC invasion through induction of MMPs. Conversely, others have found that IL-33 may instead inhibit metastasis through inducing CD40L expression on tumour infiltrating lymphocytes (171). Here, rIL-33 treatment reduced pulmonary and liver metastases by CT26 cells when injected via the tail vein and into the splenic capsule respectively, and concomitant anti-CD40L treatment significantly promoted tumour burden and reduced IFNγ expression in CD4+ T cells, NK cells and CD8+ T cells in IL-33 treated mice. Altogether, these studies suggest functional divergence of IL-33 when acting on tumour cells and the immune infiltrate, where direct action of IL-33 on tumour cells induces desmoplasia and tumour invasion, while IL-33-mediated stimulation of tumour infiltrating lymphocytes may conversely limit CRC metastasis. Therefore, the overall effect of IL-33 on CRC metastasis may possibly depend on the relative immunogenicity of the different CRC subtypes.
In the past decade checkpoint inhibitors have shown unprecedented efficacy against a broad range of cancer types (172). In the tumour microenvironment where antigen stimulation persists, effector T cells proceed to functional exhaustion (173). These exhausted T cells have significantly impaired effector functions, and are characterised by increased and sustained expression of an array of inhibitory checkpoint receptors, the best studied being cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), programmed cell death protein 1 (PD-1) and lymphocyte-activation gene 3 (LAG-3) (174, 175). Ligation with their cognate ligands, expressed on tumour cells, effectively blunts T cell effector function and promotes tumour survival (174). This can be reversed clinically through the use of checkpoint inhibitors, e.g. monoclonal antibodies that block the inhibitory receptor-ligand interaction between T cells and tumours, thereby revitalizing exhausted T cells to effectively mediate anti-tumour functions (176, 177). However despite their great success, checkpoint inhibitors are currently largely ineffective in human MSS CRC, which show a less robust T cell response compared to MSI-high CRC (4, 63, 64, 178). Importantly, MDSCs have been shown to play critical roles in treatment-resistance across several modalities of cancer treatment including chemotherapy, radiotherapy and immunotherapy (checkpoint inhibitors) in a broad range of cancers including CRC (4, 179–182). Mechanistically, MDSCs suppress anti-tumoral T cells through multiple pathways (Figure 5) thereby limiting the efficacy of checkpoint inhibitors. Antibody-mediated blockade of IL-25 signaling effectively reduced immunosuppressive tumour M-MDSCs and enhanced IFNγ expression in tumour infiltrating CD4+ and CD8+ T cells resulting in reduced tumour burden in Apc1322T/+ mice (81). This raises the possibility that concomitant IL-25-signalling blockade may potentiate checkpoint inhibitor treatment through suppressing MDSCs thereby enhancing immunotherapeutic efficacy against CRC. This could be coupled with using IL-25 as a biomarker, in particular in CRC patients with high tumour IL-25 expression, as elevated tumour IL-25 is associated with poor survival in CRC patients (80, 81) and these patients may be more responsive to IL-25 signaling blockade. Similarly, IL-33 may promote intestinal tumorigenesis in the Apcmin/+ model potentially through immunosuppressive ST2+ Tregs and mast cells (84, 91) and IL-33 blockade may similarly liberate anti-tumour immunity and improve checkpoint inhibitor efficacy. In particular, blocking IL-33 may allow preferential depletion of the likely pro-tumoral ST2+ Treg subset, which may prove to be more beneficial than other methods that target Tregs nonspecifically, especially as unlike in most cancers Tregs are associated with good prognosis in human CRC patients (137).
Conversely, conflicting reports have shown that both IL-33 and IL-25 may either promote or inhibit CAC (Table 1) (80, 82, 90, 92, 96, 97), and therefore caution should be taken when considering IL-25 or IL-33-based therapy in this cohort. Another strategy would be to target IL-25 or IL-33 during early stages of IBD, to delay or prevent progression into intestinal cancer while resolving the colitis. This would be particularly useful in the management of Crohn’s disease (CD) as unlike ulcerative colitis (UC), CD affects the whole alimentary tract and cannot be cured through pancolectomy. Nevertheless, early treatment in UC may improve patient quality of life and reduce morbidity through delaying the need for colectomy and the requirement of a stoma. A recent study found that in UC patients, there is decreased expression of 8 miRNAs of the miR-378 family which targets IL-33 mRNA, and is associated with an almost 4-fold increase in IL-33 mRNA. Of these, miR-378a-3p was further decreased in active UC patients compared those under remission and overexpression of this miRNA reduced IL-33 expression in vitro (183). Importantly, both IL-33 and IL-25 have been shown to have dual roles in IBD similar to in CAC (184, 185), and therefore miRNA-based therapy may become an attractive therapeutic option in the future to fine-tune IL-33 or IL-25 expression levels in the management of colitis and CRC in different subsets of patients.
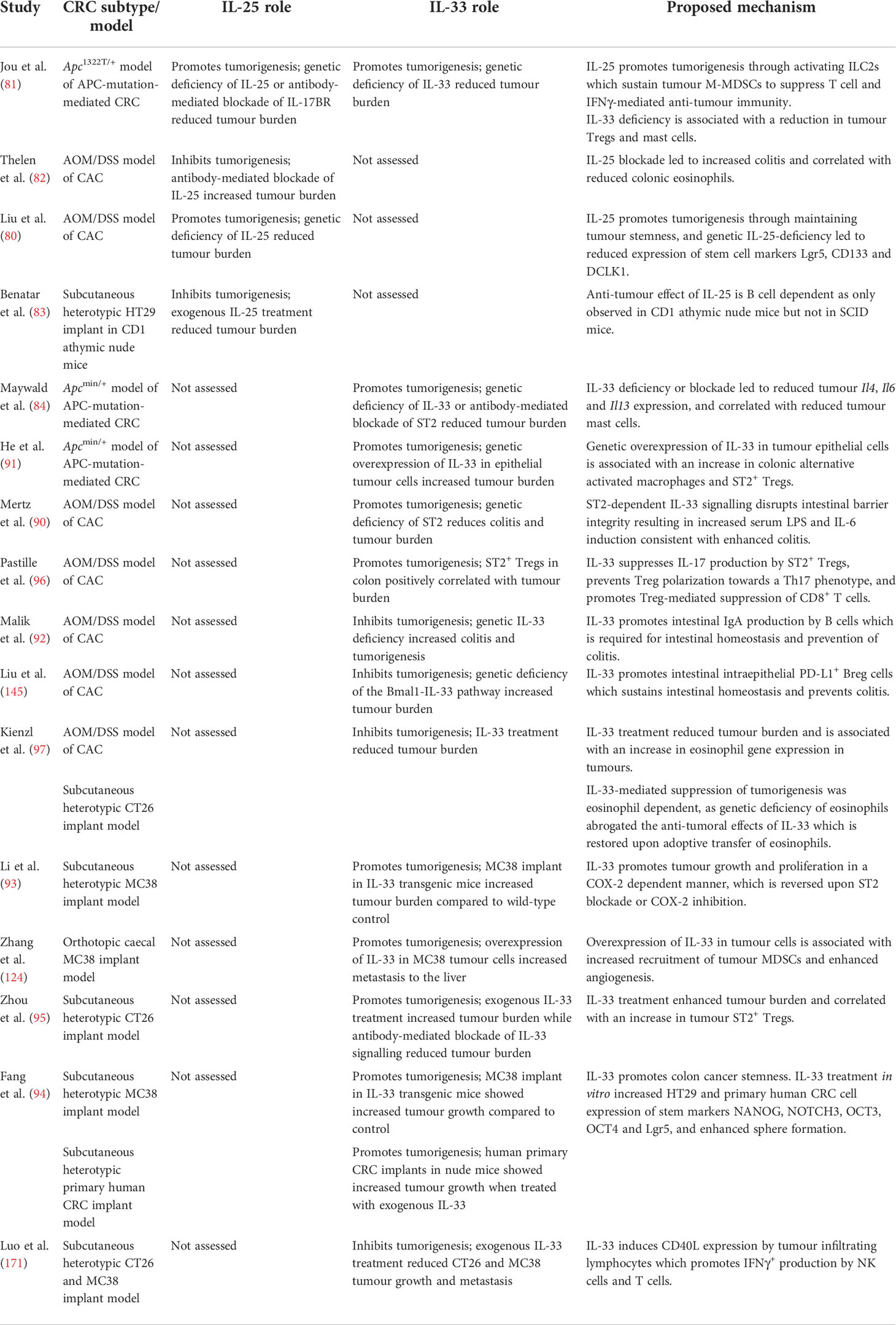
Table 1 Summary of animal studies illustrating the pro and anti-tumorigenic roles of IL-25 and IL-33 in different models of CRC.
Finally, IL-33 and IL-25 contribute to many processes involved with CRC pathogenesis, for example in shaping the intestinal immune milieu and CRC tumour microenvironment, as well as their involvement in angiogenesis, desmoplasia, tumour invasion, metastasis and in sustaining CSCs. This broad range of functions opens avenues for potential combination therapies with other existing cancer treatments, such as chemotherapy, radiotherapy or targeted therapy. For instance, chemotherapy-induced cancer cell death results in release of tumour antigens (186), and concomitant IL-25 blockade may inhibit MDSCs and liberate anti-tumour T cells to recognize and eliminate tumours. On the other hand, IL-33 promotes angiogenesis in CRC in a VEGF-independent manner (124), and therefore simultaneous blockade of IL-33 together with VEGF inhibitors (currently used clinically) may show potentiation. In the metastatic setting, targeting IL-33 and IL-25 may also prove beneficial given their role in CRC invasion and metastasis, and may also potentially be combined with existing therapies in the future for metastatic disease.
Due to their prominent role in regulating the intestinal immune response, IL-33 and IL-25 have become attractive targets in CRC. Emerging studies in the past decade have found IL-33 and IL-25 to either promote or inhibit CRC under different settings (Figures 3 and 4), and is likely dependent on the CRC subtype, tumour immunogenicity and the different immune cell populations involved (Table 1). Therefore, it is important to understand the context in which immune reactions arise in CRC and how they may differ substantially between the distinct CRC subtypes (75). As evidenced by the varying roles of IL-25 and IL-33 in different CRC subtypes and mouse models, it is critical to understand how such innate signals contribute to disease diversity. Furthermore, the additional broad range of actions of IL-33 and IL-25 affecting multiple pathways associated with CRC pathogenesis such as angiogenesis and metastasis would mean that the overall effect of these cytokines would depend on many factors including the relative contribution of each pathway in different CRC subtypes. Future advances in novel CRC models that more closely reflect the human CRC immune environment for different CRC subtypes and microbiome constitution, will further refine our understandings of IL-25 and IL-33 in CRC, and facilitate therapeutic development. Although current immunotherapeutic strategies largely rely on stimulating or engineering adaptive T cells and generally lack efficacy in CRC, it may be possible to target such upstream innate immune signals to improve therapeutic efficacy in defined subtypes. Identifying the relevant factors that govern anti-tumour immunity in specific cancer subtypes may therefore open-up novel more refined opportunities for future immunotherapy. Finally, uncovering novel biomarkers in CRC patients that predict therapeutic response to targeted therapy against IL-25 or IL-33 will be crucial in facilitating their introduction into the clinic for treating CRC in the future.
All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
This work was supported by the Medical Research Council, as part of United Kingdom Research and Innovation (also known as UK Research and Innovation) U105178805.
We thank the McKenzie lab for feedback on this review article. Parts of Figure 1, 2, 4 and 5 were created using cartoon templates by Servier Medical Art, licensed under a Creative Commons Attribution 3.0 Unported License (https://smart.servier.com/).
ANJM is on the scientific advisory board of SinoMab. ANJM developed antibodies against IL-25R which MRC has licenced to SinoMab.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer J Clin (2018) 68(6):394–424. doi: 10.3322/caac.21492
2. Arnold M, Sierra MS, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global patterns and trends in colorectal cancer incidence and mortality. Gut (2017) 66(4):683–91. doi: 10.1136/gutjnl-2015-310912
3. Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell (1990) 61(5):759–67. doi: 10.1016/0092-8674(90)90186-i
4. Overman MJ, Ernstoff MS, Morse MA. Where we stand with immunotherapy in colorectal cancer: Deficient mismatch repair, proficient mismatch repair, and toxicity management. Am Soc Clin Oncol Educ Book (2018) 38:239–47. doi: 10.1200/edbk_200821
5. Munkholm P. Review article: The incidence and prevalence of colorectal cancer in inflammatory bowel disease. Aliment Pharmacol Ther (2003) 18 Suppl 2:1–5. doi: 10.1046/j.1365-2036.18.s2.2.x
6. Robles AI, Traverso G, Zhang M, Roberts NJ, Khan MA, Joseph C, et al. Whole-exome sequencing analyses of inflammatory bowel disease-associated colorectal cancers. Gastroenterology (2016) 150(4):931–43. doi: 10.1053/j.gastro.2015.12.036
7. Vighi G, Marcucci F, Sensi L, Di Cara G, Frati F. Allergy and the gastrointestinal system. Clin Exp Immunol (2008) 153(Suppl 1):3–6. doi: 10.1111/j.1365-2249.2008.03713.x
8. Jain N, Walker WA. Diet and host-microbial crosstalk in postnatal intestinal immune homeostasis. Nat Rev Gastroenterol Hepatol (2015) 12(1):14–25. doi: 10.1038/nrgastro.2014.153
9. Zheng D, Liwinski T, Elinav E. Interaction between microbiota and immunity in health and disease. Cell Res (2020) 30(6):492–506. doi: 10.1038/s41422-020-0332-7
10. Vojdani A. A potential link between environmental triggers and autoimmunity. Autoimmune Dis (2014) 2014:437231. doi: 10.1155/2014/437231
11. Siracusa F, Schaltenberg N, Villablanca EJ, Huber S, Gagliani N. Dietary habits and intestinal immunity: From food intake to Cd4+ Th cells. Front Immunol (2019) 9:3177(3177). doi: 10.3389/fimmu.2018.03177
12. Ticinesi A, Lauretani F, Tana C, Nouvenne A, Ridolo E, Meschi T. Exercise and immune system as modulators of intestinal microbiome: Implications for the gut-muscle axis hypothesis. Exerc Immunol Rev (2019) 25:84–95.
13. Esterházy D, Canesso MCC, Mesin L, Muller PA, de Castro TBR, Lockhart A, et al. Compartmentalized gut lymph node drainage dictates adaptive immune responses. Nature (2019) 569(7754):126–30. doi: 10.1038/s41586-019-1125-3
14. Williams Drake W, Hajishengallis G, Moutsopoulos Niki M. Regional specification of oral mucosal immunity. Sci Immunol (2022) 7(72):eabp8632. doi: 10.1126/sciimmunol.abp8632
15. Peterson LW, Artis D. Intestinal epithelial cells: Regulators of barrier function and immune homeostasis. Nat Rev Immunol (2014) 14(3):141–53. doi: 10.1038/nri3608
16. Walker JA, Barlow JL, McKenzie ANJ. Innate lymphoid cells — how did we miss them? Nat Rev Immunol (2013) 13(2):75–87. doi: 10.1038/nri3349
17. Markman JL, Shiao SL. Impact of the immune system and immunotherapy in colorectal cancer. J Gastrointest Oncol (2015) 6(2):208–23. doi: 10.3978/j.issn.2078-6891.2014.077
18. Seyedian SS, Nokhostin F, Malamir MD. A review of the diagnosis, prevention, and treatment methods of inflammatory bowel disease. J Med Life (2019) 12(2):113–22. doi: 10.25122/jml-2018-0075
19. de Mattos BRR, Garcia MPG, Nogueira JB, Paiatto LN, Albuquerque CG, Souza CL, et al. Inflammatory bowel disease: An overview of immune mechanisms and biological treatments. Mediators Inflamm (2015) 2015:493012–. doi: 10.1155/2015/493012
20. Gieseck RL, Wilson MS, Wynn TA. Type 2 immunity in tissue repair and fibrosis. Nat Rev Immunol (2018) 18(1):62–76. doi: 10.1038/nri.2017.90
21. Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TKA, et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature (2010) 464(7293):1367–70. doi: 10.1038/nature08900
22. Allen JE, Sutherland TE. Host protective roles of type 2 immunity: Parasite killing and tissue repair, flip sides of the same coin. Semin Immunol (2014) 26(4):329–40. doi: 10.1016/j.smim.2014.06.003
23. Roan F, Obata-Ninomiya K, Ziegler SF. Epithelial cell–derived cytokines: More than just signaling the alarm. J Clin Invest (2019) 129(4):1441–51. doi: 10.1172/JCI124606
24. Divekar R, Kita H. Recent advances in epithelium-derived cytokines (Il-33, il-25, and thymic stromal lymphopoietin) and allergic inflammation. Curr Opin Allergy Clin Immunol (2015) 15(1):98–103. doi: 10.1097/ACI.0000000000000133
25. Rickel EA, Siegel LA, Yoon B-RP, Rottman JB, Kugler DG, Swart DA, et al. Identification of functional roles for both il-17rb and il-17ra in mediating il-25-Induced activities. J Immunol (2008) 181(6):4299–310. doi: 10.4049/jimmunol.181.6.4299
26. Maezawa Y, Nakajima H, Suzuki K, Tamachi T, Ikeda K, Inoue J, et al. Involvement of tnf receptor-associated factor 6 in il-25 receptor signaling. J Immunol (2006) 176(2):1013–8. doi: 10.4049/jimmunol.176.2.1013
27. Swaidani S, Bulek K, Kang Z, Liu C, Lu Y, Yin W, et al. The critical role of epithelial-derived Act1 in il-17- and il-25-Mediated pulmonary inflammation. J Immunol (2009) 182(3):1631–40. doi: 10.4049/jimmunol.182.3.1631
28. Wu L, Zepp JA, Qian W, Martin BN, Ouyang W, Yin W, et al. A novel il-25 signaling pathway through Stat5. J Immunol (Baltimore Md 1950) (2015) 194(9):4528–34. doi: 10.4049/jimmunol.1402760
29. Wong CK, Li PW, Lam CW. Intracellular jnk, P38 mapk and nf-kappab regulate il-25 induced release of cytokines and chemokines from costimulated T helper lymphocytes. Immunol Lett (2007) 112(2):82–91. doi: 10.1016/j.imlet.2007.07.002
30. von Moltke J, Ji M, Liang H-E, Locksley RM. Tuft-Cell-Derived il-25 regulates an intestinal Ilc2-epithelial response circuit. Nature (2016) 529(7585):221–5. doi: 10.1038/nature16161
31. Howitt MR, Lavoie S, Michaud M, Blum AM, Tran SV, Weinstock JV, et al. Tuft cells, taste-chemosensory cells, orchestrate parasite type 2 immunity in the gut. Science (2016) 351(6279):1329–33. doi: 10.1126/science.aaf1648
32. Ualiyeva S, Hallen N, Kanaoka Y, Ledderose C, Matsumoto I, Junger Wolfgang G, et al. Airway brush cells generate cysteinyl leukotrienes through the atp sensor P2y2. Sci Immunol (2020) 5(43):eaax7224. doi: 10.1126/sciimmunol.aax7224
33. Rana BMJ, Jou E, Barlow JL, Rodriguez-Rodriguez N, Walker JA, Knox C, et al. A stromal cell niche sustains Ilc2-mediated type-2 conditioning in adipose tissue. J Exp Med (2019) 216(9):1999–2009. doi: 10.1084/jem.20190689
34. Hodzic Z, Schill EM, Bolock AM, Good M. Il-33 and the intestine: The good, the bad, and the inflammatory. Cytokine (2017) 100:1–10. doi: 10.1016/j.cyto.2017.06.017
35. Moussion C, Ortega N, Girard J-P. The il-1-Like cytokine il-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: A novel 'Alarmin'? PloS One (2008) 3(10):e3331–e. doi: 10.1371/journal.pone.0003331
36. Lefrançais E, Cayrol C. Mechanisms of il-33 processing and secretion: Differences and similarities between il-1 family members. Eur Cytokine Netw (2012) 23(4):120–7. doi: 10.1684/ecn.2012.0320
37. Chan BCL, Lam CWK, Tam L-S, Wong CK. Il33: Roles in allergic inflammation and therapeutic perspectives. Front Immunol (2019) 10:364(364). doi: 10.3389/fimmu.2019.00364
38. Ali S, Mohs A, Thomas M, Klare J, Ross R, Schmitz ML, et al. The dual function cytokine il-33 interacts with the transcription factor nf-Kb to dampen nf-Kb-Stimulated gene transcription. J Immunol (2011) 187(4):1609–16. doi: 10.4049/jimmunol.1003080
39. Cayrol C, Girard J-P. The il-1-Like cytokine il-33 is inactivated after maturation by caspase-1. Proc Natl Acad Sci (2009) 106(22):9021–6. doi: 10.1073/pnas.0812690106
40. Zhao M, Ren K, Xiong X, Xin Y, Zou Y, Maynard JC, et al. Epithelial Stat6 O-glcnacylation drives a concerted anti-helminth alarmin response dependent on tuft cell hyperplasia and gasdermin c. Immunity (2022) 55(4):623–38.e5. doi: 10.1016/j.immuni.2022.03.009
41. Lefrançais E, Duval A, Mirey E, Roga S, Espinosa E, Cayrol C, et al. Central domain of il-33 is cleaved by mast cell proteases for potent activation of group-2 innate lymphoid cells. Proc Natl Acad Sci (2014) 111(43):15502–7. doi: 10.1073/pnas.1410700111
42. Cohen ES, Scott IC, Majithiya JB, Rapley L, Kemp BP, England E, et al. Oxidation of the alarmin il-33 regulates St2-dependent inflammation. Nat Commun (2015) 6(1):8327. doi: 10.1038/ncomms9327
43. Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. Il-33, an interleukin-1-Like cytokine that signals Via the il-1 receptor-related protein St2 and induces T helper type 2-associated cytokines. Immunity (2005) 23(5):479–90. doi: 10.1016/j.immuni.2005.09.015
44. Liu X, Hammel M, He Y, Tainer JA, Jeng U-S, Zhang L, et al. Structural insights into the interaction of il-33 with its receptors. Proc Natl Acad Sci (2013) 110(37):14918–23. doi: 10.1073/pnas.1308651110
45. Fournié J-J, Poupot M. The pro-tumorigenic il-33 involved in antitumor immunity: A yin and yang cytokine. Front Immunol (2018) 9:2506(2506). doi: 10.3389/fimmu.2018.02506
46. Pinto SM, Subbannayya Y, Rex DAB, Raju R, Chatterjee O, Advani J, et al. A network map of il-33 signaling pathway. J Cell Commun Signal (2018) 12(3):615–24. doi: 10.1007/s12079-018-0464-4
47. Barlow JL, Peel S, Fox J, Panova V, Hardman CS, Camelo A, et al. Il-33 is more potent than il-25 in provoking Il-13–producing nuocytes (Type 2 innate lymphoid cells) and airway contraction. J Allergy Clin Immunol (2013) 132(4):933–41. doi: 10.1016/j.jaci.2013.05.012
48. Kondo Y, Yoshimoto T, Yasuda K, Futatsugi-Yumikura S, Morimoto M, Hayashi N, et al. Administration of il-33 induces airway hyperresponsiveness and goblet cell hyperplasia in the lungs in the absence of adaptive immune system. Int Immunol (2008) 20(6):791–800. doi: 10.1093/intimm/dxn037
49. Camelo A, Rosignoli G, Ohne Y, Stewart RA, Overed-Sayer C, Sleeman MA, et al. Il-33, il-25, and tslp induce a distinct phenotypic and activation profile in human type 2 innate lymphoid cells. Blood Adv (2017) 1(10):577–89. doi: 10.1182/bloodadvances.2016002352
50. Smithgall MD, Comeau MR, Yoon BR, Kaufman D, Armitage R, Smith DE. Il-33 amplifies both Th1- and Th2-type responses through its activity on human basophils, allergen-reactive Th2 cells, inkt and nk cells. Int Immunol (2008) 20(8):1019–30. doi: 10.1093/intimm/dxn060
51. Cho KA, Suh JW, Sohn JH, Park JW, Lee H, Kang JL, et al. Il-33 induces Th17-mediated airway inflammation via mast cells in ovalbumin-challenged mice. Am J Physiol Lung Cell Mol Physiol (2012) 302(4):L429–40. doi: 10.1152/ajplung.00252.2011
52. Schiering C, Krausgruber T, Chomka A, Fröhlich A, Adelmann K, Wohlfert EA, et al. The alarmin il-33 promotes regulatory T-cell function in the intestine. Nature (2014) 513(7519):564–8. doi: 10.1038/nature13577
53. Pascual-Reguant A, Bayat Sarmadi J, Baumann C, Noster R, Cirera-Salinas D, Curato C, et al. T(H)17 cells express St2 and are controlled by the alarmin il-33 in the small intestine. Mucosal Immunol (2017) 10(6):1431–42. doi: 10.1038/mi.2017.5
54. Kleinschek MA, Owyang AM, Joyce-Shaikh B, Langrish CL, Chen Y, Gorman DM, et al. Il-25 regulates Th17 function in autoimmune inflammation. J Exp Med (2007) 204(1):161–70. doi: 10.1084/jem.20061738
55. Su J, Chen T, Ji XY, Liu C, Yadav PK, Wu R, et al. Il-25 downregulates Th1/Th17 immune response in an il-10-Dependent manner in inflammatory bowel disease. Inflammation Bowel Dis (2013) 19(4):720–8. doi: 10.1097/MIB.0b013e3182802a76
56. Xu M, Lu H, Lee YH, Wu Y, Liu K, Shi Y, et al. An interleukin-25-Mediated autoregulatory circuit in keratinocytes plays a pivotal role in psoriatic skin inflammation. Immunity (2018) 48(4):787–98.e4. doi: 10.1016/j.immuni.2018.03.019
57. Xu M, Dong C. Il-25 in allergic inflammation. Immunol Rev (2017) 278(1):185–91. doi: 10.1111/imr.12558
58. Deng C, Peng N, Tang Y, Yu N, Wang C, Cai X, et al. Roles of il-25 in type 2 inflammation and autoimmune pathogenesis. Front Immunol (2021) 12:691559. doi: 10.3389/fimmu.2021.691559
59. Gowhari Shabgah A, Amir A, Gardanova ZR, Olegovna Zekiy A, Thangavelu L, Ebrahimi Nik M, et al. Interleukin-25: New perspective and state-of-the-Art in cancer prognosis and treatment approaches. Cancer Med (2021) 10(15):5191–202. doi: 10.1002/cam4.4060
60. Liew FY, Girard JP, Turnquist HR. Interleukin-33 in health and disease. Nat Rev Immunol (2016) 16(11):676–89. doi: 10.1038/nri.2016.95
61. Larsen KM, Minaya MK, Vaish V, Peña MMO. The role of il-33/St2 pathway in tumorigenesis. Int J Mol Sci (2018) 19(9):2676. doi: 10.3390/ijms19092676
62. Kalyan A, Kircher S, Shah H, Mulcahy M, Benson A. Updates on immunotherapy for colorectal cancer. J Gastrointest Oncol (2018) 9(1):160–9. doi: 10.21037/jgo.2018.01.17
63. Müller MF, Ibrahim AEK, Arends MJ. Molecular pathological classification of colorectal cancer. Virchows Archiv (2016) 469(2):125–34. doi: 10.1007/s00428-016-1956-3
64. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. Pd-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med (2015) 372(26):2509–20. doi: 10.1056/NEJMoa1500596
65. Eng C, Kim TW, Bendell J, Argilés G, Tebbutt NC, Di Bartolomeo M, et al. Atezolizumab with or without cobimetinib versus regorafenib in previously treated metastatic colorectal cancer (Imblaze370): A multicentre, open-label, phase 3, randomised, controlled trial. Lancet Oncol (2019) 20(6):849–61. doi: 10.1016/s1470-2045(19)30027-0
66. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr., Kinzler KW. Cancer genome landscapes. Sci (New York NY) (2013) 339(6127):1546–58. doi: 10.1126/science.1235122
67. MacDonald BT, Tamai K, He X. Wnt/Beta-catenin signaling: Components, mechanisms, and diseases. Dev Cell (2009) 17(1):9–26. doi: 10.1016/j.devcel.2009.06.016
68. Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell (1996) 87(2):159–70. doi: 10.1016/s0092-8674(00)81333-1
69. Yaeger R, Shah MA, Miller VA, Kelsen JR, Wang K, Heins ZJ, et al. Genomic alterations observed in colitis-associated cancers are distinct from those found in sporadic colorectal cancers and vary by type of inflammatory bowel disease. Gastroenterology (2016) 151(2):278–87.e6. doi: 10.1053/j.gastro.2016.04.001
70. Cercek A, Lumish M, Sinopoli J, Weiss J, Shia J, Lamendola-Essel M, et al. Pd-1 blockade in mismatch repair–deficient, locally advanced rectal cancer. New Engl J Med (2022) 386(25):2363–76. doi: 10.1056/NEJMoa2201445
71. Picard E, Verschoor CP, Ma GW, Pawelec G. Relationships between immune landscapes, genetic subtypes and responses to immunotherapy in colorectal cancer. Front Immunol (2020) 11:369. doi: 10.3389/fimmu.2020.00369
72. Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology (2010) 138(6):2073–87.e3. doi: 10.1053/j.gastro.2009.12.064
73. Xiao Y, Freeman GJ. The microsatellite instable subset of colorectal cancer is a particularly good candidate for checkpoint blockade immunotherapy. Cancer Discovery (2015) 5(1):16–8. doi: 10.1158/2159-8290.CD-14-1397
74. André T, Shiu K-K, Kim TW, Jensen BV, Jensen LH, Punt C, et al. Pembrolizumab in Microsatellite-Instability–high advanced colorectal cancer. New Engl J Med (2020) 383(23):2207–18. doi: 10.1056/NEJMoa2017699
75. Soh JS, Jo SI, Lee H, Do E-j, Hwang SW, Park SH, et al. Immunoprofiling of colitis-associated and sporadic colorectal cancer and its clinical significance. Sci Rep (2019) 9(1):6833. doi: 10.1038/s41598-019-42986-1
76. Jackstadt R, Sansom OJ. Mouse models of intestinal cancer. J Pathol (2016) 238(2):141–51. doi: 10.1002/path.4645
77. Bürtin F, Mullins CS, Linnebacher M. Mouse models of colorectal cancer: Past, present and future perspectives. World J Gastroenterol (2020) 26(13):1394–426. doi: 10.3748/wjg.v26.i13.1394
78. Al-Samadi A, Moossavi S, Salem A, Sotoudeh M, Tuovinen SM, Konttinen YT, et al. Distinctive expression pattern of interleukin-17 cytokine family members in colorectal cancer. Tumor Biol (2016) 37(2):1609–15. doi: 10.1007/s13277-015-3941-x
79. Liu Y, Sun X, Zhao X, An L, Wang Z, Jiang J, et al. Expression and location of il-17a, e, f and their receptors in colorectal adenocarcinoma: Comparison with benign intestinal disease. Pathol Res Pract (2018) 214(4):482–91. doi: 10.1016/j.prp.2018.03.011
80. Liu J, Qian B, Zhou L, Shen G, Tan Y, Liu S, et al. Il25 enhanced colitis-associated tumorigenesis in mice by upregulating transcription factor Gli1. Front Immunol (2022) 13:837262. doi: 10.3389/fimmu.2022.837262
81. Jou E, Rodriguez-Rodriguez N, Ferreira AF, Jolin HE, Clark PA, Sawmynaden K, et al. An innate il-25-Ilc2-Mdsc axis creates a cancer-permissive microenvironment for apc mutation-driven intestinal tumorigenesis. Sci Immunol (2022) 7(72):eabn0175. doi: 10.1126/sciimmunol.abn0175
82. Thelen TD, Green RM, Ziegler SF. Acute blockade of il-25 in a colitis associated colon cancer model leads to increased tumor burden. Sci Rep (2016) 6:25643. doi: 10.1038/srep25643
83. Benatar T, Cao MY, Lee Y, Lightfoot J, Feng N, Gu X, et al. Il-17e, a proinflammatory cytokine, has antitumor efficacy against several tumor types in vivo. Cancer Immunol Immunother (2010) 59(6):805–17. doi: 10.1007/s00262-009-0802-8
84. Maywald RL, Doerner SK, Pastorelli L, De Salvo C, Benton SM, Dawson EP, et al. Il-33 activates tumor stroma to promote intestinal polyposis. Proc Natl Acad Sci U.S.A. (2015) 112(19):E2487–96. doi: 10.1073/pnas.1422445112
85. Islam MS, Horiguchi K, Iino S, Kaji N, Mikawa S, Hori M, et al. Epidermal growth factor is a critical regulator of the cytokine il-33 in intestinal epithelial cells. Br J Pharmacol (2016) 173(16):2532–42. doi: 10.1111/bph.13535
86. Cui G, Qi H, Gundersen MD, Yang H, Christiansen I, Sørbye SW, et al. Dynamics of the il-33/St2 network in the progression of human colorectal adenoma to sporadic colorectal cancer. Cancer Immunol Immunother (2015) 64(2):181–90. doi: 10.1007/s00262-014-1624-x
87. Cui G, Yuan A, Li Z, Goll R, Florholmen J. St2 and regulatory T cells in the colorectal Adenoma/Carcinoma microenvironment: Implications for diseases progression and prognosis. Sci Rep (2020) 10(1):5892. doi: 10.1038/s41598-020-62502-0
88. Van der Jeught K, Sun Y, Fang Y, Zhou Z, Jiang H, Yu T, et al. St2 as checkpoint target for colorectal cancer immunotherapy. JCI Insight (2020) 5(9):e136073. doi: 10.1172/jci.insight.136073
89. O'Donnell C, Mahmoud A, Keane J, Murphy C, White D, Carey S, et al. An antitumorigenic role for the il-33 receptor, St2l, in colon cancer. Br J Cancer (2016) 114(1):37–43. doi: 10.1038/bjc.2015.433
90. Mertz KD, Mager LF, Wasmer M-H, Thiesler T, Koelzer VH, Ruzzante G, et al. The il-33/St2 pathway contributes to intestinal tumorigenesis in humans and mice. Oncoimmunology (2015) 5(1):e1062966–e. doi: 10.1080/2162402X.2015.1062966
91. He Z, Chen L, Souto FO, Canasto-Chibuque C, Bongers G, Deshpande M, et al. Epithelial-derived il-33 promotes intestinal tumorigenesis in apc (Min/+) mice. Sci Rep (2017) 7(1):5520–. doi: 10.1038/s41598-017-05716-z
92. Malik A, Sharma D, Zhu Q, Karki R, Guy CS, Vogel P, et al. Il-33 regulates the iga-microbiota axis to restrain il-1α-Dependent colitis and tumorigenesis. J Clin Invest (2016) 126(12):4469–81. doi: 10.1172/JCI88625
93. Li Y, Shi J, Qi S, Zhang J, Peng D, Chen Z, et al. Il-33 facilitates proliferation of colorectal cancer dependent on cox2/Pge2. J Exp Clin Cancer Res (2018) 37(1):196. doi: 10.1186/s13046-018-0839-7
94. Fang M, Li Y, Huang K, Qi S, Zhang J, Zgodzinski W, et al. Il33 promotes colon cancer cell stemness Via jnk activation and macrophage recruitment. Cancer Res (2017) 77(10):2735–45. doi: 10.1158/0008-5472.Can-16-1602
95. Zhou Y, Ji Y, Wang H, Zhang H, Zhou H. Il-33 promotes the development of colorectal cancer through inducing tumor-infiltrating St2l(+) regulatory T cells in mice. Technol Cancer Res Treat (2018) 17:1533033818780091. doi: 10.1177/1533033818780091
96. Pastille E, Wasmer M-H, Adamczyk A, Vu VP, Mager LF, Phuong NNT, et al. The il-33/St2 pathway shapes the regulatory T cell phenotype to promote intestinal cancer. Mucosal Immunol (2019) 12(4):990–1003. doi: 10.1038/s41385-019-0176-y
97. Kienzl M, Hasenoehrl C, Valadez-Cosmes P, Maitz K, Sarsembayeva A, Sturm E, et al. Il-33 reduces tumor growth in models of colorectal cancer with the help of eosinophils. Oncoimmunology (2020) 9(1):1776059–. doi: 10.1080/2162402X.2020.1776059
98. Eberl G, Colonna M, Di Santo JP, McKenzie AN. Innate lymphoid cells. innate lymphoid cells: A new paradigm in immunology. Science (2015) 348(6237):aaa6566. doi: 10.1126/science.aaa6566
99. Fallon PG, Ballantyne SJ, Mangan NE, Barlow JL, Dasvarma A, Hewett DR, et al. Identification of an interleukin (Il)-25-Dependent cell population that provides il-4, il-5, and il-13 at the onset of helminth expulsion. J Exp Med (2006) 203(4):1105–16. doi: 10.1084/jem.20051615
100. Ricardo-Gonzalez RR, Van Dyken SJ, Schneider C, Lee J, Nussbaum JC, Liang H-E, et al. Tissue signals imprint Ilc2 identity with anticipatory function. Nat Immunol (2018) 19(10):1093–9. doi: 10.1038/s41590-018-0201-4
101. Wang S, Qu Y, Xia P, Chen Y, Zhu X, Zhang J, et al. Transdifferentiation of tumor infiltrating innate lymphoid cells during progression of colorectal cancer. Cell Res (2020) 30(7):610–22. doi: 10.1038/s41422-020-0312-y
102. Carrega P, Orecchia P, Quatrini L, Tumino N, Venè R, Benelli R, et al. Characterisation of innate lymphoid cell subsets infiltrating colorectal carcinoma. Gut (2020) 69(12):2261–63. doi: 10.1136/gutjnl-2020-320908
103. Qi J, Crinier A, Escalière B, Ye Y, Wang Z, Zhang T, et al. Single-cell transcriptomic landscape reveals tumor specific innate lymphoid cells associated with colorectal cancer progression. Cell Rep Med (2021) 2(8):100353. doi: 10.1016/j.xcrm.2021.100353
104. Wang L, Wang Y, Song Z, Chu J, Qu X. Deficiency of interferon-gamma or its receptor promotes colorectal cancer development. J Interferon Cytokine Res (2015) 35(4):273–80. doi: 10.1089/jir.2014.0132
105. Zhang C, Hou D, Wei H, Zhao M, Yang L, Liu Q, et al. Lack of interferon-Γ receptor results in a microenvironment favorable for intestinal tumorigenesis. Oncotarget (2016) 7(27):42099–109. doi: 10.18632/oncotarget.9867
106. Miret JJ, Kirschmeier P, Koyama S, Zhu M, Li YY, Naito Y, et al. Suppression of myeloid cell arginase activity leads to therapeutic response in a nsclc mouse model by activating anti-tumor immunity. J ImmunoTher Cancer (2019) 7(1):32. doi: 10.1186/s40425-019-0504-5
107. Vasquez-Dunddel D, Pan F, Zeng Q, Gorbounov M, Albesiano E, Fu J, et al. Stat3 regulates arginase-I in myeloid-derived suppressor cells from cancer patients. J Clin Invest (2013) 123(4):1580–9. doi: 10.1172/jci60083
108. Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P, et al. Hif-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med (2010) 207(11):2439–53. doi: 10.1084/jem.20100587
109. Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, et al. L-arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell (2016) 167(3):829–42.e13. doi: 10.1016/j.cell.2016.09.031
110. Raber PL, Thevenot P, Sierra R, Wyczechowska D, Halle D, Ramirez ME, et al. Subpopulations of myeloid-derived suppressor cells impair T cell responses through independent nitric oxide-related pathways. Int J Cancer (2014) 134(12):2853–64. doi: 10.1002/ijc.28622
111. Ohl K, Tenbrock K. Reactive oxygen species as regulators of mdsc-mediated immune suppression. Front Immunol (2018) 9:2499. doi: 10.3389/fimmu.2018.02499
112. Ballbach M, Dannert A, Singh A, Siegmund DM, Handgretinger R, Piali L, et al. Expression of checkpoint molecules on myeloid-derived suppressor cells. Immunol Lett (2017) 192:1–6. doi: 10.1016/j.imlet.2017.10.001
113. Yu J, Du W, Yan F, Wang Y, Li H, Cao S, et al. Myeloid-derived suppressor cells suppress antitumor immune responses through ido expression and correlate with lymph node metastasis in patients with breast cancer. J Immunol (2013) 190(7):3783–97. doi: 10.4049/jimmunol.1201449
114. Huang B, Pan PY, Li Q, Sato AI, Levy DE, Bromberg J, et al. Gr-1+Cd115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res (2006) 66(2):1123–31. doi: 10.1158/0008-5472.Can-05-1299
115. Naito Y, Saito K, Shiiba K, Ohuchi A, Saigenji K, Nagura H, et al. Cd8+ T cells infiltrated within cancer cell nests as a prognostic factor in human colorectal cancer. Cancer Res (1998) 58(16):3491–4.
116. Galon J, Mlecnik B, Bindea G, Angell HK, Berger A, Lagorce C, et al. Towards the introduction of the 'Immunoscore' in the classification of malignant tumours. J Pathol (2014) 232(2):199–209. doi: 10.1002/path.4287
117. Zlobec I, Karamitopoulou E, Terracciano L, Piscuoglio S, Iezzi G, Muraro MG, et al. Tia-1 cytotoxic granule-associated rna binding protein improves the prognostic performance of Cd8 in mismatch repair-proficient colorectal cancer. PloS One (2010) 5(12):e14282. doi: 10.1371/journal.pone.0014282
118. Pagès F, Berger A, Camus M, Sanchez-Cabo F, Costes A, Molidor R, et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. N Engl J Med (2005) 353(25):2654–66. doi: 10.1056/NEJMoa051424
119. Prall F, Dührkop T, Weirich V, Ostwald C, Lenz P, Nizze H, et al. Prognostic role of Cd8+ tumor-infiltrating lymphocytes in stage iii colorectal cancer with and without microsatellite instability. Hum Pathol (2004) 35(7):808–16. doi: 10.1016/j.humpath.2004.01.022
120. Chiba T, Ohtani H, Mizoi T, Naito Y, Sato E, Nagura H, et al. Intraepithelial Cd8+ T-Cell-Count becomes a prognostic factor after a longer follow-up period in human colorectal carcinoma: Possible association with suppression of micrometastasis. Br J Cancer (2004) 91(9):1711–7. doi: 10.1038/sj.bjc.6602201
121. Ling A, Lundberg IV, Eklöf V, Wikberg ML, Öberg Å, Edin S, et al. The infiltration, and prognostic importance, of Th1 lymphocytes vary in molecular subgroups of colorectal cancer. J Pathol Clin Res (2015) 2(1):21–31. doi: 10.1002/cjp2.31
122. Huang Q, Jacquelot N, Preaudet A, Hediyeh-Zadeh S, Souza-Fonseca-Guimaraes F, McKenzie ANJ, et al. Type 2 innate lymphoid cells protect against colorectal cancer progression and predict improved patient survival. Cancers (Basel) (2021) 13(3):559. doi: 10.3390/cancers13030559
123. Xiao P, Wan X, Cui B, Liu Y, Qiu C, Rong J, et al. Interleukin 33 in tumor microenvironment is crucial for the accumulation and function of myeloid-derived suppressor cells. Oncoimmunology (2016) 5(1):e1063772. doi: 10.1080/2162402x.2015.1063772
124. Zhang Y, Davis C, Shah S, Hughes D, Ryan JC, Altomare D, et al. Il-33 promotes growth and liver metastasis of colorectal cancer in mice by remodeling the tumor microenvironment and inducing angiogenesis. Mol Carcinog (2017) 56(1):272–87. doi: 10.1002/mc.22491
125. Zhong W, Myers JS, Wang F, Wang K, Lucas J, Rosfjord E, et al. Comparison of the molecular and cellular phenotypes of common mouse syngeneic models with human tumors. BMC Genomics (2020) 21(1):2. doi: 10.1186/s12864-019-6344-3
126. Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell (2015) 160(1-2):48–61. doi: 10.1016/j.cell.2014.12.033
127. Castle JC, Loewer M, Boegel S, de Graaf J, Bender C, Tadmor AD, et al. Immunomic, genomic and transcriptomic characterization of Ct26 colorectal carcinoma. BMC Genomics (2014) 15(1):190. doi: 10.1186/1471-2164-15-190
128. Gounaris E, Erdman SE, Restaino C, Gurish MF, Friend DS, Gounari F, et al. Mast cells are an essential hematopoietic component for polyp development. Proc Natl Acad Sci USA (2007) 104(50):19977–82. doi: 10.1073/pnas.0704620104
129. Bodduluri SR, Mathis S, Maturu P, Krishnan E, Satpathy SR, Chilton PM, et al. Mast cell-dependent Cd8(+) T-cell recruitment mediates immune surveillance of intestinal tumors in Apc(Min/+) mice. Cancer Immunol Res (2018) 6(3):332–47. doi: 10.1158/2326-6066.Cir-17-0424
130. Iwanaga K, Nakamura T, Maeda S, Aritake K, Hori M, Urade Y, et al. Mast cell-derived prostaglandin D2 inhibits colitis and colitis-associated colon cancer in mice. Cancer Res (2014) 74(11):3011–9. doi: 10.1158/0008-5472.Can-13-2792
131. Eissmann MF, Buchert M, Ernst M. Il33 and mast cells–the key regulators of immune responses in gastrointestinal cancers? Front Immunol (2020) 11:1389. doi: 10.3389/fimmu.2020.01389
132. Blatner NR, Bonertz A, Beckhove P, Cheon EC, Krantz SB, Strouch M, et al. In colorectal cancer mast cells contribute to systemic regulatory T-cell dysfunction. Proc Natl Acad Sci USA (2010) 107(14):6430–5. doi: 10.1073/pnas.0913683107
133. Wu X, Zou Y, He X, Yuan R, Chen Y, Lan N, et al. Tumor-infiltrating mast cells in colorectal cancer as a poor prognostic factor. Int J Surg Pathol (2013) 21(2):111–20. doi: 10.1177/1066896912448836
134. Tan S-Y, Fan Y, Luo H-S, Shen Z-X, Guo Y, Zhao L-J. Prognostic significance of cell infiltrations of immunosurveillance in colorectal cancer. World J Gastroenterol (2005) 11(8):1210–4. doi: 10.3748/wjg.v11.i8.1210
135. Pittoni P, Colombo MP. The dark side of mast cell-targeted therapy in prostate cancer. Cancer Res (2012) 72(4):831–5. doi: 10.1158/0008-5472.Can-11-3110
136. Ohue Y, Nishikawa H. Regulatory T (Treg) cells in cancer: Can treg cells be a new therapeutic target? Cancer Sci (2019) 110(7):2080–9. doi: 10.1111/cas.14069
137. Shang B, Liu Y, Jiang S-j, Liu Y. Prognostic value of tumor-infiltrating Foxp3+ regulatory T cells in cancers: A systematic review and meta-analysis. Sci Rep (2015) 5:15179. doi: 10.1038/srep15179
138. Sears CL, Garrett WS. Microbes, microbiota, and colon cancer. Cell Host Microbe (2014) 15(3):317–28. doi: 10.1016/j.chom.2014.02.007
139. Ivleva EA, Grivennikov SI. Microbiota-driven mechanisms at different stages of cancer development. Neoplasia (2022) 32:100829. doi: 10.1016/j.neo.2022.100829
140. Zhang X, Kelaria S, Kerstetter J, Wang J. The functional and prognostic implications of regulatory T cells in colorectal carcinoma. J Gastrointest Oncol (2015) 6(3):307–13. doi: 10.3978/j.issn.2078-6891.2015.017
141. Pastille E, Bardini K, Fleissner D, Adamczyk A, Frede A, Wadwa M, et al. Transient ablation of regulatory T cells improves antitumor immunity in colitis-associated colon cancer. Cancer Res (2014) 74(16):4258–69. doi: 10.1158/0008-5472.Can-13-3065
142. Szeponik L, Akeus P, Rodin W, Raghavan S, Quiding-Järbrink M. Regulatory T cells specifically suppress conventional Cd8αβ T cells in intestinal tumors of Apc(Min/+) mice. Cancer Immunol Immunother CII (2020) 69(7):1279–92. doi: 10.1007/s00262-020-02540-9
143. Obata-Ninomiya K, de Jesus Carrion S, Hu A, Ziegler SF. Emerging role for thymic stromal lymphopoietin-responsive regulatory T cells in colorectal cancer progression in humans and mice. Sci Transl Med (2022) 14(645):eabl6960. doi: 10.1126/scitranslmed.abl6960
144. Shevyrev D, Tereshchenko V. Treg heterogeneity, function, and homeostasis. Front Immunol (2019) 10:3100. doi: 10.3389/fimmu.2019.03100
145. Liu JL, Wang CY, Cheng TY, Rixiati Y, Ji C, Deng M, et al. Circadian clock disruption suppresses Pdl1(+) intraepithelial b cells in experimental colitis and colitis-associated colorectal cancer. Cell Mol Gastroenterol Hepatol (2021) 12(1):251–76. doi: 10.1016/j.jcmgh.2021.02.008
146. Wang X, Asami S, Kitamura D. A novel cancer immunotherapy using tumor-infiltrating b cells in the apcmin/+ mouse model. PloS One (2021) 16(1):e0245608. doi: 10.1371/journal.pone.0245608
147. Morita H, Arae K, Unno H, Toyama S, Motomura K, Matsuda A, et al. Il-25 and il-33 contribute to development of eosinophilic airway inflammation in epicutaneously antigen-sensitized mice. PloS One (2015) 10(7):e0134226. doi: 10.1371/journal.pone.0134226
148. Reichman H, Itan M, Rozenberg P, Yarmolovski T, Brazowski E, Varol C, et al. Activated eosinophils exert antitumorigenic activities in colorectal cancer. Cancer Immunol Res (2019) 7(3):388–400. doi: 10.1158/2326-6066.Cir-18-0494
149. Batlle E, Clevers H. Cancer stem cells revisited. Nat Med (2017) 23(10):1124–34. doi: 10.1038/nm.4409
150. Nakanishi Y, Seno H, Fukuoka A, Ueo T, Yamaga Y, Maruno T, et al. Dclk1 distinguishes between tumor and normal stem cells in the intestine. Nat Genet (2013) 45(1):98–103. doi: 10.1038/ng.2481
151. Park SY, Kim JY, Choi JH, Kim JH, Lee CJ, Singh P, et al. Inhibition of Lef1-mediated Dclk1 by niclosamide attenuates colorectal cancer stemness. Clin Cancer Res (2019) 25(4):1415–29. doi: 10.1158/1078-0432.Ccr-18-1232
152. Goto N, Fukuda A, Yamaga Y, Yoshikawa T, Maruno T, Maekawa H, et al. Lineage tracing and targeting of Il17rb<Sup>+</Sup> tuft cell-like human colorectal cancer stem cells. Proc Natl Acad Sci (2019) 116(26):12996–3005. doi: 10.1073/pnas.1900251116
153. Westphalen CB, Asfaha S, Hayakawa Y, Takemoto Y, Lukin DJ, Nuber AH, et al. Long-lived intestinal tuft cells serve as colon cancer-initiating cells. J Clin Invest (2014) 124(3):1283–95. doi: 10.1172/JCI73434
154. George ML, Tutton MG, Janssen F, Arnaout A, Abulafi AM, Eccles SA, et al. Vegf-a, vegf-c, and vegf-d in colorectal cancer progression. Neoplasia (2001) 3(5):420–7. doi: 10.1038/sj.neo.7900186
155. Goodlad RA, Ryan AJ, Wedge SR, Pyrah IT, Alferez D, Poulsom R, et al. Inhibiting vascular endothelial growth factor receptor-2 signaling reduces tumor burden in the apcmin/+ mouse model of early intestinal cancer. Carcinogenesis (2006) 27(10):2133–9. doi: 10.1093/carcin/bgl113
156. Guba M, Seeliger H, Kleespies A, Jauch KW, Bruns C. Vascular endothelial growth factor in colorectal cancer. Int J Colorectal Dis (2004) 19(6):510–7. doi: 10.1007/s00384-003-0576-y
157. Waldner MJ, Wirtz S, Jefremow A, Warntjen M, Neufert C, Atreya R, et al. Vegf receptor signaling links inflammation and tumorigenesis in colitis-associated cancer. J Exp Med (2010) 207(13):2855–68. doi: 10.1084/jem.20100438
158. Xie Y-H, Chen Y-X, Fang J-Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduction Targeted Ther (2020) 5(1):22. doi: 10.1038/s41392-020-0116-z
159. Corrigan CJ, Wang W, Meng Q, Fang C, Wu H, Reay V, et al. T-Helper cell type 2 (Th2) memory T cell-potentiating cytokine il-25 has the potential to promote angiogenesis in asthma. Proc Natl Acad Sci USA (2011) 108(4):1579–84. doi: 10.1073/pnas.1014241108
160. Yao X, Wang W, Li Y, Huang P, Zhang Q, Wang J, et al. Il-25 induces airways angiogenesis and expression of multiple angiogenic factors in a murine asthma model. Respir Res (2015) 16(1):39–. doi: 10.1186/s12931-015-0197-3
161. Toth M, Fridman R. Assessment of gelatinases (Mmp-2 and mmp-9 by gelatin zymography. Methods Mol Med (2001) 57:163–74. doi: 10.1385/1-59259-136-1:163
162. Huang H. Matrix metalloproteinase-9 (Mmp-9) as a cancer biomarker and mmp-9 biosensors: Recent advances. Sensors (Basel) (2018) 18(10):3249. doi: 10.3390/s18103249
163. Yang B, Tang F, Zhang B, Zhao Y, Feng J, Rao Z. Matrix metalloproteinase-9 overexpression is closely related to poor prognosis in patients with colon cancer. World J Surg Oncol (2014) 12:24. doi: 10.1186/1477-7819-12-24
164. Bendardaf R, Buhmeida A, Hilska M, Laato M, Syrjänen S, Syrjänen K, et al. Mmp-9 (Gelatinase b) expression is associated with disease-free survival and disease-specific survival in colorectal cancer patients. Cancer Invest (2010) 28(1):38–43. doi: 10.3109/07357900802672761
165. Garg P, Sarma D, Jeppsson S, Patel NR, Gewirtz AT, Merlin D, et al. Matrix metalloproteinase-9 functions as a tumor suppressor in colitis-associated cancer. Cancer Res (2010) 70(2):792–801. doi: 10.1158/0008-5472.CAN-09-3166
166. Sinnamon MJ, Carter KJ, Fingleton B, Matrisian LM. Matrix metalloproteinase-9 contributes to intestinal tumourigenesis in the adenomatous polyposis coli multiple intestinal neoplasia mouse. Int J Exp Pathol (2008) 89(6):466–75. doi: 10.1111/j.1365-2613.2008.00621.x
167. Dong W, Li H, Zhang Y, Yang H, Guo M, Li L, et al. Matrix metalloproteinase 2 promotes cell growth and invasion in colorectal cancer. Acta Biochim Biophys Sin (Shanghai) (2011) 43(11):840–8. doi: 10.1093/abbs/gmr085
168. Shin N, Son GM, Shin DH, Kwon MS, Park BS, Kim HS, et al. Cancer-associated fibroblasts and desmoplastic reactions related to cancer invasiveness in patients with colorectal cancer. Ann Coloproctol (2019) 35(1):36–46. doi: 10.3393/ac.2018.09.10
169. Landskron G, de la Fuente López M, Dubois-Camacho K, Díaz-Jiménez D, Orellana-Serradell O, Romero D, et al. Interleukin 33/St2 axis components are associated to desmoplasia, a metastasis-related factor in colorectal cancer. Front Immunol (2019) 10:1394. doi: 10.3389/fimmu.2019.01394
170. Liu X, Zhu L, Lu X, Bian H, Wu X, Yang W, et al. Il-33/St2 pathway contributes to metastasis of human colorectal cancer. Biochem Biophys Res Commun (2014) 453(3):486–92. doi: 10.1016/j.bbrc.2014.09.106
171. Luo P, Deng S, Ye H, Yu X, Deng Q, Zhang Y, et al. The il-33/St2 pathway suppresses murine colon cancer growth and metastasis by upregulating Cd40 l signaling. BioMed Pharmacother (2020) 127:110232. doi: 10.1016/j.biopha.2020.110232
172. Vaddepally RK, Kharel P, Pandey R, Garje R, Chandra AB. Review of indications of fda-approved immune checkpoint inhibitors per nccn guidelines with the level of evidence. Cancers (Basel) (2020) 12(3):738. doi: 10.3390/cancers12030738
173. McLane LM, Abdel-Hakeem MS, Wherry EJ. Cd8 T cell exhaustion during chronic viral infection and cancer. Annu Rev Immunol (2019) 37(1):457–95. doi: 10.1146/annurev-immunol-041015-055318
174. Zarour HM. Reversing T-cell dysfunction and exhaustion in cancer. Clin Cancer Res (2016) 22(8):1856–64. doi: 10.1158/1078-0432.Ccr-15-1849
175. Andrews LP, Cillo AR, Karapetyan L, Kirkwood JM, Workman CJ, Vignali DAA. Molecular pathways and mechanisms of lag-3 in cancer therapy. Clin Cancer Res (2022) clincanres.2390.2022:OF1–10. doi: 10.1158/1078-0432.CCR-21-2390
176. Granier C, De Guillebon E, Blanc C, Roussel H, Badoual C, Colin E, et al. Mechanisms of action and rationale for the use of checkpoint inhibitors in cancer. ESMO Open (2017) 2(2):e000213. doi: 10.1136/esmoopen-2017-000213
177. Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, et al. T-Cell invigoration to tumour burden ratio associated with anti-Pd-1 response. Nature (2017) 545(7652):60–5. doi: 10.1038/nature22079
178. Hermel DJ, Sigal D. The emerging role of checkpoint inhibition in microsatellite stable colorectal cancer. J Pers Med (2019) 9(1):5. doi: 10.3390/jpm9010005
179. Weber R, Fleming V, Hu X, Nagibin V, Groth C, Altevogt P, et al. Myeloid-derived suppressor cells hinder the anti-cancer activity of immune checkpoint inhibitors. Front Immunol (2018) 9:1310. doi: 10.3389/fimmu.2018.01310
180. Hou A, Hou K, Huang Q, Lei Y, Chen W. Targeting myeloid-derived suppressor cell, a promising strategy to overcome resistance to immune checkpoint inhibitors. Front Immunol (2020) 11:783(783). doi: 10.3389/fimmu.2020.00783
181. Bruchard M, Mignot G, Derangère V, Chalmin F, Chevriaux A, Végran F, et al. Chemotherapy-triggered cathepsin b release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat Med (2013) 19(1):57–64. doi: 10.1038/nm.2999
182. Kang C, Jeong S-Y, Song SY, Choi EK. The emerging role of myeloid-derived suppressor cells in radiotherapy. Radiat Oncol J (2020) 38(1):1–10. doi: 10.3857/roj.2019.00640
183. Dubois-Camacho K, Diaz-Jimenez D, de la Fuente M, Quera R, Simian D, Martínez M, et al. Inhibition of mir-378a-3p by inflammation enhances il-33 levels: A novel mechanism of alarmin modulation in ulcerative colitis. Front Immunol (2019) 10:2449. doi: 10.3389/fimmu.2019.02449
184. Chen J, He Y, Tu L, Duan L. Dual immune functions of il-33 in inflammatory bowel disease. Histol Histopathol (2020) 35(2):137–46. doi: 10.14670/hh-18-149
185. Chen J, Wang Y, Shen L, Xiu Y, Wang B. Could il-25 be a potential therapeutic target for intestinal inflammatory diseases? Cytokine Growth Factor Rev (2022) S1359–6101(22):00051–X. doi: 10.1016/j.cytogfr.2022.07.001
Keywords: IL-25, IL-33, colorectal cancer, mouse model, microenvironment, cytokine
Citation: Jou E, Rodriguez-Rodriguez N and McKenzie ANJ (2022) Emerging roles for IL-25 and IL-33 in colorectal cancer tumorigenesis. Front. Immunol. 13:981479. doi: 10.3389/fimmu.2022.981479
Received: 29 June 2022; Accepted: 15 September 2022;
Published: 03 October 2022.
Edited by:
Erwan Mortier, Centre National de la Recherche Scientifique (CNRS), FranceReviewed by:
Philippe Krebs, University of Bern, SwitzerlandCopyright © 2022 Jou, Rodriguez-Rodriguez and McKenzie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrew N. J. McKenzie, YW5tQG1yYy1sbWIuY2FtLmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.