 Shubhabrata Majumdar
Shubhabrata Majumdar Yong Lin
Yong Lin Matthew L. Bettini
Matthew L. Bettini- 1Immunology Graduate Program, Baylor College of Medicine, Houston, TX, United States
- 2Department of Pathology, University of Utah, Salt Lake City, UT, United States
Type-1 Diabetes (T1D) is a complex polygenic autoimmune disorder involving T-cell driven beta-cell destruction leading to hyperglycemia. There is no cure for T1D and patients rely on exogenous insulin administration for disease management. T1D is associated with specific disease susceptible alleles. However, the predisposition to disease development is not solely predicted by them. This is best exemplified by the observation that a monozygotic twin has just a 35% chance of developing T1D after their twin’s diagnosis. This makes a strong case for environmental triggers playing an important role in T1D incidence. Multiple studies indicate that commensal gut microbiota and environmental factors that alter their composition might exacerbate or protect against T1D onset. In this review, we discuss recent literature highlighting microbial species associated with T1D. We explore mechanistic studies which propose how some of these microbial species can modulate adaptive immune responses in T1D, with an emphasis on T-cell responses. We cover topics ranging from gut-thymus and gut-pancreas communication, microbial regulation of peripheral tolerance, to molecular mimicry of islet antigens by microbial peptides. In light of the accumulating evidence on commensal influences in neonatal thymocyte development, we also speculate on the link between molecular mimicry and thymic selection in the context of T1D pathogenesis. Finally, we explore how these observations could inform future therapeutic approaches in this disease.
Introduction
Type 1 Diabetes (T1D) is a chronic autoimmune disorder resulting from T cell-mediated destruction of the insulin-producing β cells found in the pancreatic islets (1). Like many other autoimmune disorders, T1D is caused by the combined influence of genetic predisposition and environmental factors. More than 50 genetic loci have been linked to an increased risk of developing T1D (2, 3). Not every at-risk individual will become diabetic however, and environmental factors appear to play a significant role in T1D development (4). This can be best demonstrated in monozygotic twins where a twin has approximately 35% chance of developing T1D if the other sibling is affected (5–7). Similarly, in the genetically identical non-obese diabetic (NOD) mouse strain, not all mice develop autoimmune diabetes (8). Furthermore, the incidence of T1D has increased dramatically over the past decades, strongly indicating that environmental factors are accelerating the development of T1D (9).
Similar increase in other autoimmune and inflammatory disorders is also observed (10). The general improvement of living standards over the past century is believed to have contributed to the increased incidence of these disorders. Termed the “hygiene hypothesis”, it is believed that “clean” environments lead to improper training of the developing immune system in early life, resulting in aberrant immune responses to foreign- and self-components (11). Hence, much research has gone into how various environmental and lifestyle factors, such as birthing method, diet, infection, and antibiotic use contribute to the risk for developing T1D. The results of these efforts have pointed to the alterations in the gut microbiota as a critical variable in whether a genetically susceptible individual will go on to develop T1D (4).
In this review, we will discuss the current understanding of how alterations in gut microbiota result in diabetogenic T cell activation and T1D. We first discuss factors in healthy gut development, followed by a summary of findings from human studies that identified common features of gut alteration associated with increased or decreased risk of developing T1D. We then move onto findings in animal studies, where precise manipulations in the gut microbiota and the immune system provided mechanistic insights into how gut microbial alterations result in diabetogenic T cell activation and autoimmune diabetes. As microbial antigens can share similar epitopes with diabetogenic autoantigens, we also discuss the potential role of microbial antigens in shaping diabetogenic T cell development in light of the reported link between gut microbiota and thymic selection. Furthermore, we discuss potential therapeutic interventions based on these findings and what work still needs to be done to fill in the gap in knowledge. Finally, we propose distinguishing genetic and environmental factors within the emerging concept of T1D endotypes, where there can be multiple different sets of etiological pathways resulting in T1D (12).
Association of early life microbial gut colonization with T1D
The first three years of life are crucial for the establishment of a healthy gut microbiome. This early life period undergoes waves of microbial colonization which gradually diversify and stabilize into a mature adult microbiome. After analyzing stool sample 16s rRNA sequences collected from European and US children as part of The Environmental Determinants of Diabetes in the Young (TEDDY) study, Stewart et al. have identified three distinct phases of microbiome progression: a developmental phase (3-4 mo. age), a transitional phase (15-30 mo. age) and a stable phase (≥31 mo. age) (13). Immediately after birth, the intestinal environment favors the initial expansion of facultative anaerobes like Staphylococcus, Streptococcus, Escherichia coli, and Enterobacteria that are fast colonizers and good survivors in an oxygen rich environment (14, 15). In the developmental phase, with the introduction of maternal milk, the infant gut is dominated by members of the Actinobacteria and Firmicutes phyla as they are better able to utilize and survive on milk oligosaccharides. Bifidobacterium spp. were the dominant taxa at the genus level during this time. At the species level, B. bifidum, B. breve, B. dentium, Lactobacillus rhamnosus and Staphylococcus epidermidis were observed at higher levels in breast-fed individuals (13). When compared across different geographical regions, the microbial profiles are very similar during this early period with breast milk being the major diet source (16). With the introduction of solid food after around 4-6 months age and the subsequent reduction in breast-feeding, there is a gradual increase in microbial diversity and richness. Microbial taxa feeding on milk oligosaccharides become less dominant and allow for the diversification of the microbiome with different taxa that are able to utilize nutrients supplied by the local diet (13, 16, 17). Functional metagenomic studies show a decline in enzymes like L-lactate dehydrogenase involved in milk fermentation and a corresponding increase in fiber metabolism genes like transketolase (17). By the third year, the microbiome matures and reaches a more stable state with a high alpha diversity and dominance by members of the Firmicutes phylum (13).
Several studies in humans and rodent T1D models have suggested that the microbiota is altered in T1D patients in comparison to healthy individuals. T1D patients seem to have a lower Firmicutes/Bacteroidetes ratio and reduced microbial diversity (18–20). Higher levels of Bacteroides species have been reported in T1D cases across different studies on Finnish, Spanish and US populations (18, 19, 21). Pre-diabetic children seropositive for autoimmune antibodies also had higher levels of Bacteroides (22). Concomitantly, there is an observed reduction in the abundance of short-chain fatty acid (SCFA) producing bacteria in T1D patients. Lower levels of Firmicutes and Actinobacteria members have been reported in T1D cases including, Lactobacillus and Bifidobacterium species which are dominant in breast-fed children (19, 21, 23). In accordance with this observation, some studies have suggested that infants that have not been breast-fed are at a higher risk of developing T1D than breast-fed infants, although there is no strong association with duration of breastfeeding (24–26). T1D patients also have lower levels of Clostridium cluster IV and XIVa members that are known to produce SCFAs (18). Lower levels of Prevotella, which are good SCFA producers and found abundantly in populations on a plant-based diet, have also been reported in T1D cases (19, 21). Although these observations describe altered microbiota in T1D patients, they may not be indicative of causation and might be a consequence of the altered immune system under autoimmune conditions. To address the issue of causation, longitudinal studies were required to look at the microbiota of healthy T1D genetically susceptible and pre-diabetic individuals from early time points. One such study analyzed stool samples from a cohort of 33 Finnish and Estonian infants genetically pre-disposed to T1D recruited as part of the Pathogenesis of Type 1 Diabetes - Testing the Hygiene Hypothesis (DIABIMMUNE) study. They observed a significant reduction in alpha-diversity of microbial species in infants who developed T1D with a trending overabundance of pathobiont containing genera like Blautia, Ruminococcus and Streptococcus (20). In the TEDDY study, healthy subjects had more Lactobacillus rhamnosus, Bifidobacterium dentium, Streptococcus thermophilus and Lactococcus lactis whereas sero-converters and T1D patients had higher Streptococcus spp., Bifidobacterium pseudocatenulatum, Roseburia hominis, Alistipes shahii and Bacteroides vulgatus (17). Higher levels of Parabacteroides spp. levels and lower Akkermansia spp. levels were also reported in T1D patients recruited in the TEDDY study (13). Table 1 provides a summary of important commensal bacteria that are associated with T1D incidence along with their proposed mechanisms.

Table 1 Association of Gut Commensal Bacteria with T1D incidence and proposed mechanisms.
Maintenance of gut-barrier integrity and its role in T1D
One of the major functions of the host immune-microbial cross-talk is maintenance of the gut barrier integrity. Loss of gut barrier integrity can lead to bacterial translocation and leakage of gut luminal products into sterile immune compartments causing inflammation and non-homeostatic activation of the host immune system. In order to prevent this, the host immune system has coevolved with beneficial gut microbiota to maintain homeostatic conditions where the luminal side of the barrier can harbor commensal microbiota while maintaining controlled exchange of luminal products across the epithelium. The barrier site is stratified into different levels of protection and can be divided into an outer luminal mucus layer and an inner intestinal epithelial barrier (IEB) (46). The mucus layer acts as a buffer zone for harboring microbial communities while preventing their direct contact with the epithelial layer (47). This layer is primarily composed of mucins that are special glycoproteins with O-linked glycans secreted by the intestinal goblet cells (48). The mucus layer also contains a milieu of antimicrobial peptides (AMPs) like defensins, cathelicidins and RegIIIγ that directly keep microbial communities in check and prevent their contact with the epithelium (46). These peptides are produced by the intestinal epithelial cells (IECs) (49). The immune cells are located along the epithelium in the lamina propria and in specialized lymphoid structures called Peyer’s patches. Intestinal dendritic cells (DCs) can randomly sample bacterial components near the epithelium and interact with B and T-cells in the Peyer’s patches to induce IgA production (46, 50). IgA are transcytosed across the epithelium and bind to specific bacteria to prevent their translocation across the barrier (50). The composition of the mucus layer acts as a source of nutrition for the microbiota, and along with the AMPs shape the microbial communities. The microbiota, through their cell wall components and metabolites, provide feedback to the host, predominantly in a MyD88 and inflammasome dependent manner (51, 52). This influences the production of tight junction proteins, mucins and AMPs by the IECs.
Early loss of gut barrier integrity with low-grade intestinal inflammation has been observed in T1D diagnosed cases (53). It has been suggested to be a contributing factor to the initiation of autoimmune pathogenesis as studies have shown that loss of gut barrier integrity precedes T1D onset. In T1D susceptible patients with islet autoimmunity, increased intestinal permeability was observed before clinical onset of diabetes (54). Loss of gut barrier integrity has been shown to occur prior to insulitis in the spontaneous autoimmune diabetes model in BioBreeding (BB) rats (55, 56). The increased permeability is attributed to disassembly of tight junctions through upregulation of zonulin (57, 58). Loss of barrier integrity is also associated with altered architecture of the mucus layer as seen in the NOD mice. It involved decreased expression of immune-regulatory mucins Muc1 and Muc3 and increased inflammatory Muc4 (59). This can be attributed to a reduction of SCFA producing intestinal bacteria in T1D patients as SCFAs like butyrate have been shown to enhance tight junction formation and mucin production in the epithelial cells (17, 18, 22, 60–62). Butyrate production as result of metabolic cooperation between Bacteroides thetaiotaomicron and Faecalibacterium prausnitzii (Clostridium cluster IV) also impacted goblet cell maturation and mucin glycosylation in gnotobiotic rodents (33). Acetate produced by Bifidobacterium longum infantis was involved in protecting mice from E. coli O157:H7 enteropathogenesis by preventing the translocation of Shiga toxin across the IEB (39). Similarly, Bifidobacterium dentium was also shown to modify goblet cells and increase mucin synthesis through acetate production (41).
Production of AMPs like cathelicidins is altered in NOD mice and T1D patients (63, 64). Cathelicidins are constitutively produced by intestinal epithelial cells and their expression is restricted to the pre-weaning period in mice (65). Interestingly, pancreatic endocrine cells also express Cathelicidin-related antimicrobial peptide (CRAMP) under the influence of gut microbiota (64). Defects in CRAMP production were observed both in the pancreas and colon of NOD mice (63, 64). Defective CRAMP production was shown to be associated with altered intestinal microbiota, enhanced Type I interferon expression and pathological imprinting of the immune system. Early life CRAMP treatment increased beneficial bacteria in the gut, reduced Type I interferon levels and reduced diabetes incidence (63). Similarly, the RegIIIγ has also been implicated to have a protective role in T1D (66). RegIIIγ is produced by intestinal Paneth cells under the influence of Toll-like receptor (TLR) signaling. Akkermansia muciniphila mediated upregulation of RegIIIγ and mucin production has been suggested as a possible mechanism for T1D protection in NOD mice (42, 43). Bifidobacterium breve, which has a negative correlation with T1D incidence, was also shown to enhance RegIIIγ expression in a TLR dependent manner (13, 17, 40).
Microbiota can strengthen gut barrier function through cross-talk with the innate immune system. This has been elucidated in mice colonized with segmented filamented bacteria (SFB). SFB induces IL-23 expression in CX3CR1+ myeloid cells leading to IL-22 secretion by ILC3s in the lamina propria (30). IL-22 is known for its role in promoting epithelial cell growth, AMP and mucin production (67). This was one of the proposed explanations for a study where SFB colonization in NOD mice showed protection from diabetes development (31). Along the same lines, a recent study reported low levels of IL-23 and IL-22 production as a result of intestinal inflammation in T1D mouse models. In parallel, there was an observed reduction in SFB abundance and loss of gut integrity with decreased expression of tight junction proteins and Muc2 (32).
The studies highlighted in this section provide an understanding of the importance of gut-barrier integrity maintenance in T1D. Gut commensals that are negatively associated with T1D incidences, play an important role in the maintenance of gut barrier integrity through regulation of tight junction proteins, mucin formation, AMP production and modulation of the host mucosal immune cells.
Loss of gut-barrier integrity and the establishment of an inflammatory gut environment
As mentioned earlier, T1D is a polygenic autoimmune disorder. Among the genetic loci conferring the highest risk are those localized in the HLA genes (68). While various components of the immune system are known to play a role in this disease, T1D is considered a T-cell-mediated disorder where activated diabetogenic T cells directly result in the destruction of islet β cells. The association between gut dysbiosis and T1D is well established, but how the commensal changes influence the immune environment within the gut that eventually result in the activation of the diabetogenic T cell population has only recently been investigated. Much insight has been gained with various T1D animal models such as the spontaneously diabetic NOD mouse model, streptozotocin (STZ)-induced diabetic mouse model, and the BB rat. In this section, we will discuss the current understanding of how dysbiosis and the compromised epithelial barrier establishes an inflammatory gut environment.
As the activation of T cells depends on the presentation of the cognate antigens by professional antigen-presenting cells in an inflammatory context, recent work has highlighted the role of the gut innate immune system in breaking T cell tolerance against islet beta cells during gut dysbiosis. Using various knockout animal models, it has been shown that innate signaling proteins such as MyD88 and TRIF, which control microbe-sensing through pattern-recognition receptors, can greatly impact diabetes incidence (69, 70). MyD88 deficiency protected the NOD mouse almost completely from diabetes by 30 weeks of age (69). Interestingly, when MyD88 deficiency was generated in germ-free mouse, the diabetes rate was similar to wild-type NOD mouse. These observations pointed to two important insights: 1. autoimmune diabetes is genetically determined in the NOD mouse, and 2. microbiota-immune interactions determine the rate at which diabetes occurs. The latter forms the basis of the balanced signal hypothesis, which suggests that immune-microbiota interaction induces both pro-inflammatory and anti-inflammatory signals and that the relative strength of each signal affects the disease risk. Indeed, TLR’s influence on diabetes incidence is receptor specific. TLR2 knockout protected the NOD mice from diabetes, but the protection was reversed in the germ-free setting, indicating that TLR2 interaction with the microbiota promotes diabetes (71). In contrast, targeting signaling through TLR4 appeared to provide protection from diabetes in this model (71). To complicate matters further, subtypes of the TLR4 ligand LPS (E. coli-derived vs Bacteroides-derived) possess either stimulatory or inhibitory activity when bound to TLR4 and may have opposing effects on T1D risk (72). Thus, interactions between gut microbiota and the innate immune system, mediated by various pattern recognition receptors, influences the immune context in which autoreactive T cells see its cognate antigen and may accelerate or delay disease. These findings suggest that in dysbiosis, the changing microbial diversity in the gut may shift the balance of signals toward a pro-inflammatory state, leading to compromised gut integrity. Infiltration of gut microbes through the leaky gut then activates the innate immune cells residing below the epithelium, thereby establishes an inflammatory environment in which diabetogenic T cells are likely to be activated. Interestingly, a study looking at a large cohort of Korean patients suggested that individuals with Crohn’s disease, characterized by gut dysbiosis and inflammation, are at a higher risk of developing T1D (73).
Neutrophils are a subset of innate granulocytes critically important in early immune responses to inflammation and infection. It stands to reason that neutrophils would be involved in the early immune response to invading gut commensal in the leaky gut. Current data point to the involvement of neutrophils in early stages of islet autoimmunity. Recently diagnosed T1D patients exhibited a reduction of neutrophils in circulation (74–76). This observation was associated with the accumulation of neutrophils in the pancreas, suggesting a role for neutrophils in the pathogenesis in T1D (74). In agreement, a separate study detected elevated levels of the neutrophil chemoattractant IL-8 in the pancreas of T1D patients (77). Additionally, elevated levels of circulating neutrophil granule enzymes (neutrophil elastase and proteinase 3) were detected in recently diagnosed T1D patients and in autoantibody-negative at-risk individuals (78). The same study also found that elevated circulating neutrophil granule enzymes were closely associated with increased neutrophil extracellular traps (NET) formation, suggesting the involvement of NETosis in T1D pathogenesis. Similar results were obtained in a large scale study in which NET-forming neutrophils were identified in the pancreas of T1D patients and pre-symptomatic at-risk individuals (79). Collectively, these data indicate neutrophils are involved in the early stages of human T1D. In contrast, another study did not find increases in serum levels of neutrophil elastase and proteinase 3 although the study subjects were diagnosed with T1D within three years, as opposed to within one year in the other aforementioned studies (80). This difference in criteria for patient inclusion may explain the discrepancy between the studies.
More recent work also identified an association between NET production and leaky gut in newly diagnosed T1D patients (81). Six of the eight markers of NET production examined in this study showed an increase in T1D patients, and protein arginine deiminase type 4 (PAD4) levels in particular appear to be the best predictor of gut leakage and T1D diagnosis. Furthermore, zonulin and LPS were also increased in T1D patients, indicating the association between leaky gut, infiltration of microbes and microbial products, and neutrophil activation. Neutrophil and NET formation have similarly been found in the NOD mouse model (82–85). Infiltration of neutrophils into the NOD pancreas can be detected as early as 2 weeks of age (82). In contrast, no neutrophil infiltration was detected in the B6 mouse strain which is not genetically susceptible for autoimmune diabetes. Importantly, a recent report described a direct link between gut leakage and NET formation where the induction of gut leakage by dextran sulfate sodium (DSS) treatment in the NOD mouse increased NET production and accelerated diabetes (83). However, in PAD4 knockout NOD mice, in which PAD4-mediated formation of NETs was blocked, diabetes incidence was dramatically reduced. Furthermore, T1D patient-derived NETs can induce the activation of dendritic cells (DCs), increasing its expression of HLA and co-stimulatory molecules as well as production of inflammatory cytokines (86). These changes would provide an ideal activating environment for autoreactive T cells in T1D.
Enteric infections are also associated with compromised gut barrier integrity and accelerated T1D (87, 88). Citrobacter rodentium (C. rodentium) infection is a colonic infection model that induces gut leakage. Infection with C. rodentium in the NOD and STZ-induced diabetes mouse model accelerated diabetes incidence (88). It was found that C. rodentium infection reduced antimicrobial peptide CRAMP production in the intestine, resulting in gut leakage and increased activation of macrophages and CD86+ cDCs in the mesenteric lymph node (mLN). In CRAMP knockout mice, C. rodentium infection further exacerbated the diabetes incidence.
Overall, these results suggest that homeostatic innate immune responses maintain a healthy gut environment and resists diabetes development, whereas innate immune response toward infiltrating microbiota due to compromised gut barrier promotes an inflammatory environment favorable to the activation of self-reactive lymphocytes (Figure 1).
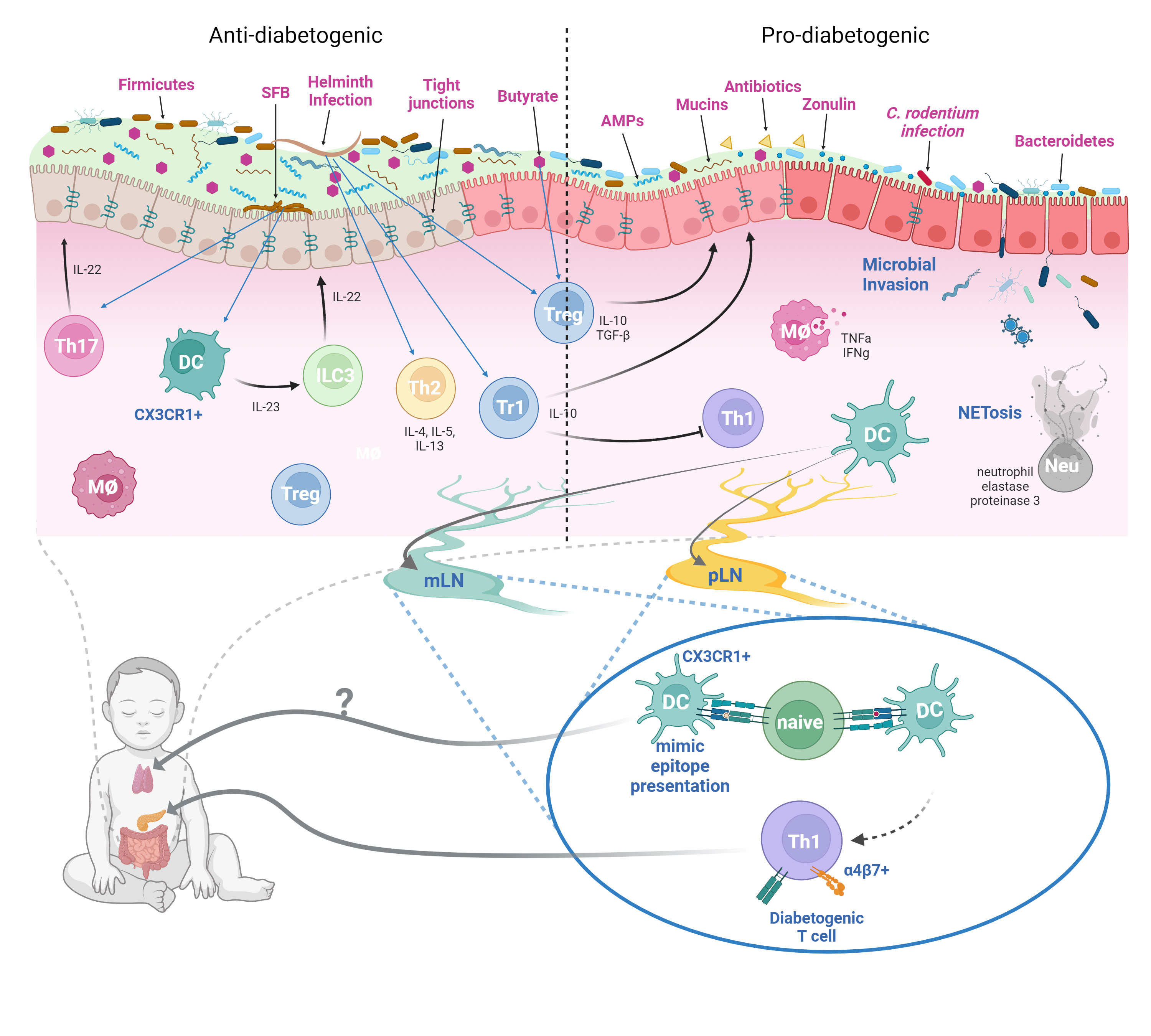
Figure 1 Dysbiosis, Compromised Gut Barrier, and T1D Risk. In a healthy gut microenvironment, most microbial species are not in direct contact with the epithelial barrier. The mucus layer serves as a barrier between the commensal microbes and the epithelium. Contained within the mucus layer are various innate immune components such as the antimicrobial peptides (AMPs) such as defensins, cathelicidins, and RegIIIγ. The adaptive immune system also supplies the mucus layer with IgA antibodies that bind to commensal microbes and prevent their adherence to the epithelium. However, certain commensals such as the SFB can directly adhere to the epithelium and stimulate Th17 response. SFB-induced immunity appears to protect NOD mice from autoimmune diabetes through enhancing intestinal barrier conferred by Type 3 immune cytokine IL-22. A high proportion of Firmicutes to Bacteroidetes ratio seems to protect against T1D presumably due to Firmicutes being a major butyrate producer. The SCFA butyrate can improve barrier integrity by enhancing barrier function of tight junctions, improve mucin production, and induction of intestinal Tregs. Helminth infections, which are more common prior to the improved hygienic practices in the past century, may also decrease T1D incidence through the induction of a type 2 response and the generation of regulatory Foxp3+ and Tr1 cells. In dysbiosis, altered Firmicute/Bacteroidetes ratio may diminish SCFA production, reducing intestinal barrier function, and promote leaky gut syndrome. Infections by certain microbes such as C. rodentium can also enhance gut permeability. Additionally, antibiotic usage can also reduce commensal diversity and lead to dysbiosis. The resulting depletion of the mucus layer and the compromised tight junction may allow infiltration of microbes through the epithelium and induce innate immune cell activation. Production of inflammatory cytokines by innate cells and NETs by neutrophils further exacerbates gut permeability. The inflammatory gut environment promotes the activation of diabetogenic T cells in the mLN and the pLN, which then traffic to the pancreas and cause islet damage. Furthermore, β-cell antigen mimics found in the gut microbes and enteric pathogens can also potentially activate diabetogenic T cells and contribute to disease. Whether these mimics can be trafficked to the thymus to mediate T cell central tolerance is unclear (figure created with BioRender.com).
Gut-pancreas immune axis in T1D
While pancreatic lymph node (pLN) is critical for T1D development in the NOD mouse, it wasn’t clear whether other lymphoid tissues can also serve as activation site of diabetogenic T cells as well (89). Initial report suggesting mesenteric lymph node (mLN) as the initial site of diabetogenic T cell activation used adoptive transfer of lymphocytes from various lymphoid organs of NOD mice at different ages to determine which age and organ had the most diabetogenic potential (90). In the young donor mice (3-weeks old), it was found that lymphocytes with diabetogenic potential were found almost exclusively in the mLNs. In contrast, by 6-weeks old, most of the diabetogenic T cells are found in the pLNs, with mLN-derived lymphocytes exhibiting only intermediate diabetogenic potential. Additionally, the expression of the gut-homing integrin LPAM (α4β7 integrin) was found in islet-infiltrating lymphocytes, indicating gut mucosal origin (91). Furthermore, NOD mice treated at an early age with an antibody blocking the mucosal addressin cell adhesion molecule-1 (MAdCAM-1) were almost completely protected from diabetes at 32-weeks old (9% diabetic compared to 80% in the control group) (92). Thus, a link between mLN and the pancreas was established. Subsequent report further established that antigens derived from the GI tract are able to reach both the mLN and the pLN and activate their cognate T cells (93). Moreover, disruption of the gut barrier allowed for the translocation of bacteria to the pLN, which activated the innate receptor NOD2 in immune cells that contributed to diabetes in the STZ-induced diabetes model (94). Overall, these data indicate inflammatory gut environment can promote activation of diabetogenic T cells in the mLN and pLN, resulting in islet autoimmunity and diabetes (Figure 1).
Regulation of T cell phenotype by the gut commensal microbiota
The interactions between the gut commensal microbiota, the epithelium, and the immune system promotes proper barrier function in steady state conditions. The commensals protect against colonization of pathogenic microbes through competition for space and nutrients but also strengthen the barrier integrity by influencing T cell differentiation (95). T cell response in the healthy gut is typically associated with the generation of homeostatic Th17 cells and Tregs, which can result in the production of various cytokines that enhance tissue barrier function. For example, the commensal SFB can adhere to the epithelium and trigger the secretion of serum amyloid A and result in IL-17 production by SFB-specific Th17 cells (30, 31, 96). IL-17 is known to act on the epithelium to enhance barrier integrity by controlling the localization of the tight junction protein occludin and enhancing production of AMPs (97–99). In contrast to homeostatic Th17 response, IL-23-driven Th17 response seems to promote autoinflammatory and autoimmune conditions such as seen in experimental autoimmune encephalomyelitis and inflammatory bowel disease. Data suggest the pathology is partly a result of IL-23 mediated conversion of Th17 to Th1 cells (100).
Gut commensals also drive Treg development. The intestinal Treg population is enriched for clones that recognize commensal microbiota (101, 102). In agreement, germ-free mice given commensal microbiota showed an expansion and activation of Tregs in the colonic lamina propria (103). In neonatal mice, the colonizing microbiota drives the formation of a population of Rorγt+ Tregs near weaning (104). The accumulation of these Tregs is associated with the dramatic shift in commensal population as a result of diet change from milk to solid food and the availability of dietary metabolites such as SCFAs and retinoic acid. These Tregs are critical to prevent the pathological imprinting of the immune system that increases susceptibility to colitis and allergic conditions (104). Microbiota can also regulate Treg development through their surface components and metabolites such as SCFAs. For example, capsular polysaccharide A (PSA) from the human commensal Bacteroides fragilis can stimulate dendritic cells to promote the differentiation of naïve T cells into IL-10-producing Tregs (105, 106). Feeding mice PSA enhanced generation of these Tregs and conferred protection against a mouse model of Multiple Sclerosis (107). Similarly, SCFAs generated from fermentation of dietary fibers has been shown to promote Treg generation in the gut (108–110). The SCFA butyrate appears to enhance Treg generation by enhancing histone H3 acetylation in the Foxp3 promoter and the conserved non-coding regions (108, 110). Butyrate also suppresses inflammatory cytokine production in DCs, resulting in a more tolerogenic, Treg-promoting environment (111, 112). Other commensal derived SCFA propionate also promoted histone acetylation at the Foxp3 locus and result in Treg generation (108). Overall, these data indicate the significant role of commensal microbiota in influencing intestinal T cell phenotype. Given that β-cell specific T cells can be activated in gut-draining lymph nodes, it is very possible that these T cells are subjected to similar phenotypic regulation by the gut microbiota and the disruption of which may result in increased risk for T1D.
Microbial regulation of thymocyte development
Central tolerance is established in the thymus during T-cell development. Developing CD4+ CD8+ double positive thymocytes are positively selected in the thymic cortex on the basis of their ability to recognizing non-agonist peptides in the context of host MHC presented by the cortical thymic epithelial cells (cTECs). Positively selected thymocytes mature into CD4+ and CD8+ single positive thymocytes and migrate into the thymic medulla where they undergo further selection mechanisms (113–115). In the medulla, thymocytes are exposed to a wide repertoire of self-antigens presented by medullary resident and migratory antigen-presenting cells (APCs). The medullary thymic epithelial cells (mTECs) can present tissue-restricted antigens in an autoimmune regulator (AIRE) and Fezf2 dependent manner (116, 117). Thymic resident CD8+ DCs can acquire antigens from mTECs and cross-present to developing CD8+ thymocytes (118). Migratory APCs including Sirpα+ CD11b+ cDC2s, CD11c+ B220+ CCR9+ pDCs and B-cells, can transport peripheral self-antigens to the thymus through blood circulation under homeostatic conditions (119–121). High affinity agonistic interactions of developing thymocytes with self-antigens in the thymus leads to deletion as a mechanism to weed out potentially auto-reactive T-cells (114, 122–124). In addition, as an extra layer of protection, developing CD4+ thymocytes with an intermediate affinity for self-antigens can be converted into FoxP3+ Treg cells that can suppress auto-reactivity in the periphery (124).
Central tolerance plays a major role in preventing autoimmunity as mutations in AIRE can lead to multi-organ autoimmunity termed as autoimmune polyendocrinopathy candidiasis ectodermal dystrophy (APECED). In T1D, there is a genetic pre-disposition to develop diabetes in individuals bearing specific high-risk HLA haplotypes and length of the variable number of tandem repeats (VNTRs) of the insulin promoter. HLA-DQ8 is one such MHC haplotype whose peptide binding groove is unfavorable for tight binding of insulin peptide B:9-23. On the other hand, individuals with shorter VNTRs might generate less insulin peptides for presentation to the developing thymocytes and lead to thymic escape of autoreactive T-cells (125–127). The pancreas also produces many post-translationally modified antigens like hybrid insulin peptides (HIPs) that might not be presented to the developing thymocytes. HIP activated T-cells have been identified in T1D patients and can initiate T1D pathogenesis as seen in NOD mice (128, 129). So, bolstering central tolerance by enhancing presentation of islet specific antigens in the thymus is one potential mechanism of tackling T1D.
Recent evidences suggest that intestinal microbiota can modulate T-cell selection in the thymus. TCR sequencing of intestinal and thymic Tregs in TCRmini mice, revealed significant repertoire overlap and resulted in identification of microbiota specific Treg TCRs in the thymus and gut (102). Antibiotic treatment altered both the thymic and intestinal Treg repertoire, suggesting intestinal microbiota might influence thymic selection of microbiota specific Treg cells. In line with this observation, microbial colonization of neonatal mouse skin resulted in a wave of migration of microbial antigen specific Tregs into the skin that mediated immune tolerance in the skin (130). Transient FTY720 treatment in the neonatal time window prevented accumulation of the Tregs in the skin while there was an increased accumulation of these Tregs in the thymus. This indirectly suggested that microbial colonization of the skin might influence microbial antigen specific Treg development in the thymus. Interestingly, colonization of adult skin with microbiota or blocking Treg migration in the neonatal period, resulted in no immune tolerance to the microbial antigen. This suggested that there might be a specific neonatal time window when the commensal microbiota can influence thymic development of microbiota specific T-cells (130). Recently, we were able to detect microbial 16SrRNA signatures in the thymus and demonstrate migration of CX3CR1+ DCs from the gut to the thymus to induce expansion of SFB antigen specific CD4+ thymocytes in young SFB colonized mice but not in adult mice. The migration of CX3CR1+ DCs is dependent on the microbiota as antibiotic treatment showed a decrease in thymic DC subsets (131). In another study, colonic pDCs were shown to migrate into the neonatal thymus in a Bacteroides fragilis dependent manner and influence development of PLZF+ innate lymphocytes (132).
Thymic development of MAIT (mucosal associated invariant T) cells can also be controlled through microbial metabolites (133). MAIT cells are evolutionarily conserved T cells with an invariant TCRα (Vα7.2–Jα33 in humans, and Vα19–Jα33 in mice) chain that associates with a limited repertoire of β-chains. They recognize and respond to bacterial or fungal Vitamin B2 precursors presented on the MHC Class Ib molecule MR1 (134, 135). MAIT cells can be detected in blood, liver and various mucosal barrier tissues (134). MAIT cells respond and migrate into infected or inflamed tissues to control pathogen load through effector cytokines like IFNγ, TNFα and IL-17 and direct lysis of infected cells through granzyme B (134, 135). On the other hand, under homeostatic conditions, mucosal RORγt+ MAIT17 cells play an important role in maintenance of gut barrier integrity through the production of IL17 and IL22 (136). Germ-free mice bear a reduced number of RORγt+ MAIT17 cells with an overall skewing towards IFNγ production and reduced IL-17 production. Upon co-housing with SPF mice, MAIT17 cell numbers are rescued in a TCR-dependent manner. The effect was mediated through thymic capture and presentation of the bacterial vitamin B2 synthesis by-product 5-OP-RU [5-(2-oxopropylideneamino)-6-D-ribitylaminouracil (5-OP-RU)] when applied topically on mouse skin or when GF mice were colonized by a vitamin B2 proficient commensal strain Enterococcus hirae (133). This has potential implications for T1D, as RORγt+ MAIT17 cells have been shown to be important for maintenance of gut barrier integrity and play a protective role in T1D (136).
Overall, these observations provide support for a novel concept for the regulation of thymic T-cell development by commensal microflora. Although this concept has not been explored directly in the context of T1D yet, we speculate that the gut-thymus axis might play a role in T1D and other autoimmune disorders through the recruitment of microbiota specific thymic Tregs which are important for maintenance of tolerogenic conditions in the gut. The gut-thymus axis could also influence T1D pathogenesis by regulating the thymic development of islet specific cross-reactive T-cells through molecular mimicry.
Molecular mimicry in T1D
Thymic selection is not foolproof and healthy individuals do have self-antigen specific T-cells in peripheral circulation and tissues (27, 137, 138). Under homeostasis, they do not initiate pathogenesis due to tolerogenic conditions maintained by Treg cells and a lack of pro-inflammatory cues (139). However, inflammatory conditions caused by infection or gut-barrier leakiness may cause a breakdown of tolerance, including enhanced antigen processing and presentation of APCs. In the previous sections, we have discussed how T1D patients are associated with microbial dysbiosis and a leaky gut that indirectly leads to activation of islet specific T-cells in the pLN (17, 54, 93, 94). Furthermore, gut microbial peptides may directly activate cross-reactive islet specific T-cells through structural and functional homology to islet specific antigens. This is termed as molecular mimicry. Molecular mimicry as a mechanism for T1D induction was proposed decades ago through the identification of sequence similarity of glutamate de-carboxylase (GAD), an enzyme produced by islet β-cells, and the P2C protein of the Coxsackie virus (140). It was suggested that T and B lymphocytes activated during the initial viral infection are able to cross-react with islet specific GAD and initiate β-cell destruction (141). Other viral molecular mimicry candidates as potential initiators for T1D had also been proposed including the human cytomegalovirus (hCMV), rotavirus, varicella and measles virus (142–144). Although these studies identify T-cell cross-reactivity to viral peptides, there is a no direct evidence showing the ability of viral molecular mimics to initiate T1D pathogenesis. Newer studies have also identified T1D specific molecular mimicry candidates among the gut commensal species. One particular study in NOD mice, identified an IGRP (islet-specific glucose-6-phosphatase catalytic subunit-related protein) peptide mimic in Fusobacteria expressing a magnesium transporter (Mgt). They were able to demonstrate activation of IGRP specific CD8 T-cells and initiation of diabetes in vivo upon transfer of T-cells activated by the mimic into NOD mice. Super-colonization by Fusobacteria resulted in accelerated diabetes in their NY8.3 TCR(tg) NOD mice (45). The cation efflux transporter ZnT8 was identified as an auto-antigen in T1D patients (145). Healthy individuals can have ZnT8 specific CD8+ T-cells in circulation and substantial fraction of them were shown to bear an antigen experienced phenotype that suggested cross-priming of these T-cells. A bacterial peptide mimic derived from Bacteroides stercoris was shown to be a mimic of the ZnT8186-194 peptide and was shown to cross-react with ZnT8186-194 specific T-cells. Interestingly, the mimic peptide had a stronger agonist potency than the ZnT8186-194 peptide (27). The gut commensal Parabacteroides distasonis has been positively correlated with T1D cases (13, 146). P. distasonis colonization in NOD mice has been demonstrated to accelerate diabetes. A molecular mimic of the insulin peptide B:9-23, derived from P. distasonis, previously identified in a DIABIMMUNE study, was shown to activate insulin specific T-cells ex vivo with similar potency (29, 147). An integrase derived peptide expressed by several Bacteroides species was found to encode a low avidity mimetope of IGRP206-214 that can recruit IGRP specific CD8+ T-cells to the GALTs and activate them. Surprisingly, in this study, these diabetogenic CD8+ T-cells recruited to the gut were able to suppress colitis through cytotoxic killing of gut DCs presenting the bacterial mimetope (28). Although not entirely focused on T1D, this study provides strong evidence for priming of cross-reactive diabetogenic T-cells in the GALTs. Overall, all these examples support the possibility of bacterial peptide mimics priming pathogenic autoreactive T-cells and accelerating T1D progression. However, more mechanistic evidence is required to support the role of molecular mimicry as an initiator of T1D pathogenesis.
Environmental factors and T cell phenotype in T1D
Chemical-induced gut leakage
As previously mentioned, DSS-driven gut leakage in the NOD mouse promotes the formation of NETs by neutrophils and accelerates diabetes (83). When co-cultured with autologous splenocytes, NETs drove the proliferation of memory Th1 and Th17 cells in the NOD mouse. Additionally, DSS-induced acceleration of T1D is associated with an increased the ratio of Th1-to-Th2 cells in the pLN. Th1 cytokines TNFα and IFNγ was also increased as a result of DSS-induced gut permeability. In contrast, the reduced gut permeability, NET formation, and diabetes incidence in PAD4 knockout mice was associated with reduced Th1-to-Th2 cell ratio. While β-cell specific T cell populations were not evaluated in this context, the general shift toward Th1 phenotype is consistent with the idea that autoimmune diabetes is a Th1-driven disease. In the same study, authors also found a dramatic increase in integrin α4β7+ T cells in the pLN, indicating a gut-primed origin. Importantly, dendritic cell activation by NETs derived from T1D patients also promoted the generation of IFNγ-producing Th1 cells (86).
Infection
Infections have long been associated with T1D risk. Whether the infection promotes or inhibits disease depends on the type of infection. Enteric infection by C. rodentium in the NOD mouse resulted in enhanced gut permeability, inflammation, and accelerated diabetes (87, 88, 148). This change was associated with increased frequency of inflammatory M1 macrophages, activated cDCs, and IFNγ-producing CD4 Th1 and CD8 Tc1 cells (87, 88, 148). Consistent with previous finding, increase in α4β7+ CD4 and CD8 T cells were observed, indicating gut-associated T cell priming. Importantly, diabetogenic CD4 T cell clone BDC2.5 showed greater proliferation in the pLN when transferred into NOD mice pre-infected with C. rodentium (148). Similarly, C. rodentium infection in the NOD.8.3 transgenic mice that bears a diabetogenic CD8 T cell receptor specific for the islet antigen IGRP resulted in increased activation of the CD8 T cell population as indicated by increased CD44 expression (87, 148).
In contrast, various murine parasitic infections including Helignosomoides polygyrus (H. polygyrus), Strongyloides venezuelensis (S. venezuelensis), Taenia crassiceps (T. crassiceps), and Schistosoma mansoni (S. mansoni.) reduced the incidence of T1D in mice (149–152). As mentioned earlier, the ‘hygiene hypothesis’ posits that increased cleanliness and improved healthcare in developed countries is a major contributor to the increased incidence of various inflammatory and autoimmune conditions. One proposed mechanism of the ‘hygiene hypothesis’ is the reduced incidence of helminth infections (153). Helminth infection-induced type 2 immune responses are associated with the production of effector cytokines that enhance tissue barrier integrity. Additionally, helminth-induced peripheral Treg may have bystander effects that reduce risk for autoinflammatory and autoimmune diseases such as T1D (154–156). Infection with S. mansoni or treatment with S. mansoni-derived soluble extracts can provide protection from T1D in the NOD mouse (149, 157, 158). S. mansoni-derived ribonuclease ω-1 driven Th2 immune response and Treg generation appear to be at least in part responsible for the disease protection (159, 160). Additionally, S. mansoni proteins have been reported to reduce colonic inflammation caused by trinitrobenzene sulfonic acid, suggesting that S. mansoni may also modulate T1D through reducing intestinal inflammation and gut leakage (161). Likewise, infection of the parasite S. venezuelensis ameliorated insulitis and was associated with a Th2 polarized response as well as increased IL-5 and IL-10 expression in splenocytes (150). Infection by the parasite H. polygyrus similarly protected NOD mice from T1D through blocking a Th1 response and inducing a Th2 response (162). Surprisingly, in IL-4 knockout NOD mice, where Th2 response is blocked, H. polygyrus infection still conferred protection from diabetes (163). This was due to the production of IL-10 by β-cell-specific effector T cells in the infected mice. These data suggest H. polygyrus mediates protection of T1D through shifting from Th1 to Th2 response as well as induction of Tr1-like Tregs that are Foxp3-IL-10+. Overall, Helminth infections seem to confer protection from T1D through shifting the adaptive immune response and the production of immunosuppressive cytokines.
Antibiotics
Accumulating evidence indicates alteration of gut microbiota through antibiotic usage affects T1D risk. In the NOD mouse, maternal antibiotic treatment had mixed results in the diabetes incidence of the offspring. In one study, treatment of the mother with a mixture of antibiotics (metronidazole, gentamicin, and polymyxin) accelerated diabetes incidence at 20 weeks (164). The treatment reduced gut microbiota diversity in the offspring and altered their T cell population. Specifically, frequency of CD8 T cells was increased in the mLN of the offspring from mother treated with antibiotics. CD4+CD62L- T cells also increased in the Peyer’s patch of the antibiotics group, suggesting enhanced T cell activation. Frequency of Foxp3+ T cells were unchanged. No further characterization of β-cell antigen-specific T cell responses were noted, therefore it is unknown whether the maternal antibiotics treatment affected the activation state of those cells. In contrast, another study found that treating NOD mothers with neomycin, polymyxin B, and streptomycin, conferred protection from diabetes in the offspring (165). The offspring’s gut microbiota showed decrease in proteobacteria and increase in G+ Firmicutes families Lachnospiraceae and Coriobacteriacea. In the immune compartment, the antibiotics group showed a higher frequency of tolerogenic APCs that resulted increased Foxp3+ Tregs and reduced T-bet+ and IFNγ+ CD4 and CD8 T cells. Importantly, co-culturing the BDC2.5 and NY8.3 diabetogenic T cell clones with the APCs from the antibiotics group reduced their activation and proliferation when compared to the controls. Similarly, co-transfer of NY8.3 T cells with the APCs into NOD.scid mouse demonstrated a delay in diabetes when the APCs were from the antibiotics-treated group. The differences in antibiotics used might explain the discrepancies seen in these two studies. Another possibility is that the baseline microbiota composition could be different between these two labs.
Studies on the effect of post-natal early life antibiotic treatment in the NOD mouse generally resulted in an acceleration of diabetes incidence (166–171). Comparison of unperturbed gut microbiota between the NOD mouse and the diabetes-resistant non-obese resistant (NOR) mouse showed that the NOD mouse naturally had less diabetes-protective bacteria (166). This difference was exacerbated with prolonged antibiotics treatment, which led to decreased in microbial diversity and protective species (eg. Desulfovibrio spp., Prevotella spp., B. acidifaciens). Furthermore, antibiotic treatment led to reduction in the metagenome of gene clusters involved in SCFA synthesis, which are associated with T1D protection (170). Immunologically, the gut alteration was associated with decreased IL-17+ CD4 T cells in the gut-lymphoid tissues. Interestingly, an increase in IFNγ+ CD4 T cells was seen only in Vancomycin-treated group and not in Neomycin-treated group, despite both groups exhibiting accelerated diabetes to similar degrees (166). Shorter-term therapeutic-dose antibiotic treatment likewise found accelerated diabetes in the antibiotics group (167, 169). Microbiota alterations resulted in reduced Bifidobacterium and the butyrate-producers S24-7, Clostridiales, Lactobacillus, Oscillospira, and Ruminococcus (167, 171, 172). In agreement, cecal butyrate levels were decreased with the short course of antibiotics (169). Dramatic downregulation of serum amyloid A, a known promoter of Th17 response, was noted in the ileal gene expression profile of the antibiotics group, consistent with the observed decreased of Th17 and Treg proportions in the lamina propria. Subsequent work supplementing Vancomycin antibiotics treatment with a butyrate-rich diet ameliorated the antibiotic-accelerated T1D and its associated induction of IFNγ+ Th1 and Tc1 cells (170). A different rescue approach via maternal cecal microbiota transfer to antibiotics-treated neonates also rescued the offspring from antibiotics-accelerated T1D and associated gut microbiota changes (171). Antibiotics treatment also decreased bacterial bile acid metabolites, which are important in the maintenance of RORγ+ Tregs in the gut (173).
Diet
Various formulations have been studied in animal models to determine the properties of diabetogenic and anti-diabetogenic diets. In the diabetes-prone BB rat model, it was reported that rats fed a cereal diet exhibited a high rate of diabetes (174). In terms of T cell phenotype, cereal diet is associated with increased activation of CD3+ and CD8α+ lymphocytes in the gut and an increased ratio of Ifng to Il4 mRNA. In contrast, rats fed a low-antigen hydrolyzed casein (HC) diet had approximately 50% reduction in diabetes incidence. Examination of the intestinal tissue identified an increase in CRAMP expression in the HC diet group when compared to the cereal diet group. CRAMP’s essential role in maintaining gut microbiota homeostasis may have contributed to the reduced diabetes incidence (175). Similar results were obtained in the diabetes-prone LEW.1AR1-iddm rat model when fed either the cereal or HC diet, with the HC diet group exhibiting significantly reduced diabetes and Ifng to Il4 mRNA when compared to the cereal group (176).
Therapeutic interventions
Based on the common features of gut commensal alterations seen in T1D, various approaches have been attempted to correct these deficits. General strategies include 1. replenishing or enhancing protective microbial species, 2. enhancing resistance to gut barrier disruption, 3. suppressing innate inflammatory response, and 4. deviating immune response toward Th-2/17 and Treg phenotypes. Modification of the gut microbiota by the probiotic VSL#3, which is enriched in the Lactobacillaceae family of bacteria, has shown benefits against T1D in the NOD mouse (177). The protective effect was mediated by an increase in indoleamine 2,3-dioxygenase (IDO) and IL-33 in the intestine. IDO is known for enhancing a tolerogenic phenotype in DCs which suppresses T cell activation while also promote Treg generation (178, 179). Intestinal IL-33 can drive production of TSLP and retinoic acid in intestinal epithelium, which are known to promote Treg generation (180–183). In addition, IL-33 can promote a Th2 immune response and may help shift the immune cells from the more diabetogenic Th1 response (184, 185). Supplementation with the probiotic IRT5 (Lactobacillus acidophilus, Lactobacillus casei, Lactobacillus reuteri, Bifidobacterium bifidium, and Streptococcus thermophiles) similarly saw a decrease in diabetes incidence (186). The effect was associated with reduced Th1 skewing and increased Treg frequency in the mLN and the lamina propria of the small intestine. Besides replenishing protective microbes through probiotic supplementation, the prebiotic approach aims at enhancing SCFA generation through increased consumption of dietary fiber such as inulin (187). Increased dietary fiber fermentation by intestinal microbes can then increase the SCFA output in the gut. In the NOD mouse and the STZ-induced diabetes model, inulin intake ameliorated diabetes (187). The inulin-treated group showed an overall decrease in CD4 T cell frequency in the mLN and a slight increase in the Treg frequency. Increased levels of AMPs α-defensin and RegIIIγ were also noted. As previously mentioned, AMPs are important in maintaining gut microbial stability and provide an additional barrier layer between the microbes and the gut epithelium, effectively enhancing barrier integrity (188). Supplementation with the yeast-derived prebiotic β-glucan also delayed diabetes and reduced insulitis (189). The shifting of the colonic immune system toward Th17 and Tregs appears to mediate the protection conferred by β-glucan. To suppress innate inflammatory response, one group tried to degrade T1D-promoting NETs in the intestine of NOD mice via oral administration of staphylococcal nuclease (SNase)-producing Lactococcus lactis (190). SNase treatment degraded NETs and reduced inflammation in the intestine and pancreas. IFNγ+ and Rorγt+ CD4 T cells were also reduced in the mLN and pLN. In addition, butyrate-producing bacteria Oscillospira, Faecalibacterium and Ruminococcus were increased. Finally, decrease in zonulin levels indicated improved gut barrier function with SNase treatment.
Conclusion
The current data support the idea that gut commensal alteration is a central factor mediating environmental variables associated with developing T1D in at-risk individuals. Dysbiosis and enhanced gut permeability are associated with development of T1D in both humans and the NOD mouse model. Reduction of SCFA-producing bacteria species are generally associated with increased T1D risk. Indeed, particular diets and enteric pathogens known to decrease SCFA-producing species are associated higher risk of T1D development. Reduced availability of SCFAs such as butyrate, the main source of energy for colonocytes, likely impaired their ability to maintain barrier integrity as well as reduced generation of Tregs in the gut. Invasion of microbes into the tissue via compromised gut barrier results in immune cell activation in both the gut and the pancreas, leading to activation of the innate immune system and establishment of a pro-Th1 adaptive immune response in at-risk individuals. As summarized in this review, this plausible sequence of events leading to T1D is supported by the observation that enhancing gut barrier function through manipulation of the gut microbiota, suppression of neutrophil activation, or deviation of the adaptive immune response towards Th2/Th17/Treg all conferred protection against T1D. While this is a plausible sequence of events leading to T1D, lack in genetic diversity in animal models limits its translatability toward the human disease. Particularly, given the heterogeneity in human T1D, the relative importance of each step in the sequence with respect to the emerging concept of T1D endotypes is unclear (12).
It may be beneficial to distinguish genetics-driven endotypes and environment-driven endotypes. HLA variations confer the greatest genetic risk in T1D susceptibility. HLA-DR3 and HLA-DR4 are strongly associated with distinct islet autoantibodies, indicating potentially distinct genetics-driven endotypes(191, 192). While antibiotics can drive accelerated T1D in animal models, the observation that different antibiotics were associated with dramatically different T cell phenotypes may indicate distinct environment-driven endotypes. Similarly, regional differences in commensal microbiota composition may be considered a distinct category of environmental-driven endotype. However, mutual regulation and feedbacks make it difficult to draw a clear line between genetics- and environment-driven endotypes as HLA differences have been reported to influence the gut microbiome (193). A recent report indicated that individuals carrying at-risk HLA haplotypes seem to have a distinct core microbiome and beta diversity compared to the general population (194). This raises an interesting question: to what degree is the dysbiotic gut environment shaped by the HLA variants? Future investigations in this direction may provide new insights into the rate at which at-risk individuals become diabetic.
Another related area worthy of further exploration is the role of gut microbiota in shaping the diabetogenic T cell repertoire. As discussed in this review, our own work has recently identified a connection between the gut microbiota and thymocyte development (131). Our data indicate that antigens derived from the gut microbiota can be transferred to the thymus via migratory DCs trafficking between the gut and the thymus, resulting in the expansion of developing T cells specific for the microbiota. Selection of gut microbiota-specific T cell repertoire by the risk-conferring MHC variants and the propensity of this T cell repertoire for Th1/2/17 and Treg differentiation in the periphery in response to the microbiota may provide a mechanistic explanation for the distinct microbiome observed in individuals carrying at-risk HLA haplotypes. Furthermore, how this process regulates T cell response toward commensals carrying mimics of diabetogenic epitopes would be insightful as well.
Author contributions
YL and SM contributed equally to this work. YL, SM, and MB wrote and edited the article. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the National Institute of Diabetes and Digestive and Kidney Diseases (grant R01-DK114456) and National Institute of Allergy and Infectious Diseases (R01-AI136963).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Katsarou A, Gudbjörnsdottir S, Rawshani A, Dabelea D, Bonifacio E, Anderson BJ, et al. Type 1 diabetes mellitus. Nat Rev Dis Prim (2017) 3:1–18. doi: 10.1038/nrdp.2017.16
2. Zhu M, Xu K, Chen Y, Gu Y, Zhang M, Luo F, et al. Identification of novel t1d risk loci and their association with age and islet function at diagnosis in autoantibody-positive t1d individuals: based on a two-stage genome-wide association study. Diabetes Care (2019) 42(8):1414–21. doi: 10.2337/dc18-2023
3. Pociot F, Lernmark Å. Genetic risk factors for type 1 diabetes. Lancet (2016) 387(10035):2331–9. doi: 10.1016/S0140-6736(16)30582-7
4. Dedrick S, Sundaresh B, Huang Q, Brady C, Yoo T, Cronin C, et al. The role of gut microbiota and environmental factors in type 1 diabetes pathogenesis. Front Endocrinol (Lausanne) (2020) 11:78/full(February). doi: 10.3389/fendo.2020.00078/full
5. Kaprio J, Tuomilehto J, Koskenvuo M, Romanov K, Reunanen A, Eriksson J, et al. Concordance for type 1 (insulin-dependent) and type 2 (non-insulin-dependent) diabetes mellitus in a population-based cohort of twins in Finland. Diabetologia (1992) 35(11):1060–7. doi: 10.1007/BF02221682
6. Barnett AH, Eff C, Leslie RDG, Pyke DA. Diabetes in identical twins. Diabetologia (1981) 20(2):87–93. doi: 10.1007/BF00262007
7. Redondo MJ, Yu L, Hawa M, Mackenzie T, Pyke DA, Eisenbarth GS, et al. Heterogeneity of type i diabetes: analysis of monozygotic twins in great britain and the united states. Diabetologia (2001) 44(3):354–62. doi: 10.1007/s001250051626
8. Pearson JA, Wong FS, Wen L. The importance of the non obese diabetic (NOD) mouse model in autoimmune diabetes. J Autoimmun (2016) 66:76–88. doi: 10.1016/j.jaut.2015.08.019
9. Knip M. Pathogenesis of type 1 diabetes: Implications for incidence trends. Horm Res Paediatr (2011) 76(s1):57–64. doi: 10.1159/000329169
10. Kondrashova A, Seiskari T, Ilonen J, Knip M, Hyöty H. The ‘Hygiene hypothesis’ and the sharp gradient in the incidence of autoimmune and allergic diseases between Russian karelia and Finland. APMIS (2013) 121(6):478–93. doi: 10.1111/apm.12023
11. Bach J-F. The hygiene hypothesis in autoimmunity: the role of pathogens and commensals. Nat Rev Immunol (2018) 18(2):105–20. doi: 10.1038/nri.2017.111
12. Battaglia M, Ahmed S, Anderson MS, Atkinson MA, Becker D, Bingley PJ, et al. Introducing the endotype concept to address the challenge of disease heterogeneity in type 1 diabetes. Diabetes Care (2020) 43(1):5–12. doi: 10.2337/dc19-0880
13. Stewart CJ, Ajami NJ, O’Brien JL, Hutchinson DS, Smith DP, Wong MC, et al. Temporal development of the gut microbiome in early childhood from the TEDDY study. Nature (2018) 562(7728):583–8. doi: 10.1038/s41586-018-0617-x
14. Palmer C, Bik EM, DiGiulio DB, Relman DA, Brown PO. Development of the human infant intestinal microbiota. PloS Biol (2007) 5(7):e177. doi: 10.1371/journal.pbio.0050177
15. Guittar J, Shade A, Litchman E. Trait-based community assembly and succession of the infant gut microbiome. Nat Commun (2019) 10(1):1–11. doi: 10.1038/s41467-019-08377-w
16. De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, et al. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci USA (2010) 107(33):14691–6. doi: 10.1073/pnas.1005963107
17. Vatanen T, Franzosa EA, Schwager R, Tripathi S, Arthur TD, Vehik K, et al. The human gut microbiome in early-onset type 1 diabetes from the TEDDY study. Nature (2018) 562(7728):589–94. doi: 10.1038/s41586-018-0620-2
18. De Goffau MC, Fuentes S, Van Den Bogert B, Honkanen H, De Vos WM, Welling GW, et al. Aberrant gut microbiota composition at the onset of type 1 diabetes in young children. Diabetologia (2014) 57(8):1569–77. doi: 10.1007/s00125-014-3274-0
19. Alkanani AK, Hara N, Gottlieb PA, Ir D, Robertson CE, Wagner BD, et al. Alterations in intestinal microbiota correlate with susceptibility to type 1 diabetes. Diabetes (2015) 64(10):3510–20. doi: 10.2337/db14-1847
20. Kostic AD, Gevers D, Siljander H, Vatanen T, Hyötyläinen T, Hämäläinen AM, et al. The dynamics of the human infant gut microbiome in development and in progression toward type 1 diabetes. Cell Host Microbe (2015) 17(2):260–73. doi: 10.1016/j.chom.2015.01.001
21. Murri M, Leiva I, Gomez-Zumaquero JM, Tinahones FJ, Cardona F, Soriguer F, et al. Gut microbiota in children with type 1 diabetes differs from that in healthy children: A case-control study. BMC Med (2013) 11(1):1–12. doi: 10.1186/1741-7015-11-46
22. De Goffau MC, Luopajärvi K, Knip M, Ilonen J, Ruohtula T, Härkönen T, et al. Fecal microbiota composition differs between children with b-cell autoimmunity and those without. Diabetes (2013) 62:1238–44. doi: 10.2337/db12-0526
23. Henrick BM, Rodriguez L, Lakshmikanth T, Pou C, Henckel E, Arzoomand A, et al. Bifidobacteria-mediated immune system imprinting early in life. Cell (2021) 184(15):3884–98.e11. doi: 10.1016/j.cell.2021.05.030
24. Lund-Blix NA, Sander SD, Størdal K, Nybo Andersen AM, Rønningen KS, Joner G, et al. Infant Feeding and Risk of Type 1 Diabetes in Two Large Scandinavian Birth Cohorts. Diabetes Care [Internet] (2017) 40(7):920–7.
25. Çiçekli İ, Durusoy R. Breastfeeding, nutrition and type 1 diabetes: a case-control study in Izmir, Turkey. Int Breastfeed J (2022) 17(1):1–11.
26. Virtanen SM, Kenward MG, Erkkola M, Kautiainen S, Kronberg-Kippilä C, Hakulinen T. Age at introduction of new foods and advanced beta cell autoimmunity in young children with HLA-conferred susceptibility to type 1 diabetes. Diabetologia [Internet]. (2006) 49(7):1512–21.
27. Culina S, Lalanne AI, Afonso G, Cerosaletti K, Pinto S, Sebastiani G, et al. Islet-reactive CD8 + T cell frequencies in the pancreas, but not in blood, distinguish type 1 diabetic patients from healthy donors. Sci Immunol (2018) 3(20):1–16. doi: 10.1126/sciimmunol.aao4013
28. Hebbandi Nanjundappa R, Ronchi F, Wang J, Clemente-Casares X, Yamanouchi J, Sokke Umeshappa C, et al. A gut microbial mimic that hijacks diabetogenic autoreactivity to suppress colitis. Cell (2017) 171(3):655–667.e17. doi: 10.1016/j.cell.2017.09.022
29. Girdhar K, Huang Q, Chow I-T, Brady C, Raisingani A, Autissier P, et al. A gut microbial peptide and molecular mimicry in the pathogenesis of type 1 diabetes. bioRxiv (2022) 350801. doi: 10.1101/2020.10.22.350801v2
30. Sano T, Huang W, Hall JA, Yang Y, Chen A, Gavzy SJ, et al. An il-23r/il-22 circuit regulates epithelial serum amyloid a to promote local effector th17 responses. Cell (2015) 163(2):381–93. doi: 10.1016/j.cell.2015.08.061
31. Kriegel MA, Sefik E, Hill JA, Wu HJ, Benoist C, Mathis D. Naturally transmitted segmented filamentous bacteria segregate with diabetes protection in nonobese diabetic mice. Proc Natl Acad Sci U.S.A. (2011) 108(28):11548–53. doi: 10.1073/pnas.1108924108
32. Rouland M, Beaudoin L, Rouxel O, Bertrand L, Cagninacci L, Saffarian A, et al. Gut mucosa alterations and loss of segmented filamentous bacteria in type 1 diabetes are associated with inflammation rather than hyperglycaemia. Gut (2022) 71(2):296–308. doi: 10.1136/gutjnl-2020-323664
33. Wrzosek L, Miquel S, Noordine ML, Bouet S, Chevalier-Curt MJ, Robert V, et al. Bacteroides thetaiotaomicron and faecalibacterium prausnitzii influence the production of mucus glycans and the development of goblet cells in the colonic epithelium of a gnotobiotic model rodent. BMC Biol (2013) 11(1):1–13. doi: 10.1186/1741-7007-11-61
34. De Groot PF, Belzer C, Aydin Ö, Levin E, Levels JH, Aalvink S, et al. Distinct fecal and oral microbiota composition in human type 1 diabetes, an observational study. PloS One (2017) 12(12):e0188475. doi: 10.1371/journal.pone.0188475
35. Patel N, Robert ME. Frontiers in celiac disease: where autoimmunity and environment meet. Am J Surg Pathol (2022) 46(1):E43–54. doi: 10.1097/PAS.0000000000001639
36. Hati S, Patel M, Mishra BK, Das S. Short-chain fatty acid and vitamin production potentials of lactobacillus isolated from fermented foods of khasi tribes, meghalaya, India. Ann Microbiol (2019) 69(11):1191–9. doi: 10.1007/s13213-019-01500-8
37. Lau K, Benitez P, Ardissone A, Wilson TD, Collins EL, Lorca G, et al. Inhibition of type 1 diabetes correlated to a lactobacillus johnsonii n6.2-mediated th17 bias. J Immunol (2011) 186(6):3538–46. doi: 10.4049/jimmunol.1001864
38. Valladares R, Sankar D, Li N, Williams E, Lai KK, Abdelgeliel AS, et al. Lactobacillus johnsonii n6.2 mitigates the development of type 1 diabetes in bb-dp rats. PloS One (2010) 5(5):e10507. doi: 10.1371/journal.pone.0010507
39. Fukuda S, Toh H, Hase K, Oshima K, Nakanishi Y, Yoshimura K, et al. Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nat (2011) 469(7331):543–7. doi: 10.1038/nature09646
40. Natividad JMM, Hayes CL, Motta JP, Jury J, Galipeau HJ, Philip V, et al. Differential induction of antimicrobial regiii by the intestinal microbiota and bifidobacterium breve ncc2950. Appl Environ Microbiol (2013) 79(24):7745. doi: 10.1128/AEM.02470-13
41. Engevik MA, Luk B, Chang-Graham AL, Hall A, Herrmann B, Ruan W, et al. Bifidobacterium dentium fortifies the intestinal mucus layer via autophagy and calcium signaling pathways. MBio (2019) 10(3):e01087–19. doi: 10.1128/mBio.01087-19
42. Hänninen A, Toivonen R, Pöysti S, Belzer C, Plovier H, Ouwerkerk JP, et al. Akkermansia muciniphila induces gut microbiota remodelling and controls islet autoimmunity in NOD mice. Gut (2018) 67(8):1445–53. doi: 10.1136/gutjnl-2017-314508
43. Everard A, Belzer C, Geurts L, Ouwerkerk JP, Druart C, Bindels LB, et al. Cross-talk between akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc Natl Acad Sci U.S.A. (2013) 110(22):9066–71. doi: 10.1073/pnas.1219451110
44. Hansen CHF, Krych L, Nielsen DS, Vogensen FK, Hansen LH, Sørensen SJ, et al. Early life treatment with vancomycin propagates akkermansia muciniphila and reduces diabetes incidence in the NOD mouse. Diabetologia (2012) 55(8):2285–94. doi: 10.1007/s00125-012-2564-7
45. Tai N, Peng J, Liu F, Gulden E, Hu Y, Zhang X, et al. Microbial antigen mimics activate diabetogenic CD8 T cells in NOD mice. J Exp Med (2016) 213(10):2129–46. doi: 10.1084/jem.20160526
46. Hooper LV, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science (2012) 336(6086):1268–73. doi: 10.1126/science.1223490
47. Johansson MEV, Phillipson M, Petersson J, Velcich A, Holm L, Hansson GC. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci U S A (2008) 105(39):15064–9. doi: 10.1073/pnas.0803124105
48. Paone P, Cani PD. Mucus barrier, mucins and gut microbiota: the expected slimy partners? Gut (2020) 69(12):2232–43. doi: 10.1136/gutjnl-2020-322260
49. Zhao Y, Chen F, Wu W, Sun M, Bilotta AJ, Yao S, et al. GPR43 mediates microbiota metabolite SCFA regulation of antimicrobial peptide expression in intestinal epithelial cells via activation of mTOR and STAT3. Mucosal Immunol (2018) 11(3):752–62. doi: 10.1038/mi.2017.118
50. Pabst O, Slack E. IgA and the intestinal microbiota: The importance of being specific. Mucosal Immunol (2020) 13(1):12–21. doi: 10.1038/s41385-019-0227-4
51. Burgueño JF, Abreu MT. Epithelial toll-like receptors and their role in gut homeostasis and disease. Nat Rev Gastroenterol Hepatol (2020) 17(5):263–78. doi: 10.1038/s41575-019-0261-4
52. Zmora N, Levy M, Pevsner-Fishcer M, Elinav E. Inflammasomes and intestinal inflammation. Mucosal Immunol (2017) 10(4):865–83. doi: 10.1038/mi.2017.19
53. Mønsted MØ, Falck ND, Pedersen K, Buschard K, Holm LJ, Haupt-Jorgensen M. Intestinal permeability in type 1 diabetes: An updated comprehensive overview. J Autoimmun (2021) 122:102674. doi: 10.1016/j.jaut.2021.102674
54. Bosi E, Molteni L, Radaelli MG, Folini L, Fermo I, Bazzigaluppi E, et al. Increased intestinal permeability precedes clinical onset of type 1 diabetes. Diabetologia (2006) 49(12):2824–7. doi: 10.1007/s00125-006-0465-3
55. Meddings JB, Jarand J, Urbanski SJ, Hardin J, Gall DG. Increased gastrointestinal permeability is an early lesion in the spontaneously diabetic BB rat. Am J Physiol Gastrointest Liver Physiol (1999) 276(4 39-4):G951–957. doi: 10.1152/ajpgi.1999.276.4.G951
56. Graham S, Courtois P, Malaisse WJ, Rozing J, Scott FW, Mowat AMI. Enteropathy precedes type 1 diabetes in the BB rat. Gut (2004) 53(10):1437–44. doi: 10.1136/gut.2004.042481
57. Watts T, Berti I, Sapone A, Gerarduzzi T, Not T, Zielke R, et al. Role of the intestinal tight junction modulator zonulin in the pathogenesis of type I diabetes in BB diabetic-prone rats. Proc Natl Acad Sci U.S.A. (2005) 102(8):2916–21. doi: 10.1073/pnas.0500178102
58. Sapone A, De Magistris L, Pietzak M, Clemente MG, Tripathi A, Cucca F, et al. Zonulin upregulation is associated with increased gut permeability in subjects with type 1 diabetes and their relatives. Diabetes (2006) 55(5):1443–9. doi: 10.2337/db05-1593
59. Sorini C, Cosorich I, Lo Conte M, De Giorgi L, Facciotti F, Lucianò R, et al. Loss of gut barrier integrity triggers activation of islet-reactive T cells and autoimmune diabetes. Proc Natl Acad Sci (2019) 116(30):15140–9. doi: 10.1073/pnas.1814558116
60. Valenzano MC, DiGuilio K, Mercado J, Teter M, To J, Ferraro B, et al. Remodeling of tight junctions and enhancement of barrier integrity of the caco-2 intestinal epithelial cell layer by micronutrients. PloS One (2015) 10(7):e0133926. doi: 10.1371/journal.pone.0133926
61. Gaudier E, Jarry A, Blottière HM, De Coppet P, Buisine MP, Aubert JP, et al. Butyrate specifically modulates MUC gene expression in intestinal epithelial goblet cells deprived of glucose. Am J Physiol Gastrointest Liver Physiol (2004) 287(6 50-6):1168–74. doi: 10.1152/ajpgi.00219.2004
62. Peng L, Li ZR, Green RS, Holzman IR, Lin J. Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase in caco-2 cell monolayers. J Nutr (2009) 139(9):1619–25. doi: 10.3945/jn.109.104638
63. Liang W, Enée E, Andre-Vallee C, Falcone M, Sun J, Diana J. Intestinal cathelicidin antimicrobial peptide shapes a protective neonatal gut microbiota against pancreatic autoimmunity. Gastroenterology (2022) 162(4):1288–1302.e16. doi: 10.1053/j.gastro.2021.12.272
64. Sun J, Furio L, Mecheri R, van der Does AM, Lundeberg E, Saveanu L, et al. Pancreatic β-cells limit autoimmune diabetes via an immunoregulatory antimicrobial peptide expressed under the influence of the gut microbiota. Immunity (2015) 43(2):304–17. doi: 10.1016/j.immuni.2015.07.013
65. Ménard S, Förster V, Lotz M, Gütle D, Duerr CU, Gallo RL, et al. Developmental switch of intestinal antimicrobial peptide expression. J Exp Med (2008) 205(1):183–93. doi: 10.1084/jem.20071022
66. Xia F, Cao H, Du J, Liu X, Liu Y, Xiang M. Reg3g overexpression promotes β cell regeneration and induces immune tolerance in nonobese-diabetic mouse model. J Leukoc Biol (2016) 99(6):1131–40. doi: 10.1189/jlb.3A0815-371RRR
67. Keir ME, Yi T, Lu TT, Ghilardi N. The role of IL-22 in intestinal health and disease. J Exp Med (2020) 217(3): e20192195 (1–9). doi: 10.1084/jem.20192195
68. Rich SS, Concannon P, Erlich H, Julier C, Morahan G, Nerup J, et al. The type 1 diabetes genetics consortium. Ann N Y Acad Sci (2006) 1079(1):1–8. doi: 10.1196/annals.1375.001
69. Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, et al. Innate immunity and intestinal microbiota in the development of type 1 diabetes. Nature (2008) 455(7216):1109–13. doi: 10.1038/nature07336
70. Gülden E, Chao C, Tai N, Pearson JA, Peng J, Majewska-Szczepanik M, et al. TRIF deficiency protects non-obese diabetic mice from type 1 diabetes by modulating the gut microbiota and dendritic cells. J Autoimmun (2018) 93(June):57–65. doi: 10.1016/j.jaut.2018.06.003
71. Burrows MP, Volchkov P, Kobayashi KS, Chervonsky AV. Microbiota regulates type 1 diabetes through toll-like receptors. Proc Natl Acad Sci (2015) 112(32):9973–7. doi: 10.1073/pnas.1508740112
72. Vatanen T, Kostic AD, D’Hennezel E, Siljander H, Franzosa EA, Yassour M, et al. Variation in microbiome LPS immunogenicity contributes to autoimmunity in humans. Cell (2016) 165(4):842–53. doi: 10.1016/j.cell.2016.04.007
73. Ae Kang E, Han K, Chun J, Soh H, Park S, Im JP, et al. Increased Risk of Diabetes in Inflammatory Bowel Disease Patients: A Nationwide Population-Based Study in Korea. J Clin Med (2019) 8(3):343.
74. Valle A, Giamporcaro GM, Scavini M, Stabilini A, Grogan P, Bianconi E, et al. Reduction of circulating neutrophils precedes and accompanies type 1 diabetes. Diabetes (2013) 62(6):2072–7. doi: 10.2337/db12-1345
75. Harsunen M, Puff R, D’Orlando O, Giannopoulou E, Lachmann L, Beyerlein A, et al. Reduced blood leukocyte and neutrophil numbers in the pathogenesis of type 1 diabetes. Horm Metab Res (2013) 45(06):467–70. doi: 10.1055/s-0032-1331226
76. Huang J, Xiao Y, Zheng P, Zhou W, Wang Y, Huang G, et al. Distinct neutrophil counts and functions in newly diagnosed type 1 diabetes, latent autoimmune diabetes in adults, and type 2 diabetes. Diabetes Metab Res Rev (2019) 35(1):e3064. doi: 10.1002/dmrr.3064
77. Planas R, Carrillo J, Sanchez A, Ruiz de Villa MC, Nuñez F, Verdaguer J, et al. Gene expression profiles for the human pancreas and purified islets in Type 1 diabetes: new findings at clinical onset and in long-standing diabetes. Clin Exp Immunol (2009) 159(1):23–44.
78. Wang Y, Xiao Y, Zhong L, Ye D, Zhang J, Tu Y, et al. Increased neutrophil elastase and proteinase 3 and augmented netosis are closely associated with β-cell autoimmunity in patients with type 1 diabetes. Diabetes (2014) 63(12):4239–48. doi: 10.2337/db14-0480
79. Vecchio F, Lo Buono N, Stabilini A, Nigi L, Dufort MJ, Geyer S, et al. Abnormal neutrophil signature in the blood and pancreas of presymptomatic and symptomatic type 1 diabetes. JCI Insight (2018) 3(18):1–17. doi: 10.1172/jci.insight.122146
80. Qin J, Fu S, Speake C, Greenbaum CJ, Odegard JM. NETosis-associated serum biomarkers are reduced in type 1 diabetes in association with neutrophil count. Clin Exp Immunol (2016) 184(3):318–22. doi: 10.1111/cei.12783
81. You Q, He DM, Shu GF, Cao B, Xia YQ, Xing Y, et al. Increased formation of neutrophil extracellular traps is associated with gut leakage in patients with type 1 but not type 2 diabetes. J Diabetes (2019) 11(8):665–73. doi: 10.1111/1753-0407.12892
82. Garciafigueroa Y, Phillips BE, Engman C, Trucco M, Giannoukakis N. Neutrophil-associated inflammatory changes in the pre-diabetic pancreas of early-age nod mice. Front Endocrinol (Lausanne) (2021) 12:565981/full(March). doi: 10.3389/fendo.2021.565981/full
83. You Q, Shen Y, Wu Y, Li Y, Liu C, Huang F, et al. Neutrophil extracellular traps caused by gut leakage trigger the autoimmune response in nonobese diabetic mice. Front Immunol (2022) 12:711423/full(January). doi: 10.3389/fimmu.2021.711423/full
84. Popp SK, Vecchio F, Brown DJ, Fukuda R, Suzuki Y, Takeda Y, et al. Circulating platelet-neutrophil aggregates characterize the development of type 1 diabetes in humans and NOD mice. JCI Insight (2022) 7(2):1–16. doi: 10.1172/jci.insight.153993
85. Shen Y, You Q, Wu Y, Wu J. Inhibition of PAD4-mediated NET formation by cl-amidine prevents diabetes development in nonobese diabetic mice. Eur J Pharmacol (2022) 916(November 2021):174623. doi: 10.1016/j.ejphar.2021.174623
86. Parackova Z, Zentsova I, Vrabcova P, Klocperk A, Sumnik Z, Pruhova S, et al. Neutrophil extracellular trap induced dendritic cell activation leads to th1 polarization in type 1 diabetes. Front Immunol (2020) 11(April):1–9. doi: 10.3389/fimmu.2020.00661
87. Lee AS, Gibson DL, Zhang Y, Sham HP, Vallance BA, Dutz JP. Gut barrier disruption by an enteric bacterial pathogen accelerates insulitis in NOD mice. Diabetologia (2010) 53(4):741–8. doi: 10.1007/s00125-009-1626-y
88. Jia L, Li J, Zhang M, Liu H, Ren Z, Dong XL, et al. Cathelicidin-related antimicrobial peptide protects against enteric pathogen-accelerated type 1 diabetes in mice. Theranostics (2022) 12(7):3438–55. doi: 10.7150/thno.61433
89. Gagnerault M-C, Luan JJ, Lotton C, Lepault F. Pancreatic lymph nodes are required for priming of β cell reactive t cells in nod mice. J Exp Med (2002) 196(3):369–77. doi: 10.1084/jem.20011353
90. Jaakkola I, Jalkanen S, Hänninen A. Diabetogenic t cells are primed both in pancreatic and gut-associated lymph nodes in nod mice. Eur J Immunol (2003) 33(12):3255–64. doi: 10.1002/eji.200324405
91. Hanninen A, Salmi M, Simell O, Jalkanen S. Mucosa-associated (beta 7-integrinhigh) lymphocytes accumulate early in the pancreas of NOD mice and show aberrant recirculation behavior. Diabetes (1996) 45(9):1173–80. doi: 10.2337/diabetes.45.9.1173
92. Hänninen A, Jaakkola I, Jalkanen S. Mucosal addressin is required for the development of diabetes in nonobese diabetic mice. J Immunol (1998) 160(12):6018–25.
93. Turley SJ, Lee J-W, Dutton-Swain N, Mathis D, Benoist C. Endocrine self and gut non-self intersect in the pancreatic lymph nodes. Proc Natl Acad Sci (2005) 102(49):17729–33. doi: 10.1073/pnas.0509006102
94. Costa FRC, Françozo MCS, de Oliveira GG, Ignacio A, Castoldi A, Zamboni DS, et al. Gut microbiota translocation to the pancreatic lymph nodes triggers NOD2 activation and contributes to T1D onset. J Exp Med (2016) 213(7):1223–39. doi: 10.1084/jem.20150744
95. Kamada N, Chen GY, Inohara N, Núñez G. Control of pathogens and pathobionts by the gut microbiota. Nat Immunol (2013) 14(7):685–90. doi: 10.1038/ni.2608
96. Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell (2009) 139(3):485–98. doi: 10.1016/j.cell.2009.09.033
97. Peric M, Koglin S, Kim S, Morizane S, Besch R, Prinz JC, et al. IL-17A enhances vitamin D3-induced expression of cathelicidin antimicrobial peptide in human keratinocytes. J Immunol (2008) 181(12):8504–12. doi: 10.4049/jimmunol.181.12.8504
98. Lee JS, Tato CM, Joyce-Shaikh B, Gulen MF, Gulan F, Cayatte C, et al. Interleukin-23-independent il-17 production regulates intestinal epithelial permeability. Immunity (2015) 43(4):727–38. doi: 10.1016/j.immuni.2015.09.003
99. Archer NK, Adappa ND, Palmer JN, Cohen NA, Harro JM, Lee SK, et al. Interleukin-17a (il-17a) and il-17f are critical for antimicrobial peptide production and clearance of staphylococcus aureus nasal colonization. Infect Immun (2016) 84(12):3575–83. doi: 10.1128/IAI.00596-16
100. Harbour SN, Maynard CL, Zindl CL, Schoeb TR, Weaver CT. Th17 cells give rise to Th1 cells that are required for the pathogenesis of colitis. Proc Natl Acad Sci U.S.A. (2015) 112(22):7061–6. doi: 10.1073/pnas.1415675112
101. Kuczma MP, Szurek EA, Cebula A, Chassaing B, Jung Y-J, Kang S-M, et al. Commensal epitopes drive differentiation of colonic Tregs. Sci Adv (2020) 6(16):eaaz3186. doi: 10.1126/sciadv.aaz3186
102. Cebula A, Seweryn M, Rempala GA, Pabla SS, McIndoe RA, Denning TL, et al. Thymus-derived regulatory T cells contribute to tolerance to commensal microbiota. Nature (2013) 497(7448):258–62. doi: 10.1038/nature12079
103. Geuking MB, Cahenzli J, Lawson MAE, Ng DCK, Slack E, Hapfelmeier S, et al. Intestinal bacterial colonization induces mutualistic regulatory T cell responses. Immunity (2011) 34(5):794–806. doi: 10.1016/j.immuni.2011.03.021
104. Al Nabhani Z, Dulauroy S, Marques R, Cousu C, Al Bounny S, Déjardin F, et al. A weaning reaction to microbiota is required for resistance to immunopathologies in the adult. Immunity (2019) 50(5):1276–1288.e5. doi: 10.1016/j.immuni.2019.02.014
105. Round JL, Mazmanian SK. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci USA (2010) 107(27):12204–9. doi: 10.1073/pnas.0909122107
106. Telesford KM, Yan W, Ochoa-Reparaz J, Pant A, Kircher C, Christy MA, et al. A commensal symbiotic factor derived from bacteroides fragilis promotes human CD39(+)Foxp3(+) T cells and treg function. Gut Microbes (2015) 6(4):234–42. doi: 10.1080/19490976.2015.1056973
107. Ochoa-Repáraz J, Mielcarz DW, Wang Y, Begum-Haque S, Dasgupta S, Kasper DL, et al. A polysaccharide from the human commensal bacteroides fragilis protects against CNS demyelinating disease. Mucosal Immunol (2010) 3(5):487–95. doi: 10.1038/mi.2010.29
108. Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, DeRoos P, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature (2013) 504(7480):451–5. doi: 10.1038/nature12726
109. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly-Y M, et al. The microbial metabolites, short-chain fatty acids, regulate colonic treg cell homeostasis. Science (2013) 341(6145):569–73. doi: 10.1126/science.1241165
110. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature (2013) 504(7480):446–50. doi: 10.1038/nature12721
111. Kaisar MMM, Pelgrom LR, van der Ham AJ, Yazdanbakhsh M, Everts B. Butyrate conditions human dendritic cells to prime type 1 regulatory t cells via both histone deacetylase inhibition and g protein-coupled receptor 109a signaling. Front Immunol (2017) 8:1429/full(OCT). doi: 10.3389/fimmu.2017.01429/full
112. Nastasi C, Fredholm S, Willerslev-Olsen A, Hansen M, Bonefeld CM, Geisler C, et al. Butyrate and propionate inhibit antigen-specific CD8+ T cell activation by suppressing IL-12 production by antigen-presenting cells. Sci Rep (2017) 7(1):14516. doi: 10.1038/s41598-017-15099-w
113. Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, Carbone FR. T Cell receptor antagonist peptides induce positive selection. Cell (1994) 76:17–27. doi: 10.1016/0092-8674(94)90169-4
114. Klein L, Kyewski B, Allen PM, Hogquist KA. Positive and negative selection of the T cell repertoire: what thymocytes see (and don't see). Nat Rev Immunol. (2014) 14(6):377–91. doi: 10.1038/nri3667
115. Hogquist KA, Jameson SC. The self-obsession of T cells: how TCR signaling thresholds affect fate 'decisions' and effector function. Nat Immunol. (2014) 15(9):815–23. doi: 10.1038/ni.2938
116. Takaba H, Morishita Y, Tomofuji Y, Danks L, Nitta T, Komatsu N, et al. Fezf2 orchestrates a thymic program of self-antigen expression for immune tolerance. Cell (2015) 163(4):975–87. doi: 10.1016/j.cell.2015.10.013
117. Anderson MS, Su MA. AIRE expands: new roles in immune tolerance and beyond (2016). Available at: www.nature.com/nri.
118. Hubert FX, Kinkel SA, Davey GM, Phipson B, Mueller SN, Liston A, et al. Aire regulates the transfer of antigen from mTECs to dendritic cells for induction of thymic tolerance. Blood (2011) 118(9):2462–72. doi: 10.1182/blood-2010-06-286393
119. Li J, Park J, Foss D, Goldschneider I. Thymus-homing peripheral dendritic cells constitute two of the three major subsets of dendritic cells in the steady-state thymus. J Exp Med (2009) 206(3):607–22. doi: 10.1084/jem.20082232
120. Baba T, Nakamoto Y, Mukaida N. Crucial contribution of thymic sirp + conventional dendritic cells to central tolerance against blood-borne antigens in a ccr2-dependent manner. J Immunol (2009) 183(5):3053–63. doi: 10.4049/jimmunol.0900438
121. Hadeiba H, Lahl K, Edalati A, Oderup C, Habtezion A, Pachynski R, et al. Plasmacytoid dendritic cells transport peripheral antigens to the thymus to promote central tolerance. Immunity (2012) 36(3):438–50. doi: 10.1016/j.immuni.2012.01.017
122. Mccaughtry TM, Baldwin TA, Wilken MS, Hogquist KA. Clonal deletion of thymocytes can occur in the cortex with no involvement of the medulla. J Exp Med (2008) 205(11):2575–84. doi: 10.1084/jem.20080866
123. Hogquist KA, Jameson SC, Bevan MJ. Strong agonist ligands for the T cell receptor do not mediate positive selection of functional CD8+T cells. Immunity (1995) 3(1):79–86. doi: 10.1016/1074-7613(95)90160-4
124. Klein L, Kyewski B, Allen PM, Hogquist KA. Positive and negative selection of the T cell repertoire: What thymocytes see (and don’t see). Nat Rev Immunol (2014) 14:377–91. doi: 10.1038/nri3667
125. Chentoufi AA, Polychronakos C. Insulin expression levels in the thymus modulate insulin-specific autoreactive t-cell tolerance. Diabetes (2002) 51(5):1383–90. doi: 10.2337/diabetes.51.5.1383
126. Vafiadis P, Bennett ST, Todd JA, Nadeau J, Grabs R, Goodyer CG, et al. Insulin expression in human thymus is modulated by INS VNTR alleles at the IDDM2 locus. Nat Genet (1997) 15(3):289–92. doi: 10.1038/ng0397-289
127. Sarugeri E, Dozio N, Belloni C, Meschi F, Pastore MR, Bonifacio E. Autoimmune responses to the beta cell autoantigen, insulin, and the INS VNTR-IDDM2 locus. Clin Exp Immunol. (1998) 114(3):370–6. doi: 10.1046/j.1365-2249.1998.00744.x
128. Baker RL, Rihanek M, Hohenstein AC, Nakayama M, Michels A, Gottlieb PA, et al. Hybrid insulin peptides are autoantigens in type 1 diabetes. Diabetes (2019) 68(9):1830–40. doi: 10.2337/db19-0128
129. Baker RL, Jamison BL, Wiles TA, Lindsay RS, Barbour G, Bradley B, et al. CD4 T cells reactive to hybrid insulin peptides are indicators of disease activity in the nod mouse. Diabetes (2018) 67(9):1836–46. doi: 10.2337/db18-0200
130. Scharschmidt TC, Vasquez KS, Truong H-A, Gearty SV, Pauli ML, Nosbaum A, et al. A wave of regulatory t cells into neonatal skin mediates tolerance to commensal microbes. Immunity (2015) 43(5):1011–21. doi: 10.1016/j.immuni.2015.10.016
131. Zegarra-Ruiz DF, Kim DV, Norwood K, Kim M, Wu W-JH, Saldana-Morales FB, et al. Thymic development of gut-microbiota-specific T cells. Nature (2021) 594(7863):413–7. doi: 10.1038/s41586-021-03531-1
132. Ennamorati M, Vasudevan C, Clerkin K, Halvorsen S, Verma S, Ibrahim S, et al. Intestinal microbes influence development of thymic lymphocytes in early life. Proc Natl Acad Sci (2020) 117(5):2570–8. doi: 10.1073/pnas.1915047117
133. Legoux F, Bellet D, Daviaud C, El Morr Y, Darbois A, Niort K, et al. Microbial metabolites control the thymic development of mucosal-associated invariant T cells. Science (2019) 366(6464):494–9. doi: 10.1126/science.aaw2719
134. Nel I, Bertrand L, Toubal A, Lehuen A. MAIT cells, guardians of skin and mucosa? Mucosal Immunol (2021) 14(4):803–14. doi: 10.1038/s41385-021-00391-w
135. Toubal A, Nel I, Lotersztajn S, Lehuen A. Mucosal-associated invariant T cells and disease. Nat Rev Immunol (2019) 19(10):643–57. doi: 10.1038/s41577-019-0191-y
136. Rouxel O, Da silva J, Beaudoin L, Nel I, Tard C, Cagninacci L, et al. Cytotoxic and regulatory roles of mucosal-associated invariant T cells in type 1 diabetes. Nat Immunol (2017) 18(12):1321–31. doi: 10.1038/ni.3854
137. Danke NA, Koelle DM, Yee C, Beheray S, Kwok WW. Autoreactive T cells in healthy individuals. J Immunol (2004) 172(10):5967–72. doi: 10.4049/jimmunol.172.10.5967
138. Bender C, Rodriguez-Calvo T, Amirian N, Coppieters KT, von Herrath MG. The healthy exocrine pancreas contains preproinsulin-specific CD8 T cells that attack islets in type 1 diabetes. Sci Adv (2020) 6(42):5586–602. doi: 10.1126/sciadv.abc5586
139. Cebula A, Kuczma M, Szurek E, Pietrzak M, Savage N, Elhefnawy WR, et al. Dormant pathogenic CD4+ T cells are prevalent in the peripheral repertoire of healthy mice. Nat Commun (2019) 10(1):1–15. doi: 10.1038/s41467-019-12820-3
140. Kaufman DL, Erlander MG, Clare-Salzler M, Atkinson MA, Maclaren NK, Tobin AJ. Autoimmunity to two forms of glutamate decarboxylase in insulin-dependent diabetes mellitus. J Clin Invest (1992) 89(1):283–92. doi: 10.1172/JCI115573
141. Atkinson MA, Bowman MA, Campbell L, Darrow BL, Kaufman DL, Maclaren NK. Cellular immunity to a determinant common to glutamate decarboxylase and coxsackie virus in insulin-dependent diabetes. J Clin Invest (1994) 94(5):2125–9. doi: 10.1172/JCI117567
142. Hiemstra HS, Schloot NC, Van Veelen PA, Willemen SJM, Franken KLMC, Van Rood JJ, et al. Cytomegalovirus in autoimmunity: T cell crossreactivity to viral antigen and autoantigen glutamic acid decarboxylase. Proc Natl Acad Sci U S A (2001) 98(7):3988–91. doi: 10.1073/pnas.071050898
143. Honeyman MC, Stone NL, Falk BA, Nepom G, Harrison LC. Evidence for molecular mimicry between human T cell epitopes in rotavirus and pancreatic islet autoantigens. J Immunol (2010) 184(4):2204–10. doi: 10.4049/jimmunol.0900709
144. Meziane FZ, Dali-Sahi M, Dennouni-Medjati N, Boulenouar H, Kachekouche Y, Benslama Y, et al. Molecular mimicry between varicella, measles virus and Hsp60 in type 1 diabetes associated HLA-DR3/DR4 molecules. Diabetes Metab Syndr Clin Res Rev (2020) 14(6):1783–9. doi: 10.1016/j.dsx.2020.08.009
145. Wenzlau JM, Juhl K, Yu L, Moua O, Sarkar SA, Gottlieb P, et al. The cation efflux transporter ZnT8 (Slc30A8) is a major autoantigen in human type 1 diabetes. Proc Natl Acad Sci USA (2007) 104(43):17040–5. doi: 10.1073/pnas.0705894104
146. Matos J, Matos I, Calha M, Santos P, Duarte I, Cardoso Y, et al. Insights from bacteroides species in children with type 1 diabetes. Microorganisms (2021) 9(7):1436. doi: 10.3390/microorganisms9071436
147. García AR, Paterou A, Lee M, Sławiński H, Wicker LS, Todd JA, et al. Peripheral tolerance to insulin is encoded by mimicry in the microbiome. bioRxiv (2019), 881433. doi: 10.1101/2019.12.18.881433v1
148. Pöysti S, Toivonen R, Takeda A, Silojärvi S, Yatkin E, Miyasaka M, et al. Infection with the enteric pathogen C. rodentium promotes islet-specific autoimmunity by activating a lymphatic route from the gut to pancreatic lymph node. Mucosal Immunol. (2022) 15(3):471–479. doi: 10.1038/s41385-022-00490-2
149. Tang C, Gao Y, Wang L, Zhu Y, Pan Q, Zhang R, et al. Role of regulatory T cells in schistosoma-mediated protection against type 1 diabetes. Mol Cell Endocrinol (2019) 491(April):110434. doi: 10.1016/j.mce.2019.04.014
150. Peres RS, Chiuso-Minicucci F, da Rosa LC, Domingues A, Zorzella-Pezavento SFG, França TGD, et al. Previous contact with strongyloides venezuelensis contributed to prevent insulitis in MLD-STZ diabetes. Exp Parasitol (2013) 134(2):183–9. doi: 10.1016/j.exppara.2013.03.007
151. Osada Y, Yamada S, Nabeshima A, Yamagishi Y, Ishiwata K, Nakae S, et al. Heligmosomoides polygyrus infection reduces severity of type 1 diabetes induced by multiple low-dose streptozotocin in mice via STAT6- and IL-10-independent mechanisms. Exp Parasitol (2013) 135(2):388–96. doi: 10.1016/j.exppara.2013.08.003
152. Espinoza-Jiménez A, Rivera-Montoya I, Cárdenas-Arreola R, Morán L, Terrazas LI. Taenia crassiceps infection attenuates multiple low-dose streptozotocin-induced diabetes. J Biomed Biotechnol (2010) 2010:850541. doi: 10.1155/2010/850541
153. Maizels RM, McSorley HJ, Smyth DJ. Helminths in the hygiene hypothesis: sooner or later? Clin Exp Immunol (2014) 177(1):38–46. doi: 10.1111/cei.12353
154. White MPJ, Smyth DJ, Cook L, Ziegler SF, Levings MK, Maizels RM. The parasite cytokine mimic hp-TGM potently replicates the regulatory effects of TGF-β on murine CD4+ T cells. Immunol Cell Biol (2021) 99(8):848–64. doi: 10.1111/imcb.12479
155. Johnston CJC, Smyth DJ, Kodali RB, White MPJ, Harcus Y, Filbey KJ, et al. A structurally distinct TGF-β mimic from an intestinal helminth parasite potently induces regulatory T cells. Nat Commun (2017) 8(1):1741. doi: 10.1038/s41467-017-01886-6
156. Cook L, Reid KT, Häkkinen E, de Bie B, Tanaka S, Smyth DJ, et al. Induction of stable human FOXP3+ tregs by a parasite-derived TGF-β mimic. Immunol Cell Biol (2021) 99(8):833–47. doi: 10.1111/imcb.12475
157. Zaccone P, Burton O, Miller N, Jones FM, Dunne DW, Cooke A. Schistosoma mansoni egg antigens induce treg that participate in diabetes prevention in NOD mice. Eur J Immunol (2009) 39(4):1098–107. doi: 10.1002/eji.200838871
158. Osada Y, Fujiyama T, Kamimura N, Kaji T, Nakae S, Sudo K, et al. Dual genetic absence of STAT6 and IL-10 does not abrogate anti-hyperglycemic effects of schistosoma mansoni in streptozotocin-treated diabetic mice. Exp Parasitol (2017) 177:1–12. doi: 10.1016/j.exppara.2017.03.008
159. Everts B, Hussaarts L, Driessen NN, Meevissen MHJ, Schramm G, van der Ham AJ, et al. Schistosome-derived omega-1 drives Th2 polarization by suppressing protein synthesis following internalization by the mannose receptor. J Exp Med (2012) 209(10):1753–67. doi: 10.1084/jem.20111381
160. Baumgart M, Tompkins F, Leng J, Hesse M. Naturally occurring cd4 + foxp3 + regulatory t cells are an essential, il-10-independent part of the immunoregulatory network in schistosoma mansoni egg-induced inflammation. J Immunol (2006) 176(9):5374–87. doi: 10.4049/jimmunol.176.9.5374
161. Moreels TG, Nieuwendijk RJ, De Man JG, De Winter BY, Herman AG, Van Marck EA, et al. Concurrent infection with schistosoma mansoni attenuates inflammation induced changes in colonic morphology, cytokine levels, and smooth muscle contractility of trinitrobenzene sulphonic acid induced colitis in rats. Gut (2004) 53(1):99–107. doi: 10.1136/gut.53.1.99
162. Saunders KA, Raine T, Cooke A, Lawrence CE. Inhibition of autoimmune type 1 diabetes by gastrointestinal helminth infection. Infect Immun (2007) 75(1):397–407. doi: 10.1128/IAI.00664-06
163. Mishra PK, Patel N, Wu W, Bleich D, Gause WC. Prevention of type 1 diabetes through infection with an intestinal nematode parasite requires IL-10 in the absence of a Th2-type response. Mucosal Immunol (2013) 6(2):297–308. doi: 10.1038/mi.2012.71
164. Tormo-Badia N, Håkansson Å, Vasudevan K, Molin G, Ahrné S, Cilio CM. Antibiotic treatment of pregnant non-obese diabetic mice leads to altered gut microbiota and intestinal immunological changes in the offspring. Scand J Immunol (2014) 80(4):250–60. doi: 10.1111/sji.12205
165. Hu Y, Peng J, Tai N, Hu C, Zhang X, Wong FS, et al. Maternal antibiotic treatment protects offspring from diabetes development in nonobese diabetic mice by generation of tolerogenic apcs. J Immunol (2015) 195(9):4176–84. doi: 10.4049/jimmunol.1500884
166. Brown K, Godovannyi A, Ma C, Zhang Y, Ahmadi-Vand Z, Dai C, et al. Prolonged antibiotic treatment induces a diabetogenic intestinal microbiome that accelerates diabetes in NOD mice. ISME J (2016) 10(2):321–32. doi: 10.1038/ismej.2015.114
167. Livanos AE, Greiner TU, Vangay P, Pathmasiri W, Stewart D, McRitchie S, et al. Antibiotic-mediated gut microbiome perturbation accelerates development of type 1 diabetes in mice. Nat Microbiol (2016) 1(11):16140. doi: 10.1038/nmicrobiol.2016.140
168. Fahey JR, Lyons BL, Olekszak HL, Mourino AJ, Ratiu JJ, Racine JJ, et al. Antibiotic-associated manipulation of the gut microbiota and phenotypic restoration in NOD mice. Comp Med (2017) 67(4):335–43.
169. Zhang X-S, Li J, Krautkramer KA, Badri M, Battaglia T, Borbet TC, et al. Antibiotic-induced acceleration of type 1 diabetes alters maturation of innate intestinal immunity. Elife (2018) 7:1–37. doi: 10.7554/eLife.37816
170. Jia L, Cao M, Chen H, Zhang M, Dong X, Ren Z, et al. Butyrate ameliorates antibiotic-driven type 1 diabetes in the female offspring of nonobese diabetic mice. J Agric Food Chem (2020) 68(10):3112–20. doi: 10.1021/acs.jafc.9b07701
171. Zhang X-S, Yin YS, Wang J, Battaglia T, Krautkramer K, Li WV, et al. Maternal cecal microbiota transfer rescues early-life antibiotic-induced enhancement of type 1 diabetes in mice. Cell Host Microbe (2021) 29(8):1249–65. doi: 10.1016/j.chom.2021.06.014
172. Yu J, Xiang J-Y, Xiang H, Xie Q. Cecal butyrate (not propionate) was connected with metabolism-related chemicals of mice, based on the different effects of the two inonotus obliquus extracts on obesity and their mechanisms. ACS Omega (2020) 5(27):16690–700. doi: 10.1021/acsomega.0c01566
173. Song X, Sun X, Oh SF, Wu M, Zhang Y, Zheng W, et al. Microbial bile acid metabolites modulate gut RORγ+ regulatory T cell homeostasis. Nature (2020) 577(7790):410–5. doi: 10.1038/s41586-019-1865-0
174. Patrick C, Wang G-S, Lefebvre DE, Crookshank JA, Sonier B, Eberhard C, et al. Promotion of autoimmune diabetes by cereal diet in the presence or absence of microbes associated with gut immune activation, regulatory imbalance, and altered cathelicidin antimicrobial peptide. Diabetes (2013) 62(6):2036–47. doi: 10.2337/db12-1243
175. Yoshimura T, McLean MH, Dzutsev AK, Yao X, Chen K, Huang J, et al. The antimicrobial peptide cramp is essential for colon homeostasis by maintaining microbiota balance. J Immunol (2018) 200(6):2174–85. doi: 10.4049/jimmunol.1602073
176. Crookshank JA, Patrick C, Wang G-S, Ariana Noel J, Scott FW. Gut immune deficits in LEW.1AR1- iddm rats partially overcome by feeding a diabetes-protective diet. Immunology (2015) 145(3):417–28. doi: 10.1111/imm.12457
177. Dolpady J, Sorini C, Di Pietro C, Cosorich I, Ferrarese R, Saita D, et al. Oral probiotic vsl3 prevents autoimmune diabetes by modulating microbiota and promoting indoleamine 2,3-dioxygenase-enriched tolerogenic intestinal environment. J Diabetes Res (2016) 1–12. doi: 10.1155/2016/7569431
178. Hill M, Tanguy-Royer S, Royer P, Chauveau C, Asghar K, Tesson L, et al. IDO expands human CD4+CD25high regulatory T cells by promoting maturation of LPS-treated dendritic cells. Eur J Immunol (2007) 37(11):3054–62. doi: 10.1002/eji.200636704
179. Park M-J, Park K-S, Park H-S, Cho M-L, Hwang S-Y, Min S-Y, et al. A distinct tolerogenic subset of splenic IDO+CD11b+ dendritic cells from orally tolerized mice is responsible for induction of systemic immune tolerance and suppression of collagen-induced arthritis. Cell Immunol (2012) 278(1–2):45–54. doi: 10.1016/j.cellimm.2012.06.009
180. Duan L, Chen J, Zhang H, Yang H, Zhu P, Xiong A, et al. Interleukin-33 ameliorates experimental colitis through promoting th2/foxp3+ regulatory t-cell responses in mice. Mol Med (2012) 18(5):753–61. doi: 10.2119/molmed.2011.00428
181. Leichner TM, Satake A, Harrison VS, Tanaka Y, Archambault AS, Kim BS, et al. Skin-derived TSLP systemically expands regulatory T cells. J Autoimmun (2017) 79:39–52. doi: 10.1016/j.jaut.2017.01.003
182. Holder BS, Grant CR, Liberal R, Ma Y, Heneghan MA, Mieli-Vergani G, et al. Retinoic acid stabilizes antigen-specific regulatory T-cell function in autoimmune hepatitis type 2. J Autoimmun (2014) 53(C):26–32. doi: 10.1016/j.jaut.2014.02.001
183. Raverdeau M, Christofi M, Malara A, Wilk MM, Misiak A, Kuffova L, et al. Retinoic acid-induced autoantigen-specific type 1 regulatory T cells suppress autoimmunity. EMBO Rep (2019) 20(5):1–13. doi: 10.15252/embr.201847121
184. Murakami-Satsutani N, Ito T, Nakanishi T, Inagaki N, Tanaka A, Vien PTX, et al. IL-33 promotes the induction and maintenance of Th2 immune responses by enhancing the function of OX40 ligand. Allergol Int (2014) 63(3):443–55. doi: 10.2332/allergolint.13-OA-0672
185. Komai-Koma M, Xu D, Li Y, McKenzie ANJ, McInnes IB, Liew FY. IL-33 is a chemoattractant for human Th2 cells. Eur J Immunol (2007) 37(10):2779–86. doi: 10.1002/eji.200737547
186. Kim TK, Lee J-C, Im S-H, Lee M-S. Amelioration of autoimmune diabetes of NOD mice by immunomodulating probiotics. Front Immunol (2020) 11:1832/full(September). doi: 10.3389/fimmu.2020.01832/full
187. Zou J, Reddivari L, Shi Z, Li S, Wang Y, Bretin A, et al. Inulin fermentable fiber ameliorates type i diabetes via il22 and short-chain fatty acids in experimental models. Cmgh (2021) 12(3):983–1000. doi: 10.1016/j.jcmgh.2021.04.014
188. Vaishnava S, Yamamoto M, Severson KM, Ruhn KA, Yu X, Koren O, et al. The antibacterial lectin regiiiγ promotes the spatial segregation of microbiota and host in the intestine. Science (2011) 334(6053):255–8. doi: 10.1126/science.1209791
189. Taylor HB, Vasu C. Impact of prebiotic β-glucan treatment at juvenile age on the gut microbiota composition and the eventual type 1 diabetes onset in non-obese diabetic mice. Front Nutr (2021) 8:769341/full(November). doi: 10.3389/fnut.2021.769341/full
190. Liang Y, Wang X, He D, You Q, Zhang T, Dong W, et al. Ameliorating gut microenvironment through staphylococcal nuclease-mediated intestinal NETs degradation for prevention of type 1 diabetes in NOD mice. Life Sci (2019) 221(January):301–10. doi: 10.1016/j.lfs.2019.02.034
191. Krischer JP, Lynch KF, Schatz DA, Ilonen J, Lernmark Å, Hagopian WA, et al. The 6 year incidence of diabetes-associated autoantibodies in genetically at-risk children: the TEDDY study. Diabetologia (2015) 58(5):980–7. doi: 10.1007/s00125-015-3514-y
192. Ilonen J, Hammais A, Laine A-P, Lempainen J, Vaarala O, Veijola R, et al. Patterns of β-cell autoantibody appearance and genetic associations during the first years of life. Diabetes (2013) 62(10):3636–40. doi: 10.2337/db13-0300
193. Andeweg SP, Keşmir C, Dutilh BE. Quantifying the impact of human leukocyte antigen on the human gut microbiota. mSphere (2021) 6(4): e00476–21. doi: 10.1128/mSphere.00476-21
Keywords: mimicry, T1D (type 1 diabetes), microbiota, T-cell, Th-17, Treg cells, tolerance
Citation: Majumdar S, Lin Y and Bettini ML (2022) Host-microbiota interactions shaping T-cell response and tolerance in type 1 diabetes. Front. Immunol. 13:974178. doi: 10.3389/fimmu.2022.974178
Received: 20 June 2022; Accepted: 25 July 2022;
Published: 18 August 2022.
Edited by:
Hubert M. Tse, University of Alabama at Birmingham, United StatesReviewed by:
David Wagner, University of Colorado Anschutz Medical Campus, United StatesShannon Wallet, University of North Carolina at Chapel Hill, United States
Copyright © 2022 Majumdar, Lin and Bettini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matthew L. Bettini, TWF0dC5CZXR0aW5pQHBhdGgudXRhaC5lZHU=