 Yu Huang1,2
Yu Huang1,2 Shuyu Fang1Ting Zeng1Junjie Chen1Lu Yang1Gan Sun1Rongxin Dai1,3Yunfei An1,3Xuemei Tang1,3Ying Dou1,2
Shuyu Fang1Ting Zeng1Junjie Chen1Lu Yang1Gan Sun1Rongxin Dai1,3Yunfei An1,3Xuemei Tang1,3Ying Dou1,2 Xiaodong Zhao1*
Xiaodong Zhao1* Lina Zhou1*
Lina Zhou1*- 1National Clinical Research Center for Child Health and Disorders, Ministry of Education Key Laboratory of Child Development and Disorders, Chongqing Key Laboratory of Child Infection and Immunity, Children’s Hospital of Chongqing Medical University, Chongqing, China
- 2Department of Hematological Oncology, Children’s Hospital of Chongqing Medical University, Chongqing, China
- 3Department of Rheumatism and Immunology, Children’s Hospital of Chongqing Medical University, Chongqing, China
Background: Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome is a rare disorder of the immune regulatory system caused by forkhead box P3 (FOXP3) mutations. Abnormal numbers or functions of regulatory T (Treg) cells account for the various autoimmune symptoms. We aimed to explore the molecular genetics and phenotypic spectra of patients with atypical IPEX syndrome in China.
Methods: We analyzed the molecular, clinical and immune phenotype characteristics of five Chinese patients with FOXP3 mutations.
Results: We summarized the molecular and phenotypic features of five patients with FOXP3 mutations, including two novel mutations. Four of the five patients displayed atypical phenotypes, and one developed immune-related peripheral neuropathy. Three of the five patients showed normal frequencies of Treg cells, but the proportions of subsets of Treg cells, CD4+ T cells and B cells were out of balance.
Conclusions: Our report broadens the understanding of the clinical features of atypical IPEX syndrome. Our detailed analyses of the immunological characteristics of these patients enhance the understanding of the possible mechanisms underlying the clinical manifestations.
Introduction
Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome (OMIM #304790) is a rare monogenic primary immunodeficiency disease caused by mutations of the X-linked gene forkhead box P3 (FOXP3) (1–3). IPEX syndrome is characterized by the classical triad of early-onset intractable enteropathy, type 1 diabetes mellitus (T1DM) and chronic dermatitis. Other autoimmune symptoms can also develop, such as hypothyroidism and antibody-mediated cytopenia (4–6). IPEX patients who do not receive treatments mostly fail to thrive and die within the early years of life (7). Allogeneic hematopoietic stem cell transplantation (HSCT) and immunosuppression (IS) are currently available, proven treatments (8).
The incidence of IPEX syndrome is less than one in a million, and to date, more than seventy pathogenic mutations of FOXP3 have been described in over 200 patients (6, 7, 9–12). The correlation of FOXP3 genotype and clinical phenotype in IPEX syndrome is not straightforward because the same mutation in different individuals may result in different clinical presentations (7, 13). With the development of gene sequencing technology, an increasing number of atypical cases of IPEX syndrome have been reported, including cases with late-onset symptoms, single-organ involvement, mild disease phenotypes or rare clinical features (14, 15). Patients with such atypical symptoms are often first misdiagnosed before a correct diagnosis is made.
Here, we report the clinical and immunologic features of five patients from China with different FOXP3 mutations, including two novel mutations. Among the five patients, four presented with atypical phenotypes; one suffered from immune-related peripheral neuropathies, which have rarely been reported (16); and three had normal percentages of regulatory T (Treg) cells but significantly elevated CD25-Foxp3+CD4+ T-cell (CD25-Foxp3+) levels relative to the healthy controls.
Materials and methods
Patients
The patients enrolled in this study were suspected to have mutations in FOXP3. All patients were admitted to the Children’s Hospital of Chongqing Medical University. Hemizygous FOXP3 mutations were identified in five patients using high-throughput sequencing. Informed consent was obtained from the patients’ guardians. This study was conducted in accordance with the tenets of the Declaration of Helsinki and was approved by the ethics committee of Children’s Hospital of Chongqing Medical University.
PCR for Sanger sequencing
Genomic DNA was extracted from whole blood cells using the QIAamp DNA Mini Kit (Qiagen, Germany) according to the manufacturer’s instructions, and then polymerase chain reaction (PCR) was performed to amplify Foxp3. The primer sequences used to amplify the mutations in patient 2 (P2), P3, and P4 were F: 5′-TCCCCATTCAGAGCATTGAGC-3′ and R: 5′-AATGTGAGGTTAGGTTCCCTGC-3′. The primer sequences used to amplify the mutation in P5 were F: 5′-CCCAGAACAACCATACCCACA-3′ and R: 5′-ATCCCGTTCCTCCCCAATGT-3′. F and R represent forward and reverse, respectively. The PCR products were sequenced by Sangon Biotech (Shanghai, China).
Immunophenotyping
Immunophenotyping of lymphocyte subpopulations was performed with the following antibodies: CD3-PerCP (clone: HIT3a, BioLegend), CD4-FITC (clone: RPA-T4, BioLegend), CD8-BV510 (clone: RPA-T8, BioLegend), CD45RA-PE-Cy7 (clone: HI100, BioLegend), CD27-APC (clone: M-T271, BioLegend), TCR aβ-PE (clone: IP2b, BioLegend), TCR γδ-BV421 (clone: B1, BioLegend), CD19-APC (clone: HIB19, BioLegend), CD27-V450 (clone: M-T271, BioLegend), IgD-AF488 (clone: IA6-2, BioLegend), CD24-PE (clone: ML5, BioLegend), and CD38-PerC P (clone: HIT2, BioLegend). The samples were acquired on a FACSCanto II flow cytometer, and the data were analyzed by FlowJo. All reference values were obtained from our recent study on peripheral lymphocyte phenotyping (17).
Flow cytometric analysis of CD4+ T-cell and B-cell subsets
To analyze Treg cells, peripheral blood mononuclear cells (PBMCs) were first stained with CD4-PE-Cy7 (clone: RPA-T4, BioLegend), CD25-BV421 (clone: BC96, BioLegend), and CD45RA-FITC (clone: HI100, BD Biosciences) antibodies. Next, the cells were fixed and permeabilized using the eBioscience Intracellular Fixation and Permeabilization kit (Thermo Fisher Scientific). Finally, the cells were stained with FOXP3-PE (clone: PCH101, eBioscience/ThermoFisher Scientific) and CD152-APC (clone: BNI3, BD Biosciences) antibodies.
Circulating follicular helper T (cTfh) cells, circulating follicular regulatory T (cTfr) cells, subsets of T helper (Th) cells and cTfh cells were quantified in 50 μL whole blood in three separate experiments. Whole blood cells were stained with CD3-PerCP (clone: HIT3a, BioLegend), CD4-PE-Cy7 (clone: RPA-T4, BioLegend), CD45RO-APC (clone: UCHL1, BD Biosciences), CD45RA-FITC (clone: HI100, BD Biosciences), CXCR5-BV421 (clone: J25ID4, BioLegend), CD25-APC (clone: MT271, BioLegend), CD127-PE (clone: A019D5, BioLegend), CXCR3-APC (clone: 1C6, BD Biosciences), and CCR6-PE (clone: G034E3, BioLegend) antibodies for 30 min at 4°C.
For characterization of circulating B-cell subsets, PBMCs were stained with CD19-PerCP-Cy5.5 (clone: SJ25C1, BioLegend), CD27-PE-Cy7 (clone: MT271, BioLegend), and IgM-APC (clone: G20-127, BD Biosciences) antibodies. The samples were acquired on a FACSCanto II flow cytometer, and the data were analyzed by FlowJo.
Conservation and pathogenicity analysis of mutated amino acids
Conservation analysis of amino acid sequences was performed by Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/). Pathogenicity analysis of mutated amino acids was performed by MutationTaster (http://www.mutationtaster.org/), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2), Provean (http://provean.jcvi.org/index.php), SIFT (http://sift.jcvi.org), and Fathmm (http://fathmm.biocompute.org.uk).
Structural analysis of Foxp3
The crystal structure of Foxp3 (PDB: 4wk8), which was determined by X-ray diffraction at a resolution of 3.40 Å, was used as the template. The structural impact of mutations was analyzed by PyMOL. The mutant residues and certain nearby residues are depicted. For a clear demonstration of the interresidue relationship, some residues are highlighted in specific colors with the computed labeled hydrogen bonds.
Statistical analysis
Data were analyzed using the unpaired t test. All statistical analyses were conducted in GraphPad Prism 8 software (GraphPad Software, Inc., San Diego, CA). P ≤ 0.05 was considered to indicate a significant difference.
Results
Clinical characteristics
Five FOXP3 mutations were identified in five patients by next-generation gene sequencing and confirmed by Sanger sequencing (Figure 1A). One (the mutation in P1) of the five mutations was located in the proline rich N-terminal (PRR) domain of FOXP3, three (the mutations in P2, P3, and P4) were located in the leucine zipper (LZ) domain, and one (the mutation in P5) was found in the C-terminal FKH domain (Figure 1B). The patients were from five different families, and all of them were male. In all kindreds, the mothers of the patients were carriers. In particular, P4’s mother had two miscarriages (IV 1, 2) for unknown reasons. In kindred 5, II10 died of diarrhea (Figure 1C).
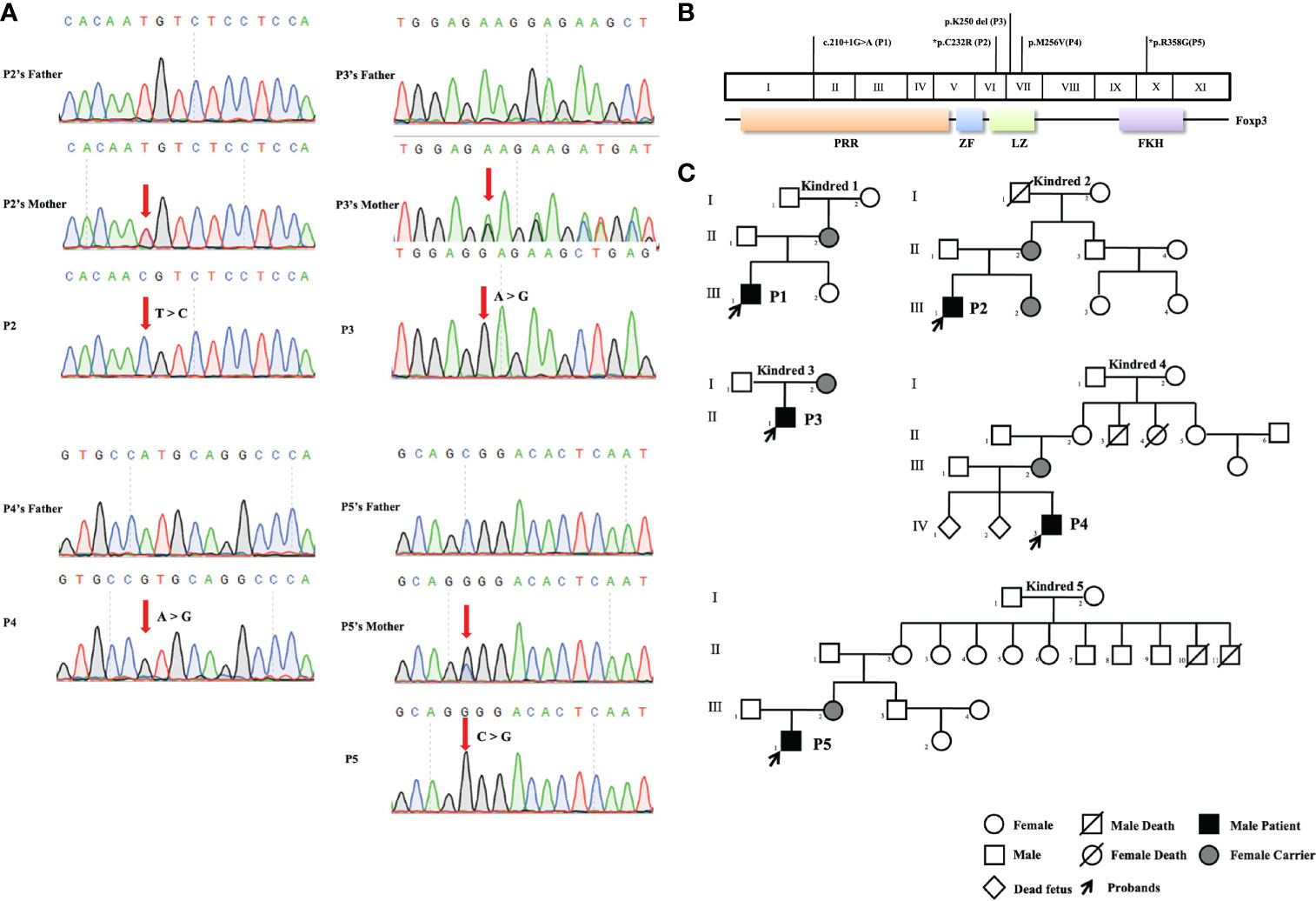
Figure 1 Pedigrees and pathogenic FOXP3 mutations of the five patients. (A) Sanger sequencing confirmed the hemizygous FOXP3 mutations of the patients. The red arrows indicate the locations of mutations. (B) The FOXP3 coding region is shown, with the pathogenic mutations carried by the five patients in this study. *Refers to novel mutations. The other mutations have been previously reported. (C) Pedigrees of the five families with FOXP3 mutations. Each family is designated by a number (1 to 5). Black indicates patients, and the probands are indicated by arrows.
There were two novel mutations: the mutation in P2 (c.694T>C, p.C232R) and the mutation in P5 (c.1072C>G, p.R358G). For the novel mutations, we performed pathogenicity analysis. The two mutated amino acids were highly conserved on multiple sequence alignment (Figure 2A) and predicted to cause disease by several algorithms (MutationTaster, PolyPhen-2, Provean, SIFT, and Fathmm) (Figure 2B). Structural analysis of the p.R358G mutant showed that the replacement of hydrophilic arginine with hydrophobic glycine disrupted the formation of hydrogen bonds with glutamate. As a result, the mutation may destabilize the protein structure or directly disrupt the functional site. The p.C232R mutation did not affect the hydrogen bonds between the amino acids (Figure 2B).
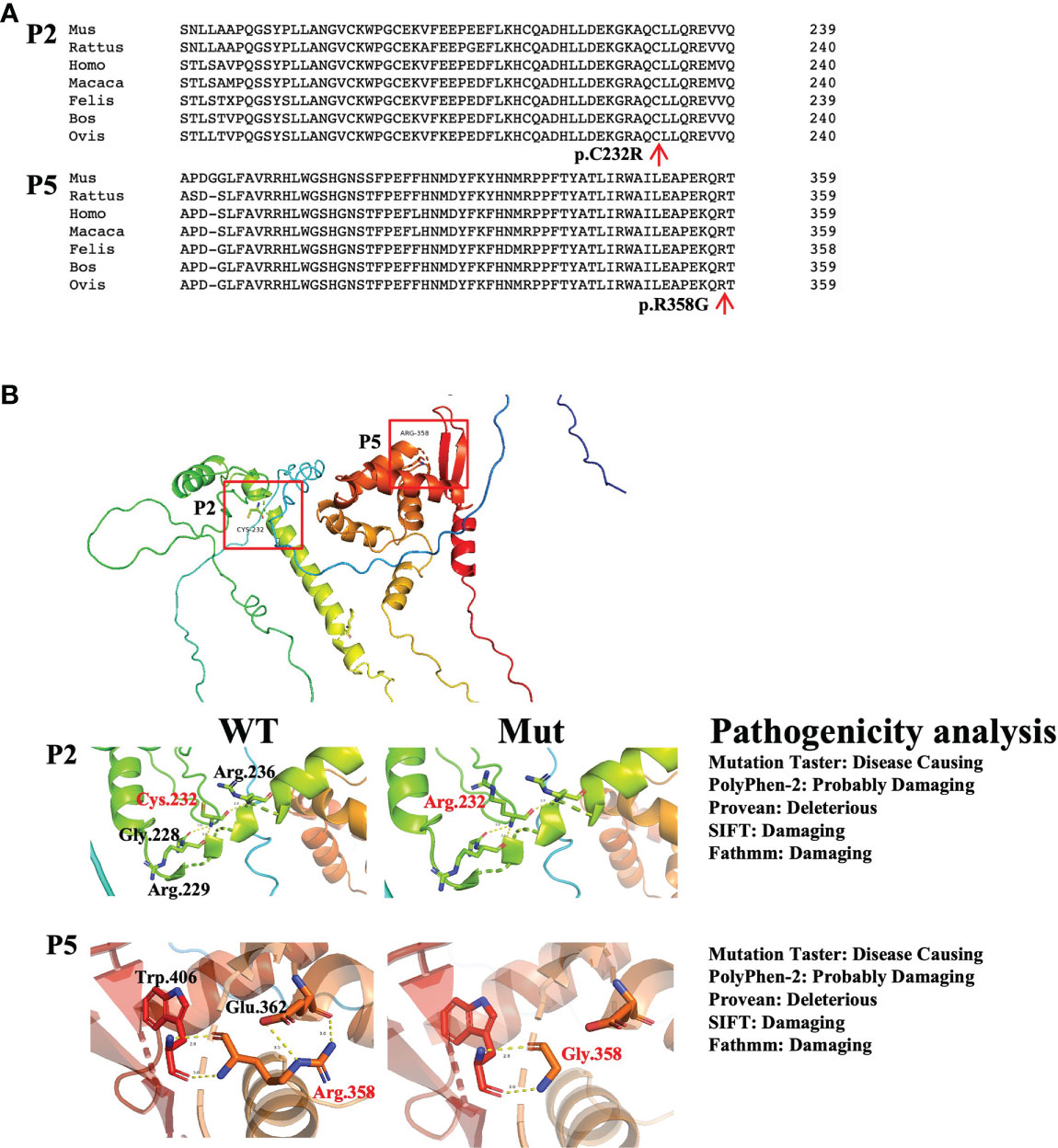
Figure 2 Pathogenicity analysis of the novel mutations. (A) Conserved analysis of amino acid sequences in different species by Clustal Omega. The red arrows refer to mutated amino acid sites. (B) The left panel shows the structural impacts of the mutations on the FoxP3 structure (PDB: 4wk8), which were analyzed by PyMOL. Hydrogen bonds are shown as dotted lines. Red indicates mutated amino acids, and black indicates amino acids around the mutations. The right panel shows the results of the pathogenicity analysis of novel mutations by MutationTaster, PolyPhen-2, Provean, SIFT and Fathmm.
The clinical characteristics of the patients are summarized in Table 1. The five patients were aged from 5 y 2 m to 14 y 5 m, and of note, the age of onset of three patients (P1, P2, and P4) was after 4 y. Diabetes was the initial symptom in P2 and P4, while the remaining patients first presented with diarrhea (P1), eczema (P5) and anemia (P3). As the disease progressed, P3 developed severe and refractory diarrhea, P1 developed chronic gastritis with eosinophil infiltration, and P5 presented with chronic diarrhea. Two patients had thyroid-related complications, including chronic thyroiditis (P2) and hypothyroidism (P4). In addition, P5 was diagnosed with congenital thyroid deficiency when he was born, which may occur in utero. Dermatitis was found in all the patients except for P2. Autoimmune hemolytic anemia was found in P1 and P3; additionally, P3 had thrombocytopenia. Three patients (P1, P2, and P3) presented with lymphadenopathy, and P1 also had hepatosplenomegaly. Failure to thrive and pneumonia were common in all the patients, and P3 had a concurrent severe fungal infection.
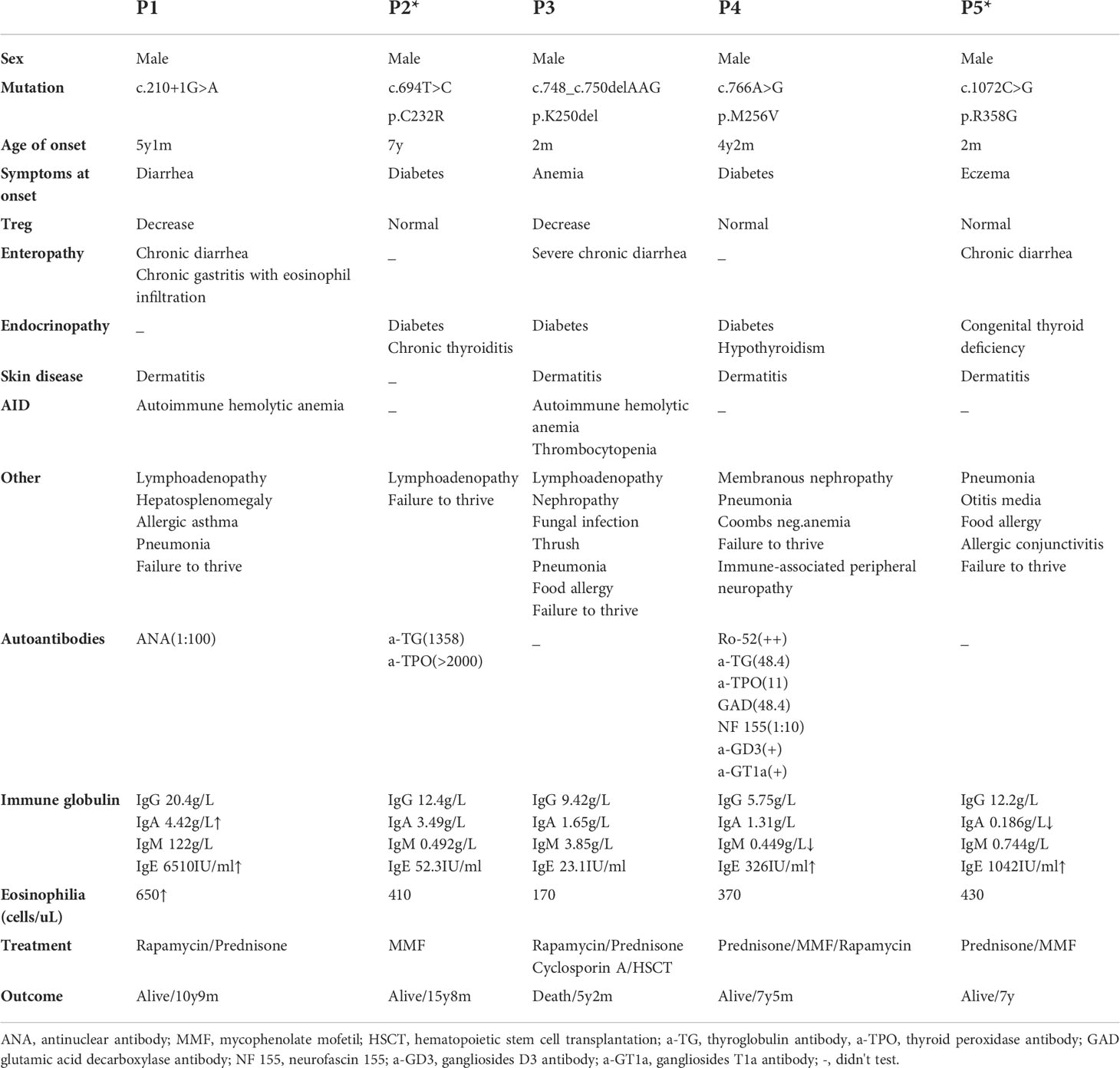
Table 1 Summary of the clinical, immunologic, and molecular findings in patients with IPEX syndrome.
Surprisingly, P4 experienced gross motor skill regression at the age of 6 y 9 m. Neurofascin 155, anti-ganglioside D3 antibody and anti-ganglioside T1a antibody were detected in his serum. No obvious abnormalities were found through either electroencephalogram or cranial MRI. These examinations indicated dysfunction of motor and sensory conduction. Thus, P4 was diagnosed with immune-associated peripheral neuropathy at the age of 7 y.
Immunosuppressors such as rapamycin and mycophenolate mofetil were used in all of the patients, and four of the five patients (except P2) received prednisone treatment. P3 received cyclosporin A and hematopoietic stem cell transplantation but died of severe bowel rejection, while the remaining patients’ symptoms were controllable to a certain degree.
Laboratory findings
Some laboratory findings are presented in Table 1. Immunoglobulin analyses revealed that P5 had decreased IgA levels, and he developed relatively severe respiratory infections. Three of the five patients (P1, P4, and P5) showed elevated IgE levels, and P1 also had eosinophilia. Different autoimmune antibodies were found in P1, P2 and P4. P2 and P4 had autoantibodies affecting the thyroid, and related clinical symptoms were also present. Some autoimmune peripheral neuropathy-related antibodies were detected in P4, as described above.
Evaluation of Treg-cell subsets
Flow cytometric analysis revealed that three patients (P2, P4, and P5) showed normal frequencies of Treg cells (Figure 3A), but higher (P<0.01) percentages of CD25-Foxp3+ cells were observed relative to the age-matched normal controls (Figure 3B).
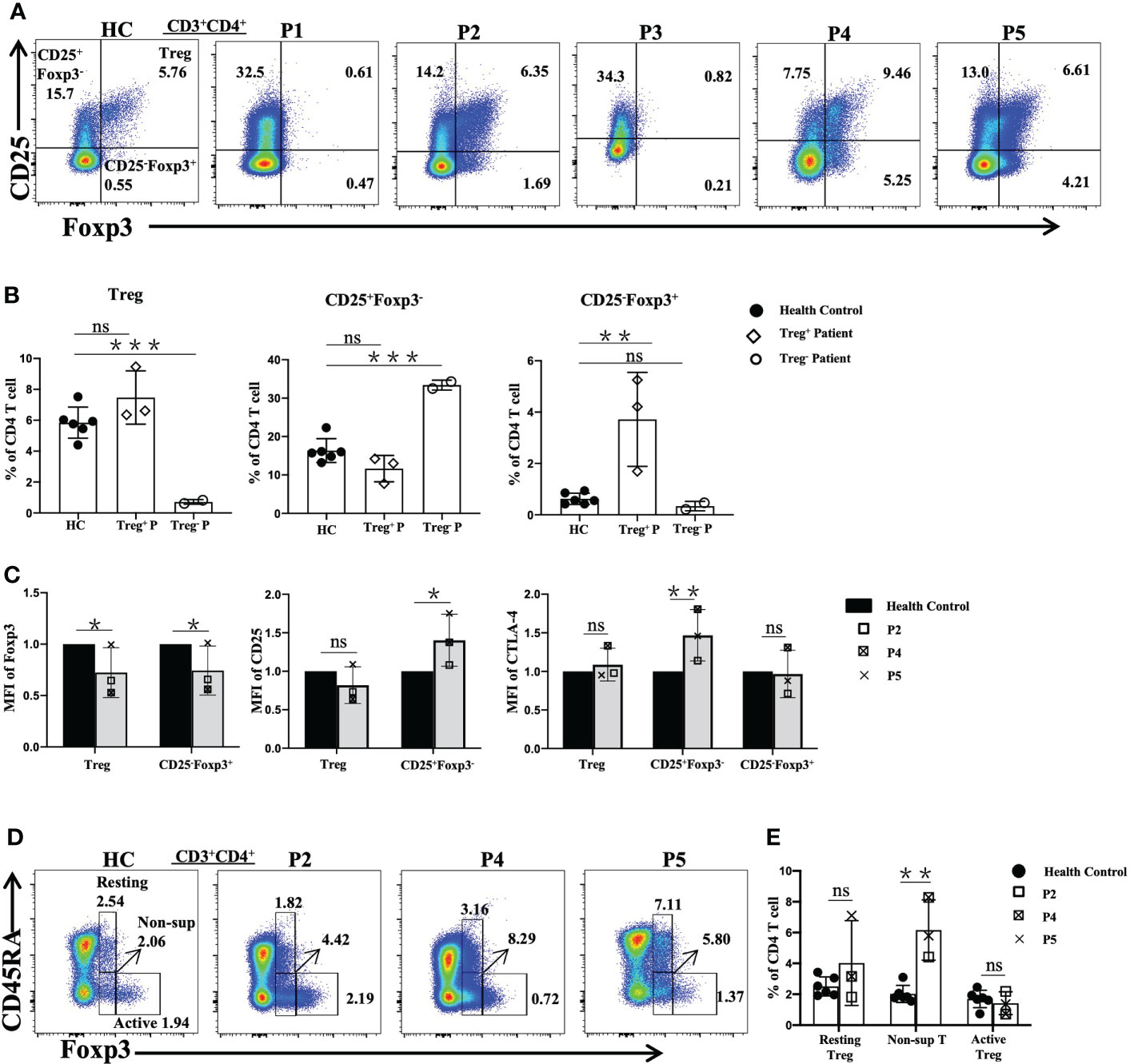
Figure 3 Flow cytometric analysis of Treg-cell subsets in patients with FOXP3 mutations. (A) and (B) The percentages of Treg (CD4+CD25+Foxp3+), CD25+Foxp3-CD4+ T and CD25+Foxp3-CD4+ T cells among CD4+ T cells. (C) The mean fluorescence index of Foxp3, CD25 and CTLA-4 in different cells. (D) and (E) The percentages of Treg-cell subsets, namely, resting Treg (CD45RA+Foxp3+), non-suppressive T (CD45RA-Foxp3+) and active Treg (CD45RA-Foxp3++) cells among CD4+ T cells. Treg+ Patient, patient with normal numbers of Treg cells; Treg- Patient, patient without Treg cells. *P<0.05; **P<0.01; ***P<0.001; ns, no significance.
Furthermore, the mean fluorescence intensity (MFI) values of FOXP3, CD25 and cytotoxic T lymphocyte-associated protein 4 (CTLA-4) in Treg, CD25+Foxp3-CD4+T (CD25+Foxp3-) and CD25-Foxp3+ cells were assessed by flow cytometry in the patients with normal Treg cells and the normal controls (Figure 3C). Compared with the values in the normal controls, the FOXP3 MFI values of Treg cells and CD25-Foxp3+ cells were lower, while the CD25 MFI value was higher (P<0.05) in the CD25+Foxp3- cells of the patients, but there was no difference in Treg cells. The MFI value of surface CTLA-4 of Treg and CD25-Foxp3+ cells of the patients was similar to that of the normal controls, while the MFI value of CTLA-4 of CD25+Foxp3- cells was higher in the patients than in the healthy controls (P<0.01).
Treg cells were subdivided into three subpopulations based on the expression of Foxp3 and CD45RA, including resting Treg cells (CD45RA+Foxp3low), active Treg cells (CD45RA-Foxp3high) and non-suppressive T cells (CD45RA-Foxp3low) (18). Compared to the normal controls, P2, P4 and P5 showed significantly increased percentages of non-suppressive T cells, while the percentages of active Treg cells of P4 and P5 were reduced. However, P2 showed relatively normal resting Treg and active Treg cell proportions (Figures 3D, E).
Skewing of peripheral T-cell subsets
Peripheral blood lymphocyte subsets were analyzed in all the patients (Table 2). When the tests were performed, all the patients were in the active phase of autoimmune disease and P3 also had pneumonia. Only two (P2 and P5) of the five patients received immunosuppressive therapy, and P1 was treated with prednisone. P2 and P4 showed slightly increased proportions of T cells, while the frequency of T cells in P3 was decreased. P5 displayed an increased percentage of CD8+ T cells and CD4+ T cells. Further analyses of T-cell subsets revealed that P2 had an increased proportion of naive CD8+ T cells. However, the percentages and numbers of central memory CD8+ T cells, effector memory CD8+ T cells, effector memory CD4+ T cells, TCRαβ+ double-negative T cells and natural killer (NK) cells in P2 were decreased. The proportions of central memory CD4+ T cells and TCRαβ+ double-negative T cells were higher in P3 than in the healthy controls. However, the ratios of CD4 naive T cells and γδ T cells in P3 were decreased. The percentage and number of NK cells in P5 were slightly higher than those in the normal controls.
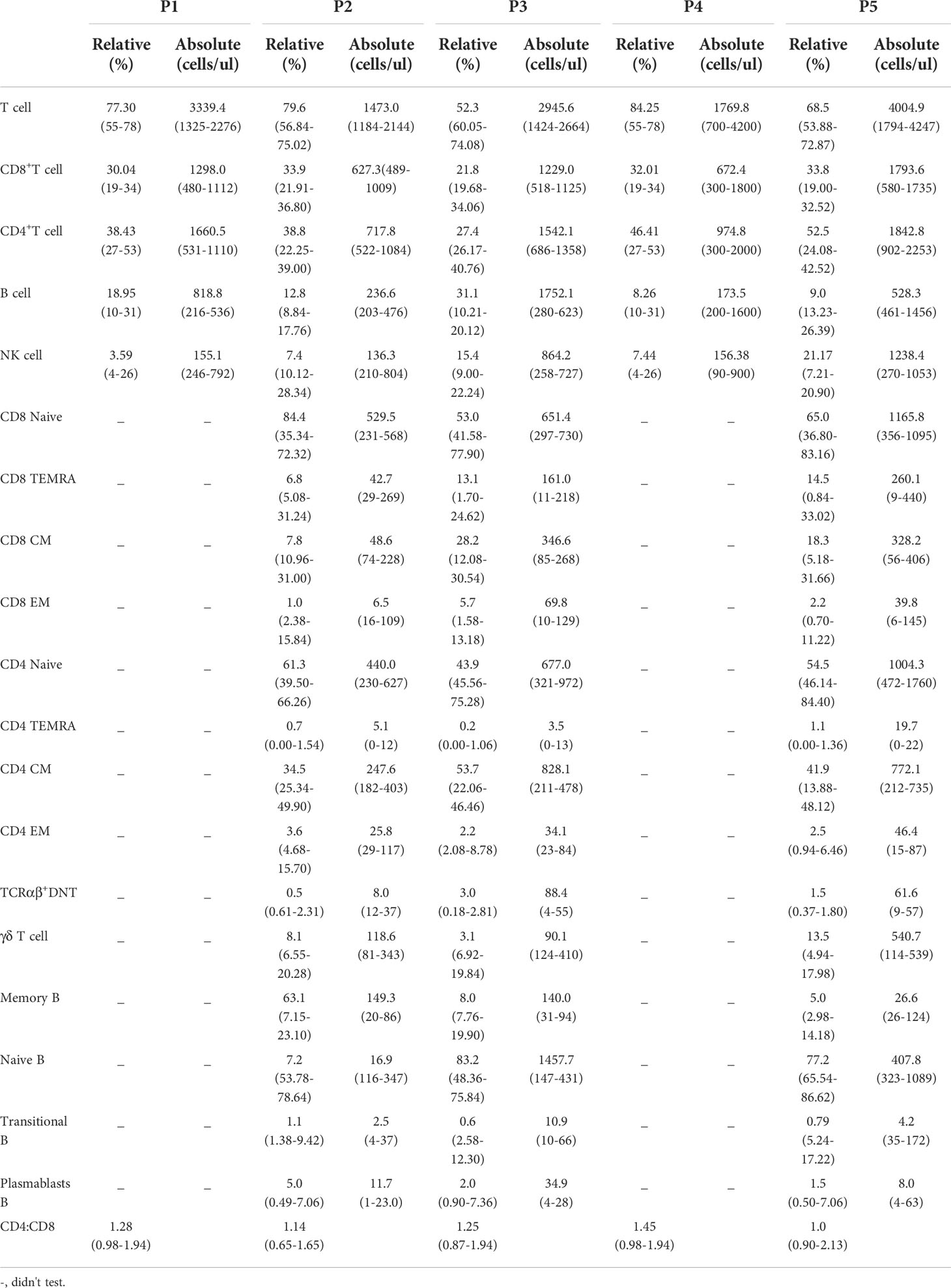
Table 2 Lymphocyte classification of patients with IPEX syndrome.
In particular, we analyzed different subsets of CD4+ T cells in P2, P3 and P5. We observed that the proportions of cTfh cells in P3 and P5 were increased (P<0.01) (Figures 4A, B) and that the frequency of cTfr cells in P2 was higher than that in the healthy controls (Figures 4C, D). The ratio of cTfh cells to cTfr cells in P5 was higher than that in healthy controls (Figure 4E). Further analysis of cTfh-cell subsets showed that the levels of Th17-like cTfh and Th1/17-like cTfh cells in P3 were increased. However, the levels of Th1-like cTfh cells in P2, P3 and P5 were reduced (P<0.05). The level of Th2-like cTfh cells was also reduced in P3 but was increased in P2 (Figures 4F, G). When we analyzed CD4+ T subsets according to cytokine secretion, we found that P3 had higher proportions of Th17 cells and Th1/17 cells, but the ratios of Th1 and Th2 cells were decreased compared to those in the normal controls. P5 showed increased Th17-cell and reduced Th1-cell levels (Figures 4H, I).
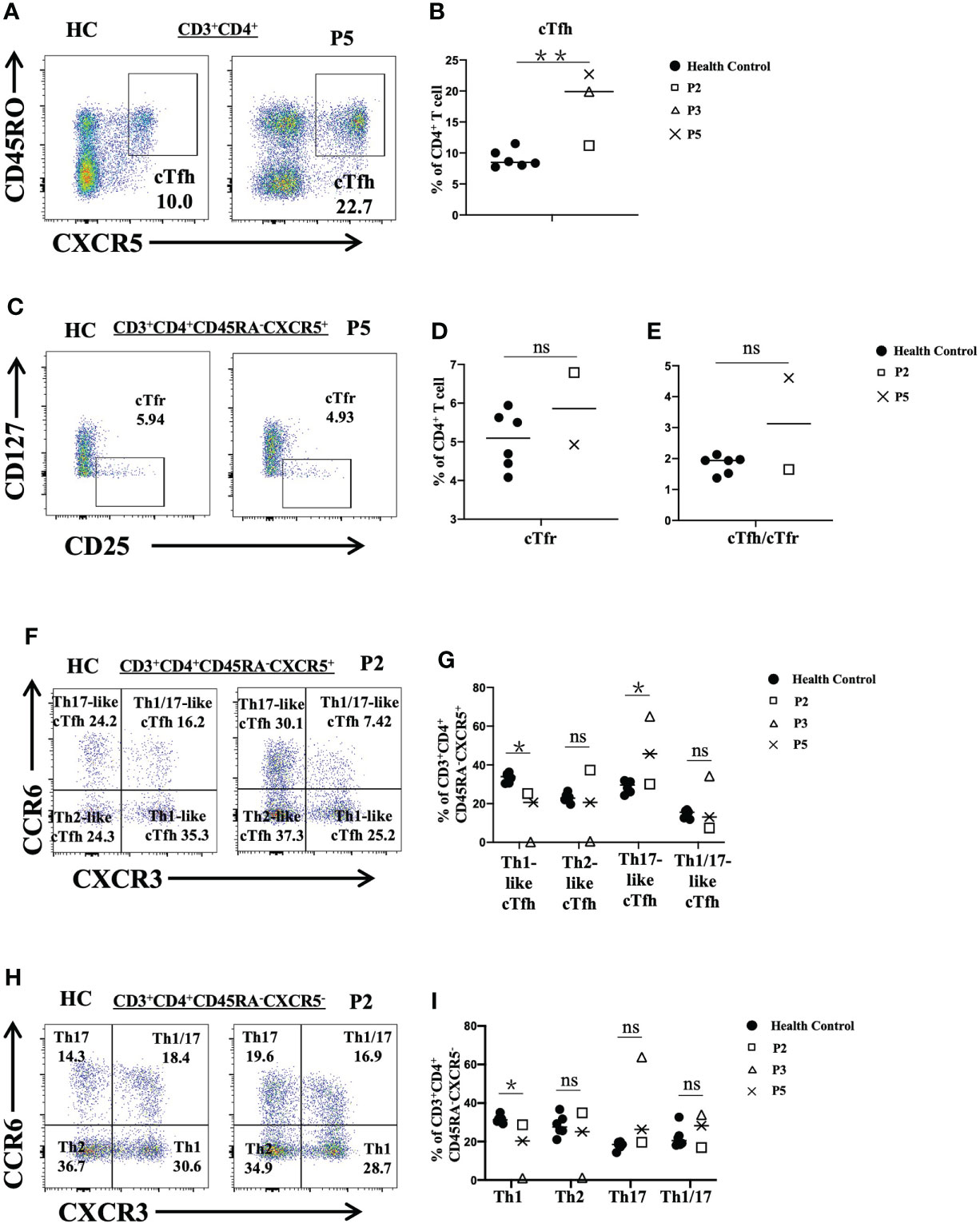
Figure 4 Flow cytometric analysis of peripheral CD4+ T-cell subsets in patients with FOXP3 deficiency. (A) and (B) The percentage of cTfh cells (CD3+CD4+CD45RA-CXCR5+) among CD4+ T cells. (C) and (D) The frequency of cTfr cells (CD3+CD4+CD127-CD25+) among CD4+ T cells. (E) The ratio of cTfh cells to cTfr cells. (F) and (G) The percentages of cTfh-cell subsets: Th1-like cTfh cells (CXCR3+CCR6-), Th2-like cTfh cells (CXCR3-CCR6-), and Th17-like cTfh cells (CXCR3-CCR6+). (H) and (I) The frequency of CD4+ T-cell (CD3+CD4+CD45RA-CXCR5-) subsets: Th1 cells (CXCR3+CCR6-), Th2 cells (CXCR3-CCR6-), and Th17 cells (CXCR3-CCR6+). *P<0.05; **P<0.01; ns, no significance.
Abnormal B-cell phenotypes
We also analyzed peripheral blood B lymphocyte subsets. The total frequencies and numbers of B cells were increased in P3, while the frequencies of B cells in P4 and P5 were decreased. Further analyses revealed that the frequencies and numbers of memory B cells in P2 and the frequencies and numbers of naive B cells in P3 were higher than the reference values. However, the levels of naive B cells in P2 and the levels of transitional B cells in P2, P3 and P5 were reduced (Table 2). Furthermore, P1, P3 and P4 had increased proportions of IgMhi B cells and decreased proportions of switched memory B (smB) cells (Figures 5A, B). P2 had decreased frequencies of IgMhi B cells and marginal zone-like B (MZ-like) cells, but the proportion of smB cells was increased (Figure 5B).
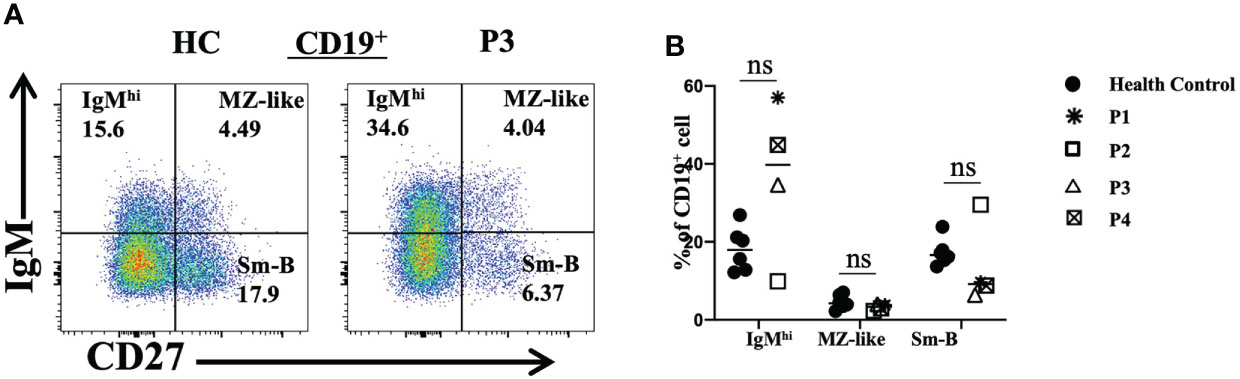
Figure 5 Flow cytometric analysis of peripheral B-cell subsets in patients with FOXP3 deficiency. (A) and (B) The percentages of IgMhi B cells (CD19+IgM+CD27-), MZ-like B cells (CD19+IgM+CD27+) and smB cells (CD19+IgM-CD27+) among B cells *P<0.05; ns, no significance.
Discussion
In this study, we described in detail the clinical and immunological features of five patients with different FOXP3 mutations. The FOXP3 gene encodes the Foxp3 protein, which is essential for the development and regulatory function of Treg cells (19, 20). Treg cells are often involved in maintaining tolerance, preventing inflammatory diseases, regulating immunity against infections, restraining tumor and anti-autoimmune responses, and so on (21).
As the clinical manifestations of IPEX patients vary, it is not surprising that four of our 5 patients (except P3) showed atypical clinical features, including unreported symptoms, late-onset of the disease, a single system affected and, unusually, the absence of severe early-onset diarrhea in our patients. Decreased numbers of Treg cells are often reported in IPEX patients, but in our study, three patients showed normal numbers of Treg cells and aberrant levels of Treg-cell subsets.
Early-onset intractable enteropathy, T1DM and chronic dermatitis are the classical triad of IPEX syndrome. However, four of our patients (except P3) only developed one of these symptoms as the main symptom and none of them had early-onset intractable diarrhea. In particular, late onset of symptoms was found in three patients (P1, P2, and P4), as most of the IPEX patients developed symptoms within 1 year of age. Three of the five mutations identified in these patients have been reported before (6, 22), but the clinical manifestations and examination results were not identical.
In particular, P4 developed immune-related peripheral neuropathy in the course of the disease, which has rarely been reported (16). P4 had the same mutation as one newly reported patient with atypical late-onset severe gastritis. The previously reported patient had autoimmune hepatitis and gastritis as his main clinical symptoms, while P4 showed severe diabetes and nephropathy (22). As the disease progressed, P4 showed gross motor skill regression and was diagnosed with immune-associated peripheral neuropathy, as related examinations indicated dysfunction of motor and sensory conduction. Neurological disease in IPEX patients has rarely been reported, except in one patient with progressive cerebellar atrophy, which is not considered to be associated with immune dysfunction (16).
The c.210+1G>A (P1) mutation has been described in detail in three patients; all of them had kidney involvement, but our patient did not. The autoimmune diseases of the reported patients included T1DM, thyroiditis and hepatitis (23, 24), but the most prominent symptom in our patient (P1) was autoimmune hemolytic anemia. Therefore, atypical clinical manifestations and different clinical manifestations caused by the same mutation may delay the diagnosis and treatment of the disease.
Two of the five mutations identified in our study had not been previously reported, and the two patients with these mutations showed atypical manifestations. The mutation in P2 was located in the LZ domain of FOXP3; disruption of the LZ domain is predicted to block transcriptional regulation of target genes and may be associated with severe autoimmune manifestations (25). As a result, P2 developed T1DM and chronic thyroiditis as the main clinical symptoms. The FKH domain interacts with many other proteins and assists in Treg transcription, but it does not affect the numbers of Treg cells (25). Hence, mutations in the FKH domain are probably associated with a milder clinical phenotype, as we found that dermatitis was the main feature of P5. Interestingly, P5 showed congenital thyroid deficiency, which has not been reported before.
The frequencies of Treg cells in three of our patients were normal, but these patients showed relatively decreased frequencies of CD25+Foxp3- cells and obviously increased frequencies of CD25-Foxp3+ cells. CD25-Foxp3+ cells lose their immunosuppressive function (26), which might explain the autoimmune manifestations of these three patients. In addition, the expression of Foxp3 in Treg and CD25-Foxp3+ cells were decreased, which can explain the loss of suppressive function and contribute to the clinical features. Treg cells usually highly expressed CD25. We found that the expression of CD25 was decreased in two of the three patients with Treg cells. As previous studies have shown, CD25 can absorb IL-2 as a cytokine sink, which deprives conventional CD4+ T cells of IL-2 and thereby contributes to the suppressive function of Treg cells. As a result, conventional CD4+ T cells are restricted from expanding and differentiating into effector T cells (6, 27). Thus, decreased expression of CD25 in mutated Treg cells may disrupt their function and differentiation.
Resting Treg cells are a stable Treg-cell phenotype, and active Treg cells have high suppressive activity. Non-suppressive T cells lack suppressive activity and can secrete inflammatory cytokines such as IL-2 and IFN-γ; thus, the majority of these cells act as activated T cells (18). In our study, P4 and P5 showed imbalances in Treg subsets. Thus, we speculate that the functions of Treg cells were disrupted in P4 and P5, although the cell numbers were in the normal range, and the increased number of non-suppressive T cells may also have contributed to the autoimmune disease the patients experienced. In addition, an increase in non-suppressive T cell numbers can lead to the production of more IL-2, which may in turn reduce the expression of CD25.
Treg cells can gain an effector phenotype concomitant with loss of Foxp3 expression. These cells may adapt to their environment and gain expression of transcription factors and chemokine receptors. Phenotypic changes in cells are normally associated with Th1, Th2, Th17, and cTfh cells (28). Previous studies have indicated that cTfh cells are associated with humoral autoimmunity and are found in several monogenic immunodeficiencies (29–31). P3 and P5 had elevated levels of cTfh cells, and they both had severe autoimmune diseases. When CD4+ T- and cTfh-cell subsets were analyzed, only P3 displayed extreme subset skewing. P3 showed elevated Th17-cell and Th17-like-cTfh cell levels. Th17 cells can secrete IL-17, which is highly associated with enteritis (32), and this may account for P3’s intractable diarrhea. cTfr cells constitute approximately 30% of non-suppressive T cells in healthy people (33). Thus, the increased proportion of cTfr cells in P2 along with elevated non-suppressive T cells might account for the loss of function of Treg cells.
A previous study revealed that some patients showed increased levels of transitional B cells and naive B cells (34); however, detailed analysis of B-cell subsets in three patients showed decreased levels of transitional B cells and inconsistent changes in naive B cells and memory B cells. cTfh cells offer critical help in B-cell class switching and affinity maturation (35), and two of the three patients showed an increased cTfh-cell frequency. Thus, we speculate that the autoimmune diseases of these patients may have influenced the cTfh-cell balance and B-cell tolerance. A more in-depth assessment of B-cell compartments may resolve these conflicting findings.
IS and HSCT are the main treatments for IPEX patients. Four of our patients received IS-inducing therapy, including MMF and rapamycin. Only P3 received HSCT for severe enteropathy, but unfortunately, he died of severe graft-versus-host-disease. P1 and P5 did not develop autoimmune disease after administration of IS-inducing treatment and prednisone. However, previous studies indicate that receipt of IS-inducing therapy or HSCT does not influence overall survival; however, less organ impairment before HSCT is related to a higher chance of survival (8). In addition, although current immunosuppressive regimens have limitations, rapamycin has been demonstrated to be the most beneficial IS-inducing therapy (36–38).
In conclusion, we described the molecular, clinical, and immune characteristics of five patients with FOXP3 mutations. We report the molecular genetics, clinical heterogeneity, and immunological abnormalities of IPEX patients, which may help us improve the diagnosis and treatment of patients with atypical manifestations. However, due to the limited number of cases, the mechanisms underlying the heterogeneity of clinical features, immune abnormalities and outcomes need to be further studied.
Data availability statement
The original contributions presented in the study are included in the article/supplementary files. Further inquiries can be directed to the corresponding authors.
Ethics statement
The studies involving human participants were reviewed and approved by Institutional Review Board of Children’s Hospital of Chongqing Medical University. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author contributions
YH, LZ, and XZ designed experiments and analyzed the data. YH wrote the first draft of the manuscript and performed the experiments. SF, TZ, JC, LY, and GS assisted with basic immunological analysis. RD, YA, XT, and YD contributed to scientific discussion, data interpretation, and revision of the manuscript. LZ and XZ supervised the research and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the General Basic Research Project from the Ministry of Education Key Laboratory of Child Development and Disorders (No. GBRP-202119).
Acknowledgments
We thank the patients and their families for participating in this study. We also thank the doctors, nurses, and other health care providers at the Children’s Hospital of Chongqing Medical University.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Wildin RS, Freitas A. Ipex and foxp3: Clinical and research perspectives. J Autoimmun (2005) 25 Suppl:56–62. doi: 10.1016/j.jaut.2005.04.008
2. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, x-linked syndrome (ipex) is caused by mutations of foxp3. Nat Genet (2001) 27(1):20–1. doi: 10.1038/83713
3. Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, et al. X-Linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet (2001) 27(1):18–20. doi: 10.1038/83707
4. Bacchetta R, Barzaghi F, Roncarolo MG. From ipex syndrome to foxp3 mutation: A lesson on immune dysregulation. Ann N Y Acad Sci (2018) 1417(1):5–22. doi: 10.1111/nyas.13011
5. Duclaux-Loras R, Charbit-Henrion F, Neven B, Nowak J, Collardeau-Frachon S, Malcus C, et al. Clinical heterogeneity of immune dysregulation, polyendocrinopathy, enteropathy, x-linked syndrome: A french multicenter retrospective study. Clin Transl Gastroenterol (2018) 9(10):201. doi: 10.1038/s41424-018-0064-x
6. Gambineri E, Ciullini Mannurita S, Hagin D, Vignoli M, Anover-Sombke S, DeBoer S, et al. Clinical, immunological, and molecular heterogeneity of 173 patients with the phenotype of immune dysregulation, polyendocrinopathy, enteropathy, x-linked (ipex) syndrome. Front Immunol (2018) 9:2411. doi: 10.3389/fimmu.2018.02411
7. Park JH, Lee KH, Jeon B, Ochs HD, Lee JS, Gee HY, et al. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (Ipex) syndrome: A systematic review. Autoimmun Rev (2020) 19(6):102526. doi: 10.1016/j.autrev.2020.102526
8. Barzaghi F, Amaya Hernandez LC, Neven B, Ricci S, Kucuk ZY, Bleesing JJ, et al. Long-term follow-up of ipex syndrome patients after different therapeutic strategies: An international multicenter retrospective study. J Allergy Clin Immunol (2018) 141(3):1036–49.e5. doi: 10.1016/j.jaci.2017.10.041
9. Alzyoud R, Alansari S, Maaitah H, AlDossari H, Monies D, Al-Mayouf SM. Familial clustering of juvenile psoriatic arthritis associated with a hemizygous Foxp3 mutation. Curr Rheumatol Rep (2021) 23(8):64. doi: 10.1007/s11926-021-01026-6
10. Rais A, Mekki N, Fedhila F, Alosaimi MF, Ben Khaled M, Zameli A, et al. Case report: Foxp3 mutation in a patient presenting with Alps. Front Immunol (2021) 12:692107. doi: 10.3389/fimmu.2021.692107
11. Steffin D, Bhar S, Fishman DS, Rider NL, Naik-Mathuria B, Martinez C, et al. Gastric adenocarcinoma in the setting of ipex syndrome. Case Rep Immunol (2021) 2021:9967198. doi: 10.1155/2021/9967198
12. Agakidis C, Agakidou E, Sarafidis K, Papoulidis I, Xinias I, Farmaki E. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome associated with a novel mutation of Foxp3 gene. Front Pediatr (2019) 7:20. doi: 10.3389/fped.2019.00020
13. Karaguzel G, Polat R, Abul MH, Cebi AH, Orhan F. Immune dysregulation, polyendocrinopathy, enteropathy, x-linked (ipex) syndrome in two siblings; same mutation but different clinical manifestations at onset. J Clin Res Pediatr Endocrinol (2021). doi: 10.4274/jcrpe.galenos.2021.2021.0005
14. Consonni F, Ciullini Mannurita S, Gambineri E. Atypical presentations of ipex: Expect the unexpected. Front Pediatr (2021) 9:643094. doi: 10.3389/fped.2021.643094
15. Ge T, Wang Y, Che Y, Xiao Y, Zhang T. Atypical late-onset immune dysregulation, polyendocrinopathy, enteropathy, x-linked syndrome with intractable diarrhea: a case report. Front Pediatr (2017) 5:267. doi: 10.3389/fped.2017.00267
16. Rim J, Byler M, Soldatos A, Notarangelo L, Leibovitch E, Jacobson S, et al. Opinion and special articles: Cerebellar ataxia and liver failure complicating ipex syndrome. Neurology (2021) 96(6):e956–e9. doi: 10.1212/wnl.0000000000011195
17. Ding Y, Zhou L, Xia Y, Wang W, Wang Y, Li L, et al. Reference values for peripheral blood lymphocyte subsets of healthy children in China. J Allergy Clin Immunol (2018) 142(3):970–3.e8. doi: 10.1016/j.jaci.2018.04.022
18. Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, et al. Functional delineation and differentiation dynamics of human cd4+ t cells expressing the foxp3 transcription factor. Immunity (2009) 30(6):899–911. doi: 10.1016/j.immuni.2009.03.019
19. Yagi H, Nomura T, Nakamura K, Yamazaki S, Kitawaki T, Hori S, et al. Crucial role of foxp3 in the development and function of human cd25+cd4+ regulatory t cells. Int Immunol (2004) 16(11):1643–56. doi: 10.1093/intimm/dxh165
20. Hori S, Nomura T, Sakaguchi S. Control of regulatory t cell development by the transcription factor foxp3. Science (2003) 299(5609):1057–61. doi: 10.1126/science.1079490
21. Sakaguchi S, Miyara M, Costantino CM, Hafler DA. Foxp3+ regulatory T cells in the human immune system. Nat Rev Immunol (2010) 10(7):490–500. doi: 10.1038/nri2785
22. Fang Y, Luo Y, Lou J, Chen J. Atypical late-onset severe gastritis in immune dysregulation, polyendocrinopathy, enteropathy, and x-linked (ipex) syndrome: 2 case reports. Med (Baltimore) (2021) 100(3):e24318. doi: 10.1097/md.0000000000024318
23. Tsuda M, Torgerson TR, Selmi C, Gambineri E, Carneiro-Sampaio M, Mannurita SC, et al. The spectrum of autoantibodies in ipex syndrome is broad and includes anti-mitochondrial autoantibodies. J Autoimmun (2010) 35(3):265–8. doi: 10.1016/j.jaut.2010.06.017
24. Bae KW, Kim BE, Choi JH, Lee JH, Park YS, Kim GH, et al. A novel mutation and unusual clinical features in a patient with immune dysregulation, polyendocrinopathy, enteropathy, x-linked (ipex) syndrome. Eur J Pediatr (2011) 170(12):1611–5. doi: 10.1007/s00431-011-1588-1
25. d’Hennezel E, Bin Dhuban K, Torgerson T, Piccirillo CA. The immunogenetics of immune dysregulation, polyendocrinopathy, enteropathy, x linked (ipex) syndrome. J Med Genet (2012) 49(5):291–302. doi: 10.1136/jmedgenet-2012-100759
26. Zohouri M, Mehdipour F, Razmkhah M, Faghih Z, Ghaderi A. Cd4(+)Cd25(-)Foxp3(+) T cells: A distinct subset or a heterogeneous population? Int Rev Immunol (2021) 40(4):307–16. doi: 10.1080/08830185.2020.1797005
27. Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ. Cd4+Cd25+Foxp3+ regulatory t cells induce cytokine deprivation-mediated apoptosis of effector cd4+ t cells. Nat Immunol (2007) 8(12):1353–62. doi: 10.1038/ni1536
28. Campbell DJ, Koch MA. Phenotypical and functional specialization of foxp3+ regulatory t cells. Nat Rev Immunol (2011) 11(2):119–30. doi: 10.1038/nri2916
29. Vinuesa CG, Linterman MA, Yu D, MacLennan IC. Follicular helper t cells. Annu Rev Immunol (2016) 34:335–68. doi: 10.1146/annurev-immunol-041015-055605
30. Ma CS, Wong N, Rao G, Avery DT, Torpy J, Hambridge T, et al. Monogenic mutations differentially affect the quantity and quality of t follicular helper cells in patients with human primary immunodeficiencies. J Allergy Clin Immunol (2015) 136(4):993–1006.e1. doi: 10.1016/j.jaci.2015.05.036
31. He J, Tsai LM, Leong YA, Hu X, Ma CS, Chevalier N, et al. Circulating precursor ccr7(lo)pd-1(hi) cxcr5+ cd4+ t cells indicate tfh cell activity and promote antibody responses upon antigen reexposure. Immunity (2013) 39(4):770–81. doi: 10.1016/j.immuni.2013.09.007
32. Yang J, Sundrud MS, Skepner J, Yamagata T. Targeting Th17 cells in autoimmune diseases. Trends Pharmacol Sci (2014) 35(10):493–500. doi: 10.1016/j.tips.2014.07.006
33. Sakaguchi S, Mikami N, Wing JB, Tanaka A, Ichiyama K, Ohkura N. Regulatory T cells and human disease. Annu Rev Immunol (2020) 38:541–66. doi: 10.1146/annurev-immunol-042718-041717
34. Shamriz O, Patel K, Marsh RA, Bleesing J, Joshi AY, Lucas L, et al. Hypogammaglobulinemia with decreased class-switched b-cells and dysregulated T-Follicular-Helper cells in ipex syndrome. Clin Immunol (2018) 197:219–23. doi: 10.1016/j.clim.2018.10.005
35. Rolf J, Bell SE, Kovesdi D, Janas ML, Soond DR, Webb LM, et al. Phosphoinositide 3-kinase activity in t cells regulates the magnitude of the germinal center reaction. J Immunol (2010) 185(7):4042–52. doi: 10.4049/jimmunol.1001730
36. Passerini L, Barzaghi F, Curto R, Sartirana C, Barera G, Tucci F, et al. Treatment with rapamycin can restore regulatory t-cell function in ipex patients. J Allergy Clin Immunol (2020) 145(4):1262–71.e13. doi: 10.1016/j.jaci.2019.11.043
37. Yong PL, Russo P, Sullivan KE. Use of sirolimus in ipex and ipex-like children. J Clin Immunol (2008) 28(5):581–7. doi: 10.1007/s10875-008-9196-1
Keywords: IPEX syndrome, primary immunodeficiency disease, FOXP3 mutations, regulatory T cells, atypical phenotypes
Citation: Huang Y, Fang S, Zeng T, Chen J, Yang L, Sun G, Dai R, An Y, Tang X, Dou Y, Zhao X and Zhou L (2022) Clinical and immunological characteristics of five patients with immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome in China–expanding the atypical phenotypes. Front. Immunol. 13:972746. doi: 10.3389/fimmu.2022.972746
Received: 19 June 2022; Accepted: 05 August 2022;
Published: 24 August 2022.
Edited by:
Rosa Bacchetta, Stanford University, United StatesCopyright © 2022 Huang, Fang, Zeng, Chen, Yang, Sun, Dai, An, Tang, Dou, Zhao and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaodong Zhao, emhhb3hkNTMwQGFsaXl1bi5jb20=; Lina Zhou, cmFjaGVsemhvdTg4QHFxLmNvbQ==