 William Donelan
William Donelan Paul R. Dominguez-Gutierrez
Paul R. Dominguez-Gutierrez Sergei Kusmartsev
Sergei Kusmartsev
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Immunol. , 27 September 2022
Sec. Cancer Immunity and Immunotherapy
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.971278
This article is part of the Research Topic Tumor Microenvironment, Inflammation, and Resistance to Immunotherapies View all 15 articles
Hyaluronan (HA) is known to be a prominent component of the extracellular matrix in tumors, and many solid cancers are characterized by aberrant HA metabolism resulting in increased production in tumor tissue. HA has been implicated in regulating a variety of cellular functions in tumor cells and tumor-associated stromal cells, suggesting that altered HA metabolism can influence tumor growth and malignancy at multiple levels. Importantly, increased HA production in cancer is associated with enhanced HA degradation due to high levels of expression and activity of hyaluronidases (Hyal). Understanding the complex molecular and cellular mechanisms involved in abnormal HA metabolism and catabolism in solid cancers could have important implications for the design of future cancer therapeutic approaches. It appears that extensive crosstalk between immune cells and HA-enriched stroma contributes to tumor growth and progression in several ways. Specifically, the interaction of tumor-recruited Hyal2-expressing myeloid-derived suppressor cells (MDSCs) of bone marrow origin with HA-producing cancer-associated fibroblasts and epithelial tumor cells results in enhanced HA degradation and accumulation of small pro-inflammatory HA fragments, which further drives cancer-related inflammation. In addition, hyaluronan-enriched stroma supports the transition of tumor-recruited Hyal2+MDSCs to the PD-L1+ tumor-associated macrophages leading to the formation of an immunosuppressive and tolerogenic tumor microenvironment. In this review, we aim to discuss the contribution of tumor-associated HA to cancer inflammation, angiogenesis, and tumor-associated immune suppression. We also highlight the recent findings related to the enhanced HA degradation in the tumor microenvironment.
Hyaluronan, also called hyaluronic acid (HA), is a member of the glycosaminoglycan family of polysaccharides synthesized at the cell surface. Distributed widely through vertebrate connective, epithelial, and neural tissues, interactions between HA and the extracellular matrix (ECM) regulate cellular processes involved in embryonic development, tissue healing, inflammation, and tumor progression (1–3). HA is synthesized as an unbranched polymer of repeating disaccharides of glucuronic acid and N-acetylglucosamine. Normal physiological HA polymers consist of 2,000-25,000 disaccharides with molecular masses in the range of 106 – 107 kDa and polymer lengths of 2-25µm. HA is synthesized at the cell membrane as an unmodified polysaccharide by one of three hyaluronan synthases (HAS): HAS1, HAS2, or HAS3.
These transmembrane enzymes initiate synthesis on the inner side of the cell membrane and extrude HA onto the outer cell surface or into the ECM. HAS2 produces the largest HA polymers and is responsible for the majority of HA synthesis. Cell surface HA binding proteins, such as CD44 and RHAMM, anchor the matrix to the cell (4, 5). The degradation of HA is catalyzed by hyaluronidase enzymes (6, 7). Hyal1 and Hyal2 are the major hyaluronidases that are expressed in most tissues and hydrolyze the linkage between N-acetylglucosamine and glucuronic acid to generate HA fragments (8). Cell surface GPI-anchored Hyal2 degrades high molecular weight HA into intermediate molecular weight fragments of 20 kDa. In a concerted effort, HA fragments are further hydrolyzed by Hyal1 in endo-lysosomal compartments generating oligosaccharides.
HA has numerous biological functions both structurally and in terms of cell signaling. HA is an important constituent of the ECM that plays a role in the lubrication of joints and maintaining connective tissue integrity (1, 3). The biophysical properties of HA such as high hydration capacity contribute to tissue homeostasis and the structural integrity of the interstitial space which is critical for cellular remodeling. The transmembrane protein CD44 is considered the principal receptor for HA (2). CD44 expression is upregulated by growth factors and pro-inflammatory cytokines such as IL-1, epidermal growth factor (EGF), and transforming growth factor-beta (TGF-β). HA-CD44 interactions regulate key cellular functions including cell-cell adhesion, cellular migration, and receptor-mediated HA internalization and degradation. RHAMM is another key HA receptor that is present at the cell surface and also has isoforms that are localized to the cytoplasm and nucleus. HA-RHAMM interactions mediate cellular migration, cytoskeleton rearrangement, and intracellular signal transduction. HA-induced signal transduction via interactions with CD44 and RHAMM is mediated through a variety of cell signaling pathways including protein kinase C, focal adhesion kinase (FAK), mitogen-activated protein kinases (MAPKs), nuclear factor NF-κB, RAS, phosphatidylinositol kinase (PI3K), tyrosine kinases, and cytoskeletal components (1, 2). Under normal homeostatic conditions, the synthesis and degradation of HA are well balanced, but a shift towards increased degradation of HA occurs during pathological conditions (2).
Multiple studies have demonstrated that cancers are associated with elevated levels of HA, and human tumors typically have higher HA concentrations than in healthy tissues (2, 3). Human breast, lung, prostate, ovarian, nephroblastomas, and colon cancer are considered to be enriched with HA (1, 2). In these tumors, HA may support tumor growth by stimulating anchorage-independent growth and proliferation of tumor cells. Elevated HA levels have been identified in the urine of patients with bladder carcinomas (9). Not surprisingly, elevated expression of hyaluronan synthases have been identified in bladder cancer tissue and HAS1 is a predictor of tumor recurrence and disease-specific survival (10, 11). In vivo studies have confirmed that highly invasive and aggressive breast cancer cell lines express significantly elevated levels of HAS2 (12, 13). Additionally, clinical studies and experiments using mouse models have further found that expression of HAS2 in breast cancer tissues is associated with metastasis and reduced overall survival (14, 15). Enhanced HA synthesis and degradation are evident in the microenvironment of several tumor types and affect tumor growth, cell motility, and metastasis (2, 3).
In experimental tumor models, the overexpression of HA synthases leads to increased HA levels and accelerated tumor growth in vivo. The level of HA expression by tumors also correlates well with metastatic progression. For example, high levels of HA are found in the tumor stroma of patients with prostate cancer and are associated with metastasis (16, 17). Supporting these tumor-promoting functions of enhanced HA synthesis, experimental evidence demonstrates that suppression of HAS, reduction of HA synthesis using inhibitors, or use of HA binding molecules have been shown to impair tumor growth and reduce metastasis in several in vitro and in vivo models (3). Abnormal activation of CD44 and RHAMM cell signaling pathways can lead to malignant behavior, and HA interactions with cell surface receptors CD44 and RHAMM promote tumor progression by affecting cell proliferation, migration, and angiogenesis (2).
HA turnover is key to understanding the role of HA metabolism in tumor progression (1–3). HA is continually turned over in normal tissues, but the degradation products are rapidly cleared. In contrast, several types of cancer, including breast, prostate, bladder, melanoma, and lung cancers, are characterized by enhanced HA degradation (2, 3 and Figure 1). It is important to understand that HA degradation in the tumor microenvironment is a complex process that involves not only the tumor but also tumor-associated cells. The tumor stroma, which consists of an extracellular matrix and many non-cancer cell types contains an abundance of degraded HA compared to adjacent normal tissue. Bladder cancer progression is associated with enhanced expression of both Hyal2 and Hyal1 (11, 18), and Hyal2 gene expression is significantly increased in patients with progressive versus non-progressive bladder cancer. Elevated hyaluronidase expression and activity in tumor tissues lead to the accumulation of small HA fragments with low molecular weight.
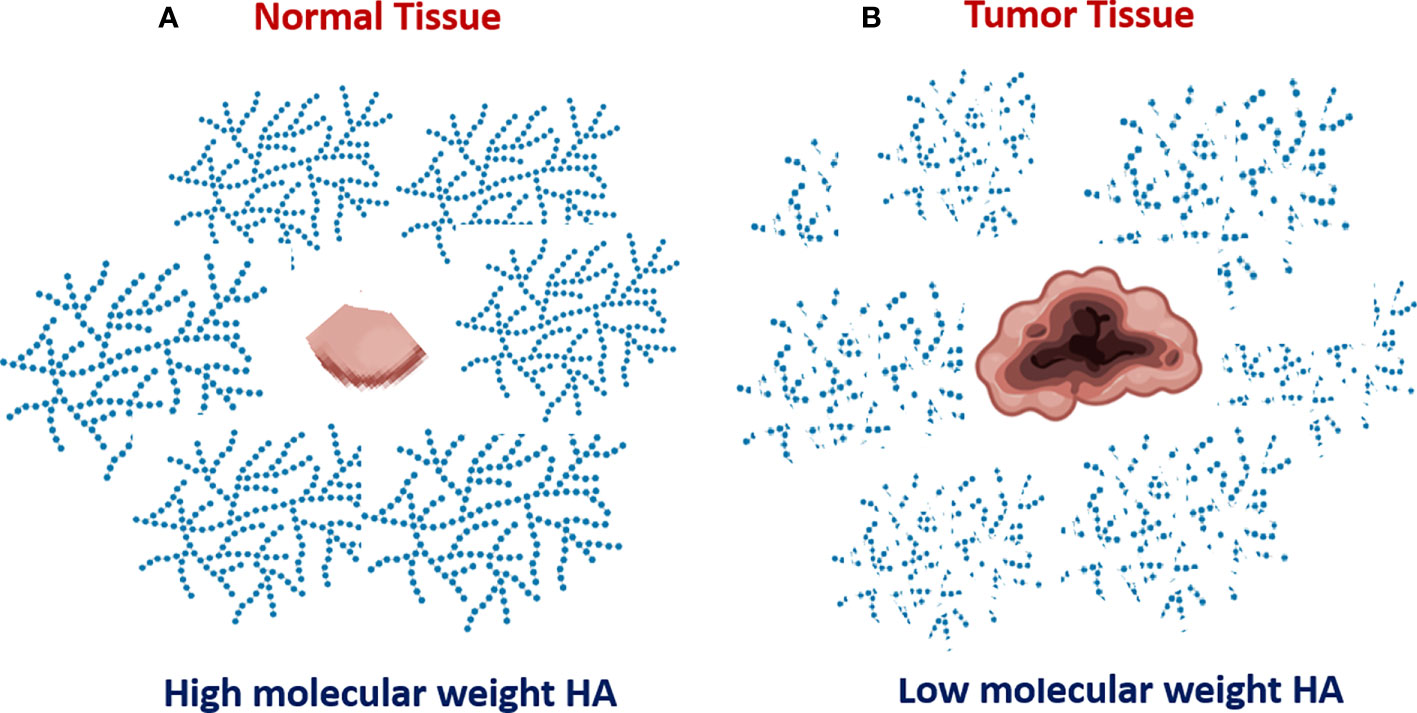
Figure 1 Increased degradation of extracellular hyaluronan in tumor tissue. Normal non-malignant bladder tissue produce HA with mostly high molecular weight (A). In contrast, the tumor bladder tissue is characterized by enhanced HA degradation and accumulation of tumor-associated HA with low molecular weight (B).
The size of HA fragments is important for biological activity: high molecular-weight hyaluronan (HMW-HA), a major component of the extracellular matrix, is anti-oncogenic, which exerts anti-inflammatory and wound-healing activities. Evidence of anti-oncogenic properties of HMW-HA comes from the fact that naked mole rats show unusual resistance to cancer and the maximal lifespan of these mice exceeds 30 years (19). Analysis of HA produced by fibroblasts from naked mole rats revealed that its molecular size is five-time higher than human or mouse HA. Naked mole-rat tissues contain larger HA polymers and less detectable fragmentation than tissues of the more tumor-susceptible mouse. Knocking down the expression of hyaluronan synthase or overexpressing the HA-degrading enzyme, Hyal2, naked mole-rat cells become susceptible to malignant transformation and readily form tumors. A recent study demonstrated that at the molecular level, HMW-HA-mediated signaling through the CD44 receptor engages the tumor-suppressive Hippo pathway (20). Thus, recruitment of the polarity-regulating kinase PAR1b by the CD44 intracellular domain results in disruption of the Hippo signaling-inhibitory PAR1b-MST complex. Once liberated from PAR1b, MST activates Hippo signaling.
In contrast, the low-molecular-weight hyaluronan (LMW-HA) is pro-oncogenic. Thus, the HA fragments with low molecular weight produced in a hyaluronidase-dependent manner, inhibited Hippo signaling by competing with HMW-HA for CD44 binding (20). Tumor growth frequently is associated with enhanced HA degradation and the accumulation of small HA fragments with low molecular weight. The LMW-HA has been shown to promote tumor growth in a multifaceted manner by stimulating cancer inflammation, angiogenesis, and spreading of metastatic tumor cells (21–26).
Cancer-related inflammation is one of the hallmarks of tumor growth (27). Increased production of various inflammatory factors (chemokines, cytokines, bioactive lipids, etc) in tumor tissue supports constant mobilization and recruitment of immunosuppressive cells, precursors of endothelial cells, and cancer-associated fibroblasts which are needed for tumor growth. Increased degradation of hyaluronan in cancer tissue leads to the accumulation of HA fragments with small molecular weight, which is associated with elevated production of multiple cytokines, chemokines, and pro-angiogenic factors (1–3, 18, 28). Mechanistically, the LMW-HA elicits pro-inflammatory responses by modulating the toll-like receptor-4 (TLR-4) and activating the nuclear factor kappa B (NF-kB). In contrast, the HMW-HA manifests an anti-inflammatory effect by inhibiting NF-kB activation (29). During inflammation, HA can be degraded by hyaluronidases or cleaved by reactive oxygen species (ROS) (30–32). HA uptake and fragmentation by macrophages are thought to be important for the resolution of inflammation (24). The molecular weight of HA directly influences the immune macrophage activation (33, 34). According to these studies, the high-molecular-weight HA greater than 1,000 kDa exerts antiangiogenic, immunosuppressive, and anti-inflammatory effects that are important in wound healing, embryogenesis, and ovulation. In contrast, both medium-molecular weight HA and low-molecular-weight HA are pro-inflammatory, pro-angiogenic, and pro-tumor (34). HA added to LPS stimulated chondrocytes responded differently according to the HA molecular weight (32). Lee et al. demonstrated that HA fragments with molecular weight less than 50 kDa significantly increased iNOS production while medium MW-HA (1000 kDa) did not affect iNOS; however, HMW-HA (5000 kDa) significantly reduced iNOS in LPS-stimulated chondrocytes. A similar result was observed in LPS- stimulated macrophages where LMW-HA led to increases in iNOS, TNF-α, IL-6, IL-1β, TGF-β1, IL-10, IL-11, CCL2, and Arg1; however, in unstimulated macrophages, IL-10 significantly up-regulated by HMW-HA (24). Furthermore, LMW-HA increases several cytokines such as MMP-12, plasminogen activator inhibitor-1 (35, 36), CCL2 (MCP-1), CCL3 (MIP-1a), CCL4 (MIP-1β), keratinocyte chemoattractant, interleukins IL-8, and IL-12 by macrophages (37–39). Additionally, LMW-HA elicits the irreversible phenotypic and functional maturation of human dendritic cells (40, 41). In the model for acute lung injury to epithelium causes the production of inflammatory cytokines and chemokines resulting in the influx of neutrophils filled by macrophages to the site with further increase of cytokines and modulation of the extracellular matrix such as HA, collagen, fibronectin. Mechanistically, the low molecular weight HA fragments bind to the CD44, RHAMM, and Toll-like receptors (5, 26, 42–44). Stimulation of TLR2/TLR4 results in enhanced production of inflammatory cytokines and chemokines. In addition, LMW-HA is a potent stimulant of arachidonic acid release in a time- and dose-dependent manner, inducing cPLA2α, ERK1/2, p38, and JNK phosphorylation, as well as activated COX2 expression and PGE2 production in primary human monocytes, murine RAW 264.7, and wild type bone marrow-derived macrophages. Specific cPLA2α inhibitors blocked HA-induced arachidonic acid release and PGE2 production in all of these cells (26). It is interesting to note that HMW-HA can significantly diminish TLR4, TLR2, MyD88, and NF-kB expression (45).
Multiple studies have demonstrated that both HMW-HA and LMW-HA are potent regulators of angiogenesis signaling, mainly by influencing endothelial cell (EC) behavior. One of the first studies demonstrated that partial degradation products of HA produced by the action of testicular hyaluronidase induced an angiogenic response, such as the formation of new blood vessels in the chick chorioallantoic membrane. Further fractionation of the digestion products showed that the activity was restricted to LMW-HA fragments between 4 and 25 disaccharides in length (46). More recently published studies indicate that LMW-HA stimulates vascular EC proliferation, migration, and tubule formation in vitro, as well as in various in vivo models of angiogenesis. In contrast, the HMW-HA displays anti-angiogenic properties by inhibiting EC proliferation, motility, and sprout formation (47–50). Since CD44 as well as RHAMM, two main receptors for HA, are present on the surface of the endothelial cells, both anti-RHAMM and anti-CD44 antibodies blocked the EC’s ability to form tubule-like structures in matrigel. Both CD44 and RHAMM stimulated by HA oligomers create a complex with ERK 1/2, which leads to the constitutive activation of ERK 1/2 and increased cell motility of invasive breast cancer cells (49). Similar ERK 1/2 pathway activation by LMW-HA has also been demonstrated in other tumor cell lines including ECs, such as human umbilical vein ECs (HUVECs), human microvessels endothelial cells, and human pulmonary artery ECs (49). Additionally, LMW-HA promotes the proliferation of HUVECs and ECs via ezrin, a linkage protein between the plasma membrane and actin skeleton that interacts with the cellular C-terminus of the CD44 receptor. In wound healing, CXCL12 stimulates angiogenesis by activating CXCR4 present on the EC surface and significantly increased cell migration, and induced faster-wound closure in wound closure assays. By pre-incubating with HMW-HA, the wound closure rate was significantly increased. In vitro studies have shown that CXCR4 activation by CXCL12 was significantly increased in HUVECs pretreated with HMW-HA; however, LMW-HA pre-incubation blocked CXCL12 signaling.
It appears that enhanced HA degradation is associated with tumor cell spreading. For example, accumulation of LMW-HA in tumor interstitial fluid correlates with lymphatic invasion and lymph node metastasis (51). Analysis of 176 serum specimens from breast cancer patients revealed (52) that level of serum LMW-HA but not total HA significantly correlated with lymph nodes metastasis, suggesting that serum LMW-HA may represent an indicator for metastasis development and prognosis for breast cancer progression. In addition, LMW-HA has been reported to induce cancer cell invasion by enhancing cancer cell motility (53).
Mechanistically, the HA receptors CD44s and RHAMM serve as mediators of HA-dependent development of metastasis (54). These receptors contribute to tumor progression via major pathways including the MAPK (MEK1, ERK1, 2) and SRC/FAK pathways that promote expression of an oncogenic transcriptome required for tumor cell survival, migration invasion, proliferation, and resistance to apoptosis (54).
CD44 co-localizes with HAS enzymes in lipid rafts where it is clustered by high molecular weight HA polymers and functions as a co-receptor for growth factor receptors to reduce the activation threshold of oncogenic driver signaling networks. For example, these small HA fragments limit oncogenic pathway activation and reverse drug resistance in CD133-positive highly tumorigenic subpopulations of ovarian carcinoma cells (55).
Another HA receptor, RHAMM is consistently overexpressed in many tumors, and its high expression is linked to the progression of multiple solid cancers. Increased expression of RHAMM is associated with castration-resistant disease in patients with prostatic metastases and elevated levels of both HA and RHAMM area are associated with a likelihood of biochemical failure in at-risk cancer patients after prostatectomy (56). Overexpression of RHAMM in breast primary cancer was linked to distant metastases (57), whereas increased RHAMM expression in colorectal cancers at the invasive front of primary tumors is linked to frequent lymphatic invasion, high tumor grade, and nodal metastasis (58).
The fragmented forms of HA occur in abundance in various malignancies. These small hyaluronan oligomers are assumed to be largely a result of increased hyaluronidase (Hyal) expression/activity (30). Functional perturbations of HA synthesis in cancer and degradation have revealed active roles of both the HA synthases and Hyals in epithelial tumor cells, stroma, tumor vascular formation, and resistance to chemotherapy (59). Six hyaluronidase-like gene sequences have been identified in humans: Hyal1-6 (6, 7), and two recently discovered enzymes with hyaluronidase activity, termed HYBID (KIAA1199; CEMIP) and TMEM (60, 61). Hyal1 and Hyal2 are the major hyaluronidases expressed in human somatic tissues. It has been proposed that Hyal1 and Hyal2 work together in a concerted effort (6, 62, 63). Hyal2 is a rate-limiting, glycosylphosphatidylinositol-linked (GPI-linked) enzyme that is anchored to the plasma membrane. It cleaves extracellular HMW-HA into intermediate size, 20-kDa HA fragments, or about 50 disaccharide units. The Hyal2-generated HA fragments are internalized, delivered to endosomes, and ultimately to lysosomes, where Hyal1 degrades the 20-kDa fragments to very small tetra-saccharides.
Importantly, both Hyal1 and Hyal2 contribute to intracellular and extracellular catabolism of hyaluronic acid, respectively, in a CD44-dependent manner (64). Several studies have shown that the HA can be internalized by normal, non-malignant cells (chondrocytes, macrophages, keratinocytes, etc.) for degradation and that the endocytosis is mediated via cell surface HA receptors. In cancer, the epithelial tumor cells frequently express the membrane-bound Hyal2 and can break down secreted HA into intermediate 20 kDa fragments. Analysis of tumor tissues from cancer patients and experimental animal tumors revealed that tumor cells frequently work together with tumor-recruited myeloid cells including myeloid-derived suppressor cells (MDSCs) and tumor-associated macrophages (TAMs) to break down the extracellular HA into small HA fragments (18, 65, 66). It appears, that tumor-associated myeloid cells have significant amounts of internalized HA and display higher levels of Hyal1 expression as compared to tumor cells. Accordingly, the tumor-associated myeloid cells, and particularly TAMs, are more efficient in breaking down the HMW-HA into small pro-inflammatory and pro-angiogenic HA fragments.
Tumor stroma supports tumor growth in multiple ways including simulation of proliferation, migration, invasion, promoting cancer-related inflammation, tumor angiogenesis, and also by contributing to tumor-associated immune suppression, resistance to cancer immunotherapy, and chemotherapy (67–72). Major cellular components of tumor stroma include cancer-associated fibroblasts (CAFs), TAMs, and mesenchymal and endothelial cells.
In addition to cells, the tumor stroma also has a highly complex extracellular component such as ECM, which comprises collagens, glycans, proteoglycans, and various secreted proteins such as growth and angiogenic factors, eicosanoids, chemokines, etc (73, 74). The state of tumor stroma is highly dynamic and is constantly being remodeled via the recruitment of myeloid cell subsets as well as precursors of endothelial and mesenchymal cells. HA is one of the major ECM components in tumor stroma. HA has been implicated in regulating a variety of cellular functions in both tumor cells and tumor-associated stromal cells, suggesting that altered HA levels can influence tumor growth and malignancy at multiple levels. Previously published studies have demonstrated that HA increases the proliferation rate of tumor cells in vitro and promotes cell survival under anchorage-independent conditions. Stromal HA accumulation is associated with a low immune response and poor prognosis in some cancers (75, 76) Due to the abnormal production of HA and its enhanced degradation in tumor tissues, it is plausible that aberrant HA metabolism in the tumor microenvironment is highly relevant for tumor growth and progression, specifically through stimulation of cancer inflammation, tumor angiogenesis, and modulation of the anti-tumor immune response. HA has frequently been implicated in monocyte/macrophage trafficking and activation. Pathology studies of human cancer specimens suggested that increased numbers of macrophages were correlated with HA accumulation in tumors (77). Also, it has been shown that TAMs preferentially infiltrate tumor tissues in an HA-dependent manner and concomitantly enhance neovascularization and tumor growth (78).
Recently we showed that HA-enriched tumor stroma directly contributes to the development of the immunosuppressive tumor microenvironment by supporting the formation of PD-L1-expressing macrophages (65). Thus, analysis of organotypic tumor tissue-slice cultures, from mice with implanted syngeneic tumors as well as from cancer patients, revealed that tumor-recruited myeloid cells directly interact with stromal cells to form the large PD-L1-expressing cell congregates. Using genetically modified tumor cells, we found that both epithelial tumor cells and vimentin-positive CAFs represent the major sources of HA in the tumor microenvironment. Furthermore, similar cell clusters comprised of HA-producing fibroblast-like cells and F4/80+PD-L1+ macrophages were detected in tumor-draining, but not in distant lymph nodes. Taking together, these findings indicate that the formation of multiple large HA-enriched stromal clusters that support the development of PD-L1-expressing antigen-presenting cells in the tumor microenvironment and draining lymph nodes could contribute to the immune escape and resistance to immunotherapy in cancer.
There are multiple pieces of evidence in various cancer types demonstrating that tumor-associated myeloid cells play pivotal roles in the formation of the immunosuppressive and tolerogenic tumor microenvironment that promotes immune escape and contributes to resistance to cancer immunotherapy. Tumors promote mobilization of myeloid-derived suppressor cells (MDSCs) using various mechanisms primarily from bone marrow and spleen (79, 80). Upon entering tumor tissue MDSCs directly interact with tumor cells. However, the mechanisms of these interactions are not fully understood. It appears that tumor-associated HA mediates the crosstalk between tumor-recruited Hyal2+ MDSCs, tumor cells, and stroma (64). Thus, Hyal2-expressing myeloid cells directly contact HA-producing tumor cells and CAFs. In a non-activated state, the HA-degrading enzyme Hyal2 in myeloid cells resides predominantly in intracellular space. However, upon activation in the tumor microenvironment Hyal2 translocate to the cell membrane, thus enabling the degradation of extracellular HA (Figure 2). The tumor-associated Hyal2-expressing myeloid cells have been detected in close contact with HA-producing CAFs and epithelial tumor cells, leading to enhanced degradation of tumor-associated fragmentation of HA in the tumor microenvironment. It should be noted that Hyal2 activity in myeloid cells is regulated by CD44 signaling and IL-1beta (18).
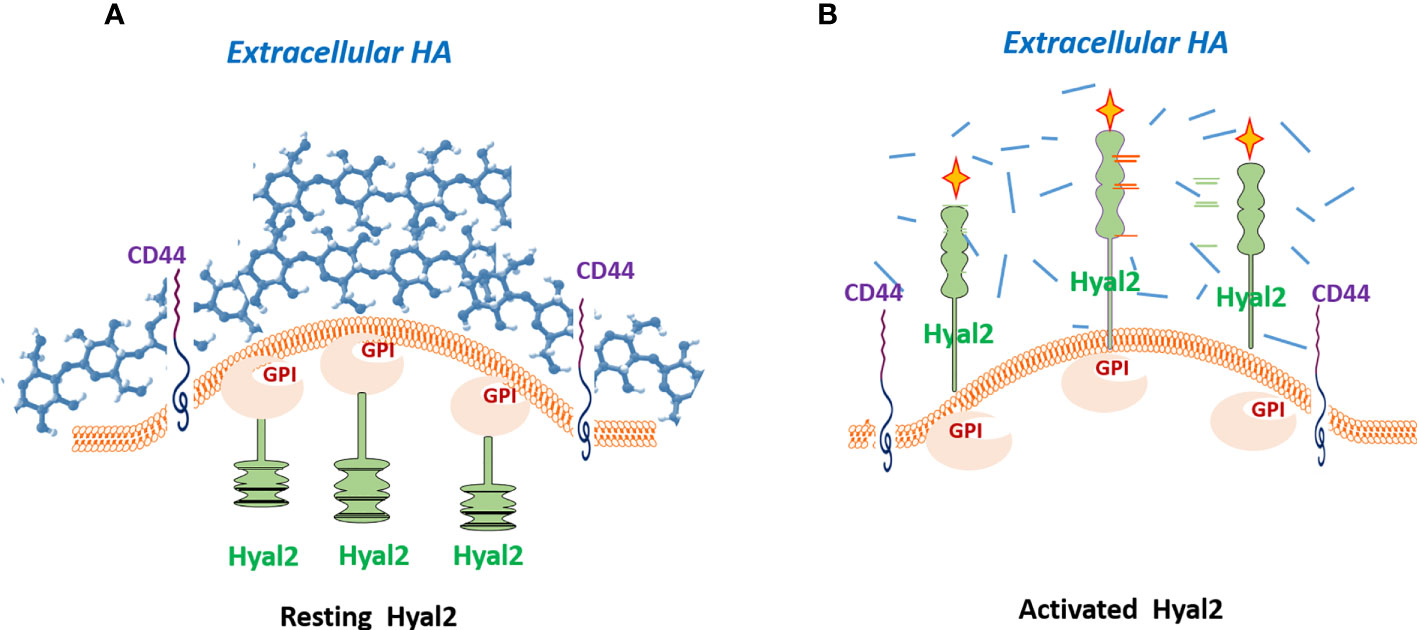
Figure 2 Hyal2 translocation is required for the degradation of extracellular hyaluronan. In a quiescent state, the Hyal2 molecule in myeloid cells resides predominantly in intracellular space (A). However, activation of the CD44 signaling pathway results in translocation of Hyal2 to the cellular membrane, enabling the degradation of extracellular HA (B). Depicted yellow crosses indicate the activated status of the enzyme Hyal2.
The increased numbers of Hyal2+ MDSCs have been detected in peripheral blood and tumor tissue in patients with bladder and kidney cancers (18, 66). In humans this co-express marker of myeloid cells CD11b and the marker of monocytic MDSCs CD33 (Hyl2+CD33+CD11b+). A similar subset of Hyal2-expressing MDSCs (Hyal2+Gr-1+CD11b+) has been found in tumor-bearing mice (65). Mobilization and tumor recruitment of Hyal2+ myeloid cells results in enhanced HA degradation in tumor tissue, leading to the accumulation of small HA fragments (~20 kDa) in the tumor microenvironment. In addition, the tumor-infiltrating myeloid cells show significantly higher expression of Hyal1 than tumor cells and have the ability to degrade HA into much smaller fragments with a molecular weight of less than 5 kDa (63). The smallest HA fragments exert the strongest pro-inflammatory and pro-angiogenic activities (2, 3, 7, 45).
Importantly, the tumor-recruited Hyal2+ MDSCs also co-express the immunosuppressive ligand PD-L1 (64, 65). In mouse tumor models, upon contact with HA-producing tumor cells or CAFs, Hyal2+ MDSCs can proliferate and differentiate into immunosuppressive macrophages, forming the large PD-L1+ myeloid cell clusters (Figures 3 and 4). Interestingly, during the transition of Hyal2+MDSCs into TAMs, the activity of HA-degrading activity of Hyal2 is reduced, while expression of immunosuppressive ligand PD-L1 is markedly up-regulated. It is still unclear how the degradation of extracellular HA promotes differentiation of Hyal2+Gr-1+PD-L1+ MDSCs into more mature immunosuppressive F4/80+PD-L1+ tumor-associated macrophages. It is plausible that under pro-inflammatory conditions in the tumor microenvironment small HA fragments promote the transition of MDSCs into activated immunosuppressive PD-L1+ macrophages. Collectively, these data indicate that tumor-recruited myeloid cells contribute to tumor growth through degradation of tumor-associated HA, promoting cancer-related inflammation, tumor angiogenesis, and tumor-associated immune suppression.
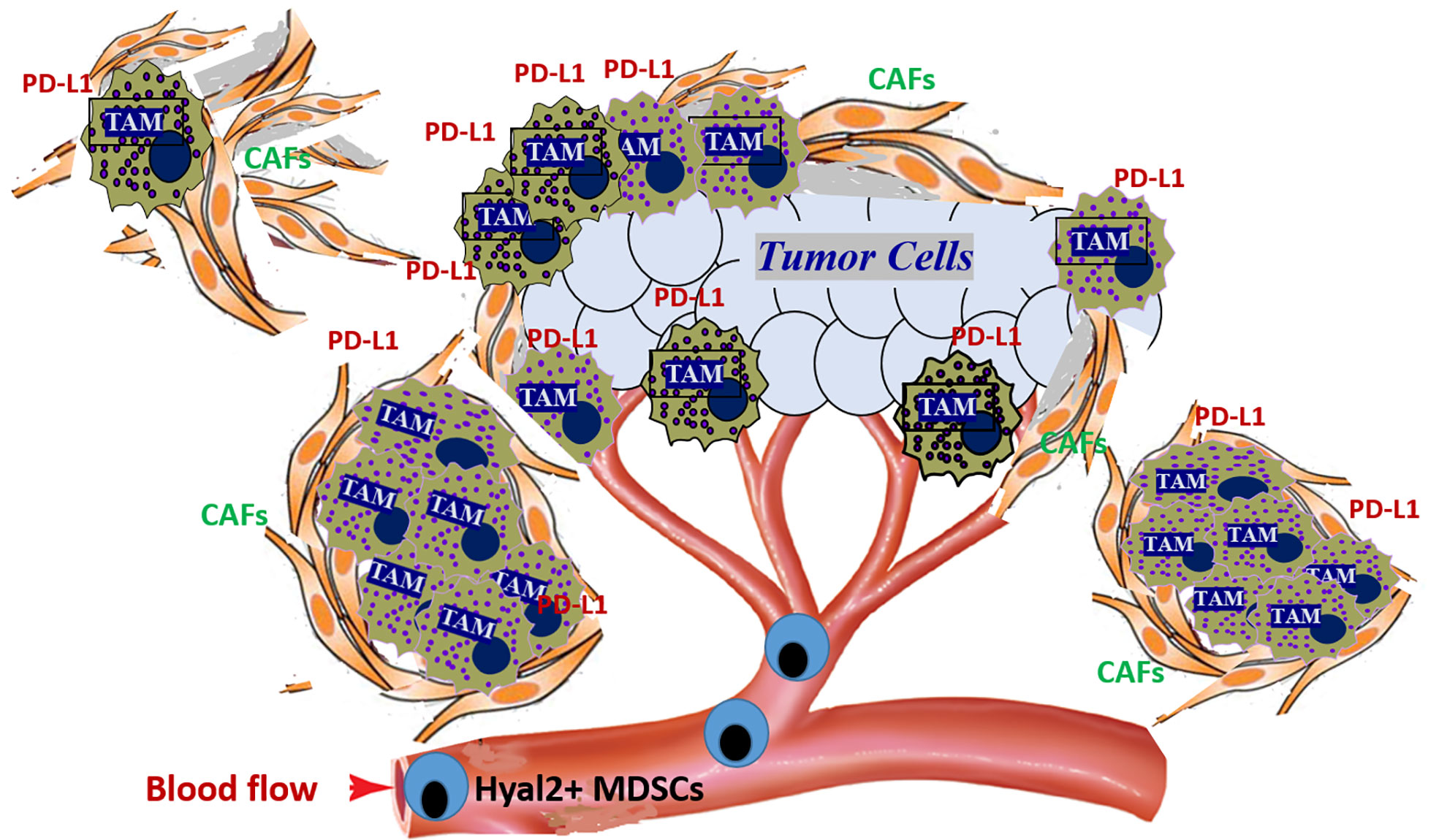
Figure 3 Tumor-recruited Hyal2+ MDSCs directly contribute to the development of the immunosuppressive tumor microenvironment by forming PD-L1+ macrophage clusters in the hyaluronan-enriched stroma. Tumors constantly secrete significant amounts of chemokines that attract the Hyal2+ MDSCs from bone marrow. Once recruited to the tumor, Hyal2-expressing myeloid cells start degradation of extracellular HA in the tumor microenvironment. Direct interaction of tumor-recruited myeloid cells with HA-producing cancer-associated fibroblasts (CAFs) and epithelial tumor cells leads to the accumulation of small HA pro-inflammatory and pro-angiogenic fragments. Hyal2+ MDSCs differentiate into immunosuppressive tumor-associated PD-L1+ macrophages (PD-L1+ TAMs), forming large PD-L1-expressing cell clusters in HA-enriched tumor stroma.
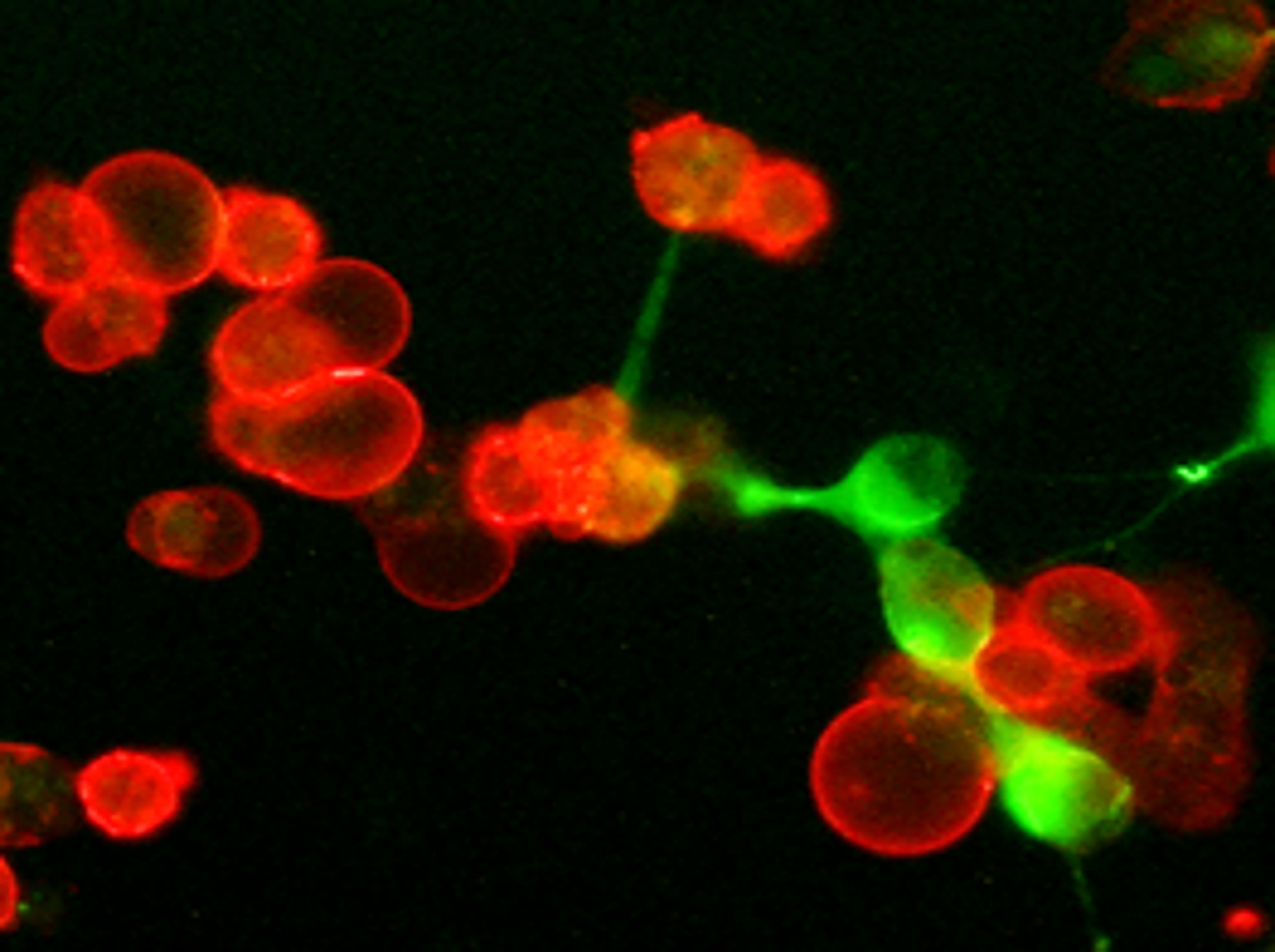
Figure 4 Tumor-associated hyaluronan directly supports the development of immunosuppressive PD-L1+ macrophages in the tumor microenvironment. Freshly prepared tumor tissue slices were cultured for twenty-four hours. Then cultures were fixed with 4% formaldehyde and stained PD-L1 (red) and HA (green). Representative IF image is shown.
Understanding the complex molecular and cellular mechanisms involved in abnormal HA metabolism and catabolism in solid cancers could have important implications for the design of future cancer therapeutic approaches. It appears that extensive crosstalk between tumor-associated myeloid cells and HA-enriched stroma contributes to the tumor growth and progression in several ways. Specifically, the interaction of tumor-recruited Hyal2-expressing myeloid cells with HA-producing stromal cells results in enhanced HA degradation and accumulation of small pro-inflammatory and pro-angiogenic HA fragments (18). HA-enriched stroma directly supports the development of PD-L1-expressing macrophages, thus contributing to the formation of the immunosuppressive, tolerogenic microenvironment by creating a PD-L1 shield and preventing T cell-mediated immune response via the PD1/PD-L1 pathway (65, 66). Constant mobilization of bone marrow-derived Hyal2+ MDSCs, CAFs, and endothelial cells contribute to the highly dynamic development of tumor stroma, enhances degradation of tumor-associated HA, and further promotes the tumor progression through stimulation of cancer-related inflammation, tumor angiogenesis, and tumor-associated immune suppression. Therefore, the normalization of HA metabolism in the tumor microenvironment could potentially provide a strong beneficial step for improving the efficacy of existing approaches to treat cancer, particularly for cancer immunotherapy.
The original contributions presented in the study are included in the article/supplementary materials. Further inquiries can be directed to the corresponding author.
WD wrote review materials related to HA metabolism in normal and malignant tissues. PD-G wrote review sections related to the roles of LMW-HA in inflammation and angiogenesis. SK composed the article, wrote review materials related to mechanisms of HA degradation in tumor microenvironment and stroma, wrote abstract, conclusion and prepared Figures. All authors contributed to the article and approved the submitted version.
This work was supported by James and Ester King Biomedical Research Program, Florida Health Department, award 8JKO5 and 1923 Fund to SK.
SK is a founder of K-Lab Therapeutics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Toole BP. Hyaluronan: from extracellular glue to pericellular cue. Nat Rev Cancer (2004) 4:528–39. doi: 10.1038/nrc1391
2. Girish KS, Kemparaju K. The magic glue hyaluronan and its eraser hyaluronidase: A biological overview. Life Sci (2007) 80:1921–43. doi: 10.1016/j.lfs.2007.02.037
3. Schmaus A, Bauer J, Sleeman JP. Sugars in the microenvironment: the sticky problem of HA turnover in tumors. Cancer Metastasis Rev (2014) 33:1059–79. doi: 10.1007/s10555-014-9532-2
4. Knudson W, Bartnik E, Knudson CB. Assembly of pericellular matrices by COS-7 cells transfected with CD44 lymphocyte-homing receptor genes. Proc Natl Acad Sci USA (1993) 90:4003–7. doi: 10.1073/pnas.90.9.4003
5. Turley EA, Noble PW, Bourguignon LYW. Signaling properties of hyaluronan receptors. J Biol Chem (2002) 277:4589–92. doi: 10.1074/jbc.R100038200
6. Csoka AB, Frost GI, Stern R. The six hyaluronidase-like genes in the human and mouse genomes. Matrix Biol (2001) 20:499–508. doi: 10.1016/S0945-053X(01)00172-X
7. Csóka AB, Scherer SW, Stern R. Expression analysis of six paralogous human hyaluronidase genes clustered on chromosomes 3p21 and 7q31. Genomics (1999) 60:356–61. doi: 10.1006/geno.1999.5876
8. Chanmee T, Ontong P, Itano N. Hyaluronan: A modulator of the tumor microenvironment. Cancer Lett (2016) 375:20–30. doi: 10.1016/j.canlet.2016.02.031
9. Lokeshwar VB, Schroeder GL, Selzer MG, Hautmann SH, Posey JT, Duncan RC, et al. Bladder tumor markers for monitoring recurrence and screening comparison of hyaluronic acid-hyaluronidase and BTA-stat tests. Cancer (2002) 95:61–72. doi: 10.1002/cncr.10652
10. Golshani R, Hautmann SH, Estrella V, Cohen BL, Kyle CC, Manoharan M, et al. HAS1 expression in bladder cancer and its relation to urinary HA test. Int J Cancer (2007) 120:1712–20. doi: 10.1002/ijc.22222
11. Kramer MW, Escudero DO, Lokeshwar SD, Golshani R, Ekwenna OO, Acosta K, et al. Association of hyaluronic acid family members (HAS1, HAS2, and HYAL-1) with bladder cancer diagnosis and prognosis. Cancer (2011) 117:1197–209. doi: 10.1002/cncr.25565
12. Li Y, Li L, Brown TJ, Heldin P. Silencing of hyaluronan synthase 2 suppresses the malignant phenotype of invasive breast cancer cells. Int J Cancer (2007) 120:2557–67. doi: 10.1002/ijc.22550
13. Udabage L, Brownlee GR, Nilsson SK, Brown TJ. The over-expression of HAS2, hyal-2 and CD44 is implicated in the invasiveness of breast cancer. Exp Cell Res (2005) 310:205–17. doi: 10.1016/j.yexcr.2005.07.026
14. Lien H-C, Lee Y-H, Jeng Y-M, Lin C-H, Lu Y-S, Yao Y-T. Differential expression of hyaluronan synthase 2 in breast carcinoma and its biological significance. Histopathology (2014) 65:328–39. doi: 10.1111/his.12390
15. Okuda H, Kobayashi A, Xia B, Watabe M, Pai SK, Hirota S, et al. Hyaluronan synthase HAS2 promotes tumor progression in bone by stimulating the interaction of breast cancer stem-like cells with macrophages and stromal cells. Cancer Res (2012) 72:537–47. doi: 10.1158/0008-5472.CAN-11-1678
16. Posey JT, Soloway MS, Ekici S, Sofer M, Civantos F, Duncan RC, et al. Evaluation of the prognostic potential of hyaluronic acid and hyaluronidase (HYAL1) for prostate cancer. Cancer Res (2003) 63:2638–44. Availabloe at: https://aacrjournals.org/cancerres/article/63/10/2638/509916/Evaluation-ofthe-Prognostic-Potential-of
17. Lipponen P, Aaltomaa S, Tammi R, Tammi M, Agren U, Kosma VM. High stromal hyaluronan level is associated with poor differentiation and metastasis in prostate cancer. Eur J Cancer (2001) 37:849–56. doi: 10.1016/S0959-8049(00)00448-2
18. Dominguez-Gutierrez PR, Kwenda EP, Donelan W, O’Malley P, Crispen PL, Kusmartsev S. Hyal2 expression in tumor-associated myeloid cells mediates cancer-related inflammation in bladder cancer. Cancer Res (2021) 81:648–57. doi: 10.1158/0008-5472.CAN-20-1144
19. Tian X, Azpurua J, Hine C, Vaidya A, Myakishev-Rempel M, Ablaeva J, et al. High-molecular-mass hyaluronan mediates the cancer resistance of the naked mole rat. Nature (2013) 499:346–9. doi: 10.1038/nature12234
20. Ooki T, Murata-Kamiya N, Takahashi-Kanemitsu A, Wu W, Hatakeyama M. High-molecular-weight hyaluronan is a hippo pathway ligand directing cell density-dependent growth inhibition via PAR1b. Dev Cell (2019) 49:590–604.e9. doi: 10.1016/j.devcel.2019.04.018
21. Bourguignon LYW, Wong G, Earle CA, Xia W. Interaction of low molecular weight hyaluronan with CD44 and toll-like receptors promotes the actin filament-associated protein 110-actin binding and MyD88-NFκB signaling leading to proinflammatory cytokine/chemokine production and breast tumor invasion. Cytoskeleton (Hoboken) (2011) 68:671–93. doi: 10.1002/cm.20544
22. Jiang D, Liang J, Noble PW. Hyaluronan as an immune regulator in human diseases. Physiol Rev (2011) 91(1):221–64. doi: 10.1152/physrev.00052.2009
23. Tolg C, Messam BJ, McCarthy JB, Nelson AC, Turley EA. Hyaluronan functions in wound repair that are captured to fuel breast cancer progression. Biomolecules (2021) 11(11):1551. doi: 10.3390/biom11111551
24. Lee BM, Park SJ, Noh I, Kim C-H. The effects of the molecular weights of hyaluronic acid on the immune responses. Biomater Res (2021) 25(1):27. doi: 10.1186/s40824-021-00228-4
25. Liu M, Tolg C, Turley E. Dissecting the dual nature of hyaluronan in the tumor microenvironment. Front Immunol (2019) 10:947. doi: 10.3389/fimmu.2019.00947
26. Sokolowska M, Chen LY, Eberlein M, Martinez-Anton A, Liu Y, Alsaaty S , et al. Low molecular weight hyaluronan activates cytosolic phospholipase A2α and eicosanoid production in monocytes and macrophage. Biol Chem (2014) 289(7):4470–88. doi: 10.1074/jbc.M113.515106
27. Mantovani A, Allavena P, Sica A and Balkwill F. Cancer-related inflammation. Nature (2008) 454:436–44. doi: 10.1038/nature07205
28. Witschen PM, Chafee TS, Brady NJ, Huggins DN, Knutson TP, LaRue RS, et al. Tumor cell associated hyluronan-CD44 signaling promotes pro-tumor inflammation in breast cancer. Cancers (2020) 12(5)1325. doi: 10.3390/cancers12051325
29. Campo GM, Avenoso A, Campo S, D'Ascola A, Nastasi G, Calatroni A. Molecular size hyaluronan differently modulates toll-like receptor-4 in LPS-induced inflammation in mouse chondrocytes. Biochimie (2010) 92(2):204–15. doi: 10.1016/j.biochi.2009.10.006
30. Stern R. Hyaluronidases in cancer biology. Semin Cancer Biol (2008) 18:275–80. doi: 10.1016/j.semcancer.2008.03.017
31. Yang J-M, Kim H-J, Cho B-K. Preparation of antioxidant hydrogel contact lenses based on interpenetrating hyaluronic acid network. Polymer (Korea) (2020) 44(1):21–9. pk. doi: 10.7317/pk.2020.44.1.21
32. Bracke KR, Dentener MA, Papakonstantinou E, Vernooy JHJ, Demoor T, Pauwels NS, et al. Enhanced deposition of low-molecular-weight hyaluronan in lungs of cigarette smoke-exposed mice. Am J Respir Cell Mol Biol (2010) 42(6):753–61. doi: 10.1165/rcmb.2008-0424OC
33. Rayahin JE, Buhrman JS, Zhang Y, Koh TJ, Gemeinhart RA. High and low molecular weight hyaluronic acid differentially influence macrophage activation. ACS Biomater Sci Eng (2015) 1(7):481–93. doi: 10.1021/acsbiomaterials.5b00181
34. D’Agostino A, Stellavato A, Corsuto L, Diana P, Filosa R, La Gatta A, et al. Is molecular size a discriminating factor in hyaluronan interaction with human cells? Carbohydr Polym (2017) 157:21–30. doi: 10.1016/j.carbpol.2016.07.125
35. Horton MR, Shapiro S, Bao C, Lowenstein CJ, Noble PW. Induction and regulation of macrophage metalloelastase by hyaluronan fragments in mouse macrophages. J Immunol (1999) 162(7):4171–6. Available at: https://www.jimmunol.org/content/162/7/4171
36. Horton MR, Olman MA, Bao C, White KE, Choi AM, Chin BY, et al. Regulation of plasminogen activator inhibitor-1 and urokinase by hyaluronan fragments in mouse macrophages. Am J Physiol Lung Cell Mol Physiol (2000) 279(4):L707–15. doi: 10.1152/ajplung.2000.279.4.L707
37. Hodge-Dufour J, Noble PW, Horton MR, Bao C, Wysoka M, Burdick MD, et al. Induction of IL-12 and chemokines by hyaluronan requires adhesion-dependent priming of resident but not elicited macrophages. J Immunol (1997) 159(5):2492–500. Available at: https://www.jimmunol.org/content/159/5/2492.long
38. Horton MR, Burdick MD, Strieter RM, Bao C, Noble PW. Regulation of hyaluronan-induced chemokine gene expression by IL-10 and IFN-gamma in mouse macrophages. J Immunol (1998) 160(6):3023–30. Available at: https://www.jimmunol.org/content/160/6/3023
39. McKee CM, Penno MB, Cowman M, Burdick MD, Strieter RM, Bao C, et al. Hyaluronan (HA) fragments induce chemokine gene expression in alveolar macrophages. the role of HA size and CD44. J Clin Invest (1996) 98(10):2403–13. doi: 10.1172/JCI119054.
40. Termeer C, Benedix F, Sleeman J, Fieber C, Voith U, Ahrens T, et al. Oligosaccharides of hyaluronan activate dendritic cells via toll-like receptor 4. J Exp Med (2002) 195(1):99–111. doi: 10.1084/jem.20001858
41. Termeer CC, Hennies J, Voith U, Ahrens T, Weiss JM, Prehm P, et al. Oligosaccharides of hyaluronan are potent activators of dendritic cells. J Immunol (2000) 165(4):1863–70. doi: 10.4049/jimmunol.165.4.1863
42. Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, et al. Regulation of lung injury and repair by toll-like receptors and hyaluronan. Nat Med (2005) 11(11):1173–9. doi: 10.1038/nm1315
43. Taylor KR, Yamasaki K, Radek KA, Nardo AD, Goodarzi H, Golenbock D, et al. Recognition of hyaluronan released in sterile injury involves a unique receptor complex dependent on toll-like receptor 4, CD44, and MD-2. J Biol Chem (2007) 282(25):18265–75. doi: 10.1074/jbc.M606352200
44. Taylor KR, Trowbridge JM, Rudisill JA, Termeer CC, Simon JC, Gallo RL. Hyaluronan fragments stimulate endothelial recognition of injury through TLR4. J Biol Chem (2004) 279(17):17079–84. doi: 10.1074/jbc.M310859200
45. Campo GM, Avenoso A, Nastasi G, Micali A, Prestipino V, Vaccaro M, et al. Hyaluronan reduces inflammation in experimental arthritis by modulating TLR-2 and TLR-4 cartilage expression. Biochim Biophys Acta (2011) 1812(9):1170–81. doi: 10.1016/j.bbadis.2011.06.006
46. West DC, Hampson IN, Arnold F, Kumar S. Angiogenesis induced by degradation products of hyaluronic acid. Science. (1985) 228(4705):1324–6. doi: 10.1126/science.2408340
47. Slevin M, Krupinski J, Gaffney J, Matou S, West D, Delisser H, et al. Hyaluronan-mediated angiogenesis in vascular disease: Uncovering RHAMM and CD44 receptor signaling pathways. Matrix Biol (2007) 26(1):58–68. doi: 10.1016/j.matbio.2006.08.261
48. Silva AL, Babo PS, Rodrigues MT, Gonçalves AI, Novoa-Carballal R, Pires RA , et al. Hyaluronic acid oligomer immobilization as angiogenic trigger for the neovascularization of TE constructs. ACS Appl Bio Mater (2021) 4(8):6023–35. doi: 10.1021/acsabm.1c00291
49. Lokeshwar VB, Selzer MG. Differences in hyaluronic acid-mediated functions and signaling in arterial, microvessel, and vein-derived human endothelial cells. J Biol Chem (2000) 275(36):27641–9. doi: 10.1074/jbc.M003084200
50. Fuchs K, Hippe A, Schmaus A, Homey B, Sleeman JP, Orian-Rousseau V. Opposing effects of high- and low-molecular weight hyaluronan on CXCL12-induced CXCR4 signaling depend on CD44. Cell Death Dis (2013) 4:e819. doi: 10.1038/cddis.2013.364
51. Schmaus A, Klusmeier S, Rothley M, Dimmler A, Sipos B, Faller G, et al. Accumulation of small hyaluronan oligosaccharides in tumour interstitial fluid correlates with lymphatic invasion and lymph node metastasis. Br J Cancer (2014) 111(3):559–67. doi: 10.1038/bjc.2014.332
52. Wu M, Cao M, He Y, Liu Y, Yang C, Du Y, et al. A novel role of low molecular weight hyaluronan in breast cancer metastasis. FASEB J (2015) 29:1290–8. doi: 10.1096/fj.14-259978
53. Sugahara KN, Murai T, Nishinakamura H, Kawashima H, Saya H, Miyasaka M, et al. Hyaluronan oligosaccharides induce CD44 cleavage and promote cell migration in CD44-expressing tumor cells. J Biol Chem (2003) 281:5861–8. doi: 10.1074/jbc.M506740200
54. Turley EA, Wood DK, McCarthy JB. Carcinoma cell hyaluronan as a “portable” cancerized pro-metastatic microenvironment. Cancer Res (2016) 76(9):2507–12. doi: 10.1158/0008-5472.CAN-15-3114
55. Slomiany MG, Dai L, Tolliver LB, Grass GD, Zeng Y, Toole BP. Inhibition o functional hyaluronan-CD44 interactions in CD133-positive primary human ovarian carcinoma cells by small hyaluronan oligosaccharides. Clin Cancer Res (2009) 15:7593–601. doi: 10.1158/1078-0432.CCR-09-2317
56. Rizzardi AE, Vogel RI, Koopmeiners JS, Forster CL, Marston LO, Rosener NK, et al. Elevated hyaluronan and hyaluronan-mediated motility receptor are associated with biochemical failure in patients with intermediate-grade prostate tumors. Cancer (2014) 120:1800–9. doi: 10.1002/cncr.28646
57. Wang C, Thor AD, Moore DH 2nd, Zhao Y, Kerschmann R, Stern R., et al. The overexpression of RHAMM, hyaluronan-binding protein that regulates RAS signaling, correlates with overexpression of mitogen-activated protein kinase and is significant parameter in breast cancer progression. Clin Cancer Res (1998) 4:367–576. Available at: https://aacrjournals.org/clincancerres/article/4/3/567/7638/The-overexpression-of-RHAMM-a-hyaluronan-binding
58. Koelzer VH, Huber B, Mele V, Iezzi G, Trippel M, Karamitopoulou E, et al. Expression of the hyaluronan-mediated motility receptor RHAMM in tumor budding cells identifies aggressive colon cancers. Hum Pathol (2015) 46(11):1573–8. doi: 10.1016/j.humpath.2015.07.010
59. Passi A, Vigetti D, Buraschi S, Iozzo RV. Dissecting the role of hyaluronan synthases in the tumor microenvironment. FEBS J (2019) 286(15):2937–49. doi: 10.1111/febs.14847
60. Nagaoka A, Yoshida H, Nakamura S, Morikawa T, Kawabata K, Kobayashi M, et al. Regulation of hyaluronan (HA) metabolism mediated by HYBID (hyaluronan-binding protein involved in HA depolymerization, KIAA1199) and HA synthases in growth factor-stimulated fibroblasts. J Biol Chem (2015) 290:30910–3092. doi: 10.1074/jbc.M115.673566
61. Tobisawa Y, Fujita N, Yamamoto H, Ohyama C, Irie F, Yamaguchi YJ. The cell surface hyaluronidase TMEM2 is essential for systemic hyaluronan catabolism and turnover. J Biol Chem (2021) 297(5):101281. doi: 10.1016/j.jbc.2021.101281
62. Stern R. Devising a pathway for hyaluronan catabolism: Are we there yet glycobiology. Glycobiology (2003) 13:105–15. doi: 10.1093/glycob/cwg112
63. Stern R. Hyaluronan catabolism: a new metabolic pathway. Eur J Cell Biol (2004) 83:317–25. doi: 10.1078/0171-9335-00392
64. Harada H, Takahashi M. CD44-dependent intracellular and extracellular catabolism of hyaluronic acid by hyaluronidase-1 and 2. J Biol Chem (2007) 282(8):5597–607. doi: 10.1074/jbc.M608358200
65. Dominguez-Gutierrez PR, Kwenda E, Donelan W, Miranda M, Doty A, O’Malley P, et al. Detection of PD-L1-expressing myeloid cell clusters in the hyaluronan-enriched stroma. J Immunol (2022) 208:2829–36. doi: 10.4049/jimmunol.2100026
66. Kusmartsev S, Kwenda E, Dominguez-Gutierrez PR, Crispen PL, O’Malley P. PD-L1+ and Hyal2+ myeloid cells in renal cell carcinoma: A case report. J Kidney Cancer VHL (2022) V.9(2):1–6. doi: 10.15586/jkcvhl.v9i2.208.
67. Kraman M, Bambrough PJ, Arnold JN, Roberts EW, Magiera L, Jones JO, et al. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science (2010) 330:827–30. doi: 10.1126/science.1195300
68. Liguori M, Solinas G, Germano G, Mantovani A, Allavena P. Tumor-associated macrophages as incessant builders and destroyers of the cancer stroma. Cancers (2011) 3(4):3740–61. doi: 10.3390/cancers3043740
69. Turley SJ, Cremasco V, Astarita JL. Immunological hallmarks of stromal cells in the tumour microenvironment. Nat Rev Immunol (2015) 15(11):669–82. doi: 10.1038/nri3902
70. Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer (2020) 20:174–86. doi: 10.1038/s41568-019-0238-1
71. Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer (2016) 16:582–98. doi: 10.1038/nrc.2016.73
72. Xiang H, Ramil CP, Hai J, Zhang C, Wang H, Watkins AA, et al. Cancer-associated fibroblasts promote immunosuppression by inducing ROS-generating monocytic MDSCs in lung squamous cell carcinoma. Cancer Immunol Res (2020) 8(4):436–50. doi: 10.1158/2326-6066.CIR-19-0507
73. Brownfield DG, Venugopalan G, Lo A, Mori H, Tanner K, Fletcher DA, et al. Patterned collagen fibers orient branching mammary epithelium through distinct signaling modules. Curr Biol (2013) 3(8):703–9. doi: 10.1016/j.cub.2013.03.032
74. Toole BP, Slomiany MG. Hyaluronan: A constitutive regulator of chemoresistance and malignancy in cancer cells. Semin Cancer Biol (2008) 4):244–50. doi: 10.1016/j.semcancer.2008.03.009
75. Tahkola K, Ahtiainenm M, Mecklin JP, Kellokumpu I, Laukkarinen J, Tammi M, et al. Stromal hyaluronan accumulation is associated with low immune response and poor prognosis in pancreatic cancer. Sci Rep (2021) 11(1):12216. doi: 10.1038/s41598-021-91796-x
76. Wu W, Chen L, Wang Y, Jin J, Xie X, Zhang J. Hyaluronic acid predicts poor prognosis in breast cancer patients: A protocol for systematic review and meta-analysis. Medicine (2020) 99(22):e20438. doi: 10.1097/MD.0000000000020438
77. Tiainen S, Tumelius R, Rilla K, Hamalainen K, Tammi M, Tammi R, et al. High numbers of macrophages, especially M2-like (CD163-positive), correlate with hyaluronan accumulation and poor outcome in breast cancer. Histopathology (2015) 66:873–83. doi: 10.1111/his.12607
78. Kobayashi N, Miyoshi S, Mikami T, Koyama H, Kitazawa M, Takeoka M, et al. Hyaluronan deficiency in tumor stroma impairs macrophage traficking and tumor neovascularization. Cancer Res (2010) 70:7073–83. doi: 10.1158/0008-5472.CAN-09-4687
79. Gabrilovich D, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol (2012) 12(4):253–68. doi: 10.1038/nri3175
Keywords: tumor microenvironment, HYAL2, hyaluronan degradation, PD-L1, MDSC, tumor-associated macrophages, cancer inflammation, tumor-associated immune suppression
Citation: Donelan W, Dominguez-Gutierrez PR and Kusmartsev S (2022) Deregulated hyaluronan metabolism in the tumor microenvironment drives cancer inflammation and tumor-associated immune suppression. Front. Immunol. 13:971278. doi: 10.3389/fimmu.2022.971278
Received: 16 June 2022; Accepted: 07 September 2022;
Published: 27 September 2022.
Edited by:
Julie Decock, Qatar Biomedical Research Institute, QatarReviewed by:
James McCarthy, University of Minnesota Twin Cities, United StatesCopyright © 2022 Donelan, Dominguez-Gutierrez and Kusmartsev. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sergei Kusmartsev, c2FrdXM2MUB1ZmwuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.