 Han Cheng
Han Cheng Lingling Chen1,2†
Lingling Chen1,2† Jin Hou
Jin Hou Xiaojun Yang
Xiaojun Yang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 24 October 2022
Sec. Inflammation
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.967989
This article is part of the Research TopicNovel Perspectives on the NLRP3 InflammasomeView all 5 articles
Radiotherapy is one of the mainstream treatment modalities for several malignancies. However, radiation-induced injury to surrounding normal tissues limits its efficacy. The NLRP3 inflammasome is an essential mechanism of innate immunity that reacts to challenges from endogenous danger signals and pathological microbes. A growing body of evidence has demonstrated a key role of NLRP3 inflammasome in the pathogenesis of radiation-induced tissue injury. Despite accumulating evidence, the potential value of the NLRP3 inflammasome in the management of radiation-induced tissue injury is not adequately recognized. We conducted a literature review to characterize the relationship between NLRP3 inflammasome and radiation injury. By analyzing recent evidence, we identify NLRP3 inflammasome as one of the executioners of radiation-induced injury, since it responds to the challenges of radiation, induces cell pyroptosis and tissue dysfunction, and initiates non-resolving inflammation and fibrosis. Based on these concepts, we propose early intervention/prevention strategies targeting NLRP3 inflammasome in a radiation context, which may help resolve imperative clinical problems.
Radiotherapy is an effective treatment modality for various types of tumors. However, radiation-induced injury to normal tissues is an unavoidable adverse effect of radiotherapy. Despite ongoing advances in radiotherapy techniques that enable precise targeting of lesions, tissues adjacent to the irradiated field are liable to be affected by ionizing radiation. Typical examples of the adverse effects of radiotherapy include occurrence of carotid stenosis following head and neck radiotherapy, cardiovascular injury after thoracic radiotherapy, and gastrointestinal injury after pelvic and abdominal irradiation (1–6). In addition, indirect damage caused by the out-off field effects effect (capability of inducing similar responses in non-irradiated tissues) of radiation cannot be overlooked. For example, patients receiving radiotherapy to the head and neck region were reported to have a higher incidence of diarrhea (7). Similarly, irradiation of rat tongue was found to induce small intestine injury (8). These adverse effects may necessitate reduction in RT doses, limiting tumor control (9). Therefore, in-depth characterization of the mechanism of radiation injury and exploration of more effective management strategies are key imperatives.
The pathophysiological processes of different stages of radiation injury are well recognized. Initially, radiation causes DNA damage in cells, resulting in double-strand DNA breaks (DSBs) (10). Radiation-induced damage to DNA may occur directly via interacting with DNA molecules and causing DSBs, or indirectly by generating free radicals, such as reactive oxygen and nitrogen species (ROS, RNS), to cause base modification, and eventually leading to DSBs (11). The occurrence of DSBs induces the DNA-damage response (DDR), which initiates DNA repair by activating the MRN (Mre11-Rad50-Nbs1)-ATM (ataxia telangiectasia mutated)-H2AX (histone variant 2AX) signaling (11, 12). The transducer protein ATM, as well as ROS and other inflammatory stimuli, are capable of activating nuclear factor kappa-B (NF-κB) in a variety of manners, in order to better prepare cells for the stress (10, 13). DDR can also induce apoptosis or senescence if the damage cannot be repaired, meanwhile heightening immune surveillance for later scavenging of the remains (11). Moreover, other forms of cell death may still occur, irreparably damaged cells acutely generate robust amounts of pro-inflammatory factors, attracting and activating immune cells to the irradiated area. These accumulated inflammatory cells may potentiate tissue injury, establishing non-resolving inflammation and aberrant tissue remodeling, resulting in tissue injury (14–17). The intricate molecular mechanisms of radiation injury are yet to be fully elucidated. However, recent evidence suggests that ionizing radiation induces activation of inflammasome which functions as the “executioner” for radiation, mediating certain kinds of tissue injuries.
Inflammasomes are macromolecular complexes that react to challenges such as exogenous or endogenous danger signals and pathological microbes, and mediate the maturation and release of interleukin-1β (IL-1β) and interleukin-18 (IL-18) as well as induction of pyroptotic cell death. The activation patterns and downstream responses of inflammasome make it an essential part of innate immunity, acting against various types of infections and injuries. Beyond host defensive response, recent studies pointed out the participation of several types of inflammasomes, including nucleotide-binding oligomerization domain (NOD)-like receptors, pyrin domain-containing protein 3 (NLRP3) and absent in melanoma 2 (AIM2) inflammasomes, in the pathology of radiation-induced normal tissue injury (18).
Among all inflammasomes so far identified, the nature of NLRP3 inflammasome is the most well characterized, which can be activated through several different mechanisms, namely the canonical, non-canonical and alternative activation (19). The canonical activation of NLRP3 inflammasome requires a two-step process, including a priming signal and an activation signal. Priming involves various pathogen-associated molecular patterns (PAMPs) (like bacterial LPS), damage-associated molecular patterns (DAMPs) (like eATP, uric acid, mtDNA, etc) and their recognition by TLRs, or through cytokines (like IL-1 and TNF-α) and their signalings (20). Priming leads to the activation of NF-κB, thereby increasing the transcription of inflammasome components (like NLRP3) and pro-IL-1β (18, 21, 22). The activation signals are most commonly induced by NLRP3 agonists, such as K+ efflux, Ca2+ signals, ROS, mitochondrial dysfunction and its released contents, and lysosomal rupture (also the released cathepsins). Upon activation, NLRP3 oligomerizes into a macromolecular inflammasome complex via recruiting adaptor protein apoptosis-associated speck-like protein containing a CARD (ASC) and effector molecule pro-caspase (cysteinyl aspartate specific proteinase)-1 (23–25). Activated inflammasome mediates the maturation of IL-1β and IL-18, as well as induces pyroptosis by cleaving its effector gasdermin-D (GSDMD) (26, 27). The non-canonical NLRP3 inflammasome activation entails human caspase-4/5 and murine caspase-11. Once lipopolysaccharide (LPS) from Gram-negative bacteria enters the cytoplasm, the aforementioned inflammatory caspases will undergo autoproteolysis and further activate GSDMD to form membrane pores, thereby inducing K+ efflux and triggering NLRP3 (19, 28). The alternative NLRP3 inflammasome activation shares no similarity with its canonical or non-canonical counterparts, which follows a complex TLR4–TRIF–RIPK1–FADD–CASP8 signaling (29).
The connections between radiation injury and inflammasome are particularly intriguing since regulation of inflammasome has been extensively studied and applied, suggesting a possibility to manipulate the complex in the context of radiation injury. For example, early intervention against radiation-induced inflammasome may alleviate tissue injury, deterring the establishment of chronic inflammation (30).
In the quest for more effective management strategies of radiation injury, this review seeks to make sense of the underlying mechanism of radiation-induced inflammasome activation and to explore the characteristics of such significant pathology.
As previously described, ionizing radiation induces DSBs, DNA damage response, and oxidative stress. DSBs and DDR upregulate multi-functional signaling pathways such as NF-κB, leading to intensified immune response. Free radicals such as ROS and RNS are the major effectors of radiation damage which mediate the oxidation of biomolecules such as DNA, protein, lipids, and the regulation of several signaling pathways. With aggravation of tissue injury, some stressed and dying cells release contents that denote tissue injury to initiate damage-control-responses (10). In the development of these processes, a variety of signals are detected by the sensor protein NLRP3, leading to its activation.
It has long been accepted that ionizing radiation causes tissue injury by disrupting the balance of reduction/oxidation system, characterized by over-production of free radicals and the induction of oxidative stress. ROS and RNS are the main types of free radicals produced on exposure to ionizing radiation, which are normally counteracted by the antioxidant system (10).
Upon irradiation, ROS is immediately produced from water radiolysis (31, 32). Moreover, ROS also possesses a self-amplifying cycle. By perturbing the mitochondrial electron transfer chain (ETC), ROS along with other free radicals can further inflict mitochondrial dysfunction, leading to enhanced generation of ROS (10). More importantly, ROS is widely accepted as a potent stimulant for NLRP3 inflammasome, which also serves significant roles in radiation biology. Radiation-induced reductive/oxidative enzymes (such as NOX, COX-2, NOS, and LOXs) produce ROS in a continuous manner, which repeatedly perturbs mitochondria and persistently provides stimulants, thereby maintaining NLRP3 inflammasome activation long after exposure to ionizing radiation (23, 31–35).
According to a study investigating the links between oxidative stress and NLRP3 inflammasome, increased ROS concentration results in the dissociation of thioredoxin-interacting protein (TXNIP) from oxidized thioredoxin-1 (Trx-1), followed by interaction of TXNIP with NLRP3, resulting in activation of the latter (36, 37). Moreover, as mentioned above, impaired mitochondria subsequently release more ROS into the cytosol, binding to NLRP3 and promoting its activation (38, 39).
It has been demonstrated that radiation directly activates the NLRP3 inflammasome in human monocyte-like cells (THP-1 cells) in a ROS-dependent manner. Eliminating ROS with N-acetylcysteine (NAC) was found to ameliorate NLRP3 activation and the release of IL-1β and IL-18 in vitro (40). Moreover, in in vivo conditions, radiation-induced NLRP3 inflammasome activation is accompanied by tissue oxidative stress, characterized by increased ROS levels, either generated by radiation or from perturbed mitochondria, leading to a hyper-oxidative state. Clearing free radicals with antioxidants, or through inducing antioxidant signaling (like Nrf2 signaling), can ameliorate inflammasome activation and alleviate animal radiation response (8, 41–43). Research above reveals that ROS-induced activation of NLRP3 inflammasome plays a critical role in the initiation and development of radiation-induced injury, thus NLRP3 may be a therapeutic target for radiation-induced injury.
In addition to ROS, other DAMPs are also generated in irradiated tissues. Ionizing radiation is known to inflict tissue injury and induce cell death, which is more constantly observed in actively dividing cells such as hematopoietic and epithelial cells (44). Lytic cell death leads to the release of cell contents, some of which serve as DAMPs and mediate the initiation of inflammation (17).
In recent studies, uric acid was shown to serve as a mediator of radiation-induced NLRP3 inflammasome activation in immune cells, mainly through endocytosis and damaging the lysosome membrane, resulting in the release of cathepsin B into cytosol, then triggering NLRP3 inflammasome (24, 44). Other DAMPs, such as extracellular ATP and mitochondrial DNA (mtDNA), may also contribute to this pathology. ATP released from damaged cells binds to the P2X7 receptor on immune cells, thereby inducing pannexin-1-dependent K+ efflux as well as the influx of extracellular DAMPs into the cytosol, leading to activation of NLRP3 inflammasome (23, 24, 39). Contents released from damaged mitochondria also may possess NLRP3 activating property, especially oxidized mtDNA, which exhibits a powerful stimulatory effect (23, 37, 45). These findings suggest that IR-induced damage may activate inflammasomes via a variety of mechanisms.
The main effectors of this process are immune cells, such as macrophages, dendritic cells, NK cells, T cells, and B cells, which lead to the secretion of pro-inflammatory factors, cell death, and release of cell contents (44, 46). Unsuccessful removal of these byproducts triggers the inflammatory cascade leading to tissue injury and chronic inflammation. A schematic illustration of the current understanding and potential mechanisms (eATP and mtDNA) of radiation-induced NLRP3 inflammasome activation is presented in Figure 1.
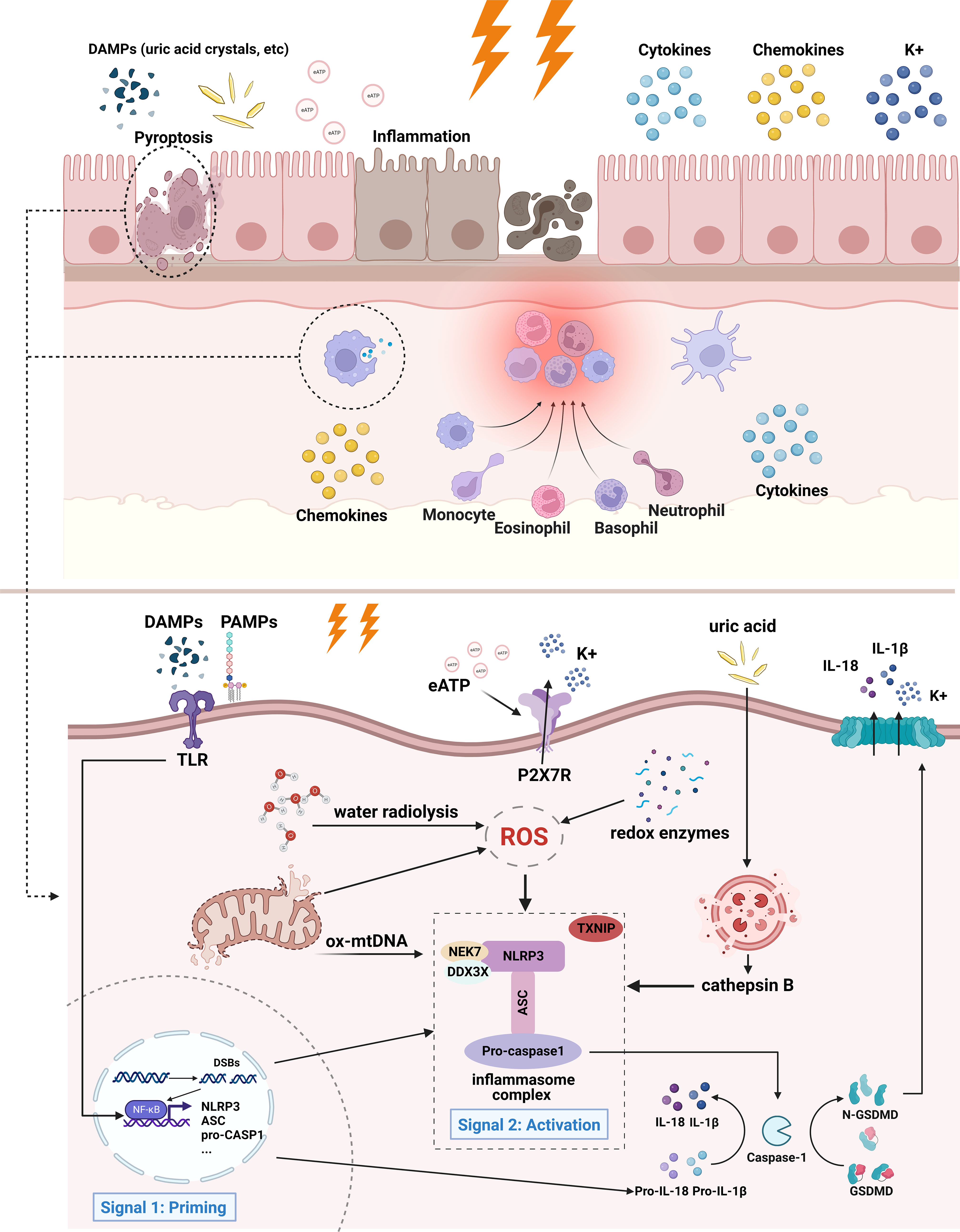
Figure 1 Mechanism of radiation-induced NLRP3 inflammasome activation. In the case of intestinal epithelial cells and infiltrating macrophages, irradiated cells initiate inflammatory responses and eventually undergo cell death. Adjacent cells may also be affected by inflammation. Specifically, extracellular PAMPs and DAMPs act as priming signals, binding to and inducing TLR signaling, thereby promoting the activation of NF-κB and the transcription of inflammasome components and pro-IL-1β. The activation signals entail ROS, K+ efflux, release of mitochondrial DNA, lysosomal rupture and cathepsin release, etc. ROS is produced from the radiolysis of water, redox enzymes, and dysfunctional mitochondria. Lysosomal membrane may be destabilized by particulate or crystalline structures (like uric acid crystal) and then lead to lysosomal rupture, releasing cathepsin B. Mitochondrial dysfunction gives rise to the release of mtDNA and cardiolipin. These activation signals stimulate NLRP3 inflammasome and lead to its oligomerization. Activated NLRP3 inflammasome mediates the maturation of IL-1β and IL-18, as well as induces pyroptosis by cleaving GSDMD. The downstream effects of NLRP3 inflammasome activation are characterized by the production of cytokines, chemokines, and recruitment of immune cells, followed by cell death. Of notes, the contribution of mtDNA and extracellular ATP to radiation-induced inflammasome has not yet been confirmed, therefore they are illustrated with fading arrows.
It is worth mentioning that the exact mechanisms of radiation-induced inflammasome activation may vary in different tissues and cells, thus may lead to divergent conclusions. Hence, we sorted and summarized the studies that investigated this process to better illustrate the differences in the physiological processes (Tables 1, 2).
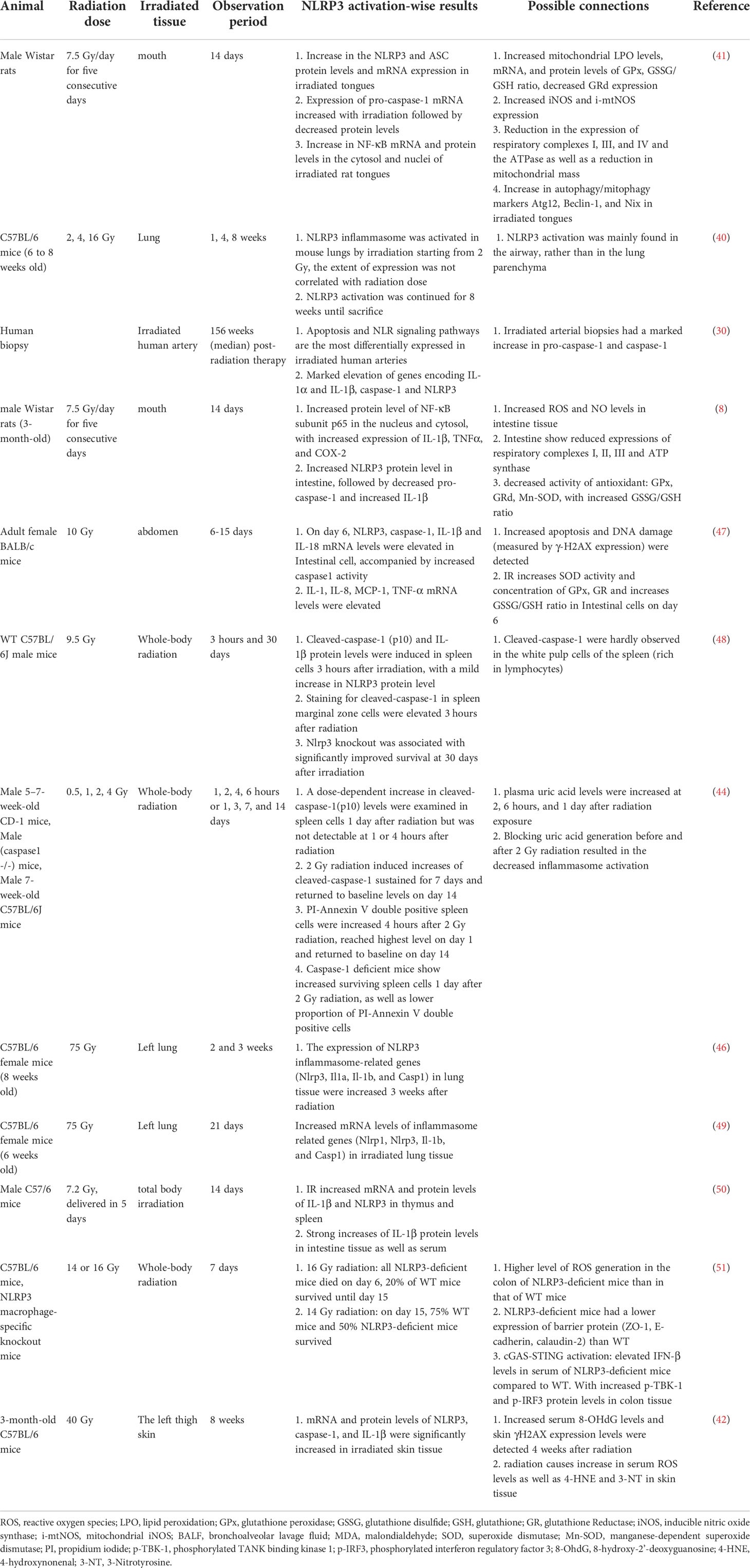
Table 1 In vivo studies involving radiation-induced NLRP3 inflammasome activation.

Table 2 In vitro studies involving radiation-induced NLRP3 inflammasome activation.
The main manifestations of radiation injury are often two-fold. Firstly, in the acute stage, the irradiated tissues exhibit typical inflammatory responses. The clinical examples include radiation-induced pneumonitis (40), oral mucositis (41), and enteropathy (55, 56). The late stage of radiation injury is often related to chronic inflammation and fibrotic pathology, such as pulmonary fibrosis (40), osteoradionecrosis of the jaws (which is also considered a fibrotic lesion) (57, 58), and radiation-induced vasculopathy (30, 59, 60). The two-stage clinical manifestations suggest the existence of a dynamic and progressive pathology, in which NLRP3 inflammasome may have certain contributions.
Cell death is a common result of inflammasome (over) activation. Regulated-cell-death, like apoptosis and pyroptosis, was discovered to play a part in the pathogenesis and progression of radiation-induced injuries (34, 48). Pyroptosis is well known to lie downstream of inflammasome activation and is executed by GSDMD. Inflammasome-activated caspase-1 (also other inflammatory caspases like -4, -5, -11) cleaves GSDMD into N- and C-terminal fragments. GSDMD-N then translocate to inner leaflet of membrane and bind to phospholipids, oligomerize in membranes to form pores, allowing the release of cell contents like mature IL-1β, IL-18, TNF-α and HMGB1, followed by pyroptotic cell death. GSDMD pores may further enhance NLRP3 inflammasome activation by promoting K+ efflux, forming a positive-feedback (61–63). Moreover, with research in cell death continuing to abound, it has been recognized that apoptosis may also be induced by inflammasomes. On the one hand, in cells with low expression of GSDMD, caspase-1 initiates apoptosis either through activating Bid (an extrinsic apoptosis pathway mediator) or through processing caspase-7, rather than inducing pyroptosis (28). On the other hand, ASC specks may also recruit and activate caspase-8 and undergo apoptosis (64). It has also recently been demonstrated that apoptosis may enhance NLRP3 inflammasome activation by cleaving pannexin-1 to induce K+ efflux (65). To recapitulate briefly, both pyroptosis and apoptosis lie downstream of inflammasome activation, depending on the expression of GSDMD, and both may contribute to further inflammasome assembling (28).
Cell death caused by inflammasome activation may present diverse results among various types of tissues. In macrophages, for example, radiation acutely activates NLRP3 inflammasome signaling both in vivo and in vitro. Low dose (<5 Gy) radiation stimulates macrophages to secrete proinflammatory molecules while high dose (>10 Gy) promotes its pyroptosis (48), which may justify immune cell loss and myeloid suppression after extreme radiation exposure or high-dose RT (66–68). Moreover, the epithelial tissues, such as oral mucosa, gut mucosa, as well as lung epithelium, are particularly susceptible to ionizing radiation. Inflammasome activation and pyroptosis have been observed in these tissues after exposure to radiation (47, 48, 53, 69). Excessive cell death in epithelium may lead to the breakdown of barriers such as skin, gut mucosal barrier, and alveolar epithelial barrier. Compromised barriers allow pathogenic microbes to invade inflammatory tissue, exacerbating immune response and tissue damage (17, 70). LPS from Gram-negative bacteria may also contribute to the inflammasome cascade through non-canonical NLRP3 inflammasome activation, thereby leading to even deteriorated conditions (29). Equally important, immoderate cell death in vascular endothelial cells causes vascular dysfunction, leading to increased permeability, impaired vascular tone, and altered blood homeostasis, aggravating already severe damage caused by radiation (10, 53).
In the background of radiation injury, cell death should be contained within a reasonable range to minimize normal tissue injury, since excessive cell death leads to tissue dysfunction of various kinds, adding to more unwanted events. Targeting solely against apoptosis or pyroptosis, however, may not fulfill the designated goal, since the two cell death machineries compete to be performed (64). Given that both pyroptosis and apoptosis lie downstream of inflammasome activation, it is therefore preferable to target this upstream pro-inflammatory signaling. In other words, early preventive measures are necessary to prevent irreversible damage to cells, which will be discussed in the later section.
Inflammasome activation enables the maturation and release of IL-1β and IL-18, thus functioning as an immune regulator in radiation-induced injury. Both IL-1β and IL-18 are multi-functional immune modulator and inflammatory amplifier, mediating the initiation of innate and adaptive immune response (17, 71). Cascaded inflammation may lead to excessive production of numerous proinflammatory cytokines (like IL-1, IL-6, TNF-α, etc) and chemokines, together with protracted radiation stimulation, may give rise to the establishment of chronic inflammation and tissue injury (14, 72).
It is widely accepted that the chronic phase of radiation-induced tissue injury involves damage repair and tissue remodeling. However, when accompanied with persistent oxidative stress and non-resolving inflammation, damaged tissues may not heal properly and result in fibrotic lesions (14, 73). Radiation-induced fibrosis (RIF) is characterized by increased collagen deposition, poor vascularity, and scarring (57). The current understanding on the development of RIF entails the augment of TGF-β signaling, vascular injury and hypoxia, chronic inflammation, and finally, the activation of myofibroblasts that mediates aberrant tissue remodeling (74).
The contributions of NLRP3 inflammasome to various types of fibrosis are gradually recognized (75). Mainly, IL-1β and IL-18 possess the capability to directly induce collagen synthesis in fibroblasts, or via interacting with SMAD signaling and promote epithelial-mesenchymal-transition, as well as inducing TGF-β through activating NF-κB (75, 76). NLRP3 is also demonstrated to directly participate in fibrosis by augmenting TGF-β signaling, independently of its inflammasome property. Though most studies on the pro-fibrosis property of inflammasome were conducted on the model of chronic kidney disease or liver fibrosis (77, 78), we may well extrapolate the potential contribution of inflammasome to RIF based on these understandings.
TGF-β, a prominent pro-fibrosis mediator, which activates fibroblasts through the SMAD signaling, is in the spotlight of studies on RIF (79, 80). The production and the function of TGF-β relate to inflammasome activation in several respects. As is discussed afore, ROS is acutely and persistently produced upon irradiation, which not only activates NLRP3 inflammasome, but is also discovered to promote the production and enhance the signaling of TGF-β (81). Moreover, the inflammasome-mediated vascular injury and dysfunction may help establish a hypoxic environment, which further augments the production of free radicals. A hypoxic state also leads to increased HIF-1a signaling and promotes various pro-fibrotic mediators (74). Potentiated local inflammation as well contributes to fibrogenic processes (14).
Further research is recommended to unravel the exact role of NLRP3 inflammasome in RIF, as regulatory strategies of the complex are widely studied and practiced. In case there is a strong connection between NLRP3 inflammasome and RIF, the latter may be prevented by targeting the former.
Although we have stressed on the detrimental aspects of inflammasome-induced response, the beneficial aspects of inflammation should not be neglected. For instance, inflammasome-induced cell death may assist the clearance of severely damaged cells. Moreover, the attracted immune cells also serve to protect tissues from barrier breakdown and potential infections, and scavenge dead cells and initiate tissue repair and remodeling. They only have a detrimental effect if the balance of inflammation is out of control. Hence, in-depth characterization of these machineries and designing of strategies specifically against undesired injuries is of vital importance.
As previously explained and analyzed, NLRP3 inflammasome plays a key role in radiation injury, as it is activated by radiation, and then mediates at least a certain range of injuries. In other words, radiation is the one that gives the order, it is the inflammasome that responds and kills—functioning as the executioner. Fortunately, there exist ample options to target NLRP3 inflammasome and restrain it from causing tissue injuries.
Radiotherapy dose is directly related to the achievement of desired local-regional control of cancer. However, severe adverse effects of radiation are the main reason for reducing the radiotherapy dose, which is especially the case when the tolerance of “organ at risk” is taken into account (9, 82). For example, if the unwanted effects (indicative symptoms or pathological changes for endpoints) of organ at risk arrive too early, the designated RT dose may not be accomplished, leading to increased risk of recurrence. Moreover, even in the absence of acute symptoms, radiologists need to be cautious when considering the late adverse effects of organs that are sensitive to radiation, restraining the efficacy of radiotherapy. Hence, strategies for counteracting the adverse effects of radiation should be explored in order to achieve a higher upper limit of radiotherapy dose.
We here propose the necessity of early intervention against inflammasome in radiation injury. The definitions of the adjective “early” are two-fold, comprising both macroscopic and microscopic perspectives. Firstly, by referring to early intervention at the clinical level, we intend to emphasize the active prevention of radiation injury, rather than passively dealing with end-stage problems. Studies focused on other NLRP3-associated clinical problems, such as post-myocardial infarction fibrosis and autoimmune diseases, have discovered that inhibition of NLRP3 inflammasome in the early phases of diseases may reduce the occurrence of severe, late-stage lesions (83, 84). Moreover, at the molecular level, the activation and downstream effects of NLRP3 inflammasome are sequential, consecutive processes, which suggests that if the inhibition is designated at the upstream of the pathway, such as transcriptional (priming) repression, sensor protein inhibition, and removal of stimulants, the entire signalling cascade may be suppressed. For example, inhibiting the activation of inflammasome signalling was shown to reduce macrophage pyroptosis (48), as well as preserve the integrity of the epithelial barrier (50, 73), thereby reducing the occurrence of tissue dysfunction.
Conventional targeting against NLRP3 inflammasome include stimulant removal, transcriptional regulation (41, 49, 50, 54), activation inhibition (84, 85), effector protein (namely caspase-1) targeting (86), and product targeting (mostly against IL-1β and GSDMD) (30, 87, 88). The representative therapeutics based on these concepts are briefly summarized in Table 3.
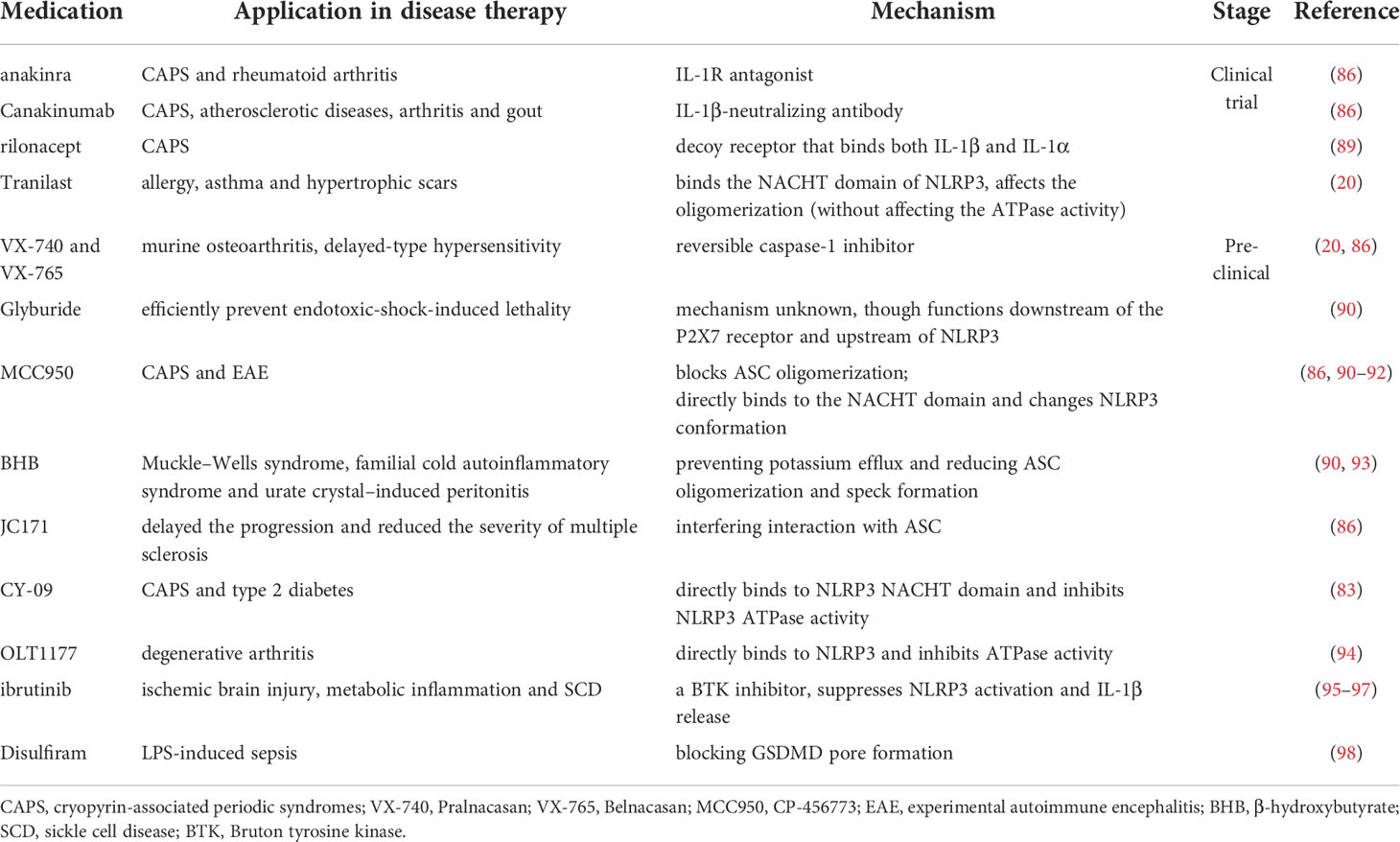
Table 3 Conventional therapeutic strategies for regulating the NLRP3 inflammasome.
Early intervention against NLRP3 inflammasome may lead to better outcomes. When solely considering the manipulation of NLRP3 inflammasome, it is noteworthy that these strategies may yield various results, as they are designed for different stages of the signaling cascade. First and foremost, elimination of ROS with antioxidants leads to suppression of the entire pathway accompanied by reduction in free radical-induced oxidative damage, which is the most desired situation (8, 52, 99). Moreover, modifying the NLRP3 inflammasome via transcriptional regulation or direct inhibition of activation (suppressing NLRP3 protein function) may generate similar outcomes, as they both prevent the oligomerization of the inflammasome complex. However, the rest of the mentioned strategies bear extra unwanted effects, since different types of inflammasomes share an identical set of downstream signaling. For example, inhibition of caspase-1 may interrupt the proper functioning of other inflammasomes (such as AIM2 and NLRC4 inflammasomes) as well, increasing the susceptibility to infection. Specific targeting of IL-1β may lead to increased incidence of infection too, as observed in the CANTOS trial (86, 100). Playing downstream of inflammasome activation, pyroptosis may be precluded via disrupting the upstream signaling. However, manipulation of the effector for pyroptosis, GSDMD, may not redeem the dying cells, as other programmed cell death pathways may substitute pyroptosis leading to cell death (64). To summarize these machineries, it is obvious that the more upstream the interventions are aimed at, the greater the outcomes may yield. Our proposition on early intervention against NLRP3 inflammasome is illustrated in Figure 2.
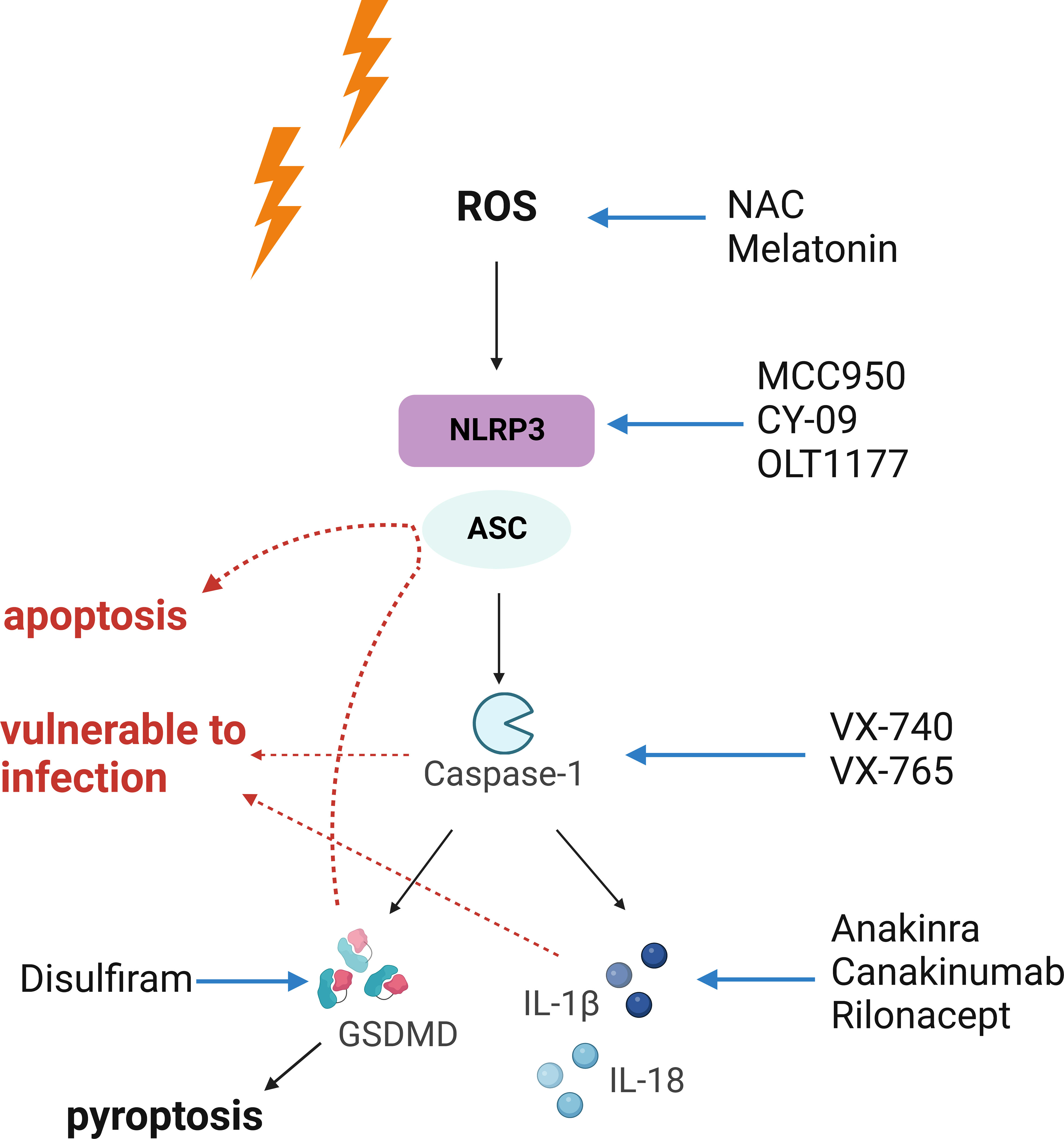
Figure 2 The necessity of early intervention against radiation-induced injury. The NLRP3 inflammasome acts as an executioner in radiation-induced tissue injury. Upon activation, it triggers a cascaded reaction, which indicates that interventions targeting upstream of the signaling may yield the most desirable outcomes. In comparison, the results of downstream intervention may be accompanied with unwanted effects. Red arrows indicate unwanted effects of strategies suppressing pyroptosis, caspase-1, or IL-1β. Blockade of pyroptosis may lead to activation of apoptosis or other death pathways. Also, caspase-1 is required for the proper function of other inflammasomes, and blockade at this point may shut down the entire inflammasome signaling, leaving the organism vulnerable to infection. Suppressing IL-1β also increases the risk of infection. Both Figures 1, 2 were created with BioRender.com.
NF-κB is a multi-functional regulator that controls diverse cellular processes like immune response, proliferation and cell survival, etc. Various PAMPs, DAMPs, and some endogenous molecules may give rise to NF-κB activation through triggering TLRs, TNFR1 and IL-1R signaling (101). In respect of radiation-induced response, NF-κB upregulates diverse pro-survival (such as antioxidant enzymes, anti-apoptotic proteins and growth factors) and pro-inflammatory (various cytokines, chemokines and adhesion molecules) genes expression (102), thus exhibiting either protective and damaging effects.
NF-κB contributes to activation of NLRP3 inflammasome by providing NLRP3 components and pro-IL-1β. Targeting NF-κB signaling may disrupt priming and ameliorate NLRP3 inflammasome activation. Targeting IRAK4, a mediator of TLR-NF-κB signaling, may partially suppress NLRP3 transcription (24). Resveratrol, a natural non-flavonoid polyphenolic as well as an antioxidant, is shown to mitigate radiation-induced NF-κB activation through regulating SIRT1 (50, 54). Similarly, Rosiglitazone, an agonist of PPARγ, is able to suppress the expression and release of NLRP3, caspase-1, and, IL-1β through attenuating NF-κB signaling (35, 103).
Post-transcriptional regulation of NLRP3 is mainly mediated by a group of miRNA and long non-coding RNA (lncRNA). miR-133b, miR-20b, miR-223 and others mediate the silencing of the NLRP3 gene. lncRNAs interact with miRNAs and can either promote or attenuate inflammasome signaling depending on tissues and pathologies (23, 83). Moreover, NLRP3 inflammasome has also been targeted post-translationally through regulating its phosphorylation, ubiquitination, and their reverse processes. For instance, targeting NLRP3 phosphorylase like JNK1 and promoting NLRP3 degradation via deubiquitylation inhibitor (23, 86) has been applied in inflammatory diseases, and is also a potential therapeutic strategy in radiation injury.
NLRP3 inflammasome has a complex regulatory mechanism, which may interact with key proteins involved in other aspects of cellular physiology. It is therefore possible to regulate NLRP3 inflammasome through modulating these intersecting pathways.
The competition of DDX3X between NLRP3 inflammasome and stress granules (SGs) provides an option to regulate NLRP3 inflammasome through modulating the formation of the latter. DDX3X is a necessary component of the NLRP3 inflammasome, which is also involved in the initiation of SGs assembly. After formation of SGs, DDX3X is sequestered within the complex, leaving NLRP3 inflammasome inactivated (104–106). Such interaction is promising for designing therapeutic strategies. For instance, pharmacological induction of SGs was shown to protect cochlea cells against ototoxic drugs challenge (107), and a similar strategy may be explored and applied in radioprotection as well.
Autophagy is also known to interact with NLRP3 inflammasome, and therefore, may serve as a potential regulatory strategy for radiation-induced inflammasome as well. Autophagy plays a particularly important role in the recycling and removal of damaged cell components (108). In the context of radiation injury, the clearance of damaged mitochondria through autophagy or mitophagy (a form of autophagy for selective removal of dysfunctional or redundant mitochondria) may avoid NLRP3 inflammasome activation (83, 108–110). Correspondingly, in a recent study, induction of autophagy was found to ameliorate radiation-induced enteropathy by promoting phagocytosis of the NLRP3 inflammasome, which further confirmed our hypothesis (111).
Dysbiosis of resident microbiota may contribute to enhanced immune response (112, 113). Moreover, barrier breakdown in radiation injury may allow the translocation of pathogenic microbes and increased infiltration of immune cells, which further exacerbates tissue damage (39, 114–116). Inspired by these observations, researchers have utilized commensal microbiota against radiation injury and obtained fruitful outcomes. Microbiota transplantation has been shown to alleviate radiation injury of the intestinal and oral mucosa; the results also indicated that transplanted microbiota can regulate immune response and further influence the outcomes of irradiated tissue (117, 118). Though the exact relationship between radiation injury and NLRP3 inflammasome has not yet been illustrated, fecal microbiota transplantation was found to inhibit the expression of inflammasomes components (NLRP3, ASC, caspase-1, and IL-1β) in rat brain, thus ameliorating stress-induced depression-like behaviors (119, 120). The effectiveness of fecal microbiota transplantation in alleviating radiation injury and suppressing NLRP3 inflammasome points towards a connection between commensal microbiota and radiation-induced inflammasome. Efforts should be made to better comprehend their interactions and design pertinent therapeutic strategies.
Mesenchymal stem cell (MSC) therapy has emerged as a promising therapeutic modality for multiple diseases considering their convenient isolation and culture, low immunogenicity, regenerative and multiple differentiation abilities, and potent immunomodulatory capacities. MSC therapy has been shown to attenuate radiation-induced brain damage by suppressing microglia pyroptosis, reducing ROS production, and NLRP3 inflammasome activation (121). Also, modified MSCs serve as a practical assistant in gene therapy for radiation injury, which efficiently deliver target genes to the injured sites and alleviate radiation injury (122). However, MSCs may transform into malignant cells; hence, it is necessary to evaluate potential side effects and to prevent them in advance.
Exosomes have been applied to the treatment of radiation injury as vectors for therapeutic agents (123). Moreover, recent evidence also suggests that the secretions of exosomes interact with inflammasome pathology (124, 125), which may be involved in MSCs-mediated therapies for radiation injury (126). As a therapeutic carrier, exosomes are considered safer than cell therapy because of their lower immunogenicity. Moreover, exosomes have almost no cytotoxicity, better storage stability, antiserum aggregation ability, and biological activity (123). Based on the above-mentioned advantages, exosomes are potential promising carriers for delivery of various therapeutic loads, such as siRNA and miRNAs against inflammasome components, to the desired target sites.
As discussed in this review, NLRP3 inflammasome is one of the executioners of radiation-induced tissue injury, which mediates a range of common radiation-induced illnesses. With ongoing advances in the research on radiation-induced inflammasome, the prospects of targeting inflammasome as a preventive measure against radiation injury appear practicable. However, there are some unsolved questions in this field. For example, the concepts this review proposes mainly focuses on the detrimental aspects of the inflammasome in radiation injury; however, the inflammasome is also responsible for tumor immunity, pathogen clearance, and initiating tissue repair in some cases (100). More importantly, due to the diverse expression patterns of the NLRP3 inflammasome, its exact roles among different forms of injuries may vary. Proper targeting of inflammasomes should be elaborate and precise, which is why there is a need for further in-depth research.
Radiotherapy is an effective treatment modality for various malignancies in clinical practice. Nonetheless, the side effects of ionizing radiation restrict its use to some extent. This is particularly the case when high doses are required for the treatment of advanced, unresectable malignancies. More importantly, the currently used measures for protecting normal tissue are generally passive and are typically implemented after the development of injuries. In between prevention, mitigation, and treatment, there is no doubt that the first one stands out as the most ideal strategy (127).
However, the reality of radioprotectants is frustrating. According to the 2008 ASCO guidelines for the clinical use of radiotherapy protectants, only dexrazoxane, doxorubicin, amifostine, and palifermin are recommended under several specific circumstances (128). This is because of the inherent challenges in the development of an effective and harmless radioprotectant. Till date, no clinical trials have been conducted in the context of radiation-induced inflammation, suggesting the inadequacy of research in this field.
Based on the analysis of the mechanism and function of the NLRP3 inflammasome in radiation injury, this review may provide insights for developing better therapeutic strategies against radiation-induced injury to normal tissues, as well as highlight the critical role of this machinery. In-depth characterization of the physiology of radiation injury and its counteracting response is crucial for the further advancement of radiotherapy.
HC, LC, MH, and JH contributed to design, drafted, and critically revised the manuscript. ZC and XY contributed to conception, design, and critically revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Nature Science Foundation of China (No. 82173458) and College Students’ Innovative Entrepreneurial Training Plan Program (No. X202012121296 & No. X202012121293).
The illustrations this review presented were created with BioRender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.

1. Texakalidis P, Giannopoulos S, Tsouknidas I, Song S, Rivet DJ, Reiter ER, et al. Prevalence of carotid stenosis following radiotherapy for head and neck cancer: A systematic review and meta-analysis. Head Neck (2020) 42:1077–88. doi: 10.1002/hed.26102
2. Liao W, Zheng Y, Bi S, Zhang B, Xiong Y, Li Y, et al. Carotid stenosis prevalence after radiotherapy in nasopharyngeal carcinoma: A meta-analysis. Radiother Oncol (2019) 133:167–75. doi: 10.1016/j.radonc.2018.11.013
3. Banfill K, Giuliani M, Aznar M, Franks K, McWilliam A, Schmitt M, et al. Cardiac toxicity of thoracic radiotherapy: Existing evidence and future directions. J Thorac Oncol (2021) 16:216–27. doi: 10.1016/j.jtho.2020.11.002
4. Lawrie TA, Green JT, Beresford M, Wedlake L, Burden S, Davidson SE, et al. Interventions to reduce acute and late adverse gastrointestinal effects of pelvic radiotherapy for primary pelvic cancers. Cochrane Database Syst Rev (2018) 1:CD012529. doi: 10.1002/14651858.CD012529.pub2
5. Mukesh M, Harris E, Jena R, Evans P, Coles C. Relationship between irradiated breast volume and late normal tissue complications: A systematic review. Radiother Oncol (2012) 104:1–10. doi: 10.1016/j.radonc.2012.04.025
6. Lambrecht M, Eekers DBP, Alapetite C, Burnet NG, Calugaru V, Coremans IEM, et al. Radiation dose constraints for organs at risk in neuro-oncology; the European particle therapy network consensus. Radiother Oncol (2018) 128:26–36. doi: 10.1016/j.radonc.2018.05.001
7. Sonis S, Elting L, Keefe D, Nguyen H, Grunberg S, Randolph-Jackson P, et al. TRIAD burden of illness investigators. unanticipated frequency and consequences of regimen-related diarrhea in patients being treated with radiation or chemoradiation regimens for cancers of the head and neck or lung. Support Care Cancer (2015) 23:433–9. doi: 10.1007/s00520-014-2395-9
8. Fernández-Gil B, Moneim AEA, Ortiz F, Shen Y-Q, Soto-Mercado V, Mendivil-Perez M, et al. Melatonin protects rats from radiotherapy-induced small intestine toxicity. PloS One (2017) 12:e0174474. doi: 10.1371/journal.pone.0174474
9. Dörr W, Herrmann T, Baumann M. Application of organ tolerance dose-constraints in clinical studies in radiation oncology. Strahlenther Onkol (2014) 190:621–7. doi: 10.1007/s00066-014-0613-5
10. Baselet B, Sonveaux P, Baatout S, Aerts A. Pathological effects of ionizing radiation: endothelial activation and dysfunction. Cell Mol Life Sci (2019) 76:699–728. doi: 10.1007/s00018-018-2956-z
11. Kavanagh JN, Redmond KM, Schettino G, Prise KM. DNA Double strand break repair: A radiation perspective. Antioxid Redox Signal (2013) 18:2458–72. doi: 10.1089/ars.2012.5151
12. Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell (2010) 40:179–204. doi: 10.1016/j.molcel.2010.09.019
13. Hellweg CE, Spitta LF, Koch K, Chishti AA, Henschenmacher B, Diegeler S, et al. The role of the nuclear factor κB pathway in the cellular response to low and high linear energy transfer radiation. Int J Mol Sci (2018) 19:2220. doi: 10.3390/ijms19082220
14. Cytlak UM, Dyer DP, Honeychurch J, Williams KJ, Travis MA, Illidge TM. Immunomodulation by radiotherapy in tumour control and normal tissue toxicity. Nat Rev Immunol (2021) 22(2):124–38. doi: 10.1038/s41577-021-00568-1
15. Santivasi WL, Xia F. Ionizing radiation-induced DNA damage, response, and repair. Antioxid Redox Signal (2014) 21:251–9. doi: 10.1089/ars.2013.5668
16. Frey B, Hehlgans S, Rödel F, Gaipl US. Modulation of inflammation by low and high doses of ionizing radiation: Implications for benign and malign diseases. Cancer Lett (2015) 368:230–7. doi: 10.1016/j.canlet.2015.04.010
17. François A, Milliat F, Guipaud O, Benderitter M. Inflammation and immunity in radiation damage to the gut mucosa. BioMed Res Int (2013) 2013:123241. doi: 10.1155/2013/123241
18. Karki R, Kanneganti T-D. Diverging inflammasome signals in tumorigenesis and potential targeting. Nat Rev Cancer (2019) 19:197–214. doi: 10.1038/s41568-019-0123-y
19. Lee K-H, Kang T-B. The molecular links between cell death and inflammasome. Cells (2019) 8:1057. doi: 10.3390/cells8091057
20. Swanson KV, Deng M, Ting JP-Y. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol (2019) 19:477–89. doi: 10.1038/s41577-019-0165-0
21. Zhong Y, Kinio A, Saleh M. Functions of NOD-like receptors in human diseases. Front Immunol (2013) 4:333. doi: 10.3389/fimmu.2013.00333
22. Franchi L, Muñoz-Planillo R, Núñez G. Sensing and reacting to microbes through the inflammasomes. Nat Immunol (2012) 13:325–32. doi: 10.1038/ni.2231
23. Zheng D, Liwinski T, Elinav E. Inflammasome activation and regulation: toward a better understanding of complex mechanisms. Cell Discovery (2020) 6:36. doi: 10.1038/s41421-020-0167-x
24. He Y, Hara H, Núñez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci (2016) 41:1012–21. doi: 10.1016/j.tibs.2016.09.002
25. Liu D, Zeng X, Li X, Cui C, Hou R, Guo Z, et al. Advances in the molecular mechanisms of NLRP3 inflammasome activators and inactivators. Biochem Pharmacol (2020) 175:113863. doi: 10.1016/j.bcp.2020.113863
26. Tang D, Kang R, Berghe Vanden T, Vandenabeele P, Kroemer G. The molecular machinery of regulated cell death. Cell Res (2019) 29(5):347–64. doi: 10.1038/s41422-019-0164-5
27. Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 inflammasome: An overview of mechanisms of activation and regulation. Int J Mol Sci (2019) 20:3328. doi: 10.3390/ijms20133328
28. Tsuchiya K. Inflammasome-associated cell death: Pyroptosis, apoptosis, and physiological implications. Microbiol Immunol (2020) 64:252–69. doi: 10.1111/1348-0421.12771
29. Huang Y, Xu W, Zhou R. NLRP3 inflammasome activation and cell death. Cell Mol Immunol (2021) 18:2114–27. doi: 10.1038/s41423-021-00740-6
30. Christersdottir T, Pirault J, Gisterå A, Bergman O, Gallina AL, Baumgartner R, et al. Prevention of radiotherapy-induced arterial inflammation by interleukin-1 blockade. Eur Heart J (2019) 40:2495–503. doi: 10.1093/eurheartj/ehz206
31. Hekim N, Cetin Z, Nikitaki Z, Cort A, Saygili EI. Radiation triggering immune response and inflammation. Cancer Lett (2015) 368:156–63. doi: 10.1016/j.canlet.2015.04.016
32. Wei J, Wang B, Wang H, Meng L, Zhao Q, Li X, et al. Radiation-induced normal tissue damage: Oxidative stress and epigenetic mechanisms. Oxid Med Cell Longev (2019) 2019:3010342. doi: 10.1155/2019/3010342
33. Huang S, Che J, Chu Q, Zhang P. The role of NLRP3 inflammasome in radiation-induced cardiovascular injury. Front Cell Dev Biol (2020) 8:140. doi: 10.3389/fcell.2020.00140
34. Yahyapour R, Motevaseli E, Rezaeyan A, Abdollahi H, Farhood B, Cheki M, et al. Reduction-oxidation (redox) system in radiation-induced normal tissue injury: Molecular mechanisms and implications in radiation therapeutics. Clin Transl Oncol (2018) 20:975–88. doi: 10.1007/s12094-017-1828-6
35. Farhood B, Ashrafizadeh M, khodamoradi E, Hoseini-Ghahfarokhi M, Afrashi S, Musa AE, et al. Targeting of cellular redox metabolism for mitigation of radiation injury. Life Sci (2020) 250:117570. doi: 10.1016/j.lfs.2020.117570
36. Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol (2010) 11:136–40. doi: 10.1038/ni.1831
37. Abderrazak A, Syrovets T, Couchie D, El Hadri K, Friguet B, Simmet T, et al. NLRP3 inflammasome: From a danger signal sensor to a regulatory node of oxidative stress and inflammatory diseases. Redox Biol (2015) 4:296–307. doi: 10.1016/j.redox.2015.01.008
38. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature (2011) 469:221–5. doi: 10.1038/nature09663
39. Wei J, Wang H, Wang H, Wang B, Meng L, Xin Y, et al. The role of NLRP3 inflammasome activation in radiation damage. BioMed Pharmacother (2019) 118:109217. doi: 10.1016/j.biopha.2019.109217
40. Li X, Gong Y, Li D, Xiang L, Ou Y, Jiang L, et al. Low-dose radiation therapy promotes radiation pneumonitis by activating NLRP3 inflammasome. Int J Radiat Oncol Biol Phys (2020) 107:804–14. doi: 10.1016/j.ijrobp.2020.02.643
41. Ortiz F, Acuña-Castroviejo D, Doerrier C, Dayoub JC, López LC, Venegas C, et al. Melatonin blunts the mitochondrial/NLRP3 connection and protects against radiation-induced oral mucositis. J Pineal Res (2015) 58:34–49. doi: 10.1111/jpi.12191
42. Wei J, Zhao Q, Zhang Y, Shi W, Wang H, Zheng Z, et al. Sulforaphane-mediated Nrf2 activation prevents radiation-induced skin injury through inhibiting the oxidative-Stress-Activated DNA damage and NLRP3 inflammasome. Antioxid (Basel) (2021) 10:1850. doi: 10.3390/antiox10111850
43. Huang S, Huang Y, Lin W, Wang L, Yang Y, Li P, et al. Sitagliptin alleviates radiation-induced intestinal injury by activating NRF2-antioxidant axis, mitigating NLRP3 inf–lammasome activation, and reversing gut microbiota disorder. Oxid Med Cell Longev (2022) 2022:2586305. doi: 10.1155/2022/2586305
44. Stoecklein VM, Osuka A, Ishikawa S, Lederer MR, Wanke-Jellinek L, Lederer JA. Radiation exposure induces inflammasome pathway activation in immune cells. J Immunol (2015) 194:1178–89. doi: 10.4049/jimmunol.1303051
45. Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity (2012) 36:401–14. doi: 10.1016/j.immuni.2012.01.009
46. Sohn S-H, Lee JM, Park S, Yoo H, Kang JW, Shin D, et al. The inflammasome accelerates radiation-induced lung inflammation and fibrosis in mice. Environ Toxicol Pharmacol (2015) 39:917–26. doi: 10.1016/j.etap.2015.02.019
47. Wu D, Han R, Deng S, Liu T, Zhang T, Xie H, et al. Protective effects of flagellin a N/C against radiation-induced NLR pyrin domain containing 3 inflammasome-dependent pyroptosis in intestinal cells. Int J Radiat Oncol Biol Phys (2018) 101:107–17. doi: 10.1016/j.ijrobp.2018.01.035
48. Liu Y, Chen J, Zhang Z, Ma X, Chen Y, Du X, et al. NLRP3 inflammasome activation mediates radiation-induced pyroptosis in bone marrow-derived macrophages. Cell Death Dis (2017) 8:e2579–9. doi: 10.1038/cddis.2016.460
49. Shin D, Lee G, Sohn S-H, Park S, Jung K-H, Lee JM, et al. Regulatory T cells contribute to the inhibition of radiation-induced acute lung inflammation via bee venom phospholipase A2 in mice. Toxins (Basel) (2016) 8:E131. doi: 10.3390/toxins8050131
50. Sun H, Cai H, Fu Y, Wang Q, Ji K, Du L, et al. The protection effect of resveratrol against radiation-induced inflammatory bowel disease via NLRP-3 inflammasome repression in mice. Dose Response (2020) 18:1559325820931292. doi: 10.1177/1559325820931292
51. Wu T, Gao J, Liu W, Cui J, Yang M, Guo W, et al. NLRP3 protects mice from radiation-induced colon and skin damage via attenuating cGAS-STING signaling. Toxicol Appl Pharmacol (2021) 418:115495. doi: 10.1016/j.taap.2021.115495
52. Chatterjee S, Pietrofesa RA, Park K, Tao J-Q, Carabe-Fernandez A, Berman AT, et al. LGM2605 reduces space radiation-induced NLRP3 inflammasome activation and damage in In vitro lung vascular networks. Int J Mol Sci (2019) 20:E176. doi: 10.3390/ijms20010176
53. Smith AO, Ju W, Adzraku SY, wenyi L, Yuting C, Qiao J, et al. Gamma radiation induce inflammasome signaling and pyroptosis in microvascular endothelial cells. J Inflamm Res (2021) 14:3277–88. doi: 10.2147/JIR.S318812
54. Fu Y, Wang Y, Du L, Xu C, Cao J, Fan T, et al. Resveratrol inhibits ionising irradiation-induced inflammation in MSCs by activating SIRT1 and limiting NLRP-3 inflammasome activation. Int J Mol Sci (2013) 14:14105–18. doi: 10.3390/ijms140714105
55. Wang J, Boerma M, Fu Q, Hauer-Jensen M. Significance of endothelial dysfunction in the pathogenesis of early and delayed radiation enteropathy. World J Gastroenterol (2007) 13:3047–55. doi: 10.3748/wjg.v13.i22.3047
56. Hauer-Jensen M, Denham JW, Andreyev HJN. Radiation enteropathy–pathogenesis, treatment and prevention. Nat Rev Gastroenterol Hepatol (2014) 11:470–9. doi: 10.1038/nrgastro.2014.46
57. Delanian S, Lefaix J-L. The radiation-induced fibroatrophic process: therapeutic perspective via the antioxidant pathway. Radiother Oncol (2004) 73:119–31. doi: 10.1016/j.radonc.2004.08.021
58. He Y, Ma C, Hou J, Li X, Peng X, Wang H, et al. Chinese Expert group consensus on diagnosis and clinical management of osteoradionecrosis of the mandible. Int J Oral Maxillofac Surg (2020) 49:411–9. doi: 10.1016/j.ijom.2019.06.015
59. Mezzaroma E, Mikkelsen RB, Toldo S, Mauro AG, Sharma K, Marchetti C, et al. Role of interleukin-1 in radiation-induced cardiomyopathy. Mol Med (2015) 21(1):210–8. doi: 10.2119/molmed.2014.00243
60. Weintraub NL, Jones WK, Manka D. Understanding radiation-induced vascular disease. J Am Coll Cardiol (2010) 55:1237–9. doi: 10.1016/j.jacc.2009.11.053
61. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: Recommendations of the nomenclature committee on cell death 2018. Cell Death Differ (2018) 25:486–541. doi: 10.1038/s41418-017-0012-4
62. Shi J, Gao W, Shao F. Pyroptosis: Gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci (2017) 42:245–54. doi: 10.1016/j.tibs.2016.10.004
63. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin d causes pyroptosis by forming membrane pores. Nature (2016) 535:153–8. doi: 10.1038/nature18629
64. Zhang P, Liu Y, Hu L, Huang K, Hong M, Wang Y, et al. NLRC4 inflammasome–dependent cell death occurs by a complementary series of three death pathways and determines lethality in mice. Sci Adv (2021) 7:eabi9471. doi: 10.1126/sciadv.abi9471
65. Chen KW, Demarco B, Heilig R, Shkarina K, Boettcher A, Farady CJ, et al. Extrinsic and intrinsic apoptosis activate pannexin-1 to drive NLRP3 inflammasome assembly. EMBO J (2019) 38:e101638. doi: 10.15252/embj.2019101638
66. Wu T, Liu W, Fan T, Zhong H, Zhou H, Guo W, et al. 5-androstenediol prevents radiation injury in mice by promoting NF-κB signaling and inhibiting AIM2 inflammasome activation. BioMed Pharmacother (2020) 121:109597. doi: 10.1016/j.biopha.2019.109597
67. Goswami S, Haldar C. Melatonin as a possible antidote to UV radiation induced cutaneous damages and immune-suppression: An overview. J Photochem Photobiol B (2015) 153:281–8. doi: 10.1016/j.jphotobiol.2015.10.006
68. Bartolini D, Wang Y, Zhang J, Giustarini D, Rossi R, Wang GY, et al. A seleno-hormetine protects bone marrow hematopoietic cells against ionizing radiation-induced toxicities. PloS One (2019) 14:e0205626. doi: 10.1371/journal.pone.0205626
69. Hu B, Jin C, Li H-B, Tong J, Ouyang X, Cetinbas NM, et al. The DNA-sensing AIM2 inflammasome controls radiation-induced cell death and tissue injury. Science (2016) 354:765–8. doi: 10.1126/science.aaf7532
70. Wu D-M, He M, Zhao Y-Y, Deng S-H, Liu T, Zhang T, et al. Increased susceptibility of irradiated mice to aspergillus fumigatus infection via NLRP3/GSDMD pathway in pulmonary bronchial epithelia. Cell Commun Signal (2022) 20:98. doi: 10.1186/s12964-022-00907-2
71. Keyel PA. How is inflammation initiated? Individual influences of IL-1, IL-18 and HMGB1. Cytokine (2014) 69:136–45. doi: 10.1016/j.cyto.2014.03.007
72. Kim JH, Jenrow KA, Brown SL. Mechanisms of radiation-induced normal tissue toxicity and implications for future clinical trials. Radiat Oncol J (2014) 32:103–15. doi: 10.3857/roj.2014.32.3.103
73. Gao J, Peng S, Shan X, Deng G, Shen L, Sun J, et al. Inhibition of AIM2 inflammasome-mediated pyroptosis by andrographolide contributes to amelioration of radiation-induced lung inflammation and fibrosis. Cell Death Dis (2019) 10:957. doi: 10.1038/s41419-019-2195-8
74. Wang B, Wei J, Meng L, Wang H, Qu C, Chen X, et al. Advances in pathogenic mechanisms and management of radiation-induced fibrosis. Biomed Pharmacother (2020) 121:109560. doi: 10.1016/j.biopha.2019.109560
75. Zhang W-J, Chen S-J, Zhou S-C, Wu S-Z, Wang H. Inflammasomes and fibrosis. Front Immunol (2021) 12:643149. doi: 10.3389/fimmu.2021.643149
76. Borthwick LA. The IL-1 cytokine family and its role in inflammation and fibrosis in the lung. Semin Immunopathol (2016) 38:517–34. doi: 10.1007/s00281-016-0559-z
77. Chi H-H, Hua K-F, Lin Y-C, Chu C-L, Hsieh C-Y, Hsu Y-J, et al. IL-36 signaling facilitates activation of the NLRP3 inflammasome and IL-23/IL-17 axis in renal inflammation and fibrosis. J Am Soc Nephrol: JASN (2017) 28:2022. doi: 10.1681/ASN.2016080840
78. Zhang L-L, Huang S, Ma X-X, Zhang W-Y, Wang D, Jin S-Y, et al. Angiotensin(1-7) attenuated angiotensin II-induced hepatocyte EMT by inhibiting NOX-derived H2O2-activated NLRP3 inflammasome/IL-1β/Smad circuit. Free Radic Biol Med (2016) 97:531–43. doi: 10.1016/j.freeradbiomed.2016.07.014
79. Straub JM, New J, Hamilton CD, Lominska C, Shnayder Y, Thomas SM. Radiation-induced fibrosis: mechanisms and implications for therapy. J Cancer Res Clin Oncol (2015) 141:1985–94. doi: 10.1007/s00432-015-1974-6
80. Caja L, Dituri F, Mancarella S, Caballero-Diaz D, Moustakas A, Giannelli G, et al. TGF-β and the tissue microenvironment: Relevance in fibrosis and cancer. Int J Mol Sci (2018) 19:1294. doi: 10.3390/ijms19051294
81. Richter K, Kietzmann T. Reactive oxygen species and fibrosis: further evidence of a significant liaison. Cell Tissue Res (2016) 365:591–605. doi: 10.1007/s00441-016-2445-3
82. Emami B. Tolerance of normal tissue to therapeutic radiation. Rep Radiother Oncol (2013) 1:123–7. doi: 10.1016/0360-3016(91)90171-y
83. Shen H-H, Yang Y-X, Meng X, Luo X-Y, Li X-M, Shuai Z-W, et al. NLRP3: A promising therapeutic target for autoimmune diseases. Autoimmun Rev (2018) 17:694–702. doi: 10.1016/j.autrev.2018.01.020
84. Gao R, Shi H, Chang S, Gao Y, Li X, Lv C, et al. The selective NLRP3-inflammasome inhibitor MCC950 reduces myocardial fibrosis and improves cardiac remodeling in a mouse model of myocardial infarction. Int Immunophar (2019) 74:105575. doi: 10.1016/j.intimp.2019.04.022
85. Coll RC, Robertson AAB, Chae JJ, Higgins SC, Muñoz-Planillo R, Inserra MC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med (2015) 21:248–55. doi: 10.1038/nm.3806
86. Mangan MSJ, Olhava EJ, Roush WR, Seidel HM, Glick GD, Latz E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discovery (2018) 17:588–606. doi: 10.1038/nrd.2018.97
87. Mezzaroma E, Mikkelsen RB, Toldo S, Mauro AG, Sharma K, Marchetti C, et al. Role of interleukin-1 in radiation-induced cardiomyopathy. Mol Med (2015) 21:210–8. doi: 10.2119/molmed.2014.00243
88. Hu JJ, Liu X, Xia S, Zhang Z, Zhang Y, Zhao J, et al. FDA-Approved disulfiram inhibits pyroptosis by blocking gasdermin d pore formation. Nat Immunol (2020) 21:736–45. doi: 10.1038/s41590-020-0669-6
89. Dinarello CA, Simon A, van der Meer JWM. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discovery (2012) 11:633–52. doi: 10.1038/nrd3800
90. Guo H, Callaway JB, Ting JP-Y. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat Med (2015) 21:677–87. doi: 10.1038/nm.3893
91. Coll RC, Robertson AAB, Chae JJ, Higgins SC, Muñoz-Planillo R, Inserra MC, et al. A small molecule inhibitior of the NLRP3 inflammasome is a potential therapeutic for inflammatory diseases. Nat Med (2015) 21:248–55. doi: 10.1038/nm.3806
92. Coll RC, Hill JR, Day CJ, Zamoshnikova A, Boucher D, Massey NL, et al. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat Chem Biol (2019) 15:556–9. doi: 10.1038/s41589-019-0277-7
93. Youm Y-H, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, Kim D, et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med (2015) 21:263–9. doi: 10.1038/nm.3804
94. Marchetti C, Swartzwelter B, Gamboni F, Neff CP, Richter K, Azam T, et al. OLT1177, a β-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proc Natl Acad Sci USA (2018) 115:E1530–9. doi: 10.1073/pnas.1716095115
95. Vogel S, Arora T, Wang X, Mendelsohn L, Nichols J, Allen D, et al. The platelet NLRP3 inflammasome is upregulated in sickle cell disease via HMGB1/TLR4 and bruton tyrosine kinase. Blood Adv (2018) 2:2672–80. doi: 10.1182/bloodadvances.2018021709
96. Ito M, Shichita T, Okada M, Komine R, Noguchi Y, Yoshimura A, et al. Bruton’s tyrosine kinase is essential for NLRP3 inflammasome activation and contributes to ischaemic brain injury. Nat Commun (2015) 6:7360. doi: 10.1038/ncomms8360
97. Purvis GSD, Collino M, Aranda-Tavio H, Chiazza F, O’Riordan CE, Zeboudj L, et al. Inhibition of bruton’s TK regulates macrophage NF-κB and NLRP3 inflammasome activation in metabolic inflammation. Br J Pharmacol (2020) 177:4416–32. doi: 10.1111/bph.15182
98. Hu JJ, Liu X, Xia S, Zhang Z, Zhang Y, Zhao J, et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin d pore formation. Nat Immunol (2020) 21(7):736–45. doi: 10.1038/s41590-020-0669-6
99. Xu P, Xu Y, Hu B, Wang J, Pan R, Murugan M, et al. Extracellular ATP enhances radiation-induced brain injury through microglial activation and paracrine signaling via P2X7 receptor. Brain Behav Immun (2015) 50:87–100. doi: 10.1016/j.bbi.2015.06.020
100. Li Y, Huang H, Liu B, Zhang Y, Pan X, Yu X-Y, et al. Inflammasomes as therapeutic targets in human diseases. Signal Transduct Target Ther (2021) 6:247. doi: 10.1038/s41392-021-00650-z
101. Afonina IS, Zhong Z, Karin M, Beyaert R. Limiting inflammation–the negative regulation of NF-κB and the NLRP3 inflammasome. Nat Immunol (2017) 18:861–9. doi: 10.1038/ni.3772
102. Hellweg CE. The nuclear factor κB pathway: A link to the immune system in the radiation response. Cancer Lett (2015) 368:275–89. doi: 10.1016/j.canlet.2015.02.019
103. Hu L, Chen H, Zhang X, Feng Z, Zhang H, Meng Q. Rosiglitazone ameliorates radiation-induced intestinal inflammation in rats by inhibiting NLRP3 inflammasome and TNF-α production. J Radiat Res (2020) 61:842–50. doi: 10.1093/jrr/rraa062
104. Samir P, Kesavardhana S, Patmore DM, Gingras S, Malireddi RKS, Karki R, et al. DDX3X acts as a live-or-die checkpoint in stressed cells by regulating NLRP3 inflammasome. Nature (2019) 573:590–4. doi: 10.1038/s41586-019-1551-2
105. Mitoma H, Hanabuchi S, Kim T, Bao M, Zhang Z, Sugimoto N, et al. The DHX33 RNA helicase senses cytosolic RNA and activates the NLRP3 inflammasome. Immunity (2013) 39:123–35. doi: 10.1016/j.immuni.2013.07.001
106. Soto-Rifo R, Ohlmann T. The role of the DEAD-box RNA helicase DDX3 in mRNA metabolism. Wiley Interdiscip Rev RNA (2013) 4:369–85. doi: 10.1002/wrna.1165
107. Gonçalves AC, Towers ER, Haq N, Porco JA, Pelletier J, Dawson SJ, et al. Drug-induced stress granule formation protects sensory hair cells in mouse cochlear explants during ototoxicity. Sci Rep (2019) 9:12501. doi: 10.1038/s41598-019-48393-w
108. Biasizzo M, Kopitar-Jerala N. Interplay between NLRP3 inflammasome and autophagy. Front Immunol (2020) 11:591803. doi: 10.3389/fimmu.2020.591803
109. Poudel B, Gurung P. An update on cell intrinsic negative regulators of the NLRP3 inflammasome. J Leukoc Biol (2018) 103(6):1165–77. doi: 10.1002/JLB.3MIR0917-350R
110. Xu Y, Shen J, Ran Z. Emerging views of mitophagy in immunity and autoimmune diseases. Autophagy (2020) 16:3–17. doi: 10.1080/15548627.2019.1603547
111. Wu D, Li J, Shen R, Li J, Yu Y, Li L, et al. Autophagy induced by micheliolide alleviates acute irradiation-induced intestinal injury via inhibition of the NLRP3 inflammasome. Front Pharmacol (2022) 12:773150. doi: 10.3389/fphar.2021.773150
112. Kiang JG, Jiao W, Cary LH, Mog SR, Elliott TB, Pellmar TC, et al. Wound trauma increases radiation-induced mortality by activation of iNOS pathway and elevation of cytokine concentrations and bacterial infection. Radiat Res (2010) 173:319–32. doi: 10.1667/RR1892.1
113. Bruckner L, Gigliotti F, Wright T, Harmsen A, Notter RH, Chess P, et al. Pneumocystis carinii infection sensitizes lung to radiation-induced injury after syngeneic marrow transplantation: role of CD4+ T cells. Am J Physiol Lung Cell Mol Physiol (2006) 290:L1087–1096. doi: 10.1152/ajplung.00441.2005
114. Guipaud O, Jaillet C, Clément-Colmou K, François A, Supiot S, Milliat F. The importance of the vascular endothelial barrier in the immune-inflammatory response induced by radiotherapy. Br J Radiol (2018) 91:20170762. doi: 10.1259/bjr.20170762
115. Yang D, Xing Y, Song X, Qian Y. The impact of lung microbiota dysbiosis on inflammation. Immunology (2020) 159:156–66. doi: 10.1111/imm.13139
116. Davis BK, Wen H, Ting JP-Y. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol (2011) 29:707–35. doi: 10.1146/annurev-immunol-031210-101405
117. Cui M, Xiao H, Li Y, Zhou L, Zhao S, Luo D, et al. Faecal microbiota transplantation protects against radiation-induced toxicity. EMBO Mol Med (2017) 9:448–61. doi: 10.15252/emmm.201606932
118. Xia C, Jiang C, Li W, Wei J, Hong H, Li J, et al. A phase II randomized clinical trial and mechanistic studies using improved probiotics to prevent oral mucositis induced by concurrent radiotherapy and chemotherapy in nasopharyngeal carcinoma. Front Immunol (2021) 12:618150. doi: 10.3389/fimmu.2021.618150
119. Rao J, Qiao Y, Xie R, Lin L, Jiang J, Wang C, et al. Fecal microbiota transplantation ameliorates stress-induced depression-like behaviors associated with the inhibition of glial and NLRP3 inflammasome in rat brain. J Psychiatr Res (2021) 137:147–57. doi: 10.1016/j.jpsychires.2021.02.057
120. Rao J, Xie R, Lin L, Jiang J, Du L, Zeng X, et al. Fecal microbiota transplantation ameliorates gut microbiota imbalance and intestinal barrier damage in rats with stress-induced depressive-like behavior. Eur J Neurosci (2021) 53:3598–611. doi: 10.1111/ejn.15192
121. Liao H, Wang H, Rong X, Li E, Xu R-H, Peng Y. Mesenchymal stem cells attenuate radiation-induced brain injury by inhibiting microglia pyroptosis. BioMed Res Int (2017) 2017:1948985. doi: 10.1155/2017/1948985
122. Xue J, Li X, Lu Y, Gan L, Zhou L, Wang Y, et al. Gene-modified mesenchymal stem cells protect against radiation-induced lung injury. Mol Ther (2013) 21:456–65. doi: 10.1038/mt.2012.183
123. Dai S, Wen Y, Luo P, Ma L, Liu Y, Ai J, et al. Therapeutic implications of exosomes in the treatment of radiation injury. Burns Trauma (2022) 10:tkab043. doi: 10.1093/burnst/tkab043
124. Noonin C, Thongboonkerd V. Exosome-inflammasome crosstalk and their roles in inflammatory responses. Theranostics (2021) 11:4436–51. doi: 10.7150/thno.54004
125. Cypryk W, Nyman TA, Matikainen S. From inflammasome to exosome–does extracellular vesicle secretion constitute an inflammasome-dependent immune response? Front Immunol (2018) 9:2188. doi: 10.3389/fimmu.2018.02188
126. Liu Y, Lou G, Li A, Zhang T, Qi J, Ye D, et al. AMSC-derived exosomes alleviate lipopolysaccharide/d-galactosamine-induced acute liver failure by miR-17-mediated reduction of TXNIP/NLRP3 inflammasome activation in macrophages. EBioMedicine (2018) 36:140–50. doi: 10.1016/j.ebiom.2018.08.054
127. Prasanna PGS, Stone HB, Wong RS, Capala J, Bernhard EJ, Vikram B, et al. Normal tissue protection for improving radiotherapy: Where are the gaps? Transl Cancer Res (2012) 1:35–48. doi: 10.3978/j.issn.2218-676X.2012.05.05
Keywords: NLRP3 inflammasome, inflammasome activation, radiation injury, therapeutic target, ROS, pyroptosis
Citation: Cheng H, Chen L, Huang M, Hou J, Chen Z and Yang X (2022) Hunting down NLRP3 inflammasome: An executioner of radiation-induced injury. Front. Immunol. 13:967989. doi: 10.3389/fimmu.2022.967989
Received: 13 June 2022; Accepted: 05 October 2022;
Published: 24 October 2022.
Edited by:
Ying Liang, University of Kentucky, United StatesReviewed by:
Venkata Surya Narayana Murty Darisipudi, University Medical Center Greifswald, GermanyCopyright © 2022 Cheng, Chen, Huang, Hou, Chen and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaojun Yang, eWFuZ3hpYW9qdW5AaS5zbXUuZWR1LmNu; Zhifeng Chen, Y3pmMjAwN0BzbXUuZWR1LmNu
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.