 Zhongkun Zhang
Zhongkun Zhang Siyu Yao2†
Siyu Yao2† Yingwen Hu
Yingwen Hu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 08 August 2022
Sec. Vaccines and Molecular Therapeutics
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.967505
Immunotherapy is revolutionizing the clinical management of patients with different cancer types by sensitizing autologous or allogenic immune cells to the tumor microenvironment which eventually leads to tumor cell lysis without rapidly killing normal cells. Although immunotherapy has been widely demonstrated to be superior to chemotherapies, only a few populations of patients with specific cancer types respond to such treatment due to the failure of systemic immune activation. In addition, severe immune-related adverse events are rapidly observed when patients with very few responses are given higher doses of such therapies. Recent advances of lipid-based nanoparticles (NPs) development have made it possible to deliver not only small molecules but also mRNAs to achieve systemic anticancer immunity through cytotoxic immune cell activation, checkpoint blockade, and chimeric antigen receptor cell therapies, etc. This review summarized recent development and applications of LNPs in anticancer immunotherapy. The diversity of lipid-based NPs would encapsulate payloads with different structures and molecular weights to achieve optimal antitumor immunity through multiple mechanisms of action. The discussion about the components of lipid-based NPs and their immunologic payloads in this review hopefully shed more light on the future direction of anticancer immunotherapy.
Over the past decade, many immunotherapeutic agents have been developed against various types of cancer in clinic trials (Figure 1). The history of cancer immunotherapy started with using attenuated bacteria, tuberculosis vaccine Bacille Calmette-Guérin (BCG), to prevent the recurrence of invasive bladder cancer (1). Later, Dunn et al. suggested that lymphocytes played a role of immunosurveillance in tumor microenvironments (TMEs) (2). However, at the time the lack of methods to detect tumor-specific antigens and the difficulties to culture lymphocytes in vitro limited its application in the cancer immunotherapy. The first anticancer immunotherapeutic agent was used T cell growth factor interleukin 2 (IL-2), where IL-2 treatment showed significant tumor suppression by enhancing T-cell activation in patients with metastatic kidney cancer and melanoma (3). In the late 1990s, the production of monoclonal antibodies (mAbs) using hybridomas has been well established which led to the development of antibody-based therapeutics targeting tumor specific surface markers (4). Recently, there have been thriving efforts in the immunostimulatory small molecules, immune checkpoint inhibitors (ICIs) targeting immune cells, autologous T cells or natural killer (NK) cells expressing chimeric antigen receptors (CARs), and mRNAs expressing tumor antigens or CARs for cancer immunotherapy (5–7). Among these, small molecules, ICIs, and mRNA therapies are used as stand-alone treatments for many solid tumors such as melanoma, non-small cell lung cancer (NSCLC), and urothelial carcinoma (5, 8), whereas CAR T cell therapies exhibit distinct clinical responses in patients with blood cancer (9).
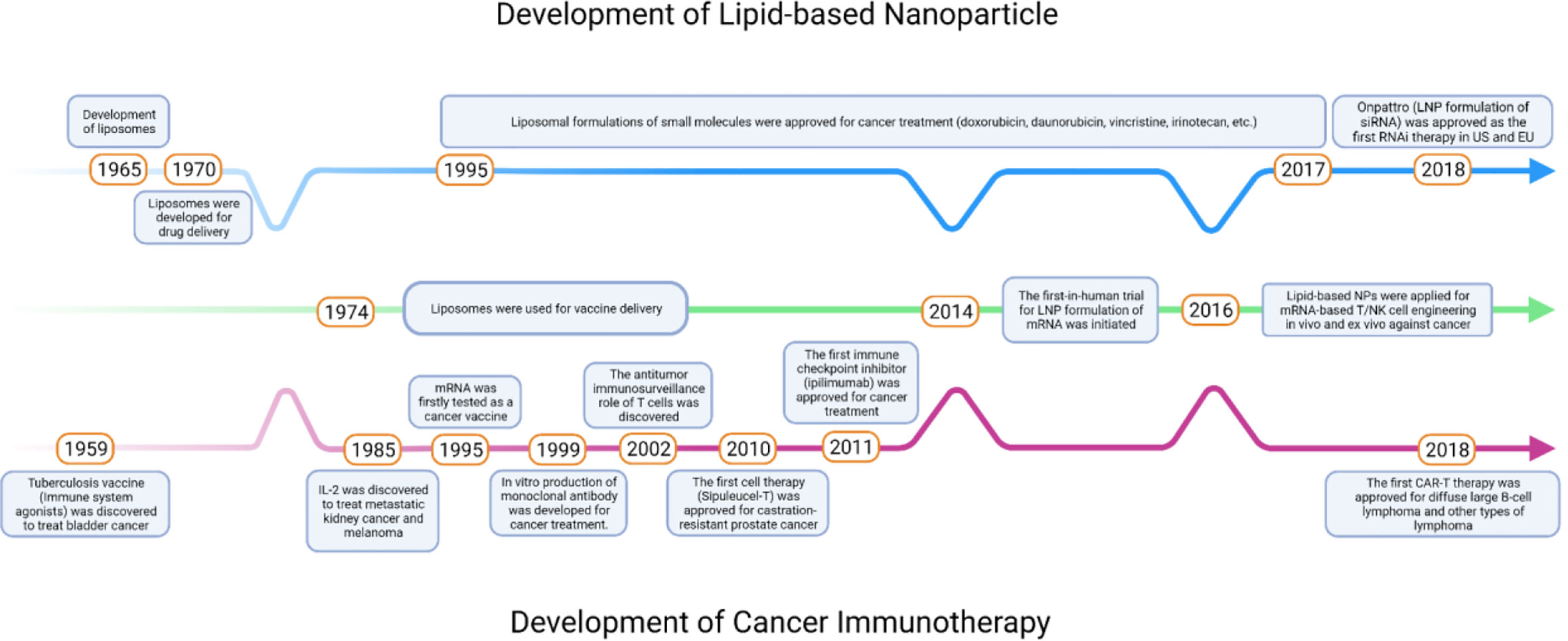
Figure 1 Timeline of key milestones throughout the development of lipid-based NPs and cancer immunotherapies.
Despite the great promise of immunotherapy against cancer, there also have been significant limitations with these immunotherapies: Poor water solubility, high immune-mediated adverse events (irAEs), and loss of bioactivity after long-term administration have limited the immunostimulatory efficacy of small molecules therapeutics (10). Unfortunately, these drawbacks are also interactional. Poor water solubility and off-target effects may lose the bioactivity of drugs and reduce the responses in patients’ bodies, and patients may develop irAEs when physicians decide to increase the dose. Therefore, the main challenges for cancer immunotherapies can be attributed to the lack of delivery systems which can keep the therapeutic payloads accessible to their targets. Lipid-based nanoparticles (NPs), including liposomes, lipid nanoparticles (LNPs), and nanoemulsions (NEs), have been developed as promising platforms to deliver a variety of therapeutic agents (11, 12) (Figures 1, 2). Compared to other nanosized delivery systems, lipid-based NPs are superior in minimizing systemic toxicity while maintaining high solubility in the water phase that neither polymeric NPs nor inorganic NPs could overcome for clinical use (13), and these advantages make lipid-based NPs the most common type of FDA-approved nanomedicines (14). Different structural designs endow lipid-based NPs with enhanced physical stabilities and the capacities of loading cargos with different sizes and hydrophobicity. From the 1960s to the 1970s, liposomes were first developed to deliver active pharmaceutical ingredients with simple vesicular formulations composed of an aqueous interior core with lipid bilayers (15–17). Later, different designs of LNPs were introduced with similar liposomal structures but resemble more micelle-like structures. The more complex internal lipid architectures in LNPs make them more suitable for encapsulating genetic payloads with long-term stability (18). NEs are biphasic systems in which oil and water phases are mixed in the form of nano-sized droplets (19). While water-in-oil NEs are important ingredients for topical applications, oil-in-water NEs were developed and preferred as an alternative system to liposomes or LNPs with micelle-like structures but covered by oil droplets (20). NEs are thermodynamically unstable compared with liposomes and LNPs which result in phase separation during long-term storage. However, NEs can be optimized to be kinetically stable with the help of surfactants and helper lipids to make the NEs more potent for encapsulating hydrophobic drugs (21). This review will discuss in detail the components of lipid-based NPs and provide an overview of the current applications of LNPs in delivering anticancer immunotherapies which may shed light on the future direction of anticancer immunotherapy.
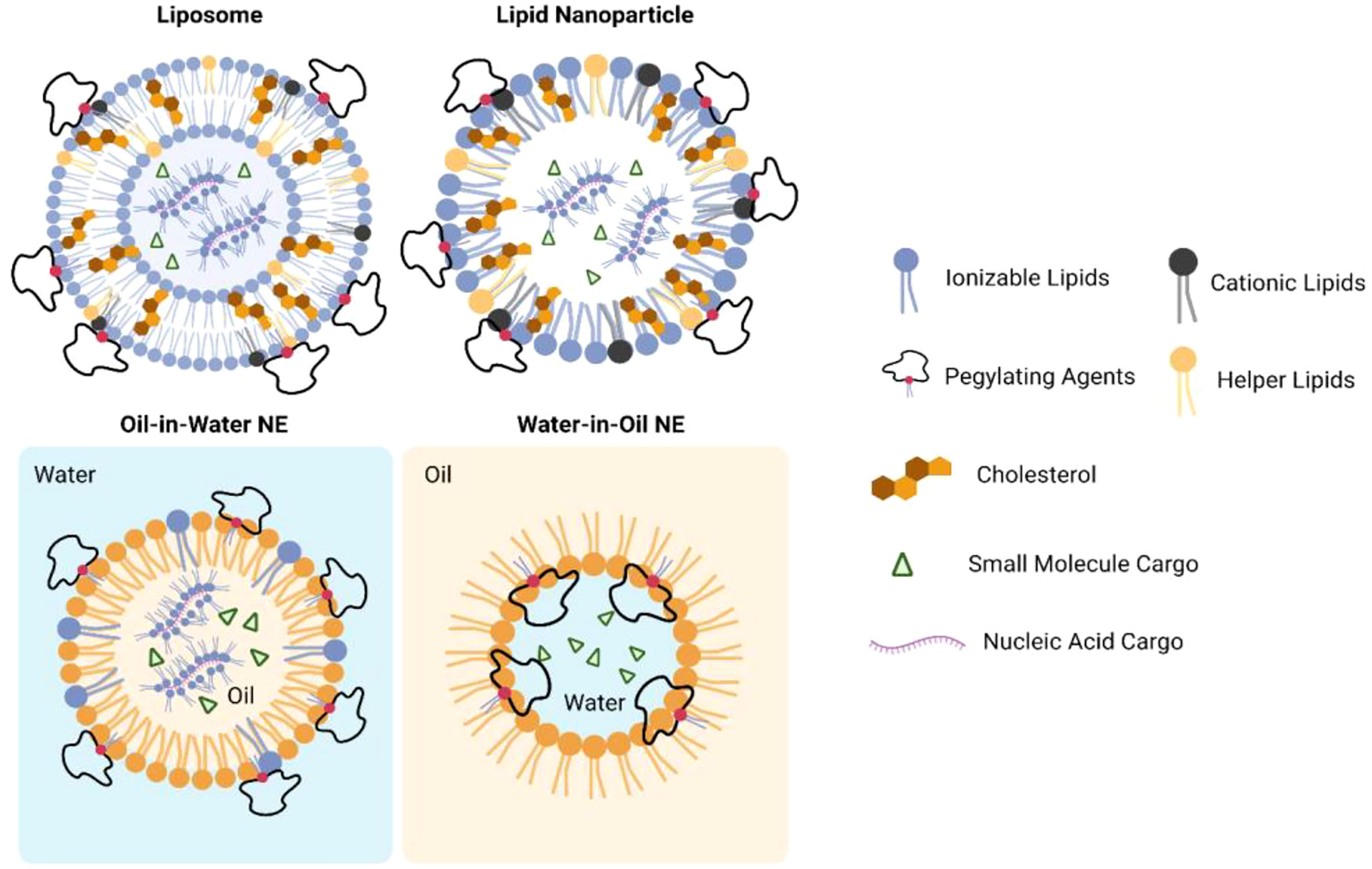
Figure 2 Structures of lipid-based nanoparticles when encapsulating small molecule or nuclei acid cargos.
Lipid-based NPs exhibit various types of structure. Majority of the lipid-based NPs exhibited near spherical shape with one or more lipid external layers (Figure 2). Although liposomes, LNPs, and NEs may exhibit different internal architectures, typical lipid-based NPs are composed of cationic lipids or ionizable lipids with tertiary or quaternary amines to encapsulate anionic payloads; helper lipids also were used to stabilize the lipid layer and facilitate membrane fusion; polyethylene glycol (PEG) lipids or surfactants were added to improve colloidal stability for long-term storage and prevent rapid degradation of payloads when administered into systemic fluids (18, 22). In addition, NEs would also include an oil phase which may be tri-, di-, or mono-acylglycerols, vegetable oils, mineral oils, free fatty acids, etc. (23). Representative structures of each component in lipid-based NPs are shown in Figure S1.
Drug encapsulation in lipid-based NPs was initially relied on the passive methods using zwitterionic lipids. It showed relatively poor encapsulation efficiencies (<40%) and low transfection potency for gene delivery (24, 25). The development of N-[1-(2,3-dioleyloxy) propyl]-N,N,N-trimethylammonium chloride (DOTMA) significantly enhanced the anionic drug encapsulation through electrostatic interactions (26). However, this first generation of such “lipoplexes” is highly unstable with broad size distributions and therefore have not been proven effective for in vivo delivery purposes. 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP), a biodegradable analogue of DOTMA, was then developed for lipid-based NPs gene delivery (27). Dimethyldioctadecylammonium bromide (DDA), a quaternary ammonium lipid, was later used in a gene delivery system as well as an immune adjuvant due to its special immunogenicity (28). The first trial of lipid-based NPs containing a cationic lipid was to encapsulate plasmid DNA (pDNA) using dioctadecyl-dimethyl-ammonium chloride (DODAC) (29). The author demonstrated that the injection of lipid/pDNA particles showed the halflife (t1/2) of pDNA was extended to 7 h in mice compared to naked pDNA, which had a t1/2 of less than 10 min. The prolonged circulation time of lipid/pDNA particles in mice resulted in dose accumulation in distal tumors over 24 h with minor accumulation in liver and spleen (30). With the development of lipid-based NPs, DOTMA and DOTAP have been successfully applied in mRNA delivery as cancer vaccines (31–34).
Ionizable lipids were a series of lipids with native pKa values below 7. It was designed to enhance anionic drug encapsulation and minimize systemic toxicities by cationic lipids due to permanent positive charges. Lipid-based NPs embedded with ionizable lipids could load anionic payloads at pH values below the pKa of ionizable lipids when it is positively charged. Then the drug-loaded lipid-based NPs are suitable for systemic delivery with a neutral exterior depending on physiological pH values (35). When trapped in endosomes after cellular uptake, where pH is lower than physiological pH, ionizable lipids are protonated and facilitate membrane destabilization which eventually releases payloads from lipid-based NPs into the intracellular compartment (35, 36). Researches showed that the optimal pKa value for ionizable lipids is around 6.5 for nucleic acid delivery in the murine FVII model (37). Aside from the pKa value, the linker between the ionizable group and alkyl chains is another factor for optimized gene delivery, where ketal linker was demonstrated to be more potent than ester and alkoxy linkers (38). Therefore, the potency of gene delivery relies considerably on the optimization of ionizable lipids in lipid-based NPs.
1,2-dioleoyl-3-dimethylammonium propane (DODAP) and 1,2-dioleyloxy-N,N-dimethyl-3-aminopropane (DODMA) were firstly developed for antisense oligonucleotides (ASOs) delivery with significantly increased circulation half-lives of ASOs in vivo through i.v. administration (39). Later, Dilinoleyl-DMA (DLinDMA), a dilinoleoyl analogue of DODMA, was introduced for silencing apolipoprotein B (ApoB) in the liver following i.v. administration (40). In another study using a murine factor VII (FVII) model for hepatic gene delivery, 2,2-dilinoleyl-4-(2-dimethylaminoethyl)- (1,3)-dioxolane (DLin-KC2-DMA, or KC2) demonstrated higher potency in delivering FVII siRNA compared with DLinDMA (38). Recent lipid screening study identified a new candidate heptatriaconta-6,9,28,31-tetraen-19-yl-4-(dimethylamino)butanoate (DLin-MC3-DMA or MC3) (37). MC3 exhibited superior siRNA delivery targeting TTR mRNA with ED50 of 0.005mg/kg (37). Under the hot trend of gene therapy, more ionizable lipids have been introduced for gene delivery specifically. Recent studies revealed that new classes of ionizable lipids have better gene delivery efficacy and pharmacokinetics than MC3, as summarized in Table S1 (41–54).
Helper lipids determine the shape of lipid-based NPs based on their different polymorphic phase tendencies, which also play an important role in intracellular delivery (55, 56). For example, lipids with the large hydrophilic heads are “cone-shaped” which tend to form micelle-like structures. Lipids with cylindrical geometry, such as phosphatidylcholine (PC), are compatible with bilayer structures. Lipids containing unsaturated acyl chains, such as unsaturated phosphatidylethanolamine (PE), have fixed geometry and are likely to form the inverted hexagonal (HII) phase (57).
Lipid bilayers formed by helper lipids can be stabilized by cholesterol where cholesterol can fill the geometry gaps between phospholipids (58). The inclusion of cholesterol in lipid-based NPs results in great stability in the presence of serum proteins. In addition, cholesterol is also able to facilitate membrane fusion which favors delivering nucleic acid cargos (59), while high percentages of cholesterol in lipid-based NP formulation may end up destabilizing lipid layers (60). Therefore, the trade-off between colloidal stability and transfection activity needs to be optimized when combining cholesterol and phospholipids in lipid-based NPs formulation.
Fusogenic oils are special components in NEs which is made of fatty acids, lipid substitutes, waxes, oil-soluble vitamins, and other lipophilic components. The viscosity, refractive index, density, phase behavior, and interfacial tension of the oils greatly determine the stability and functional properties of NEs (61). Other function of oils in NE-based drug delivery is to solubilize hydrophobic agents such as poor water-soluble chemotherapies and immunomodulatory agents (62, 63). It has been reported that some of the fusogenic oils (such as medium-chain oils, long-chain oils, and squalene) may stimulate innate immune system by activating toll-like receptors (TLRs) (64–66). There is no direct evidence showing that oil-induced innate immune activation could lead to antitumor responses in vitro or in vivo. Nonetheless, preclinical immunogenicity studies are recommended when developing nanomedicines containing fusogenic oils.
PEG lipids or surfactants are the most commonly applied non-ionic hydrophilic stabilizer for lipid-based NPs (67, 68). PEG lipids reduce the tendency of particle aggregation by steric stabilization, thereby enhancing formulation stability and prolonging the blood circulation time by reducing clearance mediated by the kidneys and mononuclear phagocyte system (MPS) (67, 68). 1,2-dimyristoyl-rac-glycero-3-methoxypolyethylene glycol-2000 (PEG2000-DMG) and 1,2-distearoyl-rac-glycero-3-methoxypolyethylene glycol-2000 (PEG2000-DSG) have been widely applied in lipid-based NPs for gene delivery (68). It also showed that LNPs containing PEG2000-DMG exhibit higher delivery efficiency in vivo than LNPs containing PEG2000-DSG, though PEG2000-DSG could prolong circulation times for LNPs in blood compared to PEG2000-DMG (68). Such differences may be attributed to the dissociation of PEG2000-DMG within LNPs compared with PEG2000-DSG, which could better facilitate cellular uptake and endosomal escape (68). In NEs, PEG lipids work as an emulsifier to improve the particle stability when the oil and water phase are mixed to yield a temporary emulsion (19). It should be noted that higher amount of PEG lipids in the formulation may encounter potential side effects such as PEG-specific immunogenicity, changes in pharmacokinetic behavior, potential toxicities of PEG lipids or side-products, etc. (67). In addition, too much of PEG lipids also may jeopardize the intracellular cargo release for gene delivery (69). Therefore, scientists in the field usually use less than 10% (molar ratio) of PEG lipids in their formulation (70).
Lipid-based NPs were firstly applied to encapsulate chemotherapies as anticancer therapeutics. Conventional anticancer chemotherapies were approved for cancer treatment based on their direct cytotoxic damage to cell proliferation, accumulating evidence indicates that the host immune system also plays an important role during the therapeutic process of chemotherapies which eventually leads to antitumor responses (71).Here are some examples of using Lipid-based NPs for the delivery of chemotherapeutics. Among all the chemotherapies approved or under clinical investigation, doxorubicin has been demonstrated to preferentially deplete myeloid-derived suppressor cells (MDSCs) in a murine breast cancer model (72). In addition, several studies revealed that doxorubicin could trigger dendritic cells (DCs) to release IL-1β which further activate IL-17 producing γδ T cells and recruit anti-tumoral IFN-γ expressing CD8+ T cells into TME (73–75). Similar to doxorubicin, daunorubicin and mitoxantrone treatments also stimulate the innate immune system and eventually lead to a major influx of myeloid and lymphoid cells into TME (76–78). Vincristine can upregulate PD-L1 and sensitize tumor antigen presenting in TME which can be a sensitizer before PD-L1 blockade therapy or DC-based immunotherapy (79, 80). Similar ability was also observed in irinotecan which could deplete regulatory T cells (Tregs), upregulate major histocompatibility complex 1 (MHC1) and PD-L1, turning TME more sensitive to immunotherapy (81). Treatment with topotecan, another camptothecin derivative to irinotecan, also has upregulated MHCI and Fas, making topotecan-treated tumors additive to effector T cell killing (82). In vitro exposure of cisplatin to melanoma cell lines resulted in the expression of T-cell chemokines such as CCL5, CXCL9, and CXCL10 (83). In clinical trials, a significant increase of tumor-infiltrating lymphocytes (TILs) was observed in 7 out of 21 breast cancer patients who received the first dose of paclitaxel (84). Other in vivo studies reported the increase of tumor MHCI and paclitaxel-dependent CD8+/CD4+ T cells infiltrated in a murine ovarian cancer model (85). Paclitaxel was also demonstrated to preferentially deplete Tregs (86), promote MDSCs into a more DC-like phenotype (87), activate TLR4, and shift macrophage polarization to a pro-inflammatory phenotype (88). Lastly, treatment of oxaliplatin in a murine colorectal cancer model resulted in upregulated tumor MHCI, and decreased immune suppressor cells (Tregs, MDSCs, and tumor-associated macrophages, TAMs) (89). Interestingly, the liposomal formulation of oxaliplatin exhibited superior antitumor immunity compared to free oxaliplatin, suggesting that accurate delivery of chemotherapies to TME would trigger better antitumor immunity (89).
Many evidence from chemotherapy-based immunomodulation revealed that successful antitumor immunity requires cross-function between innate and adaptive immune activations. Antigen-presenting cells (APCs, either dendritic cells (DCs) or macrophages) serve to early detect cancers by sensing tumor-associated antigens or pattern-/damage-associated molecular patterns (PAMPs/DAMPs) through pattern recognition receptors (PRRs) (90). Activated APCs will trigger pro-inflammatory cytokines and chemokines to employ the adaptive immune system where antigen-specific T cells will be activated to direct tumor cell killing (Figure 3) (91). Antitumor adjuvants have been developed as tumor antigen simulators, ligands for PRRs or other PAMP/DAMP-associated pathways. Agonists for TLRs, NOD-like receptors (NLRs), and RIG-I-like receptors (RLRs) are the adjuvants used to induce pro-inflammatory immune responses which favor anti-tumor activities. Among TLR agonists, Pam3Csk4, a TLR1/2 agonist, was successfully developed using LNPs with OVA mRNA against a murine lymphoma model (92). Poly (I:C), a TLR3 agonist, was capable to be incorporated in not only lipid-based NPs (93, 94) but also inorganic NPs (95). TLR7/8/9 agonists are widely studied as antitumor adjuvants. Imidaziquinoline-like TLR 7/8 agonists, including imiquimod and resiquimod (R848), was loaded into the liposomal formulation to prolong its retention time in blood circulation (96). When considering the poor water solubility of TLR7/8 agonists, NEs formulation is also a suitable delivery platform to improve drug solubility thus the drug loading (63). Synthetic TLR9 agonists are oligonucleotides (ODNs) with cytosine-phosphate-guanine (CpG) motifs. CpG ODNs can be easily incorporated into tumor-targeting lipid-based NPs with cationic or ionizable lipids through electrostatic interactions (97–100). Agonists for RLRs are also nucleic acid-based dsRNAs (101). Studies have shown that dsRNA targeting RLRs could be encapsulated into lipid-based NPs and polymeric NPs for systemic delivery in murine pancreatic cancer and breast cancer models (102, 103). In a study comparing the antitumor immunity carried out by liposomal TLR agonists and NLR agonists, NLR agonists also exhibit early antitumor activity (104). However, the antitumor activity of NOD1 agonist was not associated with DC-driven adaptive immune responses, suggesting that NOD1 activation would trigger an alternative cytotoxic adaptive immune response against tumor which differs from TLR agonists. Another liposomal-based innate immune agonist, mifamurtide, has been demonstrated to target both TLR4 and NOD2 which synergize NF-kB activation and pro-inflammatory tumoricidal activities (105). Stimulator of interferon genes (STING) is another type of PRR which located in the endoplasmic reticulum. Activated STING would potentiate type I interferons with other pro-inflammatory cytokines which may enhance antitumor immunity (106). Cyclic dinucleotides (CDNs), including cyclic guano-sine monophosphate–adenosine monophosphate (cGAMP), are common agonists for STING receptors which can be encapsulated into lipid-based NPs to potentiate systemic antitumor immunity (107–110). Since STING agonists require intracellular delivery to the endoplasmic reticulum, lipid-based NPs delivery system requires an optimized fusogenic activity for efficient endosomal escape.
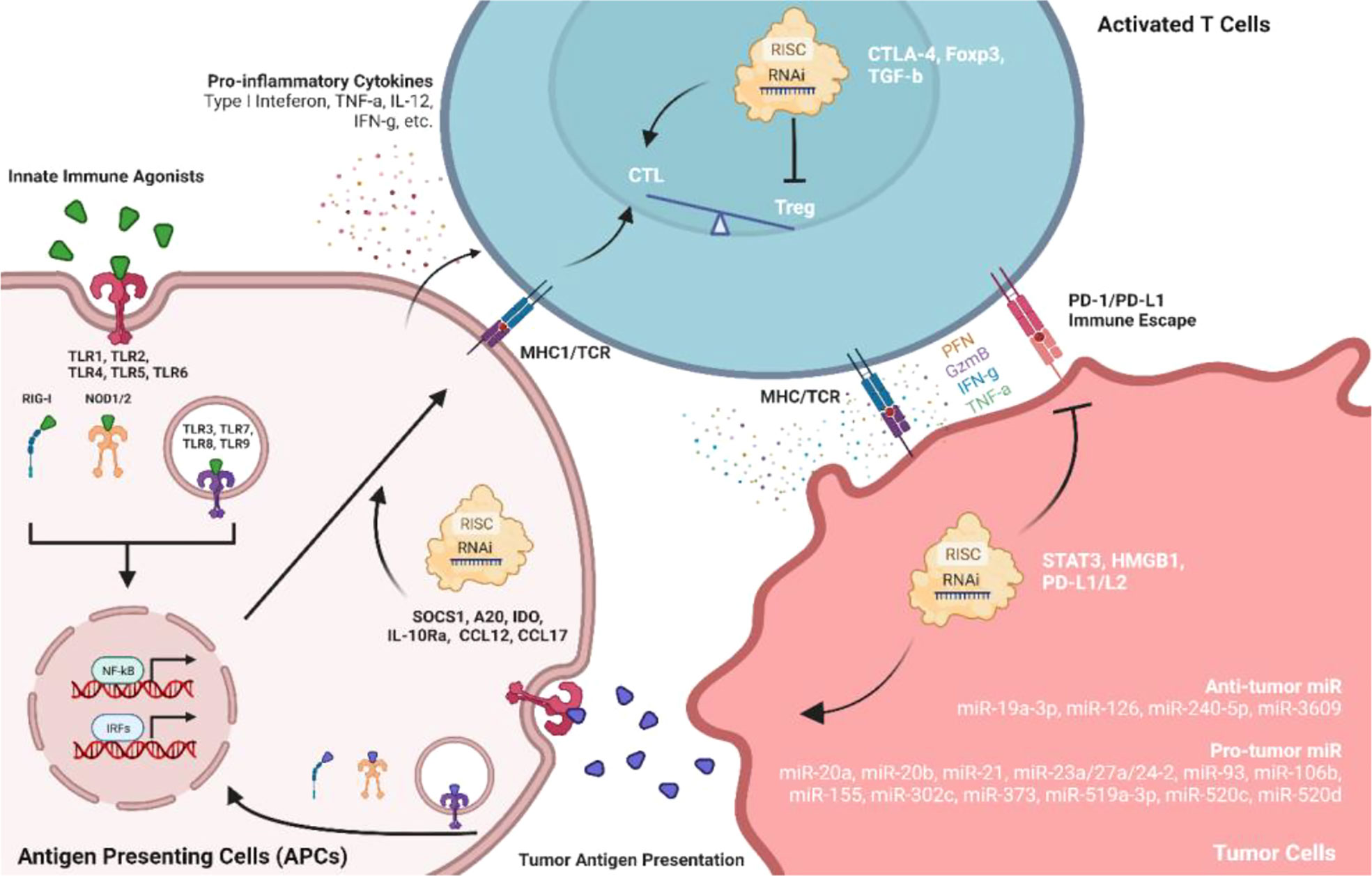
Figure 3 Role of innate immune agonists and RNAi in facilitating antitumor immune responses among APCs, T cells, and TMEs. Agonists of pattern recognition receptors (PRRs) on antigen-presenting cells can directly activate the immune system against tumors. Meanwhile, RNAi therapeutics targeting immunomodulatory factors or non-coding RNAs may also facilitate antitumor immunity.
RNAi technologies (siRNA, shRNA, miRNA, ASO, etc.) can induce specific gene regulation which became a new therapeutic area in infectious diseases, neurodegenerative disorders, cancer, and other rare diseases. Besides direct targeting of specific oncogenes, RNAi could also potentiate antitumor immune responses by downregulating immune-suppressive proteins (Figure 3). Studies reported that elevated activity of signal transducer and activator of transcription 3 (STAT3) is positively associated with tumor progression. Knockdown of STAT3 using siRNA successfully inhibits tumor growth with increased tumor infiltration of CD8+ T cell and NKT cell in murine melanoma models (111, 112). STAT3 downregulation also significantly increased pro-inflammatory cytokine production such as IFN-g, IL-12, and TNF-a (112). Suppressor of cytokine signaling 1 (SOCS1) negatively regulates antigen presentation process. Downregulation of SOCS1 by siRNA enhanced antigen-specific DC maturation with elevated expression of IFN-g, IL-12, and decreased IL-4 which potentiate Th1 cell differentiation and antitumor immunity (113). Similar activities were also observed using siRNA to downregulate A20 and indoleamine 2, 3-dioxygenase (IDO) in DCs (114–117). A20 siRNA could inhibit CD4+ CD25+ Tregs suppression which further enhanced CD8+ T cells immune responses in TMEs (118). Programmed dead ligand 1 (PD-L1) is another exciting target that facilitates tumor immune escape from host immune systems. siRNA against PD-L1 can preferentially transfer CD11c+ PD-L1+ tolerogenic DCs into potent stimulators of CTLs in TMEs through PD-L1 downregulation and multiple TLRs activation (119). The antitumor efficacy of siRNA against PD-L1 can be enhanced by co-inhibiting with CTLA-4 (120). Another study also indicated that siRNA against PD-L1 or PD-L1 improved effector functions of tumor-specific CD4+ and CD8+ T cells in vitro (121), suggesting that PD-L1/PD-L2 siRNA is an exciting strategy to inhibit the immune suppressive tumor-specific T cells. High-mobility group box 1 (HMGB1) is highly expressed in tumor cells and associated with tumor proliferation. Knockdown of tumor cell HMGB1 by shRNA did not inhibit tumor cell growth, whereas CD8+ T cell sensitized by tumor-specific IFN-g and TNF-a can be activated with attenuated functional Tregs (122). Since Foxp3 contributes to Treg development and activation, knockdown of Foxp3 on Tregs by shRNA successfully suppressed tumor growth (123). Transforming growth factor-beta (TGF-b) and IL-10 expressed by tumors could also trigger Tregs activation. siRNA against TGF-b decreased Treg level and enhanced DC-based cancer vaccine in mice with “cold” melanoma tumor (124), and siRNA targeting Interleukin-10 receptor alpha (IL-10Ra) initiated tumor-specific CD8+ T cell immune responses (125). When used in combination, siRNA against TGF-b/IL-10Ra significantly upregulated MHC1, CD40, CD80, CD86, and CCR7 in DCs which induced the strongest antitumor effects in immune-resistant murine models (125). Direct downregulation of chemokines such as CCL22 and CCL17 by siRNA also can decrease Treg recruited by monocyte-derived DCs (MoDCs) with lower CD4+/CD8+ ratios. Furthermore, intratumoral injection of MoDCs treated with siRNA against CCL22/CCL17 significantly reduced the Tregs population and increased CD8+ infiltration into TME (126).
miRNAs also indirectly facilitate the interaction between tumors and host immune systems which can be targeted for either downregulation or replacement by RNAi. Oncogenic miRNAs have been reported to promote tumor progression by inducing M2-like TAM in TMEs (miR-155 (127), miR-21 (128), miR-23a/27a/24-2 cluster (127)) and eliminating NK-mediated killing (miR-20a, miR-93, miR-520d, miR-106b, miR-373, miR-302c, miR-520c, miR-20b, miR-519a-3p (129, 130)), while tumor-suppressive miRNAs could inhibit tumor growth by shifting M2- to M1-TAM (miR-19a-3p (131)), inducing pro-inflammatory cytokines (miR-240-5p (132)), blocking immune checkpoint (miR-3609 (133)), and preventing the recruitment of immune suppressive cells (miR-126 (134)). Although current miRNA therapeutics against cancer remain focusing on the oncogenes which directly affect cell proliferation, embedding miRNA sequence with such antitumor immunomodulatory effects could also be an exciting strategy for RNAi based cancer immunotherapy.
Most of the immunotherapies against cancer rely on the delivery of tumor-associated antigens (TAAs) and tumor-specific antigens (TSAs). Neoantigens expressed in the mutated tumor microenvironment would allow the development of personalized cancer vaccines with patient-specific neoepitopes. Tumor antigens against cancer mainly depend on the delivery of tumor antigen peptides or coding mRNAs (8, 135). Delivery of long synthetic peptides from TAAs/TSAs using liposomes can greatly protect the peptides from degradation with more access to the APCs (135). Several studies have shown that liposomes encapsulating T cell epitopes against LHR hormone (136), Pan HLA-DR epitope peptides (137), HER2/neu peptide (138), and KRAS G12 mutant peptides (139) could stimulate CD4+ T cell responses against tumors. When embedding cationic lipids or external Fc receptor ligands, liposomes with tumor antigens could show greater cell uptake by APCs[141,145]. However, the identification and manufacturing of tumor antigen peptides are time-consuming. Compared with tumor antigens in the forms of pDNA or peptides, mRNA has emerged as a new potent and flexible platform for cancer immunotherapies (7). mRNA therapies can encode protein sequences and stimulate innate immune systems. More importantly, large-scale production of mRNA is relatively simple and inexpensive in a cell-free environment, and mRNA can be easily encapsulated into lipid-based NPs through electrostatic interaction when mixing the lipids and mRNAs stock solutions, all of which make the manufacturing process of mRNA in a standardized and controlled condition (8).
mRNA vaccines could stimulate an antigen-specific immune response when the encoded antigen is translated to proteins in the cytosol of APCs. The expressed proteins are processed by APCs and presented on MHC1 to CD8+ T cells, stimulating cell-mediated immune responses (Figure 4). Reports also showed that the fusion of mRNA-encoded antigen to MHCII trafficking signals derived from lysosomal proteins could also induce supportive CD4+ T helper cell response which is crucial in cancer immunotherapy (140, 141). In addition, the immunogenicity of mRNA structures may activate signals through TLR3, TLR7, and TLR8 which enhance innate immune responses (142). From this aspect, co-administration of mRNA/lipid-based NPs with adjuvants may enhance the stimulation of immune responses (143). For example, co-delivering the nucleoside-modified mRNA with a TLR4 agonist (monophosphoryl lipid A; MPLA) inside DOTAP–cholesterol mRNA lipoplexes induced innate immunity and allowed high antigen expression in vivo (143). LNPs encapsulating mRNA encoding OVA also showed significant tumor prevention when combined with TLR1/2 agonist (92). Intramuscular immunization of OVA mRNA using Pam3-LNP in mice resulted in high expression of tumor antigens with enhanced cellular immune stimulation (92). In addition, the efficiency of mRNA–LNPs encoding interleukin-12, an example of cytokines with anticancer activity, was examined by the group Lai et al. for the suppression of tumor growth in transgenic mouse models of hepatocellular carcinoma (HCC) (144). miRNA target sites can be incorporated in modified mRNAs encoding toxic or apoptotic proteins like caspase or p53 upregulated modulator of apoptosis (PUMA) (145). The presence of miRNA binding sites will allow the targeting of miRNAs that are present only in healthy cells and then enable these cells to recognize and degrade toxic mRNA. It was found that intratumoral administration of LNPs loaded with these miRNA–mRNA combination sequences in mice prevented the expression of toxic proteins from the mRNA of healthy cells but selectively triggered apoptosis in tumor cells without causing systemic toxicity (145).
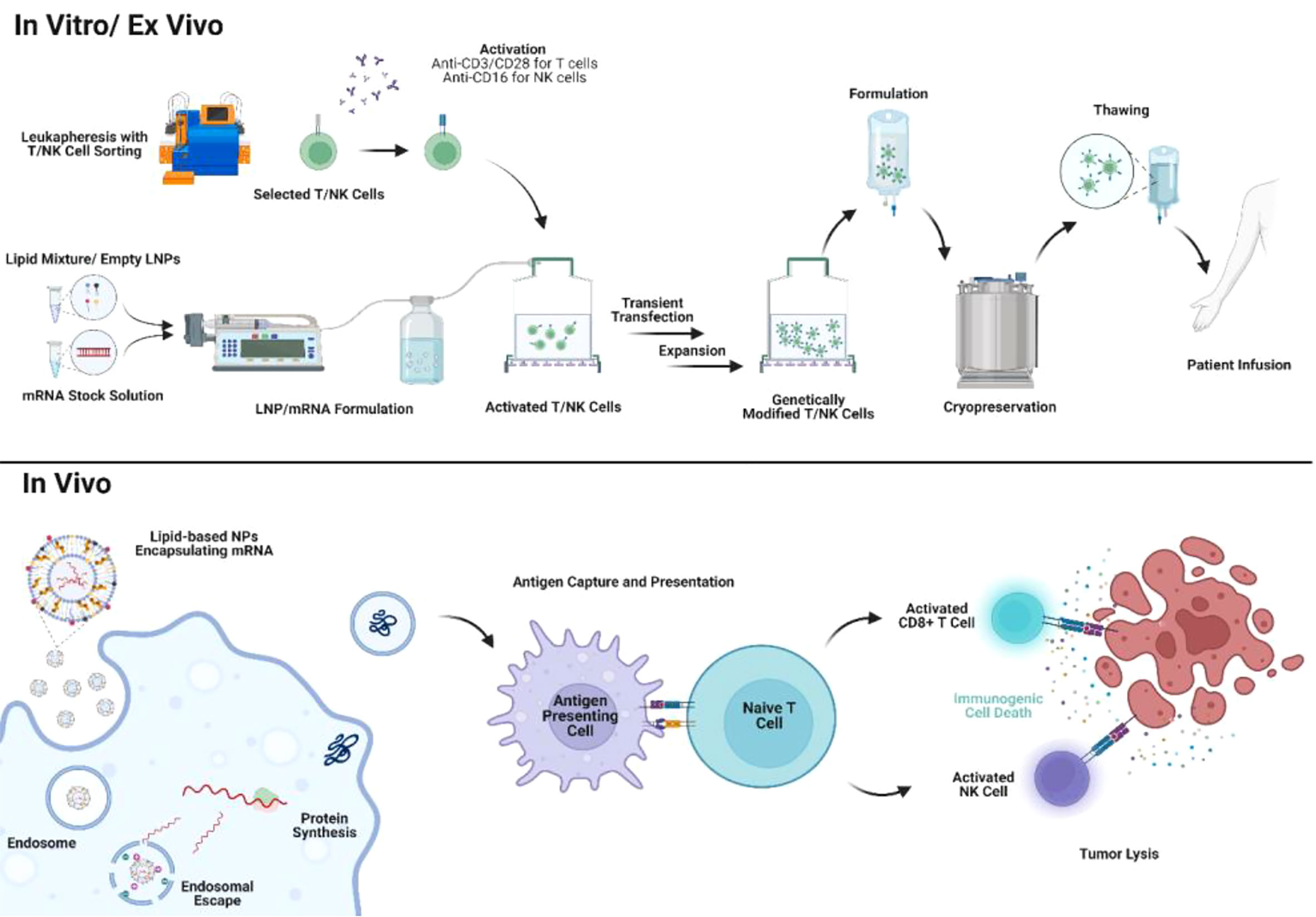
Figure 4 Application of mRNA/lipid-based NPs in mRNA therapies and cell therapies. mRNA/lipid-based NPs can be utilized for engineering cell therapy ex vivo with shortened manufacturing timeline and reduced physiological side effects. In addition, mRNA/lipid-based NPs can also be directly delivered in vivo to either engineer immune cells or as exogenous tumor antigens.
To induce a strong cytotoxic CD8 T cell response, Oberli et al. developed LNPs for the delivery of an mRNA vaccine encoding the model immunology protein, ovalbumin (OVA) (146). The authors identified an optimum formulation that contained an ionizable lipid (cKK-E12) and an additive (sodium lauryl sulfate). The optimal formulation showed increased T cell response upon reducing the molar ratio of cKK-E12 from 35% to 10%. Immunization of model mice with transgenic OVA-expressing tumor or with aggressive B16F10 melanoma using the formulated mRNA vaccine encoding the corresponding antigens resulted in strong CD8 T cell immunity activation in addition to slow tumor growth, shrinkage of tumor, and consequently, extended survival of treated mice (146). However, the increased CD8 T cell responses with decreased cKK-E12 suggest that ionizable lipids or cationic lipids themselves may have effects on directing T cell functions regardless of their main roles in binding with mRNAs.
The safety, immunogenicity, and tolerability of the first personalized IVAC MUTANOME (BioNTech RNA Pharmaceuticals GmbH), a poly-neoepitope-coding RNA vaccine, have been evaluated in phase I clinical trials (NCT02035956) targeting mutant neoantigens for the treatment of patients with melanoma. A strong immune response against the vaccine antigens was observed. T cell response was also generated against 60% of the 125 selected neoepitopes with no adverse drug reactions, indicating good tolerability of the vaccine by enrolled patients (147). Many other personalized mRNA cancer vaccines encoding different antigens have been formulated in lipid nanosystems and have already entered clinical stages (NCT03897881, NCT02316457, NCT03313778, NCT03480152, NCT03323398) (148, 149). In a phase II study using mRNA-4157 and pembrolizumab treating melanoma patients (NCT03897881), 14 out of 16 patients in the mRNA-4157 monotherapy group remained disease-free during the study, with a median follow-up time of 8 months. In the combination group, the overall response rate in the cohort (human papillomavirus-negative, immune checkpoint inhibitor-naive, head and neck squamous cell carcinomas) was 50% and the median progression-free survival was 9.8 months (150, 151). mRNA-4157 used for the treatment of gastrointestinal cancers is also in progress (NCT03480152) (152). In another phase I/II trial (NCT03323398), the safety and efficacy of mRNA encoding human OX40L (mRNA-2416) was investigated in combination with durvalumab for the treatment of ovarian cancers and other solid tumors (153). mRNA-2416 was intratumorally administered every 2 weeks for up to 12 doses with four dose levels from 1 to 8 mg. The injected lesions showed an increase in OX40L expression and enhanced T cell activation (153). In a phase I dose-escalation study (NCT03739931), mRNA-2752 encoding human OX40L, IL-23, and IL36y was designed to induce a pro-inflammatory TME and simultaneously strengthen T cell expansion as well as memory responses. mRNA-2752 was intratumorally administered every 2 weeks for up to seven doses, alone or in combination with the infusion of durvalumab. In the 22 patients (monotherapy: n = 15; combination: n = 7), six had stable disease, one had partial responses with 52% tumor reduction and five showed tumor shrinkage in treated and/or untreated sites (154, 155).
In a phase I study (NCT02410733), FixVac, a complex of RNA/lipoplexes against malignant melanoma TAAs New York-ESO 1, tyrosinase, melanoma-associated antigen A3 (MAGE-A3), and trans-membrane phosphatase with tensin homology (TPTE) showed the metabolic activity of the spleen increased post the sixth immunization, indicating the targeted delivery of an mRNA-LNP encoding four non-mutated melanoma antigens and activation of resident immune cells (156). Within the same study, a combination of FixVac with an anti-programmed cell death protein 1 (PD1) antibody augmented the antitumor effect of FixVac, resulting in an over 35% tumor regression rate in immune checkpoint inhibitor-experienced patients (156). The results suggest that LNP-based mRNA therapies can be of general utility for non-mutant TAAs in patients who experienced ICI treatments.
The development of cell therapies can be traced back to vaccination with DCs including TAAs mRNA transfected by cationic lipids in the late 1990s (157). However, TAA mRNA transfection is inefficient with only cationic lipids. With optimized lipid-based NP formulation, scientists are now able to successfully engineer DCs as cancer vaccines in vitro and in vivo (158, 159). Another study indicated that in vivo DC engineering by directly administering lipid-based NPs with mRNA encoding antitumor immune epitopes exhibited similar antitumor efficacy compared with in vitro DC engineering before infusion (160). Same as APCs, in vivo reprogramming macrophages with lipid-based NPs containing mRNA encoding M1-polarization factors also exhibit antitumor activity in multiple murine cancer models (161). Macrophages could horizontally transfer genes into TMEs (162). In other words, it is promising to develop lipid-based NPs containing mRNA or RNAi which could co-target macrophages and tumor cells.
Recently, personalized adoptive cell therapy showed great promise against non-solid tumors in clinical trials. Adoptive cell therapy includes TIL therapy, engineered T cell receptor therapy (TCR-T), CAR-T cell therapy, and NK cell therapy. Despite their tremendous potential, adoptive cell therapies also raised concerns regarding the unwanted immunological side effects and insertional mutagenesis in the human genome due to the use of viral vectors for ex vivo cell engineering (163, 164). In addition, complicated manufacturing protocols and high costs can also impede the application of CAR-T in a broader patient population (163, 164). Therefore, novel ex vivo transfection technologies are needed for a safer and affordable adoptive cell therapy (165–167). Lipid-based NPs containing coding DNA or mRNA showed outstanding efficacy in transient transfection in preclinical studies. Lipid-based NPs encapsulating mRNA can be formulated by simple rapid mixing where a lipid stock in the ethanol phase is mixed with RNA in the aqueous phase through microfluidics or syringe pumps (168). mRNA/lipid-based NPs could be developed instantly when activated T cells or NK cells are ready for cell engineering in vitro or ex vivo. Therefore, the rationale for selecting the components of lipid-based NPs for cell engineering can mainly focus on its transfection efficiency of payloads. In addition, lipid-based NPs are generally considered with low cytotoxicity (169). Therefore, the processes of gene transfection and T/NK cell activation could be simultaneous (Figure 4). The straightforward formulation and low toxicity of lipid-based NPs encapsulating mRNA would greatly reduce the manufacturing cost and time for engineering cell therapies. McKinlay et al. first developed a combinatorial library in screening efficient mRNA transfection approach to lymphocytes (170). Soon, Billingsley et al. reported the development of ionizable LNP encapsulating CAR for ex vivo T cell engineering which first demonstrated the ex vivo engineered CAR-T by LNPs exhibited similar tumor-killing activity compared with lentivirus-engineered CAR-T (171). Lipid-based NPs/mRNA transfection strategy is also used in NK cell engineering. Chandrasekaran et al. first reported the development of super NK cells engineered by liposomes containing apoptosis-inducing ligand TRAIL (172). TRAIL-engineered NK cells exhibited strong tumor-killing activity by inducing apoptosis in tumor-draining lymph nodes in vivo (172). Another study also reported that liposomes can co-deliver CAR mRNA and paclitaxel to NK cells (173). CAR-engineered NK cells showed significant cytotoxicity to HER2+/CD19+ tumors in vivo with simultaneously paclitaxel release (173). Lipid-based NPs also demonstrated efficient CAR engineering to NK cells with preserved cell viability and minimal changes in NK phenotype and function compared with the traditional electroporation method (174).
The main efforts of lipid-based NPs in clinical trials against cancer are to deliver chemotherapies and therapeutic nucleic acids as discussed in previous sections. However, the number of lipid-based NPs formulations successfully approved in the market is still limited, suggesting that the development of lipid-based NPs remains certain barrier in the translation from preclinical animal models to humans.
Non-specific gene regulation by lipid-based NPs may take place when lipid-based NPs are used for gene delivery. It is noticeable that although the structure of lipid-based NPs is almost similar to those of lipid nanoparticles or micelles expressed by cells, the synthesized functional lipids, such as cationic or ionizable lipids, are still considered exogenous lipid-like materials which may be potentially recognized by PRRs on the cell surface. The high amount of exogenous functional lipids within lipid-based NPs may further trigger unwanted cell cycle arrest, cellular metabolism, or immune responses, which would disrupt the targeted gene regulation by nucleic acid payloads in lipid-based NPs (175, 176). When lipid-based NPs are delivered to immune cells, multiple vehicle-based immune activations may be triggered. Not only functional lipids may serve as ligands for inflammatory signals (175), but also lipid solvents, such as ethanol and chloroform, during lipid-based NP manufacturing would modulate the function of immature innate immune cells (177). In addition, PEG lipids could induce the secretion of anti-PEG immunoglobulins by B cells (178). Although the effect of anti-PEG immunoglobulins on tumor microenvironment is yet to be determined, the anti-PEG immunoglobulins would increase the clearance of lipid-based NPs in systemic fluids which would decrease the antitumor potency. Non-specific gene regulation and immune activation may potentially reduce the therapeutic efficacy whereas increasing the dose of lipid-based NPs to patients would eventually result in toxicities or irAEs. To minimize the non-specific gene regulation and immune activation by lipid-based NPs, the off-target effects of functional lipids should be characterized, and the manufacturing procedures should be optimized to purify the excipients in lipid-based NPs.
Although lipid-based NPs are believed to passively target and accumulate in tumors through the enhanced permeability and retention (EPR) effect, a growing body of research has revealed that lipid-based NPs, without any ligand-modification for active targeting, mainly accumulate in the liver upon systemic administration (179). Such effects are mainly due to discontinuous vasculature, decreased blood flow rates, and an abundance of phagocytic cell types in the liver microenvironment, which might cause liver toxicities and loss of in-situ therapeutic efficacy (179). Therefore, lipid-based NPs should be further engineered to specifically target tissues or cell populations to reduce liver accumulation and increase therapeutic efficacy. Table 1.
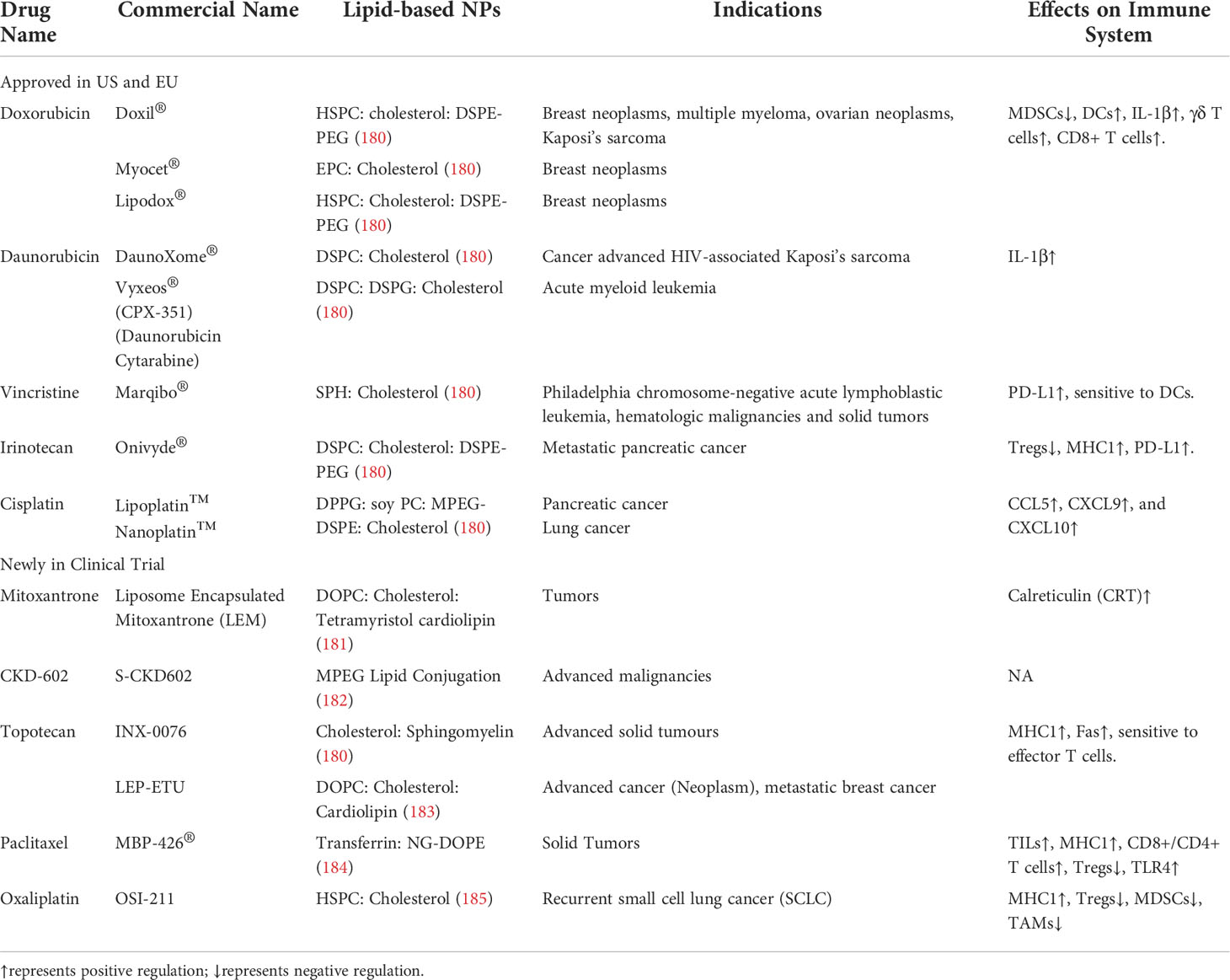
Table 1 Cases of immunological regulation of chemotherapies using lipid-based NPs as delivery system.
Lipid-based NPs represent the most advanced and widely applicable delivery vehicles for small molecules and nucleic acids. In cancer immunotherapy, lipid-based NPs not only could deliver small molecules and mRNA therapies in vivo to achieve the enormous antitumor activity but also are capable of ex vivo engineering cancer cell therapies with comparable efficiency compared to other non-viral or viral vectors. However, the formulation and large-scale manufacturing process of lipid-based NPs need to be optimized from industrial aspects. Depending on the specific drug payloads and applications, different helper lipids and cationic/ionizable lipids need to be selected to address specific challenges to immunotherapeutic delivery. For small-molecule chemotherapies or ligands for surface PPRs, lipid-based NPs with high colloidal stability should be designed to prolong their systemic retention time. Therefore, lipids with higher Tm are preferred due to their long-term stability in storage and the systemic fluid. For RNAi, mRNA, and ligands targeting intracellular PPRs, lipid-based NPs should be designed to satisfy membrane fusion and endosomal escape, such as embedding PE-based lipids and decreasing PEG-lipids, where the stability for lipid-based NPs may be compromised. Fortunately, the challenges for colloidal stability and in vivo side effects by lipid-based NPs may be bypassed in the application of cell therapies where lipid-based NPs containing mRNAs can be formulated and transfected into T/NK cells ex vivo in a real-time manner. However, due to the transient gene transfection by lipid-based NPs/mRNA, the durability of tumor-killing activities by engineered T/NK cells need to be further evaluated thoroughly. In addition, there are stilling remaining concerns for the delivery of lipid-based NPs, such as immunogenicity and tissue accumulation. Therefore, efforts should also be made on developing lipid-based NPs targeting specific organs or cell-populations as well as utilizing biodegradable formulation components with low immunogenicity (186, 187).
Moreover, the therapeutic payloads need to be designed for optimal lipid-based NP delivery efficiency and antitumor immunity. Small molecule drugs can be designed with ionizable structures to be stably encapsulated into the lipid-based NPs, and their mechanism of actions should be well identified without off-target effects such as inducing immunosuppression or autoreactive immune disorders (188). Nucleic acid drugs can be chemically modified to enhance their stabilities, and their sequence should be optimized to avoid off-target gene regulations (189). For mRNA drugs specifically, the translation efficiency can be increased by engineering their coding sequences, 5’ cap, 3’ poly(A) tail, and untranslated regions (UTRs) (18). Beside the single drug delivery, multiple immunostimulatory factors or epitopes could be delivered together within one NPs to achieve the optimal antitumor activity. For example, PPR agonists could be co-delivered with mRNA encoding TAAs to maximize tumor-specific antigen presentation; mRNA encoding Th1 epitopes in combination with RNAi silencing immune-suppressive genes may enhance CD8+ T cell and NK cell activation; Co-delivering of mRNA encoding CAR and chemotherapies in T/NK cells may also increase tumor killing activities. It is also noticeable that cancer immunotherapies only showed potency in patients with “hot” tumors where immune cells can rapidly infiltrate into TME. Systemic immunostimulatory agents, such as PPR agonists, may be involved in developing immunotherapies for a broader patient population. With the optimized formulation and proper payload recipes, lipid-based NP medicines would not only expand cancer immunotherapies to a broader range of patient populations but also improve health care in other diseases.
Conceptualization, ZZ; writing—original draft preparation, ZZ and SY; writing—review and editing, ZZ, SY, YH, XZ, and RL. All authors have read and agreed to the published version of the manuscript.
All figures are created with https://biorender.com/.
YH and XZ are employed by The Whiteoak Group, Inc.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.967505/full#supplementary-material
1. Morales A, Eidinger D, Bruce AW. Intracavitary bacillus calmette-guerin in the treatment of superficial bladder tumors. J Urol (1976) 116:180–2. doi: 10.1016/S0022-5347(17)58737-6
2. Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol (2002) 3:991–8. doi: 10.1038/ni1102-991
3. Rosenberg SA, Lotze MT, Muul LM, Leitman S, Chang AE, Ettinghausen SE, et al. Observations on the systemic administration of autologous lymphokine-activated killer cells and recombinant interleukin-2 to patients with metastatic cancer. N Engl J Med (1985) 313:1485–92. doi: 10.1056/NEJM198512053132327
4. Milstein C. The hybridoma revolution: an offshoot of basic research. BioEssays (1999) 21:966–73. doi: 10.1002/(SICI)1521-1878(199911)21:11<966::AID-BIES9>3.0.CO;2-Z
5. Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell (2015) 27:450–61. doi: 10.1016/j.ccell.2015.03.001
7. Pastor F, Berraondo P, Etxeberria I, Frederick J, Sahin U, Gilboa E, et al. An RNA toolbox for cancer immunotherapy. Nat Rev Drug Discovery (2018) 17:751–67. doi: 10.1038/nrd.2018.132
8. Guevara ML, Persano F, Persano S. Advances in lipid nanoparticles for mrna-based cancer immunotherapy. Front Chem (2020) 8:589959. doi: 10.3389/fchem.2020.589959
9. June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science (2018) 359:1361–5. doi: 10.1126/science.aar6711
10. Feng X, Xu W, Li Z, Song W, Ding J, Chen X. Immunomodulatory nanosystems. Adv Sci (2019) 6:1900101. doi: 10.1002/advs.201900101
11. Xu L, Wang X, Liu Y, Yang G, Falconer RJ, Zhao C-X. Lipid nanoparticles for drug delivery. Adv NanoBiomed Res (2022) 2:2100109. doi: 10.1002/anbr.202100109
12. Tenchov R, Bird R, Curtze AE, Zhou Q. Lipid nanoparticles─from liposomes to mrna vaccine delivery, a landscape of research diversity and advancement. ACS Nano (2021) 15:16982–7015. doi: 10.1021/acsnano.1c04996
13. Mitchell MJ, Billingsley MM, Haley RM, Wechsler ME, Peppas NA, Langer R. Engineering precision nanoparticles for drug delivery. Nat Rev Drug Discovery (2021) 20:101–24. doi: 10.1038/s41573-020-0090-8
14. Anselmo AC, Mitragotri S. Nanoparticles in the clinic: An update. Bioeng Transl Med (2019) 4:e10143. doi: 10.1002/btm2.10143
15. Ostro Mj, Giacomoni D, Lavelle D, Paxton W, Dray S. Evidence for translation of rabbit globin mRNA after liposomemediated insertion into a human cell line. Nature (1978) 274:921–3. doi: 10.1038/274921a0
16. Dimitriadis Gj. Translation of rabbit globin mRNA introduced by liposomes into mouse lymphocytes. Nature (1978) 274:923–4. doi: 10.1038/274923a0
17. Bangham AD, Horne RW. Negative staining of phospholipids and their structural modification by surface-active agents as observed in the electron microscope. J Mol Biol (1964) 8(5):660–8. doi: 10.1016/S0022-2836(64)80115-7
18. Hou X, Zaks T, Langer R, Dong Y. Lipid nanoparticles for mRNA delivery. Nat Rev Mater (2021) 6:1078–94. doi: 10.1038/s41578-021-00358-0
19. Jaiswal M, Dudhe R, Sharma PK. Nanoemulsion: an advanced mode of drug delivery system. 3 Biotech (2015) 5:123–7. doi: 10.1007/s13205-014-0214-0
20. McClements DJ. Edible nanoemulsions: fabrication, properties, and functional performance. Soft Matter (2011) 7:2297–316. doi: 10.1039/C0SM00549E
21. Sutradhar KB, Amin ML. Nanoemulsions: increasing possibilities in drug delivery. Eur J Nanomed (2013) 5:97–110. doi: 10.1515/ejnm-2013-0001
22. Cullis PR, Hope MJ. Lipid nanoparticle systems for enabling gene therapies. Mol Ther (2017) 25:1467–75. doi: 10.1016/j.ymthe.2017.03.013
23. Kumar M, Bishnoi RS, Shukla AK, Jain CP. Techniques for formulation of nanoemulsion drug delivery system: a review. Prev Nutr Food Sci (2019) 24:225–34. doi: 10.3746/pnf.2019.24.3.225
24. Fraley R, Straubinger RM, Rule G, Springer EL, Papahadjopoulos D. Liposome-mediated delivery of deoxyribonucleic acid to cells: enhanced efficiency of delivery by changes in lipid composition and incubation conditions. Biochemistry (1981) 20:6978–87. doi: 10.1021/bi00527a031
25. Fraley R, Subramani S, Berg P, Papahadjopoulos D. Introduction of liposome-encapsulated SV40 DNA into cells. J Biol Chem (1980) 255:10431–5. doi: 10.1016/S0021-9258(19)70482-7
26. Felgner PL, Gadek TR, Holm M, Roman R, Chan HW, Wenz M, et al. Lipofection: a highly efficient, lipid-mediated DNA-transfection procedure. Proc Natl Acad Sci (1987) 84:7413–7. doi: 10.1073/pnas.84.21.7413
27. Felgner J, Martin M, Tsai Y, Felgner PL. Cationic lipid-mediated transfection in mammalian cells: “Lipofection”. J Tissue Cult Methods (1993) 15:63–8. doi: 10.1007/BF01667363
28. Hilgers LAT, Snippe H. DDA as an immunological adjuvant. Res Immunol (1992) 143:494–503. doi: 10.1016/0923-2494(92)80060-X
29. Wheeler JJ, Palmer L, Ossanlou M, MacLachlan I, Graham RW, Zhang YP, et al. Stabilized plasmid-lipid particles: construction and characterization. Gene Ther (1999) 6:271–81. doi: 10.1038/sj.gt.3300821
30. Ambegia E, Ansell S, Cullis P, Heyes J, Palmer L, MacLachlan I. Stabilized plasmid–lipid particles containing PEG-diacylglycerols exhibit extended circulation lifetimes and tumor selective gene expression. Biochim Biophys Acta - Biomembr (2005) 1669:155–63. doi: 10.1016/j.bbamem.2005.02.001
31. Kranz LM, Diken M, Haas H, Kreiter S, Loquai C, Reuter KC, et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature (2016) 534:396–401. doi: 10.1038/nature18300
32. Yang J, Arya S, Lung P, Lin Q, Huang J, Li Q. Hybrid nanovaccine for the co-delivery of the mRNA antigen and adjuvant. Nanoscale (2019) 11:21782–9. doi: 10.1039/C9NR05475H
33. Lei S, Zhang X, Men K, Gao Y, Yang X, Wu S, et al. Efficient colorectal cancer gene therapy with il-15 mrna nanoformulation. Mol Pharm (2020) 17:3378–91. doi: 10.1021/acs.molpharmaceut.0c00451
34. Zhang R, Men K, Zhang X, Huang R, Tian Y, Zhou B, et al. Delivery of a modified mRNA encoding IL-22 binding protein (IL-22BP) for colon cancer gene therapy. J BioMed Nanotechnol (2018) 14:1239–51. doi: 10.1166/jbn.2018.2577
35. Bailey AL, Cullis PR. Modulation of membrane fusion by asymmetric transbilayer distributions of amino lipids. Biochemistry (1994) 33:12573–80. doi: 10.1021/bi00208a007
36. Meng C, Chen Z, Li G, Welte T, Shen H. Nanoplatforms for mRNA therapeutics. Adv Ther (2021) 4:2000099. doi: 10.1002/adtp.202000099
37. Jayaraman M, Ansell SM, Mui BL, Tam YK, Chen J, Du X, et al. Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo**. Angew Chemie (2012) 124:8657–61. doi: 10.1002/ange.201203263
38. Semple SC, Akinc A, Chen J, Sandhu AP, Mui BL, Cho CK, et al. Rational design of cationic lipids for siRNA delivery. Nat Biotechnol (2010) 28:172–6. doi: 10.1038/nbt.1602
39. Semple SC, Klimuk SK, Harasym TO, Dos Santos N, Ansell SM, Wong KF, et al. Efficient encapsulation of antisense oligonucleotides in lipid vesicles using ionizable aminolipids: formation of novel small multilamellar vesicle structures. Biochim Biophys Acta - Biomembr (2001) 1510:152–66. doi: 10.1016/S0005-2736(00)00343-6
40. Zimmermann TS, Lee ACH, Akinc A, Bramlage B, Bumcrot D, Fedoruk MN, et al. RNAi-mediated gene silencing in non-human primates. Nature (2006) 441:111–4. doi: 10.1038/nature04688
41. Maier MA, Jayaraman M, Matsuda S, Liu J, Barros S, Querbes W, et al. Biodegradable lipids enabling rapidly eliminated lipid nanoparticles for systemic delivery of rnai therapeutics. Mol Ther (2013) 21:1570–8. doi: 10.1038/mt.2013.124
42. Hassett KJ, Benenato KE, Jacquinet E, Lee A, Woods A, Yuzhakov O, et al. Optimization of lipid nanoparticles for intramuscular administration of mrna vaccines. Mol Ther - Nucleic Acids (2019) 15:1–11. doi: 10.1016/j.omtn.2019.01.013
43. Sabnis S, Kumarasinghe ES, Salerno T, Mihai C, Ketova T, Senn JJ, et al. A novel amino lipid series for mrna delivery: improved endosomal escape and sustained pharmacology and safety in non-human primates. Mol Ther (2018) 26:1509–19. doi: 10.1016/j.ymthe.2018.03.010
44. Miao L, Li L, Huang Y, Delcassian D, Chahal J, Han J, et al. Delivery of mRNA vaccines with heterocyclic lipids increases anti-tumor efficacy by STING-mediated immune cell activation. Nat Biotechnol (2019) 37:1174–85. doi: 10.1038/s41587-019-0247-3
45. Ramaswamy S, Tonnu N, Tachikawa K, Limphong P, Vega JB, Karmali PP, et al. Systemic delivery of factor IX messenger RNA for protein replacement therapy. Proc Natl Acad Sci (2017) 114:E1941–50. doi: 10.1073/pnas.1619653114
46. Finn JD, Smith AR, Patel MC, Shaw L, Youniss MR, van Heteren J, et al. A single administration of crispr/cas9 lipid nanoparticles achieves robust and persistent in vivo genome editing. Cell Rep (2018) 22:2227–35. doi: 10.1016/j.celrep.2018.02.014
47. Saadati F, Cammarone S, Ciufolini MA. A route to lipid ALC-0315: a key component of a COVID-19 mRNA vaccine. Chem – A Eur J (2022) 2022:e202200906. doi: 10.1002/chem.202200906
48. Li L, Wang R, Wilcox D, Sarthy A, Lin X, Huang X, et al. Developing lipid nanoparticle-based sirna therapeutics for hepatocellular carcinoma using an integrated approach. Mol Cancer Ther (2013) 12:2308–18. doi: 10.1158/1535-7163.MCT-12-0983-T
49. Love KT, Mahon KP, Levins CG, Whitehead KA, Querbes W, Dorkin JR, et al. Lipid-like materials for low-dose, in vivo gene silencing. Proc Natl Acad Sci (2010) 107:1864–9. doi: 10.1073/pnas.0910603106
50. Dong Y, Love KT, Dorkin JR, Sirirungruang S, Zhang Y, Chen D, et al. Lipopeptide nanoparticles for potent and selective siRNA delivery in rodents and nonhuman primates. Proc Natl Acad Sci (2014) 111:3955–60. doi: 10.1073/pnas.1322937111
51. Li B, Luo X, Deng B, Wang J, McComb DW, Shi Y, et al. An orthogonal array optimization of lipid-like nanoparticles for mrna delivery in vivo. Nano Lett (2015) 15:8099–107. doi: 10.1021/acs.nanolett.5b03528
52. Zhang X, Zhao W, Nguyen GN, Zhang C, Zeng C, Yan J, et al. Functionalized lipid-like nanoparticles for in vivo mRNA delivery and base editing. Sci Adv (2020) 6:eabc2315. doi: 10.1126/sciadv.abc2315
53. Akita H, Ishiba R, Togashi R, Tange K, Nakai Y, Hatakeyama H, et al. A neutral lipid envelope-type nanoparticle composed of a pH-activated and vitamin e-scaffold lipid-like material as a platform for a gene carrier targeting renal cell carcinoma. J Control Release (2015) 200:97–105. doi: 10.1016/j.jconrel.2014.12.029
54. Conway A, Mendel M, Kim K, McGovern K, Boyko A, Zhang L, et al. Non-viral delivery of zinc finger nuclease mrna enables highly efficient in vivo genome editing of multiple therapeutic gene targets. Mol Ther (2019) 27:866–77. doi: 10.1016/j.ymthe.2019.03.003
55. Hafez IM, Cullis PR. Roles of lipid polymorphism in intracellular delivery. Adv Drug Delivery Rev (2001) 47:139–48. doi: 10.1016/S0169-409X(01)00103-X
56. Xue Y, Feng J, Liu Y, Che J, Bai G, Dong X, et al. A synthetic carrier of nucleic acids structured as a neutral phospholipid envelope tightly assembled on polyplex surface. Adv Healthc Mater (2020) 9:1901705. doi: 10.1002/adhm.201901705
57. Cullis P, De Kruijff B. The polymorphic phase behaviour of phosphatidylethanolamines of natural and synthetic origin. A 31P NMR study. Biochim Biophys Acta - Biomembr (1978) 513:31–42. doi: 10.1016/0005-2736(78)90109-8
58. Sakurai F. Effects of erythrocytes and serum proteins on lung accumulation of lipoplexes containing cholesterol or DOPE as a helper lipid in the single-pass rat lung perfusion system. Eur J Pharm Biopharm (2001) 52:165–72. doi: 10.1016/S0939-6411(01)00165-5
59. Dabkowska AP, Barlow DJ, Hughes AV, Campbell RA, Quinn PJ, Lawrence MJ. The effect of neutral helper lipids on the structure of cationic lipid monolayers. J R Soc Interface (2012) 9:548–61. doi: 10.1098/rsif.2011.0356
60. Tenchov BG, MacDonald RC, Siegel DP. Cubic phases in phosphatidylcholine-cholesterol mixtures: cholesterol as membrane “fusogen. Biophys J (2006) 91:2508–16. doi: 10.1529/biophysj.106.083766
61. Aswathanarayan JB, Vittal RR. Nanoemulsions and their potential applications in food industry. Front Sustain Food Syst (2019) 3:95. doi: 10.3389/fsufs.2019.00095
62. Ma Y, Liu D, Wang D, Wang Y, Fu Q, Fallon JK, et al. Combinational delivery of hydrophobic and hydrophilic anticancer drugs in single nanoemulsions to treat mdr in cancer. Mol Pharm (2014) 11:2623–30. doi: 10.1021/mp400778r
63. Zhang Z, Kuo JC-T, Zhang C, Huang Y, Zhou Z, Lee RJ. A squalene-based nanoemulsion for therapeutic delivery of resiquimod. Pharmaceutics (2021) 13:2060. doi: 10.3390/pharmaceutics13122060
64. Olthof ED, Gülich AF, Renne MF, Landman S, Joosten LAB, Roelofs HMJ, et al. Immune activation by medium-chain triglyceride-containing lipid emulsions is not modulated by n-3 lipids or toll-like receptor 4. Toxicol Vitr (2015) 29:1851–8. doi: 10.1016/j.tiv.2015.07.004
65. Hidalgo MA, Carretta MD, Burgos RA. Long chain fatty acids as modulators of immune cells function: contribution of ffa1 and ffa4 receptors. Front Physiol (2021) 12:668330. doi: 10.3389/fphys.2021.668330
66. Kim EH, Woodruff MC, Grigoryan L, Maier B, Lee SH, Mandal P, et al. Squalene emulsion-based vaccine adjuvants stimulate CD8 T cell, but not antibody responses, through a RIPK3-dependent pathway. Elife (2020) 9:e52687. doi: 10.7554/eLife.52687
67. Knop K, Hoogenboom R, Fischer D, Schubert US. Poly(ethylene glycol) in drug delivery: pros and cons as well as potential alternatives. Angew Chemie Int Ed (2010) 49:6288–308. doi: 10.1002/anie.200902672
68. Zhu X, Tao W, Liu D, Wu J, Guo Z, Ji X, et al. Surface de-pegylation controls nanoparticle-mediated sirna delivery in vitro and in vivo. Theranostics (2017) 7:1990–2002. doi: 10.7150/thno.18136
69. Song LY, Ahkong QF, Rong Q, Wang Z, Ansell S, Hope MJ, et al. Characterization of the inhibitory effect of PEG-lipid conjugates on the intracellular delivery of plasmid and antisense DNA mediated by cationic lipid liposomes. Biochim Biophys Acta - Biomembr (2002) 1558:1–13. doi: 10.1016/S0005-2736(01)00399-6
70. García-Pinel B, Porras-Alcalá C, Ortega-Rodríguez A, Sarabia F, Prados J, Melguizo C, et al. Lipid-based nanoparticles: application and recent advances in cancer treatment. Nanomaterials (2019) 9:638. doi: 10.3390/nano9040638
71. Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, et al. Toll-like receptor 4–dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med (2007) 13:1050–9. doi: 10.1038/nm1622
72. Alizadeh D, Trad M, Hanke NT, Larmonier CB, Janikashvili N, Bonnotte B, et al. Doxorubicin eliminates myeloid-derived suppressor cells and enhances the efficacy of adoptive t-cell transfer in breast cancer. Cancer Res (2014) 74:104–18. doi: 10.1158/0008-5472.CAN-13-1545
73. Ma Y, Aymeric L, Locher C, Mattarollo SR, Delahaye NF, Pereira P, et al. Contribution of IL-17–producing γδ T cells to the efficacy of anticancer chemotherapy. J Exp Med (2011) 208:491–503. doi: 10.1084/jem.20100269
74. Eralp Y, Wang X, Wang J-P, Maughan MF, Polo JM, Lachman LB. Doxorubicin and paclitaxel enhance the antitumor efficacy of vaccines directed against HER 2/neuin a murine mammary carcinoma model. Breast Cancer Res (2004) 6:R275. doi: 10.1186/bcr787
75. Panis C, Lemos LGT, Victorino VJ, Herrera ACSA, Campos FC, Colado Simão AN, et al. Immunological effects of taxol and adryamicin in breast cancer patients. Cancer Immunol Immunother (2012) 61:481–8. doi: 10.1007/s00262-011-1117-0
76. Imai H, Dansako H, Ueda Y, Satoh S, Kato N. Daunorubicin, a topoisomerase II poison, suppresses viral production of hepatitis b virus by inducing cGAS-dependent innate immune response. Biochem Biophys Res Commun (2018) 504:672–8. doi: 10.1016/j.bbrc.2018.08.195
77. Sauter KAD, Wood LJ, Wong J, Iordanov M, Magun BE. Doxorubicin and daunorubicin induce processing and release of interleukin-1β through activation of the NLRP3 inflammasome. Cancer Biol Ther (2011) 11:1008–16. doi: 10.4161/cbt.11.12.15540
78. Cao C, Han Y, Ren Y, Wang Y. Mitoxantrone-mediated apoptotic b16-f1 cells induce specific anti-tumor immune response. Cell Mol Immunol (2009) 6:469–75. doi: 10.1038/cmi.2009.59
79. Shin JY, Lee SK, Kang CD, Chung JS, Lee EY, Seo SY, et al. Antitumor effect of intratumoral administration of dendritic cell combination with vincristine chemotherapy in a murine fibrosarcoma model. Histol Histopathol (2003) 18:435–47. doi: 10.14670/HH-18.435
80. Wei T, Li M, Zhu Z, Xiong H, Shen H, Zhang H, et al. Vincristine upregulates PD-L1 and increases the efficacy of PD-L1 blockade therapy in diffuse large b-cell lymphoma. J Cancer Res Clin Oncol (2021) 147:691–701. doi: 10.1007/s00432-020-03446-w
81. Iwai T, Sugimoto M, Wakita D, Yorozu K, Kurasawa M, Yamamoto K. Topoisomerase I inhibitor, irinotecan, depletes regulatory T cells and up-regulates MHC class I and PD-L1 expression, resulting in a supra-additive antitumor effect when combined with anti-PD-L1 antibodies. Oncotarget (2018) 9:31411–21. doi: 10.18632/oncotarget.25830
82. Wan S, Pestka S, Jubin RG, Lyu YL, Tsai Y-C, Liu LF. Chemotherapeutics and radiation stimulate MHC class I expression through elevated interferon-beta signaling in breast cancer cells. PLoS One (2012) 7:e32542. doi: 10.1371/journal.pone.0032542
83. Hong M, Puaux A-L, Huang C, Loumagne L, Tow C, Mackay C, et al. Chemotherapy induces intratumoral expression of chemokines in cutaneous melanoma, favoring t-cell infiltration and tumor control. Cancer Res (2011) 71:6997–7009. doi: 10.1158/0008-5472.CAN-11-1466
84. Demaria S, Volm MD, Shapiro RL, Yee HT, Oratz R, Formenti SC, et al. Development of tumor-infiltrating lymphocytes in breast cancer after neoadjuvant paclitaxel chemotherapy. Clin Cancer Res (2001) 7:3025–30.
85. Peng J, Hamanishi J, Matsumura N, Abiko K, Murat K, Baba T, et al. Chemotherapy induces programmed cell death-ligand 1 overexpression via the nuclear factor-κb to foster an immunosuppressive tumor microenvironment in ovarian cancer. Cancer Res (2015) 75:5034–45. doi: 10.1158/0008-5472.CAN-14-3098
86. Zhang L, Dermawan K, Jin M, Liu R, Zheng H, Xu L, et al. Differential impairment of regulatory T cells rather than effector T cells by paclitaxel-based chemotherapy. Clin Immunol (2008) 129:219–29. doi: 10.1016/j.clim.2008.07.013
87. Michels T, Shurin GV, Naiditch H, Sevko A, Umansky V, Shurin MR. Paclitaxel promotes differentiation of myeloid-derived suppressor cells into dendritic cells in vitro in a TLR4-independent manner. J Immunotoxicol (2012) 9:292–300. doi: 10.3109/1547691X.2011.642418
88. Cullis J, Siolas D, Avanzi A, Barui S, Maitra A, Bar-Sagi D. Macropinocytosis of nab-paclitaxel drives macrophage activation in pancreatic cancer. Cancer Immunol Res (2017) 5:182–90. doi: 10.1158/2326-6066.CIR-16-0125
89. Shimizu T, Abu Lila AS, Nishio M, Doi Y, Ando H, Ukawa M, et al. Modulation of antitumor immunity contributes to the enhanced therapeutic efficacy of liposomal oxaliplatin in mouse model. Cancer Sci (2017) 108:1864–9. doi: 10.1111/cas.13305
90. Rameshbabu S, Labadie BW, Argulian A, Patnaik A. Targeting innate immunity in cancer therapy. Vaccines (2021) 9:138. doi: 10.3390/vaccines9020138
91. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity (2013) 39:1–10. doi: 10.1016/j.immuni.2013.07.012
92. Lee K, Kim SY, Seo Y, Kim MH, Chang J, Lee H. Adjuvant incorporated lipid nanoparticles for enhanced mRNA-mediated cancer immunotherapy. Biomater Sci (2020) 8:1101–5. doi: 10.1039/C9BM01564G
93. Varypataki EM, van der Maaden K, Bouwstra J, Ossendorp F, Jiskoot W. Cationic liposomes loaded with a synthetic long peptide and poly(i:c): a defined adjuvanted vaccine for induction of antigen-specific t cell cytotoxicity. AAPS J (2015) 17:216–26. doi: 10.1208/s12248-014-9686-4
94. Burke ML, de VM, Pleasance J, Neeland M, Elhay M, Harrison P, et al. Innate immune pathways in afferent lymph following vaccination with poly(I:C)-containing liposomes. Innate Immun (2014) 20:501–10. doi: 10.1177/1753425913501213
95. Colapicchioni V, Palchetti S, Pozzi D, Marini ES, Riccioli A, Ziparo E, et al. Killing cancer cells using nanotechnology: novel poly(I:C) loaded liposome–silica hybrid nanoparticles. J Mater Chem B (2015) 3:7408–16. doi: 10.1039/C5TB01383F
96. Pauli G, Chao P-H, Qin Z, Böttger R, Lee SE, Li S-D. Liposomal resiquimod for enhanced immunotherapy of peritoneal metastases of colorectal cancer. Pharmaceutics (2021) 13:1696. doi: 10.3390/pharmaceutics13101696
97. Munakata L, Tanimoto Y, Osa A, Meng J, Haseda Y, Naito Y, et al. Lipid nanoparticles of type-a CpG D35 suppress tumor growth by changing tumor immune-microenvironment and activate CD8 T cells in mice. J Control Release (2019) 313:106–19. doi: 10.1016/j.jconrel.2019.09.011
98. Chen W, Jiang M, Yu W, Xu Z, Liu X, Jia Q, et al. CpG-based nanovaccines for cancer immunotherapy. Int J Nanomed (2021) 2021:5281–99. doi: 10.2147/IJN.S317626
99. Zhang Z, Kuo JC-T, Yao S, Zhang C, Khan H, Lee RJ. CpG oligodeoxynucleotides for anticancer monotherapy from preclinical stages to clinical trials. Pharmaceutics (2021) 14:73. doi: 10.3390/pharmaceutics14010073
100. Wilson KD, de Jong SD, Kazem M, Lall R, Hope MJ, Cullis PR, et al. The combination of stabilized plasmid lipid particles and lipid nanoparticle encapsulated CpG containing oligodeoxynucleotides as a systemic genetic vaccine. J Gene Med (2009) 11:14–25. doi: 10.1002/jgm.1267
101. Rehwinkel J, Gack MU. RIG-i-like receptors: their regulation and roles in RNA sensing. Nat Rev Immunol (2020) 20:537–51. doi: 10.1038/s41577-020-0288-3
102. Elion DL, Jacobson ME, Hicks DJ, Rahman B, Sanchez V, Gonzales-Ericsson PI, et al. Therapeutically active rig-i agonist induces immunogenic tumor cell killing in breast cancers. Cancer Res (2018) 78:6183–95. doi: 10.1158/0008-5472.CAN-18-0730
103. Das M, Shen L, Liu Q, Goodwin TJ, Huang L. Nanoparticle delivery of rig-i agonist enables effective and safe adjuvant therapy in pancreatic cancer. Mol Ther (2019) 27:507–17. doi: 10.1016/j.ymthe.2018.11.012
104. Jacoberger-Foissac C, Saliba H, Wantz M, Seguin C, Flacher V, Frisch B, et al. Liposomes as tunable platform to decipher the antitumor immune response triggered by TLR and NLR agonists. Eur J Pharm Biopharm (2020) 152:348–57. doi: 10.1016/j.ejpb.2020.05.026
105. Ando K, Mori K, Corradini N, Redini F, Heymann D. Mifamurtide for the treatment of nonmetastatic osteosarcoma. Expert Opin Pharmacother (2011) 12:285–92. doi: 10.1517/14656566.2011.543129
106. Zhou Q, Zhou Y, Li T, Ge Z. Nanoparticle-mediated sting agonist delivery for enhanced cancer immunotherapy. Macromol Biosci (2021) 21:2100133. doi: 10.1002/mabi.202100133
107. Miyabe H, Hyodo M, Nakamura T, Sato Y, Hayakawa Y, Harashima H. A new adjuvant delivery system ‘cyclic di-GMP/YSK05 liposome’ for cancer immunotherapy. J Control Release (2014) 184:20–7. doi: 10.1016/j.jconrel.2014.04.004
108. Koshy ST, Cheung AS, Gu L, Graveline AR, Mooney DJ. Liposomal delivery enhances immune activation by sting agonists for cancer immunotherapy. Adv Biosyst (2017) 1:1600013. doi: 10.1002/adbi.201600013
109. Cheng N, Watkins-Schulz R, Junkins RD, David CN, Johnson BM, Montgomery SA, et al. A nanoparticle-incorporated STING activator enhances antitumor immunity in PD-L1–insensitive models of triple-negative breast cancer. JCI Insight (2018) 3(22):e120638. doi: 10.1172/jci.insight.120638
110. Wilson DR, Sen R, Sunshine JC, Pardoll DM, Green JJ, Kim YJ. Biodegradable STING agonist nanoparticles for enhanced cancer immunotherapy. Nanomed Nanotech Biol Med (2018) 14:237–46. doi: 10.1016/j.nano.2017.10.013
111. Alshamsan A, Haddadi A, Hamdy S, Samuel J, El-Kadi AOS, Uludağ H, et al. STAT3 silencing in dendritic cells by sirna polyplexes encapsulated in plga nanoparticles for the modulation of anticancer immune response. Mol Pharm (2010) 7:1643–54. doi: 10.1021/mp100067u
112. Alshamsan A, Hamdy S, Haddadi A, Samuel J, El-Kadi AOS, Uludağ H, et al. STAT3 knockdown in B16 melanoma by siRNA lipopolyplexes induces bystander immune response in vitro and in vivo. Transl Oncol (2011) 4:178–88. doi: 10.1593/tlo.11100
113. Akita H, Kogure K, Moriguchi R, Nakamura Y, Higashi T, Nakamura T, et al. Nanoparticles for ex vivo siRNA delivery to dendritic cells for cancer vaccines: Programmed endosomal escape and dissociation☆. J Control Release (2010) 143:311–7. doi: 10.1016/j.jconrel.2010.01.012
114. Zhang X, Su Y, Song H, Yu Z, Zhang B, Chen H. Attenuated A20 expression of acute myeloid leukemia-derived dendritic cells increased the anti-leukemia immune response of autologous cytolytic T cells. Leuk Res (2014) 38:673–81. doi: 10.1016/j.leukres.2014.03.011
115. Warashina S, Nakamura T, Harashima H. A20 silencing by lipid envelope-type nanoparticles enhances the efficiency of lipopolysaccharide-activated dendritic cells. Biol Pharm Bull (2011) 34:1348–51. doi: 10.1248/bpb.34.1348
116. Chen D, Koropatnick J, Jiang N, Zheng X, Zhang X, Wang H, et al. Targeted siRNA silencing of indoleamine 2, 3-dioxygenase in antigen-presenting cells using mannose-conjugated liposomes. J Immunother (2014) 37:123–34. doi: 10.1097/CJI.0000000000000022
117. Huang T-T, Yen M-C, Lin C-C, Weng T-Y, Chen Y-L, Lin C-M, et al. Skin delivery of short hairpin RNA of indoleamine 2,3 dioxygenase induces antitumor immunity against orthotopic and metastatic liver cancer. Cancer Sci (2011) 102:2214–20. doi: 10.1111/j.1349-7006.2011.02094.x
118. Song X-T, Kabler KE, Shen L, Rollins L, Huang XF, Chen S-Y. A20 is an antigen presentation attenuator, and its inhibition overcomes regulatory T cell–mediated suppression. Nat Med (2008) 14:258–65. doi: 10.1038/nm1721
119. Cubillos-Ruiz JR, Engle X, Scarlett UK, Martinez D, Barber A, Elgueta R, et al. Polyethylenimine-based siRNA nanocomplexes reprogram tumor-associated dendritic cells via TLR5 to elicit therapeutic antitumor immunity. J Clin Invest (2009) 119(8):2231–44. doi: 10.1172/JCI37716
120. Liang L, Ge K, Zhang F, Ge Y. The suppressive effect of co-inhibiting PD-1 and CTLA-4 expression on H22 hepatomas in mice. Cell Mol Biol Lett (2018) 23:58. doi: 10.1186/s11658-018-0122-0
121. Iwamura K, Kato T, Miyahara Y, Naota H, Mineno J, Ikeda H, et al. siRNA-mediated silencing of PD-1 ligands enhances tumor-specific human T-cell effector functions. Gene Ther (2012) 19:959–66. doi: 10.1038/gt.2011.185
122. Liu Z, Falo LD, You Z. Knockdown of HMGB1 in tumor cells attenuates their ability to induce regulatory t cells and uncovers naturally acquired cd8 t cell-dependent antitumor immunity. J Immunol (2011) 187:118–25. doi: 10.4049/jimmunol.1003378
123. Tsai B-Y, Suen J-L, Chiang B-L. Lentiviral-mediated Foxp3 RNAi suppresses tumor growth of regulatory T cell-like leukemia in a murine tumor model. Gene Ther (2010) 17:972–9. doi: 10.1038/gt.2010.38
124. Conroy H, Galvin KC, Higgins SC, Mills KHG. Gene silencing of TGF-β1 enhances antitumor immunity induced with a dendritic cell vaccine by reducing tumor-associated regulatory T cells. Cancer Immunol Immunother (2012) 61:425–31. doi: 10.1007/s00262-011-1188-y
125. Ahn Y-H, Hong S-O, Kim JH, Noh KH, Song K-H, Lee Y-H, et al. The siRNA cocktail targeting interleukin 10 receptor and transforming growth factor-β receptor on dendritic cells potentiates tumour antigen-specific CD8+ T cell immunity. Clin Exp Immunol (2015) 181:164–78. doi: 10.1111/cei.12620
126. Kang S, Xie J, Ma S, Liao W, Zhang J, Luo R. Targeted knock down of CCL22 and CCL17 by siRNA during DC differentiation and maturation affects the recruitment of T subsets. Immunobiology (2010) 215:153–62. doi: 10.1016/j.imbio.2009.03.001
127. Ma S, Liu M, Xu Z, Li Y, Guo H, Ge Y, et al. A double feedback loop mediated by microRNA-23a/27a/24-2 regulates M1 versus M2 macrophage polarization and thus regulates cancer progression. Oncotarget (2016) 7:13502–19. doi: 10.18632/oncotarget.6284
128. Sahraei M, Chaube B, Liu Y, Sun J, Kaplan A, Price NL, et al. Suppressing miR-21 activity in tumor-associated macrophages promotes an antitumor immune response. J Clin Invest (2019) 129:5518–36. doi: 10.1172/JCI127125
129. Stern-Ginossar N, Gur C, Biton M, Horwitz E, Elboim M, Stanietsky N, et al. Human microRNAs regulate stress-induced immune responses mediated by the receptor NKG2D. Nat Immunol (2008) 9:1065–73. doi: 10.1038/ni.1642
130. Codo P, Weller M, Meister G, Szabo E, Steinle A, Wolter M, et al. MicroRNA-mediated down-regulation of NKG2D ligands contributes to glioma immune escape. Oncotarget (2014) 5:7651–62. doi: 10.18632/oncotarget.2287
131. Yang J, Zhang Z, Chen C, Liu Y, Si Q, Chuang T-H, et al. MicroRNA-19a-3p inhibits breast cancer progression and metastasis by inducing macrophage polarization through downregulated expression of fra-1 proto-oncogene. Oncogene (2014) 33:3014–23. doi: 10.1038/onc.2013.258
132. Hong BS, Ryu HS, Kim N, Kim J, Lee E, Moon H, et al. Tumor suppressor microRNA-204-5p regulates growth, metastasis, and immune microenvironment remodeling in breast cancer. Cancer Res (2019) 79(7):1520–34. doi: 10.1158/0008-5472.CAN-18-0891
133. Li D, Wang X, Yang M, Kan Q, Duan Z. miR3609 sensitizes breast cancer cells to adriamycin by blocking the programmed death-ligand 1 immune checkpoint. Exp Cell Res (2019) 380:20–8. doi: 10.1016/j.yexcr.2019.03.025
134. Zhang Y, Yang P, Sun T, Li D, Xu X, Rui Y, et al. miR-126 and miR-126* repress recruitment of mesenchymal stem cells and inflammatory monocytes to inhibit breast cancer metastasis. Nat Cell Biol (2013) 15:284–94. doi: 10.1038/ncb2690
135. Stephens AJ, Burgess-Brown NA, Jiang S. Beyond just peptide antigens: the complex world of peptide-based cancer vaccines. Front Immunol (2021) 12:696791. doi: 10.3389/fimmu.2021.696791
136. Rueda F, Eich C, Cordobilla B, Domingo P, Acosta G, Albericio F, et al. Effect of TLR ligands co-encapsulated with multiepitopic antigen in nanoliposomes targeted to human DCs via fc receptor for cancer vaccines. Immunobiology (2017) 222:989–97. doi: 10.1016/j.imbio.2017.06.002
137. Zamani P, Navashenaq JG, Nikpoor AR, Hatamipour M, Oskuee RK, Badiee A, et al. MPL nano-liposomal vaccine containing P5 HER2/neu-derived peptide pulsed PADRE as an effective vaccine in a mice TUBO model of breast cancer. J Control Release (2019) 303:223–36. doi: 10.1016/j.jconrel.2019.04.019
138. Zamani P, Teymouri M, Nikpoor AR, Navashenaq JG, Gholizadeh Z, Darban SA, et al. Nanoliposomal vaccine containing long multi-epitope peptide E75-AE36 pulsed PADRE-induced effective immune response in mice TUBO model of breast cancer. Eur J Cancer (2020) 129:80–96. doi: 10.1016/j.ejca.2020.01.010
139. Arbelaez CA, Estrada J, Gessner MA, Glaus C, Morales AB, Mohn D, et al. A nanoparticle vaccine that targets neoantigen peptides to lymphoid tissues elicits robust antitumor T cell responses. NPJ Vaccines (2020) 5:106. doi: 10.1038/s41541-020-00253-9
140. Melhem NM, Liu XD, Boczkowski D, Gilboa E, Barratt-Boyes SM. Robust CD4+ and CD8+ T cell responses to SIV using mRNA-transfected DC expressing autologous viral Ag. Eur J Immunol (2007) 37:2164–73. doi: 10.1002/eji.200636782
141. Van Nuffel AM, Benteyn D, Wilgenhof S, Pierret L, Corthals J, Heirman C, et al. Dendritic cells loaded with mrna encoding full-length tumor antigens prime cd4+ and cd8+ t cells in melanoma patients. Mol Ther (2012) 20:1063–74. doi: 10.1038/mt.2012.11
142. Urban-Wojciuk Z, Khan MM, Oyler BL, Fåhraeus R, Marek-Trzonkowska N, Nita-Lazar A, et al. The role of TLRs in anti-cancer immunity and tumor rejection. Front Immunol (2019) 10:2388. doi: 10.3389/fimmu.2019.02388
143. Verbeke R, Lentacker I, Wayteck L, Breckpot K, Van Bockstal M, Descamps B, et al. Co-Delivery of nucleoside-modified mRNA and TLR agonists for cancer immunotherapy: Restoring the immunogenicity of immunosilent mRNA. J Control Release (2017) 266:287–300. doi: 10.1016/j.jconrel.2017.09.041
144. Lai I, Swaminathan S, Baylot V, Mosley A, Dhanasekaran R, Gabay M, et al. Lipid nanoparticles that deliver IL-12 messenger RNA suppress tumorigenesis in MYC oncogene-driven hepatocellular carcinoma. J Immunother Cancer (2018) 6:125. doi: 10.1186/s40425-018-0431-x
145. Jain R, Frederick JP, Huang EY, Burke KE, Mauger DM, Andrianova EA, et al. MicroRNAs enable mRNA therapeutics to selectively program cancer cells to self-destruct. Nucleic Acid Ther (2018) 28:285–96. doi: 10.1089/nat.2018.0734
146. Oberli MA, Reichmuth AM, Dorkin JR, Mitchell MJ, Fenton OS, Jaklenec A, et al. Lipid nanoparticle assisted mrna delivery for potent cancer immunotherapy. Nano Lett (2017) 17:1326–35. doi: 10.1021/acs.nanolett.6b03329
147. Miller M, Sahin U, Derhovanessian E, Kloke B-P, Simon P, Bukur V, et al. IVAC MUTANOME: A first-in-human phase I clinical trial targeting individual mutant neoantigens for the treatment of melanoma. Ann Oncol (2017) 28:xi1–2. doi: 10.1093/annonc/mdx712.003
148. Gómez-Aguado I, Rodríguez-Castejón J, Vicente-Pascual M, Rodríguez-Gascón A, Solinís MÁ, del Pozo-Rodríguez A. Nanomedicines to deliver mRNA: State of the art and future perspectives. Nanomaterials (2020) 10:364. doi: 10.3390/nano10020364
149. Samaridou E, Heyes J, Lutwyche P. Lipid nanoparticles for nucleic acid delivery: Current perspectives. Adv Drug Delivery Rev (2020) 154–155:37–63. doi: 10.1016/j.addr.2020.06.002
150. Bauman J, Burris H, Clarke J, Patel M, Cho D, Gutierrez M, et al. 798 safety, tolerability, and immunogenicity of mRNA-4157 in combination with pembrolizumab in subjects with unresectable solid tumors (KEYNOTE-603): an update. J Immunother Cancer (2020) 8:A846–6. doi: 10.1136/jitc-2020-SITC2020.0798
151. Burris HA, Patel MR, Cho DC, Clarke JM, Gutierrez M, Zaks TZ, et al. A phase I multicenter study to assess the safety, tolerability, and immunogenicity of mRNA-4157 alone in patients with resected solid tumors and in combination with pembrolizumab in patients with unresectable solid tumors. J Clin Oncol (2019) 37:2523–3. doi: 10.1200/JCO.2019.37.15_suppl.2523
152. Cafri G, Gartner JJ, Zaks T, Hopson K, Levin N, Paria BC, et al. mRNA vaccine–induced neoantigen-specific T cell immunity in patients with gastrointestinal cancer. J Clin Invest (2020) 130:5976–88. doi: 10.1172/JCI134915
153. Jimeno A, Gupta S, Sullivan R, Do KT, Akerley WL, Wang D, et al. Abstract CT032: A phase 1/2, open-label, multicenter, dose escalation and efficacy study of mRNA-2416, a lipid nanoparticle encapsulated mRNA encoding human OX40L, for intratumoral injection alone or in combination with durvalumab for patients with advanc. Tumor Biol Am Assoc Cancer Res (2020) 80(16_Supplement):CT032–2. doi: 10.1158/1538-7445.AM2020-CT032
154. Bauer T, Patel M, Jimeno A, Wang D, McDermott J, Zacharek S, et al. Abstract CT210: A phase I, open-label, multicenter, dose escalation study of mRNA-2752, a lipid nanoparticle encapsulating mRNAs encoding human OX40L, IL-23, and IL-36γ, for intratumoral injection alone and in combination with immune checkpoint blockade. Clin Trials Am Assoc Cancer Res (2019) 79(13_Supplement):CT210–0. doi: 10.1158/1538-7445.AM2019-CT210
155. Patel MR, Bauer TM, Jimeno A, Wang D, LoRusso P, Do KT, et al. A phase I study of mRNA-2752, a lipid nanoparticle encapsulating mRNAs encoding human OX40L, IL-23, and IL-36γ, for intratumoral (iTu) injection alone and in combination with durvalumab. J Clin Oncol (2020) 38:3092–2. doi: 10.1200/JCO.2020.38.15_suppl.3092
156. Sahin U, Oehm P, Derhovanessian E, Jabulowsky RA, Vormehr M, Gold M, et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature (2020) 585:107–12. doi: 10.1038/s41586-020-2537-9
157. Arthur JF, Butterfield LH, Roth MD, Bui LA, Kiertscher SM, Lau R, et al. A comparison of gene transfer methods in human dendritic cells. Cancer Gene Ther (1997) 4:17–25.
158. Hobo W, Novobrantseva TI, Fredrix H, Wong J, Milstein S, Epstein-Barash H, et al. Improving dendritic cell vaccine immunogenicity by silencing PD-1 ligands using siRNA-lipid nanoparticles combined with antigen mRNA electroporation. Cancer Immunol Immunother (2013) 62:285–97. doi: 10.1007/s00262-012-1334-1
159. Garu A, Moku G, Gulla SK, Chaudhuri A. Genetic immunization with in vivo dendritic cell-targeting liposomal dna vaccine carrier induces long-lasting antitumor immune response. Mol Ther (2016) 24:385–97. doi: 10.1038/mt.2015.215
160. Firdessa-Fite R, Creusot RJ. Nanoparticles versus dendritic cells as vehicles to deliver mRNA encoding multiple epitopes for immunotherapy. Mol Ther - Methods Clin Dev (2020) 16:50–62. doi: 10.1016/j.omtm.2019.10.015
161. Zhang F, Parayath NN, Ene CI, Stephan SB, Koehne AL, Coon ME, et al. Genetic programming of macrophages to perform anti-tumor functions using targeted mRNA nanocarriers. Nat Commun (2019) 10:3974. doi: 10.1038/s41467-019-11911-5
162. Wayne EC, Long C, Haney MJ, Batrakova EV, Leisner TM, Parise LV, et al. Targeted delivery of sirna lipoplexes to cancer cells using macrophage transient horizontal gene transfer. Adv Sci (2019) 6:1900582. doi: 10.1002/advs.201900582
163. Zhao Z, Chen Y, Francisco NM, Zhang Y, Wu M. The application of CAR-T cell therapy in hematological malignancies: advantages and challenges. Acta Pharm Sin B (2018) 8:539–51. doi: 10.1016/j.apsb.2018.03.001
164. Hartmann J, Schüßler-Lenz M, Bondanza A, Buchholz CJ. Clinical development of CAR T cells–challenges and opportunities in translating innovative treatment concepts. EMBO Mol Med (2017) 9:1183–97. doi: 10.15252/emmm.201607485
165. Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood (2016) 127:3321–30. doi: 10.1182/blood-2016-04-703751
166. Xue Y, Che J, Ji X, Li Y, Xie J, Chen X. Recent advances in biomaterial-boosted adoptive cell therapy. Chem Soc Rev (2022) 51:1766–94. doi: 10.1039/D1CS00786F
167. Xue Y, Baig R, Dong Y. Recent advances of biomaterials in stem cell therapies. Nanotechnology (2022) 33:132501. doi: 10.1088/1361-6528/ac4520
168. Kim J, Eygeris Y, Gupta M, Sahay G. Self-assembled mRNA vaccines. Adv Drug Delivery Rev (2021) 170:83–112. doi: 10.1016/j.addr.2020.12.014
169. Najahi-Missaoui W, Arnold RD, Cummings BS. Safe nanoparticles: are we there yet? Int J Mol Sci (2020) 22:385. doi: 10.3390/ijms22010385
170. McKinlay CJ, Benner NL, Haabeth OA, Waymouth RM, Wender PA. Enhanced mRNA delivery into lymphocytes enabled by lipid-varied libraries of charge-altering releasable transporters. Proc Natl Acad Sci (2018) 115:E5859–66. doi: 10.1073/pnas.1805358115
171. Billingsley MM, Singh N, Ravikumar P, Zhang R, June CH, Mitchell MJ. Ionizable lipid nanoparticle-mediated mRNA delivery for human CAR T cell engineering. Nano Lett (2020) 20:1578–89. doi: 10.1021/acs.nanolett.9b04246
172. Chandrasekaran S, Chan MF, Li J, King MR. Super natural killer cells that target metastases in the tumor draining lymph nodes. Biomaterials (2016) 77:66–76. doi: 10.1016/j.biomaterials.2015.11.001
173. Siegler EL, Kim YJ, Chen X, Siriwon N, Mac J, Rohrs JA, et al. Combination cancer therapy using chimeric antigen receptor-engineered natural killer cells as drug carriers. Mol Ther (2017) 25:2607–19. doi: 10.1016/j.ymthe.2017.08.010
174. Wilk AJ, Weidenbacher NL-B, Vergara R, Haabeth OAW, Levy R, Waymouth RM, et al. Charge-altering releasable transporters enable phenotypic manipulation of natural killer cells for cancer immunotherapy. Blood Adv (2020) 4:4244–55. doi: 10.1182/bloodadvances.2020002355
175. Dowds CM, Kornell S-C, Blumberg RS, Zeissig S. Lipid antigens in immunity. bchm (2014) 395:61–81. doi: 10.1515/hsz-2013-0220
176. Hamsanathan S, Gurkar AU. Lipids as regulators of cellular senescence. Front Physiol (2022) 13:796850. doi: 10.3389/fphys.2022.796850
177. Barr T, Helms C, Grant K, Messaoudi I. Opposing effects of alcohol on the immune system. Prog Neuropsychopharmacol Biol Psychiatry (2016) 65:242–51. doi: 10.1016/j.pnpbp.2015.09.001
178. Chen B-M, Cheng T-L, Roffler SR. Polyethylene glycol immunogenicity: theoretical, clinical, and practical aspects of anti-polyethylene glycol antibodies. ACS Nano (2021) 15:14022–48. doi: 10.1021/acsnano.1c05922
179. Sago CD, Krupczak BR, Lokugamage MP, Gan Z, Dahlman JE. Cell subtypes within the liver microenvironment differentially interact with lipid nanoparticles. Cell Mol Bioeng (2019) 12:389–97. doi: 10.1007/s12195-019-00573-4
180. Bulbake U, Doppalapudi S, Kommineni N, Khan W. Liposomal formulations in clinical use: an updated review. Pharmaceutics (2017) 9:12. doi: 10.3390/pharmaceutics9020012
181. Ahmad A, Wang Y-F, Ahmad I. Separation of liposome-entrapped mitoxantrone from nonliposomal mitoxantrone in plasma: pharmacokinetics in mice. Methods Enzymol (2005) 391:176–85. doi: 10.1016/S0076-6879(05)91010-0
182. Zamboni WC, Strychor S, Joseph E, Walsh DR, Zamboni BA, Parise RA, et al. Plasma, tumor, and tissue disposition of stealth liposomal ckd-602 (s-ckd602) and nonliposomal ckd-602 in mice bearing a375 human melanoma xenografts. Clin Cancer Res (2007) 13:7217–23. doi: 10.1158/1078-0432.CCR-07-1035
183. Yeh M-K, Chang H-I, Cheng M-Y. Clinical development of liposome based drugs: formulation, characterization, and therapeutic efficacy. Int J Nanomed (2011) 7:49–60. doi: 10.2147/IJN.S26766
184. Senzer NN, Matsuno K, Yamagata N, Fujisawa T, Wasserman E, Sutherland W, et al. Abstract C36: MBP-426, a novel liposome-encapsulated oxaliplatin, in combination with 5-FU/leucovorin (LV): Phase I results of a phase I/II study in gastro-esophageal adenocarcinoma, with pharmacokinetics. clinical trials. Am Assoc Cancer Res (2009) 8(12_Supplement):C36. doi: 10.1158/1535-7163.TARG-09-C36
185. Tomkinson B, Bendele R, Giles FJ, Brown E, Gray A, Hart K, et al. OSI-211, a novel liposomal topoisomerase I inhibitor, is active in SCID mouse models of human AML and ALL. Leuk Res (2003) 27:1039–50. doi: 10.1016/S0145-2126(03)00092-4
186. Liu S, Cheng Q, Wei T, Yu X, Johnson LT, Farbiak L, et al. Membrane-destabilizing ionizable phospholipids for organ-selective mRNA delivery and CRISPR–cas gene editing. Nat Mater (2021) 20:701–10. doi: 10.1038/s41563-020-00886-0
187. Dahlman JE, Kauffman KJ, Xing Y, Shaw TE, Mir FF, Dlott CC, et al. Barcoded nanoparticles for high throughput in vivo discovery of targeted therapeutics. Proc Natl Acad Sci (2017) 114:2060–5. doi: 10.1073/pnas.1620874114
188. Ickenstein LM, Garidel P. Lipid-based nanoparticle formulations for small molecules and RNA drugs. Expert Opin Drug Deliv (2019) 16:1205–26. doi: 10.1080/17425247.2019.1669558
Keywords: lipid-based nanoparticle, drug delivery, cancer immunotherapy, cell and gene therapy, nanomedicine
Citation: Zhang Z, Yao S, Hu Y, Zhao X and Lee RJ (2022) Application of lipid-based nanoparticles in cancer immunotherapy. Front. Immunol. 13:967505. doi: 10.3389/fimmu.2022.967505
Received: 13 June 2022; Accepted: 19 July 2022;
Published: 08 August 2022.
Edited by:
Anne C. Moore, University College Cork, IrelandCopyright © 2022 Zhang, Yao, Hu, Zhao and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert J. Lee, bGVlLjEzMzlAb3N1LmVkdQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.