
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 08 September 2022
Sec. Inflammation
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.962986
This article is part of the Research TopicImmune-Mediated Inflammation in Metabolic Kidney Diseases and Therapeutic InterventionsView all 12 articles
 Qixia Shen1,2,3,4†
Qixia Shen1,2,3,4† Lisha Teng1,2,3,4†
Lisha Teng1,2,3,4† Yucheng Wang1,2,3,4†
Yucheng Wang1,2,3,4† Luying Guo1,2,3,4Feng Xu5
Luying Guo1,2,3,4Feng Xu5 Hongfeng Huang1,2,3,4Wenqing Xie1,2,3,4Qin Zhou1,2,3,4Ying Chen1,2,3,4
Hongfeng Huang1,2,3,4Wenqing Xie1,2,3,4Qin Zhou1,2,3,4Ying Chen1,2,3,4 Junwen Wang6Youying Mao7
Junwen Wang6Youying Mao7 Jianghua Chen1,2,3,4
Jianghua Chen1,2,3,4 Hong Jiang1,2,3,4*
Hong Jiang1,2,3,4*Focal segmental glomerulosclerosis (FSGS) has an over 30% risk of recurrence after kidney transplantation (Ktx) and is associated with an extremely high risk of graft loss. However, mechanisms remain largely unclear. Thus, this study identifies novel genes related to the recurrence of FSGS (rFSGS). Whole genome-wide sequencing and next-generation RNA sequencing were used to identify the candidate mutant genes associated with rFSGS in peripheral blood mononuclear cells (PBMCs) from patients with biopsy-confirmed rFSGS after KTx. To confirm the functional role of the identified gene with the MDH2 c.26C >T mutation, a homozygous MDH2 c.26C >T mutation in HMy2.CIR cell line was induced by CRISPR/Cas9 and co-cultured with podocytes, mesangial cells, or HK2 cells, respectively, to detect the potential pathogenicity of the c.26C >T variant in MDH2. A total of 32 nonsynonymous single nucleotide polymorphisms (SNPs) and 610 differentially expressed genes (DEGs) related to rFSGS were identified. DEGs are mainly enriched in the immune and metabolomic-related pathways. A variant in MDH2, c.26C >T, was found in all patients with rFSGS, which was also accompanied by lower levels of mRNA expression in PBMCs from relapsed patients compared with patients with remission after KTx. Functionally, co-cultures of HMy2.CIR cells overexpressing the mutant MDH2 significantly inhibited the expression of synaptopodin, podocin, and F-actin by podocytes compared with those co-cultured with WT HMy2.CIR cells or podocytes alone. We identified that MDH2 is a novel rFSGS susceptibility gene in patients with recurrence of FSGS after KTx. Mutation of the MDH2 c.26C >T variant may contribute to progressive podocyte injury in rFSGS patients.
Idiopathic FSGS is the most common cause of nephrotic syndrome in which approximately 50% of patients progress to end-stage renal disease and require dialysis or kidney transplantation (KTx) within 10–20 years (1, 2). Unfortunately, in FSGS patients receiving KTx, more than 30% of cases experience recurrence of FSGS (rFSGS) with poor allograft survival and loss of re-transplantation opportunity due to the high risk of rFSGS (3–5). Several major risk factors for recurrence include younger age at diagnosis, rapid progression to end-stage renal disease, being of whiteethnicity, the loss of previous allografts due to recurrence, heavy proteinuria before the transplant, and receiving pretransplant bilateral nephrectomy (6). The most widely accepted mechanism of rFSGS after KTx involves a circulating factor. The most supportive of this theory is a rare and striking case. In that case, proteinuria rapidly developed after the kidney was transplanted into a recipient with primary FSGS. A kidney graft biopsy showed podocyte foot process effacement. Then, the doctors removed the transplanted kidney on day 14 and transplanted it into another patient with end-stage renal disease due to diabetes. Strikingly, the allografted kidney functioned well and no proteinuria was observed (7). There are many circulating factors associated with rFSGS, including soluble urokinase-type plasminogen activator receptors (8–10), cardiotrophin-like cytokine factor-1 (11), apolipoprotein A-I (12, 13), and CD40-CD40L (10, 14, 15). The release of permeability factors is potentially related to disordered circulating PBMCs (16–19). However, whether genetic variants or mutations contribute to the development of rFSGS remains unexplored.
Increasing evidence shows that integrated DNA and RNA profiling is a useful technique for genomics research to uncover the molecular mechanisms of disease and to explore the link between genotype and phenotype. RNA sequencing data can provide an orthogonal verification of DNA variant calls and can be used to prioritize expressed candidates, which are more likely to exert biological effects (20). In this study, we used high-throughput sequencing of both DNA and RNA from FSGS patients accompanied by rFSGS or remission after KTx and healthy controls (HCs) to identify genes that are potentially involved in susceptibility to rFSGS. Additionally, the functional role of the identified gene related to rFSGS was also investigated.
Three healthy kidney donors (HC group) and six adult patients with biopsies confirming primary FSGS were enrolled in this study. Of them, three recurred after KTx within one month (RC group) and three had remission for as long as 2 to 5 years (RM group). In those with rFSGS, renal follow-up biopsy was also performed. Peripheral blood from healthy individuals and patients with rFSGS or remission before and after KTx was collected from the Kidney Disease Center, The First Affiliated Hospital of Zhejiang University, China. This study was approved by the Medical Ethics Committee of The First Affiliated Hospital of Zhejiang University and the ethical batch number for it is 2021IIT065. Informed consent was obtained from all individual participants.
PBMCs were isolated from 5 to 10 ml peripheral blood of KTx recipients and healthy individuals by Ficoll–Hypaque density gradient centrifugation at diagnosis of FSGS and stored at −20°C for future use. The genomic DNA was generated using the NEB Next® Ultra DNA Library Prep Kit for Illumina® (NEB, USA). The TRIzol Reagent (Invitrogen) and the NEBNext® UltraTM RNA Library Prep Kit for Illumina® (NEB, USA) were used for total RNA extraction. A NanoDrop™ 2000 (Thermo Scientific, Waltham, MA) spectrophotometer was used for a purity check, followed by agarose gel electrophoresis for degradation and contamination monitoring. A total amount of DNA of >700 ng or RNA >1 μg was at least required for library preparation and sequencing.
NEBNext adaptor hybridization and electrophoresis were performed to select a specified length of DNA fragment. For RNA extracted, the first and second strand cDNA and NEBNext Adaptor hybridization were performed, successively. A specially designed PCR protocol was used to amplify the tagged DNA and add sequencing indexes. Then, the AMPure XP system (Beckman Coulter, Beverly, USA) was used for fragment purification. A cBot Cluster Generation System using a HiSeq 2500 PE Cluster Kit (Illumina) was used for cluster generation. Finally, the library sequencing was performed by the Illumina Hiseq 2500 platform (Illumina Inc., San Diego, CA, USA). Paired-end reads of 150 bp and 125 bp/150 bp were generated for DNA and RNA sequencing, respectively.
We first removed the adapter in all reads and trimmed low-quality bases with a quality score below 20. Then, the clean reads were mapped to the hg19 reference genome by BWA (http://bio-bwa.sourceforge.net/). Picard was used to mark and remove duplicate reads that were generated by PCR-duplication. Per-sample BAM files were preprocessed as described in the GATK Best Practices (https://gatk.broadinstitute.org/hc/en-us). SNPs were called for every sample by the GATK variant discovery pipeline with haplotypeCaller mode. All mutations and variants were annotated according to annovar (http://annovar.openbioinformatics.org/en/latest/) and the annotation refGene in the UCSC Genome Browser. Synonymous and nonsense mutations were excluded using annotation (ANNOVAR/ONCOTATOR/SIFT).
RNA sequencing data were analyzed following the protocol reported previously (21). In brief, sequence reads were aligned to the genome using HISAT2 first. Then, Stringtie was used to assemble and quantify expressed genes and transcripts. Then, differential expression analysis was performed by the ballgown package of R software. Transcripts and genes statistically different between groups were identified by stattest using R software. TPM was used for abundance estimates.
Genes and transcripts with p-value <0.05, |log2Fold Change (FC)| >1 were selected as DEGs for further Kegg enrichment analysis by DAVID and GO enrichment analysis by Metascape, two web-based systems that incorporate information from different resources to detect the biological themes out of the candidate genes. Only pathways with Benjamini–Hochberg corrected p-value (PBH) <0.05 were kept as significantly enriched.
To describe the overlap between any given pair of pathways, two measurements were computed, the Jaccard Coefficient (JC) = (A ∩ B)/(A U B) and the Overlap Coefficient (OC) = (A ∩ B)/min(|A|, |B|), where A and B are the lists of genes included in the two tested pathways (22). To construct the pathway crosstalk, we selected a set of pathways for crosstalk analysis. Only pathways with a PBH value of <0.05 were used. Meanwhile, the pathways containing fewer than three candidate genes were removed because pathways with too few genes may have insufficient biological information. Next, pathway pairs with fewer than two overlapped genes were removed. The overlap of all pathway pairs was then calculated and ranked according to their JC and OC values. The selected pathway crosstalk was visualized with the software Cytoscape.
Cultured cells were lysed in RIPA Lysis Buffer (Beyotime, Shanghai, PR China) with a proteinase inhibitor cocktail, and SDS-PAGE and western blotting were performed. Results were quantified with ImageJ (National Institutes of Health, Bethesda, MD, USA). Antibodies were used to detect MDH2 (ab181873; Abcam, diluted in 1:10,000), α-tubulin (1:2,500, T9026, Sigma), synaptopodin antibody (1:1,000, ab224491, Abcam), and β-actin (1:2,000, sc-69879, Santa). The membranes were washed three times with TBST, incubated with HRP-conjugated secondary antibodies for 1 h at room temperature, visualized using a chemiluminescent substrate (Millipore, Billerica, MA), and finally analyzed using a ChemiDoc MP (Bio-Rad, Hercules, CA, USA).
Frozen cell samples in nitrogen liquid were thawed on ice, then metabolites were extracted and stored at −80°C prior to the LC–MS analysis. Pooled quality control (QC) samples were also prepared by combining 10 μl of each extraction mixture. All samples were analyzed using a TripleTOF 5600 Plus high-resolution tandem mass spectrometer (SCIEX, Warrington, UK) with both positive and negative ion modes. Chromatographic separation was performed using an ultra-performance liquid chromatography (UPLC) system (SCIEX, UK). An ACQUITY UPLC T3 column (100 mm ∗ 2.1 mm, 1.8 μm, Waters, UK) was used for the reversed-phase separation. The TripleTOF 5600 Plus system was used to detect metabolites eluted from the column. The MS data were acquired in the IDA mode. A QC sample was analyzed every 10 samples to evaluate the stability of the LC–MS.
The acquired LC–MS data pretreatment was performed using XCMS software. Raw data files were converted into mzXML format and then processed using the XCMS, CAMERA, and metaX toolbox included in the R software. Each ion was identified by the comprehensive information of retention time and m/z. The intensity of each peak was preprocessed using metaX. The open-access databases, KEGG and HMDB, were used to annotate the metabolites. Features that were detected in <50% of QC samples or 80% of test samples were removed, and values for missing peaks were extrapolated with the k‐nearest neighbor algorithm. Then, PCA, data normalization, and QC-robust spline batch correction were performed. The P-value was analyzed by Student t‐tests, which were adjusted for multiple tests using an FDR (Benjamini–Hochberg). We also conducted the supervised PLS‐DA using the metaX to variable discriminant profiling statistical method to identify more specific differences between the groups. The VIP cut‐off value of 1.0 was set to select important features.
Because B cells play an important role in rFSGS (4, 23–27) and HMy2.CIR is a human B lymphoblastoid cell line, to confirm the functional role of the identified gene with the MDH2 c.26C >T mutation, a homozygous MDH2 c.26C >T mutation in HMy2.CIR cell line was induced by CRISPR/Cas9 and co-cultured with podocytes, mesangial cells, or HK2 cells, respectively. Briefly, WT and MDH2 c.26C >T mutant Human HMy2.CIR cells were bought from Cyagen (Guangzhou, China). The HMy2.CIR cells were cultured in IMDM (12440053, Gibco), HK2 cells in DMEM/F12(D8437, Sigma), human mesangial cells (HMC) in DMEM/HG (D6429, Sigma) containing 10% FBS (12103C, Sigma) and 1% penicillin–streptomycin (15070063, Gibco), incubated at 37°C in a humidified atmosphere containing 5% CO2, and routinely passaged every 2 or 3 days. Podocytes were first grown under proliferation-permissive conditions in the presence of 33°C in a humidified atmosphere of 5% CO2. Growth media consists of RPMI 1640 (R8758, Sigma) containing 10% FBS and 1% penicillin-streptomycin supplemented with 100× Insulin-Trans-Sel-G (41400045, Gibco). For differentiation, they were switched to type I collagen-coated (354236, Corning) culture dishes deprived of ITS-G and half FBS for 7–9 days at 37°C. For Western blot, 3 × 104 podocytes, HMC, or HK2 cells were seeded in 6-well plates until 70% converged and then co-cultured with 5 × 104 WT HMy2.CIR or mutant HMy2.CIR cells for 48 h. For immunofluorescence, 104 podocytes or HMC were seeded in 24-well plates until 70% converged and then co-cultured with 104 WT HMy2.CIR or mutant HMy2.CIR cells for 48 h.
Approximately 2 × 105 serum-starved WT HMy2.CIR or mutant HMy2.CIR cells were washed with phosphate buffer saline before being fixed with 70% cold ethanol and kept at 4°C for 3.5 h. Phosphate buffer saline was used to wash cells again and resuspend cells. Cells were subjected to propidium iodide staining with a Cell Cycle and Apoptosis Analysis Kit (C1052, Beyotime), and the DNA content was measured by a fluorescence-activated cell sorting instrument (BD FACSCanto II) and analyzed by Modfit LT5.0.
Total RNA was extracted from HMC and HK2 cells by Trizol reagent (Invitrogen, USA). cDNA was prepared using the PrimeScriptTM RT reagent Kit with gDNA Eraser (No. RR047A, TaKaRa, Japan) as per the protocol of the manufacturer. Real-Time PCR was run using SYBR Green and CFX96™ Real-Time PCR Detection Systems (Bio-Rad, CA, USA) using the primers in Table S1. The mRNA levels of selected genes were calculated after normalization to GAPDH by using the 2−ΔΔCt method as per the protocol of the manufacturer.
Coverslips containing podocytes were fixed with 4% paraformaldehyde for 15 min and blocked in PBS containing 0.1% Triton X-100 and 3% bovine serum albumin(BSA) for 30 min at room temperature prior to incubation with synaptopodin antibody (1:100, sc-21537, Santa), NPHS2 antibody (1:100, ab50339, Abcam), Megsin (1:100, bs-0815R, Bioss) overnight in a humidified chamber at 4°C. Slides were incubated with secondary antibody Alexa Fluor 594-conjugated anti-goat IgG (1:500), Alexa Fluor 594-conjugated anti-rabbit IgG (1:500), and FITC-phalloidin (1:100) (P5282, Sigma) for 1 h at 37°C to stain the cytoskeleton. Sections were then examined by immunofluorescence microscopy (Leica DMLB, Wetzlar, Germany).
Results are displayed as means ± SEM. Statistical analysis using Student’s t‐tests was performed with GraphPad Prism 7 (GraphPad Inc, San Diego, CA, USA). p < 0.05 was considered statistically significant.
A total of six FSGS patients after KTx were enrolled in this study, including three with rFSGS and three with remission. In addition, three healthy kidney donors were also included as a normal control. Graft biopsy was performed in all patients. Clinically, proteinuria recurred in all patients with rFSGS but not in those with remission, which is shown in Table S2. All patients with FSGS showed severe FSGS with extensive tubulointerestial fibrosis as shown in renal biopsied kidneys before KTx (Figure 1). In patients with rFSGS, KTx did not improve renal dysfunction, with similar FSGS pathology found in the grafted kidneys as before KTx (Table S2; Figure 1). In contrast, in patients with FSGS remission, progressive renal injury was largely improved after KTx. During follow-up, these patients did not develop significant edema and proteinuria. All patients declared non-familial FSGS history.
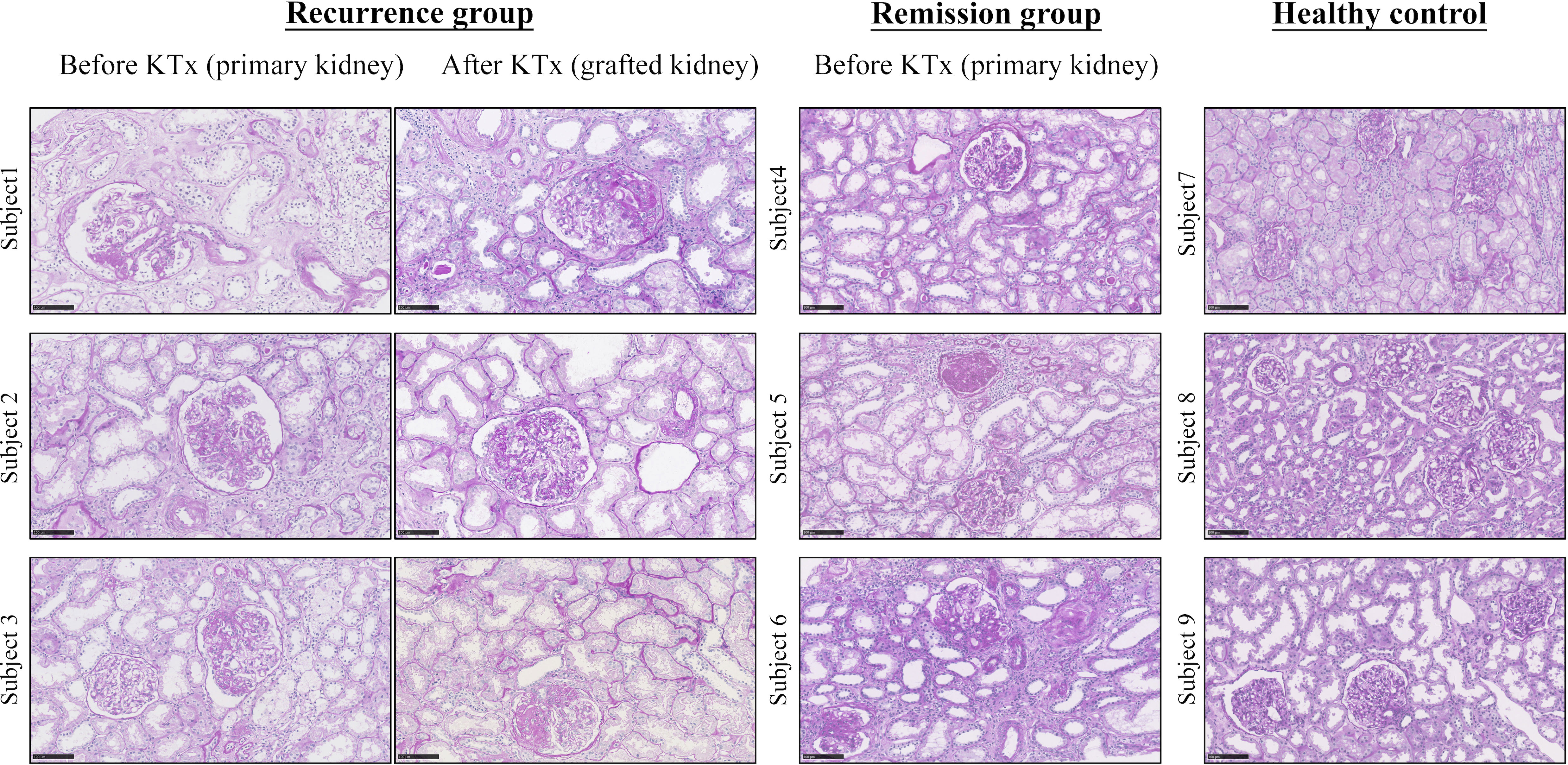
Figure 1 Representative pathology from FSGS patients before and after KTx by Periodic acid-Schiff staining. Note that focal segmental glomerular sclerosis with fibrosis is apparent in all FSGS patients before KTX and is also found in those with recurrent FSGS patients after KTx. Because KTx largely improved progressive renal injury in those FSGS patients, no renal biopsy was performed in the remission patients after KTx based on the ethical reasons. The healthy control kidneys are obtained from unmatched donor kidneys. Scale bar: 100um. KTx, kidney transplantation.
We next performed both whole-genome sequencing and transcriptomic analysis to identify the potential genetic variants responsible for rFSGS. Genome sequences of hg19 were treated as references. Figure 2 showed the number and type of SNPs presented in exons from ≥2 recurrent or remission subjects but absent in any healthy controls. We therefore identified 1,012 and 1,360 somatic SNPs in recurrent or remission subjects, respectively. Of them, 39 and 62 were nonsynonymous SNPs presented in three recurrent or remission patients, respectively. Only nonsynonymous SNP variants presented in all three recurrence patients but in ≤1 remission patient and none of the HCs were regarded as associated with rFSGS after KTx. Thus, a total of 32 nonsynonymous SNP variants mapped to exons across 29 unique genes were selected (Table S3). The pairwise comparison was conducted for the transcriptional patterns of PBMCs from three groups (Figures 3A–C). Approximately 353 DEGs between the recurrence and remission groups (Figure 3D), as well as 300 DEGs between the recurrence group and HCs, excluding those also differentially expressed between remission subjects and HCs (Figures 3D), were deemed as genes related to rFSGS after KT.
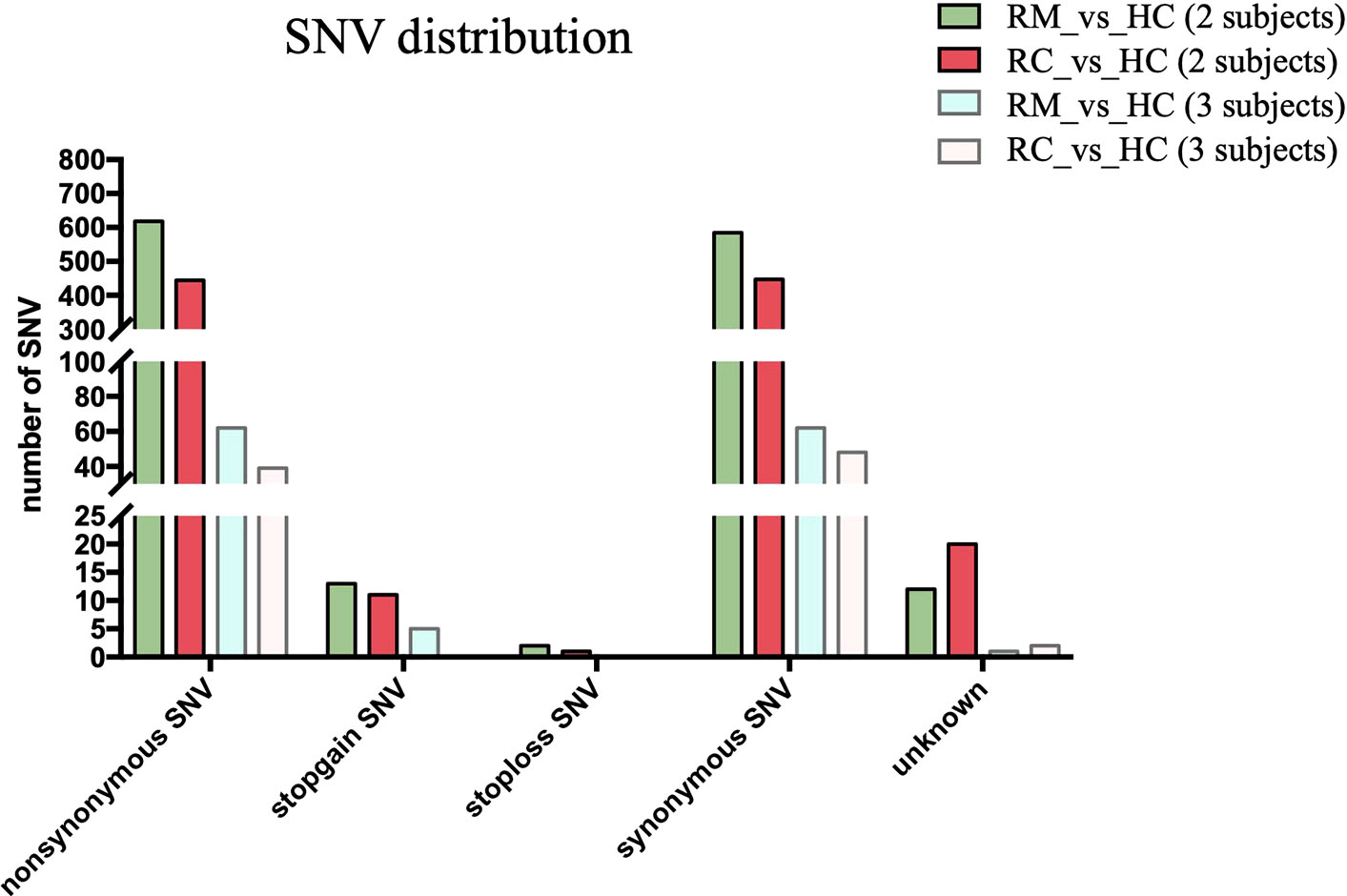
Figure 2 Identification of SNPs related to recurrence of FSGS after kidney transplantation. Bar plot showing summary of different types of SNP variants mapped to exons identified in PBMCs from recurrence (n≥2) and remission (n≥2) subjects excluding those also identified in HCs. RC, recurrence; RM, remission; HC, healthy control.
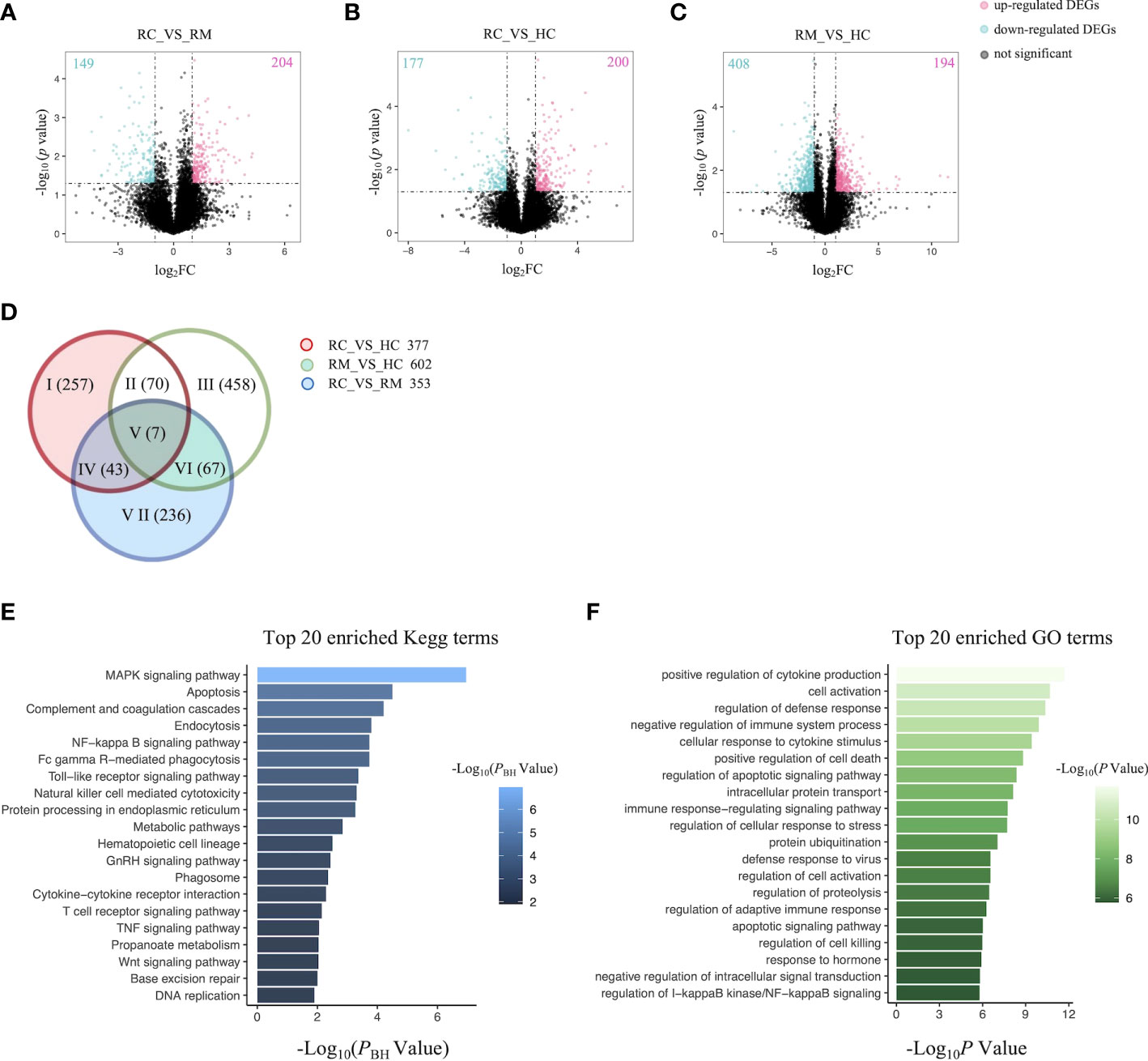
Figure 3 Identification of DEGs related to recurrence of FSGS after kidney transplantation.Volcano plot showing DEGs between (A) RC group and RM group; (B) RC group and HC group; (C) RM group and HC group. Each dot represents a gene. Red dots represent up-regulate genes, green dots represent down regulated genes. The numbers marked in the upper left and upper right corner represents the number of DEGs. (D) Venn diagram showing overlap of DEGs identified by pairwise comparison among the three groups. Top 20 enriched (E) Kegg terms and (F) GO terms. RC, recurrence; RM, remission; HC, healthy control.
The functional features of the rFSGS-related genes were examined by Kegg pathway enrichment on DAVID and GO terms enrichment on Metascape. A total of 65 significantly kegg pathways were enriched by DAVID. The top 20 enriched Kegg pathways and GO terms are shown in Figures 3E, F, respectively. Of 65 enriched Kegg pathways, 34 with three or more DEGs overlapping with one another were extracted (Table S4). All the pairs formed by these pathways were utilized to build the pathway crosstalk and were analyzed according to the average scores of coefficients JC and OC. Results shown in Figure 4A revealed the immune-related crosstalk pathways, including MAPK signaling pathways, complement, and coagulation cascades, NF-kappa B signaling pathway, Toll-like receptor signaling pathway, TNF signaling pathway, Wnt signaling pathway, etc., and the metabolism-related crosstalk pathways, such as citrate cycle, pyrimidine metabolism, galactose metabolism, etc. Of note, these immune and metabolism-related pathways were also widely connected to form the signaling pathways network. Among the top 20 enriched GO terms, positive regulation of cytokine production, cell activation, negative regulation of immune system processes, and cellular response to cytokine stimulus are ranked at the top. This is supportive of our hypothesis that circulating factors released by disturbed PBMCs may be causative of rFSGS.
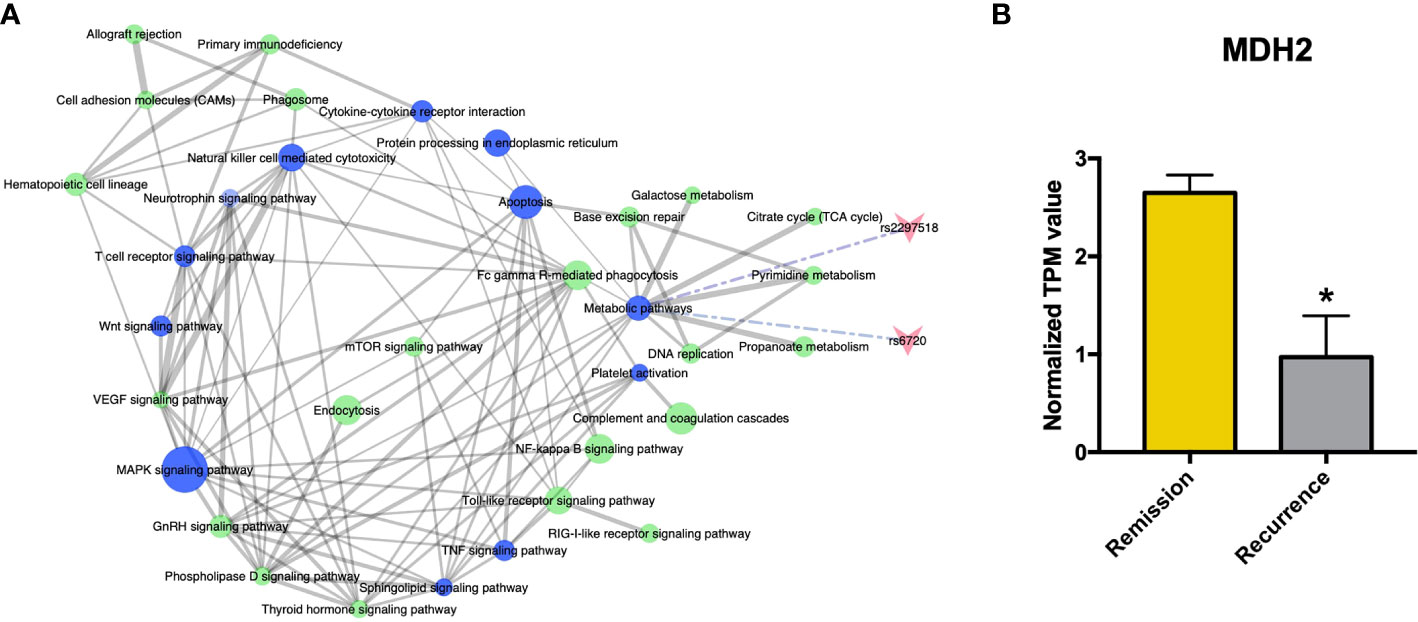
Figure 4 Crosstalk analysis among significantly enriched pathways suggested homozygous c.26C>T variant in MDH2 as a potential pathogenic mutation. (A) Pathway crosstalk among rFSGS related pathways. Triangular arrow represent SNPs, blue nodes represents pathways involving SNPs, green nodes represent pathways without SNPs involvement. Solid line represents crosstalk between each pair, Edge-width corresponds to the score of specific pathway pair. Larger edge-width indicates higher score. Dotted line represents SNPs enrichment. (B) mRNA expression level of MDH2 in remission group and recurrence group. *p <0.05.
There were two rFSGS related SNPs-mapped genes enriched in the crosstalk pathways (Table S3; Figure 4A), including rs6720 (mapped to MDH2, C >T, homozygote) and rs2297518 (mapped to NOS2, G >A, heterozygote). Both SNPs uniquely existed in all three recurrent recipients but in none of the remisson recipients or healthy donors. Furthermore, MDH2 was confirmed to be a DEG between the recurrence group and the remission group (Figure 4B), whereas NOS2 was not. MDH2 is a gene that encodes the mitochondrial malate dehydrogenase enzyme involved in the Krebs cycle. Thus, the rs6720 variant in MDH2 may be associated with the disorder in metabolic pathways and the Krebs cycle (Table S3).
Human PBMCs exist in a heterogeneous pool comprised of 70% T cells, 10% B cells, and another 10% of monocytes. Analysis of the kidney biopsy samples from children with idiopathic nephrotic syndrome revealed a significantly higher number of glomerular CD20+ B cell infiltration in FSGS patients (23). B-cell depleting antibodies like Rituximab and Ofatumumab are effective in rFSGS (4, 24–27), which indicates that the recurrence of FSGS may be B-cell related. HMy2.CIR, a human B lymphoblastoid cell line, was thus chosen to detect the effect of the homozygous c.26C >T variant on MDH2 protein expression. Indeed, substantially lower levels of MDH2 protein were detected in HMy2.CIR cells carrying the c.26C >T mutation than in WT controls (Figure 5A). Moreover, we also found the proliferation rate of mutant HMy2.CIR cells were significantly lower than that of WT cells, as the percentage of S phase was significantly reduced in mutant HMy2.CIR cells compared to WT cells (Figures S1A, B). Next, we performed a metabolomic analysis to examine whether the mutation of MDH2 in HMy2.CIR cells may lead to metabolic alterations. PLS-DA was applied in the data analysis and the score plot showed that the metabolomic profile of mutant cells was significantly altered when compared to the WT cells (Figures 5B, C). Furthermore, metabolic changes could be observed by Kegg enrichment of the differential metabolites (Figure S2). As shown in Figure 5D, the ratio of malate to citrate was significantly increased in mutant cells than in WT control, and the ratio of fumarate, a direct precursor of malate, to citrate was also higher in mutant cells than in WT control, although no statistical significance was detected. In contrast, mutation of MDH2 resulted in a decrease in the ratio between succinate and citrate without statistical significance (Figure 5D).
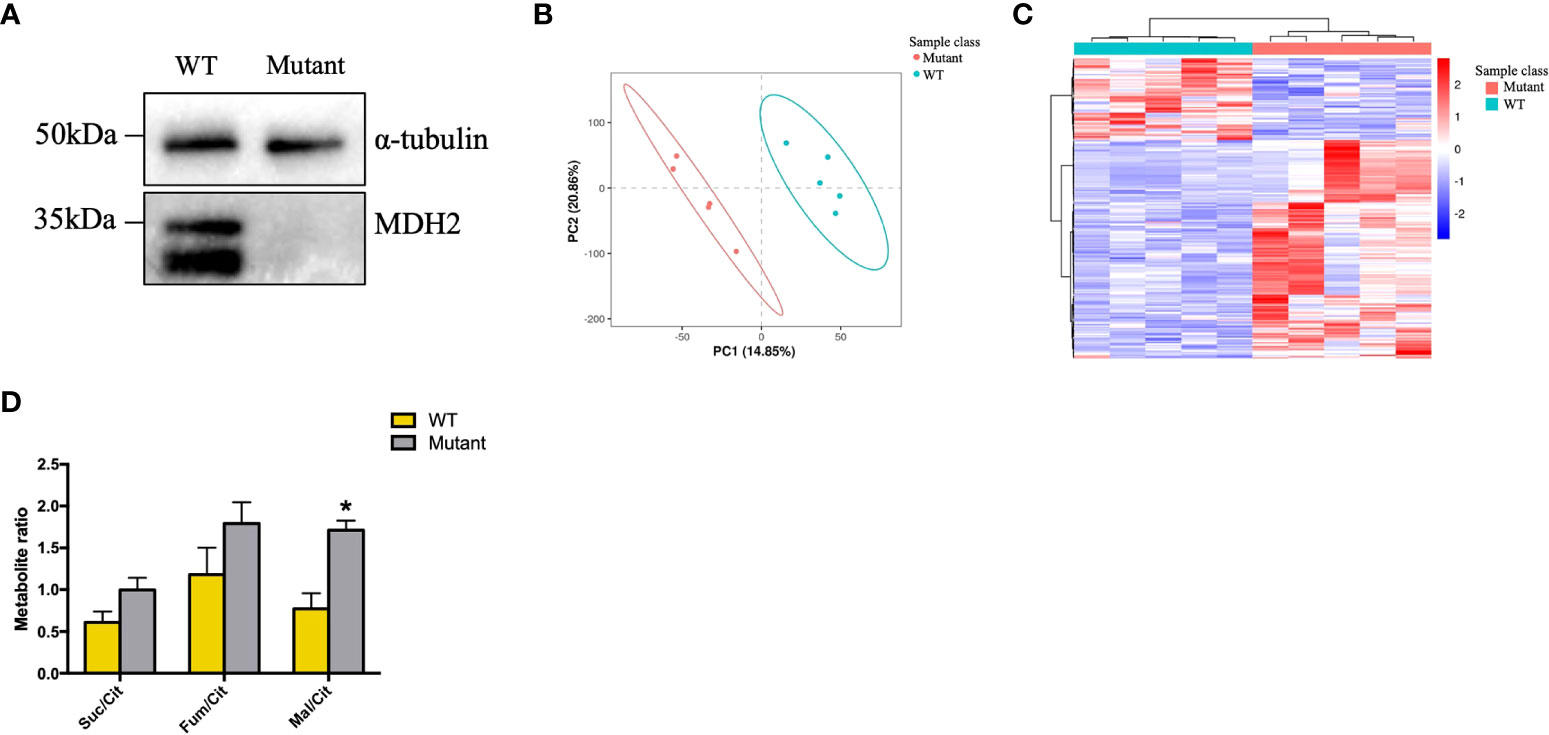
Figure 5 rs6720 leads to loss of MDH2 expression in HMy2.CIR cells and disturbed crebs cycle. (A) Western blot analysis with anti-MDH2 antibodies in rs6720 mutant and WT HMy2.CIR cells. a-tubulin was used as loading controls. (B) The scores plot of PLS-DA model of the LS-MS (positive model) spectral data between the selected PCs. Each point in the graph represents a sample. Different colors represent different groups, and different color circles represent different PCA groups. (C) Results of clustering patterns of metabolites differential expression. Red represents upregulation, blue represents downregulation. Different color bars represent different groups. (D) Metabolite ratios assessed by liquid chromatographic tandem-mass spectrometry in mutant and WT HMу2.CIR cells. Suc, succinate; Cit, citrate; Mal, malate; *p<0.05.
Because podocyte injury is a recognized cause of FSGS (28), we next examined the functional role of mutant MDH2 in podocyte injury by co-culturing mutant or WT HMy2.CIR cells with human podocytes, HMC, and HK2 cells, respectively. Immunofluorescence and western blot analysis showed that cells cultured with mutant HMy2.CIR resulted in podocyte injury as demonstrated by reducing podocyte-specific markers including expression of F-actin, synaptopodin, and podocin (Figure 6). This is in accordance with the biopsy results under the electron microscope. Podocyte foot process effacement was observed in all rFSGS patients, ranging from partially to diffusely (Table S2). In contrast, no significant changes were found in the co-culture of mutant HMy2.CIR with HMC (Figures S3A, B) and HK2 cells (Figure S4), which showed no alteration in expression of megsin (a HMC marker) that is involved in the renal tissues of various glomerular diseases (29) and expression of IL1β, RANTES, MCP-1, and TIM-1 by HK2 cells. Taken together, findings from these studies implied the restricted pathogenicity of c.26C> T variants in MDH2 to podocytes.
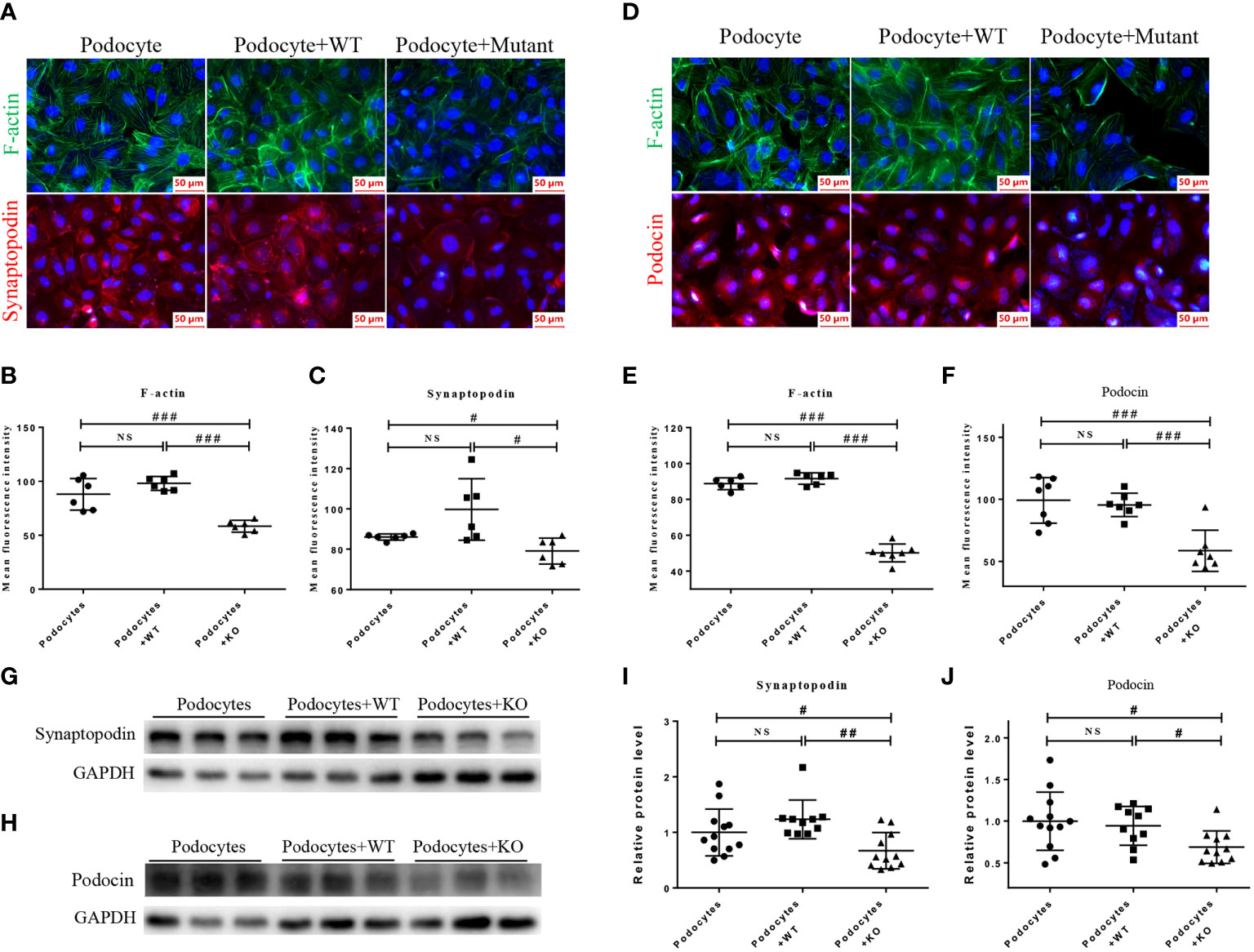
Figure 6 HMy2.CIR cells carrying rs6720 mutation in MDH2 caused injury to podocytes. (A–C) Representative pictures and quantification of immunofluorescence staining of F-actin (green), synaptopodin (red) and DAPI (blue) in podocytes before and after WT or mutant HMy2.CIR cell exposure. (D–F) Representative pictures and quantification of immunofluorescence staining of F-actin (green) and podocin (red) in podocytes before and after WT or mutant HMy2.CIR cell exposure. Six random fields were taken from each coverslip (mean ± SD, n=6). Scale bar: 50um. Western blot and statistics for Synaptopodin (G, I) and Podocin (H, J) from podocytes before and after WT or mutant HMy2.CIR cell exposure (mean ± SD, n=9-12). #p < 0.05; ##p<0.01; ###p<0.001; NS, no significance.
FSGS is a pathologic rather than a diagnostic CKD, which can be caused by rare, highly penetrant mutations in a number of genes (30). Injury to the podocytes plays a central role in the pathogenesis of FSGS (31, 32). Increasing evidence shows that mutations in genes related to FSGS are found in podocytes, including those involved in slit diaphragm structure and function, actin cytoskeleton, or cell signaling apparatus (33, 34). By integrated genomic and transcriptomic analysis, we reported here that rFSGS patients carried a homozygous c.26C> T mutation in the MDH2 gene in PBMCs with metabolic and immune dysfunction. However, the pathogenic role of mutant MDH2 in the PBMCs of rFSGS patients remains largely unknown. MDH2 encodes the enzyme malate dehydrogenase, mitochondrial in humans, which catalyzes the reversible oxidation of malate to oxaloacetate by utilizing the NAD-NADH cofactor system in the Krebs cycle. Despite the assumption that loss-of-function mutations affecting this key enzyme appear to be typically incompatible with life, pathogenic MDH2 disorder has been reported in several studies. A loss-of-function germline mutation in MDH2 was observed in paraganglioma (35). Bi-allelic mutations in MDH2 were also reported to cause the early-onset of severe encephalopathy (36). A prognostic model based on nine signature glycolytic genes, including MDH2, can accurately predict the prognosis of brain glioma patients. Among them, the mutation rate of HDAC4 and MDH2 genes was the highest at 8% (37). MDH2 was also overexpressed in endometrial carcinoma tissues and was related to the grade of the cancer (38). In renal cell carcinoma, inhibition of the glutamine-MDH2 axis suppresses in vitro tumor phenotypes in an L-2-HG-dependent manner (39). Decreased protein levels of malate dehydrogenase 2 were detected in a mouse model of methylmalonic aciduria, an inborn metabolic disorder of propionate catabolism that may lead to metabolic stroke and renal insufficiency (40). It was believed that disruption of enzymatic activities like malate dehydrogenase 2 could lead to the accumulation of specific metabolites, which give rise to epigenetic changes in the genome that cause a characteristic hypermethylated phenotype (41).
Functionally, we also uncovered that the mutation in the MDH2 gene was able to specifically cause podocyte injury. This was demonstrated by the finding that co-culture of human HMy2.CIR cells with rs6720 point mutation in MDH2 with podocytes were able to cause podocyte injury by inhibiting the expression of F-actin, synaptopodin, and podocin. Whereas no injuries were evidenced in by co-culturing the HMy2.CIR cells with other kidney cell types including human mesangial cells (HMC) and tubular epithelial cells (HK2). Thus, results from this study uncovered the restricted pathogenicity of MDH2 c.26C> T variants to podocyte injury specifically.
It should be pointed out that there are several limitations to this study. First, although integrated genomic and transcriptomic analysis was performed, this study is limited by the number of patients enrolled. Further studies on rs6720 in MDH2 leading to rFSGS in a large-scale cohort are necessary to confirm our findings. Second, though MDH2 abrogation leads to increased levels of malate/citrate ratios, as was proved in HMy2.CIR cells, we have not observed the absence of malate accumulation in rFSGS patients. Furthermore, the mechanisms underlying the pathogenicity of HMy2.CIR cells carrying homozygous c.26C> T mutations in podocyte injury has not been clarified. We speculated that the abnormal protein expression caused by the c.26C> T mutation in MDH2 led to the disorder of the TCA cycle and disturbance of cell metabolism, then the production of abnormal circulating factors by immune cells. These abnormal circulating factors resulted in podocyte injury, which is presented as inhibition of the expression of F-actin, synaptopodin, and podocin. Our next step is to include more rFSGS cases and screen out the possible circulation factors. The mechanism underlying podocyte injury should then be further investigated.
In summary, by using integrated genomic and transcriptomic analysis, we identified for the first time a bi-allelic c.26C >T variant in MDH2 in patients with rFSGS. Furthermore, we also found that this mutation is pathogenic and can cause podocyte injury specifically.
The data presented in the study are deposited in the SRA repository, accession number PRJNA842944, and the GEO repository, GSE212610.
The studies involving human participants were reviewed and approved by the Medical Ethics Committee of The First Affiliated Hospital of Zhejiang University. The patients/participants provided their written informed consent to participate in this study.
QS and HJ conceived and designed the experiments and drafted the manuscript. QS, HH, WX, YC, JC, and QZ collected human samples. QS, LT, LG, and FX performed the experiments and data analysis. QS, LT, and HJ interpreted the results of the experiments. QS, JW, YM, and HJ edited and revised the manuscript. QS and LT prepared the figures. HJ and QS approved the final version of the manuscript. All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
This work was partially supported by grants from NSFC U21A20350, 82070767, 81970651, and 82070729.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.962986/full#supplementary-material
Supplementary Table 1 | list of primer sequences used in this study.
Supplementary Table 2 | Characteristics of included subjects.
Supplementary Table 3 | list of non-synonymous SNPs related to rFSGS.
Supplementary Table 4 | Pathways included for crosstalk analysis.
1. Shoji J, Mii A, Terasaki M, Shimizu A. Update on recurrent focal segmental glomerulosclerosis in kidney transplantation. Nephron (2020) 144 Suppl 1:65–70.
2. Maisonneuve P, Agodoa L, Gellert R, Stewart JH, Buccianti G, Lowenfels AB, et al. Distribution of primary renal diseases leading to end-stage renal failure in the United States, Europe, and Australia/New Zealand: Results from an international comparative study. Am J Kidney Dis (2000) 35(1):157–65.
3. Artero M, Biava C, Amend W, Tomlanovich S, Vincenti F. Recurrent focal glomerulosclerosis: Natural history and response to therapy. Am J Med (1992) 92(4):375–83.
4. Uffing A, Perez-Saez MJ, Mazzali M, Manfro RC, Bauer AC, de Sottomaior Drumond F, et al. Recurrence of FSGS after kidney transplantation in adults. Clin J Am Soc Nephrol (2020) 15(2):247–56.
5. Savin VJ, Sharma R, Sharma M, McCarthy ET, Swan SK, Ellis E, et al. Circulating factor associated with increased glomerular permeability to albumin in recurrent focal segmental glomerulosclerosis. N Engl J Med (1996) 334(14):878–83.
6. Rudnicki M. FSGS recurrence in adults after renal transplantation. BioMed Res Int (2016) 2016:3295618. doi: 10.1155/2016/3295618
7. Gallon L, Leventhal J, Skaro A, Kanwar Y, Alvarado A. Resolution of recurrent focal segmental glomerulosclerosis after retransplantation. N Engl J Med (2012) 366(17):1648–9. doi: 10.1056/NEJMc1202500
8. Wei C, El Hindi S, Li J, Fornoni A, Goes N, Sageshima J, et al. Circulating urokinase receptor as a cause of focal segmental glomerulosclerosis. Nat Med (2011) 17(8):952–60. doi: 10.1038/nm.2411
9. Wei C, Moller CC, Altintas MM, Li J, Schwarz K, Zacchigna S, et al. Modification of kidney barrier function by the urokinase receptor. Nat Med (2008) 14(1):55–63. doi: 10.1038/nm1696
10. Wei C, Sigdel TK, Sarwal MM, Reiser J. Circulating CD40 autoantibody and suPAR synergy drives glomerular injury. Ann Transl Med (2015) 3(19):300.
11. Savin VJ, Sharma M, Zhou J, Gennochi D, Fields T, Sharma R, et al. Renal and hematological effects of CLCF-1, a b-Cell-Stimulating cytokine of the IL-6 family. J Immunol Res (2015) 2015:714964. doi: 10.1155/2015/714964
12. Lopez-Hellin J, Cantarell C, Jimeno L, Sanchez-Fructuoso A, Puig-Gay N, Guirado L, et al. A form of apolipoprotein a-I is found specifically in relapses of focal segmental glomerulosclerosis following transplantation. Am J Transpl (2013) 13(2):493–500. doi: 10.1111/j.1600-6143.2012.04338.x
13. Puig-Gay N, Jacobs-Cacha C, Sellares J, Guirado L, Gonzalez Roncero F, Jimenez C, et al. Apolipoprotein a-ib as a biomarker of focal segmental glomerulosclerosis recurrence after kidney transplantation: Diagnostic performance and assessment of its prognostic value - a multi-centre cohort study. Transpl Int (2019) 32(3):313–22. doi: 10.1111/tri.13372
14. Delville M, Sigdel TK, Wei C, Li J, Hsieh SC, Fornoni A, et al. A circulating antibody panel for pretransplant prediction of FSGS recurrence after kidney transplantation. Sci Transl Med (2014) 6(256):256ra136. doi: 10.1126/scitranslmed.3008538
15. Doublier S, Zennaro C, Musante L, Spatola T, Candiano G, Bruschi M, et al. Soluble CD40 ligand directly alters glomerular permeability and may act as a circulating permeability factor in FSGS. PloS One (2017) 12(11):e0188045. doi: 10.1371/journal.pone.0188045
16. Tao J, Mariani L, Eddy S, Maecker H, Kambham N, Mehta K, et al. JAK-STAT signaling is activated in the kidney and peripheral blood cells of patients with focal segmental glomerulosclerosis. Kidney Int (2018) 94(4):795–808. doi: 10.1016/j.kint.2018.05.022
17. Maruyama K, Tomizawa S, Shimabukuro N, Fukuda T, Johshita T, Kuroume T. Effect of supernatants derived from T lymphocyte culture in minimal change nephrotic syndrome on rat kidney capillaries. Nephron (1989) 51(1):73–6. doi: 10.1159/000185246
18. Koyama A, Fujisaki M, Kobayashi M, Igarashi M, Narita M. A glomerular permeability factor produced by human T cell hybridomas. Kidney Int (1991) 40(3):453–60. doi: 10.1038/ki.1991.232
19. Zhang X, Herr F, Vernochet A, Lorenzo HK, Beaudreuil S, Durrbach A. CASK, the soluble glomerular permeability factor, is secreted by macrophages in patients with recurrent focal and segmental glomerulo-sclerosis. Front Immunol (2020) 11:875. doi: 10.3389/fimmu.2020.00875
20. Reuter JA, Spacek DV, Pai RK, Snyder MP. Simul-seq: combined DNA and RNA sequencing for whole-genome and transcriptome profiling. Nat Methods (2016) 13(11):953–8. doi: 10.1038/nmeth.4028
21. Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and ballgown. Nat Protoc (2016) 11(9):1650–67. doi: 10.1038/nprot.2016.095
22. Hu Y, Pan Z, Hu Y, Zhang L, Wang J. Network and pathway-based analyses of genes associated with parkinson's disease. Mol Neurobiol (2017) 54(6):4452–65. doi: 10.1007/s12035-016-9998-8
23. Benz K, Buttner M, Dittrich K, Campean V, Dotsch J, Amann K. Characterisation of renal immune cell infiltrates in children with nephrotic syndrome. Pediatr Nephrol (2010) 25(7):1291–8. doi: 10.1007/s00467-010-1507-0
24. Pescovitz MD, Book BK, Sidner RA. Resolution of recurrent focal segmental glomerulosclerosis proteinuria after rituximab treatment. N Engl J Med (2006) 354(18):1961–3. doi: 10.1056/NEJMc055495
25. Alasfar S, Matar D, Montgomery RA, Desai N, Lonze B, Vujjini V, et al. Rituximab and therapeutic plasma exchange in recurrent focal segmental glomerulosclerosis postkidney transplantation. Transplantation (2018) 102(3):e115–e20. doi: 10.1097/TP.0000000000002008
26. Wang CS, Liverman RS, Garro R, George RP, Glumova A, Karp A, et al. Ofatumumab for the treatment of childhood nephrotic syndrome. Pediatr Nephrol (2017) 32(5):835–41. doi: 10.1007/s00467-017-3621-8
27. Bernard J, Bruel A, Allain-Launay E, Dantal J, Roussey G. Ofatumumab in post-transplantation recurrence of a pediatric steroid-resistant idiopathic nephrotic syndrome. Pediatr Transpl (2018) 22(4):e13175. doi: 10.1111/petr.13175
28. Fogo AB. Causes and pathogenesis of focal segmental glomerulosclerosis. Nat Rev Nephrol (2015) 11(2):76–87. doi: 10.1038/nrneph.2014.216
29. Suzuki D, Miyata T, Nangaku M, Takano H, Saotome N, Toyoda M, et al. Expression of megsin mRNA, a novel mesangium-predominant gene, in the renal tissues of various glomerular diseases. J Am Soc Nephrol (1999) 10(12):2606–13. doi: 10.1681/ASN.V10122606
30. Pollak MR. Familial FSGS. Adv Chronic Kidney Dis (2014) 21(5):422–5. doi: 10.1053/j.ackd.2014.06.001
31. D'Agati VD. Podocyte injury in focal segmental glomerulosclerosis: Lessons from animal models (a play in five acts). Kidney Int (2008) 73(4):399–406. doi: 10.1038/sj.ki.5002655
32. Barisoni L, Kriz W, Mundel P, D'Agati V. The dysregulated podocyte phenotype: a novel concept in the pathogenesis of collapsing idiopathic focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol (1999) 10(1):51–61. doi: 10.1681/ASN.V10151
33. De Vriese AS, Sethi S, Nath KA, Glassock RJ, Fervenza FC. Differentiating primary, genetic, and secondary FSGS in adults: A clinicopathologic approach. J Am Soc Nephrol (2018) 29(3):759–74. doi: 10.1681/ASN.2017090958
34. Rood IM, Deegens JK, Wetzels JF. Genetic causes of focal segmental glomerulosclerosis: Implications for clinical practice. Nephrol Dial Transpl (2012) 27(3):882–90. doi: 10.1093/ndt/gfr771
35. Cascon A, Comino-Mendez I, Curras-Freixes M, de Cubas AA, Contreras L, Richter S, et al. Whole-exome sequencing identifies MDH2 as a new familial paraganglioma gene. J Natl Cancer Inst (2015) 107(5). doi: 10.1093/jnci/djv053
36. Ait-El-Mkadem S, Dayem-Quere M, Gusic M, Chaussenot A, Bannwarth S, Francois B, et al. Mutations in MDH2, encoding a Krebs cycle enzyme, cause early-onset severe encephalopathy. Am J Hum Genet (2017) 100(1):151–9. doi: 10.1016/j.ajhg.2016.11.014
37. Bingxiang X, Panxing W, Lu F, Xiuyou Y, Chao D. A prognostic model for brain glioma patients based on 9 signature glycolytic genes. BioMed Res Int (2021) 2021:6680066. doi: 10.1155/2021/6680066
38. Zhuang Y, Xiang J, Bao W, Sun Y, Wang L, Tan M, et al. MDH2 stimulated by estrogen-GPR30 pathway down-regulated PTEN expression promoting the proliferation and invasion of cells in endometrial cancer. Transl Oncol (2017) 10(2):203–10. doi: 10.1016/j.tranon.2017.01.009
39. Shelar S, Shim EH, Brinkley GJ, Kundu A, Carobbio F, Poston T, et al. Biochemical and epigenetic insights into l-2-Hydroxyglutarate, a potential therapeutic target in renal cancer. Clin Cancer Res (2018) 24(24):6433–46. doi: 10.1158/1078-0432.CCR-18-1727
40. Wongkittichote P, Cunningham G, Summar ML, Pumbo E, Forny P, Baumgartner MR, et al. Tricarboxylic acid cycle enzyme activities in a mouse model of methylmalonic aciduria. Mol Genet Metab (2019) 128(4):444–51. doi: 10.1016/j.ymgme.2019.10.007
Keywords: recurrent focal segmental glomerulosclerosis, kidney transplantation, whole genome sequencing, next-generation RNA sequencing, MDH2
Citation: Shen Q, Teng L, Wang Y, Guo L, Xu F, Huang H, Xie W, Zhou Q, Chen Y, Wang J, Mao Y, Chen J and Jiang H (2022) Integrated genomic, transcriptomic and metabolomic analysis reveals MDH2 mutation-induced metabolic disorder in recurrent focal segmental glomerulosclerosis. Front. Immunol. 13:962986. doi: 10.3389/fimmu.2022.962986
Received: 07 June 2022; Accepted: 29 July 2022;
Published: 08 September 2022.
Edited by:
Liang Ma, Sichuan University, ChinaReviewed by:
Ming Wang, Southern Medical University, ChinaCopyright © 2022 Shen, Teng, Wang, Guo, Xu, Huang, Xie, Zhou, Chen, Wang, Mao, Chen and Jiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hong Jiang, amlhbmdob25nOTYxMTA2QHpqdS5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.