 Chenglong Liu
Chenglong Liu Mengxuan Yang1
Mengxuan Yang1 Di Zhu
Di Zhu- 1Minhang Hospital and Department of Pharmacology, School of Pharmacy, Fudan University, Shanghai, China
- 2New Drug Evaluation Center, Shandong Academy of Pharmaceutical Science, Jinan, China
- 3Department of Laboratory Medicine, Sixth Affiliated Hospital of Yangzhou University, Yangzhou, China
- 4Department of Laboratory Medicine, Affiliated Taixing Hospital of Bengbu Medical College, Taizhou, China
- 5Shanghai Engineering Research Center of ImmunoTherapeutics, Fudan University, Shanghai, China
Immune checkpoint therapy via PD-1 antibodies has shown exciting clinical value and robust therapeutic potential in clinical practice. It can significantly improve progression-free survival and overall survival. Following surgery, radiotherapy, chemotherapy, and targeted therapy, cancer treatment has now entered the age of immunotherapy. Although cancer immunotherapy has shown remarkable efficacy, it also suffers from limitations such as irAEs, cytokine storm, low response rate, etc. In this review, we discuss the basic classification, research progress, and limitations of cancer immunotherapy. Besides, by combining cancer immunotherapy resistance mechanism with analysis of combination therapy, we give our insights into the development of new anticancer immunotherapy strategies.
Introduction
Cancer immune surveillance is an important process by which the immune system can identify and eliminate nascent tumor cells (1). Normally, when tumor cells invade healthy tissue, the immune system can recognize and eliminate them based on tumor-associated antigens (TAAs). However, tumor cells can evade the immune system through a variety of mechanisms called immune escape (2). There are four main mechanisms: 1) decreasing immunogenicity by down-regulating surface antigen expression; 2) up-regulating immune checkpoints on the surface for suppressing T-cell activity; 3) recruiting suppressor immune cells such as myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Treg) as well as cytokines to form a suppressive immune microenvironment; 4) releasing acidic and toxic metabolites that inhibit the activity of immune cells in the tumor microenvironment (3).
Cancer is the second-leading cause of human death after cardiovascular and cerebrovascular diseases, and the number of patients continues to increase. Cancer treatment has progressed from surgical resection, radiation therapy, chemotherapy, and targeted drug therapy to immunotherapy. Cancer immunotherapy reactivates the body’s immune system to produce anticancer effects and thus kills and eliminates tumor cells. Immunotherapy is a promising treatment. Different from traditional therapy, immunotherapy uses some cytokines, chemokines, and immune cells to reshape the tumor microenvironment, which can lead to robust effects and prevent recurrence (4, 5). The emergence of immunotherapy has changed the standard and concept of tumor treatment. This article focuses on the latest clinical progress in cancer immunotherapy, including monoclonal antibodies (mAbs), small molecule drugs, adoptive cell therapy, oncolytic viruses, and cancer vaccines (Figure 1). We discuss limitations, immune resistance, and combination strategies in this review and hope to give a promising outlook for the future development of cancer immunotherapy.
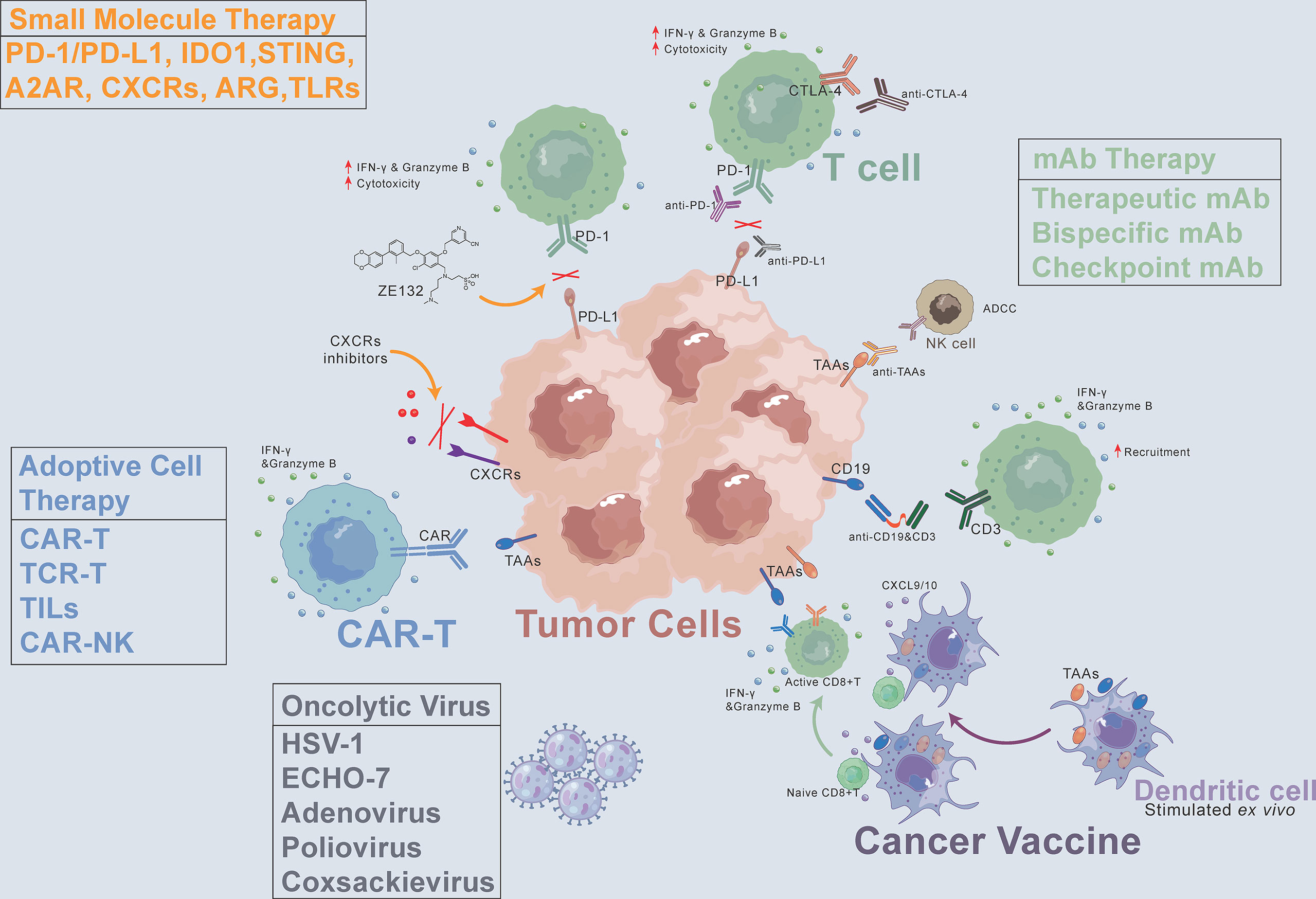
Figure 1 Cancer immunotherapy methods, including monoclonal antibodies (mAbs), small molecule drugs, adoptive cell therapy, oncolytic virus, and cancer vaccines. CAR, chimeric antigen receptor. CXCR, C-X-C motif chemokine receptor. TAAs, tumor-associated antigens. ADCC, antibody-dependent cell-mediated cytotoxicity. PD-1, programmed death-1. PD-L1, programmed death-ligand 1. CTLA-4, cytotoxic T-lymphocyte-associated protein 4.
Monoclonal antibody therapy
Therapeutic mAbs
mAbs are immunoglobulins (Ig) which commonly include two Fab terminals binding to targets and an Fc terminal binding to receptors on the surface of immune cells. All mAbs exert their function by direct targeting via Fab terminals. Additionally, Fc-Fc receptor (FcR) interaction can modulate their modes of action (MOA) (6, 7). The main Fc-mediated effector functions are classified into complement-dependent cytotoxicity (CDC), antibody-dependent cell-mediated cytotoxicity (ADCC), and antibody-dependent cellular phagocytosis (ADCP). CDC is attributed to the Fc interaction with complement component C1q, followed by the activation of the complement system leading to the downstream immune responses on different immune cells (8, 9). ADCC and ADCP are two mechanisms mediated by the direct interaction of Fc and FcγR. ADCC is mainly attributed to NK cells activated by the interaction of FcγRIIIa with the mAb’s Fc part. ADCP is mediated by FcγIIa-activated macrophages, which can phagocytose antibody-bounded tumor cells, leading to the elimination of tumor cells (8, 9). In other MOAs, mAbs are used to bind and block, such as soluble antigens (e.g., α- tumor necrosis factor (TNF-α)) and disease-dependent pathological mediators (e.g., vascular endothelial growth factor (VEGF)). Since rituximab targeting CD20 was first approved for Non-Hodgkin’s lymphoma (NHL) in 1997, the US Food and Drug Administration (FDA) has approved a variety of therapeutic monoclonal antibodies, which can target CD19, HER-2, VEGFA, EGFR, and CD52, etc. (Table 1).
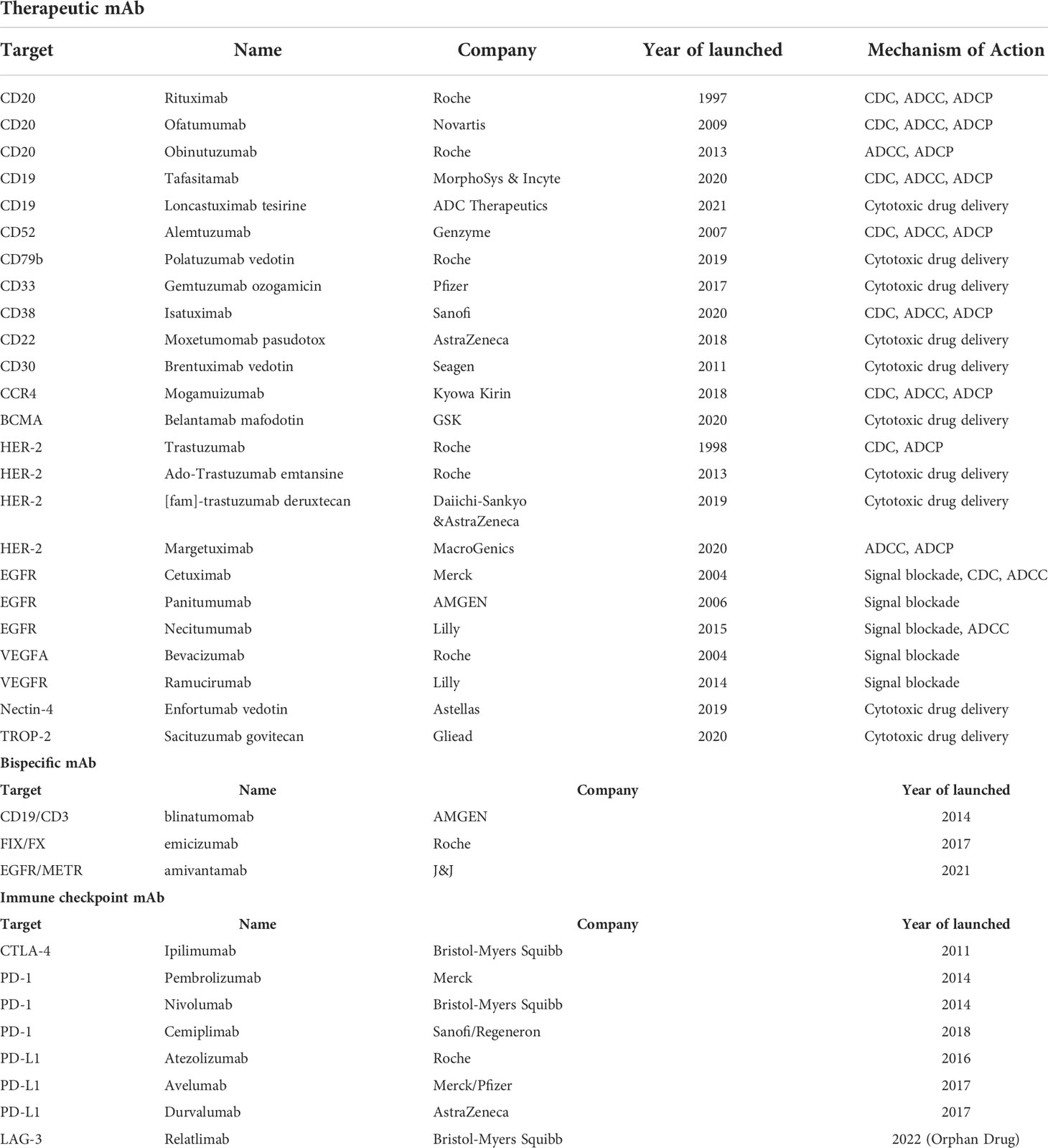
Table 1 FDA-approved mAbs (Up to March 2022).
Besides non-conjugated mAbs targeting ‘naked’ antigens, antibody-drug conjugates (ADCs) have shown promising therapeutic effects. ADCs show direct cytotoxicity based on their payloads, which can be ingested through the endocytosis of receptor-bound ADCs. Among the ADCs approved by the FDA, the indications of targets including CD22, CD30, CD33, CD79b, and BCMA are hematological tumors. Besides, HER2, Nectin-4, and Trop-2 are indicated for solid tumors. In terms of target accessibility, solid tumors are more obstructive than hematological tumors. The microenvironment of solid tumors and other factors make it difficult for mAbs to penetrate. In this regard, the accessibility of hematological tumors is better, which is the key factor why therapeutic mAbs will make breakthroughs in the field of hematological tumors first. But now, ADCs also show promising results for the treatment of solid tumors after the optimization of antibodies, linkers, and payloads. In a phase II clinical trial for HER-2-overexpressing or HER-2-mutated NSCLC (NCT03505710), the results showed that the ORR of Enhertu ([fam]-trastuzumab deruxtecan, an HER-2 ADC) was 61.9% and the median PFS was 14 months. In a phase I clinical trial of triple-negative breast cancer (TNBC), the initial objective response rate (ORR) was 43%, the complete or partial response (CR/PR) was confirmed in 5 patients, and the disease control rate (DCR) was 95% among 21 evaluable patients treated with datopotamab deruxtecan (a Trop-2 ADC).
The structure of mAb determines its MOA. Fc-engineering methods are used to endow therapeutic mAbs with stronger antitumor and immune activation abilities, which are achieved through amino acid mutation and glycosylation modification. Tafasitamab is a therapeutic mAb targeting CD-19 with upregulated MOA activity through Fc-related modifications. S239D and I332E mutations were performed in tafasitamab to enhance ADCC and ADCP. In the RE-MIND study, the ORR of tafasitamb combined with lenalidomide was 67.1%, and the CR was 39.5%, which was much higher than that of the control group treated with lenalidomide singly.
In several patients, the mAb-induced severe or partially life-threatening side effects were caused by a cytokine storm. In some cases caused by anti-CD20 mAb rituximab, it is assumed that the excessive activation of the complement system and the subsequent lysis of the targeted CD20+ cells, as well as the Fc-FcγR interactions with recruited macrophages, lead to a strong cytokine secretion (10, 11). While the side effects of mAbs therapy can be significantly less toxic than that of traditional chemotherapy, mAbs can still pose a significant risk to patients. Using the Fc-engineering strategy to reduce the immunogenicity of mAbs will provide new ideas for future development. Due to the large molecular weight, mAbs can only be administered by injection, which will lead to poor compliance for patients who require long-term treatment. Compared to mAbs, nanobody without Fc terminal has higher tissue permeability and lower production cost, which makes it become the key to succeed in mAbs development. In addition to being used singly, therapeutic antibodies are often combined with chemotherapy drugs and targeted therapy drugs. MAbs therapy will always be an important concept for tumor treatment. Further analyses will contribute to the design of safer therapeutic mAbs with fewer side effects and higher efficacy profiles in the future.
Bispecific mAbs
Bispecific mAbs (bsAbs) can bind multiple targets at the same time and have a better antitumor effect. Compared with ordinary mAbs, bsAbs offer better stability, higher specificity, and fewer side effects. They offer significant effects in clinical treatment. BsAbs are divided into two types: those that target multiple TAAs and those that engage T cells. They can produce multiple stimulations or inhibition effects, or recruit and activate more immune cells to eliminate tumor cells. Blinatumomab produced by AMGEN is the first FDA-approved bsAb that can specifically target the CD19 of tumor cells and the CD3 of T cells (Table 1). The clinical results of blinatumomab show that the response rate of patients after treatment reaches 72%, and the average life expectancy is more than nine months. Currently, amivantamab (targeting EGFR/METR) has also been approved for the treatment of non-small cell lung cancer (NSCLC) with EGFR exon 20 insertion mutations. Another approved bsAb, emicizumab, is being used to treat hemophilia. In addition to the three bsAbs already on the market, clinical studies of nearly 100 bsAbs are ongoing, which are mainly in the field of tumor therapy (12, 13). Among the bsAbs under clinical research, MEDI5752 developed by AstraZeneca is a monovalent bsAb that can target both PD-1 and CTLA-4. The results of the clinical trial (NCT03530397) have shown that MEDI5752 exhibits promising antitumor activity and durable clinical benefit in the treatment of patients with advanced solid tumors who are not eligible for standard therapy, with an objective response rate (ORR) of 19.8% and a median duration of response (DOR) of 17.5 months (AACR 2022, Abstract#CT016). AFM13 developed by Affirmed can simultaneously bind to CD30 of lymphoma cells and CD16A of natural killer (NK) cells to kill lymphoma cells without costimulatory signals. The results of the clinical trial (NCT03192202) of AFM13 have shown that 53% of patients had a complete response (CR), 37% had a partial response (PR), and progression-free survival (PFS) and overall survival (OS) were 58% and 79%, respectively (AACR 2022, Abstract#CT003).
Although bsAb is a very promising immunotherapy treatment, there are still problems. The manufacturing of bsAbs is time-consuming and costly. There are bsAb-specific byproducts, such as mispaired products, undesired fragments, and higher levels of aggregates. Additional purification strategies are needed to be designed to obtain products of high purity. At the same time, more clinical trials are needed to explore the optimal route of administration and optimal dose to increase the concentration in target tissues and reduce systemic side effects (14). In addition, bsAbs targeting solid tumors are very challenging because of the adverse effects on normal tissues or other complicated factors such as inadequate penetration (12).
Immune Checkpoint mAbs
There are immune checkpoints on the surface of T cells that can regulate the immune system. They play a negative regulatory role to prevent excessive activation of T cells to avoid autoimmune damage. However, tumor cells can use these immune checkpoints to suppress the immune response, thus performing immune escape and allowing tumor cells to escape the clearance of the immune system (15). Immune checkpoint mAbs can restore the relevant functions of T cells by blocking immune checkpoints and releasing the “brake” of the immune system (16). More than ten immune checkpoints have been discovered, and CTLA-4 and PD-1 are the most widely studied (Table 1).
Cytotoxic T lymphocyte-associated antigen-4 (CTLA-4) is a member of the CD28-B7 Ig superfamily. It is expressed on the surface of activated T cells and can act as an immune checkpoint to downregulate immune responses, thereby inhibiting the proliferation and activation of T cells (17, 18). In 2014, the FDA approved ipilimumab, a mAb targeting CTLA-4, for the treatment of melanoma; it significantly improved patient survival (19). Lynch and colleagues improved PFS in patients with NSCLC using ipilimumab in combination with paclitaxel and carboplatin (20). In addition to CTLA-4, programmed death-1 (PD-1) is another immune checkpoint molecule expressed on the surface of T cells. Its ligand (programmed cell death ligand 1 (PD-L1)) is expressed on the surface of various tumor cells (15, 21). mAb targeting the PD-1/PD-L1 pathway can relieve immunosuppression to enhance T cell activity and kill tumor cells. In 2014, the FDA approved pembrolizumab for the treatment of multiple cancers, including NSCLC, melanoma, and bladder cancer (16, 22). In current clinical use, PD-1/PD-L1 mAbs combined with chemotherapy or targeted therapy have achieved remarkable results. A phase III clinical trial of NSCLC (NCT02998528) with nivolumab combined with chemotherapy was promising, and there were event-free survival (EFS) and pathological complete response (pCR) dual-positive outcomes (AACR 2022, Abstract#CT012). AstraZeneca announced the results of a clinical trial (NCT03899610) combining durvalumab and tremelimumab in advanced epithelial ovarian cancer (targets PD-L1 and CTLA-4, respectively): the ORR was 86.7%, and the ratio of TIL, CD8, and CD8/Foxp3 in TME was significantly increased (AACR 2022, Abstract#CT010). Fc-engineering strategies are also performed in immune checkpoint mAbs. Theoretically, since PD-L1 is expressed on tumor cells, retaining ADCC activity of mAbs can simultaneously utilize the killing effect of NK cells to enhance the anti-tumor effect. This provides a new idea for us to use immune checkpoint mAbs to exert new MOAs. Only avelumab, a PD-L1 mAb, is designed with strong ADCC activity currently. Other immune checkpoints expressed on tumor cells can also learn from the design strategy of avelumab, which may greatly improve antitumor activity. For PD-1 mAbs (e.g., Durvalumab), removing FcγR affinity is beneficial to attenuate the ADCC effect, which is beneficial to preclude FcγR1 mediated binding to macrophages/myeloid-derived suppressor cells (MDSCs)-a potential mechanism by which PD-1-bound T cells may be cleared.
More immune checkpoints continue to be discovered, such as TIM-3, LAG-3, and TIGIT. LAG-3 can bind its canonical ligand (MHC-II) to downregulate T cell activity. A phase II/III clinical trial (NCT03470922) demonstrated that the median PFS of the relatlimab plus nivolumab group was 10.12 (6.37 to 15.74), which was over 2-fold compared to the nivolumab group (4.63 (3.38-5.62)). Currently, Opdualag (nivolumab+relatlimab) is the first LAG-3 antibody therapy approved by the FDA and the first innovative cancer immunotherapy approved for a new immune checkpoint in nearly 10 years. LAG-3 antibody is the third immune checkpoint inhibitor approved for marketing after CTLA4 and PD-1 antibodies (23).. In some preclinical studies, anti-TIM-3 therapy can improve anti-tumor efficacy, and combination therapy with anti-PD-1 or anti-PD-L1 can significantly reduce tumor burden and improve anti-tumor immune responses (24). Several antibodies targeting TIM-3 are currently being tested in clinical trials singly or in combination to treat acute myeloid leukemia or solid tumors (NCT04150029, NCT03680508, and NCT03099109). BsAbs targeting two immune checkpoints (PD-1&CTLA-4, PD-1&LAG-3, and PD-1&TIM-3) simultaneously have also been developed. In light of the positive clinical efficacy already noted in combination therapy targeting immune checkpoints, the outcomes of clinical trials with bsABs are promising.
In terms of adverse reactions, immune checkpoint therapy does not cause cytotoxic reactions such as myelosuppression, vomiting, and alopecia, but it can cause immune-related adverse events (irAEs) due to the activation of T cells, which can be reduced by glucocorticoids and disappear after drug discontinuation. Most irAEs are always reversible (25, 26). The overall incidence of irAEs was lower than that of chemotherapy-induced adverse events (27). Most irAEs are grades 1/2, while grades 3/4 irAEs are less frequent (28, 29). Common irAEs include cutaneous toxicity and endocrinological disturbance, while less common but serious irAEs include pulmonary toxicity, renal toxicity, hepatitis, and gastrointestinal disturbance. Rare irAEs include type 1 diabetes, cardiac, neurological, and hematologic-related toxicity (30, 31). Besides, immune checkpoint therapy only has significant effects in some patients. The premise of its effect is that the expression level of immune checkpoints is relatively high in patients. Therefore, it is necessary to carry out genetic screening of patients and apply immune checkpoint therapy to eligible patients.
Small molecule drug immunotherapy
Small molecule targeting PD-1/PD-L1
Immune escape is an important means for tumor cells to escape from being eliminated. Due to the abnormal immune surveillance mediated by immune checkpoints, tumor cells form immune escape and then obtain unlimited proliferation ability, thus leading to tumorigenesis. MAbs therapy suffers from poor tissue penetration, a long half-life, and high production costs. Thus, researchers are trying to develop small molecule inhibitors targeting immune checkpoints. Most inhibitors are currently in the early development stage (Table 2). CA-170 developed by Aurigene and Curis has made the fastest progress and entered phase II clinical trial (CTRI/2017/12/011026) (32). CA-170 targets PD-1/PD-L1 and VISTA pathways, thus leading to the proliferation and activation of T cells to produce cytokines such as IFN-γ to kill tumor cells (33). CA-170 can effectively inhibit melanoma and colon cancer in rodent models, and CA-170 is superior to mAbs in terms of safety (34–36). In clinical studies, CA-170 has the best effect on NSCLC and Hodgkin lymphoma with a total clinical benefit rate of 70% and 77.8%, respectively (37). AUNP12 was reported by Aurigene and Pierre Fabre in 2014. It is the first polypeptide PD-1/PD-L1 inhibitor and has a structure similar to the extracellular domain of PD-1 (38). The EC50 of peripheral blood mononuclear cells (PBMCs) proliferation rescue experiments reached 0.41 nM (38, 39). The in vivo experiments also showed that AUNP-12 can inhibit tumor growth and metastasis. AUNP-12 can inhibit B16F10 and 4T1 tumors in rodent models, and the tumor growth inhibition rate (TGI) of the B16F10 model reached 44% (40). In 2015-2018, BMS successively published a series of patents, and the IC50 of compounds detected by HTRF was generally less than 1 nM (41). In 2021, Liu et al. reported a small molecule inhibitor-ZE132, of which the affinity KD was 19.36 nM. ZE132 can specifically act on PD-L1 and has good antitumor efficacy in a variety of syngeneic mouse models (42).
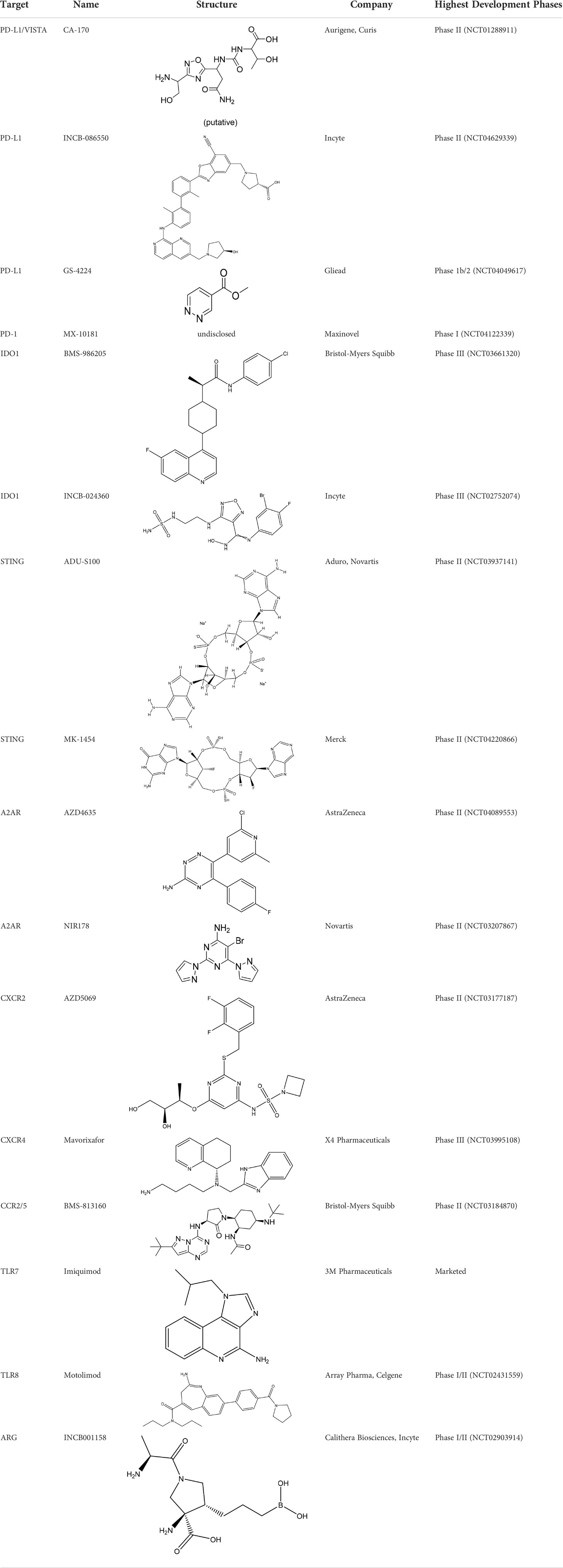
Table 2 Summary of major marketed and clinically reported small molecule immunotherapy drugs (Up to March 2022).
Small molecule inhibitors have lower binding affinity than that of mAbs, and they are prone to off-target effects, which may even bring unknown off-target toxicity. The interaction between PD-1 to PD-L1 is a protein-protein interaction. The contact interfaces of PD1/PD-L1 are large, highly flat, and hydrophobic, which makes it difficult to design compounds and develop small molecule inhibitors. Nevertheless, small molecule inhibitors have mature R&D pipelines, better tissue permeability, and controllable pharmacokinetic properties, which can help to avoid the defects of mAbs.
IDO1 inhibitors
Indoleamine 2,3-dioxygenase1 (IDO1) is a 45 kDa hemoglobin oxidase and is a key enzyme in the metabolism of the L-tryptophan-kynurenine pathway. IDO1 plays an important regulatory role in the process of immune regulation (43, 44). Functionally, IDO1 plays a key role in carcinogenesis and cancer immune escape by catalyzing the initial step of canine urinary ammonia pathway. IDO1 is overexpressed in tumor cells and antigen-presenting cells (APC). It is conducive to the formation of an immunosuppressive tumor microenvironment and is closely related to the poor prognosis of various cancers (45). Inhibition of IDO1 can activate antitumor immune responses in rodent tumor models (43). A variety of IDO1 inhibitors have entered clinical studies. BMS-986205 and epacadostat have made the fastest progress (Table 2). BMS-986205, developed by BMS, is currently in phase III clinical trial (NCT03661320) in combination with nivolumab, gemcitabine, and cisplatin in bladder cancer. In addition, there are two phase II clinical trials ongoing for bladder cancer (NCT03519256) and HNSCC (NCT03854032).
Developed by Incyte, epacadostat is one of the most well-studied IDO1 inhibitors. It shows good efficacy in mouse melanoma models and is well-tolerated (46). However, the results of the clinical trial (NCT02752074) showed that the combination of epacadostat and pembrolizumab in the treatment of melanoma did not meet the main clinical outcomes, and Incyte stopped their phase III trials (47).
The development of IDO1 inhibitors is not going well, and some clinical trials have failed. On one hand, the reason for the failure of ECHO-301 (epacadostat plus pembrolizumab) may be that the pharmacodynamic indicators are not applicable or the drug combination strategy is not matched. On the other hand, the reason may be that the exact regulatory mechanism of IDO1 in physiology and pathology or its impact on the tumor microenvironment are not well understood. The TDO pathway can play a potential compensatory role after epacadostat treatment, causing tumor immunosuppressive effects (48). However, the immune-enhancing function of IDO1 inhibitors has been verified, and IDO1 inhibitors still have the potential for development. In the future, the combination therapy of IDO1 inhibitors with other antitumor drugs should be further explored, which has important implications for the success of clinical development.
Other small molecule drugs
Stimulators of interferon genes (STING) is an immunostimulatory target and an important adaptor protein anchored in the endoplasmic reticulum that senses foreign DNA invasion. Now the STING signaling pathway has become a new target for cancer and autoimmune diseases. Experiments have shown that the activation of STING pathway can induce antitumor effects. A variety of drugs such as ADU-S100 are under clinical studies (Table 2) (49, 50). The clinical application of STING agonists is mainly focused on intratumoral injections, and it is unclear whether systemic administration is safe.
In addition, inhibitors of A2A adenosine receptor (A2AR), chemokine receptors, toll-like receptors (TLRs), arginase 1 (ARG), and other targets are in clinical development and are expected to provide more choices for antitumor drugs (Table 2) (51–54). Many projects have entered phase II/III clinical trials.
Polypeptide inhibitors, which can combine the characteristics of antibodies and small-molecule drugs, are important directions for the development of inhibitors. On one hand, they have similar affinity and specificity as antibodies. On the other hand, they have good tissue penetration and provide tunable pharmacokinetic half-life and renal clearance route to avoid hepatic and gastrointestinal toxicities due to their small molecular weight.
Small molecules are agents with a low molecular weight that are capable of modulation of intracellular targets. And small molecules are promised to improve the therapeutic management of solid tumors due to their easy administration, high bioavailability, and favorable safety profile. Given these characteristics, the development of small molecule-based strategies in cancer immunotherapy has attracted widespread interest. Although small-molecule drugs targeting the extracellular or intracellular pathways of adaptive immunity or innate immunity have been developed, most of them are in the early stage of clinical trials, and more basic experiments and clinical trials are needed to elucidate their mechanisms, clinical efficacy, and pharmacokinetics. Nevertheless, small-molecule inhibitors may be an effective replacement and supplement for mAbs, and they will remain an important part of tumor immunotherapy in the future.
Adoptive cell therapy
CAR-T
The chimeric antigen receptor (CAR) is a genetically modified and synthesized chimeric antigen receptor. It is a membrane protein composed of different protein domains in series. It is flexible and offers specific antigen recognition. Patient-derived T cells modified by CAR in vitro can recognize tumor antigens and exert antitumor effects without MHC restrictions in vivo (55).
CAR-T therapy is a revolutionary approach to cancer therapy. CAR-T therapy has made breakthroughs in lymphomas, mainly targeting CD19. In 2017, the FDA approved two CAR-T products targeting CD19 (Kymriah and Yescarta, Table 3) (56, 57). The first generation of CAR contains CD3ξ, and the second generation adds a costimulatory domain CD28 or 4-1BB based on CD3ξ. Through March 2022, the FDA has approved five CAR-T products, all of which are second-generation CARs with indications focused on lymphoma (58, 59). The third-generation CAR uses lentivirus as a transfection vector, and the intracellular segment of the CAR can have two or more costimulatory signals. However, some studies have shown that the killing activity of the third-generation CAR-T cells is not significantly improved. This may be because the activation signal generated by one co-stimulatory molecule of ITAM already reaches the threshold of T lymphocyte activation signal. Simply increasing the number of ITAM will not further enhance the activation effect of CAR-T.
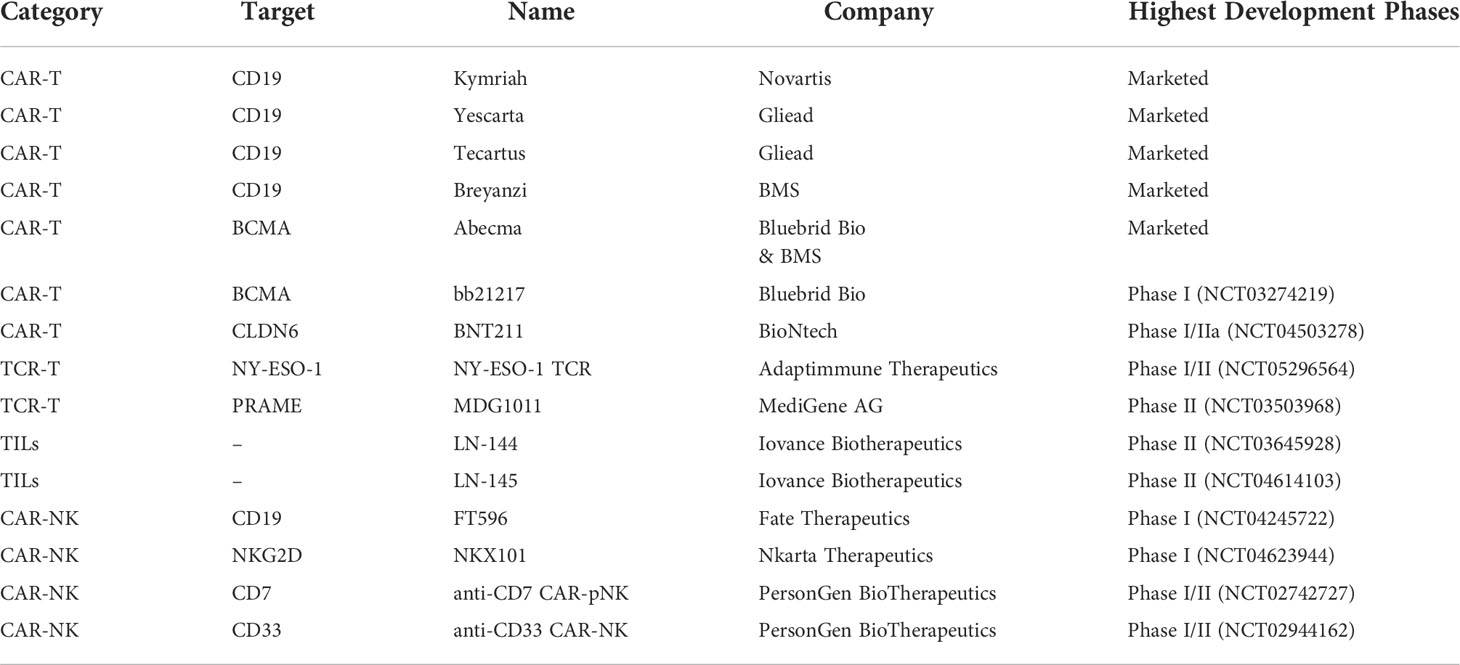
Table 3 Summary of major marketed and clinically reported adoptive cell therapy (Up to March 2022).
New ideas for CAR design are now emerging to improve efficacy. Dual-target CAR-T cells can independently identify target antigens and address the off-target effect. CD19/CD22 CAR-T and CD123/CLL1 CAR-T have shown significant antitumor activity and are currently in clinical studies, some of which have entered phase II/III (Table 3) (60, 61). According to EXUMA Biotech, targeting CD3 T cells by subcutaneous injection of a self-inactivating lentiviral vector encoding a CAR targeting CD19 resulted in the successful generation of corresponding CAR-T cells in vivo and showed significant effects in mice (AACR 2022 Abstract #3294/11). This provides a new opportunity to overcome the challenges of production time, scale, and cost of adoptive cell therapies.
For solid tumors, Hegde et al. constructed TanCAR-T that could enhance T cell function and reduce antigen escape by facilitating crosstalk between HER2-ScFv and IL-13Rα2, thus increasing CD28 expression. The data of TanCAR-T showed good efficacy in a mouse glioblastoma model (62). In 2022, Grosskopf et al. published a delivery method for hydrogel that can improve the efficacy of treatment of solid tumors by injection into areas near the tumor (63). BioNTech announced the results of the first human clinical trial (NCT04503278) of BNT211—a new generation of CAR-T therapy targeting solid tumors. The combination of CAR-T targeting CLDN6 and mRNA vaccine CARVac for CLDN6 can effectively enhance the efficacy and provide new ideas for the treatment of solid tumors (AACR 2022, Abstract #CT002). In addition, combination therapy with immune checkpoint inhibitors may also enhance the efficacy of CAR-T for solid tumors (64).
However, there are several limitations to the application of this technology. Firstly, the expression of CAR mediated by retroviral or lentiviral vectors may have an impact on the gene expression of T cells, which may produce unpredictable results. So, a comprehensive safety assessment of CAR-T cells is required before application. Secondly, the proliferation of CAR-T cells can only be achieved after induction and activation. Therefore, whether the large-scale expansion of T cells in vitro can maintain immune activity is an important factor. Thirdly, necessary technical processes are required for different patients, which may take high costs and long periods. In addition, immunosuppressive TME and efficiency of delivery to the tumor site are also major barriers to a successful CAR-T therapy. In the future, innovations in CAR design, transduction methodologies, and allogeneic CAR-T are bound to lead to improved responses and transform the treatment of patients with cancer.
TCR-T
Various new methods have been developed to enhance the antitumor efficacy of immune system, including targeting new antigens, using new engineering or modifying TCR, and creating safety switches for internal suicide genes. By transferring the exogenous TCR gene that specifically recognizes TAAs into T cells, TCR-T can be constructed to improve the affinity to TAAs and exert an MHC-dependent antitumor effect (65). Compared with CAR-T therapy, TCR-T therapy has a better safety profile due to its MHC restriction, which can alleviate adverse reactions such as cytokine storms. The TCR-T category currently in clinical trials is mainly targeting NY-ESO-1. NY-ESO-1 TCR produced by Adaptimmune Therapeutics is currently in phase I/II clinical trials (Table 3).
MART TCR-T, gp100 TCR-T, and TCR-T targeting MAGE-A3 or MAGE-A4 have achieved positive results in clinical trials. However, safe use in the clinic should consider the type of antigen and TCR affinity (66, 67). In a clinical trial of nine patients treated with TCR-T, 56% (5/9) of patients experienced an OR, one of which was a CR. However, three of nine (44%) patients experienced severe neurologic toxicities, including two deaths. The cause of death, in part, may be a cross-reaction of TCR-T with a similar epitope of MAGE-A12 in brain.
While targeting NY-ESO-1, MAGEA3, and other TAAs is an attractive strategy for the application of ACT for the treatment of solid cancers, caution must be taken to ensure a lack of cross-reactivity with vital normal tissues. In addition, modification of the CDR region of TCR must be performed with caution. Because the modified receptors, similar to those produced after immunization in HLA-transgenic mice, are not negatively selected in the thymus and may be potentially reactive to unrelated normal host proteins. There is a need to develop better screening methods to avoid such toxicity in the future. As more antigen-specific TCRs are identified, more data will become available to better understand how to use TCR-T to treat patients. Immunosuppressive TME also limits the efficacy of TCR-T therapy. Combination therapy targeting TME may be a potential strategy to improve the efficacy of TCR-T immunotherapy.
TILs
Tumor-infiltrating lymphocytes (TILs) are immune cells that exist in tumor tissues and can specifically respond to TAAs. Using TILs is an effective treatment for many cancers. The first clinical pilot study using TILs was reported in 1988 for metastatic melanoma. The result demonstrated partial response in 2 patients and partial regression in 1 patient. Tumor-specific cytolytic activity was observed in 5 patients (68). In another study by Rosenberg et al., three sequential clinical trials about TILs were performed. Objective response rates in the three trials were 49%, 52%, and 72%, respectively. A study showed that 22% of all patients achieved complete tumor regression and 19% of the patients were disease-free for more than three years (69). The OR from patients treated with standard TILs is greater than 50% and many of these patients experiencing durable CRs beyond 5 years (70–72). The effort to extend TIL therapy for the treatment of other solid cancers is ongoing. Galon et al. studied TILs in patients with colorectal cancer by gene expression profiling and in situ immunohistochemical staining (73). The results suggested that TILs act as a valuable prognostic tool in the prediction of patient survival, and the results gave convincing information regarding tumor recurrence and survival in patients with early-stage colorectal cancer.
TILs therapy mainly works by isolating TILs from tumor tissues, amplifying them in vitro with high doses of IL-2, and then injecting them into patients (68, 74). Iovance’s LN-144 therapy has achieved a disease control rate (DCR) of 80% and ORR of 38% for stage IIIc/IV melanoma patients (Table 3). More notably, patients who are not responding to immune checkpoint inhibitors still benefit. Multiple clinical trials of TILs for various types of solid tumors are currently ongoing, thus showing therapeutic potential for malignancies such as melanoma, lung, and colorectal cancers (75). TILs therapy is separate from natural lymphocytes isolated from tumor tissues and it can recognize a variety of different targets with no cytokine storms reported. Thus, TILs therapy is safer than TCR-T and CAR-T therapies and more effective in solid tumors.
However, several issues have emerged that need to be addressed. Firstly, there is an urgent need to identify alternative and predictive biomarkers to better select appropriate patients for TILs treatment to improve response rates and duration. Secondly, TILs are needed to be improved memory and effector characteristics for longer persistence and enhanced antitumor activity. In addition, although TCR-T and CAR-T therapies show very competitive performance, they can only target a single TAA or a limited array of TAAs. By contrast, TILs can recognize a panoply of unknown TAAs, which ultimately demonstrates that TILs therapy has a bright future, especially with approaches that promote TAA release and enhance T-cell persistence. At last, we also need more investigations on combination approaches that can improve long-term efficacies and reduce the cost to a more affordable level.
CAR-NK
NK cells play an important role in innate immunity. CAR-NK is a therapy like CAR-T, which uses CAR to modify NK cells. CAR-NK can be activated by targeting TAAs to release cytotoxic cytokines such as granzyme to kill tumor cells (76). CAR-NK is currently still in preclinical or clinical research, which mainly targets CD19, NKG2D, CD7, or CD33, etc. (Table 3).
In a phase I/IIa clinical trial, 11 patients with non-Hodgkin’s lymphoma and chronic lymphocytic leukemia were treated with CD19 CAR-NK. And seven patients experienced CR without serious adverse reactions (77). In 2020, NEJM published a CAR-NK treatment for hematologic tumors using cord blood-derived CAR-NK targeting CD19 that achieved complete remission in seven patients, all without a cytokine storm or neurotoxic response. Moreover, one year after treatment, CAR-NK cells are still present in the patient’s body, which is especially important for long-term antitumor therapy (77). NKG2D is an activating receptor of NK cells, which is involved in the recognition of virus-infected cells and the killing of tumor cells. In a phase I clinical trial of NKX101 (allogeneic CAR-NK cells targeting NKG2D), 3 of 5 patients treated with high doses (1.5 billion×3 and 1 billion×3) achieved CR without serious adverse reactions (NCT04623944). At AACR 2022, Senti Bio announced the results of a preclinical study of CAR-NK with a genetic circuit that secretes IL-15 in a controlled manner to improve efficacy in the treatment of solid tumors (AACR 2022 Abstract #584).
Compared with CAR-T, CAR-NK usually produces IFN-γ and GM-CSF, thus it is less likely to produce cytokine storm. CAR-NK is widely available and can be derived from allogeneic delivery without need of HLA matching. However, some factors limit the wide use of CAR-NK. The manufacturing process of CAR-NK can be further simplified and optimized. Current CARs are designed for CAR-T and they are not the best for application to NK cells. CAR design for optimal NK cell activation and cytotoxicity needs to be improved. Secondly, CAR-NK’s unspecific killing function needs to be combined with CAR-derived specific killing. In addition, limited proliferation and inhibition of the tumor microenvironment limit the clinical development of CAR-NK (78).
The lack of in vivo durability of infused cells in the absence of cytokine is one of the major drawbacks of CAR-NK therapy. Modified CAR-NK which can secret IL-2/IL-15 has demonstrated good results in some preclinical research (79). In addition, the induction of a memory-like phenotype of CAR-NKs with a cocktail of cytokines (IL-12, IL-15, and IL-18) resulted in improved responses to B-cell lymphomas in vitro and in vivo (80, 81). Immunosuppressive TME and efficiency of delivery to tumor site are also major barriers to successful CAR-NK therapy. With more pre- and clinical data in further, CAR-NK therapy may lead to revolutionary advances in tumor immunotherapy. In addition, combined therapy which includes immune checkpoints blockade and targeted therapy may provide a new direction for CAR-NK-based immunotherapy.
Oncolytic virus
Oncolytic viruses (OV) therapy is a new type of antitumor therapy, which can target tumor cells and replicate in cells to kill tumor cells. OV has become the forefront of tumor biotherapy and it is increasingly common. OV can be obtained through natural or genetic engineering, mainly including herpes virus, adenovirus, and pox virus (82). OV exerts its antitumor effects mainly by selectively replicating within tumor cells and eventually leads to tumor cell lysis. The release of TAAs after lysis can activate the immune system to eliminate tumor cells. The release of cytokines by tumor cells infected with OV can eliminate metastatic tumor cells (83, 84). In 2015, AMGEN’s T-VEC became the first OV therapy on the market with an indication of melanoma, thus marking the maturity of this technology (Table 4). Researchers are currently using various techniques to enhance the antitumor effects of OV therapy including replacing some viral genes with oncogenes or integrating TAAs genes into the OV genome to promote the production of specific immune responses (85). In addition, the combination with immune checkpoint therapy has also become an important research direction. The clinical results of CG Oncology’s OV therapy CG0070 in combination with Keytruda show 89% CR (AACR 2022 Abstract#CT036) (86).
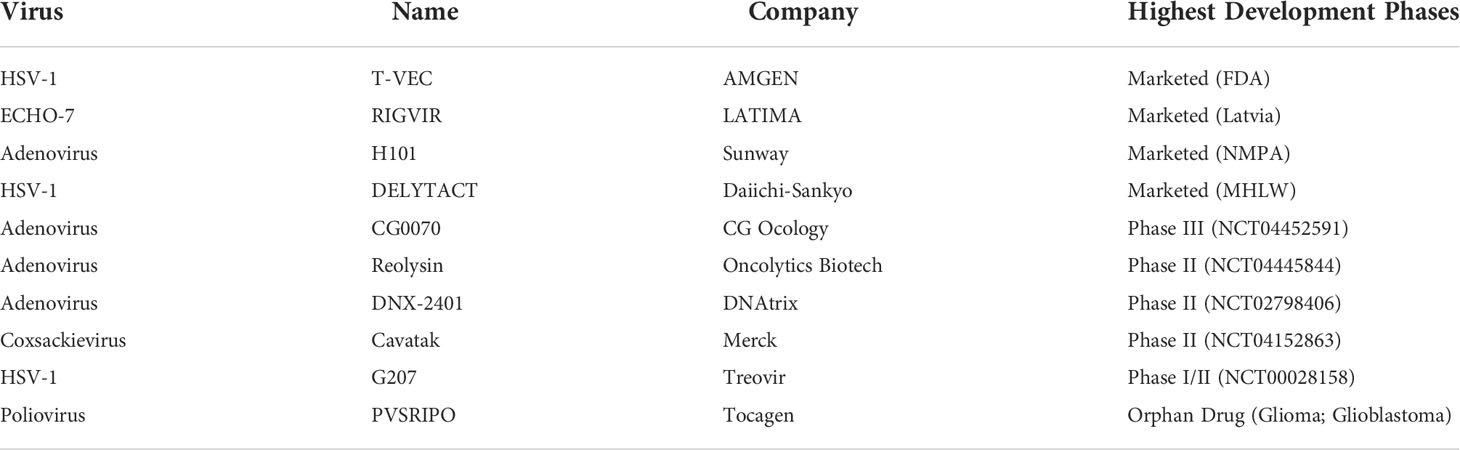
Table 4 Summary of marketed and clinically reported oncolytic virus (Up to April 2022).
OV therapy is efficacious and safe, and it is a very promising tool for tumor immunotherapy (87–89). However, its mode of administration is currently limited to intra-tumoral injection, which has limitations in clinical use. Intratumoral administration is expensive and difficult, especially in cases of malignant gliomas. Some of the novel approaches involve the use of nanoparticles, complex viral particle ligands, and immuno-modulatory agents to deliver the virus into tumor. Alternatively, delivery of OV via nanoparticles using a technologically complex image-guided delivery system has also been considered (90).. In the future, OV therapy is expected to make exciting progress by solving the problem of drug delivery and combining with other immunotherapy methods
Cancer vaccines
Preventive cancer vaccines
The immunoprevention of cancer and cancer recurrence has received extensive attention; preventative cancer vaccines have made more progress in preventing cancer than in eliminating established cancer. Nevertheless, preventing tumors obviously impacts survival. Preventive cancer vaccines mainly refer to vaccines against viruses with high carcinogenic relevance. HBV and HPV vaccines are the main representatives. The pathogenesis of HBV-associated hepatocellular carcinoma is well supported by the literature (91, 92). A variety of new HBV vaccines are now on market, such as Hepacare, HEPLISAV-B, and PreHevbrio, which expand the efficiency and scope of protection. HPV vaccines mainly include bivalent (Cervarix), quadrivalent (Gardasil), and nine-valent (Gardasil9), thus focusing on the protection of subtypes 16 and 18 used to prevent cervical cancer, vaginal cancer, and vulvar cancer caused by HPV. Due to the complex pathogenesis of tumors, this method can only be used as an auxiliary preventive method. This type of vaccine can only be used to prevent viral infection—not tumorigenesis.
Therapeutic cancer vaccines
A better understanding of the breadth of TAAs, the development of natural immune response, and new antigen delivery technologies will help to improve vaccine design. Current mature therapeutic vaccines include dendritic cell (DC) vaccine, which has antitumor effects by inducing the patient’s monocytes to become DCs ex vivo by TAAs stimulation. The cells are then infused back into the patient to stimulate the activation and expansion of cytotoxic T lymphocytes (CTLs). DC vaccine can offer long-term immune memory and can prevent tumor recurrence. Provenge is the first DC vaccine approved by the FDA for castrate-resistant prostate cancer (Table 5). The DC vaccine Ilixadencel was granted orphan drug status by the FDA in 2021 for the treatment of patients with soft tissue sarcoma. Aivita Biomedical’s DC vaccine AV-GBM-1 clinical trial (NCT03400917) results show a 28% increase in 15-month OS for glioblastoma patients. With the development of sequencing technology and bioinformatics, more and more tumor antigens have been discovered and can be used to distinguish tumor cells from normal cells. A personalized vaccine designed in this way is an important development direction for cancer vaccines in the future (93). Multiple studies are reporting that personalized vaccines have good efficacy in the treatment of melanoma (94, 95). A combination with immune checkpoints is also an important research direction and can show better efficacy than a single vaccine therapy (96). In addition to DC vaccines, therapeutic vaccines include tumor cell vaccines, DNA/mRNA vaccines, and peptide vaccines (97).

Table 5 Research progress of therapeutic cancer vaccines (Up to April 2022).
DC vaccines suffer from limited cell sources, long preparation periods, and high costs. However, their advantages include low side effects, good tolerance, and long-term immunological memory, which still give them broad market prospects.
The key to the development of the cancer vaccine is the need to identify the appropriate biomarkers and optimize the combination of treatments to improve their effectiveness in patients. The research on vaccines has been advancing in the past few decades, and many different characterized cancer vaccines are now available. However, there are still some problems that must be solved, including suitable tumor antigen and adjuvant components, suitable delivery modes, and effective methods to overcome immune attack. Although neoantigens are the best option for antitumor immunotherapy, the problem of obtaining individualized neoantigens hinders the application of cancer vaccines. This is mainly due to inherent alterations in tumor cells and the formation of an immunosuppressive TIME. Several approaches have been developed to overcome difficulties, including the use of immunostimulatory adjuvants, in combination with ACT and ICB.
Mechanisms in cancer immunotherapy resistance
Cancer immunotherapies, such as immune checkpoint blockade (ICB) and adoptive cell therapies (ACT), are effective for patients with various cancers (98). However, the response rate of cancer immunotherapies is still limited due to the lack of immunogenic antigens and various immune-resistant mechanisms (99). Understanding the immune resistance mechanisms is essential to improve the efficacy of current cancer immunotherapies.
Primary resistance and adaptive resistance
Patients who have primary resistance to cancer immunotherapies do not respond to the initial therapy. Adaptive resistance refers to the mechanism by which tumor cells can be recognized by the immune system, but it can adapt to immune attack to protect itself as the tumor progresses. The mechanism of adaptive resistance may include primary resistance, and the mechanism of primary resistance may also be the result of adaptive resistance.
The most fundamental reason why tumor cells cannot be recognized by T cells and thus lead to non-response to immunotherapy is the lack of tumor antigens. In addition, cancer cells may have tumor antigens, but the change in the antigen presentation mechanism can also result in the occurrence of immune resistance (100).
In tumor cell-intrinsic factors, insufficient tumor antigenicity and neoantigens contribute to primary and adaptive resistance. Tumor cells can evade specific immune recognition by T cells by downregulating the expression of TAAs, TSAs, and surface MHC. Tumor cells with relatively weak immunogenicity can escape the surveillance of the immune system and selectively proliferate. After the immune selection process, the immunogenicity of the tumor is getting weaker and weaker. The emergence of neoantigens can inhibit tumor progression, whereas poorly immunogenic tumors lack response to PD-1/PD-L1 blockade. Deletion of neoantigens is responsible for primary resistance to immunotherapy in triple-negative breast cancer (TNBC) (101). LINK-A, a lncRNA that can degrade phospholipase C by ubiquitin ligases, has a negative correlation with cytotoxic T lymphocytes infiltration in TNBC (102). It is currently believed that the higher the tumor mutation burden (TMB), the more neoantigens are produced, and the stronger T cell response are. Clinically, melanoma, renal cell carcinoma, and NSCLC with high TMB have a better response to anti-PD-1 therapy, while pancreatic cancer and prostate cancer with low TMB are less effective (103, 104).
In tumor cell-intrinsic factors, tumor signaling pathways can produce immunosuppressive components, or alter some gene expression to affect the efficacy of ICB. Oncogenic signaling through the MAPK pathway results in the production of VEGF and IL-8, which have inhibitory effects on T cell recruitment and function (105). Activation of AKT signaling through PTEN loss was also correlated with reduced CD8+ T cells in tumors and a poor response to anti-PD-1 in melanoma patients (106). IFN-γ signaling pathway in TIME activates JAK-STAT signaling, which can induce PD-L1 expression (107). Wnt/β-catenin signaling pathway is closely related to the occurrence and development of various tumors (108). Studies have shown that Wnt/β-catenin signaling in melanoma cells can prevent antitumor responses by interfering with the recruitment of BATF3-expressing DCs (109, 110).
In tumor-intrinsic factors, immunosuppressive metabolism in TIME can suppress immune response. Various metabolisms in tumor may cause immune resistance. Tumor cells preferentially utilize glycolysis to produce ATPs and molecules necessary for cell division such as nucleic acids, while reducing mitochondrial activity to decrease the production of reactive oxygen species (ROSs) for survival (Warburg effect) (111). Enhanced glycolysis in melanoma cells is associated with reduced infiltration of CD8+ T cells in tumors and resistance to in vitro T cell lysis and in vivo pericyte therapy, partially due to increased production of immunosuppressive lactate (112).
In addition, tumor cell-extrinsic mechanisms that lead to primary and adaptive resistance involve components other than tumor cells within TIME. Tregs reduce the expression of MHC-II molecules by secreting the inhibitory cytokine IL-10, which can affect DC maturation and suppress immune responses (113). MDSCs can express CD11b and CD33 to promote blood vessel growth, tumor invasion, and metastasis. CXCR2 can induce MDSCs to infiltrate tumors and mediate immune resistance (114). Tumor-associated macrophages (TAMs) can also affect immunotherapy responses. Several reports have discussed the role of macrophages in mediating therapeutic resistance in cancer (115–117).
Acquired resistance
A hallmark of cancer immunotherapy has been the induction of long-lasting tumor responses. However, patients who once responded to ICB sometimes relapse due to acquired resistance. Schachter et al. showed that 1/4 to 1/3 of patients with metastatic melanoma who received anti-PD-1 or anti-CTLA-4 therapy relapsed after ongoing treatment, even if they were effective against immunotherapy (118). The possible mechanisms of acquired resistance mainly include B2M mutation and loss of HLA heterozygosity, changes in tumor target antigens, and up-regulation of alternative immune checkpoints. There is evidence for each of these mechanisms can lead to acquired resistance to ICB or ACT.
B2M plays an important role in MHC-I antigen-presenting, antigen recognition, and T cell infiltration (119). Mutated B2M gene affects normal folding and transport of MHC-I, resulting in resistance to ICB (120). Sade-Feldman et al. analyzed post-treatment biopsy specimens from 17 metastatic melanoma patients with ICB treated, and they found the percentage of heterozygous deletions and point mutations of B2M was 9.4%, which suggested that B2M loss may be a common mechanism of resistance to targeted CTLA-4 or PD-1 therapy (121). GAO et al. showed that mutations in Janus kinase 1 (JAK1), JAK2, and B2M in tumor samples after immunotherapy may be the mechanisms of acquired resistance to anti-PD-L1 therapy in melanoma patients (122).
Additional evidence of loss of antigen-presenting machinery leading to acquired resistance to cancer immunotherapy is provided by a case of a patient with metastatic colorectal carcinoma who responded to TILs ACT. The TILs recognized mutated KRAS G12D presented by HLA-C*08:02 resulting in an objective antitumor response, followed by an isolated relapse in a lesion that had lost HLA-C*08:02 in chromosome 6 (123). Therefore, acquired resistance to ICB and ACT could be mediated through genetic mechanisms that altered antigen-presenting machinery and IFN-γ signaling.
Cytotoxicity T cells are specific for cancer cells that express their cognate antigen, but cancer cells may develop acquired resistance through decreased expression or mutations in these antigens. T cells turned on by checkpoint blockade therapy primarily recognize mutational neoantigens (104, 124). Gene deletions, mutations, or epigenetic alterations can lead to a decrease in MHC-presented mutational neoantigens and acquired resistance. One study found that the main cause of resistance to CD19 CAR-T cells for acute lymphoblastic leukemia was the loss of target antigens, which is mainly caused by antigen escape and lineage conversion (125, 126).
After immune checkpoint treatment, due to compensatory effects, the expression of other immune checkpoints is elevated, which in turn causes acquired resistance. TIM-3 is a negative immune checkpoint. It was found that TIM-3 was highly expressed in T-cells from animals that were resistant to anti-PD-1 treatment, which confirmed that the main mechanism of resistance to anti-PD-1 immunotherapy is the selective activation of a new immune checkpoint (23). In addition to TIM-3, other known alternative immune checkpoints are LAG-3, TIGIT, and VISTA, etc. Several clinical trials are currently undergoing to test antibodies against these immune checkpoints, both as monotherapy and combination therapy strategies, to provide additional clinical benefits (127).
Great advances occurred in the field of cancer immunotherapy due to years of mechanism exploration and clinical application development. However, to date, the benefits have been limited to a small number of patients with certain cancer types. In addition, thanks to more successful immunotherapy treatments, we now have a large proportion of patients who initially respond but eventually relapse. The mechanisms of immunotherapy resistance are complicated, and we are likely just observing the tip of the iceberg. To bring clinical benefit to the majority of patients, we need to have a comprehensive understanding of the tumor cell-intrinsic and -extrinsic factors that lead to immunotherapy resistance. These mechanisms can lead to primary, adaptive, and acquired resistance to immunotherapy. Elucidating these mechanisms will provide important clues to overcome resistance to immunotherapy.
Combination strategies for cancer immunotherapy
To enhance the effectiveness of cancer immunotherapy and overcome immunotherapy resistance, combination therapy has become a hot topic of current research (128, 129). Currently, ICB is the most used cancer immunotherapy in clinical combination.
Combination of different ICBs
An example of enhanced efficacy with combination therapy is the use of anti-CTLA-4 and anti-PD-1, which results in higher response rates and improved survival in melanoma patients (130, 131). In the phase III trial in patients with unresectable or metastatic melanoma, the five-year survival rate of the combination group (nivolumab plus ipilimumab) reached 52%, and the five-year survival rate of the nivolumab group and ipilimumab group was 44% and 26% respectively (131, 132).
In addition, blockades of TIM-3, LAG-3, and TIGIT are receiving increasing attention. In the treatment of hepatocellular carcinoma, it was found that blocking both TIM-3 and PD-1 can completely reverse the exhausted state of T cells and has a significant antitumor effect. However, blocking TIM-3 or PD-1 alone only partially restored the function of T cells (133). Both LAG3 and PD-1 can transmit co-inhibitory signals and blocking both LAG3 and PD-1 can play an immune synergistic effect by enhancing CD8+ T cell function and clearing Treg (134). TIGIT is mainly expressed on activated T cells and NK cells, which mediates immunosuppressive signal. TIGIT blockade synergizes with anti-PD-1 can enhance CD8+ T cell function and promote tumor regression (135).
Combination with chemotherapy and radiotherapy
Previously, it was believed that chemotherapy could lead to immunosuppression by affecting the number or function of lymphocytes. But in-depth studies have found that some chemotherapies can enhance tumor immunogenicity (136). Some studies believe that chemotherapy can enhance the antitumor immune response, among which pembrolizumab combined with chemotherapy has been approved by the FDA. Liposome doxorubicin combined with immunotherapy produces synergistic antitumor effects in mice, and more mice achieve complete tumor remission and prolonged survival (137). High-frequency and low-dose chemotherapy can effectively activate CTLs and inhibit immunosuppressive cells in TIME, which can promote efficacy and solve the problem of immune resistance (138). In an in-situ CRC-bearing mouse model with ineffective anti-PD-L1 treatment, the proportion of TILs was significantly increased after combination with oxaliplatin. At the same time, the combination of oxaliplatin and a novel PD-L1 blocker (PD-L1 Trap) significantly prolonged the survival of tumor-bearing mice (139). Chemotherapy combined with anti-PD-1 is used as a first-line treatment for advanced NSCLC, which has significantly more clinical benefits than a single agent (140).
Radiotherapy promotes the release of TAAs, TSAs, or damage associated molecular pattern molecules (DAMPs), which can enhance the immunogenicity of tumor cells and promote the recruitment and infiltration of immune cells. This relationship is the rationale for combination with immunotherapies. In a study of mouse model, radiotherapy combined with anti-PD-1 treatment reversed immune resistance (141). In the treatment of metastatic melanoma, radiotherapy combined with anti-CTLA-4 and anti-PD-1 therapy may become a new idea in combined immunotherapies (142). Pilones et al. reported that anti-CTLA-4 combined with radiotherapy effectively inhibited the lung metastasis of breast cancer in a mouse model (143). Deselm et al. found that radiotherapy made it more effective for CAR-T cells in a mouse model of pancreatic cancer, and the tumor cells that did not express the CAR target were also killed by CAR-T cells (144).
Combination with targeted therapy
Targeting intracellular signaling pathways with small molecule inhibitors is effective in rapidly reducing tumor volume. However, many of these drugs do not have durable effects, mainly due to the emergence of other compensatory pathways (145, 146). Emerging strategies to enhance immunotherapy response are being developed based on novel insights into T cells and overall immune function. Pembrolizumab combined with BRAF inhibitors shows synergistic antitumor activity and prolongs response time in mice with metastatic melanoma (147).
Tumor angiogenesis has an important relationship with tumor immunity. VEGF is related to the generation and regulation of MDSCs, so anti-angiogenic therapy combined with immunotherapy has a synergistic effect. Preclinical studies have shown that the combination of VEGFR inhibitor Axitinib and anti-CTLA-4 can enhance the antigen-presenting ability of DC to promote T cell proliferation in a mouse melanoma model (148). Data from clinical trials with the combination of ipilimumab and VEGF inhibitor bevacizumab showed that more than 30% of patients in the combination group observed a significant increase in CCR7+ CD8+ T cells, compared to only 6% of patients in the ipilimumab group (149). Studies have shown that sunitinib can reverse tumor-induced immunosuppression by reducing MDSCs (150). Various targeted therapies, such as EGFR, ALK, ROS1, and MEK inhibitors, are being clinically tested in combination with ICBs.
ACT combined with targeted therapy is also an innovative immunotherapeutic approach. Li et al. combine CAIX-specific CAR-T cells and sunitinib, which induces a potent antitumor response in an experimental model of metastatic RCC (151).
Other combination immunotherapies
The functional inhibition of CAR-T by PD-1/PD-L1 has been well established, which also provides the basis for the combination of PD-1/PD-L1 blockade and CAR-T. Existing preclinical studies have shown that CAR-T cells plus PD-1/PD-L1 blockade can effectively enhance the antitumor effect (152, 153).
IDO1 inhibitors have been in active clinical investigation and preliminary results suggest that IDO1 inhibitors produce additive efficacy when combined with cancer immunotherapies despite low activity as a single agent. Results from a phase I/II clinical trial, which combined IDO1 inhibitor epacadostat and Ipilimumab for the treatment of metastatic melanoma, showed an objective response and no tumor progression in some patients (154). In addition, IDO1 inhibitors combined with ICBs are also tested in various clinical trials (NCT03519256, NCT03854032, and NCT03661320). Combinations of type I interferons, TLR inhibitors, or STING agonists have also shown promise in preclinical models (155–157).
T-VEC can selectively replicate within tumors and produce granulocyte-macrophage colony-stimulating factor (GM-CSF), which triggers DC differentiation and enhancement of antigen presentation. This makes OVs susceptible to immunotherapy. OV therapy CG0070 in combination with Keytruda showed 89% CR in a clinical trial (AACR 2022 Abstract#CT036) (86).
Current combined strategies are complex because the potential combination approaches far exceed the available human and technical resources. There is an urgent need for us to test these combinations in appropriate preclinical models and to accelerate clinical translation through novel approaches to clinical trial design.
Discussion
Cancer occurrence and development is a complex process. Various immune-evasion mechanisms can counteract the body’s immune response, which becomes more complex as cancer progresses. Cancer immunotherapy can kill and eliminate tumor cells through the immune system, thus becoming another revolutionary treatment after surgical resection, radiotherapy, chemotherapy, and targeted therapy.
Various cancer immunotherapies have shown promising clinical efficacy. However, cancer immunotherapy still faces many problems and challenges. MAbs therapy is a very promising treatment for immunotherapy, which has been repeatedly demonstrated in clinical use. However, due to the immunogenicity, mAbs can cause irAEs, which requires strict monitoring in clinical use. The production process of mAbs is time-consuming and costly, and new purification strategies are needed for higher purity of mAbs. These problems are determined by the nature of the antibody itself, and we believe these problems will partly be solved with new design strategies and further optimization. The overall immune response rate of patients treated with ICB is not high, and there is a need to find reliable and effective biomarkers for precise and personalized immunotherapy. In combination with chemotherapy, mAbs have generated success against advanced-stage cancers, which previously had poor outcomes. In addition, combinations with different mAbs also showed a strong anti-tumor effect. Combination therapy may provide new opportunities for mAbs to reduce the side effects and improve the therapeutic effect in the future. Conjugation of cytotoxic agents to mAb allows for specific delivery of payloads to tumors, while multispecific antibodies grant novel mechanisms that increase specificity and facilitate delivery to historically intractable compartments. Besides, Fc- engineering mAbs can endow mAbs with stronger antitumor and immune activation ability through the incorporation of amino acid and glycan changes. With an increased understanding of immunobiology and the continued development of molecular biological methods, the possibilities for mAbs therapy are bounded only by the scope of human ingenuity.
Small molecule inhibitors for cancer immunotherapy always occupy an important position, although the sales of mAbs are far ahead. Small molecule inhibitors have mature R&D pipelines and the production process of small molecule inhibitors is more controllable than mAbs, which can help reduce costs. The controllable pharmacokinetic properties can help reduce the impact of side effects, and the good tissue permeability makes small molecule inhibitors useful for solid tumor immunotherapy. Small molecule inhibitors will always be an effective replacement and supplement for mAbs. Currently, a new form of small molecule inhibitor, proteolysis targeting chimeras (PROTAC) is tested in (pre-)clinical, such as IDO1 PROTACs. But many issues need to be addressed especially on whether it is a safe approach or whether there is a saturation in the degradation of proteins that may limit their effectiveness (158, 159).
ACT can be a potent new addition to the toolbox for cancer immunotherapy. However, many TCR-T/CAR-T clinical trials have been hampered by off-target effects and safety concerns (160, 161). While timely intervention is effective in most adverse events, side-effect management of ACT must be held in the whole process of ACT treatment. If tumor antigens are blocked by the self-secretion of tumor cells, they cannot be recognized by the immune system. Rationally designed strategies to identify candidate neoantigens and evaluate their immunogenicity are valuable for boosting the safety and efficacy of ACT. At present, the successful ACT therapy is mainly used in the treatment of hematological tumors. In solid tumors, getting CAR-T cells to infiltrate the tumor is a challenge, which can be compounded by the immunosuppressive TME. ACT combined with small molecule immunomodulator targeting immunosuppressive TIME may be effective for solid tumors.
The major challenge in oncolytic virus therapy is the targeted delivery of the virus into the tumor. In most cases, systemic administration does not work well because of preexisting immunity. Some novel approaches involve the use of nanoparticles, complex viral particle ligands, and immuno-modulatory agents to deliver OVs into the tumor. Alternatively, delivery of OVs via a nanoparticles using technologically complex image-guided delivery system has also been considered (90). Immune response after OVs infection suppresses the replication of the virus thereby posing a hindrance to the effective functioning of OVs therapy. Therefore, increasing anticancer treatments and consequently patient prognosis through contributions from molecular biology, immunology, genomics, and bioinformatics will provide a strong foundation for OVs’ potential clinical success in the future.
For preventive cancer vaccines, the successes of Gardasil are exciting. The next step is perhaps to look for other important tumorigenic antigens, possibly other viruses, to expand protection for people. In addition, for therapeutic cancer vaccines, an improved antitumor immune response is still in high demand because of the unsatisfactory clinical performance of the vaccine in tumor inhibition and regression. Personalized vaccine design and appropriate combined therapy could represent the best approach to increase the efficacy of cancer vaccines.
Compared with traditional chemoradiotherapy and targeted therapy, immunotherapy has significant advantages. Under the in-depth study of anti-tumor immune response mechanism, great progress has been made in the field of tumor immunotherapy. With the widespread application of immunotherapy, the occurrence of immune resistance has become an unavoidable problem. We are still at a very early stage of understanding the mechanisms of this immune resistance. By understanding mechanisms of immune resistance, we can enable immunotherapy to provide more survival benefits for cancer patients.
Compared with single-drug therapy, combination strategy for immunotherapy has a greater clinical effect. Clinical trials have shown that immunotherapeutic anticancer drugs, which include ICBs, ACT, chemotherapy, targeted therapy, etc., are important components of the combination. A few combination therapies have been approved by the FDA to improve clinical efficacy of cancer immunotherapy. With increasing research in identifying reliable biomarkers in guiding clinical immuno-oncology decisions, more convincing and effective combination strategies are expected.
As the development of tumor immunology, bioinformatics, and sequencing technologies, more and more mechanisms in TME will continue to be revealed. This will further the development of cancer immunotherapy and pave the way for effective cancer treatments in the future.
Author contributions
CL prepared the figures and tables and prepared the manuscript; MY and MC contributed the idea and edited the manuscript; DZ participated in this study and contributed project administration and funding acquisition; DZ contributed the idea, oversaw the process and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The current study was supported by projects on the Science and Technology Commission of Shanghai (18ZR1403900) (DZ), the National Natural Science Foundation of China (81872895) (DZ), a project on joint translational research in the School of Pharmacy and Minhang Hospital (RO-MY201712) (DZ), and Jinan Science and Technology Bureau, Innovation team for the development and evaluation of new drugs for oncology immunotherapy (2020GXRC041 to DZ).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Swann JB, Smyth MJ. Immune surveillance of tumors. J Clin Invest (2007) 117:1137–46. doi: 10.1172/JCI31405
2. Tan AC, Bagley SJ, Wen PY, Lim M, Platten M, Colman H, et al. Systematic review of combinations of targeted or immunotherapy in advanced solid tumors. J Immunother Cancer (2021) 9:e002459. doi: 10.1136/jitc-2021-002459
3. Chang CH, Qiu J, O'Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell (2015) 162:1229–41. doi: 10.1016/j.cell.2015.08.016
4. Sanmamed MF, Chen L. A paradigm shift in cancer immunotherapy: From enhancement to normalization. Cell (2018) 175:313–26. doi: 10.1016/j.cell.2018.09.035
5. Yang Y. Cancer immunotherapy: harnessing the immune system to battle cancer. J Clin Invest (2015) 125:3335–7. doi: 10.1172/JCI83871
6. Brandsma AM, Jacobino SR, Meyer S, ten Broeke T, Leusen JH. Fc receptor inside-out signaling and possible impact on antibody therapy. Immunol Rev (2015) 268:74–87. doi: 10.1111/imr.12332
7. Teige I, Mårtensson L, Frendéus BL. Targeting the antibody checkpoints to enhance cancer immunotherapy-focus on FcγRIIB. Front Immunol (2019) 10:481. doi: 10.3389/fimmu.2019.00481
8. Strohl WR. Optimization of fc-mediated effector functions of monoclonal antibodies. Curr Opin Biotechnol (2009) 20:685–91. doi: 10.1016/j.copbio.2009.10.011
9. Brennan FR, Morton LD, Spindeldreher S, Kiessling A, Allenspach R, Hey A, et al. Safety and immunotoxicity assessment of immunomodulatory monoclonal antibodies. MAbs (2010) 2:233–55. doi: 10.4161/mabs.2.3.11782
10. Kulkarni HS, Kasi PM. Rituximab and cytokine release syndrome. Case Rep Oncol (2012) 5:134–41. doi: 10.1159/000337577
11. Makino K, Nakata J, Kawachi S, Hayashi T, Nakajima A, Yokoyama M. Treatment strategy for reducing the risk of rituximab-induced cytokine release syndrome in patients with intravascular large b-cell lymphoma: a case report and review of the literature. J Med Case Rep (2013) 7:280. doi: 10.1186/1752-1947-7-280
12. Labrijn AF, Janmaat ML, Reichert JM, Parren P. Bispecific antibodies: a mechanistic review of the pipeline. Nat Rev Drug Discovery (2019) 18:585–608. doi: 10.1038/s41573-019-0028-1
13. Krishnamurthy A, Jimeno A. Bispecific antibodies for cancer therapy: A review. Pharmacol Ther (2018) 185:122–34. doi: 10.1016/j.pharmthera.2017.12.002
14. Elgundi Z, Reslan M, Cruz E, Sifniotis V, Kayser V. The state-of-play and future of antibody therapeutics. Adv Drug Delivery Rev (2017) 122:2–19. doi: 10.1016/j.addr.2016.11.004
15. Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U.S.A. (2002) 99:12293–7. doi: 10.1073/pnas.192461099
16. Wei G, Zhang H, Zhao H, Wang J, Wu N, Li L, et al. Emerging immune checkpoints in the tumor microenvironment: Implications for cancer immunotherapy. Cancer Lett (2021) 511:68–76. doi: 10.1016/j.canlet.2021.04.021
17. Buchbinder EI, Desai A. CTLA-4 and PD-1 pathways: Similarities, differences, and implications of their inhibition. Am J Clin Oncol (2016) 39:98–106. doi: 10.1097/COC.0000000000000239
18. de Miguel M, Calvo E. Clinical challenges of immune checkpoint inhibitors. Cancer Cell (2020) 38:326–33. doi: 10.1016/j.ccell.2020.07.004
19. Syn NL, Teng MWL, Mok TSK, Soo RA. De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol (2017) 18:e731–41. doi: 10.1016/S1470-2045(17)30607-1
20. Lynch TJ, Bondarenko I, Luft A, Serwatowski P, Barlesi F, Chacko R, et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line treatment in stage IIIB/IV non-small-cell lung cancer: results from a randomized, double-blind, multicenter phase II study. J Clin Oncol (2012) 30:2046–54. doi: 10.1016/j.yonc.2012.07.009
21. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med (2002) 8:793–800. doi: 10.1038/nm730
22. Singh S, Hassan D, Aldawsari HM, Molugulu N, Shukla R, Kesharwani P. Immune checkpoint inhibitors: a promising anticancer therapy. Drug Discovery Today (2020) 25:223–9. doi: 10.1016/j.drudis.2019.11.003
23. Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun (2016) 7:10501. doi: 10.1038/ncomms10501
24. Ngiow SF, von Scheidt B, Akiba H, Yagita H, Teng MW, Smyth MJ. Anti-TIM3 antibody promotes T cell IFN-γ-mediated antitumor immunity and suppresses established tumors. Cancer Res (2011) 71:3540–51. doi: 10.1158/0008-5472.CAN-11-0096
25. Fadel F, El Karoui K, Knebelmann B. Anti-CTLA4 antibody-induced lupus nephritis. N Engl J Med (2009) 361:211–2. doi: 10.1056/NEJMc0904283
26. Boutros C, Tarhini A, Routier E, Lambotte O, Ladurie FL, Carbonnel F, et al. Safety profiles of anti-CTLA-4 and anti-PD-1 antibodies alone and in combination. Nat Rev Clin Oncol (2016) 13:473–86. doi: 10.1038/nrclinonc.2016.58
27. Martins F, Sofiya L, Sykiotis GP, Lamine F, Maillard M, Fraga M, et al. Adverse effects of immune-checkpoint inhibitors: epidemiology, management and surveillance. Nat Rev Clin Oncol (2019) 16:563–80. doi: 10.1038/s41571-019-0218-0
28. Wang DY, Salem JE, Cohen JV, Chandra S, Menzer C, Ye F, et al. Fatal toxic effects associated with immune checkpoint inhibitors: A systematic review and meta-analysis. JAMA Oncol (2018) 4:1721–8. doi: 10.1001/jamaoncol.2018.3923
29. Raschi E, Gatti M, Gelsomino F, Ardizzoni A, Poluzzi E, De Ponti F. Lessons to be learnt from real-world studies on immune-related adverse events with checkpoint inhibitors: A clinical perspective from pharmacovigilance. Target Oncol (2020) 15:449–66. doi: 10.1007/s11523-020-00738-6
30. Kumar V, Chaudhary N, Garg M, Floudas CS, Soni P, Chandra AB. Current diagnosis and management of immune related adverse events (irAEs) induced by immune checkpoint inhibitor therapy. Front Pharmacol (2017) 8:49. doi: 10.3389/fphar.2017.00311
31. Schneider BJ, Naidoo J, Santomasso BD, Lacchetti C, Adkins S, Anadkat M, et al. Management of immune-related adverse events in patients treated with immune checkpoint inhibitor therapy: ASCO guideline update. J Clin Oncol (2021) 39:4073–126. doi: 10.1200/jco.21.01440
32. Lin X, Lu X, Luo G, Xiang H. Progress in PD-1/PD-L1 pathway inhibitors: From biomacromolecules to small molecules. Eur J Med Chem (2020) 186:111876. doi: 10.1016/j.ejmech.2019.111876
33. Dawkins JB, Wang J, Maniati E, Heward JA, Koniali L, Kocher HM, et al. Reduced expression of histone methyltransferases KMT2C and KMT2D correlates with improved outcome in pancreatic ductal adenocarcinoma. Cancer Res (2016) 76:4861–71. doi: 10.1158/0008-5472.CAN-16-0481
34. Carretero-González A, Lora D, Ghanem I, Zugazagoitia J, Castellano D, Sepúlveda JM, et al. Analysis of response rate with ANTI PD1/PD-L1 monoclonal antibodies in advanced solid tumors: a meta-analysis of randomized clinical trials. Oncotarget (2018) 9:8706–15. doi: 10.18632/oncotarget.24283
35. Sasikumar PG, Nair R, Muralidhara N, Sudarshan SS. 1,3,4-OXADIAZOLE AND 1,3,4-THIADIAZOLE DERIVATIVES AS IMMUNOMODULATORS. In Aurigene discovery technologies limited. (2015) WO2015033301.
36. Sasikumar PG, Nair R, Muralidhara N, Sudarshan SS. 1,2,4-OXADIAZOLE DERIVATIVES AS IMMUNOMODULATORS. In Aurigene discovery technologies limited. (2015) WO2015033299.
37. Park JJ, Thi EP, Carpio VH, Bi Y, Cole AG, Dorsey BD, et al. Checkpoint inhibition through small molecule-induced internalization of programmed death-ligand 1. Nat Commun (2021) 12:1222. doi: 10.1038/s41467-021-21410-1
38. Sasikumar PGN, Ramachandra M, Vadlamani S, Kumar V, Koteswara R, Satyam L, et al. Immunosuppression modulating compounds. In Aurigene discovery technologies limited. (2011) US2011318373.
39. Sasikumar PG, Ramachandra RK, Adurthi S, Dhudashiya AA, Vadlamani S, Vemula K, et al. A rationally designed peptide antagonist of the PD-1 signaling pathway as an immunomodulatory agent for cancer therapy, mol. Cancer Ther (2019) 18:1081–91. doi: 10.1158/1535-7163.MCT-18-0737
40. Sasikumar PG, Satyam LK, Shrimali RK, Subbarao K, Ramachandra R, Vadlamani S, et al. Abstract 2850: Demonstration of anti-tumor efficacy in multiple preclinical cancer models using a novel peptide inhibitor (Aurigene-012) of the PD1 signaling pathway. Cancer Res (2012) 72:2850–0. doi: 10.1158/1538-7445.AM2012-2850
41. Yeung K-S, Grant-Young KA, Zhu J, Saulnier MG, Frennesson DB, Langley DR, et al. Biaryl compounds useful as immunomodulators. In Bristol-Myers Squibb company. (2018) WO2018044963.
42. Liu C, Zhou F, Yan Z, Shen L, Zhang X, He F, et al. Discovery of a novel, potent and selective small-molecule inhibitor of PD-1/PD-L1 interaction with robust in vivo anti-tumour efficacy. Br J Pharmacol (2021) 178:2651–70. doi: 10.1111/bph.15457
43. Le Naour J, Galluzzi L, Zitvogel L, Kroemer G, Vacchelli E. Trial watch: IDO inhibitors in cancer therapy. Oncoimmunology (2020) 9:1777625. doi: 10.1080/2162402X.2020.1777625
44. Cheong JE, Sun L. Targeting the IDO1/TDO2-KYN-AhR pathway for cancer immunotherapy - challenges and opportunities. Trends Pharmacol Sci (2018) 39:307–25. doi: 10.1016/j.tips.2017.11.007
45. Uyttenhove C, Pilotte L, Théate I, Stroobant V, Colau D, Parmentier N, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med (2003) 9:1269–74. doi: 10.1038/nm934
46. Yue EW, Sparks R, Polam P, Modi D, Douty B, Wayland B, et al. INCB24360 (Epacadostat), a highly potent and selective indoleamine-2,3-dioxygenase 1 (IDO1) inhibitor for immuno-oncology. ACS Med Chem Lett (2017) 8:486–91. doi: 10.1021/acsmedchemlett.6b00391
47. Companies scaling back IDO1 inhibitor trials. Cancer Discovery (2018) 8:Of5. doi: 10.1158/2159-8290.CD-ND2018-007
48. Prendergast GC, Malachowski WJ, Mondal A, Scherle P, Muller AJ. Indoleamine 2,3-dioxygenase and its therapeutic inhibition in cancer. Int Rev Cell Mol Biol (2018) 336:175–203. doi: 10.1016/bs.ircmb.2017.07.004
49. Fu J, Kanne DB, Leong M, Glickman LH, McWhirter SM, Lemmens E, et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci Transl Med (2015) 7:283ra252. doi: 10.1126/scitranslmed.aaa4306
50. Liu Y, Lu X, Qin N, Qiao Y, Xing S, Liu W, et al. STING, a promising target for small molecular immune modulator: A review. Eur J Med Chem (2021) 211:113113. doi: 10.1016/j.ejmech.2020.113113
51. Lim EA, Bendell JC, Falchook GS, Bauer TM, Drake CG, Choe JH, et al. Phase 1a/b, open-label, multicenter study of AZD4635 (an adenosine 2A receptor antagonist) as monotherapy or combined with durvalumab, in patients with solid tumors. Clin Cancer Res (2022) 31:CCR-22-0612. doi: 10.1158/1078-0432.CCR-22-0612
52. Chiappori AA, Creelan B, Tanvetyanon T, Gray JE, Haura EB, Thapa R, et al. Phase I study of taminadenant (PBF509/NIR178), an adenosine 2A receptor antagonist, with or without spartalizumab (PDR001), in patients with advanced non-small cell lung cancer. Clin Cancer Res (2022) 28:2313–20. doi: 10.1158/1078-0432.CCR-21-2742
53. Jia HX, He YL. Efficacy and safety of imiquimod 5% cream for basal cell carcinoma: a meta-analysis of randomized controlled trial. J Dermatolog Treat (2020) 31:831–8. doi: 10.1080/09546634.2019.1638883
54. Choueiri TK, Atkins MB, Rose TL, Alter RS, Ju Y, Niland K, et al. A phase 1b trial of the CXCR4 inhibitor mavorixafor and nivolumab in advanced renal cell carcinoma patients with no prior response to nivolumab monotherapy. Invest New Drugs (2021) 39:1019–27. doi: 10.1007/s10637-020-01058-2
55. Huang X, Yang Y. Driving an improved CAR for cancer immunotherapy. J Clin Invest (2016) 126:2795–8. doi: 10.1172/JCI88959
56. Bach PB, Giralt SA, Saltz LB. FDA Approval of tisagenlecleucel: Promise and complexities of a $475 000 cancer drug. Jama (2017) 318:1861–2. doi: 10.1001/jama.2017.15218
57. FDA Approves second CAR T-cell therapy. Cancer Discovery (2018) 8:5–6. doi: 10.1158/2159-8290.CD-NB2017-155
58. Abreu TR, Fonseca NA, Gonçalves N, Moreira JN. Current challenges and emerging opportunities of CAR-T cell therapies. J Control Release (2020) 319:246–61. doi: 10.1016/j.jconrel.2019.12.047
59. Tang J, Hubbard-Lucey VM, Pearce L, O'Donnell-Tormey J, Shalabi A. The global landscape of cancer cell therapy. Nat Rev Drug Discovery (2018) 17:465–6. doi: 10.1038/nrd.2018.74
60. Dai H, Wu Z, Jia H, Tong C, Guo Y, Ti D, et al. Bispecific CAR-T cells targeting both CD19 and CD22 for therapy of adults with relapsed or refractory b cell acute lymphoblastic leukemia. J Hematol Oncol (2020) 13:30. doi: 10.1186/s13045-020-00856-8
61. Zhao J, Song Y, Liu D. Clinical trials of dual-target CAR T cells, donor-derived CAR T cells, and universal CAR T cells for acute lymphoid leukemia. J Hematol Oncol (2019) 12:17. doi: 10.1186/s13045-019-0705-x
62. Hegde M, Mukherjee M, Grada Z, Pignata A, Landi D, Navai SA, et al. Tandem CAR T cells targeting HER2 and IL13Rα2 mitigate tumor antigen escape. J Clin Invest (2016) 126:3036–52. doi: 10.1172/JCI83416
63. Grosskopf AK, Labanieh L, Klysz DD, Roth GA, Xu P, Adebowale O, et al. Delivery of CAR-T cells in a transient injectable stimulatory hydrogel niche improves treatment of solid tumors. Sci Adv (2022) 8:eabn8264. doi: 10.1126/sciadv.abn8264
64. Gargett T, Yu W, Dotti G, Yvon ES, Christo SN, Hayball JD, et al. GD2-specific CAR T cells undergo potent activation and deletion following antigen encounter but can be protected from activation-induced cell death by PD-1 blockade. Mol Ther (2016) 24:1135–49. doi: 10.1038/mt.2016.63
65. Ping Y, Liu C, Zhang Y. T-Cell receptor-engineered T cells for cancer treatment: current status and future directions. Protein Cell (2018) 9:254–66. doi: 10.1007/s13238-016-0367-1
66. June CH, O'Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science (2018) 359:1361–5. doi: 10.1126/science.aar6711
67. Shafer P, Kelly LM, Hoyos V. Cancer therapy with TCR-engineered T cells: Current strategies. Challenges Prospects Front Immunol (2022) 13:835762. doi: 10.3389/fimmu.2022.835762
68. Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, Toy ST, et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. a preliminary report. N Engl J Med (1988) 319:1676–80. doi: 10.1056/NEJM198812223192527
69. Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res (2011) 17:4550–7. doi: 10.1158/1078-0432.Ccr-11-0116
70. Dudley ME, Wunderlich JR, Yang JC, Sherry RM, Topalian SL, Restifo NP, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol (2005) 23:2346–57. doi: 10.1200/JCO.2005.00.240
71. Pilon-Thomas S, Kuhn L, Ellwanger S, Janssen W, Royster E, Marzban S, et al. Efficacy of adoptive cell transfer of tumor-infiltrating lymphocytes after lymphopenia induction for metastatic melanoma. J Immunother (2012) 35:615–20. doi: 10.1097/CJI.0b013e31826e8f5f
72. Goff SL, Smith FO, Klapper JA, Sherry R, Wunderlich JR, Steinberg SM, et al. Tumor infiltrating lymphocyte therapy for metastatic melanoma: analysis of tumors resected for TIL. J Immunother (2010) 33:840–7. doi: 10.1097/CJI.0b013e3181f05b91
73. Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science (2006) 313:1960–4. doi: 10.1126/science.1129139
74. Sanmamed MF, Nie X, Desai SS, Villaroel-Espindola F, Badri T, Zhao D, et al. A burned-out CD8(+) T-cell subset expands in the tumor microenvironment and curbs cancer immunotherapy. Cancer Discovery (2021) 11:1700–15. doi: 10.1158/2159-8290.CD-20-0962
75. Hall M, Liu H, Malafa M, Centeno B, Hodul PJ, Pimiento J, et al. Expansion of tumor-infiltrating lymphocytes (TIL) from human pancreatic tumors. J Immunother Cancer (2016) 4:61. doi: 10.1186/s40425-016-0164-7
76. Wang W, Jiang J, Wu C. CAR-NK for tumor immunotherapy: Clinical transformation and future prospects. Cancer Lett (2020) 472:175–80. doi: 10.1016/j.canlet.2019.11.033
77. Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med (2020) 382:545–53. doi: 10.1056/NEJMoa1910607
78. Hu Y, Tian Z, Zhang C. Natural killer cell-based immunotherapy for cancer: Advances and prospects. Engineering (2019) 5:106–14. doi: 10.1016/j.eng.2018.11.015
79. Liu M, Meng Y, Zhang L, Han Z, Feng X. High-efficient generation of natural killer cells from peripheral blood with preferable cell vitality and enhanced cytotoxicity by combination of IL-2, IL-15 and IL-18. Biochem Biophys Res Commun (2021) 534:149–56. doi: 10.1016/j.bbrc.2020.12.012
80. Romee R, Rosario M, Berrien-Elliott MM, Wagner JA, Jewell BA, Schappe T, et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med (2016) 8:357ra123. doi: 10.1126/scitranslmed.aaf2341
81. Ni J, Miller M, Stojanovic A, Garbi N, Cerwenka A. Sustained effector function of IL-12/15/18-preactivated NK cells against established tumors. J Exp Med (2012) 209:2351–65. doi: 10.1084/jem.20120944
82. Bommareddy PK, Shettigar M, Kaufman HL. Integrating oncolytic viruses in combination cancer immunotherapy. Nat Rev Immunol (2018) 18:498–513. doi: 10.1038/s41577-018-0014-6
83. Raja J, Ludwig JM, Gettinger SN, Schalper KA, Kim HS. Oncolytic virus immunotherapy: future prospects for oncology. J Immunother Cancer (2018) 6:140. doi: 10.1186/s40425-018-0458-z
84. Hemminki O, Dos Santos JM, Hemminki A. Oncolytic viruses for cancer immunotherapy. J Hematol Oncol (2020) 13:84. doi: 10.1186/s13045-020-00922-1
85. Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discovery (2015) 14:642–62. doi: 10.1038/nrd4663
86. Rosewell Shaw A, Suzuki M. Oncolytic viruses partner With T-cell therapy for solid tumor treatment. Front Immunol (2018) 9:2103. doi: 10.3389/fimmu.2018.02103
87. Ylösmäki E, Cerullo V. Design and application of oncolytic viruses for cancer immunotherapy. Curr Opin Biotechnol (2020) 65:25–36. doi: 10.1016/j.copbio.2019.11.016
88. Macedo N, Miller DM, Haq R, Kaufman HL. Clinical landscape of oncolytic virus research in 2020. J Immunother Cancer (2020) 8:e001486. doi: 10.1136/jitc-2020-001486
89. Watanabe N, McKenna MK, Rosewell Shaw A, Suzuki M. Clinical CAR-T cell and oncolytic virotherapy for cancer treatment. Mol Ther (2021) 29(2):505–20. doi: 10.1016/j.ymthe.2020.10.023
90. Roy DG, Bell JC. Cell carriers for oncolytic viruses: current challenges and future directions. Oncolytic Virother (2013) 2:47–56. doi: 10.2147/OV.S36623
91. Gao Q, Zhu H, Dong L, Shi W, Chen R, Song Z, et al. Integrated proteogenomic characterization of HBV-related hepatocellular carcinoma. Cell (2019) 179:1240. doi: 10.1016/j.cell.2019.10.038
92. Ren L, Zeng M, Tang Z, Li M, Wang X, Xu Y, et al. The antiresection activity of the X protein encoded by hepatitis virus b. Hepatology (2019) 69:2546–61. doi: 10.1002/hep.30571
93. Chen F, Zou Z, Du J, Su S, Shao J, Meng F, et al. Neoantigen identification strategies enable personalized immunotherapy in refractory solid tumors. J Clin Invest (2019) 129:2056–70. doi: 10.1172/JCI99538
94. Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature (2017) 547:217–21. doi: 10.1038/nature22991
95. Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Löwer M, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature (2017) 547:222–6. doi: 10.1038/nature23003
96. Herrera ZM, Ramos TC. Pilot study of a novel combination of two therapeutic vaccines in advanced non-small-cell lung cancer patients. Cancer Immunol Immunother (2014) 63:737–47. doi: 10.1007/s00262-014-1552-9
97. Saxena M, van der Burg SH, Melief CJM, Bhardwaj N. Therapeutic cancer vaccines. Nat Rev Cancer (2021) 21:360–78. doi: 10.1038/s41568-021-00346-0
98. Couzin-Frankel J. Breakthrough of the year 2013. Cancer immunotherapy Sci (2013) 342:1432–3. doi: 10.1126/science.342.6165.1432
99. Yaguchi T, Kawakami Y. Cancer-induced heterogeneous immunosuppressive tumor microenvironments and their personalized modulation. Int Immunol (2016) 28:393–9. doi: 10.1093/intimm/dxw030
100. Liu D, Jenkins RW, Sullivan RJ. Mechanisms of resistance to immune checkpoint blockade. Am J Clin Dermatol (2019) 20:41–54. doi: 10.1007/s40257-018-0389-y
101. Jia H, Truica CI, Wang B, Wang Y, Ren X, Harvey HA, et al. Immunotherapy for triple-negative breast cancer: Existing challenges and exciting prospects. Drug Resist Update (2017) 32:1–15. doi: 10.1016/j.drup.2017.07.002
102. Hu Q, Ye Y, Chan LC, Li Y, Liang K, Lin A, et al. Oncogenic lncRNA downregulates cancer cell antigen presentation and intrinsic tumor suppression. Nat Immunol (2019) 20:835–51. doi: 10.1038/s41590-019-0400-7
103. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science (2015) 348:124–8. doi: 10.1126/science.aaa1348
104. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science (2015) 348:69–74. doi: 10.1126/science.aaa4971
105. Liu C, Peng W, Xu C, Lou Y, Zhang M, Wargo JA, et al. BRAF inhibition increases tumor infiltration by T cells and enhances the antitumor activity of adoptive immunotherapy in mice. Clin Cancer Res (2013) 19:393–403. doi: 10.1158/1078-0432.CCR-12-1626
106. Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discovery (2016) 6:202–16. doi: 10.1158/2159-8290.Cd-15-0283
107. Zhang X, Zeng Y, Qu Q, Zhu J, Liu Z, Ning W, et al. PD-L1 induced by IFN-γ from tumor-associated macrophages via the JAK/STAT3 and PI3K/AKT signaling pathways promoted progression of lung cancer. Int J Clin Oncol (2017) 22:1026–33. doi: 10.1007/s10147-017-1161-7
108. Steinhart Z, Pavlovic Z, Chandrashekhar M, Hart T, Wang X, Zhang X, et al. Genome-wide CRISPR screens reveal a wnt-FZD5 signaling circuit as a druggable vulnerability of RNF43-mutant pancreatic tumors. Nat Med (2017) 23:60–8. doi: 10.1038/nm.4219
109. Spranger S, Dai D, Horton B, Gajewski TF. Tumor-residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell (2017) 31:711–723.e714. doi: 10.1016/j.ccell.2017.04.003
110. Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature (2015) 523:231–5. doi: 10.1038/nature14404
111. Satoh K, Yachida S, Sugimoto M, Oshima M, Nakagawa T, Akamoto S, et al. Global metabolic reprogramming of colorectal cancer occurs at adenoma stage and is induced by MYC. Proc Natl Acad Sci U.S.A. (2017) 114:E7697–e7706. doi: 10.1073/pnas.1710366114
112. Cascone T, McKenzie JA, Mbofung RM, Punt S, Wang Z, Xu C, et al. Increased tumor glycolysis characterizes immune resistance to adoptive T cell therapy. Cell Metab (2018) 27:977–987.e974. doi: 10.1016/j.cmet.2018.02.024
113. Commeren DL, Van Soest PL, Karimi K, Löwenberg B, Cornelissen JJ, Braakman E. Paradoxical effects of interleukin-10 on the maturation of murine myeloid dendritic cells. Immunology (2003) 110:188–96. doi: 10.1046/j.1365-2567.2003.01730.x
114. Highfill SL, Cui Y, Giles AJ, Smith JP, Zhang H, Morse E, et al. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci Transl Med (2014) 6:237ra267. doi: 10.1126/scitranslmed.3007974
115. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol (2017) 14:399–416. doi: 10.1038/nrclinonc.2016.217
116. Kuang DM, Zhao Q, Peng C, Xu J, Zhang JP, Wu C, et al. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J Exp Med (2009) 206:1327–37. doi: 10.1084/jem.20082173
117. Kryczek I, Zou L, Rodriguez P, Zhu G, Wei S, Mottram P, et al. B7-H4 expression identifies a novel suppressive macrophage population in human ovarian carcinoma. J Exp Med (2006) 203:871–81. doi: 10.1084/jem.20050930
118. Schachter J, Ribas A, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus ipilimumab for advanced melanoma: final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006). Lancet (2017) 390:1853–62. doi: 10.1016/S0140-6736(17)31601-X
119. Yaguchi T, Kobayashi A, Inozume T, Morii K, Nagumo H, Nishio H, et al. Human PBMC-transferred murine MHC class I/II-deficient NOG mice enable long-term evaluation of human immune responses. Cell Mol Immunol (2018) 15:953–62. doi: 10.1038/cmi.2017.106
120. Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med (2016) 375:819–29. doi: 10.1056/NEJMoa1604958
121. Sade-Feldman M, Jiao YJ, Chen JH, Rooney MS, Barzily-Rokni M, Eliane JP, et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun (2017) 8:1136. doi: 10.1038/s41467-017-01062-w
122. Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, et al. Loss of IFN-γ pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell (2016) 167:397–404.e399. doi: 10.1016/j.cell.2016.08.069
123. Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, Jia L, et al. T-Cell transfer therapy targeting mutant KRAS in cancer. N Engl J Med (2016) 375:2255–62. doi: 10.1056/NEJMoa1609279
124. van Rooij N, van Buuren MM, Philips D, Velds A, Toebes M, Heemskerk B, et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol (2013) 31:e439–442. doi: 10.1200/JCO.2012.47.7521
125. Explaining resistance to CAR T cells. Cancer Discovery (2018) 8:784–5. doi: 10.1158/2159-8290.CD-NB2018-065
126. Majzner RG, Heitzeneder S, Mackall CL. Harnessing the immunotherapy revolution for the treatment of childhood cancers. Cancer Cell (2017) 31:476–85. doi: 10.1016/j.ccell.2017.03.002
127. Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: Co-inhibitory receptors with specialized functions in immune regulation. Immunity (2016) 44:989–1004. doi: 10.1016/j.immuni.2016.05.001
128. Ninomiya S, Narala N, Huye L, Yagyu S, Savoldo B, Dotti G, et al. Tumor indoleamine 2,3-dioxygenase (IDO) inhibits CD19-CAR T cells and is downregulated by lymphodepleting drugs. Blood (2015) 125:3905–16. doi: 10.1182/blood-2015-01-621474
129. Moon EK, Wang LC, Dolfi DV, Wilson CB, Ranganathan R, Sun J, et al. Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin Cancer Res (2014) 20:4262–73. doi: 10.1158/1078-0432.CCR-13-2627
130. Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med (2015) 372:2006–17. doi: 10.1056/NEJMoa1414428
131. Larkin J, Hodi FS, Wolchok JD. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med (2015) 373:1270–1. doi: 10.1056/NEJMc1509660
132. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Lao CD, et al. Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med (2019) 381:1535–46. doi: 10.1056/NEJMc1509660
133. Liu F, Zeng G, Zhou S, He X, Sun N, Zhu X, et al. Blocking Tim-3 or/and PD-1 reverses dysfunction of tumor-infiltrating lymphocytes in HBV-related hepatocellular carcinoma. Bull Cancer (2018) 105:493–501. doi: 10.1016/j.bulcan.2018.01.018
134. Huang RY, Eppolito C, Lele S, Shrikant P, Matsuzaki J, Odunsi K. LAG3 and PD1 co-inhibitory molecules collaborate to limit CD8+ T cell signaling and dampen antitumor immunity in a murine ovarian cancer model. Oncotarget (2015) 6:27359–77. doi: 10.18632/oncotarget.4751
135. Johnston RJ, Comps-Agrar L, Hackney J, Yu X, Huseni M, Yang Y, et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell (2014) 26:923–37. doi: 10.1016/j.ccell.2014.10.018
136. Pauken KE, Wherry EJ. Overcoming T cell exhaustion in infection and cancer. Trends Immunol (2015) 36:265–76. doi: 10.1016/j.it.2015.02.008
137. Rios-Doria J, Durham N, Wetzel L, Rothstein R, Chesebrough J, Holoweckyj N, et al. Doxil synergizes with cancer immunotherapies to enhance antitumor responses in syngeneic mouse models. Neoplasia (2015) 17:661–70. doi: 10.1016/j.neo.2015.08.004
138. Banissi C, Ghiringhelli F, Chen L, Carpentier AF. Treg depletion with a low-dose metronomic temozolomide regimen in a rat glioma model. Cancer Immunol Immunother (2009) 58:1627–34. doi: 10.1007/s00262-009-0671-1
139. Song W, Shen L, Wang Y, Liu Q, Goodwin TJ, Li J, et al. Synergistic and low adverse effect cancer immunotherapy by immunogenic chemotherapy and locally expressed PD-L1 trap. Nat Commun (2018) 9:2237. doi: 10.1038/s41467-018-04605-x
140. Langer CJ, Gadgeel SM, Borghaei H, Papadimitrakopoulou VA, Patnaik A, Powell SF, et al. Carboplatin and pemetrexed with or without pembrolizumab for advanced, non-squamous non-small-cell lung cancer: a randomised, phase 2 cohort of the open-label KEYNOTE-021 study. Lancet Oncol (2016) 17:1497–508. doi: 10.1016/S1470-2045(16)30498-3
141. Deng L, Liang H, Burnette B, Beckett M, Darga T, Weichselbaum RR, et al. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J Clin Invest (2014) 124:687–95. doi: 10.1172/JCI67313
142. Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature (2015) 520:373–7. doi: 10.1038/nature14292
143. Pilones KA, Aryankalayil J, Babb JS, Demaria S. Invariant natural killer T cells regulate anti-tumor immunity by controlling the population of dendritic cells in tumor and draining lymph nodes. J Immunother Cancer (2014) 2:37. doi: 10.1186/s40425-014-0037-x
144. DeSelm C, Palomba ML, Yahalom J, Hamieh M, Eyquem J, Rajasekhar VK, et al. Low-dose radiation conditioning enables CAR T cells to mitigate antigen escape. Mol Ther (2018) 26:2542–52. doi: 10.1016/j.ymthe.2018.09.008
145. Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med (2012) 367:1694–703. doi: 10.1056/NEJMoa1210093
146. Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med (2004) 350:2129–39. doi: 10.1056/NEJMoa040938
147. Cooper ZA, Juneja VR, Sage PT, Frederick DT, Piris A, Mitra D, et al. Response to BRAF inhibition in melanoma is enhanced when combined with immune checkpoint blockade. Cancer Immunol Res (2014) 2:643–54. doi: 10.1158/2326-6066.CIR-13-0215
148. Du Four S, Maenhout SK, Niclou SP, Thielemans K, Neyns B, Aerts JL. Combined VEGFR and CTLA-4 blockade increases the antigen-presenting function of intratumoral DCs and reduces the suppressive capacity of intratumoral MDSCs. Am J Cancer Res (2016) 6:2514–31.
149. Wu X, Li J, Connolly EM, Liao X, Ouyang J, Giobbie-Hurder A, et al. Combined anti-VEGF and anti-CTLA-4 therapy elicits humoral immunity to galectin-1 which is associated with favorable clinical outcomes. Cancer Immunol Res (2017) 5:446–54. doi: 10.1158/2326-6066.CIR-16-0385
150. Li W, Zhan M, Quan YY, Wang H, Hua SN, Li Y, et al. Modulating the tumor immune microenvironment with sunitinib malate supports the rationale for combined treatment with immunotherapy. Int Immunopharmacol (2020) 81:106227. doi: 10.1016/j.intimp.2020.106227
151. Li H, Ding J, Lu M, Liu H, Miao Y, Li L, et al. CAIX-specific CAR-T cells and sunitinib show synergistic effects against metastatic renal cancer models. J Immunother (2020) 43:16–28. doi: 10.1097/CJI.0000000000000301
152. Suarez ER, Chang de K, Sun J, Sui J, Freeman GJ, Signoretti S, et al. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget (2016) 7:34341–55. doi: 10.18632/oncotarget.9114
153. John LB, Devaud C, Duong CP, Yong CS, Beavis PA, Haynes NM, et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res (2013) 19:5636–46. doi: 10.1158/1078-0432.CCR-13-0458
154. Nanda VGY, Peng W, Hwu P, Davies MA, Ciliberto G, Fattore L, et al. Melanoma and immunotherapy bridge 2015 : Naples, italy. 1-5 December 2015. J Transl Med (2016) 14:65. doi: 10.1186/s12967-016-0791-2
155. Takeda Y, Kataoka K, Yamagishi J, Ogawa S, Seya T, Matsumoto M. A TLR3-specific adjuvant relieves innate resistance to PD-L1 blockade without cytokine toxicity in tumor vaccine immunotherapy. Cell Rep (2017) 19:1874–87. doi: 10.1016/j.celrep.2017.05.015
156. Ager CR, Reilley MJ, Nicholas C, Bartkowiak T, Jaiswal AR, Curran MA. Intratumoral STING activation with T-cell checkpoint modulation generates systemic antitumor immunity. Cancer Immunol Res (2017) 5:676–84. doi: 10.1158/2326-6066.CIR-17-0049
157. Liang Y, Tang H, Guo J, Qiu X, Yang Z, Ren Z, et al. Targeting IFNα to tumor by anti-PD-L1 creates feedforward antitumor responses to overcome checkpoint blockade resistance. Nat Commun (2018) 9:4586. doi: 10.1038/s41467-018-06890-y
158. Hu B, Zhou Y, Sun D, Yang Y, Liu Y, Li X, et al. PROTACs: New method to degrade transcription regulating proteins. Eur J Med Chem (2020) 207:112698. doi: 10.1016/j.ejmech.2020.112698
159. Hu M, Zhou W, Wang Y, Yao D, Ye T, Yao Y, et al. Discovery of the first potent proteolysis targeting chimera (PROTAC) degrader of indoleamine 2,3-dioxygenase 1. Acta Pharm Sin B (2020) 10:1943–53. doi: 10.1016/j.apsb.2020.02.010
160. Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother (2013) 36:133–51. doi: 10.1097/CJI.0b013e3182829747
Keywords: immunotherapy, tumor microenvironment, immune checkpoint inhibitors, CAR-T, cancer vaccines
Citation: Liu C, Yang M, Zhang D, Chen M and Zhu D (2022) Clinical cancer immunotherapy: Current progress and prospects. Front. Immunol. 13:961805. doi: 10.3389/fimmu.2022.961805
Received: 05 June 2022; Accepted: 26 September 2022;
Published: 11 October 2022.
Edited by:
Catherine Sautes-Fridman, INSERM U1138 Centre de Recherche des Cordeliers (CRC), FranceReviewed by:
Hongji Zhang, University of Virginia, United StatesFiras Hamdan, University of Maryland, Baltimore, United States
Kohichi Takada, Sapporo Medical University, Japan
Copyright © 2022 Liu, Yang, Zhang, Chen and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Di Zhu, emh1ZGlAZnVkYW4uZWR1LmNu
†ORCID: Chenglong Liu, orcid.org/0000-0002-1657-6683
Di Zhu, orcid.org/0000-0003-4622-4600