 Haein Huh1
Haein Huh1 Ding-Wen Chen
Ding-Wen Chen Jonathan Blay
Jonathan Blay- 1School of Pharmacy, University of Waterloo, Waterloo, ON, Canada
- 2Department of Pathology, Dalhousie University, Halifax, NS, Canada
Introduction: Colorectal cancer and other adult solid cancers pose a significant challenge for successful treatment because the tumor microenvironment both hinders the action of conventional therapeutics and suppresses the immune activities of infiltrating leukocytes. The immune suppression is largely the effect of enhanced local mediators such as purine nucleosides and eicosanoids. Genetic approaches have the promise of interfering with these mechanisms of local immunosuppression to allow both intrinsic and therapeutic immunological anticancer processes. Bacterial phages offer a novel means of enabling access into tissues for therapeutic genetic manipulations.
Methods: We generated spheroids of fibroblastic and CRC cancer cells to model the 3-dimensional stromal and parenchymal components of colorectal tumours. We used these to examine the access and effects of both wildtype (WT) and epidermal growth factor (EGF)-presenting bacteriophage λ (WT- λ and EGF-λ) as a means of delivery of targeted genetic interventions in solid cancers. We used both confocal microscopy of spheroids exposed to AF488-tagged phages, and the recovery of viable phages as measured by plaque-forming assays to evaluate access; and measures of mitochondrial enzyme activity and cellular ATP to evaluate the outcome on the constituent cells.
Results: Using flourescence-tagged derivatives of these bacteriophages (AF488-WT-λ and AF488-EGF-λ) we showed that phage entry into these tumour microenvironments was possible and that the EGF ligand enabled efficient and persistent uptake into the cancer cell mass. EGF-λ became localized in the intracellular portion of cancer cells and was subjected to subsequent cellular processing. The targeted λ phage had no independent effect upon mature tumour spheroids, but interfered with the early formation and growth of cancer tissues without the need for addition of a toxic payload, suggesting that it might have beneficial effects by itself in addition to any genetic intervention delivered to the tumour. Interference with spheroid formation persisted over the duration of culture.
Discussion: We conclude that targeted phage technology is a feasible strategy to facilitate delivery into colorectal cancer tumour tissue (and by extension other solid carcinomas) and provides an appropriate delivery vehicle for a gene therapeutic that can reduce local immunosuppression and/or deliver an additional direct anticancer activity.
Introduction
The local tumor microenvironment (TME) of a solid tumor acts to suppress anti-tumor immune responses. This is the consequence both of abnormal tissue architecture and altered cellular metabolism, particularly a tendency toward regional hypoxia as a result of the compromised vasculature (1–3).
Two of the most significant families of local mediators in this context are those deriving from purine and eicosanoid metabolism (4, 5). These two networks overlap in their impact on the factors that determine the anti-tumor immune response (6). In the case of purine metabolism, extracellular nucleotides and nucleosides released as a result of altered metabolism and enzyme levels have substantial effects on both the tumor cells and the anti-tumor immune response. The tumor microenvironment has high concentrations of adenosine (7), produced both by alteration of cellular metabolic pathways due to local hypoxia (1) and the dephosphorylation of ATP released by dying cells (8). The adenosine is produced as a result of the ectonucleotidase activities of CD73 and CD39, which may be more highly expressed in cancer (8, 9). Adenosine production is facilitated by a variety of cells within the TME, including tumor cells, regulatory T cells, Th17 cells, myeloid cells, resident mesenchymal stem cells and stromal cells (8, 10, 11).
Whereas extracellular ATP has positive effects against the tumor, adenosine has substantial pro-tumor actions (12, 13). Adenosine enhances tumor growth by acting on the cancer cells (9, 14, 15), stroma (9) and tumor vasculature (9, 16). It also favors metastasis by promoting epithelial-mesenchymal transition, migration, and invasion in cancer cells (17). We have shown that the chemokine receptor CXCR4, which regulates leukocytic and stem cell homing and cancer metastasis (18–20), and the multifunctional regulator DPP4, which controls the CXCR4 ligand CXCL12 (21, 22) are regulated by adenosine (23, 24) and selected eicosanoids (25) through pathways that we have identified (26–28).
Much of adenosine’s impact though is due to substantial local immunosuppressive actions (12). The tumor immunosuppressive effects of adenosine apply to both adaptive (29) and innate immunity (30). Adenosine-producing Treg cells are one of the several mechanisms of tumor escape from the immune system (31). Adenosine is regarded to constitute a metabolic immune checkpoint (32).
Suppression of the adenosine’s effects may be a valuable adjunct to immunotherapies in improving cancer immunotherapy (33). Targeting adenosine pathways may be useful for both solid tumours (34) and lymphocytic leukemias for which the levels of extracellular adenosine are raised (35). The actions of adenosine may be blocked either by reducing its production or by interfering with the discrete receptors through which adenosine acts on different cells, including A2a, A2b and A3 receptors (11, 36–42). The conventional approach of using systemic agents to antagonize adenosine receptors or ectonucleotidases raises a major concern, however. This is a primitive host regulatory network, and adenosine and its receptors are involved in control mechanisms throughout the body, including critical cardiovascular and central nervous system functions (16, 43). We therefore favor locally-directly ablation of excess adenosine production to remove both the stimulatory effects on the tumor population (9, 14, 15) and immunosuppression (28, 29, 39, 42). Gene therapeutic approaches are well established but have their limitations. We favor exploiting a different and novel platform, based on common inhabitants of the gut microbiome.
Bacteriophages (‘phages’) offer an alternative antibacterial strategy to conventional antimicrobial drugs and may be of enhanced value in the current antimicrobial drug-resistant era (44–48). They may be useful in combination with existing antibacterial agents (46, 49). Furthermore, their ease of manipulation and greater understanding of the properties that favor phage therapy success have raised the possibility of uses beyond infectious disease (50–52).
With the advent of phage display systems (53, 54) a wide range of macromolecular ligands may be fused to the lytic phage capsid allowing for targeted delivery of therapeutic payloads (52, 55). This technology has been applied in the design of diverse therapeutic agents (56–60) and against a range of clinical objectives (60–64). It is especially useful in the field of cancer biology, in which many delivery systems are challenged by imprecise targeting and poor penetration into the tumor interstitium (65–68).
The complex pathophysiology of the tumor microenvironment has hindered the efficient delivery of high molecular weight, targeted antineoplastic agents. Systemically-administered, targeted anti-cancer drugs need to enter the tumor vasculature network, extravasate efficiently into the interstitial space and reach a majority of the tumor cells at a pharmacologically-effective dose (66, 69–74). Achieving this has proven challenging due to the high tumor interstitial fluid pressure and an irregular vasculature that leads to tumor heterogeneity with hypoxic regions (75). Gradients of nutrients and oxygen concentrations exist in the microenvironment, resulting in more active and proliferative cells being situated closer to the vasculature. However, the tumor microenvironment becomes hypoxic (<5 mmHg pO2) at a distance of 70-80 µm from a blood vessel, and anoxic at 150 µm, rendering cells in these distal regions progressively more resistant to drugs that affect the cell cycle or depend on redox mechanisms (76, 77). There are also physical barriers within the tumor interstitium that may impede penetration of therapeutic drugs, such as an abnormal extracellular matrix (ECM). The fibrous protein collagen, in particular, increases in deposition with tumor progression (78, 79). Collectively, these factors constrain many anticancer agents to peripheral areas of the tumor with limited access to cells in the inner regions (67, 77, 80–83).
Phages offer advantages over both current viral and non-viral systems of targeted delivery. Their inability to multiply within human cells vastly favors phages over modified human pathogenic viruses. Moreover, in contrast to the latter oncolytic viruses, phages are unlikely to undergo alterations in their tropism, which is a major concern with the exploitation of viruses for anticancer therapy (84). In addition, phages have the ability to penetrate through epithelium and endothelium and accumulate in various mammalian tissues and organs (85, 86) as well as the ability to bypass the blood-brain barrier (85) and the gut mucosal barrier (87, 88). Vectorial transcytosis across epithelial cells has been described whereby phages can cross an epithelial cell in an apical-to-basal direction (89, 90).
The potential utility for phages is increased further as they can be engineered to enhance their application and efficacy profile (55). Tropism for specific cells can be conferred by decoration of cell-targeting ligands to phage capsid surface proteins (53). In this strategy, the gene of interest is translationally fused to a capsid gene of the phage to produce a projecting targeting ligand (91). This same phage display approach was also used early on in cancer therapy to uncover tumor-homing peptides from a library of phages (54, 92).
The cellular barriers, fluid dynamics and structure of the ECM are different between normal tissues and the tissue of a solid tumor (79, 80, 93). Therefore, there is a need to delineate the distinct capacities of phages to penetrate in an intact and functional form through both the authentic tumor interstitium and the neoplastic parenchyma. In this study we use multicellular cellular aggregates (commonly known as ‘spheroids’) to reproduce the two different local tissue environments that the phage will need to navigate in order to deliver a cancer therapeutic payload in a patient.
Colorectal adenocarcinoma (CRC) is the most common form of large bowel cancer in humans. It afflicts older patients but is showing an increasing facility to affect younger individuals, with a 7%/year increase among 15-29 year-olds in Canada (94). Our goal in this study is to understand the potential for phage to enter and deliver a genetic therapeutic to confer an antineoplastic hit in CRC. The ability to target this cancer with a strategy that does not depend on conventional mutagenic cytoxic agents also reduces the chance of secondary cancers that occur in younger patients many years post-CRC chemotherapy (95).
Firstly, we generated a spheroid model using fibroblasts, where the cellular arrangement and secreted ECM recapitulate the tight connective tissue stroma that surrounds and separates areas of neoplastic cells. Secondly, we generated human colorectal adenocarcinoma cell spheroids to model the structure of the CRC cancer parenchyma. We investigated phage access in these 3-dimensional tissue models of the CRC stroma and parenchyma with λ phage, both unmodified and with epidermal growth factor (EGF) displayed as a targeting ligand for the EGF receptor on CRC tumor cells. We first sought to understand whether unmodified phage λ can access and diffuse within multicellular spheroids generated from NIH3T3 mouse fibroblasts, representing the stroma in a solid tumor microenvironment. EGF-displaying phages were then studied to observe their interaction and targeted effect within spheroids formed from HT-29 cells, human colon adenocarcinoma cells that we have studied extensively for their abilities to respond to anticancer drugs, natural products and mediators in the tumor microenvironment (22, 23, 25, 27, 96, 97) and which are known to overexpress EGFR (98, 99).
Methods
Spheroid culture
NIH3T3 and HT-29 cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, VA. USA), have been confirmed for genotype, and cultures were regularly tested for mycoplasma using a PCR-based approach and found to be negative for contamination. Cells were grown routinely in T25 cell culture flasks (Nunclon Delta™, ThermoFisher, Mississauga, ON) with Dulbecco’s modified Eagle medium (DMEM, HyClone™, ThermoFisher) containing 4mM L-glutamine and 4500mg/L glucose, 1mM sodium pyruvate, and 5% v/v fetal bovine serum (FBS) and incubated in a humidified atmosphere at 37°C and 90% air/10% CO2. Culture medium was replaced every 3 d. At 90% confluency, NIH3T3 or HT-29 monolayers were trypsinized (0.25% trypsin-EDTA) into single cell suspensions, rinsed and cells were counted with a hematocytometer (Fisher Scientific).
Spheroids were grown as previously described (97). Unless otherwise stated, NIH3T3 fibroblast spheroids were generated from 3 x 104 cells for initial seeding and were cultured for a total of 10 d with medium supplementation as needed. HT-29 colon adenocarcinoma spheroids were generated from 5 x 103 cells and were cultured for 7 d. Spheroids were observed with a bright field inverted microscope (Nikon® TE200), and images were captured using a Micropublisher 5.0 RTV camera with QCapture Pro™ 7 software.
Generation of EGF-displaying λ bacteriophages
Phage λ possesses approximately 415 copies of the major coat protein gpE and between 405-420 copies of gpD on the capsid surface, of which gpD is robustly tolerant of peptide fusions (53, 100). Amber suppressor strains (SupD, E, F) of W3101 E. coli were constructed as described (101) (Supplementary Table S1). Briefly, these isogenic suppressor strains of E. coli were transformed with a multicopy plasmid pHH1 (pPL451-gpD::EGF) to allow for the expression of D::EGF. The plasmid pPL451-gpD::eGFP was as described, whereby a multicopy plasmid pPL451 contained the D::eGFP sequence linked by an in-frame short linker of amino acids TSGSGSGSGSGT and a KpnI cut site (101, 102). In order to construct the multicopy plasmid pHH1, the sequence encoding eGFP was exchanged for the gene sequence of human epidermal growth factor [BC093731.1 (103)]. This encoded a translated C-terminal EGF translational fusion with the gpD capsid gene.
Isogenic W3101 SupD and SupF amber suppressor E. coli cells were transformed by the multicopy plasmid [pPL451-gpD::EGF] by the calcium heat shock system. The D::EGF fusion is under the control of a temperature-sensitive λ CI857 repressor that induces gene expression at 37 °C and also confers ampicillin resistance. Transformed colonies were selected on LB ampicillin agar plates. BB4 (supE, supF) E. coli is a double amber suppressor in which λF7 phage does not rely on the fusion plasmid for a functional copy of gene D. This was used as a positive control for efficiency of plating and as a negative control for EGF decoration.
EGFHi phages – λF7 phage with maximally decorated surfaces – were generated at higher temperatures of 37°C using SupD E. coli amber suppressor host for propagation (Supplementary Table S2). Conversely, EGFLo were generated with SupF E. coli strains, resulting in reduced complementation from the plasmid expressing the D translational fusion with overall minimal surface decoration. Previous fluorometric analysis of gpD::eGFP decorated phages estimated approximately 147 molecules per phage for the maximal fusion (SupD, 37°C), and 89 molecules per phage generated from SupF at 37°C, from which we can infer that the decoration of EGFLo phages will be approximately 40% less than EGFHi phages (101).
Transformed SupD and SupF E. coli cells were grown in LB broth over night at 37°C. 10-fold dilutions of primary lysates were prepared in 1 ml of TN buffer and were added to 0.3 ml of the transformed cells (grown to OD600 = 0.4 at 37°C), then incubated overnight at 37°C. The next day, 10 ml of TN buffer were added on to each plate and incubated for 24 h at 4°C. The top agar was scraped and transferred to a conical tube and was centrifuged at 23,000 x g at 4°C for 20 min (Avanti J-E Centrifuge, Beckman Coulter, Mississauga Canada). Negative control λF7 phages generated with a double suppressor BB4 E. coli host that did not have surface decoration (wildtype, WT) were prepared in parallel. Phage lysates were filtered through a sterile 0.2 µm syringe filter to remove cellular debris, followed by polyethylene glycol (PEG)-precipitation (Sigma-Aldrich). Precipitated phages were resuspended in DMEM supplemented by 5% FBS and were further purified through a gel chromatography. Phage samples were standardized to a concentration of 109 PFU/ml. Unless otherwise stated, 100 µl of each sample was added to each spheroid for a total of 108 PFU.
Plating assays showed a decline in phage infectivity of bacterial targets with time of incubation at 37°C (Supplementary Table S3). However, significant (~30%) infectivity was retained for both native and EGF-displaying λF7 even at the longest time period of 48 h at 37°C. Experimental data were corrected for the change in intrinsic infectivity at 37°C as noted.
λ phage titration and standard plaque-forming unit assays
Soft-agar overlay technique was used on fresh BB4 E. coli cells, as this strain generates the highest titers of λF7. A liquid culture of BB4 (300 µl) was added to 3 ml of molten top agar (Bacto Tryptone and Bacto Agar from Difco Laboratories, Sparks, MD), as well as a 100 µl volume of phage dilution in suspension.
Fluorophore labeling of λ phage and visualization using confocal microscopy
The primary amines of wildtype λ phage or EGF-displaying phage capsids were conjugated with Alexa Fluor® 488 (Thermo Fisher) according to manufacturer’s instructions. Briefly, 1012 PFU of PEG-precipitated phages were resuspended in 95 µl of 0.1-M NaHCO3 buffer (pH 8.3) and were incubated with 5 µl of dye (10 mg/ml in dimethyl sulfoxide, DMSO) at room temperature for 1 h with brief vortexing every 10 min. The volume was then brought up to 1 ml and was centrifuged for 30 min at 23,000 x g at 4 °C and washed once. The unbound dye in the supernatant was replaced with DMEM and 5% FBS to resuspend the Alexa Fluor® 488-conjugated phages. 108 PFU of phages were added to HT-29 spheroids and the spheroids incubated at 37 °C for the times shown. HT-29 spheroids were stained with 10 µM of CellTracker Red CMPTX dye™ (Molecular Probes, Invitrogen) for 1 h at 37°C prior to visualization. Spheroids were washed thoroughly with phosphate-buffered saline (PBS; 137mM NaCl, 24.8mM Tris-HCL, 5mM KCL, 0.7mM Na2HPO4, 0.5mM MgSO4 and 1mM CaCl2; pH 7.2) then transferred onto a glass bottomed 24-well plate with No.0 coverslip (MatTek, Ashland, MA). Spheroids were imaged using Zeiss LSM710 laser scanning confocal microscope (Carl Zeiss AG, Oberkochen, Baden-Wü, Germany) with 20X objective. Z-stack images were captured using Zen 2009 software (Carl Zeiss AG) from the top of the sphere to the flat base of each spheroid.
Spheroid uptake and cellular internalization assay
Prior to the application of phages, HT-29 spheroids were treated with 5 mM of NH4Cl for 45 min at 23°C to prevent subsequent lysosomal degradation of internalized phages (this was not done in other assays). Spheroids were washed with PBS and treated with 108 PFU of phages at 37°C. Spheroids were then washed thoroughly (a total of 7 times) with PBS to remove unbound phages, and incubated with glycine (200 mM, pH 2.0) for 10 min at 4°C to inactivate the phages in situ. Following two further washes with PBS at 4°C the spheroids were completely dissociated in 100 μL of 0.25% EDTA-trypsin for 20 min at 37°C and pipetted thoroughly to dissociate the cells. Suspended cells were pelleted twice by centrifugation at 7,000 x g for 10 min, separating out the phages recovered from the intercellular spaces of the spheroids, which were recovered as the supernatant. Pelleted cells were then resuspended in 100 μL of 5 mM EDTA and subjected to 3 freeze/thaw cycles to release internalized phage particles. Residual debris was removed by further centrifugation and the supernatant set aside as the “intracellular” fraction. Phages were quantified for each fraction using the plaque assay as described above, and corrected for any intrinsic loss of infectivity due to incubation at 37°C.
Assessment of phage λ on spheroid growth
The impacts of EGF-λ and WT λ phages on the growth of HT-29 spheroids were assessed with brightfield illumination (Leica DM2000, 10X objective, Micropublisher 5.0 RTV camera) using QCapture Pro™ 7 Software. No phages were added to the control spheroids. Three separate conditions were tested with HT-29 cells and two different concentrations (104 or 108 PFU) of phages:
A. Phages were added to HT-29 cells at the time of addition onto agarose, with a pre-incubation of 4°C for 60 min, following spheroid growth to d 7;
B. Phages were added to HT-29 spheroids during the spheroid formation process after 48 h and the fate of spheroids followed out to d 20;
C. Phages were added to fully-mature spheroids at d 11 and the results followed to d 16.
MTT assay on monolayer cells
The response of HT-29 cells in monolayer culture to phages as a measure of direct cytotoxicity was assayed by the MTT assay (96). Briefly, cells were seeded in DMEM supplemented with 5% FBS in 96-well plates and incubated at 37°C, 10% CO2 for 72 h to allow for initial adherence. The monolayers were then treated with either 108 or 104 PFU of EGF-decorated or WT phages for 24 h at 37°C, 10% CO2. After addition of MTT the plate was incubated on a rotary shaker at 37°C for 1 h to allow for the metabolic conversion of MTT into formazan. The medium was then removed and formazan dissolved with 100 μl of DMSO per well for 30 min at room temperature. The absorbance of the plate was measured at A492 nm on a SpectraMax M5 Microplate Reader (Molecular Devices, San Jose, USA) with subtraction of the A492 of blank wells.
ATP assay on spheroids
The effect of phages on HT-29 spheroids as a measure of presumed cytotoxicity was carried out using the CellTiter-Glo® 3D (Promega) kit for 3D cell cultures according to the manufacturer’s directions. This test quantitates ATP within multicellular spheroids as an indication of the putative number of viable cells. Briefly, 4 mM of diaminocyclohexane triacetic acid was added to spheroids in multiwall plates at a 1:1 ratio with the culture medium, the contents were mixed vigorously for 5 min on an orbital shaker and then incubated at room temperature for additional 15 min. Half of the samples were transferred to a separate container and diluted by 10-fold in MilliQ H2O with repeated mixing to ensure homogeneity. Diluted samples were then transferred to a fresh assay well. An equal volume of CellTiter-Glo® 3D Reagent was added, and the contents were mixed for an additional 2 min on an orbital shaker. Finally, the plates were incubated at room temperature for 10 min to stabilize the luminescent signal, which was then recorded on the SpectraMax M5 Microplate Reader (Molecular Devices) using the luminescence mode.
Results
Phage λ infiltrates fibroblast spheroids
We first examined the ability of native (wildtype, WT) λ phages to interact with a tissue model of the tumor stroma, using Alexa AF488-tagged WT-λ phages and observing their time-dependent association with NIH3T3 fibroblast spheroids. The NIH3T3 spheroids were grown to 250-350 μm in diameter – up to the size threshold for which there may be authentic complicating effects of hypoxia in tissue due to inadequate perfusion of oxygen. AF488-WT-λ phages adhered to the fibroblast spheroids and accumulated to be resistant to washing, in a time-dependent manner (Figure 1). No significant fluorescence was visible after 30 min (Figure 1A), but by 6 h AF488-WT-λ particles were associated with the surface cells of the spheroid (Figure 1D), and by 24 h, the fluorescent phage showed a much more extensive association with the outer cell layers of the spheroid (Figure 1G). There was no significant fluorescence when NIH3T3 spheroids were exposed to either untagged WT λ phage or AF488 fluorophore alone, (Figures 1B, C, E, F, H, I respectively) indicating that the observed fluorescence reflected the tagged phage. Visualization of the dense core regions of the large spheroid was limited using confocal microscopy but infiltration of AF488-WT-λ phage between cells that were tightly juxtaposed within the spheroid was evident. AF488-WT-λ phage particles were evident on the peripheral cells even after spheroids had been thoroughly washed to discard weakly-bound phages, suggesting either high binding affinity or phage entrapment.
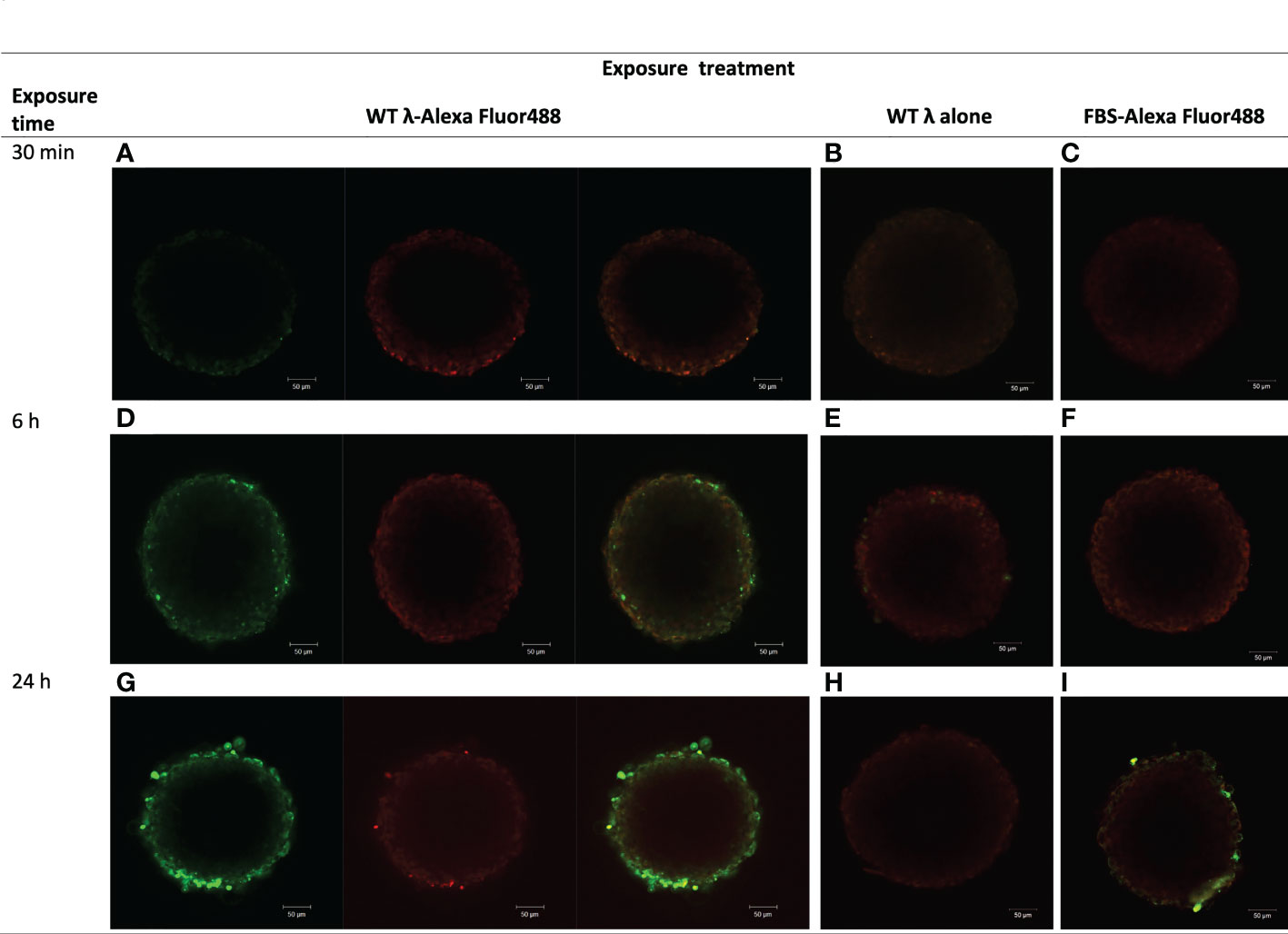
Figure 1 λ phages adhere to and accumulate in NIH3T3 fibroblast spheroids. NIH3T3 spheroids (d7, 250–350 μm in diameter) were exposed to either WT λ phages tagged with Alexa Fluor488 (A, D, G), untagged λ phages (B, E, H), or FBS tagged with Alexa Fluor488 (C, F, I) for the times shown. Spheroids were washed to discard unbound phages or FBS and tagging dye, and counterstained with CellTracker™ Red prior to imaging by confocal microscopy. Images are from a z plane 64 μm below the surface of the spheroids.
Examination of the localization of AF488-WT-λ phage particles at the earlier 30 min timepoint confirmed lack of association with the spheroid (Figure 2A). Analysis for the 6 h timepoint using either 3-dimensional reconstruction (Figure 2B) or orthogonal views of the spheroids (Figure 3A) showed that the initial association was punctate and discrete but extended over the spheroid surface. After 24 h (Figures 2C, 3B) these punctate accumulations had enlarged to become similar in size to individual cells, suggesting cellular uptake of the phage. To exclude the possibility that this was not simply the result of phagocytic uptake of protein particulates by the fibroblasts, we compared with AF488-tagged FBS. Tagged FBS also associated with the spheroid surface (Figures 2D, 3C) but was seen as larger conglomerates that were distributed irregularly across the spheroid surface and seemed often to be associated with dead cells or debris being extruded from the spheroid. We therefore characterize this as more nonspecific binding, in contrast to that of phage particles, which appeared more to involve specific interactions with viable fibroblasts.
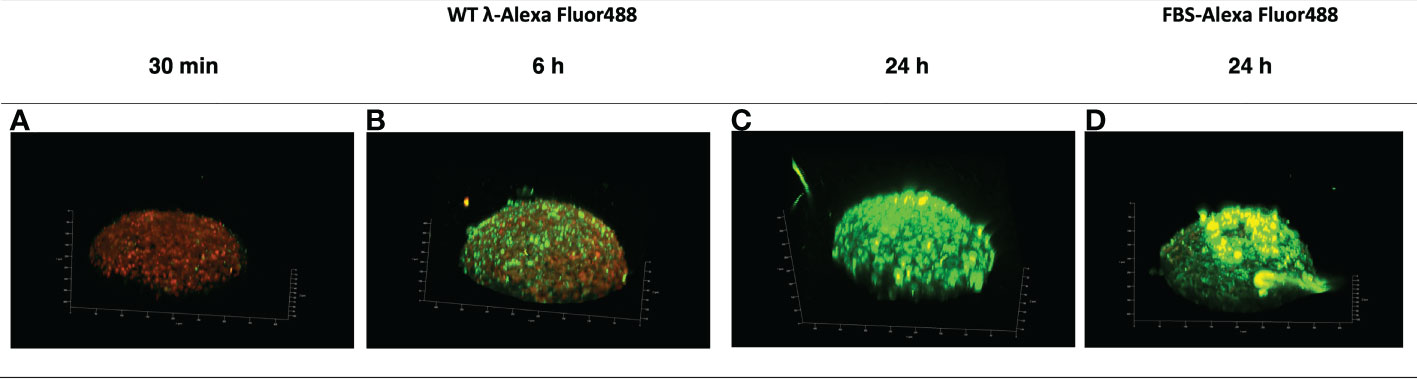
Figure 2 Accumulation of λ phages versus FBS at the surface of NIH3T3 fibroblast spheroids. NIH3T3 spheroids (d7, 250–350 μm in diameter) were exposed to either WT λ phages tagged with Alexa Fluor488 (A–C) or FBS tagged with Alexa Fluor488 (D) for the times shown. Spheroids were washed to discard unbound phages or FBS, and counterstained with CellTracker™ Red prior to imaging by confocal microscopy. The 3D reconstruction is shown.
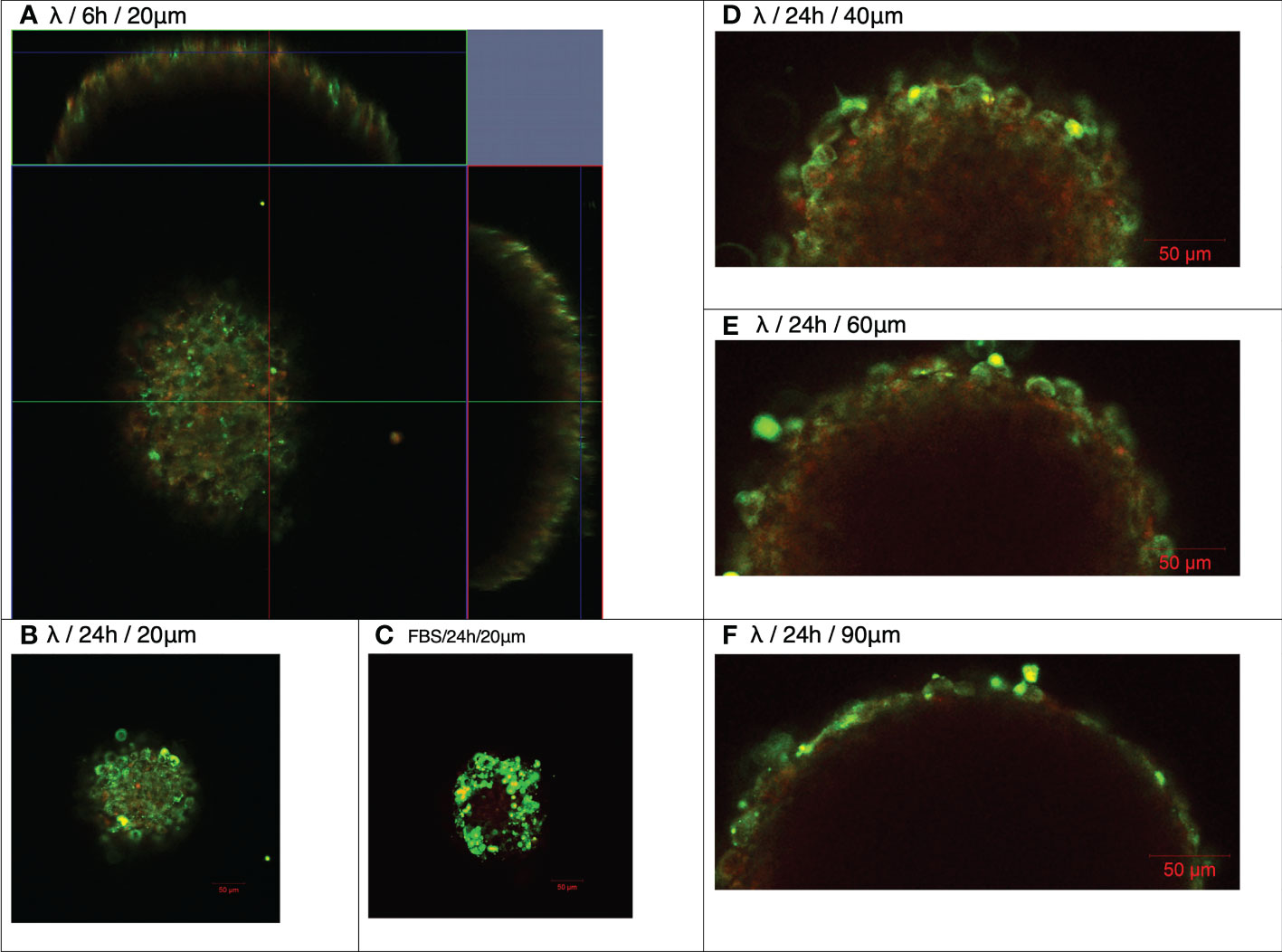
Figure 3 Association and penetration of λ phages with NIH3T3 fibroblast spheroids. NIH3T3 spheroids (d7, 250–350 μm in diameter) were exposed to either WT λ phages tagged with Alexa Fluor488 (A,B, D–F) or FBS tagged with Alexa Fluor488 (C) for the times shown. Spheroids were washed to discard unbound phages or FBS, and counterstained with CellTracker™ Red prior to imaging by confocal microscopy at the Z planes indicated (μm).
Closer observation suggested that AF488-WT-λ was progressively taken up by cells at or near the spheroid surface over the 24 h incubation period. Optical slices at 20, 40, 60 and 90 μm depths (Figures 3B, D–F) showed individual cells that had taken up phage, in some cases retaining a cytosolic distribution, as well as vesicular structures below the surface cell layer that perhaps extended 2-3 cells deep, and a deeper, more diffuse fluorescence. Measurements of the inward radial positions of discrete fluorescent particles at deeper positions showed that the deepest vesicular structures were at: 60 μm, 20.7 ± 4.2 μm; 90 μm, 13.9 ± 4.0 μm (mean ± SD, n=4). This suggests that WT-λ packaged in cellular structures is mostly in the more surface cells after 24 h, predominantly within the first 1-3 superficial cellular layers, but that released phage or processed fragments may diffuse further. The degree of inward access over longer time periods needs further study.
Phage λ tolerates gpD::EGF fusions in capsid assembly
λF7 phage harbors an amber Dam15 mutation in the gene encoding the gpD capsid protein; generation of λF7 in poor or Sup— E. coli amber suppressor hosts would lead to a greater relative complementation by the D::EGF fusion, resulting in increased incorporation of gpD::EGF molecules on the λ surface. The ability of the Dam15 mutation to be complemented by the D::EGF fusion was dependent on the tolerance of the translational allele of gpD produced by each E. coli suppressor strain within the phage capsid. Functionality of each translational allele was assessed by restored λ Dam15 plating efficiency on each strain relative to the double suppressor (supE supF) host.
The translational fusion D::EGF was placed under an inducible strong promoter, λ pL, which is under the control of a temperature-sensitive λ CI857 repressor. Under this governance, D::EGF is repressed at 30 °C (very low expression level) and induced at 37°C. The plating efficiency in the Sup- strain increased by a 103-fold when D::EGF was expressed in trans, indicating that the fusion is able to compensate partially for the Dam15 mutation and be incorporated into the capsid to form a viable phage (Supplementary Table S2). Isogenic suppressor strains of W3101 (supD, supF) substitute an amino acid in place of the amber stop signal generating a readthrough translational allele of gpD. SupD and SupF isogenic amber suppressor strains will substitute serine (gpDQ68S) and tyrosine (gpDQ68Y), respectively, in place of the stop codon at the 68th codon. The gpDQ68S translational allele is very poorly tolerated in capsid formation as demonstrated by very poor plating efficiencies of λ Dam15 on the SupD derivative (101). We similarly noted poor suppression of the Dam15 mutation by SupD in this study, where the efficiency of plating of SupD was observed to be marginally lower than that of Sup- control. Complementation of the Dam15 mutation by D::EGF fusion however, was able to restore plating in SupD hosts by more than 103-fold. In contrast, SupF is highly efficient at suppressing the Dam15 mutation, and as such, no further increase in plating efficiency was noted by the expression of the D::EGF fusion.
EGF-targeted λ phage infiltrate human colon carcinoma spheroids
Having established that the λ phage is capable of penetrating connective tissue stroma and ECM, and that EGF may be coupled to gpD capsid protein as a targeting ligand, we examined the interaction of EGF-targeted λ phage with HT-29 spheroids. HT-29 cells form tight spheroids that can resist the cytotoxicity of even small-molecular-weight compounds (97). They are therefore ideal to investigate the accumulation of λ phages in a 3-dimensional human tumor model in which the neoplastic epithelial cells form tight cell-cell contacts. Furthermore, the receptor for EGF is an appropriate target as it is overexpressed in HT-29 cells and has been implicated in this type of carcinoma. The two targeted λ phage preparations produced had two degrees of EGF display: EGFLo and EGFHi λ variants, which had decorations of approximately 90 and 147 EGF molecules/surface, respectively.
First, HT-29 colon carcinoma spheroids were treated with 108 PFU of AF488-tagged EGFHi and WT (EGF–) λ phages for up to 24 h. Both AF488-EGFHi and AF488-WT λ adhered to the HT-29 cells and accumulated so as to be resistant to rigorous washing in a time-dependent manner (Figure 4), indicating that both native and EGF-decorated phages could infiltrate the outer cellular layers of these dense CRC spheroids as with the fibroblastic stroma model.
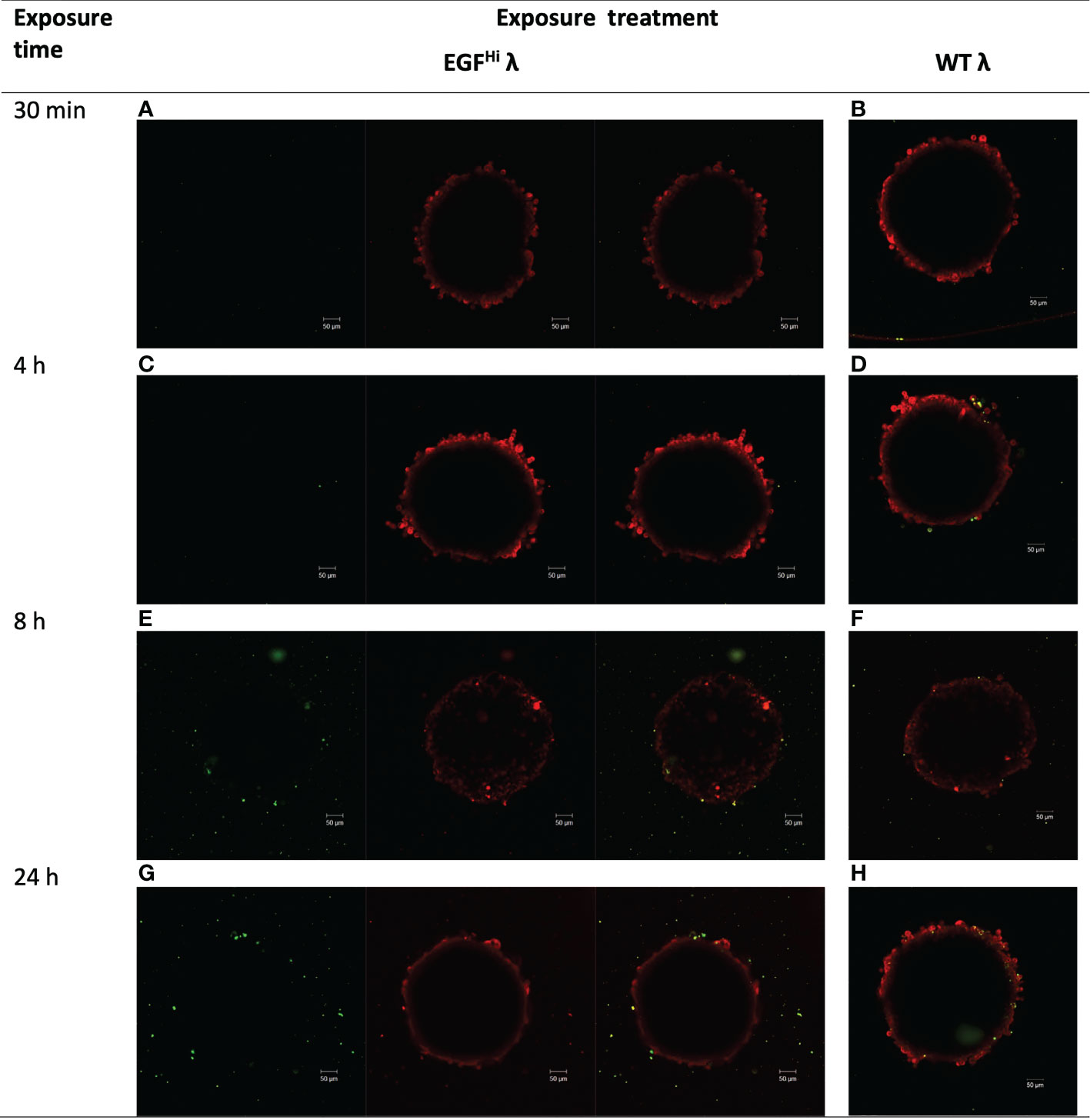
Figure 4 λ phages accumulate in HT-29 colorectal cancer spheroids, and the accumulation/distribution is altered by EGF displayed on the capsid surface. HT-29 spheroids (d6, ~300 μm diameter) were treated with WT λ or EGFHi λ phages, each tagged with Alexa Fluor488 (green). Spheroids were washed to discard unbound phages and counterstained with CellTracker™ Red prior to imaging by confocal microscopy. Panels show the images at the successive timepoints noted, for EGFHi λ (panels A, C, E, G) or WT λ (panels B, D, F, H). Images are from a z plane 64 μm below the surface of the spheroids, except for panel E (42 μm).
With AF488-WT λ there was punctate interaction with surface HT-29 cells by 4 h but no substantial increase thereafter and no focal accumulation (Figure 4D). The EGF targeting of the AF488-EGFHi-λ was associated with a delay in the association of phages with the cancer spheroid, which only became significant and clearly observable after 8 h (Figure 4E). However, the interaction of AF488-EGFHi-λ with spheroids continued and reached a higher level at 24 h (Figure 4G). As well, there were abundant small structures separate from the spheroid itself but which sedimented along with the spheroids in the washes (Figure 4G).The association of these particles with the vicinity of the spheroid itself suggested that they might be AF488-EGFHi-λ bound to EGFR on small membrane vesicles shed from the HT-29 cells, either as part of their maturation process or as a consequence of the initial interaction of the AF488-EGFHi-λ with intact, viable cells at the spheroid surface.
In view of the difficulty in evaluating spheroid uptake against this background of subcellular material, we adopted a quantitative approach to evaluate phage uptake into HT-29 cells in spheroids, and the impact of EGF targeting of the λ phage.
EGF-targeted phage λ accumulate in human colon carcinoma spheroids
Following exposure of mature spheroids over the same timeframe as in Figure 4, we extensively washed the spheroids to remove weakly-bound phages and then used a 2-stage extraction process to recover the phage particles either (i) constrained within intercellular spaces or (ii) taken up or embedded inside cells. Intact, functional λ phages were enumerated for PFU, and we corrected data for the loss of phage infectivity due to treatment duration at 37 °C.
We found that the association of WT λ within the looser surface cell layers of HT-29 spheroids was of low affinity and phages were removed by rigorous washing (Figure 5). The maximum persistent accumulation of phages was negligible, only 0.11% of input at the 8 h timepoint, the highest accumulation reached. We therefore conclude that the association of WT λ with the cancer spheroid, although rapid, is a weak association and therefore unlikely to offer great promise to infiltrative therapeutic strategies using native (untargeted) phages.
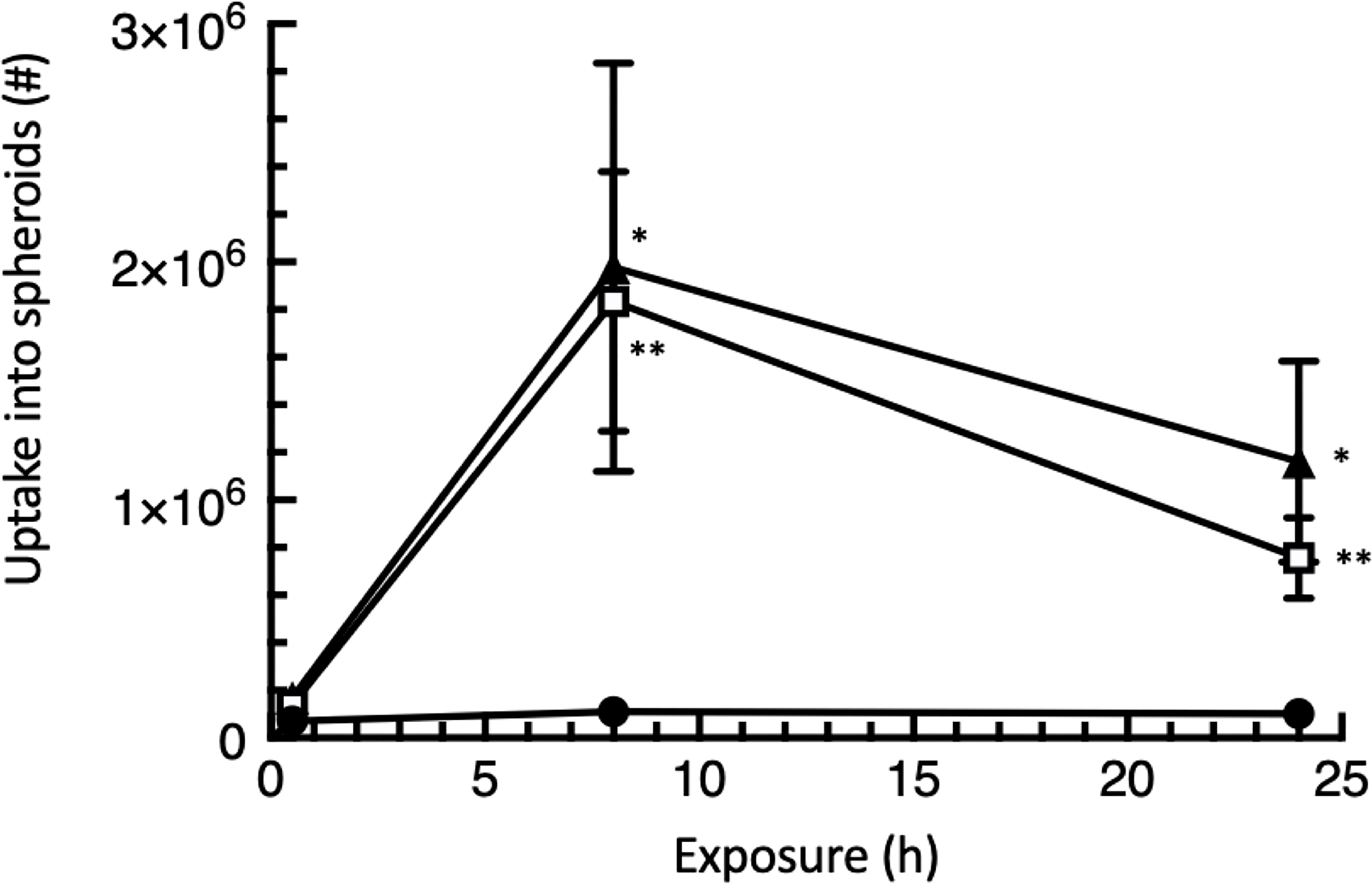
Figure 5 EGF decoration of λ phage substantially enhances penetration of into HT-29 colorectal cancer spheroids. HT-29 spheroids were exposed to 108 phages for the times shown and the total that had infiltrated into the spheroids then recovered by fully dissociating the spheroids and subjecting the cells to complete lysis. Each of the data points has been adjusted to allow for the loss of inherent phage infectivity due to the treatment duration. The data mean values ± SEM, calculated from 3 independent experiments each with 3 replicates. An uptake of 2 x 106 phage corresponds to 2% of input. The symbols represent l, WT λ; □, EGFLo λ; s, EGFHi λ. Significant differences from WT: * P<0.05; ** P<0.01.
However, authentic persistent infiltration of both EGFHi and EGFLo-λ phage particles was readily observed (Figure 5), showing that the EGF targeting strategy was successful to enable persistent uptake. EGF-targeted phages were recovered at 16.8 to 18.1-fold (8 h) or 7.4 to 11.4-fold (24 h) compared to the uptake of WT λ. The uptake of EGFHi-λ was only marginally greater than that of EGFLo-λ in these experiments. This suggests that the lower decoration level (less than 100 EGF molecules on each surface/phage) which is readily attainable, is sufficient for the outcomes seen here.
Our quantitative measurements of penetration gave lower values for EGF-targeted phage at 24 h compared with 8 h, in contrast with observations in microscopy (Figure 4). However in the approach here, we were measuring functional λ phages using the PFU assay rather than the fluorescent tag, so we speculate that the finding reflects a loss of phage functional integrity with time, within the cell mass of the cancer spheroid.
EGF-targeted phage λ is taken up intracellularly within HT-29 carcinoma cells
The two-phase extraction of extensively-washed spheroids allowed us to distinguish between phages recovered from tissue interstitial (intercellular) spaces and those present within the constituent cells (intracellular), accessible only by cellular disruption. Our approach quantified intact, functional phages as defined by their ability to form plaques in bacterial monolayers, corrected for the decline in intrinsic activity due to incubation at 37°C. Table 1 shows the results normalized relative to WT, for both EGFHi-λ and EGFLo-λ.
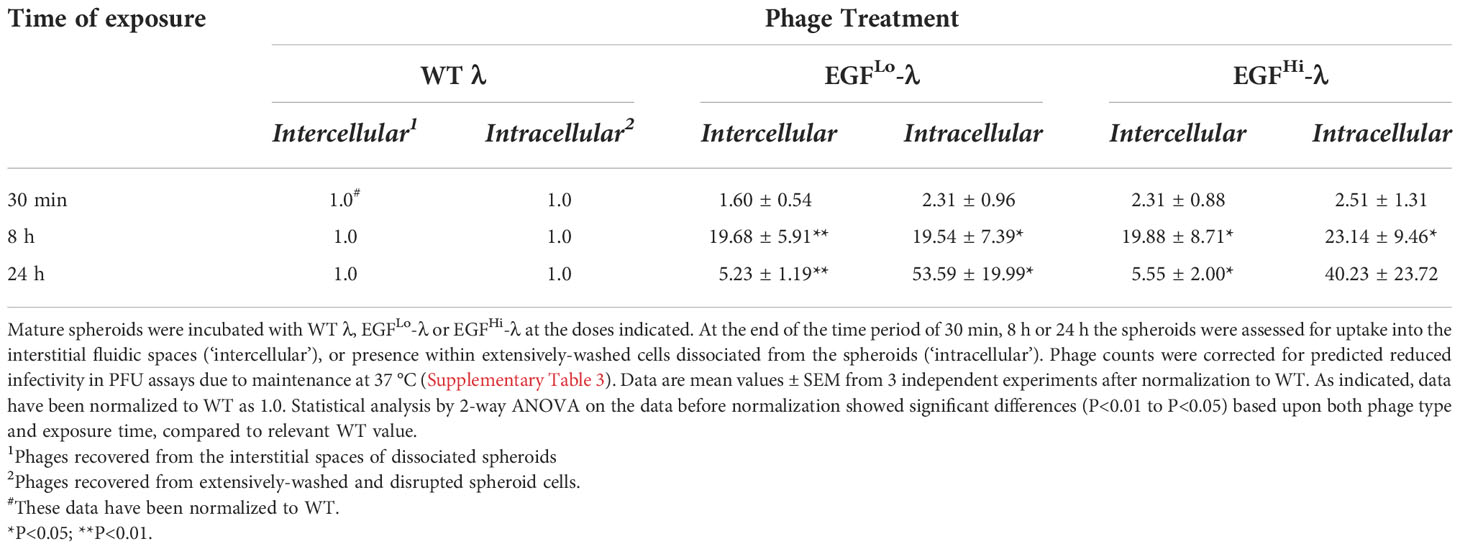
Table 1 EGF targeting of λ phage enhances uptake into HT-29 colorectal cancer spheroids and facilitates cellular entry.
The data again show that in addition to much greater preferential uptake, there is a preferential shift to the intracellular compartment with EGF decoration. For EGFLo-λ there is no shift in the distribution within the spheroid at 8 h, but at 24 h the distribution of liganded phage is biased more than 10-fold in favor of being in the intracellular compartment, showing active cellular uptake compared to the native phage. The same substantial difference is seen at 24 h for EGFHi-λ, and given the 16% higher value it may be that with the greater EGF decoration the shift to the intracellular compartment is occurring earlier at ~8 h, although further work is needed to see if this reaches statistical significance. Nevertheless, these data provide convincing evidence for the cellular uptake of EGF-targeted λ phage after entry into HT-29 CRC spheroids, and that active entry begins soon after 8h of initial exposure, which is the main phase of phage entry into the spheroid.
EGF-λ phages are not directly cytotoxic to HT-29 cells
Given the ability of EGF-λ phages to both penetrate HT-29 cancer spheroids and enter cancer cells we wished to know whether the targeted carrier phage was directly cytotoxic to these cancer cells. We first evaluated both cytotoxicity and potential interference with cell metabolism using the MTT assay, which measures mitochondrial succinate dehydrogenase (SDH) activity (96). HT-29 cells growing in a monolayer were exposed to either low (104 PFU) or high (108 PFU) concentrations of both WT and EGF-targeted λ phages for 24 h and SDH activity then evaluated. As indicated in Figure 6, there was no loss of viability (or mitochondrial function) due to direct exposure of HT-29 cells with any of these preparations, and no significant differences between the results with the EGF-decorated and WT λ phages. We therefore conclude that these phages, without payload, are not directly cytotoxic to the HT-29 cells, regardless of whether they have the EGF ligand attached, or not.
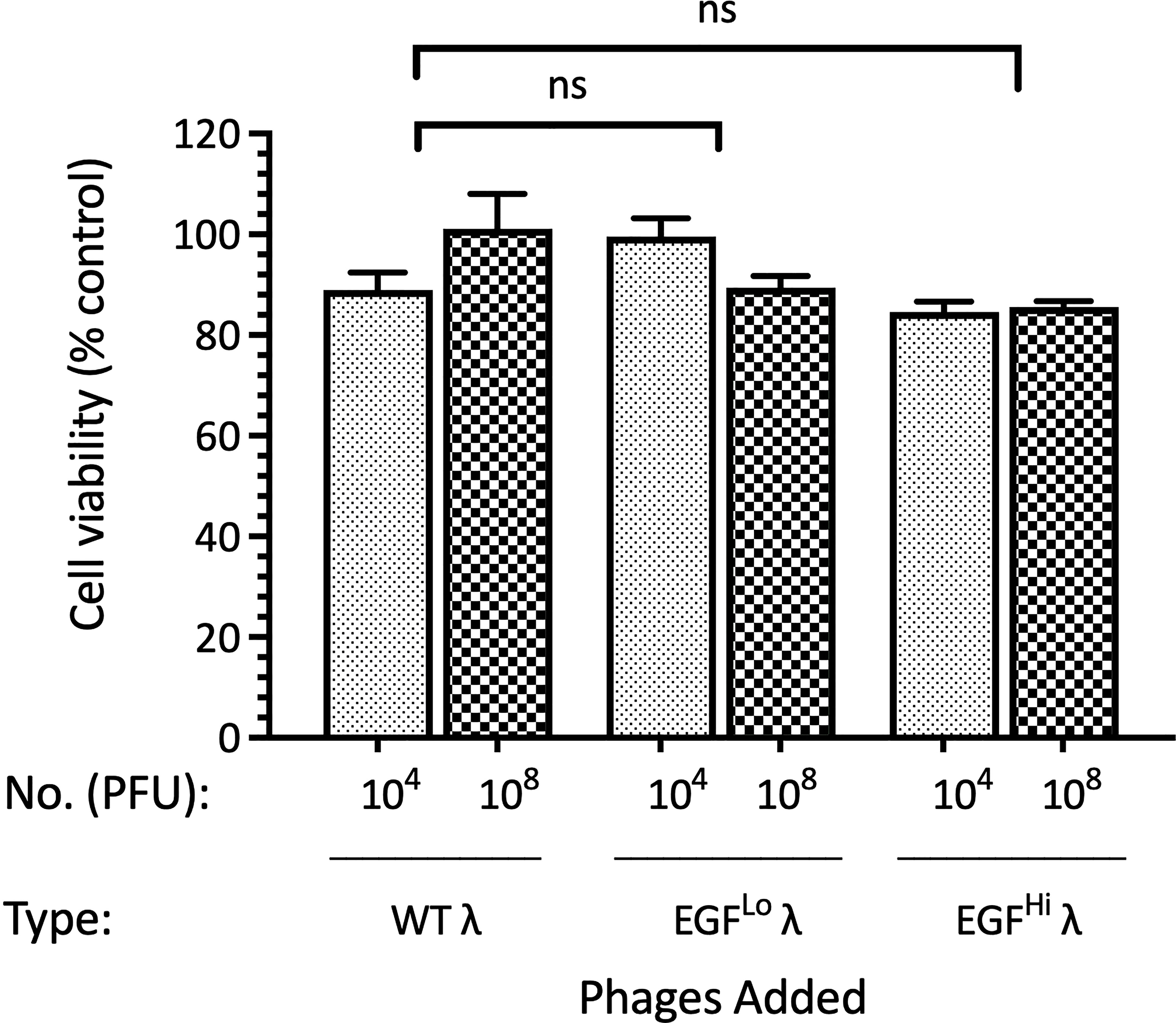
Figure 6 WT and EGF λ phages are not cytoxic for HT-29 cells grown in monolayer culture. HT-29 cells were established in monolayer for 72 h and then treated with 104 or 108 PFU of WT, EGFLo and EGFHi λ phages for a further 24 h. Cell viability was then assayed by the MTT assay and expressed relative to untreated cells. Data are expressed as mean ± SEM of 3 independent experiments, each with 3 culture replicates. Neither comparison with controls or comparison of EGF-displaying λ phages with WT (shown) showed a statistically significant difference. ns, not significant.
EGF-λ phages lead to a reduction in the ATP of HT-29 cells in spheroids
Despite there being no evidence for direct toxicity of the phage preparations against the cells in monolayer, we used a commercial assay of putative cytotoxicity to evaluate the apparent viability of the spheroid cell population after exposure to the phages. The assay measures total cellular ATP. While variability was high in these assays we noted a significant effect of EGFHi phages in depressing the cellular ATP levels in HT-29 spheroids, but no effect that reached significance for WT phages (Figure 7).
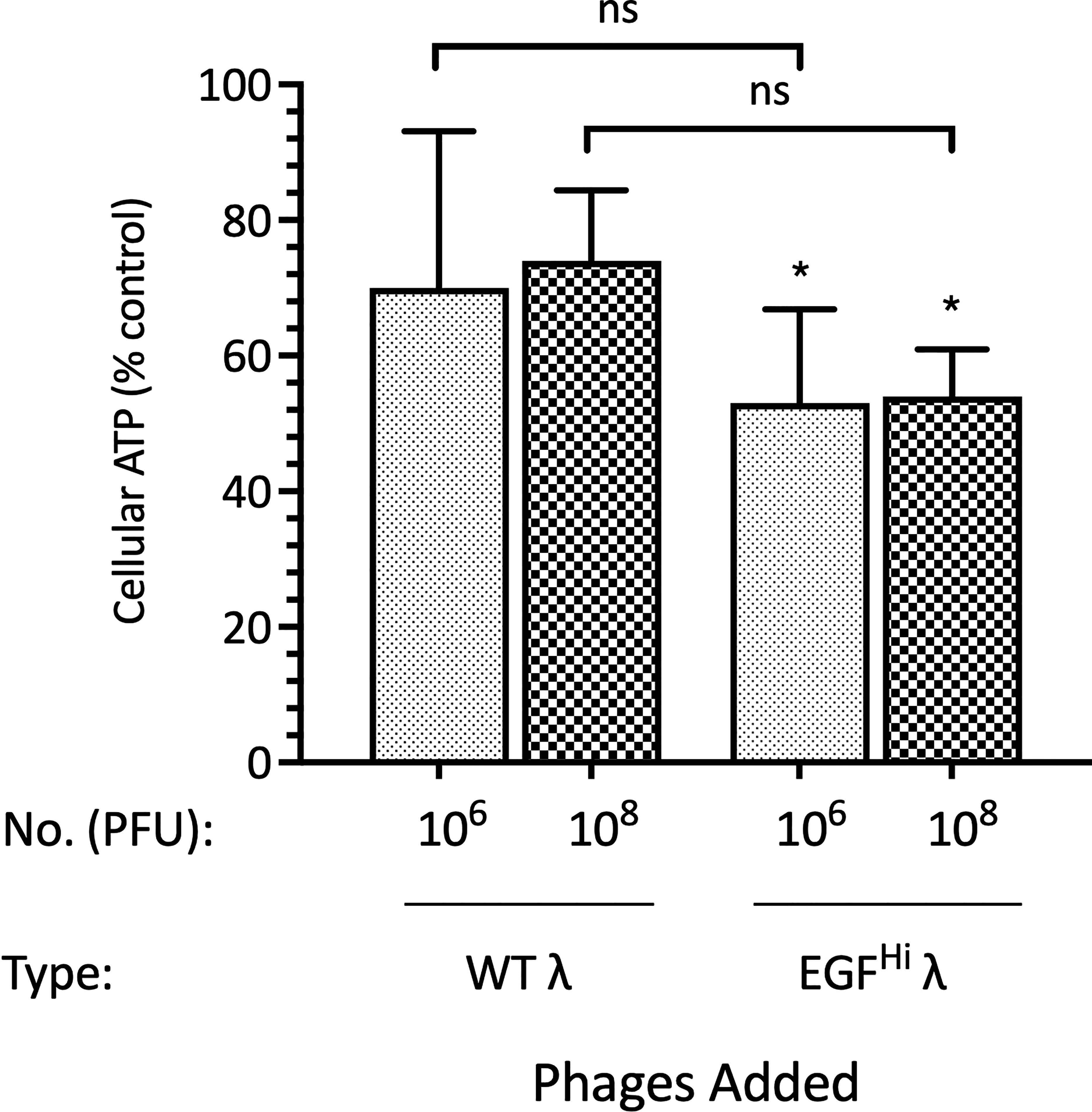
Figure 7 EGF decoration of λ phages leads to a reduction in the ATP of HT-29 cells in spheroids. HT-29 spheroids (d7) were treated with 106 or 108 PFU of WT λ or EGFHi λ phages for 24 h. The ATP content of spheroids was then determined. Data are expressed relative to spheroids in standard growth medium, as mean ± SEM of 3 independent experiments, each with 3 culture replicates. Significantly difference from control, * P<0.10; ns, not significant.
EGF-targeted λ phages inhibit early stages of spheroid growth and formation
A. Treatment during initial aggregation of the cancer cells
In our first approach we included phages at the time of seeding of HT-29 cells onto agarose. The initially-dispersed cells normally settle onto the non-adherent agarose and over the initial 2 d aggregate into a shallow disc, which subsequently folds in and merges from the periphery to form a fully intact spheroid around 7 d (97). We added WT-λ, EGFLo-λ or EGFHi-λ phages at either 104 or 108 PFU/culture at the time of seeding and observed the outcome (Table 2; Figure 8).
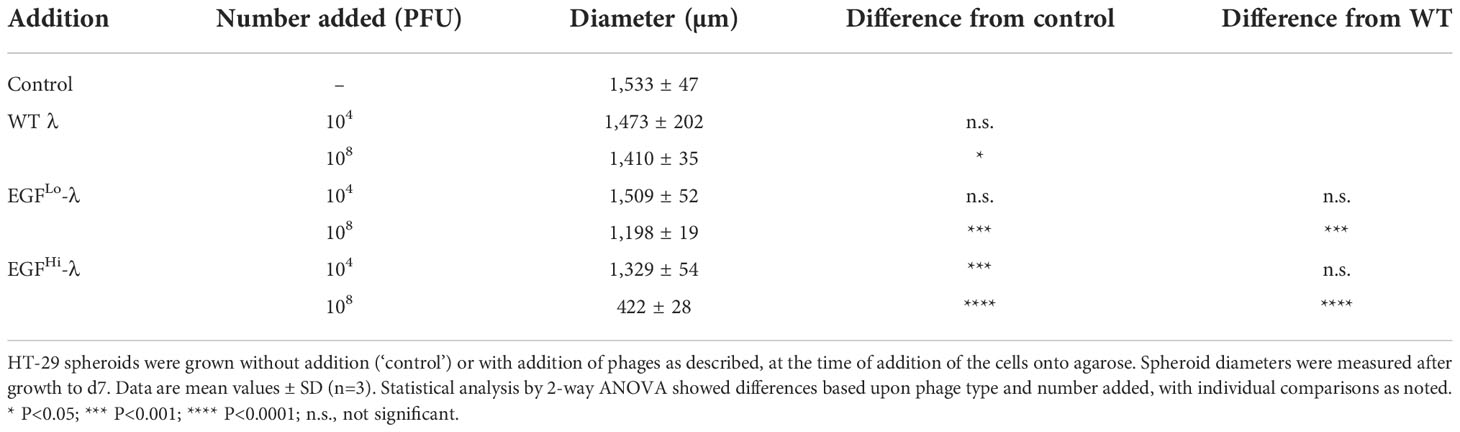
Table 2 EGF-targeted λ phage inhibit the growth of HT-29 colorectal cancer spheroids.
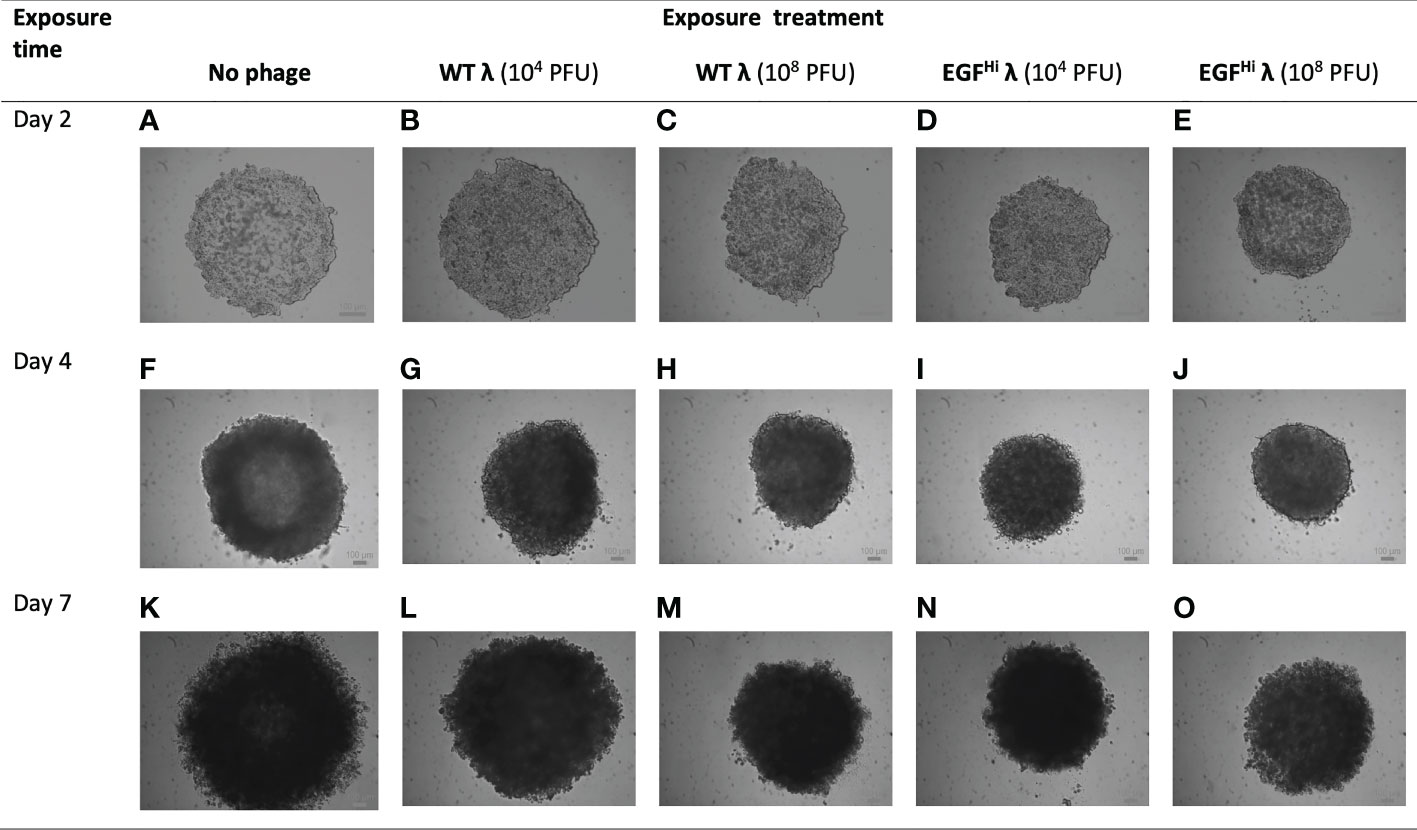
Figure 8 Addition of WT λ and EGFHi λ particles at initial cell aggregation reduce the size but do not compromise spheroid formation. WT λ (B, C, G, H, L, M) and EGFHi λ (D, E, I, J, N, O) phages at two doses (104 or 108 PFU) were added to the agarose surface at the same time as seeding with HT-29 cells. Initial cell aggregation and resultant spheroid formation were imaged up to d7 with a bright-field microscope, showing full-thickness spheroids and compared to controls (A, F, K). Representative photographs from one series in 3 independent experiments. Individual scale bars are shown.
Relative to the control situation with no phage added, the highest addition of WT-λ and EGFLo-λ significantly reduced spheroid diameter at d 7 (Table 2).The effect was more noticeable with EGFHi-λ, with significant reductions in ultimate spheroid size being achieved with additions of 104 or 108 PFU of EGFHi-λ. This reduction in size was also observable in microscopy (Figure 8). The intermediate stage of spheroid formation, in which the aggregate has a cross-sectional profile similar to that of an erythrocyte, can be seen in the d 4 no phage control (Figure 8F). Again, low concentrations (104 PFU) of WT λ had no effect on spheroid formation but higher concentrations (108 PFU) of WT λ and both concentrations of EGFHi-λ phages interfered with initial aggregate formation and subsequent growth, leading to a smaller spheroid at d 7. The measured diameter ranges respectively were: d 4, 732-772 μm vs 949-1025 μm; d 7, 823-968 μm vs 1210-1309 μm). All of the spheroids retained their typical mature final phenotype, even though sizes varied, confirming that any deviation spheroid evolution process would not be due to λ phage effects on initial cell aggregation.
B. Treatment after initial aggregation of the cancer cells
We next added our λ preparations (WT and EGFHi) at 108 PFU to partially-formed (d 2) HT-29 spheroids, so that the phages encountered the cells post aggregation as the spheroids were developing further (Figure 9). We compared effects with the natural evolution of the spheroids (control condition) over a full period of 22 d, the longest that we routinely maintain these spheroids in original culture.
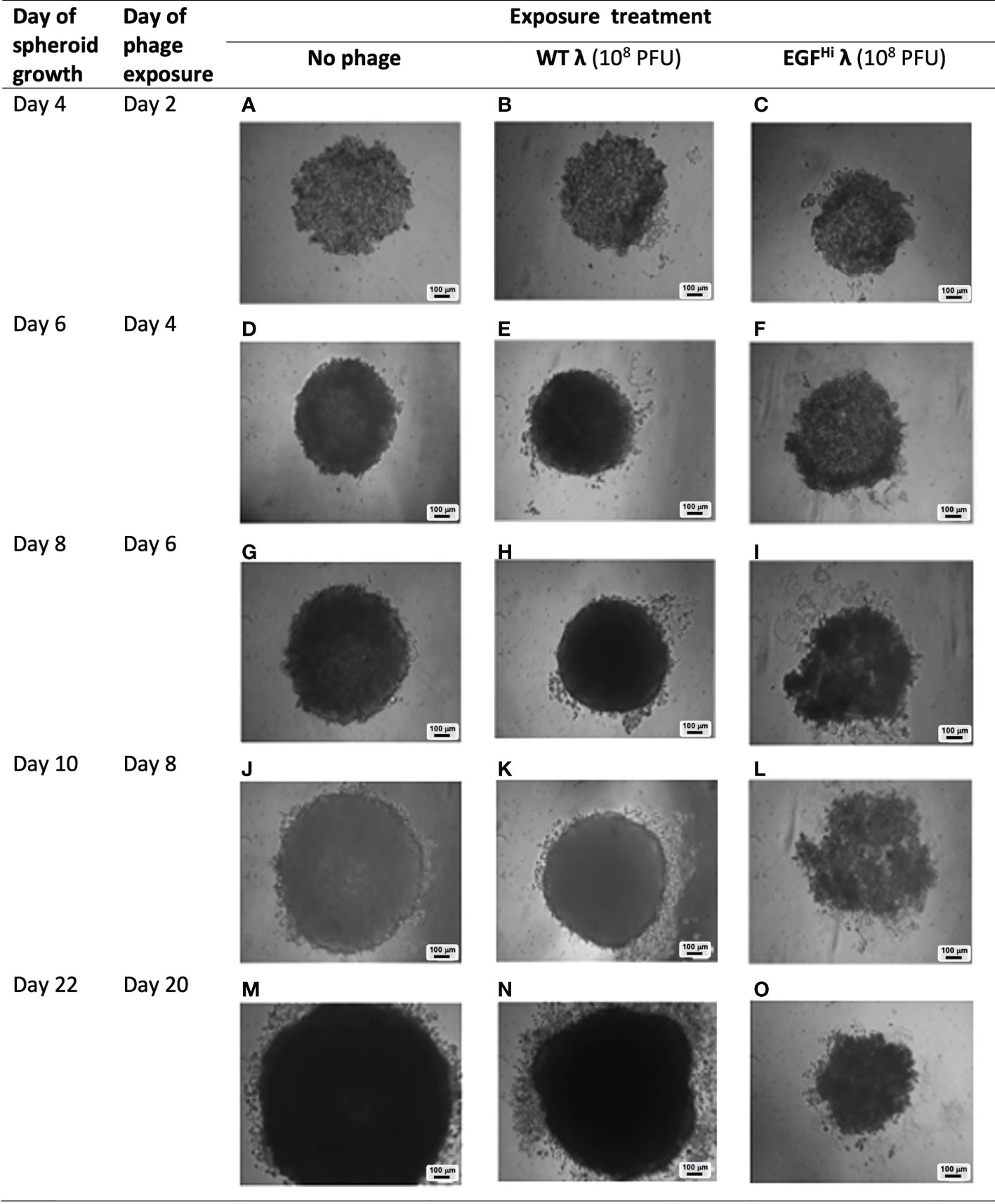
Figure 9 Persistence of effect of EGF-decorated phages on spheroid development. WT λ (B, E, H, K, N) or EGFHi λ (C, F, I, L, O) phages (108 PFU) were added to partially-formed HT29 spheroids at 48 h after seeding and followed for a further 20d as shown, and compared to controls (A, D, G, J, M). The precise morphologic findings were varied over 3 independent experiments but this shows a representative example of how the EGF λ phages had a persist disruptive effect on spheroid formation that was clearly evident out to d8 after phage addition and led to the ultimate formation of a significantly compromised spheroid. This was not evident with WT λ, or with λ phages with lower EGF decoration (EGFLo λ, not shown). Images are brightfield of spheroids in situ on agarose, and scale bars are shown.
We observed no consistent change to spheroid formation over the first 4 d of phage exposure with either of our λ preparations (Figure 9). At d 6 there was change apparent in spheroids exposed to EGFHi-λ, which began to exhibit an irregular morphology and be noticeably less dense than other spheroids. By d 8 the HT-29 spheroids treated with EGFHi-λ showed clearly aberrant formation, with the spheroidal integrity lost, cell clumps dissociating and sparse regions in the cellular aggregates. By d 20, the disruption in spheroid formation had resulted in a spheroid that was markedly smaller in diameter than the control spheroid, and less dense, although had regained more of its usual spheroidal appearance. In contrast, neither the WT λ (Figure 9), EGFLo-λ, nor lower doses (104 PFU) of the phage preparations (data not shown) exerted any observable effect on the final spheroids.
C. Treatment of fully mature spheroids with phages
Finally, we added our phage preparations to fully-mature spheroids grown to d 11 (Figure 10). The HT-29 spheroids continued to expand modestly over this period as expected and began to show early signs of disintegration as is typical for these spheroids (their sizes being such that the central portions of the spheroids are hypoxic). However, in no case (WT λ or EGFHi-λ; 104 PFU or 108 PFU) was there a consistent change in the integrity or size of the spheroids above the general spheroid variability. Thus, neither native nor EGF-decorated λ phages appear to affect mature spheroids.
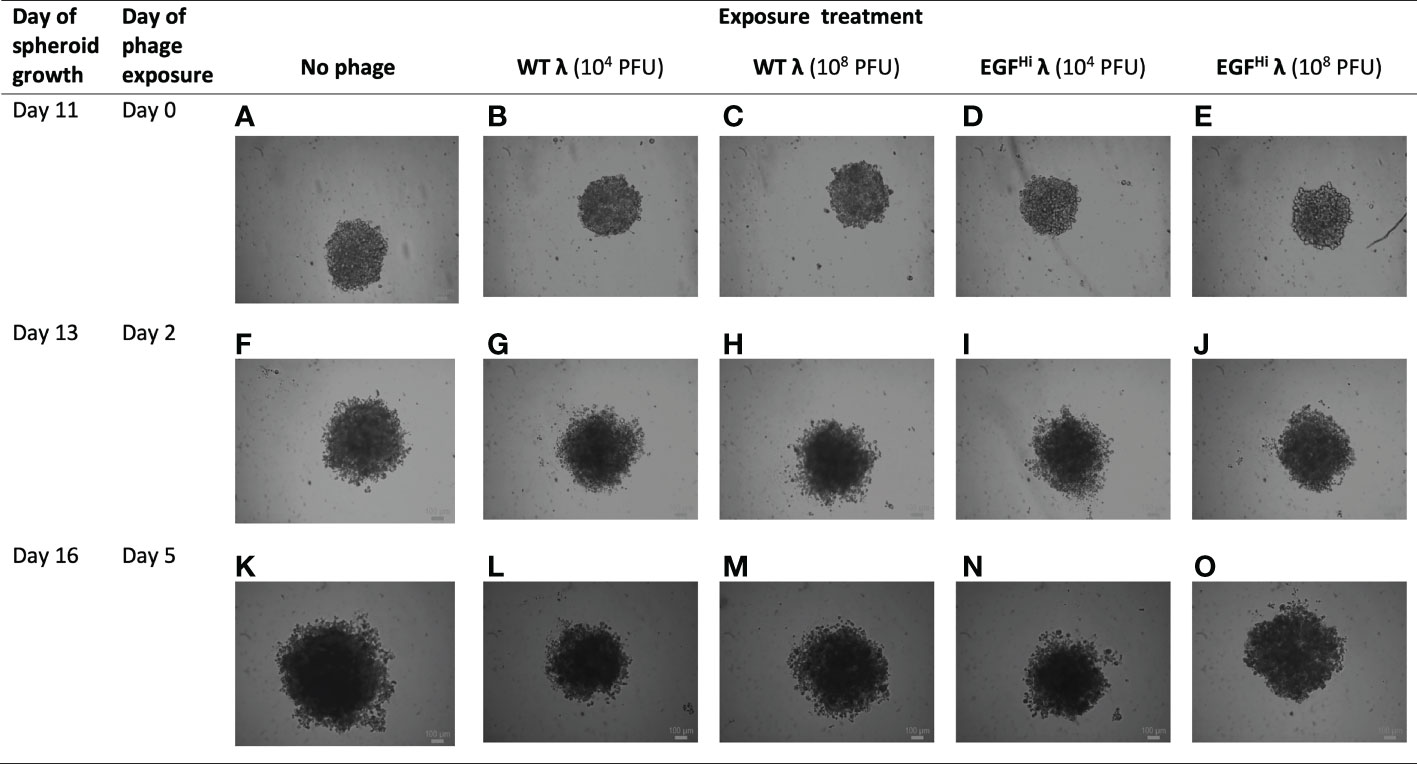
Figure 10 Addition of λ phages to fully-formed HT-29 spheroids does not impede tumor expansion. WT λ (B, C, G, H, L, M) and EGFHi λ (D, E, I, J, N, O) phages at two doses (104 or 108 PFU) were added to fully-formed but small dimension HT-29 spheroids at day 11 after seeding. Spheroid development was imaged for a further 5 d. with a bright-field microscope, showing the full-thickness spheroids and compared to controls (A, F, K). Representative photographs from one series in 3 independent experiments. Scale bar: 100 μm.
Discussion
Given the substantial advantages of phage technology for the design of novel therapeutics (104) we investigate the use of engineered bacteriophages in cancer therapeutic applications. In particular, we see the potential for interference in some of the behaviors of infiltrating immune effector cells as well as neoplastic cells that favor metastasis and are influenced by the features of the 3-dimensional tumor environment, such as low-molecular-weight tissue mediators (22, 23, 25, 27, 96, 97). For this to be done we need first to determine whether phage constructs can find their way into the dense tissue of a tumor and interact with cancer cells to deliver therapeutic cargos.
We approached this question in stages using multicellular spheroids to generate representative 3-dimensional models of the stroma and tumor tissues (97, 105). We first used spheroids of NIH3T3 cells, which are a multipotent mesenchymal cell line with predominantly fibroblast character and serve well as a model for stromal components of the tumor capsule and septa that might impede phage penetration. We found that WT λ phages (covalently tagged with a fluorochrome) adhered to spheroids and became bound or entrapped within the outer cellular layers and matrix of the model stroma. Confocal images showed λ phage particles within periphery of the fibroblast spheroids at times beginning at 6 h, a time course of ingress that would be acceptable for therapeutic purposes. Over the relatively short timeframe of 24 h, there was evidence for entry of the tagged particles in a concentrated form in the outer 2-3 cellular layers, and potential dissemination to deeper levels that needs to be further studied.
Fibroblastic cells, which tightly adhere to each other in the spheroid as they do in stromal tissue, would therefore not constitute a barrier to access of λ phage. The ability of phage particles to penetrate through solid tumours in patients will depend on a variety of factors including size and morphology of the phage and biochemical properties of the phage capsid that may affect its interactions with the ECM (106, 107). Whether there are productive interactions between λ phages and the ECM formed by the fibroblasts is a matter of further investigation; but if so this would favor local accumulation of phages in a tumor.
We next studied phage infiltration into spheroids modeling the neoplastic parenchyma, which is a greater barrier due to tighter associations between epithelial cells than fibroblasts (97). We also enabled cancer cell-selective targeting by expressing EGF at the capsid surface to allow binding to the EGF receptor (EGFR). The EGFR is a transmembrane receptor tyrosine kinase in the ErbB family of receptors and is most highly expressed on epithelial cells. It has been implicated in the progression of wide range of epithelial tumours, for which increased EGFR activity enhances growth, invasion and migration (108, 109). Although EGF : EGFR interaction is essential for normal maintenance and renewal of epithelial cell populations, the overexpression and hyperactivation of EGFR contributes to tumor progression, rendering it an important biomarker and target for anticancer therapeutics (110). EGF has been used in directing other types of vectors for a targeted approach in gene therapy applications (111, 112). Human EGF has been shown to retain its ability to bind to EGFR when fused to fd filamentous phage (112) and the display of EGF on phage particles can direct the phage vectors to recognize EGFR-expressing cancer cells (113). EGF itself is well suited as a ligand for targeting due to its stability. The displayed growth factor is unlikely to have unfolded in the conditions used in this study as recombinant human EGF unfolds only under extreme pH conditions (pH < 4 and pH > 8) or temperatures higher than those encountered here (114). In this study we displayed EGF by translational fusion to λ phage gpD capsid proteins using the dual-phage display system we previously described (101).
We used HT-29 CRC cells to model colorectal cancer tissue. HT-29 cells overexpress EGFR and are representative of wildtype-KRAS and BRAF-mutated tumours, which have been reported in approximately 5-15% of patients with colorectal cancers (115, 116). We demonstrated the time-dependent trapping of λ phages (both WT and with EGF displayed) in the peripheral layers of HT-29 spheroids. Phages accumulated to a lesser degree within HT-29 spheroids than in NIH3T3 spheroids after 24 h of treatment, which could be expected due to the tight adherence junctions between the epithelial cells comprising the HT-29 spheroids (65). Surface decoration with EGF slowed the initial penetration into HT-29 spheroids over the first 8 h, most likely due to immediate capture of the EGF ligand by EGFR on the most outer surface. The greater size of the EGF-decorated phage, which for the EGFHi-λ preparation reflects a substantially greater molecular mass (ΔMr ~ 890K) is also likely relevant. However, decoration with the 6-kDa growth factor or a comparable small polypeptide ligand is preferable to decoration with a larger targeting ligand. We have shown that phages maximally decorated by a large protein such as eGFP (33-kDa) can be as large as three times greater in diameter than WT phages (101). However, this does not indicate a concern in terms of overall access; the accumulation of EGF-decorated phage was greater than WT and involves an active process.
Quantitative analysis with rigorous washing of the spheroids showed that stable penetration of WT λ phages was in fact limited, and EGF decoration greatly enhanced and stabilized the accumulation within the cancer spheroid. In these studies, there was very little difference between uptake of the EGFLo and EGFHi variants, suggesting that the lower decoration level of less than 100 EGF molecules/phage is sufficient for efficient entry into the spheroid for delivery of a toxic payload. Higher ligand densities may encounter limits due to receptor saturation, as we have found in other contexts (14, 117).
The numbers of EGFLo/Hi-λ phages recovered in this approach declined from 8 h to 24 h, in contrast with the observations from confocal microscopy, for which there was an increase in the fluorescence intensity from tagged EGFHi-λ over this period. The quantitative method measures intact, functional phage by plaque-forming assay, whereas microscopy simply depends on sufficient residual macromolecular material to retain the fluorescent tag in situ. Hence, the observations can be rationalized if the EGF-λ becomes taken up and at least partially degraded by cells inside the spheroid after ~8 h.
Separation of EGF-λ phages from intercellular and intracellular compartments confirmed a ~20-fold overall increased uptake compared with WT λ. Moreover, by 24 h there was enrichment in the intracellular fraction by a factor of 10.2 for EGFLo-λ and 7.2 for EGFHi-λ. This clearly shows cellular uptake between 8 - 24 h and is consistent with the suggestion that a substantial fraction of the uploaded dose has undergone at least partial degradation or other inactivation that is seen in measured plaque-forming ability.
Our results are consistent with observations in other systems. The finding that EGF-λ were internalized more readily than untargeted phage agrees with the results of Kassner and colleagues who observed a similar distinction between EGF-displaying M13 phage and WT phage in transfection efficiency of COS-1 cells (113). Menon and colleagues showed that 15-60 min may be sufficient for cellular uptake of PEP-2 phages displaying ligands into ECV304 cells (118).
We wished to know whether λ phage, either WT or with EGF as a targeting ligand, had inherent cytotoxic activity or instead are likely to be a neutral vehicle for delivery of focused genetic payloads. We found that λ phages, with or without EGF decoration, were not cytotoxic for HT-29 cells in monolayers, nor had any effect on overall mitochondrial function, as assessed using an acute viability assay with succinate dehydrogenase as a readout. This is consistent with the finding that other phages have no cytotoxic effect on these cells (119). We did find evidence for an effect using an assay that is presumed to reflect viability in spheroids (see Figure 7). However, interpreting this as a loss of viability of near 50% over 24 h is both biochemically implausible given the ligand and inconsistent with the MTT assay (Figure 6) and subsequent timed morphological analyses (Figures 8–10 and Table 2). We think it more likely that this reflects changes in energy utilization in response to phage exposure, which we have shown involves uptake and likely degradation of phages. The assay itself measures ATP levels and as these mature spheroids have substantial hypoxia and low glucose levels, replenishment of ATP may be a limiting factor.
Changes in the ATP assay raised the possibility that EGF-λ might interfere with development of neoplastic tissue, as modeled with our spheroids, independently of any direct cytotoxicity. This might interact beneficially with any anticancer therapeutics, either those encoded in the phage itself or provided independently in combination treatment. For example, partial dissociation of the cell population might facilitate drug access or reduce the resistance associated with hypoxia or cell adhesion mechanisms. We therefore examined the effect of additions at different times on spheroid evolution, which is itself a mimic of the growth of microdeposits.
Addition of WT or EGFHi-λ phage to fully mature spheroids had no effect on spheroid expansion or appearance, suggesting it would be unlikely for this approach to have a direct beneficial effect on established tumours, without a toxic payload. However, the addition of EGFHi-λ at the time of cell seeding (comparable to initiation of secondary deposits from initial disseminated cells) interfered with full development of cancer spheroids. Furthermore, addition of EGFHi-λ 48 h after cell seeding on agarose, after cell aggregation and when the early cell clusters are proliferating and organizing into larger clusters, had a variable but at times dramatic effect on the fate of the spheroid. A slowly-developing effect on spheroid integrity led to significant dissociation of the cell mass over about a week, and the impact of this event was retained over a 3-week time course, leading to a much reduced and poorly-formed cell mass. This raises the possibility of a beneficial additive or synergistic effect of this delivery system as part of an antineoplastic strategy, interfering with the early seeding and expansion of microdeposits in regional establishments of secondary deposits. Collectively, our data also exclude concerns that the approach might in some way have a proneoplastic action that would make it difficult to use in an anti-cancer context.
Our findings suggest in principle that EGF-displayed/targeted λ phages may be valuable in delivery of toxic therapeutics of interventions that may release activity of local anti-tumor responses or immune checkpoint inhibitors. Our EGF-λ phages may themselves be able to disrupt early stages of CRC tumor metastasis formation. The latter effect might be due to the particular nature of the EGF signal delivered through the EGFR by the phage-EGF construct or may be a property of the λ phage itself when held in place at the cancer cell surface. Binding of EGFR by EGF-targeted phage may trigger inhibitory events similar to those for cetuximab and other mAbs that bind to the extracellular domain of EGFR (120–122). It is further possible that the effect of EGF-λ may depend on first taking up the decorated complex into the cancer cell, which will be a requirement for future delivery of a gene therapeutic. Phages can be taken up into mammalian cells via caveosomes and/or clathrin-coated vesicles, which are able to accommodate particles of at least 500 and 200 nm, respectively (123). Our data confirm that there is cellular uptake, based upon the need for cell lysis to recover a significant proportion of phages after incubation, and the loss of functionality most likely due to enzyme degradation. Alternatively, the phage itself, with or without partial activation of the EGFR, may interfere with aspects of the extracellular milieu such as ECM elaboration or structure, that can impact on tumor development.
In summary, we have shown that: (i) λ phages are able to pass through fibroblast and ECM layers that represent the stromal components of a tumor, (ii) enter and accumulate in dense tissue representing the CRC neoplastic parenchyma, (iii) EGF ligand, covalently bound to gpD capsid protein, favors entry within the cancer cell population, (iv) EGF-λ enters CRC cells in a model tumor at more than 50-fold the relative abundance of WT phage, (v) EGF-λ has the potential to interfere with the early formation and growth of cancer populations without the need for a toxic payload, and (vi) the effect of a single early exposure to EGF-λ persisted for the entire tissue progression (to d20) in our model CRC tumor system. We have therefore shown in principle the utility of targeted phage technology for infiltration of colorectal cancer tumor tissue (and by extension other solid carcinomas). The intermolecular mechanisms remain to be fully elucidated, but the approach should be translatable to patients in vivo and can be extended by insertion of a gene therapeutic to be locally expressed following cellular uptake of the phages. Such an intervention may enable new direct cytotoxic effects and/or dampen down immune escape mechanisms to allow greater host anti-tumor activity or facilitate the success of cell-based immunotherapies.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Author contributions
RS and JB contributed to conception and design of the study. HH carried out experiments with assistance of D-WC and input from the other authors. Analysis of confocal images was by MF. The first draft of the manuscript was prepared by HH and extensively revised by JB and MF with contributions from the other authors. Statistical analysis was by HH and JB. All authors contributed to manuscript revision, read, and approved the submitted version.
Acknowledgments
Supported by grants to JB from the Canadian Institutes for Health Research (CIHR; FRN 84361), the Natural Sciences and Engineering Research Council of Canada (NSERC; RGPIN 121624), and the University of Waterloo Research Incentive Fund (RIF); and to RS from NSERC (RGPIN 214684). JB is a Senior Scientist of the Beatrice Hunter Cancer Research Institute.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.957233/full#supplementary-material
References
1. Vaupel P, Multhoff G. Accomplices of the hypoxic tumor microenvironment compromising antitumor immunity: Adenosine, lactate, acidosis, vascular endothelial growth factor, potassium ions, and phosphatidylserine. Front Immunol (2017) 8:1887. doi: 10.3389/fimmu.2017.01887
2. Blay J. Adenosine and tumor microenvironment. In: Encyclopedia of cancer. Heidelberg: SpringerBerlin (2012). p. 49–52.
3. Chen G, Wu K, Li H, Xia D, He T. Role of hypoxia in the tumor microenvironment and targeted therapy. Front Oncol (2022) 12:961637. doi: 10.3389/fonc.2022.961637
4. Wang D, Cabalag CS, Clemons NJ, DuBois RN. Cyclooxygenases and prostaglandins in tumor immunology and microenvironment of gastrointestinal cancer. Gastroenterology (2021) 161(6):1813–29. doi: 10.1053/j.gastro.2021.09.059
5. Kaur T, Weadick B, Mace TA, Desai K, Odom H, Govindarajan R. Nucleoside transporters and immunosuppressive adenosine signaling in the tumor microenvironment: Potential therapeutic opportunities. Pharmacol Ther (2022) 22:108300. doi: 10.1016/j.pharmthera.2022.108300
6. Hussain M, Javeed A, Ashraf M, Yuzhu H, Mukhtar MM. Multilevel pharmacological manipulation of adenosine-prostaglandin E(2)/cAMP nexus in the tumor microenvironment: a 'two hit' therapeutic opportunity. Pharmacol Res (2013) 73:8–19. doi: 10.1016/j.phrs.2013.04.006
7. Blay J, White TD, Hoskin DW. The extracellular fluid of solid carcinomas contains immunosuppressive concentrations of adenosine. Cancer Res (1997) 57(13):2602–5.
8. Ghiringhelli F, Bruchard M, Chalmin F, Rebe C. Production of adenosine by ectonucleotidases: a key factor in tumor immunoescape. J BioMed Biotechnol (2012) 2012:473712. doi: 10.1155/2012/473712
9. Allard B, Turcotte M, Stagg J. CD73-generated adenosine: orchestrating the tumor-stroma interplay to promote cancer growth. J BioMed Biotechnol (2012) 2012:485156. doi: 10.1155/2012/485156
10. Morello S, Pinto A, Blandizzi C, Antonioli L. Myeloid cells in the tumor microenvironment: Role of adenosine. Oncoimmunology (2016) 5(3):e1108515. doi: 10.1080/2162402X.2015.1108515
11. Arab S, Alizadeh A, Asgharzade S. Tumor-resident adenosine-producing mesenchymal stem cells as a potential target for cancer treatment. Clin Exp Med (2021) 21(2):205–13. doi: 10.1007/s10238-020-00674-9
12. de Andrade Mello P, Coutinho-Silva R, Savio LEB. Multifaceted effects of extracellular adenosine triphosphate and adenosine in the tumor-host interaction and therapeutic perspectives. Front Immunol (2017) 8:1526. doi: 10.3389/fimmu.2017.01526
13. Feng LL, Cai YQ, Zhu MC, Xing LJ, Wang X. The yin and yang functions of extracellular ATP and adenosine in tumor immunity. Cancer Cell Int (2020) 20:110. doi: 10.1186/s12935-020-01195-x
14. Mujoomdar M, Bennett A, Hoskin D, Blay J. Adenosine stimulation of proliferation of breast carcinoma cell lines: evaluation of the [3H]thymidine assay system and modulatory effects of the cellular microenvironment in vitro. J Cell Physiol (2004) 201(3):429–38. doi: 10.1002/jcp.20089
15. Mujoomdar M, Hoskin D, Blay J. Adenosine stimulation of the proliferation of colorectal carcinoma cell lines. roles of cell density and adenosine metabolism. Biochem Pharmacol (2003) 66(9):1737–47. doi: 10.1016/s0006-2952(03)00548-3
16. Tabrizchi R, Bedi S. Pharmacology of adenosine receptors in the vasculature. Pharmacol Ther (2001) 91(2):133–47. doi: 10.1016/s0163-7258(01)00152-8
17. Alvarez CL, Troncoso MF, Espelt MV. Extracellular ATP and adenosine in tumor microenvironment: Roles in epithelial-mesenchymal transition, cell migration, and invasion. J Cell Physiol (2022) 237(1):389–400. doi: 10.1002/jcp.30580
18. Burger JA, Burkle A. The CXCR4 chemokine receptor in acute and chronic leukaemia: a marrow homing receptor and potential therapeutic target. Br J Haematol (2007) 137(4):288–96. doi: 10.1111/j.1365-2141.2007.06590.x
19. Gelmini S, Mangoni M, Serio M, Romagnani P, Lazzeri E. The critical role of SDF-1/CXCR4 axis in cancer and cancer stem cells metastasis. J Endocrinol Invest (2008) 31(9):809–19. doi: 10.1007/BF03349262
20. Mortezaee K. CXCL12/CXCR4 axis in the microenvironment of solid tumors: A critical mediator of metastasis. Life Sci (2020) 249:117534. doi: 10.1016/j.lfs.2020.117534
21. Blay J. CD26/DPPIV in cancer progression and spread. In: Encyclopedia of cancer, 2nd ed. Heidelberg: Springer Verlag (2009). p. 541–4.
22. Cutler MJ, Lowthers EL, Richard CL, Hajducek DM, Spagnuolo PA, Blay J. Chemotherapeutic agents attenuate CXCL12-mediated migration of colon cancer cells by selecting for CXCR4-negative cells and increasing peptidase CD26. BMC Cancer (2015) 15:882. doi: 10.1186/s12885-015-1702-2
23. Richard CL, Tan EY, Blay J. Adenosine upregulates CXCR4 and enhances the proliferative and migratory responses of human carcinoma cells to CXCL12/SDF-1alpha. Int J Cancer (2006) 119(9):2044–53. doi: 10.1002/ijc.22084
24. Tan EY, Mujoomdar M, Blay J. Adenosine down-regulates the surface expression of dipeptidyl peptidase IV on HT-29 human colorectal carcinoma cells: implications for cancer cell behavior. Am J Pathol (2004) 165(1):319–30. doi: 10.1016/S0002-9440(10)63299-3
25. Richard CL, Lowthers EL, Blay J. 15-Deoxy-delta(12,14)-prostaglandin J(2) down-regulates CXCR4 on carcinoma cells through PPARgamma- and NFkappaB-mediated pathways. Exp Cell Res (2007) 313(16):3446–58. doi: 10.1016/j.yexcr.2007.06.027
26. Richard CL, Blay J. CXCR4 in cancer and its regulation by PPARgamma. PPAR Res (2008) 2008:769413. doi: 10.1155/2008/769413
27. Tan EY, Richard CL, Zhang H, Hoskin DW, Blay J. Adenosine downregulates DPPIV on HT-29 colon cancer cells by stimulating protein tyrosine phosphatase(s) and reducing ERK1/2 activity via a novel pathway. Am J Physiol Cell Physiol (2006) 291(3):C433–44. doi: 10.1152/ajpcell.00238.2005
28. Zhang H, Conrad DM, Butler JJ, Zhao C, Blay J, Hoskin DW. Adenosine acts through A2 receptors to inhibit IL-2-induced tyrosine phosphorylation of STAT5 in T lymphocytes: role of cyclic adenosine 3',5'-monophosphate and phosphatases. J Immunol (2004) 173(2):932–44. doi: 10.4049/jimmunol.173.2.932
29. Butler JJ, Mader JS, Watson CL, Zhang H, Blay J, Hoskin DW. Adenosine inhibits activation-induced T cell expression of CD2 and CD28 co-stimulatory molecules: role of interleukin-2 and cyclic AMP signaling pathways. J Cell Biochem (2003) 89(5):975–91. doi: 10.1002/jcb.10562
30. Strakhova R, Cadassou O, Cros-Perrial E, Jordheim LP. Regulation of tumor infiltrated innate immune cells by adenosine. Purinergic Signal (2020) 16(3):289–95. doi: 10.1007/s11302-020-09701-6
31. Whiteside TL, Mandapathil M, Szczepanski M, Szajnik M. Mechanisms of tumor escape from the immune system: adenosine-producing treg, exosomes and tumor-associated TLRs. Bull Cancer (2011) 98(2):E25–31. doi: 10.1684/bdc.2010.1294
32. Ohta A. A metabolic immune checkpoint: Adenosine in tumor microenvironment. Front Immunol (2016) 7:109. doi: 10.3389/fimmu.2016.00109
33. Arab S, Hadjati J. Adenosine blockage in tumor microenvironment and improvement of cancer immunotherapy. Immune Netw (2019) 19(4):e23. doi: 10.4110/in.2019.19.e23
34. Soleimani A, Bahreyni A, Roshan MK, Soltani A, Ryzhikov M, Shafiee M, et al. Therapeutic potency of pharmacological adenosine receptors agonist/antagonist on cancer cell apoptosis in tumor microenvironment, current status, and perspectives. J Cell Physiol (2019) 234(3):2329–36. doi: 10.1002/jcp.27249
35. Cai Y, Feng L, Wang X. Targeting the tumor promoting effects of adenosine in chronic lymphocytic leukemia. Crit Rev Oncol Hematol (2018) 126:24–31. doi: 10.1016/j.critrevonc.2018.03.022
36. Ohana G, Bar-Yehuda S, Barer F, Fishman P. Differential effect of adenosine on tumor and normal cell growth: focus on the A3 adenosine receptor. J Cell Physiol (2001) 186(1):19–23. doi: 10.1002/1097-4652(200101)186:1<19::AID-JCP1011>3
37. Helms RS, Powell JD. Rethinking the adenosine-A2AR checkpoint: implications for enhancing anti-tumor immunotherapy. Curr Opin Pharmacol (2020) 53:77–83. doi: 10.1016/j.coph.2020.07.003
38. Baraldi PG, Tabrizi MA, Romagnoli R, Fruttarolo F, Merighi S, Varani K, et al. Pyrazolo[4,3-e]1,2,4-triazolo[1,5-c]pyrimidine ligands, new tools to characterize A3 adenosine receptors in human tumor cell lines. Curr Med Chem (2005) 12(11):1319–29. doi: 10.2174/0929867054020963
39. Hoskin DW, Mader JS, Furlong SJ, Conrad DM, Blay J. Inhibition of T cell and natural killer cell function by adenosine and its contribution to immune evasion by tumor cells (Review). Int J Oncol (2008) 32(3):527–35.
40. Sepulveda C, Palomo I, Fuentes E. Role of adenosine A2b receptor overexpression in tumor progression. Life Sci (2016) 166:92–9. doi: 10.1016/j.lfs.2016.10.008
41. Zhang J, Yan W, Duan W, Wuthrich K, Cheng J. Tumor immunotherapy using A2A adenosine receptor antagonists. Pharm (Basel) (2020) 13(9):14. doi: 10.3390/ph13090237
42. MacKenzie WM, Hoskin DW, Blay J. Adenosine inhibits the adhesion of anti-CD3-activated killer lymphocytes to adenocarcinoma cells through an A3 receptor. Cancer Res (1994) 54(13):3521–6.
43. Sousa JB, Diniz C. The adenosinergic system as a therapeutic target in the vasculature: New ligands and challenges. Molecules (2017) 22(5):27. doi: 10.3390/molecules22050752
44. Hatfull GF, Dedrick RM, Schooley RT. Phage therapy for antibiotic-resistant bacterial infections. Annu Rev Med (2022) 73:197–211. doi: 10.1146/annurev-med-080219-122208
45. Aranaga C, Pantoja LD, Martinez EA, Falco A. Phage therapy in the era of multidrug resistance in bacteria: A systematic review. Int J Mol Sci (2022) 23(9):20. doi: 10.3390/ijms23094577
46. Liu C, Hong Q, Chang RYK, Kwok PCL, Chan HK. Phage-antibiotic therapy as a promising strategy to combat multidrug-resistant infections and to enhance antimicrobial efficiency. Antibiot (Basel) (2022) 11(5):22. doi: 10.3390/antibiotics11050570.
47. Marongiu L, Burkard M, Lauer UM, Hoelzle LE, Venturelli S. Reassessment of historical clinical trials supports the effectiveness of phage therapy. Clin Microbiol Rev (2022):e0006222. doi: 10.1128/cmr.00062-22
48. Stacey HJ, De Soir S, Jones JD. The safety and efficacy of phage therapy: A systematic review of clinical and safety trials. Antibiot (Basel) (2022) 11(10):13. doi: 10.3390/antibiotics11101340.
49. Diallo K, Dublanchet A. Benefits of combined phage-antibiotic therapy for the control of antibiotic-resistant bacteria: A literature review. Antibiot (Basel) (2022) 11(7):11. doi: 10.3390/antibiotics11070839.
50. Xu HM, Xu WM, Zhang L. Current status of phage therapy against infectious diseases and potential application beyond infectious diseases. Int J Clin Pract (2022) 2022:4913146. doi: 10.1155/2022/4913146
51. Glonti T, Pirnay JP. In vitro techniques and measurements of phage characteristics that are important for phage therapy success. Viruses (2022) 14(7):23. doi: 10.3390/v14071490
52. Hosseinidoust Z. Phage-mediated gene therapy. Curr Gene Ther (2017) 17(2):120–6. doi: 10.2174/1566523217666170510151940
53. Beghetto E, Gargano N. Lambda-display: a powerful tool for antigen discovery. Molecules (2011) 16(4):3089–105. doi: 10.3390/molecules16043089
54. Pasqualini R, Ruoslahti E. Organ targeting in vivo using phage display peptide libraries. Nature (1996) 380(6572):364–6. doi: 10.1038/380364a0
55. Nicastro J, Sheldon K, Slavcev RA. Bacteriophage lambda display systems: developments and applications. Appl Microbiol Biotechnol (2014) 98(7):2853–66. doi: 10.1007/s00253-014-5521-1
56. Alizadeh AA, Morris MB, Church WB, Yaqoubi S, Dastmalchi S. A mechanistic perspective, clinical applications, and phage-display-assisted discovery of TNFalpha inhibitors. Drug Discovery Today (2022) 27(2):503–18. doi: 10.1016/j.drudis.2021.09.024
57. Aloisio A, Nistico N, Mimmi S, Maisano D, Vecchio E, Fiume G, et al. Phage-displayed peptides for targeting tyrosine kinase membrane receptors in cancer therapy. Viruses (2021) 13(4):19. doi: 10.3390/v13040649.
58. Ch'ng ACW, Lam P, Alassiri M, Lim TS. Application of phage display for T-cell receptor discovery. Biotechnol Adv (2022) 54:107870. doi: 10.1016/j.biotechadv.2021.107870
59. Ledsgaard L, Ljungars A, Rimbault C, Sorensen CV, Tulika T, Wade J, et al. Advances in antibody phage display technology. Drug Discov Today (2022) 27(8):2151–69. doi: 10.1016/j.drudis.2022.05.002
60. Li L, Wu S, Si Y, Li H, Yin X, Peng D. Single-chain fragment variable produced by phage display technology: Construction, selection, mutation, expression, and recent applications in food safety. Compr Rev Food Sci Food Saf (2022) 21(5):4354–77. doi: 10.1111/1541-4337.13018
61. Roth KDR, Wenzel EV, Ruschig M, Steinke S, Langreder N, Heine PA, et al. Developing recombinant antibodies by phage display against infectious diseases and toxins for diagnostics and therapy. Front Cell Infect Microbiol (2021) 11:697876. doi: 10.3389/fcimb.2021.697876
62. Sokullu E, Gauthier MS, Coulombe B. Discovery of antivirals using phage display. Viruses (2021) 13(6):26. doi: 10.3390/v13061120.
63. Zhang K, Tang Y, Chen Q, Liu Y. The screening of therapeutic peptides for anti-inflammation through phage display technology. Int J Mol Sci (2022) 23(15)16. doi: 10.3390/ijms23158554.
64. Zhang X, Zhang X, Gao H, Qing G. Phage display derived peptides for alzheimer's disease therapy and diagnosis. Theranostics (2022) 12(5):2041–62. doi: 10.7150/thno.68636
65. Choi IK, Strauss R, Richter M, Yun CO, Lieber A. Strategies to increase drug penetration in solid tumors. Front Oncol (2013) 3:193. doi: 10.3389/fonc.2013.00193
66. Dreher MR, Liu W, Michelich CR, Dewhirst MW, Yuan F, Chilkoti A. Tumor vascular permeability, accumulation, and penetration of macromolecular drug carriers. J Natl Cancer Inst (2006) 98(5):335–44. doi: 10.1093/jnci/djj070
67. Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat Rev Cancer (2006) 6(8):583–92. doi: 10.1038/nrc1893
68. Manivannan AC, Dhandapani R, Velmurugan P, Thangavelu S, Paramasivam R, Ragunathan L, et al. Phage in cancer treatment - biology of therapeutic phage and screening of tumor targeting peptide. Expert Opin Drug Deliv (2022) 19(7):873–82. doi: 10.1080/17425247.2022.2094363
69. Sriraman SK, Aryasomayajula B, Torchilin VP. Barriers to drug delivery in solid tumors. Tissue Barriers (2014) 2:e29528. doi: 10.4161/tisb.29528
70. Ying X, Wen H, Lu WL, Du J, Guo J, Tian W, et al. Dual-targeting daunorubicin liposomes improve the therapeutic efficacy of brain glioma in animals. J Control Release (2010) 141(2):183–92. doi: 10.1016/j.jconrel.2009.09.020
71. Harrison EB, Azam SH, Pecot CV. Targeting accessories to the crime: Nanoparticle nucleic acid delivery to the tumor microenvironment. Front Pharmacol (2018) 9:307. doi: 10.3389/fphar.2018.00307
72. Liu Y, Zhou J, Li Q, Li L, Jia Y, Geng F, et al. Tumor microenvironment remodeling-based penetration strategies to amplify nanodrug accessibility to tumor parenchyma. Adv Drug Delivery Rev (2021) 172:80–103. doi: 10.1016/j.addr.2021.02.019
73. Maeng J, Lee K. Protein transduction domain of translationally controlled tumor protein: characterization and application in drug delivery. Drug Deliv (2022) 29(1):3009–21. doi: 10.1080/10717544.2022.2122636
74. Shaw TK, Paul P. Recent approaches and success of liposome-based nano drug carriers for the treatment of brain tumor. Curr Drug Deliv (2022) 19(8):815–29. doi: 10.2174/1567201818666211213102308
75. Mueller-Klieser W. Three-dimensional cell cultures: from molecular mechanisms to clinical applications. Am J Physiol (1997) 273(4):C1109–23. doi: 10.1152/ajpcell.1997.273.4.C1109
76. Helmlinger G, Yuan F, Dellian M, Jain RK. Interstitial pH and pO2 gradients in solid tumors in vivo: high-resolution measurements reveal a lack of correlation. Nat Med (1997) 3(2):177–82. doi: 10.1038/nm0297-177
77. Vukovic V, Tannock IF. Influence of low pH on cytotoxicity of paclitaxel, mitoxantrone and topotecan. Br J Cancer (1997) 75(8):1167–72. doi: 10.1038/bjc.1997.201
78. Cox TR, Bird D, Baker AM, Barker HE, Ho MW, Lang G, et al. LOX-mediated collagen crosslinking is responsible for fibrosis-enhanced metastasis. Cancer Res (2013) 73(6):1721–32. doi: 10.1158/0008-5472.CAN-12-2233
79. Provenzano PP, Inman DR, Eliceiri KW, Knittel JG, Yan L, Rueden CT, et al. Collagen density promotes mammary tumor initiation and progression. BMC Med (2008) 6:11. doi: 10.1186/1741-7015-6-11
80. Jain RK. Vascular and interstitial barriers to delivery of therapeutic agents in tumors. Cancer Metastasis Rev (1990) 9(3):253–66. doi: 10.1007/BF00046364
81. Jain RK. Delivery of molecular and cellular medicine to solid tumors. Adv Drug Deliv Rev (2012) 64(Suppl):353–65. doi: 10.1016/j.addr.2012.09.011
82. Seynhaeve AL, Hoving S, Schipper D, Vermeulen CE, de Wiel-Ambagtsheer G, van Tiel ST, et al. Tumor necrosis factor alpha mediates homogeneous distribution of liposomes in murine melanoma that contributes to a better tumor response. Cancer Res (2007) 67(19):9455–62. doi: 10.1158/0008-5472.CAN-07-1599
83. Tredan O, Galmarini CM, Patel K, Tannock IF. Drug resistance and the solid tumor microenvironment. J Natl Cancer Inst (2007) 99(19):1441–54. doi: 10.1093/jnci/djm135
84. Raja J, Ludwig JM, Gettinger SN, Schalper KA, Kim HS. Oncolytic virus immunotherapy: future prospects for oncology. J Immunother Cancer (2018) 6(1):140. doi: 10.1186/s40425-018-0458-z
85. Dubos RJ, Straus JH, Pierce C. The multiplication of bacteriophage in vivo and its protective effect against an experimental infection with shigella dysenteriae. J Exp Med (1943) 78(3):161–8. doi: 10.1084/jem.78.3.161
86. Huh H, Wong S, St Jean J, Slavcev R. Bacteriophage interactions with mammalian tissue: Therapeutic applications. Adv Drug Deliv Rev (2019) 145:4–17. doi: 10.1016/j.addr.2019.01.003
87. Majewska J, Beta W, Lecion D, Hodyra-Stefaniak K, Klopot A, Kazmierczak Z, et al. Oral application of T4 phage induces weak antibody production in the gut and in the blood. Viruses (2015) 7(8):4783–99. doi: 10.3390/v7082845
88. Miedzybrodzki R, Klak M, Jonczyk-Matysiak E, Bubak B, Wojcik A, Kaszowska M, et al. Means to facilitate the overcoming of gastric juice barrier by a therapeutic staphylococcal bacteriophage A5/80. Front Microbiol (2017) 8:467. doi: 10.3389/fmicb.2017.00467
89. Nguyen S, Baker K, Padman BS, Patwa R, Dunstan RA, Weston TA, et al. Bacteriophage transcytosis provides a mechanism to cross epithelial cell layers. mBio (2017) 8(6). doi: 10.1128/mBio.01874-17
90. Nguyen S, Baker K, Padman BS, Patwa R, Dunstan RA, Weston TA, et al. Bacteriophage transcytosis provides a mechanism to cross epithelial cell layers. mBio (2018) 9(1). doi: 10.1128/mBio.02207-17
91. Smith GP. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science (1985) 228(4705):1315–7. doi: 10.1126/science.4001944
92. Poul MA, Becerril B, Nielsen UB, Morisson P, Marks JD. Selection of tumor-specific internalizing human antibodies from phage libraries. J Mol Biol (2000) 301(5):1149–61. doi: 10.1006/jmbi.2000.4026
93. Alkasalias T, Moyano-Galceran L, Arsenian-Henriksson M, Lehti K. Fibroblasts in the tumor microenvironment: Shield or spear? Int J Mol Sci (2018) 19(5):21. doi: 10.3390/ijms19051532
94. Patel P, De P. Trends in colorectal cancer incidence and related lifestyle risk factors in 15-49-year-olds in Canada, 1969-2010. Cancer Epidemiol (2016) 42:90–100. doi: 10.1016/j.canep.2016.03.009
95. Shapiro S, Hughes G, Al-Obaidi MJ, O'Reilly E, Ramesh S, Smith J, et al. Acute myeloid leukaemia secondary to treatment with capecitabine for metastatic colorectal cancer. Eur J Haematol (2007) 78(6):543–4. doi: 10.1111/j.1600-0609.2007.00864.x
96. Lefort EC, Blay J. The dietary flavonoid apigenin enhances the activities of the anti-metastatic protein CD26 on human colon carcinoma cells. Clin Exp Metastasis (2011) 28(4):337–49. doi: 10.1007/s10585-010-9364-6
97. Lowthers EL, Richard CL, Blay J. Differential sensitivity to short-chain ceramide analogues of human intestinal carcinoma cells grown in tumor spheroids versus monolayer culture. In Vitro Cell Dev Biol Anim (2003) 39(8-9):340–2. doi: 10.1290/1543-706X(2003)039<0340:DSTSCA>2.0.CO;2
98. Rubin Grandis J, Melhem MF, Gooding WE, Day R, Holst VA, Wagener MM, et al. Levels of TGF-alpha and EGFR protein in head and neck squamous cell carcinoma and patient survival. J Natl Cancer Inst (1998) 90(11):824–32. doi: 10.1093/jnci/90.11.824
99. Yang JL, Qu XJ, Russell PJ, Goldstein D. Regulation of epidermal growth factor receptor in human colon cancer cell lines by interferon alpha. Gut (2004) 53(1):123–9. doi: 10.1136/gut.53.1.123
100. Mikawa YG, Maruyama IN, Brenner S. Surface display of proteins on bacteriophage lambda heads. J Mol Biol (1996) 262(1):21–30. doi: 10.1006/jmbi.1996.0495
101. Nicastro J, Sheldon K, El-Zarkout FA, Sokolenko S, Aucoin MG, Slavcev R. Construction and analysis of a genetically tuneable lytic phage display system. Appl Microbiol Biotechnol (2013) 97(17):7791–804. doi: 10.1007/s00253-013-4898-6
102. Sokolenko S, Nicastro J, Slavcev R, Aucoin MG. Graphical analysis of flow cytometer data for characterizing controlled fluorescent protein display on lambda phage. Cytometry A. (2012) 81(12):1031–9. doi: 10.1002/cyto.a.22211
103. Strausberg RL, Feingold EA, Grouse LH, Derge JG, Klausner RD, Collins FS, et al. Generation and initial analysis of more than 15,000 full-length human and mouse cDNA sequences. Proc Natl Acad Sci U S A. (2002) 99(26):16899–903. doi: 10.1073/pnas.242603899
104. Nicastro J, Blay J, Khazaei Z, Lam P, Slavcev RA, Wong S. Bacteriophage applications - historical perspective and future potential. 1st. Cham: Springer International Publishing (2016), ISBN: 97833194579182211-9353. doi: 10.1007/978-3-319-45791-8
105. Barbone D, Van Dam L, Follo C, Jithesh PV, Zhang SD, Richards WG, et al. Analysis of gene expression in 3D spheroids highlights a survival role for ASS1 in mesothelioma. PloS One (2016) 11(3):e0150044. doi: 10.1371/journal.pone.0150044
106. Barr JJ, Auro R, Furlan M, Whiteson KL, Erb ML, Pogliano J, et al. Bacteriophage adhering to mucus provide a non-host-derived immunity. Proc Natl Acad Sci U S A. (2013) 110(26):10771–6. doi: 10.1073/pnas.1305923110
107. Yata T, Lee EL, Suwan K, Syed N, Asavarut P, Hajitou A. Modulation of extracellular matrix in cancer is associated with enhanced tumor cell targeting by bacteriophage vectors. Mol Cancer (2015) 14:110. doi: 10.1186/s12943-015-0383-4
108. Emlet DR, Gupta P, Holgado-Madruga M, Del Vecchio CA, Mitra SS, Han SY, et al. Targeting a glioblastoma cancer stem-cell population defined by EGF receptor variant III. Cancer Res (2014) 74(4):1238–49. doi: 10.1158/0008-5472.CAN-13-1407
109. Yu D, Hung MC. Overexpression of ErbB2 in cancer and ErbB2-targeting strategies. Oncogene (2000) 19(53):6115–21. doi: 10.1038/sj.onc.1203972
110. Seshacharyulu P, Ponnusamy MP, Haridas D, Jain M, Ganti AK, Batra SK. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin Ther Targets (2012) 16(1):15–31. doi: 10.1517/14728222.2011.648617
111. Roth JA, Cristiano RJ. Gene therapy for cancer: what have we done and where are we going? J Natl Cancer Inst (1997) 89(1):21–39. doi: 10.1093/jnci/89.1.21.
112. Souriau C, Fort P, Roux P, Hartley O, Lefranc MP, Weill M. A simple luciferase assay for signal transduction activity detection of epidermal growth factor displayed on phage. Nucleic Acids Res (1997) 25(8):1585–90. doi: 10.1093/nar/25.8.1585
113. Kassner PD, Burg MA, Baird A, Larocca D. Genetic selection of phage engineered for receptor-mediated gene transfer to mammalian cells. Biochem Biophys Res Commun (1999) 264(3):921–8. doi: 10.1006/bbrc.1999.1603
114. Yang CH, Wu PC, Huang YB, Tsai YH. A new approach for determining the stability of recombinant human epidermal growth factor by thermal Fourier transform infrared (FTIR) microspectroscopy. J Biomol Struct Dyn (2004) 22(1):101–10. doi: 10.1080/07391102.2004.10506985
115. Xu CF, Solomon E. Mutations of the BRCA1 gene in human cancer. Semin Cancer Biol (1996) 7(1):33–40. doi: 10.1006/scbi.1996.0005
116. Yuen ST, Davies H, Chan TL, Ho JW, Bignell GR, Cox C, et al. Similarity of the phenotypic patterns associated with BRAF and KRAS mutations in colorectal neoplasia. Cancer Res (2002) 62(22):6451–5.
117. Blay J, Brown KD. Epidermal growth factor promotes the chemotactic migration of cultured rat intestinal epithelial cells. J Cell Physiol (1985) 124(1):107–12. doi: 10.1002/jcp.1041240117
118. Ivanenkov VV, Felici F, Menon AG. Targeted delivery of multivalent phage display vectors into mammalian cells. Biochim Biophys Acta (1999) 1448(3):463–72. doi: 10.1016/s0167-4889(98)00163-3
119. Porayath C, Salim A, Palillam Veedu A, Babu P, Nair B, Madhavan A, et al. Characterization of the bacteriophages binding to human matrix molecules. Int J Biol Macromol (2018) 110:608–15. doi: 10.1016/j.ijbiomac.2017.12.052
120. Berger C, Madshus IH, Stang E. Cetuximab in combination with anti-human IgG antibodies efficiently down-regulates the EGF receptor by macropinocytosis. Exp Cell Res (2012) 318(20):2578–91. doi: 10.1016/j.yexcr.2012.09.001
121. Gill GN, Kawamoto T, Cochet C, Le A, Sato JD, Masui H, et al. Monoclonal anti-epidermal growth factor receptor antibodies which are inhibitors of epidermal growth factor binding and antagonists of epidermal growth factor binding and antagonists of epidermal growth factor-stimulated tyrosine protein kinase activity. J Biol Chem (1984) 259(12):7755–60.
122. Harding J, Burtness B. Cetuximab: an epidermal growth factor receptor chemeric human-murine monoclonal antibody. Drugs Today (Barc) (2005) 41(2):107–27. doi: 10.1358/dot.2005.41.2.882662
Keywords: phage therapy, epidermal growth factor receptors, colorectal cancer, spheroids, tumor microenvironment
Citation: Huh H, Chen D-W, Foldvari M, Slavcev R and Blay J (2022) EGFR-targeted bacteriophage lambda penetrates model stromal and colorectal carcinoma tissues, is taken up into carcinoma cells, and interferes with 3-dimensional tumor formation. Front. Immunol. 13:957233. doi: 10.3389/fimmu.2022.957233
Received: 30 May 2022; Accepted: 11 November 2022;
Published: 16 December 2022.
Edited by:
Yi Zhang, First Affiliated Hospital of Zhengzhou University, ChinaReviewed by:
Grzegorz Wegrzyn, University of Gdansk, PolandAlicja Wegrzyn, Institute of Biochemistry and Biophysics (PAN), Poland
Copyright © 2022 Huh, Chen, Foldvari, Slavcev and Blay. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jonathan Blay, am9uYXRoYW4uYmxheUB1d2F0ZXJsb28uY2E=; Roderick Slavcev, cm9kZXJpY2suc2xhdmNldkB1d2F0ZXJsb28uY2E=
†Present address: Ding-Wen Chen, Comprehensive Bone Marrow Failure Center, Children’s Hospital of Philadelphia, Philadelphia, PA, United States