 Elsa Anes
Elsa Anes David Pires
David Pires Manoj Mandal
Manoj Mandal José Miguel Azevedo-Pereira
José Miguel Azevedo-Pereira
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol., 03 August 2022
Sec. Microbial Immunology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.955407
This article is part of the Research TopicWomen in Microbial ImmunologyView all 3 articles
Cathepsins were first described, as endolysosomal proteolytic enzymes in reference to the organelles where they degrade the bulk of endogenous and exogenous substrates in a slightly acidic environment. These substrates include pathogens internalized via endocytosis and/or marked for destruction by autophagy. However, the role of cathepsins during infection far exceeds that of direct digestion of the pathogen. Cathepsins have been extensively investigated in the context of tumour associated immune cells and chronic inflammation. Several cathepsin-dependent immune responses develop in the endocytic pathway while others take place in the cytosol, the nucleus, or in the extracellular space. In this review we highlight the spatial localization of cathepsins and their implications in immune activation and resolution pathways during infection.
The term cathepsin (CTS) was initially used to refer to eleven human lysosomal proteases namely CTSs B, C (J), F, H, K, L, O, S, V (L2), X (P,Y,Z), and W (lymphopain) [reviewed in (1–3)]. They all belong to the group of cysteine cathepsins named after the presence of a cysteine amino acid residue on their catalytic site responsible for hydrolysis of peptide bonds (3). In addition to cysteine cathepsins, aspartic cathepsins D and E and serine cathepsins A and G, were also introduced into the lysosomal CTS family (4, 5).
CTS are synthesized as procathepsins and are targeted to the lumen of the endoplasmic reticulum (ER) via a signal peptide. They are later modified in the Golgi, being tagged for lysosome sorting, usually via mannose-6-phosphate receptors (MPR) (6–8). The tagged procathepsins are either directly or indirectly sorted to endosomes/lysosomes after escaping MPR and being secreted out of the cell (9–11). Around 5% of all CTSs are secreted out of the cell by the regular biosynthetic/secretory pathway (12).
Innate immune cells, such as macrophages, are able to rescue some of these extracellular CTSs to the lysosomes by expressing the cation-independent mannose 6-phosphate scavenger receptor (CI-MPR) (7, 8). There, in the low pH of the late endocytic vesicles they are processed and activated to the mature form (13).
Indeed, immune cells such as macrophages, neutrophils, natural killer cells or cytotoxic CD8+ T-lymphocytes, can store CTSs either in endocytic lytic granules or in secretory lysosomes, where exocytosis leads to delivery of CTS or their processed products to the extracellular environment (14, 15).
Lysosomal enzymes were also found in less common locations, such as the cytosol and the nucleus (16–20). The cytosolic release of mature CTS is observed as a consequence of controlled lysosomal membrane permeabilization (LMP) or as a result of a more drastic damage (15, 21). Regarding the trafficking of CTS to the nucleus, this is mostly a diversion from the biosynthetic pathway, through mechanisms involving alternative translation initiation of the nascent protein lacking a signal peptide targeting the ER (22). Another described mechanism is exon skipping that generates truncated CTS with modified signal sequences, enabling the retention in the cytosol (23) or their nuclear targeting (24).
In this mini review, we will present recent advances in the understanding of the spatial localization of CTS and their implications during immune responses to infections. A general schematic representation is depicted in Figure 1.
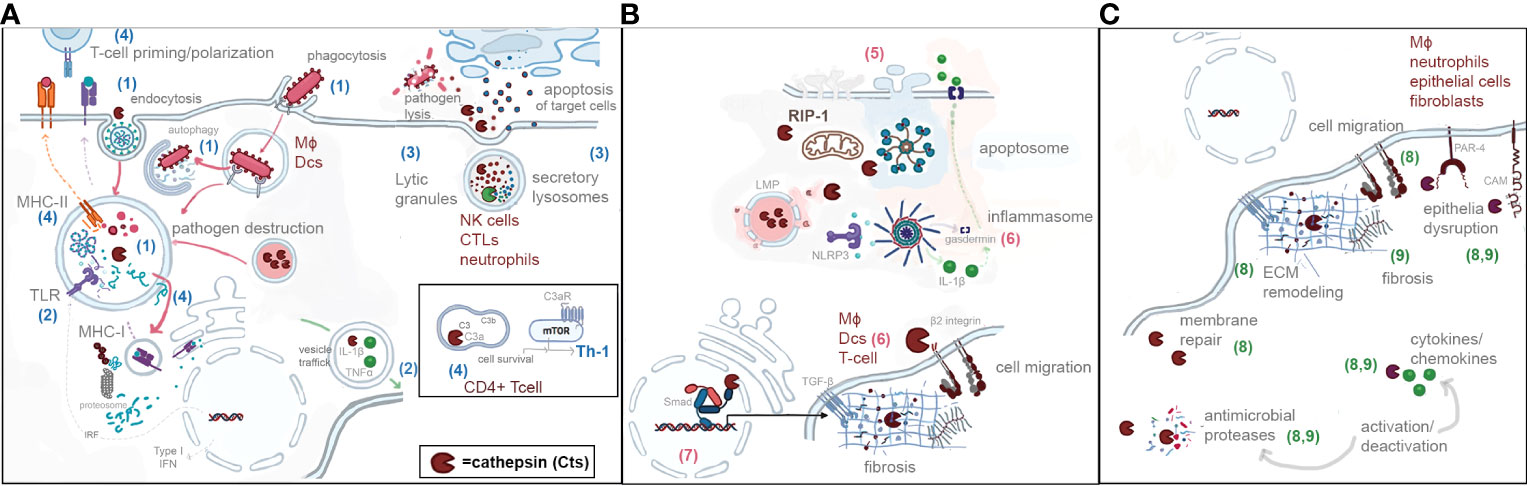
Figure 1 Schematic representation of the spatial localization of cathepsins and their roles in endocytic pathway (A) (blue numbers from 1 to 4), (B) cytosol and nucleus (pink numbers from 5 to 7), and (C) extracellular environment (green numbers 8 and 9). (1) Phagocytosis/endocytosis/autophagy. (2) Pattern recognition/cytokine activation. (3) Activation of proteases. (4) T-cell priming and polarization. (5) Programmed cell dead. (6) Inflammation. (7) Regulation of transcription. (8) ECM remodeling. (9) ECM inflammation during infection.
CTS perform major roles in phagocytosis/endocytosis and autophagy which are important cell autonomous immune mechanisms common to all cells (25). These innate mechanisms are prominent in professional phagocytes such as macrophages, and neutrophils, which constitute the first line of defense against pathogens. CTS mediate the destruction of theses pathogens due to their proteolytic activity at low pH, within the reducing environment of endolysosomes (26). It is not surprising that intracellular pathogens evolved virulence determinants to subvert the microbicidal mechanisms mediated by endolysosomal CTS as is the case for Mycobacterium tuberculosis (27–29), as well as for Salmonella, Brucella, Legionella or Chlamydia (30–33) or Francisella novicida (34). Autophagy intercepts the endolysosomal pathway (34–36) and may drive free cytosolic pathogens for destruction in lysosomes (37) or pathogens contained in vesicles (25, 38); both processes involving their entrapment in septin cages (39).
Indirectly, CTS regulate autophagy and may compromise intracellular pathogen clearance with direct implications on inflammation resolution and cell homeostasis (40–42). CTS S is required for autophagolysosome fusion events and its depletion results in accumulation of defective autophagosomes (43). CTS B suppresses the activity of a transcription factor required for expression of autophagy-related proteins (Atgs) by digestion of a calcium channel in the lysosomes (34). Also, the stimulation of the autophagy protein microtubule associated protein 1A/1B light chain 3 (LC3) is compromised by CTS K downregulation of endosomal TLR9 (44).
Innate immune receptors, such as Toll-like receptors (TLRs), detect pathogen-associated or cell damage associated signatures (PAMPS or DAMPS) leading to secretion of inflammatory cytokines such as IL-1β and TNFα. CTS interfere with both mechanisms. CTSs B, L, F or S, by cleaving and processing the ectodomains of endosomal TLRs such as 3 and 9, allow recognition of nucleic acids from endocytosed pathogens (44–48).The ectodomain cleavage represents a strategy to restrict receptor activation to endolysosomal compartments and prevent TLRs from responding to self nucleic acids (48).
CTS activity was demonstrated to either directly activate or inhibit inflammatory cytokines. While spatial localization in endosomes was not clarified, CTS B has been shown to be required for posttranslational processing and trafficking of TNFα (49) containing vesicles, and their secretion in response to TLRs 2, 4 and 9 stimulation (50). IL-1β is a potent inflammatory cytokine that needs to be tightly controlled (51, 52). Several CTS are involved in IL-1β processing in the cytosol. However, in monocytes, which are professional IL-1β producers, caspase-1 and pro- IL-1β coexists with CTS within special secretory endolysosomes (53, 54). This colocalization in vesicles located in the periphery of the cell suggests a less acidic and degradative environment (55) and seems to provide a regulatory mechanism of CTS over the amount of caspase-1 and IL-1β that are secreted by monocytes. In conventional endolysosomes, IL-1β and their precursors are normally degraded. Thus, the lysosomal pathway mediates IL-1β secretion but also provides a shutdown mechanism when IL-1β secretion is no longer needed (53, 54). Moreover, the autophagic removal of IL-1β cell activators, such as intracellular DAMPs, NLRP3 inflammasome components, and cytokines, in lysosomes contributes for deactivating the inflammatory responses (56). Cathepsins as degrading proteases in lysosomes are major players in this inflammation resolution.
During innate immune responses, neutrophils are cells involved in extracellular and intracellular pathogen clearance. Their effector functions depend on the activation of azurophil granules, serine proteases such as CTS G, granzymes, and elastase, all synthesised as inactive zymogens and activated by CTS C (57). Regulation of these neutrophil serine proteases activation is tightly controlled by sustained inhibition of CTS C through its natural inhibitor, cystatins (57).
In natural killer cells (NK cells) or in cytotoxic T lymphocytes (CTL), CTS C is responsible for the activation of progranzymes, generating granzymes A and B in secretory lysosomes (58). After immune activation they are processed and delivered out of the cell, where they induce apoptotic death of infected cells (58). CTS B is particularly relevant for protecting CTLs from their cytotoxic cargo (59). In addition, granzyme B is involved in regulating the function and maintenance of T helper cell populations (60). In regulatory T lymphocytes, activated granzymes can eliminate autologous effector cells by apoptosis, indicating an important role accomplished by Treg cells in exerting their anti-proliferative effects leading to immune resolution (61).
During adaptive immune responses, T lymphocyte priming by antigen-presenting cells (APCs) requires the recognition of processed antigenic peptides bound to major histocompatibility complex (MHC). CTS are crucial for the generation of these antigenic peptides from exogenous antigens in the endocytic pathway, and thus for CD4+ T lymphocytes priming. Furthermore, MHC class II requires CTS mediated proteolysis for degradation of the invariant chain (Ii) that blocks MHC class II molecule peptide binding site (62, 63). CTSs S, F, and L are cysteine proteases particularly implicated in these processes, with Cts S and F major players in in APCs and the last in thymocytes (29, 42, 64–67). CTS have also been shown to generate antigenic peptide motifs that favor particular T lymphocyte polarization, such as Th2 to Th1, in a mouse model of leishmaniasis (68).
CTS also impact MHC class I-mediated antigen presentation. While MHC class I molecules usually present cytosolic peptide antigens, exogenous pathogen antigens can be presented by this complex via cross-presentation. Exogenous antigens captured by dendritic cells are initially processed in the endocytic pathway by CTS S followed by their final processing in the cytosol before being presented to CD8+ T lymphocytes (69).
In addition to T cell priming, CTS were found to regulate T lymphocyte polarization independent of APCs (70). The complement system integrates innate and adaptive responses and could influence the magnitude of T cell activation (70). Naive CD4+ T lymphocytes store C3 in endosomes that can be cleaved by CTS L generating C3a and C3b. The C3aR-mediated intracellular signaling induces low levels of mechanistic target of rapamycin (mTOR) activation that regulate T cell survival (70). During infection, downstream signaling pathways of mTOR facilitates Th1 cell polarization from naive T cells (70, 71).
CTS, as stated before, may be released into the cytosol by controlled or uncontrolled lysosomal membrane permeabilization (LMP), leading to lysosomal dependent cell death (reviewed in (16, 21)). Extensive permeabilization leads to necrosis (72) while a less drastic release induces apoptosis (16, 73–77). Stringent controlled release of CTS will allow the cells to survive and physiologic responses to CTS either in the cytosol or in the nucleus (16, 78–80). In the case of Salmonella infection a control of necrotic cell death was found associated with accumulation of active cathepsins in the nucleus (20). The additional control of cathepsin activity in these compartments depends on the balance and expression of natural inhibitors (78–80). For instance, it was demonstrated that the cytosolic inhibitor Spi2A protected memory CD8+ T lymphocytes from lysosomal breakdown and cell death by inhibiting CTS B activity (81). Spi2A is a serine protease inhibitor with an unusual role inhibiting cysteine cathepsins after lysosomal permeabilization (81). Consequently, this extends the lifespan of memory T cells.
The B-cell lymphoma-2 (Bcl-2) family proteins regulate the mitochondrial pathway of apoptosis. Interestingly, this family includes proteins with anti-apoptotic (e.g., Bcl-2 and Bcl-xL) and pro-apoptotic (e.g., Bax, Bak and Bid) activities and CTSs have direct roles in regulating several members of Bcl-2 proteins. For example, CTSs B, D, and L induce the activation of Bid, resulting in its translocation to mitochondria resulting in cytochrome C release and caspase activation. Moreover, CTS degrade anti-apoptotic proteins Bcl-2, Bcl-xL, Mcl-1, and XIAP (X-linked inhibitor of apoptosis), promoting apoptosis (82, 83).). In T lymphocytes, CTS D degrades Bax, triggering apoptosis via release of cystatin C and AIF (apoptosis-inducing factor) which directly activates caspase-8 (84–86). Finally, additional CTS (e.g. C, F, H, K, L, O, S, V, W, and X) also function as mediators of lysosomal cell death either in immune and non-immune cells ls (10).
Other forms of programmed cell death lead to inflammation through cell lysis as is the case of necroptosis and pyroptosis. Necroptosis requires the kinase activity of receptor-interacting serine/threonine kinase1 (Rip1), a protein that is cleaved by CTS B and S thus controlling inflammatory cell death (17). Pyroptosis is mediated by gasdermin, a pore forming protein dependent on inflammasome activation (87, 88). After LMP, CTS B and L are major inflammasome inducers that may lead to this form of cell death therefore enhancing the inflammatory responses (89).
As stated, CTS released to the cytosol following LMP are relevant activators of inflammasomes, structures involved in innate immune responses (18, 90). Among inflammasomes the NLRP3 inflammasome is a major complex of assembled proteins in response to LMP, DAMPS or PAMPS (91–94). It is required for caspase-1 activation in the cytosol that in turn cleaves pro-IL-1β to their inflammatory mature form (90). Although CTS B and L have been associated with NLRP3 inflammasome activation, several siRNA experiments implicated CTS S and X (Z), particularly in contexts were they may compensate the lake of activity of CTS B and L (18, 95).
Inflammation is concomitant with migration of immune cells into tissues, such as lymphocytes and macrophages. CTS X is highly expressed in immune cells namely macrophages, dendritic cells and T lymphocytes (79). Its function has been associated to inflammatory responses such as cell adhesion, cell migration and phagocytosis. Some of these processes are the result of CTS X activation of transmembrane surface proteins, β2 integrins. (96–98). To do so CTS X cleaves the last four amino-acids contained in the cytosolic part of C-terminal region of β2 integrins, either Mac-1 receptor in macrophages and dendritic cells, or LFA-1 in T lymphocytes. Activation of Mac-1 enhances adhesion of macrophages and dendritic cells to extracellular matrix (ECM), improving phagocytosis and subsequent maturation of dendritic cells, a process essential for antigen processing and presentation (96). Activation of LFA-1 causes proliferation and tissue homing of T lymphocytes characteristic of acute and chronic inflammations (98).
CTS traffic to the nucleus has been associated to activation of transcription factors that control cell proliferation and differentiation (19, 22). Among transcription factors, CDP/Cux/Cut is activated by CTS L enabling accelerated cell cycle progression and carcinogenesis (19). Nuclear activity of CTS L was associated to an abnormal nuclear trafficking of the full length protein when stefinB, a CTS L inhibitor, is absent (19).
CTSs K and S were shown to interfere with nuclear membrane transport and control TGF-β signaling, leading to ECM synthesis required for cell growth and tissue fibrosis that often occurs during infections (99). They modulate the nuclear import of Smad proteins transcription factors that in turn regulate the expression of profibrotic genes such as collagen and fibronectin. In opposition, CTS B and L in nuclear membrane inhibit the effects of CTS K and S leading to decreased TGF-β signaling (99). This fibrotic pathological response may indeed be mitigated by extracellular CTS while promoting ECM degradation and helping tissue repair (100).
CTS emerge therefore as relevant players in the extracellular space as full degrading enzymes of ECM components, but the paradigm is now changing to enzymes that can specifically modify other extracellular proteins. Their secretion and activity are often dysregulated during inflammatory responses including infection [recently reviewed in (28)].
The structure of the ECM is dynamic and depends on the equilibrium between synthesis and degradation of a multitude of proteins (collagens, fibronectin, elastins), growth factors, proteoglycans, among others (11). ECM is vital to cell support and tissue integrity and has a series of regulatory functions. CTSs K, S, and V possess strong collagenolytic and elastolytic activities suggesting their involvement in ECM remodeling (2, 11). The best studied is CTS K that degrades type I collagens being essential for normal bone resorption (101). Other targets of CTS are cell adhesion contacts, influencing epithelial barriers, and cell adhesion to ECM, leading to changes in cell growth, cell migration, angiogenesis (102) and tissue repair (11, 103, 104). CTSs B and L have been shown to be released by lysosomal exocytosis playing a role in repair of the plasma membrane (105). CTS B, released from keratinocytes, attaches to cell surface where it is known to be involved in keratinocyte migration by degrading components of ECM during wound healing (106).
CTS secretion to ECM is usually high during infection. Microorganisms are sensed by innate immune receptors in mucosal cells that respond with an increased secretion of a myriad of proteases including antimicrobial peptides and CTS all having antimicrobial effects (28, 104, 107). This is the case of CTS K, highly expressed in intestinal Goblet cells, or CTS G, secreted from Paneth cells, that contributes to pathogen and microbiota control, and epithelial barrier repair (104, 107). In bronchial mucosa a protective effect was attributed to CTSs B and L (28) while CTS S, expressed mainly in macrophages, may favor the motility of cilia by preventing unspecific binding with airway circulating proteins (6).
However, the proteolytic activity of CTS may also aid infections by cleaving viral envelope proteins activating their receptor-binding or fusogenic activities, thus favoring viral infection (108–111). In chronic inflammatory conditions high concentrations of CTSs B, L, and S have been shown to cleave and inactivate several proteases, impairing their antimicrobial properties (2, 112, 113). The extensive destruction of lung parenchyma in tuberculosis is related with high levels of CTSs K, S, and V (114). CTSs G and D favor autolysis inside tuberculosis granulomas contributing to their liquefaction and disruption thus facilitating pathogen dissemination (28, 115).
The ECM breakdown products produced by extracellular proteases, including cathepsins, may act themselves as DAMPS, leading to the activation of NLRP3 inflammasomes exacerbating tissue inflammatory responses (116).
Extracellular CTS are also able to process cytokines and chemokines. CTSs L, S, and K, were shown to activate the glutamate-leucin-arginine motif (ELR) CXC ELR and inactivate non-ELR (CXCL9–12) chemokines thereby contributing to leukocyte recruitment during protective or pathological inflammation (117). CTS L secreted from fibroblasts and CTS G secreted from macrophages, neutrophils, and epithelial cells are activators of IL-8 (CXCL8). IL-8 acts both as a strong neutrophil chemoattractant, and as a proinflammatory cytokine (118, 119). In addition, CTS G activates IL-1β and TNFα as well as various signaling receptors (120). In contrast, CTS G can reduce dramatically the activity of IL-6 in fluids from inflammatory sites (121).
In the extracellular space CTS are able to cleave ectodomains of receptors and cell adhesion molecules at the cell surface, influencing by this mechanism several signaling pathways (122, 123). CTSs L and S secreted from macrophages were shown to shed CAM adhesion proteins and receptor tyrosine kinases (123). Dysbiosis-induced disruption of the epithelial barrier was found to be related with ectodomain activation of protease-activated receptor 4 (PAR 4) by neutrophil CTS G (124).
Cathepsins spatial localization is associated with distinct key roles of immune responses, with strong implications for infection control and inflammation resolution. Thus, CTS manipulations within these spatial contexts constitute potential targets for the development of new therapeutic strategies to fight infections, particular for those pathogens that developed drug resistance mechanisms to conventional treatments. The enhancement of their activity in situations where pathogen survival relies on their inhibition (e.g., drugs targeting autophagy) may help pathogen eradication from infected cells. Conversely, when infection results in poor antigen presentation, manipulation of CTS activity may improve the adaptive response and vaccine efficacy. Pathological inflammation is often a consequence of an infection. Targeting the control of inflammatory pathways may help to prevent or resolve tissue destruction and fibrotic events. There is still plenty to be investigated in this very promising area of research to fight the increasing threat of infections.
Conceptualization and writing: EA. Review and editing: DP, JA-P, and MM. Image: EA. Supervision and funding acquisition: EA. All authors contributed to the article and approved the submitted version.
This study was supported by grants from the National Foundation for Science, FCT Fundação para a Ciência e Tecnologia – Portugal, PTDC/SAU-INF/28182/2017 to EA, UIDB/04138/2020 (to IMed-ULisboa).
ADEIM-FFUL (Associação para o Desenvolvimento do Ensino e Investigação em Microbiologia).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Patel S, Homaei A, El-Seedi HR, Akhtar N. Cathepsins: Proteases that are vital for survival but can also be fatal. BioMed Pharmacother (2018) 105:526–32. doi: 10.1016/j.biopha.2018.05.148
2. Lecaille F, Lalmanach G, Andrault P-M. Antimicrobial proteins and peptides in human lung diseases: A friend and foe partnership with host proteases. Biochimie (2016) 122:151–68. doi: 10.1016/j.biochi.2015.08.014
3. Turk V, Stoka V, Vasiljeva O, Renko M, Sun T, Turk B, et al. Cysteine cathepsins: from structure, function and regulation to new frontiers. Biochim Biophys Acta (2012) 1824:68–88. doi: 10.1016/j.bbapap.2011.10.002
4. Rawlings ND, Barrett AJ, Thomas PD, Huang X, Bateman A, Finn RD. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res (2018) 46:D624–32. doi: 10.1093/nar/gkx1134
5. Rawlings ND, Waller M, Barrett AJ, Bateman A. MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res (2014) 42:D503–9. doi: 10.1093/nar/gkt953
6. Chapman HA, Riese RJ, Shi GP. Emerging roles for cysteine proteases in human biology. Annu Rev Physiol (1997) 59:63–88. doi: 10.1146/annurev.physiol.59.1.63
7. Pohlmann R, Boeker MW, Figura von K. The two mannose 6-phosphate receptors transport distinct complements of lysosomal proteins. J Biol Chem (1995) 270:27311–8. doi: 10.1074/jbc.270.45.27311
8. Collette J, Bocock JP, Ahn K, Chapman RL, Godbold G, Yeyeodu S, et al. Biosynthesis and alternate targeting of the lysosomal cysteine protease cathepsin l. Int Rev Cytol (2004) 241:1–51. doi: 10.1016/S0074-7696(04)41001-8
9. Braulke T, Bonifacino JS. Sorting of lysosomal proteins. Biochim Biophys Acta (2009) 1793:605–14. doi: 10.1016/j.bbamcr.2008.10.016
10. Yadati T, Houben T, Bitorina A, Shiri-Sverdlov R. The ins and outs of cathepsins: Physiological function and role in disease management. Cells (2020) 9:1679. doi: 10.3390/cells9071679
11. Vidak E, Javoršek U, Vizovišek M, Turk B. Cysteine cathepsins and their extracellular roles: Shaping the microenvironment. Cells (2019) 8(264):1–24. doi: 10.3390/cells8030264
12. Reiser J, Adair B, Reinheckel T. Specialized roles for cysteine cathepsins in health and disease. J Clin Invest (2010) 120:3421–31. doi: 10.1172/JCI42918
13. Savina A, Amigorena S. Phagocytosis and antigen presentation in dendritic cells. Immunol Rev (2007) 219:143–56. doi: 10.1111/j.1600-065X.2007.00552.x
14. Eder C. Mechanisms of interleukin-1beta release. Immunobiology (2009) 214:543–53. doi: 10.1016/j.imbio.2008.11.007
15. Konjar Š, Sutton VR, Hoves S, Repnik U, Yagita H, Reinheckel T, et al. Human and mouse perforin are processed in part through cleavage by the lysosomal cysteine proteinase cathepsin l. Immunology (2010) 131:257–67. doi: 10.1111/j.1365-2567.2010.03299.x
16. Reinheckel T, Tholen M. Low-level lysosomal membrane permeabilization for limited release and sublethal functions of cathepsin proteases in the cytosol and nucleus. FEBS Open Bio (2022) 12:694–707. doi: 10.1002/2211-5463.13385
17. McComb S, Shutinoski B, Thurston S, Cessford E, Kumar K, Sad S. Cathepsins limit macrophage necroptosis through cleavage of Rip1 kinase. J Immunol (2014) 192:5671–8. doi: 10.4049/jimmunol.1303380
18. Campden RI, Zhang Y. The role of lysosomal cysteine cathepsins in NLRP3 inflammasome activation. Arch Biochem Biophys (2019) 670:32–42. doi: 10.1016/j.abb.2019.02.015
19. Tamhane T, lllukkumbura R, Lu S, Maelandsmo GM, Haugen MH, Brix K. Nuclear cathepsin l activity is required for cell cycle progression of colorectal carcinoma cells. Biochimie (2016) 122:208–18. doi: 10.1016/j.biochi.2015.09.003
20. Selkrig J, Li N, Hausmann A, Mangan MSJ, Zietek M, Mateus A, et al. Spatiotemporal proteomics uncovers cathepsin-dependent macrophage cell death during salmonella infection. Nat Microbiol (2020) 5:1119–33. doi: 10.1038/s41564-020-0736-7
21. Repnik U, Stoka V, Turk V, Turk B. Lysosomes and lysosomal cathepsins in cell death. Biochim Biophys Acta (2012) 1824:22–33. doi: 10.1016/j.bbapap.2011.08.016
22. Goulet B, Baruch A, Moon N-S, Poirier M, Sansregret LL, Erickson A, et al. A cathepsin l isoform that is devoid of a signal peptide localizes to the nucleus in s phase and processes the CDP/Cux transcription factor. Mol Cell (2004) 14:207–19. doi: 10.1016/s1097-2765(04)00209-6
23. Müntener K, Zwicky R, Csucs G, Rohrer J, Baici A. Exon skipping of cathepsin b: mitochondrial targeting of a lysosomal peptidase provokes cell death. J Biol Chem (2004) 279:41012–7. doi: 10.1074/jbc.M405333200
24. Bestvater F, Dallner C, Spiess E. The c-terminal subunit of artificially truncated human cathepsin b mediates its nuclear targeting and contributes to cell viability. BMC Cell Biol (2005) 6:16–9. doi: 10.1186/1471-2121-6-16
25. Randow F, MacMicking JD, James LC. Cellular self-defense: how cell-autonomous immunity protects against pathogens. Science (2013) 340:701–6. doi: 10.1126/science.1233028
26. Savina A, Vargas P, Guermonprez P, Lennon A-M, Amigorena S. Measuring pH, ROS production, maturation, and degradation in dendritic cell phagosomes using cytofluorometry-based assays. Methods Mol Biol (2010) 595:383–402. doi: 10.1007/978-1-60761-421-0_25
27. Pires D, Marques J, Pombo JP, Carmo N, Bettencourt P, Neyrolles O, et al. Role of cathepsins in mycobacterium tuberculosis survival in human macrophages. Sci Rep (2016) 6:32247. doi: 10.1038/srep32247
28. Anes E, Azevedo-Pereira JM, Pires D. Cathepsins and their endogenous inhibitors in host defense during mycobacterium tuberculosis and HIV infection. Front Immunol (2021) 12:726984. doi: 10.3389/fimmu.2021.726984
29. Pires D, Calado M, Velez T, Mandal M, Catalão MJ, Neyrolles O, et al. Modulation of cystatin c in human macrophages improves anti-mycobacterial immune responses to mycobacterium tuberculosis infection and coinfection with HIV. Front Immunol (2021) 12:742822. doi: 10.3389/fimmu.2021.742822
30. Hilbi H, Haas A. Secretive bacterial pathogens and the secretory pathway. Traffic (2012) 13:1187–97. doi: 10.1111/j.1600-0854.2012.01344.x
31. Khweek AA, Amer A. Replication of legionella pneumophila in human cells: Why are we susceptible? Front Microbiol (2010) 1:133. doi: 10.3389/fmicb.2010.00133
32. LaRock DL, Chaudhary A, Miller SI. Salmonellae interactions with host processes. Nat Rev Microbiol (2015) 13:191–205. doi: 10.1038/nrmicro3420
33. Sachdeva K, Sundaramurthy V. The interplay of host lysosomes and intracellular pathogens. Front Cell Infect Microbiol (2020) 10:595502. doi: 10.3389/fcimb.2020.595502
34. Man SM, Kanneganti T-D. Regulation of lysosomal dynamics and autophagy by CTSB/cathepsin b. Autophagy (2016) 12:2504–5. doi: 10.1080/15548627.2016.1239679
35. Birgisdottir ÅB, Johansen T. Autophagy and endocytosis - interconnections and interdependencies. J Cell Sci (2020) 133(10)JCS228114:1–16. doi: 10.1242/jcs.228114
36. Zhao YG, Zhang H. Autophagosome maturation: An epic journey from the ER to lysosomes. J Cell Biol (2019) 218:757–70. doi: 10.1083/jcb.201810099
37. Mostowy S, Shenoy AR. The cytoskeleton in cell-autonomous immunity: structural determinants of host defence. Nat Rev Immunol (2015) 15:559–73. doi: 10.1038/nri3877
38. Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and mycobacterium tuberculosis survival in infected macrophages. Cell (2004) 119:753–66. doi: 10.1016/j.cell.2004.11.038
39. Mostowy S, Cossart P. Septins: the fourth component of the cytoskeleton. Nat Rev Mol Cell Biol (2012) 13:183–94. doi: 10.1038/nrm3284
40. Tizon B, Sahoo S, Yu H, Gauthier S, Kumar AR, Mohan P, et al. Induction of autophagy by cystatin c: a mechanism that protects murine primary cortical neurons and neuronal cell lines. PLoS One (2010) 5:e9819. doi: 10.1371/journal.pone.0009819
41. Pawar K, Sharbati J, Einspanier R, Sharbati S. Mycobacterium bovis BCG interferes with miR-3619-5p control of cathepsin s in the process of autophagy. Front Cell Infect Microbiol (2016) 6:27. doi: 10.3389/fcimb.2016.00027
42. Pires D, Bernard EM, Pombo JP, Carmo N, Fialho C, Gutierrez MG, et al. Mycobacterium tuberculosis modulates miR-106b-5p to control cathepsin s expression resulting in higher pathogen survival and poor T-cell activation. Front Immunol (2017) 8:1819. doi: 10.3389/fimmu.2017.01819
43. Yang M, Liu J, Shao J, Qin Y, Ji Q, Zhang X, et al. Cathepsin s-mediated autophagic flux in tumor-associated macrophages accelerate tumor development by promoting M2 polarization. Mol Cancer (2014) 13:43–15. doi: 10.1186/1476-4598-13-43
44. Wei W, Ren J, Yin W, Ding H, Lu Q, Tan L, et al. Inhibition of ctsk modulates periodontitis with arthritis via downregulation of TLR9 and autophagy. Cell Prolif (2020) 53:e12722. doi: 10.1111/cpr.12722
45. Wei T, Gong J, Jamitzky F, Heckl WM, Stark RW, Rössle SC. Homology modeling of human toll-like receptors TLR7, 8, and 9 ligand-binding domains. Protein Sci (2009) 18:1684–91. doi: 10.1002/pro.186
46. Garcia-Cattaneo A, Gobert F-X, Müller M, Toscano F, Flores M, Lescure A, et al. Cleavage of toll-like receptor 3 by cathepsins b and h is essential for signaling. Proc Natl Acad Sci USA (2012) 109:9053–8. doi: 10.1073/pnas.1115091109
47. Matsumoto F, Saitoh S-I, Fukui R, Kobayashi T, Tanimura N, Konno K, et al. Cathepsins are required for toll-like receptor 9 responses. Biochem Biophys Res Commun (2008) 367:693–9. doi: 10.1016/j.bbrc.2007.12.130
48. Ewald SE, Lee BL, Lau L, Wickliffe KE, Shi G-P, Chapman HA, et al. The ectodomain of toll-like receptor 9 is cleaved to generate a functional receptor. Nature (2008) 456:658–62. doi: 10.1038/nature07405
49. Ha S-D, Martins A, Khazaie K, Han J, Chan BMC, Kim SO. Cathepsin b is involved in the trafficking of TNF-α-Containing vesicles to the plasma membrane in macrophages. J Immunol (2008) 181:690–7. doi: 10.4049/jimmunol.181.1.690
50. Murray RZ, Kay JG, Sangermani DG, Stow JL. A role for the phagosome in cytokine secretion. Science (2005) 310:1492–5. doi: 10.1126/science.1120225
51. Sitia R, Rubartelli A. Evolution, role in inflammation, and redox control of leaderless secretory proteins. J Biol Chem (2020) 295:7799–811. doi: 10.1074/jbc.REV119.008907
52. Orlowski GM, Sharma S, Colbert JD, Bogyo M, Robertson SA, Kataoka H, et al. Frontline science: Multiple cathepsins promote inflammasome-independent, particle-induced cell death during NLRP3-dependent IL-1β activation. J Leukoc Biol (2017) 102:7–17. doi: 10.1189/jlb.3HI0316-152R
53. Andrei C, Dazzi C, Lotti L, Torrisi MR, Chimini G, Rubartelli A. The secretory route of the leaderless protein interleukin 1beta involves exocytosis of endolysosome-related vesicles. Mol Biol Cell (1999) 10:1463–75. doi: 10.1091/mbc.10.5.1463
54. Keller M, Rüegg A, Werner S, Beer H-D. Active caspase-1 is a regulator of unconventional protein secretion. Cell (2008) 132:818–31. doi: 10.1016/j.cell.2007.12.040
55. Johnson DE, Ostrowski P, Jaumouillé V, Grinstein S. The position of lysosomes within the cell determines their luminal pH. J Cell Biol (2016) 212:677–92. doi: 10.1083/jcb.201507112
56. Biasizzo M, Kopitar-Jerala N. Interplay between NLRP3 inflammasome and autophagy. Front Immunol (2020) 11:591803. doi: 10.3389/fimmu.2020.591803
57. Méthot N, Rubin J, Guay D, Beaulieu C, Ethier D, Reddy TJ, et al. Inhibition of the activation of multiple serine proteases with a cathepsin c inhibitor requires sustained exposure to prevent pro-enzyme processing. J Biol Chem (2007) 282:20836–46. doi: 10.1074/jbc.M702615200
58. Perišić Nanut M, Sabotič J, Jewett A, Kos J. Cysteine cathepsins as regulators of the cytotoxicity of NK and T cells. Front Immunol (2014) 5:1459. doi: 10.3389/fimmu.2017.01459
59. Balaji KN, Schaschke N, Machleidt W, Catalfamo M, Henkart PA. Surface cathepsin b protects cytotoxic lymphocytes from self-destruction after degranulation. J Exp Med (2002) 196:493–503. doi: 10.1084/jem.20011836
60. Devadas S, Das J, Liu C, Zhang L, Roberts AI, Pan Z, et al. Granzyme b is critical for T cell receptor-induced cell death of type 2 helper T cells. Immunity (2006) 25:237–47. doi: 10.1016/j.immuni.2006.06.011
61. Grossman WJ, Verbsky JW, Barchet W, Colonna M, Atkinson JP, Ley TJ. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity (2004) 21:589–601. doi: 10.1016/j.immuni.2004.09.002
62. Riese RJ, Mitchell RN, Villadangos JA, Shi GP, Palmer JT, Karp ER, et al. Cathepsin s activity regulates antigen presentation and immunity. J Clin Invest (1998) 101:2351–63. doi: 10.1172/JCI1158
63. Riese RJ, Chapman HA. Cathepsins and compartmentalization in antigen presentation. Curr Opin Immunol (2000) 12:107–13. doi: 10.1016/s0952-7915(99)00058-8
64. Hsing LC, Rudensky AY. The lysosomal cysteine proteases in MHC class II antigen presentation. Immunol Rev (2005) 207:229–41. doi: 10.1111/j.0105-2896.2005.00310.x
65. Deussing J, Roth W, Saftig P, Peters C, Ploegh HL, Villadangos JA. Cathepsins b and d are dispensable for major histocompatibility complex class II-mediated antigen presentation. Proc Natl Acad Sci USA (1998) 95:4516–21. doi: 10.1073/pnas.95.8.4516
66. Driessen C, Bryant RAR, Lennon-Duménil A-M, Villadangos JA, Bryant PW, Shi G-P, et al. Cathepsin s controls the trafficking and maturation of mhc class II molecules in dendritic cells. J Cell Biol (1999) 147:775–90. doi: 10.1083/jcb.147.4.775
67. Shi GP, Bryant RA, Riese R, Verhelst S, Driessen C, Li Z, et al. Role for cathepsin f in invariant chain processing and major histocompatibility complex class II peptide loading by macrophages. J Exp Med (2000) 191:1177–86. doi: 10.1084/jem.191.7.1177
68. Maekawa Y, Himeno K, Ishikawa H, Hisaeda H, Sakai T, Dainichi T, et al. Switch of CD4+ T cell differentiation from Th2 to Th1 by treatment with cathepsin b inhibitor in experimental leishmaniasis. J Immunol (1998) 161:2120–7.
69. Shen L, Sigal LJ, Boes M, Rock KL. Important role of cathepsin s in generating peptides for TAP-independent MHC class I crosspresentation In vivo. Immunity (2004) 21:155–65. doi: 10.1016/j.immuni.2004.07.004
70. Reis ES, Mastellos DC, Hajishengallis G, Lambris JD. New insights into the immune functions of complement. Nat Rev Immunol (2019) 19:503–16. doi: 10.1038/s41577-019-0168-x
71. Liszewski MK, Kolev M, Le Friec G, Leung M, Bertram PG, Fara AF, et al. Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity (2013) 39:1143–57. doi: 10.1016/j.immuni.2013.10.018
72. Hentze H, Lin XY, Choi MSK, Porter AG. Critical role for cathepsin b in mediating caspase-1-dependent interleukin-18 maturation and caspase-1-independent necrosis triggered by the microbial toxin nigericin. Cell Death Differ (2003) 10:956–68. doi: 10.1038/sj.cdd.4401264
73. Laforge M, Petit F, Estaquier J, Senik A. Commitment to apoptosis in CD4(+) T lymphocytes productively infected with human immunodeficiency virus type 1 is initiated by lysosomal membrane permeabilization, itself induced by the isolated expression of the viral protein nef. J Virol (2007) 81:11426–40. doi: 10.1128/JVI.00597-07
74. Guicciardi ME, Leist M, Gores GJ. Lysosomes in cell death. Oncogene (2004) 23:2881–90. doi: 10.1038/sj.onc.1207512
75. Roberg K, Ollinger K. Oxidative stress causes relocation of the lysosomal enzyme cathepsin d with ensuing apoptosis in neonatal rat cardiomyocytes. Am J Pathol (1998) 152:1151–6.
76. Kågedal K, Zhao M, Svensson I, Brunk UT. Sphingosine-induced apoptosis is dependent on lysosomal proteases. Biochem J (2001) 359:335–43. doi: 10.1042/0264-6021:3590335
77. Bewley MA, Marriott HM, Tulone C, Francis SE, Mitchell TJ, Read RC, et al. A cardinal role for cathepsin d in Co-ordinating the host-mediated apoptosis of macrophages and killing of pneumococci. PLoS Pathog (2011) 7:e1001262. doi: 10.1371/journal.ppat.1001262
78. Kopitar-Jerala N. The role of cystatins in cells of the immune system. FEBS Lett (2006) 580:6295–301. doi: 10.1016/j.febslet.2006.10.055
80. Shah A, Bano B. Cystatins in health and diseases. Int J Pept Res Ther (2008) 15:85–6. doi: 10.1007/s10989-008-9160-1
81. Byrne SM, Aucher A, Alyahya S, Elder M, Olson ST, Davis DM, et al. Cathepsin b controls the persistence of memory CD8+ T lymphocytes. J Immunol (2012) 189:1133–43. doi: 10.4049/jimmunol.1003406
82. Chwieralski CE, Welte T, Bühling F. Cathepsin-regulated apoptosis. Apoptosis (2006) 11:143–9. doi: 10.1007/s10495-006-3486-y
83. Droga-Mazovec G, Bojic L, Petelin A, Ivanova S, Romih R, Repnik U, et al. Cysteine cathepsins trigger caspase-dependent cell death through cleavage of bid and antiapoptotic bcl-2 homologues. J Biol Chem (2008) 283:19140–50. doi: 10.1074/jbc.M802513200
84. Conus S, Simon H-U. Cathepsins and their involvement in immune responses. Swiss Med Wkly (2010) 140:w13042. doi: 10.4414/smw.2010.13042
85. Bidère N, Lorenzo HK, Carmona S, Laforge M, Harper F, Dumont C, et al. Cathepsin d triggers bax activation, resulting in selective apoptosis-inducing factor (AIF) relocation in T lymphocytes entering the early commitment phase to apoptosis. J Biol Chem (2003) 278:31401–11. doi: 10.1074/jbc.M301911200
86. Zi M, Xu Y. Involvement of cystatin c in immunity and apoptosis. Immunol Lett (2018) 196:80–90. doi: 10.1016/j.imlet.2018.01.006
87. Feng S, Fox D, Man SM. Mechanisms of gasdermin family members in inflammasome signaling and cell death. J Mol Biol (2018) 430:3068–80. doi: 10.1016/j.jmb.2018.07.002
88. Swanson KV, Deng M, Ting JP-Y. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol (2019) 29:301–489. doi: 10.1038/s41577-019-0165-0
89. Brojatsch J, Lima H, Palliser D, Jacobson LS, Muehlbauer SM, Furtado R, et al. Distinct cathepsins control necrotic cell death mediated by pyroptosis inducers and lysosome-destabilizing agents. Cell Cycle (2015) 14:964–72. doi: 10.4161/15384101.2014.991194
90. Tschopp J, Martinon F, Burns K. NALPs: a novel protein family involved in inflammation. Nat Rev Mol Cell Biol (2003) 4:95–104. doi: 10.1038/nrm1019
91. Amaral EP, Riteau N, Moayeri M, Maier N, Mayer-Barber KD, Pereira RM, et al. Lysosomal cathepsin release is required for NLRP3-inflammasome activation by mycobacterium tuberculosis in infected macrophages. Front Immunol (2018) 9:1427. doi: 10.3389/fimmu.2018.01427
92. Danilchanka O, Pires D, Anes E, Niederweis M. The mycobacterium tuberculosis outer membrane channel protein CpnT confers susceptibility to toxic molecules. Antimicrob Agents Chemother (2015) 59:2328–36. doi: 10.1128/AAC.04222-14
93. Duncan JA, Gao X, Huang MT-H, O'Connor BP, Thomas CE, Willingham SB, et al. Neisseria gonorrhoeae activates the proteinase cathepsin b to mediate the signaling activities of the NLRP3 and ASC-containing inflammasome. J Immunol (2009) 182:6460–9. doi: 10.4049/jimmunol.0802696
94. Mishra BB, Moura-Alves P, Sonawane A, Hacohen N, Griffiths G, Moita LF, et al. Mycobacterium tuberculosis protein ESAT-6 is a potent activator of the NLRP3/ASC inflammasome. Cellular Microbiol. (2010) 12:1046–63. doi: 10.1111/j.1462-5822.2010.01450.x
95. Orlowski GM, Colbert JD, Sharma S, Bogyo M, Robertson SA, Rock KL. Multiple cathepsins promote pro-IL-1β synthesis and NLRP3-mediated IL-1β activation. J Immunol (2015) 195:1685–97. doi: 10.4049/jimmunol.1500509
96. Kos J, Jevnikar Z, Obermajer N. The role of cathepsin X in cell signaling. Cell Adh Migr (2009) 3:164–6. doi: 10.4161/cam.3.2.7403
97. Obermajer N, Repnik U, Jevnikar Z, Turk B, Kreft M, Kos J. Cysteine protease cathepsin X modulates immune response via activation of beta2 integrins. Immunology (2008) 124:76–88. doi: 10.1111/j.1365-2567.2007.02740.x
98. Jevnikar Z, Obermajer N, Bogyo M, Kos J. The role of cathepsin X in the migration and invasiveness of T lymphocytes. J Cell Sci (2008) 121:2652–61. doi: 10.1242/jcs.023721
99. Zhang X, Zhou Y, Yu X, Huang Q, Fang W, Li J, et al. Differential roles of cysteinyl cathepsins in TGF-β signaling and tissue fibrosis. iScience (2019) 19:607–22. doi: 10.1016/j.isci.2019.08.014
100. Obermajer N, Jevnikar Z, Doljak B, Kos J. Role of cysteine cathepsins in matrix degradation and cell signalling. Connect Tissue Res (2008) 49:193–6. doi: 10.1080/03008200802143158
101. Aguda AH, Panwar P, Du X, Nguyen NT, Brayer GD, Brömme D. Structural basis of collagen fiber degradation by cathepsin K. Proc Natl Acad Sci USA (2014) 111:17474–9. doi: 10.1073/pnas.1414126111
102. Gocheva V, Zeng W, Ke D, Klimstra D, Reinheckel T, Peters C, et al. Distinct roles for cysteine cathepsin genes in multistage tumorigenesis. Genes Dev (2006) 20:543–56. doi: 10.1101/gad.1407406
103. Li X, Wu Z, Ni J, Liu Y, Meng J, Yu W, et al. Cathepsin b regulates collagen expression by fibroblasts via prolonging TLR2/NF-κB activation. Oxid Med Cell Longev (2016) 2016:7894247. doi: 10.1155/2016/7894247
104. Zamolodchikova TS, Shcherbakov IT, Khrennikov BN, Shoibonov BB, Svirshchevskaya EV. Cathepsin G in the immune defense of the human duodenum: New sources for biosynthesis. Hum Physiol (2017) 43:326–33. doi: 10.1134/S0362119717020177
105. Castro-Gomes T, Corrotte M, Tam C, Andrews NW. Plasma membrane repair is regulated extracellularly by proteases released from lysosomes. PLoS One (2016) 11:e0152583. doi: 10.1371/journal.pone.0152583
106. Büth H, Luigi Buttigieg P, Ostafe R, Rehders M, Dannenmann SR, Schaschke N, et al. Cathepsin b is essential for regeneration of scratch-wounded normal human epidermal keratinocytes. Eur J Cell Biol (2007) 86:747–61. doi: 10.1016/j.ejcb.2007.03.009
107. Sina C, Lipinski S, Gavrilova O, Aden K, Rehman A, Till A, et al. Extracellular cathepsin K exerts antimicrobial activity and is protective against chronic intestinal inflammation in mice. Gut (2013) 62:520–30. doi: 10.1136/gutjnl-2011-300076
108. Zhao M-M, Yang W-L, Yang F-Y, Zhang L, Huang W-J, Hou W, et al. Cathepsin l plays a key role in SARS-CoV-2 infection in humans and humanized mice and is a promising target for new drug development. Signal Transduct Target Ther (2021) 6:134–12. doi: 10.1038/s41392-021-00558-8
109. Kubo Y, Hayashi H, Matsuyama T, Sato H, Yamamoto N. Retrovirus entry by endocytosis and cathepsin proteases. Adv Virol (2012) 2012:1–14. doi: 10.1155/2012/640894
110. Simmons G, Gosalia DN, Rennekamp AJ, Reeves JD, Diamond SL, Bates P. Inhibitors of cathepsin l prevent severe acute respiratory syndrome coronavirus entry. Proc Natl Acad Sci USA (2005) 102:11876–81. doi: 10.1073/pnas.0505577102
111. Huang I-C, Bosch BJ, Li F, Li W, Lee KH, Ghiran S, et al. SARS coronavirus, but not human coronavirus NL63, utilizes cathepsin l to infect ACE2-expressing cells *. J Biol Chem (2006) 281:3198–203. doi: 10.1074/jbc.M508381200
112. Rogan MP, Geraghty P, Greene CM, O'Neill SJ, Taggart CC, McElvaney NG. Antimicrobial proteins and polypeptides in pulmonary innate defence. Respir Res (2006) 7:29–11. doi: 10.1186/1465-9921-7-29
113. Taggart CC, Greene CM, Smith SG, Levine RL, McCray PB, O'Neill S, et al. Inactivation of human beta-defensins 2 and 3 by elastolytic cathepsins. J Immunol (2003) 171:931–7. doi: 10.4049/jimmunol.171.2.931
114. Squeglia F, Ruggiero A, Berisio R. Collagen degradation in tuberculosis pathogenesis: the biochemical consequences of hosting an undesired guest. Biochem J (2018) 475:3123–40. doi: 10.1042/BCJ20180482
115. Reece ST, Loddenkemper C, Askew DJ, Zedler U, Schommer-Leitner S, Stein M, et al. Serine protease activity contributes to control of mycobacterium tuberculosis in hypoxic lung granulomas in mice. J Clin Invest (2010) 120:3365–76. doi: 10.1172/JCI42796
116. de Haan JJ, Smeets MB, Pasterkamp G, Arslan F. Danger signals in the initiation of the inflammatory response after myocardial infarction. Mediators Inflammation (2013) 2013:206039. doi: 10.1155/2013/206039
117. Repnik U, Starr AE, Overall CM, Turk B. Cysteine cathepsins activate ELR chemokines and inactivate non-ELR chemokines. J Biol Chem (2015) 290:13800–11. doi: 10.1074/jbc.M115.638395
118. Ohashi K, Naruto M, Nakaki T, Sano E. Identification of interleukin-8 converting enzyme as cathepsin l. Biochim Biophys Acta (2003) 1649:30–9. doi: 10.1016/s1570-9639(03)00152-3
119. Padrines M, Wolf M, Walz A, Baggiolini M. Interleukin-8 processing by neutrophil elastase, cathepsin G and proteinase-3. FEBS Lett (1994) 352:231–5. doi: 10.1016/0014-5793(94)00952-x
120. Meyer-Hoffert U. Neutrophil-derived serine proteases modulate innate immune responses. Front Biosci (Landmark Ed) (2009) 14:3409–18. doi: 10.2741/3462
121. Bank U, Küpper B, Reinhold D, Hoffmann T, Ansorge S. Evidence for a crucial role of neutrophil-derived serine proteases in the inactivation of interleukin-6 at sites of inflammation. FEBS Lett (1999) 461:235–40. doi: 10.1016/s0014-5793(99)01466-0
122. Sambrano GR, Huang W, Faruqi T, Mahrus S, Craik C, Coughlin SR. Cathepsin G activates protease-activated receptor-4 in human platelets. J Biol Chem (2000) 275:6819–23. doi: 10.1074/jbc.275.10.6819
123. Sobotič B, Vizovišek M, Vidmar R, Van Damme P, Gocheva V, Joyce JA, et al. Proteomic identification of cysteine cathepsin substrates shed from the surface of cancer cells. Mol Cell Proteomics (2015) 14:2213–28. doi: 10.1074/mcp.M114.044628
Keywords: cathepsins, infection, inflammation, innate immunity, adaptive immunity, immune resolution
Citation: Anes E, Pires D, Mandal M and Azevedo-Pereira JM (2022) Spatial localization of cathepsins: Implications in immune activation and resolution during infections. Front. Immunol. 13:955407. doi: 10.3389/fimmu.2022.955407
Received: 28 May 2022; Accepted: 14 July 2022;
Published: 03 August 2022.
Edited by:
Xiao-Lian Zhang, Wuhan University, ChinaReviewed by:
Jinfeng Miao, Nanjing Agricultural University, ChinaCopyright © 2022 Anes, Pires, Mandal and Azevedo-Pereira. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elsa Anes, ZWFuZXNAZmYudWxpc2JvYS5wdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.