
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 26 August 2022
Sec. Autoimmune and Autoinflammatory Disorders
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.953730
This article is part of the Research TopicThe Role of Monocytes/Macrophages in Autoimmunity and AutoinflammationView all 19 articles
 Hiroto Tsuboi*
Hiroto Tsuboi* Seiji Segawa
Seiji Segawa Mizuki YagishitaHirofumi TokoFumika HondaAyako KitadaHaruka MikiAyako OhyamaShinya Hagiwara
Mizuki YagishitaHirofumi TokoFumika HondaAyako KitadaHaruka MikiAyako OhyamaShinya Hagiwara Yuya Kondo
Yuya Kondo Isao MatsumotoTakayuki Sumida
Isao MatsumotoTakayuki SumidaAdult onset Still disease (AOSD) is a systemic inflammatory disorder characterized by skin rash, spiking fever, arthritis, sore throat, lymphadenopathy, and hepatosplenomegaly. Although the etiology of this disease has not been fully clarified, both innate and acquired immune responses could contribute to its pathogenesis. Hyperactivation of macrophages and neutrophils along with low activation of natural killer (NK) cells in innate immunity, as well as hyperactivation of Th1 and Th17 cells, whereas low activation of regulatory T cells (Tregs) in acquired immunity are involved in the pathogenic process of AOSD. In innate immunity, activation of monocytes/macrophages might play central roles in the development of AOSD and macrophage activation syndrome (MAS), a severe life-threating complication of AOSD. Regarding the activation mechanisms of monocytes/macrophages in AOSD, in addition to type II interferon (IFN) stimulation, several pathways have recently been identified, such as the pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs)-pattern recognition receptors (PRRs) axis, and neutrophil extracellular traps (NETs)-DNA. These stimulations on monocytes/macrophages cause activation of the nucleotide-binding oligomerization domain, leucine-rich repeat, and pyrin domain (NLRP) 3 inflammasomes, which trigger capase-1 activation, resulting in conversion of pro-IL-1β and pro-IL-18 into mature forms. Thereafter, IL-1β and IL-18 produced by activated monocytes/macrophages contribute to various clinical features in AOSD. We identified placenta-specific 8 (PLAC8) as a specifically increased molecule in monocytes of active AOSD, which correlated with serum levels of CRP, ferritin, IL-1β, and IL-18. Interestingly, PLAC8 could suppress the synthesis of pro-IL-1β and pro-IL-18 via enhanced autophagy; thus, PLAC8 seems to be a regulatory molecule in AOSD. These findings for the activation mechanisms of monocytes/macrophages could shed light on the pathogenesis and development of a novel therapeutic strategy for AOSD.
Adult onset Still disease (AOSD) is a rare systemic inflammatory disorder of unknown etiology that occurs at the age of 16 years or older and is characterized by skin rash, spiking fever, arthritis, sore throat, lymphadenopathy, and hepatosplenomegaly (1–3).
A previous nationwide epidemiologic survey of adult Still disease (ASD) in Japan revealed an estimated prevalence of ASD of 3.9 per 100,000 (4). Analysis of 169 ASD patients showed a mean age at onset of 46 years, and only 8 patients (4.8%) developed ASD when they were aged younger than 16 years, whilst the other 158 patients (95.2%) had AOSD. Of the 146 patients with available data on the clinical course, 58 (39.7%) and 50 (34.2%) patients showed monocyclic and polycyclic systemic patterns, whilst 15 (10.3%) and 23 (15.8%) patients showed monocyclic and polycyclic systemic patterns with chronic articular involvement, respectively. Regarding the complications, disseminated intravascular coagulation (DIC) and macrophage activation syndrome (MAS), which are severe life-threating complications of AOSD, were noted in 8 (6.3%) and 19 (15.0%) of 127 patients, respectively. At the last medical examination, 145 of 164 patients (88.4%) had achieved remission, whilst 66 of 169 patients (39.1%) experienced relapse during the observation period. Importantly, lymphadenopathy and MAS were significantly associated with increased risk of relapse. Regarding treatment, oral glucocorticoids were administered to 96% of the patients, whilst methotrexate and biologic agents were administered to 41% and 16% of the patients in that survey, respectively (4). In addition, a recent comprehensive systematic literature review (5) including the nationwide epidemiologic survey in Japan described above (4), showed that the estimated prevalence of AOSD was between 0.73 and 6.77 per 100,000 individuals, and for clinical course, monocyclic systemic/self-limited was 21.1-64.3%, polycyclic systemic/intermittent was 9.3-50.0%, and chronic articular was 11.9-55.6%, respectively. The review also reported that 1.7-23.5% of patients with AOSD developed MAS, and the mortality rate of patients with AOSD was 2.3-16% (5). From these findings, prevention of relapse and MAS seem to be the current unmet medical needs in the management of AOSD.
Intravenous tocilizumab (TCZ), a monoclonal antibody against the IL-6 receptor, has been officially approved for AOSD in Japan on the basis of promising results from a double-blind, randomized, placebo-controlled phase-III trial (6). In that trial, treatment responses such as American College of Rheumatology (ACR) 20 and ACR50 in the TCZ group were double those in the placebo group, and TCZ had a significantly stronger glucocorticoid-sparing effect than that of the placebo; moreover, the TCZ interval could be prolonged after disease control in several patients without flare (6). On the other hand, the possibility that TCZ could trigger the development of MAS in AOSD patients has been proposed (7). Although TCZ could be a hopeful treatment strategy against the prevention of relapse in AOSD, which is one of the unmet medical needs in the management of AOSD, TCZ does not seem to be a complete solution for the inhibition of MAS. For another promising biological agent, anakinra which is a recombinant human IL-1 receptor antagonist also has been examined for treatment of AOSD, while it has not been officially approved for AOSD in Japan. A systematic literature review, in which 15 manuscripts (one open randomized multicenter trial and 14 observational single-arm retrospective studies) were analyzed, showed that the majority of AOSD patients treated with anakinra could achieve a complete remission also in monotherapy, and the treatment with anakinra was associated with an important corticosteroids-sparing effect (8). Interestingly, a single center experience and systematic literature review clarified that the majority but not all of pediatric MAS patients associated with systemic juvenile idiopathic arthritis (sJIA) or autoinflammatory diseases (46 out of 50 reported cases) achieved remission by treatment with anakinra (9). Thus, revealing the accurate and comprehensive mechanisms for activation of monocytes/macrophages is necessary for the development of a new radical therapeutic strategy against AOSD complicated with MAS.
In this review, we survey the pathogenic process of AOSD, including the genetic background, triggers, innate and acquired immune systems, high production of ferritin, and proinflammatory cytokines. In particular, we focus on the activation mechanisms of monocytes/macrophages in the innate immune responses, which might play central roles in the development of AOSD and MAS.
Table 1 summarizes the pathogenic process of AOSD including the genetic background, triggers, innate and acquired immune systems, high production of ferritin, and proinflammatory cytokines.
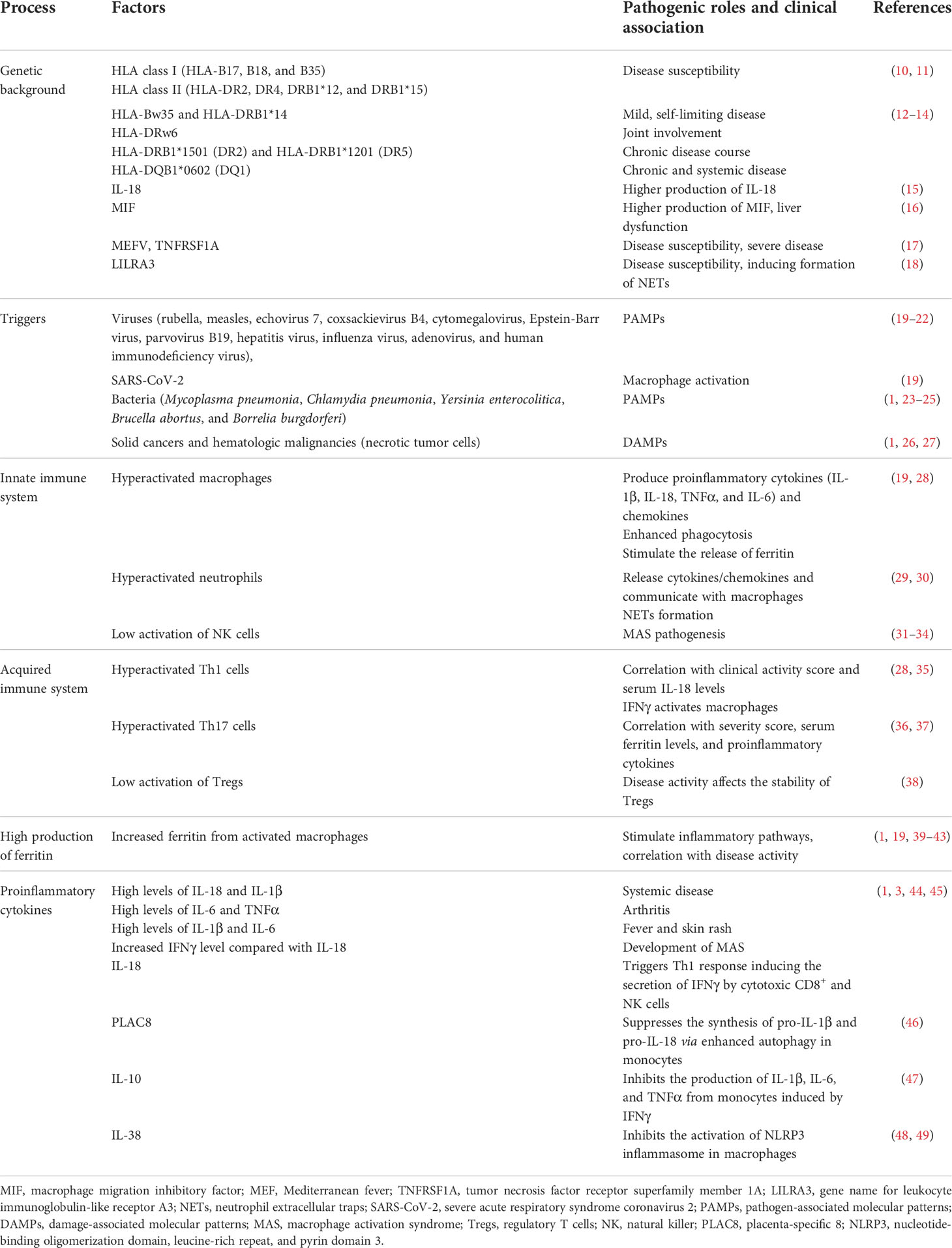
Table 1 Pathogenic process of AOSD.
A genetic association between AOSD and human leukocyte antigen (HLA), including both HLA class I (HLA-B17, B18, and B35) and HLA class II (HLA-DR2, DR4, DRB1*12, and DRB1*15), has been reported (10, 11). Moreover, some associations of the HLA type with the clinical characteristics of AOSD, such as HLA-Bw35 and HLA-DRB1*14 with a mild, self-limiting disease; HLA-DRw6 with joint involvement; HLA-DRB1*1501 (DR2) and HLA-DRB1*1201 (DR5) with a chronic disease course; and HLA-DQB1*0602 (DQ1) with chronic and systemic disease, have also been shown (12–14). Interestingly, polymorphisms in both the IL-18 gene and the macrophage migration inhibitory factor (MIF) gene have been shown to contribute to the disease susceptibility via higher production of IL-18 and MIF (15, 16). For known hereditary periodic fever syndrome genes, a previous study showed that association with 3 rare variants in the Mediterranean fever (MEFV) gene, which encodes pyrin, and mutations in the tumor necrosis factor receptor superfamily member 1A (TNFRSF1A) gene were significant and that these genetic factors were associated with a severe disease course of AOSD (17). More recently, functional leukocyte immunoglobulin-like receptor A3 (LIR-A3; gene name LILRA3) has been identified as a novel genetic risk factor for the development of AOSD, and functional LIR-A3 might play a pathogenic role by inducing formation of neutrophil extracellular traps (NETs) (18).
A large number of viruses, including the rubella, measles, echovirus 7, coxsackievirus B4, cytomegalovirus and Epstein-Barr, parvovirus B19, hepatitis, influenza, adenovirus, and human immunodeficiency viruses, have been suggested to trigger AOSD pathogenesis via activation of the aberrant response of the immune system as so-called pathogen-associated molecular patterns (PAMPs) (19–22). More recently, attention has been focused on an association with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and the common features of macrophage activation in coronavirus disease 2019 (COVID-19) and AOSD (19). In addition, some bacteria such as Mycoplasma pneumoniae, Chlamydia pneumoniae, Yersinia enterocolitica, Brucella abortus, and Borrelia burgdorferi have also been considered to be involved (1, 23–25). Other than these infectious pathogens, solid cancers and hematologic malignancies have been proposed as possible triggers of AOSD (1, 26). Damage-associated molecular patterns (DAMPs) typically released by necrotic tumor cells are recognized by innate immune receptors such as toll-like receptors (TLRs) and could stimulate innate immune responses within the tumor immune microenvironment (27), which might contribute to the development of AOSD. However, no disease-specific unique pathogenic trigger has been clearly identified, suggesting the possibility that multiple environmental triggers play a role in AOSD (1).
Activation of innate immune cells including macrophages and neutrophils plays a major pathogenic role in the development of AOSD (1). The activation mechanisms of monocytes/macrophages will be described in a later section. Hyperactivated macrophages could produce proinflammatory cytokines such as IL-1β, IL-18, TNFα and IL-6, as well as chemokines; enhanced phagocytosis; and stimulated the release of ferritin (19, 28). As for neutrophils, interaction between various PAMPs and DAMPs and a variety of specific receptors including pattern-recognition receptors (PRRs), such as TLRs and C-type lectin domain family 5-member A (CLEC5A), and Fc receptors on neutrophils promote neutrophil recruitment and activation (29). Activated neutrophils release more cytokines/chemokines and communicate with macrophages in the innate immune system. Neutrophil activation and mediator release form a positive-feedback loop that enhances neutrophil recruitment and amplifies inflammatory responses, acting as an axis of pathogenesis in AOSD (29). A previous study demonstrated that IL-18, a pivotal cytokine of AOSD, induces NETs by enhancing calcium influx into neutrophils (29). The protein components of NETs, like S100 proteins, act as ligands of TLR4 or receptors for AGEs (advanced glycation end products) (RAGE) to accelerate neutrophils and activate the release of proinflammatory cytokines, contributing to the systemic inflammation of AOSD (30). Thus, NETs formation can promote cytokine storms by linking neutrophils and macrophages, as described in a later section (29).
Contrastively, it was reported that the cytotoxic function of natural killer (NK) cells was defective in AOSD patients (31). The proinflammatory cytokines produced during AOSD, mainly IL-18, have been reported to decrease NK cell activity (32, 33). Importantly, impairment of NK cell cytotoxicity has been shown to play some roles in MAS pathogenesis (34).
A previous report showed that interferon (IFN) γ-producing helper T (Th) cells and the Th1/Th2 ratio in peripheral blood were significantly higher in patients with AOSD than in healthy controls (HCs) and that the percentages of IFNγ-producing Th cells and the Th1/Th2 ratio in peripheral blood correlated significantly with the clinical activity score and serum IL-18 levels in patients with AOSD (35). Importantly, IFNγ is known to increase the production of cytokines and chemokines, phagocytosis, and the intracellular killing of microbial pathogens by macrophages (28). Moreover, high frequencies of circulating Th17 cells were reported in active AOSD patients and correlated with the severity score, serum ferritin levels, and proinflammatory cytokines including IL-1β, IL-6, and IL-18 (36, 37). In contrast, a recent study clarified that the proportion of regulatory T cells (Tregs) was significantly lower in patients with acute AOSD patients than in the HCs and that the expression levels of IFNγ, IL-17, and IL-4 in Tregs were significantly increased, whilst the suppressive function of Tregs was impaired in patients with acute AOSD (38). These results suggested that the disease activity might affect the stability of Tregs in AOSD (38).
Ferritin is an intracellular iron storage protein including 24 subunits: heavy (H) subunits and light (L) subunits, according to their molecular weight (1). It is well known that ferritin is a characteristic mediator of AOSD (19). In AOSD, H-ferritin and the number of macrophages expressing H-ferritin have been shown to be increased, suggesting a pathogenic role (39–42). Importantly, elevated H-ferritin expressions in the lymph nodes and skin were correlated with the severity of AOSD (41, 42). Ferritin synthesis is regulated, in addition to iron availability, by different cytokines such as IL-1β and IL-6, which are overexpressed in AOSD patients (1, 19). Moreover, ferritin can stimulate inflammatory pathways to amplify the inflammatory process, supporting a hypothesis that ferritin may not only act as a bystander of the acute-phase reaction (43). Thus, increased production of ferritin from macrophage activation might correlate with the disease activity of AOSD and might serve as an activity indicator of this disease (19).
Heme oxygenase-1 (HO-1), an inducible heme-degrading enzyme, is expressed by macrophages and endothelial cells in response to various stresses. Interestingly, it was reported that among patients with hemophagocytic syndrome (HPS) and AOSD, serum HO-1 correlated closely with serum ferritin but not CRP or lactate dehydrogenase (LDH) levels (50). These findings indicated an association between HO-1 and hyperferritinemia in patients with HPS and AOSD.
Moreover, several isoforms of ferritin have been described, one of which is glycosylated ferritin. A previous report revealed that glycosylated ferritin was significantly lower in AOSD patients than in the control patients including infection, liver disease, systemic inflammatory disease, fever of unknown origin, and neoplasia. The study concluded that increased ferritin and decreased glycosylated ferritin levels could be powerful diagnostic markers of AOSD (51).
The immune system activation described above leads to the production of several proinflammatory cytokines including IL-1β, IL-18, TNFα, IL-6, IFNγ, and IL-17 from activated macrophages, neutrophils, and Th1 and Th17 cells (1). Importantly, different cytokine profiles might be responsible for the distinct clinical manifestation of AOSD (3). For example, high levels of IL-18 and IL-1β are detected in systemic disease; high levels of IL-6 and TNFα, in arthritis; and high levels of IL-1β and IL-6, in fever and skin rash, respectively (1, 3). Regarding MAS, one of the most severe complications of AOSD, a previous study of sJIA showed that an increased plasma IFNγ level in comparison with IL-18 might raise suspicion about the development of MAS in sJIA (44). In addition, IL-6 has also been associated with MAS (3). Regarding the association between IL-18 and IFNγ, IL-18 might also trigger a Th1 response, thereby inducing the secretion of IFNγ by cytotoxic CD8+ and NK cells (45). Although these correlations between the cytokine profiles and clinical features of AOSD might be clinically informative, serum cytokines are not routinely examined and might not widely affect our daily practice of individual AOSD patients (3).
In innate immunity, activation of monocytes/macrophages might play central roles in the development of AOSD and MAS, as described above. Regarding the activation mechanisms of monocytes/macrophages in AOSD, several pathways such as the PAMPs and DAMPs-PRRs axis and NETs-DNA have recently been identified (1, 19), in addition to type II IFN stimulation (28). Moreover, several inhibitory molecules that work against activation of monocytes/macrophages have been also reported. Figure 1 summarizes the activation mechanisms of monocytes/macrophages in AOSD.
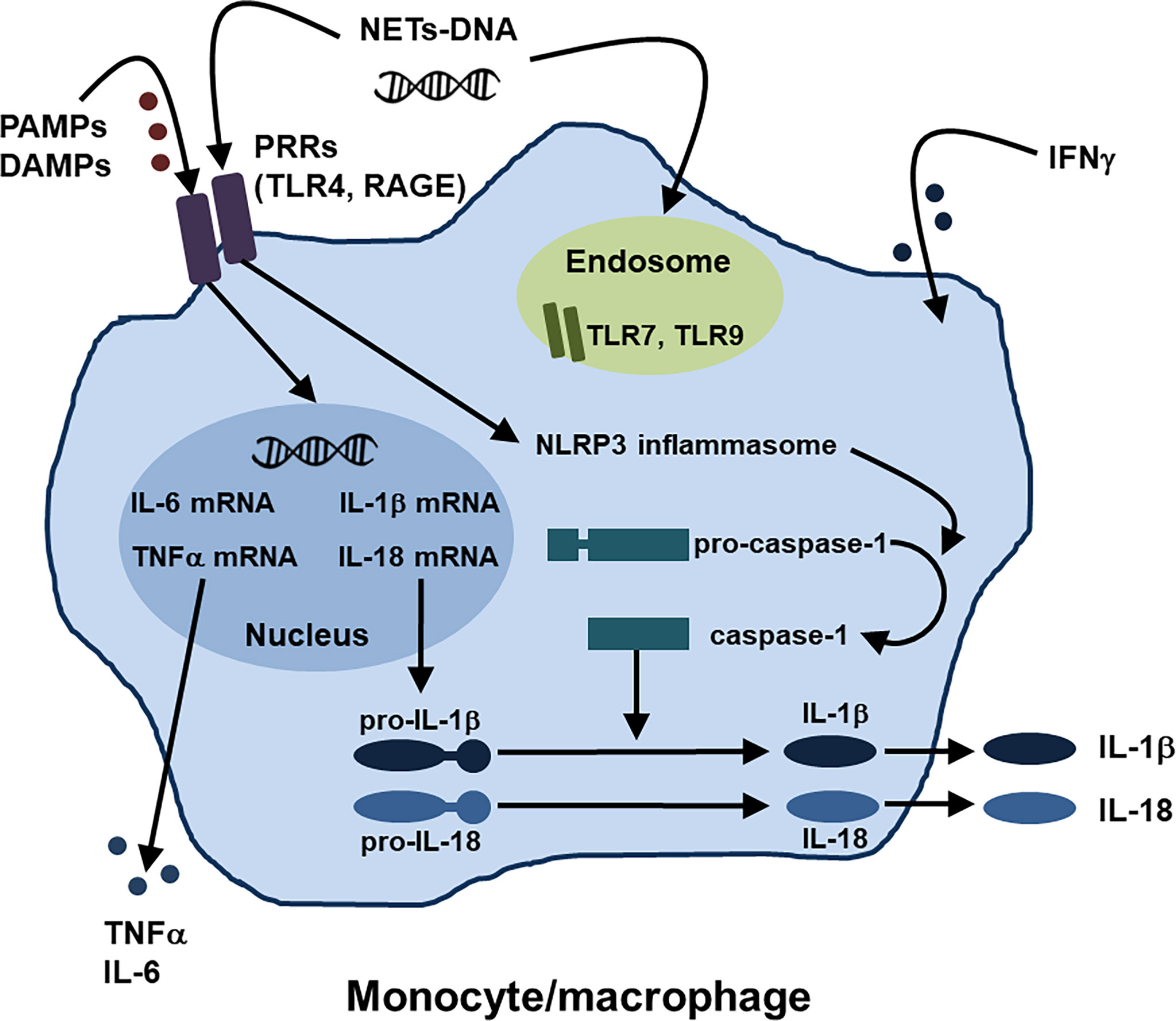
Figure 1 Activation mechanisms of monocytes/macrophages in AOSD. Regarding the activation mechanisms of monocytes/macrophages in AOSD, the PAMPs and DAMPs-PRRs axis and NETs-DNA have been identified, in addition to IFNγ stimulation. NETs, neutrophil extracellular traps; PAMPs, pathogen-associated molecular patterns; DAMPs, damage-associated molecular patterns; PRRs, pattern recognition receptors; TLR, toll-like receptor; receptor for AGEs (advanced glycation end products) RAGE; NLRP, nucleotide-binding oligomerization domain, leucine-rich repeat, and pyrin domain.
We previously compared the gene expression pattern of peripheral monocytes of active-AOSD, inactive-AOSD patients (the same patients in the active and inactive phases), and HCs by DNA microarray to identify genes specifically associated with the active phase of the disease (46). The gene expression patterns in the 3 groups (active-AOSD, inactive-AOSD, and HC) showed distinct clusters specific to each group in principal components analysis (PCA). We identified 68 genes as active-AOSD-specific highly expressed genes, and focused on placenta-specific 8 (PLAC8) among them. Importantly, the PLAC8 mRNA expression levels were significantly higher in the active-AOSD patients than in the HCs or the inactive-AOSD patients, as well as than in patients with rheumatoid arthritis (RA), Sjögren syndrome (SS), systemic lupus erythematosus (SLE), or polymyositis (PM)/dermatomyositis (DM), indicating that the upregulation of PLAC8 mRNA in monocytes was specific in the patients with active-AOSD. Interestingly, in patients with AOSD, the expression of PLAC8 mRNA in peripheral monocytes was significantly correlated with serum CRP and ferritin levels. Moreover, serum IL-1β and IL-18 levels, but not IL-6 and TNFα levels, significantly correlated with PLAC8 mRNA expression levels in the AOSD patients. Thus, these results suggested that the expression levels of PLAC8 mRNA in peripheral monocytes are an activity or severity marker for AOSD (46).
Regarding the function of PLAC8, we indicated that upregulated PLAC8 acted on the synthesis of inactive precursors of IL-1β and IL-18 and suppressed the production of IL-1β and IL-18 through enhanced autophagy which was independent of caspase-1. Thus, PLAC8 seems to be a regulatory molecule in AOSD. Figure 2 summarizes the proposed suppressive function of PLAC8 in IL-1β and IL-18 production (46).
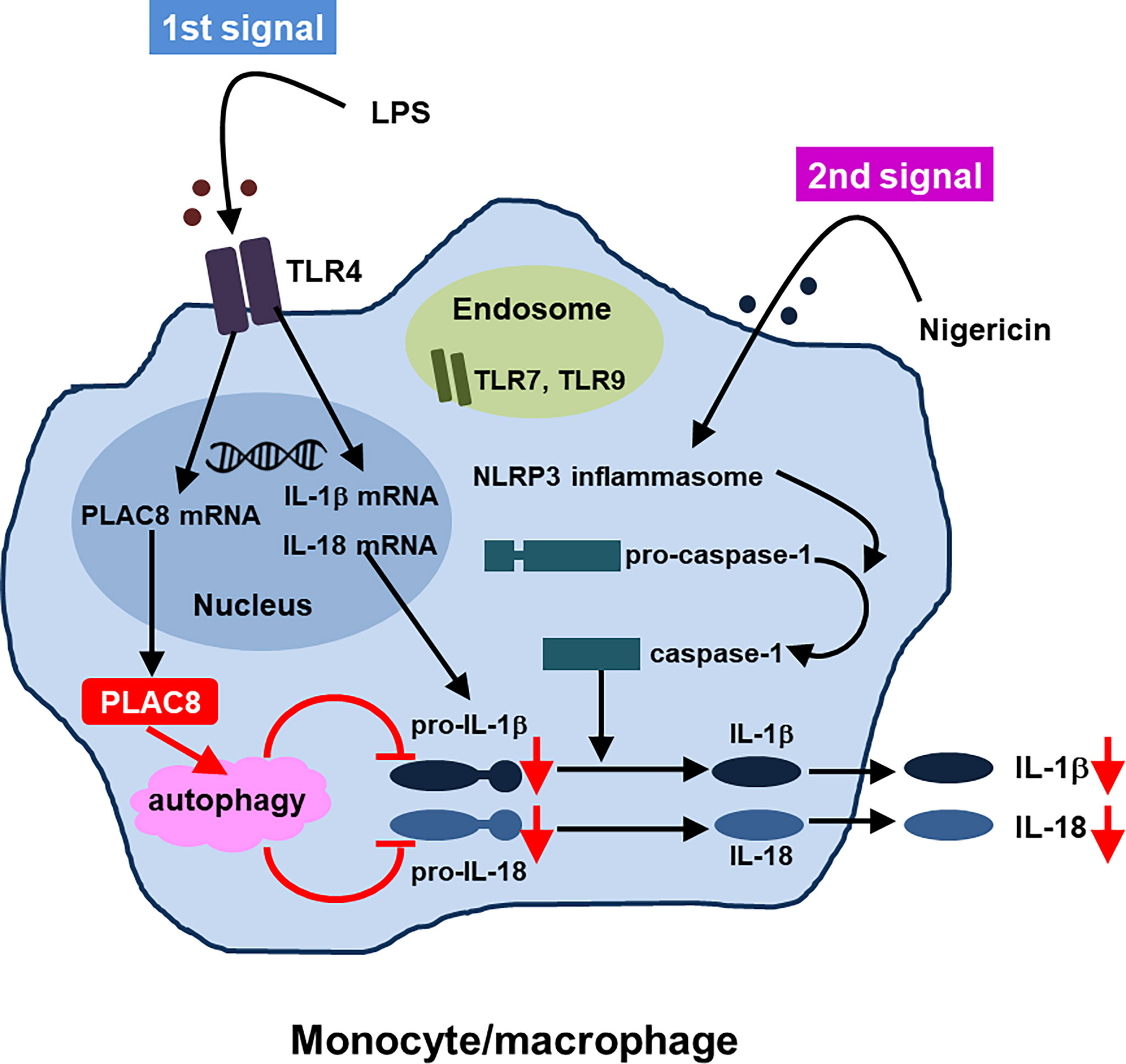
Figure 2 Proposed function of PLAC8 in IL-1β and IL-18 production (reference 46, modification)A schema illustrating how PLAC8 suppresses IL-1β and IL-18 production via enhancement of autophagy. Two steps might be needed for inhibition of IL-1β and IL-18 production by PLAC8 in primary monocytes. The first step is the upregulation of PLAC8, pro-IL-1β and pro-IL-18 in monocytes through LPS stimulation. The second step is the inhibition of pro-IL-1β and pro-IL-18 through the enhancement of autophagy by upregulated PLAC8, which is independent of caspase-1.
Monocytes/macrophages could be activated through the recognition of various PAMPs and DAMPs by different types of PRRs such as TLRs, resulting in activation of inflammasomes responsible for pro-IL-1β and pro-IL-18 activation (1, 19). The nucleotide-binding oligomerization domain, leucine-rich repeat, and pyrin domain (NLRP) can form multimeric protein complexes in response to stimuli (19). NLRP3 inflammasomes trigger caspase-1 activation to convert pro-IL-1β and pro-IL-18 into mature IL-1β and IL-18, which contribute to the pathogenesis of AOSD (1, 19).
As for the roles of TLRs and their ligands in activation of monocytes/macrophages in AOSD, interaction between various DAMPs and TLRs, including S100 proteins with TLR4, high mobility group box-1 (HMGB1) with TLR4 and RAGE, and nucleic acids with TLR7, has been identified (19, 52).
As described in the innate immune system section, a previous study demonstrated that IL-18, a pivotal cytokine of AOSD, induces NETs by enhancing the calcium influx into neutrophils (29). Interestingly, Hu et al. showed that NETs-DNA complexes were significantly increased in the circulation of patients with AOSD when compared with that of HCs and that NETs-DNA from AOSD patients activated macrophages and increased the expression of IL-1β, IL-6, and TNFα via activation of the NLRP3 inflammasome (53). The authors hypothesized that AOSD neutrophils spontaneously release NETs-DNA, leading to an enhanced proinflammatory potential mainly via TLR9 (53). These findings indicated a novel link between neutrophils and macrophages by NETs formation in AOSD.
IFNγ, type II IFN, would be the prototypic “macrophage-activating factor” which could increase cytokine and chemokine production, phagocytosis, and the intracellular killing of microbial pathogens by macrophages, thus playing important pathogenic roles in the development of AOSD and MAS (28, 54)
A recent study showed that peripheral monocytes expressed IL-10 receptors as well as IL-6 receptors and gp130 (47). As expected, IFNγ enhanced the expression levels of proinflammatory cytokines including IL-1β, IL-6, and TNFα from monocytes (47). Interestingly, IL-10 clearly inhibited the production of these cytokines induced by IFNγ stimulation (47). Thus, IL-10 seems to have an inhibitory effect on proinflammatory cytokine production by monocytes.
IL-38 is a new member of the IL-1 family with multiple functions involved in infection and immunity. A recent study revealed that LPS upregulated IL-38 and its receptor IL-36 receptor, and IL-38 shifted macrophages from a M1 to M2 phenotype, as well as IL-38 dampened LPS induced activation of NLRP3 inflammasome in mouse peritoneal macrophages (48). Thus, IL-38 can significantly inhibit the activation of NLRP3 inflammasome, resulting in a potent anti-inflammatory activity (49).
Various factors including the genetic background, triggers, innate and acquired immune systems, high production of ferritin, and proinflammatory cytokines could contribute to the pathogenic process of AOSD. Among innate immunity, activation of monocytes/macrophages might play central roles in the development of AOSD and MAS. Regarding the activation mechanisms of monocytes/macrophages in AOSD, several pathways such as the PAMPs and DAMPs-PRRs axis and NETs-DNA have been identified, in addition to type II IFN stimulation. We identified PLAC8 as a specifically increased molecule in monocytes of active AOSD, which correlated with serum levels of CRP, ferritin, IL-1β, and IL-18. Interestingly, PLAC8 could suppress the synthesis of pro-IL-1β and pro-IL-18 via enhanced autophagy; thus, PLAC8 seems to be a regulatory molecule in AOSD. These findings related to the activation mechanisms of monocytes/macrophages could shed light on the pathogenesis of AOSD and thus lead to the development of a novel therapeutic strategy for the disease.
Each author took part in the design of the study, collection of the data, and writing of the manuscript, and all agree to accept equal responsibility for the accuracy of its contents of this paper. All authors contributed to the article and approved the submitted version.
This work was supported by the Research Program for Intractable Diseases, Health and Labor Sciences Research Grants from the Ministry of Health, Labor and Welfare, Japan and Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology and Japan Society for the Promotion of Science.
We thank Flaminia Miyamasu, Medical English Communications Center, University of Tsukuba, for grammatical editing.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Giacomelli R, Ruscitti P, Shoenfeld Y. A comprehensive review on adult onset Still's disease. J Autoimmun (2018) 93:24–36. doi: 10.1016/j.jaut.2018.07.018
2. Mimura T, Kondo Y, Ohta A, Iwamoto M, Ota A, Okamoto N, et al. Evidence-based clinical practice guideline for adult Still's disease. Mod Rheumatol (2018) 28:736–57. doi: 10.1080/14397595.2018.1465633
3. Mitrovic S, Fautrel B. Clinical phenotypes of adult-onset Still's disease: New insights from pathophysiology and literature findings. J Clin Med (2021) 10:2633. doi: 10.3390/jcm10122633
4. Asanuma YF, Mimura T, Tsuboi H, Noma H, Miyoshi F, Yamamoto K, et al. Nationwide epidemiological survey of 169 patients with adult Still's disease in Japan. Mod Rheumatol (2015) 25:393–400. doi: 10.3109/14397595.2014.974881
5. Efthimiou P, Kontzias A, Hur P, Rodha K, Ramakrishna GS, Nakasato P. Adult-onset Still's disease in focus: Clinical manifestations, diagnosis, treatment, and unmet needs in the era of targeted therapies. Semin Arthritis Rheum (2021) 51:858–74. doi: 10.1016/j.semarthrit.2021.06.004
6. Kaneko Y, Kameda H, Ikeda K, Ishii T, Murakami K, Takamatsu H, et al. Tocilizumab in patients with adult-onset Still's disease refractory to glucocorticoid treatment: a randomised, double-blind, placebo-controlled phase III trial. Ann Rheum Dis (2018) 77:1720–9. doi: 10.1136/annrheumdis-2018-213920
7. Kaneko Y. Interluekin-6 inhibitors for the treatment of adult-onset Still's disease. Mod Rheumatol (2022) 32:12–5. doi: 10.1093/mr/roab004
8. Giacomelli R, Sota J, Ruscitti P, Campochiaro C, Colafrancesco S, Dagna L, et al. The treatment of adult-onset Still's disease with anakinra, a recombinant human IL-1 receptor antagonist: A systematic review of literature. Clin Exp Rheumatol (2021) 39:187–95. doi: 10.55563/clinexprheumatol/fsq5vq
9. Sönmez HE, Demir S, Bilginer Y, Özen S. Anakinra treatment in macrophage activation syndrome: A single center experience and systemic review of literature. Clin Rheumatol (2018) 37:3329–35. doi: 10.1007/s10067-018-4095-1
10. Pouchot J, Sampalis JS, Beaudet F, Carette S, Décary F, Salusinsky-Sternbach M, et al. Adult Still's disease: Manifestations, disease course, and outcome in 62 patients. Med (Baltimore) (1991) 70:118–36. doi: 10.1097/00005792-199103000-00004
11. Joung CI, Lee HS, Lee SW, Kim CG, Song YH, Jun JB, et al. Association between HLA-DR B1 and clinical features of adult onset Still's disease in Korea. Clin Exp Rheumatol (2003) 21:489–92.
12. Terkeltaub R, Esdaile JM, Décary F, Harth M, Lister J, Lapointe N. HLA-Bw35 and prognosis in adult Still's disease. Arthritis Rheum (1981) 24:1469–72. doi: 10.1002/art.1780241203
13. Wouters JM, Reekers P, van de Putte LB. Adult-onset Still's disease. disease course and HLA associations. Arthritis Rheum (1986) 29:415–8. doi: 10.1002/art.1780290316
14. Fujii T, Nojima T, Yasuoka H, Satoh S, Nakamura K, Kuwana M, et al. Cytokine and immunogenetic profiles in Japanese patients with adult Still's disease. association with chronic articular disease. Rheumatol (Oxford) (2001) 40:1398–404. doi: 10.1093/rheumatology/40.12.1398
15. Sugiura T, Maeno N, Kawaguchi Y, Takei S, Imanaka H, Kawano Y, et al. A promoter haplotype of the interleukin-18 gene is associated with juvenile idiopathic arthritis in the Japanese population. Arthritis Res Ther (2006) 8:R60. doi: 10.1186/ar1930
16. Wang FF, Huang XF, Shen N, Leng L, Bucala R, Chen SL, et al. A genetic role for macrophage migration inhibitory factor (MIF) in adult-onset Still's disease. Arthritis Res Ther (2013) 15:R65. doi: 10.1186/ar4239
17. Sighart R, Rech J, Hueber A, Blank N, Löhr S, Reis A, et al. Evidence for genetic overlap between adult onset Still's disease and hereditary periodic fever syndromes. Rheumatol Int (2018) 38:111–20. doi: 10.1007/s00296-017-3885-0
18. Wang M, Liu M, Jia J, Shi H, Teng J, Liu H, et al. Association of the leukocyte immunoglobulin-like receptor A3 gene with neutrophil activation and disease susceptibility in adult-onset Still's disease. Arthritis Rheumatol (2021) 73:1033–43. doi: 10.1002/art.41635
19. Chen PK, Chen DY. An update on the pathogenic role of macrophages in adult-onset Still's disease and its implication in clinical manifestations and novel therapeutics. J Immunol Res (2021) 2021:8998358. doi: 10.1155/2021/8998358
20. Wouters JM, van der Veen J, van de Putte LB, de Rooij DJ. Adult onset Still's disease and viral infections. Ann Rheum Dis (1988) 47:764–7. doi: 10.1136/ard.47.9.764
21. Escudero FJ, Len O, Falcó V, de Sevilla TF, Sellas A. Rubella infection in adult onset Still's disease. Ann Rheum Dis (2000) 59:493. doi: 10.1136/ard.59.6.490c
22. DelVecchio S, Skidmore P. Adult-onset Still's disease presenting as fever of unknown origin in a patient with HIV infection. Clin Infect Dis (2008) 46:e41–43. doi: 10.1086/526785
23. Perez C, Artola V. Adult Still's disease associated with mycoplasma pneumoniae infection. Clin Infect Dis (2001) 32:E105–6. doi: 10.1086/319342
24. Matsuura-Otsuki Y, Hanafusa T, Igawa K, Sato H, Nishizawa A, Yokozeki H. Macrophage activation syndrome triggered by disseminated tuberculosis with tuberculous gumma in a patient with adult-onset Still's disease and good's syndrome. Eur J Dermatol (2016) 26:309–11. doi: 10.1684/ejd.2016.2745
25. De Clerck KF, Van Offel JF, Vlieghe E, Van Marck E, Stevens WJ. Bartonella endocarditis mimicking adult Still's disease. Acta Clin Belg (2008) 63:190–2. doi: 10.1179/acb.2008.030
26. Liozon E, Ly KH, Vidal-Cathala E, Fauchais AL. Adult-onset Still's disease as a manifestation of malignancy: Report of a patient with melanoma and literature review. Rev Med Interne (2014) 35:60–4. doi: 10.1016/j.revmed.2013.02.014
27. Yanai H, Hangai S, Taniguchi T. Damage-associated molecular patterns and toll-like receptors in the tumor immune microenvironment. Int Immunol (2021) 33:841–6. doi: 10.1093/intimm/dxab050
28. Di Cola I, Ruscitti P, Giacomelli R, Cipriani P. The pathogenic role of interferons in the hyperinflammatory response on adult-onset Still's disease and macrophage activation syndrome: Paving the way towards new therapeutic targets. J Clin Med (2021) 10:1164. doi: 10.3390/jcm10061164
29. Kim JW, Ahn MH, Jung JY, Suh CH, Kim HA. An update on the pathogenic role of neutrophils in systemic juvenile idiopathic arthritis and adult-onset Still's disease. Int J Mol Sci (2021) 22:13038. doi: 10.3390/ijms222313038
30. Wang S, Song R, Wang Z, Jing Z, Wang S, Ma J. S100A8/A9 in inflammation. Front Immunol (2018) 9:1298. doi: 10.3389/fimmu.2018.01298
31. Lee SJ, Cho YN, Kim TJ, Park SC, Park DJ, Jin HM, et al. Natural killer T cell deficiency in active adult-onset Still's disease: Correlation of deficiency of natural killer T cells with dysfunction of natural killer cells. Arthritis Rheum (2012) 64:2868–77. doi: 10.1002/art.34514
32. Chaix J, Tessmer MS, Hoebe K, Fuséri N, Ryffel B, Dalod M, et al. Cutting edge: Priming of NK cells by IL-18. J Immunol (2008) 181:1627–31. doi: 10.4049/jimmunol.181.3.1627
33. Kaplanski G. Interleukin-18: Biological properties and role in disease pathogenesis. Immunol Rev (2018) 281:138–53. doi: 10.1111/imr.12616
34. Ruscitti P, Cipriani P, Di Benedetto P, Liakouli V, Carubbi F, Berardicurti O, et al. Advances in immunopathogenesis of macrophage activation syndrome during rheumatic inflammatory diseases: toward new therapeutic targets? Expert Rev Clin Immunol (2017) 13:1041–7. doi: 10.1080/1744666X.2017.1372194
35. Chen DY, Lan JL, Lin FJ, Hsieh TY, Wen MC. Predominance of Th1 cytokine in peripheral blood and pathological tissues of patients with active untreated adult onset Still's disease. Ann Rheum Dis (2004) 63:1300–6. doi: 10.1136/ard.2003.013680
36. Chen DY, Chen YM, Lan JL, Lin CC, Chen HH, Hsieh CW. Potential role of Th17 cells in the pathogenesis of adult-onset Still's disease. Rheumatol (Oxford) (2010) 49:2305–12. doi: 10.1093/rheumatology/keq284
37. Waite JC, Skokos D. Th17 response and inflammatory autoimmune diseases. Int J Inflam (2012) 819467:2012. doi: 10.1155/2012/819467
38. Shimojima Y, Ichikawa T, Kishida D, Takamatsu R, Sekijima Y. Circulating regulatory T cells in adult-onset Still's disease: Focusing on their plasticity and stability. Clin Exp Immunol (2021) 206:184–95. doi: 10.1111/cei.13648
39. Ruscitti P, Cipriani P, Di Benedetto P, Ciccia F, Liakouli V, Carubbi F, et al. Increased level of h-ferritin and its imbalance with l-ferritin, in bone marrow and liver of patients with adult onset Still's disease, developing macrophage activation syndrome, correlate with the severity of the disease. Autoimmune Rev (2015) 14:429–37. doi: 10.1016/j.autrev.2015.01.004
40. Ruscitti P, Cipriani P, Di Benedetto P, Liakouli V, Berardicurti O, Carubbi F, et al. H-ferritin and proinflammatory cytokines are increased in the bone marrow of patients affected by macrophage activation syndrome. Clin Exp Immunol (2018) 191:220–8. doi: 10.1111/cei.13057
41. Ruscitti P, Cipriani P, Ciccia F, Di Benedetto P, Liakouli V, Berardicurti O, et al. H-ferritin and CD68(+) /H-ferritin(+) monocytes/macrophages are increased in the skin of adult-onset Still's disease patients and correlate with the multi-visceral involvement of the disease. Clin Exp Immunol (2016) 186:30–8. doi: 10.1111/cei.12826
42. Ruscitti P, Ciccia F, Cipriani P, Guggino G, Di Benedetto P, Rizzo A, et al. The CD68(+)/H-ferritin(+) cells colonize the lymph nodes of the patients with adult onset Still's disease and are associated with increased extracellular level of h-ferritin in the same tissue: correlation with disease severity and implication for pathogenesis. Clin Exp Immunol (2016) 183:397–404. doi: 10.1111/cei.12738
43. Ruscitti P, Berardicurti O, Barile A, Cipriani P, Shoenfeld Y, Iagnocco A, et al. Severe COVID-19 and related hyperferritinaemia: more than an innocent bystander? Ann Rheum Dis (2020) 79:1515–6. doi: 10.1136/annrheumdis-2020-217618
44. Put K, Avau A, Brisse E, Mitera T, Put S, Proost P, et al. Cytokines in systemic juvenile idiopathic arthritis and haemophagocytic lymphohistiocytosis: tipping the balance between interleukin-18 and interferon-γ. Rheumatol (Oxford) (2015) 54:1507–17. doi: 10.1093/rheumatology/keu524
45. Colafrancesco S, Priori R, Alessandri C, Perricone C, Pendolino M, Picarelli G, et al. IL-18 serum level in adult onset Still's disease: A marker of disease activity. Int J Inflam (2012) 2012:156890. doi: 10.1155/2012/156890
46. Segawa S, Kondo Y, Nakai Y, Iizuka A, Kaneko S, Yokosawa M, et al. Placenta specific 8 suppresses IL-18 production through regulation of autophagy and is associated with adult Still disease. J Immunol (2018) 201:3534–45. doi: 10.4049/jimmunol.1800667
47. Aizaki Y, Yazawa H, Sato K, Mimura T. Dual effects of interleukin-10 on natural killer cells and monocytes and the implications for adult-onset Still's disease. Clin Exp Rheumatol (2021) 39 Suppl 132:22–9. doi: 10.55563/clinexprheumatol/2vdem0
48. Ge Y, Chen J, Hu Y, Chen X, Huang M. IL-38 alleviates inflammation in sepsis in mice by inhibiting macrophage apoptosis and activation of the NLRP3 inflammasome. Mediators Inflammation (2021), 2021:6370911–13. doi: 10.1155/2021/6370911
49. Li Z, Ding Y, Peng Y, Yu J, Pan C, Cai Y, et al. Effects of IL-38 on macrophages and myocardial ischemic injury. Front Immunol (2022) 13:894002. doi: 10.3389/fimmu.2022.894002
50. Kirino Y, Takeno M, Iwasaki M, Ueda A, Ohno S, Shirai A, et al. Increased serum HO-1 in hemophagocytic syndrome and adult-onset Still's disease: use in the differential diagnosis of hyperferritinemia. Arthritis Res Ther (2005) 7:R616–624. doi: 10.1186/ar1721
51. Fautrel B, Le Moël G, Saint-Marcoux B, Taupin P, Vignes S, Rozenberg S, et al. Diagnostic value of ferritin and glycosylated ferritin in adult onset Still's disease. J Rheumatol (2001) 28:322–9.
52. Jung JY, Kim JW, Suh CH, Kim HA. Roles of interactions between toll-like receptors and their endogenous ligands in the pathogenesis of systemic juvenile idiopathic arthritis and adult-onset Still's disease. Front Immunol (2020) 11:583513. doi: 10.3389/fimmu.2020.583513
53. Hu Q, Shi H, Zeng T, Liu H, Su Y, Cheng X, et al. Increased neutrophil extracellular traps activate NLRP3 and inflammatory macrophages in adult-onset Still's disease. Arthritis Res Ther (2019) 21:9. doi: 10.1186/s13075-018-1800-z
Keywords: adult-onset still disease, monocytes/macrophages, inflammasome, placenta-specific 8, IL-1β, IL-18
Citation: Tsuboi H, Segawa S, Yagishita M, Toko H, Honda F, Kitada A, Miki H, Ohyama A, Hagiwara S, Kondo Y, Matsumoto I and Sumida T (2022) Activation mechanisms of monocytes/macrophages in adult-onset Still disease. Front. Immunol. 13:953730. doi: 10.3389/fimmu.2022.953730
Received: 26 May 2022; Accepted: 02 August 2022;
Published: 26 August 2022.
Edited by:
Atsushi Kawakami, Nagasaki University, JapanCopyright © 2022 Tsuboi, Segawa, Yagishita, Toko, Honda, Kitada, Miki, Ohyama, Hagiwara, Kondo, Matsumoto and Sumida. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hiroto Tsuboi, SGlyb3RvLVRzdWJvaUBtZC50c3VrdWJhLmFjLmpw
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.