 Jie Lin1
Jie Lin1 XueLian Tan
XueLian Tan- 1State Key Laboratory of Oral Diseases and National Clinical Research Center for Oral Diseases and Department of Endodontics, West China Hospital of Stomatology, Sichuan University, Chengdu, China
- 2Myeloma Center, Winthrop P. Rockefeller Cancer Institute, Department of Internal Medicine, University of Arkansas for Medical Sciences (UAMS), Little Rock, AR, United States
- 3Division of Hematology and Oncology, Department of Internal Medicine, University of Iowa, Iowa, IA, United States
Porphyromonas gingivalis (P. gingivalis) is a Gram-negative anaerobic pathogen that is involved in the pathogenesis of periodontitis and systemic diseases. P. gingivalis has recently been detected in rheumatoid arthritis (RA), cardiovascular disease, and tumors, as well as Alzheimer’s disease (AD), and the presence of P. gingivalis in these diseases are correlated with poor prognosis. Macrophages are major innate immune cells which modulate immune responses against pathogens, however, multiple bacteria have evolved abilities to evade or even subvert the macrophages’ immune response, in which subsequently promote the diseases’ initiation and progression. P. gingivalis as a keystone pathogen of periodontitis has received increasing attention for the onset and development of systemic diseases. P. gingivalis induces macrophage polarization and inflammasome activation. It also causes immune response evasion which plays important roles in promoting inflammatory diseases, autoimmune diseases, and tumor development. In this review, we summarize recent discoveries on the interaction of P. gingivalis and macrophages in relevant disease development and progression, such as periodontitis, atherosclerosis, RA, AD, and cancers, aiming to provide an in-depth mechanistic understanding of this interaction and potential therapeutic strategies.
Introduction
The innate immune system is the first line of defense against pathogenic organisms. Macrophages are a critical component of this innate immune response and are integral for initiating and sustaining the adaptive immune response. Macrophages were first discovered in the late 19th century (1) and are known to shape the host immune response to bacterial infection through phagocytosis, antigen presentation, and cytokine release (2). Macrophages are highly plastic cells of the innate immune system and play central roles in immunity against microbes, despite all these, macrophages also contribute to a wide array of pathologies depending on specific microenvironments. In atherosclerosis, macrophages are major players that form foam cells and mediate plaque stability (3). Microglial activation (the resident macrophages in the central nervous system) is a salient feature of neuroinflammation that is prominent in almost all neurodegenerative diseases (4). Tumor-associated macrophages (TAMs) are one of tumor-infiltrating immune cells suppressing immune surveillance. TAMs participate in tumor angiogenesis by secreting pro-angiogenic factors, including tumor necrosis factor-alpha (TNF-α), Interleukin-1β(IL-1β), C-C motif chemokine ligand 2 (CCL-2), and matrix metalloproteases (MMPs) (5), which are essential for metastasis and development of late-stage cancers (6).
P. gingivalis is an oral colonizing pathogen that requires hemin and iron for its growth and virulence. It is a Gram-negative anaerobic pathogen, producing numerous virulence factors such as fimbriae, capsules, lipopolysaccharide (LPS), lipoteichoic acids, gingipains, and outer membrane vesicles (OMVs)for its survival in hosts. Although P. gingivalis comprises only 0.8% of total clones in active human periodontitis, it is capable of remodeling benign microbiota into dysbiotic ones and is a putative keystone pathogen in the progression of periodontitis (7). Although the role of P. gingivalis acting as an oral pathogen is well-known, the effects of P. gingivalis extend beyond the oral cavity. In Alzheimer’s disease (AD), P. gingivalis induced peripheral amyloid β protein (Aβ) influx, aggravating the progress of AD (8). In patients with clinical and subclinical rheumatoid arthritis (RA), the number of P. gingivalis was increased (9); transcriptome of human blood samples also show that 14 periodontitis-associated pathways were significantly expressed during RA pathogenesis in RA patients and human gingival tissues from periodontitis patients (9). Furthermore, P. gingivalis infection are related to the production of anticitrullinated protein antibody (ACPA) in RA (10). Patients diagnosed with esophageal squamous cell carcinoma have poorer prognosis when P. gingivalis is detected. Higher levels of P. gingivalis have been revealed in infiltrating tumoral tissues than in normal ones (11, 12). These observations strongly suggest that P. gingivalis may play critical roles in systemic diseases.
P. gingivalis’ role in systemic diseases were discovered, but the mechanisms of how P. gingivalis participate in these processes are rarely reported. We reviewed recent studies about P. gingivalis associated diseases and proposed that macrophages may serve as a communication network linking P. gingivalis infection and systemic diseases. P. gingivalis can invade and survive in resident macrophages, P. gingivalis-infected macrophages were observed to injure distant organs by producing cytotoxic extracellular vesicles in animal study (13). P. gingivalis uses complex strategies to evade the major antimicrobial mechanisms of macrophages, including pyroptosis (14), uncoupled inflammation and immune response, and disrupting phagosome-lysosome maturation (15). In this review, we will discuss how P. gingivalis acts on macrophages, evading the antimicrobial mechanisms of macrophages, and influences the polarization of macrophages in different microenvironments. The role of activation of NOD-LRR and pyrin domain-containing protein 3 (NLRP3) inflammasomes in macrophages by P. gingivalis will also be discussed. P. gingivalis will promote the polarization of macrophages and inflammasome activation if it is not eliminated by the hosts’ immune response. Thus, the survival and mechanisms of P. gingivalis from the hosts’ immune response will need to be studied and addressed. We also addressed that macrophages are a double-edged sword in response to microbial infection, its phenotype changes are essential in the processes of these diseases. An in-depth understanding of P. gingivalis-macrophage interaction with novel mechanistic insights into the pathogenesis of P. gingivalis with associated systemic diseases may be important for designing therapeutic strategies.
Macrophage polarization in P. gingivalis-associated diseases
Macrophage polarization is the process in which macrophages are activated at a certain point in space and time and form different macrophage subtypes according to their environments (16). There are two major macrophage phenotypes, classically activated M1 macrophages and alternatively activated M2 macrophages (17). Different macrophage phenotypes have distinct functions. M1 macrophages can be polarized by granulocyte-macrophage colony-stimulating factor (GM-CSF), LPS, IFN-γ, and toll-like receptor (TLR) ligands, while M2 macrophages are polarized by macrophage colony-stimulating factor (M-CSF), IL-4, IL-13, IL-10, and immune complexes (18). M1 macrophages upregulate the expression of costimulatory molecules such as CD86 on the cell surface (18). M2 macrophages exhibit anti-inflammatory properties and express high levels of CD206, arginase-1 (Arg-1), IL-10, and transforming growth factor-beta (TGF-β), which negatively regulate M1 macrophage activity and contribute to the wound healing process (16, 18, 19).
M1 macrophages have different metabolic profile compared to M2 macrophages. M1 macrophages utilize glycolysis metabolisms while in M2 macrophages the tricarboxylic acid (TCA) cycle has primacy over glycolysis (20). Metabolic reprogramming influences macrophage polarization (21, 22). In M1 macrophages the TCA cycle are broken in two places: after citrate and after succinate, which leads to citrate and succinate accumulation. Citrate was involved in producing NO, ROS, and prostaglandins (20). 24h after macrophages infected by P. gingivalis or its OMVs, will increase expression of glycolytic genes (e.g., Glut-1, Hk1/2, Pfkfb, and Pkfl) and will decrease TCA genes (e.g., Fh1, Pck2, and Suclg2) (23). P. gingivalis infection impairs TCA and suppresses α-KG production by down-regulation of Idh1/2 and Gpt1/2, while it induces the accumulation of succinate (19). α-KG is a key metabolite that induces M2 macrophage polarization, prolylhydroxylases utilize α-KG as a substrate to destabilize HIF-1α.HIF-1α degradation suppresses glycolysis, decreasing M1 macrophage (24). Also, α-KG restricts M1 macrophage activation by downregulating the NF-κB pathway. It is a co-stimulator with Jmjd3. With their combination, they will regulate the trimethylation of histone H3, lysine 27 (H3K27me3) on promoter regions of genes that define the M2 phenotype, thus promoting M2 macrophage activation (25)(Figure 1). In addition, P. gingivalis activates TLR2 and TLR4 and elicits the expression of TNF-α in macrophages (26). TNF-α is an anti-M2 macrophage factor that blocks M2 macrophage polarization on two levels: through its direct effects on macrophages and the indirect effects of TNF-α on IL-13 production by other innate cell types (17).
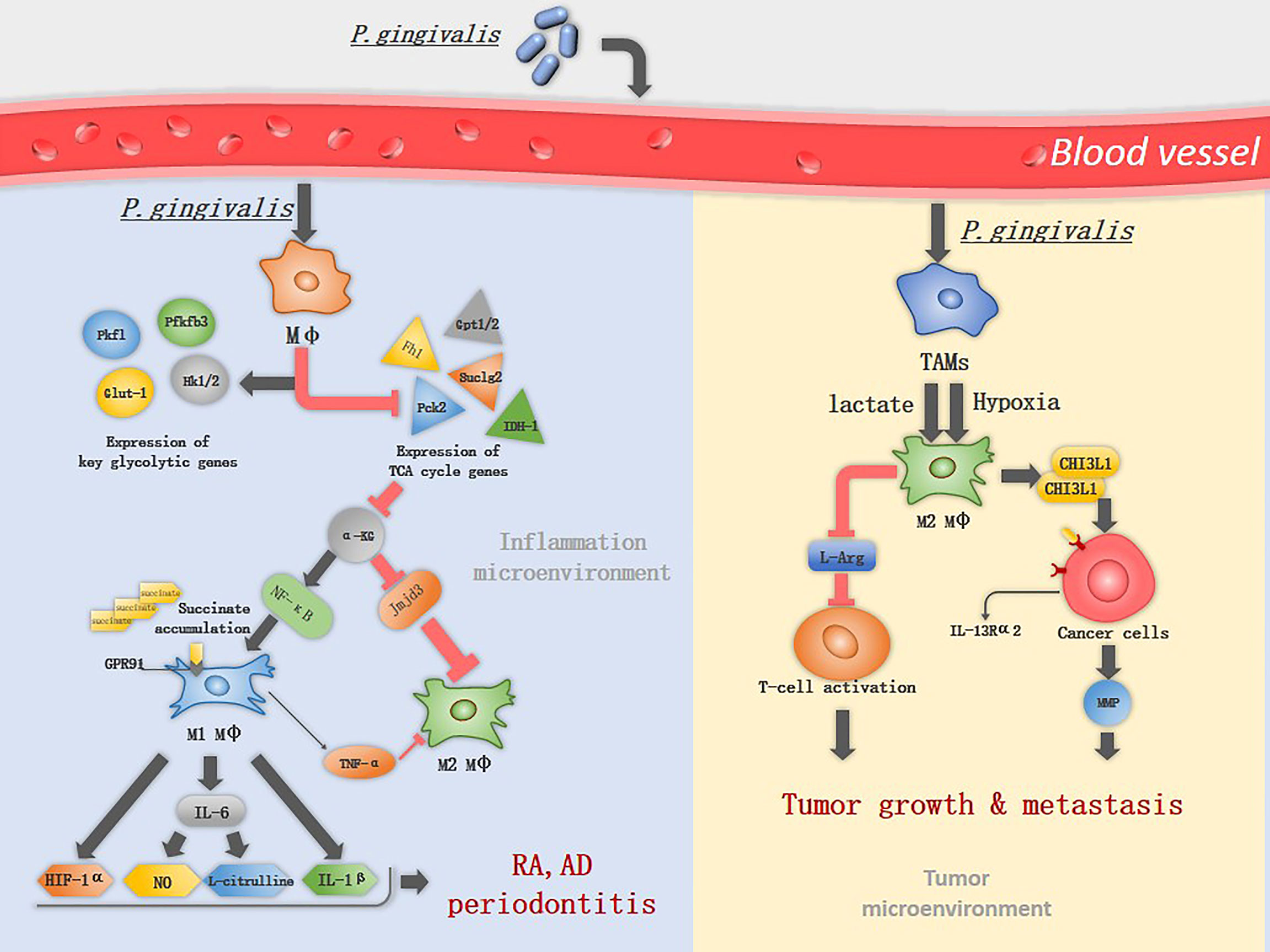
Figure 1 The mechanism of P. gingivalis in inducing macrophage polarization. P. gingivalis interacts with macrophages and increases the expression of key glycolytic genes, while inhibits the expression of TCA associated genes. α-KG is an important immunomodulator of M2 macrophage activation and can be suppressed by P. gingivalis through down-regulation of Idh1/2 and Gpt1/2, therefor induce classical M1 macrophage activation. M1 macrophages upregulate the expression of GPR91 and produce TNF-α, IL-1β, IL-6, NO, L-citrulline and HIF-1α, promoting the progress of RA and periodontitis. In TME, lactate and hypoxia promote TAM polarization into M2-like macrophages, which deplete L-Arg resulting in suppression of cytotoxic T-cell activation. M2 macrophages promote tumor metastasis by secreting CHI3L1 protein and MMPs, the CHI3L1 protein interacts with IL-13Rα2 expressing on the cancer cells, leading to upregulation of MMPs.
An imbalanced M1/M2 ratio was discovered in various systemic diseases and tumors (27). Below we have listed well-studied examples such as periodontitis, RA, AD, and tumors, to reflect the interaction between P. gingivalis and macrophages.
Periodontitis
Periodontitis is the sixth-most prevalent disease in the world, affecting many adults (28). Bacterial infection and immunological disorder are important elements of periodontitis. Gram-negative anaerobic bacteria that colonize subgingiva initiate the progression of periodontitis, and the host inflammatory response inflicts irreversible damage to the periodontal tissues (29). P. gingivalis is a putative keystone pathogen with the ability to impair innate immunity and transform a normally symbiotic microbiota into a dysbiotic state in periodontal tissue (7). Macrophages participate in the initiation of inflammation and are the major immune cells producing pro-inflammatory cytokines and mediating alveolar bone resorption in periodontitis (30). P. gingivalis induces M1macrophages by downregulating α-KG production (19), and M1 macrophages were the dominant phenotype for the gingival infiltration in response to P. gingivalis infection (31). Compared with healthy gingival tissues, the number of M1 macrophages and the M1/M2 ratio were enhanced (32). The production of pro-inflammatory cytokines characterizes M1 macrophages however, excessive or sustained pro-inflammatory cytokine production promotes periodontal destruction (33), and decreasing the M1/M2 ratio in mouse periodontitis models was proved effective in preventing alveolar bone loss (34). The newly developed nanocomposites which inhibit M1 macrophages’ polarization simultaneously initiate M2 macrophage polarization exhibiting a favorable repairment in periodontal soft tissue and decreased local periodontal inflammation in rat models, suggesting that the phenotypic switch of M1 to M2 might be a critical mechanism in mediating periodontal tissue repair.
Rheumatoid arthritis
Rheumatoid arthritis (RA) is an autoimmune disease characterized by synovial inflammation and joint erosion, affecting 1–2% of the population worldwide (35). Research shows that oral microbiota dysbiosis is a high-risk factor for subclinical RA, P. gingivalis as a keystone pathogen inducing dysbiosis (9, 10), its DNA was found in the synovial tissue recently (36). In fact, peptidylarginine deiminase (PPAD) produced by P. gingivalis is able to citrullinate both endogenous and human proteins. Citrulline-specific autoimmunity is a key feature of RA, suggesting P. gingivalis may contribute to the development of RA (37). P. gingivalis exposure leads to anti-cyclic citrullinated peptide 2 (anti-CCP2) production, and subsequent bone resorption also confirms this hypothesis (38). Study has showed increased macrophages in the inflamed synovial membrane, and exhibit the potential of macrophages to be an early hallmark of active RA (39).Considering that macrophages are major cells responding to microorganism infection, it is very likely that the P. gingivalis-macrophages interaction is an important mechanism in RA.
Macrophages are predominantly the M1 phenotype in RA (16), which is known to produce many inflammatory cytokines that promote RA progression, including TNF-α, IL-1β, IL-12, IL-18, and IL-23 (40). Among these cytokines, TNF-α plays a major role and leads to the development of chronic polyarthritis, TNF-α and IL-6 together induce the differentiation of osteoclasts in a RANKL-independent manner (41, 42). P. gingivalis induces the M1 macrophage activation. The metabolic remodeling in M1 macrophages fuels the production of lactate and succinate, acidifying the extracellular space, which promotes the formation of a low-glucose and high-lactate microenvironments, a typical microenvironments in RA (43). Succinate accumulation leads to HIF-1α activation via inhibition of prolyl hydroxylases, HIF-1α stabilization and activation facilitate the metabolic shift from OXPHOS to glycolysis, therefore, sustaining the inflammatory phenotype of M1 macrophage (44). Extracellular accumulation of succinate activates the G protein-coupled receptor 91(GPR91) on macrophages, is involved in pro-angiogenesis of RA, facilitating immune cell extravasation into the synovium (45). iNOS is highly expressed in M1 macrophages, which is then catalyzed by L-Arg into NO and L-citrulline, L-citrulline is commonly accepted as a biomarker in RA (40) (Figure 1). Transforming M1 into M2 macrophages resulted in the reduction of clinical arthritis scores (40), suggesting that altered macrophages phenotype is a viable therapeutic option in RA.
Alzheimer’s disease
Alzheimer’s disease (AD) is characterized by diminished cognitive function, specifically dysfunction of memory and judgment. The presence of extracellular Aβ marks pathologically AD. An anti-infectious agent in AD was proposed around 30 years ago on the basis of the discovery of herpes simplex virus 1 (HSV1) DNA in brain tissue in a high proportion of older people (46), and recently neuroinflammation has been suggested as a vital player in AD (47).
The role of P. gingivalis in neuroinflammation was discovered in recent studies, and small-molecule inhibitors targeting gingipains blocked P.gingivalis-induced neurodegeneration significantly (48, 49). Microglia are resident macrophages in the central nervous system constituting 5–10% of total brain cells, which changes their phenotype when stimulated by cytokines or LPS (50). The microglia-P.gingivalis interaction in the brain is related to the process of neuroinflammation. In rat brain, P. gingivalis LPS was discovered to activate microglia and increase the expression of TNF-α, IL-1β, IL-6, higher levels of the CD86 marker, and iNOS. INOS and CD86 are the landmarks of M1 microglia, were also found in P. gingivalis infected microglia (51, 52), illustrating that P. gingivalis LPS stimulates the M1 activation of microglia. It was observed in animal models that P. gingivalis trigger the polarization of M1 macrophages via TLR4/NF-κB signaling pathway (52). In peripheral circulation, macrophages are one of the pools for Aβ (53), P. gingivalis activates the NF-κB/cathepsin B pathway, promoting the generation of Aβ by macrophages (54), then peripheral Aβ is transferred into the brain by advanced glycation end products (RAGE) expressed on cerebral endothelial cells (8). In vitro cell experiment confirmed that Aβ could increase the expression of iNOS whereas it downregulated Arg-1 in microglia, inducing the M1 microglia (55).
Pro-inflammation cytokines released by M1 microglia can aggravate AD, leading to synaptic dysfunction, neuronal death and inhibition of neurogenesis (47). Microglia with M1 phenotype have decreased phagocytosis of Aβ, and extracellular Aβ is an important characteristic of AD (56). The shift of M1 microglia to M2 phenotype showed decreased expression of pro-inflammation cytokines, and an increased level of the triggering receptor expressed on myeloid cells (TREM2) on the surface of microglia, which plays an essential role in the clearance of Aβ (57). TREM2 overexpression induces microglial polarization towards the M2 phenotype by suppressing the NF-kB pathway, attenuates the cognitive impairment (50).
Tumors
During early carcinogenesis, TAMs exhibit a higher degree of similarity to M1 macrophages. M1 macrophages exert anti-tumor functions, including directly mediating cytotoxicity and antibody-dependent cell-mediated cytotoxicity to kill tumor cells (58), therefore it can efficiently recognize and destroy cancer cells. However, in the later stages of carcinogenesis, various factors in tumor microenvironments (TME), such as low pH and hypoxia promote TAM polarization into M2-like macrophages (11). TAMs with numerous M2 macrophage characteristics are linked to poor prognosis in cancer (17). M2 macrophages produce growth factors, protease and vascular endothelial growth factors (59). They also suppress T-cell functions by depleting L-arginine (L-Arg) and L-tryptophan from the TME via the expression of arginase 1 (Arg1) and indoleamine 2,3-dioxygenase (IDO), respectively (6). L-Arg is an essential amino acid for the re-expression of T-cell receptors following antigen engagement on T cells (5). M2 macrophages promote tumor metastasis by secreting chitinase 3-like protein 1 (CHI3L1) protein. The CHI3L1 protein interacts with interleukin-13 receptor α2 chain (IL-13Rα2) on the cancer cells, leading to upregulation of MMPs (5) (Figure 1).
Increasing evidence support an association between P. gingivalis and colorectal carcinoma, oral and esophageal squamous cell cancer, pancreatic cancer (60–62). One report indicated that in TME, P. gingivalis could increase the M2/M1 ratio and upregulate the expression of genes encoding for protumor molecules in TAMs, but the specific mechanism is yet unclear (11). M1 macrophages utilize glycolysis to generate adenosine triphosphate (ATP), resulting in lactate accumulation. In TME, lactate is not only a metabolic by-product, it stimulates histone lactylation which increases the expression and genes associated with M2 macrophage polarization (63).Stimulation with LPS, 4h later, inflammatory response genes (for example, Nos2) were induced, while Arg1 levels were markedly increased 24–48 h after M1 polarization. The late-phase switch to M2-like phenotype originated from increased histone lactylation (64). This may partly explain the function of P. gingivalis in TME.
Inflammasome activation in macrophages induced by P. gingivalis is implicated in multiple diseases
Inflammasomes are large, multiprotein complexes localized in the cytoplasm of the cell that initiate proteolytic processing of pro-inflammatory cytokines pro-IL-1β and pro-IL-18 into mature inflammatory cytokines. Inflammasome activation plays a key role in innate immunity and is involved in some inflammatory diseases and tumors (3).
Typical inflammasomes are constructed of pro-caspase-1, nucleotide-binding domain (NBD), and leucine-rich repeats (LRRs), called the NBD-LRR (NLR) superfamily that is responsible for the recognition of pathogen- associated molecular patterns (PAMPs) or other signals (65) and adapter molecule apoptosis-associated speck-like protein containing a caspase activation and recruitment domain (ASC) (Figure 2). Among these, caspase-1 cleaves pro-IL-1β and pro-IL-18, it also mediates their maturation and excretion (66). Inflammasome activation consists of two steps, an initial “cell priming” and a second “triggering” event, resulting in the proteolytic maturation and secretion of IL-1β (2).
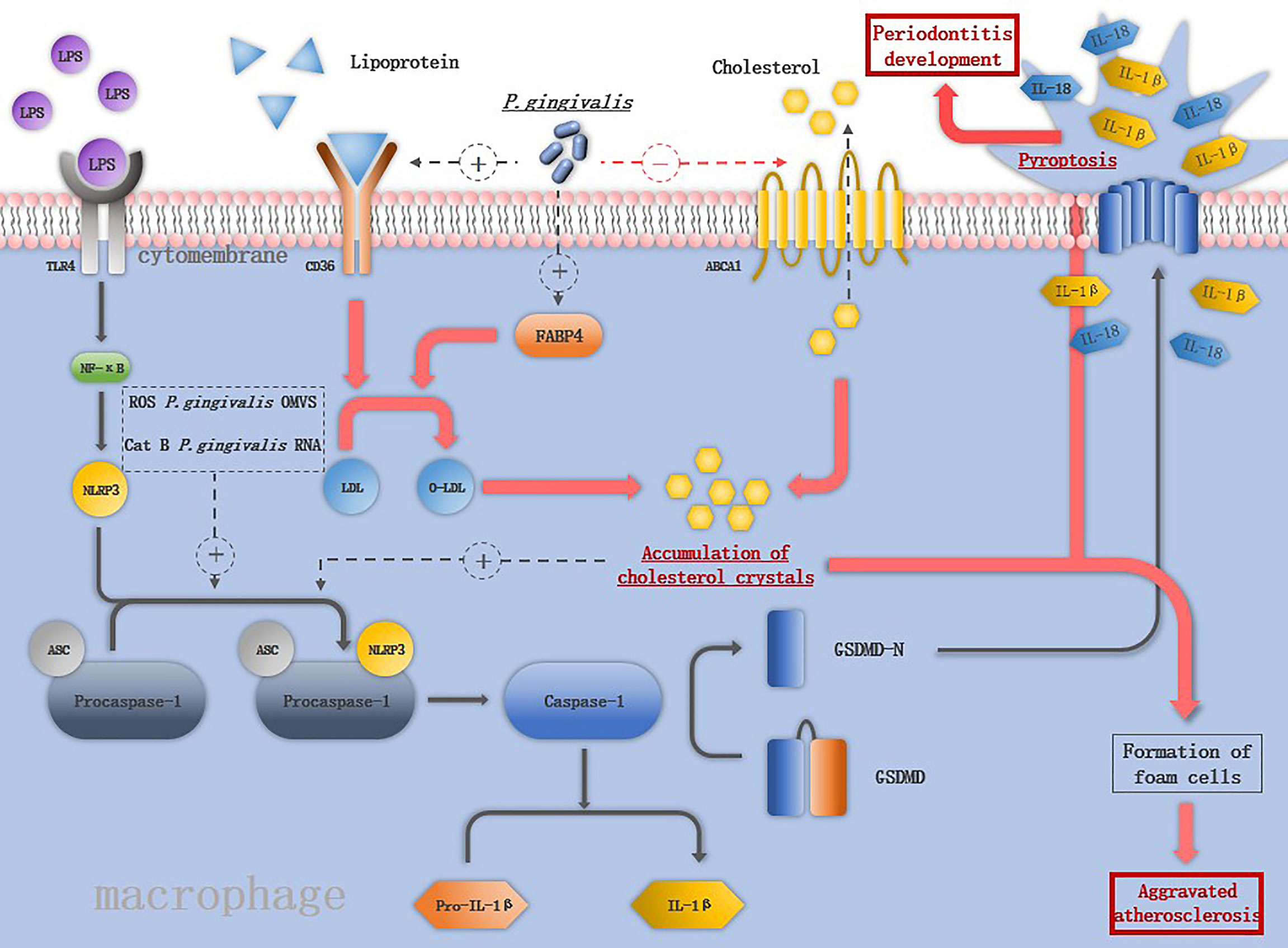
Figure 2 The mechanism of P. gingivalis in activating the inflammasomes in macrophage: LPS binds to TLR4 and activates NF-macrophage:increases the expression of keys are assembled in response to the second stimuli such as ROS, CatB, P. gingivalis OMVS and P. gingivalis RNA. The activation of NLRP3 inflammasome induces the maturation of IL-1ression of keys are assembled in response to the second stimuli such as ROS, Cs macrophage pyroptosis. Pore formation in the cell membrane leads to the release of IL-1β and IL-18 and promotes the destruction of periodontal tissue. P. gingivalis increases the expression of CD36 and FABP4, promoting the internalization of o-LDL and intracellular conversion of o-LDL to cholesterol crystals, which is able to activate NLRP3 inflammasomes. P. gingivalis also downregulates the ABCA1 on macrophages to promote cholesterol accumulation. The accumulation of cholesterol together with the releases of cellular components by pyroptosis contribute to the formation of foam cells, the hallmark cells of atherosclerosis.
Periodontitis
Various cytokines produced by macrophages have long been believed to promote the development of periodontitis; inflammasome activation induces the maturation of two vital pro-inflammatory cytokines- IL-1β and IL-18 (67). Upregulation of inflammasome components caspase-1, NLRP3, and absent in melanoma 2 (AIM2) in gingival epithelial cells and macrophages of periodontitis patients suggests its role (68). NLRP3 inflammasomes is the most well-studied inflammasomes, experiments in mice models proved that alveolar bone loss induced by P. gingivalis infection was suppressed significantly in NLRP3-KO mice (69). The activation of NLRP3 inflammasome needs two steps, firstly NF-κB signaling is activated and then NLRP3 inflammasomes are assembled in response to the second stimuli (70).
As important pro-inflammation cytokines source, macrophages interact with P. gingivalis activating NLRP3 inflammasomes. Compared to S. mitis, P. gingivalis promotes more robust secretion of IL-1β in macrophages by both NLRP3-caspase-1 canonical signaling pathway and NLRP3-caspase-4 signaling pathway (71). P. gingivalis induces the M1 macrophages accompanied by the release of HIF-1α, under hypoxia condition, augmented NLRP3 mRNA expression and more robust caspase-1 activation was detected in macrophages (72). Live P. gingivalis upregulates the miR-155 in macrophages to promote macrophage NLRP3 inflammasome activation (14). The virulence factors of P. gingivalis play different roles in inducing macrophage inflammasome activation. The OMVs of P. gingivalis included prime signal and the second signal. Both were needed to trigger the signaling cascade to activate macrophage inflammasomes and secretion of IL-1β (73), while gingipains apparently have a paradoxical role in activating inflammasomes, namely, to enhance caspase-1 activation and conversely, to cause proteolytic depletion of caspase-1 and IL-1β (23). NLRP3 inflammasome activation induce the macrophages pyroptosis, pyroptosis was initially described as a form of programmed cell death dependent on caspase-1, which was first described in 1992 in macrophages infected with Shigella flexneri (74). The prototypical form of pyroptosis is triggered by activation of pro-inflammatory caspases (caspase-1, -4, and -5 in humans and caspase-1 and -11 in mice) (75). Terminal cell lysis is then mediated through cleavage of gasdermin D (GSDMD) by one of these caspases (14, 76). Pore formation in the cell membrane leads to the release of IL-1β and IL-18 and promotes the destruction of periodontal tissue (76) (Figure 2). Pyroptosis may be an antimicrobial response of macrophages to eliminate intracellular P. gingivalis, it can also cause tissue injury, accelerate bacterial dissemination, and inhibit bacterial clearance from tissues (2).
Atherosclerosis
Atherosclerosis, a complex multi-factorial chronic inflammatory disease, is characterized by the formation of atherosclerotic plaques. Macrophage numbers increase up to 20-fold within mouse aortas during atherogenesis (77), and the presence of cholesterol-engorged macrophage foam cells in atherosclerotic plaques is a hallmark of atherosclerosis (39). According to the newest hypotheses, inflammatory processes and lipid metabolism imbalance jointly contribute to the formation of atherosclerotic plaques in the arterial wall (78). P. gingivalis DNA was detected in atherosclerotic plaque in subjects with periodontitis. Clinical studies and animal models have reported that stimulation with P. gingivalis accelerates atherosclerosis (79, 80). In vitro research has shown that P. gingivalis can increase the expression of cell adhesion molecules, pro-inflammatory cytokines, and chemokines in endothelial cells, which have crucial roles in the recruitment of monocytes to the vascular endothelium and the subsequent formation of atherosclerotic plaques (81). The macrophages-P. gingivalis interaction in atherosclerosis is related to its initiation and progression.
A key feature of atherosclerosis is lipoprotein ingestion and accumulation by arterial macrophages via CD36. P. gingivalis increases the expression of CD36 and fatty acid binding protein 4 (FABP4) on macrophages, promoting the internalization of oxidized LDL (o-LDL) and intracellular conversion of o-LDL to cholesterol crystals (82, 83). Compared to nonatherosclerotic vessels, the mRNA level of NLRP3 inflammasome-related genes is significantly increased in human atherosclerotic plaques (84). NLRP3 inflammasome activation plays an important role in pathophysiology of atherosclerosis (Figure 2), and their silence was reported to cause the stabilization of atherosclerotic plaque (78). Cholesterol crystals in macrophages can damage the phagolysosomes and induce the release of cathepsin B, and along with P. gingivalis-induced ROS production in macrophages are able to activate NLRP3 inflammasomes. IL-1β is released through NLRP3 inflammasome activation and promotes the rupture of atherosclerotic plaques (70). Macrophages turn into foam cells in four steps: uptake of lipoproteins by CD36, hydrolysis of cholesterol esters, efflux of free cholesterol regulated by cholesterol transporters ATP-binding cassettes A1 and G1 (ABCA1 and ABCG1), and re-esterification of cytosolic cholesterol (85, 86). P. gingivalis downregulates the ABCA1 on macrophages and promotes cholesterol accumulation. IL-1β is a key cytokine in atherosclerosis, and IL-1β-/- mice were reported to have a 30% reduction in the size of atherosclerotic plaques compared with the control group, in addition, monoclonal antibodies against IL-1β inhibit plaque formation in apo E-/- mice (87). P. gingivalis activates the NLRP3 inflammasomes followed by macrophage pyroptosis, and macrophage pyroptosis releases cellular components into the plaque milieu, which is thought to contribute to the formation of the lipid-rich, acellular necrotic core, which characterizes vulnerable plaques (88).
Macrophage immune response suppressed by P. gingivalis promotes disease progression
Survival of P. gingivalis from macrophages is an important prerequisite for P. gingivalis to start its promotion in the M1 macrophage polarization and activation of the inflammasomes. P. gingivalis can suppress macrophage immune responses by various mechanisms (Figure 3). Firstly, the capsule of P. gingivalis is able to reduce the ability of macrophage to phagocytose P. gingivalis, and the hemagglutinin/adhesion domain of gingipains can cleave the LPS receptor CD14 from the surface of macrophages, resulted in a lower ability to phagocytize bacteria (89, 90). When phagocytosed by macrophages, P. gingivalis is able to exit macrophages therefor avoiding being killed (91). Besides, P. gingivalis utilizes the complement C5a receptor 1-toll-like receptor 2 (C5aR1-TLR2) pathway to subvert immune response and suppresses phagolysosomal maturation, thus promotes intracellular survival (15). P. gingivalis releases sialidase which increases complement receptor 3 (CR3) activation in macrophages, then P. gingivalis interacts with CR3 and activates downstream extracellular signal-regulated kinases (ERK) 1/2, which reduces the level of IL-12p70 and inhibits IL-12-mediated clearance of pathogens (92). The inability of macrophages to clear P. gingivalis is associated with diverse diseases.
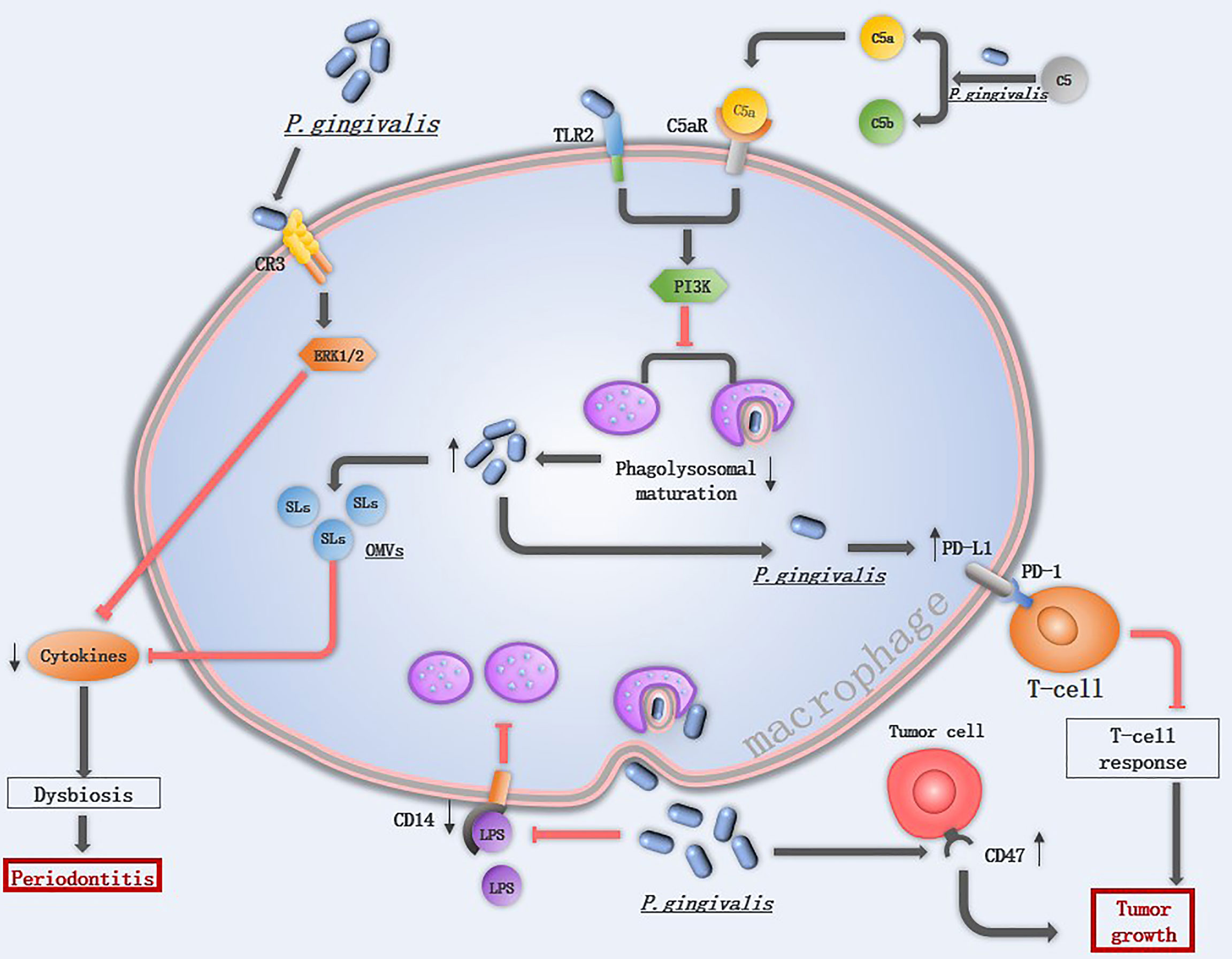
Figure 3 The mechanism of P. gingivalis in suppressing macrophage immune response: Extracellular P. gingivalis increases CR3 activation in macrophages while breakdowns C5 into C5a. The interaction of P. gingivalis with CR3 activates ERK 1/2, which reduces the cytokines level. The hemagglutinin/adhesion domain of gingipains cleave the LPS receptor CD14 from the surface of macrophages, resulted in a lower ability to phagocytize bacteria. The PD-L1 in macrophages is upregulated by P. gingivalis, which binds to PD-1 on T-cells and inhibits T-cell immune surveillance. CD47 is a phagocytosis inhibitor expressed on almost all cancer cells, it is regulated by P. gingivalis and protects cancer cells for macrophage phagocytosis. Once P. gingivalis enters into macrophages, intracellular P. gingivalis secretes OMVs containing SLs reduce the levels of pro-inflammatory cytokines, and initiates C5aR-TLR2 crosstalk signaling, activate the downstream PI3K pathway to suppress phagolysosomal maturation, avoiding being killed.
Periodontitis
Toll-like receptors (TLRs) represent a conserved family of receptors involved in the detection of pathogen-associated molecular patterns (PAMPs) and the cellular response to bacterial invasion (89). TLR2 plays an important role in P. gingivalis infection. TLR2-deficient mice failed to induce alveolar bone resorption, and P. gingivalis cannot be detected in the gingival tissue of most Tlr2−/− mice at 24 h following oral challenge (15). Once TLR2 is activated, P. gingivalis initiates downstream phosphatidylinositol 3-kinase (PI3K) instead of myeloid differentiation factor 88 (MyD88)signaling pathway to escape immune clearance, TLR2- PI3K signaling suppressing the phagolysosomal maturation thereby escaping intracellular killing in macrophages (93). An in vitro experiment also demonstrated that P. gingivalis displayed a significant cycle of entering, exiting, and re-entering in macrophages to avoid being killed (91), implicating that low abundance of P. gingivalis may still induce severe tissue damage. Gingival tissues from healthy individuals and those with periodontitis contain sphingolipids (SLs), an amphipathic lipid that is essential for P. gingivalis to survive under oxidative stress. SLs can be transported to macrophages from P. gingivalis by OMVs and SLs reduce the levels of pro-inflammatory cytokines, therefore limiting the immune response to P. gingivalis (94).
Tumors
P. gingivalis is able to promote cancer development by inhibiting apoptosis and accelerating gingival epithelial cell proliferation in the oral cavity. In addition to this, P. gingivalis also influences the immune response to oral cancer (11). One of the hallmark features of cancer is evasion of immune destruction. P. gingivalis and its LPS suppress the macrophage immune response to tumor cells mainly through upregulating PD-L1 in macrophages via the TLR-4 pathway (95). The peptidoglycan of P. gingivalis can induce PD-L1 expression on various cancer cells by a receptor-interacting protein kinase 2 (RIP2)-dependent mechanism or NOD1, NOD2 and MAPK dependent signaling pathways (96). Programmed cell death 1 (PD-1) is expressed on activated T cells, B cells, monocytes, and macrophages (97), the interaction of PD-L1 with its receptor PD-1 inhibits T-cell responses, and a blockade of this interaction has been proven to be an effective immunotherapy for several different cancers (98). CD47, a “don’t eat me” signal, is a phagocytosis inhibitor expressed on almost all cancer cells, and the ability of P. gingivalis to upregulate the expression level of cd47 in cancer cells may be another mechanism to accelerate tumor development (11).
Discussion
The innate immune system is the first line of defense against pathogenic organisms. Macrophages are a critical component of this innate immune response and are integral for initiating and sustaining the adaptive immune response. However, macrophages are a double-edged sword in the progression of host immune responses. In this review, we discussed the recently discovered mechanisms by which P. gingivalis induces macrophage polarization, activates the inflammasomes, evades the macrophage immune responses, showing that P. gingivalis induces the onset and progression of diseases not only by secreting various virulence factors but also by evading and subverting host innate immune responses.
However, there are still some limitations, in that the microenvironments surrounding macrophages is complex and they can be exposed to various cytokines, even those with opposite effects. The same molecular can play different roles in different microenvironments. For example, while HIF-1α is critical for M1 macrophage polarization, the same regulator participate in lactate-mediated M2 polarizations macrophage (24), hinting a role in both M1 and M2 macrophage polarizations, depending on the cues from the microenvironments. Due to the complex interplay of cytokines, metabolites and limited studies about the macrophages-P gingivalis interaction in tumor microenvironments, future research is expected to decipher it. Although the complexity of M1/M2 ratio, transforming the microglial polarization phenotype in mice models suggested reduced arthritis and learning and memory cognitive deficits (56, 99, 100) proved its therapeutic potentials. Considering the ability of P. gingivalis to induce macrophages M1 polarizations, blocking the macrophages-P gingivalis interaction may be a potential therapeutic method. In addition, although inflammasome activation is associated with various diseases and is discovered in P. gingivalis-infected macrophages, studies have also reported that P. gingivalis suppressed the inflammasome activation at the level of the second signal (101). This hint that the ability of P. gingivalis to activate inflammasomes is complex, and it’s necessary to uncover the function of various virulence factors in inflammasome activation. Besides, the divergent roles of NLRP3 inflammasomes are complex. It carries out both the pro-tumorigenic and anti-tumorigenic functions, host defenses against microorganisms such as Candida albicans and detrimental effects in other metabolic diseases including diabetes, obesity and atherosclerosis (102). However, the inhibition of IL-1β released by NLRP3 inflammasomes reduces cardiovascular burden in clinical data and mice studies, suggesting that modulating NLRP3 inflammasomes or the downstream cytokines is still a promising therapeutic direction in the future.
In this review, we summarized current studies about the macrophages phenotype changes infected by P. gingivalis and its systemic influence, although a more insightful mechanism of macrophages- P. gingivalis interaction is needed, and the mechanism of how P. gingivalis invades various tissues waiting to be determined, targeting macrophages in diverse diseases, reprogramming their phenotype and function, or clearing P. gingivalis and its virulence factors to block the macrophage-P. gingivalis interaction may be alternative therapeutic approach for inflammatory diseases, tumors, and autoimmune diseases. Considering P. gingivalis is an oral colonizing pathogen, sustaining a good oral hygiene or treating periodontitis may be beneficial to curing some systemic diseases.
Author Contributions
JL, DH, HX, FZ, and XT wrote and revised the manuscript. All authors contributed to the article and approved the submitted version
Funding
This work was supported by grants to XT from the National Natural Science Foundation of China (Grant No. 82001037) and the Research and Develop Program, West China Hospital of Stomatology, Sichuan University (Grant No. RD-02-202007), and grants to Dr. Tan from the Natural Science Foundation of Sichuan Province (Grant No. 2022NSFSC0751); grants to FZ from the National Cancer Institute (R01 CA236814) and US Department of Defense (DoD; CA180190), and funding from Myeloma Crowd Research Initiative Award (to FZ) and Riney Family Multiple Myeloma Research Program Fund (to FZ). And grants to DH from the National Natural Science Foundation of China (Grant No. 81970936).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity (2014) 41(1):21–35. doi: 10.1016/j.immuni.2014.06.013
2. Cecil JD, O’Brien-Simpson NM, Lenzo JC, Holden JA, Singleton W, Perez-Gonzalez A, et al. Outer membrane vesicles prime and activate macrophage inflammasomes and cytokine secretion In vitro and in vivo. Front Immunol (2017) 8:1017. doi: 10.3389/fimmu.2017.01017
3. Biswas SK, Mantovani A. Orchestration of metabolism by macrophages. Cell Metab (2012) 15(4):432–7. doi: 10.1016/j.cmet.2011.11.013
4. Li Q, Barres BA. Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol (2018) 18(4):225–42. doi: 10.1038/nri.2017.125
5. Fu LQ, Du WL, Cai MH, Yao JY, Zhao YY, Mou XZ. The roles of tumor-associated macrophages in tumor angiogenesis and metastasis. Cell Immunol (2020) 353:104119. doi: 10.1016/j.cellimm.2020.104119
6. Vakkila J, Lotze MT. Inflammation and necrosis promote tumour growth. Nat Rev Immunol (2004) 4(8):641–8. doi: 10.1038/nri1415
7. Hajishengallis G, Darveau RP, Curtis MA. The keystone-pathogen hypothesis. Nat Rev Microbiol (2012) 10(10):717–25. doi: 10.1038/nrmicro2873
8. Zeng F, Liu Y, Huang W, Qing H, Kadowaki T, Kashiwazaki H, et al. Receptor for advanced glycation end products up-regulation in cerebral endothelial cells mediates cerebrovascular-related amyloid beta accumulation after porphyromonas gingivalis infection. J Neurochem (2021) 158(3):724–36. doi: 10.1111/jnc.15096
9. Cheng Z, Do T, Mankia K, Meade J, Hunt L, Clerehugh V, et al. Dysbiosis in the oral microbiomes of anti-CCP positive individuals at risk of developing rheumatoid arthritis. Ann Rheum Dis (2021) 80(2):162–8. doi: 10.1136/annrheumdis-2020-216972
10. Jenning M, Marklein B, Ytterberg J, Zubarev RA, Joshua V, van Schaardenburg D, et al. Bacterial citrullinated epitopes generated by porphyromonas gingivalis infection-a missing link for ACPA production. Ann Rheum Dis (2020) 79(9):1194–202. doi: 10.1136/annrheumdis-2019-216919
11. Liu S, Zhou X, Peng X, Li M, Ren B, Cheng G, et al. Porphyromonas gingivalis promotes immunoevasion of oral cancer by protecting cancer from macrophage attack. J Immunol (2020) 205(1):282–9. doi: 10.4049/jimmunol.1901138
12. Olsen I, Yilmaz O. Possible role of porphyromonas gingivalis in orodigestive cancers. J Oral Microbiol (2019) 11(1):1563410. doi: 10.1080/20002297.2018.1563410
13. Yoshida K, Yoshida K, Fujiwara N, Seyama M, Ono K, Kawai H, et al. Extracellular vesicles of p. gingivalis-infected macrophages induce lung injury. Biochim Biophys Acta Mol Basis Dis (2021) 1867(11):166236. doi: 10.1016/j.bbadis.2021.166236
14. Li C, Yin W, Yu N, Zhang D, Zhao H, Liu J, et al. miR-155 promotes macrophage pyroptosis induced by porphyromonas gingivalis through regulating the NLRP3 inflammasome. Oral Dis (2019) 25(8):2030–9. doi: 10.1111/odi.13198
15. Lamont RJ, Koo H, Hajishengallis G. The oral microbiota: dynamic communities and host interactions. Nat Rev Microbiol (2018) 16(12):745–59. doi: 10.1038/s41579-018-0089-x
16. Yunna C, Mengru H, Lei W, Weidong C. Macrophage M1/M2 polarization. Eur J Pharmacol (2020) 877:173090. doi: 10.1016/j.ejphar.2020.173090
17. Murray PJ. Macrophage polarization. Annu Rev Physiol (2017) 79:541–66. doi: 10.1146/annurevphysiol-022516-034339
18. Belfield LA, Bennett JH, Abate W, Jackson SK. Exposure to porphyromonas gingivalis LPS during macrophage polarisation leads to diminished inflammatory cytokine production. Arch Oral Biol (2017) 81:41–7. doi: 10.1016/j.archoralbio.2017.04.021
19. Yu S, Ding L, Liang D, Luo L. Porphyromonas gingivalis inhibits M2 activation of macrophages by suppressing alpha-ketoglutarate production in mice. Mol Oral Microbiol (2018) 33(5):388–95. doi: 10.1111/omi.12241
20. O’Neill LA, Pearce EJ. Immunometabolism governs dendritic cell and macrophage function. J Exp Med (2016) 213(1):15–23. doi: 10.1084/jem.20151570
21. Artyomov MN, Sergushichev A, Schilling JD. Integrating immunometabolism and macrophage diversity. Semin Immunol (2016) 28(5):417–24. doi: 10.1016/j.smim.2016.10.004
22. Holland R, McIntosh AL, Finucane OM, Mela V, Rubio-Araiz A, Timmons G, et al. Inflammatory microglia are glycolytic and iron retentive and typify the microglia in APP/PS1 mice. Brain Behav Immun (2018) 68:183–96. doi: 10.1016/j.bbi.2017.10.017
23. Fleetwood AJ, Lee MKS, Singleton W, Achuthan A, Lee MC, O’Brien-Simpson NM, et al. Metabolic remodeling, inflammasome activation, and pyroptosis in macrophages stimulated by porphyromonas gingivalis and its outer membrane vesicles. Front Cell Infect Microbiol (2017) 7:351. doi: 10.3389/fcimb.2017.00351
24. Mehla K, Singh PK. Metabolic regulation of macrophage polarization in cancer. Trends Cancer (2019) 5(12):822–34. doi: 10.1016/j.trecan.2019.10.007
25. Liu P-S, Wang H, Li X, Chao T, Teav T, Christen S, et al. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat Immunol (2017) 18(9):985–94. doi: 10.1038/ni.3796
26. Meka SRK, Younis T, Reich E, Elayyan J, Kumar A, Merquiol E, et al. TNFalpha expression by porphyromonas gingivalis-stimulated macrophages relies on Sirt1 cleavage. J Periodontal Res (2021) 56(3):535–46. doi: 10.1111/jre.12853
27. Wynn T, Chawla A, Pollard JJN. Macrophage biology in development, homeostasis and disease. Nature (2013) 496(7446):445–55. doi: 10.1038/nature12034
28. Slots J. Periodontitis: facts, fallacies and the future. Periodontol 2000 (2017) 75(1):7–23. doi: 10.1111/prd.12221
29. Suvan J, Leira Y, Moreno Sancho FM, Graziani F, Derks J, Tomasi C. Subgingival instrumentation for treatment of periodontitis. a systematic review. J Clin Periodontol (2020) 47 Suppl 22:155–75. doi: 10.1111/jcpe.13245
30. Sun X, Gao J, Meng X, Lu X, Zhang L, Chen R. Polarized macrophages in periodontitis: Characteristics, function, and molecular signaling. Front Immunol (2021) 12:763334. doi: 10.3389/fimmu.2021.763334
31. Gmiterek A, Klopot A, Wojtowicz H, Trindade SC, Olczak M, Olczak T. Immune response of macrophages induced by porphyromonas gingivalis requires HmuY protein. Immunobiology (2016) 221(12):1382–94. doi: 10.1016/j.imbio.2016.07.007
32. Yu T, Zhao L, Huang X, Ma C, Wang Y, Zhang J, et al. Enhanced activity of the macrophage M1/M2 phenotypes and phenotypic switch to M1 in periodontal infection. J Periodontol (2016) 87(9):1092–102. doi: 10.1902/jop.2016.160081
33. Zhang J, Wang CM, Zhang P, Wang X, Chen J, Yang J, et al. Expression of programmed death 1 ligand 1 on periodontal tissue cells as a possible protective feedback mechanism against periodontal tissue destruction. Mol Med Rep (2016) 13(3):2423–30. doi: 10.3892/mmr.2016.4824
34. Zhuang Z, Yoshizawa-Smith S, Glowacki A, Maltos K, Pacheco C, Shehabeldin M, et al. Induction of M2 macrophages prevents bone loss in murine periodontitis models. J Dent Res (2019) 98(2):200–8. doi: 10.1177/0022034518805984
35. Siouti E, Andreakos E. The many facets of macrophages in rheumatoid arthritis. Biochem Pharmacol (2019) 165:152–69. doi: 10.1016/j.bcp.2019.03.029
36. Totaro MC, Cattani P, Ria F, Tolusso B, Gremese E, Fedele AL, et al. Porphyromonas gingivalis and the pathogenesis of rheumatoid arthritis: analysis of various compartments including the synovial tissue. Arthritis Res Ther (2013) 15(3):R66. doi: 10.1186/ar4243
37. Lundberg K, Wegner N, Yucel-Lindberg T, Venables PJ. Periodontitis in RA-the citrullinated enolase connection. Nat Rev Rheumatol (2010) 6(12):727–30. doi: 10.1038/nrrheum.2010.139
38. Courbon G, Rinaudo-Gaujous M, Blasco-Baque V, Auger I, Caire R, Mijola L, et al. Porphyromonas gingivalis experimentally induces periodontis and an anti-CCP2-associated arthritis in the rat. Ann Rheum Dis (2019) 78(5):594–9. doi: 10.1136/annrheumdis-2018-213697
39. Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol (2018) 233(9):6425–40. doi: 10.1002/jcp.26429
40. Yang X, Chang Y, Wei W. Emerging role of targeting macrophages in rheumatoid arthritis: Focus on polarization, metabolism and apoptosis. Cell Prolif (2020) 53(7):e12854. doi: 10.1111/cpr.12854
41. Degboe Y, Rauwel B, Baron M, Boyer JF, Ruyssen-Witrand A, Constantin A, et al. Polarization of rheumatoid macrophages by TNF targeting through an IL-10/STAT3 mechanism. Front Immunol (2019) 10:3. doi: 10.3389/fimmu.2019.00003
42. Yokota K, Sato K, Miyazaki T, Aizaki Y, Tanaka S, Sekikawa M, et al. Characterization and function of tumor necrosis factor and interleukin-6-Induced osteoclasts in rheumatoid arthritis. Arthritis Rheumatol (2021) 73(7):1145–54. doi: 10.1002/art.41666
43. Pucino V, Certo M, Bulusu V, Cucchi D, Goldmann K, Pontarini E, et al. Lactate buildup at the site of chronic inflammation promotes disease by inducing CD4 T cell metabolic rewiring. Cell Metab (2019) 30(6):1055–74. doi: 10.1016/j.cmet.2019.10.004
44. Gomes MTR, Guimarães ES, Marinho FV, Macedo I, Aguiar ERGR, Barber GN, et al. STING regulates metabolic reprogramming in macrophages via HIF-1α during brucella infection. PloS Pathog (2021) 17(5):e1009597. doi: 10.1371/journal.ppat.1009597
45. Li Y, Liu Y, Wang C, Xia W-R, Zheng J-Y, Yang J, et al. Succinate induces synovial angiogenesis in rheumatoid arthritis through metabolic remodeling and HIF-1α/VEGF axis. Free Radic Biol Med (2018) 126:1–14. doi: 10.1016/j.freeradbiomed.2018.07.009
46. Itzhaki RF, Golde TE, Heneka MT, Readhead B. Do infections have a role in the pathogenesis of Alzheimer disease? Nat Rev Neurol (2020) 16(4):193–7. doi: 10.1038/s41582-020-0323-9
47. Leng F, Edison P. Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat Rev Neurol (2021) 17(3):157–72. doi: 10.1038/s41582-020-00435-y
48. Olsen I, Singhrao SK. Interaction between genetic factors, porphyromonas gingivalis and microglia to promote alzheimer’s disease. J Oral Microbiol (2020) 12(1):1820834. doi: 10.1080/20002297.2020.1820834
49. Dominy SS, Lynch C, Ermini F, Benedyk M, Marczyk A, Konradi A, et al. Porphyromonas gingivalis in alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv (2019) 5(1):eaau3333. doi: 10.1126/sciadv.aau3333
50. Wu M, Liao M, Huang R, Chen C, Tian T, Wang H, et al. Hippocampal overexpression of TREM2 ameliorates high fat diet induced cognitive impairment and modulates phenotypic polarization of the microglia. Genes Dis (2022) 9(2):401–14. doi: 10.1016/j.gendis.2020.05.005
51. Tran VTA, Kang YJ, Kim H-K, Kim H-R, Cho H. Oral pathogenic bacteria-inducing neurodegenerative microgliosis in human neural cell platform. Int J Mol Sci (2021) 22(13):6925. doi: 10.3390/ijms22136925
52. Zhang J, Yu C, Zhang X, Chen H, Dong J, Lu W, et al. Porphyromonas gingivalis lipopolysaccharide induces cognitive dysfunction, mediated by neuronal inflammation via activation of the TLR4 signaling pathway in C57BL/6 mice. J Neuroinflammation (2018) 15(1):37. doi: 10.1186/s12974-017-1052-x
53. Costa MJF, de Araujo IDT, da Rocha Alves L, da Silva RL, Dos Santos Calderon P, Borges BCD, et al. Relationship of porphyromonas gingivalis and alzheimer’s disease: a systematic review of pre-clinical studies. Clin Oral Investig (2021) 25(3):797–806. doi: 10.1007/s00784-020-03764-w
54. Nie R, Wu Z, Ni J, Zeng F, Yu W, Zhang Y, et al. Porphyromonas gingivalis infection induces amyloid-β accumulation in monocytes/macrophages. J Alzheimers Dis (2019) 72(2):479–94. doi: 10.3233/JAD-190298
55. Grieco M, De Caris MG, Maggi E, Armeli F, Coccurello R, Bisogno T, et al. Fatty acid amide hydrolase (FAAH) inhibition modulates amyloid-Beta-Induced microglia polarization. Int J Mol Sci (2021) 22(14):7711. doi: 10.3390/ijms22147711
56. Liang C, Zou T, Zhang M, Fan W, Zhang T, Jiang Y, et al. MicroRNA-146a switches microglial phenotypes to resist the pathological processes and cognitive degradation of alzheimer’s disease. Theranostics (2021) 11(9):4103–21. doi: 10.7150/thno.53418
57. Sanjay, Shin JH, Park M, Lee HJ. Cyanidin-3-O-Glucoside regulates the M1/M2 polarization of microglia via PPARgamma and Abeta42 phagocytosis through TREM2 in an alzheimer’s disease model. Mol Neurobiol (2022). doi: 10.1007/s12035-022-02873-9
58. Pan Y, Yu Y, Wang X, Zhang T. Tumor-associated macrophages in tumor immunity. Front Immunol (2020) 11:583084. doi: 10.3389/fimmu.2020.583084
59. Weng YS, Tseng HY, Chen YA, Shen PC, Al Haq AT, Chen LM, et al. MCT-1/miR-34a/IL-6/IL-6R signaling axis promotes EMT progression, cancer stemness and M2 macrophage polarization in triple-negative breast cancer. Mol Cancer (2019) 18(1):42. doi: 10.1186/s12943-019-0988-0
60. Wang X, Jia Y, Wen L, Mu W, Wu X, Liu T, et al. Promotes colorectal carcinoma by activating the hematopoietic inflammasome. Cancer Res (2021) 81(10):2745–59. doi: 10.1158/0008-5472.CAN-20-3827
61. Qi Y-J, Jiao Y-L, Chen P, Kong J-Y, Gu B-L, Liu K, et al. Porphyromonas gingivalis promotes progression of esophageal squamous cell cancer via TGFβ-dependent Smad/YAP/TAZ signaling. PloS Biol (2020) 18(9):e3000825. doi: 10.1371/journal.pbio.3000825
62. Mohammed H, Varoni EM, Cochis A, Cordaro M, Gallenzi P, Patini R, et al. Oral dysbiosis in pancreatic cancer and liver cirrhosis: A review of the literature. Biomedicines (2018) 6(4):115. doi: 10.3390/biomedicines6040115
63. Diskin C, Ryan TAJ, O’Neill LAJ. Modification of proteins by metabolites in immunity. Immunity (2021) 54(1):19–31. doi: 10.1016/j.immuni.2020.09.014
64. Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, et al. Metabolic regulation of gene expression by histone lactylation. Nature (2019) 574(7779):575–80. doi: 10.1038/s41586-019-1678-1
65. Vladimer GI, Marty-Roix R, Ghosh S, Weng D, Lien E. Inflammasomes and host defenses against bacterial infections. Curr Opin Microbiol (2013) 16(1):23–31. doi: 10.1016/j.mib.2012.11.008
66. Olsen I, Yilmaz Ö. Modulation of inflammasome activity by porphyromonas gingivalis in periodontitis and associated systemic diseases. J Oral Microbiol (2016) 8:30385. doi: 10.3402/jom.v8.30385
67. Champaiboon C, Poolgesorn M, Wisitrasameewong W, Sa-Ard-Iam N, Rerkyen P, Mahanonda R. Differential inflammasome activation by porphyromonas gingivalis and cholesterol crystals in human macrophages and coronary artery endothelial cells. Atherosclerosis (2014) 235(1):38–44. doi: 10.1016/j.atherosclerosis.2014.04.007
68. Park E, Na H, Song Y, Shin S, Kim Y, Chung JJI, et al. Activation of NLRP3 and AIM2 inflammasomes by porphyromonas gingivalis infection. Infect Immun (2014) 82(1):112–23. doi: 10.1128/IAI.00862-13
69. Yamaguchi Y, Kurita-Ochiai T, Kobayashi R, Suzuki T, Ando T. Regulation of the NLRP3 inflammasome in porphyromonas gingivalis-accelerated periodontal disease. Inflammation Res (2017) 66(1):59–65. doi: 10.1007/s00011-016-0992-4
70. Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med (2015) 21(7):677–87. doi: 10.1038/nm.3893
71. Ding P-H, Yang M-X, Wang N-N, Jin L-J, Dong Y, Cai X, et al. Porphyromonas gingivalis -induced NLRP3 inflammasome activation and its downstream interleukin-1β release depend on caspase-4. Front Microbiol (2020) 11:1881. doi: 10.3389/fmicb.2020.01881
72. Folco EJ, Sukhova GK, Quillard T, Libby P. Moderate hypoxia potentiates interleukin-1β production in activated human macrophages. Circ Res (2014) 115(10):875–83. doi: 10.1161/CIRCRESAHA.115.304437
73. Cecil JD, O’Brien-Simpson NM, Lenzo JC, Holden JA, Singleton W, Perez-Gonzalez A, et al. Outer membrane vesicles prime and activate macrophage inflammasomes and cytokine secretion and and cytokine secretion In vitro and in vivo. Front Immunol (2017) 8:1017. doi: 10.3389/fimmu.2017.01017
74. Zychlinsky A, Prevost M, Sansonetti PJN. Shigella flexneri induces apoptosis in infected macrophages. Nat Rev Immunol (1992) 358(6382):167–9. doi: 10.1038/358167a0
75. Siegel R. Caspases at the crossroads of immune-cell life and death. (2006) 6(4):308–17. doi: 10.1038/nri1809
76. Robinson N, Ganesan R, Hegedus C, Kovacs K, Kufer TA, Virag L. Programmed necrotic cell death of macrophages: Focus on pyroptosis, necroptosis, and parthanatos. Redox Biol (2019) 26:101239. doi: 10.1016/j.redox.2019.101239
77. Barrett TJ. Macrophages in atherosclerosis regression. Arterioscler Thromb Vasc Biol (2020) 40(1):20–33. doi: 10.1161/ATVBAHA.119.312802
78. Hoseini Z, Sepahvand F, Rashidi B, Sahebkar A, Masoudifar A, Mirzaei H. NLRP3 inflammasome: Its regulation and involvement in atherosclerosis. J Cell Physiol (2018) 233(3):2116–32. doi: 10.1002/jcp.25930
79. Schenkein HA, Papapanou PN, Genco R, Sanz M. Mechanisms underlying the association between periodontitis and atherosclerotic disease. Periodontol 2000 (2020) 83(1):90–106. doi: 10.1111/prd.12304
80. Gibson FC, Genco CA. Porphyromonas gingivalis mediated periodontal disease and atherosclerosis: Disparate diseases with commonalities in pathogenesis through TLRs. Curr Pharm Design (2007) 13(36):3665–75. doi: 10.2174/138161207783018554
81. Wu Y, Xu W, Hou J, Liu Y, Li R, Liu J, et al. Porphyromonas gingivalis-induced MIF regulates intercellular adhesion molecule-1 expression in EA.hy926 cells and monocyte-endothelial cell adhesion through the receptors CD74 and CXCR4. Inflammation (2019) 42(3):874–83. doi: 10.1007/s10753-018-0942-0
82. Yang Y, He X, Xia S, Liu F, Luo L. Porphyromonas gingivalis facilitated the foam cell formation via lysosomal integral membrane protein 2 (LIMP2). J Periodontal Res (2021) 56(2):265–74. doi: 10.1111/jre.12812
83. Kim DJ, Rho JH, Woo BH, Joo JY, Lee JY, Song JM, et al. Periodontal pathogens modulate lipid flux via fatty acid binding protein 4. J Dent Res (2019) 98(13):1511–20. doi: 10.1177/0022034519880824
84. Paramel Varghese G, Folkersen L, Strawbridge RJ, Halvorsen B, Yndestad A, Ranheim T, et al. NLRP3 inflammasome expression and activation in human atherosclerosis. J Am Heart Assoc (2016) 5(5):e003031. doi: 10.1161/JAHA.115.003031
85. Rho JH, Kim HJ, Joo JY, Lee JY, Lee JH, Park HR. Periodontal pathogens promote foam cell formation by blocking lipid efflux. J Dent Res (2021) 100(12):1367–77. doi: 10.1177/00220345211008811
86. Wang R, Wu W, Li W, Huang S, Li Z, Liu R, et al. Activation of NLRP3 inflammasome promotes foam cell formation in vascular smooth muscle cells and atherogenesis via HMGB1. J Am Heart Assoc (2018) 7(19):e008596. doi: 10.1161/JAHA.118.008596
87. Grebe A, Hoss F, Latz E. NLRP3 inflammasome and the IL-1 pathway in atherosclerosis. Circ Res (2018) 122(12):1722–40. doi: 10.1161/CIRCRESAHA.118.311362
88. Koelwyn GJ, Corr EM, Erbay E, Moore KJ. Regulation of macrophage immunometabolism in atherosclerosis. Nat Immunol (2018) 19(6):526–37. doi: 10.1038/s41590-018-0113-3
89. Wilensky A, Tzach-Nahman R, Potempa J, Shapira L, Nussbaum G. Porphyromonas gingivalis gingipains selectively reduce CD14 expression, leading to macrophage hyporesponsiveness to bacterial infection. J Innate Immun (2015) 7(2):127–35. doi: 10.1159/000365970
90. Singh A, Wyant T, Anaya-Bergman C, Aduse-Opoku J, Brunner J, Laine ML, et al. The capsule of porphyromonas gingivalis leads to a reduction in the host inflammatory response, evasion of phagocytosis, and increase in virulence. Infect Immun (2011) 79(11):4533–42. doi: 10.1128/IAI.0501611
91. Werheim ER, Senior KG, Shaffer CA, Cuadra GA. Oral pathogen porphyromonas gingivalis can escape phagocytosis of mammalian macrophages. Microorganisms (2020) 8(9):1432. doi: 10.3390/microorganisms8091432
92. Gao SG, Yang JQ, Ma ZK, Yuan X, Zhao C, Wang GC, et al. Preoperative serum immunoglobulin G and a antibodies to porphyromonas gingivalis are potential serum biomarkers for the diagnosis and prognosis of esophageal squamous cell carcinoma. BMC Cancer (2018) 18(1):17. doi: 10.1186/s12885017-3905-1
93. Makkawi H, Hoch S, Burns E, Hosur K, Hajishengallis G, Kirschning CJ, et al. Porphyromonas gingivalis stimulates TLR2-PI3K signaling to escape immune clearance and induce bone resorption independently of MyD88. Front Cell Infect Microbiol (2017) 7:359. doi: 10.3389/fcimb.2017.00359
94. Rocha F, Moye Z, Ottenberg G, Tang P, Campopiano D, Gibson F, et al. Porphyromonas gingivalis sphingolipid synthesis limits the host inflammatory response. J Dent Res (2020) 99(5):568–76. doi: 10.1177/0022034520908784
95. Yu J, Lin Y, Xiong X, Li K, Yao Z, Dong H, et al. Detection of exosomal PD-L1 RNA in saliva of patients with periodontitis. Front Genet (2019) 10:202. doi: 10.3389/fgene.2019.00202
96. Adel-Khattab D, Groeger S, Domann E, Chakraborty T, Lochnit G, Meyle J. Porphyromonas gingivalis induced up-regulation of PD-L1 in colon carcinoma cells. Mol Oral Microbiol (2021) 36(3):172–81. doi: 10.1111/omi.12332
97. Groeger S, Jarzina F, Mamat U, Meyle J. Induction of B7-H1 receptor by bacterial cells fractions of porphyromonas gingivalis on human oral epithelial cells: B7-H1 induction by porphyromonas gingivalis fractions. Immunobiology (2017) 222(2):137–47. doi: 10.1016/j.imbio.2016.10.011
98. Shen X, Zhao B. Efficacy of PD-1 or PD-L1 inhibitors and PD-L1 expression status in cancer: meta-analysis. BMJ (2018) 362:k3529. doi: 10.1136/bmj.k3529
99. Ni L, Zhuge F, Yang S, Ma L, Zheng A, Zhao Y, et al. Hydrolyzed chicken meat extract attenuates neuroinflammation and cognitive impairment in middle-aged mouse by regulating M1/M2 microglial polarization. J Agric Food Chem (2021) 69(34):9800–12. doi: 10.1021/acs.jafc.1c03541
100. Ge G, Bai J, Wang Q, Liang X, Tao H, Chen H, et al. Punicalagin ameliorates collagen-induced arthritis by downregulating M1 macrophage and pyroptosis via NF-κB signaling pathway. Sci China Life Sci (2022) 65(3):588–603. doi: 10.1007/s11427-020-1939-1
101. Taxman DJ, Swanson KV, Broglie PM, Wen H, Holley-Guthrie E, Huang MT-H, et al. Porphyromonas gingivalis mediates inflammasome repression in polymicrobial cultures through a novel mechanism involving reduced endocytosis. J Biol Chem (2012) 287(39):32791–9. doi: 10.1074/jbc.M112.401737
Keywords: Porphyromonas gingivalis, macrophages, immune escape, systemic disease, polarization, inflammasome
Citation: Lin J, Huang D, Xu H, Zhan F and Tan X (2022) Macrophages: A communication network linking Porphyromonas gingivalis infection and associated systemic diseases. Front. Immunol. 13:952040. doi: 10.3389/fimmu.2022.952040
Received: 24 May 2022; Accepted: 30 June 2022;
Published: 27 July 2022.
Edited by:
Jean-louis Mege, Aix-Marseille Université, FranceReviewed by:
Kalina Atanasova, University of Florida, United StatesSoraya Mezouar, Aix-Marseille University, France
Xavier Heim, Aix Marseille Université, France
Copyright © 2022 Lin, Huang, Xu, Zhan and Tan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: XueLian Tan, dGFueHVlbGlhbkBzY3UuZWR1LmNu; Fenghuang Zhan, RlpoYW5AdWFtcy5lZHU=