 Masaki Shimizu
Masaki Shimizu Syuji Takei
Syuji Takei Masaaki Mori
Masaaki Mori Akihiro Yachie
Akihiro Yachie- 1Department of Child Health and Development, Graduate School of Medical and Dental Sciences, Tokyo Medical and Dental University, Tokyo, Japan
- 2Department of Pediatrics, Field of Developmental Medicine, Graduate School of Medical and Dental Sciences, Kagoshima University, Kagoshima, Japan
- 3Department of Lifetime Clinical Immunology, Graduate School of Medical and Dental Sciences, Tokyo Medical and Dental University, Tokyo, Japan
- 4Division of Medical Safety, Kanazawa University Hospital, Kanazawa, Japan
Interleukin (IL)-18 is a pleiotropic, pro-inflammatory cytokine involved in the regulation of innate and adaptive immune responses. IL-18 has attracted increasing attention as a key mediator in autoinflammatory diseases associated with the development of macrophage activation syndrome (MAS) including systemic juvenile idiopathic arthritis and adult-onset Still’s disease. In these diseases, dysregulation of inflammasome activity and overproduction of IL-18 might be associated with the development of MAS by inducing natural killer cell dysfunction. Serum IL-18 levels are high in patients with these diseases and therefore are useful for the diagnosis and monitoring of disease activity. In contrast, a recent study revealed the overproduction of IL-18 was present in cases of autoinflammation without susceptibility to MAS such as pyogenic sterile arthritis, pyoderma gangrenosum, and acne (PAPA) syndrome. The pathogenic and causative roles of IL-18 remain unclear in these autoinflammatory diseases. Further investigations are necessary to clarify the role of IL-18 and its importance as a therapeutic target in the pathogenesis of autoinflammatory diseases.
Introduction
Interleukin (IL)-18 is a pleiotropic, pro-inflammatory cytokine involved in the regulation of innate and adaptive immune responses (1). IL-18, originally identified as an interferon (IFN)-γ inducing factor, was isolated from the serum of mice pretreated with Proprionibacterium acnes, which stimulated Kupffer cells after stimulation with intraperitoneal lipopolysaccharide (2, 3). IL-18 is expressed in a wide range of cells including intestinal epithelial cells, keratinocytes, astrocytes, and endothelial cells, although the main cellular sources of IL-18 are macrophages, Kupffer cells, and dendric cells (2–7). Pro IL-18, an inactive precursor of IL-18, is cleaved by caspase-1, which is activated by the inflammasome, an intracellular multimolecular complex that plays an important role in innate immune responses as a sensor for pathogen-associated and danger-associated molecular patterns. It is then secreted as active, mature IL-18. The processing of IL-18 is mediated by the NLRP3 and the NLRC4 inflammasomes in immune cells (8, 9) and the NLRP6 and Nlrp9b inflammasomes in intestinal epithelial cells (10, 11). Secreted IL-18 binds to the IL-18 receptor, and initiates cell signaling via MyD88, IRAK, and TRAF6, which subsequently leads to the activation of the transcription factor NF-κB. The biological activity of IL-18 is regulated by IL-18 binding protein (IL-18BP) (12), which has a high affinity for IL-18 and is abundantly present in serum, with a greater than 20-fold molar excess compared with IL-18. IL-18BP binds to active IL-18 to form an inactive IL-18-IL-18BP complex. IL-18BP prevents active IL-18 from binding to IL-18 receptor and IL-18 function including IFN-γ production (12, 13). IL-18BP binds to IL-37 and forms an IL-18BP-IL-37-IL-18 receptor β subunit complex, inhibiting the formation of a functional receptor with an IL-18R α chain (14).
Immunological diseases can be divided into two major categories, autoimmune diseases and autoinflammatory diseases (15). Autoimmune diseases are characterized by a self-directed inflammation caused by aberrant adaptive immune cells responses leading to breaking self-tolerance and development of immune reactivity towards self-antigens. In contrast, autoinflammatory diseases are characterized by a self-directed tissue inflammation independent of adaptive immune cells abnormalities, where local factors at sites predisposed to disease lead to activation of innate immune cells. Proinflammatory cytokines derived from innate immune cells including macrophages and neutrophils play a pivotal role in the pathogenesis of autoinflammatory diseases. Among innate proinflammatory cytokines, IL-18 has attracted increasing attention as a key mediator in autoinflammatory diseases associated with the development of macrophage activation syndrome (MAS) including systemic juvenile idiopathic arthritis (16–25), adult-onset Still’s disease (26–30), XIAP deficiency (31, 32), NLRC4 gain of function (8, 33), neonatal onset of cytopenia, autoinflammation, rash, and episodes of hemophagocytic lymphohistiocytosis (NOCARH syndrome) (34) and purine nucleoside phosphorylase (PNP)-deficiency (35). Furthermore, a recent study reported the overproduction of IL-18 in cases of autoinflammation without susceptibility to MAS such as pyogenic sterile arthritis, pyoderma gangrenosum, and acne (PAPA) syndrome (36).
The aims of this review were to summarize the current evidence for IL-18 as a pathogenic mediator of autoinflammatory diseases and its potential utility as a biomarker for disease activity, severity, and prognosis.
IL-18 biology
IL-18 has pleiotropic biological effects (Figure 1). In the presence of IL-12 and/or IL-15, IL-18 induces IFN-γ production in macrophages, natural killer (NK) cells, dendritic cells, T cells, and B cells (37–40). In contrast, in the absence of IL-12 or IL-15, IL-18 promotes the differentiation of naïve T cells to Th2 cells, which produce IL-4 and IL-13 (41, 42). IL-18 also induces IL-4 and IL-13 production in basophils and mast cells (43). Furthermore, IL-18 induces IL-17 production by γδ T-cells (44) and promotes Th17 cells in combination with IL-23 (1). IL-18 also plays a role in T regulatory cell (Treg) differentiation during Helicobacter pylori chronic infection (45). Independent of other cytokines, IL-18 induces the expression of cell adhesion molecules, nitric oxide synthesis, and chemokine production (46–48). IL-18 activates NK cells by upregulating perforin-and FasL-dependent cytotoxicity in an IFN-γ independent manner (49).
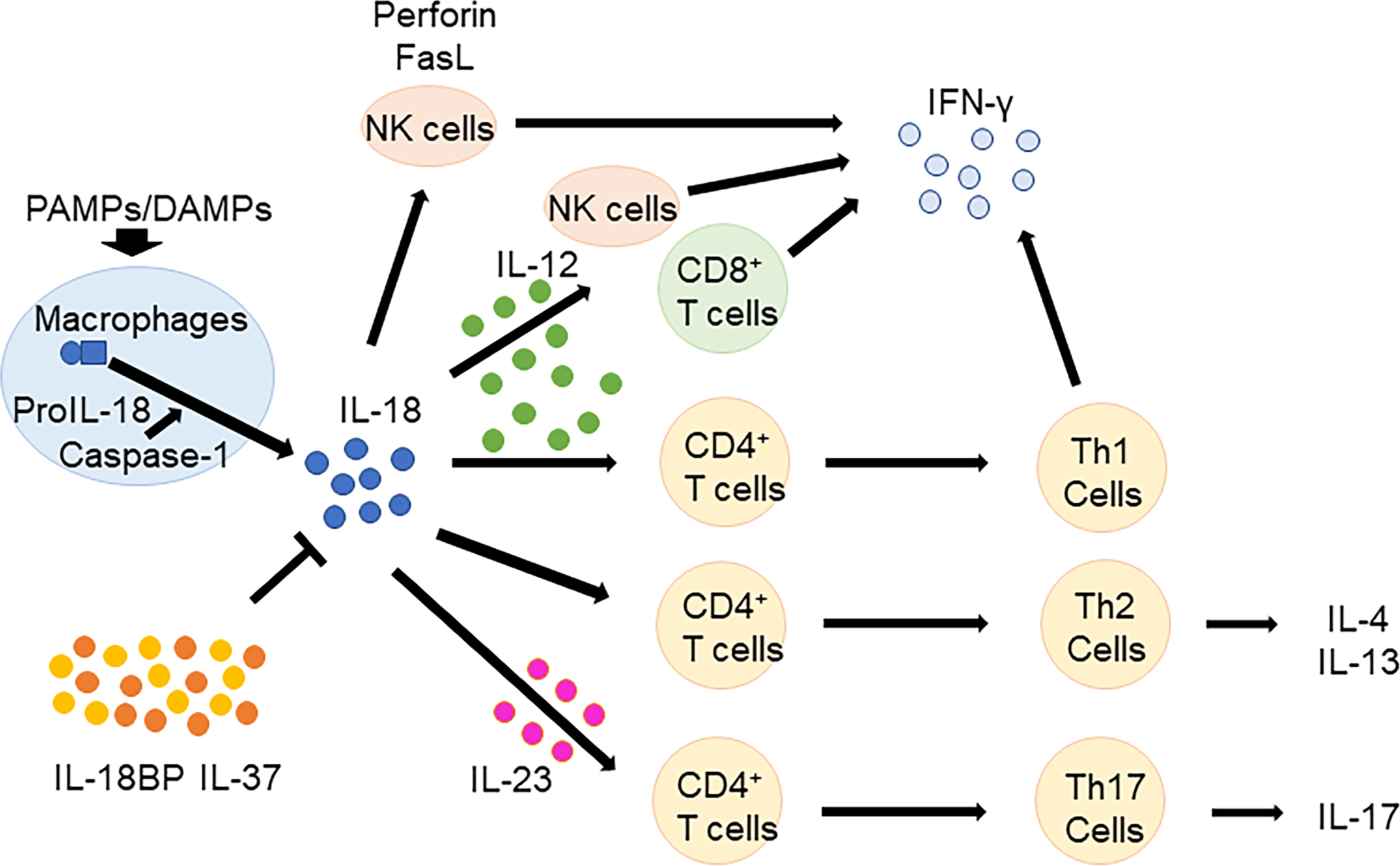
Figure 1 Biological functions of IL-18. In the presence of IL-12 and/or IL-15, IL-18 induces IFN-γ production in macrophages, NK cells, T cells, and B cells. In the absence of IL-12 or IL-15, IL-18 promotes the differentiation of naïve T cells to Th2 cells, which produce IL-4 and IL-13. Furthermore, IL-18 promotes Th17 cells in combination with IL-23. IL-18 BP and IL-37 regulates IL-18 function. IL, Interleukin; CD, cluster of differentiation; NK, natural killer; IFN, interferon; Th, T helper; BP, binding protein; PAMPs, pathogen associated molecular patterns; DAMPs, damage associated molecular patterns.
Role of IL-18 in the pathogenesis of autoinflammatory diseases
High levels of serum IL-18 levels were reported in patients with systemic juvenile idiopathic arthritis (s-JIA) (16–25) and its adult homolog adult Still’s disease (AOSD) (26–30), as well as various other inflammatory diseases including systemic lupus erythematosus (50) and Crohn’s disease (51). s-JIA and AOSD are clinically characterized by systemic features including spiking fever, skin rash, hepato/splenomegaly, generalized lymphadenopathy, serositis, and chronic arthritis, whereas arthritis can be absent in AOSD. Laboratory findings show neutrophilia, thrombocytosis, hyperferritinemia, and increased levels of inflammation markers such as C reactive protein and serum amyloid A. Recent studies revealed proinflammatory cytokines of the innate immune system, especially, IL-1, IL-6, and IL-18 play an important role in the pathogenesis of s-JIA and AOSD (52).
IL-1 upregulates endothelial adhesion molecules and facilitates the transmigration of neutrophils and other leukocytes. IL-1 activates these cells and amplifies the inflammatory response by the activation of local fibroblasts, chondrocytes, and macrophages. IL-1 also promotes osteoclast differentiation. Furthermore, IL-1 can promote proinflammatory T cell differentiation in the direction of Th17 by the action of IL-6 and other cytokines, which might play an important role in the chronic arthritis of s-JIA and AOSD (44, 53, 54). IL-1α and IL-1β bind the same receptor, namely IL-1R1, and induce the same pro-inflammatory effects including the synthesis of cytokines, interferons and chemokines; increased expression of adhesion molecules on endothelial cells and migration of immune cells into inflamed tissues, activation of adaptive immunity such as maturation and expansion of T and B cells. However, there are distinct differences in the biology of IL-1α and IL-1β. IL-1α is constitutively expressed in various cells, and its expression can be induced by inflammatory stimuli in both hematopoietic and nonhematopoietic cells (55). Not only the cleaved form of IL-1α but also IL-1α precursor (pro-IL-1α) are biologically equally active. Inflammasomes and caspase-1 have no direct effects in cleaving pro-IL-1α. ProIL-1α without the processing of maturation is released by necrotic cells and acts as an alarmin (56). In contrast, IL-1β is absent at steady state and its expression is induced upon stimulation in innate myelomonocytic cells. Pro-IL-1β is biologically inactive and cannot work as an alarmin. The mature form of IL-1β cleaved by caspase-1 is biologically active and has a function as a soluble mediator.
IL-6 induces systemic inflammation by promoting nerve cell differentiation, the dysregulated production of pain mediators in neurons, acute phase protein production in hepatocytes, dysregulation of effector and regulatory B lymphocytes and antibody production in B cells, and dysregulation of the T cell balance. IL-6 also plays an important role in local inflammation in inflamed joints by promoting osteoclast differentiation and activation, the dysregulated production of inflammatory mediators, bone homeostasis, proliferation of fibroblasts and other cells, and matrix and bone degradation. These effects of IL-6 are closely associated with the clinical symptoms of s-JIA and AOSD including joint destruction, bone loss and growth impairment, pain, fatigue, anemia, and fever (57).
The importance of IL-1 and IL-6 in the pathogenesis of s-JIA and AOSD was proven by the dramatic effects of IL-1 inhibitors (anakinra, rilonacept, and canakinumab) and IL-6 inhibitors (tocilizumab [TCZ], sarilumab) in patients with s-JIA and AOSD (58–62). However, MAS, secondary hemophagocytic lymphohistiocytosis followed by rheumatic diseases, occurs even in patients with s-JIA and AOSD receiving IL-1 and IL-6 inhibitors (63–67).
MAS is a potentially life-threatening complication clinically characterized by sustained high fever, hepato/splenomegaly, and central nervous system dysfunction (68). Laboratory findings showed cytopenia in all three blood cell lines, hepatic dysfunction, coagulopathy, hyperferritinemia, NK cell dysfunction, and the presence of hemophagocytic macrophages in bone marrow (68). MAS is a complication in 7%–14% of children with s-JIA (69, 70) and 10%–15% of patients with AOSD (71, 72). MAS may occur subclinically in another 30%–40% of patients (73).
We reported that serum IL-18 levels were increased in patients with s-JIA and AOSD, and these were further elevated in patients with MAS (16, 17, 20, 22, 23, 26). The marked elevation of serum IL-18 levels seems to be specific to MAS with s-JIA and AOSD, and patients with secondary HLH caused by other diseases had lower serum IL-18 levels compared with sJIA related MAS (17, 22). Furthermore, we reported there were two distinct groups of patients with s-JIA/AOSD having specific clinical features based on serum IL-6 and IL-18 levels (20, 26). s-JIA/AOSD patients with an IL-6–dominant pattern (s-JIA:IL-18/IL-6 <1000, AOSD IL-18/IL-6 <5000) had more severe joint disease, whereas those with an IL-18–dominant pattern (s-JIA:IL-18/IL-6 >1000) had a more severe systemic disease and developed MAS (20, 26).
Recently, patients with gain-of-function mutations in the NLRC4 gene have been identified (8, 33). Gain-of-function mutations in NLRC4 caused sustained caspase-1 cleavage and the production of active IL-18 (8, 33). Patients with gain-of-function mutations in NLRC4 can be clinically characterized by early-onset, recurrent MAS and inflammatory bowel disease, and very high IL-18 levels similar to those in patients with MAS associated with s-JIA and AOSD (8, 33). The discovery of this new disease revealed a close association between IL-18 and the development of MAS. On the other hand, some patients with gain-of-function mutations in NLRC4 showed different clinical symptoms like cryopyrin-associated periodic syndrome not associated with MAS, despite high serum levels of IL-18 (74).
X-linked inhibitor of apoptosis (XIAP) deficiency with XIAP/BIRC4 gene mutations is a primary immune deficiency clinically characterized by HLH, inflammatory bowel disease, and splenomegaly (75). XIAP belongs to the inhibitor of apoptosis family of proteins and inhibits caspases 3, 7, and 9, and has an antiapoptotic role (76–78). XIAP regulates multiple immune pathways including NOD1 and NOD2 signaling, Dectin1 signaling, TNF-receptor signaling, and the NLRP3 inflammasome. Therefore, the loss of XIAP induces the dysregulation of inflammasome activity and overproduction of inflammasome-activated cytokines (31). We previously reported serum IL-18 levels were highly elevated in XIAP deficient patients with HLH and that this elevation was sustained in the inactive phase after recovery from HLH (31). These findings indicate that NLRC4 and XIAP play an important role in regulating IL-18, and that the dysregulation of inflammasome activity and overproduction of IL-18 are closely associated with the development of MAS.
NK cell dysfunction is a characteristic finding of HLH (79) and was observed in patients with s-JIA (80–84). IL-18 increases NK cell activity. However, exposure to high levels of IL-18 can induce NK cell death (81–84). A previous report showed that NK cell dysfunction in s-JIA was closely associated with a defect in IL-18 receptor β phosphorylation (81). Furthermore, Ohya et al. recently demonstrated the impaired phosphorylation of MAPK p38 and NFκB p65 in NK cells following rIL-18 stimulation in patients with active s-JIA (83). These findings indicate that IL-18 exposure might induce NK cell dysfunction and reduce the number of NK cells. We previously reported infants born to mothers with AOSD can develop MAS and show extremely elevated levels of IL-18 (85). Furthermore, infants born to mothers with AOSD showed increased serum IL-18 levels, transmitted from the mother to the infant, as well as the transient impairment of NK cell functions (86). Furthermore, we reported that NK cell activation by exogenous IL-18 stimulation was impaired in patients with active s-JIA and high serum levels of IL-18 (82). Interestingly, NK cell activation was restored by IL-18 stimulation in patients with s-JIA after treatment began and serum IL-18 levels had decreased. Furthermore, Ohya et al. reported that the impaired phosphorylation of MAPK p38 and NF-κB p65 in NK cells following rIL-18 stimulation was recovered to normal levels with improvements in the patients’ clinical condition after treatment began (83). These findings indicate NK cell exhaustion and secondary transient NK cell dysfunction induced by exposure to high serum IL-18 levels may be closely associated with MAS development in s-JIA. However, NK cells were normal in number and function in patients with gain-of-function mutations in NLRC4 and XIAP deficiency. These findings indicate IL-18 overproduction might be associated with MAS development by inducing other mechanisms besides NK cell dysfunctions.
Recently, a case of IL-18BP deficiency with fulminant hepatitis caused by hepatitis A virus infection was reported (87). Furthermore, IL-18BPKO mice receiving repeated TLR9 stimulation developed MAS with severe hepatic inflammatory damage, which was reversed by the treatment of IL-18BP (88). These findings indicate that IL-18 might be closely associated with severe liver injury in s-JIA/AOSD and related MAS. In patients with s-JIA and AOSD, IL-18 expression was increased in reticuloendothelial cells in the liver, lymph nodes, and inflamed tissues (89–91). A previous report showed IL-17-expressing γ/δ T cells might play an important role in the chronic arthritis of s-JIA (44). Furthermore, a recent report showed a shift in Th17 responses from Tregs in acute disease to effector T cells in chronic disease in patients with s-JIA (54). IL-18 induced IL-17 production by γδ T-cells (44) and promoted Th17 in combination with IL-23 (1, 42). s-JIA patients with chronic arthritis had sustained elevated serum IL-18 levels (23). Taken together, these findings indicate IL-18 might play a role in the chronic arthritis of s-JIA through the persistent activation of IL-17 responses.
Pathogenic mechanisms of IL-18 in the pathogenesis of s-JIA and MAS were summarized in Figure 2. Overproduction of IL-1, IL-6 and IL-18 is a hallmark of active phase of s-JIA. In vivo exposure highly elevated IL-6 and IL-18 levels and induced NK cell dysfunction and decreased the number of cells. Defective NK cells are unable to contract immune responses, leading to massive expansion and activation of CD8+ T cells and overproduction of IFN-γ. Furthermore, inadequate production of IL-10, a regulatory cytokine to counter-regulate IFN-γ, might be related to the development of MAS. IFN-γ plays a pivotal role in the development of MAS. IFN-γ binds to the receptors on macrophages and activates them. Activated macrophages release even more proinflammatory cytokines including TNF-α, IL-1 and IL-6, leading to the cytokine storm. High serum IFN-γ levels are found in patients with primary HLH and secondary HLH including MAS (92–96). Furthermore, clinical symptoms of HLH have been shown to be inhibited by anti-IFN-γ antibody treatments in patents with HLH as well as animal models of HLH (97–104).
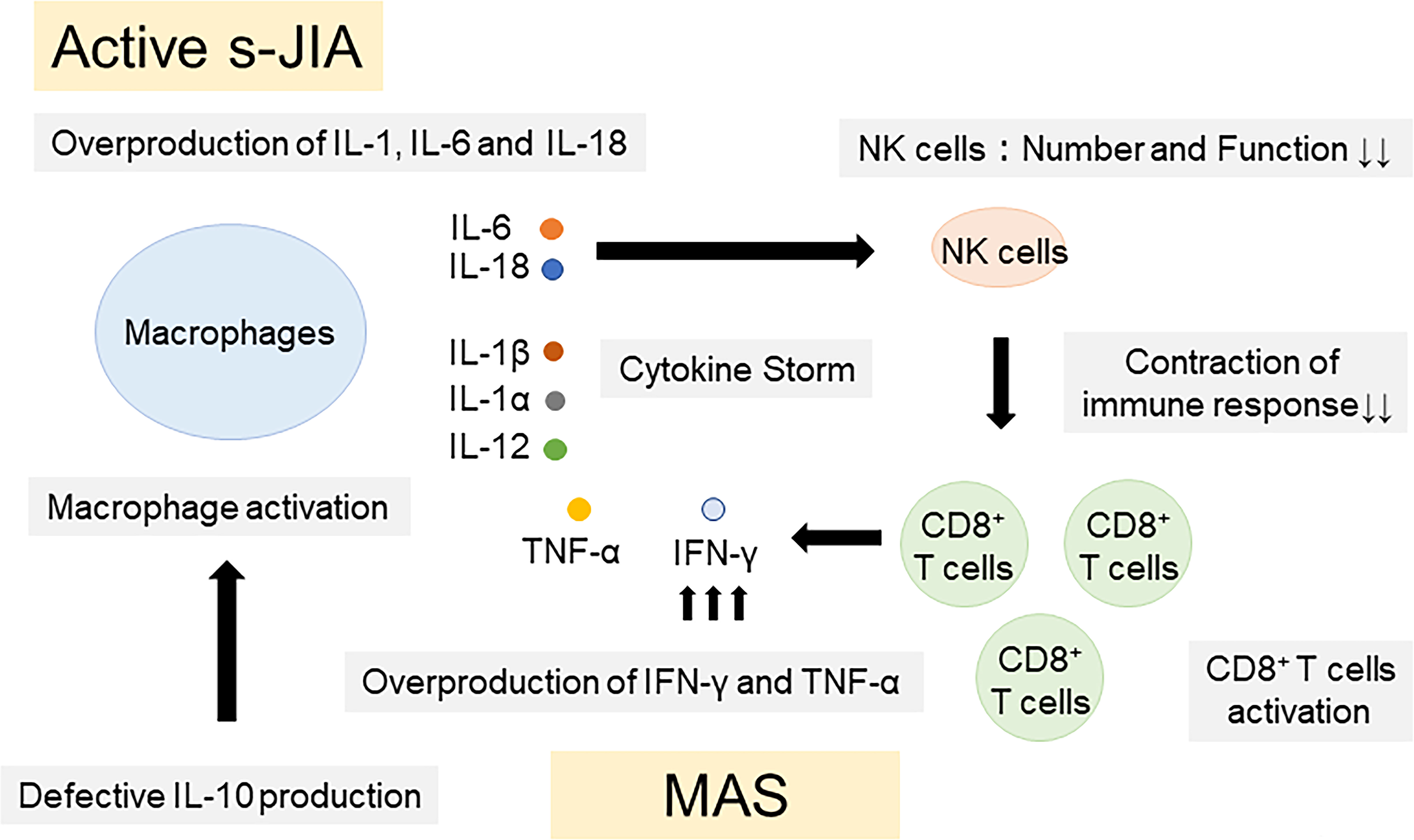
Figure 2 Pathogenic mechanisms of IL-18 in the pathogenesis of s-JIA and MAS. Overproduction of IL-1, IL-6 and IL-18 is a hallmark of active phase of s-JIA. In vivo exposure highly elevated IL-6 and IL-18 levels and induced NK cell dysfunction and decreased the number of cells. Defective NK cells are unable to contract immune responses, leading to massive expansion and activation of CD8+ T cells and overproduction of IFN-γ and TNF-α. Furthermore, inadequate production of IL-10, a regulatory cytokine to counter-regulate IFN-γ, might be related to the development of MAS. IL, Interleukin; TNF, tumor necrosis factor; IFN, Interferon; CD, cluster of differentiation; NK, natural killer; s-JIA, systemic juvenile idiopathic arthritis; MAS, macrophage activation syndrome.
Recently, a novel hematological and autoinflammatory syndrome clinically characterized by neonatal-onset cytopenia with dyshematopoiesis, autoinflammation, rash and HLH (NOCARH syndrome) was identified (34). The pathogenic variant, p.R186C at the C-terminus of cell division control protein 42 (CDC42) was specifically associated with this syndrome (34). Patients with this variant in CDC42 showed sustained extreme elevation of serum IL-18 levels, likely predisposing to development of MAS (34, 105). More recently, additional 2 different pathogenic variants at the C-terminus of CDC42, pC188Y and p*192Cext*24 were proposed to cause IL-1 blocking sensitive autoinflammation (105). Although CDC42 mutations had been associated with neurodevelopmental diseases (106–110), variants at the CDC42 C-terminus can affect the localization and function of CDC42 protein, and cause autoinflammation. Arg186 and Arg 187 in C-terminal diarginine motif of CDC42 is important for the binding of CDC42 to phosphatidylinositol 4,5-bisphosphonate (PIP2), whose interaction of CDC42 is critical in mediating the PIP2-induced actin assembly (111). Aberrant actin depolymerlization can activate the inflammasome and modulate innate immune functions (112, 113). The functional effect of actin polymerization defects caused by variants at the CDC42 C-terminus might lead to autoinflammation.
Purine nucleoside phosphorylase (PNP)-deficiency is a primary immune deficiency with PNP gene mutation, clinically characterized by a progressive combined immunodeficiency, neurologic symptoms including developmental delay, spasticity, ataxia, and pyramidal signs, and autoimmune manifestations including hemolytic anemia (114–118). PNP is one of the enzymes involved in purine salvage. Thus, PNP deficiency leads to intracellular accumulation of deoxyguanosine triphostate (dGTP) which induces toxic effects for neurons and T lymphocytes (114–117). Recently, a patient with PNP-deficiency whose clinical manifestation was highly suggestive for sJIA complicated with MAS has been reported (35). High serum IL-18 levels have been described in patients with PNP-deficiency (35, 118). PNP-deficiency is associated with overproduction of IL-18 and they are predisposed to develop MAS, although it remains unclear how PNP-deficiency affects overproduction of IL-18 and induces MAS development.
PAPA syndrome caused by PSTPIP1 gene mutations is clinically characterized by pyogenic sterile arthritis, pyoderma gangrenosum, and acne (36). Mutations of the PSTPIP1 gene induce increased binding to pyrin and the activation of NLRP3 inflammasomes (119, 120). PSTPIP1 interacts with the actin cytoskeleton by regulating the Wiskott-Aldrich Syndrome protein (119, 120). Thus, mutations of the PSTPIP1 gene can alter innate immune cell motility. Recently, Stones et al. reported serum IL-18 levels were highly increased in patients with PAPA syndrome similar to those in patients with MAS associated with s-JIA and AOSD (36). Serum IL-18 levels were significantly elevated in some patients with familial Mediterranean fever (FMF), although they were not significantly elevated in most patients with FMF (36, 121). A recent report revealed FMF patients carrying M694I and E148Q mutations in the MEFV gene had significantly higher levels of serum IL-18 compared with those carrying M694I, but not E148Q, and those carrying E148Q, but not M694I (121). Although the specific mechanism driving IL-18 through the pyrin inflammasome remains unknown, the dysregulation of interactions between pyrin and PSTPIP1 caused by MEFV and pstpip1 gene mutations might induce the overproduction of IL-18.
MAS is not a complication in patients with PAPA syndrome. Therefore, IL-18 may be elevated in PAPA syndrome without increasing the risk of MAS development. The possible reasons why IL-18 significantly elevated in PAPA syndrome without increasing the risk of MAS are 1) high enough IL-18BP level in serum, 2) differences in the inflammatory milieu of s-JIA and PAPA syndrome, and 3) differences in the cellular source of IL-18. Furthermore, elevated levels of IL-18 alone might not be able to induce MAS development. IL-18 has been studied in murine models of HLH/MAS and IL-18 blocking could not improve their survivals (88). IL-10 counter-regulates IFN-γ. In a mouse model of MAS, IL-10 blocking exacerbated the disease activity of MAS (122). A previous study revealed IL-10 production in B lymphocytes was impaired, and plasma IL-10 levels plasma were not relatively elevated compared with other proinflammatory cytokines in patients with s-JIA (123). These findings indicate that other pathogenic pathways including aberrant production of IL-10 production might contribute to the pathogenesis of MAS in addition to IL-18 overproduction induced autoinflammation.
To summarize, autoinflammatory diseases characterized by high levels of IL-18 were shown in Table 1.
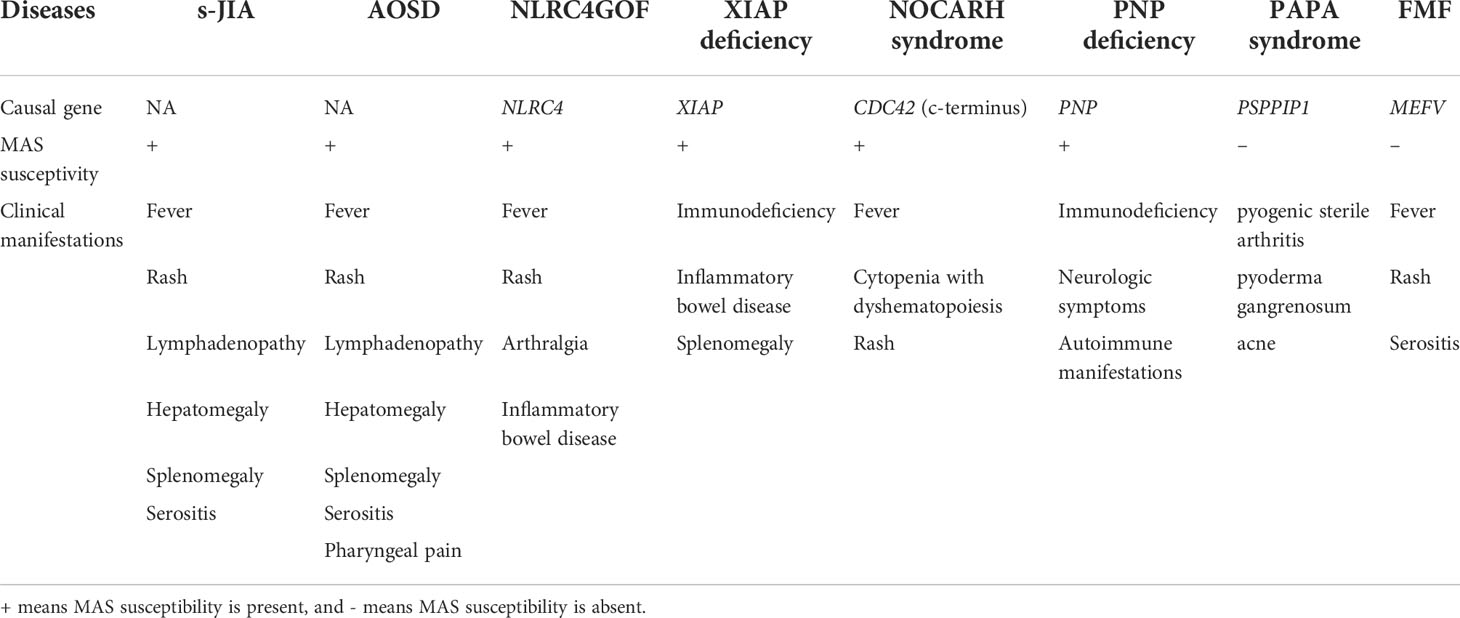
Table 1 Autoinflammatory diseases characterized by high levels of IL-18.
Clinical application of serum IL-18 levels as a biomarker for autoinflammatory diseases
Serum IL-18 levels are significantly and highly increased in active s-JIA (16–26), which can be useful for the differentiation of s-JIA from other inflammatory diseases (17, 23). We compared serum IL-18 levels in s-JIA and other inflammatory diseases (23) and found they were significantly elevated in patients with s-JIA compared with Kawasaki disease (KD), TRAPS, other subtypes of JIA, SLE, Juvenile dermatomyositis (JDM), and leukemia. The cut-off values of serum IL-18 levels for the differentiation of s-JIA from KD, FMF, TRAPS, other subtypes of JIA, SLE, JDM, and leukemia were 4560, 4800, 1685, 1728, 2400, 2125, and 2240 pg/mL, respectively. The cut-off value of serum IL-18 levels for the differentiation s-JIA from all other diseases was 4800 pg/ml.
Serum IL-18 levels are closely correlated with the disease course of s-JIA and are useful as a diagnostic laboratory criterion for remission in s-JIA. There are three types of disease course in s-JIA: 1) monocyclic course; no disease flare occurs; 2) chronic course; disease flare frequently occurs with steroid withdrawal; and 3) polycyclic pattern; disease flare repeatedly occurs after remission is achieved in each flare. We longitudinally measured serum IL-18 levels in s-JIA patients from the active phase through to the inactive phase or remission (23). In patients with a monocyclic course, serum IL-18 levels smoothly decreased to <1000 pg/mL in the inactive phase and sustained low levels in the remission phase. In contrast, in patients with a chronic pattern, serum IL-18 levels were sustained at >1000 pg/mL, even in the inactive phase. In patients with a polycyclic pattern, serum IL-18 levels were elevated at >1000 pg/mL during disease flares, although these levels were normalized in the inactive phase. The cut-off value of serum IL-18 levels for the diagnosis of remission in s-JIA was 595 pg/mL. These results indicate that the monitoring of serum IL-18 levels is useful to predict the disease course and assess remission in s-JIA.
Previous reports showed that TCZ could mask the clinical features and laboratory findings of patients with s-JIA (64–67). We compared serum IL-18 levels between s-JIA patients receiving TCZ and not receiving TCZ (23, 64) and found no statistically significant differences between the groups. Thus, the monitoring of serum IL-18 levels is useful for the assessment of disease activity in s-JIA, and in patients with s-JIA receiving TCZ.
Serum IL-18 levels are also elevated in patients with s-JIA–associated MAS (16, 17, 20–26). We compared serum IL-18 levels in s-JIA patients with active disease who later developed MAS and those who did not (20). Serum IL-18 levels in the former group were significantly higher than in the latter group in the active phase prior to the development of MAS as well as in the MAS phase. In contrast, there was no significant difference in serum IL-18 levels in the former group whether measured before or during MAS. The cut-off value for serum IL-18 levels to predict MAS development was 47,750 pg/ml. We compared the accuracy of serum biomarkers including neopterin, IL-18, CXCL9, and soluble tumor necrosis factor receptor type I (sTNF-RI) and II for the diagnosis of MAS complicating s-JIA (124). Serum neopterin levels might be the most useful marker to diagnose the transition to MAS from active-phase s-JIA.
In addition to serum total IL-18 levels, recent reports revealed IL-18BP-unbound, bioactive, and free IL-18 levels in serum were significantly elevated in patients with s-JIA and AOSD (24, 25, 29). Serum free IL-18 levels were significantly correlated with the disease activity of s-JIA and AOSD (24, 25, 29). Furthermore, these levels increased further in MAS associated s-JIA/AOSD (24, 25, 29). Most patients with s-JIA showed chronically elevated total serum IL-18 levels (24, 25). In contrast, elevation of free IL-18 in serum was largely restricted to s-JIA patients with MAS (25). Furthermore, serum free IL-18 levels reflected the disease activity of MAS (25). From these findings, serum free IL-18 levels might be useful as a biomarker of disease activity of MAS compared to total IL-18 levels.
Serum IL-18 levels are also useful for the prediction of treatment outcomes in patients with s-JIA treated with canakinumab (CAN) in combination with serum interferon (IFN)-γ levels and chemokine (C-X-C motif) ligand 9 (CXCL9) levels (21). Recently, Hinze et al. reported responders to canakinumab had higher serum IL-18 and IFN-γ levels and lower CXCL9 levels at baseline; that is, higher IL-18:CXCL9 and IFN-γ:CXCL9 ratios at baseline were associated with a better clinical response to CAN treatment in s-JIA (21). These ratios might have significant accuracy for the prediction of treatment responses.
We compared serum cytokine profiles and kinetics in patients with AOSD with those in patients with s-JIA (26). The cytokine patterns associated with s-JIA and AOSD shared common features including a predominant increase in IL-18. Interestingly, as well as s-JIA, patients with AOSD were classified into two subgroups based on serum IL-6 and IL-18 levels. The number of patients with arthritis was significantly higher in the IL-6-dominant group. These findings support the hypothesis that s-JIA and AOSD share a disease category. Serum IL-18 levels are significantly and highly increased in AOSD (26–30) and these levels clearly reflect the disease activity of AOSD. The cut-off value of serum IL-18 levels for the differentiation of AOSD from other febrile diseases was 5,000pg/mL (28). However, it is still unclear whether serum IL-18 levels are closely correlated with the disease course of AOSD, and whether they are also useful as a diagnostic laboratory criterion for remission in AOSD. Further larger studies may help to define the true diagnostic value of IL-18 as a biomarker of AOSD.
Serum IL-18 levels are highly increased in patients with PAPA syndrome (36). In contrast to s-JIA and AOSD, serum IL-18 levels did not correlate with clinical symptoms and acute phase reactants in patients with PAPA syndrome (36). However, there were clear differences in serum IL-18 levels between PSTPIP1 mutation–positive patients and PSTPIP1 mutation–negative patients (36). These findings indicate that the elevation of IL-18 correlates with PSTPIP1 mutations rather than clinical features. From these results, serum IL-18 might be useful to distinguish between patients carrying PSTPIP1 mutations from patients with suspicious clinical findings regardless of its pathogenic role.
IL-18 as a therapeutic target
Free IL-18 might be a therapeutic target in s-JIA and AOSD. Tadekinig alfa, human recombinant IL-18BP, was effective in patients with MAS associated s-JIA (125), XIAP deficiency (32), and NLRC4 gain-of-function (33). Furthermore, a case report of one patient with AOSD treated with tadekinig alfa showed serum free IL-18 levels were undetectable 2 h after the administration of IL-18BP, even though the levels were high before the subcutaneous injection (126). Furthermore, serum free IL-18 levels remained low during disease remission under tadekinig alfa therapy, and then increased again after tadekinig alfa was discontinued, in parallel to relapse phases. A phase II clinical trial of tadekinig alfa in patients with refractory AOSD (127) reported about half of the patients had a normal body temperature with CRP levels decreased to 50% of their baseline levels or <5 mg/L 3 weeks after starting tadekinig alfa. Further larger studies including randomized control studies are required to confirm the clinical effect of tadekinig alfa on IL-18 driven diseases such as s-JIA and AOSD.
Conclusions
IL-18 plays an important role in autoinflammatory diseases, in particular diseases associated with MAS including s-JIA, AOSD, XIAP deficiency, and NLRC4 gain-of-function. IL-18 inhibition is likely to be effective for these diseases. In contrast, the overproduction of IL-18 was observed in PAPA syndrome without susceptibility to MAS. The pathogenic and causative roles of IL-18 remain unclear in each disease. Further investigations are necessary to clarify the role of IL-18 and its importance as a therapeutic target in the pathogenesis of autoinflammatory diseases.
Author contributions
MS drafted the paper; ST, MM, and AY supervised and reviewed the paper. All authors read and approved the final version of the manuscript.
Funding
This work was supported by the Japan Society for the Promotion of Science (JAPS) KAKENHI (18K07786).
Acknowledgments
We thank Harumi Matsukawa for technical assistance.
Conflict of interest
Tokyo Medical and Dental University received unrestricted research grants for the salary of MM from Abbvie Japan, Asahi-Kasei, Ayumi Pharmaceutical Corporation, Chugai Pharmaceutical Co., Ltd., CSL Behring K.K., Japan Blood Products Organization, Nippon Kayaku and UCB Japan Co. Ltd., while TMDU paid the salary of MM.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Kaplanski G. Interleukin-18: Biological properties and role in disease pathogenesis. Immunol Rev (2018) 281:138–53. doi: 10.1111/imr.12616
2. Nakamura K, Okamura H, Nagata K, Komatsu T, Tamura T. Purification of a factor which provides a costimulatory signal for gamma interferon production. Infect Immun (1993) 61:64–70. doi: 10.1128/iai.61.1.64-70.1993
3. Okamura H, Tsutsi H, Komatsu T, Yutsudo M, Hakura A, Tanimoto T, et al. Cloning of a new cytokine that induces IFN-gamma production by T cells. Nature (1995) 378:88–91. doi: 10.1038/378088a0
4. Puren AJ, Fantuzzi G, Dinarello CA. Gene expression, synthesis and secretion of IL-1b and IL-18 are differentially regulated in human blood mononuclear cells and mouse spleen cells. Proc Natl Acad Sci U S A (1999) 96:2256–61. doi: 10.1073/pnas.96.5.2256
5. Stoll S, Mueller G, Kurimoto M, Saloga J, Tanimoto T, Yamauchi H, et al. Production of IL-18 (IFNg–inducing factor) messenger RNA and functional protein by murine keratinocytes. J Immunol (1997) 159:298–302.
6. Takeuchi M, Nishizaki Y, Sano O, Ohta T, Ikeda M, Kurimoto M. Immunohistochemical and immuno-electron-microscopic detection of interferon-gamma-inducing factor ("interleukin-18") in mouse intestinal epithelial cells. Cell Tissue Res (1997) 289:499–503. doi: 10.1007/s004410050895
7. Johnson EA, Guignet MA, Dao TL, Hamilton TA, Kan RK. Interleukin-18 expression increases in response to neurovascular damage following soman-induced status epilepticus in rats. J Inflammation (Lond) (2015) 12:43. doi: 10.1186/s12950-015-0089-9
8. Canna SW, de Jesus AA, Gouni S, Brooks SR, Marrero B, Liu Y, et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat Genet (2014) 46:1140–6. doi: 10.1038/ng.3089
9. Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature (2006) 440:237–41. doi: 10.1038/nature04516
10. Levy M, Thaiss CA, Zeevi D, Dohnalová L, Zilberman-Schapira G, Mahdi JA, et al. Microbiota-modulated metabolites shape the intestinal microenvironment by regulating NLRP6 inflammasome signaling. Cell (2015) 163:1428–43. doi: 10.1016/j.cell.2015.10.048
11. Zhu S, Ding S, Wang P, Wei Z, Pan W, Palm NW, et al. Nlrp9b inflammasome restricts rotavirus infection in intestinal epithelial cells. Nature (2017) 546:667–70. doi: 10.1038/nature22967
12. Novick D, Kim SH, Fantuzzi G, Reznikov LL, Dinarello CA, Rubinstein M. Interleukin-18 binding protein: A novel modulator of the Th1 cytokine response. Immunity (1999) 10:127–36. doi: 10.1016/S1074-7613(00)80013-8
13. Aizawa Y, Akita K, Taniai M, Torigoe K, Mori T, Nishida Y, et al. Cloning and expression of interleukin-18 binding protein. FEBS Lett (1999) 445:338–42. doi: 10.1016/S0014-5793(99)00148-9
14. Bufler P, Azam T, Gamboni-Robertson F, Reznikov LL, Kumar S, Dinarello CA, et al. A complex of the IL-1 homologue IL-1F7b and IL-18-binding protein reduces IL-18 activity. Proc Natl Acad Sci USA (2002) 99:13723–8. doi: 10.1073/pnas.212519099
15. McGonagle D, McDermott MF. A proposed classification of the immunological diseases. PloS Med (2006) 3:e297. doi: 10.1371/journal.pmed.0030297
16. Maeno N, Takei S, Nomura Y, Imanaka H, Hokonohara M, Miyata K. Highly elevated serum levels of interleukin-18 in systemic juvenile idiopathic arthritis but not in other juvenile idiopathic arthritis subtypes or in Kawasaki disease: Comment on the article by kawashima et al. Arthritis Rheumatol (2002) 46:2539–41. doi: 10.1002/art.10389
17. Shimizu M, Yokoyama T, Yamada K, Kaneda H, Wada H, Wada T, et al. Distinct cytokine profiles of systemic-onset juvenile idiopathic arthritis-associated macrophage activation syndrome with particular emphasis on the role of interleukin-18 in its pathogenesis. Rheumatol (Oxford) (2010) 49(9):1645–53. doi: 10.1093/rheumatology/keq133
18. Jelusić M, Lukić IK, Tambić-Bukovac L, Dubravcić K, Malcić I, Rudan I, et al. Interleukin-18 as a mediator of systemic juvenile idiopathic arthritis. Clin Rheumatol (2007) 26:1332–4. doi: 10.1007/s10067-006-0474-0
19. Chen O, Shan N, Zhu X, Wang Y, Ren P, Wei D, et al. The imbalance of IL-18/IL-18BP in patients with systemic juvenile idiopathic arthritis. Acta Biochim Biophys Sin (2013) 45:339–41. doi: 10.1093/abbs/gmt007
20. Shimizu M, Nakagishi Y, Inoue N, Mizuta M, Ko G, Saikawa Y, et al. Interleukin-18 for predicting the development of macrophage activation syndrome in systemic juvenile idiopathic arthritis. Clin Immunol (2015) 160:277–81. doi: 10.1016/j.clim.2015.06.005
21. Hinze T, Kessel C, Hinze CH, Seibert J, Gram H, Foell D. A dysregulated interleukin-18-interferon-γ-CXCL9 axis impacts treatment response to canakinumab in systemic juvenile idiopathic arthritis. Rheumatol (Oxford) (2021) 60:5165–74. doi: 10.1093/rheumatology/keab113
22. Mizuta M, Shimizu M, Irabu H, Usami M, Inoue N, Nakagishi Y, et al. Comparison of serum cytokine profiles in macrophage activation syndrome complicating different background rheumatic diseases in children. Rheumatol (Oxford) (2021) 60:231–8. doi: 10.1093/rheumatology/keaa299
23. Mizuta M, Shimizu M, Inoue N, Ikawa Y, Nakagishi Y, Yasuoka R, et al. Clinical significance of interleukin-18 for the diagnosis and prediction of disease course in systemic juvenile idiopathic arthritis. Rheumatol (Oxford) (2021) 60:2421–6. doi: 10.1093/rheumatology/keaa634
24. Yasin S, Fall N, Brown RA, Henderlight M, Canna SW, Girard-Guyonvarc'h C, et al. IL-18 as a biomarker linking systemic juvenile idiopathic arthritis and macrophage activation syndrome. Rheumatol (Oxford) (2020) 59(2):361–6. doi: 10.1093/rheumatology/kez282
25. Weiss ES, Girard-Guyonvarc'h C, Holzinger D, de Jesus AA, Tariq Z, Picarsic J, et al. Interleukin-18 diagnostically distinguishes and pathogenically promotes human and murine macrophage activation syndrome. Blood (2018) 131:1442–55. doi: 10.1182/blood-2017-12-820852
26. Inoue N, Shimizu M, Tsunoda S, Kawano M, Matsumura M, Yachie A. Cytokine profile in adult-onset still’s disease: Comparison with systemic juvenile idiopathic arthritis. Clin Immunol Orlando Fla (2016) 169:8–13. doi: 10.1016/j.clim.2016.05.010
27. Kawaguchi Y, Terajima H, Harigai M, Hara M, Kamatani N. Interleukin-18 as a novel diagnostic marker and indicator of disease severity in adult-onset still’s disease. Arthritis Rheumatol (2001) 44:1716–7. doi: 10.1002/1529-0131(200107)44:7<1716::AID-ART298>3.0.CO;2-I
28. Kudela H, Drynda S, Lux A, Horneff G, Kekow J. Comparative study of interleukin-18 (IL-18) serum levels in adult onset still's disease (AOSD) and systemic onset juvenile idiopathic arthritis (sJIA) and its use as a biomarker for diagnosis and evaluation of disease activity. BMC Rheumatol (2019) 3:4. doi: 10.1186/s41927-019-0053-z
29. Girard C, Rech J, Brown M, Allali D, Roux-Lombard P, Spertini F, et al. Elevated serum levels of free interleukin-18 in adult-onset still's disease. Rheumatol (Oxford) (2016) 55:2237–47. doi: 10.1093/rheumatology/kew300
30. Shiga T, Nozaki Y, Tomita D, Kishimoto K, Hirooka Y, Kinoshita K, et al. Usefulness of interleukin-18 as a diagnostic biomarker to differentiate adult-onset still's disease With/Without macrophage activation syndrome from other secondary hemophagocytic lymphohistiocytosis in adults. Front Immunol (2021) 12:750114. doi: 10.3389/fimmu.2021.750114
31. Wada T, Kanegane H, Ohta K, Katoh F, Imamura T, Nakazawa Y, et al. Sustained elevation of serum interleukin-18 and its association with hemophagocytic lymphohistiocytosis in XIAP deficiency. Cytokine (2014) 65:74–8. doi: 10.1016/j.cyto.2013.09.007
32. Geerlinks AV, Dvorak AM. XIAP deficiency treatment consortium. A case of XIAP deficiency successfully managed with tadekinig Alfa(rhIL-18BP). J Clin Immunol (2022) 42:901–3. doi: 10.1007/s10875-022-01236-2
33. Canna SW, Girard C, Malle L, de Jesus A, Romberg N, Kelsen J, et al. Life-threatening NLRC4-associated hyperinflammation successfully treated with IL-18 inhibition. J Allergy Clin Immunol (2017) 139(5):1698–701. doi: 10.1016/j.jaci.2016.10.022
34. Lam MT, Coppola S, Krumbach OHF, Prencipe G, Insalaco A, Cifaldi, et al. A novel disorder involving dyshematopoiesis, inflammation, and HLH due to aberrant CDC42 function. J Exp Med (2019) 216:2778–99. doi: 10.1084/jem.20190147
35. Arduini A, Marasco E, Marucci G, Pardeo M, Insalaco A, Caiello I, et al. An unusual presentation of purine nucleoside phosphorylase deficiency mimicking systemic juvenile idiopathic arthritis complicated by macrophage activation syndrome. Pediatr Rheumatol Online J (2019) 17:25. doi: 10.1186/s12969-019-0328-3
36. Stone DL, Ombrello A, Arostegui JI, Schneider C, Dang V, de Jesus A, et al. Excess serum interleukin-18 distinguishes patients with pathogenic mutations in PSTPIP1. Arthritis Rheumatol (2022) 74(2):353–7. doi: 10.1002/art.41976
37. Yoshimoto T, Takeda K, Tanaka T, Ohkusu K, Kashiwamura S, Okamura H, et al. IL-12 up-regulates IL-18 receptor expression on T cells, Th1 cells, and b cells: synergism with IL-18 for IFNgamma production. J Immunol (1998) 161:3400–7.
38. Strengell M, Matikainen S, Siren J, Lehtonen A, Foster D, Julkunen I, et al. IL-21 in synergy with IL-15 or IL-18 enhances IFN-gamma production in human NK and T cells. J Immunol (2003) 170:5464–9. doi: 10.4049/jimmunol.170.11.5464
39. Munder M, Mallo M, Eichmann K, et al. Murine macrophages secrete interferon gamma upon combined stimulation with interleukin (IL)-12 and IL-18: a novel pathway of autocrine macrophage activation. J Exp Med (1998) 187:2103–8. doi: 10.1084/jem.187.12.2103
40. Schindler H, Lutz MB, Rollinghoff M, et al. The production of IFN-gamma by IL-12/IL-18-activated macrophages requires STAT4 signaling and is inhibited by IL-4. J Immunol (2001) 166:3075–82. doi: 10.4049/jimmunol.166.5.3075
41. Hoshino T, Wiltrout RH, Young HA. IL-18 is a potent coinducer of IL-13 in NK and T cells: A new potential role for IL-18 in modulating the immune response. J Immunol (1999) 162:5070–7.
42. Yoshimoto T, Mizutani H, Tsutsui H, et al. IL-18 induction of IgE: Dependence on CD4+ T cells, IL-4 and STAT6. Nat Immunol (2000) 1:132–7. doi: 10.1038/77811
43. Yoshimoto T, Tsutsui H, Tominaga K, Hoshino K, Okamura H, Akira S, et al. IL-18, although antiallergic when administered with IL-12, stimulates IL-4andhistamine release by basophils. Proc Natl Acad Sci USA (1999) 96:13962–6. doi: 10.1073/pnas.96.24.13962
44. Kessel C, Lippitz K, Weinhage T, Hinze C, Wittkowski H, Holzinger D, et al. Proinflammatory cytokine environments can drive interleukin-17 overexpression by γ/δ T cells in systemic juvenile idiopathic arthritis. Arthritis Rheumatol (2017) 69:1480–94. doi: 10.1002/art.40099
45. Oertli M, Sundquist M, Hitzler I, Engler DB, Arnold IC, Reuter S, et al. DC-Derived IL-18drives treg differentiation, murine helicobacter pylori-specific immune tolerance, and asthma protection. J Clin Invest (2012) 122:1082–96. doi: 10.1172/JCI61029
46. Leung BP, Culshaw S, Gracie JA, Hunter D, Canetti CA, Campbell C, et al. A role for IL-18 in neutrophil activation. J Immunol (2001) 167:2879–86. doi: 10.4049/jimmunol.167.5.2879
47. Gracie JA, Forsey RJ, Chan WL, Gilmour A, Leung BP, Greer MR, et al. A proinflammatory role for IL-18 in rheumatoid arthritis. J Clin Invest (1999) 104:1393–401. doi: 10.1172/JCI7317
48. Vidal-Vanaclocha F, Fantuzzi G, Mendoza L, Fuentes AM, Anasagasti MJ, Martín J, et al. IL-18 regulates IL-1beta-Dependent hepatic melanoma metastasis via vascular cell adhesion molecule-1. Proc Natl Acad Sci USA (2000) 97:734–9. doi: 10.1073/pnas.97.2.734
49. Tsutsui H, Nakanishi K, Matsui K, Higashino K, Okamura H, Miyazawa Y, et al. IFN-gamma-inducing factor up-regulates fas ligand-mediated cytotoxic activity of murine natural killer cell clones. J Immunol (1996) 157:3967–73.
50. Mende R, Vincent FB, Kandane-Rathnayake R, Koelmeyer R, Lin E, Chang J, et al. Analysis of serum interleukin (IL)-1β and IL-18 in systemic lupus erythematosus. Front Immunol (2018) 9:1250. doi: 10.3389/fimmu.2018.01250
51. Leach ST, Messina I, Lemberg DA, Novick D, Rubenstein M, Day AS. Local and systemic interleukin-18 and interleukin-18-binding protein in children with inflammatory bowel disease. Inflammation Bowel Dis (2008) 14:68–74. doi: 10.1002/ibd.20272
52. Mellins ED, Macaubas C, Grom AA. Pathogenesis of systemic juvenile idiopathic arthritis: Some answers, more questions. Nat Rev Rheumatol (2011) 7:416–26. doi: 10.1038/nrrheum.2011.68
53. Nigrovic PA. Review: Is there a window of opportunity for treatment of systemic juvenile idiopathic arthritis? Arthritis Rheumatol (2014) 66:1405–13. doi: 10.1002/art.38615
54. Henderson LA, Hoyt KJ, Lee PY, Rao DA, Jonsson AH, Nguyen JP, et al. Th17 reprogramming of T cells in systemic juvenile idiopathic arthritis. JCI Insight (2020) 5:e132508. doi: 10.1172/jci.insight.132508
55. Rider P, Voronov E, Dinarello CA, Apte RN, Cohen I. Alarmins: feel the stress. J Immunol (2017) 198(4):1395–402. doi: 10.4049/jimmunol.1601342
56. Kim B, Lee Y, Kim E, Kwak A, Ryoo S, Bae SH, et al. The interleukin-1alpha precursor is biologically active and is likely a key alarmin in the IL-1 family of cytokines. Front Immunol (2013) 4:391. doi: 10.3389/fimmu.2013.00391
57. Choy EH, De Benedetti F, Takeuchi T, Hashizume M, John MR, Kishimoto T. Translating IL-6 biology into effective treatments. Nat Rev Rheumatol (2020) 16:335–45. doi: 10.1038/s41584-020-0419-z
58. Quartier P, Allantaz F, Cimaz R, Pillet P, Messiaen C, Bardin C, et al. A multicentre, randomised, double-blind, placebo-controlled trial with the interleukin-1 receptor antagonist anakinra in patients with systemic-onset juvenile idiopathic arthritis (ANAJIS trial). Ann Rheum Dis (2011) 70:747–54. doi: 10.1136/ard.2010.134254
59. Ilowite NT, Prather K, Lokhnygina Y, Schanberg LE, Elder M, Milojevic D, et al. Randomized, double-blind, placebo-controlled trial of the efficacy and safety of rilonacept in the treatment of systemic juvenile idiopathic arthritis. Arthritis Rheumatol (2014) 66:2570–9. doi: 10.1002/art.38699
60. Ruperto N, Brunner HI, Quartier P, Constantin T, Wulffraat N, Horneff G, et al. Two randomized trials of canakinumab in systemic juvenile idiopathic arthritis. N Engl J Med (2012) 367:2396–406. doi: 10.1056/NEJMoa1205099
61. Yokota S, Imagawa T, Mori M, Miyamae T, Aihara Y, Takei S, et al. Efficacy and safety of tocilizumab in patients with systemic-onset juvenile idiopathic arthritis: A randomised, double-blind, placebo-controlled, withdrawal phase III trial. Lancet (2008) 371:998–1006. doi: 10.1016/S0140-6736(08)60454-7
62. De Benedetti F, Brunner HI, Ruperto N, Kenwright A, Wright S, Calvo I, et al. Randomized trial of tocilizumab in systemic juvenile idiopathic arthritis. N Engl J Med (2012) 367(25):2385–95. doi: 10.1056/NEJMoa1112802
63. Grom AA, Ilowite NT, Pascual V, Brunner HI, Martini A, Lovell D, et al. Rate and clinical presentation of macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis treated with canakinumab. Arthritis Rheumatol (2016) 68:218–28. doi: 10.1002/art.39407
64. Shimizu M, Nakagishi Y, Kasai K, Yamasaki Y, Miyoshi M, Takei S, et al. Tocilizumab masks the clinical symptoms of systemic juvenile idiopathic arthritis-associated macrophage activation syndrome: The diagnostic significance of interleukin-18 and interleukin-6. Cytokine (2012) 58:287–94. doi: 10.1016/j.cyto.2012.02.006
65. Yokota S, Itoh Y, Morio T, Sumitomo N, Daimaru K, Minota S. Macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis under treatment with tocilizumab. J Rheumatol (2015) 42:712–22. doi: 10.3899/jrheum.140288
66. Schulert GS, Minoia F, Bohnsack J, Cron RQ, Hashad S, KonÉ-Paut I, et al. Effect of biologic therapy on clinical and laboratory features of macrophage activation syndrome associated with systemic juvenile idiopathic arthritis. Arthritis Care Res (Hoboken) (2018) 70:409–19. doi: 10.1002/acr.23277
67. Shimizu M, Mizuta M, Okamoto N, Yasumi T, Iwata N, Umebayashi H, et al. Tocilizumab modifies clinical and laboratory features of macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. Pediatr Rheumatol Online J (2020) 18:2. doi: 10.1186/s12969-020-0399-1
68. Shimizu M. Macrophage activation syndrome in systemic juvenile idiopathic arthritis. Immunol Med (2021) 44:237–45. doi: 10.1080/25785826.2021.1912893
69. Sawhney S, Woo P, Murray KJ. Macrophage activation syndrome: A potentially fatal complication of rheumatic disorders. Arch Dis Child (2001) 85:421–6. doi: 10.1136/adc.85.5.421
70. Moradinejad MH, Ziaee V. The incidence of macrophage activation syndrome in children with rheumatic disorders. Minerva Pediatr (2011) 63:459–66.
71. Hot A, Toh ML, Copper B, Perard L, Girard Madoux MH, Mausservey C, et al. Reactive hemophagocytic syndrome in adult-onset still disease: clinical features and long-term outcome: A case-control study of 8 patients. Med (Baltimore) (2010) 89:37–46. doi: 10.1097/MD.0b013e3181caf100
72. Fukaya S, Yasuda S, Hashimoto T, Oku K, Kataoka H, Horita T, et al. Clinical features of haemophagocytic syndrome in patients with systemic autoimmune diseases: Analysis of 30 cases. Rheumatol (Oxford) (2008) 47:1686–91. doi: 10.1093/rheumatology/ken342
73. Behrens EM, Beukelman T, Paessler M, Cron RQ., et al. Occult macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis. J Rheumatol (2007) 34:1133–8.
74. Kitamura A, Sasaki Y, Abe T, Kano H, Yasutomo K. An inherited mutation in NLRC4 causes autoinflammation in human and mice. J Exp Med (2014) 211:2385–96. doi: 10.1084/jem.20141091
75. Rigaud S, Fondaneche MC, Lambert N, Pasquier B, Mateo V, Soulas P, et al. XIAP deficiency in humans causes an X-linked lymphoproliferative syndrome. Nature (2006) 444:110–4. doi: 10.1038/nature05257
76. Filipovich AH, Zhang K, Snow AL, Marsh RA. X-Linked lymphoproliferative syndromes: Brothers or distant cousins? Blood (2010) 116:3398–408. doi: 10.1182/blood-2010-03-275909
77. Pachlopnik Schmid J, Canioni D, Moshous D, Touzot F, Mahlaoui N, Hauck F, et al. Clinical similarities and differences of patients with X-linked lymphoproliferative syndrome type 1 (XLP-1/SAP deficiency) versus type 2 (XLP-2/XIAP deficiency). Blood (2011) 117:1522–9. doi: 10.1182/blood-2010-07-298372
78. Yang X, Miyawaki T, Kanegane H. SAP and XIAP deficiency in hemophagocytic lymphohistiocytosis. Pediatr Int (2012) 54:447–54. doi: 10.1111/j.1442-200X.2012.03683.x
79. Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer (2007) 48:124–31. doi: 10.1002/pbc.21039
80. Shibatomi K, Ida H, Yamasaki S, Nakashima T, Origuchi T, Kawakami A, et al. A novel role for interleukin-18 in human natural killer cell death: High serum levels and low natural killer cell numbers in patients with systemic autoimmune diseases. Arthritis Rheumatol (2001) 44:884–92. doi: 10.1002/1529-0131(200104)44:4<884::AID-ANR145>3.0.CO;2-4
81. de Jager W, Vastert SJ, Beekman JM, Wulffraat NM, Kuis W, Coffer PJ, et al. Defective phosphorylation of interleukin-18 receptor beta causes impaired natural killer cell function in systemic-onset juvenile idiopathic arthritis. Arthritis Rheumatol (2009) 60:2782–93. doi: 10.1002/art.24750
82. Takakura M, Shimizu M, Yakoyama T, Mizuta M, Yachie A., et al. Transient natural killer cell dysfunction associated with interleukin-18 overproduction in systemic juvenile idiopathic arthritis. Pediatr Int (2018) 60:984–5. doi: 10.1111/ped.13679
83. Ohya T, Nishimura K, Murase A, Hattori S, Ohara A, Nozawa T, et al. Impaired interleukin-18 signaling in natural killer cells from patients with systemic juvenile idiopathic arthritis. ACR Open Rheumatol (2022) 4:503–10. doi: 10.1002/acr2.11426
84. Villanueva J, Lee S, Giannini EH, Graham TB, Passo MH, Filipovich A, et al. Natural killer cell dysfunction is a distinguishing feature of systemic onset juvenile rheumatoid arthritis and macrophage activation syndrome. Arthritis Res Ther (2005) 7:R30–7. doi: 10.1186/ar1453
85. Shimizu M, Kizawa T, Kato R, Suzuki T, Yachie A. Macrophage activation syndrome in neonates born to mothers with adult-onset still's disease: Perinatal effect of maternal IL-18. Clin Immunol (2019) 207:36–9. doi: 10.1016/j.clim.2019.07.005
86. Shimizu M, Sakakibara Y, Kawano M, Yachie A. Transient impairment of NK cell function in an infant born to a mother with adult-onset still's disease: Perinatal effect of maternal IL-18. Clin Immunol (2012) 143:273–4. doi: 10.1016/j.clim.2012.03.004
87. Belkaya S, Michailidis E, Korol CB, Kabbani M, Cobat A, Bastard P, et al. Inherited IL-18BP deficiency in human fulminant viral hepatitis. J Exp Med (2019) 216:1777–90. doi: 10.1084/jem.20190669
88. Chiossone L, Audonnet S, Chetaille B, Chasson L, Farnarier C, Berda-Haddad Y, et al. Protection from inflammatory organ damage in a murine model of hemophagocytic lymphohistiocytosis using treatment with IL-18 binding protein. Front Immunol (2012) 3:239. doi: 10.3389/fimmu.2012.00239
89. Pizarro TT, Michie MH, Bentz M, Woraratanadharm J, Jr SMF, Foley E, et al. IL-18, a novel immunoregulatory cytokine, is up-regulated in crohn's disease: Expression and localization in intestinal mucosal cells. J Immunol (1999) 162(11):6829–35.
90. Tanaka M, Harigai M, Kawaguchi Y, Ohta S, Sugiura T, Takagi K, et al. Mature form of interleukin 18 is expressed in rheumatoid arthritis synovial tissue and contributes to interferon-gamma production by synovial T cells. J Rheumatol (2001) 28(8):1779–87.
91. Companjen A, van der Wel L, van der Fits L, Laman J, Prens E. Elevated interleukin-18 protein expression in early active and progressive plaque-type psoriatic lesions. Eur Cytokine Netw (2004) 15(3):210–6.
92. Henter JI, Elinder G, Soder O, Hansson M, Andersson B, Andersson U. Hypercytokinemia in familial hemophagocytic lymphohistiocytosis. Blood (1991) 78:2918–22. doi: 10.1182/blood.V78.11.2918.2918
93. Osugi Y, Hara J, Tagawa S, Takai K, Hosoi G, Matsuda Y, et al. Cytokine production regulating Th1 and Th2 cytokines in hemophagocytic lymphohistiocytosis. Blood (1997) 89:4100–3. doi: 10.1182/blood.V89.11.4100
94. Xu XJ, Tang YM, Song H, Yang SL, Xu WQ, Zhao N, et al. Diagnostic accuracy of a specific cytokine pattern in hemophagocytic lymphohistiocytosis in children. J Pediatr (2012) 160:984–90. doi: 10.1016/j.jpeds.2011.11.046
95. Bracaglia C, de Graaf K, Pires Marafon D, Guilhot F, Ferlin W, Prencipe G, et al. Elevated circulating levels of interferon-γ and interferon-γ-induced chemokines characterise patients with macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. Ann Rheum Dis (2017) 76:166–72. doi: 10.1136/annrheumdis-2015-209020
96. Put K, Avau A, Brisse E, Mitera T, Put S, Proost P, et al. Inflammatory gene expression profile and defective interferon-γ and granzyme K in natural killer cells from systemic juvenile idiopathic arthritis patients. Rheumatol (Oxford) (2015) 54:1507–17. doi: 10.1093/rheumatology/keu524
97. Locatelli F, Jordan MB, Allen C, Cesaro S, Rizzari C, Rao A, et al. Emapalumab in children with primary hemophagocytic lymphohistiocytosis. N Engl J Med (2020) 382:1811–22. doi: 10.1056/NEJMoa1911326
98. Behrens EM, Canna SW, Slade K, Rao S, Kreiger PA, Paessler M, et al. Repeated TLR9 stimulation results in macrophage activation syndrome-like disease in mice. J Clin Invest (2011) 121:2264–77. doi: 10.1172/JCI43157
99. Crozat K, Hoebe K, Ugolini S, Hong NA, Janssen E, Rutschmann S, et al. Jinx, an MCMV susceptibility phenotype caused by disruption of Unc13d: A mouse model of type 3 familial hemophagocytic lymphohistiocytosis. J Exp Med (2007) 204:853–63. doi: 10.1084/jem.20062447
100. Czar MJ, Kersh EN, Mijares LA, Lanier G, Lewis J, Yap G, et al. Altered lymphocyte responses and cytokine production in mice deficient in the X-linked lymphoproliferative disease gene SH2D1A/DSHP/SAP. Pro Natl Acad Sci USA (2001) 98:7449–54. doi: 10.1073/pnas.131193098
101. Kogl T, Muller J, Jessen B, Schmitt-Graeff A, Janka G, Ehl S, et al. Hemophagocytic lymphohistiocytosis in syntaxin-11-deficient mice: T-cell exhaustion limits fatal disease. Blood (2013) 121:604–13. doi: 10.1182/blood-2012-07-441139
102. Pachlopnik Schmid J, Ho CH, Chretien F, Lefebvre JM, Pivert G, Kosco-Vilbois M, et al. Neutralization of IFNgamma defeats haemophagocytosis in LCMV-infected perforin- and Rab27a-deficient mice. EMBO Mol Med (2009) 1:112–24. doi: 10.1002/emmm.200900009
103. Prencipe G, Caiello I, Pascarella A, Grom AA, Bracaglia C, Chatel L, et al. Neutralization of IFN-gamma reverts clinical and laboratory features in a mouse model of macrophage activation syndrome. J Allergy Clin Immunol (2018) 141:1439–49. doi: 10.1016/j.jaci.2017.07.021
104. Jordan MB, Hildeman D, Kappler J, Marrack P. An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood (2004) 104:735–43. doi: 10.1182/blood-2003-10-3413
105. Coppola S, Insalaco A, Zara E, Di Rocco M, Marafon DP, Spadaro F, et al. Mutations at the c-terminus of CDC42 cause distinct hematopoietic and autoinflammatory disorders. J Allergy Clin Immunol (2022) 150:223–8. doi: 10.1016/j.jaci.2022.01.024
106. Takenouchi T, Kosaki R, Niizuma T, Hata K, Kosaki K. Macrothrombocytopenia and developmental delay with a de novo CDC42 mutation: yet another locus for thrombocytopenia and developmental delay. Am J Med Genet A (2015) 167A:2822–5. doi: 10.1002/ajmg.a.37275
107. Takenouchi T, Okamoto N, Ida S, Uehara T, Kosaki K. Further evidence of a mutation in CDC42 as a cause of a recognizable syndromic form of thrombocytopenia. Am J Med Genet A (2016) 170A:852–5. doi: 10.1002/ajmg.a.37526
108. Martinelli S, Krumbach OHF, Pantaleoni F, Coppola S, Amin E, Pannone L, et al. Functional dysregulation of CDC42 causes diverse developmental phenotypes. Am J Hum Genet (2018) 102:309–20. doi: 10.1016/j.ajhg.2017.12.015
109. Asiri A, Alwadaani D, Umair M, Alhamoudi KM, Almuhanna MH, Nasir A, et al. Pancytopenia, recurrent infection, poor wound healing, heterotopia of the brain probably associated with a candidate novel de novo CDC42 gene defect: expanding the molecular and phenotypic spectrum. Genes (2021) 12:294. doi: 10.3390/genes12020294
110. Szczawinska-Poplonyk A, Ploski R, Bernatowska E, Pac M. A novel CDC42 mutation in an 11-year old child manifesting as syndromic immunodeficiency, autoinflammation, hemophagocytic lymphohistiocytosis, and malignancy: a case report. Front Immunol (2021) 11:318. doi: 10.3389/fimmu.2020.00318
111. Chen F, Ma L, Parrini MC, Mao X, Lopez M, Wu C, et al. Cdc42 is required for PIP(2)-induced actin polymerization and early development but not for cell viability. Curr Biol (2000) 10:758–65. doi: 10.1016/S0960-9822(00)00571-6
112. Standing AS, Malinova D, Hong Y, Record J, Moulding D, Blundell MP, et al. Autoinflammatory periodic fever, immunodeficiency, and thrombocytopenia (PFIT) caused by mutation in actin-regulatory gene WDR1. J Exp Med (2017) 214:59–71. doi: 10.1084/jem.20161228
113. Kuhns DB, Fink DL, Choi U, Sweeney C, Lau K, DL P, et al. Cytoskeletal abnormalities and neutrophil dysfunction in WDR1 deficiency. Blood (2016) 128:2135–43. doi: 10.1182/blood-2016-03-706028
114. Giblett ER, Ammann AJ, Wara DW, Sandman R, Diamond LK. Nucleosidephosphorylase deficiency in a child with severely defective T-cell immunity and normal b-cell immunity. Lancet (1975) 1:1010–3. doi: 10.1016/S0140-6736(75)91950-9
115. Parvaneh N, Ashrafi MR, Yeganeh M, Pouladi N, Sayarifar F, Parvaneh L. Progressive multifocal leukoencephalopathy in purine nucleoside phosphorylase deficiency. Brain Dev (2007) 29:124–6. doi: 10.1016/j.braindev.2006.07.008
117. Wise CA, Gillum JD, Seidman CE, Lindor NM, Veile R, Bashiardes S, et al. Mutations in CD2BP1 disrupt binding to PTP PEST and are responsible for PAPA syndrome, an autoinflammatory disorder. Hum Mol Genet (2002) 11:961–9. doi: 10.1093/hmg/11.8.961
118. Yamamoto T, Moriwaki Y, Matsui K, Takahashi S, Tsutsui H, Yoshimoto T, et al. High IL-18 (interferon-gamma inducing factor) concentration in a purine nucleoside phosphorylase deficient patient. Arch Dis Child (1999) 81:179–80. doi: 10.1136/adc.81.2.179
119. Cortesio CL, Wernimont SA, Kastner DL, Cooper KM, Huttenlocher A. Impaired podosome formation and invasive migration of macrophages from patients with a PSTPIP1 mutation and PAPA syndrome. Arthritis Rheum (2010) 62:2556–8. doi: 10.1002/art.27521
120. Akkaya-Ulum YZ, Balci-Peynircioglu B, Purali N, Yilmaz E. Pyrin-PSTPIP1 colocalises at the leading edge during cell migration. Cell Biol Int (2015) 39:1384–94. doi: 10.1002/cbin.10514
121. Miyashita K, Matsuda Y, Okajima M, Toma T, Yachie A, Wada T. Role of E148Q in familial Mediterranean fever with an exon 10 mutation in MEFV. Pediatr Int (2022) 64(1):e14696. doi: 10.1111/ped.14696
122. Ouyang W, Rutz S, Crellin NK, Hymowitz SG. Regulation and functions of the IL-10 family of cytokines in inflammation and disease. Annu Rev Immunol (2011) 29:71–109. doi: 10.1146/annurev-immunol-031210-101312
123. Imbrechts M, Avau A, Vandenhaute J, Malengier-Devlies B, Put K, Mitera T, et al. Insufficient IL-10 production as a mechanism underlying thee pathogenesis of systemic juvenile idiopathic arthritis. J Immunol (2018) 201(9):2654–63. doi: 10.4049/jimmunol.1800468
124. Takakura M, Shimizu M, Irabu H, Sakumura N, Inoue N, Mizuta M, et al. Comparison of serum biomarkers for the diagnosis of macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. Clin Immunol (2019) 208:108252. doi: 10.1016/j.clim.2019.108252
125. Yasin S, Solomon K, Canna SW, Girard-Guyonvarc'h C, Gabay C, Schiffrin E, et al. IL-18 as therapeutic target in a patient with resistant systemic juvenile idiopathic arthritis and recurrent macrophage activation syndrome. Rheumatol (Oxford) (2020) 59(2):442–5. doi: 10.1093/rheumatology/kez284
126. Kiltz U, Kiefer D, Braun J, Schiffrin EJ, Girard-Guyonvarc'h C, Gabay C. Prolonged treatment with tadekinig alfa in adult-onset still's disease. Ann Rheum Dis (2020) 79:e10. doi: 10.1136/annrheumdis-2018-214496
Keywords: IL-18, systemic juvenile idiopathic arthritis, adult Still's disease, inflammasomes, NLRC4, pyogenic sterile arthritis, pyoderma gangrenosum, acne
Citation: Shimizu M, Takei S, Mori M and Yachie A (2022) Pathogenic roles and diagnostic utility of interleukin-18 in autoinflammatory diseases. Front. Immunol. 13:951535. doi: 10.3389/fimmu.2022.951535
Received: 24 May 2022; Accepted: 10 August 2022;
Published: 22 September 2022.
Edited by:
Tomoyuki Mukai, Kawasaki Medical School, JapanReviewed by:
Sinisa Savic, University of Leeds, United KingdomSoohyun Kim, Konkuk University, South Korea
Giusi Prencipe, Bambino Gesù Children’s Hospital (IRCCS), Italy
Copyright © 2022 Shimizu, Takei, Mori and Yachie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Masaki Shimizu, bXNoaW1penUucGVkQHRtZC5hYy5qcA==