
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 06 September 2022
Sec. Systems Immunology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.948419
This article is part of the Research Topic Structural modeling and computational analyses of immune system molecules View all 6 articles
 Jadson C. Santos1†
Jadson C. Santos1† Mariangela Dametto2†
Mariangela Dametto2† Ana Paula Masson3
Ana Paula Masson3 Vitor M. Faça3Rodrigo Bonacin2
Vitor M. Faça3Rodrigo Bonacin2 Eduardo A. Donadi4
Eduardo A. Donadi4 Geraldo Aleixo Passos1,5,6*
Geraldo Aleixo Passos1,5,6*The autoimmune regulator (AIRE) protein functions as a tetramer, interacting with partner proteins to form the “AIRE complex,” which relieves RNA Pol II stalling in the chromatin of medullary thymic epithelial cells (mTECs). AIRE is the primary mTEC transcriptional controller, promoting the expression of a large set of peripheral tissue antigen genes implicated in the negative selection of self-reactive thymocytes. Under normal conditions, the SIRT1 protein temporarily interacts with AIRE and deacetylates K residues of the AIRE SAND domain. Once the AIRE SAND domain is deacetylated, the binding with SIRT1 is undone, allowing the AIRE complex to proceed downstream with the RNA Pol II to the elongation phase of transcription. Considering that the in silico and in vitro binding of the AIRE SAND domain with SIRT1 provides a powerful model system for studying the dominant SAND G228W mutation mechanism, which causes the autoimmune polyglandular syndrome-1, we integrated computational molecular modeling, docking, dynamics between the whole SAND domain with SIRT1, and surface plasmon resonance using a peptide harboring the 211 to 230 residues of the SAND domain, to compare the structure and energetics of binding/release between AIRE G228 (wild-type) and W228 (mutant) SAND domain to SIRT1. We observed that the G228W mutation in the SAND domain negatively influences the AIRE-SIRT1 interaction. The disturbed interaction might cause a disruption in the binding of the AIRE SAND domain with the SIRT1 catalytic site, impairing the AIRE complex to proceed downstream with RNA Pol II.
The human autoimmune regulator (AIRE) gene spans 14 exons at chromosome 21q22.3, encoding a 1635-nucleotide mRNA and a 545-amino acid protein (1–3). Nearly 145 single-nucleotide mutations or large deletions have been observed along the AIRE gene (4, 5; Human Gene Mutation Database available at https://www.hgmd.cf.ac.uk/ac/). Some mutations in the AIRE gene are related to autoimmune polyendocrinopathy-candidiasis ectodermal dystrophy (APECED) (2, 6), also known as autoimmune polyglandular syndrome type 1 (APS-1) (OMIM entry # 240300). Most APS-1 mutations are inherited in an autosomal recessive manner, except for a group of monoallelic mutations clustered within the zinc finger domain of the first plant homeodomain (PHD1) (4, 7) and a G228W mutation at the SAND domain, which follows a dominant inheritance pattern (8–10).
The 56-kDa AIRE protein is a transcriptional regulator in medullary thymic epithelial cells (11–14), exhibiting five domains and four LXXLL motifs. The caspase activation and recruitment (CARD) domain is essential for AIRE dimerization. The nuclear localization signal (NLS) domain presents a nuclear localization signal that mediates AIRE migration from the cytoplasm to the nucleus. The Sp100, AIRE-1, NucP41/75, and DEAF-1 (SAND) domains promote protein-protein interactions and chromatin binding, which play a role in the nuclear speckle localization of AIRE, and the PHD1 and PHD2 domains function as histone code readers, interacting and supporting the process of chromatin decondensation (14–17). During its action in chromatin, AIRE interacts with several partner proteins forming the “AIRE complex,” in which SIRT1 is one of the most critical deacetylating K residues in the SAND domain, permitting the proper function of AIRE (18–22).
In addition to chromatin binding, the SAND domain may also have a regulatory role in the homomultimerization of AIRE (23, 24). The G228W SAND domain mutation was the first dominant mutation associated with the aggressiveness of APS-1 (4, 8–10); however, the reason for its dominant character and the molecular mechanism of APS-1 pathogenesis are still elusive.
Considering that i) the deacetylation of SAND domain K residues mediated by the partner SIRT1 molecule is essential for AIRE function in releasing RNA Pol II and allowing it to proceed with the elongation phase of transcription (11, 18, 25–28), and ii) the G228W mutation occurs at a position located at six amino acid residues from one of the acetylated K residues (K222), which is further deacetylated by SIRT1 (26), in this study, we hypothesized that the proximity of the G228W mutation to the K222 residue might interfere with the interaction between the SAND domain and the SIRT1 protein.
We integrated computational molecular modeling, docking, and dynamics between the whole AIRE SAND domain with the SIRT1 protein to test this hypothesis. Additionally, we evaluated the binding of a peptide harboring the 211 to 230 residues of the SAND domain with the SIRT1 protein to compare the binding/release between AIRE G228 (wild-type peptide) and W228 (mutant peptide) using surface plasmon resonance. The in silico analyses showed differences in the physical interaction between the AIRE SAND domain and the SIRT1 catalytic site for both wild-type and mutant domains. On the other hand, the surface plasmon resonance evaluation showed a stronger affinity (as seen through binding and dissociation) of the wild-type SAND domain peptide with the SIRT1 protein compared to a weaker association observed for the mutant peptide.
We used the SWISS-MODEL workspace for protein modeling (http://swissmodel.expasy.org/workspace). The amino acid sequence in the FASTA format of the SIRT1 target protein was used as input. The SWISS-MODEL algorithms performed BLASTp and provided the best homology models based on global model quality estimation (GMQE) and qualitative model energy analysis (QMEAN) statistical parameters. The GMQE provided the quality by combining the target-template alignment properties, estimating a range between 0 and 1. The scoring function of QMEAN consisted of a linear combination of structural descriptors, which is related to the high-resolution X-ray crystal structure, estimating a Z-score scheme.
Molecular docking analysis of the modeled AIRE SAND domain, wild-type or G228W mutant AIRE SAND domain, and the SIRT1 protein (PDB code 4I5I) was performed to study the stability of the protein-protein interaction. The ClusPro 2.0 protein-protein docking server (29) (available at https://cluspro.org/) was used. In the ClusPro server, the rigid body docking phase uses the PIPER docking program (https://www.schrodinger.com/products/piper), which relies on the fast Fourier transform (FFT) correlation approach. PIPER represents the interaction energy between two proteins using the formula E = w1Erep+w2Eattr+w3Eelec+w4EDARS, where E rep and E attr denote the attractive and repulsive contributions to the van der Waals interaction energy, and E elec represents the electrostatic energy. EDARS is a pairwise structure-based potential; it primarily means desolvation contributions, i.e., free energy change by removing the water molecules from the interface. The coefficients w 1, w 2, w 3, and w 4 define the molecular weights of the corresponding amino acid residues.
As mentioned above, the three-dimensional (3D) structures of the complexes formed with AIRE wild-type or with AIRE mutant peptide and SIRT1 generated in the docking section were used as initial configurations for the molecular dynamics simulations. In this work, calculations were performed to verify the stability and affinity of the complexes.
To mimic the interaction between the two peptides and SIRT1 protein, the K222 residue was acetylated using the parameters given in the AMBER Molecular Dynamics Package (https://ambermd.org/) for this residue. ACK and the connectivity library file for K222 residue were generated by tleap.
The two systems were solvated using the TIP3P water model and neutralized with four Na+ ions in the tleap tool implemented in AMBERTOOLS21 (30) (https://ambermd.org/AmberTools.php). The whole system was truncated in a box with 15 Å between the edges of the periodic box and the molecules analyzed. An 8 Å cutoff particle mesh Ewald (PME) method was used to treat the long-range electrostatic interactions. AMBER molecular dynamics software and the ff19SB force field were used to prepare the systems and run the simulations.
Before running, the complexes were subjected to minimization in two stages using the steepest descent method followed by a conjugate gradient. In the first stage, the elements, peptides, and SIRT1 were fixed, and the position of the water molecules was minimized. After that, the whole system was minimized. A restrained thermalization phase with the microcanonical ensemble (NVE) was performed, increasing the temperature by 0.1 K until it reached 300 K using the Langevin thermostat and SHAKE algorithm (https://ambermd.org/) to constrain hydrogen bonds.
The production dynamics were carried out at constant pressure (NPT ensemble) with a time step of 2 fs. The total simulation time for each complex was 2700 ns, and frames were saved every 20 ps. The analyses were performed in the last 2650 ns after the systems were equilibrated. All calculations shown in the current work were performed using AMBERTOOLS21 scripts. The visualization programs used to create the figures of the molecules were VMD for LINUXAMD64, version 1.9.3, (31) and PYMOL (https://pymol.org/2/).
The surface plasmon resonance (SPR) experiments were performed on Biacore Model T200 equipment (GE Healthcare Biosciences, Uppsala, Sweden). For the immobilization of human SIRT1 (catalog number S8846, Sigma–Aldrich, Saint Louis, MO), a CM5 sensor chip S series (GE Healthcare Biosciences) has carboxylic groups available for covalent coupling was used. All solutions used were purchased from the equipment supplier (GE Healthcare Biosciences). The procedures described here were performed automatically with the Biacore instrument using pre-established programs for binding amines in peptides or proteins.
Briefly, the carboxylic groups on the CM5 sensor chip surface were activated with EDC + NHS, forming the NHS ester reactive intermediate, which, after washing with HBS-N, readily reacted with amines dissolved in the immobilization solution. The human SIRT1 ligand was dissolved in 10 mM sodium acetate pH 4.0 plus 20 µM NAD+ (catalog number N1636, beta-nicotinamide adenine dinucleotide hydrate, Sigma–Aldrich), which was immobilized onto the Biacore sensor chip according to the manufacturer´s instructions.
To detect interactions between SIRT1 and AIRE WT or AIRE G228W mutant SAND domain peptides, different concentrations of peptides were injected onto the CM5 chip with immobilized SIRT1 protein. Peptide samples were injected sequentially from lowest to highest concentrations (20, 100, and 200 μM) with a contact time of 300 s, a 20 μl/min flow, and a dissociation time of 1200 s. The surface was regenerated between samples by injecting 2.0 M glycine pH 2.0 (30 s, 30 μl/min) followed by injection of 2.0 M NaCl (30 s, 30 μl/min). The stabilization period between injections after regeneration was 200 s.
The respective sequences of the twenty-amino acid peptide residues (Synbio Technologies, Monmouth Junction, NJ, USA) comprising positions 211 to 230 of the SAND domain of the human AIRE protein are as follows: from the C to N-terminus, AIRE wild-type (IQQVFESGGSKKCIQVGGEF) and AIRE G228W mutant (IQQVFESGGSKKCIQVGWEF). The underlined K residue represents the position where the acetyl radical was added. The experiment determined whether the K residue underlined was acetylated or not. The AIRE wild-type or AIRE G228W mutation data are available through GenBank NCBI (https://www.ncbi.nlm.nih.gov/) accession numbers (NM_000383.4): c.682G>T G [GGG] > W [TGG], coding sequence variant autoimmune regulator (NP_000374.1): p. Gly228TrpG (Gly) > W missense variant, DbSNP (https://www.ncbi.nlm.nih.gov/snp/rs121434257) and ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/variation/3313/).
Kinetic data were analyzed with Anabel Analysis of Binding Events software (32), available at (www.skscience.org/anabel). Data obtained with the different concentrations of AIRE WT or mutant peptides were combined in the same graph.
To obtain a separate 3D human AIRE SAND domain structure, we used the SwissModel tool to model proteins through homology. We used the SAND domain sequence of the human SP100 protein (PDB code 1h5p) for the modeling, which features 39.62% sequence identity to the human AIRE SAND domain. The SP100 protein SAND domain conserves the SKKCIQ amino acid sequence in the AIRE SAND domain, corresponding to the accurate interaction site between SAND and the SIRT1 protein. Thus, obtaining a 3D structure of the wild-type AIRE SAND domain and the AIRE SAND domain carrying the G228W mutation was possible. Considering that the G228W mutation is located in a loop region of the AIRE SAND domain (Figure 1), we may infer that this substitution does not cause local protein misfolding.
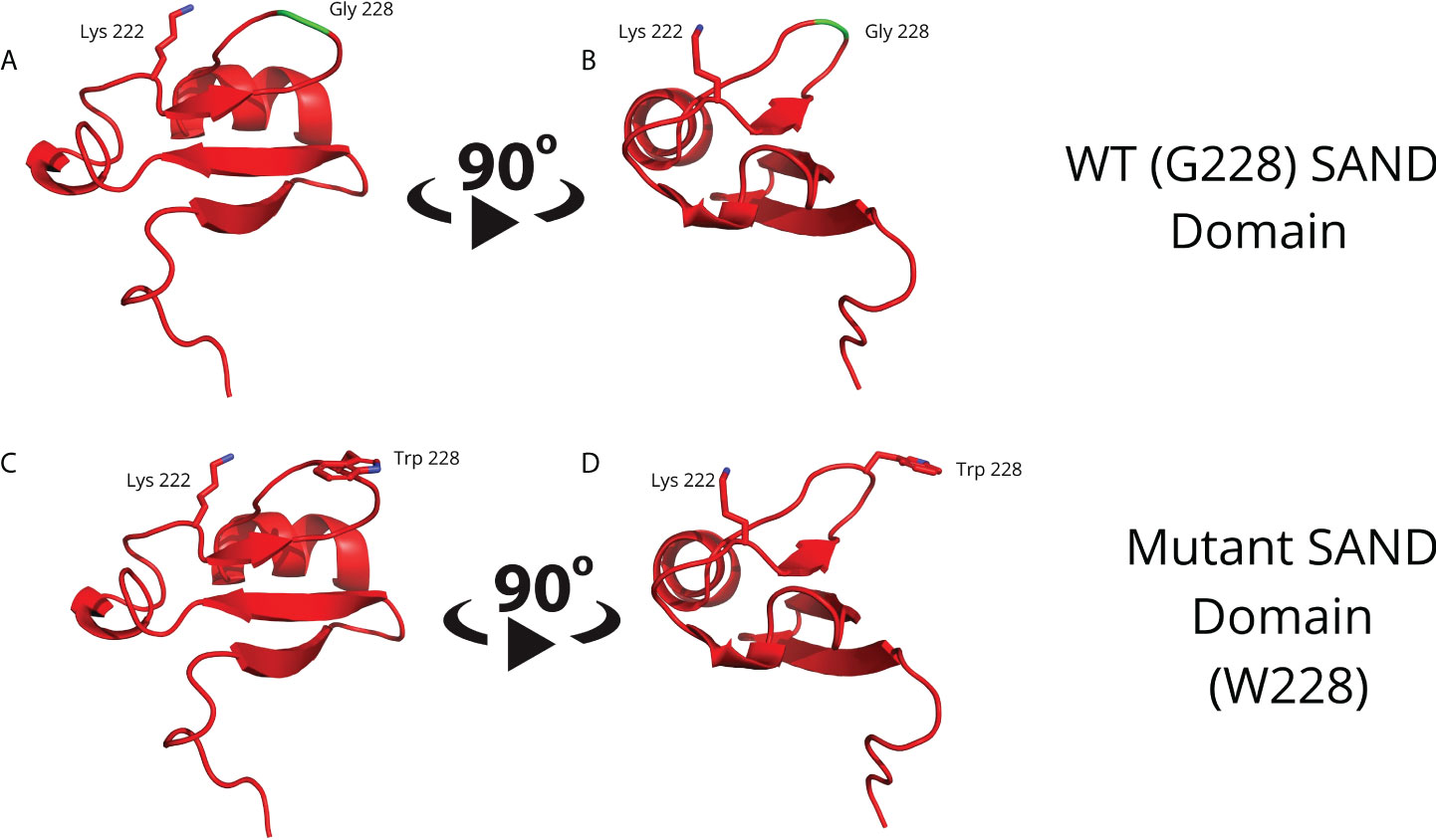
Figure 1 Molecular modeling of the G228 (Gly228) wild-type (A, B) or mutant W228 (Trp228) AIRE SAND domain (C, D) as acquired through homology modeling using the Swiss – Model with the human SP100 SAND domain as a template. The G228W mutation is located in a loop region and does not directly interfere with the formation of a secondary structure.
To obtain an interaction model between the AIRE SAND domain (wild-type or G228W mutant) and SIRT1 and assess the affinity of the mutant SAND domain, we used the ClusPro 2.0 blind molecular docking tool. The docking was performed with nonacetylated K222 to understand whether the modeled AIRE SAND domain would have an affinity for the catalytic region of the SIRT1 protein.
Through blind molecular docking, it was possible to observe an affinity between the AIRE SAND domain WT G228 or W228 mutant domain and the catalytic region/site (H363 residue) of the SIRT1 protein, even without any previous targeting or chemical modification of the K222 residue, which is a target of the SIRT1 protein (Figure 2). Important to note that when K222 is nonacetylated, the structure formed is incompatible with the expected native structure.
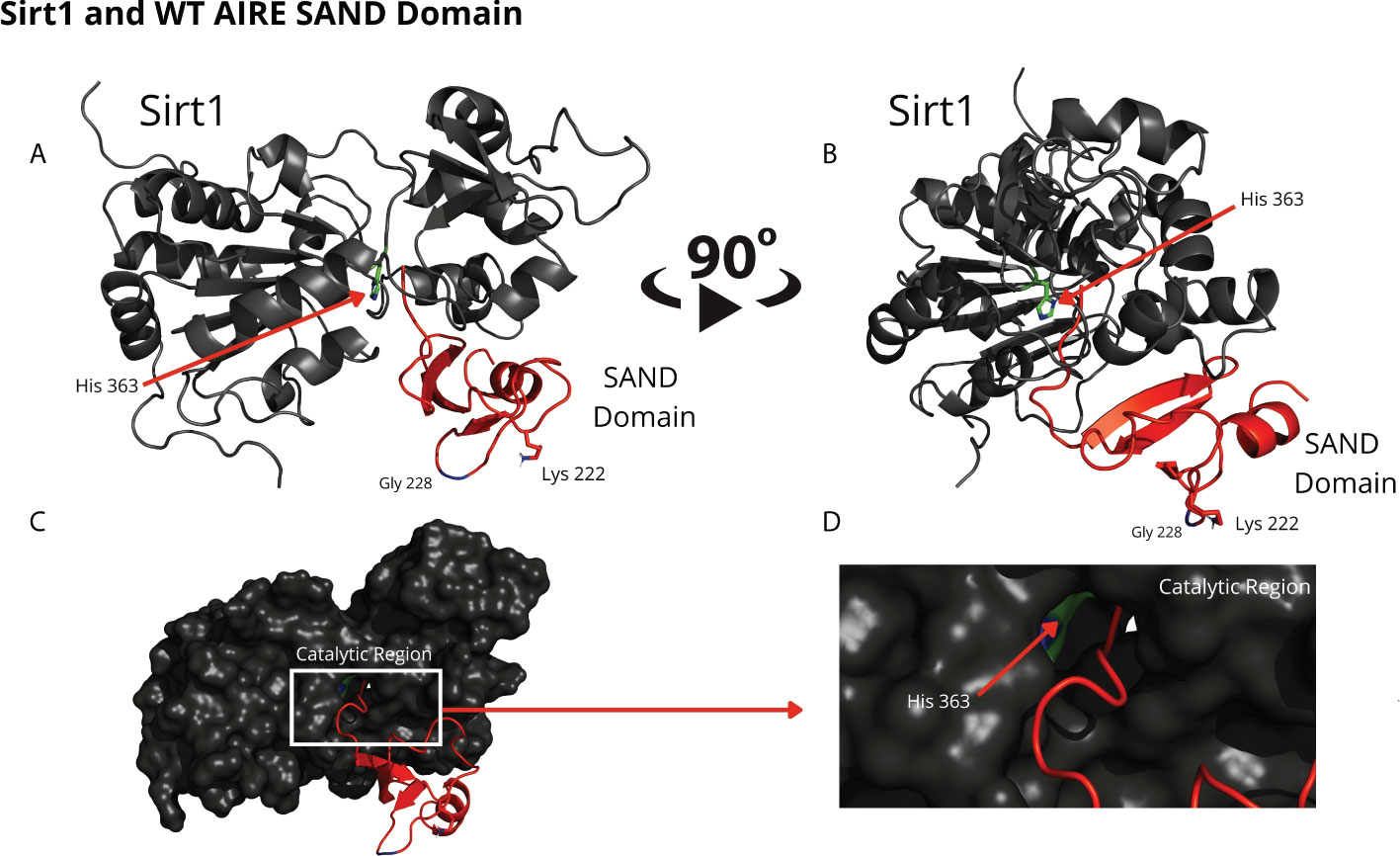
Figure 2 The blind docking output of the wild-type AIRE SAND domain shows that this domain interacts with the catalytic region of the SIRT1 protein. In (A, B) the structures of the WT SAND domain (in red) and SIRT1 (in dark gray) are represented. In this representation, the AIRE SAND domain interacts with the catalytic region through its terminal loop, and the glycine 228 (Gly228) and lysine 222 (Lys222) residues are presented with their side chains. Without the Lys222 acetylated, this formation is incompatible with the native structure of the complex formed with the complete AIRE protein, in which this loop does not exist. In (C, D) the same complex is represented with the surface of SIRT1 highlighted. (D) represents the catalytic region of SIRT1 and its catalytic residue, His363.
Comparing the structural conformations, we observed that the SAND domain mobilizes the W228 mutant residue that interacts with the SIRT1 H363 catalytic residue in the docking analyses (Figure 3).
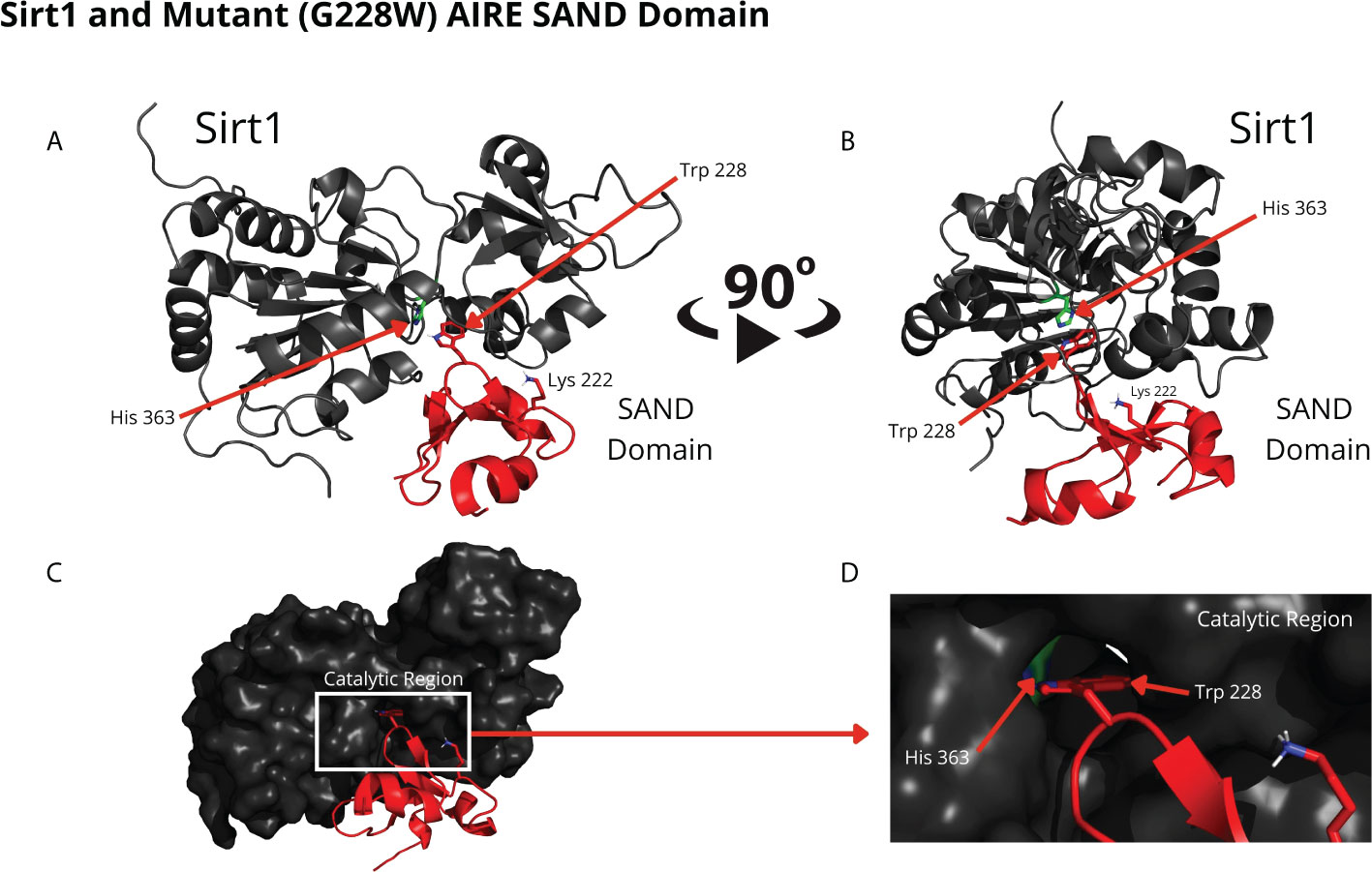
Figure 3 The blind docking output of the mutant AIRE SAND domain shows that this domain interacts with the catalytic region of the SIRT1 (His363 in green) protein through its mutant residue W228 (Trp228). The structures of the mutant SAND domain (in red) and SIRT1 (in dark gray) are represented in (A, B) In this representation, the mutant AIRE SAND domain interacts with the catalytic region through its mutant residue W228 (Trp228), and the lysine 222 (Lys222) residues are presented with its side chain. In (C, D) the same complex is represented with the surface of SIRT1 highlighted. (D) represents the catalytic region of SIRT1 and its catalytic residue, His363.
The molecular dynamics results with the AIRE SAND domain (wild-type or G228W mutant) showed that even the acetylated K222 residue at 1 µs of simulation did not allow the wild-type domain to form a stable structure, in which the K222 residue would interact with the H363 residue from the catalytic site of SIRT1. On the other hand, the G228W mutant domain exhibited a stable interaction with the SIRT1 catalytic site at the H363 residue. The analysis of the dynamics was performed to understand if K222 acetylation would be able to direct it to the catalytic residue of SIRT1.
The RMSD results showed that the WT SAND domain and SIRT1 complex is slightly less stable along the simulation time (1200 ns) than the complex formed between the SAND G228W mutant. Nevertheless, the G228W mutant domain complex remained stable in the RMSD [Figure 4, where higher angstrom distance values (Y-axis) indicate less stability]. The analysis of structures from molecular dynamics showed that even having inserted an acetyl radical at K222 residue in the WT or mutant domains, there was no targeting to the catalytic residue H363 of SIRT1 along the molecular dynamics simulation time. For molecular docking analyses, we used the SAND domain without chemical modification. Still, to understand the behavior of this domain as the SIRT1 protein recognizes it, we acetylated K222 for dynamic analysis (Figure 5).
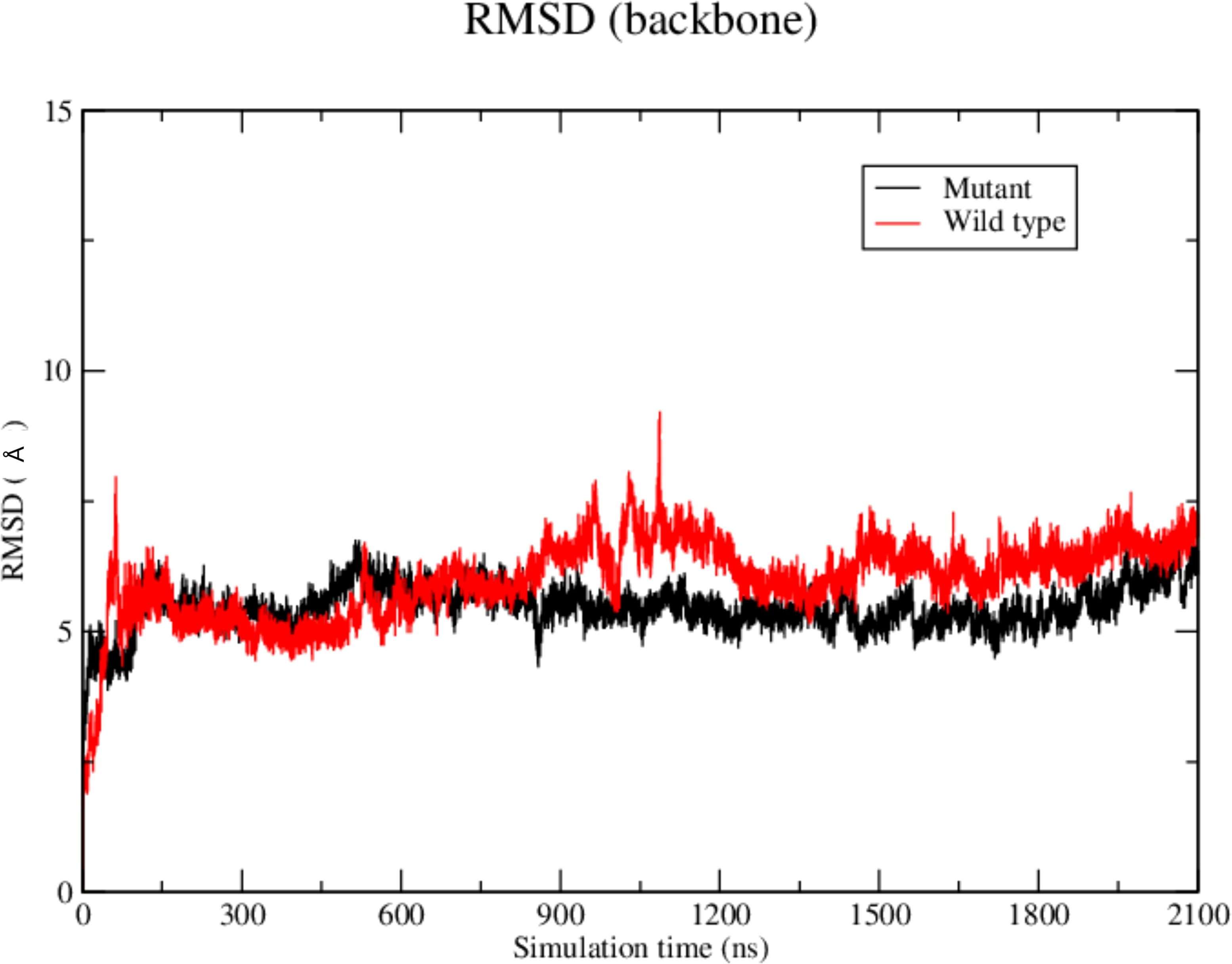
Figure 4 Root-mean square-deviation (RMSD) plot as a result of the molecular dynamics analysis of the wild-type (red) or mutant W228 (black) AIRE SAND domain with the SIRT1 protein. The RMSD plot shows that both WT and mutant SAND domains present a similar behavior along the first 600 ns. Then, the WT SAND domain exhibited a slight offset from 600 to 900 ns, and both complexes tended to stability at approximately 1200 ns.

Figure 5 Molecular dynamics structures formed between (A, B) the WT AIRE SAND domain (in red) and SIRT1 protein (in dark grey) and (C, D) between Mutant AIRE SAND domain (in red) and SIRT1 (in dark grey). (A, B) Note that the acetylated K222 residue from the AIRE SAND domain (in red) is not directed to the SIRT1 H363 catalytic residue (side chain in green) after the analyzed dynamics time (1.0 microsecond) (C, D). Considering the complex formed between the mutant AIRE SAND domain, at 1.0 microseconds of dynamics, the mutant residue W228, in the AIRE SAND domain, remained close to the catalytic H363 residue (side chain in green) of SIRT1.
Considering that, the results from molecular docking and molecular dynamics exhibited slight differences when comparing WT and mutant SAND domains during the formation of complexes with SIRT1, next, we performed surface plasmon resonance (SPR) to study the behavior of the two types of peptides when they bind to SIRT1. The SPR results indicated a differential association and dissociation dynamics between the wild-type SAND G228 or mutant SAND W228 peptides and the SIRT1 protein. The interaction kinetics showed that the wild-type SAND G228 peptide has a more significant association capacity as observed at a concentration range of 20 to 200 µM. The interaction between the wild-type G228 SAND peptide and SIRT1 was almost completely dissociated after 1200 seconds of measurement. Conversely, the mutant W228 SAND peptide was less able to associate with SIRT1 (Figure 6). These results, combined with the in silico analyses, show that the G228W mutation changes the SAND-SIRT1 interaction, which is necessary to activate the AIRE protein. Of note, SPR results were in contrast with the blind docking and molecular dynamics, i.e., instead, the WT complex was more stable and mutant less stable. The SPR results were reproduced with six different concentrations of each of the acetylated peptides, WT and mutant (range 10 to 200 µM), in addition to the control (0 µM) (Supplementary Figure 1). For clarity of the results, we chose three concentrations (20, 100 and 200 µM) in addition to the control (0 µM), whose results we plot the graph in Figure 6.
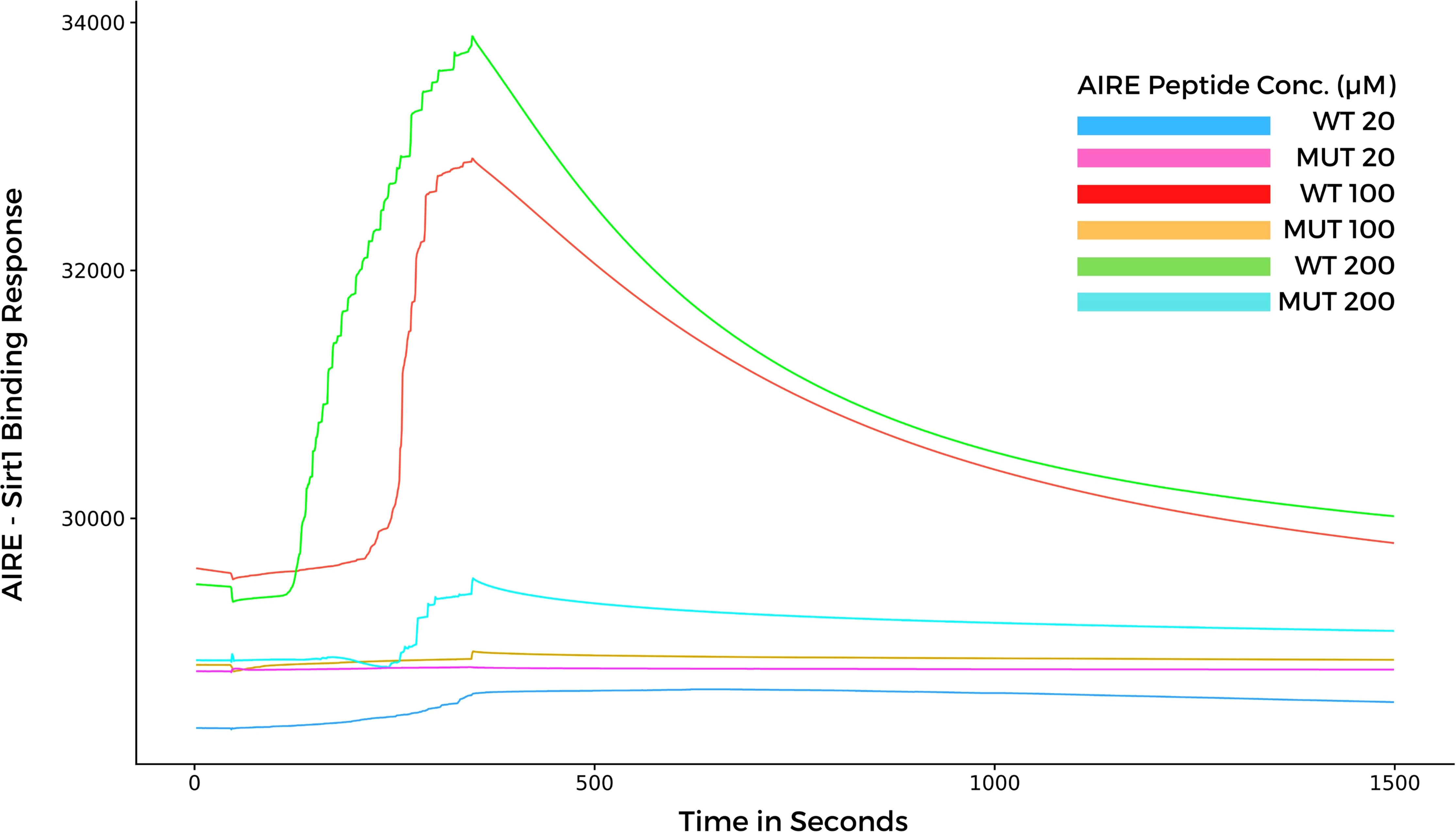
Figure 6 Surface plasmon resonance sensorgrams for binding the AIRE SAND domain with SIRT1 protein immobilized onto a Biacore sensor chip. The SAND domain peptides were assayed at various concentrations (20, 100, and 200 µM) with a contact time of 300 seconds, a 20 μl/min flow, and a dissociation time of 1200 seconds. Note that the WT AIRE SAND peptide (Gly228) associated and dissociated with SIRT1 throughout the analysis, while the mutant AIRE SAND peptide (Trp228) exhibited lesser association affinity with SIRT1 when compared to the WT peptide.
We reported that the dominant G228W mutation observed at the SAND domain of the human AIRE protein found in APS-1 patients (8) changes interaction with the deacetylase SIRT1 compared to the AIRE wild-type protein. Because of the interference of the W228 mutant residue, the kinetics of the association and dissociation processes differ compared to the wild-type, which might explain the nuclear accumulation of the AIRE complex, as observed in AIRE mutant cells. Since the mutant AIRE protein does not properly stabilize in the AIRE complex, it may abnormally accumulate in the nucleus, as previously observed (9). The binding/dissociation kinetics between AIRE-SIRT1 is crucial, and any disturbance in these processes may destabilize the catalytic action of SIRT1 for AIRE protein deacetylation. Thus, the model system employed in this work improves the understanding of the molecular mechanism of the G228W mutation.
The study of the molecular mechanisms of primary immunodeficiencies associated with the development of autoimmune disorders has contributed to understanding the mechanisms of tolerance loss to self-antigens. Although recessive AIRE mutations at the PHD1 domain may occur at a ratio of 1:1000 in the general population (10, 15), AIRE SAND G228W is the first dominant mutation associated with APS-1 (4, 8, 9). In the context of dominant inheritance, the central question is how these mutations influence the function of AIRE at the molecular level. Halonen et al. (24) and Ilmarinen et al. (9) observed that the G228W mutation quantitatively alters the nuclear location of the AIRE protein, impairing its transactivating property. Furthermore, Anderson and Su (15) demonstrated that this mutation interferes with the localization of the AIRE protein, reducing the transcription of peripheral tissue antigens (PTAs).
This study evaluated whether the G228W mutation changes the interaction with SIRT1, an essential partner protein of the AIRE complex. We accessed the 3D structure for the AIRE SAND domain by homology modeling to the human SP100 protein SAND domain, which is available in a public database. We observed that the G228W replacement, which occurs in a loop region, might not result in misfolding of the AIRE SAND 3D structure since loop regions do not present secondary structures (33, 34). The wild-type and the mutant AIRE SAND domain may not differ in their respective 3D structures, as performed by homology. This may be due to the segment’s characteristics in which the G228W mutation is present, taking the form of a flexible loop region (SAND amino acid residues 226-228). Therefore, the G228W mutation must involve another mechanism. Next, we asked whether the mutation could interfere with AIRE binding to one of its partner proteins. To initiate the downstream cascade of interactions, AIRE needs to be deacetylated, relieving RNA Pol II stalling in the chromatin. As SIRT-1 is the only deacetylase that integrates the AIRE complex, we next evaluated the binding of the AIRE SAND domain to SIRT1.
For this purpose, the structures of the wild-type SAND domain, the G228W mutant output from homology modeling, and the structure of the SIRT1 catalytic domain (PDB code 4I5I) were used. With the domains modeled, we used blind docking to understand how the complex between the SAND domains of AIRE WT and mutant would be formed with the SIRT1 protein.
We observed that the SAND domain mobilizes the W228 mutant residue that interacts with the SIRT1 H363 catalytic residue, changing with the interaction kinetics between these proteins, followed by SPR measurements. As observed in this study, due to the SAND G228W mutation, the AIRE protein might bind weakly to SIRT1. The AIRE complex, including RNA Pol II, formed with an AIRE mutant protein, might be dysfunctional and keep stalled to chromatin. The dysfunctional AIRE complex might occur due to the non-deacetylation of K222 residue, which is essential for the proper function of the AIRE protein. In addition, to signify a non-physiological situation, this finding might explain the accumulation of the AIRE complex in the nucleus of mutant cells, as previously reported (9, 24).
The AIRE protein plays a role in the cell nucleus as a tetramer (18, 19), in which the SAND domain of each subunit has K residues SIRT1 deacetylates. SIRT1 exerts its enzymatic activity, which depends on the amino acid residues near the acetylated K residues in the AIRE SAND domain (35). Accordingly, we hypothesized that the G228W mutation close to acetylated K residues could interfere with the SIRT1 interaction since it is an enzyme necessary to convert the acetylated to the nonacetylated and functional form of AIRE.
Although there is a prediction that the K222 residue is only moderately acetylated (27), previous work (26) showed that this amino acid residue is acetylated in the presence of the CBP and p300 II proteins, using mass spectrometry. Several lines of evidence stress the importance of K deacetylation in the proper interaction of AIRE with SIRT1: i) functional analyses of the G228W AIRE SAND mutation show that the subunit-containing acetylated K residue could form a hybrid tetramer unable to promote AIRE transactivation activity (9, 24), ii) AIRE mutations mimicking acetylated K243 or K253 residues in the SAND domain also reduced the transactivation activity and accumulated as nuclear bodies, whereas mutations mimicking nonacetylated K residue were functionally similar to wild-type AIRE (26), and iii) the AIRE SAND R247C mutation retains the ability to form complexes with wild-type AIRE and localize into the nucleus; however, it reduces the activation of downstream transcription through dominant-negative inhibition (36). Notably, the R247C mutation occurs close to other K residues (K243 and K253) in the SAND domain that SIRT1 can deacetylate, also harming the transactivation function of AIRE.
This approach confirmed the hypothesis raised in this study, in which we consider that the AIRE G228W mutation changes the interaction with SIRT1, mainly when evaluated using SPR analysis. Sensor-based SPR is a label-free, sensitive, precise, and in vitro tool used for determining binding kinetics between biological molecules, including antigen-antibody or protein-protein interactions, among other molecular interactions (37–40).
The complementary approaches used in this study (in silico and SPR) (30, 40) allowed us to evaluate the structure, energetic affinity, and association/dissociation kinetics of the complete AIRE SAND domain and peptides (wild-type and G228W mutation) with the SIRT1 protein. We observed that: i) both complete SAND domain can bind to the SIRT1 catalytic domain, ii) the binding kinetics of the G228W mutant peptide exhibited a weaker association when compared with its wild-type counterpart, and as a result, the wild-type SAND domain peptide bound faster and dissociated longer, and iii) taken together, these approaches provided evidence that the G228W mutation acts as a destabilizer for AIRE’s interaction with its partner SIRT1 and offers a more accurate molecular basis for clarifying how the mutant G228W AIRE protein loses its function.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
JS: raised hypothesis, discussed the analysis tools, performed molecular docking, analyzed data, interpreted the results, and wrote the manuscript. MD: discussed the analysis tools, performed molecular dynamics, analyzed data and interpreted the results. AM: performed protein binding kinetics (SPR), and interpreted the results. VF: performed protein binding kinetics (SPR), and interpreted the results. RB: performed molecular dynamics, analyzed data. ED: raised hypothesis, interpreted the results and wrote the manuscript. GP: conceived the study, raised hypothesis, organized the study pipeline, interpreted the results and wrote the manuscript. All authors contributed to the article and approved the submitted version.
This work was funded by the following Brazilian research support agencies; Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP, grant # 17/10780-4 to GAP and EAD), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, grants # 305787/2017-9 and # 311304/2021-4 to GAP, and # 302060/2019-7 to EAD), and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, Finance Code 001).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.948419/full#supplementary-material
Supplementary Figure 1 | Surface plasmon resonance sensorgram analysis of the binding between WT or G228W mutant AIRE SAND domain peptides with SIRT1 protein immobilized onto a Biacore sensor chip. Six different peptide concentrations were assayed (range 10, 15, 20, 50, 100, and 200 µM) plus the control (0 µM) for both WT and mutant peptides. Binding analysis shows a progressive increase in the association with the WT peptide, which was not observed with the mutant peptide.
1. Aaltonen J, Björses P, Sandkuijl L, Perheentupa J, Peltonen L. An autosomal locus causing autoimmune disease: autoimmune polyglandular disease type1 assigned to chromosome 21. Nat Genet (1994) 8:83–7. doi: 10.1038/ng0994-83.
2. Nagamine K, Peterson P, Scott HS, Kudoh J, Minoshima S, Heino M, et al. Positional cloning of the APECED gene. Nat Genet (1997) 17(4):393–8. doi: 10.1038/ng1297-393
3. Blechschmidt K, Schweiger M, Wertz K, Poulson R, Christensen HM, Rosenthal A, et al. The mouse aire gene: comparative genomic sequencing, gene organization, and expression. Genome Res (1999) 9:158–66. doi: 10.1101/gr.9.2.158
4. Wolff SB, Oftedal BE. Aire mutations and autoimmune diseases. In: Passos GA, editor. Thymus transcriptome and cell biology. Cham, Switzerland: Springer-Nature Switzerland AG. (2019).
5. Besnard M, Padonou F, Provin N, Giraud M, Guillonneau C. AIRE deficiency, from preclinical models to human APECED disease. Dis Model Mech (2021) 14(2):dmm046359. doi: 10.1242/dmm.046359
6. Finnish-German APECED Consortium. An autoimmune disease, APECED, caused by mutations in a novel gene featuring two PDH-type zinc-finger domains. Nat Genet (1997) 17(4):399–403. doi: 10.1038/ng1297-399
7. Oftedal BE, Hellesen A, Erichsen MM, Bratland E, Vardi A, Perheentupa J, et al. Dominant mutations in the autoimmune regulator AIRE are associated with common organ-specific autoimmune diseases. Immunity (2015) 42:1185–96. doi: 10.1016/j.immuni.2015.04.021
8. Cetani F, Barbesino G, Borsari S, Pardi E, Cianferotti E, Pinchera A, et al. A novel mutation of the autoimmune regulator gene in an Italian kindred with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy, acting in a dominant fashion and strongly cosegregating with hypothyroid autoimmune thyroiditis. J Clin Endocrinol Metab (2001) 86:4747–52. doi: 10.1210/jcem.86.10.7884
9. Ilmarinen T, Eskelin P, Halonen M, Ruppell T, Kilpikari R, Torres GD, et al. Functional analysis of SAND mutations in AIRE supports dominant inheritance of the G228W mutation. Hum Mutat (2005) 26(4):322–31. doi: 10.1002/humu.20224
10. Su MA, Giang K, Zumer K, Jiang H, Oven I, Rinn JL, et al. Mechanisms of an autoimmunity syndrome in mice caused by a dominant mutation in aire. J Clin Invest (2008) 118(5):1712–26. doi: 10.1172/JCI34523
11. Giraud M, Yoshida H, Abramson J, Rahl PB, Young RA, Mathis D, et al. Aire unleashes stalled RNA polymerase to induce ectopic gene expression in thymic epithelial cells. Proc Natl Acad Sci USA (2012) 109:535–40. doi: 10.1073/pnas.1119351109
12. Passos GA, Speck-Hernandez CA, Assis AF, Mendes-da-Cruz DA. Update on aire and thymic negative selection. Immunology (2018) 153:10–20. doi: 10.1111/imm.12831
14. Giraud M, Peterson P. The autoimmune regulator (AIRE) gene, the master activator of self-antigen expression in the thymus. In: Passos GA, editor. Thymus transcriptome and cell biology. Cham, Switzerland: Springer-Nature Switzerland AG. (2019).
15. Anderson MS, Su MA. AIRE expands: new roles in immune tolerance and beyond. Nat Rev Immunol (2016) 16:247–58. doi: 10.1038/nri.2016.9
16. Kumar PG, Laloraya M, Wang CY, Ruan QG, Davoodi-Semiromi A, Kao KJ, et al. The autoimmune regulator (AIRE) is a DNA-binding protein. J Biol Chem (2001) 276:41357–64. doi: 10.1074/jbc.M104898200
17. Perniola R, Musco G. The biophysical and biochemical properties of the autoimmune regulator (AIRE) protein. Biochim Biophys Acta (2014) 1842:326–37. doi: 10.1016/j.bbadis.2013.11.020
18. Incani F, Serra M, Meloni A, Cossu C, Saba L, Cabras T, et al. AIRE acetylation and deacetylation: effect on protein stability and transactivation activity. J BioMed Sci (2014) 27:21–85. doi: 10.1186/s12929-014-0085-z
19. Abramson J, Giraud M, Benoist C, Mathis D. Aire’s partners in the molecular control of immunological tolerance. Cell (2010) 140(1):123–35. doi: 10.1016/j.cell.2009.12.030
20. Abramson J, Husebye ES. Autoimmune regulator and self-tolerance – molecular and clinical aspects. Immunol Rev (2016) 271:127–40. doi: 10.1111/imr.12419
21. Dai H, Case AW, Riera TV, Considine T, Lee JE, Hamuro Y, et al. Crystallographic structure of a small molecule SIRT1 activator-enzyme complex. Nat Commun (2015) 6:7645. doi: 10.1038/ncomms8645
22. Glozak MA, Sengupta N, Zhang X, Seto E. Acetylation and deacetylation of non-histone proteins. Gene (2005) 363:15–23. doi: 10.1016/j.gene.2005.09.010
23. Ramsey C, Bukrinsky A, Peltonen L. Systematic mutagenesis of the functional domains of AIRE reveals their role in intracellular targeting. Hum Mol Genet (2002) 11:3299–308. doi: 10.1093/hmg/11.26.3299
24. Halonen M, Kangas H, Rüppell T, Ilmarinen T, Ollila J, Kolmer M, et al. APECED-causing mutations in AIRE reveal the functional domains of the protein. Hum Mutat (2004) 23:245–57. doi: 10.1002/humu.20003
25. Vaquero A, Scher M, Lee D, Erdjument-Bromage H, Tempst P, Reinberg D. Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol Cell (2004) 16:93105. doi: 10.1016/j.molcel.2004.08.031
26. Saare M, Rebane A, Rajashekar B, Vilo J, Peterson P. Autoimmune regulator is acetylated by transcription coactivator CBP/p300. Exp Cell Res (2012) 15:1767–78. doi: 10.1016/j.yexcr.2012.04.013
27. Chuprin A, Avin A, Goldfarb Y, Herzig Y, Levi B, Jacob A, et al. The deacetylase Sirt1 is an essential regulator of aire-mediated induction of central immunological tolerance. Nat Immunol (2015) 16:737–45. doi: 10.1038/ni.3194
28. Bheda P, Jing H, Wolberger C, Lin H. The substrate specificity of sirtuins. Annu Rev Biochem (2016) 85:405–29. doi: 10.1146/annurev-biochem-060815-014537
29. Alekseenko A, Ignatov M, Kozakov D. Protein-protein and protein-peptide docking with ClusPro server. Methods Mol Biol (2020) 2165:157–74. doi: 10.1007/978-1-0716-0708-4_9
30. Case DA, Aktulga HM, Belfon K, Ben-Shalom IY, Berryman JT, Brozell SR, et al. AMBER. San Francisco: University of California (2022). Available at: https://ambermd.org/AmberTools.php.
31. Humphrey W, Dalke A, Schulten K. VMD - visual molecular dynamics. J Molec Graphics (1996) 14:1:33–38. doi: 10.1016/0263-7855(96)00018-5
32. Krämer SD, Wöhrle J, Rath C, Roth G. Anabel: An online tool for the real-time kinetic analysis of binding events. Bioinform Biol Insights (2019) 9:13:1177932218821383. doi: 10.1177/1177932218821383
33. Donate LE, Rufino SD, Canard LH, Blundell TL. Conformational analysis and clustering of short and medium size loops connecting regular secondary structures: a database for modeling and prediction. Protein Sci (1996) 12:2600–16. doi: 10.1002/pro.5560051223
34. Bottomley MJ, Collard MW, Huggenvik JI, Liu Z, Gibson TJ, Sattler M. The SAND domain structure defines a novel DNA-binding fold in transcriptional regulation. Nat Struct Biol (2001) 8:626–33. doi: 10.1038/89675
35. Garske AL, Denu JM. SIRT1 top 40 hits: use of one-bead, one-compound acetyl-peptide libraries and quantum dots to probe deacetylase specificity. Biochemistry (2006) 45(1):94–101. doi: 10.1021/bi052015l
36. Abbott JK, Huoh YS, Reynolds PR, Yu L, Rewers M, Reddy M, et al. Dominant-negative loss of function arises from a second more frequent variant within the SAND domain of autoimmune regulator (AIRE). J Autoimmun (2018) 88:114–20. doi: 10.1016/j.jaut.2017.10.010
37. Nguyen HH, Pank J, Kang S, Kim M. Surface plasmon resonance: a versatile technique for biosensor applications. Sensors (2015) 15(5):10481–510. doi: 10.3390/s150510481
38. Wang D, Loo JF, Chen J, Yam Y, Chen S-C, He H, et al. Recent advances in surface plasmon resonance imaging sensors. Sensors (2019) 19(6):1266. doi: 10.3390/s19061266
39. Soltermann F, Struwe WB, Kukura P. Labe-free methods for optical in vitro characterization of protein-protein interactions. Phys Chem Chem Phys (2021) 23(31):16488–500. doi: 10.1039/d1cp01072g
Keywords: AIRE, G228W mutation, AIRE-SIRT1 interaction, APECED syndrome, APS1/APECED, molecular modeling, surface plasmon resonance, docking
Citation: Santos JC, Dametto M, Masson AP, Faça VM, Bonacin R, Donadi EA and Passos GA (2022) The AIRE G228W mutation disturbs the interaction of AIRE with its partner molecule SIRT1. Front. Immunol. 13:948419. doi: 10.3389/fimmu.2022.948419
Received: 19 May 2022; Accepted: 11 August 2022;
Published: 06 September 2022.
Edited by:
Peter S. Linsley, Benaroya Research Institute, United StatesReviewed by:
Karen Cerosaletti, Benaroya Research Institute, United StatesCopyright © 2022 Santos, Dametto, Masson, Faça, Bonacin, Donadi and Passos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Geraldo Aleixo Passos, cGFzc29zQHVzcC5icg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.