 Agnieszka Witalisz-Siepracka
Agnieszka Witalisz-Siepracka Klara Klein
Klara Klein Bernhard Zdársky
Bernhard Zdársky Dagmar Stoiber
Dagmar Stoiber
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 05 July 2022
Sec. Cancer Immunity and Immunotherapy
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.947568
This article is part of the Research Topic Innate Lymphocytes in Tumor Surveillance View all 10 articles
Signal transducer and activator of transcription 3 (STAT3) is a member of the Janus kinase (JAK)-STAT pathway, which is one of the key pathways contributing to cancer. STAT3 regulates transcription downstream of many cytokines including interleukin (IL)-6 and IL-10. In cancer, STAT3 is mainly described as a tumor promoter driving tumor cell proliferation, resistance to apoptosis, angiogenesis and metastasis and aberrant activation of STAT3 is associated with poor prognosis. STAT3 is also an important driver of immune evasion. Among many other immunosuppressive mechanisms, STAT3 aids tumor cells to escape natural killer (NK) cell-mediated immune surveillance. NK cells are innate lymphocytes, which can directly kill malignant cells but also regulate adaptive immune responses and contribute to the composition of the tumor microenvironment. The inborn ability to lyse transformed cells renders NK cells an attractive tool for cancer immunotherapy. Here, we provide an overview of the role of STAT3 in the dynamic interplay between NK cells and tumor cells. On the one hand, we summarize the current knowledge on how tumor cell-intrinsic STAT3 drives the evasion from NK cells. On the other hand, we describe the multiple functions of STAT3 in regulating NK-cell cytotoxicity, cytokine production and their anti-tumor responses in vivo. In light of the ongoing research on STAT3 inhibitors, we also discuss how targeting STAT3 would affect the two arms of STAT3-dependent regulation of NK cell-mediated anti-tumor immunity. Understanding the complexity of this interplay in the tumor microenvironment is crucial for future implementation of NK cell-based immunotherapies.
Natural killer (NK) cells belong to the group 1 innate lymphoid cells and are characterized by the ability to kill virally infected and malignant cells. In contrast to T cells, NK cells do not require major histocompatibility complex (MHC)-dependent priming by antigen presenting cells. The activity of NK cells is regulated by a delicate balance of germ-line encoded activating and inhibitory receptors. Upon recognition of the target cell, an NK cell releases cytotoxic granules for direct cell lysis as well as produces immunomodulatory cytokines (1, 2). In humans, these tasks are fulfilled by different subtypes of NK cells: CD56brightCD16lo/- NK cells are main producers of cytokines such as interferon (IFN) γ. In contrast, CD56dimCD16+ NK cells are highly cytotoxic, but do not produce substantial amounts of IFNγ (3). The inborn ability to lyse transformed cells renders NK cells an attractive tool for cancer immunotherapy with a potentially better safety profile compared to T cells (4, 5). Different approaches to exploit NK cells in immunotherapy are being investigated. These include adoptive transfer of cytokine-induced memory-like NK cells or chimeric antigen receptor NK (CAR-NK) cells. Currently, numerous clinical trials using such approaches are ongoing, but the efficacy of these treatments still needs to be evaluated (5, 6). In this context, understanding the complex processes employed by tumor cells to evade NK-cell immunity is crucial. These escape mechanisms include transcriptional downregulation and shedding of ligands for NK-cell activating receptors, upregulation of inhibitory ligands, as well as immune suppressive signals derived from the microenvironment (7–11). Signal transducer and activator of transcription 3 (STAT3) is constitutively activated in various cancers and plays a pivotal role in regulating all of these processes and thereby mediates the crosstalk between the tumor microenvironment and immune cells (12).
STAT3 is a member of the Janus kinase (JAK)-STAT signaling pathway, which coordinates central cellular mechanisms including differentiation, development, proliferation, immune function, or apoptosis (13, 14). The alternatively spliced STAT3 isoforms, full-length STAT3α and C-terminally-truncated STAT3β, have opposing function during tumor development. While STAT3α promotes tumor growth, STAT3β was identified as a tumor suppressor and favorable prognostic marker in cancers of different origin (15, 16). Mechanistically, JAK-STAT3 signaling is activated by diverse growth factors, peptide hormones and all interleukin (IL)-6-type cytokines including IL-6, IL-11, IL-27, IL-31, leukemia inhibitory factor (LIF), oncostatin M (OSM), ciliary neurotrophic factor (CNTF) neuropoietin (NP), cardiotrophin-1 (CT-1) and cardiotrophin-like cytokine (CLC) (17–19). IL-6 family cytokines, except for IL-31 which exerts its effects through IL-31 receptor α, induce signaling via binding to either a glycoprotein 130 (gp130) receptor β- subunit hetero- or homodimer (19, 20). Ligands bind to their cognate receptors, which undergo a conformational change, and induce subsequent activation of receptor associated JAKs (JAK1, JAK2, JAK3 and tyrosine kinase 2 (TYK2)) by autophosphorylation and/or transphosphorylation. The JAK-induced tyrosine phosphorylation of the receptor provides a docking-site for the SH2 domain of STAT3, which in turn gets phosphorylated on tyrosine 705 by JAKs (21). Activated STAT3 forms anti-parallel to parallel homo- or heterodimers with other STATs, is released from the receptor and translocates into the nucleus through interaction with importin-β1 (22, 23). To control gene expression, activated STATs target palindromic consensus sequences located in promoter and enhancer regions and in the first introns of target genes (24). Negative regulation of STAT3 occurs in the nucleus through antagonization by PIAS (protein inhibitor of activated STAT), an E3 SUMO-protein ligase, or at the receptor by SOCS (suppressor of cytokine signaling) E3 ubiquitin ligases (25, 26) (Figure 1). The transcriptional activity of STAT3 can be further regulated by phosphorylation at serine 727 (Ser727) mediated by mTOR, p38, ERK and other serine/threonine kinases. However, the exact effects of Ser727 phosphorylation have to be put in a cellular and/or promoter dependent context. Phosphorylated Ser727 promotes association with different transcription co-factors and thus activates or diminishes transcriptional responses of STAT3 (27). Moreover, it can also drive the mitochondrial metabolic activity of STAT3 and augment the electron transport chain (28). Activated STAT3 in cancer cells contributes to oxidative and glycolytic phosphorylation, survival, epithelial-to-mesenchymal transition, proliferation, metastasis, and radiation- and chemotherapy resistance (29, 30). Due to its central contribution to several hallmarks of cancer and its association with poor clinical prognosis, STAT3 represents a promising therapeutic target for cancer therapy (14, 31, 32).
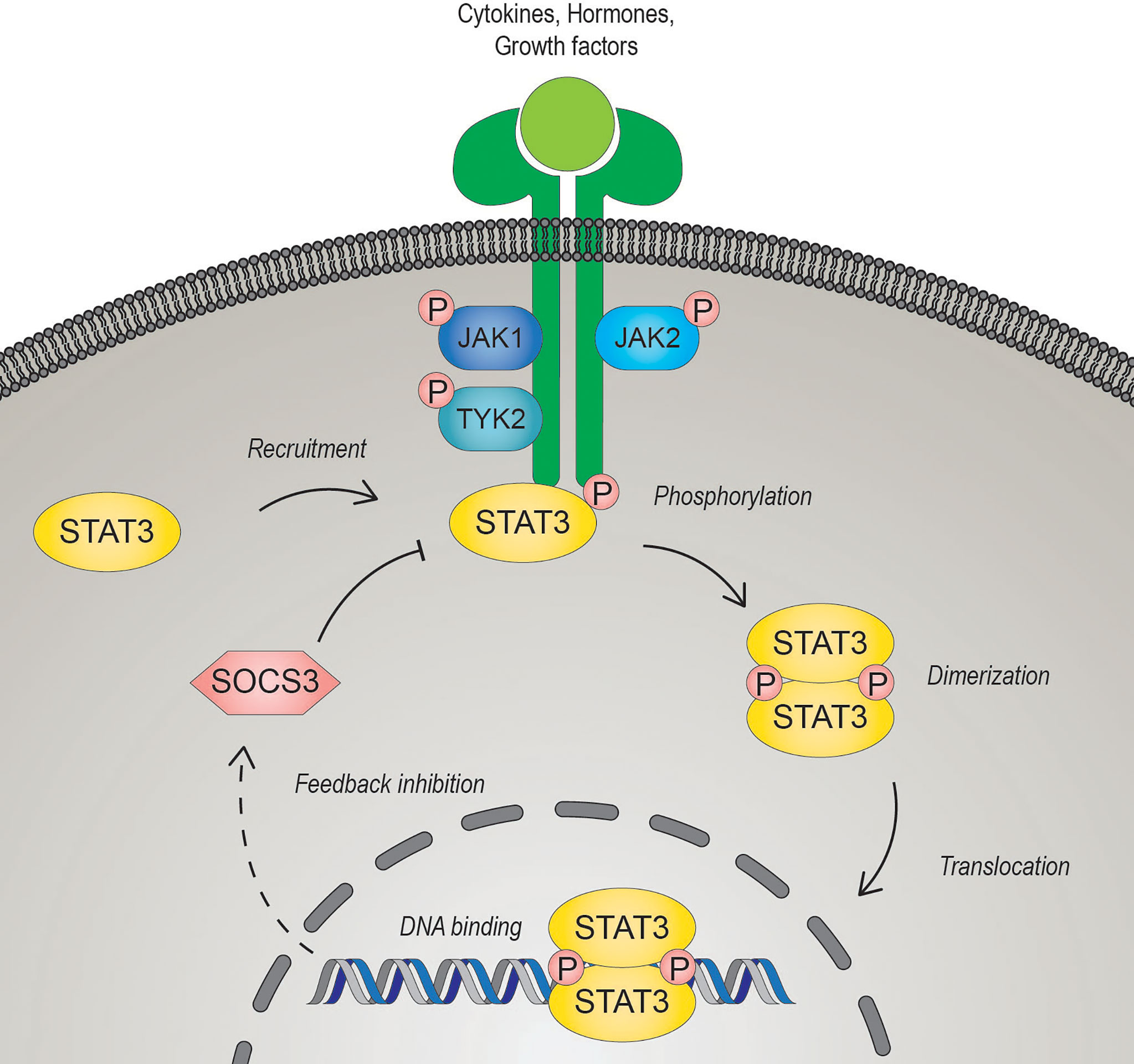
Figure 1 The JAK-STAT3 signaling pathway. Signal transducer and activator of transcription 3 (STAT3) is activated upon binding of diverse cytokines, hormones, or growth factors to their cognate receptors. Ligand-bound receptors undergo conformational changes leading to the activation of the Janus kinases (JAK). Activated JAKs trans- and/or auto- phosphorylate each other and the cytoplasmic domain of the receptor, enabling STAT3 to bind via its SRC homology 2 (SH2) domain. JAK-mediated phosphorylation of a conserved C-terminal tyrosine residue of STAT3 induces dimerization of phosphorylated STAT3 and the subsequent translocation to the nucleus to regulate gene transcription. STAT3 induces transcription of suppressor of cytokine signaling 3 (SOCS3), which can act as a negative regulator by interacting competitively with the receptor.
Inhibition of STAT3 signaling is currently explored in many clinical trials for solid and hematopoietic tumors. The direct approaches to specifically inhibit STAT3 include small molecules and decoy oligonucleotides (33). The most successful small molecule STAT3 inhibitor Napabucasin (BBI-608), which selectively binds to the DNA-binding domain of STAT3, has reached phase III trials for advanced colorectal cancer and provided excellent results as monotherapy (34). Another small molecule, TTI-101, targets the receptor binding site within the SH2 domain of STAT3 to block its recruitment and activation (35). Phase I clinical trials in advanced solid cancers including breast cancer are ongoing (NCT03195699, NCT05384119). The SH2 domain is also targeted by other small molecules, OPB- 51602, OPB-31121, OPB-111077, which are undergoing phase I/II clinical trials for solid and in case of OPB-51602 also hematopoietic tumors (reviewed in (36)). The antisense oligonucleotide, AZD9150, which is designed to target STAT3 mRNA (37), has until now reached phase I/II trials for different advanced solid cancers (e.g. NCT01839604; NCT01839604) (33). Although the specificity and potency of such antisense oligonucleotides is very promising, they face problems of efficient penetration of solid tumors and fast degradation (33). To the best of our knowledge, the STAT3 inhibitors currently tested in clinical trials have not been thoroughly studied in the context of NK cell anti-tumor responses. Here, we summarize the current knowledge on how STAT3 contributes to NK-cell fitness and tumor cell evasion from NK cells, and speculate on how targeting STAT3 may affect NK-cell tumor surveillance.
NK cells exhibit cytolytic activity towards cells that overexpress ligands for activating receptors and/or lack the expression of MHC class I and other ligands, recognized by inhibitory receptors. In healthy cells, the ligands for activating receptors are absent or expressed at very low levels. Transformed cells upregulate these ligands and become sensed by NK cells as ‘danger’ (1, 38). The activating NK-cell receptor natural killer group 2D (NKG2D) served as a paradigm in studying this mechanism. In humans, NKG2D binds to MHC class I polypeptide-related sequence A (MICA) and B (MICB) and UL16-binding proteins (ULBP1-6) (39–42). In mice, NKG2D ligands comprise of the RAE1 family (α-ϵ), H60 (a -c), and MULT1 (43–45). Upon binding of a ligand to NKG2D, the co-stimulatory molecule DAP10 is activated and the release of cytotoxic granules is induced (40). Natural cytotoxicity receptor (NCR) NKp30 recognizes the B7-H6 molecule on transformed cells and induces NK-cell activation (46). Other receptors of this family are NKp44 and NKp46, which recognize heterogenous ligands including viral and bacterial proteins (47). Further activating NK-cell receptors function more as amplifiers of NK-cell activation triggered by NKG2D or NCRs (48). An important example is DNAM-1 and its corresponding ligands CD112 and CD155 often overexpressed on tumor cells (49, 50).
NKG2D is crucially involved in NK cell-mediated tumor surveillance and is one of the best studied receptors in this context. Mice deficient in NKG2D show strong defects in immune surveillance of epithelial and lymphoid tumors (51). In line, high expression of NKG2D ligands in leukemic patients correlates with better survival (52). Absence of NKG2D ligands is also a feature of leukemic stem cells, which allows them to escape NK-cell surveillance in acute myeloid leukemia (AML) in vivo models (53). Tumor cells evade NKG2D-mediated recognition by downregulation or shedding of the ligands. Not only does it allow to hide from NK-cell cytotoxicity, but also leads to desensitization of NKG2D-mediated NK-cell activation. High levels of shed NKG2D ligands result in downregulation of NKG2D-mediated signaling (54, 55).
STAT3 has been implicated in direct transcriptional repression of NKG2D ligands. Bedel et al., revealed that inhibition or knockdown of STAT3 in the colorectal cancer cell line HT29 leads to stronger activation of NK cells and therefore killing of tumor cells in an NKG2D-dependent manner. Further, they could show that STAT3 directly binds to the MICA promoter, repressing its transcription (56). A similar mechanism has been described in multiple myeloma (MM) cell lines. Upon treatment with glycogen synthase kinase 3 (GSK-3) inhibitor, MM cell lines showed decreased STAT3 activation and reduced STAT3 binding to the MICA promoter. The effects corresponded to enhanced sensitivity of treated cell lines to NK cell-mediated lysis. Importantly, GSK-3 inhibition had no effect on MICA expression in cell lines with constitutively active STAT3. In line, the GSK-3 inhibitor was not able to reduce the activation level of constitutively active STAT3. This strongly suggests that GSK-3-induced susceptibility of MM cells to NK cells is greatly dependent on inhibiting STAT3 activation (57). In another study using colorectal cancer cells, GSK-3 inhibition significantly upregulated NKG2D ligands and increased their sensitivity to NK cells. However, in this context the dependence on STAT3 remains to be elucidated (58).
The correlative observations of low STAT3 activity and/or expression and high NKG2D ligands surface levels have also been made in other cancer entities. The adriamycin-resistant chronic myeloid leukemia (CML) cell line K562 is killed more efficiently by NK cells upon treatment with a STAT3 inhibitor and shows an upregulation of MICA and ULBP2 (59). STAT3 inhibition or silencing also enhances ULBP2 expression in parental K562 cells (60). In AML cell lines, an inverse correlation between phosphorylated STAT3 (pSTAT3) levels and MICA expression was observed after rapamycin (61) or decitabine treatment (62) but the mechanism behind it remains unclear. In hepatocellular carcinoma cell lines, a STAT3 decoy resulted in upregulation of NKG2D ligands and increase of NK cell-mediated killing (63). A layer of complexity is added by the fact that in human gastric adenocarcinoma cell lines inhibition of STAT3 results in upregulation of MICB on the cell surface as well as of the soluble ligands. This implies a potential desensitization of NK cells driven by inhibition of STAT3 (64). Although all the mentioned studies point towards a similar effect of STAT3 inhibition or silencing, the interpretation is limited, as all of the experiments where only performed in vitro. A robust in vivo xenograft model of STAT3-deficient tumor cell lines with adoptive transfer of human primary NK cells would be necessary to further elucidate the impact of STAT3 on NKG2D-mediated NK-cell surveillance.
The missing-self hypothesis, formulated in the 1980s, states that NK cells kill those cells that do not express sufficient levels of MHC I. In line, several classes of inhibitory receptors were discovered, which unleash NK-cell cytotoxicity upon downregulation of MHC I on the target cells (65, 66). These include the Ly49 family in mice and the KIR family in humans (67–69). Ly49 and KIRs sense the levels of conventional MHC I, therefore, tumor cells that downregulate MHC I molecules to escape T cell responses become targets for NK cells (70, 71). In a mouse model of carcinogen-induced Non-Small Cell Lung Cancer (NSCLC), epithelial cell-specific knockout of Stat3 led to downregulation of MHC I on transformed epithelial cells. This rendered the emerging cancer cells more susceptible to NK cell-mediated lysis (72).
STAT3 is considered as a driver of an immune suppressive tumor microenvironment. STAT3 activation is associated with high expression of tumor promoting cytokines and growth factors such as IL-10, transforming growth factor (TGF)-β and vascular endothelial growth factor (VEGF)-A. The majority of STAT3-dependent effects in the tumor microenvironment are described in the context of T cells, macrophages or dendritic cells that have been extensively reviewed by others (12, 73). However, the soluble immune suppressive modulators present in tumor microenvironment not only suppress the function of NK cells, but also impair their infiltration into the tumor (13) or even confer a switch towards pro-tumorigenic, VEGF-A-producing NK cells (74–77). The IL-10/STAT3 axis directly drives VEGF-A expression in these cells (77). TGF-β in the microenvironment also drives the conversion of cytotoxic NK cells (CD49a-CD49b+EOMES+) into ILC1 (CD49a+CD49b-EOMESint), which lose the ability to control the tumor growth (78).
In the previously mentioned, carcinogen-induced NSCLC model with epithelial cell-specific Stat3 knockout, the tumor microenvironment is enriched for proinflammatory cytokines which might contribute to enhanced NK-cell responses against the tumor (72). In support of this finding, hepatocellular carcinoma cells treated with STAT3 decoy secrete higher levels of IFNs and lower levels of immune suppressive TGF-β. Upon culture in conditioned medium derived from hepatocellular carcinoma cells pre-treated with STAT3 decoy, NK cells showed a more activated phenotype with higher expression of IFNγ, granzyme and perforin (63). In line, STAT3 in a murine melanoma cell line was shown to inhibit the expression of proinflammatory cytokines such as TNFα and IL-12 and the chemokine CCL5. Inhibition of STAT3 signaling was associated with increased levels of CCL5 and thereby enhanced lymphocyte infiltration into the tumor (79). Importantly, the conclusions were made in melanoma model overexpressing the alternatively spliced, truncated isoform of STAT3 – STAT3β, which was believed to have dominant negative functions over the full length isoform (80). However, several studies have shown that STAT3β is transcriptionally active and drives expression of its unique target genes (81–83). The potential of STAT3β to drive cytokines and chemokines that support the immune system is in accordance with the tumor suppressive potential of STAT3β-overexpressing macrophages in breast cancer (84).
Interestingly, in BCR-ABL-driven lymphoma, the deletion of STAT3 has opposite effects to those described above. STAT3-deficient B cell lymphoma shows decreased expression of proinflammatory cytokines, e.g. TNFα and chemokine CCL5. This is paralleled by a lower abundance of NK cells in the tumors. Transplantation of the lymphoma cells lacking STAT3 into mice harboring NK cells results in accelerated tumor growth, but the difference is lost in immune-deficient mice. The study postulates that targeting STAT3 in BCR-ABL-driven malignancies might impair NK-cell surveillance (85).
In summary, STAT3 activity is critically implicated in determining the outcome of cancer immunity by orchestrating the release of immunomodulating cytokines. In the majority of cases, inhibition of STAT3 signaling switches the tumor microenvironment towards immune activation (Figure 2, right).
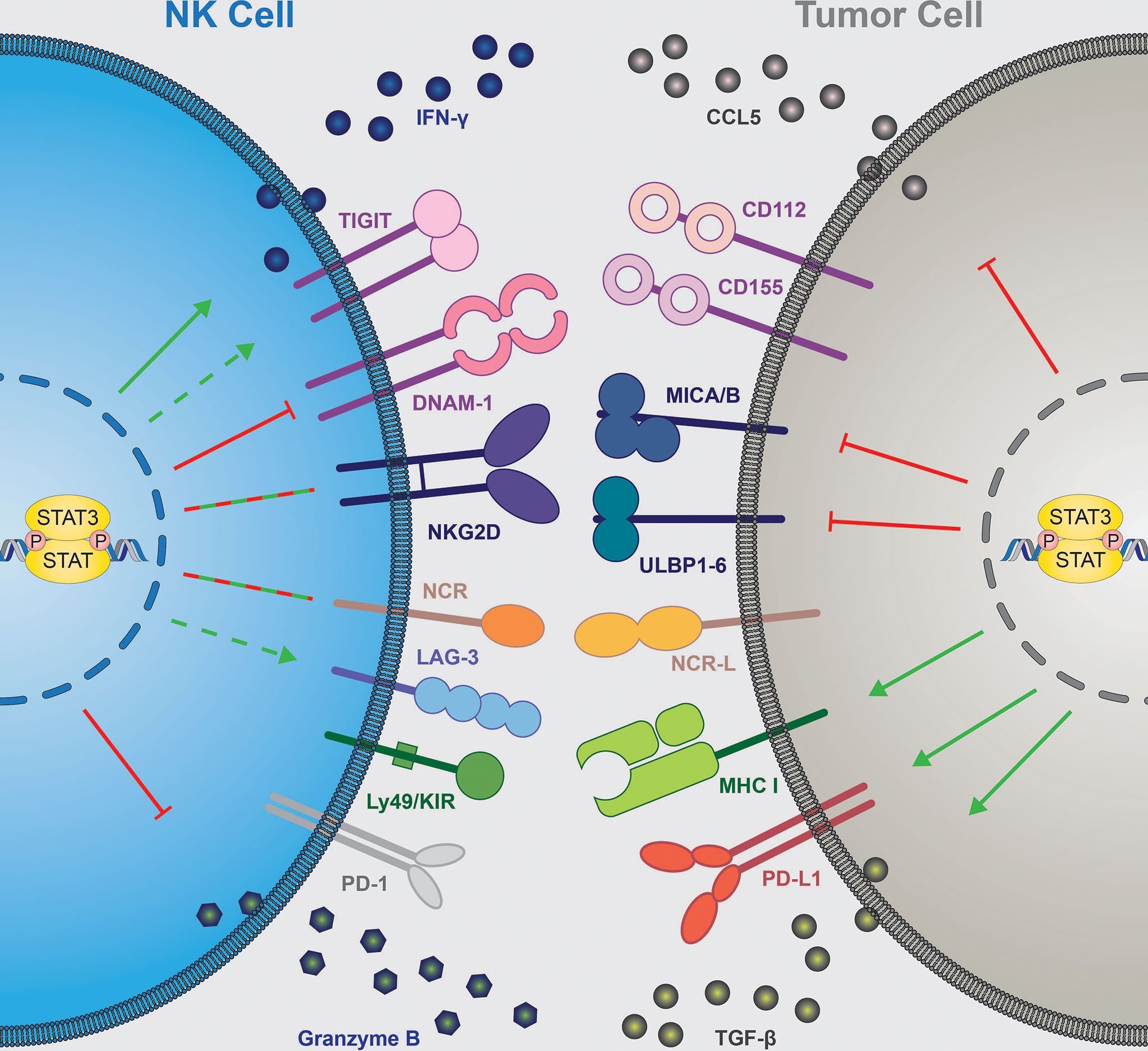
Figure 2 STAT3 contribution to NK cell-mediated tumor immune surveillance. NK cell-intrinsic STAT3 (left) inhibits expression of granzyme B and DNAM-1 ( ), increases IFNγ secretion (
), increases IFNγ secretion ( ) and seems to upregulate TIGIT and LAG-3 (
) and seems to upregulate TIGIT and LAG-3 ( ), while the effect on NCRs and NKG2D expression remain context dependent (
), while the effect on NCRs and NKG2D expression remain context dependent ( ). Tumor cell-intrinsic STAT3 (right) inhibits expression of NKG2D ligands (MICA/B, ULBPs) and NK-cell attracting chemokine CCL5 (
). Tumor cell-intrinsic STAT3 (right) inhibits expression of NKG2D ligands (MICA/B, ULBPs) and NK-cell attracting chemokine CCL5 ( ). STAT3 in tumor cells upregulates surface expression of MHC I and PD-L1 molecules and secretion of immune suppressive TGF-β (
). STAT3 in tumor cells upregulates surface expression of MHC I and PD-L1 molecules and secretion of immune suppressive TGF-β ( ). NK, natural killer; IFN, interferon; DNAM-1, DNAX accessory molecule; NKG2D, NK-cell receptor natural killer group 2D; NCR, natural cytotoxicity receptor; KIR, killer-cell immunoglobulin-like receptor; CCL5, C-C motif chemokine ligand 5; CD, cluster of differentiation; MICA/B, major histocompatibility complex class I-related sequence A/B; ULBP, UL16-binding protein; MHC I, major histocompatibility complex I; TGF-β, transforming growth factor β, STAT, signal transducer and activator of transcription. TIGIT, T cell immunoreceptor with Ig and ITIM domains; LAG-3, lymphocyte-activation gene 3; PD-(L)1, programmed cell death ligand/protein 1.
). NK, natural killer; IFN, interferon; DNAM-1, DNAX accessory molecule; NKG2D, NK-cell receptor natural killer group 2D; NCR, natural cytotoxicity receptor; KIR, killer-cell immunoglobulin-like receptor; CCL5, C-C motif chemokine ligand 5; CD, cluster of differentiation; MICA/B, major histocompatibility complex class I-related sequence A/B; ULBP, UL16-binding protein; MHC I, major histocompatibility complex I; TGF-β, transforming growth factor β, STAT, signal transducer and activator of transcription. TIGIT, T cell immunoreceptor with Ig and ITIM domains; LAG-3, lymphocyte-activation gene 3; PD-(L)1, programmed cell death ligand/protein 1.
STAT3 is activated in NK cells by a variety of cytokines, including type I IFNs, IL-2, IL-6, IL-10, IL-12, IL-15, IL-21 and IL-27, with diverse effects on NK-cell activation (86–91). NK cell-intrinsic roles of STAT3 have been analyzed in Stat3fl/fl Ncr1-iCre mice lacking STAT3 in NKp46+ cells. Stat3 deletion does not affect NK-cell development, numbers and maturation. Furthermore, NK cell-intrinsic loss of STAT3 does not impact on proliferation of NK cells in Stat3fl/fl Ncr1-iCre mice (87). However, knockdown of STAT3 in the human NK-cell line NK-92 is associated with a decreased proliferation rate, correlating with reduced cyclin D1 expression, and overexpression of STAT3 enhances human NK-cell expansion (92). In line, IL-21, which primarily activates STAT3 in NK cells, promotes human NK-cell expansion associated with an increased telomere length (93–95).
Effects of cancer cell-extrinsic STAT3 deficiency on anti-tumor immunity have initially been reported in an inducible STAT3 knockout mouse model (Stat3fl/fl Mx1-Cre mice) (96). Both Stat3fl/fl Mx1-Cre and Stat3fl/fl Ncr1-iCre mice demonstrated that lack of STAT3 enhances NK cell-mediated surveillance in different transplantable tumor models (87, 96). Treatment with the small-molecule STAT3 inhibitor CPA7 boosts anti-tumor responses against the subcutaneously injected urothelial carcinoma cell line MB49, which is largely dependent on T cells with a partial involvement of NK cells (96). NK cell-intrinsic STAT3 deficiency is sufficient to increase surveillance of melanoma and leukemia cell lines (87). Overall, these data provide evidence that STAT3 suppresses the anti-tumor activity of NK cells (13, 87, 96). The enhanced cytotoxicity of NK cells upon loss of STAT3 goes along with increased levels of the cytotoxic effector molecules perforin and granzyme B (87, 88).
STAT3 is also involved in regulating expression of activating NK-cell receptors. STAT3 decreases DNAM-1 expression on NK cells. Elevated DNAM-1 levels upon Stat3 deletion contribute to enhanced killing of DNAM-1 ligand-expressing tumor cells, e.g. B16F10 melanoma cells (87). NK cells show a STAT3-dependent upregulation of NKG2D surface levels in response to IL-10 and IL-21 stimulation, associated with enhanced NK-cell degranulation (88, 95, 97). While NK cell-intrinsic loss of STAT3 does not suffice to impact on NKG2D expression, STAT3-deficiency in the entire hematopoietic system (Stat3fl/fl Tie2-Cre mice) causes a reduction of NKG2D levels (87, 95). In contrast to a potential negative regulatory role of STAT3 on DNAM-1 expression in murine NK cells (87), these results indicate that STAT3 enhances NKG2D expression (95). However, another report demonstrated that IL-2-activated human NK cells display decreased NKG2D levels upon IL-21 stimulation (98). In addition, STAT3 activation by IL-6 and IL-8 produced by tumor cells has been reported to decrease levels of NKG2D and NKp30 on NK cells (91). Therefore, the impact of STAT3 on the regulation of NKG2D might vary depending on the specific upstream stimuli, signaling pathways and additional STAT proteins involved (13, 88). Similar to NKG2D, STAT3 has also been reported to bind to the promoter and drive the transcription of NCR1 gene encoding NKp46 (92, 95).
In line with the suppressive effect of STAT3 on NK-cell functionality reported in the murine system (87), tumor-derived cytokines, such as IL-6 and IL-8, impair human NK-cell function in a STAT3-dependent manner (91). On the contrary, another study found a positive correlation between STAT3 levels and expression of cytotoxic effector molecules and cytokines in human NK cells (92). STAT3 levels are reduced in NK cells from chronic hepatitis B virus (HBV) patients, which is associated with lower degranulation and IFNγ production (92). Early cytokine production, including IFNγ, in IL-15 primed human NK cells also requires STAT3 (99). An involvement of STAT3 in the regulation of IFNγ production has also been described in murine NK cells, where STAT3 directly binds to the Ifng promoter and contributes to cytokine-induced IFNγ production (87). Besides cytotoxic activity and production of proinflammatory cytokines, NK cells also produce immunosuppressive cytokines and have immunoregulatory functions (100, 101). STAT3 also impacts on these functions. For example, the immunosuppressive cytokines IL-10 and TGF-β are upregulated in a STAT3-dependent manner in tumor-infiltrating NK cells that are positive for the immune checkpoint CD73 (12, 102).
Hypoxia is an important feature of solid tumors that is associated with immune suppression and escape (103). Indeed, hypoxia represses NK-cell cytotoxicity by induction of SHP-1 expression, which in turn reduces STAT3 activation (104). STAT3 also interacts with one of the main players driving hypoxic response - hypoxia inducible factor 1α (HIF-1α) (105). In IL-15 primed human NK cells, HIF-1α responses rely on STAT3 (99). In line, STAT3 induces HIF-1α-mediated upregulation of miR-224, which is paralleled by reduced NKp46 expression and a dampened NK cell-mediated killing of prostate cancer cells (12, 106, 107). The exact underlying mechanisms how STAT3 activity contributes to hypoxia-driven effects on NK-cell cytotoxicity remain to be determined.
Altogether, STAT3 has complex effects on NK-cell activity, including the expression of cytotoxic granule proteins, cytokines and NK-cell receptors (Figure 2, left). Whether STAT3 has an overall beneficial or detrimental effect on NK-cell function appears to be context-dependent (13, 87, 92, 95). STAT3 induces its own negative feedback regulation, including the upregulation of SOCS3 (Figure 1). SOCS3 suppresses NK cells, as loss of SOCS3 enhances NK-cell proliferation and cytotoxicity (108). The effects of SOCS3 on STAT3 activation might depend on the upstream stimuli, which are likely to be differently susceptible to SOCS3-mediated inhibition (109, 110). This could contribute to the context-dependent effects of STAT3 in NK cells. As mentioned above, cancer cell-intrinsic alternatively spliced STAT3 isoforms have opposite roles in driving tumor progression. Since STAT1 isoforms differentially affect NK-cell functionality (111), it is attractive to speculate that also STAT3α and β have non-redundant functions in regulating NK cells. Analyzing the consequences of STAT3α or β deficiency in NK cells might improve our understanding of STAT3-dependent effects.
Another level of evidence for a crucial role of STAT3 in NK-cell biology stems from studies analyzing NK cells in patients with STAT3 mutations. Heterozygous germline STAT3 loss-of-function (LOF) mutations are found in autosomal dominant hyper IgE syndrome (HIES) patients, which display immunological deficiencies with increased susceptibility to infections linked to impaired STAT3-regulated T helper 17 (Th17)-mediated immune responses and B cell function (112–116). NK cells from HIES patients harboring STAT3 LOF mutations have decreased NKG2D levels (95). This might be associated with impaired NK cell function, however a thorough functional characterization of NK cells from HIES patients has not been published. Apart from LOF mutations, germline and somatic activating mutations of STAT3 have been described in humans with different disease characteristics (112–115, 117). Germline STAT3 gain-of-function (GOF) mutations are associated with diverse clinical manifestations, including immunodeficiencies and autoimmune diseases (112, 118–122). Haapaniemi et al. reported that NK-cell numbers are reduced in patients with germline STAT3 GOF mutations, while maturation and functionality of NK cells are unaffected (120). However, another study did not find reduced NK-cell numbers (123), indicating that the impact of STAT3 GOF mutations on immune cells, including NK cells, varies between patients (122).
An oncogenic potential of STAT3 in NK cells has been indicated by the discovery of somatic STAT3 GOF mutations, predominantly within the SH2 domain, in a subset of patients with different NK-cell malignancies, including chronic lymphoproliferative disorder of NK cells (CLPD-NK), aggressive NK-cell leukemia (ANKL) and extranodal NK/T-cell lymphomas of nasal type (NKTCL) (124–135). Apart from STAT3 GOF mutations, enhanced STAT3 phosphorylation can also be observed in NK-cell malignancies by other means, including activation of upstream JAKs or reduced expression of negative regulators of STAT3 (124, 127, 131–133, 136–139). Pro-tumorigenic effects of STAT3 on NK cells are linked to its role in proliferation and survival (133, 136, 140–142). IL-10 mediated STAT3 activation as well as somatic STAT3 GOF mutations increase expression of MYC and thereby drive metabolic activation of ANKL cells, fueling leukemia cell survival and proliferation (131). To the best of our knowledge, the effects of somatic STAT3 GOF mutations on the functionality of malignant NK cells have not been directly tested. Interestingly, STAT3 GOF mutations in CLPD-NK patients correlate with a cytotoxic CD16hiCD57-phenotype and a more symptomatic disease, characterized by anemia and severe neutropenia (127, 134, 135, 143).
Immune checkpoint inhibitors are one of the most successful approaches of immunotherapy. By targeting the inhibitory ligands (programmed cell death ligand 1 - PD-L1) or inhibitory receptors (programmed cell death protein 1 - PD-1, cytotoxic T-lymphocyte-associated protein 4 - CTLA-4) with monoclonal antibodies, the immune response against tumors can be unleashed (144). It is clear that T cells are the main drivers of immune checkpoint inhibitor responses, but a role of NK cells herein has been proposed (145). Several reports find PD-1-dependent effects of NK cells in specific tumors including MM (146), Kaposi sarcoma (147), Hodgkin lymphoma (148) and head and neck cancer (149). Other studies indicate that the expression of PD-1 in NK cells is minor or neglectable (150) and the exact function of NK cells in anti-PD-1/PD-L1 therapy remains a matter of debate (151). Importantly, vast evidence indicates the direct involvement of STAT3 in driving PD-L1 and PD-L2 expression in tumor cells (132, 152–154). For example, in T cell lymphoma STAT3 is required for induction of PD-L1 transcription by directly binding its promoter (153). Therefore, combinatorial inhibition of STAT3 and PD-L1/PD-1 axis has been explored as an attractive approach. Encouraging results from preclinical studies (33, 155, 156) led to first clinical trials combining STAT3 inhibitor (BBI-608) with anti-PD-L1 therapies in metastatic colorectal carcinoma (NCT03647839, NCT02851004) or STAT3 targeting antisense oligonucleotide (AZD9150) with anti-PD-L1 therapy in NSCLC (NCT03334617) and other solid tumors. It remains unclear whether NK cells contribute to this combination therapy outcome. Xu et al. suggested that in vitro combination of PD-L1 and STAT3 inhibition enhances NK-cell cytotoxicity against prostate cancer cell lines, but the in vivo relevance still needs to be elucidated (157).
Novel immune checkpoint molecules are currently emerging and some have entered clinical trials or have recently been approved. In contrast to the above described PD-1 and CTLA-4, the expression of novel checkpoints: T cell immunoreceptor with Ig and ITIM domains (TIGIT), lymphocyte-activation gene 3 (LAG-3) and T cell immunoglobulin and mucin-domain-containing-3 (TIM-3) is clearly shared between T and NK cells (158–161). The exact role of STAT3 in the regulation of these checkpoints has not been addressed in NK cells. In T regulatory cells, TIM-3 is strongly downregulated upon STAT3 inhibition suggesting a potential dependency of TIM-3 expression on STAT3 in other lymphocytes (162). It is attractive to speculate that LAG-3 expression in NK cells might be enhanced by STAT3 signaling. IL-12, which predominantly signals via STAT4 and STAT3, was shown to upregulate LAG-3 expression in NK cells (163). In line, STAT3 inhibition together with blockage of the STAT3-activating cytokine IL-6, downregulated TIGIT expression in the human NK92 cell line. Combination of TIGIT checkpoint inhibitor with blockage of IL-6R and STAT3 enhanced cytotoxicity of NK92 cells towards prostate cancer cells (164). Although further investigations are essential, both studies suggest a potential synergism between STAT3 targeting molecules and novel checkpoint inhibitors in driving NK responses.
STAT3 regulates immune evasion from NK cells on several different levels. It helps tumor cells to hide from NK cells by downregulating activating ligands, drives an immune suppressive environment, which in turn limits the chemoattraction and activity of NK cells, and intrinsically regulates NK-cell responses (Figure 2). Somatic GOF mutations in STAT3 have an oncogenic potential in NK cells underlining its key role in NK-cell biology. It remains unclear why the effects of STAT3 on NK-cell biology are so complex and context dependent. The discrepancy between some findings in human and mouse NK cells might come from in vitro cultures of human NK cells that do not reflect the situation in vivo with different cytokines present in the tumor microenvironment in the mouse models. Moreover, one cannot exclude that the results obtained using mice with STAT3-deficient NK cells are influenced by compensatory mechanisms, including upregulation of STAT5, which is a key driver of NK-cell survival and functionality (77, 165, 166). Not only can the STATs compensate for each other (23, 167) but also crosstalk to other signaling pathways (168). This adds another layer of complexity in understanding the role of STAT3 in NK-cell anti-tumor responses.
Inhibition of STAT3 signaling is currently explored in many clinical trials for solid and hematopoietic tumors. The direct approaches have reached clinical trials but achieving specificity over other STAT family members remains challenging (33, 169, 170). Based on the current evidence, STAT3 inhibition might not only impair tumor cell survival but also enhance their recognition by NK cells. Moreover, targeting NK cell-intrinsic STAT3 could unleash their anti-tumor responses in some tumor models (87), while the consequences on other aspects of NK-cell functionality are difficult to predict. At the moment, there is an unmet need to understand the effects of STAT3 inhibitors on NK-cell anti-tumor responses in vivo to be able to foresee the clinically relevant consequences.
It is well appreciated that combination therapies enhance the efficacy and reduce resistance compared to monotherapies. This has triggered extensive attempts in combining STAT3 inhibitors with other drugs (33). A new avenue in immunotherapy is opened by combinations of STAT3 inhibitors explored with the emerging immune checkpoint inhibitors. Importantly, targeting STAT3 in the immune system might have complex systemic effects ranging from autoimmunity to immunodeficiency as indicated by phenotypes of patients with mutations in STAT3 (112–116). In summary, targeting STAT3 might be an attractive approach in restoring NK-cell anti-tumor immunity but needs to be carefully evaluated in different tumor types and biological contexts.
AW-S, KK and BZ designed the concept and wrote the manuscript. DS designed the concept and critically reviewed the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Austrian Science Fund (FWF) under grant P32693 to DS and the Austrian Academy of Sciences (ÖAW) (DOC scholarship to KK).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Dagmar Gotthardt for critically reviewing the manuscript.
1. Koch J, Steinle A, Watzl C, Mandelboim O. Activating Natural Cytotoxicity Receptors of Natural Killer Cells in Cancer and Infection. Trends Immunol (2013) 34(4):182–91. doi: 10.1016/j.it.2013.01.003
2. Raulet DH, Vance RE. Self-Tolerance of Natural Killer Cells. Nat Rev Immunol (2006) 6(7):520–31. doi: 10.1038/nri1863
3. Freud AG, Mundy-Bosse BL, Yu J, Caligiuri MA. The Broad Spectrum of Human Natural Killer Cell Diversity. Immunity. (2017) 47(5):820–33. doi: 10.1016/j.immuni.2017.10.008
4. Pahl J, Cerwenka A. Tricking the Balance: NK Cells in Anti-Cancer Immunity. Immunobiology. (2017) 222(1):11–20. doi: 10.1016/j.imbio.2015.07.012
5. Shimasaki N, Jain A, Campana D. NK Cells for Cancer Immunotherapy. Nat Rev Drug Discov Nat Res (2020) Vol. 19:200–18. doi: 10.1038/s41573-019-0052-1
6. Berrien-elliott MM, Foltz JA, Russler-germain DA, Neal CC, Tran J, Gang M, et al. Hematopoietic Cell Transplantation Donor-Derived Memory-Like NK Cells Functionally Persist After Transfer Into Patients With Leukemia. Sci Transl Med (2022) 14:eabm1375. doi: 10.1126/scitranslmed.abm1375
7. Nowbakht P, Ionescu MCS, Rohner A, Kalberer CP, Rossy E, Mori L, et al. Ligands for Natural Killer Cell-Activating Receptors are Expressed Upon the Maturation of Normal Myelomonocytic Cells But at Low Levels in Acute Myeloid Leukemias. Blood. (2005) 105(9):3615–22. doi: 10.1182/blood-2004-07-2585
8. Kearney CJ, Ramsbottom KM, Voskoboinik I, Darcy PK, Oliaro J. Loss of DNAM-1 Ligand Expression by Acute Myeloid Leukemia Cells Renders Them Resistant to NK Cell Killing. Oncoimmunology (2016) 5(8):e1196308. doi: 10.1080/2162402X.2016.1196308
9. Vago L, Gojo I. Immune Escape and Immunotherapy of Acute Myeloid Leukemia. J Clin Invest (2020) 130(4):1552–64. doi: 10.1172/JCI129204
10. Sabry M, Lowdell MW. Tumor-Primed NK Cells: Waiting for the Green Light. Front Immunol (2013) 4. doi: 10.3389/fimmu.2013.00408
11. Okumura G, Iguchi-Manaka A, Murata R, Yamashita-Kanemaru Y, Shibuya A, Shibuya K. Tumor-Derived Soluble CD155 Inhibits DNAM-1-Mediated Antitumor Activity of Natural Killer Cells. J Exp Med (2020) 217(4):1. doi: 10.1084/jem.20191290
12. Zhang L, Kuca K, You L, Zhao Y, Musilek K, Nepovimova E, et al. Signal Transducer and Activator of Transcription 3 Signaling in Tumor Immune Evasion. Pharmacol Ther (2022) 230:107969. doi: 10.1016/j.pharmthera.2021.107969
13. Cacalano NA. Regulation of Natural Killer Cell Function by STAT3. Front Immunol. (2016) 7:128. doi: 10.3389/fimmu.2016.00128
14. Huynh J, Chand A, Gough D, Ernst M. Therapeutically Exploiting STAT3 Activity in Cancer - Using Tissue Repair as a Road Map. Nat Rev Cancer 2018/12/24 (2019) 19(2):82–96. doi: 10.1038/s41568-018-0090-8
15. Aigner P, Just V, Stoiber D. STAT3 Isoforms: Alternative Fates in Cancer? Vol. 118. Cytokine Acad Press (2019) 118:27–34. doi: 10.1016/j.cyto.2018.07.014
16. Zhang HX, Yang PL, Li EM, Xu LY. STAT3beta, a Distinct Isoform From STAT3. Int J Biochem Cell Biol (2019) 110:130–9. doi: 10.1016/j.biocel.2019.02.006
17. Johnson DE, O’Keefe RA, Grandis JR. Targeting the IL-6/JAK/STAT3 Signalling Axis in Cancer. Nat Rev Clin Oncol (2018) 15(4):234. doi: 10.1038/nrclinonc.2018.8
18. Xu J, Lin H, Wu G, Zhu M, Li M. IL-6/STAT3 Is a Promising Therapeutic Target for Hepatocellular Carcinoma. Front Oncol (2021) 11:5366. doi: 10.3389/fonc.2021.760971
19. Jones SA, Jenkins BJ. Recent Insights Into Targeting the IL-6 Cytokine Family in Inflammatory Diseases and Cancer. Nat Rev Immunol (2018) 18(12):773–89. doi: 10.1038/s41577-018-0066-7
20. Diveu C, Lelièvre E, Perret D, Lak-Hal AHL, Froger J, Guillet C, et al. GPL, a Novel Cytokine Receptor Related to GP130 and Leukemia Inhibitory Factor Receptor. J Biol Chem (2003) 278(50):49850–9. doi: 10.1074/jbc.M307286200
21. Moser B, Edtmayer S, Witalisz-Siepracka A, Stoiber D. The Ups and Downs of Stat Inhibition in Acute Myeloid Leukemia. Biomedicines. (2021) 9(8):1051. doi: 10.3390/biomedicines9081051
22. Cimica V, Chen H-C, Iyer JK, Reich NC. Dynamics of the STAT3 Transcription Factor: Nuclear Import Dependent on Ran and Importin-B1. PloS One (2011) 6(5):e20188. doi: 10.1371/journal.pone.0020188
23. Wingelhofer B, Neubauer HA, Valent P, Han X, Constantinescu SN, Gunning PT, et al. Implications of STAT3 and STAT5 Signaling on Gene Regulation and Chromatin Remodeling in Hematopoietic Cancer. Leuk 2018/05/08 (2018) 32(8):1713–26. doi: 10.1038/s41375-018-0117-x
24. Zhou Y, Chen JJ. STAT3 Plays an Important Role in DNA Replication by Turning on WDHD1. Cell Biosci (2021) 11(1):1–10. doi: 10.1186/s13578-020-00524-x
25. Chung CD, Liao J, Liu B, Rao X, Jay P, Berta P, et al. Specific Inhibition of Stat3 Signal Transduction by PIAS3. Sci (80- ). (1997) 278(5344):1803–5. doi: 10.1126/science.278.5344.1803
26. Babon JJ, Varghese LN, Nicola NA. Inhibition of IL-6 Family Cytokines by SOCS3. Semin Immunol (2014) 26(1):13. doi: 10.1016/j.smim.2013.12.004
27. Decker T, Kovarik P. Serine Phosphorylation of STATs. Oncogene (2000) 19(21):2628–37. doi: 10.1038/sj.onc.1203481
28. Wegrzyn J, Potla R, Chwae YJ, Sepuri NBV, Zhang Q, Koeck T, et al. Function of Mitochondrial Stat3 in Cellular Respiration. Science. (2009) 323(5915):793–7. doi: 10.1126/science.1164551
29. Amaya ML, Inguva A, Pei S, Jones C, Krug A, Ye H, et al. The STAT3-MYC Axis Promotes Survival of Leukemia Stem Cells by Regulating SLC1A5 and Oxidative Phosphorylation. Blood. (2022) 139(4):584–96. doi: 10.1182/blood.2021013201
30. Jin W. Role of JAK/STAT3 Signaling in the Regulation of Metastasis, the Transition of Cancer Stem Cells, and Chemoresistance of Cancer by Epithelial–Mesenchymal Transition. Cells (2020) 9(1):217. doi: 10.3390/cells9010217
31. Tolomeo M, Cascio A. The Multifaced Role of Stat3 in Cancer and its Implication for Anticancer Therapy. Int J Mol Sci (2021) 22(2):1–25. doi: 10.3390/ijms22020603
32. de Araujo ED, Keserű GM, Gunning PT, Moriggl R. Targeting STAT3 and STAT5 in Cancer. Cancers (Basel) (2020) 12(8):1–8. doi: 10.3390/cancers12082002
33. Zou S, Tong Q, Liu B, Huang W, Tian Y, Fu X. Targeting Stat3 in Cancer Immunotherapy. Mol Cancer (2020) 19(1):1–19. doi: 10.1186/s12943-020-01258-7
34. Jonker DJ, Nott L, Yoshino T, Gill S, Shapiro J, Ohtsu A, et al. Napabucasin Versus Placebo in Refractory Advanced Colorectal Cancer: A Randomised Phase 3 Trial. Lancet Gastroenterol Hepatol (2018) 3(4):263–70. doi: 10.1016/S2468-1253(18)30009-8
35. Bharadwaj U, Eckols TK, Xu X, Kasembeli MM, Chen Y, Adachi M, et al. Small-Molecule Inhibition of STAT3 in Radioresistant Head and Neck Squamous Cell Carcinoma. Oncotarget (2019) 10(16):1603. doi: 10.18632/oncotarget.8368
36. Qin JJ, Yan L, Zhang J, Zhang WD. STAT3 as a Potential Therapeutic Target in Triple Negative Breast Cancer: A Systematic Review. J Exp Clin Cancer Res (2019) 38(1):1–16. doi: 10.1186/s13046-019-1206-z
37. Hong D, Kurzrock R, Kim Y, Woessner R, Younes A, Nemunaitis J, et al. AZD9150, a Next-Generation Antisense Oligonucleotide Inhibitor of STAT3 With Early Evidence of Clinical Activity in Lymphoma and Lung Cancer. Sci Transl Med (2015) 7(314):314ra185. doi: 10.1126/scitranslmed.aac5272
38. Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, et al. Innate or Adaptive Immunity? The Example of Natural Killer Cells. Science. (2011) 331(6013):44–9. doi: 10.1126/science.1198687
39. Cosman D, Müllberg J, Sutherland CL, Chin W, Armitage R, Fanslow W, et al. ULBPs, Novel MHC Class I-Related Molecules, Bind to CMV Glycoprotein UL16 and Stimulate NK Cytotoxicity Through the NKG2D Receptor. Immunity. (2001) 14(2):123–33. doi: 10.1016/S1074-7613(01)00095-4
40. Raulet DH. Roles of the NKG2D Immunoreceptor and its Ligands. Nat Rev Immunol (2003) 3(10):781–90. doi: 10.1038/nri1199
41. Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, et al. Activation of NK Cells and T Cells by NKG2D, a Receptor for Stress-Inducible MICA. Science. (1999) 285(5428):727–9. doi: 10.1126/science.285.5428.727
42. Liu H, Wang S, Xin J, Wang J, Yao C, Zhang Z. Role of NKG2D and its Ligands in Cancer Immunotherapy. Am J Cancer Res (2019) 9(10):2064–78.
43. Cerwenka A, Bakker ABH, McClanahan T, Wagner J, Wu J, Phillips JH, et al. Retinoic Acid Early Inducible Genes Define a Ligand Family for the Activating NKG2D Receptor in Mice. Immunity. (2000) 12(6):721–7. doi: 10.1016/S1074-7613(00)80222-8
44. Diefenbach A, Jensen ER, Jamieson AM, Raulet DH. Rae1 and H60 Ligands of the NKG2D Receptor Stimulate Tumour Immunity. Nature (2001) 413(6852):165–71. doi: 10.1038/35093109
45. Takada A, Yoshida S, Kajikawa M, Miyatake Y, Tomaru U, Sakai M, et al. Two Novel NKG2D Ligands of the Mouse H60 Family With Differential Expression Patterns and Binding Affinities to NKG2D. J Immunol (2008) 180(3):1678–85. doi: 10.4049/jimmunol.180.3.1678
46. Brandt CS, Baratin M, Yi EC, Kennedy J, Gao Z, Fox B, et al. The B7 Family Member B7-H6 is a Tumor Cell Ligand for the Activating Natural Killer Cell Receptor NKp30 in Humans. J Exp Med (2009) 206(7):1495–503. doi: 10.1084/jem.20090681
47. Barrow AD, Martin CJ, Colonna M. The Natural Cytotoxicity Receptors in Health and Disease. Front Immunol (2019) 10:909. doi: 10.3389/fimmu.2019.00909
48. Sivori S, Vacca P, Del Zotto G, Munari E, Mingari MC, Moretta L. Human NK Cells: Surface Receptors, Inhibitory Checkpoints, and Translational Applications. Cell Mol Immunol (2019) 16(5):430–41. doi: 10.1038/s41423-019-0206-4
49. Shibuya A, Campbell D, Hannum C, Yssel H, Franz-Bacon K, McClanashan T, et al. DNAM-1, a Novel Adhesion Molecule Involved in the Cytolytic Function of T Lymphocytes. Immunity. (1996) 4(6):573–81. doi: 10.1016/S1074-7613(00)70060-4
50. Bottino C, Castriconi R, Pende D, Rivera P, Nanni M, Carnemolla B, et al. Identification of PVR (CD155) and Nectin-2 (CD112) as Cell Surface Ligands for the Human DNAM-1 (CD226) Activating Molecule. J Exp Med (2003) 198(4):557–67. doi: 10.1084/jem.20030788
51. Guerra N, Tan YX, Joncker NT, Choy A, Gallardo F, Xiong N, et al. NKG2D-Deficient Mice are Defective in Tumor Surveillance in Models of Spontaneous Malignancy. Immunity. (2008) 28(4):571–80. doi: 10.1016/j.immuni.2008.02.016
52. Mastaglio S, Wong E, Perera T, Ripley J, Blombery P, Smyth MJ, et al. Natural Killer Receptor Ligand Expression on Acute Myeloid Leukemia Impacts Survival and Relapse After Chemotherapy. Blood Adv (2018) 2(4):335–46. doi: 10.1182/bloodadvances.2017015230
53. Paczulla AM, Rothfelder K, Raffel S, Konantz M, Steinbacher J, Wang H, et al. Absence of NKG2D Ligands Defines Leukaemia Stem Cells and Mediates Their Immune Evasion. Nat (2019) 572(7768):254–9. doi: 10.1038/s41586-019-1410-1
54. Groh V, Wu J, Yee C, Spies T. Tumour-Derived Soluble MIC Ligands Impair Expression of NKG2D and T-Cell Activation. Nature. (2002) 419(6908):734–8. doi: 10.1038/nature01112
55. Raulet DH, Guerra N. Oncogenic Stress Sensed by the Immune System: Role of Natural Killer Cell Receptors. Nat Rev Immunol (2009) 9(8):568–80. doi: 10.1038/nri2604
56. Bedel R, Thiery-Vuillemin A, Grandclement C, Balland J, Remy-Martin JP, Kantelip B, et al. Novel Role for STAT3 in Transcriptional Regulation of NK Immune Cell Targeting Receptor MICA on Cancer Cells. Cancer Res (2011) 71(5):1615–26. doi: 10.1158/0008-5472.CAN-09-4540
57. Fionda C, Malgarini G, Soriani A, Zingoni A, Cecere F, Iannitto ML, et al. Inhibition of Glycogen Synthase Kinase-3 Increases NKG2D Ligand MICA Expression and Sensitivity to NK Cell–Mediated Cytotoxicity in Multiple Myeloma Cells: Role of STAT3. J Immunol (2013) 190(12):6662–72. doi: 10.4049/jimmunol.1201426
58. Lopez-Soto A, Huergo-Zapico L, Galvan JA, Rodrigo L, de Herreros AG, Astudillo A, et al. Epithelial-Mesenchymal Transition Induces an Antitumor Immune Response Mediated by NKG2D Receptor. J Immunol (2013) 190(8):4408–19. doi: 10.4049/jimmunol.1202950
59. Cai X, Lu X, Jia Z, Zhang X, Han W, Rong X, et al. STAT3 Contributes to NK Cell Recognition by Modulating Expression of NKG2D Ligands in Adriamycin-Resistant K562/AO2 Cells. Int J Hematol (2015) 102(5):536–43. doi: 10.1007/s12185-015-1860-7
60. Lu X, Zhu Z, Jiang L, Sun X, Jia Z, Qian S, et al. Matrine Increases NKG2D Ligand ULBP2 in K562 Cells via Inhibiting JAK/STAT3 Pathway: A Potential Mechanism Underlying the Immunotherapy of Matrine in Leukemia. Am J Transl Res (2015) 7(10):1838.
61. Zhu Z, Bai Y, Lu X, Ding J, Qi C. Rapamycin Downregulates NKG2D Ligands in Acute Myeloid Leukemia Cells via an Activation of the STAT3 Pathway: A Potential Mechanism for Rapamycin-Induced Immune Escape in Leukemia. Transl Cancer Res (2019) 8(2):473–82. doi: 10.21037/tcr.2019.03.01
62. Zhu Z, Lu X, Jiang L, Sun X, Zhou H, Jia Z, et al. STAT3 Signaling Pathway is Involved in Decitabine Induced Biological Phenotype Regulation of Acute Myeloid Leukemia Cells. Am J Transl Res (2015) 7(10):1896–907.
63. Sun X, Sui Q, Zhang C, Tian Z, Zhang J. Targeting Blockage of Stat3 in Hepatocellular Carcinoma Cells Augments Nk Cell Functions via Reverse Hepatocellular Carcinoma-Induced Immune Suppression. Mol Cancer Ther (2013) 12(12):2885–96. doi: 10.1158/1535-7163.MCT-12-1087
64. Garrido-Tapia M, Hernández CJ, Ascui G, Kramm K, Morales M, Gárate V, et al. STAT3 Inhibition by STA21 Increases Cell Surface Expression of MICB and the Release of Soluble MICB by Gastric Adenocarcinoma Cells. Immunobiology. (2017) 222(11):1043–51. doi: 10.1016/j.imbio.2017.05.009
65. Karre K, Ljunggren HG, Piontek G, Kiessling R. Selective Rejection of H-2-Deficient Lymphoma Variants Suggests Alternative Immune Defence Strategy. Nature. (1986) 319(6055):675–8. doi: 10.1038/319675a0
66. Kärre K. Natural Killer Cell Recognition of Missing Self. Nat Immunol (2008) 9(5):477–80. doi: 10.1038/ni0508-477
67. Karlhofer FM, Ribaudo RK, Yokoyama WM. MHC Class I Alloantigen Specificity of Ly-49+ IL-2-Activated Natural Killer Cells. Nature. (1992) 358(6381):66–70. doi: 10.1038/358066a0
68. Colonna M, Samaridis J. Cloning of Immunoglobulin-Superfamily Members Associated With HLA-C and HLA-B Recognition by Human Natural Killer Cells. Science. (1995) 268(5209):405–8. doi: 10.1126/science.7716543
69. Wagtmann N, Rajagopalan S, Winter CC, Peruui M, Long EO. Killer Cell Inhibitory Receptors Specific for HLA-C and HLA-B Identified by Direct Binding and by Functional Transfer. Immunity. (1995) 3(6):801–9. doi: 10.1016/1074-7613(95)90069-1
70. Diefenbach A, Raulet DH. Strategies for Target Cell Recognition by Natural Killer Cells. Immunol Rev (2001) 181:170–84. doi: 10.1034/j.1600-065X.2001.1810114.x
71. Huntington ND, Cursons J, Rautela J. The Cancer-Natural Killer Cell Immunity Cycle. Nat Rev Cancer (2020) 20(8):437–54. doi: 10.1038/s41568-020-0272-z
72. Ihara S, Kida H, Arase H, Tripathi LP, Chen YA, Kimura T, et al. Inhibitory Roles of Signal Transducer and Activator of Transcription 3 in Antitumor Immunity During Carcinogen-Induced Lung Tumorigenesis. Cancer Res (2012) 72(12):2990–9. doi: 10.1158/0008-5472.CAN-11-4062
73. Kortylewski M, Yu H. Role of Stat3 in Suppressing Anti-Tumor Immunity. Curr Opin Immunol (2008) 20(2):228. doi: 10.1016/j.coi.2008.03.010
74. Bruno A, Focaccetti C, Pagani A, Imperatori AS, Spagnoletti M, Rotolo N, et al. The Proangiogenic Phenotype of Natural Killer Cells in Patients With Non – Small. Neoplasia. (2013) 15(2):133–42. doi: 10.1593/neo.121758
75. Bruno A, Ferlazzo G, Albini A, Noonan DM. A Think Tank of TINK/TANKs: Tumor-Infiltrating/Tumor-Associated Natural Killer Cells in Tumor Progression and Angiogenesis. J Natl Cancer Inst (2014) 106(8):dju200. doi: 10.1093/jnci/dju200
76. Bruno A, Bassani B, D’Urso DG, Pitaku I, Cassinotti E, Pelosi G, et al. Angiogenin and the MMP9–TIMP2 Axis are Up-Regulated in Proangiogenic, Decidual NK-Like Cells From Patients With Colorectal Cancer. FASEB J (2018) 32(10):5365–77. doi: 10.1096/fj.201701103R
77. Gotthardt D, Putz EM, Grundschober E, Prchal-Murphy M, Straka E, Kudweis P, et al. STAT5 Is a Key Regulator in NK Cells and Acts as Molecular Switch From Tumor Surveillance to Tumor Promotion. Cancer Discovery (2016) 6(4):414–29. doi: 10.1158/2159-8290.CD-15-0732
78. Gao Y, Souza-Fonseca-Guimaraes F, Bald T, Ng SS, Young A, Ngiow SF, et al. Tumor Immunoevasion by the Conversion of Effector NK Cells Into Type 1 Innate Lymphoid Cells. Nat Immunol (2017) 18(9):1004–15. doi: 10.1038/ni.3800
79. Wang T, Niu G, Kortylewski M, Burdelya L, Shain K, Zhang S, et al. Regulation of the Innate and Adaptive Immune Responses by Stat-3 Signaling in Tumor Cells. Nat Med (2004) 10(1):48–54. doi: 10.1038/nm976
80. Caldenhoven E, Van Dijk TB, Solari R, Armstrong J, Raaijmakers JAM, Lammers JWJ, et al. Stat3β, a Splice Variant of Transcription Factor STAT3, Is a Dominant Negative Regulator of Transcription. J Biol Chem (1996) 271(22):13221–7. doi: 10.1074/jbc.271.22.13221
81. Zammarchi F, De Stanchina E, Bournazou E, Supakorndej T, Martires K, Riedel E, et al. Antitumorigenic Potential of STAT3 Alternative Splicing Modulation. Proc Natl Acad Sci U S A (2011) 108(43):17779–84. doi: 10.1073/pnas.1108482108
82. Musteanu M, Blaas L, Mair M, Schlederer M, Bilban M, Tauber S, et al. Stat3 is a Negative Regulator of Intestinal Tumor Progression in Apc(Min) Mice. Gastroenterology. (2010) 138(3):1003-11 e1-5. doi: 10.1053/j.gastro.2009.11.049
83. Maritano D, Sugrue ML, Tininini S, Dewilde S, Strobl B, Fu XP, et al. The STAT3 Isoforms α and β Have Unique and Specific Functions. Nat Immunol (2004) 5(4):401–9. doi: 10.1038/ni1052
84. Dang W, Tang H, Cao H, Wang L, Zhang X, Tian W, et al. Strategy of STAT3β Cell-Specific Expression in Macrophages Exhibits Antitumor Effects on Mouse Breast Cancer. Gene Ther (2015) 22(12):977–83. doi: 10.1038/gt.2015.70
85. Putz EM, Hoelzl MA, Baeck J, Bago-Horvath Z, Schuster C, Reichholf B, et al. Loss of STAT3 in Lymphoma Relaxes NK Cell-Mediated Tumor Surveillance. Cancers (Basel) (2014) 6(1):193–210. doi: 10.3390/cancers6010193
86. Matsui M, Kishida T, Nakano H, Yoshimoto K, Shin-Ya M, Shimada T, et al. Interleukin-27 Activates Natural Killer Cells and Suppresses NK-Resistant Head and Neck Squamous Cell Carcinoma Through Inducing Antibody-Dependent Cellular Cytotoxicity. Cancer Res (2009) 69(6):2523–30. doi: 10.1158/0008-5472.CAN-08-2793
87. Gotthardt D, Putz EM, Straka E, Kudweis P, Biaggio M, Poli V, et al. Loss of STAT3 in Murine NK Cells Enhances NK Cell-Dependent Tumor Surveillance. Blood. (2014) 124(15):2370–9. doi: 10.1182/blood-2014-03-564450
88. Gotthardt D, Sexl V. STATs in NK-Cells: The Good, the Bad, and the Ugly. Front Immunol (2017) 7:694. doi: 10.3389/fimmu.2016.00694
89. Gotthardt D, Trifinopoulos J, Sexl V, Putz EM. JAK/STAT Cytokine Signaling at the Crossroad of NK Cell Development and Maturation. Front Immunol (2019) 12:10. doi: 10.3389/fimmu.2019.02590
90. Wu Y, Tian Z, Wei H. Developmental and Functional Control of Natural Killer Cells by Cytokines. Front Immunol (2017) 8:930. doi: 10.3389/fimmu.2017.00930
91. Wu J, Gao FX, Wang C, Qin M, Han F, Xu T, et al. IL-6 and IL-8 Secreted by Tumour Cells Impair the Function of NK Cells via the STAT3 Pathway in Oesophageal Squamous Cell Carcinoma. J Exp Clin Cancer Res (2019) 38(1):321. doi: 10.1186/s13046-019-1310-0
92. Zheng B, Yang Y, Han Q, Yin C, Pan Z, Zhang J. STAT3 Directly Regulates NKp46 Transcription in NK Cells of HBeAg-Negative CHB Patients. J Leukoc Biol (2019) 106(4):987–96. doi: 10.1002/JLB.2A1118-421R
93. Wendt K, Wilk E, Buyny S, Schmidt RE, Jacobs R. Interleukin-21 Differentially Affects Human Natural Killer Cell Subsets. Immunology. (2007) 122(4):486–95. doi: 10.1111/j.1365-2567.2007.02675.x
94. Denman CJ, Senyukov VV, Somanchi SS, Phatarpekar PV, Kopp LM, Johnson JL, et al. Membrane-Bound IL-21 Promotes Sustained Ex Vivo Proliferation of Human Natural Killer Cells. PloS One (2012) 7(1):e30264. doi: 10.1371/journal.pone.0030264
95. Zhu S, Phatarpekar PV, Denman CJ, Senyukov VV, Somanchi SS, Nguyen-Jackson HT, et al. Transcription of the Activating Receptor NKG2D in Natural Killer Cells is Regulated by STAT3 Tyrosine Phosphorylation. Blood. (2014) 124(3):403–11. doi: 10.1182/blood-2013-05-499707
96. Kortylewski M, Kujawski M, Wang T, Wei S, Zhang S, Pilon-Thomas S, et al. Inhibiting Stat3 Signaling in the Hematopoietic System Elicits Multicomponent Antitumor Immunity. Nat Med (2005) 11(12):1314–21. doi: 10.1038/nm1325
97. Takaki R, Hayakawa Y, Nelson A, Sivakumar PV, Hughes S, Smyth MJ, et al. IL-21 Enhances Tumor Rejection Through a NKG2D-Dependent Mechanism. J Immunol (2005) 175(4):2167–73. doi: 10.4049/jimmunol.175.4.2167
98. Burgess SJ, Marusina AI, Pathmanathan I, Borrego F, Coligan JE. IL-21 Down-Regulates NKG2D/DAP10 Expression on Human NK and CD8+ T Cells. J Immunol (2006) 176(3):1490–7. doi: 10.4049/jimmunol.176.3.1490
99. Coulibaly A, Velásquez SY, Kassner N, Schulte J, Barbarossa MV, Lindner HA. STAT3 Governs the HIF-1α Response in IL-15 Primed Human NK Cells. Sci Rep (2021) 11(1):7023. doi: 10.1038/s41598-021-84916-0
100. Jiang Y, Yang M, Sun X, Chen X, Ma M, Yin X, et al. IL-10 + NK and TGF-β + NK Cells Play Negative Regulatory Roles in HIV Infection. BMC Infect Dis (2018) 18(1):80. doi: 10.1186/s12879-018-2991-2
101. Martinez-Espinosa I, Serrato JA, Ortiz-Quintero B. Role of IL-10-Producing Natural Killer Cells in the Regulatory Mechanisms of Inflammation During Systemic Infection. Biomolecules. (2021) 12(1):4. doi: 10.3390/biom12010004
102. Neo SY, Yang Y, Record J, Ma R, Chen X, Chen Z, et al. CD73 Immune Checkpoint Defines Regulatory NK Cells Within the Tumor Microenvironment. J Clin Invest (2020) 130(3):1185–98. doi: 10.1172/JCI128895
103. Barsoum IB, Koti M, Siemens DR, Graham CH. Mechanisms of Hypoxia-Mediated Immune Escape in Cancer. Cancer Res (2014) 74(24):7185–90. doi: 10.1158/0008-5472.CAN-14-2598
104. Teng R, Wang Y, Lv N, Zhang D, Williamson RA, Lei L, et al. Hypoxia Impairs NK Cell Cytotoxicity Through SHP-1-Mediated Attenuation of STAT3 and ERK Signaling Pathways. J Immunol Res (2020) 2020:4598476. doi: 10.1155/2020/4598476
105. Niu G, Briggs J, Deng J, Ma Y, Lee H, Kortylewski M, et al. Signal Transducer and Activator of Transcription 3 is Required for Hypoxia-Inducible Factor-1alpha RNA Expression in Both Tumor Cells and Tumor-Associated Myeloid Cells. Mol Cancer Res (2008) 6(7):1099–105. doi: 10.1158/1541-7786.MCR-07-2177
106. Chen Ch, Li Sx, Xiang Lx, Mu H, Wang Sb, Yu Ky. HIF-1α Induces Immune Escape of Prostate Cancer by Regulating NCR1/NKp46 Signaling Through miR-224. Biochem Biophys Res Commun (2018) 503(1):228–34. doi: 10.1016/j.bbrc.2018.06.007
107. Coulibaly A, Bettendorf A, Kostina E, Figueiredo AS, Velásquez SY, Bock HG, et al. Interleukin-15 Signaling in HIF-1α Regulation in Natural Killer Cells, Insights Through Mathematical Models. Front Immunol (2019) 10:2401. doi: 10.3389/fimmu.2019.02401
108. Naeimi Kararoudi M, Elmas E, Lamb M, Chakravarti N, Trikha P, Lee DA. Disruption of SOCS3 Promotes the Anti-Cancer Efficacy of Primary NK Cells. Blood (2018) 132(Supplement 1):5687–7. doi: 10.1182/blood-2018-99-116621
109. Niemand C, Nimmesgern A, Haan S, Fischer P, Schaper F, Rossaint R, et al. Activation of STAT3 by IL-6 and IL-10 in Primary Human Macrophages Is Differentially Modulated by Suppressor of Cytokine Signaling 3. J Immunol (2003) 170(6):3263–72. doi: 10.4049/jimmunol.170.6.3263
110. Wormald S, Zhang JG, Krebs DL, Mielke LA, Silver J, Alexander WS, et al. The Comparative Roles of Suppressor of Cytokine Signaling-1 and -3 in the Inhibition and Desensitization of Cytokine Signaling. J Biol Chem (2006) 281(16):11135–43. doi: 10.1074/jbc.M509595200
111. Meissl K, Simonović N, Amenitsch L, Witalisz-Siepracka A, Klein K, Lassnig C, et al. STAT1 Isoforms Differentially Regulate NK Cell Maturation and Anti-Tumor Activity. Front Immunol (2020), 11:2189. doi: 10.3389/fimmu.2020.02189
112. Haddad E. STAT3: Too Much may be Worse Than Not Enough! Blood (2015) 125(4):583–4. doi: 10.1182/blood-2014-11-610592
113. Laurence ADJ, Uhlig HH. When Half a Glass of STAT3 is Just Not Enough. Blood. (2016) 128(26):3020–1. doi: 10.1182/blood-2016-11-750539
114. Lorenzini T, Dotta L, Giacomelli M, Vairo D, Badolato R. STAT Mutations as Program Switchers: Turning Primary Immunodeficiencies Into Autoimmune Diseases. J Leukoc Biol (2017) 101(1):29–38. doi: 10.1189/jlb.5RI0516-237RR
115. Klein K, Stoiber D, Sexl V, Witalisz-siepracka A. Untwining Anti-Tumor and Immunosuppressive Effects of JAK Inhibitors-A Strategy for Hematological Malignancies? Cancers (Basel). (2021) 13(11):2611. doi: 10.3390/cancers13112611
116. Pelham SJ, Lenthall HC, Deenick EK, Tangye SG. Elucidating the Effects of Disease-Causing Mutations on STAT3 Function in Autosomal-Dominant Hyper-IgE Syndrome. J Allergy Clin Immunol (2016) 138(4):1210–3. doi: 10.1016/j.jaci.2016.04.020
117. de Araujo ED, Orlova A, Neubauer HA, Bajusz D, Seo HS, Dhe-Paganon S, et al. Structural Implications of STAT3 and STAT5 SH2 Domain Mutations. Cancers (Basel) (2019) 11(11):1757. doi: 10.3390/cancers11111757
118. Flanagan SE, Haapaniemi E, Russell MA, Caswell R, Allen HL, De Franco E, et al. Activating Germline Mutations in STAT3 Cause Early-Onset Multi-Organ Autoimmune Disease. Nat Genet (2014) 46(8):812–4. doi: 10.1038/ng.3040
119. Milner JD, Vogel TP, Forbes L, Ma CA, Stray-Pedersen A, Niemela JE, et al. Early-Onset Lymphoproliferation and Autoimmunity Caused by Germline STAT3 Gain-of-Function Mutations. Blood. (2015) 125(4):591–9. doi: 10.1182/blood-2014-09-602763
120. Haapaniemi EM, Kaustio M, Rajala HLM, Van Adrichem AJ, Kainulainen L, Glumoff V, et al. Autoimmunity, Hypogammaglobulinemia, Lymphoproliferation, and Mycobacterial Disease in Patients With Activating Mutations in STAT3. Blood. (2015) 125(4):639–48. doi: 10.1182/blood-2014-04-570101
121. Jägle S, Heeg M, Grün S, Rensing-Ehl A, Maccari ME, Klemann C, et al. Distinct Molecular Response Patterns of Activating STAT3 Mutations Associate With Penetrance of Lymphoproliferation and Autoimmunity. Clin Immunol (2020) 210:108316. doi: 10.1016/j.clim.2019.108316
122. Faletti L, Ehl S, Heeg M. Germline STAT3 Gain-of-Function Mutations in Primary Immunodeficiency: Impact on the Cellular and Clinical Phenotype. BioMed J (2021) 44(4):412–21. doi: 10.1016/j.bj.2021.03.003
123. Nabhani S, Schipp C, Miskin H, Levin C, Postovsky S, Dujovny T, et al. STAT3 Gain-of-Function Mutations Associated With Autoimmune Lymphoproliferative Syndrome Like Disease Deregulate Lymphocyte Apoptosis and can be Targeted by BH3 Mimetic Compounds. Clin Immunol (2017) 181:32–42. doi: 10.1016/j.clim.2017.05.021
124. Jerez A, Clemente MJ, Makishima H, Koskela H, LeBlanc F, Ng KP, et al. STAT3 Mutations Unify the Pathogenesis of Chronic Lymphoproliferative Disorders of NK Cells and T-Cell Large Granular Lymphocyte Leukemia. Blood. (2012) 120(15):3048–57. doi: 10.1182/blood-2012-06-435297
125. Fasan A, Kern W, Grossmann V, Haferlach C, Haferlach T, Schnittger S. STAT3 Mutations are Highly Specific for Large Granular Lymphocytic Leukemia. Leukemia. (2013) 27(7):1598–600. doi: 10.1038/leu.2012.350
126. Barilà G, Teramo A, Calabretto G, Vicenzetto C, Gasparini VR, Pavan L, et al. Stat3 Mutations Impact on Overall Survival in Large Granular Lymphocyte Leukemia: A Single-Center Experience of 205 Patients. Leukemia. (2020) 34(4):1116–24. doi: 10.1038/s41375-019-0644-0
127. Gasparini VR, Binatti A, Coppe A, Teramo A, Vicenzetto C, Calabretto G, et al. A High Definition Picture of Somatic Mutations in Chronic Lymphoproliferative Disorder of Natural Killer Cells. Blood Cancer J (2020) 10(4):42. doi: 10.1038/s41408-020-0309-2
128. Küçük C, Jiang B, Hu X, Zhang W, Chan JKC, Xiao W, et al. Activating Mutations of STAT5B and STAT3 in Lymphomas Derived From γδ-T or NK Cells. Nat Commun (2015) 6:6025. doi: 10.1038/ncomms7025
129. Lee S, Park HY, Kang SY, Kim SJ, Hwang J, Lee S, et al. Genetic Alterations of JAK/STAT Cascade and Histone Modification in Extranodal NK/T-Cell Lymphoma Nasal Type. Oncotarget. (2015) 6(19):17764–76. doi: 10.18632/oncotarget.3776
130. Dufva O, Kankainen M, Kelkka T, Sekiguchi N, Awad SA, Eldfors S, et al. Aggressive Natural Killer-Cell Leukemia Mutational Landscape and Drug Profiling Highlight JAK-STAT Signaling as Therapeutic Target. Nat Commun (2018) 9(1):1567. doi: 10.1038/s41467-018-03987-2
131. Huang L, Liu D, Wang N, Ling S, Tang Y, Wu J, et al. Integrated Genomic Analysis Identifies Deregulated JAK/STAT-MYC-Biosynthesis Axis in Aggressive NK-Cell Leukemia. Cell Res (2018) 28(2):172–86. doi: 10.1038/cr.2017.146
132. Song TL, Nairismägi ML, Laurensia Y, Lim JQ, Tan J, Li ZM, et al. Oncogenic Activation of the STAT3 Pathway Drives PD-L1 Expression in Natural Killer/T-Cell Lymphoma. Blood. (2018) 132(11):1146–58. doi: 10.1182/blood-2018-01-829424
133. Seffens A, Herrera A, Tegla C, Buus TB, Hymes KB, Ødum N, et al. STAT3 Dysregulation in Mature T and NK Cell Lymphomas. Cancers (Basel) (2019) 11(11):1711. doi: 10.3390/cancers11111711
134. Kawakami T, Sekiguchi N, Kobayashi J, Yamane T, Nishina S, Sakai H, et al. STAT3 Mutations in Natural Killer Cells are Associated With Cytopenia in Patients With Chronic Lymphoproliferative Disorder of Natural Killer Cells. Int J Hematol (2019) 109(5):563–71. doi: 10.1007/s12185-019-02625-x
135. Barilà G, Teramo A, Calabretto G, Ercolin C, Boscaro E, Trimarco V, et al. Dominant Cytotoxic NK Cell Subset Within CLPD-NK Patients Identifies a More Aggressive NK Cell Proliferation. Blood Cancer J (2018) 8(6):51. doi: 10.1038/s41408-018-0088-1
136. Coppo P, Gouilleux-Gruart V, Huang Y, Bouhlal H, Bouamar H, Bouchet S, et al. STAT3 Transcription Factor Is Constitutively Activated and is Oncogenic in Nasal-Type NK/T-Cell Lymphoma. Leukemia. (2009) 23(9):1667–78. doi: 10.1038/leu.2009.91
137. Teramo A, Gattazzo C, Passeri F, Lico A, Tasca G, Cabrelle A, et al. Intrinsic and Extrinsic Mechanisms Contribute to Maintain the JAK/STAT Pathway Aberrantly Activated in T-Type Large Granular Lymphocyte Leukemia. Blood. (2013) 121(19):3843–54. doi: 10.1182/blood-2012-07-441378
138. Barilà G, Calabretto G, Teramo A, Vicenzetto C, Gasparini VR, Semenzato G, et al. T Cell Large Granular Lymphocyte Leukemia and Chronic NK Lymphocytosis. Best Pract Res Clin Haematol (2019) 32(3):207–16. doi: 10.1016/j.beha.2019.06.006
139. Kim D, Park G, Huuhtanen J, Ghimire B, Rajala H, Moriggl R, et al. STAT3 Activation in Large Granular Lymphocyte Leukemia is Associated With Cytokine Signaling and DNA Hypermethylation. Leukemia. (2021) 35(12):3430–43. doi: 10.1038/s41375-021-01296-0
140. Yu H, Kortylewski M, Pardoll D. Crosstalk Between Cancer and Immune Cells: Role of STAT3 in the Tumour Microenvironment. Nat Rev Immunol (2007) 7(1):41–51. doi: 10.1038/nri1995
141. Epling-Burnette PK, Liu JH, Catlett-Falcone R, Turkson J, Oshiro M, Kothapalli R, et al. Inhibition of STAT3 Signaling Leads to Apoptosis of Leukemic Large Granular Lymphocytes and Decreased Mcl-1 Expression. J Clin Invest (2001) 107(3):351–61. doi: 10.1172/JCI9940
142. Lamy T, Moignet A, Loughran TP. LGL Leukemia: From Pathogenesis to Treatment. Blood. (2017) 129(9):1082–94. doi: 10.1182/blood-2016-08-692590
143. Pastoret C, Desmots F, Drillet G, Le Gallou S, Boulland ML, Thannberger A, et al. Linking the KIR Phenotype With STAT3 and TET2 Mutations to Identify Chronic Lymphoproliferative Disorders of NK Cells. Blood. (2021) 137(23):3237–50. doi: 10.1182/blood.2020006721
144. Pardoll DM. The Blockade of Immune Checkpoints in Cancer Immunotherapy. Nat Rev Cancer (2012) 12(4):252–64. doi: 10.1038/nrc3239
145. Poggi A, Zocchi MR. Natural Killer Cells and Immune-Checkpoint Inhibitor Therapy: Current Knowledge and New Challenges. Mol Ther - Oncolytics (2022) 24:26–42. doi: 10.1016/j.omto.2021.11.016
146. Benson DM, Bakan CE, Mishra A, Hofmeister CC, Efebera Y, Becknell B, et al. The PD-1/PD-L1 Axis Modulates the Natural Killer Cell Versus Multiple Myeloma Effect: A Therapeutic Target for CT-011, a Novel Monoclonal Anti–PD-1 Antibody. Blood. (2010) 116(13):2286–94. doi: 10.1182/blood-2010-02-271874
147. Beldi-Ferchiou A, Lambert M, Dogniaux S, Vély F, Vivier E, Olive D, et al. PD-1 Mediates Functional Exhaustion of Activated NK Cells in Patients With Kaposi Sarcoma. Oncotarget. (2016) 7(45):72961–77. doi: 10.18632/oncotarget.12150
148. Vari F, Arpon D, Keane C, Hertzberg MS, Talaulikar D, Jain S, et al. Immune Evasion via PD-1/PD-L1 on NK Cells and Monocyte/Macrophages is More Prominent in Hodgkin Lymphoma Than DLBCL. Blood. (2018) 131(16):1809–19. doi: 10.1182/blood-2017-07-796342
149. Concha-Benavente F, Kansy B, Moskovitz J, Moy J, Chandran U, Ferris RL. PD-L1 Mediates Dysfunction in Activated PD-1 Þ NK Cells in Head and Neck Cancer Patients. Cancer Immunol Res (2018) 6(12):1548–60. doi: 10.1158/2326-6066.CIR-18-0062
150. Judge SJ, Dunai C, Aguilar EG, Vick SC, Sturgill IR, Khuat LT, et al. Minimal PD-1 Expression in Mouse and Human NK Cells Under Diverse Conditions. J Clin Invest (2020) 130(6):3051–68. doi: 10.1172/JCI133353
151. Cho MM, Quamine AE, Olsen MR, Capitini CM. Programmed Cell Death Protein 1 on Natural Killer Cells: Fact or Fiction? J Clin Invest Am Soc Clin Invest (2020) 130:2816–9. doi: 10.1172/JCI137051
152. Atsaves V, Tsesmetzis N, Chioureas D, Kis L, Leventaki V, Drakos E, et al. PD-L1 is Commonly Expressed and Transcriptionally Regulated by STAT3 and MYC in ALK-Negative Anaplastic Large-Cell Lymphoma. Leukemia. (2017) 31(7):1633–7. doi: 10.1038/leu.2017.103
153. Marzec M, Zhang Q, Goradia A, Raghunath PN, Liu X, Paessler M, et al. Oncogenic Kinase NPM/ALK Induces Through STAT3 Expression of Immunosuppressive Protein CD274 (PD-L1, B7-H1). Proc Natl Acad Sci U S A (2008) 105(52):20852–7. doi: 10.1073/pnas.0810958105
154. Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep (2017) 19(6):1189–201. doi: 10.1016/j.celrep.2017.04.031
155. Luo F, Luo M, Rong QX, Zhang H, Chen Z, Wang F, et al. Niclosamide, an Antihelmintic Drug, Enhances Efficacy of PD-1/PD-L1 Immune Checkpoint Blockade in non-Small Cell Lung Cancer. J Immunother Cancer (2019) 7(1):245. doi: 10.1186/s40425-019-0733-7
156. Ku JM, Hong SH, Kim HI, Kim MJ, Ku S-J, Bae K-R, et al. SH003 Overcomes Drug Resistance and Immune Checkpoints by Inhibiting JAK-STAT3 Signaling in MCF7/ADR Cells. Phytomed Plus (2021) 1(4):100111. doi: 10.1016/j.phyplu.2021.100111
157. Xu LJ, Ma Q, Zhu J, Li J, Xue BX, Gao J, et al. Combined Inhibition of JAK1,2/Stat3−PD−L1 Signaling Pathway Suppresses the Immune Escape of Castration−Resistant Prostate Cancer to NK Cells in Hypoxia. Mol Med Rep (2018) 17(6):8111–20. doi: 10.3892/mmr.2018.8905
158. Stanietsky N, Simic H, Arapovic J, Toporik A, Levy O, Novik A, et al. The Interaction of TIGIT With PVR and PVRL2 Inhibits Human NK Cell Cytotoxicity. Proc Natl Acad Sci U S A (2009) 106(42):17858–63. doi: 10.1073/pnas.0903474106
159. Triebel F, Jitsukawa S, Baixeras E, Roman-Roman S, Genevee C, Viegas-Pequignot E, et al. LAG-3, a Novel Lymphocyte Activation Gene Closely Related to CD4. J Exp Med (1990) 171(5):1393–405. doi: 10.1084/jem.171.5.1393
160. Gleason MK, Lenvik TR, McCullar V, Felices M, O’Brien MS, Cooley SA, et al. Tim-3 is an Inducible Human Natural Killer Cell Receptor That Enhances Interferon Gamma Production in Response to Galectin-9. Blood. (2012) 119(13):3064–72. doi: 10.1182/blood-2011-06-360321
161. Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: Co-Inhibitory Receptors With Specialized Functions in Immune Regulation. Immunity. (2016) 44(5):989–1004. doi: 10.1016/j.immuni.2016.05.001
162. Huang L, Xu Y, Fang J, Liu W, Chen J, Liu Z, et al. Targeting STAT3 Abrogates Tim-3 Upregulation of Adaptive Resistance to PD-1 Blockade on Regulatory T Cells of Melanoma. Front Immunol (2021) 12:1120. doi: 10.3389/fimmu.2021.654749
163. Sun H, Sun C, Xiao W. Expression Regulation of Co-Inhibitory Molecules on Human Natural Killer Cells in Response to Cytokine Stimulations. Cytokine. (2014) 65(1):33–41. doi: 10.1016/j.cyto.2013.09.016
164. González-Ochoa S, Tellez-Bañuelos MC, Méndez-Clemente AS, Bravo-Cuellar A, Flores GH, Palafox-Mariscal LA, et al. Combination Blockade of the IL6R/STAT-3 Axis With TIGIT and Its Impact on the Functional Activity of NK Cells Against Prostate Cancer Cells. Wang S ed J Immunol Res (2022) 2022:1–19. doi: 10.1155/2022/1810804
165. Eckelhart E, Warsch W, Zebedin E, Simma O, Stoiber D, Kolbe T, et al. A Novel Ncr1 -Cre Mouse Reveals the Essential Role of STAT5 for NK-Cell Survival and Development. Blood. (2011) 117(5):1565–74. doi: 10.1182/blood-2010-06-291633
166. Villarino AV, Sciumè G, Davis FP, Iwata S, Zitti B, Robinson GW, et al. Subset- and Tissue-Defined STAT5 Thresholds Control Homeostasis and Function of Innate Lymphoid Cells. J Exp Med (2017) 214(10):2999–3014. doi: 10.1084/jem.20150907
167. Walker SR, Nelson EA, Yeh JE, Pinello L, Yuan G-C, Frank DA. STAT5 Outcompetes STAT3 to Regulate the Expression of the Oncogenic Transcriptional Modulator BCL6. Mol Cell Biol (2013) 33(15):2879–90. doi: 10.1128/MCB.01620-12
168. Hu X, li J, Fu M, Zhao X, Wang W. The JAK/STAT Signaling Pathway: From Bench to Clinic. Signal Transduct Target Ther (2021) 6(1):1–33. doi: 10.1038/s41392-021-00791-1
169. Beebe JD, Liu JY, Zhang JT. Two Decades of Research in Discovery of Anticancer Drugs Targeting STAT3, How Close Are We? Pharmacol Ther (2018) 191:74–91. doi: 10.1016/j.pharmthera.2018.06.006
Keywords: STAT3, NK cells, tumor immune surveillance, JAK-STAT, immunotherapy
Citation: Witalisz-Siepracka A, Klein K, Zdársky B and Stoiber D (2022) The Multifaceted Role of STAT3 in NK-Cell Tumor Surveillance. Front. Immunol. 13:947568. doi: 10.3389/fimmu.2022.947568
Received: 18 May 2022; Accepted: 13 June 2022;
Published: 05 July 2022.
Edited by:
Camille Guillerey, The University of Queensland, AustraliaReviewed by:
Holger A. Lindner, University of Heidelberg, GermanyCopyright © 2022 Witalisz-Siepracka, Klein, Zdársky and Stoiber. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dagmar Stoiber, ZGFnbWFyLnN0b2liZXJAa2wuYWMuYXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.