 Peck Y. Ong
Peck Y. Ong
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 27 July 2022
Sec. Molecular Innate Immunity
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.943640
This article is part of the Research Topic Innate Immunity: A Top Player in Inflammatory Skin Diseases? View all 8 articles
Atopic dermatitis (AD) is a chronic inflammatory skin disease with barrier defects and immune dysregulations. The pathogenesis of AD involves the physical barrier as well as epithelial cells, which are considered a vital part of the innate immunity of the skin. The importance of filaggrin mutations in the pathogenesis of AD has also been well-established with reproducible results around the world in multiple studies and ethnic groups. This protein plays an important role in skin barrier functions and further reaffirms barrier defects as one of the primary causes of AD. The main epithelial cells, keratinocytes, function as a major sentinel for the skin in detecting danger signals or microbial pathogens, and trigger downstream immune responses. In AD, these cells express TSLP, IL-33 and IL-25, which lead to downstream systemic production of type 2 cytokines. In spite of major advances in our understanding of the innate immunity of AD, recent success in the systemic therapeutics of AD have focused on targeting the products of the adaptive immunity, particularly cytokines produced by T cells. In addition to type 2 cytokines, type 17 cytokines have also been implicated in the pathogenesis of AD. The current review examines the implications of these cytokines in AD from clinical perspectives.
Atopic dermatitis (AD) is a chronic inflammatory skin disease that typically presents in early childhood. It is characterized by dry, erythematous and pruritic skin rashes. These rashes lead to a vicious cycle of itching and scratching that eventually disrupts sleep and daily activities. The poor quality of life in AD patients, as compared to other chronic conditions, has been well-documented (1). Patients with AD are conscious about their rashes and may be bullied or discriminated by peers due to their appearance (2). Adult patients may make life and career decisions based on their AD (3). Family members of AD patients are also affected due to the care and attention that are needed in these patients (4). Other co-morbidities of AD include depression, anxiety disorder, infections and other atopic diseases including food allergy, asthma and allergic rhinitis.
Looking back 20 years in our understanding of AD pathogenesis (5), which highlighted the importance of cell-mediated immunity, the current review aims to re-examine what we have learnt from the recent development in the treatments of AD. Products of cell-mediated immunity including type 2 cytokines, IL-4, IL-5 and IL-13, and type 1 cytokine, IFN-γ, were thought to be important in the acute and chronic lesions of AD, respectively. Clinical studies with anti-IL-5 monoclonal antibody (mAb) and IFN-γ in AD patients have either been ineffective or inconclusive (6). On the other hand, targeting both IL-4 and IL-13 has revolutionized the treatment of moderate to severe AD. The efficacy of blocking both IL-4 and IL-13 by dupilumab, which targets the IL-4α receptor, has been shown in both phase 2 and 3 trials (7, 8). The observation in real-world setting has further confirmed the efficacy of dupilumab (9), and established the role of IL-4 and IL-13 in the pathogenesis of AD. The approval of dupilumab led the way to more systemic treatments for AD that aim to target type 2 cytokines or type 2-associated cytokines such as IL-31. These recently-approved treatments include anti-IL-13 mAb and oral Janus kinase (JAK) inhibitors (10).
Th2 cells were thought to be the main source of IL-4 and IL-13 (5). More recent studies have uncovered other cells that are capable of producing these two cytokines and other cytokines that enhance the production of IL-4 and IL-13 (11). In addition to IL-4 and IL-13, an increased expression of IL-17A and its potential pathogenic role has also become an emerging concept in AD (12, 13). To further understand the origin of these cytokines in AD, a basic understanding of the immune system in relation to AD is needed.
“The immune system clearly exists to protect the host from infectious agents” (14). Janeway’s deduction of the innate immune response being specific for infectious non-self led to his postulation of pattern-recognition receptors and eventual discovery of toll-like receptors (TLRs) (15). These receptors act as the immune system’s sentinel for invading microbial pathogens and initiate immune response against these organisms. A classic pathway of immune response at the mucosal surface is the activation of epithelial cells by TLR, leading to the production of chemokines, which summon antigen-presenting cells and effector T cells to the site of infection (16). IFN-γ production by Th1 cells has long known to be an important mediator of cellular immunity. The importance of type 17 immunity is illustrated by STAT3 mutation in autosomal dominant hyper-IgE syndrome, which results in a lack of Th17 cell development and increased susceptibility to skin bacterial infections and other mucosal fungal opportunistic infections such as oral candidiasis and pneumocystis lung infections (17, 18). Other contributors of type 17 immunity in the skin include innate γδ T cells and innate lymphoid cell group 3 (ILC3) (19, 20). Much of Janeway’s concept has been applied to study immune responses during an actual or impending infection. The mucosal barrier was thought to be simply a physical barrier and sensor of infectious threats. More recent understanding of barrier defense presents a more complex mechanism that involves more than just the epithelial cells, but also commensal microbes and adaptive immunity. The need for an efficient barrier is not only for prevention of infections, but also to prevent collateral damage from an effector response against an invading pathogen. Excessive type 1 and type 17 responses present as inflammation that can be devastating to the host, as seen in conditions such as psoriasis and other autoimmune conditions. While stratum corneum, corneocytes, keratinocytes and epidermal molecules including filaggrin, lipids and tight junctions serve as the physical barrier against the invasion of pathogens, any compromise to this physical barrier (eg. an inherent barrier defect such as filaggrin deficiency or an immature newborn skin) would put the skin at risk for infection and inflammation. The so-called homeostatic immunity involves the interaction between adaptive immunity and skin microbiota (21). It has emerged as an important concept in maintaining and repairing skin barrier. Beneath the skin barrier is the home for many memory T cells. Indeed, adult skin contains four times as many memory T cells as there are in peripheral blood (22). Among these memory T cells are a subset known as tissue-resident memory T cells (TRM), which are non-circulating T cells that are tasked to respond rapidly to invading pathogens (23). These pathogens are captured and processed by antigen-presenting cells, and presented to T cells bearing T cell receptors (TCRs) that are specific for the pathogens in the context of major histocompatibility complex (MHC) restriction. However, it is now known that not all TCRs among TRM are specific for pathogens. Some of the TCRs are directed towards normal skin microbiota such as coagulase-negative staphylococci (CoNS). A subsets of TRM that has emerged as the main commensal-specific T cells is the CD8+ T cells that express IL-17A (Tc17) (24). TCRs on S. epidermidis-specific Tc17 are restricted to unconventional MHC class Ib molecule, rather than the classic MHC Ia molecule for CD8+ T cells. Of interest, the peptides that are recognized by these Tc17 cells come from secreted proteins of the commensal bacteria. One such peptide is N-formyl methionine from S. epidermidis. These S. epidermidis-specific Tc17 were found to express a panel of genes that are involved in tissue repair. They were also shown to promote and accelerate wound healing. Harrison et al. (25) subsequently found that S. epidermidis-specific Tc17 are capable of co-expressing the transcription factors retinoic acid-related orphan receptor-γt (RORγt) for type 17 immunity as well as GATA-binding protein 3 (GATA-3) for type 2 immunity (25). They found that these Tc17 cells express IL-5 and IL-13 mRNA without producing IL-5 and IL-13 proteins. On skin barrier breach and an encounter between these specific Tc17 and S. epidermidis leads to the production of IL-5 and IL-13 in the presence of IL-18. As type 2 immunity has been implicated to play a role in barrier repair (26), the study by Harrison et al. demonstrates the plasticity of Tc17 to switch between type 17 immunity against invading pathogens and type 2 immunity for barrier repair. Mucosal-associated invariant T (MAIT) cells are T cells expressing invariant αβ TCR that also recognizes processed peptides in the context of MHC class Ib (27). A classic example of these peptides are microbial-derived peptides from riboflavin (vitamin B12) (28). MAIT cells develop in a specific time window during the neonatal period when exposed to riboflavin-synthesizing bacteria (27). Cutaneous MAIT cells express IL-17A but not interferon-γ. These cells display IL-23R and their development can be promoted by IL-23 in the presence of riboflavin-producing bacteria. However, riboflavin-synthesizing S. epidermidis-specific MAIT cells expand in the presence of IL-18, rather than IL-23 (27). MAIT cells regulate response to tissue injury and promote tissue repair.
One of the primary defects of AD is skin barrier defects. Normal-looking AD skin has been shown to be deficient in cholesterol, fatty acid, ceramides, filaggrin and tight junctions (29, 30). The observation that AD skin is deficient in filaggrin led to the eventual confirmation of filaggrin loss-of-function mutations in AD patients (31, 32). This establishes genetic barrier defects as one of the primary causes of AD. Another well-established genetic cause of AD is atopy. Parental history of atopic diseases including allergic rhinitis or asthma are known risk factors for AD (33). AD lesions are characterized by an increased expression of type 2 cytokines including IL-4, IL-5 and IL-13 (34, 35). Normal-looking AD skin also contains subclinical inflammation with increased expression of type 2 cytokines, as compared to healthy skin (34). AD keratinocytes express an increased level TSLP, IL-33 and IL-25 (36–38). These cytokines act as alarmins that induce the production of IL-4, IL-5 and IL-13 from ILC2 and Th2 cells (Figure 1). The role of Th22 cells in AD remains to be defined. A subset of CD1a-restricted Th22 cells reside primarily in the skin (39). These cells recognize self-lipid antigen presented on CD1a or CD1a itself independent of lipid antigen (39, 40). Their expression of IL-22 likely plays a role in epithelial repair mechanisms (40). TSLP also induces Th2-expression of IL-31, which has been shown to play a major role in the itch of AD (41). The pathogenic role of IL-4 and IL-13 in AD has now been well-supported by the resolution of clinical inflammation with the use of dupilumab in moderate to severe AD patients. In addition to causing inflammation, the chronic expression of these cytokines in AD lesions is a predisposing factor for increased Staphylococcus aureus (S. aureus) colonization and infections in these patients. This has also been supported by the use of dupilumab in AD patients in whom S. aureus colonization and clinical infections were shown to decrease significantly (42–44). Plausible mechanisms by which IL-4 and IL-13 contribute to S. aureus colonization and skin infections is through their suppressive effects on skin barrier functions, expression of antimicrobial peptides (AMPs) and type 17 immunity (45–47). Although the expression of IL-17A has consistently been found to be increased in AD lesions (48, 49), the role of this cytokine in AD pathogenesis is controversial. The use of monoclonal antibodies against IL-17A or IL-12/23, which inhibits the Th1 and Th17 pathways, have failed to improve AD disease severity (50–52). These observations argue against the role of IL-17A as part of AD inflammation, rather, its presence is part of an effector response against S. aureus (47, 53). Of interest, AD lesional IL-17A expression decreases after dupilumab treatment (54), further supporting its presence is in response to an increase in type 2 inflammation and S. aureus. While recurrent skin infections are common in AD patients, invasive staphylococcal infections are relatively rare and opportunistic fungal infections are absent in AD patients (55). The increased, yet attenuated, type 17 immunity in AD likely contributes to these protections. The role of CD8+ T cells in AD has been shown in mouse models (56, 57). CD4+ T cell-depleted AD lesions were predominated by IL-17-producing CD8+ T cells (57). In human AD lesions, an increased number of CD8+ T cells has also been described (58). Interestingly, most of these CD8+ T cells were found in the epidermis (58). There is a high percentage of IL-13-producing CD8+ T cells with a smaller number of IL-17A-producing CD8+ T cells in AD lesions (58). Whether these cells co-express type 2 and type 17 transcription factors, as described by Harrison et al. (25), will need further investigations. Further studies are also needed to analyze the specificity of their TCRs for CoNS or other cutaneous commensal bacteria. CD8+ MAIT cells have been found to be a source of IL-17A in human psoriatic lesions (59), whether these cells play a role in the barrier defense of AD will be also require further studies. The interaction between commensal-specific T cells and CoNS in barrier defense is dependent on the strains of CoNS as well as their mode of growth (biofilm vs planktonic) (60). Biofilm formation by S. epidermidis potentially inhibits the interaction between these bacteria and commensal-specific T cells, leading to a decreased production of IL-17A, therefore favoring S. aureus colonization (60). S. aureus virulence is also a major factor in AD inflammation. These bacteria are capable of inducing TSLP and IL-33, rather than AMPs, from keratinocytes of AD patients (61, 62). Superantigens produced by S. aureus also induce the expression IL-31 in AD patients (63). The underlying AD inflammation and clinical severity has been shown to be driven by differences in strain-specific S. aureus and S. epidermidis colonization (64).
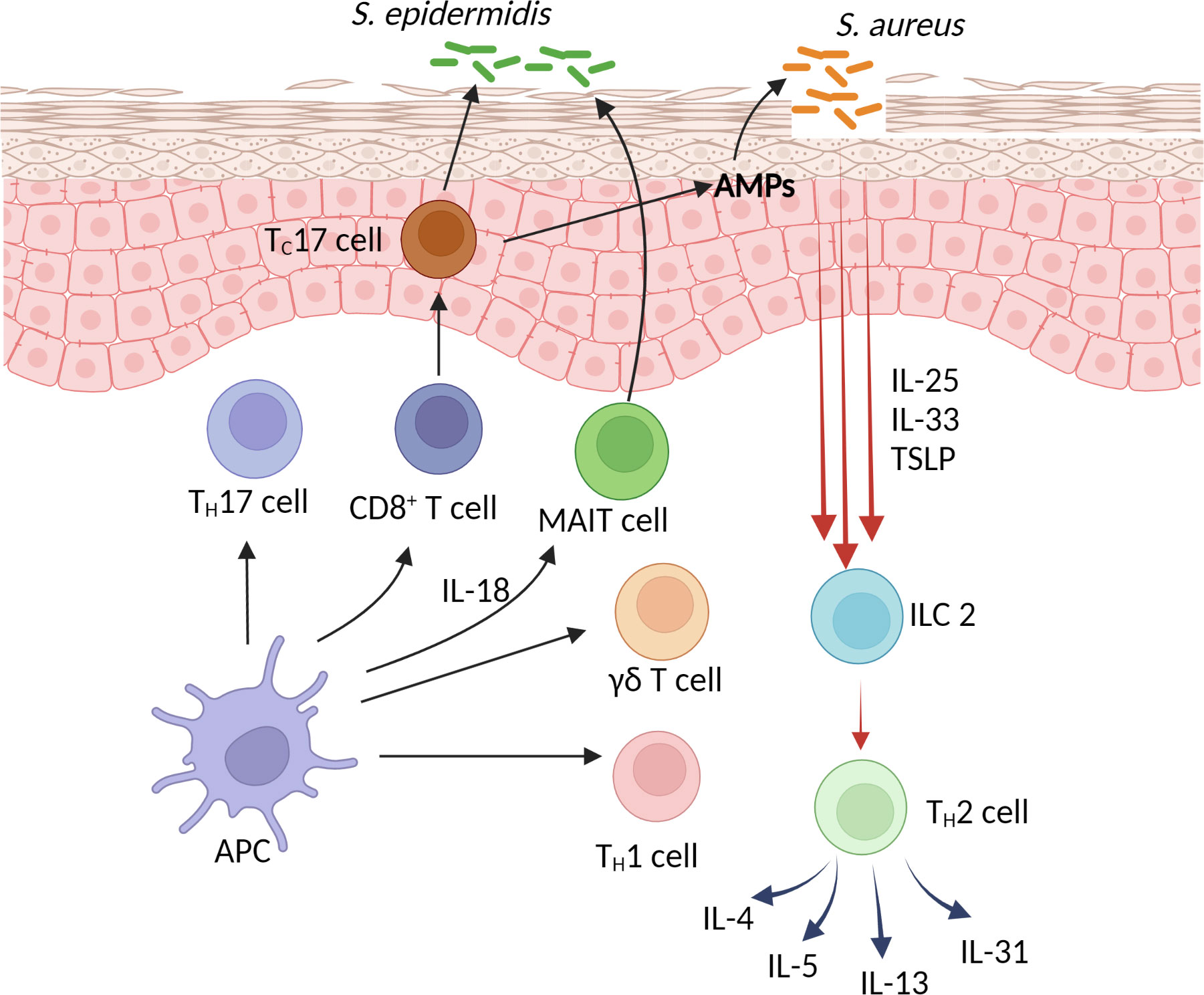
Figure 1 Skin barrier defense and the pathogenesis of atopic dermatitis.
The pathogenesis of AD remains an intriguing topic over many decades. With the recent use of mAbs in the therapy of AD has broadened our understanding of the pathogenesis of this chronic skin condition. Type 2 cytokines, IL-4 and IL-13, not only contribute to AD inflammation, these 2 cytokines also predispose to S. aureus colonization and infections in AD patients. On the other hand, AD patients appear to have an intact type 17 immunity without contributing to AD inflammation. This is supported by an absence of rare opportunistic infections and a lack of efficacy by anti-IL-17A and anti-IL-12/23 therapies in the treatment of AD. While defects in skin barrier functions and innate immunity play an important role in the pathogenesis of AD, type 2 and type 17 cytokines have traditionally been thought to be contributed by Th2 and Th17 cells, respectively. However, recent concepts in mucosal immunity show that innate cells such as ILC2 and ILC3, and innate-like lymphocytes such as Tc17, MAIT cells and γδ T cells, which have both adaptive and innate properties that recognize nonclassical MHC Ib molecule, are also important contributors of type 2 and type 17 cytokines. Therefore, both innate and adaptive immunity likely contributes equally to the pathogenesis of AD.
PO conceived the ideas, concepts, and wrote the paper.
I thank Dr. Juli Wu for her assistance with Figure 1. PO is supported in part by The Albert and Bettie Sacchi Foundation.
PO has been on advisory board for Incyte, Abbvie, Janssen, and has obtained research funding from Regeneron, Sanofi Genzyme, Leo, Incyte.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Beattie PE, Lewis-Jones MS. A comparative study of impairment of quality of life in children with skin disease and children with other chronic childhood diseases. Br J Dermatol (2006) 155(1):145–51. doi: 10.1111/j.1365-2133.2006.07185.x
2. Stingeni L, Belloni Fortina A, Baiardini I, Hansel K, Moretti D, Cipriani F. Atopic dermatitis and patient perspectives: Insights of bullying at school and career discrimination at work. J Asthma Allergy (2021) 14:919–28. doi: 10.2147/JAA.S317009
3. Foulkes AC, Warren RB. Patients' narratives. Curr Probl Dermatol (2013) 44:145–57. doi: 10.1159/000350194
4. Capozza K, Gadd H, Kelley K, Russell S, Shi V, Schwartz A. Insights from caregivers on the impact of pediatric atopic dermatitis on families: "I'm tired, overwhelmed, and feel like I'm failing as a mother". Dermatitis (2020) 31(3):223–7. doi: 10.1097/DER.0000000000000582
6. Siegels D, Heratizadeh A, Abraham S, Binnmyr J, Brockow K, Irvine AD, et al. European Academy of allergy, clinical immunology atopic dermatitis guideline group. Systemic treatments in the management of atopic dermatitis: A systematic review and meta-analysis. Allergy (2021) 76(4):1053–76. doi: 10.1111/all.14631
7. Beck LA, Thaçi D, Hamilton JD, Graham NM, Bieber T, Rocklin R, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N Engl J Med (2014) 371(2):130–9. doi: 10.1056/NEJMoa1314768
8. Simpson EL, Bieber T, Guttman-Yassky E, Beck LA, Blauvelt A, Cork MJ, et al. SOLO 1 and SOLO 2 investigators. two phase 3 trials of dupilumab versus placebo in atopic dermatitis. N Engl J Med (2016) 375(24):2335–48. doi: 10.1056/NEJMoa1610020
9. Silverberg JI, Simpson EL, Boguniewicz M, De Bruin-Weller MS, Foley P, Kataoka Y, et al. Dupilumab provides rapid and sustained clinically meaningful responses in adults with moderate-to-severe atopic dermatitis. Acta Derm Venereol (2021) 101(11):adv00585. doi: 10.2340/actadv.v101.307
10. Leung DYM, Paller AS, Guttman-Yassky E. New therapies for atopic dermatitis: How will they impact skin care? Ann Allergy Asthma Immunol (2022) 128(4):344–5. doi: 10.1016/j.anai.2022.01.023
11. Cevikbas F, Steinhoff M. IL-33: a novel danger signal system in atopic dermatitis. J Invest Dermatol (2012) 132(5):1326–9. doi: 10.1038/jid.2012.66
12. Esaki H, Brunner PM, Renert-Yuval Y, Czarnowicki T, Huynh T, Tran G, et al. Early-onset pediatric atopic dermatitis is TH2 but also TH17 polarized in skin. J Allergy Clin Immunol (2016) 138(6):1639–51. doi: 10.1016/j.jaci.2016.07.013
13. Noda S, Suárez-Fariñas M, Ungar B, Kim SJ, de Guzman Strong C, Xu H, et al. The Asian atopic dermatitis phenotype combines features of atopic dermatitis and psoriasis with increased TH17 polarization. J Allergy Clin Immunol (2015) 136(5):1254–64. doi: 10.1016/j.jaci.2015.08.015
14. Janeway CA Jr. Approaching the asymptote? evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol (1989) 54 Pt 1:1–13. doi: 10.1101/sqb.1989.054.01.003
15. Medzhitov R, Preston-Hurlburt P, Janeway CA Jr. A human homologue of the drosophila toll protein signals activation of adaptive immunity. Nature (1997) 388(6640):394–7. doi: 10.1038/41131
16. Yang D, Chertov O, Bykovskaia SN, Chen Q, Buffo MJ, Shogan J, et al. Beta-defensins: linking innate and adaptive immunity through dendritic and T cell CCR6. Science (1999) 286(5439):525–8. doi: 10.1126/science.286.5439.525
17. Holland SM, DeLeo FR, Elloumi HZ, Hsu AP, Uzel G, Brodsky N, et al. STAT3 mutations in the hyper-IgE syndrome. N Engl J Med (2007) 357(16):1608–19. doi: 10.1056/NEJMoa073687
18. Milner JD, Brenchley JM, Laurence A, Freeman AF, Hill BJ, Elias KM, et al. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature (2008) 452(7188):773–6. doi: 10.1038/nature06764
19. Papotto PH, Ribot JC, Silva-Santos B. IL-17+ γδ T cells as kick-starters of inflammation. Nat Immunol (2017) 18(6):604–11. doi: 10.1038/ni.3726
20. Valle-Noguera A, Ochoa-Ramos A, Gomez-Sánchez MJ, Cruz-Adalia A. Type 3 innate lymphoid cells as regulators of the host-pathogen interaction. Front Immunol (2021) 12:748851. doi: 10.3389/fimmu.2021.748851
21. Belkaid Y, Harrison OJ. Homeostatic immunity and the microbiota. Immunity (2017) 46(4):562–76. doi: 10.1016/j.immuni.2017.04.008
22. Clark RA, Chong B, Mirchandani N, Brinster NK, Yamanaka K, Dowgiert RK, et al. The vast majority of CLA+ T cells are resident in normal skin. J Immunol (2006) 176(7):4431–9. doi: 10.4049/jimmunol.176.7.4431
23. Ho AW, Kupper TS. T Cells and the skin: from protective immunity to inflammatory skin disorders. Nat Rev Immunol (2019) 19(8):490–502. doi: 10.1038/s41577-019-0162-3
24. Linehan JL, Harrison OJ, Han SJ, Byrd AL, Vujkovic-Cvijin I, Villarino AV, et al. Non-classical immunity controls microbiota impact on skin immunity and tissue repair. Cell (2018) 172(4):784–96. doi: 10.1016/j.cell.2017.12.033
25. Harrison OJ, Linehan JL, Shih HY, Bouladoux N, Han SJ, Smelkinson M, et al. Commensal-specific T cell plasticity promotes rapid tissue adaptation to injury. Science (2019) 363(6422):eaat6280. doi: 10.1126/science.aat6280
26. Gieseck RL 3rd, Wilson MS, Wynn TA. Type 2 immunity in tissue repair and fibrosis. Nat Rev Immunol (2018) 18(1):62–76. doi: 10.1038/nri.2017.90
27. Constantinides MG, Link VM, Tamoutounour S, Wong AC, Perez-Chaparro PJ, Han SJ, et al. MAIT cells are imprinted by the microbiota in early life and promote tissue repair. Science (2019) 366(6464):eaax6624. doi: 10.1126/science.aax6624
28. Kjer-Nielsen L, Patel O, Corbett AJ, Le Nours J, Meehan B, Liu L, et al. MR1 presents microbial vitamin b metabolites to MAIT cells. Nature (2012) 491(7426):717–23. doi: 10.1038/nature11605
29. Elias PM. Primary role of barrier dysfunction in the pathogenesis of atopic dermatitis. Exp Dermatol (2018) 27(8):847–51. doi: 10.1111/exd.13693
30. De Benedetto A, Rafaels NM, McGirt LY, Ivanov AI, Georas SN, Cheadle C, et al. Tight junction defects in patients with atopic dermatitis. J Allergy Clin Immunol (2011) 127(3):773–86.e1-7. doi: 10.1016/j.jaci.2010.10.018
31. Seguchi T, Cui CY, Kusuda S, Takahashi M, Aisu K, Tezuka T. Decreased expression of filaggrin in atopic skin. Arch Dermatol Res (1996) 288(8):442–6. doi: 10.1007/BF02505232
32. Palmer CN, Irvine AD, Terron-Kwiatkowski A, Zhao Y, Liao H, Lee SP, et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet (2006) 38(4):441–6. doi: 10.1038/ng1767
33. Ravn NH, Halling AS, Berkowitz AG, Rinnov MR, Silverberg JI, Egeberg A, et al. How does parental history of atopic disease predict the risk of atopic dermatitis in a child? a systematic review and meta-analysis. J Allergy Clin Immunol (2020) 145(4):1182–93. doi: 10.1016/j.jaci.2019.12.899
34. Hamid Q, Boguniewicz M, Leung DY. Differential in situ cytokine gene expression in acute versus chronic atopic dermatitis. J Clin Invest (1994) 94(2):870–6. doi: 10.1172/JCI117408
35. Hamid Q, Naseer T, Minshall EM, Song YL, Boguniewicz M, Leung DY. In vivo expression of IL-12 and IL-13 in atopic dermatitis. J Allergy Clin Immunol (1996) 98(1):225–31. doi: 10.1016/s0091-6749(96)70246-4
36. Soumelis V, Reche PA, Kanzler H, Yuan W, Edward G, Homey B, et al. Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nat Immunol (2002) 3(7):673–80. doi: 10.1038/ni805
37. Savinko T, Matikainen S, Saarialho-Kere U, Lehto M, Wang G, Lehtimäki S, et al. IL-33 and ST2 in atopic dermatitis: expression profiles and modulation by triggering factors. J Invest Dermatol (2012) 132(5):1392–400. doi: 10.1038/jid.2011.446
38. Hvid M, Vestergaard C, Kemp K, Christensen GB, Deleuran B, Deleuran M. IL-25 in atopic dermatitis: A possible link between inflammation and skin barrier dysfunction? J Invest Dermatol (2011) 131(1):150–7. doi: 10.1038/jid.2010.277
39. Cotton RN, Cheng TY, Wegrecki M, Le Nours J, Orgill DP, Pomahac B, et al. Human skin is colonized by T cells that recognize CD1a independently of lipid. J Clin Invest (2021) 131(1):e140706. doi: 10.1172/JCI140706
40. de Jong A, Cheng TY, Huang S, Gras S, Birkinshaw RW, Kasmar AG, et al. CD1a-autoreactive T cells recognize natural skin oils that function as headless antigens. Nat Immunol (2014) 15(2):177–85. doi: 10.1038/ni.2790
41. Takaoka A, Arai I, Sugimoto M, Yamaguchi A, Tanaka M, Nakaike S. Expression of IL-31 gene transcripts in NC/Nga mice with atopic dermatitis. Eur J Pharmacol (2005) 516(2):180–1. doi: 10.1016/j.ejphar.2005.04.040
42. Callewaert C, Nakatsuji T, Knight R, Kosciolek T, Vrbanac A, Kotol P, et al. IL-4Rα blockade by dupilumab decreases staphylococcus aureus colonization and increases microbial diversity in atopic dermatitis. J Invest Dermatol (2020) 140(1):191–202.e7. doi: 10.1016/j.jid.2019.05.024
43. Fleming P, Drucker AM. Risk of infection in patients with atopic dermatitis treated with dupilumab: A meta-analysis of randomized controlled trials. J Am Acad Dermatol (2018) 78(1):62–69.e1. doi: 10.1016/j.jaad.2017.09.052
44. Eichenfield LF, Bieber T, Beck LA, Simpson EL, Thaçi D, de Bruin-Weller M, et al. Infections in dupilumab clinical trials in atopic dermatitis: A comprehensive pooled analysis. Am J Clin Dermatol (2019) 20(3):443–56. doi: 10.1007/s40257-019-00445-7
45. Ong PY, Ohtake T, Brandt C, Strickland I, Boguniewicz M, Ganz T, et al. Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N Engl J Med (2002) 347(15):1151–60. doi: 10.1056/NEJMoa021481
46. Howell MD, Kim BE, Gao P, Grant AV, Boguniewicz M, Debenedetto A, et al. Cytokine modulation of atopic dermatitis filaggrin skin expression. J Allergy Clin Immunol (2007) 120(1):150–5. doi: 10.1016/j.jaci.2007.04.031
47. Leyva-Castillo JM, McGurk A, Geha MDR. Allergic skin inflammation and s. aureus skin colonization are mutually reinforcing. Clin Immunol (2020) 218:108511. doi: 10.1016/j.clim.2020.108511
48. Eyerich K, Pennino D, Scarponi C, Foerster S, Nasorri F, Behrendt H, et al. IL-17 in atopic eczema: Linking allergen-specific adaptive and microbial-triggered innate immune response. J Allergy Clin Immunol (2009) 123(1):59–66.e4. doi: 10.1016/j.jaci.2008.10.031
49. Mao Y, Yang C, Tang L, Liu G, Cheng L, Chen M. Increased expression of T helper 17 cells and interleukin-17 in atopic dermatitis: A systematic review and meta-analysis. Ann Palliat Med (2021) 10(12):12801–9. doi: 10.21037/apm-21-3590
50. Saeki H, Kabashima K, Tokura Y, Murata Y, Shiraishi A, Tamamura R, et al. Efficacy and safety of ustekinumab in Japanese patients with severe atopic dermatitis: a randomized, double-blind, placebo-controlled, phase II study. Br J Dermatol (2017) 177(2):419–27. doi: 10.1111/bjd.15493
51. Ungar B, Pavel AB, Li R, Kimmel G, Nia J, Hashim P, et al. Phase 2 randomized, double-blind study of IL-17 targeting with secukinumab in atopic dermatitis. J Allergy Clin Immunol (2021) 147(1):394–7. doi: 10.1016/j.jaci.2020.04.055
52. Husein-ElAhmed H, Steinhoff M. Effectiveness of ustekinumab in patients with atopic dermatitis: Analysis of real-world evidence. J Dermatolog Treat (2021) 25:1–6. doi: 10.1080/09546634.2021.1914315
53. Sugaya M. The role of Th17-related cytokines in atopic dermatitis. Int J Mol Sci (2020) 21(4):1314. doi: 10.3390/ijms21041314
54. Möbus L, Rodriguez E, Harder I, Stölzl D, Boraczynski N, Gerdes S, et al. Atopic dermatitis displays stable and dynamic skin transcriptome signatures. J Allergy Clin Immunol (2021) 147(1):213–23. doi: 10.1016/j.jaci.2020.06.012
55. Wang V, Keefer M, Ong PY. Antibiotic choice and methicillin-resistant staphylococcus aureus rate in children hospitalized for atopic dermatitis. Ann Allergy Asthma Immunol (2019) 122(3):314–7. doi: 10.1016/j.anai.2018.12.001
56. Hennino A, Vocanson M, Toussaint Y, Rodet K, Benetière J, Schmitt AM, et al. Skin-infiltrating CD8+ T cells initiate atopic dermatitis lesions. J Immunol (2007) 178(9):5571–7. doi: 10.4049/jimmunol.178.9.5571
57. Christensen GB, Hvid M, Kvist PH, Deleuran B, Deleuran M, Vestergaard C, et al. CD4+ T cell depletion changes the cytokine environment from a TH1/TH2 response to a TC17-like response in a murine model of atopic dermatitis. Int Immunopharmacol (2011) 11(9):1285–92. doi: 10.1016/j.intimp.2011.04.010
58. Hijnen D, Knol EF, Gent YY, Giovannone B, Beijn SJ, Kupper TS, et al. CD8(+) T cells in the lesional skin of atopic dermatitis and psoriasis patients are an important source of IFN-γ, IL-13, IL-17, and IL-22. J Invest Dermatol (2013) 133(4):973–9. doi: 10.1038/jid.2012.456
59. Teunissen MBM, Yeremenko NG, Baeten DLP, Chielie S, Spuls PI, de Rie MA, et al. The IL-17A-producing CD8+ T-cell population in psoriatic lesional skin comprises mucosa-associated invariant T cells and conventional T cells. J Invest Dermatol (2014) 134(12):2898–907. doi: 10.1038/jid.2014.261
60. Gonzalez T, Stevens ML, Baatyrbek Kyzy A, Alarcon R, He H, Kroner JW, et al. Biofilm propensity of staphylococcus aureus skin isolates is associated with increased atopic dermatitis severity and barrier dysfunction in the MPAACH pediatric cohort. Allergy (2021) 76(1):302–13. doi: 10.1111/all.14489
61. Vu AT, Baba T, Chen X, Le TA, Kinoshita H, Xie Y, et al. Staphylococcus aureus membrane and diacylated lipopeptide induce thymic stromal lymphopoietin in keratinocytes through the toll-like receptor 2-toll-like receptor 6 pathway. J Allergy Clin Immunol (2010) 126(5):985–93, 993.e1-3. doi: 10.1016/j.jaci.2010.09.002
62. Al Kindi A, Williams H, Matsuda K, Alkahtani AM, Saville C, Bennett H, et al. Staphylococcus aureus second immunoglobulin-binding protein drives atopic dermatitis via IL-33. J Allergy Clin Immunol (2021) 147(4):1354–1368.e3. doi: 10.1016/j.jaci.2020.09.023
63. Sonkoly E, Muller A, Lauerma AI, Pivarcsi A, Soto H, Kemeny L, et al. IL-31: a new link between T cells and pruritus in atopic skin inflammation. J Allergy Clin Immunol (2006) 117(2):411–7. doi: 10.1016/j.jaci.2005.10.033
Keywords: atopic dermatitis, S. aureus, S. epiderimidis, coagulase - negative Staphylococcus, IL-17A
Citation: Ong PY (2022) Atopic dermatitis: Is innate or adaptive immunity in control? A clinical perspective. Front. Immunol. 13:943640. doi: 10.3389/fimmu.2022.943640
Received: 14 May 2022; Accepted: 01 July 2022;
Published: 27 July 2022.
Edited by:
Paola Maura Tricarico, Institute for Maternal and Child Health Burlo Garofolo (IRCCS), ItalyReviewed by:
Patrick M. Brunner, Icahn School of Medicine at Mount Sinai, United StatesCopyright © 2022 Ong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peck Y. Ong, cHlvbmdAY2hsYS51c2MuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.