
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 31 August 2022
Sec. Parasite Immunology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.941977
This article is part of the Research Topic Rising Stars in Parasite Immunology 2021 View all 14 articles
 Yifan Wu1Megan Duffey1,2
Yifan Wu1Megan Duffey1,2 Saira Elizabeth Alex3Charlie Suarez-Reyes1Eva H. Clark1,2,3
Saira Elizabeth Alex3Charlie Suarez-Reyes1Eva H. Clark1,2,3 Jill E. Weatherhead1,2,3*
Jill E. Weatherhead1,2,3*Non-communicable diseases (NCDs) like cardiovascular disease, chronic respiratory diseases, cancers, diabetes, and neuropsychiatric diseases cause significant global morbidity and mortality which disproportionately affect those living in low resource regions including low- and middle-income countries (LMICs). In order to reduce NCD morbidity and mortality in LMIC it is imperative to understand risk factors associated with the development of NCDs. Certain infections are known risk factors for many NCDs. Several parasitic helminth infections, which occur most commonly in LMICs, have been identified as potential drivers of NCDs in parasite-endemic regions. Though understudied, the impact of helminth infections on the development of NCDs is likely related to helminth-specific factors, including species, developmental stage and disease burden. Mechanical and chemical damage induced by the helminth in combination with pathologic host immune responses contribute to the long-term inflammation that increases risk for NCD development. Robust studies from animal models and human clinical trials are needed to understand the immunologic mechanisms of helminth-induced NCDs. Understanding the complex connection between helminths and NCDs will aid in targeted public health programs to reduce helminth-induced NCDs and reduce the high rates of morbidity that affects millions of people living in parasite-endemic, LMICs globally.
Non-communicable diseases (NCDs) such as cardiovascular disease, chronic respiratory diseases, cancers, diabetes, and neuropsychiatric diseases are now the most common causes of morbidity and mortality globally, including in low- and middle-income countries (LMICs) (1). NCDs are responsible for over 71% of all deaths worldwide and 1.6 billion disability adjusted life years (DALYs) (2). The rise in NCDs is likely multifactorial due to changes in lifestyle including reduced physical activity and non-nutritious diets, increased ability to diagnose NCDs in resource limited regions around the world, lengthening of the human life expectancy and enhanced control efforts of communicable diseases (3, 4). Though NCDs are common in all countries regardless of income level, LMICs have higher NCD-related mortality rates compared to those living in high-income countries (HICs); in fact, 80% of NCD-related deaths occur in LMICs (5). Furthermore, while NCD-related mortality in HICs is associated with older age groups, NCD-related deaths in LMICs are concentrated in younger adults (the 30-69 years old age group). Strikingly, 85% of NCD-related deaths in persons 30-69 years old occur in LMIC (6). These differences between LMICs and HICs demonstrate the profound impact of social determinants of health, such as limited resources, poor health infrastructure and poverty, on global health outcomes related to NCDs and the need to identify region-specific modifiable risk factors for NCDs (7).
Generally, factors such as high blood pressure, smoke exposure, poor glucose control, and obesity are associated with increased risk of NCDs. However, major differences exist between NCD risk factors in HICs compared to LMICs. While diet composition is a standard risk factor for the development of NCDs in both HICs and LMICs, cigarette smoking is a much larger driver of NCDs in HICs compared to LMICs. In contrast, environmental drivers of NCDs (e.g., use of indoor biomass fuel and infectious diseases) are more common in LMICs than HICs. Furthermore, because infectious diseases remain more prevalent in LMICs, NCDs attributed to infections are associated with higher DALYs in LMICs compared to HICs (8).
Several infectious pathogens are recognized as substantial contributors to the development of various NCDs. Most of these pathogens are more common in LMICs than in HICs. For instance, Hepatitis B virus and Hepatitis C virus are well-known risk factors for liver cirrhosis and cancer. Human Papilloma virus (HPV) and the bacteria Helicobacter pylori can also cause cancer (9–11). Many infectious respiratory pathogens are known to result in chronic respiratory disorders including chronic obstructive respiratory disease (COPD; a potential late consequence of tuberculosis, histoplasmosis, etc.) and asthma (which can manifest after childhood Respiratory Syncytial Virus infection) (12, 13). Though less well-known, parasitic infections—from protozoa like Trypanosoma cruzi (an infection that manifests as Chagas disease, leading to the development of cardiomyopathy or GI disease in 30% of infected individuals) and Trypanosoma brucei (an infection that causes sleeping sickness, leading to chronic neurologic disease in Sub-Saharan Africa) to helminths—can also increase one’s risk for NCDs (14). This review will focus on helminth infections that drive the development of NCDs in endemic LMICs (Figure 1). Improving treatment and control of these helminth infections will be an important part of reducing global NCD morbidity and mortality.
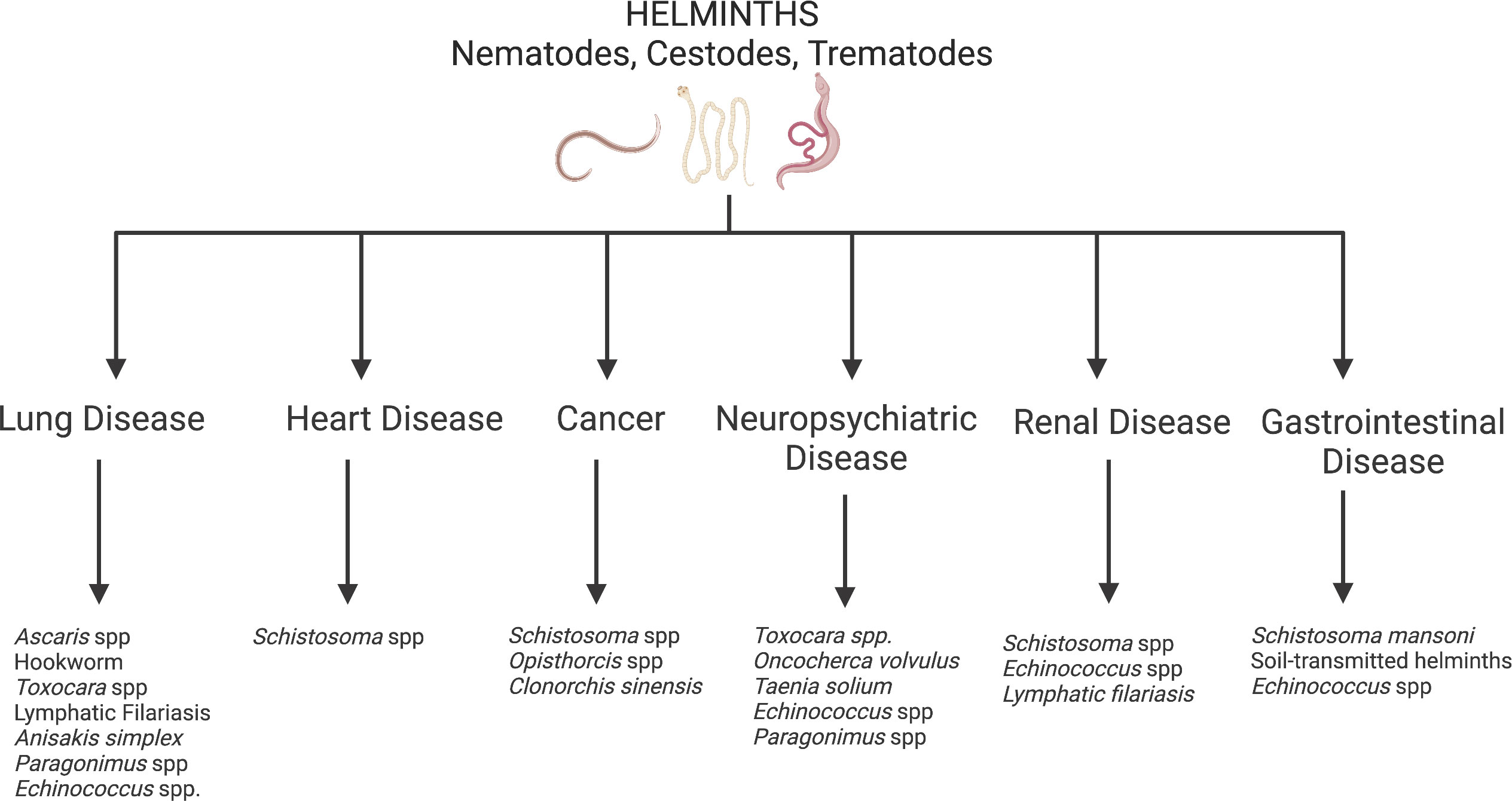
Figure 1 Helminth induced non-communicable diseases. Created with BioRender.com.
Helminths are multicellular parasitic worms that disproportionately infect persons living in poverty-stricken regions of the world. While by far more prevalent in tropical LMICs, helminths prosper in nearly all impoverished regions with climates supportive of the parasitic life cycle (15), including in the southern United States (U.S.). Helminth infections generally do not cause high mortality rates but contribute to high morbidity and subsequent DALYs. Heavy helminth infections can lead to childhood malnutrition, growth restriction and neurocognitive impairment that impact school attainment and work productivity into adulthood (16). Additionally, helminth infections cause clinical manifestations and sequelae that are unique consequences of the individual helminth’s life cycle. For this reason, several helminth infections can lead to NCDs including cardiovascular disease, lung disease, cancer and neuropsychiatric disease (Figure 1). On a community level, helminth-induced NCDs could have detrimental consequences on the economic growth in helminth endemic regions, keeping the entire community in poverty.
Ascariasis (“roundworm”), caused by either Ascaris lumbricoides or Ascaris suum, is the most common human helminth infection (17). The life cycle of Ascaris spp. begins with ingestion of Ascaris eggs from the contaminated environment. The larvae hatch in the digestive tract and migrate to the liver followed by the lungs via the systemic circulation. In the lungs, the larvae infiltrate the pulmonary parenchyma through the endovasculature and mature into late stage larvae prior to ascending the bronchotracheal tree and returning to the intestines to develop into adult worms (18). The larval migratory phase through the lungs can also cause prolonged mechanical and chemical lung damage leading to functional changes including asthma and chronic obstructive pulmonary disease (COPD). Animal models have shown that pulmonary larval migration causes a coordinated recruitment of eosinophils and neutrophils as well as type 2 T helper cells (Th2) that secrete type-2 cytokines (IL-4, IL-5, IL-13) and Th17 cells. (18, 19). These type-2 and type-17 immune responses inhibit larval development and reduce parasite burden, but lead to extreme allergic airway disease, an asthma phenotype (19, 20). Data from these mouse models have been corroborated in human clinical studies. Previous studies have not only shown an increased risk of asthma (21) but increased asthma severity and need for hospitalization in children exposed to Ascaris in endemic regions (22). Together, these data suggest ascariasis is likely a major environmental cause of asthma in endemic regions. Moreover, mice previously infected with Ascaris have increased lung volumes, lung compliance and alveolar mean linear intercept (MLI) up to 9 months post infection, representing an emphysematous COPD phenotype. Mechanistic analysis reveals enhanced secretion of matrix metalloproteinase (MMP)-12 from alveolar macrophages, a key mediator in COPD pathology. Though no human clinical studies have yet been done to evaluate the association of COPD with Ascaris larval migration, Ascaris infection in pigs–a natural Ascaris host with similar lung-body weight ratio as humans–can lead to chronic paroxysmal coughing and expiratory dyspnea, suggesting Ascaris larval migration can cause chronic lung disease.
Along with the chronic lung disease induced in Ascaris mouse models, chronic vascular damage has also been described (23). Studies of mouse lungs 9 months post infection demonstrate pulmonary vascular permeability, erythrocyte extravasation and hemosiderin-laden macrophages, as well as chronic anemia. Anemia is a common clinical manifestation in children with ascariasis classically thought to be secondary to malabsorption of nutrients during the adult intestinal stage (24). However, as mice do not develop patent adult worm infection, the chronic bleeding in the lungs was likely a source of the persistent anemia. After migration through the lungs, Ascaris larvae mature and develop into adult worms in the intestines where they live up to 1-2 years. Clinical manifestations of intestinal ascariasis are usually subtle. However, hepatobiliary disease can develop from adult worms invading the biliary tract which can result in biliary strictures, biliary cirrhosis and atrophy of the liver progressing to end stage liver disease (25).
Hookworms, Ancylostoma duodenale and Necator americanus, infect humans through skin penetration of L3 filariform larvae (infectious stage) found in the soil. L3 larvae travel in the circulatory system to the lungs and enter the alveoli, transcend the trachea and are swallowed back to the gastrointestinal tract, where they molt into L4 larvae and develop into adult worms (26, 27). While there are no clinical studies suggesting hookworm infection is a risk factor for asthma (28, 29), a mouse model of hookworm (using Nippostrongylus brasiliensis) found larval migration through the host lungs and mucosal damage increased expression of Trefil factor 2 (TFF2), a central effector molecule in asthma. TFF2 orchestrated chronic lung repair but drove IL-13 associated allergic inflammation and airway hyperreactivity while promoting lung fibrosis after N. brasiliensis larval migration (30). In a chronic mouse model of N. brasiliensis, larval migration through the lungs induced an emphysematous COPD phenotype (31) as well as persistent hemosiderin-laden macrophages further suggesting the chronic impact of acute hookworm larval migration through the host lungs. The most serious known consequence of hookworm infection is chronic iron-deficiency anemia which can have devastating outcomes in pregnant persons (i.e., pregnancy loss and premature labor) and young children (i.e., cognitive delays) (32, 33). Overwhelming evidence supports that iron deficiency anemia is a direct result of intestinal hookworms infection. Adult hookworms attach to the intestinal mucosa using buccal plates and secret anticoagulant peptides that aid in blood extravasation and digestion utilized for nutrient acquisition by the worm (34). Heavy adult hookworm burden can lead to losses of over 1 mL of blood per day depending on the species of hookworm and the worm burden (35).
Trichuriasis is caused by oral ingestion of Trichuris trichiura (“whipworm”) eggs from the contaminated environment (36). After ingestion, larvae hatch in the small intestines and develop into adult worms in the cecum where the anterior end inserts into the colonic mucosa and causes structural changes and localized inflammatory infiltration (37). Children with heavy burden of Trichuris can develop colitis similar to inflammatory bowel disease and dysentery syndromes (38, 39). Animal models with chronic trichuriasis have evidence of transmural colonic inflammation and immune profiles dominated by proinflammatory mediators interferon (IFN)-γ, interleukin (IL)-1β, STAT4, tumor necrosis factor (TNF)-α, and IL-6, mimicking both murine models of inflammatory bowel disease and human inflammatory bowel disease (40). Marked elevation of IFN-γ during chronic trichuriasis as well as the production of an IFN-γ homologue by the intestinal helminth have been linked to epithelial dysregulation (41) and progression to chronic Trichuris colitis (42). Furthermore, Trichuris infection in mice with a genetic predisposition to developing inflammatory bowel disease have accelerated progression of colitis with exaggerated mucosal inflammatory infiltration and cytokine production (43). In addition, clinical studies evaluating children with heavy burden of Trichuris infection, indicate colonic pathology with severe, chronic infiltration of inflammatory cells and mucosal destruction consistent with colitis providing supporting evidence that trichuriasis may contribute to human inflammatory bowel disease in parasite-endemic regions (44). Trichuriasis can also be associated with iron-deficiency anemia in children with heavy disease burden resulting from microhemorrhages and blood oozing around the colonic mucosal entry site as seen in Trichuris dysentery syndrome (44, 45).
Toxocariasis is a zoonotic helminth disease of dogs and cats (Toxocara canis and Toxocara cati) (46, 47). Humans, most commonly young children, become infected with Toxocara spp. after ingesting soil contaminated with Toxocara spp. eggs, or rarely when ingesting undercooked meat containing larvae. Following ingestion, L3 larvae hatch from the egg in the intestines and migrate to host tissues including the liver, lungs, heart and central nervous system (CNS) (48). Because humans are accidental hosts, Toxocara larvae cannot complete their life cycle in humans and die within host viscera overtime, inducing eosinophilic inflammation and granuloma formation (49). Toxocariasis is typically asymptomatic, but larval migration into host tissues can cause syndromes such as visceral larvae migrans (VLM), neurotoxocariasis (NT) and ocular larvae migrans (OLM).
Cases of acute NT classically present as eosinophilic meningoencephalitis, myelitis, cerebral vasculitis, or seizures. These clinical manifestations are direct results of larval migration and death within the CNS and the sequalae of the profound inflammatory immune response within tissue (50). Specifically, NT-related epilepsy in children and adults is likely due to increased concentrations of pro-inflammatory cytokines within the brain parenchyma, aberrant neurotransmitter activity and parenchymal scarring (51–53). Murine models of chronic NT demonstrate changes in mRNA expression of key pro-inflammatory cytokines (TNF-α and IL-6), inducible nitric oxide synthase, and neurotransmitters (increased norepinephrine and glutamate and decreased GABA, dopamine, and serotonin expression) compared to controls (54–56). Furthermore, Toxocara larval migration through tissue induces the formation of eosinophilic granulomas that can cause CNS scarring which become foci of seizure activity. Observed mechanisms of brain injury-measured by increased expression of glial fibrillary acidic protein (GFAP), AβPP, substance P, transforming growth factor β1 (TGF-β1), ubiquitin-proteasome system (UPS), NF-L, S100B, tTG, and p-tau, in mice infected with Toxocara–suggest a possible link between NT and chronic neurogenerative (e.g., Alzheimer’s) and neuropsychiatric diseases (57–60). Although, adequately powered clinical studies are needed to make definitive conclusions (61, 62).
Larval migration through the host lungs is associated with the development of chronic lung disease (63–66). Acute Toxocara larval migration and subsequent larval death in the lungs causes wheezing and cough due to airway hyperreactivity and excessive mucous secretion. Beyond acute disease, previous exposure to Toxocara is a risk factor for the development of chronic asthma in children (63, 64). Toxocara infection in the lungs stimulates innate and type-2 adaptive immune responses, marked by elevated IL-4, IL-5, and IL-13, which results in immunoglobulin class switching to IgE, recruitment and survival of tissue dwelling eosinophils, goblet cell metaplasia and airway hyperreactivity manifesting as asthma. High levels of circulating IgE are capable of binding to mast cells, inciting mast cell degranulation and release of pro-inflammatory mediators further contributing to allergic airway pathology (67–69). Murine models have further demonstrated that Toxocara can not only independently cause allergic airway disease but can exacerbate airway inflammation in animals with established allergic airway disease induced by ovalbumin (OVA) sensitization and OVA challenge with overt expression of IL-4, IL-5, and IL-10 (70, 71).
OLM most commonly occurs in older children (classically aged 5-10) and adults as a result of Toxocara larval migration and death within the eye. Larval death promotes sustained inflammation causing extracellular matrix remodeling and development of eosinophilic granulomas, leading to a wide range of ocular disease including chorioretinitis, vitritis, endophthalmitis, and optic neuropathy as well as retinal detachment leading to blindness (72–75). Animal models of OLM demonstrate that matrix metalloproteinases (MMPs), MMP-2 and MMP-9, and prolonged elevated concentrations of pro-inflammatory cytokines IL-6, IL-8, IL-10, and VEGF aid in recruitment of leukocytes, particularly eosinophils, and break-down fibrin in the posterior chamber extracellular matrix proteins leading to eosinophilic granuloma formation (76, 77). In the U.S. alone, OLM causes approximately 70 cases of blindness annually (78). While reports suggest OLM occurs in 6.6 cases per 100,000 persons, the true global burden of OLM remains unknown (79).
LF is mosquito-borne disease caused by Wuchereria bancrofti, Brugia malayi, and Brugia timori, though most disease (approximately 90%) is due to W. bancrofti (80). Infection occurs when infective stage larvae (L3) are injected into the skin during a mosquito blood meal and travel to the lymphatic vessel where the L3 develop into adult worms (80, 81). Adult worms in the lymphatics release microfilariae into the blood circulation. Pathology is primarily caused by the adult worms, as their presence in the lymphatic vessels can lead to long-term lymphatic inflammation resulting in significant and often disfiguring lymphedema of the lower extremities and other appendages, known as “elephantiasis.” Further, over time, filarial lymphedema is often complicated by bacterial infections of the overlying skin and difficulty with ambulation associated with significant morbidity (80, 81).
Additionally, LF can cause other chronic diseases secondary to the microfilaria stage. Microfilariae that transverse the lungs induce a severe allergic airway disease with airway hyperresponsiveness (manifesting as chronic cough and wheezing), pulmonary and peripheral eosinophilia and pulmonary infiltrates, like clinical asthma, known as pulmonary eosinophilia syndrome (82). Pulmonary function tests reveal pulmonary eosinophilia syndrome cause a restrictive lung disease pattern and may lead to pulmonary fibrosis if left untreated. The pathophysiology is thought to be related to release of filarial antigens that have homology to common allergens such as tPE γ-glutamyl transpeptidase. For instance, B. malayi infection may lead to γ-GT specific IgG1 and γ-GT specific IgE antibody expansion (83). Murine models of B. malayi have demonstrated that pulmonary eosinophilia syndrome is modulated by type-2 cytokines such as IL-4. Interestingly, this disease phenotype can be suppressed by IL-12, leading to decreased IgE, eosinophilia, and airway hyperresponsiveness, thereby downregulating filaria-induced lung immunopathology (84).
Filarial extrapulmonary pathology may additionally involve the joints and the kidneys. Arthritis (and even vasculitis) attributable to lymphatic filariasis is uncommon (mostly reported in Indian patients with W. brancrofti) but may manifest either as oligoarthritis or as polyarticular pseudo-rheumatism (85–88). Symptoms are often unresponsive to non-steroidal anti-inflammatories but improve with anti-filarial treatment (e.g., diethylcarbamazine [DEC]). The pathogenesis is thought to be related to either immune complex deposition or inflammation caused by the presence of the adult worm in the joint space. Regarding the kidneys, patients with filariasis may develop chronic kidney disease manifesting as proteinuria and nephrotic syndrome (89–92). The mechanism is likely driven by immune complex deposition in response to the presence of adult worms and microfilariae. Both the renal tubules and glomeruli may be affected. In an Indian study of 14 patients with filariasis due to W. bancrofti and proteinuria, hematuria, or chyluria, six were found to have mesangioproliferative changes, three had inflammatory cell proliferation, and two had endocapillary cell proliferation (93). Immunofluorescence of kidney tissue demonstrated mesangial deposits of IgG alone or in combination with complement 3 (C3) in patients with mesangioproliferative changes and granular deposit of IgG and C3 along the capillary wall in those with endocapillary cell proliferation (93). Another study of patients with filariasis in India due to B. malayi indicated that the glomeruli are affected more often than the tubules in symptomatic patients, however noted that proteinuria persisted even after treatment with DEC (94).
The filarial worm Onchocerca volvulus is well-known to cause vision impairment, primarily in Africa (though disease foci still exist in Yemen, Venezuela, and Brazil) (95). In 2017, more than 1 million people already had vision loss due to the disease (96). Importantly, intracytoplasmic bacteria (Wolbachia) often live symbiotically within O. volvulus. The pathology of onchocerciasis is likely due to inflammatory responses against both the microfilariae, which circulate in the subcutaneous tissue and lymphatic system and trigger a host inflammatory response (i.e., granuloma formation and eventually fibrosis) when they die, and symbiotic Wolbachia. Repeat infections lead to worse cumulative disease. The specific mechanisms that lead to ocular disease remain unclear. Study of tissue sections have demonstrated infiltrates containing plasma cells, eosinophils, and mast cells (97). Degenerating microfilariae may cause punctate keratitis. Inflammation (and possibly an autoantibody formation) (98, 99) related to microfilariae can lead to anterior uveitis and chorioretinitis. Corneal pathology is associated with increased systemic and corneal type-2 cytokines expression, illustrated by in vivo studies in which IL-4 gene knockout mice developed less severe or no O. volvulus-mediated keratitis (100). Regarding molecular mimicry, O. volvulus antigen Ov39 is cross-reactive with the retinal antigen hr44 and induces ocular inflammation in rats (99). Interestingly, corneal inflammation is not induced by extracts derived from O. volvulus depleted of Wolbachia (101), and may be related to expression of adaptor molecules such as TIRAP/Mal (101) and myeloid differentiation factor 88 (102), which is a necessary part of some toll like receptor (TLR) signaling pathways.
O. volvulus may also contribute to a form of incompletely described, but likely progressive epilepsy known as “nodding disease” (103, 104). Nodding disease has been primarily recognized among children in east Africa (particularly Tanzania, Sudan, and Uganda) though outbreaks over the last decade are reported from Uganda, Liberia, Tanzania, the Democratic Republic of Congo, and southern Sudan. Nodding disease classically manifests as episodes during which the head bobs forward repeatedly for several minutes and the individual may seem unresponsive; these episodes are sometimes associated with generalized tonic-clonic and/or absence seizures. Nodding syndrome may progress to significant cognitive disability and, eventually, death (105). No specific cerebrospinal fluid or neuroimaging abnormalities are yet associated with the disease, though some individuals are noted to have significant atrophy of the hippocampal and glia matter (106). The association between O. volvulus and nodding disease came about after one Ugandan study suggested a higher rate of epilepsy in communities with higher O. volvulus prevalence (107), however, the underlying biological mechanism for such an association remains unclear (108–110). One possibility under active investigation is that nodding syndrome may be due to autoantibodies (e.g., to leiomodin-1) produced in response to O. volvulus infection (111).
Three Schistosoma species cause most intestinal (S. japonicum and S. mansoni) and urogenital disease (S. haematobium). The host immune response to Schistosoma species eggs is responsible for the clinical manifestations of the disease syndromes. Paired adult worms reside in small veins (of the lower urinary tract for S. haematobium and of the mesenteric plexus for S. japonicum) and release Schistosoma species eggs which migrate through associated organs, thereby causing considerable irritation, prolonged inflammation, and granuloma/fibrosis development (112, 113). Schistosomiasis egg migration and the subsequent chronic immune activation can have a wide range of effects on end-organ disease.
Renal disease, ranging from asymptomatic to end-stage renal disease (ESRD), is a result of direct egg induced inflammation or Schistosoma antigen - immune complex formation within the kidney most commonly seen in S. mansoni hepatosplenic disease (114). In a Brazilian longitudinal study of 24 patients with schistosomiasis (likely S. mansoni as this is the major species found in Brazil), fifteen (68.1%) had related hepato-splenic disease, thirteen (54.1%) had nephrotic-nephritic syndrome, twenty (83.3%) had hematuria and 18 (75.0%) had hypertension. After nearly 60 months of follow up, nine patients developed ESRD (115). Approximately 5-6% of patients with hepatosplenic schistosomiasis develop glomerular involvement (114). The most common schistosomiasis glomerular disease is mesangial proliferative glomerulonephritis followed by membranoproliferative glomerulonephritis and less commonly focal and segmental glomerulosclerosis, exudative glomerulonephritis and amyloidsis (114, 116). Additionally, urogenital schistosomiasis from S. haematobium can cause lower urinary track fibrosis and calcification which can lead to renal outlet obstruction, ureter reflux, interstitial nephritis and ESRD (116).
S. mansoni’s predilection for the venous portal-mesenteric system can result in liver fibrosis, portal hypertension as well as end-stage liver disease. In hepatosplenic schistosomiasis, portal hypertension occurs from an eosinophilic granulomatous reaction to Schistosoma egg deposited in presinusoidal portal venules causing presinusoidal hepatic fibrosis typically with preserved liver function (117). Imaging of the liver often shows splenomegaly and calcified eggs along enhanced portal tracks in the liver by CT scan (118). The consequences of portal hypertension include gastric bleeding, such as esophageal varices, which is responsible for an estimated 200,000 deaths annually in sub-Sahara Africa, and ascites (119). Polarized Th2 immune responses during hepatosplenic schistosomiasis not only promotes liver fibrosis but also impairs Th1 anti-viral immunity. Co-infection with HBV and/or HCV, common in schistosomiasis-endemic regions, can accelerate liver pathology particularly advancing viral-induced hepatocellular carcinoma and liver failure (120, 121).
Sch-PH is believed to be a leading cause of pulmonary hypertension in Schistosoma endemic regions around the world and can result in right-sided heart failure (122). Sch-PH is most commonly associated with chronic hepato-splenic schistosomiasis as a result of Schistosoma mansoni infection. Approximately 5-10% of patients with hepato-splenic schistosomiasis will develop Sch-PH which can result in devastating cardiovascular disease including end-stage right ventricular heart failure (123, 124). The immunopathogenesis of Sch-PH is likely multifactorial. Hepato-splenic disease causing portopulmonary hypertension may occur due to the underlying liver disease or from Schistosoma egg embolization and inflammation induced vasculopathy with medial thickening, intimal remodeling and formation of granulomas and fibrosis (125).
S. haematobium and S. japonicum are both designated biological human carcinogens by the International Agency for Research on Cancer (126).
Urogenital schistosomiasis due to S. haematobium is common in endemic areas and complicated by the development of bladder cancer. The incidence of urogenital schistosomiasis-associated squamous cell bladder carcinoma is estimated at 3-4 cases per 100,000 (127). Several mechanisms have been implicated in the oncogenesis of Schistosoma haematobium infection (128): 1) Schistosoma antigen can increase proliferation and longevity of urothelium cells (129) 2) Elevated p53 levels have been documented in both pre-malignant and malignant lesions associated with schistosomiasis (130, 131) 3) Oncogenic mutation of the KRAS gene can be induced in urothelium exposed to whole parasite extract (132) 4) Soluble Schistosoma egg antigens (SEA) increase proliferation and oxidative stress and decrease apoptosis (133) 5) Repeated deposition of eggs in the bladder wall and migration of the eggs through the urothelium results in chronic inflammatory infiltrate (134) and parasite-induced oxygen derived free radicals, genetic mutations and the production of carcinogenic compounds (131, 133, 135) as well as 6) Epigenetic changes via hypermethylation of the host genome (136). Of note, S. haematobium may contribute to other types of cancer as well. An autopsy study from the Central Pathology Institute of Baghdad, Iraq found that, between 1939-1952, of 2276 autopsies, 174 had carcinoma and 113 had S. haematobium involvement of the bladder (137). S. haematobium was present in 3 with liver cancer, 7 with bladder cancer, 3 with prostate/genitalia cancer, 2 with intestinal/rectal cancer, and 2 with undescribed cancer types. Particularly in the case of liver cancer, co-infection with S. haematobium plus another carcinogenic organism, such as the hepatitis B virus, may amplify S. haematobium’s carcinogenic potential (138). Additionally, S. haematobium can work in concert with HPV to promote the development of cervical cancer. Proposed mechanisms include S. haematobium induced mechanical damage to the cervical epithelium and S. haematobium induced local immune modulation creating a niche for HPV proliferation (139, 140).
A less common Schistosoma species, S. japonicum, is more often associated with liver cancer and colorectal cancer (141), although this is mostly based on epidemiologic associations (142–144) and pathology data (145, 146). S. japonicum eggs, retained in the intestinal wall, cause prolonged irritation that can result in fibrosis, mucosal hyperplasia, polyp development, and adenocarcinoma formation (147, 148). Additionally, an “egg embolism” can occur leading to pathology in the liver and other organs (149). A pathology study evaluating the association between S. japonicum and liver cancer in 4,611 necropsies revealed 227 cases of hepatocellular carcinoma (HCC); 24 (10.6% of these) were associated with Schistosoma japonica. Importantly, 27% of these cases had a positive Hepatitis B surface antigen, indicating the possibility that multiple types of co-infection may synergistically contribute to carcinogenesis (150). S. japonicum likely induces multiple mechanisms that contribute to malignant transformation of colonic and/or liver tissue. These mechanisms include chronic inflammation (147, 151), carcinogenic molecules derived from S. japonicum itself (152, 153), immunomodulation (154, 155), and oncogenic mutations (156).
Two trematodes, or liver flukes, are associated with malignancy (Clonorchis sinensis and Opisthorchis viverini with cholangiocarcinoma) and, similar to schistosomiasis, are classified as biological human carcinogens by the International Agency for Research on Cancer (126, 157, 158). Liver flukes’ relative contributions to carcinogenesis are likely related to the length and severity of infection, the host’s immune status, and other environmental and host genetic factors (159). Both Clonorchis sinensis and Opisthorchis viverrini are food-borne trematodes found in East Asia; C. sinensis is endemic to southern China, Korea, eastern Russia, and northern Vietnam, whereas O. viverrini is endemic to Thailand, Lao People’s Democratic Republic, Cambodia, and central Vietnam. Though fewer than 10% of people with liver fluke infections will develop cholangiocarcinoma, the incidence of cholangiocarcinoma is significant in regions of high C. sinensis or O. viverrini prevalence. For example, the incidence of O. viverrini-associated cholangiocarcinoma is approximately 98 per 100,000 people in the highly endemic region of the Thai province of Khon Kaen, where the prevalence of O. viverrini infection ranges from 2-70% (160). A Korean study published in 1996 showed that 33% of the cholangiocarcinoma cases evaluated were positive for C. sinensis by stool examination (161). Adult flukes can inhabit the biliary track for decades and cause recurrent pyogenic cholangitis. This repeated and/or prolonged inflammation of the biliary tree may contribute to later development of chronic biliary track disease including cholangiocarcinoma, however the exact mechanism(s) by which the liver flukes contribute to carcinogenesis are not yet fully elucidated. Likely liver fluke infection generates multiple mechanisms leading to carcinogenesis, including 1) Mechanical damage from physical contact of the biliary epithelium with the parasite, 2) Inflammatory pathology related to the host-parasite immune response, and 3) Prior to chemical damage from fluke excretory-secretory products (ESPs) (158, 162, 163). A sizeable body of literature exists detailing the carcinogenic potential of ESPs. Fluke ESPs can induce proliferation (164, 165), apoptosis (166, 167), chromatin remodeling (166), and inflammation of biliary epithelial cells (168). Resulting repetitive insults to the biliary epithelium may lead to hyperplasia and adenomatous changes with subsequent malignant transformation (158).
Paragonimiasis, the lung fluke, results from ingesting metacercariae in raw or undercooked crab or crayfish. The metacercariae encyst in the duodenum, travel through the intestinal wall into the peritoneal cavity, abdominal wall and diaphragm transversing into the lungs and subsequently encapsulating into adult flukes within lung parenchyma (169). During active infection, paragonimiasis can mimic the radiographic appearance and clinical manifestations of pulmonary tuberculosis or even lung cancer, including pleural disease, solitary nodules and cavitary lesions, commonly presenting with chronic cough, chest pain and hemoptysis (170–172). However chronic lung pathology can also occur from pleuropulmonary paragonimiasis. Infection left untreated or repetitive infection with Paragonimus spp, most commonly Paragonimus westermani, can lead to the development of bronchiectasis and chronic bronchitis (173, 174). The pathophysiology of Paragonimus spp. associated chronic lung disease remains unknown. More robust animal models and human clinical studies are needed.
Although rare (occurring in approximately 0.8% cases of paragonimiasis), aberrant migration of Paragonimus spp to the brain can also lead to the development of hemorrhagic stroke and epilepsy. In cases of ectopic paragonimiasis, 30-60% occur in the brain and most commonly occurs in children (175). Adult worms migrate through the perivascular connective tissue around the jugular vein and carotid artery into the posterior circulation via the skull base foramina leading to mechanical damage secondary to parasitic migratory tracts and formation of eosinophilic granulomas with central necrosis and charcot-leyden crystals (176–178). Early disease typically presents as meningoencephalitis, vasculitis and necrotizing granulomas, manifesting clinically as epilepsy and hemiplegia. Cerebral paragonimiasis can also cause pseudoaneurysms that are often misdiagnosed as noninfectious vascular malformation in children. In 17 patients with cerebral paragonimiasis and hemorrhagic strokes 35% had evidence of pseudoaneurysm and pseudoaneurysm rupture (179). In children with chronic cerebral paragonimiasis, CNS investigation reveals perivascular granulomas formation and calcification and associated cortical and subcortical atrophy (175). In a case series of 14 children with cerebral paragonimiasis, intracranial hemorrhage and eosinophilic granulomas were commonly identified. Despite targeted therapy, two of the 14 children had persistent hemiplegia on long-term follow-up secondary to sequelae of hemorrhagic strokes (180). In addition to epilepsy and paralysis, children with cerebral paragonimiasis can also have long-term behavioral changes. If treated early with antiparasitic therapy, seizures generally improve. However, dizziness, memory loss, personality changes, and loss of fine motor function, often do not completely resolve (181).
Taenia solium (the pork tapeworm) can cause intestinal disease (“taeniasis”), which is acquired after ingestion of T. solium larvae (“cysticerci”) via infected pork or cysticercosis, due to inadvertent consumption of T. solium eggs via exposure to a person or pig with taeniasis (182, 183). Cysticercosis can develop once T. solium eggs hatch into larvae in the intestine, migrate to various host organ systems, and develop into cystic larvae within the tissue. Neurocysticercosis (NCC) is the most consequential form of cysticercosis and occurs when cysticerci develop in the CNS (including the brain, eyes, and spinal cord). NCC is a leading cause of adult-onset epilepsy worldwide (184). Most of the clinical manifestations seen in NCC are related to the host immune response to the parasite, and are dependent on the number, stage, and size of cysticerci. For instance, nearly 80% of patients diagnosed with NCC report at least one lifetime seizure (185). Seizures typically are seen in patients who have parenchymal NCC (186). They are triggered by disruption of the brain parenchyma and therefore neuronal signaling caused by certain stages of T. solium cysts, specifically as cysts naturally progress from viable to a final calcified stage (187). Viable cysts are often able to evade the immune response (187–189), thus it is the inflammatory reaction to degenerating cysts (which have lost their ability to modulate the host immune system) and mechanical obstruction of normal neuronal pathways caused by residual calcifications from old cystic lesions that provoke seizures (190). Further, the degree of cyst-induced inflammation may be directly related to frequency of seizure recurrence (191). Studies of human brain tissue sections with NCC suggested that the initial immune response (prior to peri-cyst granuloma formation) is characterized by innate and Th1 cells and cytokines, including natural killer cells, macrophages, T cells, and interleukin (IL)-12 (187). Subsequently, as granulomas develop around degenerating cysts, a chronic immune reaction develops, characterized by both type-1 and type-2 immune responses (192). Additionally, symptomatic NCC patients may produce lymphocytes primed towards Th1 responses (193) and have distinct TLR polymorphisms (194). Calcified NCC specifically has been associated with not only unique TLRs but also higher serum levels of matrix metalloproteinases (MMP)-9; the same study found MMP-9 to be associated with seizure recurrence (195).
While most patients diagnosed with NCC have parenchymal disease (and calcified parenchymal disease is more common than viable parenchymal cysts), a rare but important subset of NCC is extra-parenchymal disease (186). Extra-parenchymal disease is defined by cystic lesions that develop in the ventricles, subarachnoid space, spine, or retina. Ventricle and subarachnoid cysts can cause symptoms via mass effect on surrounding tissues, the host inflammatory response to the cyst tissue can trigger aseptic meningitis, and both the cysts themselves and scarring from accompanying inflammation can cause obstructive and/or communicating hydrocephalus that often requires invasive intervention via placement of a ventriculo-peritoneal shunt or surgical removal of cysts (196). Cerebrospinal fluid from patients with subarachnoid disease seem to have significantly elevated levels of type-1 and type-2 cytokines compared to patients with parenchymal disease (197, 198).
Cystic echinococcosis (E. granulosus) and alveolar echinococcosis (E. multilocularis) results from ingestion of Echinococcus spp eggs in contaminated soil (199–201). From the intestines the eggs hatch releasing oncospheres that penetrate the intestinal wall and travel classically to the liver or lungs and, aberrantly, to other organs like the brain where they develop into thin-walled cysts. Patients with Echinococcus cysts are typically asymptomatic until the lesions create a mass effect on surrounding tissue or rupture causing a systemic inflammatory response (201). The clinical course of echinococcosis in different organ compartments is associated with a wide spectrum of complications that can lead to chronic disease in the liver, lungs, brain and kidneys (201, 202). In a cohort of 506 patients with cystic echinococcosis, 204 developed long-term complications as a result of their illness (202).
Patients with liver lesions, from either Cystic Echinococcus or Alveolar Echinococcus, have high risk of hepatic complications. Approximately 1/3rd of all patients with hepatic disease will develop a long-term complication of the hepatobiliary system including biliary track fistulas, biliary cirrhosis, cholangitis, pancreatitis and portal hypertension with associated gastrointestinal bleeding (201, 203). Mass effect from hepatic cysts cause increased pressure on hepatic and biliary tissue leading to compression and necrosis of adjacent tissues. Bile duct damage, rupture and subsequent development of biliary fistulas are thus common. The mass effect of hepatic cysts can also reduce portal vein inflow causing portal vein thrombosis and portal hypertension as well as compression and displacement of hepatic veins leading to Budd-Chiari syndrome. Liver pathology in patients with E. multilocularis hepatobiliary cysts were found to have periportal fibrosis, perilobular fibrosis and amyloid deposition (204). Pulmonary cysts can also cause mass effect on surrounding mediastinal structures, however clinical manifestations most commonly result from acute rupture of pulmonary cysts, release of immunogenic antigens from the cystic fluid, and exaggerated type-2 immune responses leading to an asthma-like syndrome of wheezing and coughing (201). Furthermore, some mouse models have shown that the presence of Echinococcus antigens may exacerbate established allergic airway disease through increased type-2 cytokine signaling resulting in histopathologic changes consistent with asthmatic disease (205). Although rare, occurring in approximately 1% of all cases of E. multilocularis, cerebral echinococcosis is associated with high mortality, with a 10-year survival rate of 28.4%. The presence of E. multilocularis cystic fluid can have cytotoxic effects on the surrounding cerebral parenchyma resulting in inflammatory recruitment and tissue necrosis (206). Of those with cerebral echinococcosis that survive, nearly 30% will have long-term neurologic sequalae including epilepsy, vision loss and hemiplegia (207–209). Several case reports and case series suggest that echinococcosis from either E. granulosus or E. multilocularis can be associated with kidney disease. The mechanism of Echinococcus renal disease is thought to be immune-complex mediated leading to tubulointerstial nephritis, glomerulonephritis (minimal change disease, mesangioproliferative) and nephrotic syndromes (210). In a case series of patients with hepatic cysts with associated proteinuria, kidney biopsy demonstrated disease was driven by hydatid antigen and the development of immune-mediated glomerulonephritis (211–213). As the literature related to extra-hepatic echinococcosis is limited to case reports and cases series, more in depth studies are needed to determine the mechanisms of Echinococcus driven extra-hepatic chronic diseases.
Helminths, which disproportionately affect persons living in poverty within LMICs, represent a major driver of morbidity on a global scale. Their role in driving NCDs including chronic lung disease, cancer, cardiovascular disease and inflammatory bowel disease are plausible but more robust mechanistic studies and human clinical trials are required to draw definitive conclusions (Table 1). These studies are necessary to aid in uncovering risk factors associated with the rising incidence and prevalence associated with NCDs in LMIC and other helminth-endemic regions around the world. Understanding this connection between helminths and NCDs will aid in targeted public health programs to reduce helminth-induced NCDs and reduce the high rates of morbidity in these regions. In the mean-time focus on helminth prevention and control efforts are critical. Access to mass drug treatment programs, water, sanitation, and hygiene interventions and health education programs for high-risk populations, especially children and pregnant persons, will result in reduced worm burdens and subsequently reduced morbidity. Elimination efforts should also remain a public health priority as the complex mechanisms of helminth induced NCDs are being evaluated.

Table 1 Proposed mechanisms of helminth induced non-communicable diseases.
YW and JW conceptualized, wrote and edited the manuscript. MD, SA, CS-R, and EC wrote and edited the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by NIH NIAID K23 K23AI168583-01 and NIH NIAID K08 AI143968-01.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. World Health Organization. The global health observatory: Mortality and global health estimates. Available at: https://www.who.int/data/gho/data/themes/mortality-and-global-health-estimates (Accessed May 11, 2022).
2. Institute for Health Metrics and Evaluation. Non-communicable diseases. Lancet Global Burden Dis. 393:R2–3. Available at: https://www.healthdata.org/results/gbd_summaries/2019/non-communicable-diseases-level-1-causeaccessed8-9-2022.
3. Bigna JJ, Noubiap JJ. The rising burden of non-communicable diseases in sub-Saharan Africa. Lancet Glob Heal (2019) 7(10):e1295–6. doi: 10.1016/S2214-109X(19)30370-5
4. Gouda HN, Charlson F, Sorsdahl K, Ahmadzada MPHS, Ferrari AJ, Erskine H, et al. Burden of non-communicable diseases in sub-Saharan Africa, 1990-2017: results from the global burden of disease study 2017. Lancet Glob Heal (2019) 7:e1375–87. doi: 10.1016/S2214-109X(19)30374-2
5. World Healtth Organization. 2008-2013 action plan for the global strategy for the prevention and control of noncommunicable diseases (2008). Available at: http://apps.who.int/iris/bitstream/handle/10665/44009/9789241597418_eng.pdf?sequence=1 (Accessed May 6, 2022).
6. World Healtth Organization. Noncommunicable diseases: Mortality. Available at: https://www.who.int/data/gho/data/themes/topics/topic-details/GHO/ncd-mortality (Accessed May 10, 2022).
7. Niessen LW, Mohan D, Akuoku JK, Mirelman AJ, Ahmed S, Koehlmoos TP, et al. Tackling socioeconomic inequalities and non-communicable diseases in low-income and middle-income countries under the sustainable development agenda. Lancet (London England) (2018) 391(10134):2036–46. doi: 10.1016/S0140-6736(18)30482-3
8. Coates MM, Kintu A, Gupta N, Wroe EB, Adler AJ, Kwan GF, et al. Burden of non-communicable diseases from infectious causes in 2017: a modelling study. Lancet Glob Heal (2020) 8(12):e1489–98. doi: 10.1016/S2214-109X(20)30358-2
9. Morales-Sánchez A, Fuentes-Pananá EM. Human viruses and cancer. Viruses (2014) 6(10):4047–79. doi: 10.3390/V6104047
10. Lee YC, Chiang TH, Chou CK, Tu YK, Liao WC, Wu MS, et al. Association between helicobacter pylori eradication and gastric cancer incidence: A systematic review and meta-analysis. Gastroenterology (2016) 150(5):1113–24.e5. doi: 10.1053/J.GASTRO.2016.01.028
11. De Martel C, Ferlay J, Franceschi S, Vignat J, Bray F, Forman D, et al. Global burden of cancers attributable to infections in 2008: a review and synthetic analysis. Lancet Oncol (2012) 13(6):607–15. doi: 10.1016/S1470-2045(12)70137-7
12. Fan H, Fan W, Liu J, Zeng W, Zheng S, Tian H, et al. Pulmonary tuberculosis as a risk factor for chronic obstructive pulmonary disease: a systematic review and meta-analysis. Ann Transl Med (2021) 9(5):390–0. doi: 10.21037/ATM-20-4576
13. Zhou Y, Tong L, Li M, Wang Y, Li L, Yang D, et al. Recurrent wheezing and asthma after respiratory syncytial virus bronchiolitis. Front Pediatr (2021) 9:649003. doi: 10.3389/fped.2021.649003
14. Ogoina D, Onyemelukwe GC. The role of infections in the emergence of non-communicable diseases (NCDs): Compelling needs for novel strategies in the developing world. J Infect Public Health (2009) 2(1):14–29. doi: 10.1016/J.JIPH.2009.02.001
15. Hotez PJ, Molyneux D, Hotez P, Hotez PJ. NTDs V.2.0: “Blue marble health”–neglected tropical disease control and elimination in a shifting health policy landscape. PloS Negl Trop Dis (2013) 7(11):e2570. doi: 10.1371/journal.pntd.0002570
16. Weatherhead JE, Hotez PJ. Worm infections in children. Pediatr Rev (2015) 36(8):341–52. doi: 10.1542/pir.36-8-341
17. Institute for Health Metrics and Evaluation. Global burden of disease 2019 cause and risk summaries: Ascariasis. Available at: http://www.healthdata.org/results/gbd_summaries/2019/ascariasis-level-4-cause (Accessed April 13, 2021).
18. Gazzinelli-Guimarães PH, Gazzinelli-Guimarães AC, Silva FN, Mati VLT, Dhom-Lemos LC, Barbosa FS, et al. Parasitological and immunological aspects of early ascaris spp. infection in mice. Int J Parasitol (2013) 43(9):697–706. doi: 10.1016/j.ijpara.2013.02.009
19. Weatherhead JE, Porter P, Coffey A, Haydel D, Versteeg L, Zhan B, et al. Ascaris larval infection and lung invasion directly induces severe allergic airway disease in mice. Infect Immun (2018), IAI.00533–18. doi: 10.1128/IAI.00533-18
20. Gazzinelli-Guimaraes PH, De Queiroz Prado R, Ricciardi A, Bonne-Annee S, Sciurba J, Karmele EP, et al. Allergen presensitization drives an eosinophil-dependent arrest in lung-specific helminth development. J Clin Invest (2019) 129(9):3686–701. doi: 10.1172/JCI127963
21. Palmer LJ, Celedón JC, Weiss ST, Wang B, Fang Z, Xu X. Ascaris lumbricoides infection is associated with increased risk of childhood asthma and atopy in rural China. Am J Respir Crit Care Med (2002) 165(11):1489–93. doi: 10.1164/rccm.2107020
22. Hunninghake GM, Soto-Quiros ME, Avila L, Klanderman BJ, Silverman EK, Celedon JC, et al. Sensitization to ascaris lumbricoides and severity of childhood asthma in Costa Rica. J Allergy Clin Immunol (2007) 119(3):654–61. doi: 10.1016/j.jaci.2006.12.609
23. Wu Y, Li E, Knight M, Adeniyi-Ipadeola G, Song L-Z, Burns AR, et al. Transient ascaris suum larval migration induces intractable chronic pulmonary disease and anemia in mice. PloS Negl Trop Dis (2021) 15(12):1–18. doi: 10.1371/JOURNAL.PNTD.0010050
24. Rajagopal S, Hotez PJ, Bundy DAP. Micronutrient supplementation and deworming in children with geohelminth infections. PloS Negl Trop Dis (2014) 8(8):1–3. doi: 10.1371/journal.pntd.0002920
25. Khuroo MS, Rather AA, Khuroo NS, Khuroo MS. Hepatobiliary and pancreatic ascariasis. World J Gastroenterol (2016) 22(33):7507–17. doi: 10.3748/wjg.v22.i33.7507
26. Institute for Health Metrics and Evaluation. Global burden of disease 2019 cause and risk summaries: Hookworm disease. Available at: https://www.healthdata.org/results/gbd_summaries/2019/hookworm-disease-level-4-cause (Accessed May 10, 2022).
27. CDC - hookworm - biology. Available at: https://www.cdc.gov/parasites/hookworm/biology.html (Accessed May 10, 2022).
28. Leonardi-Bee J, Pritchard D, Britton J. Asthma and current intestinal parasite infection: systematic review and meta-analysis. Am J Respir Crit Care Med (2006) 174(5):514–23. doi: 10.1164/rccm.200603-331OC
29. Feary J, Venn A, Brown A, Hooi D, Falcone FH, Mortimer K, et al. Safety of hookworm infection in individuals with measurable airway responsiveness: a randomized placebo-controlled feasibility study. Clin Exp Allergy (2009) 39(7):1060–8. doi: 10.1111/J.1365-2222.2009.03187.X
30. Wills-Karp M, Rani R, Dienger K, Lewkowich I, Fox JG, Perkins C, et al. Trefoil factor 2 rapidly induces interleukin 33 to promote type 2 immunity during allergic asthma and hookworm infection. J Exp Med (2012) 209(3):607–22. doi: 10.1084/JEM.20110079
31. Marsland BJ, Kurrer M, Reissmann R, Harris NL, Kopf M. Nippostrongylus brasiliensis infection leads to the development of emphysema associated with the induction of alternatively activated macrophages. Eur J Immunol (2008) 38(2):479–88. doi: 10.1002/eji.200737827
32. Ness TE, Agrawal V, Bedard K, Ouellette L, Erickson TA, Hotez P, et al. Maternal hookworm infection and its effects on maternal health: A systematic review and meta-analysis. Am J Trop Med Hyg (2020) 103(5):1958–68. doi: 10.4269/AJTMH.20-0503
33. Loukas A, Hotez PJ, Diemert D, Yazdanbakhsh M, McCarthy JS, Correa-Oliveira R, et al. Hookworm infection. Nat Rev Dis Prim (2016) 2(1):1–18. doi: 10.1038/nrdp.2016.88
34. Pearson MS, Tribolet L, Cantacessi C, Periago MV, Valerio MA, Jariwala AR, et al. Molecular mechanisms of hookworm disease: Stealth, virulence, and vaccines. J Allergy Clin Immunol (2012) 130(1):13–21. doi: 10.1016/J.JACI.2012.05.029
35. Crompton DWT. The public health importance of hookworm disease. Parasitology (2000) 121(S1):S39–50. doi: 10.1017/S0031182000006454
36. Institute for Health Metrics and Evaluation. Global burden of disease 2019 cause and risk summaries: Trichuriasis. Available at: https://www.healthdata.org/results/gbd_summaries/2019/trichuriasis-level-4- (Accessed May 10, 2022).
37. CDC - trichuriasis - biology. Available at: https://www.cdc.gov/parasites/whipworm/biology.html (Accessed May 10, 2022).
38. Bundy DAP, Cooper ES. Trichuris and trichuriasis in humans. Adv Parasitol (1989) 28(C):107–73. doi: 10.1016/S0065-308X(08)60332-2
39. Stephenson L, Holand C, Cooper E. The public health significance of trichuris trichiura. Parasitology (2000) 121(S1):S73–95. doi: 10.1017/S0031182000006867
40. Levison SE, McLaughlin JT, Zeef LAH, Fisher P, Grencis RK, Pennock JL. Colonic transcriptional profiling in resistance and susceptibility to trichuriasis: phenotyping a chronic colitis and lessons for iatrogenic helminthosis. Inflammation Bowel Dis (2010) 16(12):2065–79. doi: 10.1002/ibd.21326
41. Artis D, Potten CS, Else KJ, Finkelman FD, Grencis RK. Trichuris muris: Host intestinal epithelial cell hyperproliferation during chronic infection is regulated by interferon-γ. Exp Parasitol (1999) 92(2):144–53. doi: 10.1006/EXPR.1999.4407
42. Grencis RK, Entwistle GM. Production of an interferon-gamma homologue by an intestinal nematode: functionally significant or interesting artefact? Parasitology (1997) 115 Suppl:S101–6. doi: 10.1017/s0031182097002114
43. Bhardwaj EK, Else KJ, Rogan MT, Warhurst G. Increased susceptibility to trichuris muris infection and exacerbation of colitis in Mdr1a-/- mice. World J Gastroenterol (2014) 20(7):1797–806. doi: 10.3748/wjg.v20.i7.1797
44. Else KJ, Keiser J, Holland CV, Grencis RK, Sattelle DB, Fujiwara RT, et al. Whipworm and roundworm infections. Nat Rev Dis Prim (2020) 6(1):1–23. doi: 10.1038/s41572-020-0171-3
45. Layrisse M, Aparcedo L, Martínez-Torres C, Roche M. Blood loss due to infection with trichuris trichiura. Am J Trop Med Hyg (1967) 16(5):613–9. doi: 10.4269/AJTMH.1967.16.613
46. Farmer A, Beltran T, Choi YS. Prevalence of toxocara species infection in the U.S.: Results from the national health and nutrition examination survey, 2011-2014. PLoS Neglected Tropical Siseases (2017) 11(7):e0005818. doi: 10.1371/journal.pntd.0005818
47. Fialho PMM, Corrêa CRS. A systematic review of toxocariasis: A neglected but high-prevalence disease in Brazil. Am J Trop Med Hyg (2016) 94(6):1193–9. doi: 10.4269/AJTMH.15-0733
48. CDC - toxocariasis - biology. Available at: https://www.cdc.gov/parasites/toxocariasis/biology.html (Accessed January 11, 2022).
49. Despommier D. Toxocariasis: Clinical aspects, epidemiology, medical ecology, and molecular aspects. Clin Microbiol Rev (2003) 16(2):265. doi: 10.1128/CMR.16.2.265-272.2003
50. Eberhardt O, Bialek R, Nägele T, Dichgans J. Eosinophilic meningomyelitis in toxocariasis: case report and review of the literature. Clin Neurol Neurosurg (2005) 107(5):432–8. doi: 10.1016/J.CLINEURO.2004.10.003
51. Khatir AA, Sepidarkish M, Rajabalizadeh MR, Moghaddam SA, Aghapour S, Mahravar S, et al. Case-control study to assess the association between epilepsy and toxocara Infection/Exposure. Microorg (2021) 9(10):2091. doi: 10.3390/MICROORGANISMS9102091
52. Luna J, Cicero CE, Rateau G, Quattrocchi G, Marin B, Bruno E, et al. Updated evidence of the association between toxocariasis and epilepsy: Systematic review and meta-analysis. PloS Negl Trop Dis (2018) 12(7):1–19. doi: 10.1371/JOURNAL.PNTD.0006665
53. Quattrocchi G, Nicoletti A, Marin B, Bruno E, Druet-Cabanac M, Preux PM. Toxocariasis and epilepsy: Systematic review and meta-analysis. PloS Negl Trop Dis (2012) 6(8):e1775. doi: 10.1371/JOURNAL.PNTD.0001775
54. van Vliet EA, Aronica E, Vezzani A, Ravizza T. Review: Neuroinflammatory pathways as treatment targets and biomarker candidates in epilepsy: emerging evidence from preclinical and clinical studies. Neuropathol Appl Neurobiol (2018) 44(1):91–111. doi: 10.1111/NAN.12444
55. Angwafor SA, Bell GS, Njamnshi AK, Singh G, Sander JW. Parasites and epilepsy: Understanding the determinants of epileptogenesis. Epilepsy Behav (2019) 92:235–44. doi: 10.1016/J.YEBEH.2018.11.033
56. Othman AA, Abdel-Aleem GA, Saied EM, Mayah WW, Elatrash AM. Biochemical and immunopathological changes in experimental neurotoxocariasis. Mol Biochem Parasitol (2010) 172(1):1–8. doi: 10.1016/j.molbiopara.2010.03.006
57. Chou CM, Lee YL, Liao CW, Huang YC, Fan CK. Enhanced expressions of neurodegeneration-associated factors, UPS impairment, and excess aβ accumulation in the hippocampus of mice with persistent cerebral toxocariasis. Parasites Vectors (2017) 10(1):1–14. doi: 10.1186/S13071-017-2578-6/FIGURES/8
58. Khademvatan S, Khajeddin N, Izadi S, Yousefi E. Investigation of anti- toxocara and anti- toxoplasma antibodies in patients with schizophrenia disorder. Schizophr Res Treat (2014) 2014:1–7. doi: 10.1155/2014/230349
59. Alvarado-Esquivel C. Toxocara infection in psychiatric inpatients: A case control seroprevalence study. PloS One (2013) 8(4):e62606. doi: 10.1371/JOURNAL.PONE.0062606
60. Liao C-W, Fan C-K, Kao T-C, et al. Brain injury-associated biomarkers of TGF-beta1, S100B, GFAP, NF-l, tTG, AbetaPP, and tau were concomitantly enhanced and the UPS was impaired during acute brain injury caused by toxocara canis in mice. BMC Infectious Diseases (2008) 8(1):1–15. doi: 10.1186/1471-2334-8-84
61. Fan CK, Holland CV, Loxton K, Barghouth U. Cerebral toxocariasis: Silent progression to neurodegenerative disorders? Clin Microbiol Rev (2015) 28(3):663–86. doi: 10.1128/CMR.00106-14
62. Waindok P, Janecek-Erfurth E, Lindenwald DL, Wilk E, Schughart K, Geffers R, et al. Toxocara canis- and toxocara cati-induced neurotoxocarosis is associated with comprehensive brain transcriptomic alterations. Microorg (2022) 10(1):177. doi: 10.3390/MICROORGANISMS10010177
63. Li L, Gao W, Yang X, Wu D, Bi H, Zhang S, et al. Asthma and toxocariasis. Ann Allergy Asthma Immunol (2014) 113(2):187–92. doi: 10.1016/j.anai.2014.05.016
64. Aghaei S, Riahi SM, Rostami A, Mohammadzadeh I, Javanian M, Tohidi E, et al. Toxocara spp. infection and risk of childhood asthma: A systematic review and meta-analysis. Acta Trop (2018) 182:298–304. doi: 10.1016/J.ACTATROPICA.2018.03.022
65. Sandra Guadalupe BG, Mario Noé MG, Gustavo Esteban PA, Gonzalez-Bobadilla NY, Clavijo-Sanchez K, Chavez-Zea AL, et al. Detection of antigens and anti-toxocara canis antibodies in children with different asthma severities. Immunity Inflammation Dis (2021) 9(2):435–42. doi: 10.1002/iid3.403
66. Fialho PMM, Correa CRS, Lescano SZ. Asthma and seroconversion from toxocara spp. infection: Which comes first? BioMed Res Int (2018) 2018:4280792. doi: 10.1155/2018/4280792
67. Ma G, Holland CV, Wang T, Hofmann A, Fan C-K, Maizels RM, et al. Human toxocariasis. Lancet Infect Dis (2018) 18(1):e14–24. doi: 10.1016/S1473-3099(17)30331-6
68. Pinelli E AC. Toxocara infection and its association with allergic manifestations. Endocrine Metab Immune Disord (2012) 12(1):33–44. doi: 10.2174/187153012799278956
69. Zibaei M, Shayesteh Z, Moradi N, Bahadory S. Human toxocara infection: Allergy and immune responses. Antiinflamm Antiallergy Agents Med Chem (2019) 18(2):82–90. doi: 10.2174/1871523018666181210115840
70. Bakhshani A, Shirvan SP, Maleki M, Haghparast A, Borji H. Evaluation of the effect of toxocara cati infection in the mouse model of allergic asthma: Exacerbation of allergic asthma symptoms and Th2 types of response. Comp Immunol Microbiol Infect Dis (2020) 71:101488. doi: 10.1016/J.CIMID.2020.101488
71. Pinelli E, Brandes S, Dormans J, Gremmer E, Van Loveren H. Infection with the roundworm toxocara canis leads to exacerbation of experimental allergic airway inflammation. Clin Exp Allergy (2008) 38(4):649–58. doi: 10.1111/j.1365-2222.2007.02908.x
72. Choi K-D, Choi J-H, Choi S-Y, Ho Jung J. Toxocara optic neuropathy: clinical features and ocular findings. Int J Ophthalmol (2018) 11(3):520–3. doi: 10.18240/ijo.2018.03.26
73. Sahu ES, Pal B, Sharma T, Biswas J. Ocular immunology and inflammation clinical profile, treatment, and visual outcome of ocular toxocara in a tertiary eye care centre. Ocul Immunol Inflamm (2016) 26(5):753–9. doi: 10.1080/09273948.2016.1249375
74. Rubinsky-Elefant G, Hirata CE, Yamamoto JH, Ferreira MU. Human toxocariasis: Diagnosis, worldwide seroprevalences and clinical expression of the systemic and ocular forms. Ann Trop Med Parasitol (2010) 104(1):3–23. doi: 10.1179/136485910X12607012373957
75. Rockey JH, Donnelly JJ, Stromberg BE, Soulsby EJL. Immunopathology of toxocara canis and ascaris suum infections of the eye: The role of the eosinophil. Investig Ophthalmol Vis Sci (1979) 18(11):1172–84.
76. Shyu LY, Chen KM, Lai SC. Matrix metalloproteinase-2 and matrix metalloproteinase-9 in mice with ocular toxocariasis. Parasitol Res (2019) 118(2):483–91. doi: 10.1007/s00436-018-06196-4
77. Wang H, Tao Y. Clinical features and prognostic factors in northern Chinese patients with peripheral granuloma type of ocular toxocariasis: A retrospective cohort study. Ocul Immunol Inflamm (2020) 29(7-8):1259–64. doi: 10.1080/09273948.2020.1804592
78. Ocular toxocariasis — united states, 2009–2010. Available at: https://www.cdc.gov/mmwr/preview/mmwrhtml/mm6022a2.htm (Accessed May 10, 2022).
79. Good B, Holland CV, Taylor MRH, Larragy J, Moriarty P, O’Regan M. Ocular toxocariasis in schoolchildren. Clin Infect Dis (2004) 39(2):173–8. doi: 10.1086/421492
80. CDC - lymphatic filariasis - biology. Available at: https://www.cdc.gov/parasites/lymphaticfilariasis/biology.html (Accessed July 15, 2022).
81. Babu S, Nutman TB. Immunopathogenesis of lymphatic filarial disease1. Springer-Verlag (2012) 34(6):847–861. doi: 10.1007/s00281-012-0346-4
82. Mehlotra RK, Hall LR, Haxhiu MA, Pearlman E. Reciprocal immunomodulatory effects of gamma interferon and interleukin-4 on filaria-induced airway hyperresponsiveness. Infect Immun (2001) 69(3):1463. doi: 10.1128/IAI.69.3.1463-1468.2001
83. Lobos E, Nutman TB, Hothersall JS, Moncad S. Elevated immunoglobulin e against recombinant brugia malayi γ-glutamyl transpeptidase in patients with bancroftian filariasis: Association with tropical pulmonary eosinophilia or putative immunity. Infect Immun (2003) 71(2):747. doi: 10.1128/IAI.71.2.747-753.2003
84. Mehlotra RK, Hall LR, Higgins AW, Dreshaj IA, Haxhiu MA, Kazura JW, et al. Interleukin-12 suppresses filaria-induced pulmonary eosinophilia, deposition of major basic protein and airway hyperresponsiveness. Parasite Immunol (1998) 20(10):455–62. doi: 10.1046/j.1365-3024.1998.00174.x
85. Sarker PC, Khandker HH, Sarker CR, Khatoon M. Clinico-laboratory profile of 45 filarial arthritis cases. Mymensingh Med J (2007) 16(2 Suppl):S7–11.
86. Dreyer G, Dreyer P, Piessens WF. Extralymphatic disease due to bancroftian filariasis. Braz J Med Biol Res (1999) 32(12):1467–72. doi: 10.1590/S0100-879X1999001200003
87. Case T, Leis B, Witte M, Way D, Bernas M, Borgs P, et al. Vascular abnormalities in experimental and human lymphatic filariasis. Lymphology (1991) 24(4):174–83.
88. Bouvet JP, Thérizol M, Auquier L. Microfilarial polyarthritis in a massive loa loa infestation. a case report. Acta Trop (1977) 34(3):281–4. doi: 10.5169/seals-312267
89. Rao RV, Anupindi L, Chatterjee A, Varghese GK, Krishnanand BR. Filarial nephritis: A cause of nephrotic syndrome. Trop Geogr Med (1993) 45(4):180.
90. Waugh DA, Alexander JH, Ibels LS. Filarial chyluria associated glomerulonephritis and therapeutic considerations in the chyluric patient. Aust N Z J Med (1980) 10(5):559–62. doi: 10.1111/J.1445-5994.1980.TB04977.X
91. Yap HK, Woo KT, Yeo PPB. The nephrotic syndrome associated with filariasis. Ann Acad Med Singapore (1982) 11(1):61–3.
92. Zheng HJ, Tao ZH, Reddy MV, Harinath BC, Piessens WF. Parasite antigens in sera and urine of patients with bancroftian and brugian filariasis detected by sandwich Elisa with monoclonal antibodies. Am J Trop Med Hyg (1987) 36(3):554–60. doi: 10.4269/AJTMH.1987.36.554
93. Rath RN, Mishra N, Sahu RN, Mohanty G, Das BK. Renal involvement in bancroftian filariasis. Natl Med J India (1991) 4(2):65–8.
94. Langhammer J, Birk HW, Zahner H. Renal disease in lymphatic filariasis: Evidence for tubular and glomerular disorders at various stages of the infection. Trop Med Int Heal (1997) 2(9):875–84. doi: 10.1046/J.1365-3156.1997.D01-404.X
95. World Healtth Organization. Onchocerciasis. In: World health organization fact sheets (2022). Available at: https://www.who.int/news-room/fact-sheets/detail/onchocerciasis.
96. Institute for Health Metrics and Evaluation. Global burden of disease study 2017. Lancet (2018) Seattle, WA: IHME. https://www.healthdata.org/sites/default/files/files/policy_report/2019/GBD_2017_Booklet.pdf.
97. Burchard GD, Buettner DW, Bierther M. Electron microscopical studies on onchocerciasis. III. the onchocerca-nodule. Tropenmed Parasitol (1979) 30(1):103–12.
98. Donnelly JJ. Autoantibody induced by experimental ocular onchocerca infection: Identification and cháracterization of autoantigens. Autoimmunity (1988) 1(3):207–16. doi: 10.3109/08916938808997165
99. McKechnie NM, Gürr W, Yamada H, Copland D, Braun G. Antigenic mimicry: Onchocerca volvulus antigen-specific T cells and ocular inflammation. Investig Ophthalmol Vis Sci (2002) 43(2):411–8.
100. Pearlman E, Lass JH, Bardenstein DS, Diaconu E, Hazlett FE Jr, Albright J, et al. Onchocerca volvulus-mediated keratitis: Cytokine production by IL-4-Deficient mice. Experimental Parasitology (1996) 84(2):274–281. doi: 10.1006/expr.1996.0113
101. Andre AS, Blackwell NM, Hall LR, Hoerauf A, Brattig NW, Volkmann L, et al. The role of endosymbiotic wolbachia bacteria in the pathogenesis of river blindness. Science (2002) 295(5561):1892–5. doi: 10.1126/SCIENCE.1068732
102. Gillette-Ferguson I, Hise AG, Sun Y, Diaconu E, McGarry HF, Taylor MJ, et al. Wolbachia- and onchocerca volvulus-induced keratitis (river blindness) is dependent on myeloid differentiation factor 88. Infect Immun (2006) 74(4):2442–5. doi: 10.1128/IAI.74.4.2442-2445.2006
103. Jilek WG, Jilek-Aall LM. The problem of epilepsy in a rural Tanzanian tribe. Afr J Med Sci (1970) 1(3):305–7.
104. Winkler AS, Friedrich K, König R, Meindl M, Helbok R, Unterberger I, et al. The head nodding syndrome-clinical classification and possible causes. Epilepsia (2008) 49:2008–15. doi: 10.1111/j.1528-1167.2008.01671.x
105. Couper J. Prevalence of childhood disability in rural KwaZulu natal. South Afr Med J (2016) 92(7):549–52.
106. Winkler AS, Friedrich K, Velicheti S, Dharsee J, Konig R, Nassri A, et al. MRI Findings in people with epilepsy and nodding syndrome in an area endemic for onchocerciasis: an observational study. Afr Health Sci (2013) 13(2):529–40. doi: 10.4314/AHS.V13I2.51
107. Kipp W, Kasoro S, Burnham G. Onchocerciasis and epilepsy in Uganda. Lancet (1994) 343(8890):183–4. doi: 10.1016/S0140-6736(94)90980-6
108. König R, Nassri A, Meindl M, Matuja W, Kindunda AR, Siegmund V, et al. The role of onchocerca volvulus in the development of epilepsy in a rural area of Tanzania. Parasitology (2010) 137(10):1559–68. doi: 10.1017/S0031182010000338
109. Sejvar JJ, Kakooza AM, Foltz JL, Makumbi I, Atai-Omoruto AD, Malimbo M, et al. Clinical, neurological, and electrophysiological features of nodding syndrome in kitgum, Uganda: an observational case series. The Lancet Neurology (2013) 12(2):166–174. doi: 10.1016/S1474-4422(12)70321-6
110. Johnson TP, Sejvar J, Nutman TB, Nath A. The pathogenesis of nodding syndrome. Annual Review of Pathology: Mechanisms of Disease (2020) 15:395–417. doi: 10.1146/annurev-pathmechdis
111. Johnson TP, Tyagi R, Lee PR, Lee M-H, Johnson KR, Kowalak J, et al. Nodding syndrome may be an autoimmune reaction to the parasitic worm onchocerca volvulus HHS public access author manuscript. Sci Transl Med (2017) 9(377):1–10. doi: 10.1126/scitranslmed.aaf6953
112. Deol AK, Fleming FM, Calvo-Urbano B, Walker M, Bucumi V, Gnandou I, et al. Schistosomiasis - assessing progress towards the 2020 and 2025 globalgoals. N Engl J Med (2019) 381(26):2519. doi: 10.1056/NEJMOA1812165
113. . CDC - schistosomiasis - resources for health professionals. Available at: https://www.cdc.gov/parasites/schistosomiasis/health_professionals/index.html (Accessed May 10, 2022).
114. da Silva GB, Duarte DB, Barros EJG, De Francesco Daher E. Schistosomiasis-associated kidney disease: A review. Asian Pacific J Trop Dis (2013) 3(1):79. doi: 10.1016/S2222-1808(13)60018-3
115. Neves PDM, Jorge LB, Cavalcante LB, Malheiros D, Woronik V, Dias C. Schistosomiasis-associated glomerulopathy: Clinical aspects, pathological characteristics, and renal outcoomes. Clin Nephrol (2020) 93(5):251–61. doi: 10.5414/CN110013
116. Barsoum RS. Schistosomiasis and the kidney. Semin Nephrol (2003) 23(1):34–41. doi: 10.1053/SNEP.2003.50003A
117. Obeid FN, Smith RF, Elliott JP, Reddy DJ, Hageman JH. Bilharzial portal hypertension. Arch Surg (1983) 118(6):702–8. doi: 10.1001/ARCHSURG.1983.01390060024005
118. Manzella A, Ohtomo K, Monzawa S, Lim JH. Schistosomiasis of the liver. Abdom Imaging (2008) 33(2):144–50. doi: 10.1007/S00261-007-9329-7
119. Tamarozzi F, Fittipaldo VA, Orth HM, Richter J, Buonfrate D, Riccardi N, et al. Diagnosis and clinical management of hepatosplenic schistosomiasis: A scoping review of the literature. PloS Negl Trop Dis (2021) 15(3):e0009191. doi: 10.1371/JOURNAL.PNTD.0009191
120. Omar HH. Impact of chronic schistosomiasis and HBV/HCV co-infection on the liver: current perspectives. Hepat Med (2019) 11:131–6. doi: 10.2147/HMER.S155962
121. Sanghvi MM, Hotez PJ, Fenwick A. Neglected tropical diseases as a cause of chronic liver disease: the case of schistosomiasis and hepatitis c Co-infections in Egypt. Liver Int (2013) 33(2):165–8. doi: 10.1111/LIV.12052
122. Ferrari TCA, Albricker ACL, Gonçalves IM, Freire CMV. Schistosome-associated pulmonary arterial hypertension: A review emphasizing pathogenesis. Front Cardiovasc Med (2021) 0:724254. doi: 10.3389/FCVM.2021.724254
123. Zeng X, Huang X, Rathinasabapathy A, Xu Z, Li K, Liu N, et al. Prevalence of schistosoma japonicum-associated pulmonary hypertension in china: An echocardiography-based assessment. Ann Am Thorac Soc (2021) 18(12):2095–8. doi: 10.1513/ANNALSATS.202012-1573RL
124. Sibomana JP, Campeche A, Carvalho-Filho RJ, Correa RA, Duani H, Guimaraes VP, et al. Schistosomiasis pulmonary arterial hypertension. Front Immunol (2020) 11. doi: 10.3389/FIMMU.2020.608883
125. Kumar R, Mickael C, Kassa B, Sanders L, Koyanagi D, Hernandez-Saavedra D, et al. Th2 CD4+ T cells are necessary and sufficient for schistosoma-pulmonary hypertension. J Am Heart Assoc (2019) 8(15):1–28. doi: 10.1161/JAHA.119.013111
126. Bouvard V, Baan R, Straif K, Grosse Y, Secretan B, El Ghissassi F, et al. A review of human carcinogens–part b: biological agents. Lancet Oncol (2009) 10(4):321–2. doi: 10.1016/S1470-2045(09)70096-8
127. Shiff C, Veltri R, Naples J, Quartey J, Otchere J, Anyan W, et al. Ultrasound verification of bladder damage is associated with known biomarkers of bladder cancer in adults chronically infected with schistosoma haematobium in Ghana. Trans R Soc Trop Med Hyg (2006) 100(9):847–54. doi: 10.1016/J.TRSTMH.2005.10.010
128. Brindley PJ, da Costa JMC, Sripa B. Why does infection with some helminths cause cancer? Trends Cancer (2015) 1(3):174–82. doi: 10.1016/j.trecan.2015.08.011
129. Botelho M, Ferreira AC, Oliveira MJ, Domingues A, Machado JC, da Costa JMC. Schistosoma haematobium total antigen induces increased proliferation, migration and invasion, and decreases apoptosis of normal epithelial cells. Int J Parasitol (2009) 39(10):1083–91. doi: 10.1016/J.IJPARA.2009.02.016
130. Chaudhary KS, Lu QL, Abel PD, Khandan Nia N, Shoma AM, El Baz MA, et al. Expression of bcl-2 and p53 oncoproteins in schistosomiasis-associated transitional and squamous cell carcinoma of urinary bladder. Br J Urol (1997) 79(1):78–84. doi: 10.1046/J.1464-410X.1997.30717.X
131. Chala B, Choi MH, Moon KC, Kim HS, Kwak C, Hong ST. Development of urinary bladder pre-neoplasia by schistosoma haematobium eggs and chemical carcinogen in mice. Korean J Parasitol (2017) 55(1):21–9. doi: 10.3347/KJP.2017.55.1.21
132. Botelho MC, Veiga I, Oliveira PA, Lopes C, Teixeira M, Correia da Costa JM, et al. Carcinogenic ability of schistosoma haematobium possibly through oncogenic mutation of KRAS gene. Adv Cancer Res Treat (2013) 2013:1–11.
133. Botelho MC, Machado JC, da Costa JMC. Schistosoma haematobium and bladder cancer: what lies beneath? Virulence (2010) 1(2):84–7. doi: 10.4161/VIRU.1.2.10487
134. Fu CL, Odegaard JI, Herbert DR, Hsieh MH. A novel mouse model of schistosoma haematobium egg-induced immunopathology. PloS Pathog (2012) 8(3):e1002605. doi: 10.1371/JOURNAL.PPAT.1002605
135. Tuffour I, Ayi I, Gwira TM, Dumashie E, Ashong Y, Appiah-Opong R. Schistosoma egg antigen induces oncogenic alterations in human prostate cells. Anal Cell Pathol (2018) 2018:1–10. doi: 10.1155/2018/4675380
136. Zhong X, Isharwal S, Naples JM, Shiff C, Veltri RW, Shao C, et al. Hypermethylation of genes detected in urine from ghanaian adults with bladder pathology associated with schistosoma haematobium infection. PloS One (2013) 8(3):e59089. doi: 10.1371/JOURNAL.PONE.0059089
137. Shamma AH. Schistosomiasis and cancer in Iraq. Am J Clin Pathol (1955) 25(11):1283–4. doi: 10.1093/AJCP/25.11.1283
138. Al-Fouadi A, Parkin DM. Cancer in Iraq: seven years’ data from the baghdad tumour registry. Int J Cancer (1984) 34(2):207–13. doi: 10.1002/IJC.2910340211
139. Savardekar LS, Balaiah D, Mali BN. Association of schistosoma haematobium and human papillomavirus in cervical cancer: a case report. Acta Cytol (2010) 54(2):205–8. doi: 10.1159/000325011
140. Kjetland EF, Ndhlovu PD, Mduluza T, Deschoolmeester V, Midzi N, Gomo E, et al. The effects of genital schistosoma haematobium on human papillomavirus and the development of cervical neoplasia after five years in a Zimbabwean population. Eur J Gynaecol Oncol (2010) 31(2):169–73.
141. Takemura Y, Kikuchi S, Inaba Y. Epidemiologic study of the relationship between schistosomiasis due to schistosoma japonicum and liver cancer/cirrhosis. Am J Trop Med Hyg (1998) 59(4):551–6. doi: 10.4269/AJTMH.1998.59.551
142. Xu Z, Su D -L. Schistosoma japonicum and colorectal cancer: an epidemiological study in the people’s republic of China. Int J Cancer (1984) 34(3):315–8. doi: 10.1002/IJC.2910340305
143. Guo W, Zheng W, Li JY, Chen JS, Blot WJ. Correlations of colon cancer mortality with dietary factors, serum markers, and schistosomiasis in China. Nutr Cancer (1993) 20(1):13–20. doi: 10.1080/01635589309514266
144. Qiu DC, Hubbard AE, Zhong B, Zhang Y, Spear RC. A matched, case-control study of the association between schistosoma japonicum and liver and colon cancers, in rural China. Ann Trop Med Parasitol (2005) 99(1):47–52. doi: 10.1179/136485905X19883
145. Johansen MV, Bøgh HO, Nansen P, Christensen NO. Schistosoma japonicum infection in the pig as a model for human schistosomiasis japonica. Acta Trop (2000) 76(2):85–99. doi: 10.1016/S0001-706X(00)00103-0
146. Hamid HKS. Schistosoma japonicum–associated colorectal cancer: A review. Am J Trop Med Hyg (2019) 100(3):501. doi: 10.4269/AJTMH.18-0807
147. Ming-Chai C, Chi-yuan C, Pei-Yu C, Jen-Chun H. Evolution of colorectai cancer in schistosomiasis: Transitional mucosal changes adjacent to large intestinal carcinoma in colectomy specimens. Cancer (1980) 46(7):1661–75. doi: 10.1002/1097-0142(19801001)46:7<1661::AID-CNCR2820460728>3.0.CO;2-O
148. Ch’en MC, Hu JCP. Pathogenesis of carcinoma of the colon and rectum in schistosomiasis japonica: a study on 90 cases. Chin Med J (1965) 84(8):513–25.
149. Ross AGP, Bartley PB, Sleigh AC, Richard Olds G, Li Y, Williams GM, et al. Schistosomiasis. N Engl J Med (2002) 346(16):1212–20. doi: 10.1056/NEJMRA012396
150. Nakashima T, Okuda K, Kojiro M, Sakamoto K, Kubo Y, Shimokawa Y. Primary liver cancer coincident with schistosomiasis japonica: A study of 24 necropsies. Cancer (1975) 36(4):1483–9. doi: 10.1002/1097-0142
151. Rosin MP, Anwar WA, Ward A. Inflammation, chromosomal instability, and cancer: the schistosomiasis model. Cancer Res (1994) 54:1929–33.
152. Long X-C, Bahgat M, Chlichlia K, Ruppel A, Li Y-L. Detection of inducible nitric oxide synthase in schistosoma japonicum and s. mansoni J Helminthol (2004) 78(1):47–50. doi: 10.1079/JOH2003202
153. Ishii A, Matsuoka H, Aji T, Hayatsu H, Wataya Y, Arimoto S, et al. Evaluation of the mutagenicity and the tumor-promoting activity of parasite extracts: Schistosoma japonicum and clonorchis sinensis. Mutat Res Toxicol (1989) 224(2):229–33. doi: 10.1016/0165-1218(89)90160-2
154. Yang Q, Qiu H, Xie H, Qi Y, Cha H, Qu J, et al. A schistosoma japonicum infection promotes the expansion of myeloid-derived suppressor cells by activating the JAK/STAT3 pathway. J Immunol (2017) 198(12):4716–27. doi: 10.4049/JIMMUNOL.1601860
155. Tang CL, Lei JH, Wang T, Lu S-J, Guan F, Liu W-Q, et al. Effect of CD4+ CD25+ regulatory T cells on the immune evasion of schistosoma japonicum. Parasitol Res (2011) 108(2):477–80. doi: 10.1007/S00436-010-2089-2
156. Zhang R, Takahashi S, Orita SI, Yoshida A, Maruyama H, Shirai T, et al. p53 gene mutations in rectal cancer associated with schistosomiasis japonica in Chinese patients. Cancer Lett (1998) 131(2):215–21. doi: 10.1016/S0304-3835(98)00154-2
157. Na BK, Pak JH, Hong SJ. Clonorchis sinensis and clonorchiasis. Acta Trop (2020) 203:1–10. doi: 10.1016/J.ACTATROPICA.2019.105309
158. Sripa B, Brindley PJ, Mulvenna J, Laha T, Smout MJ, Mairiang E, et al. The tumorigenic liver fluke opisthorchis viverrini –multiple pathways to cancer. Trends Parasitol (2012) 28(10):395. doi: 10.1016/J.PT.2012.07.006
159. Keiser J, Utzinger J. Food-borne trematodiases. Clin Microbiol Rev (2009) 22(3):466–83. doi: 10.1128/CMR.00012-09
160. Andrews RH, Sithithaworn P, Petney TN. Opisthorchis viverrini: an underestimated parasite in world health. Trends Parasitol (2008) 24(11):497–501. doi: 10.1016/J.PT.2008.08.011
161. Shin HR, Lee CU, Park HJ, Seol S-Y, Chung J-M, Choi H-C, et al. Hepatitis b and c virus, clonorchis sinensis for the risk of liver cancer: a case-control study in pusan, Korea. Int J Epidemiol (1996) 25(5):933–40. doi: 10.1093/IJE/25.5.933
162. Botelho MC, Alves H, Richter J. Wound healing and cancer progression in opisthorchis viverrini associated cholangiocarcinoma. Parasitol Res (2016) 115(7):2913–4. doi: 10.1007/S00436-016-5090-6
163. Pak JH, Lee JY, Jeon BY, Dai F, Yoo WG, Hong SJ. Cytokine production in cholangiocarcinoma cells in response to clonorchis sinensis excretory-secretory products and their putative protein components. Korean J Parasitol (2019) 57(4):379–87. doi: 10.3347/KJP.2019.57.4.379
164. Thuwajit C, Thuwajit P, Kaewkes S, Sripa B, Uchida K, Miwa M, et al. Increased cell proliferation of mouse fibroblast NIH-3T3 in vitro induced by excretory/secretory product(s) from opisthorchis viverrini. Parasitology (2004) 129(Pt 4):455–64. doi: 10.1017/S0031182004005815
165. Pak JH, Kim IK, Kim SM, Maeng S, Song KJ, Na B-K, et al. Induction of cancer-related microRNA expression profiling using excretory-secretory products of clonorchis sinensis. Parasitol Res (2014) 113(12):4447–55. doi: 10.1007/S00436-014-4127-Y
166. Kim DW, Kim JY, Moon JH, Kim K-B, Kim T-S, Hong S-J, et al. Transcriptional induction of minichromosome maintenance protein 7 (Mcm7) in human cholangiocarcinoma cells treated with clonorchis sinensis excretory-secretory products. Mol Biochem Parasitol (2010) 173(1):10–6. doi: 10.1016/J.MOLBIOPARA.2010.03.005
167. Pak JH, Moon JH, Hwang SJ, Cho SH, Seo SB, Kim TS. Proteomic analysis of differentially expressed proteins in human cholangiocarcinoma cells treated with clonorchis sinensis excretory-secretory products. J Cell Biochem (2009) 108(6):1376–88. doi: 10.1002/JCB.22368
168. Nam JH, Moon JH, Kim IK, Lee M-R, Hong S-J, Ahn JH, et al. Free radicals enzymatically triggered by clonorchis sinensis excretory-secretory products cause NF-κB-mediated inflammation in human cholangiocarcinoma cells. Int J Parasitol (2012) 42(1):103–13. doi: 10.1016/J.IJPARA.2011.11.001
169. CDC - paragonimiasis - biology. Available at: https://www.cdc.gov/parasites/paragonimus/biology.html (Accessed July 15, 2022).
170. Mukae H, Taniguchi H, Matsumoto N, Iiboshi H, Ashitani J-I, Matsukura S, et al. Clinicoradiologic features of pleuropulmonary paragonimus westermani on kyusyu island, Japan. Chest (2001) 120(2):514–20. doi: 10.1378/CHEST.120.2.514
171. Devi KR, Narain K, Bhattacharya S, Negmu K, Agatsuma T, Blair D, et al. Pleuropulmonary paragonimiasis due to paragonimus heterotremus: molecular diagnosis, prevalence of infection and clinicoradiological features in an endemic area of northeastern India. Trans R Soc Trop Med Hyg (2007) 101:786–92. doi: 10.1016/j.trstmh.2007.02.028
172. Itoh N, Tsukahara M, Yamasaki H, Morishima Y, Sugiyama H, Kurai H. Paragonimus westermani infection mimicking recurrent lung cancer: A case report. Journal of Infection and Chemotherapy (2016) 22(12):815–818. doi: 10.1016/j.jiac.2016.07.002
173. Procop GW. North American paragonimiasis (caused by paragonimus kellicotti) in the context of global paragonimiasis. Clin Microbiol Rev (2009) 22(3):415–46. doi: 10.1128/CMR.00005-08
174. Im JG, Kong Y, Shin YM, Yang SO, Song JG, Han MC, et al. Pulmonary paragonimiasis: clinical and experimental studies. Radiographics (1993) 13(3):575–86. doi: 10.1148/radiographics1338316665
175. Oh SJ. Cerebral and spinal paragonimiasis histopathological study. J Neurol Sci (1969) 9(2):205–36. doi: 10.1016/0022-510X(69)90072-0
176. Kashida Y, Niiro M, Maruyama H, Hanaya R. Cerebral paragonimiasis with hemorrhagic stroke in a developed country. J Stroke Cerebrovasc Dis (2018) 27(10):2648–9. doi: 10.1016/J.JSTROKECEREBROVASDIS.2018.05.033
177. Wang Q, Hou L, Liu L. Diagnosis and treatment of hemorrhagic cerebral paragonimiasis: Three case reports and literature review. Turk Neurosurg (2020) 30(4):624–8. doi: 10.5137/1019-5149.JTN.22666-18.3
178. Xia Y, Ju Y, Chen J, You C. Cerebral paragonimiasis: a retrospective analysis of 27 cases. J Neurosurg Pediatr (2015) 15(1):101–6. doi: 10.3171/2014.10.PEDS14208
179. Li L, Zhang Y, Zhu J, Zhai X, Cai J, He L, et al. Intracranial pseudoaneurysm caused by cerebral paragonimiasis in pediatric patients. Pediatric Neurology (2020) 109:47–51. doi: 10.1016/j.pediatrneurol.2020.03.018
180. Chen J, Chen Z, Li F, Lin J, Meng H, Feng H. Cerebral paragonimiasis that manifested as intracranial hemorrhage: Clinical article. J Neurosurg Pediatr (2010) 6(6):572–8. doi: 10.3171/2010.9.PEDS1099
181. Chen J, Chen Z, Lin J, Zhu G, Meng H, Cui G, et al. Cerebral paragonimiasis: A retrospective analysis of 89 cases. Clin Neurol Neurosurg (2013) 115(5):546–51. doi: 10.1016/j.clineuro.2012.06.025
182. CDC - DPDx - cysticercosis. Available at: https://www.cdc.gov/dpdx/cysticercosis/index.html (Accessed May 10, 2022).
183. Taeniasis and cysticercosis. Available at: https://www.who.int/health-topics/taeniasis-and-cysticercosis#tab=tab_1 (Accessed May 10, 2022).
184. Debacq G, Moyano LM, Garcia HH, Boumediene F, Marin B, Ngoungou EB, et al. Systematic review and meta-analysis estimating association of cysticercosis and neurocysticercosis with epilepsy. PloS Negl Trop Dis (2017) 11(3):1–17. doi: 10.1371/JOURNAL.PNTD.0005153
185. Carabin H, Ndimubanzi PC, Budke CM, Nguyen H, Qian Y, Cowan LD, et al. Clinical manifestations associated with neurocysticercosis: a systematic review. PloS Negl Trop Dis (2011) 5(5):1–13. doi: 10.1371/JOURNAL.PNTD.0001152
186. White AC, Coyle CM, Rajshekhar V, Singh G, Allen Hauser W, Mohanty A, et al. Diagnosis and treatment of neurocysticercosis: 2017 clinical practice guidelines by the infectious diseases society of America (IDSA) and the American society of tropical medicine and hygiene (ASTMH). Clin Infect Dis (2018) 66(8):e49–75. doi: 10.1093/CID/CIX1084
187. Restrepo BI, Llaguno P, Sandoval MA, Enciso JA, Teale JM. Analysis of immune lesions in neurocysticercosis patients: central nervous system response to helminth appears Th1-like instead of Th2. J Neuroimmunol (1998) 89(1):64–72. doi: 10.1016/S0165-5728(98)00112-X
188. Arechavaleta F, Molinari JL, Tato P. A taenia solium metacestode factor nonspecifically inhibits cytokine production. Parasitol Res (1998) 84(2):117–22. doi: 10.1007/S004360050367
189. White A, Tato P, Molinari J. Host-parasite interactions in taenia solium cysticercosis. Infect Agents Dis (1992) 1(4):185–93.
190. Singh G, Burneo JG, Sander JW. From seizures to epilepsy and its substrates: Neurocysticercosis. Epilepsia (2013) 54(5):783–92. doi: 10.1111/EPI.12159
191. Herrick JA, Maharathi B, Kim JS, Abundis GG, Garg A, Gonzales I, et al. Inflammation is a key risk factor for persistent seizures in neurocysticercosis. Ann Clin Transl Neurol (2018) 5(5):630–9. doi: 10.1002/ACN3.562
192. Restrepo BI, Alvarez JI, Castaño JA, Arias LF, Restrepo M, Trujillo J, et al. Brain granulomas in neurocysticercosis patients are associated with a Th1 and Th2 profile. Infect Immun (2001) 69(7):4554–60. doi: 10.1128/IAI.69.7.4554-4560.2001
193. Prasad A, Prasad KN, Gupta RK, Pradhan S. Increased expression of ICAM-1 among symptomatic neurocysticercosis. J Neuroimmunol (2009) 206(1-2):118–20. doi: 10.1016/J.JNEUROIM.2008.09.015
194. Verma A, Prasad KN, Gupta RK, Kumar Singh A, Kumar Nyati K, Rizwan A, et al. Toll-like receptor 4 polymorphism and its association with symptomatic neurocysticercosis. J Infect Dis (2010) 202(8):1219–25. doi: 10.1086/656395
195. Lachuriya G, Garg RK, Jain A, Singh Malhotra H, Kumar Singh A, Jain B, et al. Toll-like receptor-4 polymorphisms and serum matrix metalloproteinase-9 in newly diagnosed patients with calcified neurocysticercosis and seizures. Med (United States) (2016) 95(17):1–7. doi: 10.1097/MD.0000000000003288
196. Nash TE, O’Connell EM, Hammoud DA, Wetzler L, Ware JAM, Mahanty S. Natural history of treated subarachnoid neurocysticercosis. Am J Trop Med Hyg (2020) 102(1):78–89. doi: 10.4269/AJTMH.19-0436
197. Sáenz B, Fleury A, Chavarría A, Hernandez M, Crispin JC, Vargas-Rojas MI, et al. Neurocysticercosis: local and systemic immune-inflammatory features related to severity. Med Microbiol Immunol (2012) 201(1):73–80. doi: 10.1007/S00430-011-0207-0
198. Harrison S, Thumm L, Nash TE, Nutman TB, O’Connell EM. The local inflammatory profile and predictors of treatment success in subarachnoid neurocysticercosis. Clin Infect Dis (2021) 72(9):E326–33. doi: 10.1093/CID/CIAA1128
199. CDC - echinococcosis - biology. Available at: https://www.cdc.gov/parasites/echinococcosis/biology.html (Accessed July 15, 2022). 86
200. Echinococcosis. Available at: https://www.who.int/news-room/fact-sheets/detail/echinococcosis (Accessed July 15, 2022).
201. Eckert J, Gemmell MA, Meslin F-X, Pawłowski ZS. World health organization world organisation for animal health WHO/OIE manual on echinococcosis in humans and animals: a public health problem of global concern. Available at: http://www.oie.int (Accessed July 15, 2022).
202. Collado-Aliaga J, Romero-Alegría A, Alonso-Sardón M, Muro A, Lopez-Bernus A, Velasco-Tirado V, et al. Complications associated with initial clinical presentation of cystic echinococcosis: A 20-year cohort analysis. Am J Trop Med Hyg (2019) 101(3):628–35. doi: 10.4269/ajtmh.19-0019
203. Greco S, Cannella R, Giambelluca D, Pecoraro G, Battaglia E, Midiri M, et al. Complications of hepatic echinococcosis: multimodality imaging approach. Insights into Imaging (2019) 10(1):1–12. doi: 10.1186/s13244-019-0805-8
204. Ali-Khan Z, Rausch RL. Demonstration of amyloid and immune complex deposits in renal and hepatic parenchyma of alaskan alveolar hydatid disease patients. Ann Trop Med Parasitol (2016) 81(4):381–392. doi: 10.1080/00034983.1987.11812136
205. Ghabdian S, Parande Shirvan S, Maleki M, Borji H. Exacerbation of allergic asthma by somatic antigen of echinococcus granulosus in allergic airway inflammation in BALB/c mice. Parasit Vectors (2022) 15(1):1–9. doi: 10.1186/S13071-021-05125-2
206. Aydin Y, Barlas O, Yolas C, Hakkiu Aydin I, Ceviz A, Aladag A, et al. Alveolar hydatid disease of the brain: Report of four cases. J Neurosurg (1986) 65(1):115–9. doi: 10.3171/JNS.1986.65.1.0115
207. Algros M-P, Majo F, Bresson-Hadni S, Koch S, Godard J, Cattin F, et al. Intracerebral alveolar echinococcosis. Infection (2003) 31(1):63–5. doi: 10.1007/s15010-002-2178-y
208. Li S, Chen J, He Y, Chen J, Liao S, Deng Y, et al. Incidence and risk factors associated with the development of epilepsy in patients with intracranial alveolar echinococcosis. Epilepsy Res (2021) 174:106643. doi: 10.1016/j.eplepsyres.2021.106643
209. Qiu M-D, Hun Z-S, Liu S-C, Wen L-C, Wang S-H, Bao L-X. Intracranial alveolar echinococeosis in China: Discussion of surgical treatment. Acta Neurochir (1986) 82:123–7. doi: 10.1007/BF01456372
210. Morar R, Feldman C. Pulmonary echinococcosis. European Respiratory Journal (2003) 21(6):1069–1077. doi: 10.1183/09031936.03.00108403
211. Gelman R, Brook G, Green J, Ben-Itzhak O, Nakhoul F. Minimal change glomerulonephritis associated with hydatid disease. Clin Nephrol (2000) 53(2):152–5.
212. Ibarrola AS, Sobrini B, Guisantes J, Prado J, Diez J, Monfa JM, et al. Membranous glomerulonephritis secondary to hydatid disease. Am J Med (1981) 70(2):311–5. doi: 10.1016/0002-9343(81)90766-X
213. Covic A, Caruntu L A, Goldsmith DJ. Reversible nephrotic syndrome due to mesangiocapillary glomerulonephritis secondary to hepatic hydatid disease. Nephrol Dial Transpl (1996) 11:2074–6. doi: 10.1093/oxfordjournals.ndt.a027101
214. Oliveira FMS, Matias PH da P, Kraemer L, Gazzinelli-Guimaraes AC, Santos FV, Oliveira Amorim CC, et al. Comorbidity associated to ascaris suum infection during pulmonary fibrosis exacerbates chronic lung and liver inflammation and dysfunction but not affect the parasite cycle in mice. PloS Negl Trop Dis (2019) 13(11):1–30. doi: 10.1371/journal.pntd.0007896
215. Grencis RK, Entwistle GM. Production of an interferon-gamma homologue by an intestinal nematode: functionally significant or interesting artefact? Parasitology (1997) 115(7):101–5. doi: 10.1017/S0031182097002114
216. Ocular toxocariasis — united states, 2009–2010. Available at: https://www.cdc.gov/mmwr/preview/mmwrhtml/mm6022a2.htm (Accessed January 11, 2022).
217. Purello-D’Ambrosio F, Pastorello E, Gangemi S, Lombardo G, Ricciardi L, Fogliani O, et al. Incidence of sensitivity to anisakis simplex in a risk population of fishermen/fishmongers. Ann Allergy Asthma Immunol (2000) 84(4):439–44. doi: 10.1016/S1081-1206(10)62278-8
218. Daschner A, Cuéllar C, Valls A. Towards a differential definition of atopy: Anisakis simplex and the relationship between parasites and arthropods in respiratory allergy. Parasite Immunol (2008) 30(8):417–24. doi: 10.1111/J.1365-3024.2008.01041.X
219. Bernardini R, Mistrello G, Novembre E, Roncarolo D, Zanotta S, Lombardi E, et al. Cross-reactivity between IgE-binding proteins from anisakis simplex and dermatophagoides pteronyssinus. Int J Immunopathol Pharmacol (2005) 18(4):671–5. doi: 10.1177/039463200501800408
220. Pakasa NM, Nseka NM, Nyimi LM. Secondary collapsing glomerulopathy associated with loa loa filariasis. Am J Kidney Dis (1997) 30(6):836–9. doi: 10.1016/S0272-6386(97)90090-1
221. Buell KG, Whittaker C, Chesnais CB, Jewell PD, Pion SDS, Walker M, et al. Atypical clinical manifestations of loiasis and their relevance for endemic populations. Open Forum Infect Dis (2019) 6(11). doi: 10.1093/OFID/OFZ417
222. Andrade ZA. Schistosomiasis and liver fibrosis. Parasite Immunol (2009) 31(11):656–63. doi: 10.1111/J.1365-3024.2009.01157.X
Keywords: helminths, non-communicable diseases, cestodes, trematodes, nematodes, immunity
Citation: Wu Y, Duffey M, Alex SE, Suarez-Reyes C, Clark EH and Weatherhead JE (2022) The role of helminths in the development of non-communicable diseases. Front. Immunol. 13:941977. doi: 10.3389/fimmu.2022.941977
Received: 12 May 2022; Accepted: 01 August 2022;
Published: 31 August 2022.
Edited by:
Pedro H. Gazzinelli-Guimaraes, National Institute of Allergy and Infectious Diseases (NIH), United StatesReviewed by:
Ahmad Ali Othman, Tanta University, EgyptCopyright © 2022 Wu, Duffey, Alex, Suarez-Reyes, Clark and Weatherhead. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jill E. Weatherhead, d2VhdGhlcmhAYmNtLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.