 Weiyang Li
Weiyang Li Na Chang
Na Chang Liying Li
Liying Li- Department of Cell Biology, Municipal Laboratory for Liver Protection and Regulation of Regeneration, Capital Medical University, Beijing, China
Kupffer cells (KCs) are key regulators of liver immunity composing the principal part of hepatic macrophages even body tissue macrophages. They reside in liver sinusoids towards portal vein. The micro-environment shapes KCs unique immunosuppressive features and functions. KCs express specific surface markers that distinguish from other liver macrophages. By engulfing gut-derived foreign products and apoptotic cells without triggering excessive inflammation, KCs maintain homeostasis of liver and body. Heterogeneity of KCs has been identified in different studies. In terms of the origin, adult KCs are derived from progenitors of both embryo and adult bone marrow. Embryo-derived KCs compose the majority of KCs in healthy and maintain by self-renewal. Bone marrow monocytes replenish massively when embryo-derived KC proliferation are impaired. The phenotype of KCs is also beyond the traditional dogma of M1-M2. Functionally, KCs play central roles in pathogenesis of acute and chronic liver injury. They contribute to each pathological stage of liver disease. By initiating inflammation, regulating fibrosis, cirrhosis and tumor cell proliferation, KCs contribute to the resolution of liver injury and restoration of tissue architecture. The underlying mechanism varied by damage factors and pathology. Understanding the characteristics and functions of KCs may provide opportunities for the therapy of liver injury. Herein, we attempt to afford insights on heterogeneity and functions of KCs in liver injury using the existing findings.
Introduction
The liver acts as a crucial filtration system of body. It removes gut-derived products from the liver arterial and portal vein. To fulfil this, liver harbors the largest number of tissue macrophages, named Kupffer cells (KCs). KCs are first reported as components of vascular endothelial cells by Karl Wilhelm von Kupffer in 1876 (1). In 1974, Wisse distinguishes KCs from endothelial cells and defines them as liver sinusoids residing macrophages (2). Then, KCs are identified as F4/80+ (mouse) or CD68+ (human) macrophages that emerged in liver and maintained their number mainly by proliferation (3). In recent years, people have recognized that liver macrophages are composed by not only KCs, but also other macrophages (such as bone marrow monocyte-derived macrophages (BMMs) or capsular macrophages). Meanwhile, KC definition is updated. In the new definition, mouse KCs are liver macrophages that specifically expressing CLEC4F regardless of origins (4). Besides, CLEC2 (5), TIM4 (6) and VSIG4 (7) are also recognized as mouse KC markers. Among them, VSIG4 has recently been reported to be conservative among mouse and human (7). Accordingly, KCs can be distinguished from other macrophages (8).
In recent years, researchers pay more attention to KC features and functions, and have got many impressive results on this issue. The heterogeneity of KCs has been unveiled, especially in mouse. In this review, we summarize and present insights on heterogeneity and function of KCs (liver macrophages that express KC markers) under new definition.
KC in Homeostasis
Heterogeneity of KC Origin
KCs mainly develop in the three hemopoiesis waves (Figure 1) and the developmental program is conserved between mouse and human (9). In mouse, the first wave (primitive hemopoiesis) starts around embryonic days 7.5 (E7.5). Early erythro-myeloid progenitors (EMPs)(CSF1R+) originated from yolk sac (YS) give rise to YS-macrophages. Then, partial YS-macrophages migrate into fetal liver and develop into KCs (10). In the second wave (transient hemopoiesis), late EMPs (MYB+) seed in fetal liver and give rise to fetal monocytes, which specialize into KCs before E16.5 (11). In the third wave (definitive hemopoiesis), hematopoietic stem cells arise intra-embryonically around E10.5 and migrate into fetal liver. Then, hematopoietic stem cells give rise to KCs undergo a monocytic intermediate during perinatal period (11).
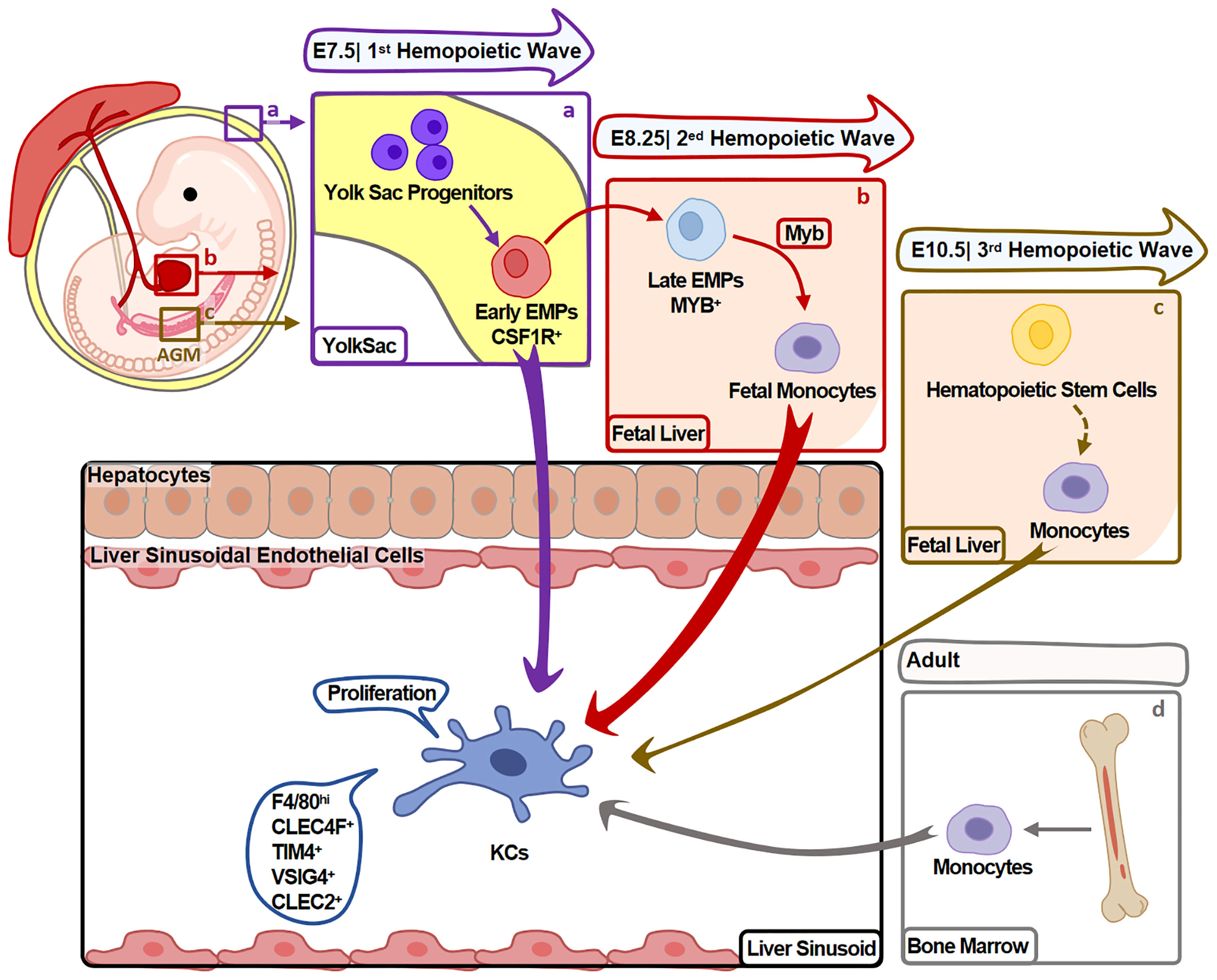
Figure 1 Heterogeneity of KC Origin. KCs mainly develop during hemopoiesis. (A) In the first hemopoietic wave (around E7.5), YS-macrophages are differentiated from EMPs in YS without experiencing monocyte stage. Partial YS-macrophages then migrate and reside in fetal liver, further specialize into KCs. (B) In the second hemopoietic wave (around E8.25), late EMPs differentiate into KCs undergo a monocytic intermediate in fetal liver. (C) In the third hemopoietic wave (around E10.5), hematopoietic stem cells originate from the aorta-gonads-mesonephros (AGM) region and migrate to fetal liver. They give rise to monocytes that differentiate into KCs during perinatal period. KCs developed in this process contribute minor to adult KCs. (D) Fourthly, in adults, BM monocytes also give rise to minor KCs. Arrow width represents the contribution of relative progenitors to KCs.
Fate-mapping studies suggest that the first two waves arise majority of KCs. Hematopoietic stem cells-derived KCs (in the third wave) contribute to a minor part of KCs, and do not replace KCs from the first two waves. These embryo-derived KCs (Em-KCs) inhabit in liver for the whole life and maintain by proliferation (11, 12).
In adults, bone marrow monocytes also contribute to maintain KC pool when Em-KCs are insufficient to maintain it (13). Studies have demonstrated that when Em-KCs are depleted by clodronate liposomes (CLs) (14, 15), diphtheria toxin (4), anti-TIM4 antibody (6) or high dose irradiation (16), monocytes are recruited and differentiate into KCs to replenish the vacancy. In patients received liver transplant, recipient macrophages are also emerged in transplanted livers (17). Therefore, monocytes might differentiate into KCs in both mouse and human. But it should be noticed that these bone marrow monocyte-derived KCs (BM-KCs) are different from BMMs in injured liver. BM-KCs express KC markers (including CLEC4F, CLEC2, VSIG4, TIM4), while BMMs mainly highly express CCR2 and show no KC marker expressions (6, 18).
Besides, KC pool remodeling schedule has been reported in two different mouse models: (a) TIM4 antibody-depleted KC mouse model. In this model, KCs are fully depleted in 2 hours followed by the replenishment of immature BM-KCs (TIM4-F4/80+). TIM4-BM-KCs are matured as TIM4+BM-KCs after 30 days (6). (b) Clec4f-DTR transgenic mouse model. Using this model, two groups provide information from distinct aspects. Martin Guilliams and colleagues report that dead Em-KCs initiate the process by activating liver sinusoidal endothelial cells (LSECs) and hepatic stellate cells (HSCs) around. Then, activated LSECs and HSCs recruit Ly6C+monocytes from circulation at day 1 post Em-KCs depletion. Monocytes then acquire KC phenotype and loss Ly6C expressions gradually in 6 days (18). Christopher Glass group describe more details in transcriptional level. They report that KC lineage determining transcription factors (LDTFs) (NR1H3, SPIC, ID3) are increased while monocyte gene (CCR2) is decreased in replenished monocytes within 12 hours post depletion. In the following 36 hours, more KC LDTFs (MAF, TFE, MAFB, TFEC, etc) gradually express accompanied by the decrease of monocyte related transcription factors (C/EBP, RUNX, SP2, etc.). At day 3 to 7, KC markers CLEC4F and TIM4 are up-regulated (19).
Three signals from liver sinusoids are reported to mediate BM-KCs differentiation. Firstly, DLL4/Notch signaling initiates monocytes differentiation by inducing the increase of KC LDTFs (18). Secondly, TGFB1/BMP9-SMAD4 signaling maintains KC phenotype. Thirdly, KC lineage transcription factor LXRa (encoded by Nr1h3) are also crucial for KC phenotypic maintenance (19). Moreover, two recent studies identified that BMP9/10-ALK1-SMAD4 signaling also regulate KC identity, proliferation and functions (7, 20). But the contribution of TGFB1 in KC identity is controversial: Christopher K. Glass group suggest that TGFB1-SMAD4 signaling maintains KC phenotype in the presence of DLL4 (19). However, Tang group deny the contribution of TGFβ1 using Tgfbr2fl/flClec4fCre mouse (20). Whether other receptors in TGFβ superfamily (e.g. BMP receptors) (20) mediate TGFβ1 function is less studied. Therefore, the significance of TGFB1 signaling in KC identification need more in-depth studies.
Collectively, the origins of KCs are complex. They are mainly originated from three hemopoiesis waves. But BM monocytes also contribute to KC pool when Em-KCs are insufficient to maintain it. The ability of BM monocytes differentiating into KCs has been validated. But the process of KC replenishment is varied and depends on KC depletion methods. The mechanism underlying monocyte-KCs differentiation still needs further study.
Heterogeneity of KC Phenotype and Function
Although the common functions of KCs have been reported as removing bacteria and apoptotic cells (21–24), antigen-presenting (25), and iron/lipid metabolism (4, 26). The consensus is that KCs are heterogeneity in phenotype and function. But no criteria are accepted for KC classification till now. We here summarize the existing findings on mouse KCs (Table 1) and human KCs will be described in part 2 (Human KCs).
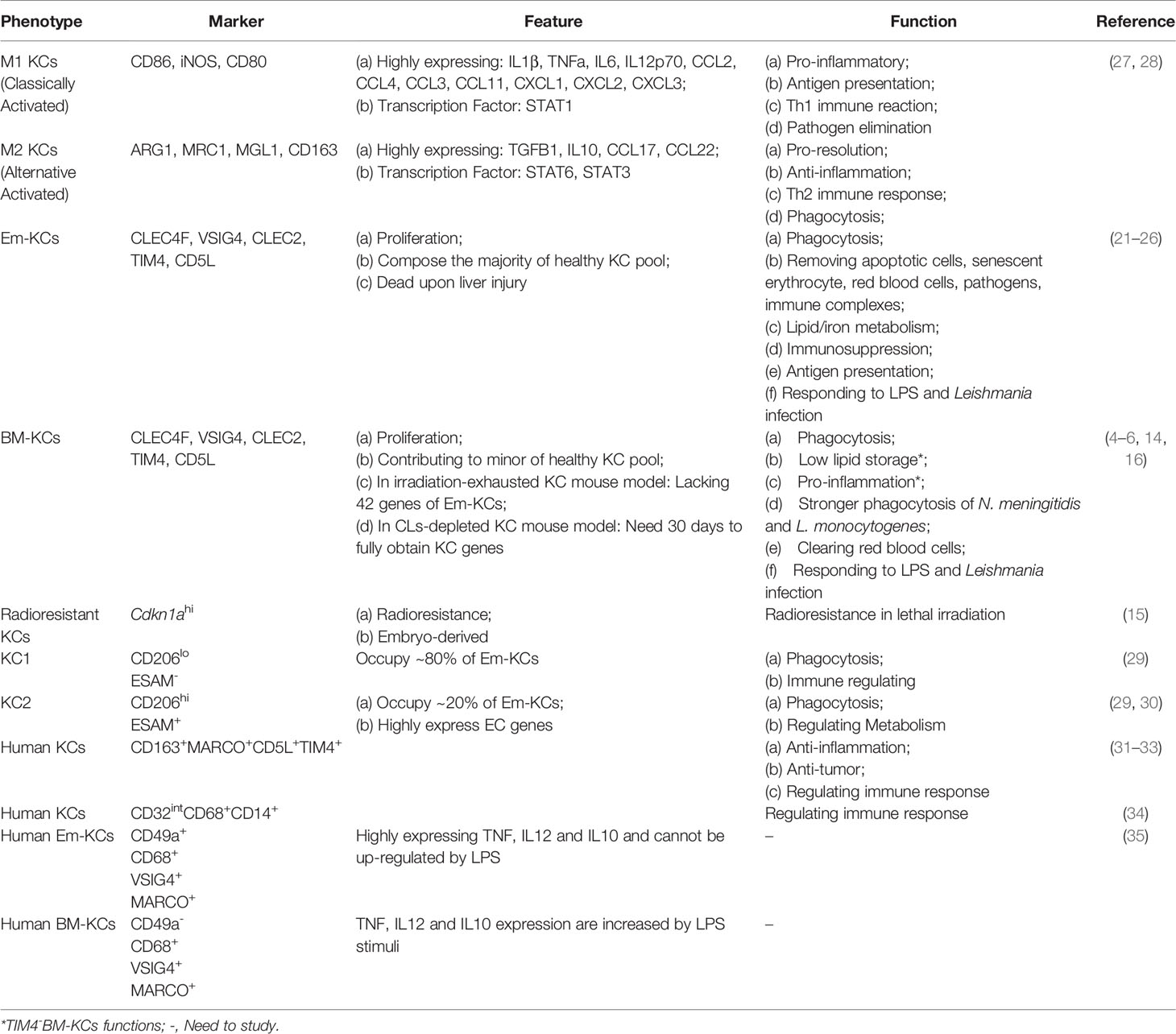
Table 1 KC heterogeneity and function in homeostasis.
Historically, KCs are classified using M1 (classical activated macrophages, pro-inflammation) and M2 (alternative activated macrophages, pro-repairing) dogma via their phenotype under different stimuli. For example, KCs are activated into M1 phenotype by lipopolysaccharide (LPS), IFN-gamma or M-CSF, characterizing by highly expressing pro-inflammatory genes IL1B, TNFα and iNOS (36). While IL4 or GM-CSF may polarize KCs into M2 type that highly express ARG1, IL10 and MRC1 (27, 28). However, later studies found that M1-M2 dichotomy is insufficient to classify KCs, especially in liver injury (37, 38). Therefore, more studies attempt to seek an accurate method to classify KCs.
Some studies use the origin of KCs to describe their heterogeneity. As described in part 1.1, KCs are composed by BM- and Em-KCs. Based on the existing studies, BM- and Em-KCs seem to be similar but not identical. First, BM- and Em-KC gene profiles are inconsistent. In irradiation-exhausted KC model, BM-KCs lack in 42 Em-KC genes related to iron homeostasis (16). While iron/lipid metabolism related genes are at similar levels in BM- and Em-KCs in Clec4f-DTR transgenic mice (4). Second, functions of BM- and Em-KCs are different. As described above, BM-KCs might be shortage in iron homeostasis for the lack of related genes compared with Em-KCs in irradiation-exhausted KC mouse model (16). In the same model, BM- and Em-KCs show different phagocytic ability based on ligand specificity exist. BM-KCs exhibit stronger ability to engulf N. meningitidis and L. monocytogenes compared to Em-KCs, while the phagocytic capacities for red blood cells and S. typhimurium clearing are similar (16). The findings in CLs-depleted KC model might explain the difference on phagocytic ability. It reports that BM-KCs need to undergo a 30 to 60 days’ “education” process to obtain similar phagocytosis capacity to Em-KCs (14). However, whether the differences between studies are caused by mouse model or experimental conditions need further studies.
Besides, Em-KCs are also considered to be heterogeneity. In irradiation-exhausted model, a cluster of radioresistant KCs (Cdkn1ahi) are identified (15). However, specific functions of the radioresistant Em-KCs need in-depth studies. Besides, two distinct Em-KC clusters have been unveiled using scRNA-seq: KC1 (CD206loESAM–, occupy ~80% of Em-KCs) and KC2 (CD206hiESAM+). KC1 shows stronger immune signature while KC2 is bias to regulate metabolism. Meanwhile, KC1 and KC2 show similar phagocytic capacities and distributions in liver (29). In recent research, KC2 is considered as doublet of KCs and LSECs via CITE-Seq analysis (7). Herein, further studies still need to explore and describe Em-KCs heterogeneity.
Finally, KC heterogeneity might also be determined by localization. In terms to the anatomical structure and transcriptional differences, hepatic lobule is heterogeneity and is separated into periportal, mid and pericentral zones (39). Based on it, the metabolic and immune zonation of hepatic lobule have been studied (40, 41). Therefore, studies are also trying to unveil KC heterogeneity based on their localization. The existing findings support the view that KCs prefer to locate in the periportal and mid zones (adhere to the portal vein) (7, 39, 40). The portal vein-adhering localization of KCs is maintained by endothelial MYD88-mediated CCL9 gradients (40) and gut microbiota derived commensal D-lactate (42).
Human KC
Similar to mouse KCs, human KCs also locate around portal vein (31) and are considered to be heterogeneity. But the research progression has been at a slow pace by the limitations of technology. Herein, we conclude the available findings for human KCs (Table 1).
First, the common feature and function of human KCs have been studied. But it has not reached agreement since the lack of unified markers to identify human KCs. In three separate scRNA-seq studies, human KCs are identified as CD163+MARCO+CD5L+TIMD4+ (31–33). Functionally, these KCs are potential to anti-inflammation, anti-tumor and regulating immune (31–33). Comparing to MARCO- macrophages, KCs express less inflammatory (TNFα) but more immunosuppressive genes (e.g. PD-L1) under LPS or IFN-gamma stimuli (31). Another study suggests that KCs are characterized as CD32intCD68+CD14+ and have potential to regulate immune response (34). In recent study, VSIG4 is considered as the best maker of human KCs (7). Therefore, human KCs remain need accurate and canonical definition.
Second, human KC heterogeneity is also reported. In accordance with mouse KCs, human KCs are also separated into Em- and BM-KCs. CD49a has been suggested to distinguish them, while Em-KCs are CD49a+ (35). Em-KCs express high levels of pro-inflammatory TNFα, IL12 and anti-inflammatory IL10, suggesting their dual role in inflammation. But LPS cannot affect these cytokines expressions in Em-KCs. In contrast, the three cytokines are at low levels in BM-KCs while can be up-regulated by LPS (35). Therefore, Em-KCs seems to be the functional cluster in homeostasis while BM-KCs function in injury.
Collectively, the following problems are exposed in current research: Firstly, human KC canonical markers are still lacking. Since human KCs are difficult to distinguish from other macrophages, their functions are also difficult to study. Secondly, human KC heterogeneity (especially origins) is hard to study due to technical limitations. Thirdly, knowledge of mouse KCs cannot be transferred directly to human KCs because of species difference. Fourthly, the individual difference of human beings poses the difficulty to study the universality feature and function of human KCs, especially under diseases.
KC in Acute Liver Injury
Acetaminophen (APAP)-Induced Acute Liver Injury
APAP is a widely used analgesic-antipyretic drug, but is also a major cause of acute liver injury. APAP overdose leads to hepatocytes apoptosis which triggers activation of immune cells including KCs (43, 44).
KC number changes dynamically in APAP liver injury. In APAP-stimulated liver, KC number reduces gradually within 48 hours. After 72 hours, residual KCs start to recover by self-renewal (45).
Functionally, KCs play protective and pro-repairing roles in APAP-liver injury, since KCs depletion leads to more serious injury and slower recovery (14) (Figure 2). The underlying mechanisms include: First, KCs promote hepatocytes survival via IL10 (46). Second, KCs facilitate tissue repair through regulating extracellular matrix (ECM) remodeling. KCs express ECM remodeling-related genes, including MMP12, MMP13, TIMP2, TIMP3 and ADAM23 (47), among which, MMP12 is well studied. MMP12 helps to restore hepatocyte proliferation and to reduce necrosis (48). KCs increase MMP12 expression (45) to degrade elastin (a major component of ECM) and facilitate liver repairing (49).
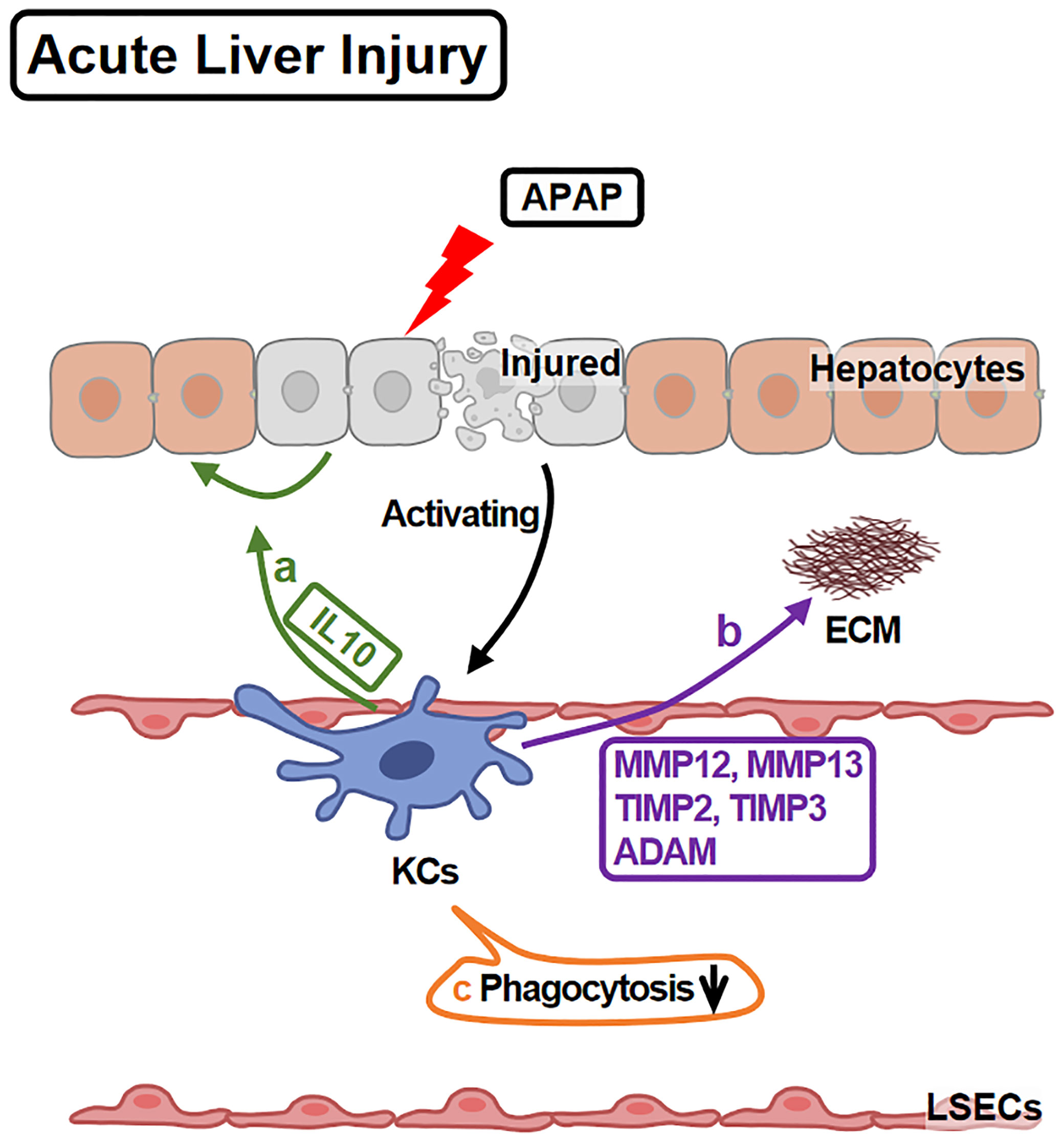
Figure 2 KCs in APAP-induced Acute Liver Injury. APAP overdose leads to hepatocyte injury and death which activate KCs. Activated KCs initiate the repairing response: (A) KCs secrete IL10 to promote injured hepatocyte regeneration and survival. (B) KCs release MMP12, MMP13, TIMP2, TIMP3 and ADAM, remodeling ECM. (C) However, KCs phagocytotic ability is injured during these processes.
Infected Liver Injury
As major component of liver immunity, KCs play important role in infected liver injury. KCs uptake and eradicate pathogens in this process, which usually causes KC death. In hepatitis B virus (HBV) induced acute liver injury, KCs are reduced in number while promote anti-virus response (50). Besides, scRNA-seq analysis show that CD206+ESAM+ Em-KCs respond to IL2 and cross-present antigens to enhance T cell mediated HBV killing (30). In vaccinia virus and murine cytomegalovirus infection, KCs are reduced and replenished by both proliferation and infiltrated monocytes. Type I IFN response is suggested to modulate monocytes-KCs differentiation (51). In adenovirus infection, KCs bind and uptake adenovirus via VSIG4 and complement C3 (52).
In Listeria monocytogenes infection, KCs engulf bacteria and cause their own necroptosis. KC necroptosis recruits monocytes, and induces aggregation of Th2 cytokines including basophil-IL4 and hepatocyte-IL33. Monocytes differentiate into KCs and obtain proliferative property under IL33/IL4 stimulation. Functionally, BM-KCs attenuate inflammation and restore liver homeostasis (53).
Liver Ischemia-Reperfusion Injury (LIRI)
LIRI refers to further aggravation of ischemic injury after blood perfusion recovery, causing by multiple risks including liver transplantation. LIRI induces the necrotic depletion of KCs (54). KC number reduces in 6 hours after LIRI while recovers at day 3 and increases at day 7. The recovered and increased KCs are immature BM-KCs featured by TIM4-lacking (6).
During LIRI, KCs mediate inflammation by IL1B, which is induced by inflammasomes NLRP3 and AIM2 (55, 56). In parallel, a CSF3+KC cluster in rat shows potential to promote inflammation by multiple cytokines and chemokines under IR challenge (57). On the other hand, KCs also regulate LIRI recovery. KCs resolve liver inflammation by TIM4-mediated IL10 up-regulation and TNFα down-regulation. Additionally, KCs also promote the recovery via TIM4-mediated efferocytosis (6).
Acute Liver Injury Induced by Other Risks
In acute carbon tetrachloride (CCl4)-liver injury, KCs are impaired resulting in the decrease of IL-6, further delayed liver regeneration (58). In LPS induced acute liver injury, KCs induce liver inflammation and hepatocyte death by up-regulating TNFα (59), although the up-regulations of both pro- and anti-inflammatory transcription factors are detected (60). Meanwhile, LPS-activated KCs increase the mortality because of the low level of CETP (61).
Collectively, KCs play a complicated dual role in acute liver injury, which might be depended on damage factors. But there are two common points: First, KC number is reduced under acute injury, which leads to monocyte infiltration. Second, KC functions are impaired in different damage. Therefore, replenishing KC number and functions might be a therapeutic strategy for acute liver injury.
KC in Chronic Liver Injury
Non-Alcoholic Steatohepatitis (NASH)
NASH is a severe liver disease that can advance to fibrosis, cirrhosis even hepatocellular carcinoma. The characters of NASH include metabolic disorders, liver inflammation and steatosis.
In NASH liver, Em-KCs are reduced with the increase of diet cholesterol content (62–64). It might be caused by NASH-induced cell apoptosis or death (5, 65). Following KC reduction, monocytes are recruited and differentiate into BM-KCs (5, 64). At the same time, residual Em-KCs expand by proliferation (63) while CD206hiESAM+ Em-KCs (KC2 mentioned in Part 1.2) maintains stable in number (29).
KCs play various roles in regulating liver injury in NASH (Figure 3). 891 genes in KCs are reported to be up-regulated under NASH. These genes are associated with functions of ECM remodeling, lipid metabolism, bacterial clearance and recruitment of circulating monocytes (62). Moreover, KCs amplify or attenuate liver inflammation. Mitochondrial DNA from apoptotic hepatocytes activates STING/NF-kB signaling pathway in KCs and leads to inflammation amplification (66). KCs also recruit monocytes and neutrophils via CCL2 and CXCL1. Monocytes aggravate inflammation while neutrophiles contribute to killing bacteria (67). KCs attenuate inflammation by removing gut microbial products via VSIG4 (68, 69).
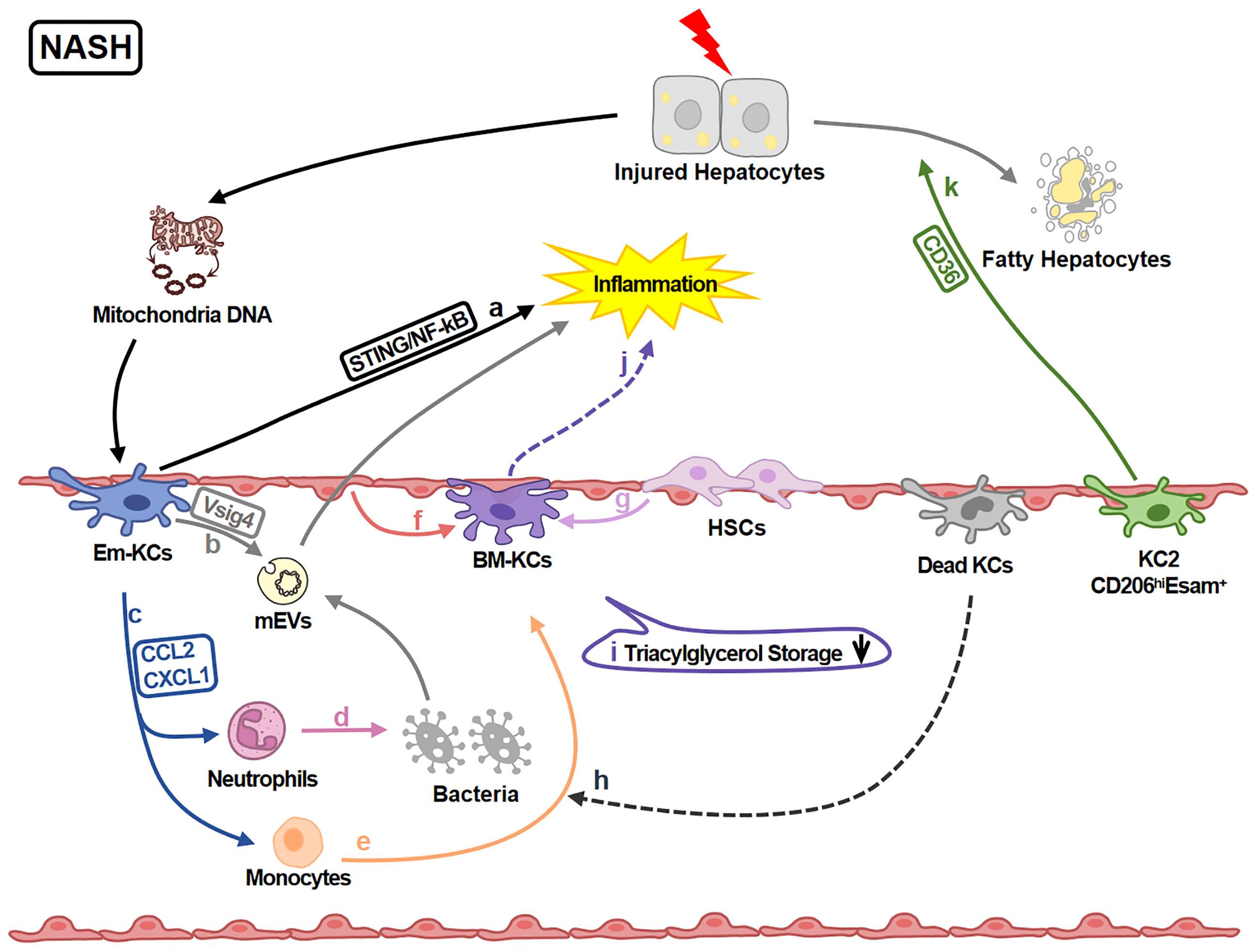
Figure 3 KCs in NASH-related Liver Injury. In NASH livers, KCs regulate inflammation and lipid metabolism. (A) KCs-STING/NF-kB signaling is activated by mitochondria DNA from injured hepatocytes and amplify the inflammation. (B) KCs inversely engulf gut mEV (microbial DNA containing extracellular vesicles) via VSIG4 and attenuate inflammation. (C) KCs also secret chemokines CCL2 and CXCL1 recruiting neutrophils and monocytes. (D) Neutrophils help killing pathogens like bacteria. (E) Monocytes can differentiate into BM-KCs. (F, G) The differentiation process is regulated by LSECs and HSCs. (H) Dead KCs are the initiators of the replenishment by opening KC niche. (I) The triacylglycerol storage capacity of BM-KCs is decreased comparing to Em-KCs. (J) Meanwhile, BM-KCs promote inflammatory response. (K) KC2 (CD206hiESAM+KCs) regulate lipid metabolism via CD36 leading to the damage of hepatocytes.
Additionally, functions of different KC sub-clusters are also reported. For example, KC2 aggravate liver injury via CD36 (29); Em-KCs are more efficient for triglyceride storage (5, 70) while BM-KCs are more inflammatory (5). It still needs further study to explore whether these differences are caused by origins, KCs impairment and/or the different state of KCs (such as immature or mature).
Therefore, KCs play multiple roles in NASH. The details of KC functions, especially based on their heterogeneity, still need deeply study.
Cholestatic Liver Injury
Cholestatic liver injury involves a variety of disorders of bile flow and/or formation. KCs are reduced in primary biliary cholangitis (a chronic cholestatic liver disease) (71). In bile duct ligation (BDL) mouse model, KC depletion results in a reduction of hepatocyte regeneration and more serious injury (72, 73). However, liver fibrosis is alleviated by KC deletion (73). In-depth study suggests that IL6 might be a potential functional molecule (72). On the other hand, BDL impairs the clearance function of KCs, resulting the reduction of gut-derived LPS clearance and the aggravation of inflammatory damage (74, 75). In parallel, KC scavengers MARCO and CD5L are reduced in human biliary atresia (76). Decreased clearance function might be a reason of the susceptibility to bacterial infection after BDL injury (74).
Toxic Liver Injury
As the major organ for detoxification, liver is easily to be injured by toxicant (such as chemicals, drugs and alcohol). In this part, we mainly focus on chronic toxic liver injury since acute toxic liver injury has been discussed in Part 3.
In CCl4-chronic liver injury model, KC number is reduced and is partially replenished by Ly6Clo monocytes during repairing (77). Functionally, KCs amplify inflammation by recruiting pro-inflammatory macrophages via TREM1 in early stage of injury (78). KCs phagocytic and pathogen killing potentials might be decreased for complement C6, MARCO and TIM4 in KCs are down-regulated (79). But in another study, KC-TIM4 is increased (80). Herein, more studies are needed to demonstrate how KC phagocytic capability changes during toxic liver injury.
In alcohol injured livers, alcohol also induces KC number decreasing (81). KCs are activated by apoptotic hepatocyte-derived mitochondrial DNA via TLR3. Activated KCs produce high level of IL1B which induces IL17A releasing from T cells (82) and promotes hepatocellular carcinoma development (83).
Chronic HBV Infection
In part 3.2, we have discussed the importance of KCs in acute HBV infection. In chronic HBV infection, KCs also play protective role in various ways. Firstly, KCs produce IL1B via activated NLRP3 (84) to attenuate the susceptibility of HBV infection (85). Secondly, KCs recognize virus and enhance natural killer cells activation via IL18 (86). Thirdly, KCs act as antigen-presenting cells to activate T cells (87). Additionally, KCs contribute to liver immune tolerance by attenuating T cell proliferation (88), attenuating B cell antibody production (89), suppressing CD8+T cell cytotoxicity and promoting Treg expansion (90). Inversely, HBV also employs different strategies to modulate KCs to favour the establishment of infection, such as impairing IL-1B production (84) and promoting IL-10 releasing (91).
Liver Fibrosis and Cirrhosis/Portal Hypertension
Liver fibrosis, a common pathological process of chronic liver injury, is characterized by the excessive deposition of ECM (especially collagen). Liver fibrosis further progresses to cirrhosis if it has not been well treated. Portal hypertension is a major cause of morbidity and mortality in cirrhosis (92). In human cirrhosis, KC number has been reported to be unchanged (33, 93) while rat KCs are reduced upon cirrhosis (94). A recent study reports that infiltrated monocytes differentiate into BM-KCs upon liver stromal cells-IL6 stimuli (95).
KCs regulate ECM deposition in various ways (Figure 4). Firstly, KCs affect collagen production by regulating HSC (the major cellular source of collagen) activities. KCs promote HSC activation and collagen production via TGFB1 (80). For another, KCs contribute to HSC survival by EGFR and TNFR1A/1B signaling (79). Secondly, KCs regulate ECM remodeling directly. KCs promote collagen cross-linking and scar formation (96) by LOXL2 (97). Inversely, KCs produce MMP9 to promote collagen resolution (98). Consistently, KC infusion shows anti-fibrotic effects (99).Thirdly, KCs also transdifferentiate into fibroblast-like cells contributing to ECM deposition (100).
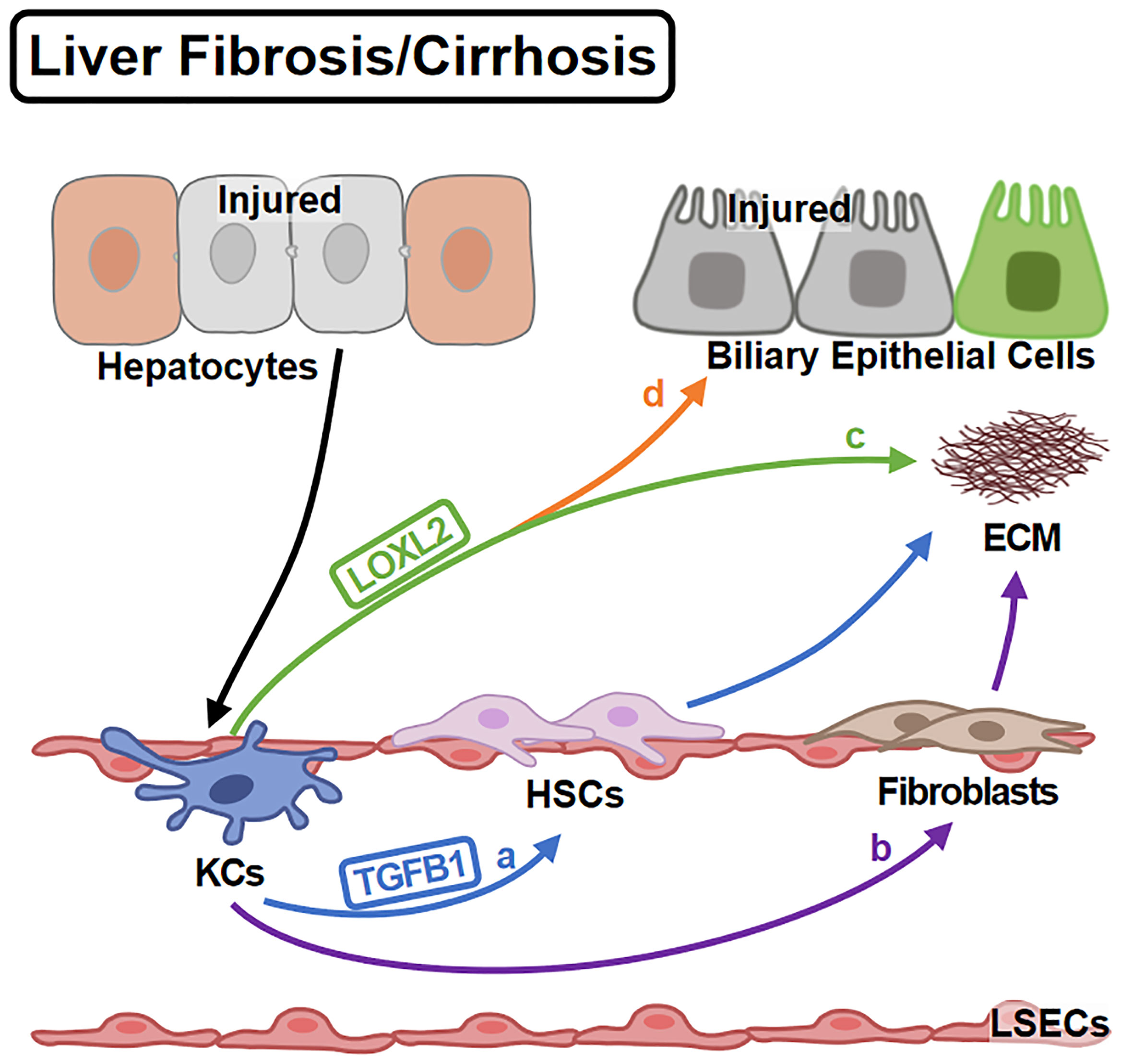
Figure 4 KCs in Liver Fibrosis/Cirrhosis. Liver fibrosis is the over-repair in chronic liver injury. KCs are activated by injured hepatocytes and promote fibrosis in different ways: (A) KCs promote HSC activation via TGFB1. (B) KCs themselves can differentiate into fibroblasts contributing to ECM deposition. On the other hand, (C) KCs mediate the cross-linking of collagen via LOXL2. (D) KCs also induce the impairment of biliary epithelial cells by LOXL2.
In human cirrhosis, phagocytic and anti-bacteria abilities of KCs are weakened (93) which might cause the susceptibility to bacterial infection and the cirrhotic death (101). At the same time, activated KCs might help increase portal venous pressure for the level of soluble CD163 (the sensitive marker of KC activation) is associated with portal venous pressure gradient (102). Activated KCs also increase portal pressure by inducing vasoconstrictor including cysteinyl leukotriene (103). On the other hand, human KCs might not contribute to fibrogenesis directly for they are away from fibrotic areas (33). The contradiction in human KCs studies might be caused by the influence of other liver macrophages, because of the lacking of canonical markers to distinguish them, especially in early studies.
Hepatocellular Carcinoma (HCC)
HCC is an inflammation associated cancer caused by multiple etiological factors. KCs are reduced in mouse (104) and human (105) HCC. Human KC reduction is caused by tumor cell-CCL2 (105). However, mouse KCs could be replenished for CLEC4F expression is unchanged (106). Herein, the change of KCs pool in HCC still needs further study.
Functionally, KCs promote neutrophil-mediated liver toxicity through IL12 (107). KCs also attenuate T cell responses thereby promote tumor growth and decrease anti-PD-1 therapeutic sensitivity (108). While up-regulating microRNA-206 expression might attenuate the tumor-promoting effect on of KCs (109). Besides, CD163+CD206+FOLR2+KCs co-localize and interact with PLVAP+ endothelial cells and immunosuppressive T cells, further maintained the immunosuppressive micro-environment (110). On the other hand, cancer cells might regulate KC activities directly since liver macrophage activity and function are affected by cancer cells (111). Cancer cells also change macrophage cytokines-releasing (112) and metabolism (113) which further promote HCC progression. Cancer cell-KCs crosstalk exacerbates HCC by initiating a vicious circle which reinforces each other.
Conclusion and Further Prospect
As one of the important components of liver, KCs perform important role in health, acute/chronic liver injury, liver fibrosis/cirrhosis and HCC. In healthy liver, KCs help to maintain liver homeostasis. In injured liver or HCC, KCs are usually reduced, but the detail and reason for reducing need in-depth study. KC functions in injured liver are complicated (Table 2). Further studies are needed to confirm the universal between different injuries. In recent years, the improvements of technology (such as scRNA-seq) provides more information and details about KC heterogeneity in health and diseases. But the conclusions are controversial and lack an accepted clustering criterion. In conclusion, it is necessary to further clarify KC functions (especially based on their heterogeneity) in liver diseases. These studies will be helpful in understanding the mechanism of liver diseases and developing new therapeutic target.
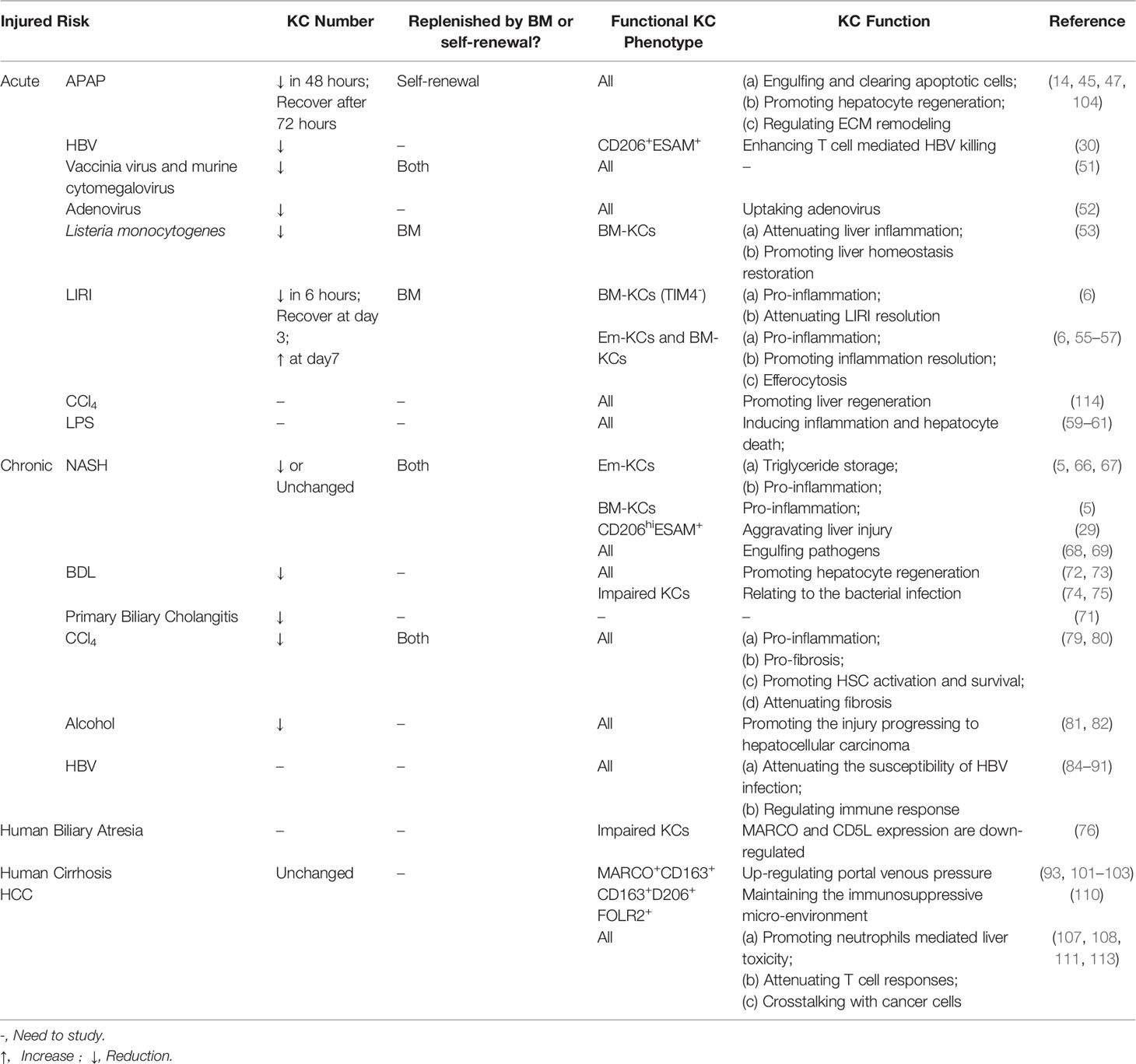
Table 2 KC heterogeneity and function in Liver injury.
Author Contributions
WL and NC drafted the manuscript and designed the figures. LL and NC made the final corrections. LL applied the funds. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the National Natural and Science Foundation of China (81970532, 82170622).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
APAP, Acetaminophen; BDL, bile duct ligation; BM-KCs, bone marrow-derived Kupffer cells; CLs, clodronate liposomes; E, embryonic days; DTR, diphtheria toxin receptor; EMP, erythro-myeloid progenitors; Em-KCs, embryo-derived Kupffer cells; HSC, hepatic stellate cell; HBV, hepatitis B virus; HCC, hepatocellular carcinoma; KCs, Kupffer cells; LDTF, lineage determining transcription factor; LPS, lipopolysaccharide; LIRI, liver ischemia-reperfusion injury; LSEC, liver sinusidal endothelial cells; NASH, non-alcoholic steatohepatitis; scRNA-seq, Single cell RNA-sequencing; YS, yolk sac.
References
1. Wake K, Decker K, Kirn A, Knook DL, Mccuskey RS, Bouwens L, et al. Cell Biology and Kinetics of Kupffer Cells in the Liver. Int Rev Cytol (1989) 118:173–229. doi: 10.1016/s0074-7696(08)60875-x
2. Wisse E. Observations on the Fine Structure and Peroxidase Cytochemistry of Normal Rat Liver Kupffer Cells. J Ultrastruct Res (1974) 46:393–426. doi: 10.1016/s0022-5320(74)90064-1
3. Basit H, Tan ML, Webster DR. “Histology, Kupffer Cell’, in StatPearls [Internet]. ed. Abai B, et al. (2021) (Treasure Island, FL: StatPearls), 1–5.
4. Scott CL, Zheng F, De Baetselier P, Martens L, Saeys Y, De Prijck S, et al. Bone Marrow-Derived Monocytes Give Rise to Self-Renewing and Fully Differentiated Kupffer Cells. Nat Commun (2016) 7:10321. doi: 10.1038/ncomms10321
5. Tran S, Baba I, Poupel L, Dussaud S, Moreau M, Gélineau A, et al. Impaired Kupffer Cell Self-Renewal Alters the Liver Response to Lipid Overload During Non-Alcoholic Steatohepatitis. Immunity (2020) 53:627–40. doi: 10.1016/j.immuni.2020.06.003
6. Ni M, Zhang J, Sosa R, Zhang H, Wang H, Jin D, et al. T-Cell Immunoglobulin and Mucin Domain-Containing Protein-4 Is Critical for Kupffer Cell Homeostatic Function in the Activation and Resolution of Liver Ischemia Reperfusion Injury. Hepatology (2021) 74:2118–32. doi: 10.1002/hep.31906
7. Guilliams M, Bonnardel J, Haest B, Vanderborght B, Wagner C, Remmerie A, et al. Spatial Proteogenomics Reveals Distinct and Evolutionarily Conserved Hepatic Macrophage Niches. Cell (2022) 185:379–96. doi: 10.1016/j.cell.2021.12.018
8. Sierro F, Evrard M, Rizzetto S, Melino M, Mitchell AJ, Florido M, et al. Liver Capsular Network of Monocyte-Derived Macrophages Restricts Hepatic Dissemination of Intraperitoneal Bacteria by Neutrophil Recruitment. Immunity (2017) 47:374–88. doi: 10.1016/j.immuni.2017.07.018
9. Godin I, Cumano A. The Hare and the Tortoise: An Embryonic Haematopoietic Race. Nat Rev Immunol (2002) 2:593–604. doi: 10.1038/nri857
10. Hoeffel G, Chen J, Lavin Y, Low D, Almeida FF, See P, et al. C-Myb+ Erythro-Myeloid Progenitor-Derived Fetal Monocytes Give Rise to Adult Tissue-Resident Macrophages. Immunity (2015) 42:665–78. doi: 10.1016/j.immuni.2015.03.011
11. Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue-Resident Macrophages Originate From Yolk-Sac-Derived Erythro-Myeloid Progenitors. Nature (2015) 518:547–51. doi: 10.1038/nature13989
12. Bertrand JY, Jalil A, Klaine M, Jung S, Cumano A, Godin I. Three Pathways to Mature Macrophages in the Early Mouse Yolk Sac. Blood (2005) 106:3004–11. doi: 10.1182/blood-2005-02-0461
13. Schulz C, Gomez PE, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, et al. Lineage of Myeloid Cells Independent of Myb and Hematopoietic Stem Cells. Science (2012) 336:86–90. doi: 10.1126/science.1219179
14. David BA, Rezende RM, Antunes MM, Santos MM, Freitas LM, Diniz AB, et al. Combination of Mass Cytometry and Imaging Analysis Reveals Origin, Location, and Functional Repopulation of Liver Myeloid Cells in Mice. Gastroenterology (2016) 151:1176–91. doi: 10.1053/j.gastro.2016.08.024
15. Soysa R, Lampert S, Yuen S, Douglass AN, Li W, Pfeffer K, et al. Fetal Origin Confers Radioresistance on Liver Macrophages via P21. J Hepatol (2019) 71:553–62. doi: 10.1016/j.jhep.2019.04.015
16. Beattie L, Sawtell A, Mann J, Frame TCM, Teal B, de Labastida Rivera F, et al. Bone Marrow-Derived and Resident Liver Macrophages Display Unique Transcriptomic Signatures But Similar Biological Functions. J Hepatol (2016) 65:758–68. doi: 10.1016/j.jhep.2016.05.037
17. Bittmann I, Bottino A, Baretton GB, Gerbes AL, Zachoval R, Rau HG, et al. The Role of Graft-Resident Kupffer Cells and Lymphocytes of Donor Type During the Time Course After Liver Transplantation–a Clinico-Pathological Study. Virchows Arch (2003) 443:541–8. doi: 10.1007/s00428-003-0861-8
18. Bonnardel J, T'Jonck W, Gaublomme D, Browaeys R, Scott CL, Martens L, et al. Stellate Cells, Hepatocytes, and Endothelial Cells Imprint the Kupffer Cell Identity on Monocytes Colonizing the Liver Macrophage Niche. Immunity (2019) 51:638–54. doi: 10.1016/j.immuni.2019.08.017
19. Sakai M, Troutman TD, Seidman JS, Ouyang Z, Spann NJ, Abe Y, et al. Liver-Derived Signals Sequentially Reprogram Myeloid Enhancers to Initiate and Maintain Kupffer Cell Identity. Immunity (2019) 51:655–70. doi: 10.1016/j.immuni.2019.09.002
20. Zhao D, Yang F, Wang Y, Li S, Li Y, Hou F, et al. ALK1 Signaling is Required for the Homeostasis of Kupffer Cells and Prevention of Bacterial Infection. J Clin Invest (2021) 132:e150489. doi: 10.1172/JCI150489
21. Balmer ML, Slack E, de Gottardi A, Lawson MA, Hapfelmeier S, Miele L, et al. The Liver may Act as a Firewall Mediating Mutualism Between the Host and its Gut Commensal Microbiota. Sci Transl Med (2014) 6:237r–66r. doi: 10.1126/scitranslmed.3008618
22. Fabriek BO, van Bruggen R, Deng DM, Ligtenberg AJ, Nazmi K, Schornagel K, et al. The Macrophage Scavenger Receptor CD163 Functions as an Innate Immune Sensor for Bacteria. Blood (2009) 113:887–92. doi: 10.1182/blood-2008-07-167064
23. Helmy KY, Katschke KJ, Gorgani NN, Kljavin NM, Elliott JM, Diehl L, et al. CRIg: A Macrophage Complement Receptor Required for Phagocytosis of Circulating Pathogens. Cell (2006) 124:915–27. doi: 10.1016/j.cell.2005.12.039
24. Jiang Y, Tang Y, Hoover C, Kondo Y, Huang D, Restagno D, et al. Kupffer Cell Receptor CLEC4F is Important for the Destruction of Desialylated Platelets in Mice. Cell Death Differ (2021) 28:3009–21. doi: 10.1038/s41418-021-00797-w
25. You Q, Cheng L, Kedl RM, Ju C. Mechanism of T Cell Tolerance Induction by Murine Hepatic Kupffer Cells. Hepatology (2008) 48:978–90. doi: 10.1002/hep.22395
26. Scott CL, Guilliams M. The Role of Kupffer Cells in Hepatic Iron and Lipid Metabolism. J Hepatol (2018) 69:1197–9. doi: 10.1016/j.jhep.2018.02.013
27. Wynn TA, Chawla A, Pollard JW. Macrophage Biology in Development, Homeostasis and Disease. Nature (2013) 496:445–55. doi: 10.1038/nature12034
28. Wang C, Ma C, Gong L, Guo Y, Fu K, Zhang Y, et al. Macrophage Polarization and Its Role in Liver Disease. Front Immunol (2021) 12:803037. doi: 10.3389/fimmu.2021.803037
29. Blériot C, Barreby E, Dunsmore G, Ballaire R, Chakarov S, Ficht X, et al. Subset of Kupffer Cells Regulates Metabolism Through the Expression of CD36. Immunity (2021) 54:2101–16. doi: 10.1016/j.immuni.2021.08.006
30. De Simone G, Andreata F, Bleriot C, Fumagalli V, Laura C, Garcia-Manteiga JM, et al. Identification of a Kupffer Cell Subset Capable of Reverting the T Cell Dysfunction Induced by Hepatocellular Priming. Immunity (2021) 54:2089–100. doi: 10.1016/j.immuni.2021.05.005
31. Macparland SA, Liu JC, Ma XZ, Innes BT, Bartczak AM, Gage BK, et al. Single Cell RNA Sequencing of Human Liver Reveals Distinct Intrahepatic Macrophage Populations. Nat Commun (2018) 9:4383. doi: 10.1038/s41467-018-06318-7
32. Aizarani N, Saviano A, Sagar, Mailly L, Durand S, Herman JS, et al. A Human Liver Cell Atlas Reveals Heterogeneity and Epithelial Progenitors. Nature (2019) 572:199–204. doi: 10.1038/s41586-019-1373-2
33. Ramachandran P, Dobie R, Wilson-Kanamori JR, Dora EF, Henderson B, Luu NT, et al. Resolving the Fibrotic Niche of Human Liver Cirrhosis at Single-Cell Level. Nature (2019) 575:512–8. doi: 10.1038/s41586-019-1631-3
34. Wu X, Hollingshead N, Roberto J, Knupp A, Kenerson H, Chen A, et al. Human Liver Macrophage Subsets Defined by CD32. Front Immunol (2020) 11:2108. doi: 10.3389/fimmu.2020.02108
35. Martrus G, Goebels H, Langeneckert AE, Kah J, Flomm F, Ziegler AE, et al. CD49a Expression Identifies a Subset of Intrahepatic Macrophages in Humans. Front Immunol (2019) 10:1247. doi: 10.3389/fimmu.2019.01247
36. Tian Z, Hou X, Liu W, Han Z, Wei L. Macrophages and Hepatocellular Carcinoma. Cell Biosci (2019) 9:79. doi: 10.1186/s13578-019-0342-7
37. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity (2014) 41:14–20. doi: 10.1016/j.immuni.2014.06.008
38. Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, et al. Transcriptome-Based Network Analysis Reveals a Spectrum Model of Human Macrophage Activation. Immunity (2014) 40:274–88. doi: 10.1016/j.immuni.2014.01.006
39. Hildebrandt F, Andersson A, Saarenpaa S, Larsson L, Van Hul N, Kanatani S, et al. Spatial Transcriptomics to Define Transcriptional Patterns of Zonation and Structural Components in the Mouse Liver. Nat Commun (2021) 12:7046. doi: 10.1038/s41467-021-27354-w
40. Gola A, Dorrington MG, Speranza E, Sala C, Shih RM, Radtke AJ, et al. Commensal-Driven Immune Zonation of the Liver Promotes Host Defence. Nature (2021) 589:131–6. doi: 10.1038/s41586-020-2977-2
41. Halpern KB, Shenhav R, Matcovitch-Natan O, Tóth B, Lemze D, Golan M, et al. Single-Cell Spatial Reconstruction Reveals Global Division of Labour in the Mammalian Liver. Nature (2017) 542:352–6. doi: 10.1038/nature21065
42. Mcdonald B, Zucoloto AZ, Yu IL, Burkhard R, Brown K, Geuking MB, et al. Programing of an Intravascular Immune Firewall by the Gut Microbiota Protects Against Pathogen Dissemination During Infection. Cell Host Microbe (2020) 28:660–8. doi: 10.1016/j.chom.2020.07.014
43. Ramachandran A, Jaeschke H. Acetaminophen Hepatotoxicity. Semin Liver Dis (2019) 39:221–34. doi: 10.1055/s-0039-1679919
44. Kolodziejczyk AA, Federici S, Zmora N, Mohapatra G, Dori-Bachash M, Hornstein S, et al. Acute Liver Failure is Regulated by MYC- and Microbiome-Dependent Programs. Nat Med (2020) 26:1899–911. doi: 10.1038/s41591-020-1102-2
45. Holt MP, Cheng L, Ju C. Identification and Characterization of Infiltrating Macrophages in Acetaminophen-Induced Liver Injury. J Leukoc Biol (2008) 84:1410–21. doi: 10.1189/jlb.0308173
46. Nguyen NT, Umbaugh DS, Sanchez-Guerrero G, Ramachandran A, Jaeschke H. Kupffer Cells Regulate Liver Recovery Through Induction of Chemokine Receptor CXCR2 on Hepatocytes After Acetaminophen Overdose in Mice. Arch Toxicol (2022) 96:305–20. doi: 10.1007/s00204-021-03183-0
47. Zigmond E, Samia-Grinberg S, Pasmanik-Chor M, Brazowski E, Shibolet O, Halpern Z, et al. Infiltrating Monocyte-Derived Macrophages and Resident Kupffer Cells Display Different Ontogeny and Functions in Acute Liver Injury. J Immunol (2014) 193:344–53. doi: 10.4049/jimmunol.1400574
48. Kopec AK, Joshi N, Cline-Fedewa H, Wojcicki AV, Ray JL, Sullivan BP, et al. Fibrin(ogen) Drives Repair After Acetaminophen-Induced Liver Injury via Leukocyte Alphambeta2 Integrin-Dependent Upregulation of Mmp12. J Hepatol (2017) 66:787–97. doi: 10.1016/j.jhep.2016.12.004
49. Pellicoro A, Aucott RL, Ramachandran P, Robson AJ, Fallowfield JA, Snowdon VK, et al. Elastin Accumulation Is Regulated at the Level of Degradation by Macrophage Metalloelastase (MMP-12) During Experimental Liver Fibrosis. Hepatology (2012) 55:1965–75. doi: 10.1002/hep.25567
50. Wu LL, Peng WH, Wu HL, Miaw SC, Yeh SH, Yang HC, et al. Lymphocyte Antigen 6 Complex, Locus C(+) Monocytes and Kupffer Cells OrchestrateLiver Immune Responses Against Hepatitis B Virus in Mice. Hepatology (2019) 69:2364–80. doi: 10.1002/hep.30510
51. Borst K, Frenz T, Spanier J, Tegtmeyer P, Chhatbar C, Skerra J, et al. Type I Interferon Receptor Signaling Delays Kupffer Cell Replenishment During Acute Fulminant Viral Hepatitis. J Hepatol (2018) 68:682–90. doi: 10.1016/j.jhep.2017.11.029
52. He JQ, Katschke KJ, Gribling P, Suto E, Lee WP, Diehl L, et al. CRIg Mediates Early Kupffer Cell Responses to Adenovirus. J Leukoc Biol (2013) 93:301–6. doi: 10.1189/jlb.0612311
53. Blériot C, Dupuis T, Jouvion G, Eberl G, Disson O, Lecuit M. Liver-Resident Macrophage Necroptosis Orchestrates Type 1 Microbicidal Inflammation and Type-2-Mediated Tissue Repair During Bacterial Infection. Immunity (2015) 42:145–58. doi: 10.1016/j.immuni.2014.12.020
54. Yue S, Zhou H, Wang X, Busuttil RW, Kupiec-Weglinski JW, Zhai Y. Prolonged Ischemia Triggers Necrotic Depletion of Tissue-Resident Macrophages To Facilitate Inflammatory Immune Activation in Liver Ischemia Reperfusion Injury. J Immunol (2017) 198:3588–95. doi: 10.4049/jimmunol.1601428
55. Kim HY, Kim SJ, Lee SM. Activation of NLRP3 and AIM2 Inflammasomes in Kupffer Cells in Hepatic Ischemia/Reperfusion. FEBS J (2015) 282:259–70. doi: 10.1111/febs.13123
56. Cai J, Zhang X, Chen P, Li Y, Liu S, Liu Q, et al. The ER Stress Sensor Inositol-Requiring Enzyme 1alpha in Kupffer Cells Promotes Hepatic Ischemia-Reperfusion Injury. J Biol Chem (2022) 298:101532. doi: 10.1016/j.jbc.2021.101532
57. Yang X, Lu D, Wang R, Lian Z, Lin Z, Zhuo J, et al. Single-Cell Profiling Reveals Distinct Immune Phenotypes That Contribute to Ischaemia-Reperfusion Injury After Steatotic Liver Transplantation. Cell Prolif (2021) 54:e13116. doi: 10.1111/cpr.13116
58. Xiang X, Feng D, Hwang S, Ren T, Wang X, Trojnar E, et al. Interleukin-22 Ameliorates Acute-on-Chronic Liver Failure by Reprogramming Impaired Regeneration Pathways in Mice. J Hepatol (2020) 72:736–45. doi: 10.1016/j.jhep.2019.11.013
59. Filliol A, Piquet-Pellorce C, Raguenes-Nicol C, Dion S, Farooq M, Lucas-Clerc C, et al. RIPK1 Protects Hepatocytes From Kupffer Cells-Mediated TNF-Induced Apoptosis in Mouse Models of PAMP-Induced Hepatitis. J Hepatol (2017) 66:1205–13. doi: 10.1016/j.jhep.2017.01.005
60. Soysa R, Bean JC, Wu X, Lampert S, Yuen S, Crispe IN. Early-Derived Murine Macrophages Temporarily Renounce Tissue Identity During Acute Systemic Inflammation. J Immunol (2021) 207:569–76. doi: 10.4049/jimmunol.2001324
61. van der Tuin S, Li Z, Berbee J, Verkouter I, Ringnalda LE, Neele AE, et al. Lipopolysaccharide Lowers Cholesteryl Ester Transfer Protein by Activating F4/80(+)Clec4f(+)Vsig4(+)Ly6C(-) Kupffer Cell Subsets. J Am Heart Assoc (2018) 7:e8105. doi: 10.1161/JAHA.117.008105
62. Mcgettigan B, Mcmahan R, Orlicky D, Burchill M, Danhorn T, Francis P, et al. Dietary Lipids Differentially Shape Nonalcoholic Steatohepatitis Progression and the Transcriptome of Kupffer Cells and Infiltrating Macrophages. Hepatology (2019) 70:67–83. doi: 10.1002/hep.30401
63. Devisscher L, Scott CL, Lefere S, Raevens S, Bogaerts E, Paridaens A, et al. Non-Alcoholic Steatohepatitis Induces Transient Changes Within the Liver Macrophage Pool. Cell Immunol (2017) 322:74–83. doi: 10.1016/j.cellimm.2017.10.006
64. Daemen S, Gainullina A, Kalugotla G, He L, Chan MM, Beals JW, et al. Dynamic Shifts in the Composition of Resident and Recruited Macrophages Influence Tissue Remodeling in NASH. Cell Rep (2021) 34:108626. doi: 10.1016/j.celrep.2020.108626
65. Seidman JS, Troutman TD, Sakai M, Gola A, Spann NJ, Bennett H, et al. Niche-Specific Reprogramming of Epigenetic Landscapes Drives Myeloid Cell Diversity in Nonalcoholic Steatohepatitis. Immunity (2020) 52:1057–74. doi: 10.1016/j.immuni.2020.04.001
66. Yu Y, Liu Y, An W, Song J, Zhang Y, Zhao X. STING-Mediated Inflammation in Kupffer Cells Contributes to Progression of Nonalcoholic Steatohepatitis. J Clin Invest (2019) 129:546–55. doi: 10.1172/JCI121842
67. Krenkel O, Puengel T, Govaere O, Abdallah AT, Mossanen JC, Kohlhepp M, et al. Therapeutic Inhibition of Inflammatory Monocyte Recruitment Reduces Steatohepatitis and Liver Fibrosis. Hepatology (2018) 67:1270–83. doi: 10.1002/hep.29544
68. Caussy C, Hsu C, Lo MT, Liu A, Bettencourt R, Ajmera VH, et al. Link Between Gut-Microbiome Derived Metabolite and Shared Gene-Effects With Hepatic Steatosis and Fibrosis in NAFLD. Hepatology (2018) 68:918–32. doi: 10.1002/hep.29892
69. Luo Z, Ji Y, Gao H, Gomes DRF, Bandyopadhyay G, Jin Z, et al. CRIg(+) Macrophages Prevent Gut Microbial DNA-Containing Extracellular Vesicle-Induced Tissue Inflammation and Insulin Resistance. Gastroenterology (2021) 160:863–74. doi: 10.1053/j.gastro.2020.10.042
70. Remmerie A, Martens L, Thoné T, Castoldi A, Seurinck R, Pavie B, et al. Osteopontin Expression Identifies a Subset of Recruited Macrophages Distinct From Kupffer Cells in the Fatty Liver. Immunity (2020) 53:641–57. doi: 10.1016/j.immuni.2020.08.004
71. Reuveni D, Brezis MR, Brazowski E, Vinestock P, Leung PSC, Thakker P, et al. Interleukin 23 Produced by Hepatic Monocyte-Derived Macrophages Is Essential for the Development of Murine Primary Biliary Cholangitis. Front Immunol (2021) 12:718841. doi: 10.3389/fimmu.2021.718841
72. Gehring S, Dickson EM, San MM, van Rooijen N, Papa EF, Harty MW, et al. Kupffer Cells Abrogate Cholestatic Liver Injury in Mice. Gastroenterology (2006) 130:810–22. doi: 10.1053/j.gastro.2005.11.015
73. Osawa Y, Seki E, Adachi M, Suetsugu A, Ito H, Moriwaki H, et al. Role of Acid Sphingomyelinase of Kupffer Cells in Cholestatic Liver Injury in Mice. Hepatology (2010) 51:237–45. doi: 10.1002/hep.23262
74. Sato K, Hall C, Glaser S, Francis H, Meng F, Alpini G. Pathogenesis of Kupffer Cells in Cholestatic Liver Injury. Am J Pathol (2016) 186:2238–47. doi: 10.1016/j.ajpath.2016.06.003
75. Remetic J, Ghallab A, Hobloss Z, Brackhagen L, Hassan R, Myllys M, et al. Loss of Bile Salt Export Pump Aggravates Lipopolysaccharide-Induced Liver Injury in Mice Due to Impaired Hepatic Endotoxin Clearance. Hepatology (2021) 75:1095–109. doi: 10.1002/hep.32289
76. Wang J, Xu Y, Chen Z, Liang J, Lin Z, Liang H, et al. Liver Immune Profiling Reveals Pathogenesis and Therapeutics for Biliary Atresia. Cell (2020) 183:1867–83. doi: 10.1016/j.cell.2020.10.048
77. Ramachandran P, Pellicoro A, Vernon MA, Boulter L, Aucott RL, Ali A, et al. Differential Ly-6C Expression Identifies the Recruited Macrophage Phenotype, Which Orchestrates the Regression of Murine Liver Fibrosis. Proc Natl Acad Sci USA (2012) 109:E3186–95. doi: 10.1073/pnas.1119964109
78. Barry-Hamilton V, Spangler R, Marshall D, Mccauley S, Rodriguez HM, Oyasu M, et al. Allosteric Inhibition of Lysyl Oxidase-Like-2 Impedes the Development of a Pathologic Microenvironment. Nat Med (2010) 16:1009–17. doi: 10.1038/nm.2208
79. Terkelsen MK, Bendixen SM, Hansen D, Scott E, Moeller AF, Nielsen R, et al. Transcriptional Dynamics of Hepatic Sinusoid-Associated Cells After Liver Injury. Hepatology (2020) 72:2119–33. doi: 10.1002/hep.31215
80. Wu H, Chen G, Wang J, Deng M, Yuan F, Gong J. TIM-4 Interference in Kupffer Cells Against CCL4-Induced Liver Fibrosis by Mediating Akt1/Mitophagy Signalling Pathway. Cell Prolif (2020) 53:e12731. doi: 10.1111/cpr.12731
81. Ambade A, Lowe P, Kodys K, Catalano D, Gyongyosi B, Cho Y, et al. Pharmacological Inhibition of CCR2/5 Signaling Prevents and Reverses Alcohol-Induced Liver Damage, Steatosis, and Inflammation in Mice. Hepatology (2019) 69:1105–21. doi: 10.1002/hep.30249
82. Lee JH, Shim YR, Seo W, Kim MH, Choi WM, Kim HH, et al. Mitochondrial Double-Stranded RNA in Exosome Promotes Interleukin-17 Production Through Toll-Like Receptor 3 in Alcohol-Associated Liver Injury. Hepatology (2020) 72:609–25. doi: 10.1002/hep.31041
83. Ma HY, Yamamoto G, Xu J, Liu X, Karin D, Kim JY, et al. IL-17 Signaling in Steatotic Hepatocytes and Macrophages Promotes Hepatocellular Carcinoma in Alcohol-Related Liver Disease. J Hepatol (2020) 72:946–59. doi: 10.1016/j.jhep.2019.12.016
84. Yu X, Lan P, Hou X, Han Q, Lu N, Li T, et al. HBV Inhibits LPS-Induced NLRP3 Inflammasome Activation and IL-1beta Production via Suppressing the NF-kappaB Pathway and ROS Production. J Hepatol (2017) 66:693–702. doi: 10.1016/j.jhep.2016.12.018
85. Watashi K, Liang G, Iwamoto M, Marusawa H, Uchida N, Daito T, et al. Interleukin-1 and Tumor Necrosis Factor-Alpha Trigger Restriction of Hepatitis Bvirus Infection via a Cytidine Deaminase Activation-Induced Cytidine Deaminase (AID). J Biol Chem (2013) 288:31715–27. doi: 10.1074/jbc.M113.501122
86. Tu Z, Bozorgzadeh A, Pierce RH, Kurtis J, Crispe IN, Orloff MS. TLR-Dependent Cross Talk Between Human Kupffer Cells and NK Cells. J Exp Med (2008) 205:233–44. doi: 10.1084/jem.20072195
87. Ebrahimkhani MR, Mohar I, Crispe IN. Cross-Presentation of Antigen by Diverse Subsets of Murine Liver Cells. Hepatology (2011) 54:1379–87. doi: 10.1002/hep.24508
88. Chen J, Wang XM, Wu XJ, Wang Y, Zhao H, Shen B, et al. Intrahepatic Levels of PD-1/PD-L Correlate With Liver Inflammation in Chronic Hepatitis B. Inflammation Res (2011) 60:47–53. doi: 10.1007/s00011-010-0233-1
89. Xu L, Yin W, Sun R, Wei H, Tian Z. Kupffer Cell-Derived IL-10 Plays a Key Role in Maintaining Humoral Immune Tolerance in Hepatitis B Virus-Persistent Mice. Hepatology (2014) 59:443–52. doi: 10.1002/hep.26668
90. Li H, Zheng HW, Chen H, Xing ZZ, You H, Cong M, et al. Hepatitis B Virus Particles Preferably Induce Kupffer Cells to Produce TGF-Beta1over Pro-Inflammatory Cytokines. Dig Liver Dis (2012) 44:328–33. doi: 10.1016/j.dld.2011.11.005
91. Faure-Dupuy S, Delphin M, Aillot L, Dimier L, Lebosse F, Fresquet J, et al. Hepatitis B Virus-Induced Modulation of Liver Macrophage Function Promotes Hepatocyte Infection. J Hepatol (2019) 71:1086–98. doi: 10.1016/j.jhep.2019.06.032
92. Tsochatzis EA, Bosch J, Burroughs AK. Liver Cirrhosis. Lancet (2014) 383:1749–61. doi: 10.1016/S0140-6736(14)60121-5
93. Pose E, Coll M, Martinez-Sanchez C, Zeng Z, Surewaard B, Catala C, et al. Programmed Death Ligand 1 Is Overexpressed in Liver Macrophages in Chronic Liver Diseases, and Its Blockade Improves the Antibacterial Activity Against Infections. Hepatology (2021) 74:296–311. doi: 10.1002/hep.31644
94. Lough J, Rosenthall L, Arzoumanian A, Goresky CA. Kupffer Cell Depletion Associated With Capillarization of Liver Sinusoids in Carbon Tetrachloride-Induced Rat Liver Cirrhosis. J Hepatol (1987) 5:190–8. doi: 10.1016/s0168-8278(87)80572-x
95. Buonomo E, Mei S, Guinn S, Leo I, Peluso M, Nolan MA, et al. Liver Stromal Cells Restrict Macrophage Maturation and Stromal IL-6 Limits the Differentiation of Cirrhosis-Linked Macrophages. J Hepatol (2022) 76:1127–37. doi: 10.1016/j.jhep.2021.12.036
96. Vadasz Z, Kessler O, Akiri G, Gengrinovitch S, Kagan HM, Baruch Y, et al. Abnormal Deposition of Collagen Around Hepatocytes in Wilson's Disease is Associated With Hepatocyte Specific Expression of Lysyl Oxidase and Lysyl Oxidase Like Protein-2. J Hepatol (2005) 43:499–507. doi: 10.1016/j.jhep.2005.02.052
97. Pollheimer MJ, Racedo S, Mikels-Vigdal A, Marshall D, Bowlus C, Lackner C, et al. Lysyl Oxidase-Like Protein 2 (LOXL2) Modulates Barrier Function in Cholangiocytes in Cholestasis. J Hepatol (2018) 69:368–77. doi: 10.1016/j.jhep.2018.04.009
98. Feng M, Ding J, Wang M, Zhang J, Zhu X, Guan W. Kupffer-Derived Matrix Metalloproteinase-9 Contributes to Liver Fibrosis Resolution. Int J Biol Sci (2018) 14:1033–40. doi: 10.7150/ijbs.25589
99. Li W, He F. Infusion of Kupffer Cells Expanded in Vitro Ameliorated Liver Fibrosis in a Murine Model of Liver Injury. Cell Transplant (2021) 30:83905350. doi: 10.1177/09636897211004090
100. Li X, Hollingshead N, Lampert S, Truong CD, Li W, Niu J, et al. A Conserved Pathway of Transdifferentiation in Murine Kupffer Cells. Eur J Immunol (2021) 51:2452–63. doi: 10.1002/eji.202049124
101. Jalan R, Fernandez J, Wiest R, Schnabl B, Moreau R, Angeli P, et al. Bacterial Infections in Cirrhosis: A Position Statement Based on the EASL Special Conference 2013. J Hepatol (2014) 60:1310–24. doi: 10.1016/j.jhep.2014.01.024
102. Holland-Fischer P, Gronbaek H, Sandahl TD, Moestrup SK, Riggio O, Ridola L, et al. Kupffer Cells Are Activated in Cirrhotic Portal Hypertension and Not Normalised by TIPS. Gut (2011) 60:1389–93. doi: 10.1136/gut.2010.234542
103. Steib CJ, Bilzer M, Op DWM, Pfeiler S, Hartmann AC, Hennenberg M, et al. Treatment With the Leukotriene Inhibitor Montelukast for 10 Days Attenuates Portal Hypertension in Rat Liver Cirrhosis. Hepatology (2010) 51:2086–96. doi: 10.1002/hep.23596
104. Lefere S, Degroote H, Van Vlierberghe H, Devisscher L. Unveiling the Depletion of Kupffer Cells in Experimental Hepatocarcinogenesis Through Liver Macrophage Subtype-Specific Markers. J Hepatol (2019) 71:631–3. doi: 10.1016/j.jhep.2019.03.016
105. Thomann S, Weiler S, Wei T, Sticht C, de la Torre C, Toth M, et al. YAP-Induced Ccl2 Expression Is Associated With a Switch in Hepatic Macrophage Identity and Vascular Remodelling in Liver Cancer. Liver Int (2021) 41:3011–23. doi: 10.1111/liv.15048
106. Kessler SM, Hoppstadter J, Hosseini K, Laggai S, Haybaeck J, Kiemer AK. Lack of Kupffer Cell Depletion in Diethylnitrosamine-Induced Hepatic Inflammation. J Hepatol (2019) 70:813–5. doi: 10.1016/j.jhep.2018.11.018
107. Siwicki M, Gort-Freitas NA, Messemaker M, Bill R, Gungabeesoon J, Engblom C, et al. Resident Kupffer Cells and Neutrophils Drive Liver Toxicity in Cancer Immunotherapy. Sci Immunol (2021) 6:i7083. doi: 10.1126/sciimmunol.abi7083
108. Sheng J, Zhang J, Wang L, Tano V, Tang J, Wang X, et al. Topological Analysis of Hepatocellular Carcinoma Tumour Microenvironment Based on Imaging Mass Cytometry Reveals Cellular Neighbourhood Regulated Reversely by Macrophages With Different Ontogeny. Gut (2021) 76:1176–91. doi: 10.1136/gutjnl-2021-324339
109. Liu N, Wang X, Steer CJ, Song G. MicroRNA-206 Promotes the Recruitment of CD8(+) T Cells by Driving M1 Polarisation of Kupffer Cells. Gut (2021) gutjnl-2021-324170. doi: 10.1136/gutjnl-2021-324170
110. Sharma A, Seow J, Dutertre CA, Pai R, Bleriot C, Mishra A, et al. Onco-Fetal Reprogramming of Endothelial Cells Drives Immunosuppressive Macrophages in Hepatocellular Carcinoma. Cell (2020) 183:377–94. doi: 10.1016/j.cell.2020.08.040
111. Chen DP, Ning WR, Jiang ZZ, Peng ZP, Zhu LY, Zhuang SM, et al. Glycolytic Activation of Peritumoral Monocytes Fosters Immune Privilege via the PFKFB3-PD-L1 Axis in Human Hepatocellular Carcinoma. J Hepatol (2019) 71:333–43. doi: 10.1016/j.jhep.2019.04.007
112. Zhang W, Zhangyuan G, Wang F, Jin K, Shen H, Zhang L, et al. The Zinc Finger Protein Miz1 Suppresses Liver Tumorigenesis by Restricting Hepatocyte-Driven Macrophage Activation and Inflammation. Immunity (2021) 54:1168–85. doi: 10.1016/j.immuni.2021.04.027
113. Li S, Yu J, Huber A, Kryczek I, Wang Z, Jiang L, et al. Metabolism Drives Macrophage Heterogeneity in the Tumor Microenvironment. Cell Rep (2022) 39:110609. doi: 10.1016/j.celrep.2022.110609
Keywords: macrophages, Kupffer cells, heterogeneity, function, liver injury
Citation: Li W, Chang N and Li L (2022) Heterogeneity and Function of Kupffer Cells in Liver Injury. Front. Immunol. 13:940867. doi: 10.3389/fimmu.2022.940867
Received: 10 May 2022; Accepted: 01 June 2022;
Published: 27 June 2022.
Edited by:
Jinhang Gao, Sichuan University, ChinaReviewed by:
Li Tang, Beijing Proteome Research Center, ChinaCamille Bleriot, Institut Gustave Roussy, France
Copyright © 2022 Li, Chang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liying Li, bGlsaXlpbmdAY2NtdS5lZHUuY24=