 Shaozhe Cai
Shaozhe Cai Ziwei Hu
Ziwei Hu Yu Chen
Yu Chen Jixin Zhong
Jixin Zhong Lingli Dong
Lingli Dong- Department of Rheumatology and Immunology, Tongji Hospital, Tongji Medical College of Huazhong University of Science and Technology, Wuhan, China
Studies have confirmed the involvement of a variety of lymphocyte subsets, including type 2 helper T lymphocytes (Th2) and IgG4+ B lymphocytes, in the pathogenesis of IgG4-related disease (IgG4-RD). Those lymphocytes contribute to the major pathogenetic features of IgG4-RD. However, they are not the only cellular components in the immunoinflammatory environment of this mysterious disease entity. Recent studies have suggested that various non-lymphocytic components, including macrophages and fibroblasts, may also play an important role in the pathogenetic process of IgG4-RD in terms of contributing to the chronic and complex progress of the disease. Therefore, the potential role of non-lymphocyte in the pathogenesis of IgG4-RD is worth discussing.
Background of IgG4-RD
IgG4-RD is a newly defined autoimmune disease of the century. Single or multiple organs/tissues may be involved simultaneously or successively in the disease progress, including salivary glands, lacrimal glands, pancreas, biliary tract, kidney, lung, retroperitoneum, and etc. The main clinical manifestations are tume-factive enlargement of the affected organs, often accompanied with increased serum IgG4 level (1). The typical pathological characteristics of IgG4-RD are dense lymphoplasmacytic infiltration, storiform fibrosis, and obliterative phlebitis (2). Increased eosinophil infiltration can also be observed. According to 2020 revised Comprehensive Diagnostic Criteria and 2019 American College of Rheumatology/European League Against Rheumatism (ACR/EULAR) classification criteria of IgG4-RD, fibrosis and lymphoplasmacytic infiltration are essential indices for the diagnosis of IgG4-RD (3, 4). The exact pathogenesis of IgG4-RD is still in the exploratory stage. Recent studies have suggested that multiple lymphocyte subsets including type 2 helper T cell (Th2), follicular helper T cell (Tfh), CD4+ cytotoxic T cell (CD4+ CTL), and B cells are involved. The interactions among these cells eventually mediate inflammation and fibrosis, contributing significantly to the pathogenesis of IgG4-RD (5–8).
Although, as an autoimmune disease, interactions between T and B cells contribute the majority of the pathogenesis in IgG4-RD (9), however, they are not the only cellular subsets involved in the real immuno-inflammatory response: residential parenchymal, mesenchymal cells, and other inflammatory cells infiltrated into the involved tissues, can respond to the stimuli derived from the local inflammatory microenvironment and further participate in the inflammatory response, which finally contribute to the histopathological features of IgG4-RD. Recent studies have also indicated that several non-lymphocytes, including macrophage and basophil, may exert important effects on the pathogenesis of IgG4-RD (10–12). Therefore, exploring the potential role of non-lymphocytes is crucial for in-depth understanding to the pathogenesis of IgG4-RD. This review will discuss this issue from the following three perspectives, based on the current researches in the field of IgG4-RD and other related conditions.
Non-T/B lymphocyte-mediated fibrosis
As mentioned above, profibrotic factors (e.g., IL-1β, TGF-β, LOXL2, PDGF, etc.) production resulting from the interactions between T (primarily CD4+ CTL) and B cells is the core fibrogenic mechanism of IgG4-RD (13). However, a growing number of researches have revealed that innate immune cells may play important roles in this process (Figure 1).
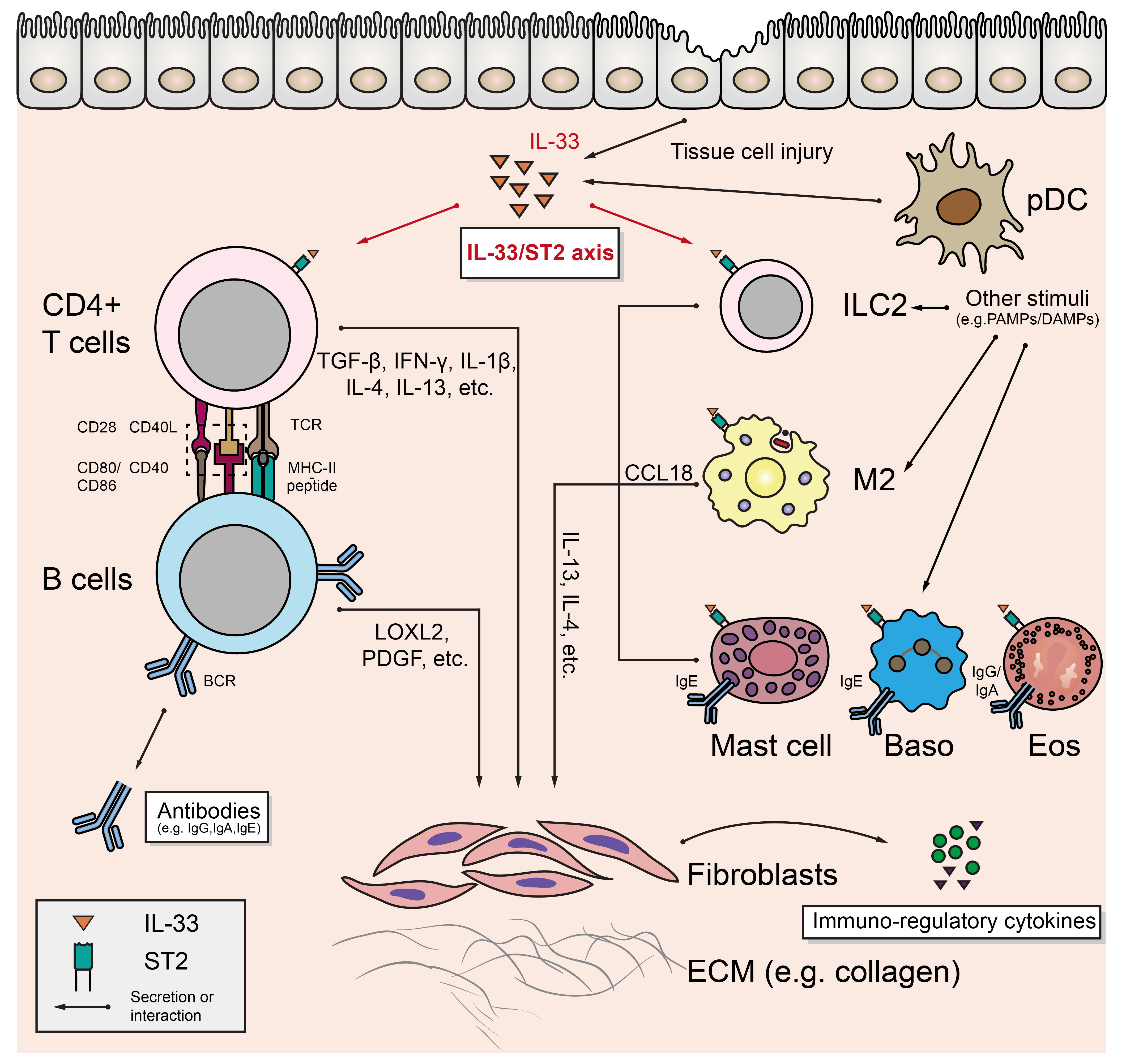
Figure 1 The underlying mechanism of fibrosis in IgG4-RD and the role of IL-33/ST2 axis.
Alternatively activated macrophage
The alternatively activated macrophage (AAM), also known as M2 macrophage, is induced by typical type 2 cytokines (such as IL-4 and IL-13), and can be found in several pathophysiological conditions such as allergy, parasitic infection, and the maintenance of metabolic homeostasis in adipose tissue (14). Ishiguro N et al. found that TLR7 (mainly located on M2 in the IgG4-RD affected tissues) activation in vitro lead to enhanced interleukin 33 (IL-33) production by M2, which was also confirmed by the finding that human Toll-like receptor 7 (huTLR7)-transgenic C57BL/6 mice had severer tissue fibrosis after receiving the treatment of resiquimod (R848) (12). M2 may also aggravate fibrosis through CC-chemokine ligand 18 (CCL18). Akiyama M et al. found that serum CCL18 level was significantly positively correlated with IgG4-RD RI (15). Furukawa S et al. found that CCL18 was co-localized with massive infiltrated M2 in IgG4-RD affected tissues, and positively associated with the fibrosis scores (16). Honda F et al. confirmed that blocking CCL8 (analog of human CCL18) in LATY136F knock-in mice (an animal model of IgG4-RD) alleviated the degree of fibrosis in affected salivary glands, and further in vitro assays revealed that CCL8 could directly stimulate collagen production in mouse fibroblasts (17). M2 can also induce plasma cell recruitment by secreting tumor necrosis factor ligand superfamily member 13(TNFSF13/APRIL) and further promote fibrogenesis (18).
Type 2 innate lymphoid cells
Innate lymphoid cells (ILCs) develop from common lymphoid progenitors (CLPs) in bone marrow. Except for the absence of antigen-specific receptor, ILCs are similar to T cells in terms of transcriptional and functional features. According to the expression of key transcription factors in their development and the pattern of cytokine secretion, ILCs can be divided into three groups: ILC1 (expresses T-bet and produces IFN-γ), ILC2 (expresses GATA3 and produces IL-4, IL-5, and IL-13), and ILC3 (expresses RORγt, and produces IL-22 or IL-17) (19, 20). Zhang P et al. found that the circulating ILC2 level in IgG4-RD patients was significantly higher than that in healthy controls and was positively correlated with the number of circulating regulatory T cells (Tregs). In addition, the expression levels of CD154, PD-1, and CXCR5 on ILC2 were also positively correlated with the level of B cells in peripheral blood, serum IgG4, and IgE (21). Although they didn’t directly demonstrate the effect of IL-13 and IL-4 secreted by ILC2 on IgG4-RD fibrosis, multiple studies suggested that IL-13 and/or IL-4 could mediate tissue fibrosis either directly (by acting on fibroblasts) or indirectly (e.g., by acting on alternatively activated macrophage) (22–24).
Mast cells, eosinophils, and basophils
Mast cells, basophils (Baso), and eosinophils (Eos) share many similarities in the expression of surface receptors and cytokines: they all express immunoglobulin Fc receptors (Eos expresses FcγRII/CD32 and FcαRI/CD89 for IgG and IgA; Baso and mast cells express high-affinity IgE Fc receptor FcϵRI) and secrete typical Th2-type immune response-related cytokines (such as IL-4 and IL-13) to accelerate the development of fibrosis after activation (25). Although understanding to the detailed mechanism of these cells in IgG4-RD is still limited, the pathological features of IgG4-RD suggest potential roles of these cells in the fibrosis of IgG4-RD: in recent studies, mild to moderate eosinophil infiltration was observed in the affected tissues, and increased peripheral blood eosinophil count and serum IgE level were detected in IgG4-RD patients (2, 26, 27), which suggests that Eos may tightly related to the pathogenesis of IgG4-RD. Increased local IgE may also activate Baso and mast cells in affected tissues, which can further produce profibrotic factors to enhance the fibrosis process in IgG4-RD.
Potential roles of IL-33/ST2 axis in IgG4-RD fibrosis
An important feature of the aforementioned cellular components is that they all express suppression of tumorigenicity 2 (ST2, also known as IL-1RL1), which is the receptor of a IL-1 family member, interleukin 33 (IL-33) (28, 29). IL-33 is constitutively and highly expressed in endothelial cells, epithelial cells, and fibroblast-like cells, and can be induced to express in several CD45+ cell subsets (e.g., macrophages) under the inflammatory stimulation (28). IL-33 can act as an alarmin to indicate the tissue injury: when cells are damaged, full-length IL-33 is released and transformed to highly active form to exert its effects as a cytokine, after being cleaved by proteases secreted by local inflammatory cells (such as chymase derived from mast cells) (28–30). Li D et al. reported that IL-33 released by bleomycin-injured lung epithelial cells induced the release of IL-13 from ILC2, and then promoted the differentiation of macrophage to IL-33 secreting M2, which further lead to the secretion of IL-13 and TGF-β, and promoted the pulmonary fibrosis (22). Besides in ILC2 and M2, the expression of profibrotic factor IL-13 can also be up-regulated in Th2 cells, mast cells, Baso, and Eos under the treatment of IL-33 (22, 31–34). These researches suggest that IL-33/ST2 axis may be the core axis connecting the fibrosis mediated by Th2-type immune responses. Despite that the potential profibrotic role of IL-33/ST2 axis has been mentioned in some studies of IgG4-RD (12, 21, 35), its main target cells and its temporal influencing order on potential target cells are unknown. In addition, although IL-33 is mostly associated with Th2-type immune responses in current studies, non-Th2-type cells, including Th1 and Treg cells, can also express ST2 and respond to IL-33 stimulation (36, 37). Therefore, the specific role of IL-33/ST2 axis in the pathogenesis of IgG4-RD still remains to be explored.
Beside promoting fibrosis, the above-mentioned immune cells may also influence the IgG4-RD immune response by other means such as secreting a variety of immunoregulatory agents: for example, IL-33 can enhance the activation of Th2 cells (32); Baso and monocytes can increase the production of IgG4 after the activation of some specific pattern recognition receptors (10, 38). Moreover, fibroblasts acting as collagen producing effector cells in this process can also exert other effects on the immunoinflammatory process of IgG4-RD (see below).
Non-profibrotic effects of fibroblasts
Fibroblasts are non-hematopoietic, non-endothelial, non-parenchymal, non-epithelial and non-mesothelial cells with mesenchymal origin. However, fibroblasts can be derived from mesothelial-, epithelial-, or endothelial-to-mesenchymal transition, and may be transformed from hematopoietic cells (39). As illustrated above, fibrosis is one of the core pathological features of IgG4-RD. The transition of fibroblasts into ECM-producing myofibroblasts is a common feature of fibrosis-related diseases (40). However, in a dynamic and complex inflammatory response, activated fibroblasts can not only functionate as a “collagen maker”, but also participate in the regulation of immune processes (Figure 2).
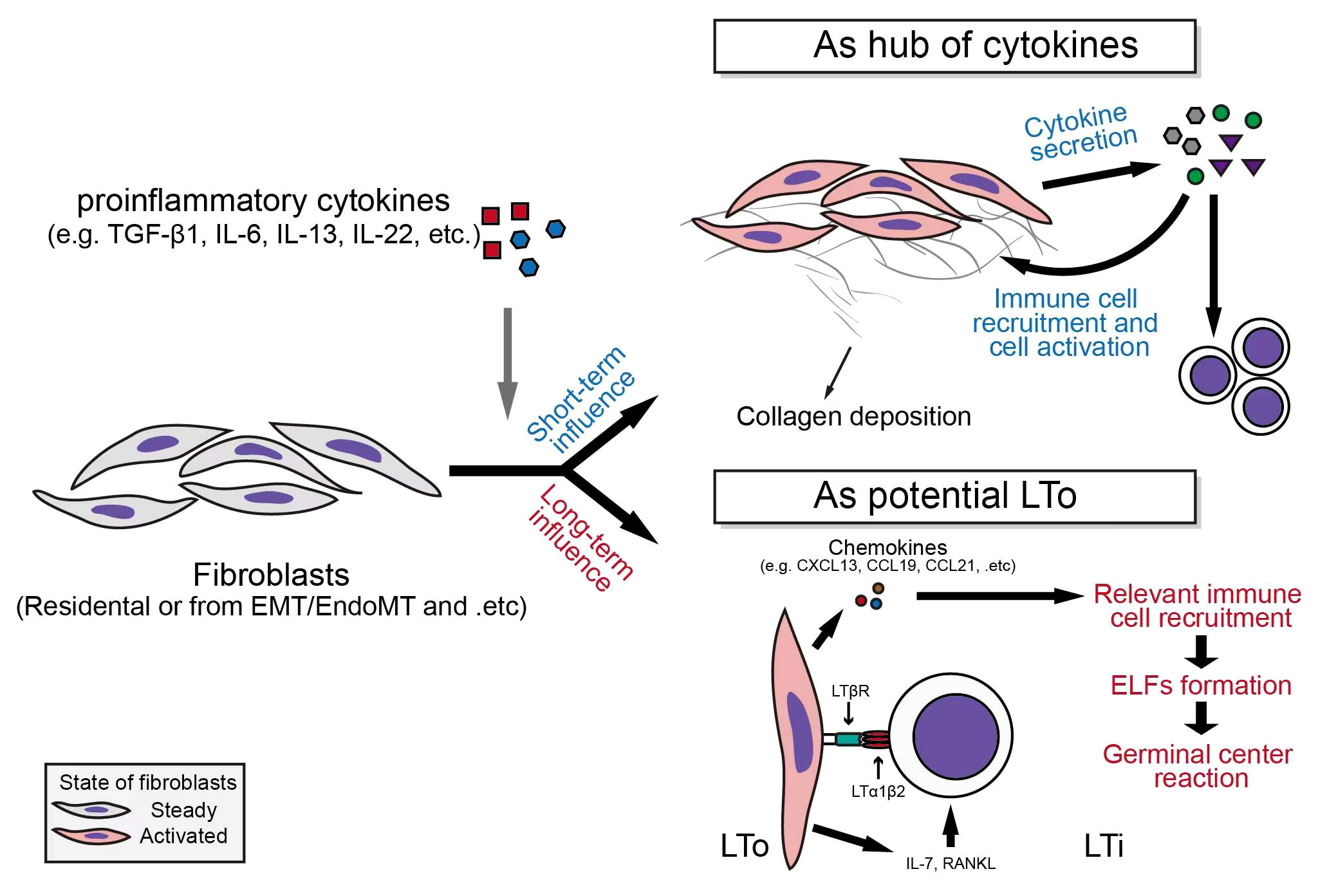
Figure 2 Fibroblasts serve as secretion centers of inflammatory factors and participate in the formation of ectopic lymphoid follicles.
Fibroblasts can serve as a hub of cytokines
Fibroblasts express a variety of cytokine receptors (e.g., CCR7, CXCR2, IL-4R, IL-6R, IFN-γR, etc.) and pattern recognition receptors, which indicate their ability to receive inflammatory signal derived from different pathological environments, and also suggests their strong potential to make response to the stimulation of these receptors (41–43). For example, in malignant tumors, cancer-associated fibroblasts (CAFs) serve as the main source of inflammatory factors including CXCL12, CXCL1, and IL-6, and participate in the regulation of immune responses in various malignant tumors (e.g., pancreatic ductal adenocarcinoma) (44). In classic fibrotic diseases, such as idiopathic pulmonary fibrosis (IPF), fibroblasts can up-regulate the expressions of IL-6, IL-8, and CCL2, under the stimulation of IL-1β (45); Furthermore, the expression of CCL5 was also up-regulated in response to CCL21 stimulation in primary lung fibroblasts derived from IPF patients (46). In Dupuytren’s disease, tumor necrosis factor α (TNFα) stimulates myofibroblasts to secrete IL-33, which in turn increase the secretion of TNFα from other immune cells, and finally forms a vicious circle (47).
The important roles of fibroblasts in autoimmune conditions have also been demonstrated in human specimens and animal models. In serum transfer induced arthritis (STIA) K/BxN mouse model, the secretion of cytokines by THY1+ fibroblasts of hip joint, including CXCL12, CCL2, CCL5 and IL-6, is significantly enhanced under the stimulation of TNFα (48). TNFα, IL-1β, and hypoxia also up-regulate the expression of cytokines, including IL-6, CXCL8 (namely, IL-8), CCL2, CXCL11, CXCL12, and VEGF, as well as the expression of cytokine receptors such as TNFRSF9 in synovial fibroblasts derived from rheumatoid arthritis (RA) patients (49, 50). In systemic sclerosis (SSc), IL-22 synergistically works with TNFα to induce the expression of IL-8 and CCL2 in skin fibroblasts, while the simultaneous stimulation of TGF-β1 and IL-17A can lead to an up-regulation of IL-6 expression in fibroblasts from affected skin by nearly a hundredfold (51, 52).
In involved tissues of IgG4-related retroperitoneal fibrosis (IgG4-RPF), the co-localization of IL-6, BAFF, IL-7, IL-12 p70 and IL-23 with αSMA is observed by Zongfei J et al, and the expression levels of both IL-6 and IL-6R are higher than those in the control tissues, suggesting the pro-inflammatory properties of fibroblasts in affected tissues of IgG4-RD patients (53). Elevated secretion of BAFF, IL-7, IL-12 p70, and IL-23 in the aortic adventitial fibroblasts (AAFs) after the activation of IL-6/IL-6R axis can also be seen (53). It is worth mentioning that the increased production of IL-7, IL-12, IL-23, and other cytokines by IgG4-RD fibroblasts can not only play an immediate role in regulating the immunoinflammatory processes via affecting the functional status of infiltrated immune cells, but also can exert a long-term impact on the progress of autoimmune diseases by participating in the formation of ectopic lymphoid follicles (ELFs) (54).
Fibroblasts may participate in the formation of ectopic lymphoid follicles (ELFs)
Ectopic lymphoid follicles, also known as tertiary lymphoid structures (TLSs), which is induced by inflammatory insults, and can contribute to the “propagation” of antigenic specific responses in local tissues (55). The developed ELF is a complex collection of white blood cells and specific mesenchymal cells, which is functionally similar to the secondary lymphoid organs (SLOs) including spleen and lymph nodes and provides a site for germinal center reaction (GC reactions) (54, 55).
The interaction between lymphoid tissue inducer (LTi) and lymphoid tissue organizer (LTo) is required for the generation of lymphoid follicles. LTis are developed from IL-7Rα+ lymphoid progenitors, and this is a process that requires the expression of retinoic acid receptor-related orphan receptor-γt (RORγt) (56). LTi cells express lymphotoxin α1β2 (LTα1β2) under the stimulation of IL-7 and RANKL (55, 56). Via binding to LTβ receptor (LTβR) expressed on LTo cells, LTα1β2 induces the release of chemokines (e.g., CXCL13, CCL19, and CCL21), as well as the expression of adhesion molecules, which further recruits immune cells, and promotes the formation of lymphoid structures (55, 57). In actual pathological environment, a variety of cells, including Th17 and Tγδ17, are involved in the formation of ELF (55). In chronic inflammation, a key step in the formation process of ELF is the transition of resident stromal cells (e.g., fibroblasts) to LTo-like phenotype after receiving the stimulation of inflammatory stimuli: for example, in synovial tissues of RA patients, the enhancement of IL-6 signal can lead to increased IL-7 production in synovial fibroblasts, which can induce LTi recruitment into the affected tissues and promote ELF formation (57–60).
TLS assists to eliminate pathogenic microorganisms when it is produced in local site of infections. However, in some autoimmune settings (e.g., Sjogren’s syndrome, systemic lupus erythematosus, etc.), the formation of TLS can aggravate the progress of autoimmune diseases through somatic hypermutation (SHM), isotype switching, and affinity maturation (56, 61). It is worth mentioning that Nayar S et al. found that during TLS formation in salivary glands in patients with Primary Sjogren’s Syndrome (pSS), the differentiation of a group of resident podoplanin (PDPN) positive mesenchymal cells into the “immunofibroblasts” that could support the early TLS formation happens before the infiltration of lymphocytes into the salivary glands, which is mainly regulated by IL-13 derived from resident ILCs and stromal cells (62). After lymphocyte infiltration, those immunofibroblasts were further phenotypically stabilized and proliferated under the influence of IL-22 and lymphotoxin (62). These studies show that innate immune cells may have already been primed for the potential chronicity of adaptive immune response before it occurs, and provide important hints for the potential mechanism of ELF formation in autoimmune diseases with mucosa containing organs’ involvement.
ELF probably plays an important role in the pathogenesis of IgG4-RD (63): there is marked fibroblast activation in IgG4-RD involved tissues, and Zongfei J et al. observed that fibroblasts in affected tissues could express IL-7 (an ELF formation promoting cytokine) after the activation of IL-6/IL-6R axis (53); Chen Y et al. found an abnormal expansion of Tfh cells in IgG4-RD patient tissues (5); In addition, infiltration of a large amount of B cells into the affected tissues, and the diversity of B cell clones suggest the possible existence of ectopic germinal center reactions in IgG4-RD (64, 65); Zhang P et al. observed the infiltration of ILC in the affected tissues of IgG4-RD patients, and the positive correlation of ILC2 with the disease severity of IgG4-RD (21). All these aforementioned components can be involved in the formation of ELF and participate in the subsequent immunoinflammatory reactions. Considering the predisposition of exo-/endocrine gland involvement (mucosa containing organs), marked fibroblast activation (potential LTo), and dense infiltration of immune cells (potential LTi) in IgG4-RD, the possible role of fibroblasts in the formation of ELF in the pathogenesis of IgG4-RD is worthy of further investigation. Besides, fibroblasts can express major histocompatibility complex class II (MHC-II) molecules under the influence of inflammatory stimuli, which leads them become the target cells of CD4+ CTLs resulting in apoptosis and the subsequent biological processes (see below) (66).
Atypical antigen presentation
Antigen presentation is an important immunological process that assists T cells in antigen recognition and activation. Antigen-presenting cells (APCs) can be grouped into professional APCs and non-professional APCs based on the expression of different MHC molecules. Professional APCs include dendritic cells (DCs), macrophages, and B cells (67). Most nucleated cells can present antigen to CD8+ T cells via MHC-I molecules, while professional APCs present antigen peptides to CD4+ T lymphocytes through their constitutively expressed MHC-II molecules (68). With the help of CD4/CD8, and other surface molecules such as CD45, the binding of T cell receptor (TCR) to antigen peptide-MHC complex lead to the phosphorylation of immunoreceptor tyrosine-based activation motif (ITAM) on the cytoplasmic tail of CD3, and initiates the downstream TCR signaling (69).
However, in some pathological conditions, the expression of MHC-II molecules can be induced on some non-professional APCs by inflammatory stimuli (namely, ectopic expression). Winau F et al. found that IFN-γ could up-regulate the expression of MHC-II in hepatic stellate cells (HSC) (70). Kato et al. found that the MHC-II expression on synovial fibroblasts was increased when co-cultured with Th1 cells (71). Even human T cells can express MHC-II molecules and present autoantigenic peptides after activation of TCR signaling (72). In addition to the aforementioned cells, cells including Eos, Baso, and epithelial cells may express MHC-II molecules under the stimulation of different inflammatory stimuli, which is well reviewed by Kambayashi T and Laufer TM (73).
In IgG4-RD, Perugino CA et al. found that CD4+ CTL mediated the apoptosis of a variety of cells in affected tissues, including T cells, B cells, acinar cells, ductal cells, and endothelial cells, and the expression level of HLA-DR in these apoptotic cells was significantly increased (66). Interestingly, those apoptotic cells are mainly non-endothelial, non-immune cells with mesenchymal origin (66). However, the regulatory mechanisms and specific roles of atypical MHC-II expression in these cells remain unknown in IgG4-RD. Considering the existence of the previously mentioned inducing factors for the atypical expression of MHC-II molecules (e.g., infiltration of immune cell subsets, and immunoinflammatory microenvironment) (7, 10, 21, 74) and the potential influence of environmental factors on IgG4-RD (75), atypical antigen presentations via MHC-II molecules may be the factor to promote or even initiate the development of IgG4-RD. In addition, inflammatory microenvironment could also up-regulate the expression of MHC-I molecules, while the dying cells induced by CD4+CTL may contribute to cross-presentation, which may be an important basis for the potential pathogenic role of CD8+ T cells in IgG4-RD (66, 76, 77) (Figure 3).
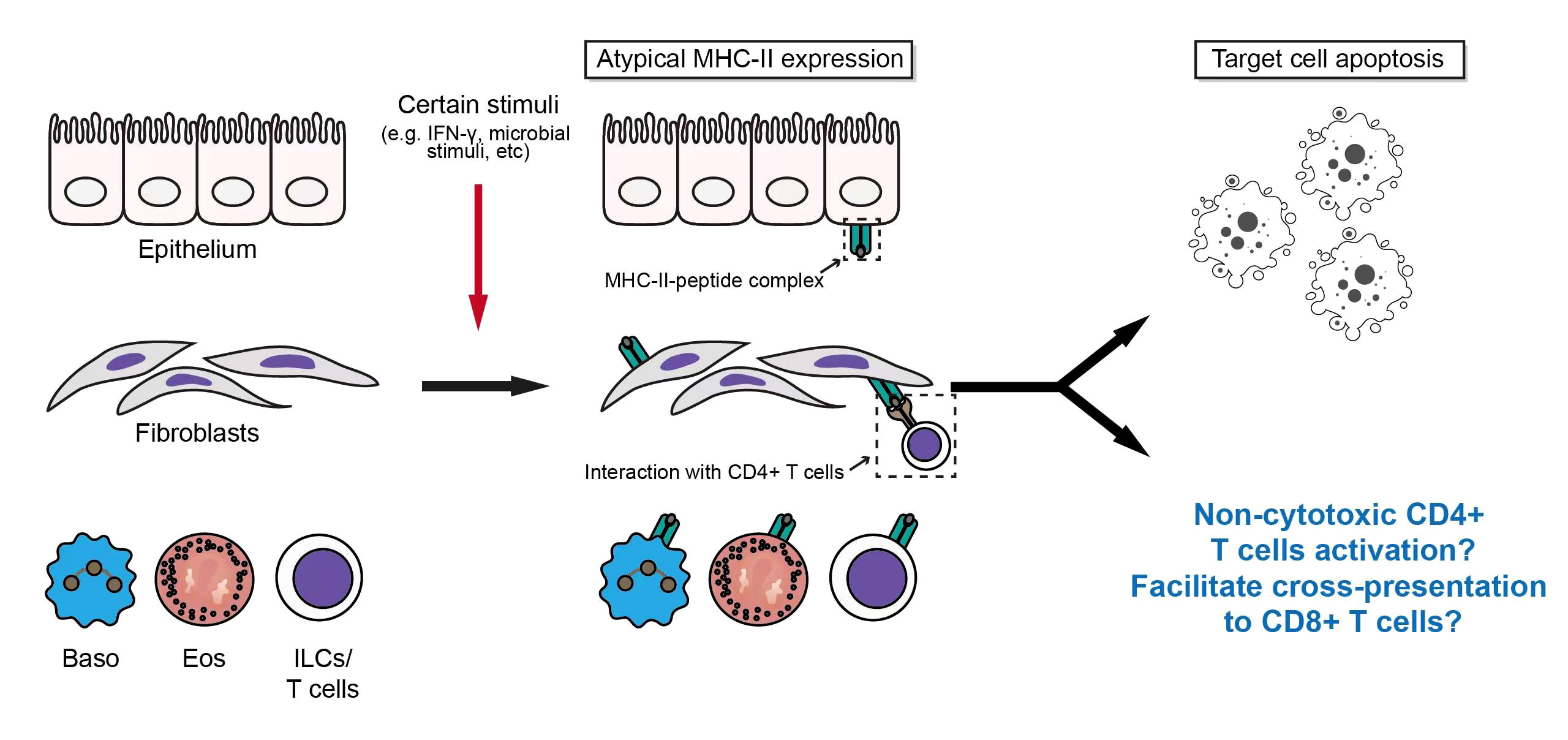
Figure 3 Induction of atypical antigen presentation and its potential effect on IgG4-RD.
Summary and prospect
Non-lymphocyte can exert important effects on the immune-inflammatory regulations in many pathological conditions, such as RA, SLE, SSc, and etc. (78–80). Current studies on IgG4-RD and basic immunological evidence suggest that non-lymphocyte may play roles in the initiation of IgG4-RD disease (such as atypical antigen presentation), disease severity (such as mediating fibrosis and participating in the regulation of immune inflammation), as well as chronic and complex progress (such as the formation of ectopic lymphoid follicles). Although more and more evidence suggest the potential role of non-lymphocyte in the pathogenesis of IgG4-RD, the research on IgG4-RD is still very limited at present in this area, mainly because IgG4-RD is a newly defined disease entity in this century, and there is no particularly suitable animal model for IgG4-RD investigation. Animal models that have been published so far include the patient serum injection model proposed by Shiokawa et al. (injecting serum IgG of IgG4-RD patients into male Balb/C neonatal mice), LATY136F knock-in mouse model identified by Yamada et al., and Human TLR7 transgenic mouse model proposed by Ishiguro N et al. (12, 81, 82). These models may assist in exploring the potential roles of non-lymphocyte in IgG4-RD in vivo. Considering the lack of appropriate animal models, the application of high-throughput techniques coupled with well-designed experiments may help further mechanistically elucidating the functional status of these cells and their association with IgG4-RD: via single-cell transcriptome sequencing (scRNA-Seq), Valenzi E et al. identified the heterogeneity of fibroblasts in patients with systemic sclerosis associated interstitial lung disease (SSC-ILD) revealing that myofibroblast differentiation and proliferation were the key pathological mechanisms promoting fibrosis in patients with SSC-ILD (83); Der E et al. found that the signal signatures of type I interferon and the fibrosis-related signaling in renal tubular cells and keratinocyte of lupus nephritis patients were differentiated from that in healthy controls (84). It is worth mentioning that detailed investigation into the pathological roles of non-lymphocytes may help provide helpful therapeutic insights, not only for IgG4-RD, but also for many other autoimmune conditions: different diseases may share some pathogenic cytokines, which have the potential to be the therapeutic targets. For example, besides in IgG4-RD, IL-33/ST2 axis has been identified to play roles in several fibrotic process related diseases (e.g., pSS), and antibodies targeting have been utilized in the clinical trials of several diseases, including atopic dermatitis, and asthma (80, 85); type I interferon can also exert pathological effects and be the therapeutic target in several autoimmune conditions (e.g. SLE) (86). In consideration of the fact that the components of non-lymphocytes may affect the pathogenesis of IgG4-RD at multiple levels, further mechanistic researches and large-scale clinical association analyses focusing on the specific roles of different non-lymphocytes in the initiation and progress of IgG4-RD will help indicate the optimal usage of target therapy, which in turn, on this basis, will deepen our understanding to the potential pathogenic roles of non-lymphocyte and related inflammatory factors in IgG4-RD.
Author contributions
SC and ZH performed literature search and drafted the manuscript. YC, JZ, and LD revised the manuscript. All author designed and reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the National Natural Science Foundation of China (81974254 and 81901651), and Tongji Hospital Clinical Research Flagship Program (No. 2019CR206).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Lanzillotta M, Mancuso G, Della-Torre E. Advances in the diagnosis and management of IgG4 related disease. BMJ (2020) 369:m1067. doi: 10.1136/bmj.m1067
2. Deshpande V, Zen Y, Chan JK, Yi EE, Sato Y, Yoshino T, et al. Consensus statement on the pathology of IgG4-related disease. Mod Pathol (2012) 25(9):1181–92. doi: 10.1038/modpathol.2012.72
3. Umehara H, Okazaki K, Kawa S, Takahashi H, Goto H, Matsui S, et al. The 2020 revised comprehensive diagnostic (RCD) criteria for IgG4-RD. Mod Rheumatol (2021) 31(3):529–33. doi: 10.1080/14397595.2020.1859710
4. Wallace ZS, Naden RP, Chari S, Choi HK, Della-Torre E, Dicaire JF, et al. The 2019 American college of Rheumatology/European league against rheumatism classification criteria for IgG4-related disease. Ann rheum dis (2020) 79(1):77–87. doi: 10.1136/annrheumdis-2019-216561
5. Chen Y, Lin W, Yang H, Wang M, Zhang P, Feng R, et al. Aberrant expansion and function of follicular helper T cell subsets in IgG4-related disease. Arthritis Rheumatol (2018) 70(11):1853–65. doi: 10.1002/art.40556
6. Della-Torre E, Rigamonti E, Perugino C, Baghai-Sain S, Sun N, Kaneko N, et al. B lymphocytes directly contribute to tissue fibrosis in patients with IgG4-related disease. J Allergy Clin Immunol (2020) 145(3):968–81.e14. doi: 10.1016/j.jaci.2019.07.004
7. Maehara T, Mattoo H, Ohta M, Mahajan VS, Moriyama M, Yamauchi M, et al. Lesional CD4+ IFN-gamma+ cytotoxic T lymphocytes in IgG4-related dacryoadenitis and sialoadenitis. Ann rheum dis (2017) 76(2):377–85. doi: 10.1136/annrheumdis-2016-209139
8. Grados A, Ebbo M, Piperoglou C, Groh M, Regent A, Samson M, et al. T Cell polarization toward TH2/TFH2 and TH17/TFH17 in patients with IgG4-related disease. Front Immunol (2017) 8:235. doi: 10.3389/fimmu.2017.00235
9. Perugino CA, Stone JH. IgG4-related disease: an update on pathophysiology and implications for clinical care. Nat Rev Rheumatol (2020) 16(12):702–14. doi: 10.1038/s41584-020-0500-7
10. Watanabe T, Yamashita K, Sakurai T, Kudo M, Shiokawa M, Uza N, et al. Toll-like receptor activation in basophils contributes to the development of IgG4-related disease. J gastroenterol (2013) 48(2):247–53. doi: 10.1007/s00535-012-0626-8
11. Furukawa S, Moriyama M, Miyake K, Nakashima H, Tanaka A, Maehara T, et al. Interleukin-33 produced by M2 macrophages and other immune cells contributes to Th2 immune reaction of IgG4-related disease. Sci Rep (2017) 7:42413. doi: 10.1038/srep42413
12. Ishiguro N, Moriyama M, Furusho K, Furukawa S, Shibata T, Murakami Y, et al. Activated M2 macrophages contribute to the pathogenesis of IgG4-related disease via toll-like receptor 7/Interleukin-33 signaling. Arthritis Rheumatol (2020) 72(1):166–78. doi: 10.1002/art.41052
13. Pillai S, Perugino C, Kaneko N. Immune mechanisms of fibrosis and inflammation in IgG4-related disease. Curr Opin Rheumatol (2020) 32(2):146–51. doi: 10.1097/BOR.0000000000000686
14. Van Dyken SJ, Locksley RM. Interleukin-4- and interleukin-13-mediated alternatively activated macrophages: roles in homeostasis and disease. Annu Rev Immunol (2013) 31:317–43. doi: 10.1146/annurev-immunol-032712-095906
15. Akiyama M, Yasuoka H, Yoshimoto K, Takeuchi T. CC-chemokine ligand 18 is a useful biomarker associated with disease activity in IgG4-related disease. Ann rheum dis (2018) 77(9):1386–7. doi: 10.1136/annrheumdis-2017-212110
16. Furukawa S, Moriyama M, Tanaka A, Maehara T, Tsuboi H, Iizuka M, et al. Preferential M2 macrophages contribute to fibrosis in IgG4-related dacryoadenitis and sialoadenitis, so-called mikulicz's disease. Clin Immunol (2015) 156(1):9–18. doi: 10.1016/j.clim.2014.10.008
17. Honda F, Tsuboi H, Ono Y, Abe S, Takahashi H, Ito K, et al. Pathogenic roles and therapeutic potential of the CCL8-CCR8 axis in a murine model of IgG4-related sialadenitis. Arthritis Res Ther (2021) 23(1):214. doi: 10.1186/s13075-021-02597-6
18. Kawakami T, Mizushima I, Yamada K, Fujii H, Ito K, Yasuno T, et al. Abundant a proliferation-inducing ligand (APRIL)-producing macrophages contribute to plasma cell accumulation in immunoglobulin G4-related disease. Nephrol Dial Transpl (2019) 34(6):960–9. doi: 10.1093/ndt/gfy296
19. Bal SM, Golebski K, Spits H. Plasticity of innate lymphoid cell subsets. Nat Rev Immunol (2020) 20(9):552–65. doi: 10.1038/s41577-020-0282-9
20. Colonna M. Innate lymphoid cells: Diversity, plasticity, and unique functions in immunity. Immunity (2018) 48(6):1104–17. doi: 10.1016/j.immuni.2018.05.013
21. Zhang P, Liu Z, Peng L, Zhou J, Wang M, Li J, et al. Phenotype, function and clinical significance of innate lymphoid cells in immunoglobulin G4-related disease. Rheumatology (Oxford) (2022) 61(5):2197–209. doi: 10.1093/rheumatology/keab610
22. Li D, Guabiraba R, Besnard AG, Komai-Koma M, Jabir MS, Zhang L, et al. IL-33 promotes ST2-dependent lung fibrosis by the induction of alternatively activated macrophages and innate lymphoid cells in mice. J Allergy Clin Immunol (2014) 134(6):1422–32.e11. doi: 10.1016/j.jaci.2014.05.011
23. Peng H, Sarwar Z, Yang XP, Peterson EL, Xu J, Janic B, et al. Profibrotic role for interleukin-4 in cardiac remodeling and dysfunction. Hypertension. (2015) 66(3):582–9. doi: 10.1161/HYPERTENSIONAHA.115.05627
24. Bellini A, Marini MA, Bianchetti L, Barczyk M, Schmidt M, Mattoli S. Interleukin (IL)-4, IL-13, and IL-17A differentially affect the profibrotic and proinflammatory functions of fibrocytes from asthmatic patients. Mucosal Immunol (2012) 5(2):140–9. doi: 10.1038/mi.2011.60
25. Stone KD, Prussin C, Metcalfe DD. IgE, mast cells, basophils, and eosinophils. J Allergy Clin Immunol (2010) 125(2 Suppl 2):S73–80. doi: 10.1016/j.jaci.2009.11.017
26. Culver EL, Sadler R, Bateman AC, Makuch M, Cargill T, Ferry B, et al. Increases in IgE, eosinophils, and mast cells can be used in diagnosis and to predict relapse of IgG4-related disease. Clin Gastroenterol Hepatol (2017) 15(9):1444–52.e6. doi: 10.1016/j.cgh.2017.02.007
27. Wallace ZS, Deshpande V, Mattoo H, Mahajan VS, Kulikova M, Pillai S, et al. IgG4-related disease: Clinical and laboratory features in one hundred twenty-five patients. Arthritis Rheumatol (2015) 67(9):2466–75. doi: 10.1002/art.39205
28. Liew FY, Girard JP, Turnquist HR. Interleukin-33 in health and disease. Nat Rev Immunol (2016) 16(11):676–89. doi: 10.1038/nri.2016.95
29. Chan BCL, Lam CWK, Tam LS, Wong CK. IL33: Roles in allergic inflammation and therapeutic perspectives. Front Immunol (2019) 10:364. doi: 10.3389/fimmu.2019.00364
30. Lott JM, Sumpter TL, Turnquist HR. New dog and new tricks: evolving roles for IL-33 in type 2 immunity. J Leukoc Biol (2015) 97(6):1037–48. doi: 10.1189/jlb.3RI1214-595R
31. Cherry WB, Yoon J, Bartemes KR, Iijima K, Kita H. A novel IL-1 family cytokine, IL-33, potently activates human eosinophils. J Allergy Clin Immunol (2008) 121(6):1484–90. doi: 10.1016/j.jaci.2008.04.005
32. Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. (2005) 23(5):479–90. doi: 10.1016/j.immuni.2005.09.015
33. Saluja R, Khan M, Church MK, Maurer M. The role of IL-33 and mast cells in allergy and inflammation. Clin Transl Allergy (2015) 5:33. doi: 10.1186/s13601-015-0076-5
34. Saluja R, Ketelaar ME, Hawro T, Church MK, Maurer M, Nawijn MC. The role of the IL-33/IL-1RL1 axis in mast cell and basophil activation in allergic disorders. Mol Immunol (2015) 63(1):80–5. doi: 10.1016/j.molimm.2014.06.018
35. Minaga K, Watanabe T, Hara A, Yoshikawa T, Kamata K, Kudo M. Plasmacytoid dendritic cells as a new therapeutic target for autoimmune pancreatitis and IgG4-related disease. Front Immunol (2021) 12:713779. doi: 10.3389/fimmu.2021.713779
36. Peine M, Marek RM, Lohning M. IL-33 in T cell differentiation, function, and immune homeostasis. Trends Immunol (2016) 37(5):321–33. doi: 10.1016/j.it.2016.03.007
37. Faustino LD, Griffith JW, Rahimi RA, Nepal K, Hamilos DL, Cho JL, et al. Interleukin-33 activates regulatory T cells to suppress innate gammadelta T cell responses in the lung. Nat Immunol (2020) 21(11):1371–83. doi: 10.1038/s41590-020-0785-3
38. Watanabe T, Yamashita K, Fujikawa S, Sakurai T, Kudo M, Shiokawa M, et al. Involvement of activation of toll-like receptors and nucleotide-binding oligomerization domain-like receptors in enhanced IgG4 responses in autoimmune pancreatitis. Arthritis rheumatism (2012) 64(3):914–24. doi: 10.1002/art.33386
39. Buechler MB, Turley SJ. A short field guide to fibroblast function in immunity. Semin Immunol (2018) 35:48–58. doi: 10.1016/j.smim.2017.11.001
40. Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med (2012) 18(7):1028–40. doi: 10.1038/nm.2807
41. Dorrier CE, Aran D, Haenelt EA, Sheehy RN, Hoi KK, Pintaric L, et al. CNS fibroblasts form a fibrotic scar in response to immune cell infiltration. Nat Neurosci (2021) 24(2):234–44. doi: 10.1038/s41593-020-00770-9
42. Proost P, Verpoest S, Van de Borne K, Schutyser E, Struyf S, Put W, et al. Synergistic induction of CXCL9 and CXCL11 by toll-like receptor ligands and interferon-gamma in fibroblasts correlates with elevated levels of CXCR3 ligands in septic arthritis synovial fluids. J Leukoc Biol (2004) 75(5):777–84. doi: 10.1189/jlb.1003524
43. Habiel DM, Hogaboam C. Heterogeneity in fibroblast proliferation and survival in idiopathic pulmonary fibrosis. Front Pharmacol (2014) 5. doi: 10.3389/fphar.2014.00002
44. Biffi G, Tuveson DA. Diversity and biology of cancer-associated fibroblasts. Physiol Rev (2021) 101(1):147–76. doi: 10.1152/physrev.00048.2019
45. Hadjicharalambous MR, Roux BT, Feghali-Bostwick CA, Murray LA, Clarke DL, Lindsay MA. Long non-coding RNAs are central regulators of the IL-1beta-Induced inflammatory response in normal and idiopathic pulmonary lung fibroblasts. Front Immunol (2018) 9:2906. doi: 10.3389/fimmu.2018.02906
46. Pierce EM, Carpenter K, Jakubzick C, Kunkel SL, Evanoff H, Flaherty KR, et al. Idiopathic pulmonary fibrosis fibroblasts migrate and proliferate to CC chemokine ligand 21. Eur Respir J (2007) 29(6):1082–93. doi: 10.1183/09031936.00122806
47. Izadi D, Layton TB, Williams L, McCann F, Cabrita M, Espirito Santo AI, et al. Identification of TNFR2 and IL-33 as therapeutic targets in localized fibrosis. Sci Adv (2019) 5(12):eaay0370. doi: 10.1126/sciadv.aay0370
48. Croft AP, Campos J, Jansen K, Turner JD, Marshall J, Attar M, et al. Distinct fibroblast subsets drive inflammation and damage in arthritis. Nature. (2019) 570(7760):246–51. doi: 10.1038/s41586-019-1263-7
49. Hitchon C, Wong K, Ma G, Reed J, Lyttle D, El-Gabalawy H. Hypoxia-induced production of stromal cell-derived factor 1 (CXCL12) and vascular endothelial growth factor by synovial fibroblasts. Arthritis Rheumatism (2002) 46(10):2587–97. doi: 10.1002/art.10520
50. Loh C, Park SH, Lee A, Yuan R, Ivashkiv LB, Kalliolias GD. TNF-induced inflammatory genes escape repression in fibroblast-like synoviocytes: transcriptomic and epigenomic analysis. Ann rheum dis (2019) 78(9):1205–14. doi: 10.1136/annrheumdis-2018-214783
51. Dufour AM, Alvarez M, Russo B, Chizzolini C. Interleukin-6 and type-I collagen production by systemic sclerosis fibroblasts are differentially regulated by interleukin-17A in the presence of transforming growth factor-beta 1. Front Immunol (2018) 9:1865. doi: 10.3389/fimmu.2018.01865
52. Brembilla NC, Dufour AM, Alvarez M, Hugues S, Montanari E, Truchetet ME, et al. IL-22 capacitates dermal fibroblast responses to TNF in scleroderma. Ann rheum dis (2016) 75(9):1697–705. doi: 10.1136/annrheumdis-2015-207477
53. Zongfei J, Rongyi C, Xiaomeng C, Lili M, Lingying M, Xiufang K, et al. In vitro IL-6/IL-6R trans-signaling in fibroblasts releases cytokines that may be linked to the pathogenesis of IgG4-related disease. Front Immunol (2020) 11:1272. doi: 10.3389/fimmu.2020.01272
54. Barone F, Gardner DH, Nayar S, Steinthal N, Buckley CD, Luther SA. Stromal fibroblasts in tertiary lymphoid structures: A novel target in chronic inflammation. Front Immunol (2016) 7:477. doi: 10.3389/fimmu.2016.00477
55. Jones GW, Jones SA. Ectopic lymphoid follicles: inducible centres for generating antigen-specific immune responses within tissues. Immunology (2016) 147(2):141–51. doi: 10.1111/imm.12554
56. van de Pavert SA, Mebius RE. New insights into the development of lymphoid tissues. Nat Rev Immunol (2010) 10(9):664–74. doi: 10.1038/nri2832
57. Lanzillotta M, Fernandez-Codina A, Culver E, Ebbo M, Martinez-Valle F, Schleinitz N, et al. Emerging therapy options for IgG4-related disease. Expert Rev Clin Immunol (2021) 17(5):471–83. doi: 10.1080/1744666X.2021.1902310
58. Buckley CD, Barone F, Nayar S, Benezech C, Caamano J. Stromal cells in chronic inflammation and tertiary lymphoid organ formation. Annu Rev Immunol (2015) 33:715–45. doi: 10.1146/annurev-immunol-032713-120252
59. Sawa S, Kamimura D, Jin GH, Morikawa H, Kamon H, Nishihara M, et al. Autoimmune arthritis associated with mutated interleukin (IL)-6 receptor gp130 is driven by STAT3/IL-7-dependent homeostatic proliferation of CD4+ T cells. J Exp Med (2006) 203(6):1459–70. doi: 10.1084/jem.20052187
60. Nayar S, Campos J, Chung MM, Navarro-Nunez L, Chachlani M, Steinthal N, et al. Bimodal expansion of the lymphatic vessels is regulated by the sequential expression of IL-7 and lymphotoxin alpha1beta2 in newly formed tertiary lymphoid structures. J Immunol (2016) 197(5):1957–67. doi: 10.4049/jimmunol.1500686
61. Bombardieri M, Lewis M, Pitzalis C. Ectopic lymphoid neogenesis in rheumatic autoimmune diseases. Nat Rev Rheumatol (2017) 13(3):141–54. doi: 10.1038/nrrheum.2016.217
62. Nayar S, Campos J, Smith CG, Iannizzotto V, Gardner DH, Mourcin F, et al. Immunofibroblasts are pivotal drivers of tertiary lymphoid structure formation and local pathology. Proc Natl Acad Sci USA (2019) 116(27):13490–7. doi: 10.1073/pnas.1905301116
63. Maehara T, Moriyama M, Nakashima H, Miyake K, Hayashida JN, Tanaka A, et al. Interleukin-21 contributes to germinal centre formation and immunoglobulin G4 production in IgG4-related dacryoadenitis and sialoadenitis, so-called mikulicz's disease. Ann rheum dis (2012) 71(12):2011–19. doi: 10.1136/annrheumdis-2012-201477
64. Jarrell JA, Baker MC, Perugino CA, Liu H, Bloom MS, Maehara T, et al. Neutralizing anti-IL-1 receptor antagonist autoantibodies induce inflammatory and fibrotic mediators in IgG4-related disease. J Allergy Clin Immunol (2022) 149(1):358–68. doi: 10.1016/j.jaci.2021.05.002
65. Culver EL, Vermeulen E, Makuch M, van Leeuwen A, Sadler R, Cargill T, et al. Increased IgG4 responses to multiple food and animal antigens indicate a polyclonal expansion and differentiation of pre-existing b cells in IgG4-related disease. Ann rheum dis (2015) 74(5):944–7. doi: 10.1136/annrheumdis-2014-206405
66. Perugino CA, Kaneko N, Maehara T, Mattoo H, Kers J, Allard-Chamard H, et al. CD4(+) and CD8(+) cytotoxic T lymphocytes may induce mesenchymal cell apoptosis in IgG4-related disease. J Allergy Clin Immunol (2021) 147(1):368–82. doi: 10.1016/j.jaci.2020.05.022
67. Schuijs MJ, Hammad H, Lambrecht BN. Professional and 'Amateur' antigen-presenting cells in type 2 immunity. Trends Immunol (2019) 40(1):22–34. doi: 10.1016/j.it.2018.11.001
68. Kotsias F, Cebrian I, Alloatti A. Antigen processing and presentation. Int Rev Cell Mol Biol (2019) 348:69–121. doi: 10.1016/bs.ircmb.2019.07.005
69. Brownlie RJ, Zamoyska R. T Cell receptor signalling networks: branched, diversified and bounded. Nat Rev Immunol (2013) 13(4):257–69. doi: 10.1038/nri3403
70. Winau F, Hegasy G, Weiskirchen R, Weber S, Cassan C, Sieling PA, et al. Ito Cells are liver-resident antigen-presenting cells for activating T cell responses. Immunity (2007) 26(1):117–29. doi: 10.1016/j.immuni.2006.11.011
71. Kato H, Endres J, Fox DA. The roles of IFN-gamma versus IL-17 in pathogenic effects of human Th17 cells on synovial fibroblasts. Mod Rheumatol (2013) 23(6):1140–50. doi: 10.3109/s10165-012-0811-x
72. Costantino CM, Spooner E, Ploegh HL, Hafler DA. Class II MHC self-antigen presentation in human b and T lymphocytes. PLoS One (2012) 7(1):e29805. doi: 10.1371/journal.pone.0029805
73. Kambayashi T, Laufer TM. Atypical MHC class II-expressing antigen-presenting cells: can anything replace a dendritic cell? Nat Rev Immunol (2014) 14(11):719–30. doi: 10.1038/nri3754
74. Lin W, Jin L, Chen H, Wu Q, Fei Y, Zheng W, et al. B cell subsets and dysfunction of regulatory b cells in IgG4-related diseases and primary sjogren's syndrome: the similarities and differences. Arthritis Res Ther (2014) 16(3):R118. doi: 10.1186/ar4571
75. Wallwork R, Perugino CA, Fu X, Harkness T, Zhang Y, Choi HK, et al. The association of smoking with immunoglobulin G4-related disease: a case-control study. Rheumatol (Oxford) (2021) 60(11):5310–7. doi: 10.1093/rheumatology/keab172
76. Zhou F. Molecular mechanisms of IFN-gamma to up-regulate MHC class I antigen processing and presentation. Int Rev Immunol (2009) 28(3-4):239–60. doi: 10.1080/08830180902978120
77. Thomas HE, Parker JL, Schreiber RD, Kay TW. IFN-gamma action on pancreatic beta cells causes class I MHC upregulation but not diabetes. J Clin Invest (1998) 102(6):1249–57. doi: 10.1172/JCI2899
78. Weyand CM, Goronzy JJ. The immunology of rheumatoid arthritis. Nat Immunol (2021) 22(1):10–8. doi: 10.1038/s41590-020-00816-x
79. Tsokos GC, Lo MS, Costa Reis P, Sullivan KE. New insights into the immunopathogenesis of systemic lupus erythematosus. Nat Rev Rheumatol (2016) 12(12):716–30. doi: 10.1038/nrrheum.2016.186
80. Kania G, Rudnik M, Distler O. Involvement of the myeloid cell compartment in fibrogenesis and systemic sclerosis. Nat Rev Rheumatol (2019) 15(5):288–302. doi: 10.1038/s41584-019-0212-z
81. Shiokawa M, Kodama Y, Kuriyama K, Yoshimura K, Tomono T, Morita T, et al. Pathogenicity of IgG in patients with IgG4-related disease. Gut (2016) 65(8):1322–32. doi: 10.1136/gutjnl-2015-310336
82. Yamada K, Zuka M, Ito K, Mizuguchi K, Kakuchi Y, Onoe T, et al. LatY136F knock-in mouse model for human IgG4-related disease. PLoS One (2018) 13(6):e0198417. doi: 10.1371/journal.pone.0198417
83. Valenzi E, Bulik M, Tabib T, Morse C, Sembrat J, Trejo Bittar H, et al. Single-cell analysis reveals fibroblast heterogeneity and myofibroblasts in systemic sclerosis-associated interstitial lung disease. Ann rheum dis (2019) 78(10):1379–87. doi: 10.1136/annrheumdis-2018-214865
84. Der E, Suryawanshi H, Morozov P, Kustagi M, Goilav B, Ranabothu S, et al. Tubular cell and keratinocyte single-cell transcriptomics applied to lupus nephritis reveal type I IFN and fibrosis relevant pathways. Nat Immunol (2019) 20(7):915–27. doi: 10.1038/s41590-019-0386-1
85. Soyfoo MS, Nicaise C. Pathophysiologic role of interleukin-33/ST2 in sjogren's syndrome. Autoimmun Rev (2021) 20(3):102756. doi: 10.1016/j.autrev.2021.102756
Keywords: IgG4-related disease, non-lymphocyte, immunoinflammatory regulation, ectopic lymphoid follicles, atypical antigen presentation
Citation: Cai S, Hu Z, Chen Y, Zhong J and Dong L (2022) Potential roles of non-lymphocytic cells in the pathogenesis of IgG4-related disease. Front. Immunol. 13:940581. doi: 10.3389/fimmu.2022.940581
Received: 10 May 2022; Accepted: 04 July 2022;
Published: 28 July 2022.
Edited by:
Shengjun Wang, Jiangsu University Affiliated People’s Hospital, ChinaReviewed by:
Hidenori Ohnishi, Gifu University, JapanSabina Islam, Massachusetts General Hospital and Harvard Medical School, United States
Copyright © 2022 Cai, Hu, Chen, Zhong and Dong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lingli Dong, dGpoZG9uZ2xsQDE2My5jb20=; Jixin Zhong, emhvbmdqaXhpbjYyMEAxNjMuY29t; Yu Chen, Y2hlbnl1MDcyMEAxMjYuY29t
†These authors have contributed equally to this work and share first authorship