
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 15 September 2022
Sec. Autoimmune and Autoinflammatory Disorders
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.940122
This article is part of the Research TopicA year in review: Discussions in Autoimmune and Autoinflammatory DisordersView all 12 articles
 Esther Guadalupe Corona-Sanchez1,2,3†
Esther Guadalupe Corona-Sanchez1,2,3† Erika Aurora Martínez-García1,2,3†
Erika Aurora Martínez-García1,2,3† Andrea Verónica Lujano-Benítez1,4
Andrea Verónica Lujano-Benítez1,4 Oscar Pizano-Martinez1,3,5
Oscar Pizano-Martinez1,3,5 Ivette Alejandra Guerra-Durán1
Ivette Alejandra Guerra-Durán1 Efrain Chavarria-Avila1,6
Efrain Chavarria-Avila1,6 Andrea Aguilar-Vazquez1,4
Andrea Aguilar-Vazquez1,4 Beatriz Teresita Martín-Márquez1,3
Beatriz Teresita Martín-Márquez1,3 Kevin Javier Arellano-Arteaga7
Kevin Javier Arellano-Arteaga7 Juan Armendariz-Borunda8,9
Juan Armendariz-Borunda8,9 Felipe Perez-Vazquez6
Felipe Perez-Vazquez6 Ignacio García-De la Torre10
Ignacio García-De la Torre10 Arcelia Llamas-García11
Arcelia Llamas-García11 Brenda Lucía Palacios-Zárate11
Brenda Lucía Palacios-Zárate11 Guillermo Toriz-González12
Guillermo Toriz-González12 Monica Vazquez-Del Mercado1,3,11*
Monica Vazquez-Del Mercado1,3,11*Idiopathic inflammatory myopathies (IIMs) are a group of rare, acquired autoimmune diseases characterized by profound muscle weakness and immune cell invasion into non-necrotic muscle. They are related to the presence of antibodies known as myositis-specific antibodies and myositis-associated antibodies, which are associated with various IIM phenotypes and the clinical prognosis. The possibility of the participation of other pathological mechanisms involved in the inflammatory response in IIM has been proposed. Such mechanisms include the overexpression of major histocompatibility complex class I in myofibers, which correlates with the activation of stress responses of the endoplasmic reticulum (ER). Taking into account the importance of the ER for the maintenance of homeostasis of the musculoskeletal system in the regulation of proteins, there is probably a relationship between immunological and non-immunological processes and autoimmunity, and an example of this might be IIM. We propose that ER stress and its relief mechanisms could be related to inflammatory mechanisms triggering a humoral response in IIM, suggesting that ER stress might be related to the triggering of IIMs and their auto-antibodies’ production.
Idiopathic inflammatory myopathies (IIMs), also known as myositis, are a group of conditions characterized by chronic inflammation of the musculoskeletal system that leads to proximal or distal muscle weakness, although other organs such as the skin, joints, heart, lungs, and gastrointestinal tract can also be affected (1). The IIM pathogenesis includes genetic, environmental, and immune factors (2, 3). To date, the immunopathological mechanisms of this group of conditions remain incompletely understood; however, they are related to inflammatory responses characterized by the infiltration of T- and B-cells in muscle tissue, the presence of myositis-specific antibodies (MSAs) myositis-associated auto-antibodies (MAAs), and ubiquitous abnormal overexpression of major histocompatibility complex class I (MHC-I) in myofibers (2) (Figure 1A). However, one of the clinical observations is that the level of muscle inflammation does not correspond to the severity of the disease or the alterations in muscle fibers in patients with IIM, so non-immunological mechanisms are involved (4). Among the non-immunological mechanisms involved in the pathogenesis of IIM are endoplasmic reticulum (ER) stress and the responses that avoid or relieve this stress, such as the unfolded protein response (UPR), ER-associated protein degradation (ERAD), and autophagy (Figure 1B). Specifically, the UPR increases the capacity of the ER to fold proteins efficiently and attenuates the general translation of proteins to reduce the load on the ER, while proteins that cannot be repaired are removed by ERAD and autophagy (5). It is important to emphasize that the ER is very sensitive to challenges that can compromise its structure, integrity, and function; such challenges include calcium (Ca2+) depletion, protein glycosylation, disulfide-bond formation, hypoxia, redox conditions, and viral infection, which can result in unfolded or misfolded protein accumulation, generating ER stress that triggers an inflammatory response (6–8) (Figure 1A). Currently, it is known that ER stress is involved in the pathogenesis of different diseases, such as obesity, diabetes, atherosclerosis, inflammatory bowel disease, Alzheimer’s disease, breast cancer, rheumatoid arthritis, Sjögren’s syndrome, and myopathies, among others (9, 10). This review will focus on current state-of-the-art research seeking to understand ER stress, focusing on UPR, ERAD, and autophagy as trigger factors in the IIM clinical phenotype pathogenesis and the possible link with MSAs.
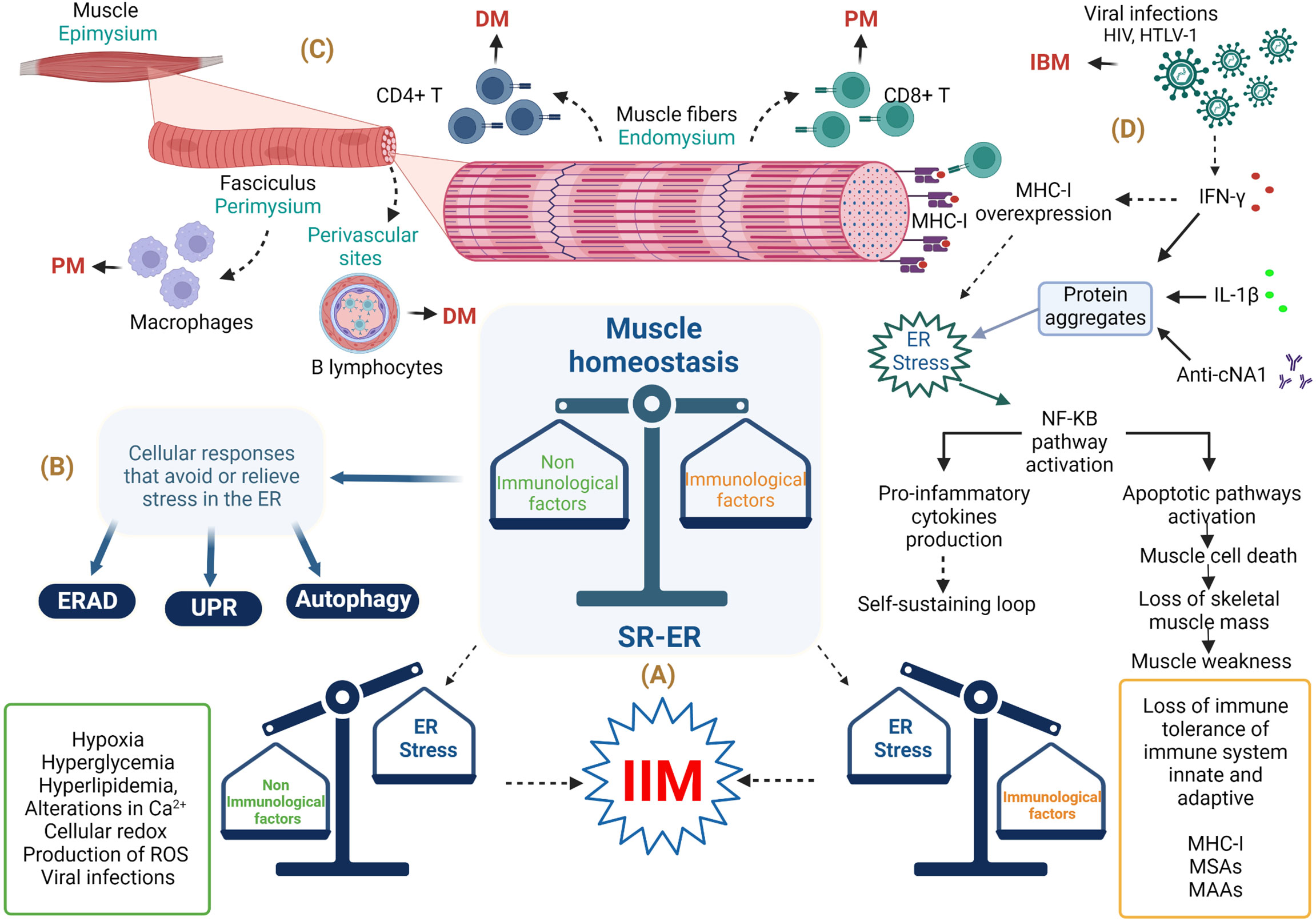
Figure 1 Involvement of endoplasmic reticulum stress in the musculoskeletal system in idiopathic inflammatory myopathies. (A) Immunopathological and non-immunopathological factors with possible repercussions on homeostasis of the musculoskeletal system. (B) ER stress relief mechanisms. (C) Histological importance in the diagnosis of IIM. (D) Involvement of immune and non-immune responses in IBM. Abbreviations: anti-cN1A, anti-cytosolic 5’-nucleotidase 1A antibodies; DM, dermatomyositis; ER, endoplasmic reticulum; ERAD, endoplasmic reticulum–associated protein degradation; HIV, human immunodeficiency virus; HTLV-1, human T lymphotropic virus; IBM, inclusion body myositis; IFN-γ, interferon gamma; IIM, idiopathic inflammatory myopathy; IL-1β, interleukin-1 beta; MHC-1, major histocompatibility complex class I; MSAs, myositis-specific antibodies; MAAs, myositis-associated auto-antibodies; NF-KB, nuclear factor κB; PM, polymyositis; ROS, reactive oxygen species; SR-ER, sarcoplasmic reticulum–endoplasmic reticulum; UPR, unfolded protein response. The figure was created with BioRender.com (agreement no. ZO24BHA4GB).
In 2017, the European League Against Rheumatism and the American College of Rheumatology (EULAR/ACR) published the most recent criteria for myositis classification for adult and juvenile IIM, which covered the following conditions: dermatomyositis (DM), amyopathic DM (ADM), juvenile DM (JDM), polymyositis (PM), immune-mediated necrotizing myopathy (IMNM), juvenile myositis (JM), and inclusion body myositis (IBM) (11). The clinical characteristics of IIM are proximal and distal muscle weakness, fatigue, fever, cutaneous features including pathognomonic rashes in DM (heliotrope, Gottron’s sign), dysphagia, increased serum muscle enzyme levels (of, e.g., aspartate aminotransferase, alanine aminotransferase, lactate dehydrogenase, and aldolase), muscle biopsy with histopathological features related to inflammation in the perimysium or perivascular areas, necrotic fibers between other characteristics, detection of conduction abnormalities by electromyography, and the presence of MSAs and MAAs (12). MSAs are used as a diagnostic tool in IIM; such auto-antibodies include anti-aminoacyl tRNA synthetase antibodies (anti-ARS), anti-nucleosome remodeling deacetylase antibodies (anti–Mi-2), anti-melanoma differentiation–associated protein 5 (anti-MDA5/CADM140), anti-nuclear matrix protein (anti-MJ/NXP2), anti-transcription intermediary factor-1 γ/α (anti-TIF-1 γ/α or p155/140, anti-hydroxymethylglutaryl coenzyme A reductase (anti-HMGCR), anti-small ubiquitin-like modifier activating (anti-SAE), anti-cytosolic 5’-nucleotidase 1A (anti-cN1A), and anti-signal recognition particle (anti-SRP) (13, 14) (Table 1). However, the anti-histidyl tRNA synthetase antibody (anti–Jo-1), an anti-ARS auto-antibody, is considered only by the EULAR/ACR 2017 guideline as a criterion for IIM classification (15). IIM treatment aims to reduce muscle inflammation and improve extra muscular manifestations, allowing patients to have a better quality of life, and it consists of high doses of oral glucocorticoids as initial treatment in combination with other conventional disease-modifying anti-rheumatic drugs or biological therapy (1).

Table 1 Stratification of Idiopathic Inflammatory Myopathies patients according to myositis-specific antibodies.
This phenotype is characterized by the presence of heliotrope, a pathognomonic cutaneous manifestation that involves the presence of a bilateral and symmetrical edema with violaceous coloration in the upper eyelids (16) as well as the presence of other cutaneous rashes, including Gottron’s sign, V sign, and Shawl’s sign (17). Histopathology shows predominant CD4+/T-cell perivascular infiltration (18). In classical adult DM, the enzyme muscle level rises significantly, but it usually carries a good clinical prognosis. This phenotype of IIM has been associated with the presence of anti–Mi-2 and anti-MJ/NXP2 (1, 13).
These patients present the same cutaneous manifestations observed in classic DM; however, there is no clinical or laboratory evidence of muscle disease (19). This phenotype of IIM has been broadly associated with anti-MDA5/CADM140 auto-antibodies and the development of rapidly progressive interstitial lung disease (13).
This DM variant includes the same features as those of classic DM; however, it develops during childhood or youth. This phenotype has a greater association with calcinosis, pericarditis, and gastrointestinal ulcers (20, 21).
PM is observed as a kind of proximal muscle weakness without dermatologic manifestation found usually in the adult population. Histopathology shows predominant CD8+/T-cell perivascular infiltration and upregulation of MHC-I molecules in muscle biopsy (18). PM patients are usually seronegative for MSAs or MAAs, and they are now considered a rare IIM subgroup; in a study of a U.K. IIM cohort of 255 patients, PM was diagnosed in 37/255 (14.5%) patients, but reclassification using the EULAR/ACR 2017 criteria led to the final diagnosis of only 9/255 (3.5%) patients, with the rest meeting criteria for IMNM, DM, overlap syndromes, etc. (22). This phenotype of IIM might be associated with anti-synthetase syndrome, and patients are usually positive for anti-ARS auto-antibodies, especially anti–Jo-1 (13, 23).
The necrotizing terminology in this context is used to describe muscle fiber necrosis with minimal leukocyte infiltrates, which could be due to genetic causes or statin-induced (24). The muscular weakness found in these patients progresses more rapidly and tends to be more severe. Another important features are the serum levels of creatine phosphokinase, which are remarkably high in this phenotype of IIM compared to other types of myositis, and the presence of anti-SRP or anti-HMGCR auto-antibodies (25, 26).
This myositis phenotype is diagnosed according to the EULAR/ACR 2017 guideline when the first symptom occurs at <18 years of age and there are no cutaneous manifestations (the major differential feature of JDM) (15).
IBM patients experience a slow progression of skeletal–muscle disease, usually after 50 years of age, with a higher prevalence in men and a predominance of distal muscle weakness. In addition, dysphagia occurs in >50% of IBM patients (1). Serum detection of anti-cN1A is highly specific for IBM (90%–95%) compared to DM or PM (5%–10%); however; this auto-antibody has also been detected in patients with systemic lupus erythematosus (0%–20%) and Sjögren’s syndrome (0%–36%) (27).
The common denominator in all IIMs is muscle inflammation (28). Muscle fibers have a cylindrical structure and consist of intercalating thick and thin filaments (myofilaments) that are organized longitudinally in sarcomeres that allow the contraction of muscle fibers (29). In particular, muscle fibers are arranged in three layers of connective tissue, as follows (a) the endomysium, which surrounds the muscle fibers; (b) the perimysium; and (c) the epimysium, which is composed of fasciculi surrounding the entire muscle (30). There are no conclusive data on the pathophysiological mechanisms related to muscle inflammation in IIM, where genetic factors such as human leukocyte antigen classes I and II (HLA-I and HLA-II) and environmental factors such as ultraviolet radiation and viral infections play a role (31–33). However, these do not fully explain the triggering of IIM, so the participation of immunopathological mechanisms related to the innate and adaptive immune systems are probably linked to IIM development (1). Nevertheless, as we have mentioned in previous paragraphs, the level of muscle inflammation does not correspond to the severity of the disease or to alterations in muscle biopsy in IIM; therefore, these pre-suppose the contribution of non-immunological mechanisms such as ER stress and altered proteins responses (UPR, ERAD, and autophagy). Based on this information, it is impossible to rule out the idea that the immunological mechanisms do not have repercussions on the non-immunological ones.
The involvement of both the innate and the adaptive immune systems has been reflected in histopathological evidence characterized by the infiltration of inflammatory cells in the muscle and skin, microvasculature affection, auto-antibody production against nuclear and cytoplasmic proteins, and inflammatory responses by interferon signature (34). For example, in DM patients, a complement-mediated micro-angiopathy that affects blood vessels of muscle tissue has been described; resulting in complement activation and membrane attack complex formation that leads to perivascular inflammation, capillary necrosis, and ischemic damage of myofibers; along with pro-inflammatory cytokines, macrophages, a high percentage of CD4+ T and B lymphocytes in perivascular sites, and dendritic plasmacytoid cells (18, 35, 36). The immune response in PM is mediated by lymphocytic infiltrates with a predominance of CD8+ T lymphocytes related to perforin and granzyme release in the endomysium as well as a lower proportion of macrophages in perivascular sites; increased expression of MHC-I; the roles of interleukin (IL)-1 and interferon-γ (IFN-γ) in myotoxicity; chronic inflammation and fibrosis via the involvement of transforming growth factor-β (TGF-β); and T-cell extravasation in muscle tissue through the involvement of chemokines like C-X-C motif chemokine ligand 8 (CXCL8), C-X-C motif chemokine ligand 9 (CXCL9), C-C motif chemokine ligand 2 (CCL2), and C-C motif chemokine ligand 9 (CCL9) (18, 36, 37) (Figure 1C). It has been observed that muscle cells also express human leukocyte antigen-G (HLA-G) in IIM due to stimulation by IFN-γ, which is synthesized locally by inflammatory cells of the disease-specific immune micro-environment, and the increase of HLA-G can interfere with cytotoxic effector functions of CD8+ T- and natural killer (NK) cells (38) because this molecule is a ligand for killer cell Ig-like receptor 2DL4 (KIR2DL4, also known as CD158d), an inhibitor receptor expressed in these cells (39). The immunopathological mechanisms involved in IMNM are mainly associated with statin prescription and the presence of anti-SRP and/or anti-HMGCR auto-antibodies; further ectopic expression of the respective auto-antigens in the myofiber surface has been reported (35, 40, 41). In addition, these auto-antibodies can induce muscle atrophy incrementally in the transcription of genes related to atrophy—such as muscle atrophy F-box (MAFbx) and tripartite motif containing 63 (TRIM63)—and reactive oxygen species (ROS) and can decrease the production of anti-inflammatory cytokines such as IL-4 and IL-13 (26, 42). Particularly, IBM is considered a complex disorder involving inflammatory and cytotoxic responses mediated by CD8+ T-cells with vacuole formation, accumulation of tubulofilamentous inclusions, and cytoplasmic accumulations of amyloid filaments, which could trigger or exacerbate ER stress responses (43). The IBM pathogenesis includes mitochondrial dysfunction as reflected by high levels of differential growth factor 15 (GDF15), a mitochondrial disease marker (44). Furthermore, in myoblasts of IBM patients, reduced adenosine triphosphate production, cellular vulnerability to oxidative stress, and reduced mitochondrial size have been documented (44). The mitochondrial damage in IBM is also associated with impaired autophagy mechanisms, and abnormal autophagy causes autophagosome accumulation with vacuole formation (27).
The cell has an integrated and interconnected signaling system that avoids or relieves ER stress through processes such as UPR, ERAD, and autophagy, which are considered to be non-immunological mechanisms associated with IIM pathogenesis (45, 46). The fundamental goal of these mechanisms is to recover ER homeostasis to preserve ER functions that are important for cell survival (47). Briefly, we describe some important mechanisms to achieve this goal.
An organelle associated with skeletal muscle homeostasis is the sarcoplasmic reticulum (SR), which is part of compartmentalization in the cytoplasm of the eukaryotic cell by an endomembrane system (ES) (48–50). The functioning of the ES can be represented as a factory, where proteins assembly begins; components are then delivered to the Golgi apparatus (GA), where the protein assembly ends, and the proteins are modified, labeled, classified, and finally packaged to be sent to their destination. The cell nucleus coordinates this entire manufacturing process in communication with ES through transport vesicles that bud from the membrane (donor) to merge with the membrane of the next compartment (acceptor), giving rise to what is known as the secretory pathway (51–53). Because there are multiple actors in the production of proteins, coordination between them is crucial to ensure protein synthesis occurs with an adequate structure and function, and one of these main actors is the ER (54, 55). The ER is involved in many cellular functions such as synthesis and processing (folding, maturation, and post-translational modification) of proteins (56–58). The ER is divided into three domains that are functionally and structurally different (56). The first domain includes a nuclear envelope, which forms using inner and outer membranes, which are continuous to the nuclear pores and function as a site where the proteins of the membrane are diffused through the nucleus and cytoplasm (56, 59). The second domain represents a smooth and rough ER (with attached ribosomes) (56). The third domain involves numerous heterotypic membrane contact sites with membranes of other organelles, such as the plasma membrane ER–plasmatic membrane, which is a classic example of an heterotypic membrane contact site and the first such example described in muscle cells related to muscle contraction through a massive influx of Ca2+ (60), ER–mitochondria, ER–peroxisomes, ER–lipid droplets, and ER–Golgi (61, 62). The secretory pathway can be explained in the following steps: (a) synthesis, anchoring, and translocation of the protein to ER lumen; (b) protein folding and quality control; and (c) protein sorting (63).
When the proteins are synthesized by ribosomes attached to the ER membrane, they are translocated to the ER while their translation is in progress (co-translational manner), and these proteins will either follow the conventional secretory pathway or exist inside the ER. If free ribosomes synthesize proteins in the cytoplasm, they can be directed to different organelles, such as the nucleus, mitochondria, or peroxisomes, and can also enter the ER when their translation is complete in a process called post-translational translocation (64). The ribosomes attached to the ER membrane are responsible for synthesis in approximately 1/3 of total cellular proteins, and it is well known that this attachment is promoted by the SRP complex, which consists of 7SL RNA and six different polypeptides that are named according to their molecular mass in kilodaltons (kDa), as follows: SRP9, SRP14, SRP19, SRP54, SRP68, and SRP72 (65–68). The SRP complex recognizes a sequence of hydrophobic amino acids in the N-terminal region of the nascent protein known as a signal sequence or leader sequence through SRP54 kDa (65, 69–74). When protein translation begins in the cytoplasm, the signal sequence is exposed, and the SRP complex recognizes it through SRP19 kDa, preventing the continuation of the translation (arrest elongation) (65, 67, 75–77). Finally, the SRP complex is attached to its receptor through a heterodimer formed by SRP68 and SRP72 kDa, allowing the nascent protein–ribosome complex to be coupled to the translocation site, which is a protein channel known as translocon (74, 76, 78–80). This event permits the continuation of protein translation with the subsequent translocation of the polypeptide to the ER lumen through translocon while the ribosome is still synthesizing it or with insertion in the ER membrane (65, 67, 81–84) (Figure 2A).
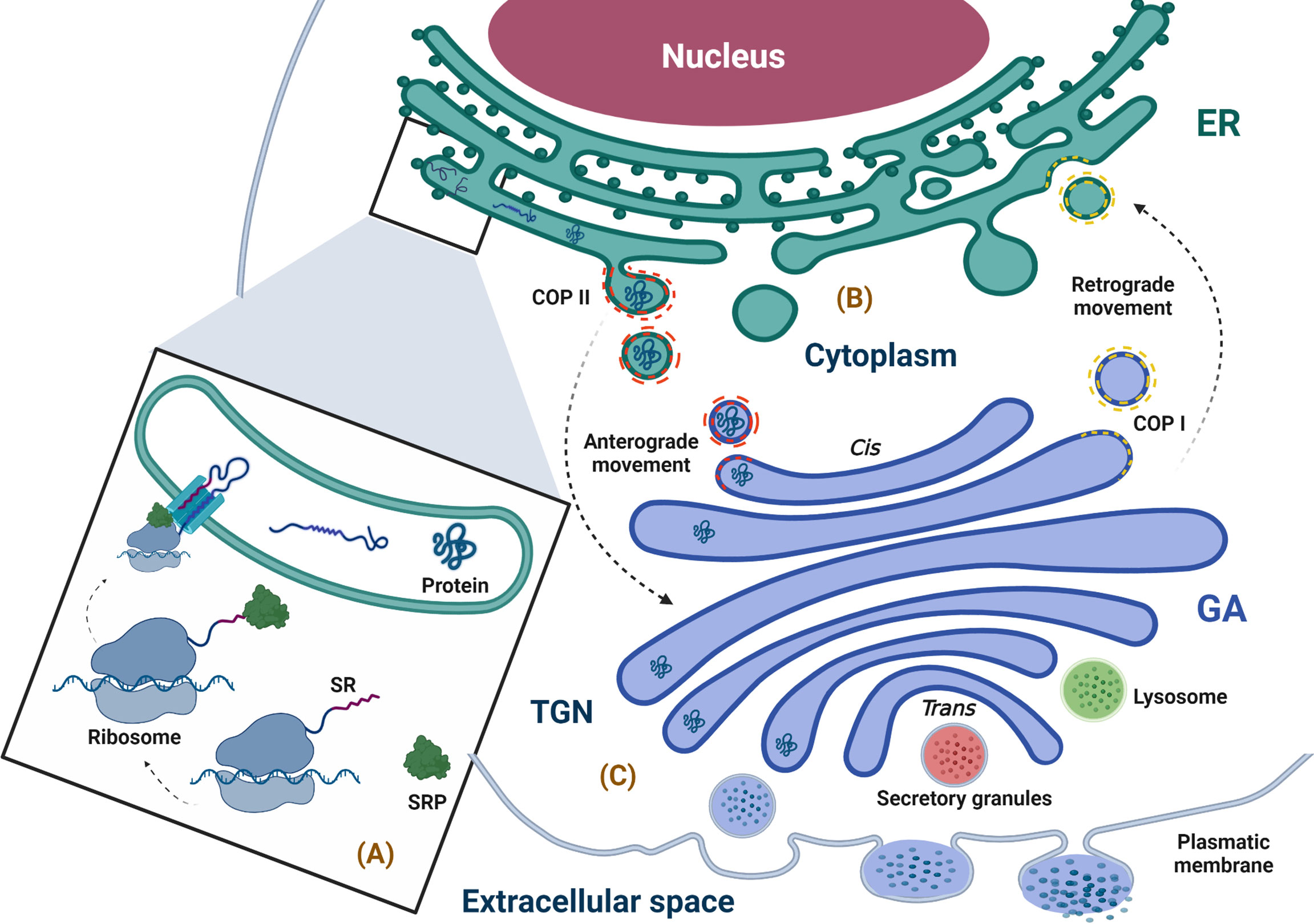
Figure 2 Physiological overview of the secretory pathway. (A) Synthesis, anchoring, and translocation of protein to the ER (ribosome binding to the ER membrane is promoted by the SRP complex, which recognizes the SR; once in the ER lumen, the ribosome–nascent protein complex, coupled to the translocon, translates the polypeptide into the lumen of the ER), (B) Protein folding and quality control (the newly synthesized proteins with correct folding and assembly continue their forward movement in COPII-coated vesicles that fuse with the GA for cargo delivery; however, the secretory pathway is bidirectional, and the circulatory pathway may stretch from the GA to ER, moving retrogradely through COPI-coated vesicles). (C) Protein sorting (when proteins arrive at the TGN, they are classified and packaged in transport vesicles to be sent to their final destination, i.e., the extracellular space). ER, endoplasmic reticulum; GA, Golgi apparatus; SR, signal recognition; SRP, signal recognition particle; TGN, trans-Golgi network. The figure was created with BioRender.com (agreement no. AM24BH3QGW).
Once in the ER, the folded protein is modified and exposed to quality control in order to achieve its native structure (proper three-dimensional conformation) through co-translational modifications such as N-glycosylation and disulfide bond formation (68, 82, 85, 86). Importantly, those proteins that undergo quality control in the ER to confirm their correct folding and assembly are packaged in a coat complex protein (COP)II–coated vesicle, which allows its transport through the secretory pathway by an anterograde movement (forward transport) to the next compartment of this pathway, i.e., the GA (48, 87). Furthermore, traffic through the secretory pathway is bidirectional; transportation from the GA to the ER is a retrograde movement allowing immature or ER-resident proteins to go back to ER through COPI-coated vesicles produced in the trans-Golgi network (TGN) and involved in traffic between GA compartments (88) (Figure 2B). In this way, the ER is not only responsible for the synthesis of proteins that follow the secretory pathway but also for their processing and maturation and is essential for shipment to its destiny (89–91).
Multiple factors assist in the folding and maturation process of newly synthesized proteins, such as ER chaperone proteins and folding enzymes (92, 93). The best-recognized ER chaperones are the glucose-regulated/immunoglobulin-binding protein of 78 kDa (BiP/GRP78), calnexin (CNX), and calreticulin (CRT) (93–101).
Within the GA, proteins continue their maturation; the N-linked glycan chains (preformed oligosaccharides) added to the peptides in the ER are structurally modified through a series of reactions that occur in a sequential manner via multiple trimming and elongation steps (102). The labeled proteins with their specific N-linked glycan chains, when they arrive to the TGN, are classified and packaged in transport vesicles to be sent to their final destination in the cellular surface, extracellular medium, or any of the compartments of the secretory pathway (53, 101, 103) (Figure 2C).
The ER is very sensitive to cellular disturbances that can disrupt the efficiency of its function, including a loss of Ca2+ homeostasis, impaired redox balance and endogenous ROS production, nutrient deprivation, virus infection, changes in protein glycosylation and autophagy defects, protein folding defects, or an increase in protein synthesis level (104). These cellular disturbances can influence protein synthesis and folding, including post-translational modifications, and lead to an accumulation of unfolded and/or misfolded proteins in the ER lumen, generating a cellular stress situation known as ER stress (6, 8, 57, 105).
The best-known response to relieve ER stress caused by the detection of unfolded and/or misfolded proteins in the ER lumen is the UPR pathway (106) (Figure 1B). UPR transduces the stress signal from the ER to the cell nucleus through three sensors, which are the transmembrane proteins inositol-requiring kinase 1 (IRE1), double-stranded RNA–activated protein kinase (PKR)-like ER kinase (PERK), and activating transcription factor 6 (ATF6) (68, 107, 108). The N-terminal luminal domain of these three sensors is responsible for detecting the accumulation of unfolded/misfolded proteins in the ER lumen (109). BiP/GRP78 is a repressor of UPR when it forms a stable complex with the luminal domain of the three sensors in unstressed cells; however, high concentrations of unfolded/misfolded proteins compete for BiP/GRP78, blocking its attachment to the three sensors (competition model) (110–114).
IRE1 is a type I transmembrane protein resident in the ER; PERK is also an ER resident type I transmembrane protein with serine/threonine kinase activity (115). Under conditions of ER stress, PERK can activate nuclear factor (NF)-κB in a manner dependent on the control of the translation mediated by the phosphorylation of eIF2α and independent of I kappa B kinase (IKK) activation, respectively (116). Finally, ATF6 is a type II transmembrane protein whose C-terminal domain is pointed toward the lumen of the ER and the N-terminal domain to the cytosol (117–119). Following stress-induced dissociation of BiP/GRP78 from ATF6 in the ER, it travels through COPII transport vesicles to the GA, where it undergoes proteolytic processing (120, 121).
ERAD is a crucial mechanism for maintaining protein quality control in the ER. It is responsible for removing misfolded proteins and redirecting them to the cytosol to be removed by the proteasome (122–124). Given the above, it is clear that the proteasome cannot process protein aggregates, so the cell has another important intracellular proteolytic system called autophagy (125).
Autophagy is an important catabolic process of cell degradation and macromolecule recycling (mainly damaged/old organelles and protein aggregates) (126). It is constitutively carried out at basal levels to maintain cellular homeostasis, but the level of autophagy can markedly increase under stress conditions as a cytoprotective response (127). Three types of autophagy are known to exist, including micro-autophagy, macro-autophagy, and chaperone-mediated autophagy, and all culminate in lysosome-mediated cargo degradation but differ in the mechanisms through which they deliver cargo to lysosomes (128). Macro-autophagy is the most studied process for the degradation of large loads, such as damaged organelles and protein aggregates; when speaking of autophagy, macro-autophagy is typically what is being referred to (129). The cellular hallmark of autophagy is the formation of double-membrane structures called autophagosomes that are responsible for transporting cargo to lysosomes (130).
To deliver cargo to the lysosome, the autophagosome fuses with the lysosome through its outer membrane, leading to the formation of an autolysosome; lysosomal enzymes first break down the inner membrane of the autophagosome to release the autophagic cargo and then break it down into its components, which are returned to the cytoplasm via the lysosomal membrane, allowing for their re-use by the cell (131). It has been reported that autophagosomes are composed of membranes from multiple sources, so the ER could be one of several sources (132).
Autophagy is mediated by a group of approximately 30 evolutionary conserved autophagy-related genes (ATGs), many of which have been detected in mammalian homologs. Among them, ATG1–10, 12–14, 16–18, 29, and 31 are essential for autophagosome formation (133). Autophagy can be selective or non-selective. Selective autophagy consists of labeling the cargo to be sequestered using ubiquitin (134). Subsequently, autophagy receptors such as ubiquitin-binding protein p62, also called sequestosome 1 (SQSTM1), recognize the marked charge and function as adapters that, by associating with certain specific proteins of the inner membrane (proteins of the ATG8 family) of the phagophore (early stage of autophagosome formation), can connect the cargo labeled with the inner membrane of the growing phagophore to allow for phagophore formation (135).
The IIM pathogenesis is associated with immunological and non-immunological mechanisms, which involve the participation of autoreactive T lymphocytes and auto-antibody production. Despite these, the literature has not explained the origin of muscle fiber damage or muscle weakness (136). To date, the most well-described example is upregulation of the expression of MHC-I in myofibers (sarcolemma surface), whose expression is not usual in normal muscle tissue (endomysial capillaries), thus stimulating the activation of ER stress responses such as an accumulation of misfolded glycoproteins and the activation of NF-κB, causing an inflammatory response via IRE1, tumor necrosis factor (TNF)-α, and TRAF2 (137, 138). It has been demonstrated that BiP/GRP78, CRT, and heat shock protein 90-α2 expression levels are augmented according to intermediate or high MHC-I expression in myositis muscle (139). In addition, elevated expression of several genes, including PERK and ATF3, in myositis, has been observed. Furthermore, the PERK signaling via transcription factor CCAAT/enhancer binding protein δ (C/EBPδ) has been associated with tumors by the action of chemokines such as CXCL8, and C-C motif chemokine ligand 20 (CCL20), which have been related to tumor-promotion with immunosuppressive properties and are triggered by activated oncogenes, nutrient deprivation, and hypoxia (140). In addition, the increased expression of PERK has been correlated with macrophage and dendritic cell infiltration into the tumor micro-environment (141). At this time, the link between IIM and cancer development is not well understood (142), although factors strongly associated with cancer development are anti-TIF-1γ and anti-MJ/NXP2 (143). In addition, our research group reported the presence of MSAs, particularly anti–TIF-1γ, in a group of women with breast cancer without clinical evidence of myositis (144). We will next address the three points of this section.
As mentioned above, the ER stress response pathway activates NF-κB. Interestingly, it has been shown that caspase-12, the main caspase associated with the apoptosis pathway, is highly expressed in mouse skeletal muscle tissues, so the activated apoptotic pathways in muscle cells could induce cell death, a loss of skeletal muscle mass, and muscle dysfunction and weakness (4).
It has also been reported that TRE-H-2Kb x mck-Tta (HT) mice with conditional overexpression of H-2Kb in muscle presented an upregulation of ER stress markers and molecular targets of UPR such as BiP/GRP78, CRT, CNX, and ATF6, in addition to pathologic features of non-specific myopathy with variations in myofiber size, numerous centronucleated fibers, and endomysial fibrosis (139). In that work, the authors also reported that an intracellular accumulation of MHC-I in the muscle of IBM patients correlated with UPR (139). Such findings suggest that MHC-I involvement in myositis is pathological because of the triggering of UPR responses instead of the facilitation of antigen presentation to CD8+ T lymphocytes. Therefore, the detection of MHC-I by immunostaining may be a diagnostic approach in IIM (145). However, although the positive detection of MHC-I has a high sensitivity for IIM, it has a low specificity, hence, it has been suggested that it can be combined with the positive detection of MHC-II (146).
In a study of our work group, we reported that recombinant human IL-1β induced phosphorylation and upregulation of SRP72 in Jurkat cells (147). Importantly, this cytokine and IL-18 are overexpressed in muscle biopsies of patients with DM and PM (148, 149). IL-1β and IL-18 are proteolytically matured by the NLRP3 inflammasome and allow the recruitment and maturation of caspase-1 (150). Another mechanism linked to the NLRP3 inflammasome is pyroptosis, a kind of cellular death, in addition to apoptosis and NETosis, which are also sources of auto-antigens related to the inflammatory response in the immunopathogenesis of autoimmune diseases such as IIM (151). In IMNM, an isolated report showed a correlation of BiP/GRP78 in muscle biopsy assessed by immunohistochemistry and serum lactate dehydrogenase along with a negative correlation with the Manual Muscle Testing–8 score (152).
Aggregates of p62/SQSTM1 protein, which normally degrade during autophagy, can be detected in the muscles of IBM and IMNM patients, suggesting dysregulation of the autophagic process of this protein (153, 154). It is known that dysregulation or excessive autophagy could lead to cell death and has been associated with several human diseases, including IBM (155). Although autophagy has not been extensively studied in other IIM phenotypes, Cappallettic et al. (156) found that, in addition to IBM, autophagy processes are also activated in PM, DM, and JDM. Girolamo et al. (157) reported a higher proportion of myofibers correlated with the presence of autophagy markers such as microtubule-associated protein light chain 3b (LC3b) and p62/SQSTM1 in muscle biopsies from patients with IMNM compared to those from patients with DM and PM.
Recent study was reported that a selected group of autophagy-related genes, such as CCL2, cyclin-dependent kinase inhibitor 1A (CDKN1A), FOS, myelocytomatosis (MYC), and TNF superfamily member 10 (TNFSF10)—whose functions are related to IFN-I signaling pathways—significantly influenced the infiltration of multiple immune cells, including B-cells, macrophages, and NK cells, in samples from DM patients compared to controls, suggesting that these genes may be potential diagnostic biomarkers for DM (158).
In addition to the detection of MHC-I by immunostaining, another diagnostic tool in IIM is the presence of MSAs. Although a direct association between MSAs production and ER stress has not been defined, some reports have touched on the subject. The SRP auto-antigen might be an example of antigen released from tissue damage (159), a target of anti-SRP antibodies (160). It has been reported that auto-antibodies against SRP54 exist in IMNM and PM patients with dilated cardiomyopathy, disease severity, and remarkably high levels of muscle enzymes (160–162). One study of DM patients documented the presence of anti-SRP72 antibodies (163). Recently, a possible association between anti-calreticulin (anti-CRT) antibodies and malignancy in IIM was reported (164).
In patients seropositive for anti-cN1A, an auto-antibody commonly associated with IBM, colocalization of the cN1A auto-antigen with p62/SQSTM1 (an autophagy-related protein considered a pathological hallmark of IBM) in perinuclear regions of myofibers was found (165). Furthermore, in vitro and in vivo passive immunization models with immunoglobulin G extracted from anti-cN1A–seropositive IBM patient serum samples have been found to exhibit higher p62/SQSTM1 expression and abundant aggregations in the cytoplasm of supplemented rhabdomyosarcoma cells. Likewise, anti-cN1A–positive IBM immunoglobulin G–injected mice showed p62/SQSTM1 aggregates in myofibers (165). Concerning the involvement of ER stress as a potent inducer of autophagy, it has been hypothesized that other MSAs, such as anti-SRP, in IMNM could alter the function of SRP in the proper elongation of polypeptide chains, the induction of ER stress, and chaperone-assisted selective autophagy of defective polypeptides (154). Interestingly, both anti-SRP and anti-HMGCR auto-antibodies might have shown a pathogenic role in vitro because they are involved in muscle fiber atrophy associated with the increase of IL-6, TNF, and ROS as well as impaired muscle regeneration by a defect of myoblast fusion due to decreased levels of IL-4 and IL-13 (166).
Inflammatory mediators such as cytokines have been reported in the muscles of IBM patients, with a correlation between the messenger RNA expression of IL-1β and amyloid precursor protein, a protein frequently observed in rimmed vacuoles associated with the IBM phenotype. Upregulation of amyloid precursor protein and β-amyloid expression in skeletal muscle after IL-1β and IFN-γ stimulation, as well as co-localization of IL-1β and β-amyloid, has also been reported (167). Additionally, IFN-γ induces ubiquitylated inclusions in a manner independent of cell type in mouse and human cells (168). Considering that viral infections, especially by human immunodeficiency virus or human T lymphotropic virus 1, are related to IBM immunopathogenesis (169), we could infer that they facilitate an autoimmune process because of the secretion of cytokines such as IFN-γ, which promotes, in turn, the MHC-I upregulation and ubiquitylated inclusions that finally trigger ER stress responses (Figure 1D).
It is interesting that protein aggregates have also been reported in other autoimmune diseases, e.g., Sjögren’s syndrome, which has also been associated with the presence of the auto-antibody anti-cN1A in up to 36% of patients (27, 170). In addition to all these mechanisms, the presence of auto-antibodies can also probably cause ER stress because in patients with lupus nephritis, the anti–double-stranded DNA antibodies (anti-dsDNA) bound to a human mesangial cell induce ER stress and activation of NF-κB via PERK/eIF2α/ATF4 (171). Finally, it is important to recall that MSAs have a pathogenic role or are an epiphenomenon in IIM (172).
Taking into account the importance of the ER for the maintenance of homeostasis of the musculoskeletal system in the regulation of proteins, there is probably a relationship between immunological, non-immunological, and infectious pathophysiological processes for the activation of ER stress and autoimmunity. An example of this might be IIMs, and, although this process is not fully understood, there are indications of the participation of cells of the immune system, auto-antibodies, viral processes, involvement of MHC-I, and cytokine signaling pathways in the activation of ER stress. However, ER stress responses have also been observed in other autoimmune diseases, as discussed above. Another special issue, which was not mentioned in this review and is important to consider in future studies, is the design of possible targeted therapies to attenuate, modulate, or eliminate ER stress in addition to the classical therapies used in IIM. Finally, it is possible to suggest that ER stress is related to the origin of autoimmune diseases and their possible consequences on auto-antibody production in IIM.
Conceived and designed the idea: EC-S, EM-G, AL-B and MV-M. Conducted the bibliographic search: EC-S, EM-G, AL-B, OP-M, IG-D, EC-A, AA-V, BM-M, KA-A, JA-B, FP-V, GT-G and MV-M. Analysis and discussion of the information: EC-S, EM-G, AL-B, OP-M, IG-D, EC-A, AA-V, BM-M, KA-A, JA-B, FP-V, IG-T, AL-G, BP-Z, GT-G and MV-M. Wrote the paper: EC-S, EM-G, AL-B, OP-M, EC-A, AA-V, and MV-M. Figure editing: EM-G, and AL-B. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Lundberg IE, Fujimoto M, Vencovsky J, Aggarwal R, Holmqvist M, Christopher-Stine L, et al. Idiopathic inflammatory myopathies. Nat Rev Dis Primers (2021) 7(1):86. doi: 10.1038/s41572-021-00321-x
2. Ceribelli A, De Santis M, Isailovic N, Gershwin ME, Selmi C. The immune response and the pathogenesis of idiopathic inflammatory myositis: A critical review. Clin Rev Allergy Immunol (2017) 52(1):58–70. doi: 10.1007/s12016-016-8527-x
3. Loredo Martinez M, Zampieri S, Franco C, Ghirardello A, Doria A, Gatto M. Nonimmune mechanisms in idiopathic inflammatory myopathies. Curr Opin Rheumatol (2020) 32(6):515–22. doi: 10.1097/BOR.0000000000000748
4. Nagaraju K, Casciola-Rosen L, Lundberg I, Rawat R, Cutting S, Thapliyal R, et al. Activation of the endoplasmic reticulum stress response in autoimmune myositis: Potential role in muscle fiber damage and dysfunction. Arthritis Rheum (2005) 52(6):1824–35. doi: 10.1002/art.21103
5. Senft D, Ronai ZA. Upr, autophagy, and mitochondria crosstalk underlies the er stress response. Trends Biochem Sci (2015) 40(3):141–8. doi: 10.1016/j.tibs.2015.01.002
6. Rao RV, Bredesen DE. Misfolded proteins, endoplasmic reticulum stress and neurodegeneration. Curr Opin Cell Biol (2004) 16(6):653–62. doi: 10.1016/j.ceb.2004.09.012
7. Prell T, Lautenschlager J, Grosskreutz J. Calcium-dependent protein folding in amyotrophic lateral sclerosis. Cell Calcium (2013) 54(2):132–43. doi: 10.1016/j.ceca.2013.05.007
8. Chaudhari N, Talwar P, Parimisetty A, Lefebvre d’Hellencourt C, Ravanan P. A molecular web: Endoplasmic reticulum stress, inflammation, and oxidative stress. Front Cell Neurosci (2014) 8:213. doi: 10.3389/fncel.2014.00213
9. Kawasaki N, Asada R, Saito A, Kanemoto S, Imaizumi K. Obesity-induced endoplasmic reticulum stress causes chronic inflammation in adipose tissue. Sci Rep (2012) 2:799. doi: 10.1038/srep00799
10. Engin F, Nguyen T, Yermalovich A, Hotamisligil GS. Aberrant islet unfolded protein response in type 2 diabetes. Sci Rep (2014) 4:4054. doi: 10.1038/srep04054
11. Lundberg IE, Tjarnlund A, Bottai M, Werth VP, Pilkington C, Visser M, et al. European League against Rheumatism/American college of rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups. Ann Rheum Dis (2017) 76(12):1955–64. doi: 10.1136/annrheumdis-2017-211468
12. Lundberg IE, Miller FW, Tjarnlund A, Bottai M. Diagnosis and classification of idiopathic inflammatory myopathies. J Intern Med (2016) 280(1):39–51. doi: 10.1111/joim.12524
13. Satoh M, Tanaka S, Ceribelli A, Calise SJ, Chan EK. A comprehensive overview on myositis-specific antibodies: New and old biomarkers in idiopathic inflammatory myopathy. Clin Rev Allergy Immunol (2017) 52(1):1–19. doi: 10.1007/s12016-015-8510-y
14. Yoo IS, Kim J. The role of autoantibodies in idiopathic inflammatory myopathies. J Rheum Dis (2019) 26(3):165–78. doi: 10.4078/jrd.2019.26.3.165
15. Lundberg IE, Tjarnlund A, Bottai M, Werth VP, Pilkington C, de Visser M, et al. European League against Rheumatism/American college of rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups. Arthritis Rheumatol (2017) 69(12):2271–82. doi: 10.1002/art.40320
16. Muro Y, Sugiura K, Akiyama M. Cutaneous manifestations in dermatomyositis: Key clinical and serological features-a comprehensive review. Clin Rev Allergy Immunol (2016) 51(3):293–302. doi: 10.1007/s12016-015-8496-5
17. Findlay AR, Goyal NA, Mozaffar T. An overview of polymyositis and dermatomyositis. Muscle Nerve (2015) 51(5):638–56. doi: 10.1002/mus.24566
18. Vattemi G, Mirabella M, Guglielmi V, Lucchini M, Tomelleri G, Ghirardello A, et al. Muscle biopsy features of idiopathic inflammatory myopathies and differential diagnosis. Auto Immun Highlights (2014) 5(3):77–85. doi: 10.1007/s13317-014-0062-2
19. Concha JSS, Tarazi M, Kushner CJ, Gaffney RG, Werth VP. The diagnosis and classification of amyopathic dermatomyositis: A historical review and assessment of existing criteria. Br J Dermatol (2019) 180(5):1001–8. doi: 10.1111/bjd.17536
20. Tansley SL, McHugh NJ, Wedderburn LR. Adult and juvenile dermatomyositis: Are the distinct clinical features explained by our current understanding of serological subgroups and pathogenic mechanisms? Arthritis Res Ther (2013) 15(2):211. doi: 10.1186/ar4198
21. Saini I, Kalaivani M, Kabra SK. Calcinosis in juvenile dermatomyositis: Frequency, risk factors and outcome. Rheumatol Int (2016) 36(7):961–5. doi: 10.1007/s00296-016-3467-6
22. Loarce-Martos J, Lilleker JB, Parker M, McHugh N, Chinoy H. Polymyositis: Is there anything left? a retrospective diagnostic review from a tertiary myositis centre. Rheumatol (Oxford) (2021) 60(7):3398–403. doi: 10.1093/rheumatology/keaa801
23. Amato AA, Griggs RC. Unicorns, dragons, polymyositis, and other mythological beasts. Neurology (2003) 61(3):288–9. doi: 10.1212/wnl.61.3.288
24. Malik A, Hayat G, Kalia JS, Guzman MA. Idiopathic inflammatory myopathies: Clinical approach and management. Front Neurol (2016) 7:64. doi: 10.3389/fneur.2016.00064
25. Mammen AL. Autoimmune myopathies: Autoantibodies, phenotypes and pathogenesis. Nat Rev Neurol (2011) 7(6):343–54. doi: 10.1038/nrneurol.2011.63
26. Allenbach Y, Benveniste O, Stenzel W, Boyer O. Immune-mediated necrotizing myopathy: Clinical features and pathogenesis. Nat Rev Rheumatol (2020) 16(12):689–701. doi: 10.1038/s41584-020-00515-9
27. Greenberg SA. Inclusion body myositis: Clinical features and pathogenesis. Nat Rev Rheumatol (2019) 15(5):257–72. doi: 10.1038/s41584-019-0186-x
28. Frontera WR, Ochala J. Skeletal muscle: A brief review of structure and function. Calcif Tissue Int (2015) 96(3):183–95. doi: 10.1007/s00223-014-9915-y
29. Rassier DE. Sarcomere mechanics in striated muscles: From molecules to sarcomeres to cells. Am J Physiol Cell Physiol (2017) 313(2):C134–C45. doi: 10.1152/ajpcell.00050.2017
30. Exeter D, Connell DA. Skeletal muscle: Functional anatomy and pathophysiology. Semin Musculoskelet Radiol (2010) 14(2):97–105. doi: 10.1055/s-0030-1253154
31. Aguilar-Vazquez A, Chavarria-Avila E, Pizano-Martinez O, Ramos-Hernandez A, Andrade-Ortega L, Rubio-Arellano ED, et al. Geographical latitude remains as an important factor for the prevalence of some myositis autoantibodies: A systematic review. Front Immunol (2021) 12:672008. doi: 10.3389/fimmu.2021.672008
32. Rothwell S, Cooper RG, Lundberg IE, Miller FW, Gregersen PK, Bowes J, et al. Dense genotyping of immune-related loci in idiopathic inflammatory myopathies confirms hla alleles as the strongest genetic risk factor and suggests different genetic background for major clinical subgroups. Ann Rheum Dis (2016) 75(8):1558–66. doi: 10.1136/annrheumdis-2015-208119
33. Adler BL, Christopher-Stine L. Triggers of inflammatory myopathy: Insights into pathogenesis. Discovery Med (2018) 25(136):75–83.
34. Hornung T, Wenzel J. Innate immune-response mechanisms in dermatomyositis: An update on pathogenesis, diagnosis and treatment. Drugs (2014) 74(9):981–98. doi: 10.1007/s40265-014-0240-6
35. Dalakas MC. Inflammatory muscle diseases. N Engl J Med (2015) 372(18):1734–47. doi: 10.1056/NEJMra1402225
36. Arahata K, Engel AG. Monoclonal antibody analysis of mononuclear cells in myopathies. I: Quantitation of subsets according to diagnosis and sites of accumulation and demonstration and counts of muscle fibers invaded by T cells. Ann Neurol (1984) 16(2):193–208. doi: 10.1002/ana.410160206
37. Dalakas MC, Hohlfeld R. Polymyositis and dermatomyositis. Lancet (2003) 362(9388):971–82. doi: 10.1016/S0140-6736(03)14368-1
38. Wiendl H, Mitsdoerffer M, Weller M. Express and protect yourself: The potential role of hla-G on muscle cells and in inflammatory myopathies. Hum Immunol (2003) 64(11):1050–6. doi: 10.1016/j.humimm.2003.07.001
39. Lanier LL. Natural killer cells fertile with receptors for hla-G? Proc Natl Acad Sci U.S.A. (1999) 96(10):5343–5. doi: 10.1073/pnas.96.10.5343
40. Mohassel P, Mammen AL. Anti-hmgcr myopathy. J Neuromuscul Dis (2018) 5(1):11–20. doi: 10.3233/JND-170282
41. Mammen AL, Chung T, Christopher-Stine L, Rosen P, Rosen A, Doering KR, et al. Autoantibodies against 3-Hydroxy-3-Methylglutaryl-Coenzyme a reductase in patients with statin-associated autoimmune myopathy. Arthritis Rheum (2011) 63(3):713–21. doi: 10.1002/art.30156
42. Pinal-Fernandez I, Casal-Dominguez M, Mammen AL. Immune-mediated necrotizing myopathy. Curr Rheumatol Rep (2018) 20(4):21. doi: 10.1007/s11926-018-0732-6
43. Dalakas MC. Mechanisms of disease: Signaling pathways and immunobiology of inflammatory myopathies. Nat Clin Pract Rheumatol (2006) 2(4):219–27. doi: 10.1038/ncprheum0140
44. Oikawa Y, Izumi R, Koide M, Hagiwara Y, Kanzaki M, Suzuki N, et al. Mitochondrial dysfunction underlying sporadic inclusion body myositis is ameliorated by the mitochondrial homing drug ma-5. PloS One (2020) 15(12):e0231064. doi: 10.1371/journal.pone.0231064
45. Chipurupalli S, Samavedam U, Robinson N. Crosstalk between er stress, autophagy and inflammation. Front Med (Lausanne) (2021) 8:758311. doi: 10.3389/fmed.2021.758311
46. Coley W, Rayavarapu S, Nagaraju K. Role of non-immune mechanisms of muscle damage in idiopathic inflammatory myopathies. Arthritis Res Ther (2012) 14(2):209. doi: 10.1186/ar3791
47. Adams CJ, Kopp MC, Larburu N, Nowak PR, Ali MMU. Structure and molecular mechanism of er stress signaling by the unfolded protein response signal activator Ire1. Front Mol Biosci (2019) 6:11. doi: 10.3389/fmolb.2019.00011
48. Gomez-Navarro N, Miller E. Protein sorting at the er-golgi interface. J Cell Biol (2016) 215(6):769–78. doi: 10.1083/jcb.201610031
49. Simpson JC. Modification of the mammalian endomembrane system in healthy and diseased cells. Int J Mol Sci (2020) 21(6):1–4. doi: 10.3390/ijms21062133
50. Rayavarapu S, Coley W, Nagaraju K. Endoplasmic reticulum stress in skeletal muscle homeostasis and disease. Curr Rheumatol Rep (2012) 14(3):238–43. doi: 10.1007/s11926-012-0247-5
51. Bonifacino JS, Glick BS. The mechanisms of vesicle budding and fusion. Cell (2004) 116(2):153–66. doi: 10.1016/s0092-8674(03)01079-1
52. Brandizzi F, Barlowe C. Organization of the er-golgi interface for membrane traffic control. Nat Rev Mol Cell Biol (2013) 14(6):382–92. doi: 10.1038/nrm3588
53. Witkos TM, Lowe M. Recognition and tethering of transport vesicles at the golgi apparatus. Curr Opin Cell Biol (2017) 47:16–23. doi: 10.1016/j.ceb.2017.02.003
54. Sorrentino V. Molecular determinants of the structural and functional organization of the sarcoplasmic reticulum. Biochim Biophys Acta (2004) 1742(1-3):113–8. doi: 10.1016/j.bbamcr.2004.08.016
55. Friedman JR, Voeltz GK. The er in 3d: A multifunctional dynamic membrane network. Trends Cell Biol (2011) 21(12):709–17. doi: 10.1016/j.tcb.2011.07.004
56. Voeltz GK, Rolls MM, Rapoport TA. Structural organization of the endoplasmic reticulum. EMBO Rep (2002) 3(10):944–50. doi: 10.1093/embo-reports/kvf202
57. Cao T, Peng B, Zhou X, Cai J, Tang Y, Luo J, et al. Integrated signaling system under endoplasmic reticulum stress in eukaryotic microorganisms. Appl Microbiol Biotechnol (2021) 105(12):4805–18. doi: 10.1007/s00253-021-11380-1
58. Unanue ER, Urano F. Endoplasmic reticulum: An interface between the immune system and metabolism. Diabetes (2014) 63(1):48–9. doi: 10.2337/db13-1478
59. English AR, Voeltz GK. Endoplasmic reticulum structure and interconnections with other organelles. Cold Spring Harb Perspect Biol (2013) 5(4):a013227. doi: 10.1101/cshperspect.a013227
60. Helle SC, Kanfer G, Kolar K, Lang A, Michel AH, Kornmann B. Organization and function of membrane contact sites. Biochim Biophys Acta (2013) 1833(11):2526–41. doi: 10.1016/j.bbamcr.2013.01.028
61. Li C, Qian T, He R, Wan C, Liu Y, Yu H. Endoplasmic reticulum-plasma membrane contact sites: Regulators, mechanisms, and physiological functions. Front Cell Dev Biol (2021) 9:627700: doi: 10.3389/fcell.2021.627700
62. Gallo A, Vannier C, Galli T. Endoplasmic reticulum-plasma membrane associations : Structures and functions. Annu Rev Cell Dev Biol (2016) 32:279–301. doi: 10.1146/annurev-cellbio-111315-125024
63. Hebert DN, Molinari M. In and out of the er: Protein folding, quality control, degradation, and related human diseases. Physiol Rev (2007) 87(4):1377–408. doi: 10.1152/physrev.00050.2006
64. Tirincsi A, Sicking M, Hadzibeganovic D, Hassdenteufel S, Lang S. The molecular biodiversity of protein targeting and protein transport related to the endoplasmic reticulum. Int J Mol Sci (2021) 23(1):1–49. doi: 10.3390/ijms23010143
65. Nyathi Y, Wilkinson BM, Pool MR. Co-Translational targeting and translocation of proteins to the endoplasmic reticulum. Biochim Biophys Acta (2013) 1833(11):2392–402. doi: 10.1016/j.bbamcr.2013.02.021
66. Walter P, Lingappa VR. Mechanism of protein translocation across the endoplasmic reticulum membrane. Annu Rev Cell Biol (1986) 2:499–516. doi: 10.1146/annurev.cb.02.110186.002435
67. Kaufman RJ. Regulation of mrna translation by protein folding in the endoplasmic reticulum. Trends Biochem Sci (2004) 29(3):152–8. doi: 10.1016/j.tibs.2004.01.004
68. Schwarz DS, Blower MD. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell Mol Life Sci (2016) 73(1):79–94. doi: 10.1007/s00018-015-2052-6
69. Kurzchalia TV, Wiedmann M, Girshovich AS, Bochkareva ES, Bielka H, Rapoport TA. The signal sequence of nascent preprolactin interacts with the 54k polypeptide of the signal recognition particle. Nature (1986) 320(6063):634–6. doi: 10.1038/320634a0
70. Krieg UC, Walter P, Johnson AE. Photocrosslinking of the signal sequence of nascent preprolactin to the 54-kilodalton polypeptide of the signal recognition particle. Proc Natl Acad Sci U.S.A. (1986) 83(22):8604–8. doi: 10.1073/pnas.83.22.8604
71. Wild K, Juaire KD, Soni K, Shanmuganathan V, Hendricks A, Segnitz B, et al. Reconstitution of the human srp system and quantitative and systematic analysis of its ribosome interactions. Nucleic Acids Res (2019) 47(6):3184–96. doi: 10.1093/nar/gky1324
72. Walter P, Blobel G. Translocation of proteins across the endoplasmic reticulum. ii. signal recognition protein (Srp) mediates the selective binding to microsomal membranes of in-Vitro-Assembled polysomes synthesizing secretory protein. J Cell Biol (1981) 91(2 Pt 1):551–6. doi: 10.1083/jcb.91.2.551
73. Rapoport TA. Transport of proteins across the endoplasmic reticulum membrane. Science (1992) 258(5084):931–6. doi: 10.1126/science.1332192
74. Dudek J, Pfeffer S, Lee PH, Jung M, Cavalie A, Helms V, et al. Protein transport into the human endoplasmic reticulum. J Mol Biol (2015) 427(6 Pt A):1159–75. doi: 10.1016/j.jmb.2014.06.011
75. Walter P, Blobel G. Translocation of proteins across the endoplasmic reticulum iii. signal recognition protein (Srp) causes signal sequence-dependent and site-specific arrest of chain elongation that is released by microsomal membranes. J Cell Biol (1981) 91(2 Pt 1):557–61. doi: 10.1083/jcb.91.2.557
76. Walter P, Johnson AE. Signal sequence recognition and protein targeting to the endoplasmic reticulum membrane. Annu Rev Cell Biol (1994) 10:87–119. doi: 10.1146/annurev.cb.10.110194.000511
77. Mary C, Scherrer A, Huck L, Lakkaraju AK, Thomas Y, Johnson AE, et al. Residues in Srp9/14 essential for elongation arrest activity of the signal recognition particle define a positively charged functional domain on one side of the protein. RNA (2010) 16(5):969–79. doi: 10.1261/rna.2040410
78. Gilmore R, Blobel G, Walter P. Protein translocation across the endoplasmic reticulum. i. detection in the microsomal membrane of a receptor for the signal recognition particle. J Cell Biol (1982) 95(2 Pt 1):463–9. doi: 10.1083/jcb.95.2.463
79. Meyer DI, Krause E, Dobberstein B. Secretory protein translocation across membranes-the role of the “Docking protein’. Nature (1982) 297(5868):647–50. doi: 10.1038/297647a0
80. Agarraberes FA, Dice JF. Protein translocation across membranes. Biochim Biophys Acta (2001) 1513(1):1–24. doi: 10.1016/s0304-4157(01)00005-3
81. Saraogi I, Shan SO. Molecular mechanism of Co-translational protein targeting by the signal recognition particle. Traffic (2011) 12(5):535–42. doi: 10.1111/j.1600-0854.2011.01171.x
82. Hetz C, Papa FR. The unfolded protein response and cell fate control. Mol Cell (2018) 69(2):169–81. doi: 10.1016/j.molcel.2017.06.017
83. Johnson AE, van Waes MA. The translocon: A dynamic gateway at the er membrane. Annu Rev Cell Dev Biol (1999) 15:799–842. doi: 10.1146/annurev.cellbio.15.1.799
84. Wang M, Kaufman RJ. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature (2016) 529(7586):326–35. doi: 10.1038/nature17041
85. Braakman I, Hebert DN. Protein folding in the endoplasmic reticulum. Cold spring. Harb Perspect Biol (2013) 5(5):a013201. doi: 10.1101/cshperspect.a013201
86. Ruggiano A, Foresti O, Carvalho P. Quality control: Er-associated degradation: Protein quality control and beyond. J Cell Biol (2014) 204(6):869–79. doi: 10.1083/jcb.201312042
87. Baumann O, Walz B. Endoplasmic reticulum of animal cells and its organization into structural and functional domains. Int Rev Cytol (2001) 205:149–214. doi: 10.1016/s0074-7696(01)05004-5
88. Adolf F, Rhiel M, Hessling B, Gao Q, Hellwig A, Bethune J, et al. Proteomic profiling of mammalian copii and copi vesicles. Cell Rep (2019) 26(1):250–65 e5. doi: 10.1016/j.celrep.2018.12.041
89. Helenius A, Marquardt T, Braakman I. The endoplasmic reticulum as a protein-folding compartment. Trends Cell Biol (1992) 2(8):227–31. doi: 10.1016/0962-8924(92)90309-b
90. Kleizen B, Braakman I. Protein folding and quality control in the endoplasmic reticulum. Curr Opin Cell Biol (2004) 16(4):343–9. doi: 10.1016/j.ceb.2004.06.012
91. Csala M, Kereszturi E, Mandl J, Banhegyi G. The endoplasmic reticulum as the extracellular space inside the cell: Role in protein folding and glycosylation. Antioxid Redox Signal (2012) 16(10):1100–8. doi: 10.1089/ars.2011.4227
92. Ellgaard L, Helenius A. Er quality control: Towards an understanding at the molecular level. Curr Opin Cell Biol (2001) 13(4):431–7. doi: 10.1016/s0955-0674(00)00233-7
93. Hartl FU, Bracher A, Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature (2011) 475(7356):324–32. doi: 10.1038/nature10317
94. Ellgaard L, McCaul N, Chatsisvili A, Braakman I. Co- and post-translational protein folding in the er. Traffic (2016) 17(6):615–38. doi: 10.1111/tra.12392
95. Hendershot L, Wei J, Gaut J, Melnick J, Aviel S, Argon Y. Inhibition of immunoglobulin folding and secretion by dominant negative bip atpase mutants. Proc Natl Acad Sci U.S.A. (1996) 93(11):5269–74. doi: 10.1073/pnas.93.11.5269
96. Lee YK, Brewer JW, Hellman R, Hendershot LM. Bip and immunoglobulin light chain cooperate to control the folding of heavy chain and ensure the fidelity of immunoglobulin assembly. Mol Biol Cell (1999) 10(7):2209–19. doi: 10.1091/mbc.10.7.2209
97. Balchin D, Hayer-Hartl M, Hartl FU. In vivo aspects of protein folding and quality control. Science (2016) 353(6294):aac4354. doi: 10.1126/science.aac4354
98. Helenius A. How n-linked oligosaccharides affect glycoprotein folding in the endoplasmic reticulum. Mol Biol Cell (1994) 5(3):253–65. doi: 10.1091/mbc.5.3.253
99. Gething MJ. Role and regulation of the er chaperone bip. Semin Cell Dev Biol (1999) 10(5):465–72. doi: 10.1006/scdb.1999.0318
100. Michalak M, Groenendyk J, Szabo E, Gold LI, Opas M. Calreticulin, a multi-process calcium-buffering chaperone of the endoplasmic reticulum. Biochem J (2009) 417(3):651–66. doi: 10.1042/BJ20081847
101. Lamriben L, Graham JB, Adams BM, Hebert DN. N-Glycan-Based er molecular chaperone and protein quality control system: The calnexin binding cycle. Traffic (2016) 17(4):308–26. doi: 10.1111/tra.12358
102. Stanley P. Golgi glycosylation. Cold Spring Harb Perspect Biol (2011) 3(4):1–13. doi: 10.1101/cshperspect.a005199
103. Lee MC, Miller EA, Goldberg J, Orci L, Schekman R. Bi-directional protein transport between the er and golgi. Annu Rev Cell Dev Biol (2004) 20:87–123. doi: 10.1146/annurev.cellbio.20.010403.105307
104. Costa CAD, Manaa WE, Duplan E, Checler F. The endoplasmic reticulum Stress/Unfolded protein response and their contributions to parkinson’s disease physiopathology. Cells (2020) 9(11):1–24. doi: 10.3390/cells9112495
105. Oakes SA, Papa FR. The role of endoplasmic reticulum stress in human pathology. Annu Rev Pathol (2015) 10:173–94. doi: 10.1146/annurev-pathol-012513-104649
106. Karagoz GE, Acosta-Alvear D, Walter P. The unfolded protein response: Detecting and responding to fluctuations in the protein-folding capacity of the endoplasmic reticulum. Cold Spring Harb Perspect Biol (2019) 11(9):1–18. doi: 10.1101/cshperspect.a033886
107. Hetz C, Zhang K, Kaufman RJ. Mechanisms, regulation and functions of the unfolded protein response. Nat Rev Mol Cell Biol (2020) 21(8):421–38. doi: 10.1038/s41580-020-0250-z
108. Walter P, Ron D. The unfolded protein response: From stress pathway to homeostatic regulation. Science (2011) 334(6059):1081–6. doi: 10.1126/science.1209038
109. Parmar VM, Schroder M. Sensing endoplasmic reticulum stress. Adv Exp Med Biol (2012) 738:153–68. doi: 10.1007/978-1-4614-1680-7_10
110. Carrara M, Prischi F, Ali MM. Upr signal activation by luminal sensor domains. Int J Mol Sci (2013) 14(3):6454–66. doi: 10.3390/ijms14036454
111. Okamura K, Kimata Y, Higashio H, Tsuru A, Kohno K. Dissociation of Kar2p/Bip from an er sensory molecule, Ire1p, triggers the unfolded protein response in yeast. Biochem Biophys Res Commun (2000) 279(2):445–50. doi: 10.1006/bbrc.2000.3987
112. Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of bip and er stress transducers in the unfolded-protein response. Nat Cell Biol (2000) 2(6):326–32. doi: 10.1038/35014014
113. Ma K, Vattem KM, Wek RC. Dimerization and release of molecular chaperone inhibition facilitate activation of eukaryotic initiation factor-2 kinase in response to endoplasmic reticulum stress. J Biol Chem (2002) 277(21):18728–35. doi: 10.1074/jbc.M200903200
114. Shen J, Chen X, Hendershot L, Prywes R. Er stress regulation of Atf6 localization by dissociation of Bip/Grp78 binding and unmasking of golgi localization signals. Dev Cell (2002) 3(1):99–111. doi: 10.1016/s1534-5807(02)00203-4
115. Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-Reticulum-Resident kinase. Nature (1999) 397(6716):271–4. doi: 10.1038/16729
116. Deng J, Lu PD, Zhang Y, Scheuner D, Kaufman RJ, Sonenberg N, et al. Translational repression mediates activation of nuclear factor kappa b by phosphorylated translation initiation factor 2. Mol Cell Biol (2004) 24(23):10161–8. doi: 10.1128/MCB.24.23.10161-10168.2004
117. Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor Atf6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell (1999) 10(11):3787–99. doi: 10.1091/mbc.10.11.3787
118. Kohno K. Stress-sensing mechanisms in the unfolded protein response: Similarities and differences between yeast and mammals. J Biochem (2010) 147(1):27–33. doi: 10.1093/jb/mvp196
119. Hillary RF, FitzGerald U. A lifetime of stress: Atf6 in development and homeostasis. J BioMed Sci (2018) 25(1):48. doi: 10.1186/s12929-018-0453-1
120. Schindler AJ, Schekman R. In vitro reconstitution of er-stress induced Atf6 transport in copii vesicles. Proc Natl Acad Sci U.S.A. (2009) 106(42):17775–80. doi: 10.1073/pnas.0910342106
121. Ye J, Rawson RB, Komuro R, Chen X, Dave UP, Prywes R, et al. Er stress induces cleavage of membrane-bound Atf6 by the same proteases that process srebps. Mol Cell (2000) 6(6):1355–64. doi: 10.1016/s1097-2765(00)00133-7
122. Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature (2006) 443(7113):780–6. doi: 10.1038/nature05291
123. Lamark T, Johansen T. Autophagy: Links with the proteasome. Curr Opin Cell Biol (2010) 22(2):192–8. doi: 10.1016/j.ceb.2009.11.002
124. Hwang J, Qi L. Quality control in the endoplasmic reticulum: Crosstalk between erad and upr pathways. Trends Biochem Sci (2018) 43(8):593–605. doi: 10.1016/j.tibs.2018.06.005
125. Venkatraman P, Wetzel R, Tanaka M, Nukina N, Goldberg AL. Eukaryotic proteasomes cannot digest polyglutamine sequences and release them during degradation of polyglutamine-containing proteins. Mol Cell (2004) 14(1):95–104. doi: 10.1016/s1097-2765(04)00151-0
126. Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol (2018) 19(6):349–64. doi: 10.1038/s41580-018-0003-4
127. Yang Z, Klionsky DJ. Mammalian autophagy: Core molecular machinery and signaling regulation. Curr Opin Cell Biol (2010) 22(2):124–31. doi: 10.1016/j.ceb.2009.11.014
128. Levine B, Kroemer G. Biological functions of autophagy genes: A disease perspective. Cell (2019) 176(1-2):11–42. doi: 10.1016/j.cell.2018.09.048
129. Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res (2014) 24(1):24–41. doi: 10.1038/cr.2013.168
130. Henderson JM, Weber C, Santovito D. Beyond self-recycling: Cell-specific role of autophagy in atherosclerosis. Cells (2021) 10(3):1–21. doi: 10.3390/cells10030625
131. Cai Y, Arikkath J, Yang L, Guo ML, Periyasamy P, Buch S. Interplay of endoplasmic reticulum stress and autophagy in neurodegenerative disorders. Autophagy (2016) 12(2):225–44. doi: 10.1080/15548627.2015.1121360
132. Mari M, Tooze SA, Reggiori F. The puzzling origin of the autophagosomal membrane. F1000 Biol Rep (2011) 3:25. doi: 10.3410/B3-25
133. Mizushima N, Yoshimori T, Ohsumi Y. The role of atg proteins in autophagosome formation. Annu Rev Cell Dev Biol (2011) 27:107–32. doi: 10.1146/annurev-cellbio-092910-154005
134. Shaid S, Brandts CH, Serve H, Dikic I. Ubiquitination and selective autophagy. Cell Death Differ (2013) 20(1):21–30. doi: 10.1038/cdd.2012.72
135. Lamark T, Svenning S, Johansen T. Regulation of selective autophagy: The P62/Sqstm1 paradigm. Essays Biochem (2017) 61(6):609–24. doi: 10.1042/EBC20170035
136. Rayavarapu S, Coley W, Kinder TB, Nagaraju K. Idiopathic inflammatory myopathies: Pathogenic mechanisms of muscle weakness. Skelet Muscle (2013) 3(1):13. doi: 10.1186/2044-5040-3-13
137. Kurdi M, Alshareef A, Bamaga AK, Fadel ZT, Alrawaili MS, Hakamy S, et al. The assessment of major histocompatibility complex (Mhc) class-I expression in different neuromuscular diseases. Degener Neurol Neuromuscul Dis (2021) 11:61–8. doi: 10.2147/DNND.S340117
138. Kaneko M, Niinuma Y, Nomura Y. Activation signal of nuclear factor-kappa b in response to endoplasmic reticulum stress is transduced Via Ire1 and tumor necrosis factor receptor-associated factor 2. Biol Pharm Bull (2003) 26(7):931–5. doi: 10.1248/bpb.26.931
139. Freret M, Drouot L, Obry A, Ahmed-Lacheheb S, Dauly C, Adriouch S, et al. Overexpression of mhc class I in muscle of lymphocyte-deficient mice causes a severe myopathy with induction of the unfolded protein response. Am J Pathol (2013) 183(3):893–904. doi: 10.1016/j.ajpath.2013.06.003
140. Sheshadri N, Poria DK, Sharan S, Hu Y, Yan C, Koparde VN, et al. Perk signaling through C/Ebpdelta contributes to er stress-induced expression of immunomodulatory and tumor promoting chemokines by cancer cells. Cell Death Dis (2021) 12(11):1038. doi: 10.1038/s41419-021-04318-y
141. Wang P, Han L, Yu M, Cao Z, Li X, Shao Y, et al. The prognostic value of perk in cancer and its relationship with immune cell infiltration. Front Mol Biosci (2021) 8:648752 doi: 10.3389/fmolb.2021.648752
142. Tiniakou E, Mammen AL. Idiopathic inflammatory myopathies and malignancy: A comprehensive review. Clin Rev Allergy Immunol (2017) 52(1):20–33. doi: 10.1007/s12016-015-8511-x
143. Fiorentino DF, Chung LS, Christopher-Stine L, Zaba L, Li S, Mammen AL, et al. Most patients with cancer-associated dermatomyositis have antibodies to nuclear matrix protein nxp-2 or transcription intermediary factor 1gamma. Arthritis Rheum (2013) 65(11):2954–62. doi: 10.1002/art.38093
144. Vazquez-Del Mercado M, Martinez-Garcia EA, Daneri-Navarro A, Gomez-Banuelos E, Martin-Marquez BT, Pizano-Martinez O, et al. Presence of anti-Tif-1gamma, anti-Ro52, anti-Ssa/Ro60 and anti-Su/Ago2 antibodies in breast cancer: A cross-sectional study. Immunopharmacol Immunotoxicol (2021) 43(3):328–33. doi: 10.1080/08923973.2021.1910833
145. Jain A, Sharma MC, Sarkar C, Bhatia R, Singh S, Handa R. Major histocompatibility complex class I and ii detection as a diagnostic tool in idiopathic inflammatory myopathies. Arch Pathol Lab Med (2007) 131(7):1070–6. doi: 10.5858/2007-131-1070-MHCCIA
146. Rodriguez Cruz PM, Luo YB, Miller J, Junckerstorff RC, Mastaglia FL, Fabian V. An analysis of the sensitivity and specificity of mhc-I and mhc-ii immunohistochemical staining in muscle biopsies for the diagnosis of inflammatory myopathies. Neuromuscul Disord (2014) 24(12):1025–35. doi: 10.1016/j.nmd.2014.06.436
147. Arana-Argaez VE, Delgado-Rizo V, Pizano-Martinez OE, Martinez-Garcia EA, Martin-Marquez BT, Munoz-Gomez A, et al. Inhibitors of mapk pathway Erk1/2 or P38 prevent the il-1{Beta}-Induced up-regulation of Srp72 autoantigen in jurkat cells. J Biol Chem (2010) 285(43):32824–33. doi: 10.1074/jbc.M110.121087
148. Lundberg I, Kratz AK, Alexanderson H, Patarroyo M. Decreased expression of interleukin-1alpha, interleukin-1beta, and cell adhesion molecules in muscle tissue following corticosteroid treatment in patients with polymyositis and dermatomyositis. Arthritis Rheum (2000) 43(2):336–48. doi: 10.1002/1529-0131(200002)43:2<336::AID-ANR13>3.0.CO;2-V
149. Tucci M, Quatraro C, Dammacco F, Silvestris F. Interleukin-18 overexpression as a hallmark of the activity of autoimmune inflammatory myopathies. Clin Exp Immunol (2006) 146(1):21–31. doi: 10.1111/j.1365-2249.2006.03180.x
150. Zheng D, Liwinski T, Elinav E. Inflammasome activation and regulation: Toward a better understanding of complex mechanisms. Cell Discovery (2020) 6:36. doi: 10.1038/s41421-020-0167-x
151. Lee KH, Kang TB. The molecular links between cell death and inflammasome. Cells (2019) 8(9):1–23. doi: 10.3390/cells8091057
152. Ma X, Gao HJ, Zhang Q, Yang MG, Bi ZJ, Ji SQ, et al. Endoplasmic reticulum stress is involved in muscular pathogenesis in idiopathic inflammatory myopathies. Front Cell Dev Biol (2022) 10:791986. doi: 10.3389/fcell.2022.791986
153. Uruha A, Goebel HH, Stenzel W. Updates on the immunopathology in idiopathic inflammatory myopathies. Curr Rheumatol Rep (2021) 23(7):56. doi: 10.1007/s11926-021-01017-7
154. Fischer N, Preusse C, Radke J, Pehl D, Allenbach Y, Schneider U, et al. Sequestosome-1 (P62) expression reveals chaperone-assisted selective autophagy in immune-mediated necrotizing myopathies. Brain Pathol (2020) 30(2):261–71. doi: 10.1111/bpa.12772
155. Nogalska A, D’Agostino C, Terracciano C, Engel WK, Askanas V. Impaired autophagy in sporadic inclusion-body myositis and in endoplasmic reticulum stress-provoked cultured human muscle fibers. Am J Pathol (2010) 177(3):1377–87. doi: 10.2353/ajpath.2010.100050
156. Cappelletti C, Galbardi B, Kapetis D, Vattemi G, Guglielmi V, Tonin P, et al. Autophagy, inflammation and innate immunity in inflammatory myopathies. PloS One (2014) 9(11):e111490. doi: 10.1371/journal.pone.0111490
157. Girolamo F, Lia A, Annese T, Giannini M, Amati A, D’Abbicco D, et al. Autophagy markers Lc3 and P62 accumulate in immune-mediated necrotizing myopathy. Muscle Nerve (2019) 60(3):315–27. doi: 10.1002/mus.26608
158. Wang L, Fang D, Liu Y. Autophagy-related genes are potential diagnostic biomarkers for dermatomyositis. Ann Transl Med (2022) 10(4):228. doi: 10.21037/atm-22-70
159. Rosen A, Casciola-Rosen L. Autoantigens as partners in initiation and propagation of autoimmune rheumatic diseases. Annu Rev Immunol (2016) 34:395–420. doi: 10.1146/annurev-immunol-032414-112205
160. Reeves WH, Nigam SK, Blobel G. Human autoantibodies reactive with the signal-recognition particle. Proc Natl Acad Sci U.S.A. (1986) 83(24):9507–11. doi: 10.1073/pnas.83.24.9507
161. Ma X, Xu L, Li Y, Bu B. Immunotherapy reversed myopathy but not cardiomyopathy in a necrotizing autoimmune myopathy patient with positive anti-srp and mda-5 autoantibodies. BMC Cardiovasc Disord (2021) 21(1):88. doi: 10.1186/s12872-021-01900-2
162. Momomura M, Miyamae T, Nozawa T, Kikuchi M, Kizawa T, Imagawa T, et al. Serum levels of anti-Srp54 antibodies reflect disease activity of necrotizing myopathy in a child treated effectively with combinatorial methylprednisolone pulses and plasma exchanges followed by intravenous cyclophosphamide. Mod Rheumatol (2014) 24(3):529–31. doi: 10.3109/14397595.2013.852852
163. Utz PJ, Hottelet M, Le TM, Kim SJ, Geiger ME, van Venrooij WJ, et al. The 72-kda component of signal recognition particle is cleaved during apoptosis. J Biol Chem (1998) 273(52):35362–70. doi: 10.1074/jbc.273.52.35362
164. Chen H, Yang H, Cheng QX, Ge YP, Peng QL, Zhang YM, et al. A novel autoantibody targeting calreticulin is associated with cancer in patients with idiopathic inflammatory myopathies. Clin Transl Immunol (2020) 9(10):e1195. doi: 10.1002/cti2.1195
165. Tawara N, Yamashita S, Zhang X, Korogi M, Zhang Z, Doki T, et al. Pathomechanisms of anti-cytosolic 5’-nucleotidase 1a autoantibodies in sporadic inclusion body myositis. Ann Neurol (2017) 81(4):512–25. doi: 10.1002/ana.24919
166. Arouche-Delaperche L, Allenbach Y, Amelin D, Preusse C, Mouly V, Mauhin W, et al. Pathogenic role of anti-signal recognition protein and anti-3-Hydroxy-3-Methylglutaryl-Coa reductase antibodies in necrotizing myopathies: Myofiber atrophy and impairment of muscle regeneration in necrotizing autoimmune myopathies. Ann Neurol (2017) 81(4):538–48. doi: 10.1002/ana.24902
167. Schmidt J, Barthel K, Wrede A, Salajegheh M, Bahr M, Dalakas MC. Interrelation of inflammation and app in sibm: Il-1 beta induces accumulation of beta-amyloid in skeletal muscle. Brain (2008) 131(Pt 5):1228–40. doi: 10.1093/brain/awn053
168. Seifert U, Bialy LP, Ebstein F, Bech-Otschir D, Voigt A, Schroter F, et al. Immunoproteasomes preserve protein homeostasis upon interferon-induced oxidative stress. Cell (2010) 142(4):613–24. doi: 10.1016/j.cell.2010.07.036
169. Dalakas MC. Inflammatory, immune, and viral aspects of inclusion-body myositis. Neurology (2006) 66(2 Suppl 1):S33–8. doi: 10.1212/01.wnl.0000192129.65677.87
170. Katsiougiannis S, Tenta R, Skopouli FN. Endoplasmic reticulum stress causes autophagy and apoptosis leading to cellular redistribution of the autoantigens Ro/Sjogren’s syndrome-related antigen a (Ssa) and La/Ssb in salivary gland epithelial cells. Clin Exp Immunol (2015) 181(2):244–52. doi: 10.1111/cei.12638
171. Zhang H, Zhao C, Wang S, Huang Y, Wang H, Zhao J, et al. Anti-dsdna antibodies induce inflammation Via endoplasmic reticulum stress in human mesangial cells. J Transl Med (2015) 13:178. doi: 10.1186/s12967-015-0536-7
Keywords: endoplasmic reticulum stress, idiopathic inflammatory myopathies, myositis specific antibodies, autophagy, myositis associated antibodies
Citation: Corona-Sanchez EG, Martínez-García EA, Lujano-Benítez AV, Pizano-Martinez O, Guerra-Durán IA, Chavarria-Avila E, Aguilar-Vazquez A, Martín-Márquez BT, Arellano-Arteaga KJ, Armendariz-Borunda J, Perez-Vazquez F, García-De la Torre I, Llamas-García A, Palacios-Zárate BL, Toriz-González G and Vazquez-Del Mercado M (2022) Autoantibodies in the pathogenesis of idiopathic inflammatory myopathies: Does the endoplasmic reticulum stress response have a role? Front. Immunol. 13:940122. doi: 10.3389/fimmu.2022.940122
Received: 10 May 2022; Accepted: 24 August 2022;
Published: 15 September 2022.
Edited by:
Janine Lamb, The University of Manchester, United KingdomReviewed by:
Maria Marcela Bravo-Zehnder, Universidad de San Sebastián, ChileCopyright © 2022 Corona-Sanchez, Martínez-García, Lujano-Benítez, Pizano-Martinez, Guerra-Durán, Chavarria-Avila, Aguilar-Vazquez, Martín-Márquez, Arellano-Arteaga, Armendariz-Borunda, Perez-Vazquez, García-De la Torre, Llamas-García, Palacios-Zárate, Toriz-González and Vazquez-Del Mercado. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Monica Vazquez-Del Mercado, ZHJhdm1lQGhvdG1haWwuY29t
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.