 Françoise Vuillier1,2
Françoise Vuillier1,2 Zhi Li1,2
Zhi Li1,2 Iain Black1,2Melania Cruciani1,2
Iain Black1,2Melania Cruciani1,2 Erminia Rubino1,2
Erminia Rubino1,2 Frédérique Michel1,2
Frédérique Michel1,2 Sandra Pellegrini1,2*
Sandra Pellegrini1,2*- 1Cytokine Signaling Unit, Department of Immunology, Institut Pasteur, Paris, France
- 2Microenvironment and Immunity Unit, Institut Pasteur, Paris, France
Regulation of innate immune responses is essential for maintenance of immune homeostasis and development of an appropriate immunity against microbial infection. We show here that miR-3614-5p, product of the TRIM25 host gene, is induced by type I interferon (IFN-I) in several human non-immune and immune cell types, in particular in primary myeloid cells. Studies in HeLa cells showed that miR-3614-5p represses both p110 and p150 ADAR1 and reduces constitutive and IFN-induced A-to-I RNA editing. In line with this, activation of innate sensors and expression of IFN-β and the pro-inflammatory IL-6 are promoted. MiR-3614-5p directly targets ADAR1 transcripts by binding to one specific site in the 3’UTR. Moreover, we could show that endogenous miR-3614-5p is associated with Ago2 and targets ADAR1 in IFN-stimulated cells. Overall, we propose that, by reducing ADAR1, IFN-I-induced miR-3614-5p contributes to lowering the activation threshold of innate sensors. Our findings provide new insights into the role of miR-3614-5p, placing it as a potential fine tuner of dsRNA metabolism, cell homeostasis and innate immunity.
Introduction
MicroRNAs (miRNAs) are non-coding RNAs of ~22 nucleotides that post-transcriptionally downregulate gene expression through translational destabilization, blockage or cleavage of mRNA targets in a specific base pairing recognition manner (1). These small RNA molecules have been found involved in every biological process and are considered important regulatory elements also in the control of innate defense against invading pathogens. Numerous miRNAs have been shown to curb or promote innate immune cell activation at multiple layers by targeting pattern recognition receptors, adaptors, positive and negative signaling components, transcription factors and regulators. Thereby these tuning molecules contribute to homeostasis, production of immune cytokines and magnitude of the inflammatory response (2).
Type I interferons (IFN-I) are innate cytokines critical for the immediate host defense against viral infection. They are induced upon cell recognition of microbial products and act by up-regulating effector molecules of cell-intrinsic antiviral immunity. Moreover, they modulate functions of many immune cell populations and orchestrate innate and adaptive antiviral responses. IFN-I also mediate protective effects in other pathological contexts, such as some cancers and multiple sclerosis, but if improperly produced they can exert damaging effects, as in some bacterial infections and autoimmune diseases (3). Pathways leading to the production of IFN-I and the signaling pathway activated by IFN-I are highly regulated and can be impacted by numerous miRNAs (4). Conversely, little is known on miRNAs whose abundance can be directly modulated by IFN-I at the level of transcription or processing. Here we report on miR-3614-5p that we found upregulated by IFN-I in several human cell types.
Present knowledge on miR-3614-5p is still limited. This miRNA was identified in a few studies exploring differential miRNA expression in cancer and was ascribed both pro- and anti-tumoral effects. MiR-3614-5p was found to be more expressed in cervical cancer (5) and in breast cancer tissues (6) with respect to adjacent normal tissues. Conversely, miR-3614-5p was proposed to attenuate proliferation and invasion of non-small cell lung cancer cells by targeting the glycolysis enzyme phosphoglycerate mutase 1 (PGAM1) (7) and the WNT pathway-related NFATC2 transcription factor (8). Another study suggested a role of miR-3614-5p in opposing hepatocarcinoma cell proliferation and migration by targeting the transcription factor YY1 (9). The analysis of data from the Cancer Genome Atlas (TCGA) revealed lower miR-3614-5p expression in colorectal cancer tissues compared to adjacent normal tissue, making it a potential prognostic marker of low survival (10).
A role of miR-3614-5p in autoimmunity was suggested by the identification of a Crohn’s disease risk single nucleotide polymorphism possibly associated with miR-3614-5p (11). A reduced level of miR-3614-5p was correlated with inflammation in the epicardial adipose tissue of coronary artery disease patients and studies in monocytic-like THP1 cells suggested a role of miR-3614-5p in downmodulating inflammatory cytokine response through targeting TRAF6 (12). Recently, the presence of miR-3614-5p and two other miRNAs (miR-125a-3p and miR-1246) in plasma exosomal RNA from dermatomyositis patients was correlated with skeletal muscle damage (13).
No unique comprehensive picture has emerged so far on the role of miR-3614-5p. Interestingly, miR-3614-5p was scored in several genome-wide surveys of miRNAs that are modulated in response to infection. MiR-3614-5p was found up-regulated in human cells latently infected with Kaposi’s sarcoma-associated herpes virus (14), in human monocyte-derived DC infected with bacterial species (15), in human macrophages infected with Dengue virus (16), and in primary monocytes infected with influenza A virus or stimulated with a TLR7/8 ligand (17).
Here we have studied the expression of miR-3614-5p in some human cell lines and in myeloid cells. Through functional analysis in HeLa cells we demonstrate that miR-3614-5p is a bona fide IFN-I-induced miRNA that, by directly targeting the two ADAR1 isoforms, takes part in the control of RNA editing.
Materials and methods
Antibodies and reagents
We used antibodies against ADAR1 (Cell Signaling Technology), Ago2 (MBL, Life Science), tubulin (Merck) and IRF9 (gift from D.E. Levy, NYU School of Medicine, NY, NY). Irrelevant mouse IgG was purchased from R&D. Recombinant IFN-α2b was a gift from D. Gewert (Wellcome Foundation, Beckenham, Kent, UK). IFN-β produced by Biogen Idec (Cambridge, MA, USA) was a gift of G. Uzé. Horseradish peroxidase (HRP)-conjugated anti-rabbit IgG and anti-mouse IgG were purchased from Jackson. GM-CSF, IL-4 and M-CSF were purchased from Miltenyi Biotec.
Isolation of monocytes and derived macrophages and dendritic cells
Peripheral blood mononuclear cells (PBMCs) and monocytes were isolated from freshly collected buffy coats as previously described (18). Freshly isolated monocytes were differentiated in macrophages or immature dendritic cells (DC). Monocytes were resuspended in complete medium RPMI 10% FCS supplemented with IL-4 (50 ng/ml) GM-CSF (100 ng/ml) for 6 days to obtain immatures monocyte-derived DC (Mo-DC) or with M-CSF (100 ng/ml) for 6 days for monocyte-derived macrophages. Medium was renewed every third day. Immature Mo-DC were analyzed for lack of CD14 and acquisition of DC-SIGN marker.
Cell lines
HEK293T, HeLa S3, HepG2 and A549 cell lines were cultured in DMEM Glutamax (Gibco) supplemented with 10% fetal calf serum. THP1, HuT78 and Jurkat cells were cultured in RPMI Glutamax (Gibco) supplemented with 10% fetal calf serum (Pan-Biotech), non essential amino acids and sodium pyruvate.
RNA extraction and RT-qPCR
Total RNA was extracted from Trizol using the miRNeasy mini kit (Qiagen, Germantown, MD, USA) following the manufacturer’s instructions. Quantification and purity were assessed by Nanodrop spectrophotometer (Nanodrop2000, Thermo Fisher Scientific). For measuring ADAR1 total or p150, TRIM25, IL-6 and IFN-β transcripts, total RNA was reverse-transcribed using high-capacity cDNA reverse transcription (RT) kit (Applied Biosystems, Thermo Fisher Scientific). Quantitative PCR (qPCR) assays were performed using the FastStart SYBR Green Master Mix (Roche) on QuantStudio 3 Real-Time PCR system (Applied Biosystems, Thermo Fisher Scientific). Transcript levels were normalized to 18S levels using the equation 2-ΔCt. See Table S1 for oligonucleotide sequences.
MiRNA expression was determined as previously described (18).
Protein analysis
Whole-cell extracts were prepared and western blots were performed as previously described (19). Signals were detected with an enhanced chemiluminescence (ECL) detection reagent (SuperSignal® WestPico Chemiluminescent Substrate, Thermo Scientific). Images were acquired with a Fuji Image Quant LAS-4000 machine. The data were analyzed by using Multi Gauge v3.2 (Fuji Image analyzing application) software. Quantified bands were normalized with respect to tubulin, as indicated in the figure legends.
Reverse transfection of mimic or miRNA inhibitor
MiRIDIAN miRNA mimic and inhibitor specific for miR-3614-5p were purchased from Horizon Discovery Ltd. MiRIDIAN miRNA Mimic Negative Control #1 and miRIDIAN miRNA Inhibitor Negative Control #1 (Horizon Discovery Ltd) were used to assess specificity of the effect driven by the specific miRNA sequences. Mimic (50 nM) and inhibitor (100 nM) reverse transfections in HeLa S3 cells were performed as previously described (18).
Analysis of RNA editing
The cDNA fragments containing the candidate editing sites were amplified for AZIN1 using the primers AZIN1-Frw and AZIN1-Rev, and for EIF2AK2 with EIF2AK2-Frw and EIF2AK2-Rev primers. PCR was performed with Phusion DNA polymerase (Thermo Scientific). A-to-I/G RNA editing was detected by sequencing the PCR products using AZIN1-seq Rev or EIF2AK2-Rev primers (Table S1). The editing event is given by a mixture of A+G peaks in the sequence chromatogram and measured calculating the ratio between the area of the peak corresponding to G and the sum of the areas of the double peaks A+G. Sequence analysis was performed with the software Bioedit. For the sequence of the primers used refer to Supplementary Table 1.
Luciferase reporter assay
For cloning the ADAR1 3’UTR in the psiCHECK2 vector (Promega, Madison, WI, USA), the first 1500 nucleotides of ADAR1 3’UTR were amplified from HeLa S3 cDNA and cloned downstream of the Renilla luciferase ORF using NotI and XhoI restriction sites. The resulting plasmid (psiCHECK2-ADAR1 3’UTR) was mutated in the binding sites of miR-3614-5p (Figure 5B) using QuikChange XL site-directed mutagenesis kit (Aligent Technologies, Santa Clara, CA, USA) and specific primers for each site (215-219mut Frw/Rev, 589-593mut Frw/Rev, 687-691mut Frw/Rev, and 1175-1179mut Frw/Rev) (Table S1). New plasmids were sequenced. Mut215, mut591, mut689, and mut1175 are mutated in a single site, mut215+1175, mut591+1175 and mut689+1175 are mutated in two sites, mut591+689+1175 are mutated in three sites. For the luciferase reporter assay, HeLa S3 cells were reverse transfected with miR-3614-5p mimic (see above). After 24 hr, plasmid transfection was carried out using FuGENE HD (Promega) according to the manufacturer’s instructions. Three biological replicates were prepared for each condition. Cells were lysed 24 hr post-transfection and analyzed with the Dual-Luciferase® Reporter Assay System (Promega) according to the manufacturer’s instructions. Firefly luciferase was measured as normalizer.
Ago2-miRNA-co-immunoprecipitation assay
Antibody-coated protein G/protein A-agarose was prepared by incubating protein G/protein A-agarose (Calbiochem) with anti-human Argonaute2 (Ago2) antibody or irrelevant mouse IgG (R&D) overnight at 4°C. HeLa S3 cells were stimulated with IFN-α2 (100 pM) for 24 hr or left untreated and lysed in lysis buffer as previously reported (20). Lysates were cleared by centrifugation (14,000 x g) for 10 min at 4°C. Supernatants were collected and pre-cleared with protein G/protein A-agarose. Cleared lysates were then incubated with the antibody-coated protein G/protein A-agarose for 3 hr at 4°C. Bound material was washed five times with lysis buffer. Finally, a fraction (10%) was used for western blotting. The remaining fraction was digested with proteinase K and used for RNA extraction.
Results
MiR-3614-5p is induced by IFN-I and targets ADAR1
Here we asked whether the expression of miR-3614-5p can be directly modulated by IFN-I. We chose myeloid cells as they are key producers and target cells of IFN-I early during immune response activation. Stem-loop RT-qPCR was performed on RNA isolated from primary monocytes, monocyte-derived dendritic cells and monocyte-derived macrophages that were stimulated or not by IFN-β (Figures 1A, B). MiR-3614-5p was found to be lowly basally expressed and induced by IFN in all subsets, most highly in undifferentiated monocytes. The level of miR-3614-5p was also compared in monocytes and the human cell lines HeLa, A549, HepG2, 293T, THP1 and in the CD4+ T cell lines HuT78 and Jurkat. As shown in Figure 1C, the miRNA was lowly expressed and induced by IFN-α to variable extent, except in 293T and Jurkat cells.
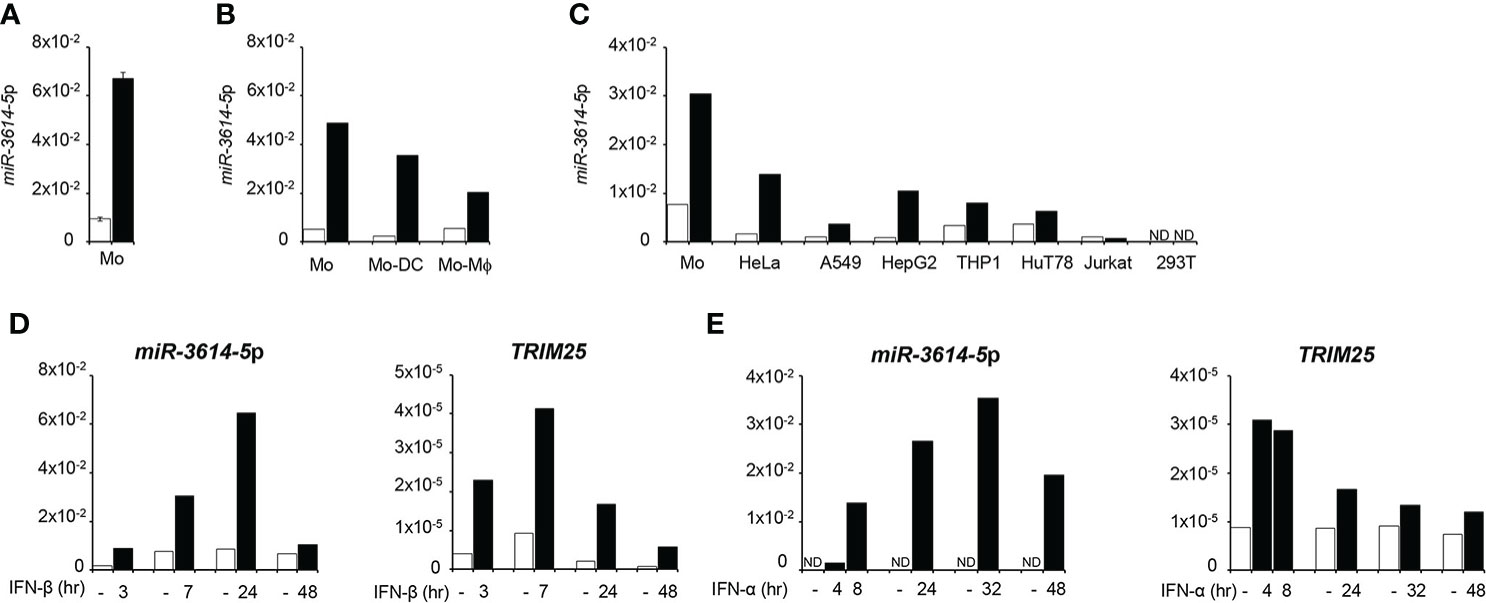
Figure 1 MiR-3614-5p is induced by type I IFN. (A) MiR-3614-5p expression in monocytes purified from two healthy donors and treated with 100pM IFN-β for 24 hr (black) or not treated (white). Results are shown as expression (2-ΔCt) relative to U6 used as housekeeping gene. Values are means ± SEM. (B) Mir-3614-5p expression in monocytes from one donor and in derived macrophages (Mo-Mϕ) and dendritic cells (Mo-DC). IFN-β stimulation and results as in A). (C) Mir-3614-5p expression in different human cell lines and in primary monocytes from a single donor (Mo). Stimulation with IFN-α (100pM) for 8 hr. Data are representative of two independent experiments. ND, not detected. (D) MiR-3614-5p (left panel) and TRIM25 (right panel) levels measured in primary monocytes stimulated with IFN-β (100pM) as indicated. Results for TRIM25 are shown as expression (2-ΔCt) relative to 18S. Data are representative of two independent experiments. (E) MiR-3614-5p (left panel) and TRIM25 (right panel) levels measured in HeLa cells stimulated with IFN-α (100pM) for the indicated times. Results expressed as in D). Data are representative of four independent experiments.
The miR-3614-5p genomic sequence resides in the TRIM25 (tripartite motif-containing protein 25) gene, a well known ISG operating in innate immune pathways (21). In particular, pri-miR-3614 is found in the last TRIM25 exon (exon 9) that comprises some coding sequences and a 3.79 kb-long 3’UTR (Figure 2). Given this location, we monitored in parallel the kinetics of induction by IFN-I of both miR-3614-5p and TRIM25. In primary monocytes and in HeLa cells, TRIM25 mRNA levels peaked 4 to 8 hr after IFN addition, while miR-3614-5p levels peaked at later times (Figures 1D, E).
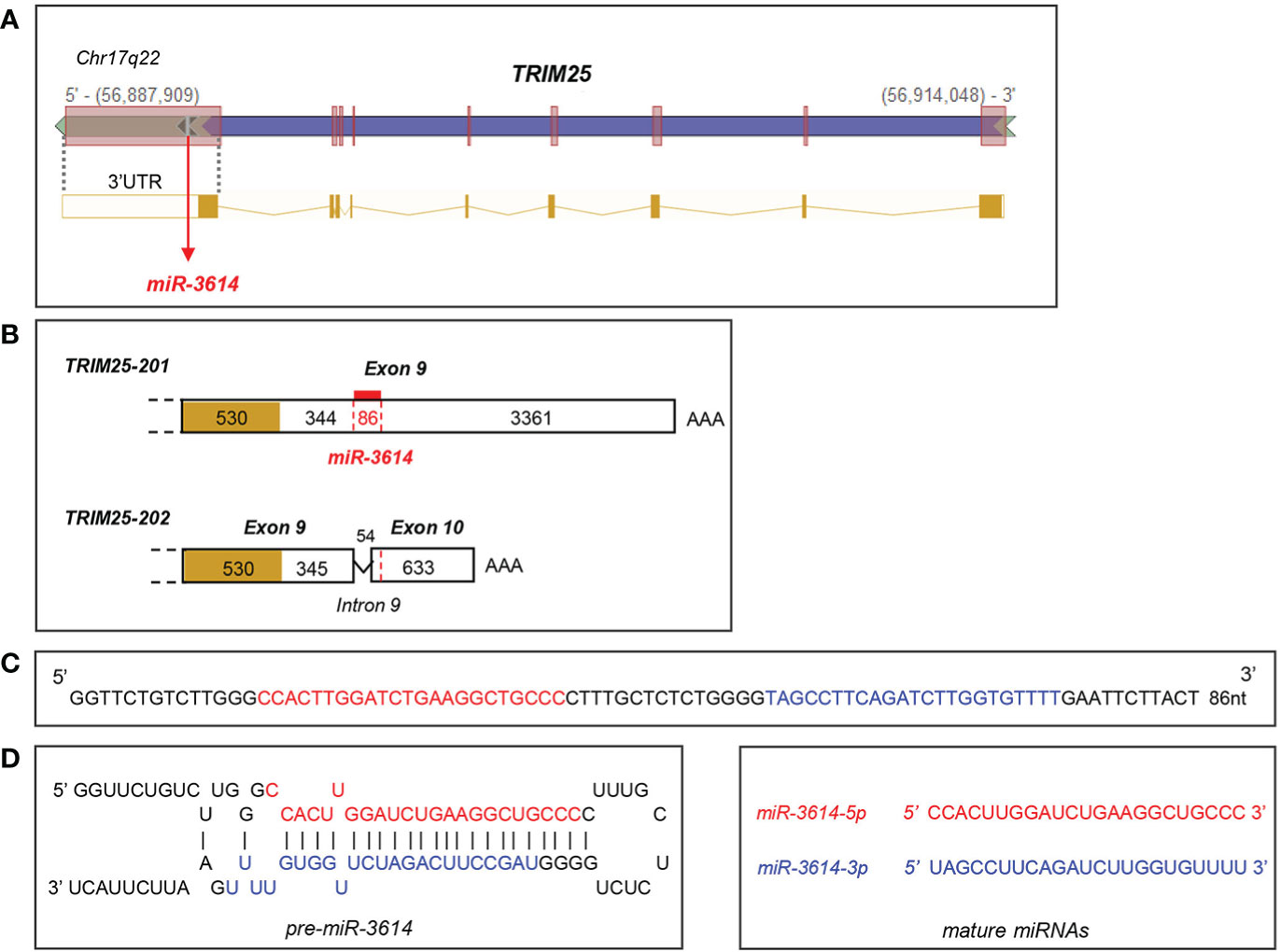
Figure 2 Localisation and sequence of miR-3614. (A) Genomic position of miR-3614 sequence in the human TRIM25 gene (from Intragenic microRNA database, miRIAD). (B) Localisation of miR-3614 in the 3’UTR of TRIM25. Only the last exon(s) of the two protein-coding transcripts, TRIM25-201 and TRIM25-202, is shown. The coding region is yellow-coloured and the 3’UTR is white-coloured. In red the 86 nt miR-3614 sequence that overlaps an intronic splice site. (C) The 86 nt miR-3614 sequence. (D) Secondary structure prediction of the miRNA precursor. MiR-3614-5p and -3p in red and blue, respectively.
We used four prediction algorithms to identify candidate gene targets of miR-3614-5p and one of the best scored hit was ADAR1, member of the adenosine deaminase acting on RNA (ADAR) family and key RNA editing enzyme preventing activation of nucleic acid sensors by self-dsRNAs (22). The ADAR1 gene specifies two transcripts generated through alternative promoters and splicing of exon 1. One transcript is constitutively expressed and encodes the ubiquitous 110-kDa protein isoform (p110) predominantly nuclear, while the other transcript is induced by IFN-I and encodes a 150-kDa protein isoform (p150) with a unique N-ter domain allowing nuclear-cytoplasmic shuttling (23). We sought to analyze the impact of miR-3614-5p on ADAR1 RNA and protein in HeLa cells. Expression of a mimic of miR-3614-5p caused a reduction of ADAR1 transcripts measured by RT-qPCR (Figure 3A). In cells treated with IFN-α, the miR-3614-5p mimic led to a substantial reduction of the inducible ADAR1 p150 transcript as well (Figure 3B). Of note, at 72 hr post-stimulation the effect of the mimic on ADAR1 had waned, possibly due to reduced editing and consequent accumulation of unedited dsRNA with production of IFN-β and induction of ADAR1 p150 (see next section). The impact of miR-3614-5p at the protein level was studied by western blot. In non-stimulated cells, a reduction of nearly 50% of both the constitutive p110 and the weakly expressed p150 isoform was observed. In IFN-α stimulated cells, the induced p150 isoform was also considerably reduced (Figure 3C).
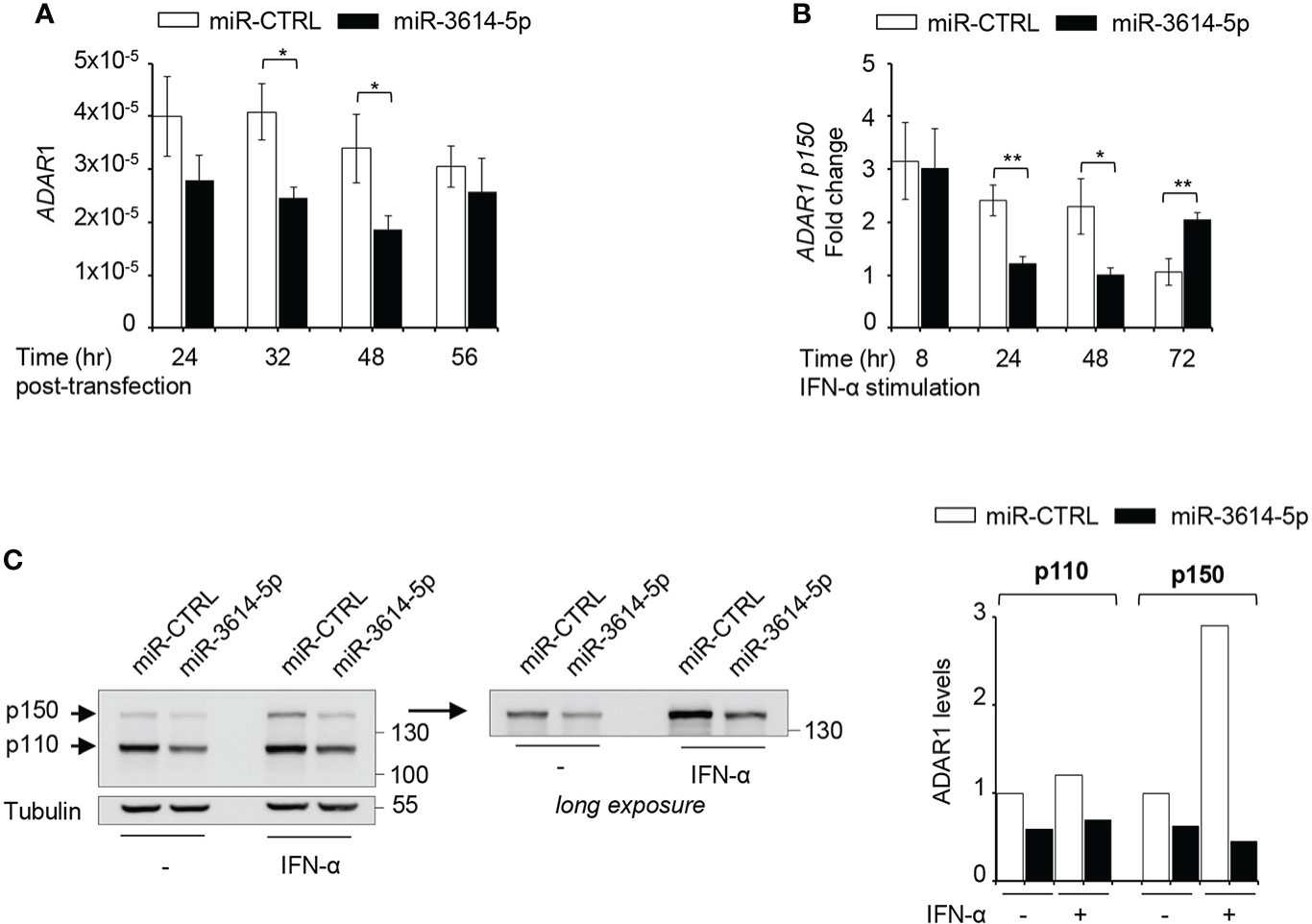
Figure 3 A miR-3614-5p mimic targets both ADAR1 isoforms. (A) A miR-3614-5p or a miR-control (miR-CTRL) mimic was retrotransfected in HeLa cells. At the indicated times post-transfection the level of total ADAR1 mRNA was measured by qPCR. Results are shown as expression (2-ΔCt) relative to 18S used as housekeeping gene. Values are means ± SEM (n=3 separate experiments). *p<0.05. (B) HeLa were retrotransfected as in A) with miR-3614-5p or a miR-control (miR-CTRL) and 48 hr later, IFN-α (100pM) was added. Relative expression of p150 ADAR1 was measured at the indicated times after stimulation. The fold change represents the ratio of IFN-stimulated cells relative to unstimulated cells. Values are means ± SEM (n=3), *p<0.05, **p<0.01. (C) HeLa cells were retrotransfected as in A). Twenty-four hr later cells were stimulated with IFN-α (100pM) for 2 days and then processed for western blot analysis. Tubulin was used as loading control. Right panel, the membrane was covered below the 130 kDa marker and exposed for longer time to better visualize the p150 ADAR1 isoform. Protein bands were quantified and normalized to tubulin. For each isoform results are expressed as ratio of miR-3614-5p-transfected cells relative to miR-CTRL-transfected cells. The value in untreated miR-CTRL-transfected cells set to 1. The data are representative of three independent experiments.
MiR-3614-5p lowers dsRNA editing
The above data showed that miR-3614-5p is an IFN-inducible miRNA that is able to target constitutive and inducible ADAR1. Given the key function of ADAR1 in adenosine to inosine (A-to-I) editing self and viral dsRNA, we reasoned that the miR-3614-5p-mediated decline in the level of steady-state ADAR1 could cause an accumulation of unedited endogenous dsRNAs, leading to aberrant activation of key sensors, like (RIG-I)-like receptors, and induction of IFN-β and pro-inflammatory cytokines. To test this possibility, we compared the level of IFN-β and IL-6 mRNAs in control and miR-3614-5p-transfected cells. Indeed, both cytokine transcripts were up-regulated in miR-3614-5p-transfected cells at late times (Figure 4A). Moreover, the transcript level of the sensors RIG-I and MDA5 - themselves ISGs - was also augmented (Figure 4A). Western blot analysis of cell lysates confirmed a 50% reduction of ADAR1 in miR-3614-5p-transfected cells with a substantial increase in the amount of the transcription factor IRF9, an ISG product (Figure 4B).
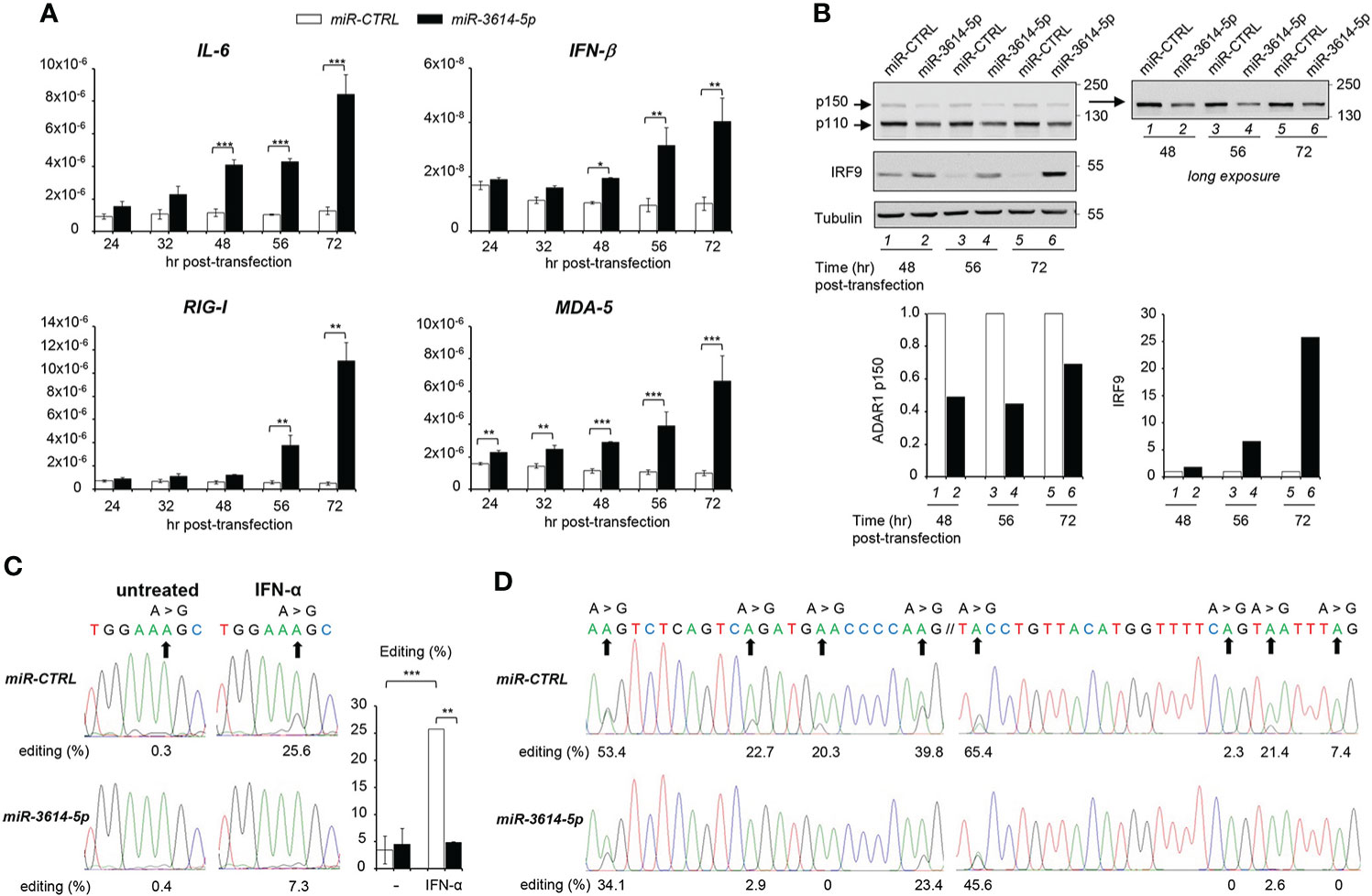
Figure 4 MiR-3614-5p impacts innate cytokine expression and RNA editing. (A) HeLa cells were retrotransfected with the mir-3614-5p or the miR-control (miR-CTRL) as mimics. The relative expression of IL-6, IFN-β, RIG-I and MDA-5 was measured at different times after retrotransfection, as indicated. Values are means ± SEM (n=3 separate experiments); *p<0.05; **p<0.01; ***p<0.001. (B) Cells retrotransfected as in A) were lysed for western blot analysis at different times post-transfection. Lysates were immunoblotted as indicated. Tubulin was used as loading control. Right panel, the membrane was covered below the 130 kDa marker and exposed for longer time to better visualize the p150 ADAR1 isoform. Protein bands were quantified and normalized to tubulin. Results are expressed as ratios of miR-3614-5p-transfected cells (black) relative to miR-CTRL-transfected cells (white). Values in miR-CTRL-transfected cells were set to 1. These data are representative of two experiments. (C) HeLa cells were retrotransfected with the mir-3614-5p or the miR-control (miR-CTRL) as mimics. Two days later cells were left untreated or treated with IFN-α (100pM) for an additional 72 hr. RNA was extracted and editing was determined, as described in Materials and Methods. The percentage of editing was measured after sequencing the AZIN1 PCR products. Percentages of editing in AZIN1 transcripts are shown below the chromatograms of a representative experiment. Right histogram, three independent experiments were quantified. Values are means ± SEM; **p<0.01; ***p<0.001. (D) HeLa cells were retrotransfected with mir-3614-5p or miR-control (miR-CTRL) as mimics and left in culture for 56 hr. RNA was extracted and editing was determined, as described in Materials and Methods. The percentage of editing was measured after sequencing the EIF2AK2 PCR products. Two regions of an editing box located in 3’UTR of EIF2AK2 transcripts are represented. The two sequences have their genomic counterpart localized at chr2: 37,328,008-37,328,100 (24). Their percentages of editing are shown below the chromatograms. The data are representative of two independent experiments.
To demonstrate that miR-3614-5p impinges on ADAR1-mediated editing, we measured editing of antizyme inhibitor 1 RNA (AZIN1), a well known ADAR1 substrate that is commonly used as an editing activity read-out (25). In the absence of IFN, AZIN1 editing was barely detectable in control unstimulated cells, so that the effect of miR-3614-5p could not be evaluated in baseline condition. Conversely, after IFN stimulation the level of AZIN1 editing was increased from 0.3% to 25.6% in control cells. However, in miR-3614-5p-transfected cells the increase was much less, from 0.4% to 7.3% (Figure 4C).
Having shown that mir-3614-5p impacts editing in IFN stimulated conditions, we asked whether miR-3614-5p could affect constitutive editing. Zhu et al. (24) reported on three clusters of constitutive A-to-I editing sites that are located in the 3’UTR of EIF2AK2 (PKR) and were named editing boxes. Working with HeLa cells, the authors validated these sites as being edited by ADAR1 and quantified the editing ratios for each of them. In line with this, we tested whether miR-3614-5p could reduce the level of editing at these same sites. We measured A-to-I editing in two EIF2AK2 regions 56 hr after retrotransfection of control or miR-3614-5p mimic (Figure 4D). The percentages of editing in control samples were remarkably similar to those reported in (24). Importantly, we found that the expression of miR-3614-5p decreased editing and this whatever the level of editing (high, intermediate or low) was in the control. Thus, by targeting ADAR1, miR-3614-5p can impact editing in resting and in IFN-stimulated conditions. Altogether, these data suggest that miR-3614-5p is involved in RNA metabolism, thus contributing to regulation of the innate immune response.
MiR-3614-5p directly targets the 3’UTR of ADAR1
Next we sought to validate a direct impact of miR-3614-5p on ADAR1 3’UTR. The two ADAR1 transcripts - encoding p110 and p150 isoforms - possess an identical 3’UTR, except for 10 additional nucleotides at the very 3’ end of the p150-encoding transcript. The whole 3’UTR was scrutinized using miRNA target prediction algorithms. Canonical miRNA targeting involves nucleotides 2-7 at the 5’ end of the miRNA. This “seed” sequence pairs with complementary nucleotides in the 3’UTR of mRNA (1). Complementarity with the 3’end of the miRNA can compensate for imperfect matches within the seed sequence. We scored four candidate miR-3614-5p target sites, all located in the 5’ half of the 3’UTR. Each site was named according to the position of the seed-pairing nucleotide. According to Agarwal et al. (26), site 215 and site 1175 are canonical sites, while site 591 and site 689 are non canonical 5-mer sites (Figure 5A).
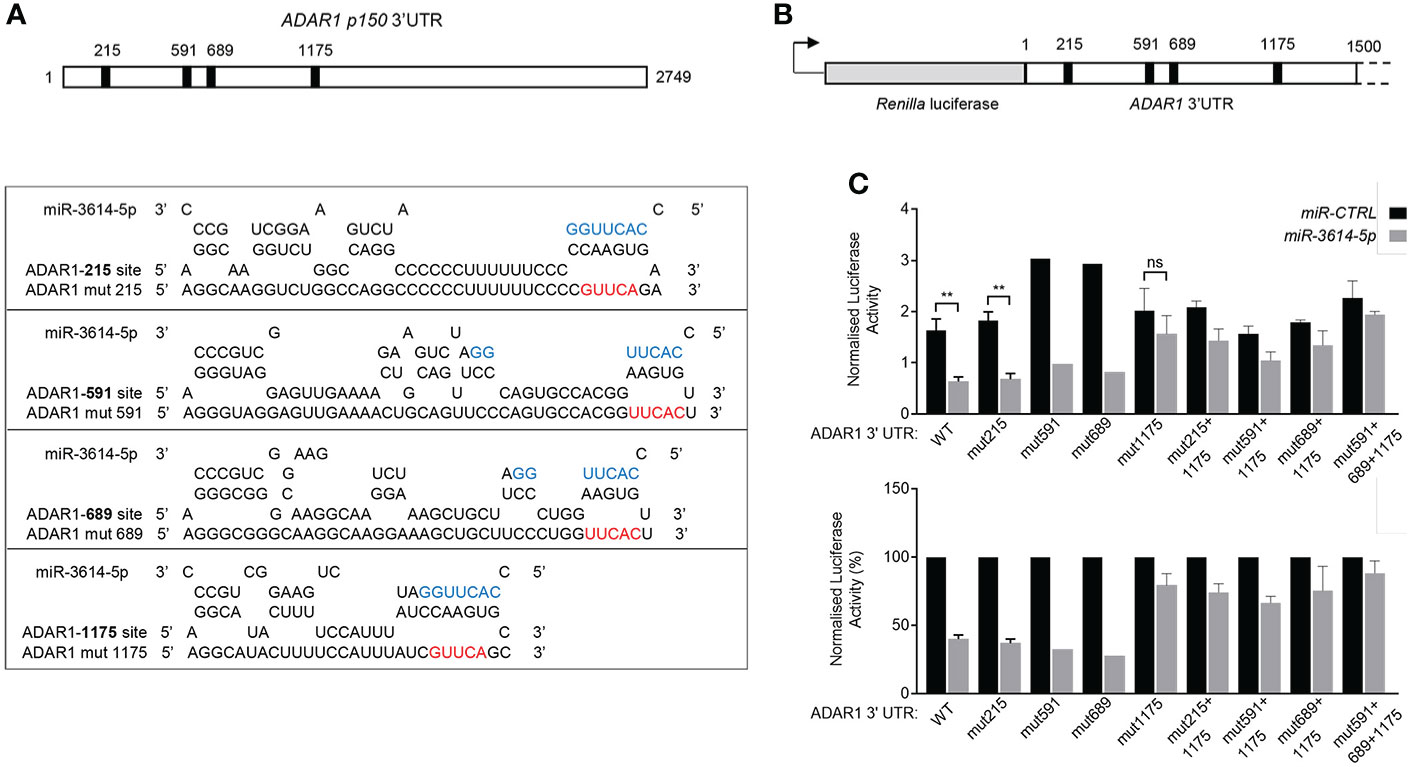
Figure 5 MiR-3614-5p targets ADAR1 3’UTR through one major binding site. (A) Schematic diagram of the 3’UTR of the p150 ADAR1 transcript. Black boxes indicate the location of the four predicted binding sites of miR-3614-5p. Site 215 matches to miR-3614-5p positions 2-8 of the seed sequence with an A opposite to position 1. Site 1175 matches to miR-3614-5p positions 2-10. Site 591 and site 689 are two non-canonical 5-mer binding sites. The RNAhybrid miRNA target prediction tool was used (https://bibiserv.cebitec.uni-bielefeld.de/rnahybrid). Mutant ADAR1 target sites (mut215, mut591, mut689, mut1175) were generated by mutating 5nt complementary to the 5’ seed region of miR-3614-5p. The seed sequence is in blue. The mutated nucleotides are in red. (B) Schematic diagram of the 1.5 kb ADAR1 3’UTR sequence containing the four predicted miR-3614-5p binding sites (black squares) cloned downstream of Renilla luciferase in the psiCHECK-2 vector. (C) Luciferase assay to assess direct interaction of miR-3614-5p with the ADAR1 3’UTR. HeLa cells were retrotransfected with the miR-3614-5p mimic or the non-targeting control mimic (50nM) and the next day transfected with the dual luciferase reporter psiCHECK-2 carrying the ADAR1 3’UTR, as in (B). Twenty four hr later Renilla luciferase activity was measured and normalized to the internal firefly luciferase control (top panel). WT and mut1175 data: mean of four experiments each in triplicate. Mut215 data: mean of 3 experiments each in triplicate. Mut591, mut689 data: one experiment in triplicate. Data on combined mutations: mean of two experiments each in triplicate. Statistics is available only for results with 3 independent experiments. Data represented as mean ± SEM. *p<0.05, **p<0.01, ns = not significant. Normalised luciferase activities (top panel) are represented as percentage of the control mimic (bottom panel). Data are represented as mean ± SEM.
To study the direct and site-specific activity of miR-3614-5p we used a reporter assay. A 1.5 kb-long fragment of the 3’UTR encompassing the four sites was subcloned downstream of Renilla luciferase in the psiCHECK-2 reporter plasmid (Figure 5B). HeLa cells were first retrotransfected with the miR-3614-5p mimic or the control mimic and then transfected with the reporter plasmid. Luciferase activity was measured 24 hr later. As shown in Figure 5C, the miR-3614-5p mimic caused nearly 60% reduction of luciferase activity.
To identify which site(s) in the 3’UTR was targeted by miR-3614-5p, we introduced mutations in each of the four sites (Figure 5A). Mutation of site 215, site 591 or site 689 had minimal effects, while mutation of site 1175 reduced the effect of miR-3614-5p (Figure 5C). Combining the mutation of sites 215, 591 or 689 with that of site 1175 did not further reduce the effect of miR-3614-5p compared to mutation of site 1175 alone, providing further evidence that the predicted sites 215, 591 and 689 are not targeted by miR-3614-5p (Figure 5C). In conclusion, these data demonstrate that miR-3614-5p is able to down-modulate expression of a reporter gene containing the ADAR1 3’UTR and that this occurs through direct binding of the miRNA to site 1175.
Endogenous miR-3614-5p is recruited to RISC and can repress ADAR1 transcripts
Having validated the effect of miR-3614-5p expressed as mimic on ADAR1, we turned to the endogenous miRNA. Argonaute2 (Ago2) is an essential component of RISC (RNA-induced silencing complex), which helps binding of miRNA to the target-site on mRNA. Hence the Ago2-associated miRNA represents the functional fraction involved in mRNA down-regulation (1). Here, we aimed to investigate the association of miR-3614-5p with RISC, using an Argonaute2-miRNA-co-immunoprecipitation assay (see Materials and Methods). We measured levels of miR-3614-5p and ADAR1 mRNA present in Ago2 immunoprecipitates from untreated and IFN-treated HeLa cells (Figures 6A, B). We could show that after IFN stimulation miR-3614-5p was particularly enriched in Ago2 antibody-coated beads with respect to control IgG beads. In addition, we detected the presence of ADAR1 mRNA in the Ago2 immunoprecipitated material, further supporting the binding of miR-3614-5p to ADAR1 (Figure 6B).
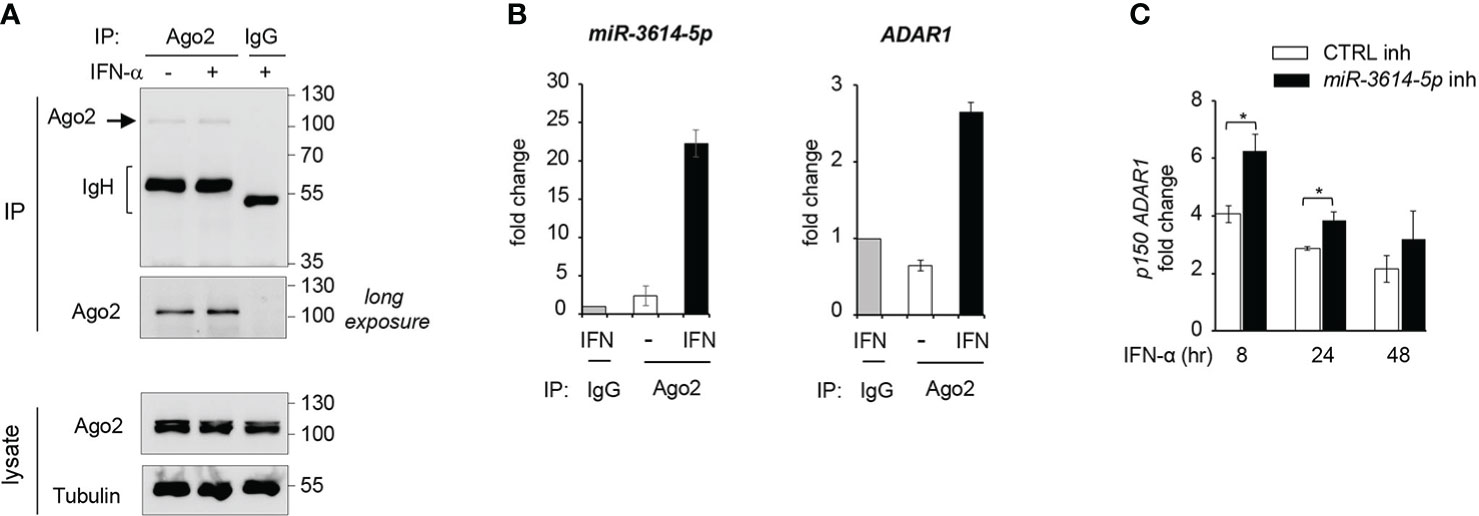
Figure 6 Endogenous miR-3614-5p is associated with Ago-2 and targets ADAR1 in IFN-stimulated cells. (A) HeLa cells were stimulated with IFN-α (100pM) for 24 hr and processed for Argonaute2-miRNA-co-immunoprecipitation. Endogenous Argonaute2 (Ago2) was immunoprecipitated (IP) from whole cell lysates with anti-Ago2 or an IgG control. Samples were analysed by western blot as indicated. (B) miR-3614-5p (left panel) or ADAR1 mRNA (right panel) was measured by qPCR in the Ago2 immunoprecipitated fraction relative to the IgG fraction, shown in A). The Ct values of miR3614-5p or ADAR1 in the IgG immunoprecipitate were substracted from the Ct values in Ago2 immunoprecipitates (IFN-treated or untreated) to generate ΔCts. The results shown are expressed as fold change and represent the expression (2-ΔCt) relative to control IgG set at 1. Data are mean +/- SEM of two independent experiments. (C) HeLa cells were transfected with the miR-3614-5p inhibitor (miR-3614-5p inh) or the control inhibitor (CTRL inh). Two days later, IFN-α (100pM) was added. Relative expression of p150 ADAR1 was measured at different times after stimulation as indicated. The fold change represents the ratio of IFN-treated cells relative to non-treated cells. Values are means ± SEM (n=3 separate experiments). *p < 0.05.
Next, we aimed to deplete endogenous miR-3614-5p from IFN-treated HeLa cells and measure the effect on ADAR p150 mRNA. For this, cells were retrotransfected with a miR-3614-5p inhibitor or a control inhibitor and then left untreated or treated with IFN for different times. As shown in Figure 6C, in cells treated with IFN-α the miR-3614-5p inhibitor led to an increase of the ADAR1 p150 transcript. The above results on the endogenous miR-3614-5p reinforce our conclusion that ADAR1 is a bona fide target of this IFN-induced miRNA.
Discussion
In this study we show that the primate-specific miR-3614-5p is induced by IFN-I in several human immune and non-immune cell types, in particular primary myeloid cells, which are known to be central in the initiation of an immune response. Using overexpression and inhibition approaches in HeLa cells, we demonstrate that one functional target of miR-3614-5p is the enzyme ADAR1. We identified a major binding site in the 3’UTR of ADAR1 transcripts through which miR-3614-5p plays its regulatory role and showed that, upon IFN stimulation, endogenous miR-3614-5p and ADAR1 are both recruited by the RISC component Ago2.
MiR-3614-5p was previously identified in cultured macrophages infected with Dengue virus (16). The authors noticed that miR-3614-5p was upregulated not only in infected cells but also in the non-infected cells of the culture, which suggests the action of a soluble mediator, i.e. IFN-I, released by infected cells. With high throughput sequencing technologies, the knowledge on miRNAs that are directly modulated by IFN-I in specific cell types/tissues and physio-pathological contexts has increased. Indeed, Rodriguez-Gala et al. (27) searched for IFN-regulated miRNAs in human primary CD4+ T cells and identified 57 miRNAs that appear weakly modulated by IFN, 24 being upregulated and 33 repressed. MiR-3614-5p was highlighted as one of the most upregulated miRNA (8 fold after 24 hr), rising from a very low threshold level. Interestingly, miR-3614-5p was found to be upregulated by IFN-β also in human skeletal muscle myoblasts (13).
The miR-3614 gene has some interesting features. The 86 nt-sequence is found only in primates (Supplementary Figure 1) and is likely the product of recent evolution. This sequence gives rise to mature miR-3614-5p and -3p, with -5p being predominant, and resides within the TRIM25 host gene, which provides expression constraint. TRIM25 and miR-3614-5p transcripts are both up-regulated by IFN-I through the IFN-responsive TRIM25 promoter (28). However, their accumulation profiles differ. In primary monocytes and HeLa cells TRIM25 mRNA peaked 4 to 8 hr after IFN addition, while miR-3614-5p peaked at later times (Figures 1D, E). The unique position of the pri-miRNA may account for the different accumulation profile. As schematized in Figure 2, pri-miR-3614 resides in the 3.79 kb-long 3’UTR of the abundant TRIM25-201 transcript. Interestingly, miR-3614 belongs to the class of SO-miRNAs (splice site overlapping miRNA), since the miR-3614 extends over a splice site [see Table 1 in (29)]. Splicing of intronic sequences at this site generates an alternative protein-coding transcript (TRIM25-202) that contains a shorter 3’UTR of 0.9 kb and is expressed at very low level. Given the unusual location of miR-3614, i.e. overlapping a splice site in the TRIM25 3’UTR, the relative amount of TRIM25 transcripts and pri-miR-3614 produced may be governed by a mechanism of competition between splicing and miRNA processing machineries (30, 31). Indeed, the depletion of the splicing factor SF3b1 in HeLa cells was found to increase the level of several mature SO-miRNAs including miR-3614-3p (29). To try to uncover a potential cross-regulation of TRIM25 and miR-3614-5p, we analyzed the effect of depleting SF3b1 on the abundance of both RNAs. Given the low level of endogenous miR-3614-5p in HeLa cells, we used a human TRIM25 expression plasmid (pSPORT6-TRIM25) that retains a portion of the 3’UTR (clone ID: 4419084, Open Biosytems). When transfected, this plasmid transcribes TRIM25 as well as miR-3614. In cells depleted of SF3b1 we did find an increase of miR-3614-5p and -3p as reported in (29), but we did not observe a change in the relative abundance of TRIM25-201 and -202 isoforms (data not shown). Hence, further work is needed to address mechanistic questions on the biogenesis of miR-3614-5p. Moreover, the expression levels of TRIM25 isoforms and miR-3614-5p will have to be assessed in different tissues/cell types and in different contexts.
TRIM25 is a RING-type E3 ligase that catalyzes ubiquitination of numerous proteins in multiple RNA-dependent pathways (21). TRIM25 plays a crucial role in the RIG-I antiviral response pathway (32), though other E3 ligases such as RIPLET can activate RIG-I (33). TRIM25 is also involved in the activation of other antiviral sensors, such as zinc finger antiviral protein or ZAP (34). Of note TRIM25 can conjugate not only ubiquitin but also ISG15, this latter being highly induced in IFN-primed cells, and TRIM25 auto-ISGylation was shown to downregulate its activity (35). TRIM25 is also known as EFP (estrogen-responsive finger protein) for its capacity to respond to estrogens via an enhancer element in the 3’UTR (36). TRIM25 constitutes a key regulator of metastatic gene signatures in breast cancer (37) and elevated TRIM25 mRNA is associated with poor prognosis in breast cancer and several other cancers (38). Since, as we propose here, miR-3614-5p, a product of TRIM25, fine tunes immune response, it will be interesting to study miR-3614-5p expression and function in these cancer contexts.
MiR-3614-5p is lowly expressed at baseline and modestly induced by IFN-I, at least in the cell types we and others have studied. This feature does not suggest direct targeting of abundant viral transcripts, but rather points to a role of miR-3614-5p in fine-tuning host genes. We show here that the two ADAR1 transcript isoforms, which share the 3’UTR, are directly targeted by miR-3614-5p, in accordance with previous work (16). The downregulation of ADAR1 by specific miRNAs has been previously reported. MiR-1, involved in myogenesis regulation (39, 40), miR-143 (34), miR-17-5p and miR-432 (41) were found to directly bind to ADAR1 3’UTR. The genes encoding miR-17-5p and miR-432 are frequently amplified in melanomas and their overexpression is coupled with reduced ADAR1 expression that has been associated with aggressive features of metastatic melanomas. Of note, the highly conserved miR-1 was shown to be induced by IFN-β in human hepatoma HuH7 cells, murine primary hepatocytes (42), and PBMC (43). Hence, it is possible that in certain contexts IFN-induced miR-1 and miR-3614-5p cooperate to reduce ADAR1 level.
ADAR1 is a dsRNA-specific deaminase that takes part in the discrimination of self vs non-self RNAs. Its dsRNA editing activity (conversion of adenosine to inosine) is most often exerted on paired Alu elements present in primate self RNAs. In any given cell type and in normal condition, the baseline level of ADAR1 sets the threshold for innate immune activation, as this enzyme restrains aberrant activation of dsRNA sensors by endogenous RNA, preventing production of IFN-I (44). Partial loss-of-function mutations in ADAR1 cause an autoinflammatory Aicardi-Goutières syndrome-related interferonopathy in humans (45). In infectious contexts, ADAR1 has been regarded as proviral for its ability to edit dsRNA and to competitively bind dsRNA, restraining activation of immune sensors such as MDA-5 and PKR, a potent inhibitor of translation (46). However, for some viruses ADAR1-mediated editing can be regarded as having an antiviral effect (47).
In cancer the influence of ADAR1 is complex. It has been shown to have mostly pro-oncogenic effects exerted through diverse and context-specific mechanisms (48). Several cancer cell lines rely on ADAR1 for survival (49). High AZIN editing in colorectal cancer was correlated with disease severity (50). However, in melanoma, loss of ADAR1 was reported to promote metastatic growth due to deregulation of several miRNAs (41). Interestingly, loss of ADAR1 was shown to sensitize tumor cells to immunotherapy and to contribute to overcome resistance to checkpoint blockade at least in part through increased IFN-I production (51).
Our functional studies in HeLa cells indicate that IFN-induced miR-3614-5p impacts on the level of ADAR1 proteins and dsRNA editing, eventually promoting activation of innate immune signaling with upregulation of IFN-β and IL-6 (scheme in Figure 7). In this view, miR-3614-5p upregulation can be interpreted as reducing the threshold of activation of innate sensors, such as RIG-I and MDA-5, in an IFN-autocrine/paracrine dependent manner, thereby facilitating an efficient antiviral response.
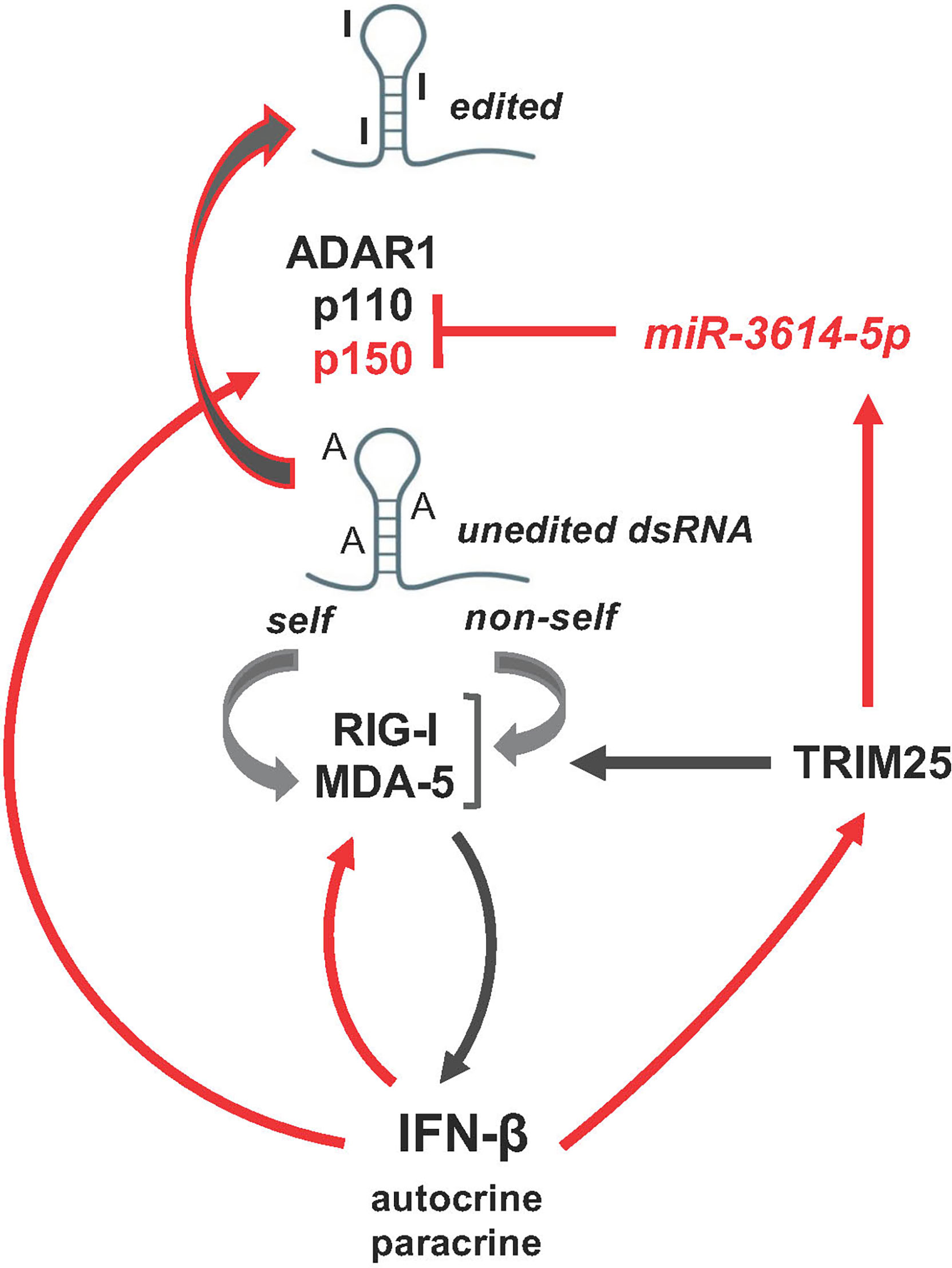
Figure 7 Schematic representation of miR-3614-5p involvement in the network of activation of the dsRNA sensors RIG-I and MDA-5 mediating induction of IFN-I and inflammatory response. Of these intricate pathways only the few actors studied here are depicted. Red arrows, transcriptional gene induction by IFN-I, which acts in an autocrine and paracrine fashion.
Having shown that ADAR1 is a direct target of miR-3614-5p adds up to the multiple regulatory mechanisms that have been shown to modulate ADAR1 levels and functions (52) and opens up questions about the level of miR-3614-5p in contexts where ADAR1 was shown to exert proviral, antiviral, oncogenic or tumor suppressor roles. Obviously, the investigation of the single miR-3614-5p-ADAR1 interaction has its limitation, since other mRNAs are expected to be targeted by this miRNA in a cell-type and context-specific manner. Since miR-3614-5p has most likely emerged in primates, research in mouse models cannot be carried out, but high-throughput technologies and computational approaches should help to fully appreciate the complexity of gene regulation by miR-3614-5p and its possible involvement in immune dysregulation in disease.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material Further inquiries can be directed to the corresponding author.
Author Contributions
FV, MC, ER, ZL, FM and SP designed the study. FV, MC, ZL and IB performed experiments. SP and FV wrote the manuscript. All authors read and provided input on the manuscript and approved the final version. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by the Fondation pour la Recherche Médicale (Equipe FRM DEQ20170336741) and institutional funds from Institut Pasteur and Institut national de la santé et de la recherche médicale (Inserm). ZL was supported by Centre national de la recherche scientifique (CNRS). ER was supported by FRM. MC was supported by the FRM grant above. IB was supported by the Erasmus+ EU programme (University of Glasgow).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.939907/full#supplementary-material.
Supplementary Figure 1 | Comparative analysis of miR-3614 in human and non-human primates. (A) Phylogenetic tree and (B) sequence alignment of miR-3614 (human chr17:56891270-56891355) in 24 primates. https://www.ensembl.org/Homo_sapiens/Location/Compara_Alignments?db=core;g=ENSG00000284542;r=17:56891270-56891355;t=ENST00000581261;time=1649078338264.264.
References
1. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell (2009) 136(2):215–33. doi: 10.1016/j.cell.2009.01.002
2. Mehta A, Baltimore D. MicroRNAs as regulatory elements in immune system logic. Nat Rev Immunol (2016) 16(5):279–94. doi: 10.1038/nri.2016.40
3. Tomasello E, Pollet E, Vu Manh TP, Uze G, Dalod M. Harnessing mechanistic knowledge on beneficial versus deleterious IFN-I effects to design innovative immunotherapies targeting cytokine activity to specific cell types. Front Immunol (2014) 5:526. doi: 10.3389/fimmu.2014.00526
4. Forster SC, Tate MD, Hertzog PJ. MicroRNA as type I interferon-regulated transcripts and modulators of the innate immune response. Front Immunol (2015) 6:334. doi: 10.3389/fimmu.2015.00334
5. Witten D, Tibshirani R, Gu SG, Fire A, Lui WO. Ultra-high throughput sequencing-based small RNA discovery and discrete statistical biomarker analysis in a collection of cervical tumours and matched controls. BMC Biol (2010) 8:58. doi: 10.1186/1741-7007-8-58
6. Persson H, Kvist A, Rego N, Staaf J, Vallon-Christersson J, Luts L, et al. Identification of new microRNAs in paired normal and tumor breast tissue suggests a dual role for the ERBB2/Her2 gene. Cancer Res (2011) 71(1):78–86. doi: 10.1158/0008-5472.CAN-10-1869
7. Li F, Yang H, Kong T, Chen S, Li P, Chen L, et al. PGAM1, regulated by miR-3614-5p, functions as an oncogene by activating transforming growth factor-beta (TGF-beta) signaling in the progression of non-small cell lung carcinoma. Cell Death Dis (2020) 11(8):710. doi: 10.1038/s41419-020-02900-4
8. Shang J, Wang Z, Chen W, Yang Z, Zheng L, Wang S, et al. Pseudogene CHIAP2 inhibits proliferation and invasion of lung adenocarcinoma cells by means of the WNT pathway. J Cell Physiol (2019) 234(8):13735–46. doi: 10.1002/jcp.28053
9. Feng Z, Ye Z, Xie J, Chen W, Li W, Xing C. Study on the mechanism of LOXL1-AS1/miR-3614-5p/YY1 signal axis in the malignant phenotype regulation of hepatocellular carcinoma. Biol Direct (2021) 16(1):24. doi: 10.1186/s13062-021-00312-8
10. Han L, Sun Y, Lu C, Ma C, Shi J, Sun D. MiR-3614-5p is a potential novel biomarker for colorectal cancer. Front Genet (2021) 12:666833. doi: 10.3389/fgene.2021.666833
11. Wohlers I, Bertram L, Lill CM. Evidence for a potential role of miR-1908-5p and miR-3614-5p in autoimmune disease risk using integrative bioinformatics. J Autoimmun (2018) 94:83–9. doi: 10.1016/j.jaut.2018.07.010
12. Huang W, Wu X, Xue Y, Zhou Y, Xiang H, Yang W, et al. MicroRNA-3614 regulates inflammatory response via targeting TRAF6-mediated MAPKs and NF-kappaB signaling in the epicardial adipose tissue with coronary artery disease. Int J Cardiol (2021) 324:152–64. doi: 10.1016/j.ijcard.2020.09.045
13. Li L, Zuo X, Liu D, Luo H, Zhang H, Peng Q, et al. Plasma exosomal RNAs has potential as both clinical biomarkers and therapeutic targets of dermatomyositis. Rheumatol (Oxford) (2021) 61(6):2672–81. doi: 10.1093/rheumatology/keab753
14. Viollet C, Davis DA, Reczko M, Ziegelbauer JM, Pezzella F, Ragoussis J, et al. Next-generation sequencing analysis reveals differential expression profiles of MiRNA-mRNA target pairs in KSHV-infected cells. PloS One (2015) 10(5):e0126439. doi: 10.1371/journal.pone.0126439
15. Siddle KJ, Tailleux L, Deschamps M, Loh YH, Deluen C, Gicquel B, et al. Bacterial infection drives the expression dynamics of microRNAs and their isomiRs. PloS Genet (2015) 11(3):e1005064. doi: 10.1371/journal.pgen.1005064
16. Diosa-Toro M, Echavarria-Consuegra L, Flipse J, Fernandez GJ, Kluiver J, van den Berg A, et al. MicroRNA profiling of human primary macrophages exposed to dengue virus identifies miRNA-3614-5p as antiviral and regulator of ADAR1 expression. PloS Negl Trop Dis (2017) 11(10):e0005981. doi: 10.1371/journal.pntd.0005981
17. Rotival M, Siddle KJ, Silvert M, Pothlichet J, Quach H, Quintana-Murci L. Population variation in miRNAs and isomiRs and their impact on human immunity to infection. Genome Biol (2020) 21(1):187. doi: 10.1186/s13059-020-02098-w
18. Rubino E, Cruciani M, Tchitchek N, Le Tortorec A, Rolland AD, Veli O, et al. Human ubiquitin-specific peptidase 18 is regulated by microRNAs via the 3'Untranslated region, a sequence duplicated in long intergenic non-coding RNA genes residing in chr22q11.21. Front Genet (2020) 11:627007. doi: 10.3389/fgene.2020.627007
19. Vuillier F, Li Z, Commere PH, Dynesen LT, Pellegrini S. USP18 and ISG15 coordinately impact on SKP2 and cell cycle progression. Sci Rep (2019) 9(1):4066. doi: 10.1038/s41598-019-39343-7
20. Goff LA, Davila J, Swerdel MR, Moore JC, Cohen RI, Wu H, et al. Ago2 immunoprecipitation identifies predicted microRNAs in human embryonic stem cells and neural precursors. PloS One (2009) 4(9):e7192. doi: 10.1371/journal.pone.0007192
21. Martin-Vicente M, Medrano LM, Resino S, Garcia-Sastre A, Martinez I. TRIM25 in the regulation of the antiviral innate immunity. Front Immunol (2017) 8:1187. doi: 10.3389/fimmu.2017.01187
22. Samuel CE. Adenosine deaminase acting on RNA (ADAR1), a suppressor of double-stranded RNA-triggered innate immune responses. J Biol Chem (2019) 294(5):1710–20. doi: 10.1074/jbc.TM118.004166
23. George CX, Samuel CE. Human RNA-specific adenosine deaminase ADAR1 transcripts possess alternative exon 1 structures that initiate from different promoters, one constitutively active and the other interferon inducible. Proc Natl Acad Sci U S A (1999) 96(8):4621–6. doi: 10.1073/pnas.96.8.4621
24. Zhu S, Xiang JF, Chen T, Chen LL, Yang L. Prediction of constitutive a-to-I editing sites from human transcriptomes in the absence of genomic sequences. BMC Genomics (2013) 14:206. doi: 10.1186/1471-2164-14-206
25. Tulluri V, Nemmara VV. Role of antizyme inhibitor proteins in cancers and beyond. Onco Targets Ther (2021) 14:667–82. doi: 10.2147/OTT.S281157
26. Agarwal V, Bell GW, Nam JW, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. Elife (2015) 4:e05005. doi: 10.7554/eLife.05005
27. Rodriguez-Galan A, Dosil SG, Gomez MJ, Fernandez-Delgado I, Fernandez-Messina L, Sanchez-Cabo F, et al. MiRNA post-transcriptional modification dynamics in T cell activation. iScience (2021) 24(6):102530. doi: 10.1016/j.isci.2021.102530
28. Nakasato N, Ikeda K, Urano T, Horie-Inoue K, Takeda S, Inoue S. A ubiquitin E3 ligase efp is up-regulated by interferons and conjugated with ISG15. Biochem Biophys Res Commun (2006) 351(2):540–6. doi: 10.1016/j.bbrc.2006.10.061
29. Pianigiani G, Licastro D, Fortugno P, Castiglia D, Petrovic I, Pagani F. Microprocessor-dependent processing of splice site overlapping microRNA exons does not result in changes in alternative splicing. RNA (2018) 24(9):1158–71. doi: 10.1261/rna.063438.117
30. Melamed Z, Levy A, Ashwal-Fluss R, Lev-Maor G, Mekahel K, Atias N, et al. Alternative splicing regulates biogenesis of miRNAs located across exon-intron junctions. Mol Cell (2013) 50(6):869–81. doi: 10.1016/j.molcel.2013.05.007
31. Mattioli C, Pianigiani G, Pagani F. A competitive regulatory mechanism discriminates between juxtaposed splice sites and pri-miRNA structures. Nucleic Acids Res (2013) 41(18):8680–91. doi: 10.1093/nar/gkt614
32. Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun L, et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-i-mediated antiviral activity. Nat (2007) 446(7138):916–20. doi: 10.1038/nature05732
33. Hayman TJ, Hsu AC, Kolesnik TB, Dagley LF, Willemsen J, Tate MD, et al. RIPLET, and not TRIM25, is required for endogenous RIG-i-dependent antiviral responses. Immunol Cell Biol (2019) 97(9):840–52. doi: 10.1111/imcb.12284
34. Ahmad S, Mu X, Yang F, Greenwald E, Park JW, Jacob E, et al. Breaching self-tolerance to alu duplex RNA underlies MDA5-mediated inflammation. Cell (2018) 172(4):797–810.e13. doi: 10.1016/j.cell.2017.12.016
35. Zou W, Wang J, Zhang DE. Negative regulation of ISG15 E3 ligase EFP through its autoISGylation. Biochem Biophys Res Commun (2007) 354(1):321–7. doi: 10.1016/j.bbrc.2006.12.210
36. Ikeda K, Orimo A, Higashi Y, Muramatsu M, Inoue S. Efp as a primary estrogen-responsive gene in human breast cancer. FEBS Lett (2000) 472(1):9–13. doi: 10.1016/S0014-5793(00)01421-6
37. Walsh LA, Alvarez MJ, Sabio EY, Reyngold M, Makarov V, Mukherjee S, et al. An integrated systems biology approach identifies TRIM25 as a key determinant of breast cancer metastasis. Cell Rep (2017) 20(7):1623–40. doi: 10.1016/j.celrep.2017.07.052
38. Uhlen M, Zhang C, Lee S, Sjostedt E, Fagerberg L, Bidkhori G, et al. A pathology atlas of the human cancer transcriptome. Science (2017) 357(6352):eaan2507. doi: 10.1126/science.aan2507
39. Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, et al. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature (2005) 433(7027):769–73. doi: 10.1038/nature03315
40. Hsieh CL, Liu H, Huang Y, Kang L, Chen HW, Chen YT, et al. ADAR1 deaminase contributes to scheduled skeletal myogenesis progression via stage-specific functions. Cell Death Differ (2014) 21(5):707–19. doi: 10.1038/cdd.2013.197
41. Nemlich Y, Greenberg E, Ortenberg R, Besser MJ, Barshack I, Jacob-Hirsch J, et al. MicroRNA-mediated loss of ADAR1 in metastatic melanoma promotes tumor growth. J Clin Invest (2013) 123(6):2703–18. doi: 10.1172/JCI62980
42. Pedersen IM, Cheng G, Wieland S, Volinia S, Croce CM, Chisari FV, et al. Interferon modulation of cellular microRNAs as an antiviral mechanism. Nature (2007) 449(7164):919–22. doi: 10.1038/nature06205
43. Scagnolari C, Zingariello P, Vecchiet J, Selvaggi C, Racciatti D, Taliani G, et al. Differential expression of interferon-induced microRNAs in patients with chronic hepatitis c virus infection treated with pegylated interferon alpha. Virol J (2010) 7:311. doi: 10.1186/1743-422X-7-311
44. George CX, Ramaswami G, Li JB, Samuel CE. Editing of cellular self-RNAs by adenosine deaminase ADAR1 suppresses innate immune stress responses. J Biol Chem (2016) 291(12):6158–68. doi: 10.1074/jbc.M115.709014
45. Crow YJ, Stetson DB. The type I interferonopathies: 10 years on. Nat Rev Immunol (2021) 20:1–13. doi: 10.1038/s41577-021-00633-9
46. Chung H, Calis JJA, Wu X, Sun T, Yu Y, Sarbanes SL, et al. Human ADAR1 prevents endogenous RNA from triggering translational shutdown. Cell (2018) 172(4):811–24.e14. doi: 10.1016/j.cell.2017.12.038
47. Pfaller CK, George CX, Samuel CE. Adenosine deaminases acting on RNA (ADARs) and viral infections. Annu Rev Virol (2021) 8(1):239–64. doi: 10.1146/annurev-virology-091919-065320
48. Baker AR, Miliotis C, Ramirez-Moya J, Marc T, Vlachos IS, Santisteban P, et al. Transcriptome profiling of ADAR1 targets in triple-negative breast cancer cells reveals mechanisms for regulating growth and invasion. Mol Cancer Res (2022) 20(6):960–71. doi: 10.1158/1541-7786.MCR-21-0604
49. Bhate A, Sun T, Li JB. ADAR1: A new target for immuno-oncology therapy. Mol Cell (2019) 73(5):866–8. doi: 10.1016/j.molcel.2019.02.021
50. Shigeyasu K, Okugawa Y, Toden S, Miyoshi J, Toiyama Y, Nagasaka T, et al. AZIN1 RNA editing confers cancer stemness and enhances oncogenic potential in colorectal cancer. JCI Insight (2018) 3(12):e99976. doi: 10.1172/jci.insight.99976
51. Ishizuka JJ, Manguso RT, Cheruiyot CK, Bi K, Panda A, Iracheta-Vellve A, et al. Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nat (2019) 565(7737):43–8. doi: 10.1038/s41586-018-0768-9
Keywords: type I interferon, microRNA, ADAR1, TRIM25, innate response
Citation: Vuillier F, Li Z, Black I, Cruciani M, Rubino E, Michel F and Pellegrini S (2022) IFN-I inducible miR-3614-5p targets ADAR1 isoforms and fine tunes innate immune activation. Front. Immunol. 13:939907. doi: 10.3389/fimmu.2022.939907
Received: 09 May 2022; Accepted: 27 June 2022;
Published: 22 July 2022.
Edited by:
Birgit Strobl, University of Veterinary Medicine Vienna, AustriaReviewed by:
Iris Behrmann, University of Luxembourg, LuxembourgCarl Walkley, University of Melbourne, Australia
Copyright © 2022 Vuillier, Li, Black, Cruciani, Rubino, Michel and Pellegrini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sandra Pellegrini, c2FuZHJhLnBlbGxlZ3JpbmlAcGFzdGV1ci5mcg==