 Xia Zhou
Xia Zhou Sohail Khan
Sohail Khan Dabing Huang
Dabing Huang Lu Li
Lu Li
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 15 September 2022
Sec. Cancer Immunity and Immunotherapy
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.938470
This article is part of the Research Topic Recent advances and challenges in cancer immunotherapies for patients with autoimmune diseases View all 11 articles
The development of immune checkpoint inhibitors is becoming a promising approach to fight cancers. Antibodies targeting immune checkpoint proteins such as CTLA-4 and PD-1 can reinvigorate endogenous antitumor T-cell responses and bring durable advantages to several malignancies. However, only a small subset of patients benefit from these checkpoint inhibitors. Identification of new immune checkpoints with the aim of combination blockade of multiple immune inhibitory pathways is becoming necessary to improve efficiency. Recently, several B7 family-related proteins, TIGIT, VSIG4, and VSIG3, which belong to the VSIG family, have attracted substantial attention as coinhibitory receptors during T-cell activation. By interacting with their corresponding ligands, these VSIG proteins inhibit T-cell responses and maintain an immune suppressive microenvironment in tumors. These results indicated that VSIG family members are becoming putative immune checkpoints in cancer immunotherapy. In this review, we summarized the function of each VSIG protein in regulating immune responses and in tumor progression, thus providing an overview of our current understanding of VSIG family members.
Immune checkpoint receptors are membrane molecules that can modulate lymphocyte activation upon encoding their cognate ligands on antigen-presenting cells or target cells. They play an essential role in controlling excessive immune responses by transmitting a stop signal to attenuate T-cell activation and maintain immune homeostasis. However, tumors always take advantage of these inhibitory pathways to escape attack from antitumor immune cells (1, 2). Various malignancies are found to confer an overall immunosuppressive tumor microenvironment by upregulating the expression of immune checkpoint receptors and their ligands. To unleash effector T-cell responses and enhance endogenous antitumor activity, therapies targeting these immunoregulatory proteins are becoming an encouraging approach. The most successful immune checkpoint blockade (ICB) therapy is anti-PD-1/PD-L1, which has been shown to confer therapeutic advantages for a variety of cancers, such as non-small cell lung carcinoma (NSCLC), malignant melanoma, kidney cancer, and liver cancer (3–8). Another well-studied immune checkpoint is cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) (1). The first immune checkpoint inhibitor (ipilimumab) targeting CTLA-4 was approved in 2011 by the Food and Drug Administration (FDA) and has been demonstrated to control tumor growth and prolong survival in melanoma (9–11). Since then, the application of ICBs has brought a groundbreaking paradigm shift in cancer treatment, particularly for advanced-stage cancers (9, 12–14). With the growing research interest in cancer immunotherapy, many new checkpoints have been identified and extensively studied in recent years, such as TIM3, TIGIT, VISTA, LAG-3, BTLA, B7-H3, B7-H4, and B7-H5 (15–19). Most of them belong to the B7 family (VISTA, B7-H3, B7-H4, B7-H5), which is characterized by typical extracellular IgV-like and IgC-like domains and is categorized as the immunoglobulin superfamily (IgSF) (20). These proteins can function as coinhibitory receptors to deliver negative signaling towards T cells upon TCR engagement, therefore inhibiting T-cell activation, expansion, and functional polarization. Recently, the V-Set and immunoglobulin (Ig) domain-containing (VSIG) family, which also belongs to IgSF and exhibits structural similarity with the B7 family proteins, has been increasingly recognized as a potential immune checkpoint contributing to tumor evasion. This family is currently comprised of eight members, including VSIG1, VSIG2, VSIG3, VSIG4, VSIG8, VSIG9, VSIG10, and VSIG10 L, which are all type I transmembrane proteins that are expressed by a variety of both immune and nonimmune cells, and many possess immunosuppressive properties. For example, VSIG3, a ligand of VISTA, is essential for its role in T-cell suppression (21, 22). Another member, VSIG4, is also well known for its potent ability to suppress T-cell responses (23). A star member of the VSIG family is VSIG9, also known as TIGIT, which has emerged as a promising cancer therapeutic target due to its apparent function in limiting antitumor T-cell and NK-cell responses (24–26). These studies suggested that the members of the VSIG family could be potent candidates for developing novel ICB therapies. However, there is still a range of related proteins in this family that have yet to be studied extensively, and the mechanism whereby VSIG family proteins inhibit immune responses is not fully understood. To attract more attention to this family, this review aims to introduce VSIG family members and their role in regulating the T-cell response in cancers.
The discovery of the VSIG protein family dates back to the 1990s. The first identified member of the VSIG family is VSIG2, initially called CTX. This gene was first cloned by Chrétien and colleagues from the cortical thymocyte of Xenopus in 1996 (27). The homolog gene of VSIG2 in chickens, mice, and humans was cloned two years later (28). It is located on chromosome 11, 11q24.2. The second member, VSIG4, was cloned and found to be localized in the pericentromeric region of the human X chromosome (29). Since then, other members showing sequence similarities with these identified VSIG proteins have been discovered, including VSIG1, VSIG3, VSIG8, VSIG9 (widely known as TIGIT), VSIG10, and VSIG10 L. The corresponding genes of those VSIG family members are present on different chromosomes in humans, with the exception of VSIG1, which is also present on the X chromosome at a different position from VSIG4 (Xq22.3 and Xq12, respectively). Of these eight members, only the structures of VSIG4 and TIGIT (RCSB-PDB ID: 5IMK and 3Q0H, respectively) were experimentally resolved through X-ray crystallization, while the rest had computationally predicted structures available. From these data, VSIG family members have been shown to have either an IgV domain, an IgC2 domain, or both (23, 26, 28–33). The IgV domain is shared by all members except for VSIG10 and VSIG10 L (UniProt Accession # Q8N0Z9 and Q86VR7), while IgC2 is a common feature of all members except for VSIG8 (UniProt Accession # P0DPA2) and TIGIT. Moreover, they are all type I transmembrane proteins with a wide range of expression. The structural composition of the extracellular domain (ECD) is highly conserved between hVSIG and mVSIG members.
Although the tissue distribution of VSIG proteins is not fully described due to antibody unavailability for some of the family members, genetic analysis and sequencing data reveal a diverse range of expression of VSIG proteins from testicular germ cells (VSIG1 and VSIG3) (32, 34) to the hair shaft (VSIG8) (35, 36) and immune cells (VSIG4 and TIGIT) (23–26, 33, 37–39), hence serving a wide array of functions in both humans and mice. A brief overview of VSIG family members and their functions, expression patterns, and roles in immunotherapy are summarized in Table 1 and Table 2 In the following context, we will discuss the role of each member of the VSIG family in detail, provide a comprehensive summary of our current understanding of these proteins, and highlight their potential as new targets for ICB therapy.
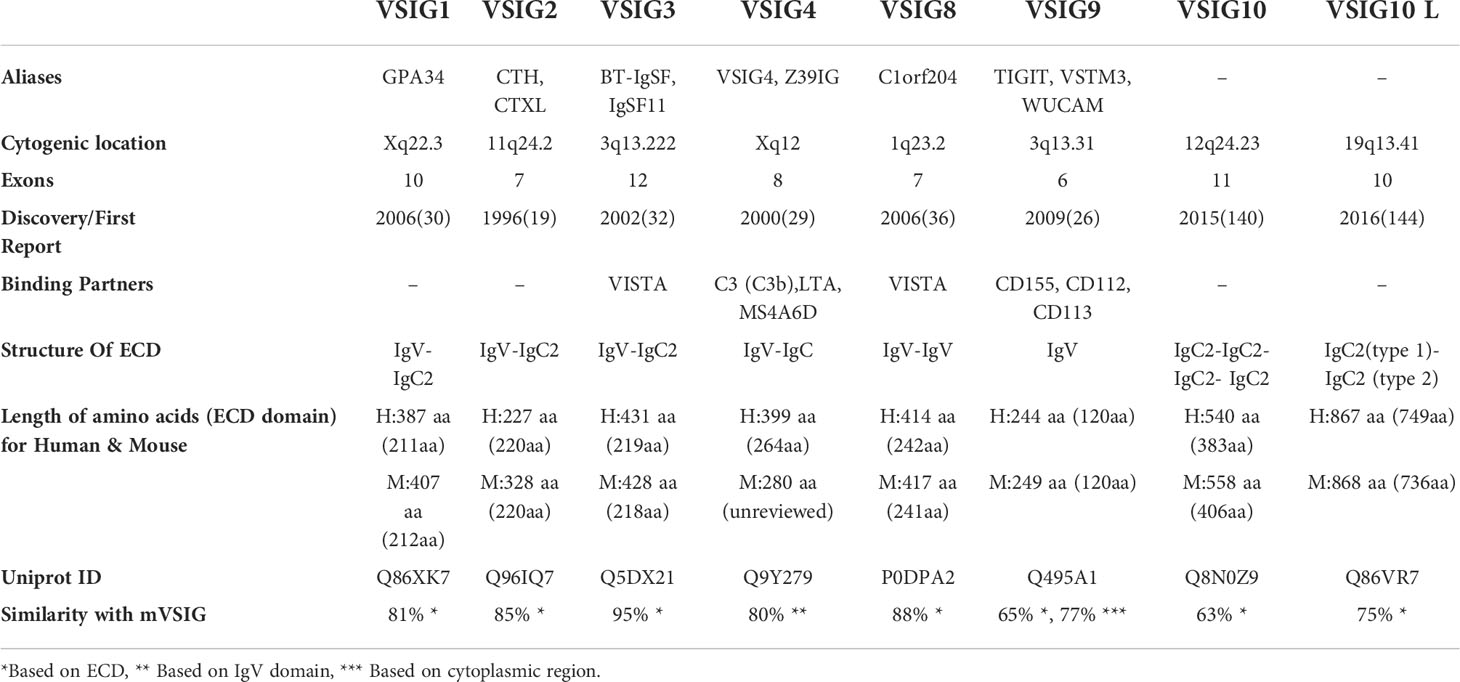
Table 1 Brief overview of VSIG family members.
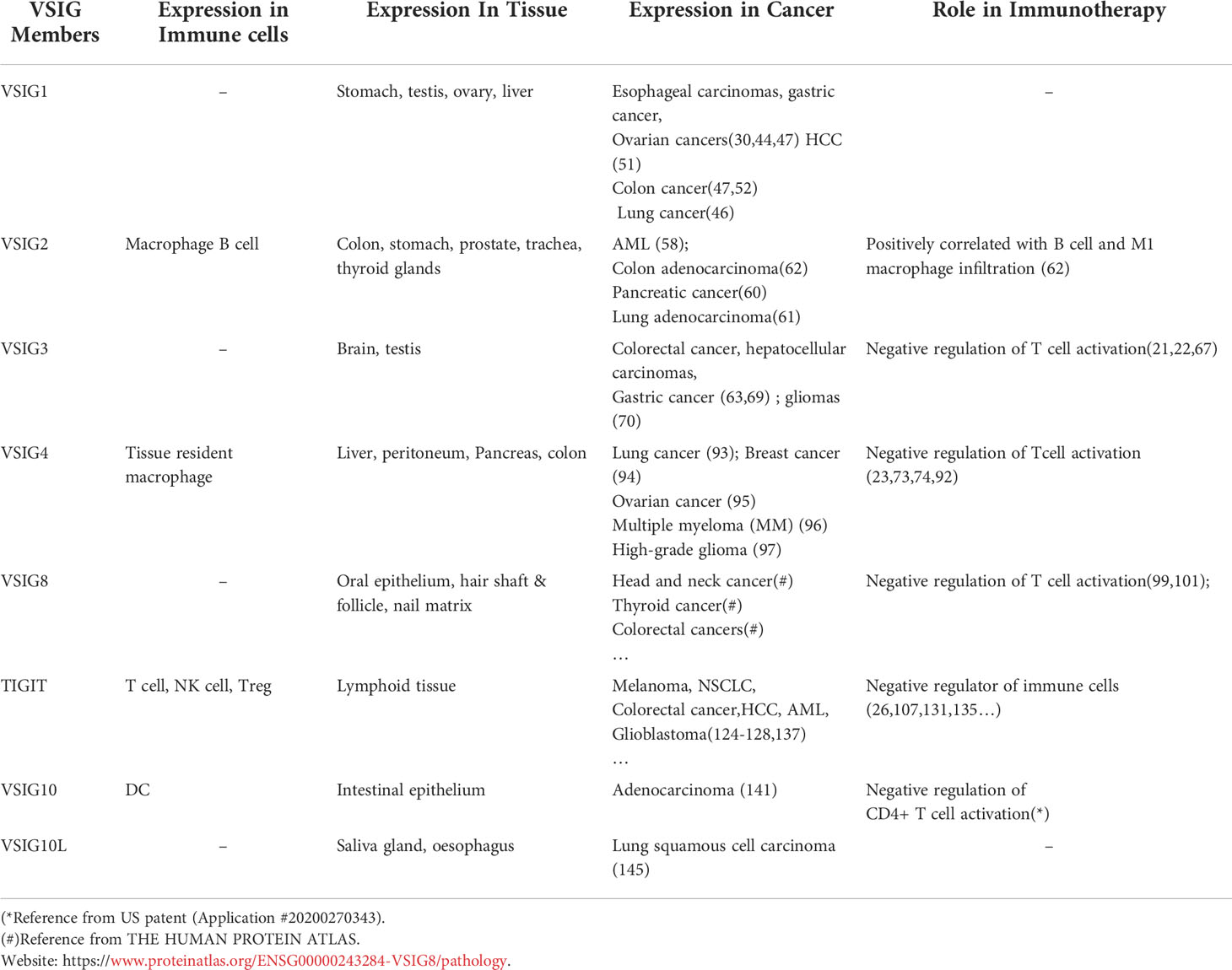
Table 2 Diverse expression and function of VSIG family members.
IgSF is a large protein superfamily of cell surface and soluble proteins involved in adhesion processes, binding, and cell recognition. Members of this superfamily are defined by structural similarities to immunoglobulins and the presence of an Ig domain (40). VSIG1 is a typical IgSF with two extracellular Ig-like domains and a short cytoplasmic domain. It is also known as the radioiodinated cell surface A33 antigen or glycoprotein A34, which was first characterized to be a tissue-restricted cell surface protein predominantly expressed in the gastric mucosa (30, 41). It was subcellularly localized in the adherens junctions of glandular epithelia and was critical for ensuring proper differentiation of glandular gastric epithelium (41). Three alternatively spliced isoforms of VSIG1, including VSIG1A, B and C, were identified in mice, with the latter being specifically expressed in the testis (41). In this tissue, VSIG1 was found to be a ZO1 (zonula occludens-1)-binding junctional adhesion molecule (JAM) localized on the surface of sperm cells to facilitate their interactions with Sertoli cells, suggesting that VSIG1 may be involved in supporting spermatogenesis (34). However, this has been challenged by a recent report showing that VSIG1 knockout mice had normal development and function of sperm cells, and whether the absence of phenotype upon VSIG1 deletion was caused by unknown compensatory mechanisms or genetic redundancy remains to be investigated (42).
Due to its abundant expression in the stomach, VSIG1 has been extensively studied in gastric cancers. In a cohort of 232 gastric adenocarcinoma samples, Chen et al. reported that VSIG1 was significantly reduced at both the mRNA and protein levels in gastric tumor tissues compared to paired noncancerous gastric mucosal tissues (43). Inoue et al. also reported a dramatic decrease in VSIG1 expression in 219 of 362 gastric cancer specimens (44). Furthermore, downregulation of VSIG1 was significantly correlated with poor overall survival and worse clinical outcome in gastric cancer patients (43–45), suggesting that VSIG1 may function as a tumor suppressor gene. In support of this, overexpression of VSIG1 diminished the proliferation and migration of multiple gastric cancer cell lines in vitro (44).
In contrast, VSIG1 seemed to be upregulated in a variety of nongastric carcinomas (30, 44, 46, 47). It was identified as a signature gene for gastric-type differentiation of serrated pathway-associated colon carcinoma (47–49) and lung adenocarcinoma (50). The coexpression of VSIG1 in the cytoplasm of hepatocytes with thyroid transcription factor 1 (TTF1) was also considered to be a potential lineage shift indicator of conventional to gastric-type hepatocellular carcinoma (HCC) (51). In the same study, Gurzu et al. further revealed that VSIG1 was strongly correlated with epithelial-mesenchymal transition (EMT) genes, such as E-cadherin and N-cadherin and VIM (51). Since VSIG1 is known to function as a JAM involved in tight junction assembly, these data implicated a potential role of VSIG1 in modulating EMT during tumor metastasis. In support of this, Bernal et al. showed that VSIG1 knockdown increased while gain of VSIG1 inhibited the migration of colon cancer cells (52). Overall, although the function of VSIG1 in modulating antitumor immune responses has not been explored thus far, given its importance as a cell surface tumor suppressor in contraining tumor growth and metastasis, targeting VSIG1 would be of great value for the treatment of different types of cancer (30).
VSIG2 is composed of an ECD of 220 aa containing IgC2 and IgV domains and a cytoplasmic tail of 63 aa (28). VSIG2 is also known as CTXL (cortical thymocyte-like protein). It was initially identified as a marker predominantly expressed on cortical thymocytes in Xenopus and was designated CTX (cortical thymocyte of Xenopus). Due to the abundant expression of VSIG2 on double-positive thymocytes of Xenopus and on recent T-cell immigrants in chickens, it was considered to be involved in T-cell development in these species (27). However, by cloning its mouse and human homologues, namely, CTM (cortical thymocyte of mouse) and CTH (crotical thymocyte of human), respectively, VSIG2 was found to be abundantly expressed in the thyroid glands, trachea, prostate, colon, and stomach but weakly expressed in the lung and bladder but not in the thymus (28). These initial data suggest that VSIG2 may be an ancestral lymphocyte receptor before the introduction of somatic rearrangement in mammals.
Although the physiopathological function of VSIG2 remains to be explored, a close association of VSIG2 with the progression of various human diseases has been demonstrated in recent years using multiomics approaches, highlighting the potential of VSIG2 as a biomarker for the diagnosis of many diseases. VSIG2 was found to be significantly upregulated in the corneal samples of Fuchs endothelial corneal dystrophy (FECD) patients (53), in the intestinal biopsy of irritable bowel syndrome (IBS-D) patients (54), in the plasma of acute tubular injury and interstitial fibrosis/tubular atrophy patients (55), and in the plasma of incident heart failure patients (56). Moreover, the single nucleotide polymorphisms (SNPs) of VSIG2 are strongly associated with serologic profile and cytokine phenotype in systemic lupus erythematosus (SLE) (57). Aberrant VSIG2 expression was also found in tumors. Heimeng et al. reported that VSIG2 expression in acute myeloid leukemia (AML) patients correlated with poor prognosis according to The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) databases (58). VSIG2 expression has been found to be significantly upregulated in patients with nonmuscle invasive bladder cancer but downregulated in patients with muscle invasive bladder cancer; thus, it could serve as a biomarker of invasiveness in bladder cancers (59). In primary lung adenocarcinoma and pancreatic cancer, VSIG2 was characterized as a member of the DNA methylation-related prognostic signature (60, 61). High expression and methylation of VSIG2 correlated with poor survival in these cancer patients (58, 60, 61).
The potential effect of VSIG2 in modulating antitumor immunity was also implicated in colorectal cancers. In a recent study, Cui Z et al. reported lower expression of VSIG2 in colon adenocarcinoma (COAD) samples, and its downregulation was associated with a poor overall survival rate in COAD patients (62). However, it appears that VSIG2 functions as a tumor suppressor gene to ensure tumor immune surveillance rather than an immune checkpoint molecule in this particular cancer type. Interestingly, VSIG2 expression positively correlated with B-cell and M1 macrophage infiltration (62). Since these cells are normally the most abundant antigen-presenting cells in tumor tissues, dissecting the role of VSIG2 in these cells may have implications for understanding the biology of T-cell activation in the tumor microenvironment.
VSIG3 contains a V-type and C2-type immunoglobulin domain, a C-terminal PDZ domain, and a transmembrane domain (63). It was first cloned in 2002 and was then indicated to be a cell adhesion molecule that mediates homophilic cellular interactions (32). VSIG3 is also known as IgSF gene 11 (IgSF11) or brain- and testis-specific IgSF (BT-IgSF) because of its abundant expression in these two organs in mammals (32, 64). hVSIG3 contains 12 exons encoding two isoforms that share 97% amino acid identity and are thus considered to be identical in function. VSIG3-mediated cell adhesion can regulate the development of neurons and excitatory synaptic transmission and the differentiation of osteoclasts, and its mutation in zebrafish was associated with impaired migration and survival of melanophores (65, 66).
In addition to its role in cell adhesion, one of the most notable functions of VSIG3 relies on its capacity to regulate immune responses. VSIG3 is reported to be a ligand for the novel B7 family immune checkpoint V-domain immunoglobulin suppressor of T-cell activation (VISTA) (21, 22, 67). VISTA is mainly expressed on naïve T cells and functions as an important regulator in maintaining T-cell tolerance through the induction of peripheral T-cell deletion (68). Truncated VSIG3 ECD containing either the IgV- or IgC2-type domain bound to human VISTA protein in a similar manner as the full-length ECD. Most importantly, the crosslinking of VSIG3 during TCR stimulation significantly inhibited T-cell activation by reducing the production of cytokines and chemokines. Blockade of VISTA significantly attenuated VSIG3-mediated T-cell inhibition, suggesting that this process is dependent on its recognition with VISTA (67). The VSIG3 and VISTA interaction was further demonstrated by Xie et al. using a coimmunoprecipitation (Co-IP) assay (21). They also revealed the crystal structure of the human VSIG3 ECD and designed a small molecule inhibitor, K284-3046, based on protein−protein docking analysis. This chemical inhibitor showed potent effects in diminishing VSIG3-mediated T-cell suppression (21).
The identification of VSIG3 as a binding partner for VISTA has important implications for tumor immunotherapy. As a well-known coinhibitory molecule for T cells, VISTA is highly expressed in myeloid cells and T cells that infiltrate into tumors and help create an immunosuppressive tumor microenvironment by enhancing Treg differentiation and inhibiting T-cell activation. In contrast, VSIG3 was highly expressed in a number of cancers, such as colorectal cancers, gastric cancer, hepatocellular carcinomas, and gliomas, but not on immune cells (63, 69). Suppression of VSIG3 by small interfering RNA (siRNA) attenuated the proliferation of gastric cancer cells in vitro, suggesting that the expression level of VSIG3 is essential for the fate of cancer cells (63). Ghouzlani et al. also found that high VSIG3 expression was related to a strong immunosuppressive microenvironment and functionally compromised T cells in glioma (70). Therefore, it is speculated that highly upregulated VSIG3 in tumor cells could reinforce immune inhibitory signals to VISTA-expressing T cells in the tumor microenvironment, generating antibodies or chemical inhibitors that specifically block the VSIG3-VISTA interaction and could increase the efficiency of VISTA-based ICB therapy (22).
VSIG4, also known as Z39Ig or CRIg, was first described in 2000 as an X chromosome-located gene (29). VSIG4 contains two Ig-like domains, one complete IgV domain and a truncated IgC domain, and shares all the conserved amino acids with known B7 family members; thus, it is also considered a B7 family-related protein (23). There exist two alternatively spliced forms of human VSIG4 protein: the long isoform of VSIG4(L) encodes both IgV and IgC2 domains, while the short isoform of VSIG4(S) encodes only a single IgV domain (37). In comparison, murine VSIG4 only shows the short isoform of a single IgV domain. Human and mouse VSIG4 share 83% sequence homology within the IgV domain (71). The IgV domain is responsible for binding to the β chain of C3b to promote phagocytosis (72). The intracellular portion of VSIG4 contains a cAMP/cGMP-dependent protein kinase phosphorylation site and a protein kinase C phosphorylation site, yet the function of these sites remains unclear (29). Notably, VSIG4 expression is restricted to tissue resident macrophages, including liver Kupffer cells (37), peritoneal macrophages (23), pancreatic macrophages (73, 74), synovial lining macrophages in the joint, and interstitial macrophages in the heart (38, 75, 76); however, the extent of expression on these macrophages can be downregulated by inflammatory cytokines (77, 78). This expression pattern suggests a role for VSIG4 in maintaining tissue homeostasis.
VSIG4 is well known as the complement receptor of the Ig superfamily (CRIg) with a high binding affinity to complement components C3b and iC3b. Upon activation, C3—the central component of the complement system—is cleaved to a small C3a fragment and a large C3b fragment by C3 convertase, and iC3b is subsequently produced by complement factor I-mediated cleavage of C3b (79, 80). Both C3b and iC3b are potent opsonins that can coat the surface of invading pathogens, apoptotic cells, or immune complexes to facilitate their clearance by the mononuclear phagocytic system. By recognizing C3b- and iC3b-tagged pathogens, accumulating evidence suggests a critical role of VSIG4 in host defense against bloodstream infections by promoting Kupffer cells to take up complement-tagged bacteria, fungi, and viruses (37, 81, 82). This function is vital to prevent the dissemination of pathogens to some vulnerable organs, such as the heart and kidney (37). However, it was also reported that VSIG4 facilitated a relatively slow clearance of circulating bacteria when compared to scavenger receptor-mediated fast clearance (83). This slow clearance of circulating bacteria may be essential to enable a timely induction of adaptive immune responses. Moreover, VSIG4 was suggested to be a pattern recognition receptor that directly binds and captures blood-borne gram-positive bacteria by recognizing lipoteichoic acid (LTA) during Staphylococcus aureus infection (84).
Structural analysis revealed that VSIG4 can also dramatically inhibit the activity of C3 and C5 convertase upon binding to C3b, thereby preventing the alternative pathway of complement activation (72). Given that inappropriate activation of complement is usually associated with unwanted and exacerbated inflammation, recombinant VSIG4-Fc protein has been exploited as a decoy receptor to efficiently alleviate a variety of inflammatory diseases by preventing excessive complement activation, such as experimental autoimmune uveoretinitis (EAU) (85), intestinal ischemia/reperfusion (IR) injury (86), type 1 diabetes (73, 74), arthritis (71, 72), and SLE (87). This complement inhibitory function could be further improved by fusing VSIG4 with the alternative pathway inhibitory domain of factor H (FH) (88). In addition, recent studies reported that VSIG4 can directly inhibit macrophage-mediated inflammation independent of complement. Huang et al. found that macrophages lacking VSIG4 showed increased activation of the NLRP3 inflammasome upon stimulation (89). VSIG4 was found to interact with the transmembrane protein MS4A6D to form a surface inhibitory signaling complex, leading to attenuated NLRP3 inflammasome activation via a JAK2-STAT3-A20 signaling cascade (89, 90). In addition, VSIG4 was also able to intervene in mitochondrial pyruvate metabolism in macrophages by activating the PI3K-Akt-STAT3 pathway, thereby resulting in reduced oxidative phosphorylation and diminished M1 polarization of macrophages (91). These data suggest the possible applications of targeting VSIG4 in treating inflammatory diseases.
As a macrophage-specific immune regulator, VSIG4 is a potent coinhibitory ligand that strongly suppresses T-cell proliferation and cytokine production. T-cell stimulation in the presence of recombinant VSIG4 caused T-cell anergy, cell cycle arrest at the G0/G1 phase (23, 73, 74, 92), and skewed differentiation of CD4+ T cells towards Foxp3+ Treg cells. Importantly, this T-cell inhibitory effect was only found with plate-bound but not soluble VSIG4 protein, suggesting that the crosslink of VSIG4 with a putative binding partner on the surface of T cells is required to deliver inhibitory signals. Indeed, VSIG4 can directly bind activated T cells without the need for serum, demonstrating the existence of a complement-independent ligand of VSIG4 on T cells, which remains to be determined. Although VSIG4 expression is restricted in tissue-resident macrophages at a steady state, several studies have reported an upregulation of VSIG4 expression in lung cancer (93), breast cancer (94), ovarian cancer (95), and multiple myeloma (MM) (96). By examining VSIG4 expression in tumor tissues, it was found to be highly enriched in tumor-associated macrophages but not in tumor cells or other immune cells (93). High expression of VSIG4 is correlated with poor prognosis of high-grade glioma (97), and its deficiency led to significantly inhibited growth of Lewis lung cancer cells (LLC) in mice (93). Based on these findings, VSIG4 is becoming an attractive macrophage-specific immune checkpoint molecule in cancer immunotherapy. Identifying the ligand of VSIG4 on T cells would be pivotal for understanding the mechanisms whereby VSIG4 modulates antitumor T-cell responses and is fundamentally important for developing high-efficacy inhibitors that aim to block VSIG4-mediated T-cell suppression in cancer Figure 1.
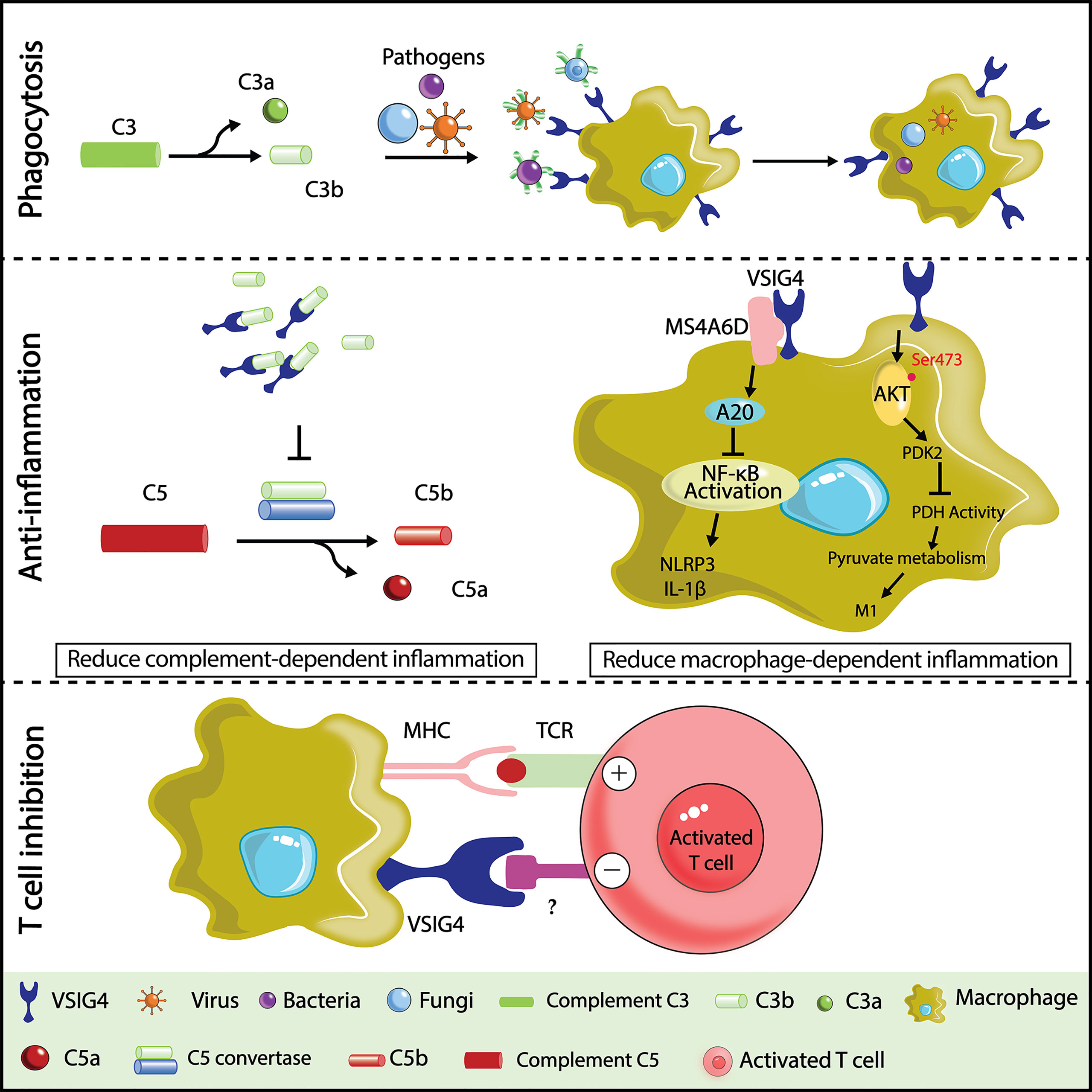
Figure 1 Illustration of VSIG4 functions. In host defense against bloodstream infections,VSIG4 recognizes C3b and helps macrophages phagocytose C3b- or iC3b-tagged pathogens (bacteria, viruses, fungi, etc.). In a variety of complement activation-dependent inflammatory diseases, VSIG4 delivers anti-inflammatory signals by binding C3b to prevent the alternative pathway of complement activation. VSIG4 also inhibits macrophage M1 activation by regulating inflammasome activation and pyruvate metabolism. VSIG4 also inhibits T-cell activation, proliferation, and IL-2 production upon binding to an unknown ligand on T cells.
VSIG8 is a relatively less explored member of the VSIG family, approximately 45 kDa in size. Mature hVSIG8 contains two Ig-V domains, which are a part of the ECD spanning 242 aa. hVSIG8 shares 88% and 89% identity with the VSIG8 of mouse and rat, respectively. It was identified through proteomic analysis of the human hair shaft (35, 36) and found to also be expressed in the oral epithelium, superficial layers of the nail matrix, and hair follicles (98). Interestingly, Wang et al. reported that immobilized recombinant VSIG8 could suppress human T-cell proliferation and cytokine production and decrease the differentiation of naïve CD4+ T cells towards Th1 cells, confirming its role as a negative regulator of T-cell responses (99). VSIG8 was later reported to be a putative binding partner of VISTA (100), and a US patent (WO2016090347 A1) also reported the interaction of VSIG8 and VISTA, demonstrating the suppressive effects of this interaction on T cells. In addition, Chen et al. demonstrated the VSIG8-VISTA interaction by ELISA, MST and Co-IP assays and confirmed its function in inhibiting human T-cell activation (101). However, a recent study using a functional ELISA suggested no interaction between human VSIG8 and VISTA (67). Similarly, George et al. generated a two-sided fusion protein that contained the ECD of both VSIG8 and OX40 L and reported no binding between this fusion protein and recombinant VISTA, although it was able to bind VISTA-expressing macrophages or tumor cells. Nevertheless, this VSIG8-Fc-OX40 L fusion protein stimulated T-cell activation and antitumor activity, possibly by blocking VSIG8-mediated inhibitory signaling (102). Future studies generating VSIG8-deficient animals and blocking antibodies will further enhance our understanding of this potential immune checkpoint molecule in cancer immunotherapy.
VSIG9, well known as TIGIT, with a full name of T-cell immunoglobulin and ITIM domain (also known as WUCAM or Vstm3), is currently one of the most attractive and promising immune checkpoint targets. TIGIT also belongs to the poliovirus receptor (PVR)/nectin family and is widely expressed on activated NK cells, CD8+ T cells, CD4+ T cells, and Treg cells (24–26, 33, 39). TIGIT was discovered in 2009 by three independent groups (25, 26, 33). One reported that TIGIT was an adhesion molecule mediating TFH (follicular T helper) and FDC (follicular DC) interactions (33), whereas the other two identified TIGIT as a coinhibitory receptor on T and NK cells (25, 26). The structure of TIGIT comprises a short intracellular domain with one immunoglobulin tyrosine tail (ITT)-like motif and one immunoreceptor tyrosine-based inhibitory motif (ITIM), a type I transmembrane domain, and an extracellular IgV domain (26, 33, 39). While there is a 58% overall sequence identity between human and mouse TIGIT (25, 26), their ITIM-containing sequences that confer immune inhibitory functions are identical. TIGIT has three ligands, including CD155, CD112, and CD113, which all belong to the PVR/nectin family receptors (25, 103, 104). Their expression features and binding affinity with TIGIT are listed in Table 3.

Table 3 Expression patterns of TIGIT ligands and their relative binding affinity and function.
Among these ligands, CD155 exhibited the highest affinity with TIGIT (104, 105). CD155 is mainly expressed on dendritic cells (DCs), T cells, B cells and macrophages. Engaging of TIGIT with CD155 has been shown to prevent excessive immune cell activation and sustain immune homeostasis (33, 106–108). Notably, there are two other PVR family receptors, CD226 and CD96, which share sequence homology with TIGIT and compete with TIGIT for CD155 binding (109–111). However, as opposed to TIGIT and CD96, CD226 acts as an activating receptor that promotes T-cell and NK-cell activation opon CD155 ligation (112–114). TIGIT binds CD112 and CD113 with lower affinity than CD155, and the functional consequences of their binding have been less characterized. CD112 is also known as the ligand for the coinhibitory receptor CD112R, which was recently discovered as an immune checkpoint receptor expressed on T cells and NK cells (109, 115). Similar to CD155, CD112, another common ligand of TIGIT, can also bind CD226 (109). The competition and balance among TIGIT, CD226, CD96, and CD112R for the same ligands is quite complex and has been extensively reviewed elsewhere (112, 116, 117). The interactions between TIGIT and its ligands with other VSIG members are summarized in Figure 2.
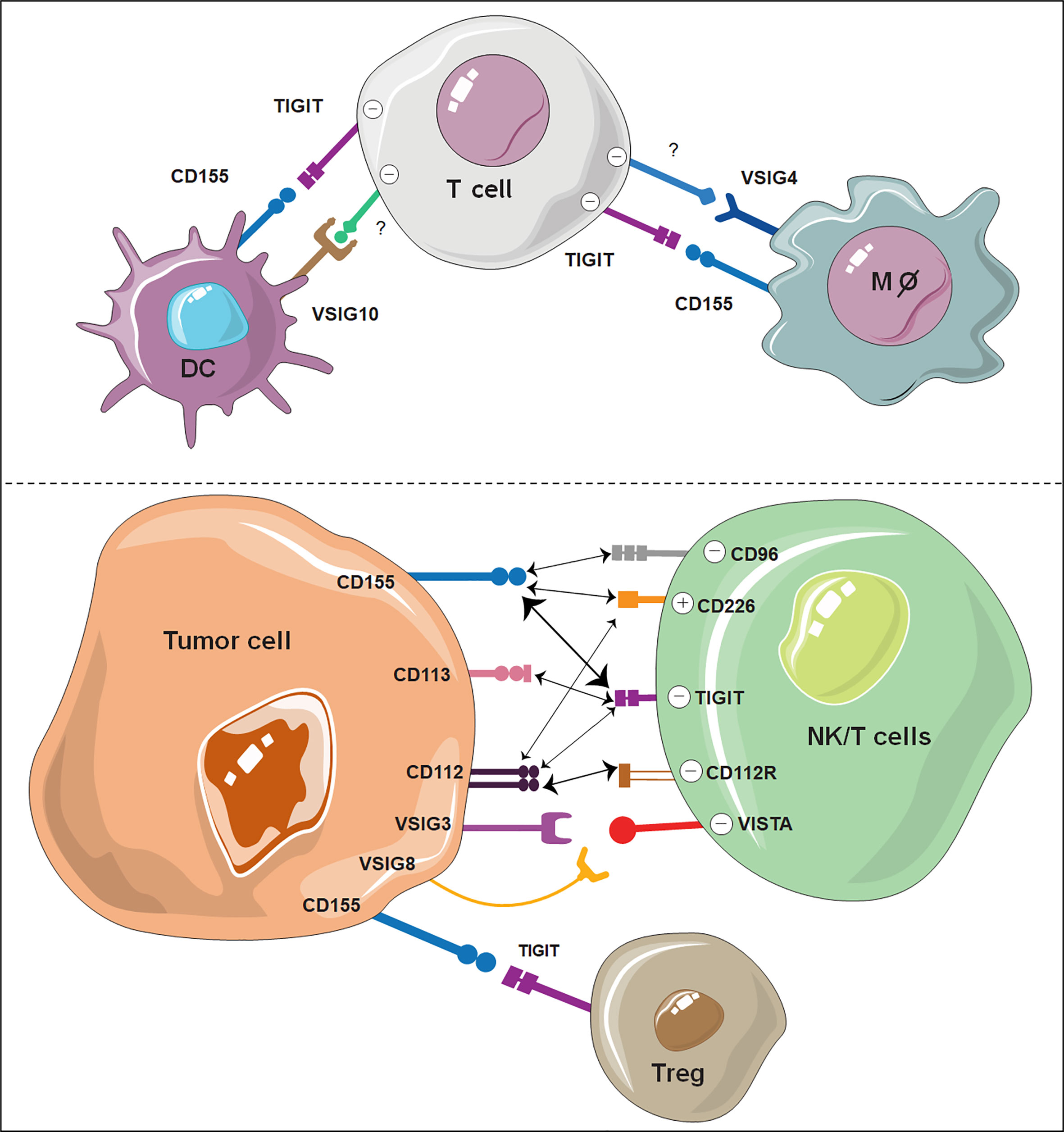
Figure 2 Illustration of VSIG family members as potential ICBs on immune cells. The interaction of TIGIT, VSIG3, VSIG8 and VSIG4 with other ligand-expressing cells shows great potential as novel immune checkpoints. VSIG10 also shows potential as a coinhibitory receptor on DCs. The width of the arrows is proportional to the relative binding affinities. The strongest interactions are between TIGIT/CD155 and CD112R/CD112. Negative (-) represents an inhibitory signal, and positive (+) represents an activating signal.
Because of the binding affinity advantage, the TIGIT-CD155 interaction prevails in TIGIT-mediated immune inhibition (26). The TIGIT-CD155 axis exerts its inhibitory effects on T and NK cells through three distinct mechanisms of action, including 1) a cell intrinsic manner by transmitting inhibitory signals to the effector cells (26); 2) a cell extrinsic manner by modulating the cytokine profile of CD155-expressing cells, such as DCs and macrophages (26, 107); and 3) by competing with CD226-mediated costimulatory signals (118). The cell intrinsic effect of TIGIT was well characterized by Inozume et al. by expressing truncated CD155 without a cytoplasmic domain, which was sufficient to suppress T-cell production of IFN-γ in a TIGIT-dependent manner (119). Similarly, agonistic anti-TIGIT mAbs were capable of dampening mouse and human T-cell proliferation and cytokine production (39, 118, 120). Following TIGIT-CD155 binding, the ITT-like motif is phosphorylated and subsequently bound to growth factor receptor-bound protein 2 (Grb2) or β-arrestin 2, which leads to the recruitment of SHIP-1 and SHP2 and abolishment of PI3K and MAPK and NF-κB signaling (121). TIGIT can also work in a cell-extrinsic manner by modulating the cytokine profile of CD155-expressing cells, such as DCs and macrophages. The altered cytokine milieu, e.g., increased IL-10 levels and decreased IL-12 levels, in turn, can lead to attenuated activation of NK and T cells (26).
The tumor microenvironment has taken advantage of the TIGIT-CD155 inhibitory pathway as an important strategy to evade immune surveillance and thus result in uncontrolled tumor growth (122, 123). TIGIT is highly expressed on CD8+ tumor-infiltrating lymphocytes (TILs) in various tumors, such as gastric cancer, colon cancer, breast cancer, melanoma, and NSCLC (124–127). TIGIT+ CD8+ TILs are dysfunctional with reduced effector cytokine production and impaired degranulation, exhibiting a typical feature of exhaustion. Blocking the TIGIT pathway can drastically reverse T-cell exhaustion. In AML, TIGIT+ CD8+ exhausted T cells were reinvigorated by knockdown of TIGIT expression (128). The prominent advantage of TIGIT over other immune checkpoints lies in its potent ability to restrain NK-cell responses. Upon binding to its ligand CD155 expressed by tumor cells, TIGIT-expressing NK cells dramatically diminish their cell cytotoxicity (129, 130). Zhang Q et al. showed that antibody blockade of TIGIT prevented NK exhaustion and unleashed antitumor immunity in an NK-cell-dependent manner, collectively leading to tumor regression (131). Moreover, TIGIT is highly expressed on Treg cells and is essential to maintain the suppressive capabilities of Tregs, which potentially inhibit a variety of immune cells by suppressing Th1 and Th17 cells (132–135). A study demonstrated that TIGIT suppresses antitumor immunity primarily via Tregs but not CD8+ T cells (135). In addition, TIGIT also functions as a ligand to skew the maturation or polarization of intratumoral myeloid cells, including DCs and macrophages, towards a state with increased IL-10 but decreased IL-12 secretion (26). This results in DC tolerance and alternative activation of macrophages, both contributing to tumor immune tolerance.
Owing to the importance of TIGIT-CD155 engagement in NK and T-cell dysfunction in tumors, developing therapeutic agents to block this pathway holds great promise for cancer immunotherapy. There is strong evidence that TIGIT blockade has a direct effect in reversing T-cell dysfunction in cancer patients. Anti-TIGIT mAb treatment has been shown to escalate the proliferation, cytokine production, and degranulation of bone marrow CD8+ T cells from MM patients and peripheral blood CD8+ T cells from melanoma patients (125, 136). Recent advances have also proposed a dual blockade of PD-1 and TIGIT as a more inspiring method for cancer immunotherapy than a single TIGIT blockade. Whereas blocking each of the PD-1/PD-L1 or TIGIT pathways does not remarkably impede the growth of CT26 tumors, a dual blockade synergizes to increase the proliferation and function of antitumor CD8+ T cells, which results in protective memory T cells, complete tumor rejection, and overall prolonged survival (126, 131). These effects have been abrogated upon CD8+ T-cell deficiency. The translational potential of dual PD-1/TIGIT inhibition has already been demonstrated; it increases the proliferation and function of intratumoral antigen-specific CD8+ T cells in melanoma patients to an extent that is much more dramatic than a single blockade (119, 125). A recent phase II study also indicated that dual PD-L1/TIGIT blockade (atezolizumab/tiragolumab) has superior clinical benefits compared to PD-L1 blockade alone in NSCLC patients, despite similar profiles of toxicity and tolerability (137). Apart from PD-1, TIGIT blockade could also synergize with other ICBs in cancer immunotherapy. For instance, TIGIT and TIM-3 inhibition in mice cooperated to promote an antitumor immune response (135); dual blockade of TIGIT and LAG3 improved the treatment efficacy in a mouse model of anti-PD-1-resistant lung cancer (138). In conclusion, TIGIT, as a new immune checkpoint, possesses great potential for cancer immunotherapy. Effective tumor control for certain types of cancer can be expected by combining anti-TIGIT with other ICB inhibitors.
VSIG10 contains four Ig-like C2-type domains in its ECD with 63% identity between hVSIG10 and mVSIG10 (139, 140). VSIG10 was highly expressed on both normal and cancer epithelial cells based on transcriptional data. Moreover, Papasotiriou et al. reported the overexpression of VSIG10 in adenocarcinoma; however, no expression was observed in melanoma, prostate, breast, or pancreatic cancer (141). Until recently, there was no report about its biological function. According to a US patent (Application #20200270343), recombinant VSIG10-Fc fusion protein was able to suppress CD4+ T-cell activation and cytokine production, pinpointing its potential as an immune checkpoint inhibitor. Interestingly, VSIG10 was also predicted to be abundantly expressed by DC subsets both in humans and mice. Growing data points toward the importance of immune checkpoints expressed on DCs in dampening the antitumor response. For instance, DCs highly express PD-L1 and have been demonstrated to be an important target of PD-L1 blocking antibodies (142). These PD-L1-expressing DCs are identified to attenuate T-cell activation and regulate its response to ICBs (143).
Hence, the generation of anti-VSIG10 antibodies, as reported in the patent, may present a promising DC-targeting ICB cancer immunotherapy. Similar to VSIG10, VSIG10 L also contains IgC2 in its ECD. VSIG10 L is normally expressed in the healthy esophagus and squamous mucosa; however, it is downregulated in esophageal adenocarcinoma and Barrett’s esophagus (144). High expression is found in lung squamous cell carcinoma (145), pointing to the dual nature of VSIG10 L in cancers. Further exploration is needed, as they could be potential biomarkers or immune checkpoints.
ICB treatment has brought a revolutionary advance for cancer therapy in the past decade. Antibodies targeting PD-1 and CTLA-4 were approved by the FDA and were proven to be effective against several cancer types. Despite tremendous success, over half of the patients remain poorly responsive to these regimens, possibly due to the involvement of multiple immune inhibitory pathways in the cancer microenvironment. Therefore, seeking new immune checkpoint molecules is becoming increasingly important for the optimization of ICB-based cancer immunotherapy. Here, we show that many VSIG family members show potent effects of T-cell inhibition in cancer, and antitumor immunity can dramatically benefit from the blockade of these molecules. The most attractive and promising member of the VSIG family is TIGIT, and its blockade has achieved great success in reinvigorating antitumor NK and T-cell responses (126, 131, 135). There are currently over 50 clinical trials underway to study the therapeutic effect, safety, and tolerability of TIGIT blockade in cancer, either using it alone or in combination with other cancer therapeutics. Apart from TIGIT, VSIG3, VSIG8, and VSIG4 also show great potential as novel immune checkpoints. As putative binding partners for the well-known coinhibitory molecule VISTA, both VSIG3 and VSIG8 were able to negatively regulate T-cell responses and can be targeted in certain cancer types in which antitumor immunity is predominantly affected by the VISTA pathway. VSIG4 is of particular interest because it is specifically expressed by tissue resident macrophages, which are becoming increasingly appreciated as critical contributors to tumor progression and metastasis. Blockade of VSIG4 to functionally reprogram macrophages thus stands out as an important complement to the current T-cell-based immunotherapy regimens. In addition, although not fully validated, VSIG10 shows potential as a coinhibitory receptor expressed by another important type of myeloid immune cell, namely, DCs, which are also largely overlooked in the field of immune checkpoint therapy. Overall, VSIG family proteins represent an important group of transmembrane receptors that emerge as immune checkpoints controlling the fates of multiple types of immune cells in tumors, spanning from myeloid cells to lymphoid cells. Therapeutically targeting these proteins could be beneficial to the current regimen of ICB treatment in cancer.
XZ and SK wrote the manuscript, LL and DH supervised the study and revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the NSFC (82003008), the China Postdoctoral Science Foundation (2021M703076 and 2020M682048), the Fundamental Research Funds for the Central Universities (WK9110000171 and WK9110000086), the Anhui Provincial Natural Science Foundation of China (2008085MH299) and the Postdoctoral Research Funding of Anhui Province in 2019 (2019B371).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
ICB, immune checkpoint blockade; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; AML, acute myeloid leukemia; TCGA, The Cancer Genome Atlas; GEO, Gene Expression Omnibus; COAD, colon adenocarcinoma; VISTA, V-domain immunoglobulin suppressor of T-cell activation; Co-IP, coimmunoprecipitation; MM, multiple myeloma; TIGIT, T-cell immunoreceptor with immunoglobulin and ITIM domain; PVR, poliovirus receptor; TFH, follicular T helper; FDC, follicular DC; ITT, immunoglobulin tyrosine tail; ITIM, immunoreceptor tyrosine-based inhibitory motif; LTA, recognizing lipoteichoic acid; ELISA, Enzyme-linked Immuno Sorbent Assay; MST, Microscale Thermophoresis; ECD, extracellular domain; TTF1, subcellular localization of thyroid transcription factor 1; SLE, systemic lupus erythematosus; NSCLC, non-small cell lung carcinoma; FECD, fuchs endothelial corneal dystrophy; JAM, junctional adhesion molecule; SNP, single nucleotide polymorphisms; EMT, epithelial-mesenchymal transition; IBS-D, intestinal biopsy of irritable bowel syndrome; HCC, hepatocellular carcinoma.
1. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12(4):252–64. doi: 10.1038/nrc3239
2. Carlino MS, Larkin J, Long GV. Immune checkpoint inhibitors in melanoma. Lancet (2021) 398(10304):1002–14. doi: 10.1016/S0140-6736(21)01206-X
3. Garon EB, Rizvi NA, Hui RN, Leighl N, Balmanoukian AS, Eder JP, et al. Pembrolizumab for the treatment of non-Small-Cell lung cancer. New Engl J Med (2015) 372(21):2018–28. doi: 10.1056/NEJMoa1501824
4. Ribas A, Hamid O, Daud A, Hodi FS, Wolchok JD, Kefford R, et al. Association of pembrolizumab with tumor response and survival among patients with advanced melanoma. Jama-Journal Am Med Assoc (2016) 315(15):1600–9. doi: 10.1001/jama.2016.4059
5. Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science (2018) 359(6382):1350. doi: 10.1126/science.aar4060
6. Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. New Engl J Med (2015) 373(19):1803–13. doi: 10.1056/NEJMoa1510665
7. Rini BI, Plimack ER, Stus V, Gafanov R, Hawkins R, Nosov D, et al. Pembrolizumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. New Engl J Med (2019) 380(12):1116–27. doi: 10.1056/NEJMoa1816714
8. Yau T, Park JW, Finn RS, Cheng AL, Mathurin P, Edeline J, et al. LBA38_PR - CheckMate 459: A randomized, multi-center phase III study of nivolumab (NIVO) vs sorafenib (SOR) as first-line (1L) treatment in patients (pts) with advanced hepatocellular carcinoma (aHCC). Ann Oncol (2019) 30:v874–5. doi: 10.1093/annonc/mdz394.029
9. Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363(8):711–23. doi: 10.1056/NEJMoa1003466
10. Specenier P. Ipilimumab in melanoma. Expert Rev Anticancer Ther (2016) 16(8):811–26. doi: 10.1080/14737140.2016.1211936
11. Robert C, Thomas L, Bondarenko I, O'Day S, Weber J, Garbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med (2011) 364(26):2517–26. doi: 10.1056/NEJMoa1104621
12. Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob JJ, Cowey CL. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. New Engl J Med (2017) 377(14):1345–56. doi: 10.1056/NEJMoa1709684
13. Motzer RJ, Tannir NM, McDermott DF, Frontera OA, Melichar B, Choueiri TK, et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. New Engl J Med (2018) 378(14):1277–90. doi: 10.1056/NEJMoa1712126
14. Hellmann MD, Ciuleanu TE, Pluzanski A, Lee JS, Otterson GA, Audigier-Valette C, et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. New Engl J Med (2018) 378(22):2093–104. doi: 10.1056/NEJMoa1801946
15. Wolf Y, Anderson AC, Kuchroo VK. TIM3 comes of age as an inhibitory receptor. Nat Rev Immunol (2020) 20(3):173–85. doi: 10.1038/s41577-019-0224-6
16. He X, Xu C. Immune checkpoint signaling and cancer immunotherapy. Cell Res (2020) 30(8):660–9. doi: 10.1038/s41422-020-0343-4
17. Kraehenbuehl L, Weng C-H, Eghbali S, Wolchok JD, Merghoub T. Enhancing immunotherapy in cancer by targeting emerging immunomodulatory pathways. Nat Rev Clin Oncol (2022) 19(1):37–50. doi: 10.1038/s41571-021-00552-7
18. Andrews LP, Yano H, Vignali DAA. Inhibitory receptors and ligands beyond PD-1, PD-L1 and CTLA-4: breakthroughs or backups. Nat Immunol (2019) 20(11):1425–34. doi: 10.1038/s41590-019-0512-0
19. Kalbasi A, Ribas A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat Rev Immunol (2020) 20(1):25–39. doi: 10.1038/s41577-019-0218-4
20. Collins M, Ling V, Carreno BM. The B7 family of immune-regulatory ligands. Genome Biol (2005) 6(6):223. doi: 10.1186/gb-2005-6-6-223
21. Xie X, Chen C, Chen W, Jiang J, Wang L, Li T, et al. Structural basis of VSIG3: The ligand for VISTA. Front Immunol (2021) 12:625808. doi: 10.3389/fimmu.2021.625808
22. Yang W, Padkjaer SB, Wang JS, Sun Z, Shan B, Yang L, et al. Construction of a versatile expression library for all human single-pass transmembrane proteins for receptor pairings by high throughput screening. J Biotechnol (2017) 260:18–30. doi: 10.1016/j.jbiotec.2017.08.023
23. Vogt L, Schmitz N, Kurrer MO, Bauer M, Hinton HI, Behnke S, et al. VSIG4, a B7 family-related protein, is a negative regulator of T cell activation. J Clin Invest (2006) 116(10):2817–26. doi: 10.1172/JCI25673
24. Manieri NA, Chiang EY, Grogan JL. TIGIT: a key inhibitor of the cancer immunity cycle. Trends Immunol (2017) 38(1):20–8. doi: 10.1016/j.it.2016.10.002
25. Stanietsky N, Simic H, Arapovic J, Toporik A, Levy O, Novik A, et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc Natl Acad Sci (2009) 106(42):17858–63. doi: 10.1073/pnas.0903474106
26. Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol (2009) 10(1):48–57. doi: 10.1038/ni.1674
27. Chrétien I, Robert J, Marcuz A, Garcia-Sanz JA, Courtet M, Du Pasquier L, et al. CTX, a novel molecule specifically expressed on the surface of cortical thymocytes in xenopus. Eur J Immunol (1996) 26(4):780–91. doi: 10.1002/eji.1830260409
28. Chretien I, Marcuz A, Courtet M, Katevuo K, Vainio O, Heath JK, et al. CTX, a xenopus thymocyte receptor, defines a molecular family conserved throughout vertebrates. Eur J Immunol (1998) 28(12):4094–104. doi: 10.1002/(SICI)1521-4141(199812)28:12<4094::AID-IMMU4094>3.0.CO;2-2
29. Langnaese K, Colleaux L, Kloos DU, Fontes M, Wieacker P. Cloning of Z39Ig, a novel gene with immunoglobulin-like domains located on human chromosome X. Biochim Et Biophys Acta-Gene Struct Expression (2000) 1492(2-3):522–5. doi: 10.1016/S0167-4781(00)00131-7
30. Scanlan MJ, Ritter G, Yin B, Williams C Jr, Cohen LS, Coplan KA, et al. Glycoprotein A34, a novel target for antibody-based cancer immunotherapy. Cancer Immun (2006) 6(2).
31. Bazzoni G. The JAM family of junctional adhesion molecules. Curr Opin Cell Biol (2003) 15(5):525–30. doi: 10.1016/S0955-0674(03)00104-2
32. Suzu S, Hayashi Y, Harumi T, Nomaguchi K, Yamada M, Hayasawa H, et al. Molecular cloning of a novel immunoglobulin superfamily gene preferentially expressed by brain and testis. Biochem Biophys Res Commun (2002) 296(5):1215–21. doi: 10.1016/S0006-291X(02)02025-9
33. Boles KS, Vermi W, Facchetti F, Fuchs A, Wilson TJ, Diacovo TG, et al. A novel molecular interaction for the adhesion of follicular CD4 T cells to follicular DC. Eur J Immunol (2009) 39(3):695–703. doi: 10.1002/eji.200839116
34. Kim E, Lee Y, Kim J-S, Song B-S, Kim S-U, Huh J-W, et al. Extracellular domain of V-set and immunoglobulin domain containing 1 (VSIG1) interacts with sertoli cell membrane protein, while its PDZ-binding motif forms a complex with ZO-1. Molecules Cells (2010) 30(5):443–8. doi: 10.1007/s10059-010-0138-4
35. Rice RH, Xia Y, Alvarado RJ, Phinney BS. Proteomic analysis of human nail plate. J Proteome Res (2010) 9(12):6752–8. doi: 10.1021/pr1009349
36. Lee YJ, Rice RH, Lee YM. Proteome analysis of human hair shaft: from protein identification to posttranslational modification. Mol Cell Proteomics (2006) 5(5):789–800. doi: 10.1074/mcp.M500278-MCP200
37. Helmy KY, Katschke KJ Jr., Gorgani NN, Kljavin NM, Elliott JM, Diehl L, et al. CRIg: a macrophage complement receptor required for phagocytosis of circulating pathogens. Cell (2006) 124(5):915–27. doi: 10.1016/j.cell.2005.12.039
38. Walker MG. Z39Ig is co-expressed with activated macrophage genes. Biochim Et Biophys Acta-Gene Struct Expression (2002) 1574(3):387–90. doi: 10.1016/S0167-4781(01)00358-X
39. Levin SD, Taft DW, Brandt CS, Bucher C, Howard ED, Chadwick EM, et al. Vstm3 is a member of the CD28 family and an important modulator of T-cell function. Eur J Immunol (2011) 41(4):902–15. doi: 10.1002/eji.201041136
40. Li Y, Guo M, Fu Z, Wang P, Zhang Y, Gao Y, et al. Immunoglobulin superfamily genes are novel prognostic biomarkers for breast cancer. Oncotarget (2017) 8(2):2444–56. doi: 10.18632/oncotarget.13683
41. Oidovsambuu O, Nyamsuren G, Liu SA, Goring W, Engel W, Adham IM. Adhesion protein VSIG1 is required for the proper differentiation of glandular gastric epithelia. PloS One (2011) 6(10):e25908. doi: 10.1371/journal.pone.0025908
42. Jung Y, Bang H, Kim YH, Park NE, Park YH, Park C, et al. V-Set and immunoglobulin domain-containing 1 (VSIG1), predominantly expressed in testicular germ cells, is dispensable for spermatogenesis and Male fertility in mice. Animals (2021) 11(4):1037. doi: 10.3390/ani11041037
43. Chen YB, Pan K, Li SP, Xia JC, Wang W, Chen JG, et al. Decreased expression of V-set and immunoglobulin domain containing 1 (VSIG1) is associated with poor prognosis in primary gastric cancer. J Surg Oncol (2012) 106(3):286–93. doi: 10.1002/jso.22150
44. Inoue Y, Matsuura S, Yoshimura K, Iwashita Y, Kahyo T, Kawase A, et al. Characterization of V-set and immunoglobulin domain containing 1 exerting a tumor suppressor function in gastric, lung, and esophageal cancer cells. Cancer Sci (2017) 108(8):1701–14. doi: 10.1111/cas.13295
45. Kovecsi A, Gurzu S, Szentirmay Z, Kovacs Z, Bara TJ, Jung I. Paradoxical expression pattern of the epithelial mesenchymal transition-related biomarkers CD44, SLUG, n-cadherin and VSIG1/Glycoprotein A34 in gastrointestinal stromal tumors. World J Gastrointest Oncol (2017) 9(11):436–43. doi: 10.4251/wjgo.v9.i11.436
46. Sata Y, Nakajima T, Fukuyo M, Matsusaka K, Hata A, Morimoto J, et al. High expression of CXCL14 is a biomarker of lung adenocarcinoma with micropapillary pattern. Cancer Sci (2020) 111(7):2588–97. doi: 10.1111/cas.14456
47. Kim JH, Kim KJ, Rhee YY, Bae JM, Cho NY, Lee HS, et al. Gastric-type expression signature in serrated pathway-associated colorectal tumors. Hum Pathol (2015) 46(5):643–56. doi: 10.1016/j.humpath.2015.01.003
48. Delker D, Kanth P, Bronner MP, Burt RW, Hagedorn C. RNA Sequencing of sessile serrated colon polyps identifies differentially expressed genes and immunohistochemical markers. Gastroenterology (2014) 146(5):S813–3. doi: 10.1371/journal.pone.0088367
49. Marra G. An “expressionistic” look at serrated precancerous colorectal lesions. Diagn Pathol (2021) 16(1):1–17. doi: 10.1186/s13000-020-01064-1
50. Snyder EL, Watanabe H, Magendantz M, Hoersch S, Chen TA, Wang DG. Nkx2-1 represses a latent gastric differentiation program in lung adenocarcinoma. Mol Cell (2013) 50(2):185–99. doi: 10.1016/j.molcel.2013.02.018
51. Gurzu S, Sugimura H, Szederjesi J, Szodorai R, Braicu C, Kobori L, et al. Interaction between cadherins, vimentin, and V-set and immunoglobulin domain containing 1 in gastric-type hepatocellular carcinoma. Histochem Cell Biol (2021) 156(4):377–90. doi: 10.1007/s00418-021-02006-8
52. Bernal C, Silvano M, Tapponnier Y, Anand S, Angulo C, Altaba ARI. Functional pro-metastatic heterogeneity revealed by spiked-scRNAseq is shaped by cancer cell interactions and restricted by VSIG1. Cell Rep (2020) 33(6):108372. doi: 10.1016/j.celrep.2020.108372
53. Wen H, Gallo RA, Huang X, Cai J, Mei S, Farooqi AA, et al. Incorporating differential gene expression analysis with predictive biomarkers to identify novel therapeutic drugs for Fuchs endothelial corneal dystrophy. J Ophthalmol (2021) p:5580595. doi: 10.1155/2021/5580595
54. Chai YN, Qin J, Li YL, Tong YL, Liu GH, Wang XR, et al. TMT proteomics analysis of intestinal tissue from patients of irritable bowel syndrome with diarrhea: Implications for multiple nutrient ingestion abnormality. J Proteomics (2021) 231:103995. doi: 10.1016/j.jprot.2020.103995
55. Schmidt IM, Mothi SS, Wilson PC, Palsson R, Srivastava A, Onul IF, et al. Circulating plasma biomarkers in biopsy-confirmed kidney disease. Clin J Am Soc Nephrol (2022) 17(1):27–37. doi: 10.2215/Cjn.09380721
56. Ferreira JP, Verdonschot J, Collier T, Wang P, Pizard A, Bar C, et al. Proteomic bioprofiles and mechanistic pathways of progression to heart failure the HOMAGE study. Circ-Heart Fail (2019) 12(5):e005897. doi: 10.1161/CIRCHEARTFAILURE.118.005897
57. Kariuki SN, Franek BS, Kumar AA, Arrington J, Mikolaitis RA, Utset TO, et al. Trait-stratified genome-wide association study identifies novel and diverse genetic associations with serologic and cytokine phenotypes in systemic lupus erythematosus. Arthritis Res Ther (2010) 12(4):1–12. doi: 10.1186/ar3101
58. Yan HM, Qu JW, Cao W, Liu Y, Zheng GF, Zhang EF, et al. Identification of prognostic genes in the acute myeloid leukemia immune microenvironment based on TCGA data analysis. Cancer Immunol Immunother (2019) 68(12):1971–8. doi: 10.1007/s00262-019-02408-7
59. He Y, Wu YX, Liu Z, Li BP, Jiang N, Xu P, et al. Identification of signature genes associated with invasiveness and the construction of a prognostic model that predicts the overall survival of bladder cancer (vol 12, 694777, 2021). Front Genet (2022) 13. doi: 10.3389/fgene.2022.880633
60. Deng GC, Sun DC, Zhou Q, Lv Y, Yan H, Han QL, et al. Identification of DNA methylation-driven genes and construction of a nomogram to predict overall survival in pancreatic cancer. BMC Genomics (2021) 22(1):791. doi: 10.1186/s12864-021-08097-w
61. Meng J, Cao L, Song H, Chen L, Qu Z. Integrated analysis of gene expression and DNA methylation datasets identified key genes and a 6-gene prognostic signature for primary lung adenocarcinoma. Genet Mol Biol (2021) 44(4):e20200465. doi: 10.1590/1678-4685-GMB-2020-0465
62. Cui Z, Li Y, He S, Wen F, Xu X, Lu L, et al. Key candidate genes - VSIG2 of colon cancer identified by weighted gene Co-expression network analysis. Cancer Manag Res (2021) 13:5739–50. doi: 10.2147/cmar.S316584
63. Watanabe T, Suda T, Tsunoda T, Uchida N, Ura K, Kato T, et al. Identification of immunoglobulin superfamily 11 (IGSF11) as a novel target for cancer immunotherapy of gastrointestinal and hepatocellular carcinomas. Cancer Sci (2005) 96(8):498–506. doi: 10.1111/j.1349-7006.2005.00073.x
64. Harada H, Suzu S, Hayashi Y, Okada S, BT-IgSF. BT-IgSF, a novel immunoglobulin superfarnily protein, functions as a cell adhesion molecule. J Cell Physiol (2005) 204(3):919–26. doi: 10.1002/jcp.20361
65. Eom DS, Inoue S, Patterson LB, Gordon TN, Slingwine R, Kondo S, et al. Melanophore migration and survival during zebrafish adult pigment stripe development require the immunoglobulin superfamily adhesion molecule Igsf11. PloS Genet (2012) 8(8):e1002899. doi: 10.1371/journal.pgen.1002899
66. Ahi EP, Sefc KM. A gene expression study of dorso-ventrally restricted pigment pattern in adult fins of neolamprologus meeli, an African cichlid species. Peerj (2017) 5:e2843. doi: 10.7717/peerj.2843
67. Wang JH, Wu GP, Manick B, Hernandez V, Renelt M, Erickson C, et al. VSIG-3 as a ligand of VISTA inhibits human T-cell function. Immunology (2019) 156(1):74–85. doi: 10.1111/imm.13001
68. ElTanbouly MA, Zhao YD, Nowak E, Li JN, Schaafsma E, Le Mercier I, et al. VISTA is a checkpoint regulator for naive T cell quiescence and peripheral tolerance. Science (2020) 367(6475):264. doi: 10.1126/science.aay0524
69. Katoh M, Katoh M. IGSF11 gene, frequently up-regulated in intestinal-type gastric cancer, encodes adhesion molecule homologous to CXADR, FLJ22415 and ESAM. Int J Oncol (2003) 23(2):525–31. doi: 10.3892/ijo.23.2.525
70. Ghouzlani A, Rafii S, Karkouri M, Lakhdar A, Badou A. The promising IgSF11 immune checkpoint is highly expressed in advanced human gliomas and associates to poor prognosis. Front Oncol (2021) 10. doi: 10.3389/fonc.2020.608609
71. Katschke KJ Jr., Helmy KY, Steffek M, Xi H, Yin J, Lee WP, et al. A novel inhibitor of the alternative pathway of complement reverses inflammation and bone destruction in experimental arthritis. J Exp Med (2007) 204(6):1319–25. doi: 10.1084/jem.20070432
72. Wiesmann C, Katschke KJ, Yin JP, Helmy KY, Steffek M, Fairbrother WJ, et al. Structure of C3b in complex with CRIg gives insights into regulation of complement activation. Nature (2006) 444(7116):217–20. doi: 10.1038/nature05263
73. Yuan X, Yang BH, Dong Y, Yamamura A, Fu W. CRIg, a tissue-resident macrophage specific immune checkpoint molecule, promotes immunological tolerance in NOD mice, via a dual role in effector and regulatory T cells. Elife (2017) 6:e29540. doi: 10.7554/eLife.29540
74. Fu W, Wojtkiewicz G, Weissleder R, Benoist C, Mathis D. Early window of diabetes determinism in NOD mice, dependent on the complement receptor CRIg, identified by noninvasive imaging. Nat Immunol (2012) 13(4):361–8. doi: 10.1038/ni.2233
75. Shin W, Jeon Y, Choi I, Kim YJ. V-Set and ig domain-containing 4 (VSIG4)-expressing hepatic F4/80(+) cells regulate oral antigen-specific responses in mouse. Eur J Immunol (2018) 48(4):632–43. doi: 10.1002/eji.201747212
76. Lee MY, Kim WJ, Kang YJ, Jung YM, Kang YM, Suk K, et al. Z39Ig is expressed on macrophages and may mediate inflammatory reactions in arthritis and atherosclerosis. J Leukoc. Biol (2006) 80(4):922–8. doi: 10.1189/jlb.0306160
77. Gorgani NN, Thathaisong U, Mukaro VR, Poungpair O, Tirimacco A, Hii CS, et al. Regulation of CRIg expression and phagocytosis in human macrophages by arachidonate, dexamethasone, and cytokines. Am J Pathol (2011) 179(3):1310–8. doi: 10.1016/j.ajpath.2011.05.021
78. Munawara U, Small AG, Quach A, Gorgani NN, Abbott CA, Ferrante A. Cytokines regulate complement receptor immunoglobulin expression and phagocytosis of candida albicans in human macrophages: A control point in anti-microbial immunity. Sci Rep (2017) 7(1):4050. doi: 10.1038/s41598-017-04325-0
79. Sahu A, Lambris JD. Structure and biology of complement protein C3, a connecting link between innate and acquired immunity. Immunol Rev (2001) 180:35–48. doi: 10.1034/j.1600-065X.2001.1800103.x
80. Pangburn MK, Schreiber RD, Muller-Eberhard HJ. Formation of the initial C3 convertase of the alternative complement pathway. acquisition of C3b-like activities by spontaneous hydrolysis of the putative thioester in native C3. J Exp Med (1981) 154(3):856–67. doi: 10.1084/jem.154.3.856
81. Sun DL, Sun P, Li HM, Zhang MS, Liu GG, Strickland AB, et al. Fungal dissemination is limited by liver macrophage filtration of the blood. Nat Commun (2019) 10:1–14. doi: 10.1038/s41467-019-12381-5
82. Jiang H, Wang Z, Serra D, Frank MM, Amalfitano A. Recombinant adenovirus vectors activate the alternative complement pathway, leading to the binding of human complement protein C3 independent of anti-ad antibodies. Mol Ther (2004) 10(6):1140–2. doi: 10.1016/j.ymthe.2004.08.015
83. Broadley SP, Plaumann A, Coletti R, Lehmann C, Wanisch A, Seidlmeier A, et al. Dual-track clearance of circulating bacteria balances rapid restoration of blood sterility with induction of adaptive immunity. Cell Host Microbe (2016) 20(1):36–48. doi: 10.1016/j.chom.2016.05.023
84. Zeng ZT, Surewaard BGJ, Wong CHY, Geoghegan JA, Jenne CN, Kubes P. CRIg functions as a macrophage pattern recognition receptor to directly bind and capture blood-borne gram-positive bacteria. Cell Host Microbe (2016) 20(1):99–106. doi: 10.1016/j.chom.2016.06.002
85. Chen M, Muckersie E, Luo C, Forrester JV, Xu HP. Inhibition of the alternative pathway of complement activation reduces inflammation in experimental autoimmune uveoretinitis. Eur J Immunol (2010) 40(10):2870–81. doi: 10.1002/eji.201040323
86. Chen J, Crispin JC, Lucca JD, Tsokos GC. A novel inhibitor of the alternative pathway of complement attenuates intestinal Ischemia/Reperfusion-induced injury. J Surg Res (2011) 167(2):E131–6. doi: 10.1016/j.jss.2009.05.041
87. Lieberman LA, Mizui M, Nalbandian A, Bosse R, Crispin JC, Tsokos GC. Complement receptor of the immunoglobulin superfamily reduces murine lupus nephritis and cutaneous disease. Clin Immunol (2015) 160(2):286–91. doi: 10.1016/j.clim.2015.05.006
88. Qiao Q, Teng XY, Wang N, Lu RQ, Guo L, Zhang X, et al. A novel CRIg-targeted complement inhibitor protects cells from complement damage. FASEB J (2014) 28(11):4986–99. doi: 10.1096/fj.14-258046
89. Huang XY, Feng ZQ, Jiang YZ, Li JL, Xiang Q, Guo S, et al. VSIG4 mediates transcriptional inhibition of Nlrp3 and il-1 beta in macrophages. Sci Adv (2019) 5(1):eaau7426. doi: 10.1126/sciadv.aau7426
90. Ji N, Wu L, Shi H, Li Q, Yu A, Yang Z. VSIG4 attenuates NLRP3 and ameliorates neuroinflammation via JAK2-STAT3-A20 pathway after intracerebral hemorrhage in mice. Neurotox Res (2022) 40(1):78–88. doi: 10.1007/s12640-021-00456-5
91. Li J, Diao B, Guo S, Huang X, Yang C, Feng Z, et al. VSIG4 inhibits proinflammatory macrophage activation by reprogramming mitochondrial pyruvate metabolism. Nat Commun (2017) 8(1):1322. doi: 10.1038/s41467-017-01327-4
92. Xu SX, Sun ZL, Li L, Liu J, He J, Song DL, et al. Induction of T cells suppression by dendritic cells transfected with VSIG4 recombinant adenovirus. Immunol Lett (2010) 128(1):46–50. doi: 10.1016/j.imlet.2009.11.003
93. Liao Y, Guo S, Chen Y, Cao D, Xu H, Yang C, et al. VSIG4 expression on macrophages facilitates lung cancer development. Lab Invest (2014) 94(7):706–15. doi: 10.1038/labinvest.2014.73
94. Sturtz LA, Deyarmin B, van Laar R, Yarina W, Shriver CD, Ellsworth RE. Gene expression differences in adipose tissue associated with breast tumorigenesis. Adipocyte (2014) 3(2):107–14. doi: 10.4161/adip.28250
95. Byun JM, Jeong DH, Choi IH, Lee DS, Kang MS, Jung KO, et al. The significance of VSIG4 expression in ovarian cancer. Int J Gynecol Cancer (2017) 27(5):872–8. doi: 10.1097/IGC.0000000000000979
96. Roh J, Jeon Y, Lee AN, Lee SM, Kim Y, Sung CO, et al. The immune checkpoint molecule V-set ig domain-containing 4 is an independent prognostic factor for multiple myeloma. Oncotarget (2017) 8(35):58122–32. doi: 10.18632/oncotarget.19468
97. Xu T, Jiang Y, Yan Y, Wang HX, Lu CY, Xu HC, et al. VSIG4 is highly expressed and correlated with poor prognosis of high-grade glioma patients. Am J Trans Res (2015) 7(6):1172–80. doi:
98. Yap EH, Rosche T, Almo S, Fiser A. Functional clustering of immunoglobulin superfamily proteins with protein-protein interaction information calibrated hidden Markov model sequence profiles. J Mol Biol (2014) 426(4):945–61. doi: 10.1016/j.jmb.2013.11.009
99. Wang J, Manick B, Renelt M, Hansen L, Wu G, Kalabokis V. VSIG-8 is a co-inhibitory ligand and an immune checkpoint molecule for human T cells. J Immunol (2018) 200(1 Supplement):47.4–4. doi:
100. Sasikumar PG, Naremaddepalli SS, Ramachandra RK, Gowda N, Yerramsetti MR, Bandireddy SR, et al. Abstract B006: Functional antagonism of VSIG8-mediated immune suppression by oral VISTA agents. Mol Cancer Ther (2018) 17(1 Supplement):B006–6. doi: 10.1158/1535-7163.TARG-17-B006
101. Chen W, Qie C, Hu X, Wang L, Jiang J, Liu W, et al. A small molecule inhibitor of VSIG-8 prevents its binding to VISTA. Invest New Drugs (2022) 40(4):690–9. doi: 10.1007/s10637-022-01244-4
102. Fromm G, Silva Sd, Johannes K, Patel A, Hornblower J, Schreiber TH. Abstract 5564: Agonist redirected checkpoint, VSIG8-Fc-OX40L, for cancer immunotherapy. Cancer Res (2018) 78(13 Supplement):5564–4. doi: 10.1158/1538-7445.AM2018-5564
103. Sanchez-Correa B, Valhondo I, Hassouneh F, Lopez-Sejas N, Pera A, Bergua JM, et al. DNAM-1 and the TIGIT/PVRIG/TACTILE axis: novel immune checkpoints for natural killer cell-based cancer immunotherapy. Cancers (2019) 11(6):877. doi: 10.3390/cancers11060877
104. Satoh-Horikawa K, Nakanishi H, Takahashi K, Miyahara M, Nishimura M, Tachibana K, et al. Nectin-3, a new member of immunoglobulin-like cell adhesion molecules that shows homophilic and heterophilic cell-cell adhesion activities. J Biol Chem (2000) 275(14):10291–9. doi: 10.1074/jbc.275.14.10291
105. Deuss FA, Gully BS, Rossjohn J, Berry R. Recognition of nectin-2 by the natural killer cell receptor T cell immunoglobulin and ITIM domain (TIGIT). J Biol Chem (2017) 292(27):11413–22. doi: 10.1074/jbc.M117.786483
106. Mendelsohn CL, Wimmer E, Racaniello VR. Cellular receptor for poliovirus: molecular cloning, nucleotide sequence, and expression of a new member of the immunoglobulin superfamily. Cell (1989) 56(5):855–65. doi: 10.1016/0092-8674(89)90690-9
107. Chen X, Lu P-H, Liu L, Fang Z-M, Duan W, Liu Z-L, et al. TIGIT negatively regulates inflammation by altering macrophage phenotype. Immunobiology (2016) 221(1):48–55. doi: 10.1016/j.imbio.2015.08.003
108. Takai Y, Irie K, Shimizu K, Sakisaka T, Ikeda W. Nectins and nectin-like molecules: roles in cell adhesion, migration, and polarization. Cancer Sci (2003) 94(8):655–67. doi: 10.1111/j.1349-7006.2003.tb01499.x
109. Tahara-Hanaoka S. Functional characterization of DNAM-1 (CD226) interaction with its (CD155) and nectin-2 (PRR-2/CD112). Int Immunol (2004) 16(4):533–8. doi: 10.1093/intimm/dxh059
110. Fuchs A, Cella M, Giurisato E, Shaw AS, Colonna M. Cutting edge: CD96 (tactile) promotes NK cell-target cell adhesion by interacting with the poliovirus receptor (CD155). J Immunol (2004) 172(7):3994–8. doi: 10.4049/jimmunol.172.7.3994
111. Seth S, Maier MK, Qiu Q, Ravens I, Kremmer E, Förster R, et al. The murine pan T cell marker CD96 is an adhesion receptor for CD155 and nectin-1. Biochem Biophys Res Commun (2007) 364(4):959–65. doi: 10.1016/j.bbrc.2007.10.102
112. Blake SJ, Dougall WC, Miles JJ, Teng MW, Smyth MJ. Molecular pathways: Targeting CD96 and TIGIT for cancer immunotherapy. Clin Cancer Res (2016) 22(21):5183–8. doi: 10.1158/1078-0432.CCR-16-0933
113. Chan CJ, Martinet L, Gilfillan S, Souza-Fonseca-Guimaraes F, Chow MT, Town L, et al. The receptors CD96 and CD226 oppose each other in the regulation of natural killer cell functions. Nat Immunol (2014) 15(5):431–8. doi: 10.1038/ni.2850
114. Fuchs A, Colonna M. The role of NK cell recognition of nectin and nectin-like proteins in tumor immunosurveillance. Semin Cancer Biol (2006) 16(5):359–66. doi: 10.1016/j.semcancer.2006.07.002
115. Zhu Y, Paniccia A, Schulick AC, Chen W, Koenig MR, Byers JT, et al. Identification of CD112R as a novel checkpoint for human T cells. J Exp Med (2016) 213(2):167–76. doi: 10.1084/jem.20150785
116. Jin HS, Park Y. Hitting the complexity of the TIGIT-CD96-CD112R-CD226 axis for next-generation cancer immunotherapy. BMB Rep (2021) 54(1):2–11. doi: 10.5483/BMBRep.2021.54.1.229
117. Chauvin JM, Zarour HM. TIGIT in cancer immunotherapy. J Immunother Cancer (2020) 8(2):e000957. doi: 10.1136/jitc-2020-000957
118. Lozano E, Dominguez-Villar M, Kuchroo V, Hafler DA. The TIGIT/CD226 axis regulates human T cell function. J Immunol (2012) 188(8):3869–75. doi: 10.4049/jimmunol.1103627
119. Inozume T, Yaguchi T, Furuta J, Harada K, Kawakami Y, Shimada S. Melanoma cells control antimelanoma CTL responses via interaction between TIGIT and CD155 in the effector phase. J Invest Dermatol (2016) 136(1):255–63. doi: 10.1038/JID.2015.404
120. Joller N, Hafler JP, Brynedal B, Kassam N, Spoerl S, Levin SD, et al. Cutting edge: TIGIT has T cell-intrinsic inhibitory functions. J Immunol (2011) 186(3):1338–42. doi: 10.4049/jimmunol.1003081
121. Liu S, Zhang H, Li M, Hu D, Li C, Ge B, et al. Recruitment of Grb2 and SHIP1 by the ITT-like motif of TIGIT suppresses granule polarization and cytotoxicity of NK cells. Cell Death Differ. (2013) 20(3):456–64. doi: 10.1038/cdd.2012.141
122. Gao J, Zheng Q, Xin N, Wang W, Zhao C. CD 155, an onco-immunologic molecule in human tumors. Cancer Sci (2017) 108(10):1934–8. doi: 10.1111/cas.13324
123. Li X-Y, Das I, Lepletier A, Addala V, Bald T, Stannard K, et al. CD155 loss enhances tumor suppression via combined host and tumor-intrinsic mechanisms. J Clin Invest (2018) 128(6):2613–25. doi: 10.1172/JCI98769
124. Johnston RJ, Comps-Agrar L, Hackney J, Yu X, Huseni M, Yang Y, et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell (2014) 26(6):923–37. doi: 10.1016/j.ccell.2014.10.018
125. Chauvin J-M, Pagliano O, Fourcade J, Sun Z, Wang H, Sander C, et al. TIGIT and PD-1 impair tumor antigen–specific CD8+ T cells in melanoma patients. J Clin Invest (2015) 125(5):2046–58. doi: 10.1172/JCI80445
126. He W, Zhang H, Han F, Chen X, Lin R, Wang W, et al. CD155T/TIGIT signaling regulates CD8+ T-cell metabolism and promotes tumor progression in human gastric cancer. Cancer Res (2017) 77(22):6375–88. doi: 10.1158/0008-5472.CAN-17-0381
127. Gil Del Alcazar CR, Huh SJ, Ekram MB, Trinh A, Liu LL, Beca F, et al. Immune escape in breast cancer during in situ to invasive carcinoma transition. Cancer Discovery (2017) 7(10):1098–115. doi: 10.1158/2159-8290.CD-17-0222
128. Kong Y, Zhu L, Schell TD, Zhang J, Claxton DF, Ehmann WC, et al. T-Cell immunoglobulin and ITIM domain (TIGIT) associates with CD8+ T-cell exhaustion and poor clinical outcome in AML patients. Clin Cancer Res (2016) 22(12):3057–66. doi: 10.1158/1078-0432.CCR-15-2626
129. Stanietsky N, Rovis TL, Glasner A, Seidel E, Tsukerman P, Yamin R, et al. Mouse TIGIT inhibits NK-cell cytotoxicity upon interaction with PVR. Eur J Immunol (2013) 43(8):2138–50. doi: 10.1002/eji.201243072
130. Li M, Xia PY, Du Y, Liu SW, Huang GL, Chen J, et al. T-Cell immunoglobulin and ITIM domain (TIGIT) Receptor/Poliovirus receptor (PVR) ligand engagement suppresses interferon-gamma production of natural killer cells via beta-arrestin 2-mediated negative signaling. J Biol Chem (2014) 289(25):17647–57. doi: 10.1074/jbc.M114.572420
131. Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W, et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat Immunol (2018) 19(7):723–32. doi: 10.1038/s41590-018-0132-0
132. Joller N, Lozano E, Burkett PR, Patel B, Xiao S, Zhu C, et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity (2014) 40(4):569–81. doi: 10.1016/j.immuni.2014.02.012
133. Fuhrman CA, Yeh WI, Seay HR, Saikumar Lakshmi P, Chopra G, Zhang L, et al. Divergent phenotypes of human regulatory T cells expressing the receptors TIGIT and CD226. J Immunol (2015) 195(1):145–55. doi: 10.4049/jimmunol.1402381
134. Koch MA, Tucker-Heard Gs Perdue NR, Killebrew JR, Urdahl KB, Campbell DJ. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol (2009) 10(6):595–602. doi: 10.1038/ni.1731
135. Kurtulus S, Sakuishi K, Ngiow S-F, Joller N, Tan DJ, Teng MW, et al. TIGIT predominantly regulates the immune response via regulatory T cells. J Clin Invest (2015) 125(11):4053–62. doi: 10.1172/JCI81187
136. Guillerey C, Harjunpää H, Carrié N, Kassem S, Teo T, Miles K, et al. TIGIT immune checkpoint blockade restores CD8+ T-cell immunity against multiple myeloma. Blood J Am Soc Hematol (2018) 132(16):1689–94. doi: 10.1182/blood-2018-01-825265
137. Rodriguez-Abreu D, Johnson ML, Hussein MA, Cobo M, Patel AJ, Secen NM, et al. Primary analysis of a randomized, double-blind, phase II study of the anti-TIGIT antibody tiragolumab (tira) plus atezolizumab (atezo) versus placebo plus atezo as first-line (1L) treatment in patients with PD-L1-selected NSCLC (CITYSCAPE). Am Soc Clin Oncol (2020) 38:15_suppl:9503–03. doi: 10.1200/JCO.2020.38.15_suppl.9503
138. Hu Y, Welsh J, Paris S, Bertolet G, Barsoumian H, Schuda L, et al. 575 dual blockade of LAG3 and TIGIT improves the treatment efficacy of a nanoparticle-mediated immunoradiation in anti-PD1 resistant lung cancer in mice. J ImmunoTher Cancer (2021) 9(Suppl 2):A604–4. doi: 10.1136/jitc-2021-SITC2021.575
139. Human VSIG10 antibody (2022). Available at: https://www.rndsystems.com/cn/products/human-vsig10-antibody-988622_mab9969.
140. Kim DS, Choi D, Hahn Y. Loss of ancestral n-glycosylation sites in conserved proteins during human evolution. Int J Mol Med (2015) 36(6):1685–92. doi: 10.3892/ijmm.2015.2362
141. Papasotiriou I, Parsonidis P, Ntanovasilis D-A, Iliopoulos AC, Beis G, Apostolou P. Identification of new targets and biomarkers in lung cancer. J Clin Oncol (2019) 37(15_suppl):e14656–6. doi: 10.1200/JCO.2019.37.15_suppl.e14656
142. Mayoux M, Roller A, Pulko V, Sammicheli S, Chen S, Sum E, et al. Dendritic cells dictate responses to PD-L1 blockade cancer immunotherapy. Sci Transl Med (2020) 12(534):eaav7431. doi: 10.1126/scitranslmed.aav7431
143. Peng Q, Qiu X, Zhang Z, Zhang S, Zhang Y, Liang Y, et al. PD-L1 on dendritic cells attenuates T cell activation and regulates response to immune checkpoint blockade. Nat Commun (2020) 11(1):4835. doi: 10.1038/s41467-020-18570-x
144. Fecteau RE, Kong J, Kresak A, Brock W, Song Y, Fujioka H, et al. Association between germline mutation in VSIG10L and familial Barrett neoplasia. JAMA Oncol (2016) 2(10):1333–9. doi: 10.1001/jamaoncol.2016.2054
Keywords: immune checkpoint, VSIG4/CRIg, VSIG, TIGIT, cancer immunotherapy, antitumor T-cell response, coinhibitory receptor
Citation: Zhou X, Khan S, Huang D and Li L (2022) V-Set and immunoglobulin domain containing (VSIG) proteins as emerging immune checkpoint targets for cancer immunotherapy. Front. Immunol. 13:938470. doi: 10.3389/fimmu.2022.938470
Received: 07 May 2022; Accepted: 29 August 2022;
Published: 15 September 2022.
Edited by:
Akira Shibuya, University of Tsukuba, JapanReviewed by:
Francesca Finetti, University of Siena, ItalyCopyright © 2022 Zhou, Khan, Huang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dabing Huang, aGRhYmluZ0B1c3RjLmVkdS5jbg==; Lu Li, bGlsdUB1c3RjLmVkdS5jbg==
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.