
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 25 August 2022
Sec. Autoimmune and Autoinflammatory Disorders
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.935275
This article is part of the Research Topic Intestinal and Liver Autoimmunity: Unique Pathogenesis, Diseases and Crosstalk View all 5 articles
 Shanshan Xiong1†
Shanshan Xiong1† Jinyu Tan1†
Jinyu Tan1† Yu Wang1
Yu Wang1 Jinshen He1
Jinshen He1 Fan Hu1
Fan Hu1 Xiaomin Wu1
Xiaomin Wu1 Zishan Liu1
Zishan Liu1 Sinan Lin1
Sinan Lin1 Xuehua Li2
Xuehua Li2 Zhihui Chen3*Ren Mao1,4*
Zhihui Chen3*Ren Mao1,4*Creeping fat is a specific feature of Crohn’s disease (CD) and is characterized by mesenteric fat wrapping around the intestine. It highly correlates with intestinal transmural inflammation, muscular hypertrophy, fibrosis, and stricture formation. However, the pathogenesis of creeping fat remains unclear. Molecular crosstalk exists between mesenteric fat and the intestine. Indeed, creeping fat contains different types of cells, including adipocytes and immune cells. These cell types can produce various cytokines, fatty acids, and growth factors, which affect the mesenteric fat function and modulate intestinal inflammation and immunity. Moreover, adipocyte progenitors can produce extracellular matrix to adapt to fat expansion. Previous studies have shown that fat fibrosis is an important feature of adipose tissue malfunction and exists in other diseases, including metabolic disorders, cancer, atrial fibrillation, and osteoarthritis. Furthermore, histological sections of CD showed fibrosis in the creeping fat. However, the role of fibrosis in the mesenteric fat of CD is not well understood. In this review, we summarized the possible mechanisms of fat fibrosis and its impact on other diseases. More specifically, we illustrated the role of various cells (adipocyte progenitors, macrophages, mast cells, and group 1 innate lymphoid cells) and molecules (including hypoxia-inducible factor 1-alpha, transforming growth factor-beta, platelet-derived growth factor, and peroxisome proliferator-activated receptor-gamma) in the pathogenesis of fat fibrosis in other diseases to understand the role of creeping fat fibrosis in CD pathogenesis. Future research will provide key information to decipher the role of fat fibrosis in creeping fat formation and intestinal damage, thereby helping us identify novel targets for the diagnosis and treatment of CD.
Crohn’s disease (CD) is a chronic inflammatory disease that can affect any part of the digestive tract, and its pathophysiological mechanism is complex (1). Within 10 years of its diagnosis, approximately 50% of the patients need surgical treatment. Moreover, the disease cannot be cured with a high disability rate, placing a heavy burden on patients, families, and society (2). In severe cases of CD, the mesenteric white adipose tissue surrounding the diseased intestinal wall extends from the mesenteric attachment and partially covers the intestinal circumference to form “creeping fat” (3, 4). Creeping fat is a unique pathological feature of CD, and its presence correlates with intestinal transmural inflammation, muscular hypertrophy, fibrosis, and stricture formation (5–7). Moreover, this structure helps recognize the site of the most severe lesions, fibrosis, and stenosis during surgery, and resection of the diseased mesentery reduces the postoperative recurrence of CD (8). However, the pathogenesis of creeping fats remains unclear.
With the rise in metabolic diseases, researchers are focusing on the function of adipose tissue, including creeping fat. There is molecular crosstalk between the mesenteric fat and the intestinal wall (9–11). Creeping fat contains diverse cell types, including adipocyte progenitors, adipocytes, and immune cells. These cells produce various cytokines, fatty acids, or growth factors, which affect mesenteric fat function, which, in turn, can modulate intestinal inflammation and immunity (12). For instance, some adipokines, including adiponectin and leptin, shape the local macrophages to mostly the M2 subtype, suggesting a protective role of mesenteric fat in CD (10). The free fatty acids secreted by the adipocytes in creeping fat modulate intestinal smooth muscle proliferation, which further promotes stricture formation (6). Moreover, adipocyte progenitors can produce extracellular matrix (ECM) to adapt to fat expansion (13). Abnormal ECM deposition, a feature of fibrosis development in adipose tissue, is associated with tissue inflammation and adipose tissue malfunction in metabolic diseases (14, 15). Indeed, fat fibrosis exists in various diseases, such as metabolic disorders, tumors, atrial fibrillation (AF), and immune diseases (Figure 1) (14). Furthermore, histological sections of CD showed fibrosis in the creeping fat (16, 17). However, the role of fat fibrosis in the pathogenesis of creeping fat has gained little attention due to the lack of an ideal preclinical model.
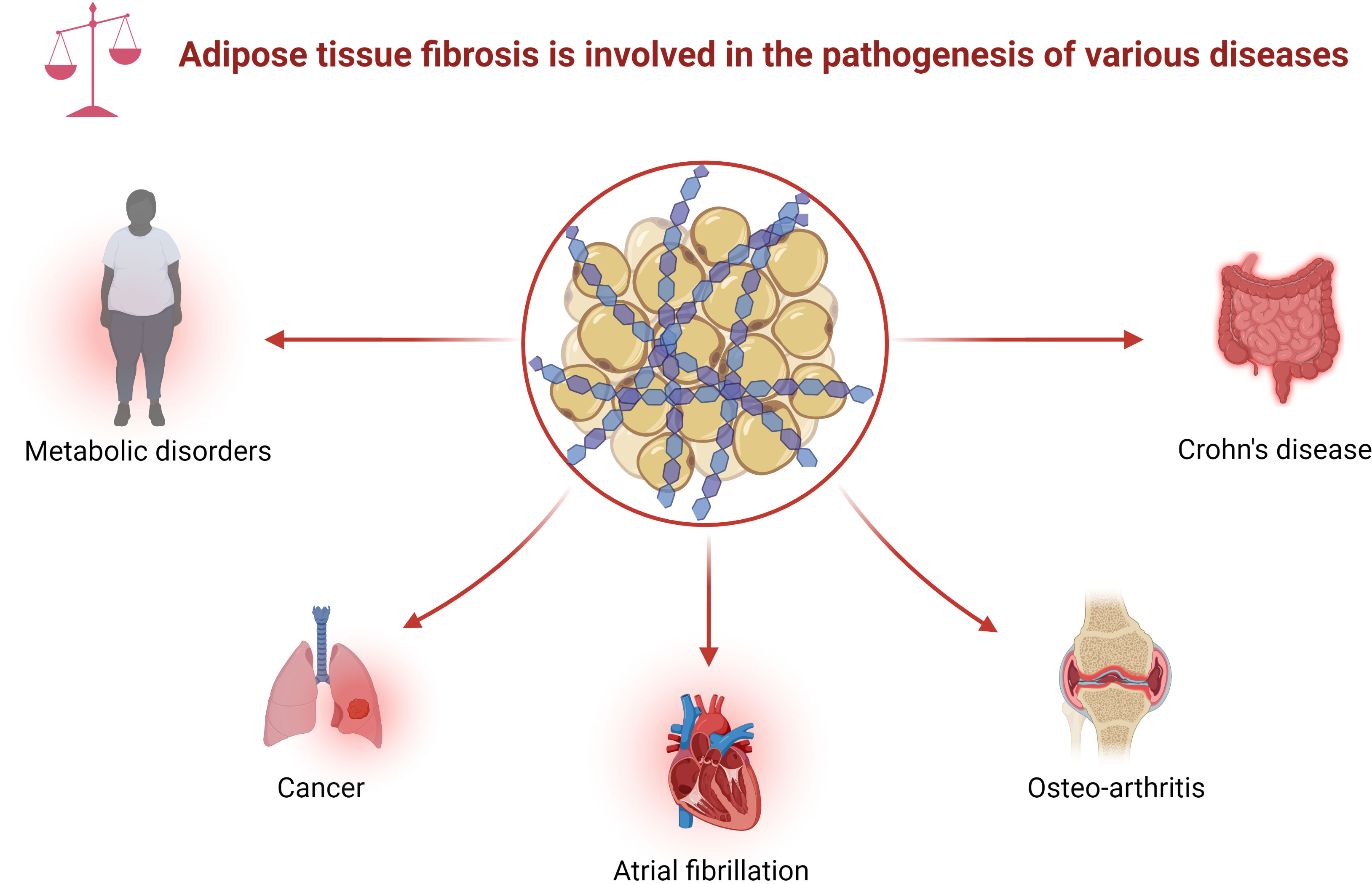
Figure 1 Adipose tissue fibrosis is involved in the pathogenesis of various diseases. Adipose tissue fibrosis, defined as excessive deposition of ECM in adipose tissue, appears in various diseases, including metabolic disorders, cancer, atrial fibrillation, osteoarthritis, and Crohn’s disease. ECM, extracellular matrix.
In this review, we aimed to discuss the possible mechanisms of adipose tissue fibrosis and its impact on other diseases. We believe that the mechanism from these disorders may be pivotal to elucidate the role of adipose tissue fibrosis in the pathogenesis of CD. It will help researchers get a comprehensive overview of the topic and provide directions for conducting future research to identify novel targets for CD diagnosis and treatment.
Adipose tissue fibrosis is the excessive deposition of ECM in adipose tissue. Although the ECM composition of adipose tissue is similar to that of other tissues, the relative content of ECM proteins may differ (18). In addition, there are significant differences in the proportion of ECM proteins between the adipose tissues of lean and obese individuals (19). The size and morphology of adipocytes also change in expanded fat, and there is no clear understanding of these specific manifestations.
The most abundant ECM proteins in adipose tissue are members of the collagen family, including collagen I, III, and V, and microfibrillar collagen IV (20). Type I collagen is the predominant one that maintains the structure and function of adipose tissue, along with other fibril-forming molecules (21). Collagen IV is the main component of the basement membrane and is necessary for adipocyte survival. It provides cellular structural support and interacts with integrins to transfer signals (22, 23). Increased collagen VI levels are observed in the fibrotic adipose tissue of both rodents and humans during metabolic challenges (24). Compared with other ECM proteins, collagen VI is more specific to adipose tissue and is critical for adipose tissue fibrosis and dysfunction (24). Collagen VI consists of three subunits, α1(VI), α2(VI), and α3(VI), which are highly regulated from the gene to the post-translational levels (25). Morphologically, collagen VI-null mice in the high-fat diet and ob/ob mutation group have an increased adipocyte cell size and reduced necrotic cell death and inflammation in adipose tissue (24). In addition, the glucose clearance rate, lipid metabolic parameters, and insulin signaling dramatically improved in collagen VI-null mice (24). These findings indicate that adipose tissue fibrosis caused by type VI collagen is associated with systemic and local metabolic disorders (23).
Moreover, endotrophin, a post-transcriptional protein-derived product of collagen VI, is overexpressed in the adipose tissue of ob/ob mice (26). Endotrophin plays a crucial role in tumor development (27, 28) and adipose tissue fibrosis and inflammation (26). Mechanistically, collagen VI and endotrophin cooperatively regulate the adipogenic and lipolytic capacity of adipocytes via the MAPK signaling pathway, regardless of their role in structural support in obesity-related metabolic diseases (29).
Collagen quantity is determined by the balance of enzymes that promote ECM synthesis and degradation. The synthesis enzymes include intracellular enzymes that participate in the processing of ECM protein precursors and extracellular inhibitors of degrading enzymes (18). Degradation enzymes include the fibrinolytic system, matrix metalloproteinase (MMP), and tissue inhibitors of MMPs (TIMPs) (18, 30). In this review, we will focus on the MMP system since its level significantly alters in the adipose tissue during adipose tissue expansion, and it also plays a role in cleaving collagen, thereby remodeling the ECM (31). Moreover, the MMP system causes adipose dysfunction and inflammation during tumor growth regulation (32).
In nutritionally induced obese mice, the mRNA expression of MMP-3, MMP-11, MMP-12, MMP-13, MMP-14, and TIMP-1 is upregulated, while that of MMP-7, MMP-9, MMP-16, MMP-24, and TIMP-4 is downregulated (32). MMP-2 and MMP-9 levels are also significantly higher in obese patients than in the controls (33–35). MMP-2 inhibitors prevent 3T3-L1 preadipocyte differentiation in a dose-dependent manner, and increased expression of MMP-2 and MMP-9 is associated with the loss of basement membrane type IV collagen (36, 37). Circulating MMP-9 also increases in obese patients and is associated with insulin resistance (38). In addition, membrane type 1 MMP (MT1-MMP, also known as MMP14) is critical in adipose tissue ECM remodeling. MMP14 activates MMP-2 and forms ternary complexes important for basement membrane remodeling during adipogenesis with TIMP-2, MMP-2, or MMP-9 (39, 40). It can also regulate the cleavage of collagen I. Furthermore, the MMP-14-null mice develop soft tissue fibrosis, which may be related to collagen renewal disorders (41). Simply put, MMP14 is crucial for adipocyte differentiation and collagen synthesis, and its absence affects the function of adipose tissue.
Myofibroblasts are pivotal for ECM production and remodeling (42). However, little is known about its role in adipose tissue fibrosis. Adipocyte progenitors can differentiate into myofibroblasts, which drive ECM synthesis in obesity (14). Platelet-derived growth factor receptor alpha (PDGFRα)+ adipocyte progenitors can obtain myofibroblast phenotypes and produce the highest levels of fibrosis markers in fibrotic adipose tissue (43). In addition, a subgroup of PDGFRα+ cells with high CD9 expression is associated with white adipose tissue fibrosis and metabolic disorders (43).
The adipose tissue of obese individuals has a significantly high number of macrophages than in control, and these macrophages are associated with inflammation and insulin resistance (44). There are two main types of macrophages: M1 and M2 (45). M1 macrophages promote inflammation, leading to insulin resistance in the adipose tissue of obese individuals (46). However, M2 macrophages can inhibit M1 macrophages, which maintain adipose tissue homeostasis (47). Furthermore, a crown-like structure represents macrophages clustering in dead and dying adipocytes (48). Studies have shown that macrophages in the crown-like structure are mainly M1, while M2 macrophages are abundant in adipocytes in fibrotic areas (49). These differences in the distribution of macrophages may be associated with differences in their functions. The co-culture of M1 macrophages with adipocytes leads to a more M2 phenotype, suggesting that inflammation and fibrosis coexist in adipose tissues (49). Interestingly, macrophage-inducible C-type lectin (Mincle) modulates macrophage function and correlates with myofibroblast activation and ECM remodeling, and mincle-knock out mice are protected against adipose tissue fibrosis (50, 51). Moreover, infiltrating macrophages in adipose tissue can release signals that attract fibroblasts and regulate adipose tissue fibrosis (14).
Apart from macrophages, mast cells are also present in adipose tissue and are associated with collagen accumulation and adipose tissue remodeling. Immature progenitor cells are released from the bone marrow and settle in vascularized tissue to mature within the blood (52, 53). The abundance of mast cells appears to increase in both animal models and patients with obesity, implicating their potential role in metabolic diseases (54–56). Furthermore, mast cells that infiltrate obese adipose tissue secrete mast cell protease 6 and induce collagen V expression, contributing to adipose tissue fibrosis and accelerating insulin resistance by inhibiting preadipocyte differentiation (57).
Recently, group 1 innate lymphoid cells (ILC1s) have been shown to be involved in the pathogenesis of adipose tissue fibrosis. Wang et al. showed that the number of ILC1 in adipose tissue increases in obese patients with type 2 diabetes and induces fat fibrosis in an interferon-γ-dependent manner (58). In addition, the reconstitution of adipose ILC1s by adoptive transfer in Prkdc-/- IL2rg-/- mice (immunodeficient mice) promotes adipose tissue fibrosis via transforming growth factor-beta 1 (TGF-β1) signaling, whereas inhibiting the accumulation of ILC1s can reduce fibrosis in adipose tissue and improve glucose tolerance (58).
In general, adipose tissue fibrosis is associated with the differentiation of adipocyte progenitors and the infiltration of various immune cells (14). Poorly differentiated mesenteric adipocytes and enriched immune cells are also confirmed in the mesenteric adipose tissue of CD patients (59, 60). Furthermore, our group previously showed that intestinal muscle cells and preadipocytes in CD patients interacted with each other and activated muscle cells could produce an ECM scaffold (6). Recently, another team highlighted the role of TLR4-mediated macrophages in mesenteric adipocyte dysfunction (61). All in all, creeping fat fibrosis is a complex process involving various cells, including fibroblasts, muscle cells, preadipocytes, and macrophages. The specific cellular mechanism of fat fibrosis still needs further investigation.
Adipose tissue expansion leads to a hypoxic status because angiogenesis fails to catch up with tissue growth. One of the key mediators of this process is hypoxia-inducible factor 1-alpha (HIF1α) (62). HIF1α cannot stimulate the expected angiogenesis program but can induce adipose tissue fibrosis and insulin resistance (63). Its transcriptional target lysyl oxidase is significantly increased in leptin-deficient ob/ob mice and crosslinks collagen I and III to form fibrillar collagen (63). Subsequently, the M1 macrophages are recruited, which release various inflammatory mediators, such as interleukin (IL)-6, monocyte chemoattractant protein 1, tumor necrosis factor-alpha, and IL-1β, and induce inflammation (64). Furthermore, a selective HIF1α inhibitor (PX-478) or a negative HIF1α mutation could suppress the formation of adipose tissue fibrosis in high-fat diet-fed mice and improve the metabolic state (65). Consistent with the findings above, Zuo et al. found that mesenteric adipose tissue contiguous with the involved intestine has a higher level of HIF1α than that in the uninvolved intestine (59). Although the exact mechanisms of the changes observed are still not clear, previous literatures on HIF1α give clues to the study of adipose tissue fibrosis in CD.
The TGF-β superfamily proteins are crucial regulators of adipose tissue remodeling. TGF-β1 and activin A belong to this superfamily and assist human adipose progenitors to acquire the myofibroblast phenotype and prevent their differentiation into adipocytes (66, 67). Likewise, when exposed to TGF-β1, murine-derived 3T3‐L1 preadipocytes synthesize more ECM proteins and reduce differentiation (68). The response to TGF-β is mediated by SMAD2, SMAD3, and SMAD4, which subsequently activate the fibrotic genes, such as collagen, fibronectin, and ECM remodeling enzymes (69). M2 macrophages in adipose tissue express high levels of TGF-β, which can be enhanced by co-culture with adipocytes. The expression of downstream effectors, such as phosphorylated SMAD, plasminogen activator inhibitor-1, and collagens, is also increased in macrophages and adipocytes (49, 70). Metformin can decrease ECM deposition in adipose tissue by activating AMPK signaling and inhibiting TGF-β1/Smad3 signaling, which improves fibrosis and prevents uncontrolled adipose tissue expansion in metabolic disorders (71). As for CD, studies have shown that the level of TGF-β in the mesenteric adipose tissue was significantly increased, with the Smad2/3 signaling pathway activated. This change also led to excessive ECM synthesis in the mesenteric adipose tissue (59, 61).
PDGF is another key fibrosis signaling molecule. It combines two conserved tyrosine kinase receptors: PDGFRα and PDGFRβ, which play important roles in the proliferative profibrotic phenotype (72). Adipocyte progenitors express both PDGFRα and PDGFRβ.
When PDGFRα signaling is activated, adipocyte progenitors synthesize ECM and function like profibrotic cells, contributing to pathological remodeling and adipose tissue dysfunction in obesity (43). PDGFα signaling typically correlates with Zfp521 overexpression (73). However, how PDGFα signal transduction converts progenitors into a fibrotic phenotype is yet to be completely elucidated. A study suggested that PDGFα may act by upregulating the mTOR mRNA translation and ribosomal biogenesis signaling pathways and control the expression of the imprinted gene network related to cell growth and tissue homeostasis (74).
Furthermore, PDGFRβ inhibits the adipogenic potential of progenitors and promotes liver and kidney fibrosis (75). However, there is no evidence of its direct role in driving adipose tissue fibrosis (75). A study showed that adiponectin-positive intradermal adipose tissue can be transformed into myofibroblasts in bleomycin-treated mice (76). Hence, it can be speculated that the reactivation of these receptors under pathological conditions is related to fibrosis or tissue dysfunction.
According to the current available literature, no relevant research exists to study the role of PDGF in CD fat fibrosis. Since PDGF has shown its exact pro-fibrotic role in other organs, future study is needed to elucidate its mechanism in mesenteric adipose tissue fibrosis of CD.
The peroxisome proliferator-activated receptor (PPAR) family comprises three members: PPAR-α, PPAR-δ, and PPAR-γ. Among them, PPAR-γ is mainly present in adipose tissue and regulates adipogenesis and lipid metabolism (77). When treated with a PPARγ agonist, diabetic db/db mice showed lower collagen expression, suggesting an anti-fibrotic capacity of PPARγ (63). Furthermore, a study revealed an association between PPARγ2 and HIF1α, since HIF1α attenuates adipogenesis and promotes white adipose tissue fibrosis in obesity by driving PPARγ S112 phosphorylation via autocrine/paracrine signaling. The blocking effect can be imitated by an antagonist of PDGFR (imatinib). Therefore, PDGFR signaling may play a key role in HIF1α activation and inhibition of PPARγ activity (78). Although there is no statistical significance, tissue concentrations of PPARγ were also increased in creeping fat adjacent to the lesion (61). It is reasonable to speculate that PPARγ may also be involved in the fibrotic process of mesenteric adipose tissue in CD patients.
There are multiple other signaling molecules involved in the process of adipose tissue fibrosis, such as connective tissue growth factor (79, 80), growth hormone (81), and myocardin-related transcription factor A (82). However, no reliable conclusions could be drawn based on the results of previous studies. Nevertheless, the findings from the above-mentioned studies show that adipose tissue fibrosis is driven by an imbalance between the fibrogenic and adipogenic potential of adipose tissue progenitors. The cellular and molecular mechanisms of adipose tissue fibrosis are represented in a pictorial form in Figure 2, providing clues to the molecular mechanism of mesenteric adipose tissue fibrosis in CD.
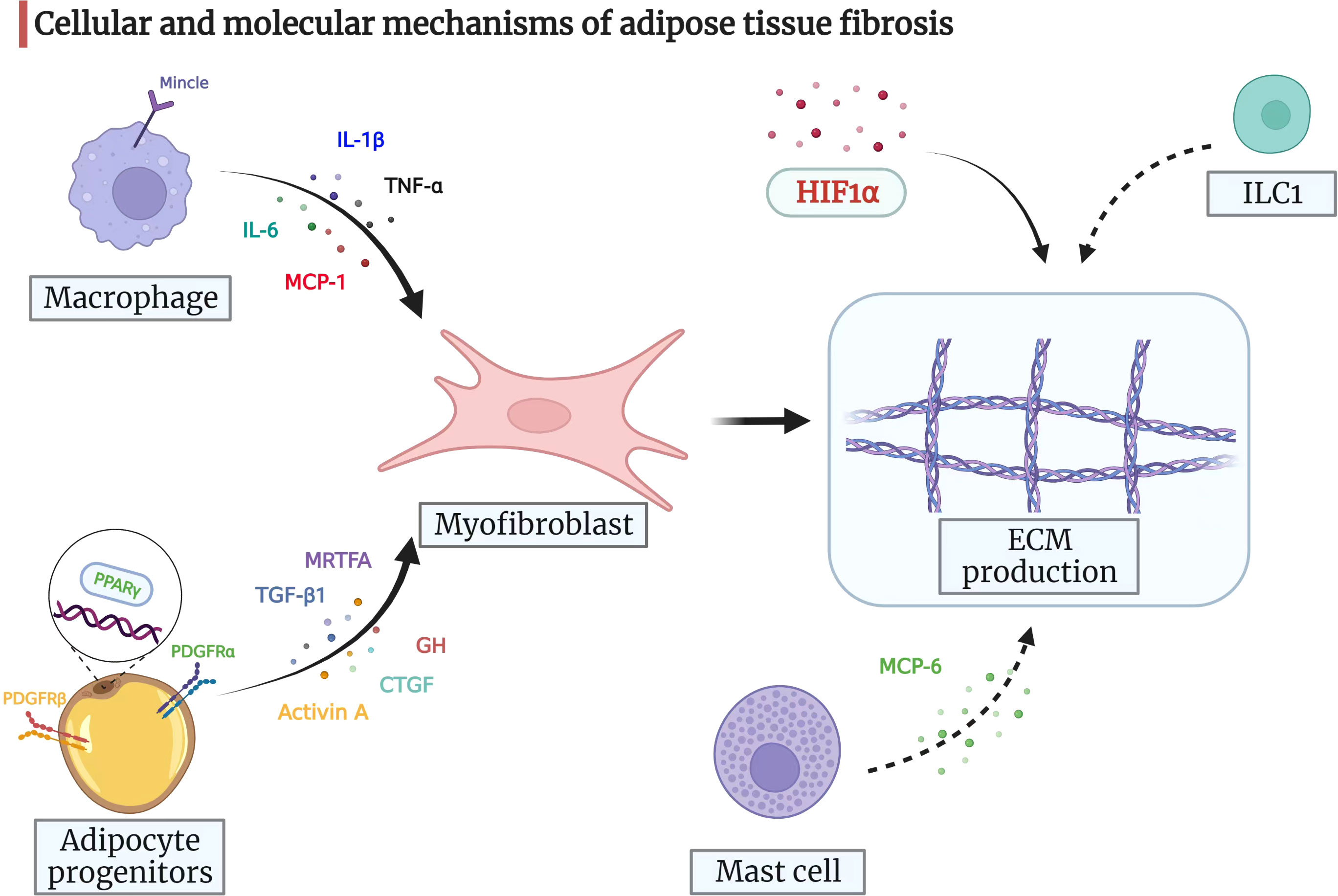
Figure 2 Cellular and molecular mechanisms of adipose tissue fibrosis. Myofibroblasts are pivotal for ECM production and remodeling. Adipocyte progenitors can differentiate into myofibroblasts, which then drive ECM synthesis. Adipocyte progenitors express both PDGFRα and PDGFRβ. When PDGFRα signaling is activated, adipocyte progenitors synthesize ECM and function as profibrotic cells. PDGFRβ inhibits the adipogenic potential of progenitors. The nuclear receptor PPARγ also regulates adipogenesis with anti-fibrotic potential. Moreover, TGF-β1, activin A, CTGF, GH, and MRTFA may drive adipose progenitors to acquire a myofibroblast phenotype and prevent differentiation into adipocytes under certain circumstances. The infiltrating macrophages in adipose tissue can release signals, such as IL-6, MCP-1, TNF-α, and IL-1β, which attract fibroblasts and regulate adipose tissue fibrosis. In addition, macrophage-inducible C-type lectin (Mincle) modulates macrophage function and correlates with myofibroblast activation and ECM remodeling. Mast cells secrete MCP-6 and induce collagen V expression, contributing to adipose tissue fibrosis and accelerating insulin resistance by inhibiting preadipocyte differentiation. ILC1 in adipose tissue induces fat fibrosis in an IFN-γ-dependent manner. HIF1α promotes adipose tissue fibrosis and is a potential therapeutic target for adipose tissue fibrosis and associated metabolic disorders. ECM, extracellular matrix; PDGF, platelet-derived growth factor; PPARγ, peroxisome proliferator-activated receptor-gamma; TGF-β1, transforming growth factor-beta 1; CTGF, connective tissue growth factor; GH, growth hormone; MRTFA, myocardin-related transcription factor A; IL-6, interleukin-6; MCP-1, monocyte chemoattractant protein-1; TNF-α, tumor necrosis factor-alpha; IL-1β: interleukin-1beta; MCP-6, mast cell protease 6; ILC1s, group 1 innate lymphoid cells; IFN-γ, interferon-gamma; HIF1α, hypoxia-inducible factor 1-alpha.
Adipose tissue fibrosis has been observed in various diseases, including obesity, cancer, and arthritis. Information from these disorders may be pivotal to understanding the role of adipose tissue fibrosis in CD pathogenesis.
Adipose tissue in obese patients exhibits excessive synthesis of fibrotic tissues, associated with phenotypic changes in preadipocyte and pro-inflammatory environments (83). Among obese patients, a subset is considered to be metabolically healthy, while the other subset might be affected by adipose tissue fibrosis (84).
Fibrosis in visceral adipose tissue may have a positive effect on metabolism. In obese patients, the degree of omental fibrosis negatively correlates with the size of the omental fat cells (19). Compared with the adipose tissue in non-diabetic subjects, the degree of omental adipose tissue fibrosis in the adipose tissue, the frequency of preadipocytes, and expression of fibrotic genes are lower and the fat cells are larger in diabetic subjects (85). Another study further supported the link between human omental fat fibrosis and metabolic outcomes. The tensile strength of adipose tissue was used as a proxy for the severity of fibrosis, and the tensile strength of omental adipose tissue decreased in obese subjects with type 2 diabetes than in healthy obese subjects (86). Omental fibrosis also has a more positive effect on lipid metabolism since it negatively correlates with circulating triglyceride levels and positively correlates with high-density lipoprotein cholesterol levels (19). These data indicate that omental fibrosis may be an adaptive phenomenon that can limit the expansion of fat cells and help reduce the negative effects of adipocyte hypertrophy on metabolism.
However, the situation is different for subcutaneous fat. When matched for body mass index, obese patients with insulin resistance show increased expression of ECM markers in subcutaneous fat than in insulin-sensitive obese subjects (87). This result was confirmed in another study on non-diabetic subjects (88). Similar conclusions have been reached by animal studies (24). Furthermore, it has been demonstrated that lacking collagen VI can improve the body’s energy homeostasis (24). This suggests that the increase in the ECM of adipose tissue, especially in subcutaneous fat, is not only caused by obesity but may also be critical for insulin resistance (87).
Therefore, omental and subcutaneous fats exert different effects on metabolism. However, inconsistent conclusions have been drawn from different studies of omental fat (14). Moreover, the influence of fat fibrosis on metabolism is very complicated and most likely related to the location of fat and other possible factors.
Obesity is associated with an increased risk of cancer development and a poor prognosis, and adipose tissue fibrosis may be a contributing factor. Both myofibroblasts and fibroblasts are more prevalent in histologically normal breast tissues closer to the edge of breast adenocarcinoma (89). Bo et al. found that obesity increases interstitial fibrosis, which stimulates breast cancer growth via altered mechanosignaling (90). This finding illustrated that obesity-associated ECM could drive the tumorigenic potential of premalignant cells (90).
Another group has investigated the interaction between tumor cells, stromal cells, and adipocytes. Collagen VI is abundantly expressed in adipocytes and can promote tumor growth at its early stages (91). Meanwhile, endotrophin, a cleavage product of the COL6α3 chain, enhances fibrosis and is associated with mammary tumor growth. These effects are partially mediated by an enhanced TGF-β signaling (27). Furthermore, Incio et al. showed that obesity-induced desmoplasia led to tumor progression and poor response to chemotherapy in pancreatic ductal adenocarcinoma (92).
Interestingly, fat fibrosis has also been associated with cancer cachexia. Increased collagen and fibers are found in the adipose tissue of cancer cachexia than in weight-stable cancer patients and controls (93). This phenomenon is associated with altered TGF-β signaling, which can affect the structure and function of adipose tissue (93).
AF is the most common type of arrhythmia. A French group obtained epicardial adipose tissue and thoracic subcutaneous fat samples from 41 patients (94). They showed that the differentially expressed genes were related to ECM remodeling, inflammation, infection, and thrombosis. However, AF was present only in a subset of the study population (94). Furthermore, Abe et al. collected left atrial appendage samples from 59 patients with AF during surgery (95). They showed that fibrotic remodeling of epicardial adipose tissue was associated with left atrial myocardial fibrosis in these patients (95). In another group, Haemers et al. collected atrial samples from 92 patients with AF, analyzed the fibrosis of subepicardial fatty infiltrates, and showed that fibrosis of the fatty infiltrates was predominant in the patients with permanent AF (96) Moreover, cytotoxic lymphocyte and adipocyte cell death may also be involved in this process (96).
Adipose tissue fibrosis is also involved in the pathogenesis of osteoarthritis. Eymard et al. obtained intra-articular and subcutaneous adipose tissues from patients with osteoarthritis during knee or hip replacement and showed that fibrosis, vascularization, and immune cell infiltration were higher in the intra-articular adipose tissue than in the subcutaneous adipose tissue (97). Moreover, the levels of cytokines, such as IL-6, IL-8, and prostaglandin E2 also increased (97). Another study focused on a mouse model of early osteoarthritis with 20 weeks of feeding a high-fat diet (98). The high-fat diet did not alter inflammation and macrophage infiltration but increased the infrapatellar fat pad fibrosis, suggesting that the intra-articular adipocyte is a distinct cell type (98). However, the underlying mechanism of how a high-fat diet increased infrapatellar fat pad fibrosis is under investigation.
To sum up, fat fibrosis correlates with the pathogenesis, disease activity, and prognosis of different kinds of diseases. Though it may be different from that in CD, this information provides useful tools, techniques, and direction to decipher the role of fat fibrosis in CD.
Since fat fibrosis is involved in various diseases, it is important to elucidate the occurrence and pathogenesis of fibrosis in the creeping fat of CD. In 2003, Geboes et al. mentioned that “fibrous strands are present in the mesenteric fat, irradiating from the intestine and surrounding thickened, hypertrophied fat lobules” (16). However, this area has received little attention to date since it is challenging to establish an ideal animal model to recapitulate creeping fat fibrosis in humans.
Our team used human CD paired samples to investigate the transcriptional signature of these “fibrous strands” (12). Compared with material from low fibrous band samples, the high fibrous band samples were enriched for mRNAs encoded by 661 genes (p < 0.05 and fold change ≥2), including the genes with known roles in fibrosis, such as COL1A1, FAP, COL6A3, COL1A2, COL5A1, and MMP2. We also created a novel mouse model using repeated colonic biopsies for functional studies (12). In this newly established model, the mucosa was injured, and fat accumulation was detected around the intestine in C57BL/6J mice, mimicking the gross features of creeping fat in CD (12). Histological analysis indicated that fibrosis extended into the mesenteric adipose tissue. Finally, we generated a 24-gene set list (including COL1A1, COL5A1, LUM, MMP2, and FAP) using both the human CD dataset and the mouse model and linked the list to inflammatory fibroblasts and treatment response (12). Although our study has expanded the knowledge on fibrosis in creeping fat, much remains to be investigated.
Ha et al. reported an altered Schaedler flora in mice with Clostridium innocuum and found mesenteric adipose tissue expansion in both dextran sulfate sodium-treated and untreated groups (99). They also successfully isolated bacteria in mesenteric adipose tissue, indicating that C. innocuum translocated to mesenteric adipose tissue and promoted adipose tissue expansion (99). Single-cell RNA sequencing showed that both profibrotic and pro-adipogenic signals were present in creeping fat (99). Therefore, bacterial translocation may play a role in the formation of creeping fat (99). Moreover, another study showed that mesenteric microbiota from CD patients promotes intestinal inflammation in mice (100).
In another study by our team, we used novel intestinal tissue and cell interaction systems to illustrate that muscle cells in CD patients could produce an ECM scaffold that triggered preadipocyte migration out of the mesenteric adipose tissue (6). This finding highlights that cell–cell interaction and activated intestinal muscle cells are important players in creeping fat formation (Figure 3) (6).
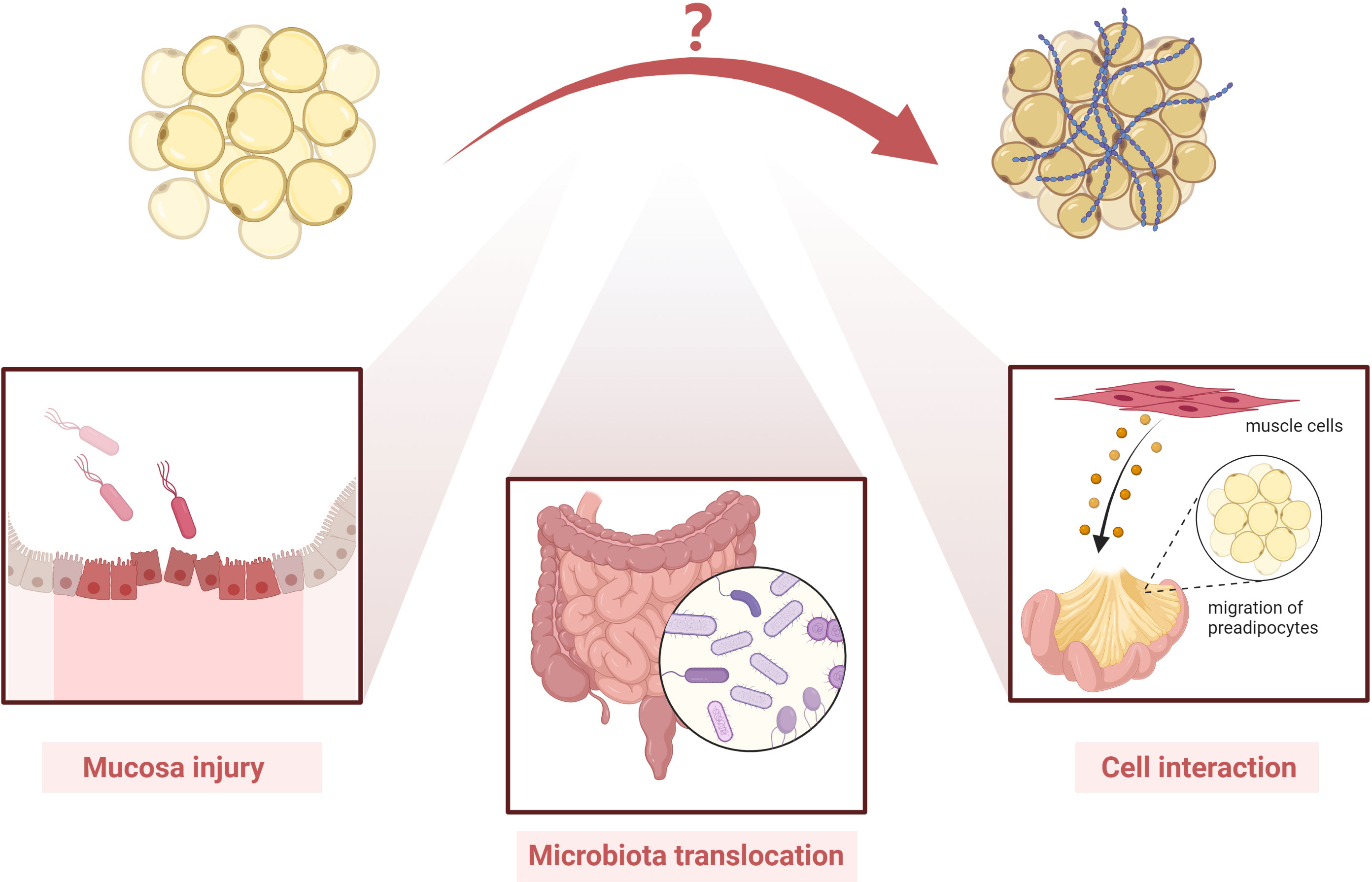
Figure 3 Possible players involved in the pathogenesis of creeping fat fibrosis. Although the pathogenesis of creeping fat fibrosis remains unclear, according to the current literature, mucosal injury, microbiota translocation, cell interaction, and activated intestinal muscle cells are important players in creeping fat formation. However, other mechanisms need to be investigated in future studies.
Recently, Zuo et al. also observed aberrant ECM remodeling in the mesenteric adipose tissue of CD. This area served as a reservoir for inflammatory cells and factors. Moreover, TLR4-mediated macrophages were shown to play a role in mesenteric ECM remodeling and thus affecting the adipocyte function. They further validated the function of macrophages and TLR-4 using in vivo and in vitro experiments (61).
All in all, various cells, including fibroblasts, smooth muscle cells, preadipocyte, and macrophages, are involved in the fat fibrosis of CD. The mechanisms need further study.
In summary, adipose tissue fibrosis in creeping fat is a complex phenomenon involving various cytokines and cellular interactions. The findings from other diseases will help us functionally investigate the latent mechanisms of creeping fat fibrosis. However, there is still a lot to unravel to completely understand this process. As a characteristic manifestation in CD, creeping fat occurs in tandem with abnormal mural and mucosal changes. The functional changes in fibrotic mesenteric fat and how these functional changes affect CD disease activity in the adjacent intestinal segments still need to be addressed. It is of great importance in future studies to investigate the specific genes, cells, and putative mechanism that play a role in the fibrotic process of creeping fat. We believe that understanding the mechanism behind adipose tissue fibrosis in creeping fat will help develop novel targets for the diagnosis and treatment of CD.
RM and ZC conceived the idea. SX and JT performed the literature search and drafted the manuscript. SX and YW drafted the figures. All authors contributed to the revision of the manuscript. RM supervised the study. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (NSFC grant Nos. 81970483, 82170537 and 82222010 to RM).
All figures are created with BioRender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
AF, atrial fibrillation; CD, Crohn’s disease; ECM, extracellular matrix; HIF1α, hypoxia-inducible factor 1-alpha; IL, interleukin; ILC1s, group 1 innate lymphoid cells; MMP, matrix metalloproteinase; PDGF, platelet-derived growth factor; PPARγ, peroxisome proliferator-activated receptor-gamma; TGF-β1, transforming growth factor-beta 1; TIMP, tissue inhibitors of MMP.
1. Martin JC, Chang C, Boschetti G, Ungaro R, Giri M, Grout JA, et al. Single-cell analysis of crohn's disease lesions identifies a pathogenic cellular module associated with resistance to anti-tnf therapy. Cell (2019) 178(6):1493–508.e20. doi: 10.1016/j.cell.2019.08.008
2. Torres J, Mehandru S, Colombel JF, Peyrin-Biroulet L. Crohn's disease. Lancet (London England) (2017) 389(10080):1741–55. doi: 10.1016/s0140-6736(16)31711-1
3. Peyrin-Biroulet L, Chamaillard M, Gonzalez F, Beclin E, Decourcelle C, Antunes L, et al. Mesenteric fat in crohn's disease: A pathogenetic hallmark or an innocent bystander? Gut (2007) 56(4):577–83. doi: 10.1136/gut.2005.082925
4. Sheehan AL, Warren BF, Gear MW, Shepherd NA. Fat-wrapping in crohn's disease: Pathological basis and relevance to surgical practice. Br J Surg (1992) 79(9):955–8. doi: 10.1002/bjs.1800790934
5. Suau R, Pardina E, Domènech E, Lorén V, Manyé J. The complex relationship between microbiota, immune response and creeping fat in crohn's disease. J Crohn's Colitis (2022) 16(3):472–89. doi: 10.1093/ecco-jcc/jjab159
6. Mao R, Doyon G, Gordon IO, Li J, Lin S, Wang J, et al. Activated intestinal muscle cells promote preadipocyte migration: A novel mechanism for creeping fat formation in crohn's disease. Gut (2022) 71(1):55–67. doi: 10.1136/gutjnl-2020-323719
7. Paeschke A, Erben U, Kredel LI, Kühl AA, Siegmund B. Role of visceral fat in colonic inflammation: From crohn's disease to diverticulitis. Curr Opin Gastroenterol (2017) 33(1):53–8. doi: 10.1097/mog.0000000000000324
8. Rivera ED, Coffey JC, Walsh D, Ehrenpreis ED. The mesentery, systemic inflammation, and crohn's disease. Inflamm Bowel Dis (2019) 25(2):226–34. doi: 10.1093/ibd/izy201
9. Zulian A, Cancello R, Micheletto G, Gentilini D, Gilardini L, Danelli P, et al. Visceral adipocytes: Old actors in obesity and new protagonists in crohn's disease? Gut (2012) 61(1):86–94. doi: 10.1136/gutjnl-2011-300391
10. Kredel LI, Batra A, Stroh T, Kühl AA, Zeitz M, Erben U, et al. Adipokines from local fat cells shape the macrophage compartment of the creeping fat in crohn's disease. Gut (2013) 62(6):852–62. doi: 10.1136/gutjnl-2011-301424
11. Weidinger C, Ziegler JF, Letizia M, Schmidt F, Siegmund B. Adipokines and their role in intestinal inflammation. Front Immunol (2018) 9:1974. doi: 10.3389/fimmu.2018.01974
12. Mao R, Kurada S, Gordon IO, Baker ME, Gandhi N, McDonald C, et al. The mesenteric fat and intestinal muscle interface: Creeping fat influencing stricture formation in crohn's disease. Inflamm Bowel Dis (2019) 25(3):421–6. doi: 10.1093/ibd/izy331
13. Jo J, Gavrilova O, Pack S, Jou W, Mullen S, Sumner AE, et al. Hypertrophy and/or hyperplasia: Dynamics of adipose tissue growth. PloS Comput Biol (2009) 5(3):e1000324. doi: 10.1371/journal.pcbi.1000324
14. Datta R, Podolsky MJ, Atabai K. Fat fibrosis: Friend or foe? JCI Insight (2018) 3(19). doi: 10.1172/jci.insight.122289
15. Sun K, Tordjman J, Clément K, Scherer PE. Fibrosis and adipose tissue dysfunction. Cell Metab (2013) 18(4):470–7. doi: 10.1016/j.cmet.2013.06.016
16. Geboes K, Fiocci C, et al. Histopathology of crohn’s disease and ulcerative colitis. In: Inflammatory bowel disease, (London, UK: Churchill Livingstone) (2003). p. 255–76.
17. Xiong S, Whitehurst CE, Li L, Heo GS, Lai CW, Jain U, et al. Reverse translation approach generates a signature of penetrating fibrosis in crohn's disease that is associated with anti-tnf response. Gut (2021) 71(7):1289–301. doi: 10.1136/gutjnl-2020-323405
18. Mariman EC, Wang P. Adipocyte extracellular matrix composition, dynamics and role in obesity. Cell Mol Life Sci (2010) 67(8):1277–92. doi: 10.1007/s00018-010-0263-4
19. Divoux A, Tordjman J, Lacasa D, Veyrie N, Hugol D, Aissat A, et al. Fibrosis in human adipose tissue: Composition, distribution, and link with lipid metabolism and fat mass loss. Diabetes (2010) 59(11):2817–25. doi: 10.2337/db10-0585
20. Gelse K, Pöschl E, Aigner T. Collagens–structure, function, and biosynthesis. Adv Drug Delivery Rev (2003) 55(12):1531–46. doi: 10.1016/j.addr.2003.08.002
21. Liu X, Xu Q, Liu W, Yao G, Zhao Y, Xu F, et al. Enhanced migration of murine fibroblast-like 3t3-L1 preadipocytes on type I collagen-coated dish is reversed by silibinin treatment. Mol Cell Biochem (2018) 441(1-2):35–62. doi: 10.1007/s11010-017-3173-z
22. Kühn K, Wiedemann H, Timpl R, Risteli J, Dieringer H, Voss T, et al. Macromolecular structure of basement membrane collagens. FEBS Lett (1981) 125(1):123–8. doi: 10.1016/0014-5793(81)81012-5
23. Reggio S, Rouault C, Poitou C, Bichet JC, Prifti E, Bouillot JL, et al. Increased basement membrane components in adipose tissue during obesity: Links with tgfβ and metabolic phenotypes. J Clin Endocrinol Metab (2016) 101(6):2578–87. doi: 10.1210/jc.2015-4304
24. Khan T, Muise ES, Iyengar P, Wang ZV, Chandalia M, Abate N, et al. Metabolic dysregulation and adipose tissue fibrosis: Role of collagen vi. Mol Cell Biol (2009) 29(6):1575–91. doi: 10.1128/mcb.01300-08
25. Chu ML, Conway D, Pan TC, Baldwin C, Mann K, Deutzmann R, et al. Amino acid sequence of the triple-helical domain of human collagen type vi. J Biol Chem (1988) 263(35):18601–6. doi: 10.1016/S0021-9258(18)37327-7.
26. Sun K, Park J, Gupta OT, Holland WL, Auerbach P, Zhang N, et al. Endotrophin triggers adipose tissue fibrosis and metabolic dysfunction. Nat Commun (2014) 5:3485. doi: 10.1038/ncomms4485
27. Park J, Scherer PE. Adipocyte-derived endotrophin promotes malignant tumor progression. J Clin Invest (2012) 122(11):4243–56. doi: 10.1172/jci63930
28. Park J, Morley TS, Scherer PE. Inhibition of endotrophin, a cleavage product of collagen vi, confers cisplatin sensitivity to tumours. EMBO Mol Med (2013) 5(6):935–48. doi: 10.1002/emmm.201202006
29. Oh J, Kim CS, Kim M, Jo W, Sung YH, Park J. Type vi collagen and its cleavage product, endotrophin, cooperatively regulate the adipogenic and lipolytic capacity of adipocytes. Metabolism (2021) 114:154430. doi: 10.1016/j.metabol.2020.154430
30. Christiaens V, Scroyen I, Lijnen HR. Role of proteolysis in development of murine adipose tissue. Thromb Haemost (2008) 99(2):290–4. doi: 10.1160/th07-10-0589
31. Feola A, Ricci S, Kouidhi S, Rizzo A, Penon A, Formisano P, et al. Multifaceted breast cancer: The molecular connection with obesity. J Cell Physiol (2017) 232(1):69–77. doi: 10.1002/jcp.25475
32. Maquoi E, Munaut C, Colige A, Collen D, Lijnen HR. Modulation of adipose tissue expression of murine matrix metalloproteinases and their tissue inhibitors with obesity. Diabetes (2002) 51(4):1093–101. doi: 10.2337/diabetes.51.4.1093
33. Bouloumié A, Sengenès C, Portolan G, Galitzky J, Lafontan M. Adipocyte produces matrix metalloproteinases 2 and 9: Involvement in adipose differentiation. Diabetes (2001) 50(9):2080–6. doi: 10.2337/diabetes.50.9.2080
34. Catalán V, Gómez-Ambrosi J, Rodríguez A, Ramírez B, Silva C, Rotellar F, et al. Increased adipose tissue expression of lipocalin-2 in obesity is related to inflammation and matrix metalloproteinase-2 and metalloproteinase-9 activities in humans. J Mol Med (Berl) (2009) 87(8):803–13. doi: 10.1007/s00109-009-0486-8
35. Derosa G, Ferrari I, D'Angelo A, Tinelli C, Salvadeo SA, Ciccarelli L, et al. Matrix metalloproteinase-2 and -9 levels in obese patients. Endothelium (2008) 15(4):219–24. doi: 10.1080/10623320802228815
36. Zeng ZS, Cohen AM, Guillem JG. Loss of basement membrane type iv collagen is associated with increased expression of metalloproteinases 2 and 9 (Mmp-2 and mmp-9) during human colorectal tumorigenesis. Carcinogenesis (1999) 20(5):749–55. doi: 10.1093/carcin/20.5.749
37. Croissandeau G, Chrétien M, Mbikay M. Involvement of matrix metalloproteinases in the adipose conversion of 3t3-L1 preadipocytes. Biochem J (2002) 364(Pt 3):739–46. doi: 10.1042/bj20011158
38. Unal R, Yao-Borengasser A, Varma V, Rasouli N, Labbate C, Kern PA, et al. Matrix metalloproteinase-9 is increased in obese subjects and decreases in response to pioglitazone. J Clin Endocrinol Metab (2010) 95(6):2993–3001. doi: 10.1210/jc.2009-2623
39. Sillat T, Saat R, Pöllänen R, Hukkanen M, Takagi M, Konttinen YT. Basement membrane collagen type iv expression by human mesenchymal stem cells during adipogenic differentiation. J Cell Mol Med (2012) 16(7):1485–95. doi: 10.1111/j.1582-4934.2011.01442.x
40. Deryugina EI, Ratnikov B, Monosov E, Postnova TI, DiScipio R, Smith JW, et al. Mt1-mmp initiates activation of pro-Mmp-2 and integrin Alphavbeta3 promotes maturation of mmp-2 in breast carcinoma cells. Exp Cell Res (2001) 263(2):209–23. doi: 10.1006/excr.2000.5118
41. Holmbeck K, Bianco P, Caterina J, Yamada S, Kromer M, Kuznetsov SA, et al. Mt1-Mmp-Deficient mice develop dwarfism, osteopenia, arthritis, and connective tissue disease due to inadequate collagen turnover. Cell (1999) 99(1):81–92. doi: 10.1016/s0092-8674(00)80064-1
42. Rieder F, Fiocchi C, Rogler G. Mechanisms, management, and treatment of fibrosis in patients with inflammatory bowel diseases. Gastroenterology (2017) 152(2):340–50.e6. doi: 10.1053/j.gastro.2016.09.047
43. Marcelin G, Ferreira A, Liu Y, Atlan M, Aron-Wisnewsky J, Pelloux V, et al. A pdgfrα-mediated switch toward Cd9(High) adipocyte progenitors controls obesity-induced adipose tissue fibrosis. Cell Metab (2017) 25(3):673–85. doi: 10.1016/j.cmet.2017.01.010
44. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest (2003) 112(12):1796–808. doi: 10.1172/jci19246
45. Thomas D, Apovian C. Macrophage functions in lean and obese adipose tissue. Metabolism (2017) 72:120–43. doi: 10.1016/j.metabol.2017.04.005
46. Zatterale F, Longo M, Naderi J, Raciti GA, Desiderio A, Miele C, et al. Chronic adipose tissue inflammation linking obesity to insulin resistance and type 2 diabetes. Front Physiol (2019) 10:1607. doi: 10.3389/fphys.2019.01607
47. Hill AA, Reid Bolus W, Hasty AH. A decade of progress in adipose tissue macrophage biology. Immunol Rev (2014) 262(1):134–52. doi: 10.1111/imr.12216
48. Cinti S, Mitchell G, Barbatelli G, Murano I, Ceresi E, Faloia E, et al. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J Lipid Res (2005) 46(11):2347–55. doi: 10.1194/jlr.M500294-JLR200
49. Spencer M, Yao-Borengasser A, Unal R, Rasouli N, Gurley CM, Zhu B, et al. Adipose tissue macrophages in insulin-resistant subjects are associated with collagen vi and fibrosis and demonstrate alternative activation. Am J Physiol Endocrinol Metab (2010) 299(6):E1016–27. doi: 10.1152/ajpendo.00329.2010
50. Tanaka M, Ikeda K, Suganami T, Komiya C, Ochi K, Shirakawa I, et al. Macrophage-inducible c-type lectin underlies obesity-induced adipose tissue fibrosis. Nat Commun (2014) 5:4982. doi: 10.1038/ncomms5982
51. Marcelin G, Silveira ALM, Martins LB, Ferreira AV, Clément K. Deciphering the cellular interplays underlying obesity-induced adipose tissue fibrosis. J Clin Invest (2019) 129(10):4032–40. doi: 10.1172/jci129192
52. Żelechowska P, Agier J, Kozłowska E, Brzezińska-Błaszczyk E. Mast cells participate in chronic low-grade inflammation within adipose tissue. Obes Rev (2018) 19(5):686–97. doi: 10.1111/obr.12670
53. Bremer AA, Devaraj S, Afify A, Jialal I. Adipose tissue dysregulation in patients with metabolic syndrome. J Clin Endocrinol Metab (2011) 96(11):E1782–8. doi: 10.1210/jc.2011-1577
54. Altintas MM, Azad A, Nayer B, Contreras G, Zaias J, Faul C, et al. Mast cells, macrophages, and crown-like structures distinguish subcutaneous from visceral fat in mice. J Lipid Res (2011) 52(3):480–8. doi: 10.1194/jlr.M011338
55. Divoux A, Moutel S, Poitou C, Lacasa D, Veyrie N, Aissat A, et al. Mast cells in human adipose tissue: Link with morbid obesity, inflammatory status, and diabetes. J Clin Endocrinol Metab (2012) 97(9):E1677–85. doi: 10.1210/jc.2012-1532
56. Liu J, Divoux A, Sun J, Zhang J, Clément K, Glickman JN, et al. Genetic deficiency and pharmacological stabilization of mast cells reduce diet-induced obesity and diabetes in mice. Nat Med (2009) 15(8):940–5. doi: 10.1038/nm.1994
57. Hirai S, Ohyane C, Kim YI, Lin S, Goto T, Takahashi N, et al. Involvement of mast cells in adipose tissue fibrosis. Am J Physiol Endocrinol Metab (2014) 306(3):E247–55. doi: 10.1152/ajpendo.00056.2013
58. Wang H, Shen L, Sun X, Liu F, Feng W, Jiang C, et al. Adipose group 1 innate lymphoid cells promote adipose tissue fibrosis and diabetes in obesity. Nat Commun (2019) 10(1):3254. doi: 10.1038/s41467-019-11270-1
59. Zuo L, Li Y, Zhu W, Shen B, Gong J, Guo Z, et al. Mesenteric adipocyte dysfunction in crohn's disease is associated with hypoxia. Inflamm Bowel Dis (2016) 22(1):114–26. doi: 10.1097/mib.0000000000000571
60. Li J, Zuo L, Geng Z, Li Q, Cheng Y, Yang Z, et al. Pygopus2 ameliorates mesenteric adipocyte poor differentiation to alleviate crohn's disease -like colitis via the Axin2/Gsk3β pathway. Cell Prolif (2022):e13292. doi: 10.1111/cpr.13292
61. Zuo L, Li J, Zhang X, Geng Z, Song X, Wang Y, et al. Aberrant mesenteric adipose extracellular matrix remodeling is involved in adipocyte dysfunction in crohn's disease: The role of tlr-4-Mediated macrophages. J Crohn's Colitis (2022):jjac087. doi: 10.1093/ecco-jcc/jjac087
62. Lee YS, Kim JW, Osborne O, Oh DY, Sasik R, Schenk S, et al. Increased adipocyte O2 consumption triggers hif-1α, causing inflammation and insulin resistance in obesity. Cell (2014) 157(6):1339–52. doi: 10.1016/j.cell.2014.05.012
63. Halberg N, Khan T, Trujillo ME, Wernstedt-Asterholm I, Attie AD, Sherwani S, et al. Hypoxia-inducible factor 1alpha induces fibrosis and insulin resistance in white adipose tissue. Mol Cell Biol (2009) 29(16):4467–83. doi: 10.1128/mcb.00192-09
64. Warbrick I, Rabkin SW. Hypoxia-inducible factor 1-alpha (Hif-1α) as a factor mediating the relationship between obesity and heart failure with preserved ejection fraction. Obes Rev (2019) 20(5):701–12. doi: 10.1111/obr.12828
65. Sun K, Halberg N, Khan M, Magalang UJ, Scherer PE. Selective inhibition of hypoxia-inducible factor 1α ameliorates adipose tissue dysfunction. Mol Cell Biol (2013) 33(5):904–17. doi: 10.1128/mcb.00951-12
66. Bourlier V, Sengenès C, Zakaroff-Girard A, Decaunes P, Wdziekonski B, Galitzky J, et al. Tgfbeta family members are key mediators in the induction of myofibroblast phenotype of human adipose tissue progenitor cells by macrophages. PloS One (2012) 7(2):e31274. doi: 10.1371/journal.pone.0031274
67. Keophiphath M, Achard V, Henegar C, Rouault C, Clément K, Lacasa D. Macrophage-secreted factors promote a profibrotic phenotype in human preadipocytes. Mol Endocrinol (2009) 23(1):11–24. doi: 10.1210/me.2008-0183
68. Gagnon AM, Chabot J, Pardasani D, Sorisky A. Extracellular matrix induced by tgfbeta impairs insulin signal transduction in 3t3-L1 preadipose cells. J Cell Physiol (1998) 175(3):370–8. doi: 10.1002/(sici)1097-4652(199806)175:3<370::Aid-jcp15>3.0.Co;2-9
69. Kaufman JM, Anslow RD. Treatment of refractory angina pectoris with nitroglycerin and graded exercise. Jama (1966) 196(2):151–5. doi: 10.1001/jama.1966.03100150097023
70. Bourlier V, Zakaroff-Girard A, Miranville A, De Barros S, Maumus M, Sengenes C, et al. Remodeling phenotype of human subcutaneous adipose tissue macrophages. Circulation (2008) 117(6):806–15. doi: 10.1161/circulationaha.107.724096
71. Luo T, Nocon A, Fry J, Sherban A, Rui X, Jiang B, et al. Ampk activation by metformin suppresses abnormal extracellular matrix remodeling in adipose tissue and ameliorates insulin resistance in obesity. Diabetes (2016) 65(8):2295–310. doi: 10.2337/db15-1122
72. He C, Medley SC, Kim J, Sun C, Kwon HR, Sakashita H, et al. Stat1 modulates tissue wasting or overgrowth downstream from pdgfrβ. Genes Dev (2017) 31(16):1666–78. doi: 10.1101/gad.300384.117
73. Sun C, Berry WL, Olson LE. Pdgfrα controls the balance of stromal and adipogenic cells during adipose tissue organogenesis. Development (2017) 144(1):83–94. doi: 10.1242/dev.135962
74. Iwayama T, Steele C, Yao L, Dozmorov MG, Karamichos D, Wren JD, et al. Pdgfrα signaling drives adipose tissue fibrosis by targeting progenitor cell plasticity. Genes Dev (2015) 29(11):1106–19. doi: 10.1101/gad.260554.115
75. Sun C, Sakashita H, Kim J, Tang Z, Upchurch GM, Yao L, et al. Mosaic mutant analysis identifies Pdgfrα/Pdgfrβ as negative regulators of adipogenesis. Cell Stem Cell (2020) 26(5):707–21.e5. doi: 10.1016/j.stem.2020.03.004
76. Marangoni RG, Korman BD, Wei J, Wood TA, Graham LV, Whitfield ML, et al. Myofibroblasts in murine cutaneous fibrosis originate from adiponectin-positive intradermal progenitors. Arthritis Rheumatol (2015) 67(4):1062–73. doi: 10.1002/art.38990
77. Tontonoz P, Spiegelman BM. Fat and beyond: The diverse biology of ppargamma. Annu Rev Biochem (2008) 77:289–312. doi: 10.1146/annurev.biochem.77.061307.091829
78. Shao M, Hepler C, Zhang Q, Shan B, Vishvanath L, Henry GH, et al. Pathologic Hif1α signaling drives adipose progenitor dysfunction in obesity. Cell Stem Cell (2021) 28(4):685–701.e7. doi: 10.1016/j.stem.2020.12.008
79. Yoshino J, Patterson BW, Klein S. Adipose tissue ctgf expression is associated with adiposity and insulin resistance in humans. Obes (Silver Spring) (2019) 27(6):957–62. doi: 10.1002/oby.22463
80. Kumar A, Ruan M, Clifton K, Syed F, Khosla S, Oursler MJ. Tgf-B mediates suppression of adipogenesis by estradiol through connective tissue growth factor induction. Endocrinology (2012) 153(1):254–63. doi: 10.1210/en.2011-1169
81. List EO, Berryman DE, Buchman M, Jensen EA, Funk K, Duran-Ortiz S, et al. Gh knockout mice have increased subcutaneous adipose tissue with decreased fibrosis and enhanced insulin sensitivity. Endocrinology (2019) 160(7):1743–56. doi: 10.1210/en.2019-00167
82. Lin JZ, Rabhi N, Farmer SR. Myocardin-related transcription factor a promotes recruitment of Itga5+ profibrotic progenitors during obesity-induced adipose tissue fibrosis. Cell Rep (2018) 23(7):1977–87. doi: 10.1016/j.celrep.2018.04.057
83. Henegar C, Tordjman J, Achard V, Lacasa D, Cremer I, Guerre-Millo M, et al. Adipose tissue transcriptomic signature highlights the pathological relevance of extracellular matrix in human obesity. Genome Biol (2008) 9(1):R14. doi: 10.1186/gb-2008-9-1-r14
84. Vishvanath L, Gupta RK. Contribution of adipogenesis to healthy adipose tissue expansion in obesity. J Clin Invest (2019) 129(10):4022–31. doi: 10.1172/jci129191
85. Muir LA, Neeley CK, Meyer KA, Baker NA, Brosius AM, Washabaugh AR, et al. Adipose tissue fibrosis, hypertrophy, and hyperplasia: Correlations with diabetes in human obesity. Obes (Silver Spring) (2016) 24(3):597–605. doi: 10.1002/oby.21377
86. Lackey DE, Burk DH, Ali MR, Mostaedi R, Smith WH, Park J, et al. Contributions of adipose tissue architectural and tensile properties toward defining healthy and unhealthy obesity. Am J Physiol Endocrinol Metab (2014) 306(3):E233–46. doi: 10.1152/ajpendo.00476.2013
87. Lawler HM, Underkofler CM, Kern PA, Erickson C, Bredbeck B, Rasouli N. Adipose tissue hypoxia, inflammation, and fibrosis in obese insulin-sensitive and obese insulin-resistant subjects. J Clin Endocrinol Metab (2016) 101(4):1422–8. doi: 10.1210/jc.2015-4125
88. Spencer M, Unal R, Zhu B, Rasouli N, McGehee RE Jr., Peterson CA, et al. Adipose tissue extracellular matrix and vascular abnormalities in obesity and insulin resistance. J Clin Endocrinol Metab (2011) 96(12):E1990–8. doi: 10.1210/jc.2011-1567
89. Trujillo KA, Heaphy CM, Mai M, Vargas KM, Jones AC, Vo P, et al. Markers of fibrosis and epithelial to mesenchymal transition demonstrate field cancerization in histologically normal tissue adjacent to breast tumors. Int J Cancer (2011) 129(6):1310–21. doi: 10.1002/ijc.25788
90. Seo BR, Bhardwaj P, Choi S, Gonzalez J, Andresen Eguiluz RC, Wang K, et al. Obesity-dependent changes in interstitial ecm mechanics promote breast tumorigenesis. Sci Transl Med (2015) 7(301):301ra130. doi: 10.1126/scitranslmed.3010467
91. Iyengar P, Espina V, Williams TW, Lin Y, Berry D, Jelicks LA, et al. Adipocyte-derived collagen vi affects early mammary tumor progression in vivo, demonstrating a critical interaction in the Tumor/Stroma microenvironment. J Clin Invest (2005) 115(5):1163–76. doi: 10.1172/jci23424
92. Incio J, Liu H, Suboj P, Chin SM, Chen IX, Pinter M, et al. Obesity-induced inflammation and desmoplasia promote pancreatic cancer progression and resistance to chemotherapy. Cancer Discovery (2016) 6(8):852–69. doi: 10.1158/2159-8290.Cd-15-1177
93. Alves MJ, Figuerêdo RG, Azevedo FF, Cavallaro DA, Neto NI, Lima JD, et al. Adipose tissue fibrosis in human cancer cachexia: The role of tgfβ pathway. BMC Cancer (2017) 17(1):190. doi: 10.1186/s12885-017-3178-8
94. Gaborit B, Venteclef N, Ancel P, Pelloux V, Gariboldi V, Leprince P, et al. Human epicardial adipose tissue has a specific transcriptomic signature depending on its anatomical peri-atrial, peri-ventricular, or peri-coronary location. Cardiovasc Res (2015) 108(1):62–73. doi: 10.1093/cvr/cvv208
95. Abe I, Teshima Y, Kondo H, Kaku H, Kira S, Ikebe Y, et al. Association of fibrotic remodeling and Cytokines/Chemokines content in epicardial adipose tissue with atrial myocardial fibrosis in patients with atrial fibrillation. Heart Rhythm (2018) 15(11):1717–27. doi: 10.1016/j.hrthm.2018.06.025
96. Haemers P, Hamdi H, Guedj K, Suffee N, Farahmand P, Popovic N, et al. Atrial fibrillation is associated with the fibrotic remodelling of adipose tissue in the subepicardium of human and sheep atria. Eur Heart J (2017) 38(1):53–61. doi: 10.1093/eurheartj/ehv625
97. Ioan-Facsinay A, Kloppenburg M. Osteoarthritis: Inflammation and fibrosis in adipose tissue of osteoarthritic joints. Nat Rev Rheumatol (2017) 13(6):325–6. doi: 10.1038/nrrheum.2017.53
98. Barboza E, Hudson J, Chang WP, Kovats S, Towner RA, Silasi-Mansat R, et al. Profibrotic infrapatellar fat pad remodeling without M1 macrophage polarization precedes knee osteoarthritis in mice with diet-induced obesity. Arthritis Rheumatol (2017) 69(6):1221–32. doi: 10.1002/art.40056
99. Ha CWY, Martin A, Sepich-Poore GD, Shi B, Wang Y, Gouin K, et al. Translocation of viable gut microbiota to mesenteric adipose drives formation of creeping fat in humans. Cell (2020) 183(3):666–83.e17. doi: 10.1016/j.cell.2020.09.009
Keywords: creeping fat, adipose tissue, fibrosis, extracellular matrix, Crohn’s disease
Citation: Xiong S, Tan J, Wang Y, He J, Hu F, Wu X, Liu Z, Lin S, Li X, Chen Z and Mao R (2022) Fibrosis in fat: From other diseases to Crohn’s disease. Front. Immunol. 13:935275. doi: 10.3389/fimmu.2022.935275
Received: 03 May 2022; Accepted: 03 August 2022;
Published: 25 August 2022.
Edited by:
Jun Shen, Shanghai Jiao Tong University, ChinaReviewed by:
Hon Wai Koon, University of California, Los Angeles, United StatesCopyright © 2022 Xiong, Tan, Wang, He, Hu, Wu, Liu, Lin, Li, Chen and Mao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ren Mao, bWFvcjVAbWFpbC5zeXN1LmVkdS5jbg==; Zhihui Chen, Y2h6aGh1aUBtYWlsLnN5c3UuZWR1LmNu
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.